JP2017081925A - サングリフェリン誘導体及びそれらの製造方法 - Google Patents
サングリフェリン誘導体及びそれらの製造方法 Download PDFInfo
- Publication number
- JP2017081925A JP2017081925A JP2016225660A JP2016225660A JP2017081925A JP 2017081925 A JP2017081925 A JP 2017081925A JP 2016225660 A JP2016225660 A JP 2016225660A JP 2016225660 A JP2016225660 A JP 2016225660A JP 2017081925 A JP2017081925 A JP 2017081925A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- formula
- pharmaceutically acceptable
- sanglifehrin
- acceptable salt
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
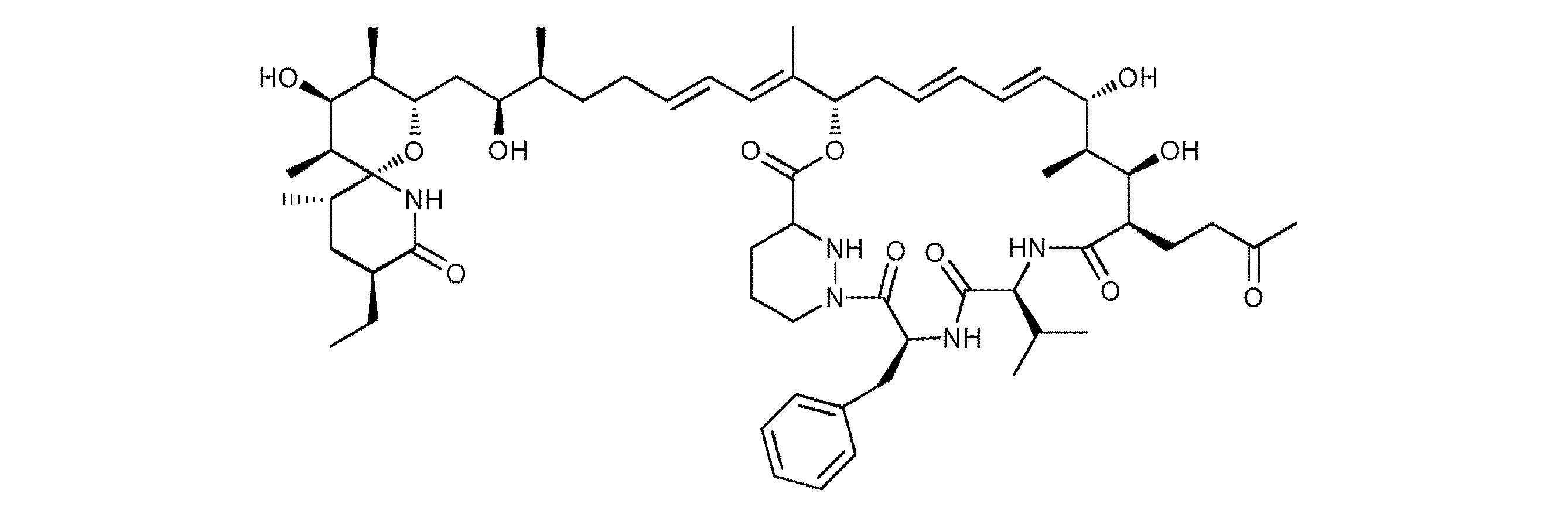
- 0 CC[C@@](C[C@@](C)[C@@]1(N2)O[C@@](CC[C@@]([C@@](C)CCC=CC=C(C)CCC=CN=C[C@@]([C@](C)[C@]([C@@](CCC(C)=*)C(N[C@@](C(C)C)C(N[C@@](Cc3ccccc3)C(N(CCC3)NC3C(OC)=O)=O)=O)=O)O)O)O)[C@](C)C=C1C)C2=O Chemical compound CC[C@@](C[C@@](C)[C@@]1(N2)O[C@@](CC[C@@]([C@@](C)CCC=CC=C(C)CCC=CN=C[C@@]([C@](C)[C@]([C@@](CCC(C)=*)C(N[C@@](C(C)C)C(N[C@@](Cc3ccccc3)C(N(CCC3)NC3C(OC)=O)=O)=O)=O)O)O)O)[C@](C)C=C1C)C2=O 0.000 description 3
- PAXWZBFQYGHPSW-CKGBYEKGSA-N CC(C)[C@@H](C(NC(C1)([C@@H]1c1ccccc1)C(N(CCC1)NC1C(O[C@@H](C/C=C/C=C/[C@@H]([C@H](C)[C@H]([C@H]1CCC(C)=O)O)O)/C(/C)=C/C=C/CC[C@H](C)[C@H](C[C@@H]([C@H](C)[C@H](C)[C@H](C)N2)O[C@@H](C)CC3(C(C)C3)C2=O)O)=O)O)=O)NC1=O Chemical compound CC(C)[C@@H](C(NC(C1)([C@@H]1c1ccccc1)C(N(CCC1)NC1C(O[C@@H](C/C=C/C=C/[C@@H]([C@H](C)[C@H]([C@H]1CCC(C)=O)O)O)/C(/C)=C/C=C/CC[C@H](C)[C@H](C[C@@H]([C@H](C)[C@H](C)[C@H](C)N2)O[C@@H](C)CC3(C(C)C3)C2=O)O)=O)O)=O)NC1=O PAXWZBFQYGHPSW-CKGBYEKGSA-N 0.000 description 1
- PHECAFXFVXYACH-KNXHTXMQSA-N CC[C@@H](C[C@H](C)[C@@]([C@H]1C)(N2)O[C@@H](C[C@@H]([C@@H](C)CC/C=C/C=C(\C)/[C@H](C/C=C/C=C/[C@@H]([C@H](C)[C@H]([C@@H](CCC(C)=O)C(N[C@@H](C(C)C)C(NC(C3)(C3c(cc3O)cc(F)c3F)C(N3NC4CCC3)=O)=O)=O)O)O)OC4=O)O)[C@H](C)[C@@H]1O)C2=O Chemical compound CC[C@@H](C[C@H](C)[C@@]([C@H]1C)(N2)O[C@@H](C[C@@H]([C@@H](C)CC/C=C/C=C(\C)/[C@H](C/C=C/C=C/[C@@H]([C@H](C)[C@H]([C@@H](CCC(C)=O)C(N[C@@H](C(C)C)C(NC(C3)(C3c(cc3O)cc(F)c3F)C(N3NC4CCC3)=O)=O)=O)O)O)OC4=O)O)[C@H](C)[C@@H]1O)C2=O PHECAFXFVXYACH-KNXHTXMQSA-N 0.000 description 1
- HILGPKUNFVJGKM-DQVCBYNKSA-N CC[C@@H](C[C@H](C)[C@@]1(N2)O[C@@H](C[C@@H]([C@@H](C)CC/C=C/C=C(\C)/[C@H](C/C=C/C=C/[C@@H]([C@H](C)[C@H]([C@@H](CCC(C)=O)C(N[C@@H](C(C)C)C(NC(C3)([C@@H]3c3cc(O)c(C)cc3)C(N3NC4CCC3)=O)O)=O)O)O)OC4=O)O)[C@H](C)C=C1C)C2=O Chemical compound CC[C@@H](C[C@H](C)[C@@]1(N2)O[C@@H](C[C@@H]([C@@H](C)CC/C=C/C=C(\C)/[C@H](C/C=C/C=C/[C@@H]([C@H](C)[C@H]([C@@H](CCC(C)=O)C(N[C@@H](C(C)C)C(NC(C3)([C@@H]3c3cc(O)c(C)cc3)C(N3NC4CCC3)=O)O)=O)O)O)OC4=O)O)[C@H](C)C=C1C)C2=O HILGPKUNFVJGKM-DQVCBYNKSA-N 0.000 description 1
Images








Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/18—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing at least two hetero rings condensed among themselves or condensed with a common carbocyclic ring system, e.g. rifamycin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/5025—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Engineering & Computer Science (AREA)
- General Engineering & Computer Science (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- Genetics & Genomics (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Abstract
Description
本発明は、例えば、ウイルス感染症、特に、C型肝炎ウイルス(HCV)及びHIVなどのRNAウ
イルスによる感染症の治療におけるシクロフィリン阻害剤として、並びに/又は、例えば
、移植拒絶反応の予防において使用するための免疫抑制剤として、並びに、例えば、炎症
性障害において使用するための抗炎症剤として有用である、サングリフェリンアナログに
関する。本発明は、医薬、特にHCV感染症を治療するための医薬、及び、免疫抑制剤若し
くは抗炎症剤として使用するための医薬における、それらの使用方法、又は、医薬として
有用なさらなる化合物の生成における中間体としてのそれらの使用方法も提供する。
(C型肝炎)
C型肝炎ウイルス(HCV)はプラス鎖のRNAウイルスであり、感染は輸血後肝炎の主な原因
である。HCVは最もよく見られる慢性の血液由来の感染であり、米国における肝臓疾患に
よる死亡の主な原因である。世界保健機関は、1億7千万人を超えるHCV感染の慢性キャリ
アがいると推定しており、これは世界の人口の約3%である。治療していないHCV感染患者
のうち、約70%〜85%は慢性HCV感染を起こし、そのため肝硬変及び肝細胞癌を起こすリス
クが高い。先進国では、肝臓癌の全症例の50〜76%及び全肝臓移植の3分の2は慢性HCV感染
によるものである(Mannsら、2007)。
への伝染の源となる。HCV感染は、関節痛(関節の疼痛)、皮膚の発疹、及び主として腎臓
に対する内臓損傷などの、非肝臓合併症を起こす。HCV感染は重大な世界的な医療の負担
であり、現在C型肝炎に利用できるワクチンはない(Straderら、2004; Jacobsonら、2007;
Mannsら、2007 Pawlotsky、2005; Zeuzem及びHermann、2002)。
現在の標準治療(SoC)は、24〜48週間のペグ化インターフェロンα(pIFNα)の皮下注
射及び抗ウイルス薬リバビリンの経口投薬である。治療の成功は持続性ウイルス陰性化(S
VR)により定義されるが、これは治療期間の最後及び6ヶ月後に血清中にHCV RNAが存在し
ないことにより定義される。SoCに対する全奏効率は、主に遺伝子型及び治療前HCV RNAレ
ベルによる。遺伝子型2及び3の患者は、遺伝子型1に感染している患者よりもSoCに対して
、より反応しそうである(Melnikova, 2008; Jacobsonら、2007)。
とができず、フルコースの完了に関する頻発する問題につながる。SoCの全臨床SVR率(ove
rall clinical SVR rate)はわずか50%ほどである(Melnikova, 2008)。耐性の発生は、治
療の失敗の潜在的なもう1つの因子である(Jacobsonら、2007)。SoCは、うつ病の過去の顕
著なエピソード又は心臓病を持つ患者など、治療の候補と考えられないいくらかの患者に
おいても禁忌である。治療の中断につながることの多いSoCの副作用には、インフルエン
ザ様疾病、発熱、疲労、血液疾患、貧血、白血球減少(leucopaenia)、血小板減少(thr
ombocytopaenia)、脱毛症、及びうつ病がある(Mannsら、2007)。
慮すると、より安全で効き目のある新しい治療が、HCV感染の治療に切実に求められてい
る。新しい治療の目的は、向上した効力、向上した毒性プロファイル、向上した耐性プロ
ファイル、向上した生活の質、及びその結果得られる患者のコンプライアンスの向上を含
む。HCVは生活環が短く、そのため、薬物療法の間の薬物耐性の発生はよく見られる。
としても知られている)が開発されつつあり、これはウイルスRNAポリメラーゼNS5B又は
ウイルスプロテアーゼNS3などのウイルスタンパク質を標的とするものである(Jacobsonら
、2007; Parfieniukら、2007)。さらに、ウイルスの標的でなくヒトのタンパク質(例え
ば、シクロフィリン)を標的とする新規の化合物も開発されつつあり、薬物療法の間の耐
性の発生率低減につながると期待されている(Mannsら、2007; Pockros, 2008; Pawlotsky
J-M, 2005)。
シクロフィリン(CyP)は、タンパク質のコンフォメーション変化と折りたたみを容易に
するペプチジル-プロリルシス-トランスイソメラーゼ活性を示す細胞タンパク質のファミ
リーである。CyPは、転写調節、免疫応答、タンパク質分泌、及びミトコンドリア機能な
どの細胞プロセスに関与している。HCVウイルスは、ヒトの感染の間その生活環のためにC
yPをリクルートする。元々は、CyPが、RNA複製を促進するHCV非構造的タンパク質NS5B RN
AポリメラーゼのRNA結合活性を刺激すると考えられていたが、CyP PPIアーゼ活性の要求
を含む、代わりの仮説もいくつか提案されてきた。A及びBを含むCyPの種々のアイソフォ
ームが、HCVの生活環に関与していると考えられている(Yangら、2008; Appelら、2006; C
hatterjiら、2009; Gaitherら、2010)。マウス(Colganら、2000)及びヒトT細胞(Braaten
及びLuban、2001)においてノックアウトを発生させる能力は、CyPAが細胞の成長及び生存
にとって任意であることを示している。類似の結果が、細菌(Herrlerら、1994)、ニュー
ロスポラ(Neurospora)(Tropschugら、1989)、及びサッカロマイセス・セレビシエ(Saccha
romyces cerevisiae)(Dolinskiら、1997)におけるCyPAホモログの分裂により観察された
。したがって、CyPの阻害は、HCV感染を治療するための新規で魅力的な宿主標的であり、
SVRを高め、耐性の出現を防止し、治療の副作用を低減する目的で、現行のSoC又はSTAT-C
/DAA薬に対する新しい潜在的な追加物である。
抑制臨床アナログDEBIO-025(Paeshuyseら、2006; Flisiakら、2008)、NIM811(Mathyら、2
008)、及びSCY-635(Hopkinsら、2009)はシクロフィリンに結合することが知られており、
シクロフィリン阻害剤としてHCV感染の治療におけるインビトロ及び臨床的な効能を示し
た(Crabbeら、2009; Flisiakら、2008; Mathyら、2008; Inoueら、2007; Ishiiら、2006;
Paeshuyseら、2006)。早期のCsAに関する耐性研究はHCV NS5B RNAポリメラーゼにおける
変異を示し、シクロフィリンBのみがHCV複製プロセスに関与するだろうということを示唆
したが(Robidaら、2007)、最近の研究はHCV複製におけるシクロフィリンAの欠くことので
きない役割を示唆した(Chatterjiら、2009; Yangら、2008)。NS5Aウイルスタンパク質に
おける変異がCsA耐性にも関連し、NS5AがCyPA及びCypBの両方とそれらの特異的ペプチジ
ル-プロリルシス/トランスイソメラーゼ(PPIアーゼ)活性に関して相互作用することを考
慮すると、ウイルスの生活環における両シクロフィリンの役割がさらに示唆される(Hanou
lleら、2009)。
ューリンに依存する。これは、HCV活性に必須な要件がCyP結合であり、カルシニューリン
結合は必要でないことを示した。DEBIO-025は、HCV治療用の臨床的に最も進んだシクロフ
ィリン阻害剤であり、最も優勢な4種のHCV遺伝子型(遺伝子型1、2、3、及び4)に対して
インビトロ及びインビボの効力を示した。耐性研究は、DEBIO-025に対する耐性を与える
変異がポリメラーゼ及びプロテアーゼの阻害剤に関して報告されていたものとは異なり、
STAT-C/DAA耐性ウイルスレプリコンとは交叉耐性が全くないことを示した。より重要であ
るが、DEBIO-025は、プロテアーゼ及びポリメラーゼの両方の阻害剤に対する耐性を与え
るエスケープ変異の発生も防止した(Crabbeら、2009)。
関連すると思われるいくつかの問題があり、例えば、療法の中止につながることがあり臨
床の投与レベルを限定してきた特定の有害事象;可変的な効能を生み出し得る可変的な薬
物動態;及び投薬問題を生み出し得る薬物-薬物相互作用の増大したリスクがある。
疲労、及び発熱があった。臨床的に最も重要なAEは、高ビリルビン血症及び血小板数の減
少(血小板減少)であった。ペグ-IFNは重度の血小板減少を起こすことがあり、DEBIO-025
と組み合わせれば重大な臨床上の問題となるだろう。ビリルビンの増加及び血小板の減少
は両方とも、NIM-811による早期の臨床試験において記載されてきた(Keら、2009)。DEBIO
-025臨床試験の間に観察された高ビリルビン血症は治療の中止の後で逆転したが、それは
16人の患者のうち4人の治療中断の原因であり、将来の試験のための投与レベルの低下の
原因であった。HCVにおけるシクロフィリン阻害剤の抗ウイルス効果は投与量に関連する
ので、投与量の低下は抗ウイルス効果の低下につながり、単独療法として投与された場合
、CsA系シクロフィリン阻害剤の後のいくつかの試験は、HCVウイルス量の低下が全くない
か、又はあまり低下しないことを示した(Lawitzら、2009; Hopkinsら、2009; Nelsonら、
2009)。DEBIO-025及びシクロスポリンAは、胆汁酸塩輸送ポンプなどの胆汁トランスポー
ター及び他の肝トランスポーター(特にMRP2/cMOAT/ABCC2)の阻害剤であることが知られて
いる(Crabbeら、2009)。胆汁トランスポーター、特にMRP2との相互作用が、DEBIO-025の
高投与レベルで見られる高ビリルビン血症の原因であり得ることが示唆された(Nelsonら
、2009、Wringら、2010)。OAT1B1及びOAT1B3(Konigら、2010)などの他の薬物トランスポ
ーターの阻害を介するCsAクラス関連の薬物-薬物相互作用(DDI)も1つの懸念となる可能
性があり、特定の組み合わせ、及びHIVなどの共感染の治療を受けるいくらかの患者にお
ける使用を潜在的に制限している(Sedenら、2010)。
基質であり、ヒトP糖タンパク質(MDR1)の基質及び阻害剤であることが知られている(Crab
beら、2009)。シクロスポリンAは、インビトロでCYP3A4の阻害剤であることも示された(N
iwaら、2007)。これは、例えば、ケトコナゾール、シメチジン、及びリファンピシンなど
、CYP3A4の基質、誘導剤、又は阻害剤である他の薬物との薬物-薬物相互作用の増大した
リスクがあり得ることを示している。さらに、P糖タンパク質による輸送を受けやすい薬
物(例えば、ジゴキシン)との相互作用も予想され、これは付随する他の疾病の治療を受
けているHCV患者に重症の薬物-薬物相互作用を起こし得る(Crabbeら、2009)。CsAは非常
に変わりやすい薬物動態を持つことも知られており、初期の製剤は1〜89%の経口バイオ
アベイラビリティを示した(Kapurtzakら、2004)。費用のかかる患者の血中濃度のモニタ
リングがないと、これは、血漿中濃度上昇による副作用の出現率の上昇をもたらすことが
あり、或いは血漿中濃度低下による臨床反応の低下をもたらすことがある。
、HCV感染に対する併用療法において使用するための、より強力で安全なCyP阻害剤を発見
し、かつ開発する必要がある。
サングリフェリンA(SfA)及びその天然の同族体は、ストレプトマイセス属(Streptomyce
s) sp.A92-308110(DSM 9954としても知られる)(国際公開第97/02285号参照)により産生さ
れる混合非リボソームペプチド/ポリケチドのクラスに属し、シクロフィリンA(CyPA)に対
するそれらの高い親和性に基づいて元々発見された。SfAは、発酵ブロス中に最も豊富に
ある成分であり、CsAに比べてCyPAに対しておよそ20倍高い親和性を示す。これは、サン
グリフェリンがHCVの治療に有用になり得るという示唆をもたらした(国際公開第2006/138
507号)。サングリフェリンは、インビトロで試験される場合、CsAよりも低い免疫抑制活
性を示すことも示された(Sanglierら、1999; Fehrら、1999)。SfAは、CyPAのCsA結合部位
に高い親和性で結合する(Kallenら、2005)。
サングリフェリンは、混合型ポリケチドシンターゼ(PKS)/非リボソームペプチドシンテ
ターゼ(NRPS)によって生合成される(国際公開第2010/034243号、Quら、2011参照)。その2
2員のマクロライド骨格は、ポリケチド炭素鎖及びトリペプチド鎖からなる。該ペプチド
鎖は、アミド結合によって連結された、1つの天然アミノ酸、バリン、並びに、2つの非
天然アミノ酸、(S)-メタ-チロシン及び(S)-ピペラジン酸からなる。(S)-メタ-チロシンを
生成するフェニルアラニンのヒドロキシル化は(NRPS上でin situで、又は、生合成前に
)、sfaAの遺伝子産物を介して起こると考えられている。
SfAの免疫抑制作用機序は、CsA、FK506、及びラパマイシンなどの他の公知のイムノフ
ィリン-結合性免疫抑制薬のものとは異なる。SfAは、CsAの標的であるカルシニューリン
のホスファターゼ活性を阻害せず(Zenkeら、2001)、その代わりに、その免疫抑制活性は
、インターロイキン-6の阻害(Hartelら、2005)、インターロイキン-12の阻害(Steinschul
teら、2003)、及びインターロイキン-2-依存性T細胞増殖の阻害(Zhang及びLiu、2001)に
よるものとされてきた。しかし、SfAがその免疫抑制効果を発揮する分子標的及び機構は
今のところ未知である。
より媒介されていると考えられている。実際、SfAの酸化開裂から誘導されたマクロ環化
合物(ヒドロキシマクロ環)はCyPAに対して強い親和性を示した(Sedraniら、2003)。X線結
晶構造データは、ヒドロキシマクロ環がCsAと同じCyPAの活性部位に結合することを示し
た。SfAのマクロ環部分に基づくアナログは免疫抑制性が欠けていることも先に示されて
おり(Sedraniら、2003)、HCV療法における潜在的な使用のための非免疫抑制CyP阻害剤の
設計の機会を与える。
症性障害、及び、呼吸器疾患の予防のような分野において使用するための、低毒性の免疫
抑制薬を開発する機会もある:クローン病、ベーチェット病、ブドウ膜炎、乾癬、アトピ
ー性皮膚炎、関節リュウマチ、腎炎症候群、再生不良性貧血、胆汁性肝硬変、喘息、肺線
維症、慢性閉塞性肺疾患(COPD)、及び、セリアック病。サングリフェリンは、免疫抑制活
性の新規の機構を有し (Zenkeら、2001)、樹状細胞ケモカインを介して作用する可能性が
あることが示されており(Immeckeら、2011)、したがって、シクロスポリンA、ラパマイシ
ン、及びFK506などの現在の臨床薬とは異なる作用機序を有する薬剤を開発する機会があ
る。
(ヒト免疫不全ウイルス(HIV))
CsA及びDEBIO-025などのシクロフィリン阻害剤は、HIV複製の阻害にも潜在的な効用を
示した。シクロフィリン阻害剤は、HIV逆転写の進行/完了の間のCyPAの機能と干渉すると
考えられている(Ptakら、2008)。しかし、臨床試験では、DEBIO-025は、それぞれ9名及び
2名の患者でHIV-1 RNAレベルを≧0.5及び>1 log10 コピー/mLに低減しただけであり、治
療された患者のうち27名はHIV-1 RNAレベルの低下を全く示さなかった(Steynら、2006)。
この後で、DEBIO-025はHCV/HIV共感染患者で試験され、HCVに対してより良い効能を示し
、HIV臨床試験は中止された(Watashiら、2010参照)。
世界中で3000万人を超える人々がHIV-1に感染しており、毎年300万の新しい症例がある
。治療選択は、高活性抗レトロウイルス療法(HAART)の導入と共に劇的に向上した(Schopm
anら、2010)。2008年までに、およそ25種の抗レトロウイルス薬がHIV-1の治療用に認可さ
れ、9種のヌクレオシド逆転写酵素阻害剤(NRTI)、4種の非-ヌクレオシド逆転写酵素阻害
剤(NNRTI)、9種のプロテアーゼ阻害剤(PI)、1種の融合阻害剤、1種のCCR5阻害剤、及び1
種のインテグラーゼ阻害剤(Shafer及びSchapiro、2008)が含まれる。しかし、これらの現
行のレジメンはいずれも、完全なウイルス排除はもたらさず、それらは重症の副作用をも
たらすことがあり、抗ウイルス耐性も未だ大きな懸念である。したがって、特に、シクロ
フィリン阻害剤の場合のように、認可された薬物がない作用機序クラスにおいて、新しい
抗ウイルス療法の必要が未だ存在する。
B型肝炎はヘパドナウイルス科のDNAウイルスであり、B型肝炎の原因因子である。HCV及
びHIVの場合とは異なり、B型肝炎ウイルスに対するシクロフィリン阻害剤の活性の発表さ
れた記述は非常に少なかった。Ptakら(2008)は、HBVに対するDebio-025の弱い活性(IC50
は4.1μM)を記載したが、Xieら(2007)はHBVに対するCsAのいくらかの活性(IC50>1.3μg/
mL)を記載した。これはHIV及びHCVとは対照的であり、HIV及びHCVではシクロフィリン阻
害剤のナノモーラーの抗ウイルス活性の報告が多数ある。
HBVは世界中で最大4億人を感染させており、ウイルス性慢性肝炎及び肝細胞癌の主な原
因である。2008年現在、HBVの治療に認可された薬物が6種ある;インターフェロンアルフ
ァ及びペグ化インターフェロンアルファ、3種のヌクレオシドアナログ(ラミブジン、エン
テカビル、及びテルビブジン)、及び1種のヌクレオチドアナログ(アデホビルジピボキシ
ル)。しかし、耐性の率の高さ、許容性の低さ、及び起こり得る副作用のため、新しい治
療選択が必要とされている(Ferirら、2008)。
ミトコンドリアの高コンダクタンス透過性遷移孔(high conductance permeability tra
nsition pores)の開口は、ミトコンドリア透過性遷移(MPT)のオンセットを開始する。こ
れは原因となる事象であり、酸化ストレス、Ca2+毒性、及び虚血/再潅流後の肝細胞にお
ける壊死及びアポトーシスをもたらす。シクロフィリン阻害剤によるシクロフィリンD(シ
クロフィリンFとしても知られる)の阻害は、透過性遷移孔の開口を遮断することが示され
ており、これらのストレスの後の細胞死を保護する。したがって、シクロフィリンD阻害
剤は、筋ジストロフィー、特にウールリッヒ型先天性筋ジストロフィー及びベスレムミオ
パチー(Millayら、2008、国際公開第2008/084368号、Palmaら、2009)、多発性硬化症(For
teら、2009)、糖尿病(Fujimotoら、2010)、筋萎縮性側索硬化症(Martin 2009)、双極性障
害(Kubotaら、2010)、アルツハイマー病(Du及びYan、2010)、ハンチントン病(Perryら、2
010)、心筋梗塞後の回復(Gomezら、2007)、及び慢性のアルコール消費(Kingら、2010)な
ど、mPTP開口が関与するとされてきた適応症において有用になり得る。
シクロフィリン阻害剤は、水痘帯状疱疹ウイルス(Ptakら、2008)、A型インフルエンザ
ウイルス(Liuら、2009)、重症急性呼吸器症候群コロナウイルス並びに他のヒト及びネコ
コロナウイルス(Chenら、2005、Ptakら、2008)、デングウイルス(Kaulら、2009)、黄熱病
ウイルス(Qingら、2009)、西ナイルウイルス(Qingら、2009)、西部馬脳炎ウイルス(Qing
ら、2009)、サイトメガロウイルス(Kawasakiら、2007)、及びワクシニアウイルス(Castro
ら、2003)などの他のウイルスに対して潜在的な活性を有し、そのためこれらのウイルス
感染の治療において潜在的な活性を有する。
癌など、他の治療分野におけるシクロフィリン阻害剤及びシクロフィリン阻害の効用に
ついても報告がある(Hanら、2009)。
サングリフェリンなどの化合物の薬物開発における問題の1つは、経口バイオアベイラ
ビリティの低下をもたらす、急速な代謝及びグルクロン酸抱合である。これが、食物によ
る影響の可能性を高め、剤形からの不完全放出をより頻繁にし、患者間のばらつきを大き
くする可能性がある。
ストロフィー、若しくは心筋梗塞後の回復の促進などの、シクロフィリンの阻害が有用に
なり得る他の疾病分野、又は、免疫抑制が有用である他の疾病分野の治療においても効用
を持ち得る、新規のシクロフィリン阻害剤及び抗炎症薬を特定する必要が未だ存在する。
好ましくは、そのようなシクロフィリン阻害剤は、下記の性質の1つ以上を含む、現在利
用可能なシクロフィリン阻害剤よりも向上した性質を有する:おそらくはP450代謝の低減
及び/又はグルクロン酸抱合の低減による、より長い半減期若しくは増大した経口バイオ
アベイラビリティ、向上した水への溶解度、HCVに対する向上した効力、低減された毒性(
肝毒性を含む)、標的臓器(例えば、HCVの場合は肝臓)に対する高い曝露及び/若しくは長
い半減期(投薬頻度の減少を可能にする)などの向上した薬理プロファイル、低減されたレ
ベルのCYP3A4の代謝及び阻害並びに低減された(Pgp)阻害(より容易な多剤組み合わせを
可能にする)によるなどの低減された薬物-薬物相互作用、高ビリルビン血症の機会低下
をもたらす、MRP2への低い結合などの、向上した副作用プロファイル、より低い免疫抑制
効果、耐性ウイルス種、特にCsA及びCsAアナログ(例えば、DEBIO-025)耐性ウイルス種に
対する向上した活性、及びより高い治療(及び/又は選択性)指数。本発明は、上記の性
質の1つ以上を有し得る新規のサングリフェリンアナログを開示する。特に、本発明は、
新規の変異合成(mutasynthetic)サングリフェリンアナログを開示し、該サングリフェ
リンアナログは、少なくともいくつかの実施態様において、例えば、増大したミクロソー
ム半減期及び/又はHCVに対する向上した効力によって示されるように、例えば、低いレ
プリコンEC50及び/又は増大した選択性指数によって示されるように、P450による代謝又
はグルクロン酸抱合が低減されている。
治療において効用を有し得る、新規の免疫抑制剤を開発する必要もある。好ましくは、そ
のような免疫抑制剤は、公知の天然のサングリフェリンよりも向上した性質を有し、該性
質には以下の1以上が含まれる:おそらくはP450代謝の低減及び/又はグルクロン酸抱合
の低減による、より長い半減期若しくは増大した経口バイオアベイラビリティ、向上した
水への溶解度、T細胞増殖アッセイで見られるような免疫抑制活性の向上した効力、低減
された毒性(肝毒性を含む)、標的臓器に対する高い曝露及び/若しくは長い半減期(投薬頻
度の減少を可能にする)などの向上した薬理プロファイル、低減されたレベルのCYP3A4の
代謝及び阻害並びに低減された(Pgp)阻害(より容易な多剤組み合わせを可能にする)に
よるなどの低減された薬物-薬物相互作用、並びに向上した副作用プロファイル。本発明
は、上記の性質の1つ以上を有し得る、新規のサングリフェリンアナログを開示する。特
に、本発明は、少なくともいくつかの実施態様において、例えば、増大したミクロソーム
半減期によって示されるように、P450による代謝又はグルクロン酸抱合が低減されており
、例えば、低いT細胞増殖IC50によって示されるように、向上した免疫抑制効力を有し得
る、新規のサングリフェリンアナログを開示する。
ナログを提供する。これらのアナログは、ストレプトマイセス属sp.A92-308110 (DSM 995
4としても知られている)などのサングリフェリン産生生物に、メタ-チロシンのアナログ
を供給することによって、或いは、より優先的には、sfaA又はsfaAのホモログが不活化さ
れているか又は欠失している、サングリフェリン産生生物の遺伝子改変誘導体にメタ-チ
ロシンアナログを供給することによって、生成することができる。この結果、本発明は、
変異合成サングリフェリンアナログ、これらの化合物の製造方法、及び、医薬におけるこ
れらの化合物の使用方法、又は、さらなる化合物の製造における中間体としての、これら
の化合物の使用方法を提供する。
サングリフェリンアナログ及びそれらの誘導体、又はそれらの医薬として許容し得る塩:
R1、R2、R3、R4及びR5は、独立に、H、F、Cl、Br、C2-6アルケニル又はC1-10アルキル
を表し、ここで、前記アルキル基の1以上の炭素原子は、O、N及びS(O)p(pは0、1又は2
を表す。)から選択されるヘテロ原子によって任意に置き換えられており、かつここで、
前記アルキル基の1以上の炭素原子は、カルボニルによって任意に置き換えられており、
かつ該アルキル基は、1以上のハロゲン原子によって任意に置換されていてもよく;
X1、X2、X3、X4及びX5は、独立に、C又はNを表し、かつ、これらの基のうちNを表すも
のについては、結合される置換基が存在せず;
ただし、R1、R3、R4及びR5が全てHを表し、かつX1、X2、X3、X4及びX5が全てCを表す場
合、R2はOHを表すことができない。)
を、それらの任意の互変異性体、又はトランスとして示されているC26、27位のC=C(サン
グリフェリンAの構造を参照)のC=C結合がシスである、それらの異性体を含めて、並びに
、
C-53ケト及びC-15ヒドロキシル基及びメタノールの組み合わせによりケタールが形成さ
れている、それらのメタノール付加物を含めて、提供する。
の互変異性体を包含し、例えば、エノール化合物が図示されている場合には、ケト化合物
を包含しており、その逆もまた同様である。
式(I)の定義に含められる具体的な互変異性体は、(i) C-53ケト基がC-15ヒドロキシル
とヘミケタールを形成するもの、又は(ii)C-15及びC-17ヒドロキシルがC-53ケトと結合し
てケタールを形成できるもの、である。全ての番号付けは、親サングリフェリンA構造の
体系を使用する。
冠詞「1つの(a)」及び「1つの(an)」は、1つ又は2つ以上(すなわち、少なくとも1つ
)の該冠詞の文法的な対象を意味するように本願明細書において使用される。例としては
、「1つのアナログ」は、1つのアナログ又は2つ以上のアナログを意味する。
本願明細書では、用語「アナログ(複数可)」は、互いに構造的に類似しているが組成
がわずかに異なる(1つの原子を他の原子で置き換える、又は特定の官能基の有無など)
化合物を意味する。
本願明細書では、用語「炎症性障害」は、炎症により引き起こされる障害のリストを意
味し、限定されないが、以下のものを含む:尋常性ざ瘡、アテローム性動脈硬化症、喘息
、自己免疫疾患(Automimmune disease)(急性散在性脳脊髄炎(ADEM)、アジソン病、円
形脱毛症、強直性脊椎炎、抗リン脂質抗体症候群(APS)、自己免疫性溶血性貧血、自己免
疫性肝炎、自己免疫性内耳疾患、水疱性類天疱瘡、セリアック病、シャーガス病、慢性閉
塞性肺疾患(COPD)、クローン病、皮膚筋炎、1型糖尿病、子宮内膜症、グッドパスチャー
症候群、グレーブス病、ギラン・バレー症候群、橋本病、化膿性汗腺炎、川崎病、IgA腎
症、特発性血小板減少性紫斑病、間質性膀胱炎、エリテマトーデス、混合性結合組織病、
モルフェア、多発性硬化症(MS)、重症筋無力症、ナルコレプシー、神経性筋強直症、尋常
性天疱瘡、悪性貧血、乾癬、乾癬性関節炎、多発性筋炎、原発性胆汁性肝硬変、関節リュ
ウマチ、統合失調症、強皮症、シェーグレン症候群、全身硬直症候群、側頭動脈炎、潰瘍
性大腸炎、血管炎、白斑、ウェゲナー肉芽腫症など)、炎症性腸疾患、骨盤内炎症性疾患
、関節リュウマチ、及び移植拒絶反応。
的に類似しているが組成がわずかに異なる(1つの原子を他の原子で置き換える、又は特
定の官能基の有無など)化合物、特にストレプトマイセス属sp.A92-308110の発酵により
生成したものを意味する。例には、サングリフェリンBなど、国際公開第97/02285号及び
国際公開第98/07743号で議論されているサングリフェリン様化合物がある。
フェリンアナログ(複数可)」は、サングリフェリンA、B、C又はDに構造的に類似している
が組成がわずかに異なる(1つ以上の原子を他の原子で置き換える、又は特定の官能基の
有無など)化合物、特に、培養物にメタ-チロシンアナログを供給する、ストレプトマイ
セス属sp.A92-308110又はその変異体の発酵により生成したものを意味する。
に類似しているが組成がわずかに異なる(1つ以上の原子を他の原子で置き換える、又は
特定の官能基の有無など)化合物、特に、式(III)に記載のものを意味する。
エンベロープウイルスを意味する。
本願明細書では、用語「HIV」は、ヒト免疫不全ウイルス、ヒト後天性免疫不全症候群
の原因因子を意味する。
吸収されるか、又は生物活性部位で利用可能になる程度又は速度を意味する。この性質は
、化合物の溶解度、消化管中の吸収速度、タンパク結合及び代謝の程度などを含むいくつ
かの因子に依存する。当業者によく知られているであろうバイオアベイラビリティの種々
の試験が本願明細書に記載される(Egorinら、2002も参照されたい)。
塩水(PBS)又は5%グルコース溶液への溶解度を意味する。水への溶解度の試験は、以下に
、「水への溶解度アッセイ」として実施例に与えられている。
得る無機又は有機の酸又は塩基から形成される従来の塩並びに四級アンモニウム酸付加塩
を含む。好適な酸性塩のより具体的な例には、塩化水素酸、臭化水素酸、硫酸、リン酸、
硝酸、過塩素酸、フマル酸、酢酸、プロピオン酸、コハク酸、グリコール酸、ギ酸、乳酸
、マレイン酸、酒石酸、クエン酸、パルモン酸(palmoic)、マロン酸、ヒドロキシマレイ
ン酸、フェニル酢酸、グルタミン酸、安息香酸、サリチル酸、フマル酸、トルエンスルホ
ン酸、メタンスルホン酸、ナフタレン-2-スルホン酸、ベンゼンスルホン酸、ヒドロキシ
ナフトエ酸、ヨウ化水素酸、リンゴ酸、ステロン酸(steroic)、タンニン酸の塩などがあ
る。塩酸塩は特に興味深い。シュウ酸などの他の酸は、それ自体医薬として許容し得るも
のではないが、本発明の化合物及びその医薬として許容し得る塩を得る際に中間体として
有用な塩の製造に有用になり得る。好適な塩基性塩のより具体的な例には、ナトリウム、
リチウム、カリウム、マグネシウム、アルミニウム、カルシウム、亜鉛、N,N'-ジベンジ
ルエチレンジアミン、クロロプロカイン、コリン、ジエタノールアミン、エチレンジアミ
ン、N-メチルグルカミン及びプロカインの塩がある。以下での本発明の化合物への言及は
、式(I)の化合物とそれらの医薬として許容し得る塩の両方を含む。
ルキルは、C1-6アルキル、例えばC1-4アルキルである。
「アルケニル」は、2個以上の炭素を含むアルキル基であって、1個以上の二重結合を
有し、不飽和であるものを意味する。
C1-4アルキル基がある。アルケニル基の例には、-CH=CH2及び-CH2CH=CH2などのC2-4アル
ケニル基がある。
用語「治療」には、予防的処置並びに治療的処置が含まれる。
本発明は、先に説明した変異合成サングリフェリンアナログ、これらの化合物の製造方
法、及び医薬におけるこれらの化合物の使用方法を提供する。
一実施態様において、該化合物は、C-53ケト及びC-15ヒドロキシル基及びメタノールの
組み合わせによりケタールが形成されている、それらのメタノール付加物である。別の実
施態様において、それはメタノール付加物ではない。
0アルキル基(例えばC1-6アルキル基)の炭素原子は、ヘテロ原子によって置き換えられて
いる。
-CH3がNによって置き換えられている場合、形成される基は-NH2である。-CH2-がNによ
って置き換えられている場合、形成される基は-NH-である。-CHR-がNによって置き換えら
れている場合、形成される基は-NR-である。したがって、R1、R2、R3、R4及びR5内の窒素
原子は第一級、第二級又は第三級の窒素原子であることができる。
-CH3がOによって置き換えられている場合、形成される基は-OHである。
ル基)の炭素原子がヘテロ原子によって置き換えられている場合、それは好適には、O、S
又はN、特にN又はO、とりわけOによって置き換えられている。
R1、R2、R3、R4及びR5のいずれか1つが、基S(O)pを含む場合、変数pは、好適には0又
は1を表す。一実施態様において、pは0を表す。別の実施態様において、pは1を表す。別
の実施態様において、pは2を表す。
キル基)が2個以上のヘテロ原子を含む場合、これらは典型的には、2個以上の炭素原子
によって隔てられるべきである。
好適には、R1、R2、R3、R4及びR5の1以上が表すことができるC1-10アルキル基(例えば
、C1-6アルキル基)の炭素原子は、ヘテロ原子によって置き換えられていないか、そうで
なければ、OH又はNH2を表す。
キル基)の炭素原子がカルボニルによって置き換えられている場合、該カルボニルは、好
適には別の炭素原子又は窒素原子に隣接して配置される。好適には、カルボニル基は硫黄
原子又は酸素原子に隣接して配置されない。
例えば、R1、R2、R3、R4及びR5の1以上が、-COC1-3アルキル、例えば、-COMeを表すこ
とができる。
C1-6アルキル)基の炭素原子は、カルボニルによって置き換えられていない。
R1、R2、R3、R4及びR5の1以上が表すことができるC1-10アルキル基(例えば、C1-6アル
キル基)は、1以上のハロゲン原子によって置換されることができる。例えば、R1、R2、R
3、R4及びR5の1以上が、-CF3を表すことができる。或いは、1以上のR1、R2、R3、R4及
びR5が、1以上の(例えば、1)Cl又はF原子によって置換されたC1-10アルキル(例えば、C
1-6アルキル)を表すことができる(例えば、-CH2CH2Cl)。
ロゲンによって置換されていない。
1以上のR1、R2、R3、R4及びR5基(複数可)がC1-10アルキル基(例えば、C1-6アルキル基
)を表す場合、好適には、該基(複数可)はC1-4アルキル(例えば、メチルなどのC1-2アルキ
ル)を表す。
一実施態様において、R1、R2、R3、R4及びR5の1以上は、C1-6アルキル(C1-2アルキル
など)又はC2-3アルケニルを表し、例えば、R1、R2、R3、R4及びR5の1以上はメチルを表
す。
好適には、R1は、H又はF、特にHを表す。
好適には、R2は、H、F、Cl、CF3、OH、NH2又はC1-6アルキル(例えば、メチル)を表す。
より好適には、R2は、H、F、OH又はNH2、特にOHを表す。
好適には、R3は、H、F、Cl、CF3、OH又はC1-6アルキル(例えば、メチル)を表す。より
好適には、R3は、H、Me又はFを表す。R3は、エチルを表すこともできる。
好適には、R4は、H又はFを表す。
好適には、R5は、H、F、Cl、CF3、OH又はC1-6アルキル(例えば、メチル)を表す。より
好適には、R5は、H又はFを表す。
好適には、R1、R2、R3、R4又はR5の1以上、より好適には、2以上(例えば、3以上)は
、Hを表さない。
好適には、R3若しくはR4又はR3及びR4は、Fを表す。別の実施態様において、R1及びR3
は、Fを表す。
一実施態様において、X1はNを表す(したがって、R1は存在しない)。別のより好ましい
実施態様において、X1はCを表す。
好適には、X3はCを表す。
好適には、X4はCを表す。
好適には、X5はCを表す。
前記化合物は、式(II)の化合物である。
下記の構造によって表されるように、本発明の好適な実施態様において、R1はHを表し
、R2はOHを表し、R3はHを表し、R4はFを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はFを表し、R4はFを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はFを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はMeを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はHを表し、R3はHを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5は
Cを表す:
、R2はNH2を表し、R3はHを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はFを表し、R3はHを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5は
Cを表す:
、R2はOHを表し、R3はHを表し、R4はFを表し、R5はHを表し、X1はNを表し、かつX2、X3、
X4及びX5はCを表し:
、R2はOHを表し、R3はHを表し、R4はFを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はFを表し、R4はFを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はFを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はMeを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はHを表し、R3はHを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5は
Cを表す:
、R2はNH2を表し、R3はHを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はFを表し、R3はHを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5は
Cを表す:
、R2はOHを表し、R3はEtを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はFを表し、R4はHを表し、R5はHを表し、かつX1、X2、X3、X4及びX5
はCを表す:
、R2はOHを表し、R3はHを表し、R4はFを表し、R5はHを表し、X1はNを表し、かつX2、X3、
X4及びX5はCを表し:
)は、トランス形態ではなく、シス形態であってもよい。
一般に、本発明の化合物は、変異合成によって製造される。
一般に、式(I)若しくは(II)のいくつかの化合物、又は医薬として許容し得るそれらの
塩の製造方法は、以下を含む:
・サングリフェリン産生株(ストレプトマイセス属sp.A92-308110(DSM 9954としても知ら
れる)など)の培養物、又は、より好ましくは、sfaA遺伝子又はsfaA遺伝子のホモログが
不活化されているか又は欠失しているサングリフェリン産生株の培養物を、発酵ブロスに
接種すること
・該発酵ブロスに、(式(III)で示される)メタ-チロシンアナログを供給すること
・サングリフェリンアナログが産生されるまで、発酵を継続させること
・サングリフェリンアナログを抽出及び単離すること
基を表す。)。
(II)の化合物について定義したとおりである。
前記供給材料は、式(III)の化合物のラセミ形態又はL-形態であることができる。
造される。式(III)の化合物に至る1つの一般的な経路は、以下のスキーム1に示されると
おりである:
O2R7)とのカップリング、b)必要に応じて、水素化及び脱保護。PG=保護基。
。式(IV)の化合物から式(III)の化合物を生成する際に、保護及び脱保護化学の利用を要
する可能性がある。これらの技術は当業者に公知であり、好適な保護基は、「グリーンの
有機合成における保護基(Greene's Protective Groups in Organic Synthesis)」(Wut
s及びGreene、第4版、2007)に記載されている。
に関して、「ボーゲルの実用有機化学の教科書(Vogel's Textbook of Practical Organic
Chemistry)」(Furnissら、1989)及び「マーチの先端有機化学(March's Advanced Organ
ic Chemistry)」(Smith及びMarch、2001)を含むが、これらに限定されない標準的な教科
書を調べることもできる。
与できる。2種(又は、それより多い)の薬剤の共投与は、使用されるそれぞれの投与量
を低減させ、それにより副作用を低減させる可能性があり、効力の向上をもたらすことが
でき、そのため、より高いSVR、及び耐性の低減をもたらすことができる。
療から採用されるHCV感染の治療のための1以上の治療薬(複数可)とともに共投与され
る。これは、インターフェロン(例えば、pIFNα及び/又はリバビリン)でもよい。
治療を対象とした薬剤)などの1以上の他の抗ウイルス薬とともに共投与され、該抗ウイ
ルス薬は以下の1以上であってよい:非-ヌクレオシドポリメラーゼ阻害剤(例えば、IDX3
75、VCH-222、BI207127、ANA598、VCH-916)、ヌクレオシド若しくはヌクレオチドポリメ
ラーゼ阻害剤(例えば、2'-C-メチルシチジン、2'-C-メチルアデノシン、R1479、PSI-6130
、R7128、R1626)、プロテアーゼ阻害剤(例えば、BILN-2061、VX-950(Telaprevir)、SCH50
3034(Boceprevir)、TMC435350、MK-7009、R7227/ITMN-191、EA-058、EA-063)又はウイル
ス侵入阻害剤(例えば、PRO 206)。
のいずれかによって製造することができる。そのような方法は、有効成分(本発明の化合
物)を、1種以上の副成分を構成する担体と混合する工程を含む。一般的に、製剤は、有効
成分を、液体担体又は微粉砕固体担体又は両方と均一かつ密接に混合し、次いで、必要な
場合、生成物を成形することにより製造される。
の酸、又は塩基、付加塩の形態にある有効成分を含む、医薬製剤の形態で経口投与される
だろう。治療すべき障害及び患者、並びに投与経路によって、組成物は種々の投与量で投
与され得る。
着色剤を含んでいてもよい、錠剤、カプセル剤、オビュール剤(ovules)、エリキシル剤
、液剤又は懸濁剤の形態で、経口的に、頬側的に又は舌下的に投与することができる。
酸ナトリウム、炭酸カルシウム、リン酸水素カルシウム及びグリシンなどの賦形剤、デン
プン(好ましくは、トウモロコシ、ジャガイモ又はタピオカデンプン)、デンプングリコー
ル酸ナトリウム、クロスカルメロースナトリウム及び特定の複合シリケートなどの崩壊剤
、及びポリビニルピロリドン、ヒドロキシプロピルメチルセルロース(HPMC)、ヒドロキシ
-プロピルセルロース(HPC)、スクロース、ゼラチン及びアカシアなどの造粒結合剤。さら
に、ステアリン酸マグネシウム、ステアリン酸、グリセリルベヘネート及びタルクなどの
滑沢剤が含まれ得る。
。この点において好ましい賦形剤には、ラクトース、デンプン、セルロース、乳糖又は高
分子量ポリエチレングリコールがある。水性懸濁剤及び/又はエリキシル剤では、本発明
の化合物は、乳化剤及び/又は懸濁化剤とともに、並びに、水、エタノール、プロピレン
グリコール及びグリセリン、並びにそれらの組み合わせなどの希釈剤とともに、様々な甘
味剤若しくは着香剤、着色物質又は色素と組み合わせることができる。
適な機械中で、結合剤(例えば、ポビドン、ゼラチン、ヒドロキシプロピルメチルセルロ
ース)、滑沢剤、不活性な希釈剤、保存剤、崩壊剤(例えば、デンプングリコール酸ナトリ
ウム、架橋ポビドン、架橋カルボキシメチルセルロースナトリウム)、界面活性剤又は分
散剤と任意に混合された、粉末又は顆粒などの自由流動形態にある有効成分を圧縮するこ
とにより製造できる。湿製錠剤は、好適な機械中で、不活性な液体希釈剤により湿らせた
粉末状化合物の混合物を成型することにより製造できる。錠剤は任意にコーティング又は
溝をつけてもよく、例えばヒドロキシプロピルメチルセルロースを種々の比率で利用して
有効成分の緩徐放出又は制御放出を与えて所望の放出プロファイルを与えるように製剤で
きる。
、カシェ剤、若しくは錠剤などの分離した単位として;散剤若しくは顆粒剤として;水性
液体若しくは非水性液体の液剤又は懸濁剤として;又は水中油型液体エマルション若しく
は油中水型液体エマルションとして提供することができる。有効成分は、ボーラス、舐剤
、又はペースト剤として提供することもできる。
る分野に従来ある他の薬剤を含んでよく、例えば、経口投与に好適なものは着香剤を含ん
でよいことを理解されたい。
好都合なことに、保存剤及び緩衝剤などの薬剤はビヒクル中に溶かすことができる。安
定性を増すために、組成物を、バイアルに充填した後に凍結でき、真空下で水が除去され
る。次いで、凍結乾燥された乾燥散剤をバイアルに密封し、付随する注射用の水のバイア
ルを供給して、使用前に液体を再構成できる。
性質及び重症度並びに対象の肉体的状態、選択された投与経路により変わるだろう。適切
な用量は当業者により容易に決定できる。
組成物は、投与の方法により、0.1重量%から、好ましくは5〜60%、より好ましくは10〜
30重量%の本発明の化合物を含み得る。
与の形態、経路、及び部位、並びに治療される特定の対象の年齢及び病態により決定され
、最終的には利用すべき適切な用量を医師が決定するだろうことを当業者は認識するだろ
う。この用量は、適切なだけ頻繁に繰り返すことができる。副作用が現れた場合、用量の
量及び/又は頻度を、通常の診療に従って、変更又は低減できる。
-医薬として使用するための、本発明の化合物
-HCV又はHIV感染などのウイルス感染(特にRNAウイルス感染)を治療するための医薬として
使用するための、抗炎症薬として使用するための、又は、臓器移植拒絶反応の予防のため
の、本発明の化合物
-本発明の化合物を、医薬として許容し得る希釈剤又は担体とともに含む、医薬組成物
-本発明の化合物を、医薬として許容し得る希釈剤又は担体とともに含み、第2の又は二次
の有効成分、特にHCV若しくはHIV感染などのウイルス感染の治療に適応される有効成分、
抗炎症薬として使用するための有効成分、又は、臓器移植拒絶反応の予防のための有効成
分をさらに含む、医薬組成物
-治療有効量の本発明の化合物を対象に投与することを含む、HCV若しくはHIV感染などの
ウイルス感染(特にRNAウイルス感染)の治療方法、抗炎症薬としての使用のための方法、
又は、臓器移植拒絶反応の予防方法
-HCV若しくはHIV感染などのウイルス感染の治療のための医薬、抗炎症薬として使用する
ための医薬、又は、臓器移植拒絶反応の予防のための医薬の製造のための、本発明の化合
物の使用
-サングリフェリン産生細菌(ストレプトマイセス属種(例えば、A92-308110)など)に、
式(III)の化合物又はそれらの塩を供給すること、及び、変異合成サングリフェリンが産
生されるように該細菌を培養することを含む、変異合成サングリフェリン(式(I)又は(II)
の化合物など)の製造方法
-前記サングリフェリン産生細菌が、sfaA遺伝子又はsfaA遺伝子ホモログが不活化されて
いるか又は欠失しているストレプトマイセス属種である、前項に記載の方法
-変異合成サングリフェリンを単離する工程をさらに含む、前二項に記載の方法。
の化合物のうちのいずれかの酸及びエステルなど)、並びに式(IV)の新規化合物も、それ
らの塩及びエステルを含めて、本発明の態様を形成する。
(材料及び方法)
(菌株及び増殖条件)
BIOT-4253及びBIOT-4370とも称されるサングリフェリン産生株ストレプトマイセス属種
A92-308110(DSM no 9954、ドイツ、ブラウンシュヴァイクのDSMZから購入)、又はその誘
導体(BIOT-4585など)を、オートミール寒天培地、MAM、ISP4、又はISP2(下記参照)上で
28℃で維持する。
ohn Innes Centreから入手したものであり、Biermanらの文献(1992)及びKieserらの文
献(2000)に記載されている。
子を、蒸留水中の20%w/v滅菌グリセロールに回収し、0.5mlのアリコートで-80℃で保存し
た。凍結胞子ストックを、シード培地SGS又はSM25-3の接種に使用した。接種されたシー
ド培地を、5.0又は2.5 cmの行程で200〜300 rpmで振盪しながら、27℃で24時間インキュ
ベートした。発酵培地SGP-2又はBT6を2.5%〜10%のシード培養物により接種し、5又は2.5
cmの行程で200〜300 rpmで振盪しながら、24℃で4〜5日間インキュベートした。次いで、
培養物を抽出のために回収した。
メチル(2S)-2-アミノ-3-(6-ヒドロキシ(2-ピリジル))プロパノアート、L-3-アミノフェ
ニルアラニンメチルエステル、L-4-メチル-メタ-チロシンメチルエステル、L-4-フルオロ
-メタ-チロシンメチルエステル及びL-4,5-ジフルオロ-メタ-チロシンメチルエステルは、
Netchem社(米国)から購入した。
DL-3-フルオロフェニルアラニン及びL-フェニルアラニンは、Sigma社(英国)から購入し
た。
DL-メタ-チロシンは、Fluorochem社(英国)から購入した。
L-メタ-チロシンは、Alfa Aesar社(英国)から購入した。
-(3-フルオロ-5-ヒドロキシフェニル)プロパノアート(10)、メチル2-アミノ-3-(2-フルオ
ロ-5-ヒドロキシフェニル)プロパノアート(11)、メチル2-アミノ-3-(2-フルオロ-3-ヒド
ロキシフェニル)プロパノアート(12)及びメチル2-アミノ-3-(2,6-ジフルオロ-3-ヒドロキ
シフェニル)プロパノアート(13)は、以下のように合成した。
℃で滴加した。該添加の後、該反応混合物を-20℃で3時間撹拌し、氷水を注意深く加え、
DCMで抽出した。該有機層を水及びブラインで洗浄し、Na2SO4で乾燥し、濾過して、濃縮
した。該残渣をシリカ上のフラッシュクロマトグラフィーにより精製し、所望の化合物8-
2を与えた。
該反応混合物を室温で一晩撹拌した。水を加え、アセトンを真空下で除去し、次いでEtOA
cで抽出し、該有機層を水及びブラインで洗浄し、Na2SO4で乾燥し、濾過して、濃縮した
。該残渣をシリカ上のフラッシュクロマトグラフィーにより精製し、所望の化合物8-3を
与えた。
物を80℃で2時間撹拌した。該黄色の反応混合物を冷却し、冷やしたEtOH(10mL)を加え、
該混合物を氷浴中で15分間冷却し、次いで30mLの氷水に注ぎ入れ、冷却し、該生成物を濾
過により回収した。該固体を真空中で乾燥し、8-4を得た。
た。該溶媒を回転蒸発により除去し、該残渣をEtOAc 50mL中に溶解し、該EtOAc溶液を水
で2回洗浄し、濃縮して8-5を与えた。
した。濾過により触媒を除去した後、該溶媒を蒸発させて生成物8-6を与えた。
8-6(210mg)の3N HCl(10mL)溶液を24時間還流した。該溶液を濃縮して乾燥させ、該残渣
を逆-コンビフラッシュにより精製して対象生成物8を与えた。
ル)プロパノアート(10)
.5M、107.3mmol)を-78℃で滴加した。それを30分間撹拌し、N,N-ジメチルホルムアミド(1
5.1mL、195.1mmol)をこの温度で加えた。それをさらに30分間撹拌し、冷浴を外した。1時
間後、該反応物を飽和塩化アンモニウム水溶液でクエンチした。該有機層を水及び飽和塩
化ナトリウム水溶液で洗浄し、乾燥し(硫酸ナトリウム)、濾過して、濃縮した。該残渣を
シリカ上のクロマトグラフィーにより精製し9-2を与えた。
で滴加した。該添加の後、該反応混合物を-20℃で3時間撹拌し、氷水を注意深く加え、DC
Mで抽出した。該有機層を水及びブラインで洗浄し、Na2SO4で乾燥し、濾過して、濃縮し
た。該残渣をシリカ上のフラッシュクロマトグラフィーにより精製して、所望の化合物9-
3を与えた。
64g、14mmol)のDCM(150mL)溶液に、室温でDBU(4.26g、28mmol)を加えた。10分後、9-3(1.
95g、14mmol)を加え、得られた混合物を室温で一晩撹拌した。該溶液をEtOAc(150mL)で希
釈し、分離して該有機層を1N HClで洗浄し、Na2SO4で乾燥し、濾過して、濃縮した。該残
渣をシリカ上のフラッシュクロマトグラフィーにより精製して、9-4を与えた。
り触媒を除去した後、該溶媒を蒸発させて、10を与えた。
10(300mg、1.4mmol)のEtOH(30mL)溶液に、NaOH水溶液(2N、4mL)を加え、該反応物を室
温で30分間撹拌した。該溶媒を除去し、該残渣を2N HClでpH=6に中和し、形成された白色
結晶を濾過により回収し、対象化合物9を与えた。
で滴加した。該添加の後、該反応物を-20℃で4時間撹拌した。次いで、氷/水をゆっくり
添加し、該層を分離し、該有機層を水及びブラインで洗浄し、Na2SO4で乾燥し、蒸発乾固
させた。該残渣を、さらに精製することなく次の工程に使用した。
、9mmol)の100mL DCM溶液に、DBU(2.8g、18mmol)を室温で加え、10分後、化合物11-2(前
工程からの粗製化合物)を加え、室温で2時間撹拌した。次いで、該溶液をDCM(50mL)で希
釈し、1N HCl(20mL)で洗浄し、Na2SO4で乾燥し、蒸発乾固させた。該残渣をシリカゲルク
ロマトグラフィー(石油エーテル/酢酸エチル=5/1)により精製し、11-3を与えた。
上で水素化した。濾過により触媒を除去した後、該溶媒を蒸発させて粗生成物を得て、こ
れを逆-コンビフラッシュにより精製し、11を白色固体として得た。
で滴加した。該添加の後、該反応物を-20℃で4時間撹拌した。氷/水をゆっくり添加した
後、該層を分離し、該有機層を水及びブラインで洗浄し、Na2SO4で乾燥し、蒸発乾固させ
た。該残渣を、さらに精製することなく次の工程に使用した。
、9mmol)の100mL DCM溶液に、DBU(2.7mL、18mmol)を室温で加え、10分後、化合物12-2(前
工程からの粗製化合物)を加え、室温で2時間撹拌した。次いで、該溶液をDCM(100mL)で希
釈し、1N HCl(30mL)で洗浄し、Na2SO4で乾燥し、蒸発乾固させた。該残渣をシリカゲルク
ロマトグラフィー(石油エーテル/酢酸エチル=5/1)により精製し、12-3を与えた。
C上で水素化した。濾過により触媒を除去した後、該溶媒を蒸発させて粗生成物を得て、
これを逆-コンビフラッシュにより精製し、所望の化合物12を白色固体として得た。
及びBnBr(2.2mL、18.5mmol)を0℃で加えた。該反応物を室温で2時間撹拌した。水(100mL)
及びEA(200mL)を加え、該有機層を、水(50mL)及びブライン(50mL)で洗浄し、Na2SO4で乾
燥し、蒸発乾固させた。該残渣をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチ
ル=10/1)により精製し、粗製の13-1を与えた。
間撹拌した。DMF(1.3g、0.018mmol)を加え、再度30分間撹拌した。次いで、冷浴を外し、
該反応混合物を室温で1時間撹拌し、その後、水でクエンチした。それを酢酸エチル(20mL
、3回)で抽出し、Na2SO4で乾燥し、蒸発乾固させた。該残渣をシリカゲルクロマトグラフ
ィー(石油エーテル/酢酸エチル=10/1)により精製し、13-2を黄色の固体として与えた。
8mg、2.2mmol)の20mL DCM溶液に、DBU(319mg、2.1mmol)を室温で加えた。10分後、化合物
13-2(500mg、2mmol)を加え、室温で2時間撹拌した。次いで、該溶液をDCM(50mL)で希釈し
、1N HCl(20mL)で洗浄し、Na2SO4で乾燥し、蒸発乾固させた。該残渣をシリカゲルクロマ
トグラフィー(石油エーテル/酢酸エチル=5/1)により精製し、13-3を黄色の油として与え
た。
化した。濾過により触媒を除去した後、該溶媒を蒸発させて粗生成物を得て、これを逆-
コンビフラッシュにより精製し、所望の化合物13を白色固体として得た。
培地の調製に使用した水は、Millipore Elix Analytical Grade Water Purification S
ystemを使用して調製した。
BIOT-4585の凍結保存胞子ストックを室温で解凍した。増殖性培養(種培養)は、胞子ス
トック4.0mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ中のSM25培地400
mLに移すことによって調製した。培養は、27℃、250rpm(5.0cm行程)で48時間行った。該
種培養から、25mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ中の250mL
の産生培地SGP2+5%HP20に移した。24℃、250rpm(2.5cm行程)での24時間の培養の後、各産
生フラスコに、1Mの塩酸中の、所望の前駆体の250mMのラセミ溶液又は125mMの鏡像異性的
に純粋な溶液を2mL、及び、250mMのDL-ピペラジン酸のメタノール溶液を2mL加えて、前駆
体のそれぞれの鏡像異性体の最終濃度を1mMとした。24℃、250rpm(2.5cm行程)で、さらに
4日間、培養を続けた。
培養ブロス(1 mL)及び酢酸エチル(1 mL)を加え、15〜30分間混合し、次いで10分間遠心
分離する。0.4 mLの有機層を回収し、蒸発乾固させ、次いで0.20 mLのアセトニトリルに
再溶解させる。
HPLC条件:
C18 Hyperclone BDS C18 カラム 3μ, 4.6 mm×150 mm
Phenomenex Analytical C18 Security Guard Cartridge(KJ0-4282)を備える
カラム温度50℃
流速1 mL/分
240 nmで紫外モニター
20 μLアリコートを注入
0分:55% B
1.0分:55% B
6.5分:100% B
10.0分:100% B
10.05分:55% B
13.0分:55% B
溶媒Bは、アセトニトリル+0.1%ギ酸である。
これらの条件下でSfAは5.5分に溶離する。
これらの条件下でSfBは6.5分に溶離する。
転しているBruker Daltonics Esquire 3000+ エレクトロスプレー質量分析器と組み合わ
せた一体型Agilent HP1100 HPLCシステムで実施する。
(HPLC条件)
C18 Hyperclone BDS C18カラム3μ, 4.6 mm×150 mm
Phenomenex Analytical C18 Security Guard Cartridge (KJ0-4282)を備える
カラム温度50℃
流速1 mL/分
210、240、及び254 nmで紫外モニター
0分:10% B
2.0分:10% B
15分:100% B
17分:100% B
17.05分:10% B
20分:10% B
溶媒Aは、水+0.1% ギ酸である。
溶媒Bは、アセトニトリル+0.1% ギ酸である。
MSは、150〜1500amuをスキャンする、スイッチングモード(ポジティブとネガティブの
間をスイッチング)で運転する。
API-2000、API-4000又はUPLC装置を使用
HPLC条件:
15用: ACQUITY UPLC BEH C18 (2.1×50 mm, 1.7 μm)
化合物5、14、16、17、18、19用 Ultimate XB-C18 (2.1×50 mm, 5 μm) カラム温度50℃
流速0.6 mL/分
0.2 分: 20% B
0.6 分: 95% B
1.1 分: 95% B
1.15 分: 20% B
1.5 分: 停止
溶媒Aは、H2O-0.025% FA-1mM NH4OACである。
溶媒Bは、ACN-0.025% FA-1mM NH4OACである。
0.3 分: 10% B
0.8 分: 95% B
2.3 分: 95% B
2.31 分: 10% B
3.5 分: 停止
溶媒Aは、H2O-0.1% FAである。
溶媒Bは、MeOH-0.1% FAである。
遺伝子型1HCVに対する抗ウイルス効能を下記のように試験できる:試験物質の添加の1
日前に、HCV遺伝子型1b I389luc-ubi-neo/NS3-3'/5.1レプリコン(Vrolijkら、2003)を含
み、細胞増殖培地[10% FCS、1%非必須アミノ酸(11140035)、1% ペニシリン/ストレプトマ
イシン(15140148)、及び2%ジェネティシン(10131027)を補ったDMEM(カタログ番号4196503
9) Invitrogen社製]中で1.3〜1.4の比で継代し、3〜4日間75mLの組織培養フラスコ(Techn
o Plastic Products社製)で増殖させたHuh5.2細胞を回収し、6500細胞/ウェル(100μL/ウ
ェル)の密度で96-ウェル組織培養マイクロタイタープレート(抗代謝効果の評価にはFalco
n、Beckton Dickinson社製、抗ウイルス効果の評価にはCulturPlate、Perkin Elmer社製)
中で、アッセイ培地(DMEM、10% FCS、1%非必須アミノ酸、1%ペニシリン/ストレプトマイ
シン)に播種した。マイクロタイタープレートを一晩インキュベートすると(37℃、5% CO2
、相対湿度95〜99%)、非コンフルエントな細胞単層を生じる。希釈系列を準備する;各希
釈系列を少なくとも二連で実施する。アッセイのセットアップ後に、マイクロタイタープ
レートを72時間(37℃、5% CO2、相対湿度95〜99%)インキュベートする。
μLの5% MTS(Promega社製)溶液と替え、1.5時間(37℃、5% CO2、相対湿度95〜99%)インキ
ュベートする。吸光度を498nmの波長で測定し(Safire2、Tecan社製)、光学濃度(OD値)を
、非処理対照のパーセンテージに変換する。
洗浄バッファを吸引し、25μLのGlo Lysis Buffer(カタログ番号E2661、Promega社製)を
加え、その後室温で5分間溶解を進行させる。その後、50μLのLuciferase Assay System(
カタログ番号E1501、Promega社製)を加え、ルシフェラーゼ発光シグナルを直ちに定量化
する(1000ms積分時間/ウェル、Safire2、Tecan社製)。相対発光単位を非処理対照のパー
センテージに変換する。
%の阻害が観察される濃度を表す。CC50(用量-反応曲線から得られる値)は、細胞の代謝活
性が非処理細胞の代謝活性の50%に低下する濃度を表す。選択指数(SI)は、化合物の治療
域を表すものであるが、CC50/EC50として計算する。
性の30%以下の低減が見られる場合に、HCVレプリコン系における真の抗ウイルス効果を惹
起すると考えられる。
結果については、実施例12を参照されたい。
ッセイ)
レプリコン細胞(遺伝子型1a(H77)及び2a(JFH-1)のサブゲノムレプリコン)を、ダルベッ
コ改変基本培地(DMEM)、10%ウシ胎仔血清(FBS)、1%ペニシリン-ストレプトマイシン(pen-
strep)、1%グルタミン、1%非必須アミノ酸、250μg/ml G418で、5%CO2のインキュベータ
ーにおいて37℃で増殖させる。すべての細胞培養試薬は、Mediatech社(ハーンドン、VA)
から購入できる。
プレートに、5x103細胞/ウェルで、播種する。その翌日に、前記培地を、5%FBSの存在下
で連続希釈された化合物を含むDMEMに替える。HCVレプリコン抗ウイルスアッセイは、段
階的な化合物希釈液において、化合物の効果を試験する。簡単に言うと、HCVレプリコン
を含む細胞を、96-ウェルプレートに播種する。試験物質をDMEM+5%FBSで連続的に希釈す
る。該希釈された化合物を、前記プレートの適切なウェルに適用する。37℃、72時間のイ
ンキュベーションの後、該細胞をプロセシングする。各ウェルからの細胞内RNAを、RNeas
y96キット(Qiagen社)を用いて抽出する。HCV RNAのレベルを、先に説明されたように(Vro
lijkら、2003)、TaqMan(登録商標)One-Step RT-PCR Master Mix Reagents(Applied Bio
systems社、フォスターシティ、CA)及びABI Prism 7900配列検出システム(Applied Biosy
stems社)を用いる、逆転写-リアルタイムPCRアッセイにより測定する。細胞傷害性効果は
、TaqMan(登録商標)Ribosomal RNA Control Reagents(Applied Biosystems社)を用いて
、細胞数の示度として測定する。次いで、HCV RNA及びリボソームRNAの量を利用して、該
当するIC50値(レプリコン複製を50%阻害する濃度)を得る。
ミクロソームにおける代謝率は、以下のように分析できる:
マウス又はヒト肝臓ミクロソームを、緩衝液C(0.1Mリン酸カリウム緩衝液、1.0mM EDTA
、pH7.4)で、2.5mg/mLの濃度まで希釈した。次いで、5μMの化合物添加溶液(ACN9.5μL中
の10mM DMSOストック溶液0.5μLが、緩衝液C 990μLに加えられる。)50μLを、ミクロソ
ーム溶液(2.5mg/mL)50μL、緩衝液C 110μLに加えることによってミクロソーム安定性試
料を調製し、よく混合した。全ての試料を、37℃でおよそ15分間プレインキュベートした
。この後、NADPH溶液(12.5mM)40μLを、穏やかにかき混ぜながら加えることにより、反応
を開始した。アリコート(40μL)を、0、15、30、45及び60分で移し、内部標準を含むACN(
120μL)でクエンチした。タンパク質を遠心分離(4000rpm、15分)により除去し、LC-MS/MS
により、試料プレートを化合物濃度について分析した。次いで、分析物の濃度を元々存在
していた量と比較する標準法により、半減期を計算した。
結果については、実施例13を参照されたい。
予め液体窒素中に保存しておいた凍結保存肝細胞を、37±1℃の振盪水浴に2分±15秒間
置く。次いで、該肝細胞を、10X体積の予め温めておいたKrebs-Henseleit重炭酸塩(KHB)
緩衝液(2000mg/Lグルコース。炭酸カルシウム及び炭酸水素ナトリウム不含。Sigma社)に
加え、穏やかにかき混ぜ、500rpmで3分間遠心分離する。遠心分離後、該上清を注意深く
除去し、10X体積の予め温めておいたKHB緩衝液を加えて細胞ペレットを再懸濁する。これ
を穏やかにかき混ぜて、500rpmで3分間遠心分離する。次いで、該上清を除去し、廃棄す
る。次いで、細胞生存率及び収率を、細胞数により決定し、これらの値を利用してヒト肝
細胞懸濁液を作成し適切な播種密度(生存細胞密度=2×106細胞/mL)にする。2Xの投薬溶液
を、予め温めておいたKHB(1%DMSO)中で調製する(200μM添加溶液:DMSO 980μLに基質スト
ック溶液(10mM)20μL、2X投薬溶液:KHB 990μLに200μM添加溶液10μL(希釈後2μM))。
×106細胞/mL)50μLを添加して、計時を開始する。次いで、プレートを37℃でインキュベ
ートする。内部標準を含むアセトニトリル100μLを、インキュベーション時間(0、15、30
、60及び120分)の終了後に各ウェルに添加し、穏やかにかき混ぜ、予め温めておいた肝細
胞溶液50μLを添加する(2×106細胞/mL)。インキュベーション終了時に、細胞生存率を測
定する。試料を、4℃、4000rpmで15分間遠心分離し、上清を超純水で2倍に希釈し、化合
物レベルをLC-MS/MSにより分析する。
水への溶解度を下記のように試験できる:サングリフェリンアナログの10 mMストック
溶液を室温で100% DMSOにおいて調製する。三連の0.01 mLのアリコートを、アンバーバイ
アル中で、pH 7.3の0.1 M PBS溶液又は100% DMSOにより0.5mLにする。得られた0.2 mM溶
液を、IKA(登録商標)vibrax VXRシェーカーで、室温で6時間振盪し、次いで得られた溶
液又は懸濁液を2 mLエッペンドルフチューブに移し、13200 rpmで30分間遠心分離する。
次いで、上清液体のアリコートを上述のLCMS法により分析する。
を、50%MeOH水溶液により40μM、16μM、4μM、1.6μM、0.4μM、0.16μM、0.04μM、及
び0.002μMに希釈することにより、検量線を作成する。次いで、基準点をMeOH:PBS 1:1に
より1:20にさらに希釈する。1:20希釈後の最終濃度は、2000nM、800nM、200nM、80nM、20
nM、8nM、2nM、及び1nMである。次いで、基準を、内部標準(ヒドロキシマクロ環、6)を
含む同体積(1:1)のACNと混合する。試料を遠心分離し(5分間、12000rpm)、次いでLC/MSに
より分析する。
二連で、1.5mLエッペンドルフチューブ中でpH7.4のPBSに希釈して、最終DMSO濃度1%で100
μMの標的濃度にする(例えば、4μLの10mM DMSOストック溶液を396μLの100mMリン酸緩衝
液に入れる)。次いで、試料チューブを室温で穏やかに4時間振盪する。試料を遠心分離し
(10分間、15000rpm)、未溶解の粒子を沈殿させる。上清を新しいチューブに移し、PBSで
希釈する(各被験物質の希釈率は、利用される分析装置での化合物のシグナルレベルによ
り確認する)。次いで、希釈した試料を、同体積(1:1)のMeOHと混合する。最後に、試料
を、LC-MS/MS分析用の内部標準(ヒドロキシマクロ環、6)を含む同体積(1:1)のACNと混
合する。
細胞透過性を下記のように試験できる:被験化合物をDMSOに溶かして10mMにし、次いで
緩衝液にさらに希釈して、最終10μM投薬濃度を与える。蛍光マーカールシファーイエロ
ーも含めて、膜統合性をモニターする。次いで、被験化合物をCaco-2細胞単層の頂端膜側
にアプライし、基底面コンパートメントへの化合物透過を測定する。これを逆方向に実施
し(基底面から頂端へ)、能動輸送を調べる。LC-MS/MSを利用して、被験化合物及び標準対
照化合物(プロパノロール及びアセブトロールなど)の両方のレベルを定量化する。
インビボアッセイを利用して、化合物のバイオアベイラビリティを測定することもでき
る。一般的に、化合物を、静脈内(i.v.)及び経口的(p.o.)の両方で試験動物(例えば、マ
ウス又はラット)に投与し、一定間隔で採血して、薬物の血漿濃度が時間とともに変動す
る様子を調査する。経時的な血漿濃度の経時変化を利用して、標準モデルを利用して化合
物の絶対的なバイオアベイラビリティをパーセンテージとして計算できる。典型的なプロ
トコルの例を以下に記載する。
薬する。5分、10分、15分、30分、45分、60分、90分、120分、180分、240分、360分、420
分、及び2880分に採血し、試料中の本発明の化合物又は親化合物の濃度をHPLCにより決定
する。次いで、血漿濃度の経時変化を利用して、血漿濃度-時間曲線下面積(AUC-体循環に
達する未変化の薬物の総量に正比例する)、最大(ピーク)血漿中薬物濃度、最大血漿中
薬物濃度が起こる時間(ピーク時間)などのキーパラメーターを得ることができるが、バイ
オアベイラビリティの正確な決定に利用できる追加の因子には下記がある:化合物の終末
半減期、全身クリアランス、定常状態分布容積、及びF%。次いで、これらのパラメーター
を、非コンパートメント法又はコンパートメント法により分析して、計算されたバイオア
ベイラビリティのパーセンテージを与えるが、この種類の方法の例に関しては、Egorinら
、2002、及び該文献の引用文献を参照されたい。
サングリフェリンアナログについて、全血を分析する。p.o.投与及びi.v.投与の両方の
ために、化合物を5%エタノール/5%クレモフォールEL/90%食塩水に配合する。3匹の雄CD1
マウスのグループに、1mg/kg i.v.又は10mg/kg p.o.で投薬する。血液試料(40μL)を、投
薬前、及び0.25、0.5、2、8、及び24時間で、伏在静脈を介して採取し、等量のdH20で希
釈し、直ちにドライアイス上に置く。試料は、分析まで-70℃で保管する。試料中の本発
明の化合物又は親化合物の濃度を、LCMSにより、以下のように測定する:20μLの血液:H2O
(1:1、v/v)/PK試料を、100ng/mLの内部標準(ヒドロキシルマクロ環、6)20μL、標準溶液/
MeOH 20μL及びACN 150μLとともに加え、1500rpmで1分間ボルテックスし、12000rpmで5
分間遠心分離する。次いで、該上清をLC-MS/MSにインジェクトする。血中濃度の経時変化
をプロットし、それを利用して、全血中濃度-時間曲線下面積(AUC-体循環に達する未変化
の薬物の総量に正比例する)を得る。可能な場合には、これらの値を利用して、経口バイ
オアベイラビリティ(F%)及び他のPKパラメータを得る。
ATCCから入手したHuh-7及びHepG2細胞を、10%ウシ胎仔血清(FBS)、1%ペニシリン-スト
レプトマイシン(pen-strep)及び1%グルタミンを含むダルベッコ改変基本培地(DMEM)で増
殖させ;一方、CEM細胞(ATCCから入手したヒトT-細胞白血病細胞)を、10%FBS、1%pen-stre
p及び1%グルタミンを含むRPMI 1640培地で増殖させる。新鮮なヒトPBMCを、少なくとも2
人の正常な選別ドナーから入手した全血から単離する。簡単に言うと、末梢血細胞を、低
速遠心分離及びPBS中での再懸濁により、2〜3回ペレット化/洗浄し、混入している血小板
を除去する。次いで、該洗浄した血液細胞を、ダルベッコリン酸塩緩衝化食塩水(D-PBS)
で1:1に希釈し、50mLの遠心チューブ内のリンパ球分離培地(LSM;Mediatech社のcellgrow;
密度1.078+/-0.002g/ml;カタログ番号85-072-CL)14mL上に層状に重ね、600Xgで30分間遠
心分離する。生じた界面からバンド化したPBMCを穏やかに吸引し、その後、低速遠心分離
によりPBSで2回洗浄する。最後の洗浄の後、トリパンブルー排除により細胞を計数し、15
%ウシ胎仔血清(FBS)、2mM L-グルタミン、4μg/mL PHA-Pを補ったRPMI 1640中に、1x107
細胞/mLで再懸濁する。該細胞を、37℃で48〜72時間インキュベートする。インキュベー
ト後、PBMCを遠心分離し、15%FBS、2mM L-グルタミン、100U/mLペニシリン、100μg/mLス
トレプトマイシン、10μg/mLゲンタマイシン、及び20U/mLリコンビナントヒトIL-2を含む
RPMI 1640に再懸濁する。
することによって評価する。培地のみを含む細胞は、細胞対照(CC)となる。Huh-7及びHep
G2細胞を、5x103細胞/ウェルの濃度で96-ウェルプレートに播種する。翌日に、培地を吸
引し、5%FBSを含む、対応培地100μLを加える。試験薬物の希釈液を、2X濃度でマイクロ
タイターチューブ内に調製し、各濃度の100μLを、標準的なフォーマットで適切なウェル
に配置する。72時間後、細胞を細胞傷害性評価のためにプロセシングする。
細胞/ウェルで、100Lの体積で蒔く。同様に、CEM細胞を1x104細胞/ウェルで蒔く。次いで
、試験薬物の2X調製物100μLを、標準的なフォーマットで適切なウェルに添加する。培養
を6〜7日間維持し、次いで、細胞傷害性測定のためにプロセシングする。
て測定する。該アッセイは、損傷した膜を有する細胞からの乳酸デヒドロゲナーゼ(dehyr
odgenase)(LDH)の放出を、蛍光定量的かつ均質なフォーマットで測定する。培地に放出さ
れるLDHは、レサズリンを蛍光性レゾルフィン生成物に変換する共役酵素アッセイを利用
して測定される。生成される蛍光の量は、溶解した細胞の数に比例する。6つの段階希釈
濃度の各化合物を該細胞に適用し、適切な場合には、TC50(細胞生存率を50%減少させる、
薬物の毒性濃度)及びTC90(細胞生存率を90%減少させる、薬物の毒性濃度)値を得る。
MDR1(P糖タンパク質1)及びMRP2トランスポーターの阻害及び活性化の評価には、Solvo
Biotechnology社のインビトロATPアーゼアッセイ(Glavinasら、2003)が利用できる。化合
物(0.1、1、10、及び100μM)を、バナジン酸塩の非存在下と存在下の両方で、MDR1又はMR
P2膜小胞と共にインキュベートし、潜在的なATPアーゼ活性化を試験する。さらに、トラ
ンスポーターATPアーゼ活性の起こりうる阻害を検出するために、ベラパミル/スルファサ
ラジンの存在下で類似のインキュベーションを実施する。ATPアーゼ活性は、無機のリン
酸塩を分光測定により定量化して測定する。活性化は、ATPアーゼ活性のバナジン酸感受
性増加から計算する。阻害は、ベラパミル/スルファサラジンに媒介されるATPアーゼ活性
の低下から決定する。
(1.1 sfaA欠失コンストラクトの構築)
sfaA(ヌクレオチド位置14396〜21362、NCBI配列受入番号FJ809786)を含むコスミドTL30
06(配列番号3)の〜7kbのEcoRV-StuIフラグメントを、EcoRV及びStuIによる消化によって
切り取り、得られた単離フラグメントを、予めEcoRVで消化され、エビアルカリホスファ
ターゼ(Roche社)で処理されたpKC1139に直接ライゲートした。このプラスミドをpSGK268
と命名した。
このクローンに含まれるsfaA遺伝子のインフレーム欠失は、Gene Bridges社により供給
されるRed/ET組換えキット(カタログ番号K006)を使用して行った。
ゼを使用するキットで供給されるFRT-PGK-gb2-neo-FRTテンプレートDNAから、ネオマイシ
ンマーカーを増幅した。得られた〜1.7kbの増幅産物を、ゲル電気泳動によって単離し、Q
iaEXレジンで該ゲルから精製した。
0μg/ml)を含むプレート上で選別した。該欠失コンストラクトの導入は、本質的に前記Ge
ne Bridges社のキットのプロトコルに従って行った。単一コロニーを2TY(アプラマイシ
ン(50μg/ml))中で一晩増殖させ、pRedET(tet)プラスミドで形質転換し、30℃でアプラ
マイシン(50μg/ml)及びテトラサイクリン(3μg/ml)で選別した。単一コロニーを使用し
て、この株の一晩培養物を、3mlの2TY(アプラマイシン(50μg/ml)及びテトラサイクリン
(3μg/ml))において30Cで調製した。この培養物0.5mlを使用して、10mlの2TY(アプラマ
イシン(50μg/ml)及びテトラサイクリン(3μg/ml))に30℃で播種し、OD600nm〜0.5まで
増殖させた。この培養物1.4mlを、2つのエッペンドルフチューブの各々に移し、10%アラ
ビノース50μLを一方のチューブに加えて、Red/ET組換えタンパク質の発現を誘導した。
チューブを37℃で〜1時間振盪した。誘導細胞及び非誘導細胞をベンチトップ遠心機でペ
レット化し、冷却滅菌水で2回洗浄した(毎回、再懸濁し、遠心分離して細胞をペレット
化する)。得られたペレットを約30〜40μLの水に懸濁し、氷上に保持した。氷上で、先
に単離した1.7kbの分断フラグメントを前記誘導及び非誘導チューブに加え、1mmのBiorad
社製エレクトロキュベットに移した。該試料を電気穿孔し(Biorad Micropulser、1.8kV、
時定数〜4ms)、2TY 1ml(抗生物質不含)を加えて混合し、細胞を該キュベットから移した
。細胞を、37℃で〜3時間、振盪しながら(1100rpm、エッペンドルフサーモミキサーコン
パクト)インキュベートし、その後、アプラマイシン(50μg/ml)及びカナマイシン(25μg/
ml)を含む2TYプレート上に蒔き、37℃で一晩インキュベートした。誘導試料プレート由来
のコロニーを、50μg/mlのカナマイシンを含む2TYプレート上に画線して純化し、カナマ
イシン耐性カセットの導入を確認した。それぞれの細菌コロニーについてのPCRを利用し
て該カセットの導入を確認した。これらの培養物からプラスミドを調製し、消化して、期
待されるプラスミドpSGK270を確認した。次いで、プラスミドをNsiIで消化して前記マー
カーフラグメントを除去し、残部を再ライゲートして、sfaAインフレーム欠失コンストラ
クト、pSGK271を作製した。
プラスミドpSGK271を、標準技術を使用して大腸菌ET12567 pUZ8002に形質転換し、アプ
ラマイシン(50μg/ml)、カナマイシン(25μg/ml)及びクロラムフェニコール(chloroamphe
nicol)(10μg/ml)を含む2TYプレート上で選別した。得られた株を、アプラマイシン(50μ
g/ml)、カナマイシン(25μg/ml)及びクロラムフェニコール(10μg/ml)を含む3mlの液体2T
Yに播種し、37℃、250rpmで、一晩インキュベートした。この培養液0.8mlを使用して、ア
プラマイシン(50μg/ml)、カナマイシン(25μg/ml)及びクロラムフェニコール(10μg/ml)
を含む、50mlのファルコンチューブ中の液体2TY 10mlに播種し、OD600nmが〜0.5に達する
まで、37℃、250rpmでインキュベートした。得られた培養物を、4℃、3500rpmで10分間遠
心分離し、2TY培地10mlによる洗浄を、各洗浄の後に遠心分離によって細胞をペレット化
しつつ、2回行った。得られたペレットを、2TY 0.5mlに再懸濁し、使用に先立って氷上に
保持した。このプロセスのタイミングは、以下に記載のストレプトマイセス胞子の調製の
完了に合わせた。
ンフルエントなプレートから、20%グリセロール〜3mlに再懸濁することにより回収した。
胞子を遠心分離し(5000rpm、10分、室温)、50mM TES緩衝液で2回洗浄し、その後、50mM T
ES緩衝液1mlに再懸濁し、2つのエッペンドルフチューブ間で分配した。これらのチューブ
に、50℃で10分間、水浴中でヒートショックを施し、その後、2TY 0.5mlを加え、エッペ
ンドルフサーモミキサーコンパクトにおいて、37℃で4〜5時間インキュベートした。
大腸菌100μL)の比で混合し、直ちにR6プレート上に拡げ、37℃のインキュベーターに移
した。およそ2時間のインキュベーションの後、これらのプレートを、ナリジクス酸を含
む滅菌水2mlで覆い、最終プレート内濃度を25μg/Lとした。プレートを37℃のインキュベ
ーターに一晩戻し、その後、アプラマイシンを含む滅菌水2mlで覆い、最終プレート内濃
度を20〜25μg/Lとした。〜4〜7日後に現れる接合完了体コロニーを、アプラマイシン(25
μg/L)及びナリジクス酸(25μg/L)を含むISP4培地にパッチし、37℃でインキュベートし
た。十分な菌糸成長が観察されたら、菌株を、アプラマイシン(25μg/L)を含むISP4培地
に37℃で再パッチし、胞子形成させた。次いで、ISP4(抗生物質不含)にパッチして、37℃
で3〜4日間インキュベートすることにより、菌株を(温度感受性プラスミドの除去を促進
するために)3回継代した。最終的に、菌株をISP4にパッチし、28℃でインキュベートして
十分に胞子形成させた(5〜7日)。胞子を回収し、28℃のISP4プレート上に連続希釈して、
単一コロニーの選別を可能にした。胞子形成した単一コロニーを、アプラマイシン(25μg
/L)を含む、又は含まないISP4プレートに二重にパッチして、プラスミドの喪失を確認し
、サングリフェリン産生に関する試験の前に、〜7日増殖させた。
よく胞子形成した菌株の、単一の〜7mmの寒天プラグを使用して、滅菌SM25-3培地7mlに
播種し、2インチ行程振盪機において、27℃、200rpmでインキュベートした。48時間の増
殖の後、この培養物0.7mlを、5%のHP20樹脂とともにSGP2培地7mlを含む滅菌ファルコンチ
ューブに移した。回収前に、1インチ行程の振盪インキュベーターで、24℃、300rpmで5日
間、培養物を増殖させた。細菌培養物0.8mlを取り出し、等分する前に、培養物全体にわ
たって樹脂を十分に分散させることを確実に行って、2mlのエッペンドルフチューブに等
分した。アセトニトリル0.8ml及びギ酸15μLを加え、該チューブを約30分間混合した。該
混合物を遠心分離によって清澄化し、該抽出物170μLをHPLCバイアルに移して、HPLCによ
り分析した。
菌株の抽出物をHPLCにより分析した。サングリフェリンA及びBを産生する菌株は、野生
型に復帰しているので、それ以上分析しなかった。サングリフェリンA及びB産生が欠如し
ている菌株は、サングリフェリン様発色団を表す、保持時間6.5分の低レベル(〜1〜2mg/L
)のピークを示した。LCMSによる分析によって、このピークは、サングリフェリンの期待
されるm/z値から-16単位の、m/z値1073を有することが示された。このピークは、メタ-ヒ
ドロキシチロシン不存在下でのフェニルアラニンの取り込みによるものと推論した。
的なsfaA変異を化学的に補完して、新規のサングリフェリン類に至る変異合成プロセスを
可能にできるかどうかを評価した。菌株をSM25-3種培地で48時間増殖させ、その後、5%樹
脂を含むSGP2産生培地に移した。さらに24時間増殖させた後、対照として使用する供給無
しの株を伴って、菌株に、2mM DLメタ-ヒドロキシチロシン(1M HCL中の0.16M溶液の100μ
Lの添加)又は2mM L-フェニルアラニンを、3連で供給した。菌株にピペコリン酸(メタノー
ル中2mM)も供給し、生成物の収率を高めた。さらに4日増殖させた後、菌株を回収し、抽
出してHPLCにより分析した。メタ-ヒドロキシチロシンが前記sfaA変異を完全に補完する
ことが示され、L-フェニルアラニンの添加は、前記の-16 amuの化合物のレベルを高めた
。さらなる調査のために、sfaA欠失変異体として菌株Biot-4585を選択した。
他の方法を使用して、sfaA欠失変異体を作製することができる。例としては、sfaA挿入
不活化変異体(WO2010/034243(その内容の全体が、引用により本願明細書に組み込まれて
いる)の実施例12など)が挙げられる。この菌株を、WO2010/034243に記載されているよう
に作製し、菌株名BIOT-4452を与えた。
使用して、ホモロジーの上流領域を増幅する。該増幅産物をT4ポリヌクレオチドキナーゼ
(NEB)で処理し、エビアルカリホスファターゼ(Roche社)で処理することにより脱リン酸化
してあるpUC18にクローン化する。得られるコンストラクトを制限消化によりチェックし
、完全に配列決定して、所望の配列が生成されており、かつポリメラーゼ増幅の間にエラ
ーが導入されていないことを確実にする。このコンストラクトをEcoRI及びNsiIで消化し
、該生成物をゲル電気泳動によって分析する。所望の配列を含むバンド(即ち、上流ホモ
ロジー〜2kb)を該ゲルから切り取り、標準的な手順(QiaEXレジン)を使用して精製する。
第2の系列のオリゴヌクレオチド:
を使用して、ホモロジーの下流領域を増幅する。該増幅産物をT4ポリヌクレオチドキナー
ゼ(NEB)で処理し、エビアルカリホスファターゼ(Roche社)で処理することによって脱リン
酸化してあるpUC18にクローン化する。得られるコンストラクトを制限消化により分析し
、完全に配列決定して、所望の配列が生成されており、かつポリメラーゼ増幅の間にエラ
ーが導入されていないことを確実にする。このコンストラクトをHindIII及びNsiIで消化
し、該生成物をゲル電気泳動により分析する。所望の配列を含むバンド(即ち下流ホモロ
ジー〜2kb)を該ゲルから切り取り、標準的な手順(QiaEXレジン)を使用して精製する。ベ
クターpKC1139をEcoRI及びHindIIIで消化して、該大きいベクターフラグメントをゲル電
気泳動により単離し、標準的な方法(QiaEXレジン)により精製する。次いで、単離された
上流及び下流のホモロジーフラグメントを、このpKC1139のフラグメントにスリーウェイ
ライゲーションでクローン化して所望のsfaA欠失コンストラクトを作製する。
成(即ち、Genscript社又は他のベンダー)を使用して、所望の配列(配列番号8)を含むコン
ストラクトを作製する。この購入されるコンストラクトをBamHI及びXbaIを使用して消化
し、関心対象の配列を切り取り、該生成物をゲル電気泳動により分析する。所望の配列を
含むバンド(〜4kb)を該ゲルから切り取り、標準的な手順を使用して精製する。ベクターp
KC1139をBamHI及びXbaIで消化し、該大きいフラグメントをゲル電気泳動により単離して
、標準的な方法により精製する。次いで、該2つの単離フラグメントを、ともにライゲー
トして、所望のsfaA欠失コンストラクトを作製する。
これらの代替的なsfaA欠失コンストラクトを、実施例セクション1.2の方法を使用して
、接合によりストレプトマイセス属sp.A92-308110(DSM9954)に導入する。
sfaAが破壊された変異体(BIOT-4452又はBIOT-4585)の胞子ストックを、MAM、ISP4、ISP
3又はISP2培地での増殖後に調製し、蒸留水中の20%w/vグリセロールに保存して、-80℃で
保管した。増殖性培養(種培養)は、胞子ストック(1%v/v)を、フォームプラグを備えた50m
Lの遠心チューブ内の種培地(SM25培地)7mLに播種することにより調製した。該培養チュー
ブを、27℃、250rpm(5cm行程)で48時間インキュベートした。該種培養から、10%(v/v)を
、フォームプラグを備えた50mLの遠心チューブ内の7mLの産生培地SGP-2に移した。培養を
、24℃、300rpm(2.5cm行程)で実施した。サングリフェリン変異合成アナログの産生のた
めに、フィード化合物(変異シントン(mutasynthon))の0.32M溶液(1N HCl溶液)0.05mLを、
各チューブに播種後24時間で加え、最終濃度を2mMとした。さらに、ピペラジン酸(メタノ
ール溶液)の0.32M溶液0.05mlを、各チューブに24時間で加え、最終濃度を2mMとした。培
養を、フィード後さらに4日間続けた。
とによって抽出した。アセトニトリル0.8mlを、ギ酸0.015mlとともに加えた。次いで、該
混合物を、Vibrax上で30分間振盪した。次いで、該チューブを、13000rpmで10分間遠心分
離し、上清0.15mlを、分析用に取り出した。抽出物を、全般的方法に記載したように分析
した。
生成物の、LCMS H+及びNa+アダクト、予想される分子量、及び保持時間とともに示す。サ
ングリフェリンAアナログに関連する主要なピークが示される。全てのケースにおいて、L
CMSピークはサングリフェリンBアナログについても見られた(Mass-18)。
発酵は、メチル2-アミノ-3-(3-フルオロ-5-ヒドロキシフェニル)プロパノアート及びDL
-ピペラジン酸を前駆体として利用して、全般的方法に記載したように行い、前駆体は両
方とも26時間で加えた。
して、清澄化したブロスから細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物の
存在量が5%未満であることを確認したアッセイの後で廃棄した。該細胞及び樹脂を、マグ
ネチックスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニ
トリル抽出物を、遠心分離により、又は重力下で沈降させることにより回収した。次いで
、該細胞及び樹脂の2回目のアセトニトリル抽出を、同じ条件下で実施した。合わせたア
セトニトリル抽出物を、減圧下で残留水性ボリューム(residual aqueous volume)まで濃
縮し、次いで、pH6に調整した。これを、酢酸エチルで2回抽出し、合わせた有機化合物を
減圧下で乾燥させ、最終粗製物(1.3g)を与えた。
グしたシリカゲルカラム(10x2cm)上にロードした。該カラムを、酢酸エチルで、次いで、
アセトンの段階的な増加(酢酸エチル中10%、20%、30%、等)で溶出させた。およそ250mLの
フラクションを集め、分析用LCにより対象化合物を同定し、合わせて乾燥させた。この物
質(278mg)をメタノール(1.8ml)に溶解し、調製用HPLCにより精製した。Waters Xterra MS
C18カラム(10ミクロン、19cm x 250mm)を使用し、溶媒を21mL/分でポンプ送りした。溶媒
Aは水、溶媒Bはアセトニトリルであった。該カラムを、インジェクション後6分間、50%B
で均一濃度で流し、その後、30分で100%Bとなる勾配で流した。純粋なフラクションをHPL
C-UVにより同定し、合わせた。これらのフラクションを減圧下で乾燥させ、対象化合物を
オフホワイトの非晶質固体として得た(20mg)。
発酵は、メチル(S)-2-アミノ-3-(3,4-ジフルオロ-5-ヒドロキシフェニル)プロパノアー
ト及びDL-ピペラジン酸を前駆体として利用して、全般的方法に記載したように行い、前
駆体は両方とも26時間で加えた。
して、清澄化したブロスから該細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物
の存在量が5%未満であることを確認したアッセイの後で廃棄した。該細胞及び樹脂を、マ
グネチックスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセト
ニトリル抽出物を、遠心分離により、又は重力下で沈降させることにより回収した。次い
で、該細胞及び樹脂の2回目のアセトニトリル抽出を、同じ条件下で実施した。合わせた
アセトニトリル抽出物を、減圧下で残留水性ボリュームまで濃縮し、次いでpH6に調整し
た。これを、酢酸エチルで2回抽出し、合わせた有機化合物を減圧下で乾燥させ、最終粗
製物(1.6g)を与えた。
たシリカゲルカラム(10x2cm)上にロードした。該カラムを、酢酸エチルで、次いで、アセ
トンの段階的な増加(酢酸エチル中10%、20%、30%、等)で溶出させた。およそ250mLのフラ
クションを集め、分析用LCにより対象化合物を同定し、合わせて乾燥させた。この物質(1
88mg)をメタノール1.8mlに溶解し、調製用HPLCにより精製した。Waters Xterra MSC18カ
ラム(10ミクロン、19cm x 250mm)を使用し、溶媒を21mL/分でポンプ送りした。溶媒Aは水
、溶媒Bはアセトニトリルであった。該カラムを、インジェクション後6分間、50%Bで均一
濃度で流し、次いで、30分で100%Bとなる勾配で流した。これらのフラクションを減圧下
で乾燥させ、対象化合物をオフホワイトの非晶質固体として得た(15mg)。
メチル(S)-2-アミノ-3-(4-フルオロ-3-ヒドロキシフェニル)プロパノアート及びDL-ピ
ペラジン酸前駆体を使用した。前駆体を27時間で加えたことを除き、全般的方法に従って
行った。
して、該清澄化ブロスから該細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物の
存在量が5%未満であることを確認したアッセイの後で廃棄した。該細胞及び樹脂を、マグ
ネチックスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニ
トリル抽出物を、遠心分離により、又は重力下で沈降させることにより回収した。次いで
、該細胞及び樹脂の2回目のアセトニトリル抽出を、同じ条件下で実施した。
pH6に調整した。これを、酢酸エチルで2回抽出し、合わせた有機化合物を減圧下で乾燥さ
せて、最終の油状粗製物を与えた(4.2g)。
たシリカゲルカラム(15x2cm)上にロードした。該カラムを、酢酸エチルで、次いで、アセ
トンの段階的な増加(酢酸エチル中10%、20%、30%、等)で溶出させた。およそ250mLのフラ
クションを集め、分析用LCにより対象化合物を同定し、合わせて乾燥させた。この物質(3
90mg)をメタノール2.4mlに溶解し、調製用HPLCにより精製した。Waters Xterra MSC18カ
ラム(10ミクロン、19cm x 250mm)を使用し、溶媒を21mL/分でポンプ送りした。溶媒Aは水
、溶媒Bはアセトニトリルであった。該カラムを、インジェクション後6分間、50%Bで均一
濃度で流し、次いで、30分で100%Bとなる勾配で流した。純粋なフラクションをHPLC-UVに
より同定し、合わせた。これらのフラクションを、減圧下で乾燥させ、対象化合物をオフ
ホワイトの非晶質固体として得た(38mg)。
BIOT-4585の凍結保存された胞子ストックを、室温で解凍した。増殖性培養(種培養)は
、胞子ストック0.4mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内のSM2
5培地400mLに移すことにより調製した。培養を、27℃、250rpm(2.5cm行程)で、48時間行
った。該種培養から、20mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内
の400mLの産生培地SGP2+5%HP20に移した。24℃、250rpm(2.5cm行程)での24時間の培養の
後、1M塩酸中のメチル(S)-2-アミノ-3-(3-ヒドロキシ-4-メチルフェニル)プロパノアート
の200mM溶液2mL及びDL-ピペラジン酸の400mMメタノール溶液2mLを、各産生フラスコに加
え、前駆体のそれぞれの鏡像異性体の最終濃度を1mMとした。培養を、24℃、250rpm(2.5c
m行程)でさらに4日間続けた。
細胞及び樹脂を清澄化ブロスから分離した。該清澄化ブロスを、対象化合物の存在量が5%
未満であることを確認したアッセイの後で廃棄した。該細胞及び樹脂を、オーバーヘッド
パドルスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニト
リル抽出物を、重力下で沈降させることにより回収した。次いで、該細胞及び樹脂の2回
目のアセトニトリル抽出を、同じ条件下で実施した。合わせたアセトニトリル抽出物を、
減圧下で残留水性ボリュームまで濃縮し、次いで、pH6に調整した。これを、酢酸エチル
で2回抽出し、合わせた有機化合物を減圧下で乾燥させ、最終粗抽出物を与えた(7.6g)。
たシリカゲルカラム(15x2cm)上にロードした。該カラムを、酢酸エチルで、次いで、アセ
トンの段階的な増加(酢酸エチル中10%、20%、30%、等)で溶出させた。およそ250mLのフラ
クションを集め、分析用LCにより対象化合物を同定し、合わせて乾燥させた。この物質(3
19mg)をメタノール2.4mlに溶解し、調製用HPLCにより精製した。Waters Xterra MSC18カ
ラム(10ミクロン、19cm x 250mm)を使用し、溶媒を21mL/分でポンプ送りした。溶媒Aは水
、溶媒Bはアセトニトリルであった。該カラムを、インジェクション後6分間、50%Bで均一
濃度で流し、次いで、30分で100%Bとなる勾配で流した。純粋なフラクションを、HPLC-UV
により同定し、合わせた。これらのフラクションを減圧下で乾燥させ、対象化合物をオフ
ホワイトの非晶質固体として得た(14.9mg)。
BIOT-4585の凍結保存された胞子ストックを、室温で解凍した。増殖性培養(種培養)は
、胞子ストック0.4mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内のSM2
5培地400mLに移すことにより調製した。培養を、27℃、250rpm(2.5cm行程)で48時間行っ
た。該種培養から、500mLを、7LのApplikonファーメンター内の4.5Lの産生培地SGP2+5%HP
20に移し、24℃、400rpm(カスケードDOTコントロール(cascade DOT control))、通気2.5L
/分及び30%DOT(カスケード撹拌コントロール(cascade agitation control))で培養した。
24時間の培養の後、1M塩酸中の(S)-2-アミノ-3-フェニルプロパン酸の667mM溶液7.5mLを
、該ファーメンターに加え、該前駆体の最終濃度を1mMとした。培養を、24℃、400rpm(カ
スケードDOTコントロール)、通気2.5L/分及び30%DOT(カスケード撹拌コントロール)で、
さらに4日間続けた。
清澄化ブロスから該細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物の存在量が
5%未満であることを確認したアッセイの後廃棄した。該細胞及び樹脂を、オーバーヘッド
パドルスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニト
リル抽出物を、重力下で沈降させることにより回収した。次いで、該細胞及び樹脂の2回
目のアセトニトリル抽出を、同じ条件下で実施したが、該2回目の抽出物は、遠心分離に
より回収した。合わせたアセトニトリル抽出物を、減圧下で残留水性ボリュームまで濃縮
し、次いで、pH6に調整した。これを、酢酸エチルで2回抽出し、合わせた有機化合物を減
圧下で乾燥させ、最終粗製物を与えた(55g)。
象化合物はメタノール/水部分に認められ、これを乾燥させた。この乾燥抽出物(48g)を酢
酸エチル30mlに溶解し、酢酸エチル1Lでコンディショニングしたシリカゲルカラム(20x5c
m)上にロードした。該カラムを、酢酸エチルで、次いで、アセトンの段階的な増加(酢酸
エチル中10%、20%、30%、等)で溶出させた。およそ250mLのフラクションを集め、分析用L
Cにより対象化合物を同定し、合わせて乾燥させた。この物質(813mg)をメタノールに溶解
し、調製用HPLCにより精製した。Waters Xterra MSC18カラム(10ミクロン、19cm x 250mm
)を使用し、溶媒を21mL/分でポンプ送りした。溶媒Aは水、溶媒Bはアセトニトリルであっ
た。該カラムを、インジェクション後6分間、50%Bで均一濃度で流し、次いで、30分で100
%Bとなる勾配で流した。純粋なフラクションをHPLC-UVにより同定し、合わせた。これら
のフラクションを減圧下で乾燥させ、対象化合物をオフホワイトの非晶質固体として得た
(34mg)。
ェリンA(化合物19)の単離)
メチル(2S)-2-アミノ-3-(3-ヒドロキシ(2-ピリジル))プロパノアート及びDL-ピペラジ
ン酸前駆体を使用した。増殖性(種)培養の間の前記インキュベーターの行程が2.5cmであ
ったことを除き、全般的方法に従って行った。
清澄化ブロスから該細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物の存在量が
5%未満であることを確認したアッセイの後廃棄した。該細胞及び樹脂を、オーバーヘッド
パドルスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニト
リル抽出物を、重力下で沈降させることにより回収した。次いで、該細胞及び樹脂の2回
目のアセトニトリル抽出を、同じ条件下で実施した。合わせたアセトニトリル抽出物を、
減圧下で残留水性ボリュームまで濃縮し、次いで、pH6に調整した。これを、酢酸エチル
で2回抽出し、合わせた有機化合物を減圧下で乾燥させ、最終粗抽出物を与えた(7g)。
シリカゲルカラム(15x2cm)上にロードした。該カラムを、酢酸エチルで、次いで、アセト
ンの段階的な増加(酢酸エチル中10%、20%、30%、等から100%アセトンへ、次いで、アセト
ン中で、1%メタノールから段階的に5%メタノールへ)で溶出させた。およそ250mLのフラク
ションを集め、分析用LCにより対象化合物を同定し、合わせて乾燥させた。この物質(204
mg)をメタノールに溶解し、調製用HPLCにより精製した。Waters Xterra MSC18カラム(10
ミクロン、19cm x 250mm)を使用し、溶媒を21mL/分でポンプ送りした。溶媒Aは水、溶媒B
はアセトニトリルであった。該カラムを、インジェクション後6分間、50%Bで均一濃度で
流し、次いで、30分で100%Bとなる勾配で流した。純粋なフラクションをHPLC-UVにより同
定し、合わせた。これらのフラクションを減圧下で乾燥させ、対象化合物をオフホワイト
の非晶質固体として得た(4mg)。
BIOT-4585の凍結保存された胞子ストックを、室温で解凍した。増殖性培養(種培養)は
、胞子ストック0.4mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内のSM2
5培地400mLに移すことにより調製した。培養を、27℃、250rpm(2.5cm行程)で、48時間行
った。該種培養から、20mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内
の400mLの産生培地SGP2+5%HP20に移した。24℃、250rpm(2.5cm行程)で24時間培養した後
、1M塩酸中の2-アミノ-3-(3-フルオロフェニル)プロパン酸の400mM溶液2mL及びDL-ピペラ
ジン酸の400mMメタノール溶液2mLを各産生フラスコに加え、該前駆体のそれぞれの鏡像異
性体の最終濃度を1mMとした。培養を、24℃、250rpm(2.5cm行程)で、さらに4日間続けた
。
清澄化ブロスから該細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物の存在量が
5%未満であることを確認したアッセイの後廃棄した。該細胞及び樹脂を、オーバーヘッド
パドルスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニト
リル抽出物を、重力下で沈降させることにより回収した。次いで、該細胞及び樹脂の2回
目のアセトニトリル抽出を、同じ条件下で実施した。3回目の抽出物を、残りの細胞及び
樹脂の混合物の遠心分離により得た。合わせたアセトニトリル抽出物を、減圧下で残留水
性ボリュームまで濃縮し、次いで、pH6に調整した。これを酢酸エチルで2回抽出し、合わ
せた有機化合物を減圧下で乾燥させ、最終粗抽出物を与えた(10.5g)。
したシリカゲルカラム(15x2cm)上にロードした。該カラムを、酢酸エチルで、次いで、ア
セトンの段階的な増加(酢酸エチル中10%、20%、30%、等)で溶出させた。およそ250mLのフ
ラクションを集め、分析用LCにより対象化合物を同定し、合わせて乾燥させた。この物質
(342mg)をメタノールに溶解し、調製用HPLCにより精製した。Waters Xterra MSC18カラム
(10ミクロン、19cm x 250mm)を使用し、溶媒を21mL/分でポンプ送りした。溶媒Aは水、溶
媒Bはアセトニトリルであった。該カラムを、インジェクション後30分間、53%Bで均一濃
度で流した。純粋なフラクションをHPLC-UVにより同定し、合わせた。これらのフラクシ
ョンを減圧下で乾燥させ、対象化合物をオフホワイトの非晶質固体として得た(6mg)。
BIOT-4585の凍結保存された胞子ストックを、室温で解凍した。増殖性培養(種培養)は
、胞子ストック0.4mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内のSM2
5培地400mLに移すことにより調製した。培養を、27℃、250rpm(2.5cm行程)で、48時間行
った。該種培養から、20mLを、フォームプラグを備えた2Lのエルレンマイヤーフラスコ内
の400mLの産生培地SGP2+5%HP20に移した。24℃、250rpm(2.5cm行程)で24時間培養した後
、1M塩酸中のメチル(S)-2-アミノ-3-(3-アミノフェニル)プロパノアートの200mM溶液2mL
を、各産生フラスコに加え、該前駆体の最終濃度を1mMとした。培養を、24℃、250rpm(2.
5cm行程)で、さらに4日間続けた。
清澄化ブロスから該細胞及び樹脂を分離した。該清澄化ブロスを、対象化合物の存在量が
5%未満であることを確認したアッセイの後廃棄した。該細胞及び樹脂を、オーバーヘッド
パドルスターラーを使用して、2倍体積のアセトニトリルで1時間撹拌した。該アセトニト
リル抽出物を、重力下で沈降させることにより回収した。次いで、該細胞及び樹脂の2回
目のアセトニトリル抽出を、同じ条件下で実施した。3回目の抽出物を、残りの細胞及び
樹脂の混合物の遠心分離により得た。
pH6に調整した。これを酢酸エチルで2回抽出し、合わせた有機化合物を減圧下で乾燥させ
、最終粗製物を与えた。
カゲルカラム上にロードする。該カラムを、有機溶媒で、極性を増大させながら溶出させ
る。およそ250mLのフラクションを集め、分析用LCにより対象化合物を同定し、合わせて
乾燥させる。この物質をメタノールに溶解し、調製用HPLCにより精製する。純粋なフラク
ションをHPLC-UVにより同定し、合わせる。これらのフラクションを減圧下で乾燥させ、
対象化合物をオフホワイトの非晶質固体として得る。
)
全般的方法に記載したように、Huh5.2細胞を用いるレプリコンアッセイで、化合物を分
析した。シクロスポリンA(1)、サングリフェリンA(5)、及びヒドロキシマクロ環(6)を、
対照として含めた。
強力であり(低いEC50によって示される)、該株化細胞に対する優れた選択性を有する(高
い選択指数によって示される)。先に記載されたサングリフェリンA(5)は、HCV阻害におい
て14、15及び17よりも効力が低く、シクロスポリンA(1)は、効力が低く、1及び5は両方と
も、選択指数が乏しい。
ヒト及びマウスの肝臓ミクロソームにおける化合物の安定性を、全般的方法に記載した
ように分析した。サングリフェリンA(5)を対照として含めた。
ムにおける安定性が、サングリフェリンA(5)に比べて高く、また、14、15及び17は全て、
マウス肝臓ミクロソームにおいて、より安定である。
限に本願明細書に組み込まれる。
明細書及び以下の請求項の全体にわたって、文脈から反対の意味が要求されない限り、
「含む(comprise)」という言葉並びに「含む(comprises)」及び「含む(comprising)」
などの変形体は、述べられた整数若しくは工程又は整数の群の包含を意味するが、他の整
数若しくは工程又は整数若しくは工程の群の排除を意味しないと理解されるだろう。
予め液体窒素中に保存しておいた凍結保存肝細胞を、37±1℃の振盪水浴に2分±15秒間置く。次いで、該肝細胞を、10X体積の予め温めておいたKrebs-Henseleit重炭酸塩(KHB)緩衝液(2000mg/Lグルコース。炭酸カルシウム及び炭酸水素ナトリウム不含。Sigma社)に加え、穏やかにかき混ぜ、500rpmで3分間遠心分離する。遠心分離後、該上清を注意深く除去し、10X体積の予め温めておいたKHB緩衝液を加えて細胞ペレットを再懸濁する。これを穏やかにかき混ぜて、500rpmで3分間遠心分離する。次いで、該上清を除去し、廃棄する。次いで、細胞生存率及び収率を、細胞数により決定し、これらの値を利用してヒト肝細胞懸濁液を作成し適切な播種密度(生存細胞密度=2×10 6 細胞/mL)にする。2Xの投薬溶液を、予め温めておいたKHB(1%DMSO)中で調製する(200μM添加溶液:DMSO 980μLに基質ストック溶液(10mM)20μL、2X投薬溶液:KHB 990μLに200μM添加溶液10μL(希釈後2μM))。
Claims (21)
- 下記式(I)若しくは式(II)の化合物、又は、それらの医薬として許容し得る塩であって
、それらの任意の互変異性体、又は、トランスとして示されているC26、27のC=C結合がシ
スである、それらの異性体を含み、かつ、C-53ケト及びC-15ヒドロキシル基及びメタノー
ルの組み合わせによりケタールが形成されている、それらのメタノール付加物を含む、前
記化合物、又は、それらの医薬として許容し得る塩:
R1、R2、R3、R4及びR5は、独立に、H、F、Cl、Br、C2-6アルケニル又はC1-10アルキル
を表し、ここで、該アルキル基の1以上の炭素原子は、O、N及びS(O)p(pは0、1又は2を表
す。)から選択されるヘテロ原子によって任意に置き換えられており、かつここで、該ア
ルキル基の1以上の炭素原子は、カルボニルによって任意に置き換えられており、かつ該
アルキル基は、1以上のハロゲン原子によって任意に置換されていてもよく;
X1、X2、X3、X4及びX5は、独立に、C又はNを表し、かつこれらの基のうちNを表すもの
については、結合される置換基が存在せず;
ただし、R1、R3、R4及びR5が全てHを表し、かつX1、X2、X3、X4及びX5が全てCを表す場
合、R2はOHを表すことができない。)。 - X1がCを表す、請求項1記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - X2がCを表す、請求項1記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - X3がCを表す、請求項1記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - X4がCを表す、請求項1記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - X5がCを表す、請求項1記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - R1、R3、R4及びR5が、独立に、H、F、Cl、CF3、OH及びC1-6アルキルから選択される、
請求項1〜6のいずれか1項記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - R2が、H、F、Cl、CF3、OH、NH2及びC1-6アルキルから選択される、請求項1〜7のいずれ
か1項記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として許容し得る塩。 - R2がOHを表す、請求項8記載の式(I)若しくは式(II)の化合物、又はそれらの医薬として
許容し得る塩。 - 以下から選択される、請求項1記載の式(I)若しくは式(II)の化合物であって、それらの
任意の互変異性体、又は、トランスとして示されているC26、27のC=C結合がシスである、
それらの異性体を含み、かつ、C-53ケト及びC-15ヒドロキシル基及びメタノールの組み合
わせによりケタールが形成されている、それらのメタノール付加物を含む、前記化合物、
又は、それらの医薬として許容し得る塩:
(上記式は以下のように表すこともできる。)
(上記式は以下のように表すこともできる。)
- 以下から選択される、請求項1記載の式(I)若しくは式(II)の化合物であって、それらの
任意の互変異性体、又は、トランスとして示されているC26、27のC=C結合がシスである、
それらの異性体を含み、かつ、C-53ケト及びC-15ヒドロキシル基及びメタノールの組み合
わせによりケタールが形成されている、それらのメタノール付加物を含む、前記化合物、
又は、それらの医薬として許容し得る塩:
- 医薬として使用するための、請求項1〜11のいずれか1項記載の化合物。
- HCV若しくはHIV感染などのウイルス感染症、又は、筋ジストロフィーを治療するための
医薬として使用するための、請求項1〜11のいずれか1項記載の化合物。 - 請求項1〜11のいずれか1項記載の化合物を、医薬として許容し得る希釈剤又は担体とと
もに含む、医薬組成物。 - 請求項1〜11のいずれか1項記載の化合物を、医薬として許容し得る希釈剤又は担体とと
もに含み、さらに第2の又は二次の有効成分を含む、医薬組成物。 - 治療有効量の請求項1〜11のいずれか1項記載の化合物を対象に投与することを含む、HC
V若しくはHIV感染などのウイルス感染症、又は、筋ジストロフィーの治療方法。 - 治療有効量の請求項1〜11のいずれか1項記載の化合物を対象に投与することを含む、炎
症性障害の治療方法。 - 炎症性障害を治療するための医薬として使用するための、請求項1〜11のいずれか1項記
載の化合物。 - ストレプトマイセス属種などのサングリフェリン産生細菌に、式(III)の化合物:
いて定義したとおりであり、かつR7は、H又はエステル形成基を表す。)又はそれらの塩
を供給する工程、及び、変異合成サングリフェリンが産生されるように該細菌を培養する
工程を含む、変異合成サングリフェリンの製造方法。 - 前記サングリフェリン産生細菌が、sfaA遺伝子又はsfaA遺伝子のホモログが不活化され
ているか又は欠失しているストレプトマイセス属種である、請求項19記載の方法。 - 前記変異合成サングリフェリンを単離する工程をさらに含む、請求項19又は20記載の方
法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB1021522.6 | 2010-12-20 | ||
| GBGB1021522.6A GB201021522D0 (en) | 2010-12-20 | 2010-12-20 | Novel compounds and methods for their production |
| GBGB1113626.4A GB201113626D0 (en) | 2011-08-08 | 2011-08-08 | Novel compounds and methods for their production |
| GB1113626.4 | 2011-08-08 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013545497A Division JP6086869B2 (ja) | 2010-12-20 | 2011-12-20 | サングリフェリン誘導体及びそれらの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017081925A true JP2017081925A (ja) | 2017-05-18 |
| JP6293852B2 JP6293852B2 (ja) | 2018-03-14 |
Family
ID=45478358
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013545497A Expired - Fee Related JP6086869B2 (ja) | 2010-12-20 | 2011-12-20 | サングリフェリン誘導体及びそれらの製造方法 |
| JP2016225660A Expired - Fee Related JP6293852B2 (ja) | 2010-12-20 | 2016-11-21 | サングリフェリン誘導体及びそれらの製造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013545497A Expired - Fee Related JP6086869B2 (ja) | 2010-12-20 | 2011-12-20 | サングリフェリン誘導体及びそれらの製造方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US9271977B2 (ja) |
| EP (2) | EP2654753B1 (ja) |
| JP (2) | JP6086869B2 (ja) |
| KR (2) | KR101935213B1 (ja) |
| CN (2) | CN107090476B (ja) |
| BR (1) | BR112013015501A2 (ja) |
| CA (2) | CA2822347C (ja) |
| EA (1) | EA022408B1 (ja) |
| ES (1) | ES2703433T3 (ja) |
| MX (1) | MX348758B (ja) |
| WO (1) | WO2012085553A1 (ja) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JO3063B1 (ar) * | 2011-03-29 | 2017-03-15 | Neurovive Pharmaceutical Ab | مركب مبتكر وطرق لانتاجه |
| CN104877006A (zh) * | 2015-06-02 | 2015-09-02 | 宁夏泰益欣生物科技有限公司 | 一种利用萨菲菌素发酵液制备萨菲菌素粗品的方法 |
| CN105695541B (zh) * | 2016-03-10 | 2019-03-15 | 苏州华赛生物工程技术有限公司 | 一种抗生素nvp018中间体的分离纯化方法 |
| AU2017361856A1 (en) | 2016-11-18 | 2019-05-23 | Neurovive Pharmaceutical Ab | Use of sanglifehrin macrocyclic analogues as anticancer compounds |
| WO2021142115A1 (en) * | 2020-01-09 | 2021-07-15 | President And Fellows Of Harvard College | Sanglifehrin analogs and uses thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11509092A (ja) * | 1995-07-04 | 1999-08-17 | ノバルティス・アクチエンゲゼルシャフト | マクロライド |
| JP2008546708A (ja) * | 2005-06-17 | 2008-12-25 | ノバルティス アクチエンゲゼルシャフト | Hcvにおけるサングリフェリンの使用 |
| WO2010034243A1 (en) * | 2008-09-24 | 2010-04-01 | Shanghai Institute Of Organic Chemistry, Chinese Academy Of Sciences | Novel gene cluster |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6124453A (en) * | 1995-07-04 | 2000-09-26 | Novartis Ag | Macrolides |
| US8450281B2 (en) | 2007-01-04 | 2013-05-28 | Debiopharm S.A. | Non-immunosuppressive cyclosporin for treatment of Ullrich congenital muscular dystrophy |
| WO2011098805A1 (en) * | 2010-02-09 | 2011-08-18 | Biotica Technology Limited | Sanglifehrin based compounds |
-
2011
- 2011-12-20 JP JP2013545497A patent/JP6086869B2/ja not_active Expired - Fee Related
- 2011-12-20 CA CA2822347A patent/CA2822347C/en active Active
- 2011-12-20 BR BR112013015501A patent/BR112013015501A2/pt not_active Application Discontinuation
- 2011-12-20 US US13/995,666 patent/US9271977B2/en active Active
- 2011-12-20 EA EA201390925A patent/EA022408B1/ru not_active IP Right Cessation
- 2011-12-20 MX MX2013007055A patent/MX348758B/es active IP Right Grant
- 2011-12-20 EP EP11808295.7A patent/EP2654753B1/en active Active
- 2011-12-20 CA CA3028414A patent/CA3028414C/en active Active
- 2011-12-20 KR KR1020187006060A patent/KR101935213B1/ko not_active Expired - Fee Related
- 2011-12-20 WO PCT/GB2011/052524 patent/WO2012085553A1/en not_active Ceased
- 2011-12-20 ES ES11808295T patent/ES2703433T3/es active Active
- 2011-12-20 KR KR1020137018964A patent/KR101900974B1/ko active Active
- 2011-12-20 CN CN201710086249.2A patent/CN107090476B/zh not_active Expired - Fee Related
- 2011-12-20 EP EP18188805.8A patent/EP3427737A1/en not_active Withdrawn
- 2011-12-20 CN CN201180068004.8A patent/CN103619336B/zh not_active Expired - Fee Related
-
2016
- 2016-11-21 JP JP2016225660A patent/JP6293852B2/ja not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH11509092A (ja) * | 1995-07-04 | 1999-08-17 | ノバルティス・アクチエンゲゼルシャフト | マクロライド |
| JP2008546708A (ja) * | 2005-06-17 | 2008-12-25 | ノバルティス アクチエンゲゼルシャフト | Hcvにおけるサングリフェリンの使用 |
| WO2010034243A1 (en) * | 2008-09-24 | 2010-04-01 | Shanghai Institute Of Organic Chemistry, Chinese Academy Of Sciences | Novel gene cluster |
Non-Patent Citations (1)
| Title |
|---|
| BIOORGANIC AND MEDICINAL CHEMISTRY LETTERS, vol. 11, JPN6015049962, 2001, pages 1609 - 1612, ISSN: 0003638307 * |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2822347C (en) | 2019-11-05 |
| ES2703433T3 (es) | 2019-03-08 |
| AU2011346823A1 (en) | 2013-07-18 |
| MX348758B (es) | 2017-06-28 |
| KR101935213B1 (ko) | 2019-01-03 |
| EP3427737A1 (en) | 2019-01-16 |
| CN107090476B (zh) | 2021-09-24 |
| EA022408B1 (ru) | 2015-12-30 |
| CA3028414C (en) | 2021-01-26 |
| EA201390925A1 (ru) | 2014-01-30 |
| AU2011346823A8 (en) | 2016-09-01 |
| US20140080837A1 (en) | 2014-03-20 |
| CN107090476A (zh) | 2017-08-25 |
| KR20140021532A (ko) | 2014-02-20 |
| MX2013007055A (es) | 2013-11-01 |
| KR20180027626A (ko) | 2018-03-14 |
| CA3028414A1 (en) | 2012-06-28 |
| KR101900974B1 (ko) | 2018-09-21 |
| EP2654753B1 (en) | 2018-10-31 |
| US9271977B2 (en) | 2016-03-01 |
| EP2654753A1 (en) | 2013-10-30 |
| CN103619336A (zh) | 2014-03-05 |
| WO2012085553A1 (en) | 2012-06-28 |
| JP6293852B2 (ja) | 2018-03-14 |
| BR112013015501A2 (pt) | 2017-05-16 |
| JP6086869B2 (ja) | 2017-03-08 |
| JP2014501752A (ja) | 2014-01-23 |
| AU2011346823B2 (en) | 2016-08-04 |
| CN103619336B (zh) | 2017-10-27 |
| CA2822347A1 (en) | 2012-06-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6118792B2 (ja) | マクロ環状化合物及びその製造方法 | |
| JP6293852B2 (ja) | サングリフェリン誘導体及びそれらの製造方法 | |
| JP5894935B2 (ja) | サングリフェリン系化合物 | |
| WO2011098808A1 (en) | Sanglifehrin based compounds | |
| CN103987377A (zh) | 新剂型 | |
| WO2011098805A1 (en) | Sanglifehrin based compounds | |
| WO2011144924A1 (en) | Amine or amid analogues of sanglifehrin | |
| AU2011346823B8 (en) | Sanglifehrin derivatives and methods for their production | |
| CN102770139B (zh) | 基于Sanglifehrin的化合物 | |
| HK1188561A (en) | Sanglifehrin derivatives and methods for their production | |
| HK1188561B (en) | Sanglifehrin derivatives and methods for their production |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20170327 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20170912 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20171127 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20171227 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20180123 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20180214 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6293852 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S531 | Written request for registration of change of domicile |
Free format text: JAPANESE INTERMEDIATE CODE: R313531 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |