JP2017100989A - ブタジエンの製造方法 - Google Patents
ブタジエンの製造方法 Download PDFInfo
- Publication number
- JP2017100989A JP2017100989A JP2015234994A JP2015234994A JP2017100989A JP 2017100989 A JP2017100989 A JP 2017100989A JP 2015234994 A JP2015234994 A JP 2015234994A JP 2015234994 A JP2015234994 A JP 2015234994A JP 2017100989 A JP2017100989 A JP 2017100989A
- Authority
- JP
- Japan
- Prior art keywords
- gas
- reactor
- raw material
- concentration
- butadiene
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/42—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with a hydrogen acceptor
- C07C5/48—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by dehydrogenation with a hydrogen acceptor with oxygen as an acceptor
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/002—Mixed oxides other than spinels, e.g. perovskite
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/24—Chromium, molybdenum or tungsten
- B01J23/31—Chromium, molybdenum or tungsten combined with bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- B01J23/85—Chromium, molybdenum or tungsten
- B01J23/88—Molybdenum
- B01J23/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/8876—Arsenic, antimony or bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/04—Mixing
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J37/00—Processes, in general, for preparing catalysts; Processes, in general, for activation of catalysts
- B01J37/08—Heat treatment
- B01J37/082—Decomposition and pyrolysis
- B01J37/088—Decomposition of a metal salt
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
- C07C11/12—Alkadienes
- C07C11/16—Alkadienes with four carbon atoms
- C07C11/167—1, 3-Butadiene
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/50—Constitutive chemical elements of heterogeneous catalysts of Group V (VA or VB) of the Periodic Table
- B01J2523/54—Bismuth
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2523/00—Constitutive chemical elements of heterogeneous catalysts
- B01J2523/60—Constitutive chemical elements of heterogeneous catalysts of Group VI (VIA or VIB) of the Periodic Table
- B01J2523/68—Molybdenum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/18—Arsenic, antimony or bismuth
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/24—Chromium, molybdenum or tungsten
- C07C2523/28—Molybdenum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/24—Chromium, molybdenum or tungsten
- C07C2523/31—Chromium, molybdenum or tungsten combined with bismuth
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/74—Iron group metals
- C07C2523/745—Iron
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/74—Iron group metals
- C07C2523/75—Cobalt
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36
- C07C2523/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/85—Chromium, molybdenum or tungsten
- C07C2523/88—Molybdenum
- C07C2523/887—Molybdenum containing in addition other metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Physics & Mathematics (AREA)
- Thermal Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Catalysts (AREA)
Abstract
Description
<反応ガス組成>
直鎖状ブテン:炭素数4の炭化水素の合計に対して、50〜100体積%
炭素数4の炭化水素:反応ガス全量に対して、5〜15体積%
O2:炭素数4の炭化水素の合計に対して、40〜120体積%
N2:炭素数4の炭化水素の合計に対して、500〜1000体積%
H2O:炭素数4の炭化水素の合計に対して、90〜900体積%
以下に、本実施形態に係る製造方法で用いられる触媒(酸化脱水素反応触媒)の好適な態様について詳述する。
(Mo)a(Bi)b(Co)c(Ni)d(Fe)e(X)f(Y)g(Z)h(Si)i(O)j …(1)
(式中、Xはマグネシウム(Mg)、カルシウム(Ca)、亜鉛(Zn)、セリウム(Ce)及びサマリウム(Sm)からなる群より選択される少なくとも1種の元素を示し、Yはナトリウム(Na)、カリウム(K)、ルビジウム(Rb)、セシウム(Cs)及びタリウム(Tl)からなる群より選択される少なくとも1種の元素を示し、Zはホウ素(B)、リン(P)、砒素(As)及びタングステン(W)からなる群より選択される少なくとも1種の元素を示す。また、a〜jはそれぞれの元素の原子比を表し、a=12のとき、b=0.5〜7、c=0〜10、d=0〜10(但しc+d=1〜10)、e=0.05〜3、f=0〜2、g=0.04〜2、h=0〜3、i=0〜48の範囲にあり、またjは他の元素の酸化状態を満足させる数値である。)
硝酸コバルト・六水和物12.3g及び硝酸鉄・九水和物5.8gを純水25.0gに加え、常温で撹拌し、溶解させた。この溶液を溶液Aと称す。
内径10.9mm、長さ300mmのステンレス製反応管に、製造例1で製造された複合酸化物触媒3.0mLを充填した。反応管には外径3.1mmのステンレス管を挿入し、挿入管の中に熱電対を設置して反応器内の温度を測定した。なお、熱媒体は電気炉を使用した。
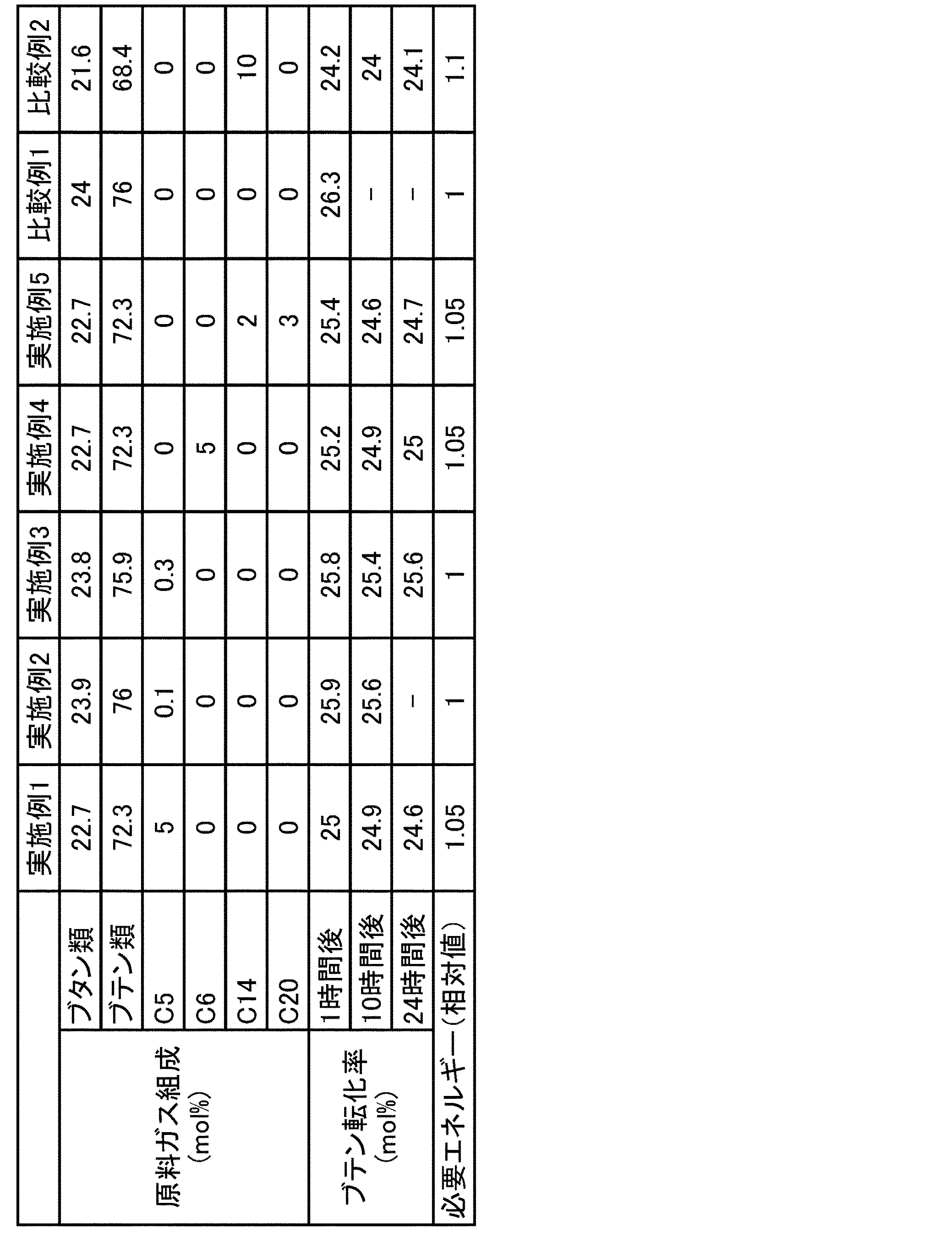
原料ガスの組成を表1に記載の組成に変更したこと以外は、実施例1と同様の条件で反応を行った。反応開始から1時間後及び10時間後に生成ガスをサンプリングし、実施例1と同様の条件で分析を行った。分析の結果、直鎖状ブテンの転化率は表1に示すとおりであった。
原料ガスの組成を表1に記載の組成に変更したこと以外は、実施例1と同様の条件で反応を行った。反応開始から1時間後、10時間後及び24時間後に生成ガスをサンプリングし、実施例1と同様の条件で分析を行った。分析の結果、直鎖状ブテンの転化率は表1に示すとおりであった。
原料ガスの組成を表1に記載の組成に変更したこと以外は、実施例1と同様の条件で反応を行った。反応開始から1時間後、10時間後及び24時間後に生成ガスをサンプリングし、実施例1と同様の条件で分析を行った。分析の結果、直鎖状ブテンの転化率は表1に示すとおりであった。
原料ガスの組成を表1に記載の組成に変更したこと以外は、実施例1と同様の条件で反応を行った。反応開始から1時間後、10時間後及び24時間後に生成ガスをサンプリングし、実施例1と同様の条件で分析を行った。分析の結果、直鎖状ブテンの転化率は表1に示すとおりであった。
原料ガスの組成を表1に記載の組成に変更したこと以外は、実施例1と同様の条件で反応を行った。反応開始後、1時間後にサンプリングし、実施例1と同様の条件で分析を行った。分析の結果、直鎖状ブテンの転化率は表1に示すとおりであった。
原料ガスの組成を表1に記載の組成に変更したこと以外は、実施例1と同様の条件で反応を行った。反応開始から1時間後、10時間後及び24時間後に生成ガスをサンプリングし、実施例1と同様の条件で分析を行った。分析の結果、直鎖状ブテンの転化率は表1に示すとおりであった。
Claims (3)
- 直鎖状ブテンを含有する原料ガスと分子状酸素を含有する酸素含有ガスとを反応器に供給し、触媒の存在下で酸化脱水素反応を行って、ブタジエンを含有する生成ガスを得る工程を備え、
前記触媒が、モリブデン及びビスマスを含有する複合酸化物を含み、
前記原料ガス中の炭素原子数5以上の炭化水素の濃度が0.05〜7.0mol%である、ブタジエンの製造方法。 - 前記原料ガス中の炭素原子数5以上の炭化水素の濃度が0.2〜6.0mol%である、請求項1に記載の製造方法。
- 前記原料ガス中の前記直鎖状ブテンの濃度が60mol%以上である、請求項1又は2に記載の製造方法。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015234994A JP6716237B2 (ja) | 2015-12-01 | 2015-12-01 | ブタジエンの製造方法 |
| PCT/JP2016/080971 WO2017094382A1 (ja) | 2015-12-01 | 2016-10-19 | ブタジエンの製造方法 |
| US15/779,615 US10647638B2 (en) | 2015-12-01 | 2016-10-19 | Method for producing butadiene |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015234994A JP6716237B2 (ja) | 2015-12-01 | 2015-12-01 | ブタジエンの製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017100989A true JP2017100989A (ja) | 2017-06-08 |
| JP6716237B2 JP6716237B2 (ja) | 2020-07-01 |
Family
ID=58796984
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015234994A Active JP6716237B2 (ja) | 2015-12-01 | 2015-12-01 | ブタジエンの製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US10647638B2 (ja) |
| JP (1) | JP6716237B2 (ja) |
| WO (1) | WO2017094382A1 (ja) |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60115532A (ja) * | 1983-11-25 | 1985-06-22 | Nippon Zeon Co Ltd | ブタジエンの製造方法 |
| JPS60126235A (ja) * | 1983-12-14 | 1985-07-05 | Nippon Zeon Co Ltd | ブタジエンの製造方法 |
| JP2003220335A (ja) * | 2001-11-21 | 2003-08-05 | Mitsubishi Chemicals Corp | 複合酸化物触媒の製造方法 |
| WO2010137595A1 (ja) * | 2009-05-29 | 2010-12-02 | 三菱化学株式会社 | 共役ジエンの製造方法 |
| JP2011006381A (ja) * | 2009-05-29 | 2011-01-13 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
| JP2012077076A (ja) * | 2010-09-10 | 2012-04-19 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
| JP2012106942A (ja) * | 2010-11-16 | 2012-06-07 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
| WO2012157495A1 (ja) * | 2011-05-19 | 2012-11-22 | 旭化成ケミカルズ株式会社 | 共役ジオレフィンの製造方法及び製造装置 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012111699A (ja) | 2010-11-22 | 2012-06-14 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
-
2015
- 2015-12-01 JP JP2015234994A patent/JP6716237B2/ja active Active
-
2016
- 2016-10-19 US US15/779,615 patent/US10647638B2/en active Active
- 2016-10-19 WO PCT/JP2016/080971 patent/WO2017094382A1/ja not_active Ceased
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60115532A (ja) * | 1983-11-25 | 1985-06-22 | Nippon Zeon Co Ltd | ブタジエンの製造方法 |
| JPS60126235A (ja) * | 1983-12-14 | 1985-07-05 | Nippon Zeon Co Ltd | ブタジエンの製造方法 |
| JP2003220335A (ja) * | 2001-11-21 | 2003-08-05 | Mitsubishi Chemicals Corp | 複合酸化物触媒の製造方法 |
| WO2010137595A1 (ja) * | 2009-05-29 | 2010-12-02 | 三菱化学株式会社 | 共役ジエンの製造方法 |
| JP2011006381A (ja) * | 2009-05-29 | 2011-01-13 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
| JP2012077076A (ja) * | 2010-09-10 | 2012-04-19 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
| JP2012106942A (ja) * | 2010-11-16 | 2012-06-07 | Mitsubishi Chemicals Corp | 共役ジエンの製造方法 |
| WO2012157495A1 (ja) * | 2011-05-19 | 2012-11-22 | 旭化成ケミカルズ株式会社 | 共役ジオレフィンの製造方法及び製造装置 |
Non-Patent Citations (1)
| Title |
|---|
| JUNG, JI CHUL ET AL.: "Effect of calcination temperature on the catalytic performance of Co9Fe3Bi1Mo12O51 in the oxidative", CATALYSIS COMMUNICATIONS, vol. 9, no. 10, JPN6016050058, 2008, pages 2059 - 2062, ISSN: 0003993413 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20180346395A1 (en) | 2018-12-06 |
| US10647638B2 (en) | 2020-05-12 |
| WO2017094382A1 (ja) | 2017-06-08 |
| JP6716237B2 (ja) | 2020-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR101741930B1 (ko) | 산화물 촉매 및 그 제조 방법, 및 불포화 알데히드, 디올레핀 및 불포화 니트릴의 제조 방법 | |
| JP5371692B2 (ja) | 共役ジオレフィンの製造方法 | |
| WO2010137595A1 (ja) | 共役ジエンの製造方法 | |
| JP2011178719A (ja) | ブタジエンの製造方法 | |
| JP5895546B2 (ja) | 複合酸化物触媒及び共役ジエンの製造方法 | |
| JP5874488B2 (ja) | 複合金属酸化物触媒及び共役ジエンの製造方法 | |
| KR101726113B1 (ko) | 부타디엔의 제조 방법 | |
| JP5767795B2 (ja) | ブタジエンの製造方法 | |
| TWI541229B (zh) | 共軛二烯之製造方法 | |
| JP6229201B2 (ja) | 複合金属酸化物触媒及び共役ジエンの製造方法 | |
| JP6716237B2 (ja) | ブタジエンの製造方法 | |
| JP7210262B2 (ja) | ブタジエンの製造方法 | |
| JP5825981B2 (ja) | 共役ジオレフィンの製造方法 | |
| JP6405857B2 (ja) | 共役ジエンの製造方法 | |
| US11452978B2 (en) | Catalytic oxidation method and method for producing conjugated diene | |
| CN105916579B (zh) | 制备丁二烯的方法 | |
| JP5750252B2 (ja) | ブタジエンの製造方法 | |
| JP2012197272A (ja) | 共役ジエンの製造方法 | |
| JP2012072076A (ja) | 共役ジオレフィンの製造方法 | |
| JP2014189543A (ja) | 共役ジエンの製造方法 | |
| JP6540422B2 (ja) | 複合酸化物触媒 | |
| JP6443074B2 (ja) | 複合金属酸化物触媒及び共役ジエンの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180410 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190312 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190513 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20191023 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20191223 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20200519 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20200610 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6716237 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R157 | Certificate of patent or utility model (correction) |
Free format text: JAPANESE INTERMEDIATE CODE: R157 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |