JP2017014502A - ポリカーボネート樹脂組成物、その製造方法、成形体 - Google Patents
ポリカーボネート樹脂組成物、その製造方法、成形体 Download PDFInfo
- Publication number
- JP2017014502A JP2017014502A JP2016128509A JP2016128509A JP2017014502A JP 2017014502 A JP2017014502 A JP 2017014502A JP 2016128509 A JP2016128509 A JP 2016128509A JP 2016128509 A JP2016128509 A JP 2016128509A JP 2017014502 A JP2017014502 A JP 2017014502A
- Authority
- JP
- Japan
- Prior art keywords
- polycarbonate resin
- compound
- resin composition
- metal
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/20—General preparatory processes
- C08G64/30—General preparatory processes using carbonates
- C08G64/302—General preparatory processes using carbonates and cyclic ethers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/02—Aliphatic polycarbonates
- C08G64/0208—Aliphatic polycarbonates saturated
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/16—Aliphatic-aromatic or araliphatic polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G64/00—Macromolecular compounds obtained by reactions forming a carbonic ester link in the main chain of the macromolecule
- C08G64/40—Post-polymerisation treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/08—Metals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/10—Metal compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/06—Ethers; Acetals; Ketals; Ortho-esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L69/00—Compositions of polycarbonates; Compositions of derivatives of polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/08—Metals
- C08K2003/0818—Alkali metal
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/08—Stabilised against heat, light or radiation or oxydation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/10—Transparent films; Clear coatings; Transparent materials
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/02—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group
- C08L2205/025—Polymer mixtures characterised by other features containing two or more polymers of the same C08L -group containing two or more polymers of the same hierarchy C08L, and differing only in parameters such as density, comonomer content, molecular weight, structure
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyesters Or Polycarbonates (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
Description
芳香族ポリカーボネート樹脂(B)と、
長周期型周期表第I族の金属の化合物及び長周期型周期表第II族の金属の化合物からなる群から選択される少なくとも1種の化合物(C)とを含有し、
前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)との合計量100重量部に対する前記化合物(C)の含有量が、該化合物(C)中の金属量で0.8重量ppm以上、かつ1000重量ppm以下であり、
示差走査熱量分析により測定されるガラス転移温度が単一であるポリカーボネート樹脂組成物。
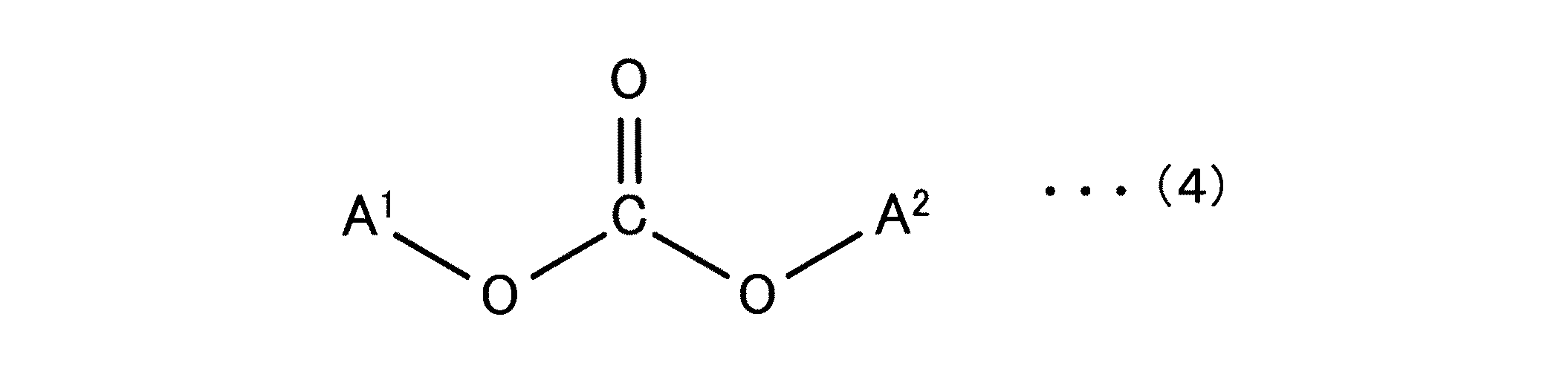
芳香族ポリカーボネート樹脂(B)と、
長周期型周期表第I族の金属の化合物及び長周期型周期表第II族の金属の化合物からなる群から選択される少なくとも1種の化合物(C)と、
クラウンエーテル化合物(D)とを含有し、
前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)との合計量100重量部に対する前記化合物(C)の含有量が、該化合物(C)中の金属量で0.8重量ppm以上1000重量ppm以下であり、
前記化合物(C)の金属換算量に対する前記クラウンエーテル化合物(D)の含有量が0.1倍モル以上10倍モル以下である、ポリカーボネート樹脂組成物。
該添加工程後に、前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)とを溶融反応させる反応工程とを有する、ポリカーボネート樹脂組成物の製造方法。
[20]全ジオールに由来する構成単位100モル%に対する下記式(1)で表される化合物に由来する構成単位の割合が50モル%を超えるポリカーボネート樹脂(A)と、
芳香族ポリカーボネート樹脂(B)と、を含有し、
下記条件(I)及び(II)を満足するポリカーボネート樹脂組成物及び該ポリカーボネート樹脂組成物を含有してなる成形体。
(I)厚さ2mmの成形体の全光線透過率が80%以上。
(II)示差走査熱量分析により測定されるガラス転移温度が単一。
ポリカーボネート樹脂(A)は、下記式(1)で表されるジヒドロキシ化合物に由来する構成単位(これを、適宜「構成単位(a)」という)を含む。ポリカーボネート樹脂(A)は、構成単位(a)のホモポリカーボネート樹脂であってもよいし、構成単位(a)以外の構成単位を共重合したポリカーボネート樹脂であってもよい。耐衝撃性により優れるという観点からは、共重合ポリカーボネート樹脂であることが好ましい。
オキシアルキレングリコール類としては、例えば、ジエチレングリコール、トリエチレングリコール、テトラエチレングリコール、ポリエチレングリコール及びポリプロピレングリコール等を採用することができる。
1族金属化合物としては、重合活性と得られるポリカーボネート樹脂(A)の色調の観点から、リチウム化合物が好ましい。
ポリカーボネート樹脂組成物を成形してなる成形体の透明性、初期Haze(ヘーズ)、耐衝撃性をより向上できるという観点からは、重合触媒は、2族金属化合物であることがより好ましい。成形体の透明性、初期Haze(ヘーズ)、耐衝撃性をさらにより向上できるという観点からは、2族金属化合物としては、マグネシウム化合物、カルシウム化合物又はバリウム化合物が好ましく、重合活性と得られるポリカーボネート樹脂(A)の色調の観点から、マグネシウム化合物及び/又はカルシウム化合物が更に好ましく、カルシウム化合物が最も好ましい。
なお、重合時に触媒として用いた化合物は、特段の精製作業を行わない場合、ポリカーボネート樹脂自体にも製造時に用いた量と同等の量が残留することとなる。
ポリカーボネート樹脂(A)は、前記式(1)で表されるジヒドロキシ化合物等のように原料として用いられるジヒドロキシ化合物と、炭酸ジエステルとを、重合触媒の存在下、エステル交換反応により重縮合させることによって得られる。
なお、ポリカーボネート樹脂組成物中の酸性化合物(E)の含有量は、ICP−MS(誘導結合プラズマ質量分析計)を用いて、酸性化合物(E)に含まれる元素の量として測定できる。
芳香族ポリカーボネート樹脂(B)としては、例えば下記一般式(7)で表される芳香族ジヒドロキシ化合物に由来する構成単位を主構成単位とするポリカーボネート樹脂がある。
4,4’−ビフェノール、2,4’−ビフェノール、3,3’−ジメチル−4,4’−ジヒドロキシ−1,1’−ビフェニル、3,3’−ジメチル−2,4’−ジヒドロキシ−1,1’−ビフェニル、3,3’−ジ−(t−ブチル)−4,4’−ジヒドロキシ−1,1’−ビフェニル、3,3’,5,5’−テトラメチル−4,4’−ジヒドロキシ−1,1’−ビフェニル、3,3’,5,5’−テトラ−(t−ブチル)−4,4’−ジヒドロキシ−1,1’−ビフェニル、2,2’,3,3’,5,5’−ヘキサメチル−4,4’−ジヒドロキシ−1,1’−ビフェニル等のビフェニル化合物。
これらの中でも好ましいジヒドロキシ化合物としては、ビス−(4−ヒドロキシ−3,5−ジメチルフェニル)メタン、ビス−(4−ヒドロキシフェニル)メタン、ビス−(4−ヒドロキシ−3−メチルフェニル)メタン、1,1−ビス−(4−ヒドロキシフェニル)エタン、2,2−ビス−(4−ヒドロキシフェニル)プロパン、2,2−ビス−(4−ヒドロキシ−3−メチルフェニル)プロパン、2,2−ビス−(4−ヒドロキシ−3,5−ジメチルフェニル)プロパン、1,1−ビス−(4−ヒドロキシフェニル)シクロヘキサン、1,1−ビス−(4−ヒドロキシ−3,3,5−トリメチルフェニル)シクロヘキサン、ビス−(4−ヒドロキシフェニル)フェニルメタン、1,1−ビス−(4−ヒドロキシフェニル)−1−フェニルエタン、1,1−ビス−(4−ヒドロキシフェニル)−1−フェニルプロパン、ビス−(4−ヒドロキシフェニル)ジフェニルメタン、2−ヒドロキシフェニル(4−ヒドロキシフェニル)メタン、2,2−(2−ヒドロキシフェニル)(4−ヒドロキシフェニル)プロパン、があげられる。
エステル交換法は、ジヒドロキシ化合物と炭酸ジエステルとを塩基性触媒、さらにはこの塩基性触媒を中和する酸性物質を添加し、溶融エステル交換縮重合を行う製造方法である。ジヒドロキシ化合物としては、前記例示のビフェニル化合物、ビスフェノール化合物があげられる。
ポリカーボネート樹脂組成物に対して配合する化合物(C)は、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)とのエステル交換反応を促進することができる。エステル交換反応は、前記樹脂組成物を作製する際に、例えばポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)との混練時における加熱により起こり、化合物(C)により促進される。その結果、樹脂組成物におけるポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)との相溶性が向上するため、樹脂組成物の透明性を高めることが可能になる。そして、高い透明性を備えつつも、生物起源物質含有率を下げることなく、耐熱性、耐湿熱性、及び耐衝撃性等の特性に優れた樹脂組成物の実現が可能になる。化合物(C)は、長周期型周期表の1族の金属及び2族の金属からなる群から選択される少なくとも一種を含む化合物であれば良く、少なくとも長周期型周期表の1族の金属の化合物を含むことが、ヘーズが低く、耐湿熱性、耐熱性が良好となるという観点から好ましい。特に、後述の通り、前記化合物(C)として、少なくとも長周期型周期表第I族の金属の化合物を含有し、前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)との合計量100重量部に対する長周期型周期表第I族の金属の化合物の含有量が金属量で0.8重量ppm以上1000重量ppm以下であることが、さらに、ヘーズが低く、耐湿熱性、耐熱性も良好となり、好色調、耐湿熱性、透明性もより向上させることができ好ましい。
したがって、本発明のポリカーボネート樹脂組成物は、長周期型周期表第I族の金属の化合物及び長周期型周期表第II族の金属の化合物を含むことが、ヘーズが低く、耐湿熱性、耐熱性も良好となり、好色調、耐湿熱性、透明性もより向上させることができるという観点から好ましい。
なお、ポリカーボネート樹脂組成物中の化合物(C)の含有量は、ICP−MS(誘導結合プラズマ質量分析計)を用いて、金属量として測定できる。
[クラウンエーテル化合物(D)]
なお、クラウンエーテル化合物(D)は、例えば、以下のとおり検出することができる。具体的には、まず、ポリカーボネート樹脂組成物を塩化メチレン等の溶媒に溶解させる。次いで、溶解物をアセトンに加え、ポリマー成分を沈殿させる。その後、アセトン可溶分を採取し、ガスクロマトグラフ質量分析計などによりアセトン可溶分中に含まれるクラウンエーテル化合物(D)を検出することができる。
ポリカーボネート樹脂組成物は、さらに酸性化合物(E)を含有することが好ましい。この酸性化合物(E)は、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)とを配合する際に添加されるものであり、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の製造時に用いられる上述の触媒失活剤を含まない概念である。これらの触媒失活剤は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の製造段階においてその効果自体が失われているためである。なお、酸性化合物(E)としては、上述の触媒失活剤と同様の物質を用いることができる。
なお、ポリカーボネート樹脂組成物中の酸性化合物(E)の含有量は、ICP−MS(誘導結合プラズマ質量分析計)を用いて、酸性化合物(E)に含まれる元素の量として測定できる。
本発明のポリカーボネート樹脂組成物は、これを成形してなる厚さ2mmの成形体の厚さ方向の全光線透過率が80%以上であることが好ましい。また、透明用途への適用性と原着時の鮮映性が良好になるという観点から、前記全光線透過率は、85%以上がより好ましく、88%以上がさらに好ましく、90%以上が特に好ましい。また、厚さ2mmの成形体のヘーズは、1%以下が好ましく、0.5%以下がより好ましく、0.3%以下がさらに好ましい。なお、全光線透過率の測定方法は、後述の実施例において説明する。ヘーズも全光線透過率と同様の方法により測定することができる。
ポリカーボネート樹脂組成物のTgは、パーキン・エルマー社製示差走査熱量計「DSC7」を用い、窒素ガス雰囲気下、25℃から200℃まで加熱速度20℃/分で昇温し、200℃で3分間保持した後、25℃まで冷却速度20℃/分で降温を行い、25℃で3分間保持した後、再度200℃まで加熱速度5℃/分で昇温した際、得られたDSC曲線からJIS‐K7121(1987年)の方法に準拠して求めたTmgの値とする。また、ガラス転移温度の単一性の評価としては、具体的には、DSC曲線のピークが単一(ガラス転移温度を示す変曲点が1つだけ現れる)である場合をガラス転移温度が単一であると評価し、DSC曲線のピークが複数(ガラス転移温度を示す変曲点が複数現れる)である場合をガラス転移温度が単一ではないと評価する。
前記ポリカーボネート樹脂組成物には、種々の添加剤を添加することができる。前記添加剤としては、染顔料、酸化防止剤、UV吸収剤、光安定剤、離型剤、熱安定剤、難燃剤、難燃助剤、無機充填剤、衝撃改良剤、加水分解抑制剤、発泡剤、核剤等があり、ポリカーボネート樹脂に通常用いられる添加剤を使用することができる。
染顔料としては、無機顔料、有機顔料、及び有機染料等の有機染顔料が挙げられる。
無機顔料としては具体的には例えば、カーボンブラック;酸化チタン、亜鉛華、弁柄、酸化クロム、鉄黒、チタンイエロー、亜鉛−鉄系ブラウン、銅−クロム系ブラック、銅−鉄系ブラック等の酸化物系顔料等;が挙げられる。
有機顔料及び有機染料等の有機染顔料としては具体的には例えばフタロシアニン系染顔料;アゾ系、チオインジゴ系、ペリノン系、ペリレン系、キナクリドン系、ジオキサジン系、イソインドリノン系、キノフタロン系等の縮合多環染顔料;アンスラキノン系、ペリノン系、ペリレン系、メチン系、キノリン系、複素環系、メチル系の染顔料等;が挙げられる。
これら染顔料は1種を単独で用いてもよく、2種以上を混合して用いてもよい。
染顔料の量は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の合計100重量部に対して、0.05重量部以上5重量部以下であることが好ましい。より好ましくは0.05重量部以上3重量部以下、さらに好ましくは0.1重量部以上2重量部以下がよい。着色剤の量が0.05重量部未満では鮮映性のある原着成形品が得られづらい。5重量部より多いと、成形品の表面粗さが大きくなり、鮮映性のある原着成形品が得られづらい。
酸化防止剤としては、樹脂に使用される一般的な酸化防止剤が使用できるが、酸化安定性、熱安定性観点から、ホスファイト系酸化防止剤、イオウ系酸化防止剤、およびフェノール系酸化防止剤が好ましい。ここで、酸化防止剤の添加量は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の合計100重量部に対し、5重量部以下が好ましい。この場合には、成形時における金型の汚染をより確実に防止し、表面外観のより優れた成形品を得ることが可能になる。同様の観点から、酸化防止剤の添加量は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の合計100重量部に対し、3重量部以下がより好ましく、2重量部以下が更に好ましい。また、酸化防止剤の添加量は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の合計100重量部に対し、0.001重量部以上が好ましい。この場合には、成形安定性に対する改良効果を十分に得ることができる。同様の観点から、酸化防止剤の添加量は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)の合計100重量部に対し、0.002重量部以上がより好ましく、0.005重量部以上が更に好ましい。
ホスファイト系酸化防止剤としては、トリフェニルホスファイト、トリス(ノニルフェニル)ホスファイト、トリス(2,4−ジ−tert−ブチルフェニル)ホスファイト、トリデシルホスファイト、トリオクチルホスファイト、トリオクタデシルホスファイト、ジデシルモノフェニルホスファイト、ジオクチルモノフェニルホスファイト、ジイソプロピルモノフェニルホスファイト、モノブチルジフェニルホスファイト、モノデシルジフェニルホスファイト、モノオクチルジフェニルホスファイト、ビス(2,6−ジ−tert−ブチル−4−メチルフェニル)ペンタエリスリトールジホスファイト、2,2−メチレンビス(4,6−ジ−tert−ブチルフェニル)オクチルホスファイト、ビス(ノニルフェニル)ペンタエリスリトールジホスファイト、ビス(2,4−ジ−tert−ブチルフェニル)ペンタエリスリトールジホスファイト、ジステアリルペンタエリスリトールジホスファイト等が挙げられる。
これらの中でも、トリスノニルフェニルホスファイト、トリス(2,4−ジ−tert−ブチルフェニル)ホスファイト、ビス(2,4−ジ−tert−ブチルフェニル)ペンタエリスリトールジホスファイト、ビス(2,6−ジ−tert−ブチル−4−メチルフェニル)ペンタエリスリトールジホスファイトが好ましく使用される。これらの化合物は、1種又は2種以上を併用することができる。
イオウ系酸化防止剤としては、例えば、ジラウリル−3,3’−チオジプロピオン酸エステル、ジトリデシル−3,3’−チオジプロピオン酸エステル、ジミリスチル−3,3’−チオジプロピオン酸エステル、ジステアリル−3,3’−チオジプロピオン酸エステル、ラウリルステアリル−3,3’−チオジプロピオン酸エステル、ペンタエリスリトールテトラキス(3−ラウリルチオプロピオネート)、ビス[2−メチル−4−(3−ラウリルチオプロピオニルオキシ)−5−tert−ブチルフェニル]スルフィド、オクタデシルジスルフィド、メルカプトベンズイミダゾール、2−メルカプト−6−メチルベンズイミダゾール、1,1’−チオビス(2−ナフトール)などをあげることができる。前記のうち、ペンタエリスリトールテトラキス(3−ラウリルチオプロピオネート)が好ましい。これらの化合物は、1種又は2種以上を併用することができる。
フェノール系酸化防止剤としては、例えばペンタエリスリトールテトラキス(3−メルカプトプロピオネート)、ペンタエリスリトールテトラキス(3−ラウリルチオプロピオネート)、グリセロール−3−ステアリルチオプロピオネート、トリエチレングリコール−ビス[3−(3−tert−ブチル−5−メチル−4−ヒドロキシフェニル)プロピオネート]、1,6−ヘキサンジオール−ビス[3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェニル)プロピオネート]、ペンタエリスリトール−テトラキス[3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェニル)プロピオネート]、オクタデシル−3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェニル)プロピオネート、1,3,5−トリメチル−2,4,6−トリス(3,5−ジ−tert−ブル−4−ヒドロキシベンジル)ベンゼン、N,N−ヘキサメチレンビス(3,5−ジ−tert−ブチル−4−ヒドロキシ−ヒドロシンナマイド)、3,5−ジ−tert−ブチル−4−ヒドロキシ−ベンジルホスホネート−ジエチルエステル、トリス(3,5−ジ−tert−ブチル−4−ヒドロキシベンジル)イソシアヌレート、4,4’−ビフェニレンジホスフィン酸テトラキス(2,4−ジ−tert−ブチルフェニル)、3,9−ビス{1,1−ジメチル−2−[β−(3−tert−ブチル−4−ヒドロキシ−5−メチルフェニル)プロピオニルオキシ]エチル}−2,4,8,10−テトラオキサスピロ(5,5)ウンデカン、2,6−ジ−tert−ブチル−p−クレゾール、2,6−ジ−tert−ブチル−4−エチルフェノール等の化合物が挙げられる。
これらの化合物の中でも、炭素数5以上のアルキル基によって1つ以上置換された芳香族モノヒドロキシ化合物が好ましく、具体的には、オクタデシル−3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェニル)プロピオネート、ペンタエリスリチル−テトラキス{3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネート}、1,6−ヘキサンジオール−ビス[3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェニル)プロピオネート]、1,3,5−トリメチル−2,4,6−トリス(3,5−ジ−tert−ブチル−4−ヒドロキシベンジル)ベンゼン等が好ましく、ペンタエリスリチル−テトラキス{3−(3,5−ジ−t−ブチル−4−ヒドロキシフェニル)プロピオネートが更に好ましい。これらの化合物は、1種又は2種以上を併用することができる。
紫外線吸収剤としては、ベンゾトリアゾール系化合物、ベンゾフェノン系化合物、トリアジン系化合物、ベンゾエート系化合物、ヒンダードアミン系化合物、サリチル酸フェニルエステル系化合物、シアノアクリレート系化合物、マロン酸エステル系化合物、シュウ酸アニリド系化合物等が挙げられる。これらは、単独又は2種以上を併用してもよい。
シアノアクリレート系化合物としては、エチル−2−シアノ−3,3−ジフェニルアクリレート、2’−エチルヘキシル−2−シアノ−3,3−ジフェニルアクリレート等が挙げられる。
マロン酸エステル系化合物としては、2−(1−アリールアルキリデン)マロン酸エステル類等が挙げられる。なかでも、マロン酸[(4−メトキシフェニル)−メチレン]−ジメチルエステル(Clariant社製、HostavinPR−25)、2−(パラメトキシベンジリデン)マロン酸ジメチルが好ましい。
シュウ酸アニリド系化合物としては、2−エチル−2’−エトキシ−オキサルアニリド(Clariant社製、SanduvorVSU)等が挙げられる。
光安定剤としては、ヒンダードアミン系光安定剤が挙げられ、その分子量は、1000以下が好ましい。この場合には、成形品の耐候性をより向上させることができる。同様の観点から光安定剤の分子量は900以下がより好ましい。また、光安定剤の分子量は300以上が好ましい。この場合には、耐熱性をより向上させることができ、成形時における金型の汚染をより確実に防止することができる。その結果、表面外観のより優れた成形品を得ることができる。同様の観点から、光安定剤の分子量は400以上がより好ましい。さらに、光安定剤は、ピペリジン構造を有する化合物であることが好ましい。ここで規定するピペリジン構造とは、飽和6員環のアミン構造となっていればよく、ピペリジン構造の一部が置換基により置換されているものも含む。置換基としては、炭素数4以下のアルキル基があげられ、特にはメチル基が好ましい。特に、ピペリジン構造を複数有する化合物が好ましく、それら複数のピペリジン構造がエステル構造により連結されている化合物が好ましい。
ポリカーボネート樹脂組成物は、成形時における離型性を付与するための離型剤として、前記ポリカーボネート樹脂100重量部に対して、多価アルコールの脂肪酸エステルを0.0001重量部以上2重量部以下含有してもよい。多価アルコールの脂肪酸エステルの量をこの範囲に調整することにより、添加効果が充分に得られ、成形加工における離型の際に、離型不良により成形品が割れることをより確実に防止することができる。さらにこの場合には、樹脂組成物の白濁や成形加工時に金型に付着する付着物の増大をより一層抑制することができる。多価アルコールの脂肪酸エステルの含有量は、0.01重量部以上、1.5重量部以下であることがより好ましく、0.1重量部以上、1重量部以下であることがさらに好ましい。
また、耐熱性及び耐湿性の観点から、多価アルコールの脂肪酸エステルとしては、全エステルがより好ましい。
また前記ポリカーボネート樹脂組成物は、本発明の効果を損なわない範囲で、例えば芳香族ポリエステル、脂肪族ポリエステル、ポリアミド、ポリスチレン、ポリオレフィン、アクリル、アモルファスポリオレフィン、ABS、ASなどの合成樹脂、ポリ乳酸、ポリブチレンスクシネートなどの生分解性樹脂などの1種又は2種以上と混練して、ポリマーアロイとしても用いることもできる。
前記ポリカーボネート樹脂組成物には、意匠性を維持できる範囲において、ガラス繊維、ガラスミルドファイバー、ガラスフレーク、ガラスビーズ、シリカ、アルミナ、酸化チタン、硫酸カルシウム粉体、石膏、石膏ウィスカー、硫酸バリウム、タルク、マイカ、ワラストナイト等の珪酸カルシウム、カーボンブラック、グラファイト、鉄粉、銅粉、二硫化モリブデン、炭化ケイ素、炭化ケイ素繊維、窒化ケイ素、窒化ケイ素繊維、黄銅繊維、ステンレス繊維、チタン酸カリウム繊維、これらのウィスカー等の無機充填剤や、木粉、竹粉、ヤシ澱粉、コルク粉、パルプ粉などの粉末状有機充填剤;架橋ポリエステル、ポリスチレン、スチレン・アクリル共重合体、尿素樹脂などのバルン状・球状有機充填剤;炭素繊維、合成繊維、天然繊維などの繊維状有機充填剤を添加することもできる。
前記ポリカーボネート樹脂組成物は、前記特定のポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)に、前記特定の化合物(C)を金属量換算で0.5重量ppm以上1000重量ppm以下添加する添加工程を行い、その後、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)とを溶融反応させる反応工程を行うことにより製造できる。反応工程においては、化合物(C)の存在により、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)とのエステル交換反応が促進され、相溶性の高い樹脂組成物が得られる。なお、ポリカーボネート樹脂(A)、芳香族ポリカーボネート樹脂(B)、化合物(C)としては、前述と同様のものを使用することができる。
ポリカーボネート樹脂組成物の混合温度は200℃〜300℃が好ましい。この場合には、反応混練に要する時間の短縮が可能になり、反応に必要となる化合物(C)の量を抑制することができる。その結果、樹脂の劣化に伴う色調が悪化をより確実に防止することができると共に、耐衝撃性や耐湿熱性などの実用面での物理特性をより向上させることができる。同様の観点から、混合温度は220℃〜280℃であることがより好ましい。
また混合時間については、前記同様の樹脂劣化をより確実に回避するという観点から無用な長大化は回避されるべきであり、化合物(C)の量や混合温度との兼ね合いとなるが、10秒以上150秒以下が好ましく、より好ましくは10秒以上25秒以下であり、これを満たすような化合物(C)の量や混合温度の条件設定が必要となる。
ポリカーボネート樹脂組成物は、射出成形法、押出成形法、圧縮成形法等の通常知られている方法で成形することができる。成形によって得られる成形体は、透明性に優れると共に、生物起源物資含有率、耐熱性、耐湿熱性、及び耐衝撃性を高いレベルでバランスよく兼ね備える。また、ポリカーボネート樹脂組成物を成形してなる成形体においては、色調、耐候性、機械的強度等の向上や、残存低分子成分や異物の低減も可能である。したがって、成形体は車両用内装部品に好適である。
以下の各製造例、実施例1−1〜1−24、比較例1−1〜比較例1−4において、ポリカーボネート樹脂(A)、芳香族ポリカーボネート樹脂(B)及び樹脂組成物の物性ないし特性の評価は次の方法により行った。
ポリカーボネート樹脂(A)又は芳香族ポリカーボネート樹脂(B)のサンプルを塩化メチレンに溶解させ、0.6g/dLの濃度のポリカーボネート樹脂溶液を調製した。森友理化工業社製ウベローデ型粘度管を用いて、温度20.0℃±0.1℃の条件で溶媒の通過時間t0と溶液の通過時間tとを測定し、次式(i)に基づいて相対粘度ηrelを算出した。次いで、相対粘度ηrelから、次式(ii)に基づいて比粘度ηspを求めた。
ηrel=t/t0 ・・・(i)
ηsp=ηrel−1 ・・・(ii)
得られた比粘度ηspを溶液の濃度c(g/dL)で割ることにより、還元粘度(ηsp/c)を求めた。この還元粘度の値が高いほど分子量が大きいことを意味する。
ポリカーボネート樹脂組成物の溶融粘度は、(株)東洋精機製作所製キャピラリーレオメータ「キャピログラフ1B」を用い、ダイス直径1mm、長さ10mm、流入角90℃、予熱時間2分、測定温度240℃にてせん断速度12.16〜6080sec-1の範囲で測定し、せん断速度(SR)91.2sec-1の時の粘度である。また、ポリカーボネート樹脂の溶融粘度の測定においては、測定に用いるポリカーボネート樹脂は予め90℃で4時間以上乾燥したものを使用する。理想粘度は、ポリカーボネート樹脂組成物の各成分の溶融粘度に配合率(重量%)を掛けて足し合わせたものであり、理想粘度に対する比率は、ポリカーボネート樹脂組成物の溶融粘度を理想粘度で割り、100をかけたものである。
ポリカーボネート樹脂組成物のTgは、パーキン・エルマー社製示差走査熱量計「DSC7」を用い、窒素ガス雰囲気下、25℃から200℃まで加熱速度20℃/分で昇温し、200℃で3分間保持した後、25℃まで冷却速度20℃/分で降温を行い、25℃で3分間保持した後、再度200℃まで加熱速度5℃/分で昇温した際、得られたDSC曲線からJIS‐K7121(1987年)の方法に準拠して求めたTmgの値である。また、ガラス転移温度の単一性の評価を行った。具体的には、DSC曲線のピークが単一である場合を単一性が「○」であると評価し、DSC曲線のピークが複数である場合を単一性が「×」であると評価した。
ポリカーボネート樹脂組成物中の金属量は、ICP−MS(誘導結合プラズマ質量分析計)を用いて、測定した。具体的には、ポリカーボネート樹脂組成物の試料約0.5gを正確に秤量し、硫酸・硝酸で加圧密閉分解した。加圧密閉分解には、マイクロウェーブ分解装置パーキン・エルマー社製MULTIWAVを使用した。分解液を適宜純水で希釈して、ICP−MS(サーモクエスト社製ELEMENT)で測定した。なお、測定したアルカリ、アルカリ土類金属はLi、Na、K、Cs、Mg、Ca、Baである。なお、後述の実施例1−1〜実施例1−24における金属量は、化合物(C)由来の金属だけでなく、ポリカーボネート樹脂(A)由来の金属(例えばCa)や芳香族ポリカーボネート樹脂(B)由来の金属(例えばCs)を含む。
ポリカーボネート樹脂組成物のペレットを、熱風乾燥機を用いて、90℃で4時間以上乾燥した。次に、乾燥したペレットを射出成形機(日本製鋼所社製J75EII型)に供給し、樹脂温度240℃、金型温度60℃、成形サイクル50秒間の条件で成形を行うことにより、射出成形板(幅100mm×長さ100mm×厚さ2mm)を得た。JIS K7136(2000年)に準拠し、日本電色工業(株)製ヘーズメータ「NDH2000」を使用し、D65光源にて、射出成形板の全光線透過率を測定した。なお、全光線透過率は、80%以上を合格とし、目視にて射出成形板が不透明であることが明かな場合には、全光線透過率の測定値の代わりに、評価結果を「不透明」とした。
楠本化成(株)製の恒温恒湿槽「HIFLEX FX224P」の設定を80℃、95%RH、又は85℃、85%RHとし、槽内に、幅100mm(又は50mm)×長さ100mm×厚さ2mmの試験片を120時間又は240時間静置して湿熱処理を施した。その後、試験片を取り出し、ヘーズを測定し、耐湿熱試験前のヘーズとの差(ΔHaze)を求めた。尚、ヘーズの測定は、日本電色工業(株)製ヘーズメータ「NDH2000」を用いてJIS−K7136(2000年)に準拠して行った。ΔHazeが大きいほど耐湿熱性が悪く、小さいほど耐湿熱性が良好であることを意味する。なお、上述の全光線透過率の測定において、目視にて射出成形板が不透明であった場合には、本試験(耐湿熱試験)の実施を省略した。
ポリカーボネート樹脂組成物のペレットを、熱風乾燥機を用いて、90℃で4時間以上乾燥した。次に、乾燥したペレットを射出成形機(日本製鋼所社製J75EII型)に供給し、樹脂温度240℃、金型温度60℃、成形サイクル50秒間の条件で成形を行うことにより、JIS−K7139(2009年)記載の多目的試験片A型を得た。得られた多目的試験片から長さ80mm、幅10mm、厚さ4mmの試験片を切削し、JIS−K7191−2(2007年)に準拠し、A法(試験片に加える曲げ応力1.80MPa)にて荷重たわみ温度を測定した。なお、本試験では、90℃以上を合格とするが、好ましくは95℃以上がよく、さらに好ましくは100℃以上がよい。
ポリカーボネート樹脂組成物のペレットを、熱風乾燥機を用いて、90℃で4時間以上乾燥した。次に、乾燥したペレットを射出成形機(日本製鋼所社製J75EII型)に供給し、樹脂温度240℃、金型温度60℃、成形サイクル50秒間の条件で成形を行うことにより、射出成形板(幅100mm×長さ100mm×厚さ2mm)を得た。得られた射出成形板を(株)島津製作所製「島津ハイドロショットHITS−P10形」を使用し、温度23℃または−20℃、ストライカ径5/8インチ、支持台径40mm、試験速度4.4m/sの条件にてハイレート試験を実施した。延性破壊率は5点評価したサンプルのうち、延性破壊した点数を評価点数で割り100を掛けて求めた。
放射線炭素14(C14)は、大気中で宇宙線により一定速度で生成され、一定の速度で失われているので(半減期:5370年)、自然界には一定量が存在している。大気中の二酸化炭素を吸収している植物はこのC14を一定量含有するが、伐採等により炭酸同化作用が起こらなくなると一定の速度で消失していくので、この性質を利用して放射性炭素年代測定法が作られている。化石燃料は、宇宙線の影響を長時間受けていないので、すべてのC14が消失している。一方、バイオ由来の化学品はC14の供給が止まってから、短時間しかたっていないので、C14の含有量はほぼ一定の値であるといえる。
次に、実施例のように、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)のブレンドの場合の生物起源物質含有率は、芳香族ポリカーボネート樹脂(B)は化石燃料由来原料より製造されたポリマーであるので、その生物起源物質含有率は0%である。実施例は重量比でブレンドしているので、それぞれのポリカーボネート樹脂のモル質量(単位:g/mol)を計算し、それぞれ重量をモル質量で除して、それよりモル分率に変換する。これに前述のポリカーボネート樹脂(A)の生物起源物質含有量とそのモル分率の積よりブレンドの生物起源物質含有量を計算する。尚、生物起源由来物質の算出に関しては、樹脂成分のみで算出し、化合物(C)、熱安定剤、離型剤等の成分については考慮しない。
以下の各製造例、実施例1−1〜1−24及び比較例1−1〜1−4で用いた化合物の略号、及び製造元は次の通りである。
・ISB:イソソルビド[ロケットフルーレ社製]
・CHDM:1,4−シクロヘキサンジメタノール[SKChemical社製]
・TCDDM:トリシクロデカンジメタノール[OXEA社製]
・BPC:2,2−ビス(4−ヒドロキシ-3−メチルフェニル)プロパン(本州化学社製)
<炭酸ジエステル>
・DPC:ジフェニルカーボネート[三菱化学(株)製]
<触媒失活剤(酸性化合物(E))>
・亜リン酸[太平化学産業(株)製](分子量82.0)
・Irganox1010:ペンタエリスリトール−テトラキス[3−(3,5−ジ−tert−ブチル−4−ヒドロキシフェニル)プロピオネート][BASF社製]
・AS2112:トリス(2,4−ジ−tert−ブチルフェニル)ホスファイト[(株)ADEKA製](分子量646.9)
<離型剤>
・E−275:エチレングリコールジステアレート[日油(株)製]
竪型攪拌反応器3器と横型攪拌反応器1器、並びに二軸押出機からなる連続重合設備を用いて、ポリカーボネート樹脂の重合を行った。具体的には、まず、ISB、CHDM、及びDPCをそれぞれタンクで溶融させ、ISBを35.2kg/hr、CHDMを14.9kg/hr、DPCを74.5kg/hr(モル比でISB/CHDM/DPC=0.700/0.300/1.010)の流量で第1竪型攪拌反応器に連続的に供給した。同時に、触媒としての酢酸カルシウム1水和物の添加量が全ジヒドロキシ化合物1molに対して1.5μmolとなるように酢酸カルシウム1水和物の水溶液を第1竪型攪拌反応器に供給した。各反応器の反応温度、内圧、滞留時間はそれぞれ、第1竪型攪拌反応器:190℃、25kPa、90分、第2竪型攪拌反応器:195℃、10kPa、45分、第3竪型攪拌反応器:210℃、3kPa、45分、第4横型攪拌反応器:225℃、0.5kPa、90分とした。得られるポリカーボネート樹脂の還元粘度が0.41dL/g〜0.43dL/gとなるように、第4横型攪拌反応器の内圧を微調整しながら運転を行った。
製造例1−1において得られたポリカーボネート樹脂(A)を「PC−A1」という。PC−A1の溶融粘度(240℃、せん断速度91.2sec-1)は720Pa・sである。
反応器への各原料の供給量を、ISBを25.4kg/hr、CHDMを25.0kg/hr、DPCを74.8kg/hr(モル比でISB/CHDM/DPC=0.500/0.500/1.006)、全ジヒドロキシ化合物1molに対する酢酸カルシウム1水和物を1.5μmolとし、得られるポリカーボネート樹脂の還元粘度を0.60dL/gから0.63dL/gとなるようにした以外は、上述の製造例1−1と同様に樹脂を作製し、ISB/CHDMのモル比が50/50mol%のポリカーボネート樹脂を得た。前記ポリカーボネート樹脂に対して、触媒失活剤として0.65重量ppmの亜リン酸(リン原子の量として0.24重量ppm)を添加した。なお、亜リン酸は次のようにして添加した。製造例1−2において得られたポリカーボネート樹脂のペレットに、亜リン酸のエタノール溶液をまぶして混合したマスターバッチを調製し、押出機の第1ベント口の手前(押出機の樹脂供給口側)から、押出機中のポリカーボネート樹脂100重量部に対して、マスターバッチを1重量部となるように供給した。
製造例1−2において得られたポリカーボネート樹脂(A)を「PC−A2」という。PC−A2の溶融粘度(240℃、せん断速度91.2sec-1)は1120Pa・sである。
温度100℃に制御された還流冷却器と撹拌翼とを具備した重合反応装置に、ISB、CHDM、及び蒸留精製して塩化物イオン濃度を10ppb以下にしたDPCを、モル比率でISB/CHDM/DPC=0.27/0.73/1.00になるように仕込み、さらに全ジヒドロキシ化合物1molに対する触媒としての酢酸カルシウム1水和物の添加量が1.5μmolとなるように、酢酸カルシウム1水和物の水溶液を仕込んだ後、窒素置換を十分に行った。続いて熱媒で加温を行い、内温が100℃になった時点で撹拌を開始し、内温が100℃になるように制御しながら内容物を融解させ均一にした。その後、昇温を開始し、40分かけて内温を210℃に調節し、内温が210℃に到達した時点でこの温度を保持するように制御すると同時に、減圧を開始し、210℃に到達してから90分かけて内圧を13.3kPa(絶対圧力、以下同様)に調節し、この圧力を保持しながら、さらに30分間保持した。重合反応とともに副生するフェノール蒸気は、還流冷却器への入口温度が100℃に制御された蒸気を冷媒として用いた還流冷却器に導き、フェノール蒸気中に若干量含まれるモノマー成分を重合反応器に戻し、凝縮しないフェノール蒸気は続いて45℃の温水を冷媒として用いた凝縮器に導いて回収した。このようにしてオリゴマー化させた内容物を、一旦大気圧にまで復圧させた後、撹拌翼および前記同様に制御された還流冷却器を具備した別の重合反応装置に移し、昇温および減圧を開始して、60分かけて内温を210℃、圧力を200Paに調節した。その後、20分かけて内温を220℃、圧力を133Pa以下に調節し、所定の撹拌動力になった時点で復圧し、重合反応装置出口より押し出される溶融状態のポリカーボネート樹脂をペレタイザーによりペレット化することにより、ペレットを得た。還元粘度は0.63dl/gであった。
このようにして、ISB/CHDMのモル比が27/73mol%のポリカーボネート樹脂を得た。製造例1−3において得られたポリカーボネート樹脂(A)を「PC−A3」という。PC−A3の溶融粘度(240℃、せん断速度91.2sec-1)は640Pa・sである。
温度100℃に制御された還流冷却器と撹拌翼とを具備した重合反応装置に、ISB、TCDDM、及び蒸留精製して塩化物イオン濃度を10ppb以下にしたDPCを、モル比率でISB/TCDDM/DPC0.70/0.30/1.00になるように仕込み、さらに全ジヒドロキシ化合物1molに対する触媒としての酢酸カルシウム1水和物の添加量が1.5μmolとなるように、酢酸カルシウム1水和物の水溶液を仕込んだ後、窒素置換を十分に行うことにより、反応装置内の酸素濃度を0.0005〜0.001体積%に調節した。続いて熱媒で加温を行い、内温が100℃になった時点で撹拌を開始し、内温が100℃になるように制御しながら内容物を融解させ均一にした。その後、昇温を開始し、40分かけて内温を210℃に調節し、内温が210℃に到達した時点でこの温度を保持するように制御すると同時に、減圧を開始し、210℃に到達してから90分で13.3kPaにして、この圧力を保持するようにしながら、さらに60分間保持した。重合反応とともに副生するフェノール蒸気は、還流冷却器への入口温度が100℃に制御された蒸気を冷媒として用いた還流冷却器に導き、フェノール蒸気中に若干量含まれるジヒドロキシ化合物や炭酸ジエステルを重合反応器に戻し、凝縮しないフェノール蒸気は続いて45℃の温水を冷媒として用いた凝縮器に導いて回収した。このようにしてオリゴマー化させた内容物を、一旦大気圧にまで復圧させた後、撹拌翼及び前記同様に制御された還流冷却器を具備した別の重合反応装置に移し、昇温及び減圧を開始して、60分かけて内温を220℃、圧力を200Paに調節した。その後、20分かけて内温を230℃、圧力を133Pa以下に調節して、所定の撹拌動力になった時点で大気圧に復圧し、内容物をストランドの形態で抜出し、回転式カッターによりカーボネート共重合体をペレット状にした。このようにして、ISB/TCDDMのモル比が70/30mol%のポリカーボネート樹脂を得た。前記ポリカーボネート樹脂に対して0.65重量ppmの亜リン酸(リン原子の量として0.24重量ppm)を添加した。なお、亜リン酸は次のようにして添加した。製造例1−1において得られたポリカーボネート樹脂のペレットに、亜リン酸のエタノール溶液をまぶして混合したマスターバッチを調製し、押出機の第1ベント口の手前(押出機の樹脂供給口側)から、押出機中のポリカーボネート樹脂100重量部に対して、マスターバッチを1重量部となるように供給した。
製造例1−4において得られたポリカーボネート樹脂(A)を「PC−A4」という。PC−A4の溶融粘度(240℃、せん断速度91.2sec-1)は1120Pa・sである。
・PC−B1:三菱エンジニアリングプラスチックス社製ノバレックス7022J(ビスフェノールA構成単位100モル%の芳香族ポリカーボネート樹脂、溶融粘度(240℃、せん断速度91.2sec-1)3260Pa・s)
・PC−B2:下記の製造例によって得られる芳香族ポリカーボネート樹脂
・PC−B3:バイエルマテリアルサイエンス社製APEC1897(ビスフェノールAと1,1−ビス(4−ヒドロキシ−3,3,5−トリメチルフェニル)シクロヘキサンとの共重合体からなる芳香族ポリカーボネート樹脂、溶融粘度(240℃、せん断速度91.2ec-1)は粘度が高すぎて測定不能)
181.8kgのBPCと57.7kgのDPCとの混合物に、炭酸セシウム水溶液を添加した。添加量は、ジヒドロキシ化合物であるBPC1mol当たりの炭酸セシウム量が2.0μmolとなるように調整した。次に、攪拌機、熱媒ジャケット、真空ポンプ、及び還流冷却器を具備した内容量400Lの第1反応器に混合物を投入した。次に、第1反応器内を圧力1.33kPa(10Torr)に減圧し、続いて、窒素で大気圧に復圧する操作を10回繰り返すことにより、第1反応器の内部を窒素置換した。その後、熱媒ジャケットに温度230℃の熱媒を通すことにより第1反応器の内温を徐々に昇温させ、混合物を溶解させた。その後、この溶融混合物を第2反応器に移送した。尚、第2反応器は、内容量が400Lであり、攪拌機、熱媒ジャケット、真空ポンプ、及び還流冷却管を具備する。回転速度60rpmに調節した撹拌機により第2反応器内の溶融混合物を撹拌すると共に、熱媒ジャケット内の温度をコントロールすることにより第2反応器の内温を220℃に保った。そして、第2反応器の内部で行われるBPCとDPCのオリゴマー化反応により副生するフェノールを留去しながら、第2反応器内の圧力を絶対圧で101.3kPa(760Torr)から13.3kPa(100Torr)まで減圧した。次に、第2反応器の内部を30rpmの回転速度で攪拌し、熱媒ジャケットにて内温を昇温し、第2反応器内を絶対圧で101.3kPaから13.3kPaまで減圧した。その後、昇温を継続し、内圧を絶対圧で13.3kPaから399Pa(3Torr)まで減圧して留出を行うことにより、フェノールを系外に除去した。さらに、昇温を続け、第2反応器内の絶対圧が70Pa(約0.5Torr)に到達後、この圧力(70Pa)を保持し、重縮合反応を行った。その際、攪拌動力に応じて攪拌回転数を10rpmとし、第2反応器内の最終的な内部温度は275℃とした。第2反応器の攪拌機が予め定めた所定の攪拌動力となったときに、重縮合反応を終了した。第2反応器での重合反応時間は310分であった。このようにして、芳香族ポリカーボネート樹脂(PC−B2)を得た。PC−B2の溶融粘度(240℃、せん断速度91.2sec-1)は3040Pa・sである。
本例においては、ポリカーボネート樹脂(A)としてPC−A1を用い、芳香族ポリカーボネート樹脂(B)としてPC−B1を用い、化合物(C)として、粉末状の炭酸水素ナトリウム(和光純薬(株)製 試薬特級)を用いた。そして、70重量部のポリカーボネート樹脂(A)と、30重量部の芳香族ポリカーボネート樹脂(B)と、金属換算(Na換算)で20重量ppmの化合物(C)とを配合し、二軸混練機(日本製鋼所社製、TEX−30α(L/D=52.5、L(mm):スクリュの長さ、D(mm):スクリュの直径))を用いて混練を行い、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)との溶融反応を行った。混練機スクリュの直径D(mm)に対するニーディングゾーンの合計長さLtの比は6であり(Lt/D=6)、混練条件は、流量:20kg/h、スクリュ回転速度:200rpm、シリンダー温度:230℃である。前記押出機に2つの真空ベント口を有しており、ベント真空度:11kPaの条件で行った。混練による溶融反応後に樹脂組成物をストランド状に押出し、水冷工程を経てペレット状にカッティングを行いポリカーボネート樹脂組成物のペレットを得た。
下記の(1)及び(2)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムの添加量(Na換算量)を20ppmから10ppmに変更した。
(2)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムの添加量(Na換算量)を20ppmから10ppmに変更した。
(2)実施例1−1と同様にしてポリカーボネート樹脂組成物を作製した後、再度、溶融混練を行う際に、触媒失活剤として、亜リン酸からなる酸性化合物(E)を添加した。添加量は、化合物(C)の添加量に対して0.5倍モルである。
(3)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムの添加量(Na換算量)を20ppmから10ppmに変更した。
(2)実施例1−1と同様にしてポリカーボネート樹脂組成物を作製した後、再度、溶融混練を行う際に、触媒失活剤として、亜リン酸からなる酸性化合物(E)を添加した。添加量は、化合物(C)の添加量に対して1倍モルである。
(3)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムの添加量(Na換算量)を20ppmから10ppmに変更した。
(2)実施例1−1と同様にしてポリカーボネート樹脂組成物を作製した後、再度、溶融混練を行う際に、触媒失活剤として、亜リン酸からなる酸性化合物(E)を添加した。添加量は、化合物(C)の添加量に対して1.5倍モルである。
(3)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムの添加量(Na換算量)を20ppmから10ppmに変更した。
(2)実施例1−1と同様にしてポリカーボネート樹脂組成物を作製した後、再度、溶融混練を行う際に、触媒失活剤として、亜リン酸からなる酸性化合物(E)を添加した。添加量は、化合物(C)の添加量に対して2倍モルである。
(3)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(5)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを濃度0.4重量%の水酸化ナトリウム水溶液に変更した。添加量(Na換算量)は100ppmである。
(2)混練機スクリュのニーディングゾーンの合計長さをL/D=6からL/D=21.5に変更した。
(3)真空ベント口を2つから1つに変更した。
(4)ベント真空度を11kPaから21kPaに変更した。
(5)混練条件における流量を20kg/hから10kg/hに変更した。
下記の(1)及び(2)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを炭酸セシウム(粉末)に変更した。添加量(Cs換算量)は、10重量ppmである。
(2)ベント真空度を11kPaから6kPaに変更した。
下記の(1)及び(2)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを炭酸セシウム(粉末)に変更した。添加量(Cs換算量)は、5重量ppmである。
(2)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを炭酸セシウム(粉末)に変更した。添加量(Cs換算量)は5重量ppmである。
(2)実施例1−1と同様にしてポリカーボネート樹脂組成物を作製した後、再度、溶融混練を行う際に、触媒失活剤として、亜リン酸からなる酸性化合物(E)を添加した。添加量は、化合物(C)の添加量に対して2倍モルである。
(3)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表1に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを塩化ナトリウム(粉末)に変更した。添加量(Na換算量)は100重量ppmである。
(2)混練機スクリュのニーディングゾーンの合計長さをL/D=6からL/D=18に変更した。
(3)ベント真空度を11kPaから21kPaに変更した。
下記の(1)及び(2)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は3重量ppmである。
(2)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(3)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は3重量ppmである。
(2)実施例1−1と同様にしてポリカーボネート樹脂組成物を作製した後、再度、溶融混練を行う際に、触媒失活剤として、亜リン酸からなる酸性化合物(E)を添加した。添加量は、化合物(C)の添加量に対して2倍モルである。
(3)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)ポリカーボネート樹脂(A)の配合割合を90重量部に変更し、芳香族ポリカーボネート樹脂(B)の配合割合を10重量部に変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は3重量ppmである。
(3)混練条件における流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)ポリカーボネート樹脂(A)の配合割合を50重量部に変更し、芳香族ポリカーボネート樹脂(B)の配合割合を50重量部に変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は3重量ppmである。
(3)混練条件における流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)ポリカーボネート樹脂(A)として用いられるPC−A1をPC−A4に変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は3重量ppmである。
(3)混練条件における流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)芳香族ポリカーボネート樹脂(B)として用いられるPC−B1をPC−B2に変更した。添加量は5重量ppmである。
(2)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は5重量ppmである。
(3)流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)芳香族ポリカーボネート樹脂(B)として用いられるPC−B1をPC−B3に変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は5重量ppmである。
(3)流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)ポリカーボネート樹脂(A)として用いられるPC−A1をPC−A2に変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウムを炭酸カリウム(粉末)に変更した。添加量(K換算量)は3重量ppmである。
(3)流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表2に示す。
(1)ポリカーボネート樹脂(A)として用いられるPC−A1をPC−A2に変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウムを水酸化カルシウム(粉末)に変更した。添加量(K換算量)は500重量ppmである。
(3)流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表3に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを塩化ナトリウム(粉末)に変更した。添加量(Na換算量)は10重量ppmである。
下記の(1)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表3に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムを酢酸リチウムに変更した。添加量(Li換算量)は10重量ppmである。
下記の(1)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表3に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムをステアリン酸リチウムに変更した。添加量(Li換算量)は3重量ppmである。
下記の(1)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表3に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムをオルトケイ酸ナトリウムに変更した。添加量(Na換算量)は10重量ppmである。
下記の(1)及び(2)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表4に示す。なお、本例のポリカーボネート樹脂組成物中のセシウム量は0.2重量ppm、カルシウム量は0.2重量ppmであった。金属量は、上述のICP−MSにより測定した。本例においては、樹脂組成物の製造時にポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)に対して別途化合物(C)を添加していないため、これらの金属量は、ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)由来のものである。本例におけるポリカーボネート樹脂組成物中に含まれる長周期型周期表第I族、第II族の金属化合物から選択される少なくとも1種類の化合物(C)の含有量は0.4重量ppmであった。ポリカーボネート樹脂(A)及び芳香族ポリカーボネート樹脂(B)由来の金属量については、後述の比較例1−2〜1−4も同様である。
(1)樹脂組成物の製造時に添加される化合物(C)の添加量を0に変更した。
(2)ベント真空度を11kPaから21kPaに変更した。
下記の(1)〜(4)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表4に示す。
(1)ポリカーボネート樹脂(A)として用いられるPC−A1をPC−A3に変更した。
(2)化合物(C)の添加量を0に変更した。
(3)流量を20kg/hから10kg/hに変更した。
(4)ベント真空度を11kPaから6kPaに変更した。
下記の(1)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表4に示す。
(1)化合物(C)として用いられる炭酸水素ナトリウムをテトラ−n−ブトキシチタン(以後、TBTと略記する)に変更した。添加量は1000重量ppmである。
下記の(1)及び(2)に示す変更点を除いては、実施例1−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製した。本例の評価結果を表4に示す。
(1)ベント真空度を6kPaから101kPaへ変更した。
(2)化合物(C)として用いられる炭酸水素ナトリウム量(Na換算量)を10重量ppmに変更した。
以下の実施例、比較例、参考例において、ポリカーボネート樹脂(A)、芳香族ポリカーボネート樹脂(B)及び樹脂組成物の物性ないし特性の評価は次の方法により行った。
上述のI−1と同様の方法により測定した。
上述のI−3と同様の方法により測定した。
上述のI−4と同様の方法により測定した。なお、後述の実施例2−1〜実施例2−7、比較例2−1〜2−4、参考例2−1〜2−2における金属量は、化合物(C)由来の金属だけでなく、ポリカーボネート樹脂(A)由来の金属(例えばCa)や芳香族ポリカーボネート樹脂(B)由来の金属(例えばCs)を含む。
上述のI−5と同様の方法により測定した。
楠本化成(株)製の恒温恒湿槽「HIFLEX FX224P」の設定を85℃、85%RHとし、槽内に、幅100mm(又は50mm)×長さ100mm×厚さ2mmの試験片を480時間静置して湿熱処理を施した。その後、試験片を取り出し、ヘーズを測定し、耐湿熱試験前のヘーズとの差(ΔHaze)を求めた。尚、ヘーズの測定は、日本電色工業(株)製ヘーズメータ「NDH2000」を用いてJIS−K7136(2000年)に準拠して行った。ΔHazeが大きいほど耐湿熱性が悪く、小さいほど耐湿熱性が良好であることを意味する。なお、上述の全光線透過率の測定において、目視にて射出成形板が不透明であった場合には、本試験(耐湿熱試験)の実施を省略した。
上述のI−7と同様の方法により測定した。なお、本試験では、90℃以上を合格とするが、好ましくは95℃以上がよく、さらに好ましくは100℃以上がよい。
上述のI−8と同様の方法により測定した。
ポリカーボネート樹脂組成物のペレットを、熱風乾燥機を用いて、90℃で4時間以上乾燥した。次に、乾燥したペレットを射出成形機(日本製鋼所社製J75EII型)に供給し、樹脂温度240℃、金型温度60℃、成形サイクル50秒間の条件で成形を行うことにより、JIS−K7139(2009年)に記載の多目的試験片A型を得た。得られた多目的試験片から長さ80mm、幅10mm、厚さ4mmの試験片を切削し、JIS−K7171(2008年)に準拠し、曲げ弾性率を測定した。
上述のI−9と同様の方法により測定した。
・炭酸カリウム(和光純薬工業社製 試薬特級)
・炭酸水素ナトリム(和光純薬工業社製 試薬特級)
・炭酸セシウム(ナカライテスク社製 試薬特級)
・18C6E(18−クラウン−6−エーテル):1,4,7,10,13,16−ヘキサオクサシクロオクタデカン(東京化成工業社製)
・15C5E(15−クラウン−5−エーテル):1,4,7,10,13−ペンタオキサシクロペンタデカン(東京化成工業社製)
・亜リン酸(和光純薬工業社製 試薬特級)
本例においては、ポリカーボネート樹脂(A)として上述のPC−A1を用い、芳香族ポリカーボネート樹脂(B)として上述のノバレックス7022J(すなわち、PC−B1)を用い、化合物(D)として18−クラウン−6−エーテルを用い、化合物(C)として炭酸カリウムを用いた。そして、70重量部のポリカーボネート樹脂(A)と、30重量部の芳香族ポリカーボネート樹脂(B)と、化合物(C)に対して1倍モルの化合物(D)と、金属換算(K換算)で2重量ppmの化合物(D)とを配合し、二軸混練機(日本製鋼所社製、TEX−30α(L/D=52.5、L(mm):スクリュの長さ、D(mm):スクリュの直径))を用いて混練を行い、ポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)との溶融反応を行った。混練機スクリュの直径D(mm)に対するニーディングゾーンの合計長さLtの比は6であり(Lt/D=6)、混練条件は、流量:10kg/h、スクリュ回転速度:200rpm、シリンダー温度:230℃である。前記押出機は2つの真空ベント口を有しており、ベント真空度:11kPaの条件で行った。混練による溶融反応後に樹脂組成物をストランド状に押出し、水冷工程を経てペレット状にカッティングを行いポリカーボネート樹脂組成物のペレットを得た。
実施例2−1において得られたポリカーボネート樹脂組成物のペレットに、酸性化合物(E)である亜リン酸の15重量%エタノール溶液を均一にまぶした。なお、酸性化合物(E)の添加量は、化合物(C)として添加した炭酸カリウム中の金属(K)量に対して2倍モルとなるように調整した。その後、エタノールを風乾により除去した。このようにして得られたペレットを実施例2−1と同様に溶融押出し、ペレット化を行った。さらに実施例2−1と同様にポリカーボネート樹脂組成物の乾燥、成形、評価を行った。その結果を表5に示す。
本例においては、化合物(D)として15−クラウン−5−エーテルを用い、化合物(C)として炭酸水素ナトリウムを用い、化合物(C)の量を金属換算(Na換算)で3重量ppmに変えた以外は、実施例1と同様にしてポリカーボネート樹脂組成物のペレットを作製し、このペレットに、酸性化合物(E)である亜リン酸の15重量%エタノール溶液を均一にまぶした。なお、酸性化合物(E)の添加量は、化合物(C)として添加した炭酸水素ナトリウム中の金属(Na)量に対して2倍モルとなるように調整した。その後、エタノールを風乾により除去した。このようにして得られたペレットを実施例2−1と同様に溶融押出し、ペレット化を行った。さらに実施例1と同様にポリカーボネート樹脂組成物の乾燥、成形、評価を行った。その結果を表5に示す。
本例においては、化合物(C)に対する化合物(D)の量を2倍モルに変更し、化合物(C)として炭酸セシウムを用い、化合物(C)の量を金属換算(Cs換算)で7重量ppmに変えた以外は、実施例1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表5に示す。
実施例2−4において得られたポリカーボネート樹脂組成物のペレットに、酸性化合物(E)である亜リン酸の15重量%エタノール溶液を均一にまぶした。なお、酸性化合物(E)の添加量は、化合物(C)として添加した炭酸セシウム中の金属(Cs)量に対して2倍モルとなるように調整した。その後、エタノールを風乾により除去した。このようにして得られたペレットを実施例2−1と同様に溶融押出し、ペレット化を行った。さらに実施例1と同様にポリカーボネート樹脂組成物の乾燥、成形、評価を行った。その結果を表5に示す。
本例においては、化合物(C)に対する化合物(D)の量を0.1倍モルに変更し、化合物(C)として炭酸リチウムを用い、化合物(C)の量を金属換算(Cs換算)で10重量ppmに変えた以外は、実施例1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表5に示す。
本例においては、化合物(C)に対する化合物(D)の量を0.1倍モルに変更し、化合物(C)としてステアリン酸リチウムを用い、化合物(C)の量を金属換算(Cs換算)で3重量ppmに変えた以外は、実施例2−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表5に示す。
本例においては、化合物(D)を添加しなかった以外は、実施例1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表6に示す。
本例においては、化合物(D)を添加せず、化合物(C)として用いる炭酸カリウムの量を金属換算で5重量ppmに変更した以外は、実施例2−1と同様に行った。その結果を表6に示す。
本例においては、化合物(D)を添加せず、化合物(C)として炭酸水素ナトリウムを用い、化合物(C)の量を金属換算(Na換算)で5重量ppmに変えた以外は、実施例2−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表6に示す。
本例においては、化合物(D)を添加せず、化合物(C)として炭酸水素ナトリウムを用い、化合物(C)の量を金属換算(Na換算)で10重量ppmに変更した以外は、実施例2−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表6に示す。
本例においては、化合物(C)及び化合物(D)を添加しなかった以外は、実施例2−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表6に示す。
本例においては、芳香族ポリカーボネート樹脂(B)の代わりにポリカーボネート樹脂(A)である上述のPC−A3を用い、化合物(C)及び化合物(D)を添加しなかった以外は、実施例2−1と同様にしてポリカーボネート樹脂組成物を作製し、これを用いて成形体(試験片)を作製し、さらに実施例2−1と同様の評価を行った。その結果を表6に示す。
Claims (19)
- 下記式(1)で表される化合物に由来する構成単位を含むポリカーボネート樹脂(A)と、
芳香族ポリカーボネート樹脂(B)と、
長周期型周期表第I族の金属の化合物及び長周期型周期表第II族の金属の化合物からなる群から選択される少なくとも1種の化合物(C)とを含有し、
前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)との合計量100重量部に対する前記化合物(C)の含有量が、該化合物(C)中の金属量で0.8重量ppm以上かつ1000重量ppm以下であり、
示差走査熱量分析により測定されるガラス転移温度が単一である、ポリカーボネート樹脂組成物。
- 下記式(1)で表される化合物に由来する構成単位を含むポリカーボネート樹脂(A)と、
芳香族ポリカーボネート樹脂(B)と、
長周期型周期表第I族の金属の化合物及び長周期型周期表第II族の金属の化合物からなる群から選択される少なくとも1種の化合物(C)と、
クラウンエーテル化合物(D)とを含有し、
前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)との合計量100重量部に対する前記化合物(C)の含有量が、該化合物(C)中の金属量で0.8重量ppm以上1000重量ppm以下であり、
前記化合物(C)の金属換算量に対する前記クラウンエーテル化合物(D)の含有量が0.1倍モル以上10倍モル以下である、ポリカーボネート樹脂組成物。
- 示差走査熱量分析により測定されるガラス転移温度が単一である、請求項2に記載のポリカーボネート樹脂組成物。
- 前記ポリカーボネート樹脂組成物を成形してなる厚さ2mmの成形体の全光線透過率が80%以上である、請求項1〜3のいずれか1項に記載のポリカーボネート樹脂組成物。
- 長周期型周期表第I族の金属及び長周期型周期表第II族の金属を含む、請求項1〜4のいずれか1項に記載のポリカーボネート樹脂組成物。
- 前記化合物(C)として、少なくとも長周期型周期表第I族の金属の化合物を含有し、前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)との合計量100重量部に対する長周期型周期表I族の金属の化合物の含有量が金属量で0.8重量ppm〜1000重量ppmである、請求項1〜5のいずれか1項に記載のポリカーボネート樹脂組成物。
- 前記長周期型周期表第I族の金属の化合物がナトリウム化合物、カリウム化合物、及びセシウム化合物からなる群から選ばれる少なくとも1種である、請求項1〜6のいずれか1項に記載のポリカーボネート樹脂組成物。
- 前記化合物(C)が、無機塩(炭酸塩を含む)、カルボン酸塩、フェノラート、ハロゲン化合物、及び水酸化化合物からなるグループより選ばれる少なくとも1種である、請求項1〜7のいずれか1項に記載のポリカーボネート樹脂組成物。
- さらに酸性化合物(E)を含有する、請求項1〜8のいずれか1項に記載のポリカーボネート樹脂組成物。
- 前記酸性化合物(E)の含有量が、前記化合物(C)中の金属の含有量に対して0.1倍モル以上5倍モル以下である、請求項9に記載のポリカーボネート樹脂組成物。
- 請求項1〜10のいずれか1項に記載のポリカーボネート樹脂組成物を含有してなる成形体。
- 下記式(1)で表される化合物に由来する構成単位を含むポリカーボネート樹脂(A)と芳香族ポリカーボネート樹脂(B)との合計量100重量部に対して、長周期型周期表第I族の金属の化合物及び長周期型周期表第II族の金属の化合物からなる群から選択される少なくとも一種の化合物(C)を金属量換算で0.5重量ppm以上1000重量ppm以下添加する添加工程と、
該添加工程後に、前記ポリカーボネート樹脂(A)と前記芳香族ポリカーボネート樹脂(B)とを溶融反応させる反応工程とを有する、ポリカーボネート樹脂組成物の製造方法。
- 前記反応工程おける溶融反応を減圧下にて行う、請求項12に記載のポリカーボネート樹脂組成物の製造方法。
- 前記反応工程における溶融反応を真空度30kPa以下という条件で行う、請求項12又は13に記載のポリカーボネート樹脂組成物の製造方法。
- 前記化合物(C)が、無機塩(炭酸塩を含む)、カルボン酸塩、フェノラート、ハロゲン化合物、水酸化化合物からなるグループより選ばれる少なくとも1種である、請求項12〜14のいずれか1項に記載のポリカーボネート樹脂組成物の製造方法。
- 前記化合物(C)がナトリウム化合物、カリウム化合物、及びセシウム化合物からなるグループより選ばれる少なくとも1種である、請求項12〜15のいずれか1項に記載のポリカーボネート樹脂組成物の製造方法。
- 前記添加工程においては、さらにクラウンエーテル化合物(D)を添加し、前記化合物(C)の金属換算量に対する前記クラウンエーテル化合物(D)の添加量が0.1倍モル以上10倍モル以下である、請求項12〜16のいずれか1項に記載のポリカーボネート樹脂組成物の製造方法。
- 前記添加工程において、さらに酸性化合物(E)を添加する、請求項12〜17のいずれか1項に記載のポリカーボネート樹脂組成物の製造方法。
- 前記酸性化合物(E)の添加量が、前記化合物(C)の金属の添加量に対して、0.1倍モル以上5倍モル以下である、請求項18に記載のポリカーボネート樹脂組成物の製造方法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015131492 | 2015-06-30 | ||
| JP2015131491 | 2015-06-30 | ||
| JP2015131491 | 2015-06-30 | ||
| JP2015131492 | 2015-06-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017014502A true JP2017014502A (ja) | 2017-01-19 |
| JP6807175B2 JP6807175B2 (ja) | 2021-01-06 |
Family
ID=57608735
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016128509A Active JP6807175B2 (ja) | 2015-06-30 | 2016-06-29 | ポリカーボネート樹脂組成物、その製造方法、成形体 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10526446B2 (ja) |
| EP (1) | EP3318603B1 (ja) |
| JP (1) | JP6807175B2 (ja) |
| KR (1) | KR102566077B1 (ja) |
| CN (2) | CN111171545B (ja) |
| WO (1) | WO2017002886A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018165341A (ja) * | 2017-03-28 | 2018-10-25 | 三菱ケミカル株式会社 | ポリカーボネート樹脂組成物、その製造方法、成形体 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11555114B2 (en) | 2017-08-02 | 2023-01-17 | Idemitsu Kosan Co., Ltd. | Method for producing polycarbonate resin composition |
| EP3805314B1 (en) * | 2018-06-08 | 2022-09-07 | Mitsubishi Chemical Corporation | Polycarbonate resin composition, molded article and multilayer body |
| WO2022080188A1 (ja) * | 2020-10-14 | 2022-04-21 | 三菱鉛筆株式会社 | 鉛筆芯 |
| CN114085511B (zh) * | 2021-11-08 | 2023-07-11 | 金发科技股份有限公司 | 一种低翘曲高冲击的聚碳酸酯组合物及其制备方法和应用 |
| CN114031918B (zh) * | 2021-11-22 | 2023-05-26 | 万华化学集团股份有限公司 | 一种低黄度指数的聚碳酸酯组合物及其制备方法与用途 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09169837A (ja) * | 1995-12-19 | 1997-06-30 | Mitsubishi Chem Corp | 芳香族ポリカーボネートの製造方法 |
| JP2011021171A (ja) * | 2008-11-28 | 2011-02-03 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂、ポリカーボネート樹脂組成物、光学フィルム及びポリカーボネート樹脂成形品 |
| WO2011071162A1 (ja) * | 2009-12-10 | 2011-06-16 | 三菱化学株式会社 | ポリカーボネート樹脂組成物並びにこれを成形して得られる成形体、フィルム、プレート及び射出成形品 |
| JP2012509382A (ja) * | 2008-11-18 | 2012-04-19 | サビック・イノベーティブ・プラスチックス・アイピー・ベスローテン・フェンノートシャップ | イソソルビドをベースとするポリカーボネートの安定化 |
| JP2013007017A (ja) * | 2010-12-16 | 2013-01-10 | Mitsubishi Engineering Plastics Corp | 難燃性ポリカーボネート樹脂組成物及びそれからなる成形品 |
| JP2014152210A (ja) * | 2013-02-06 | 2014-08-25 | Teijin Ltd | 強化ポリカーボネート樹脂組成物 |
| JP2015007248A (ja) * | 2014-09-02 | 2015-01-15 | 帝人株式会社 | ポリカーボネート樹脂ペレット |
| WO2017000154A1 (zh) * | 2015-06-30 | 2017-01-05 | 华东理工大学 | 聚碳酸酯树脂组合物、其制造方法、成形体 |
Family Cites Families (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004111106A1 (ja) | 2003-06-16 | 2004-12-23 | Teijin Limited | ポリカーボネートおよびその製造方法 |
| US7138479B2 (en) | 2003-12-31 | 2006-11-21 | General Electric Company | Aliphatic diol polycarbonates and their preparation |
| JP4759518B2 (ja) | 2004-10-14 | 2011-08-31 | 帝人株式会社 | 光弾性定数の低いポリカーボネート及びそれからなるフィルム |
| JP2009046519A (ja) | 2005-11-29 | 2009-03-05 | Mitsui Chemicals Inc | ポリカーボネート共重合体からなる透明耐熱成形体、その製造方法およびその用途 |
| US7491788B1 (en) * | 2006-05-19 | 2009-02-17 | Sabic Innovative Plastics Ip B.V. | High heat polycarbonate compositions, methods for the preparation thereof, and articles derived therefrom |
| CN101754994A (zh) * | 2007-05-17 | 2010-06-23 | 帝人株式会社 | 具有植物来源成分的聚碳酸酯的制备方法 |
| JP4774024B2 (ja) | 2007-07-12 | 2011-09-14 | 帝人株式会社 | 光ディスク基板および光ディスク |
| JP5714788B2 (ja) | 2007-09-10 | 2015-05-07 | 帝人株式会社 | プラスチックレンズおよびその製造方法 |
| JP5481784B2 (ja) * | 2007-12-12 | 2014-04-23 | 三菱化学株式会社 | ポリカーボネート樹脂組成物及びその製造方法 |
| JP5716362B2 (ja) | 2009-11-19 | 2015-05-13 | 三菱化学株式会社 | ポリカーボネート共重合体 |
| CN103319706A (zh) * | 2009-11-30 | 2013-09-25 | 三菱化学株式会社 | 聚碳酸酯树脂及其制造方法 |
| EP2511340A4 (en) | 2009-12-10 | 2015-07-08 | Mitsubishi Chem Corp | POLYCARBONATE RESIN COMPOSITION AND MOLDING PRODUCTS |
| EP2511341A4 (en) | 2009-12-10 | 2015-07-08 | Mitsubishi Chem Corp | POLYCARBONATE RESIN COMPOSITION AND MOLDING PRODUCTS |
| KR101794501B1 (ko) | 2009-12-10 | 2017-11-07 | 미쯔비시 케미컬 주식회사 | 폴리카보네이트 수지 조성물 및 성형품 |
| WO2011071165A1 (ja) | 2009-12-10 | 2011-06-16 | 三菱化学株式会社 | ポリカーボネート樹脂組成物及び成形品 |
| CN103370373B (zh) * | 2011-02-17 | 2015-08-26 | 三菱化学株式会社 | 聚碳酸酯树脂组合物 |
| KR101940339B1 (ko) * | 2011-03-31 | 2019-01-18 | 미쯔비시 케미컬 주식회사 | 폴리카보네이트 수지 조성물 및 그 성형품 |
| TWI549986B (zh) | 2011-05-19 | 2016-09-21 | 三菱瓦斯化學股份有限公司 | A high-flow polycarbonate copolymer, a method for producing a high molecular weight aromatic polycarbonate resin, and an aromatic polycarbonate compound |
| JP2013076981A (ja) | 2011-09-14 | 2013-04-25 | Mitsubishi Chemicals Corp | 位相差フィルム、並びにこれを用いた円偏光板及び画像表示装置 |
| JP2013076982A (ja) | 2011-09-14 | 2013-04-25 | Mitsubishi Chemicals Corp | 位相差フィルム、並びにこれを用いた円偏光板及び画像表示装置 |
| JP6398242B2 (ja) * | 2013-03-21 | 2018-10-03 | 三菱ケミカル株式会社 | 樹脂組成物及びそれを用いたフィルム |
-
2016
- 2016-06-29 EP EP16817994.3A patent/EP3318603B1/en active Active
- 2016-06-29 JP JP2016128509A patent/JP6807175B2/ja active Active
- 2016-06-29 CN CN202010095657.6A patent/CN111171545B/zh active Active
- 2016-06-29 CN CN201680038136.9A patent/CN107849345B/zh active Active
- 2016-06-29 KR KR1020177037530A patent/KR102566077B1/ko active Active
- 2016-06-29 WO PCT/JP2016/069357 patent/WO2017002886A1/ja not_active Ceased
-
2017
- 2017-12-28 US US15/856,721 patent/US10526446B2/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09169837A (ja) * | 1995-12-19 | 1997-06-30 | Mitsubishi Chem Corp | 芳香族ポリカーボネートの製造方法 |
| JP2012509382A (ja) * | 2008-11-18 | 2012-04-19 | サビック・イノベーティブ・プラスチックス・アイピー・ベスローテン・フェンノートシャップ | イソソルビドをベースとするポリカーボネートの安定化 |
| JP2011021171A (ja) * | 2008-11-28 | 2011-02-03 | Mitsubishi Chemicals Corp | ポリカーボネート樹脂、ポリカーボネート樹脂組成物、光学フィルム及びポリカーボネート樹脂成形品 |
| WO2011071162A1 (ja) * | 2009-12-10 | 2011-06-16 | 三菱化学株式会社 | ポリカーボネート樹脂組成物並びにこれを成形して得られる成形体、フィルム、プレート及び射出成形品 |
| JP2013007017A (ja) * | 2010-12-16 | 2013-01-10 | Mitsubishi Engineering Plastics Corp | 難燃性ポリカーボネート樹脂組成物及びそれからなる成形品 |
| JP2014152210A (ja) * | 2013-02-06 | 2014-08-25 | Teijin Ltd | 強化ポリカーボネート樹脂組成物 |
| JP2015007248A (ja) * | 2014-09-02 | 2015-01-15 | 帝人株式会社 | ポリカーボネート樹脂ペレット |
| WO2017000154A1 (zh) * | 2015-06-30 | 2017-01-05 | 华东理工大学 | 聚碳酸酯树脂组合物、其制造方法、成形体 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018165341A (ja) * | 2017-03-28 | 2018-10-25 | 三菱ケミカル株式会社 | ポリカーボネート樹脂組成物、その製造方法、成形体 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR102566077B1 (ko) | 2023-08-10 |
| US10526446B2 (en) | 2020-01-07 |
| JP6807175B2 (ja) | 2021-01-06 |
| EP3318603A1 (en) | 2018-05-09 |
| CN107849345A (zh) | 2018-03-27 |
| KR20180022712A (ko) | 2018-03-06 |
| EP3318603B1 (en) | 2020-11-18 |
| WO2017002886A1 (ja) | 2017-01-05 |
| US20180118883A1 (en) | 2018-05-03 |
| CN107849345B (zh) | 2020-03-13 |
| EP3318603A4 (en) | 2019-01-16 |
| CN111171545B (zh) | 2023-04-28 |
| CN111171545A (zh) | 2020-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102656231B (zh) | 聚碳酸酯树脂组合物以及将其成型而得到的成型体、膜、板和注射成型品 | |
| JP6714877B2 (ja) | ポリカーボネート樹脂組成物、およびその成形体 | |
| JP6807175B2 (ja) | ポリカーボネート樹脂組成物、その製造方法、成形体 | |
| CN103370373B (zh) | 聚碳酸酯树脂组合物 | |
| KR102200887B1 (ko) | 충격강도가 개선된 친환경 폴리에스테르 카보네이트 수지 조성물 및 그 제조방법 | |
| JP6671114B2 (ja) | ポリカーボネート樹脂組成物、その製造方法、成形体、及びその製造方法 | |
| WO2011071165A1 (ja) | ポリカーボネート樹脂組成物及び成形品 | |
| CN107849344B (zh) | 聚碳酸酯树脂复合物 | |
| JP7095358B2 (ja) | ポリカーボネート樹脂組成物及びその成形体 | |
| JP6950234B2 (ja) | ポリカーボネート樹脂組成物、その製造方法、成形体 | |
| CN107922720B (zh) | 聚碳酸酯树脂组合物、其制造方法、成形体 | |
| CN107709459B (zh) | 聚碳酸酯树脂组合物、其制造方法、成形体 | |
| CN107709458B (zh) | 聚碳酸酯树脂组合物、其制造方法、成形体 | |
| JP6693232B2 (ja) | ポリカーボネート樹脂組成物及び成形体 | |
| JP6642212B2 (ja) | ポリカーボネート樹脂組成物及び成形体 | |
| JP6693231B2 (ja) | ポリカーボネート樹脂組成物及び成形体 | |
| JP2018150480A (ja) | ポリカーボネート樹脂組成物及び成形体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160720 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160707 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20170519 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190402 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20191218 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200107 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200306 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200602 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20200803 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20201110 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20201207 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6807175 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |