JP2017105748A - 1,5−アンヒドロ−1−置換フェニル−d−グルシトール化合物の製造方法 - Google Patents
1,5−アンヒドロ−1−置換フェニル−d−グルシトール化合物の製造方法 Download PDFInfo
- Publication number
- JP2017105748A JP2017105748A JP2016119447A JP2016119447A JP2017105748A JP 2017105748 A JP2017105748 A JP 2017105748A JP 2016119447 A JP2016119447 A JP 2016119447A JP 2016119447 A JP2016119447 A JP 2016119447A JP 2017105748 A JP2017105748 A JP 2017105748A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- range
- formula
- compound represented
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- BGEQKRXVLNGNHH-XRIGFGBMSA-N C/[O]=C(/[C@@H]([N]#C)/[O]=[Mn]/C)\C=N Chemical compound C/[O]=C(/[C@@H]([N]#C)/[O]=[Mn]/C)\C=N BGEQKRXVLNGNHH-XRIGFGBMSA-N 0.000 description 1
- 0 CNC(*=C)c1cc(ONC)ccc1Cc1ccc(*)cc1 Chemical compound CNC(*=C)c1cc(ONC)ccc1Cc1ccc(*)cc1 0.000 description 1
Landscapes
- Saccharide Compounds (AREA)
Abstract
【課題】ナトリウム依存性グルコース共輸送体1の阻害活を有する医薬として有用な、1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の新規な製造方法の提供。
【解決手段】式(XI)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、式(II)で表される化合物を複数の工程により変換して、式(XI)で表される化合物を製造する方法。
【選択図】なし
【解決手段】式(XI)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、式(II)で表される化合物を複数の工程により変換して、式(XI)で表される化合物を製造する方法。
【選択図】なし
Description
本発明は、ナトリウム依存性グルコース共輸送体1(sodium-dependent glucose
cotransporter 1、以下、SGLT1と記載する)阻害活性を有する医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物、及びその製造中間体の製造方法に関する。さらに詳しくは、(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール、及びその製造中間体である(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトールの製造方法に関する。
cotransporter 1、以下、SGLT1と記載する)阻害活性を有する医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物、及びその製造中間体の製造方法に関する。さらに詳しくは、(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール、及びその製造中間体である(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトールの製造方法に関する。
SGLT1は、小腸粘膜上皮、腎臓近位尿細管、骨格筋、心筋、気管及び脳に発現しており、糖の細胞内への吸収・取り込みに関与する能動輸送性の糖輸送担体である。SGLT1は小腸において特に多く存在しており、食事にて摂取した炭水化物の消化により生じたグルコース及びガラクトースの細胞内への吸収に関与している。小腸に発現しているSGLT1の機能を阻害する化合物は、糖の吸収を抑制することから、糖尿病治療薬として有用であるとして医薬品開発が行われている(特許文献1〜3)。また、SGLT1を阻害する化合物は便秘症の予防または治療に有用であることが、特許文献4に開示されている。
特許文献1〜4に記載された(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(以下、化合物(I)と記載する)の製造方法は、超低温(−80〜−70℃)反応工程及びシリカゲルカラムクロマトグラフィー精製工程を含み、工業的大量生産に適した製造方法とは言えなかった。
例えば、特許文献4に開示されている化合物(I)の製造方法をスキーム1に示す。
スキーム1
例えば、特許文献4に開示されている化合物(I)の製造方法をスキーム1に示す。
スキーム1
また、特許文献1〜4に記載された(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(以下、化合物(XI)と記載する)の製造方法も、シリカゲルカラムクロマトグラフィー精製工程を含み、工業的大量生産に適した製造方法とは言えなかった。
例えば、特許文献2及び特許文献4に開示されている、化合物(XI)の製造方法をスキーム2に示す。化合物(XI)は非晶質の化合物であるが、化合物(XI)のエタノール溶媒和物は結晶として得ることができる。化合物(XI)のエタノール溶媒和物は、他の結晶形(A形、B形、C形、二水和物;特許文献2)に容易に変動することが可能である。
スキーム2
例えば、特許文献2及び特許文献4に開示されている、化合物(XI)の製造方法をスキーム2に示す。化合物(XI)は非晶質の化合物であるが、化合物(XI)のエタノール溶媒和物は結晶として得ることができる。化合物(XI)のエタノール溶媒和物は、他の結晶形(A形、B形、C形、二水和物;特許文献2)に容易に変動することが可能である。
スキーム2
Organic Letters、2012、vol.14、No.6、1480−1483.
本発明の目的は、医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の、工業的大量生産に適した製造方法を提供することである。
本発明者らは、上記課題を解決すべく鋭意検討した結果、化合物(I)を製造中間体として経由し、超低温反応工程及びシリカゲルカラムクロマトグラフィー精製工程を回避した化合物(XI)の製造方法を見出し、本発明を完成した。
すなわち、本発明は以下の通りである。
(1)式(XI)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
(1)式(XI)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
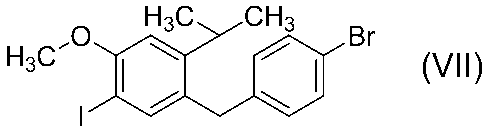
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(vi)前記式(VII)で表される化合物を式(XII)で表される化合物に変換する工程と、
(vii)前記式(XII)で表される化合物を式(XIII)で表される化合物に変換する工程と、
(viii)前記式(XIII)で表される化合物を式(I)で表される化合物に変換する工程と、
(ix)前記式(I)で表される化合物を式(XIV)で表される化合物に変換する工程と、
(x)前記式(XIV)で表される化合物を式(XV)で表される化合物に変換する工程と、
(xi)前記式(XV)で表される化合物を前記式(XI)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。
(2)式(I)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(vi)前記式(VII)で表される化合物を式(VIII)で表される化合物に変換する工程と、
(vii)前記式(VIII)で表される化合物を式(IX)で表される化合物に変換する工程と、
(viii)前記式(IX)で表される化合物を式(X)で表される化合物に変換する工程と、
(ix)前記式(X)で表される化合物を前記式(I)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。
本発明により、SGLT1阻害活性を有する医薬として有用な化合物(XI)の工業的大量生産が可能になった。
以下に、本発明の具体的な態様、置換基の定義等を説明する。
本発明において、「n」はノルマル(normal)を、「i」はイソ(iso)を、「s」及び「sec」はセカンダリー(secondary)を、「t」及び「tert」はターシャリー(tertiary)を、「c」はシクロ(cyclo)を、「o」はオルト(ortho)を、「m」はメタ(meta)を、「p」はパラ(para)を示す。「Ac」はアセチル(acetyl)を、「TMS」はトリメチルシリル(trimethylsilyl)を、「Piv」はピバロイル(pivaloyl)を示す。
以下、本発明を実施するための形態を具体的に説明する。
本発明は、例えば、以下のスキーム3〜5に示す方法によって実施することができるが、この方法により限定的に解釈されるものではない。
製造中間体である式(I)で表される化合物は、以下のスキーム3に示す方法によって得ることができる。
スキーム3
スキーム3
工程(i):
化合物(1)を不活性溶媒中、添加剤の存在下または非存在下、メチル化剤と反応させることにより、化合物(2)を得ることができる。出発原料となる化合物(1)は、市販品として入手可能である。
メチル化剤としては、例えば、塩化メチルマグネシウム、臭化メチルマグネシウム、よう化メチルマグネシウム、メチルリチウム等を使用することができる。
不活性溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
添加剤としては、例えば、塩化セリウム(III)、塩化亜鉛、塩化ランタン(III)ビス(塩化リチウム)錯体等が挙げられる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜100℃の範囲であり、より好ましくは20〜40℃の範囲である。
メチル化剤の使用量は、原料の化合物(1)に対して2〜6モル当量の範囲で使用することができ、好ましくは2.5〜4モル当量の範囲であり、より好ましくは2.5〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(1)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(2)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
化合物(1)を不活性溶媒中、添加剤の存在下または非存在下、メチル化剤と反応させることにより、化合物(2)を得ることができる。出発原料となる化合物(1)は、市販品として入手可能である。
メチル化剤としては、例えば、塩化メチルマグネシウム、臭化メチルマグネシウム、よう化メチルマグネシウム、メチルリチウム等を使用することができる。
不活性溶媒としては、例えば、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
添加剤としては、例えば、塩化セリウム(III)、塩化亜鉛、塩化ランタン(III)ビス(塩化リチウム)錯体等が挙げられる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜100℃の範囲であり、より好ましくは20〜40℃の範囲である。
メチル化剤の使用量は、原料の化合物(1)に対して2〜6モル当量の範囲で使用することができ、好ましくは2.5〜4モル当量の範囲であり、より好ましくは2.5〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(1)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(2)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
工程(ii):
化合物(2)を不活性溶媒中、または溶媒非存在下、還元剤及び酸と反応させることにより、化合物(3)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜60℃の範囲である。
還元剤の使用量は、原料の化合物(2)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1.5〜3モル当量の範囲である。
酸の使用量は、原料の化合物(2)に対して1〜20モル当量の範囲で使用することができ、好ましくは1〜10モル当量の範囲であり、より好ましくは1.5〜5モル当量の範囲である。
溶媒の使用量は、原料の化合物(2)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(3)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(3)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(2)を不活性溶媒中、または溶媒非存在下、還元剤及び酸と反応させることにより、化合物(3)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜60℃の範囲である。
還元剤の使用量は、原料の化合物(2)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1.5〜3モル当量の範囲である。
酸の使用量は、原料の化合物(2)に対して1〜20モル当量の範囲で使用することができ、好ましくは1〜10モル当量の範囲であり、より好ましくは1.5〜5モル当量の範囲である。
溶媒の使用量は、原料の化合物(2)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(3)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(3)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(iii):
化合物(3)を不活性溶媒中、活性化剤存在下、マグネシウムと反応させてグリニャール試薬(4)を調製後、4−ブロモベンズアルデヒド(5)と反応させることにより、化合物(6)を得ることができる。
活性化剤としては、例えば、よう素、1,2−ジブロモエタン、水素化ジイソブチルアルミニウム等を使用することができる。
不活性溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
グリニャール試薬(4)調製時の反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは20〜100℃の範囲であり、より好ましくは40〜80℃の範囲である。
グリニャール試薬(4)と化合物(5)の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−20〜100℃の範囲であり、より好ましくは0〜30℃の範囲である。
マグネシウムの使用量は、原料の化合物(3)に対して1〜5モル当量の範囲で使用することができ、好ましくは1〜3モル当量の範囲であり、より好ましくは1〜1.2モル当量の範囲である。
活性化剤の使用量は、原料の化合物(3)に対して0.0001〜1モル当量の範囲で使用することができ、好ましくは0.001〜0.1モル当量の範囲であり、より好ましくは0.001〜0.01モル当量の範囲である。
化合物(5)の使用量は、原料の化合物(3)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(3)及び化合物(5)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(6)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(6)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(3)を不活性溶媒中、活性化剤存在下、マグネシウムと反応させてグリニャール試薬(4)を調製後、4−ブロモベンズアルデヒド(5)と反応させることにより、化合物(6)を得ることができる。
活性化剤としては、例えば、よう素、1,2−ジブロモエタン、水素化ジイソブチルアルミニウム等を使用することができる。
不活性溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
グリニャール試薬(4)調製時の反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは20〜100℃の範囲であり、より好ましくは40〜80℃の範囲である。
グリニャール試薬(4)と化合物(5)の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−20〜100℃の範囲であり、より好ましくは0〜30℃の範囲である。
マグネシウムの使用量は、原料の化合物(3)に対して1〜5モル当量の範囲で使用することができ、好ましくは1〜3モル当量の範囲であり、より好ましくは1〜1.2モル当量の範囲である。
活性化剤の使用量は、原料の化合物(3)に対して0.0001〜1モル当量の範囲で使用することができ、好ましくは0.001〜0.1モル当量の範囲であり、より好ましくは0.001〜0.01モル当量の範囲である。
化合物(5)の使用量は、原料の化合物(3)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(3)及び化合物(5)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(6)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(6)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(iv):
化合物(6)を不活性溶媒中、または溶媒非存在下、還元剤及び酸と反応させることにより、化合物(7)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜60℃の範囲である。
還元剤の使用量は、原料の化合物(6)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜2モル当量の範囲である。
酸の使用量は、原料の化合物(6)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(6)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(7)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(7)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(6)を不活性溶媒中、または溶媒非存在下、還元剤及び酸と反応させることにより、化合物(7)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜60℃の範囲である。
還元剤の使用量は、原料の化合物(6)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜2モル当量の範囲である。
酸の使用量は、原料の化合物(6)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(6)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(7)は、蒸留、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(7)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(v):
化合物(7)を不活性溶媒中、よう素化剤と反応させることにより、化合物(8)を得ることができる。
よう素化剤としては、例えば、一塩化よう素、ピリジン一塩化よう素、よう素−過酸化水素、よう素−よう素酸カリウム−酸、よう素―硝酸アンモニウムセリウム(IV)、よう化カリウム−過酸化水素、よう化ナトリウム−N−クロロコハク酸イミド、N−ヨードコハク酸イミド、1,3−ジヨード−5,5−ジメチルヒダントイン−酸、N−ヨードサッカリン、ジクロロよう素酸ベンジルトリメチルアンモニウム−塩化亜鉛等を使用することができる。
酸としては、例えば、塩酸、硫酸、酢酸等を使用することができる。
不活性溶媒としては、例えば、メタノール、エタノール、1−プロパノール、2−プロパノール、tert−ブチルアルコール等のアルコール系溶媒、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜80℃の範囲である。
よう素化剤の使用量は、原料の化合物(7)に対して0.1〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
酸の使用量は、原料の化合物(7)に対して0.1〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.1〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(7)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(8)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
化合物(7)を不活性溶媒中、よう素化剤と反応させることにより、化合物(8)を得ることができる。
よう素化剤としては、例えば、一塩化よう素、ピリジン一塩化よう素、よう素−過酸化水素、よう素−よう素酸カリウム−酸、よう素―硝酸アンモニウムセリウム(IV)、よう化カリウム−過酸化水素、よう化ナトリウム−N−クロロコハク酸イミド、N−ヨードコハク酸イミド、1,3−ジヨード−5,5−ジメチルヒダントイン−酸、N−ヨードサッカリン、ジクロロよう素酸ベンジルトリメチルアンモニウム−塩化亜鉛等を使用することができる。
酸としては、例えば、塩酸、硫酸、酢酸等を使用することができる。
不活性溶媒としては、例えば、メタノール、エタノール、1−プロパノール、2−プロパノール、tert−ブチルアルコール等のアルコール系溶媒、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜80℃の範囲である。
よう素化剤の使用量は、原料の化合物(7)に対して0.1〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
酸の使用量は、原料の化合物(7)に対して0.1〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.1〜3モル当量の範囲である。
溶媒の使用量は、原料の化合物(7)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(8)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
工程(vi):
化合物(8)を不活性溶媒中、有機マグネシウム試薬と反応させてグリニャール試薬(9)を調製後、化合物(10)と反応させることにより、化合物(11)を得ることができる。なお、化合物(10)は、特許文献US2002/0137903A1記載の方法にて合成することができる。
有機マグネシウム試薬としては、例えば、塩化イソプロピルマグネシウム、塩化イソプロピルマグネシウム−塩化リチウム錯体、塩化2−ブチルマグネシウム−塩化リチウム錯体、塩化イソプロピルマグネシウム−ビス[2−(N,N−ジメチルアミノ)エチル]エーテル錯体等を使用することができる。
不活性溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
グリニャール試薬(9)調製時の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜20℃の範囲であり、より好ましくは−20〜0℃の範囲である。グリニャール試薬(9)と化合物(10)の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜20℃の範囲であり、より好ましくは−20〜0℃の範囲である。
有機マグネシウム試薬の使用量は、原料の化合物(8)に対して0.9〜2モル当量の範囲で使用することができ、好ましくは0.95〜1.1モル当量の範囲であり、より好ましくは0.95〜1.05モル当量の範囲である。
化合物(10)の使用量は、原料の化合物(8)に対して0.9〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(8)及び化合物(10)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(11)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、または後処理することなく反応液のまま、次の工程の原料として使用することができる。
化合物(8)を不活性溶媒中、有機マグネシウム試薬と反応させてグリニャール試薬(9)を調製後、化合物(10)と反応させることにより、化合物(11)を得ることができる。なお、化合物(10)は、特許文献US2002/0137903A1記載の方法にて合成することができる。
有機マグネシウム試薬としては、例えば、塩化イソプロピルマグネシウム、塩化イソプロピルマグネシウム−塩化リチウム錯体、塩化2−ブチルマグネシウム−塩化リチウム錯体、塩化イソプロピルマグネシウム−ビス[2−(N,N−ジメチルアミノ)エチル]エーテル錯体等を使用することができる。
不活性溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、またはこれらの混合溶媒等を使用することができる。
グリニャール試薬(9)調製時の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜20℃の範囲であり、より好ましくは−20〜0℃の範囲である。グリニャール試薬(9)と化合物(10)の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜20℃の範囲であり、より好ましくは−20〜0℃の範囲である。
有機マグネシウム試薬の使用量は、原料の化合物(8)に対して0.9〜2モル当量の範囲で使用することができ、好ましくは0.95〜1.1モル当量の範囲であり、より好ましくは0.95〜1.05モル当量の範囲である。
化合物(10)の使用量は、原料の化合物(8)に対して0.9〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(8)及び化合物(10)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(11)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、または後処理することなく反応液のまま、次の工程の原料として使用することができる。
工程(vii):
化合物(11)を酸存在下、メタノール溶媒中にて反応させることにより、化合物(12)を得ることができる。
酸としては、例えば、塩化水素、臭化水素、よう化水素、硫酸、メタンスルホン酸、パラトルエンスルホン酸、ベンゼンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜50℃の範囲である。
酸の使用量は、原料の化合物(11)に対して0.01〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
メタノールの使用量は、原料の化合物(11)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(12)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(12)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(11)を酸存在下、メタノール溶媒中にて反応させることにより、化合物(12)を得ることができる。
酸としては、例えば、塩化水素、臭化水素、よう化水素、硫酸、メタンスルホン酸、パラトルエンスルホン酸、ベンゼンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜50℃の範囲である。
酸の使用量は、原料の化合物(11)に対して0.01〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
メタノールの使用量は、原料の化合物(11)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(12)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(12)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(viii):
化合物(12)を不活性溶媒中、塩基存在下、アセチル化剤と反応させることにより、化合物(13)を得ることができる。
アセチル化剤としては、例えば、塩化アセチル、臭化アセチル、無水酢酸等を使用することができる。
塩基としては、例えば、ピリジン、コリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミン等を使用することができる。ピリジンは溶媒としても使用することができる。触媒として4−ジメチルアミノピリジンを使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜40℃の範囲である。
アセチル化剤の使用量は、原料の化合物(12)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
塩基の使用量は、原料の化合物(12)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
溶媒の使用量は、原料の化合物(12)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(13)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(13)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(12)を不活性溶媒中、塩基存在下、アセチル化剤と反応させることにより、化合物(13)を得ることができる。
アセチル化剤としては、例えば、塩化アセチル、臭化アセチル、無水酢酸等を使用することができる。
塩基としては、例えば、ピリジン、コリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミン等を使用することができる。ピリジンは溶媒としても使用することができる。触媒として4−ジメチルアミノピリジンを使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜40℃の範囲である。
アセチル化剤の使用量は、原料の化合物(12)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
塩基の使用量は、原料の化合物(12)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
溶媒の使用量は、原料の化合物(12)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(13)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(13)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(ix):
化合物(13)を不活性溶媒中、水存在下または非存在下、還元剤及び酸と反応させることにより、化合物(I)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜50℃の範囲である。
還元剤の使用量は、原料の化合物(13)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜2モル当量の範囲である。
酸の使用量は、原料の化合物(13)に対して1〜20モル当量の範囲で使用することができ、好ましくは1〜10モル当量の範囲であり、より好ましくは1〜5モル当量の範囲である。
水の使用量は、原料の化合物(13)に対して0.5〜5モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1モル当量である。
溶媒の使用量は、原料の化合物(13)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(I)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(I)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(13)を不活性溶媒中、水存在下または非存在下、還元剤及び酸と反応させることにより、化合物(I)を得ることができる。
還元剤としては、例えば、トリエチルシラン、トリイソプロピルシラン、トリフェニルシラン、tert−ブチルジメチルシラン、クロロジメチルシラン、クロロジイソプロピルシラン、ジメチルフェニルシラン、1,1,3,3−テトラメチルジシロキサン等を使用することができる。
酸としては、例えば、三フッ化ホウ素ジエチルエーテル錯体、三フッ化ホウ素テトラヒドロフラン錯体、塩化アルミニウム、トリフルオロメタンスルホン酸トリメチルシリルエステル等のルイス酸、またはトリフルオロメタンスルホン酸、メタンスルホン酸、硫酸、トリフルオロ酢酸等のブレンステッド酸を使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、酢酸、水、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜50℃の範囲である。
還元剤の使用量は、原料の化合物(13)に対して1〜10モル当量の範囲で使用することができ、好ましくは1〜5モル当量の範囲であり、より好ましくは1〜2モル当量の範囲である。
酸の使用量は、原料の化合物(13)に対して1〜20モル当量の範囲で使用することができ、好ましくは1〜10モル当量の範囲であり、より好ましくは1〜5モル当量の範囲である。
水の使用量は、原料の化合物(13)に対して0.5〜5モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1モル当量である。
溶媒の使用量は、原料の化合物(13)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(I)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(I)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
また、式(I)で表される化合物は、以下のスキーム4に示す方法によっても得ることができる。
スキーム4
スキーム4
工程(i):
ハロゲン化亜鉛を不活性溶媒中、有機リチウム試薬と反応させた後、化合物(8)と反応させることにより、ジアリール亜鉛試薬(14)を得ることができる。引き続き、化合物(14)にエーテル系溶媒を添加後、化合物(15)と反応させることにより、化合物(16)を得ることができる。
または、化合物(8)を不活性溶媒中、有機リチウム試薬と反応させた後、ハロゲン化亜鉛と臭化リチウムのジブチルエーテル溶液を作用させることにより、ジアリール亜鉛試薬(14)を得、引き続き、化合物(15)と反応させることによっても、化合物(16)を得ることができる。
ハロゲン化亜鉛としては、例えば、塩化亜鉛、臭化亜鉛、ヨウ化亜鉛を使用することができる。
不活性溶媒としては、例えば、トルエン、キシレン、ベンゼン等の炭化水素系溶媒等、及びこれら炭化水素系溶媒とテトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル等のエーテル系溶媒との混合溶媒を使用することができる。
有機リチウム試薬としては、例えば、メチルリチウム、n−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、n−ヘキシルリチウム等を使用することができる。
エーテル系溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル等を使用することができる。
化合物(15)は、特許文献5及び非特許文献1に記載の方法にて合成することができる。
ジアリール亜鉛試薬(14)調製時の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜30℃の範囲であり、より好ましくは−20〜30℃の範囲である。
ジアリール亜鉛試薬(14)と化合物(15)の反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは50〜110℃の範囲であり、より好ましくは90〜100℃の範囲である。
ハロゲン化亜鉛の使用量は、原料の化合物(8)に対して0.3〜2モル当量の範囲で使用することができ、好ましくは0.5〜1モル当量の範囲であり、より好ましくは0.5〜0.6モル当量の範囲である。
有機リチウム試薬の使用量は、原料の化合物(8)に対して0.9〜2モル当量の範囲で使用することができ、好ましくは0.9〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
臭化リチウムの使用量は、原料の化合物(8)に対して0.3〜2モル当量の範囲で使用することができ、好ましくは0.5〜1モル当量の範囲であり、より好ましくは0.5〜0.6モル当量の範囲である。
化合物(15)の使用量は、原料の化合物(8)に対して0.9〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(8)及び化合物(15)に対して0.5〜100質量倍の範囲で使用することができ、好ましくは0.5〜30質量倍の範囲であり、より好ましくは0.5〜10質量倍の範囲である。
化合物(16)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(16)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
ハロゲン化亜鉛を不活性溶媒中、有機リチウム試薬と反応させた後、化合物(8)と反応させることにより、ジアリール亜鉛試薬(14)を得ることができる。引き続き、化合物(14)にエーテル系溶媒を添加後、化合物(15)と反応させることにより、化合物(16)を得ることができる。
または、化合物(8)を不活性溶媒中、有機リチウム試薬と反応させた後、ハロゲン化亜鉛と臭化リチウムのジブチルエーテル溶液を作用させることにより、ジアリール亜鉛試薬(14)を得、引き続き、化合物(15)と反応させることによっても、化合物(16)を得ることができる。
ハロゲン化亜鉛としては、例えば、塩化亜鉛、臭化亜鉛、ヨウ化亜鉛を使用することができる。
不活性溶媒としては、例えば、トルエン、キシレン、ベンゼン等の炭化水素系溶媒等、及びこれら炭化水素系溶媒とテトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル等のエーテル系溶媒との混合溶媒を使用することができる。
有機リチウム試薬としては、例えば、メチルリチウム、n−ブチルリチウム、sec−ブチルリチウム、tert−ブチルリチウム、n−ヘキシルリチウム等を使用することができる。
エーテル系溶媒としては、例えば、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、ジブチルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル等を使用することができる。
化合物(15)は、特許文献5及び非特許文献1に記載の方法にて合成することができる。
ジアリール亜鉛試薬(14)調製時の反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは−40〜30℃の範囲であり、より好ましくは−20〜30℃の範囲である。
ジアリール亜鉛試薬(14)と化合物(15)の反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは50〜110℃の範囲であり、より好ましくは90〜100℃の範囲である。
ハロゲン化亜鉛の使用量は、原料の化合物(8)に対して0.3〜2モル当量の範囲で使用することができ、好ましくは0.5〜1モル当量の範囲であり、より好ましくは0.5〜0.6モル当量の範囲である。
有機リチウム試薬の使用量は、原料の化合物(8)に対して0.9〜2モル当量の範囲で使用することができ、好ましくは0.9〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
臭化リチウムの使用量は、原料の化合物(8)に対して0.3〜2モル当量の範囲で使用することができ、好ましくは0.5〜1モル当量の範囲であり、より好ましくは0.5〜0.6モル当量の範囲である。
化合物(15)の使用量は、原料の化合物(8)に対して0.9〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(8)及び化合物(15)に対して0.5〜100質量倍の範囲で使用することができ、好ましくは0.5〜30質量倍の範囲であり、より好ましくは0.5〜10質量倍の範囲である。
化合物(16)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。化合物(16)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(ii):
化合物(16)を塩基または酸存在下、メタノール溶媒またはメタノール−水混合溶媒中にて反応させることにより、化合物(17)を得ることができる。
塩基としては、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、リチウムメトキシド、ナトリウムメトキシド、カリウムメトキシド等を使用することができる。
酸としては、例えば、塩化水素、臭化水素、よう化水素、硫酸、メタンスルホン酸、パラトルエンスルホン酸、ベンゼンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは50〜80℃の範囲である。
塩基または酸の使用量は、原料の化合物(16)に対して0.01〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
溶媒の使用量は、原料の化合物(16)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(17)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(17)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(16)を塩基または酸存在下、メタノール溶媒またはメタノール−水混合溶媒中にて反応させることにより、化合物(17)を得ることができる。
塩基としては、例えば、水酸化リチウム、水酸化ナトリウム、水酸化カリウム、リチウムメトキシド、ナトリウムメトキシド、カリウムメトキシド等を使用することができる。
酸としては、例えば、塩化水素、臭化水素、よう化水素、硫酸、メタンスルホン酸、パラトルエンスルホン酸、ベンゼンスルホン酸、トリフルオロメタンスルホン酸、トリフルオロ酢酸等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは50〜80℃の範囲である。
塩基または酸の使用量は、原料の化合物(16)に対して0.01〜10モル当量の範囲で使用することができ、好ましくは0.1〜5モル当量の範囲であり、より好ましくは0.2〜2モル当量の範囲である。
溶媒の使用量は、原料の化合物(16)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(17)は、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(17)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
工程(iii):
化合物(17)を不活性溶媒中、塩基存在下、アセチル化剤と反応させることにより、化合物(I)を得ることができる。
アセチル化剤としては、例えば、塩化アセチル、臭化アセチル、無水酢酸等を使用することができる。
塩基としては、例えば、ピリジン、コリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミン等を使用することができる。ピリジンは溶媒としても使用することができる。触媒として4−ジメチルアミノピリジンを使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜40℃の範囲である。
アセチル化剤の使用量は、原料の化合物(17)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
塩基の使用量は、原料の化合物(17)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
溶媒の使用量は、原料の化合物(17)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(I)は、上述の通り、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(I)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
化合物(17)を不活性溶媒中、塩基存在下、アセチル化剤と反応させることにより、化合物(I)を得ることができる。
アセチル化剤としては、例えば、塩化アセチル、臭化アセチル、無水酢酸等を使用することができる。
塩基としては、例えば、ピリジン、コリジン、2,6−ルチジン、トリエチルアミン、ジイソプロピルエチルアミン等を使用することができる。ピリジンは溶媒としても使用することができる。触媒として4−ジメチルアミノピリジンを使用することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N−メチルピロリドン、ジメチルスルホキシド、またはこれらの混合溶媒等を使用することができる。
反応温度は、通常、−80℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは0〜40℃の範囲である。
アセチル化剤の使用量は、原料の化合物(17)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
塩基の使用量は、原料の化合物(17)に対して4〜30モル当量の範囲で使用することができ、好ましくは6〜20モル当量の範囲であり、より好ましくは8〜15モル当量の範囲である。
溶媒の使用量は、原料の化合物(17)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(I)は、上述の通り、再結晶による精製品のほか、リスラリー、クロマトグラフィー等の方法による精製品として得ることができ、また、未精製品として得ることもできる。化合物(I)は、反応の後処理後の濃縮残渣として精製することなく、あるいは後処理溶液として濃縮することなく、次の工程の原料として使用することができる。
式(XI)で表される化合物は、以下のスキーム5に示す方法によって得ることができる。
スキーム5
スキーム5
工程(i):
化合物(I)を不活性溶媒中、パラジウム触媒、ホスフィンリガンド及び塩基存在下、化合物(18)と反応(Heck反応)させた後、不活性溶媒中、n−ヘキシルアミンと塩を形成させて化合物(19)を得ることができる。
化合物(18)は、文献(Tetrahedron、1998、vol.54、4357−4366)記載の方法にて合成することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、水またはこれらの混合溶媒等を使用することができる。
パラジウム触媒としては、例えば、酢酸パラジウム、塩化パラジウム、パラジウム炭素、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム、ビス(トリフェニルホスフィン)パラジウムジクロリド、ビス(ベンゾニトリル)パラジウムジクロリド、ビス(アセトニトリル)パラジウムジクロリド等を使用することができる。
ホスフィンリガンドとしては、例えば、トリフェニルホスフィン、トリ−o−トリルホスフィン、トリ−tert−ブチルホスフィン、1,3−ビス(ジフェニルホスフィノ)プロパン等を使用することができる。
塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、酢酸ナトリウム、カリウム tert−ブトキシド等を使用することができる。
反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは60〜100℃の範囲である。
化合物(18)の使用量は、原料の化合物(I)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.5モル当量の範囲である。
パラジウム触媒の使用量は、原料の化合物(I)に対して0.01〜1モル当量の範囲で使用することができ、好ましくは0.05〜0.5モル当量の範囲であり、より好ましくは0.1〜0.2モル当量の範囲である。
ホスフィンリガンドの使用量は、原料の化合物(I)に対して0.01〜1モル当量の範囲で使用することができ、好ましくは0.05〜0.5モル当量の範囲であり、より好ましくは0.1〜0.3モル当量の範囲である。
塩基の使用量は、原料の化合物(I)に対して1〜10モル当量の範囲で使用することができ、好ましくは2〜6モル当量の範囲であり、より好ましくは3〜5モル当量の範囲である。
反応溶媒の使用量は、原料の化合物(I)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
n−ヘキシルアミンの使用量は、原料の化合物(I)に対して1〜2モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
n−ヘキシルアミン塩(19)を合成する時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、水またはこれらの混合溶媒等を使用することができる。
化合物(19)は、結晶化による精製品のほか、リスラリー、再結晶等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
化合物(I)を不活性溶媒中、パラジウム触媒、ホスフィンリガンド及び塩基存在下、化合物(18)と反応(Heck反応)させた後、不活性溶媒中、n−ヘキシルアミンと塩を形成させて化合物(19)を得ることができる。
化合物(18)は、文献(Tetrahedron、1998、vol.54、4357−4366)記載の方法にて合成することができる。
不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン、ヘプタン、ヘキサン、シクロヘキサン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、水またはこれらの混合溶媒等を使用することができる。
パラジウム触媒としては、例えば、酢酸パラジウム、塩化パラジウム、パラジウム炭素、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム、ビス(トリフェニルホスフィン)パラジウムジクロリド、ビス(ベンゾニトリル)パラジウムジクロリド、ビス(アセトニトリル)パラジウムジクロリド等を使用することができる。
ホスフィンリガンドとしては、例えば、トリフェニルホスフィン、トリ−o−トリルホスフィン、トリ−tert−ブチルホスフィン、1,3−ビス(ジフェニルホスフィノ)プロパン等を使用することができる。
塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、酢酸ナトリウム、カリウム tert−ブトキシド等を使用することができる。
反応温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは60〜100℃の範囲である。
化合物(18)の使用量は、原料の化合物(I)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.5モル当量の範囲である。
パラジウム触媒の使用量は、原料の化合物(I)に対して0.01〜1モル当量の範囲で使用することができ、好ましくは0.05〜0.5モル当量の範囲であり、より好ましくは0.1〜0.2モル当量の範囲である。
ホスフィンリガンドの使用量は、原料の化合物(I)に対して0.01〜1モル当量の範囲で使用することができ、好ましくは0.05〜0.5モル当量の範囲であり、より好ましくは0.1〜0.3モル当量の範囲である。
塩基の使用量は、原料の化合物(I)に対して1〜10モル当量の範囲で使用することができ、好ましくは2〜6モル当量の範囲であり、より好ましくは3〜5モル当量の範囲である。
反応溶媒の使用量は、原料の化合物(I)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
n−ヘキシルアミンの使用量は、原料の化合物(I)に対して1〜2モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
n−ヘキシルアミン塩(19)を合成する時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシド、水またはこれらの混合溶媒等を使用することができる。
化合物(19)は、結晶化による精製品のほか、リスラリー、再結晶等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
工程(ii):
化合物(19)を不活性溶媒中、酸の水溶液にて処理して化合物(19)のカルボン酸遊離体を得ることができる。化合物(20)を不活性溶媒中、塩基の水溶液にて処理して化合物(20)のアミン遊離体を得ることができる。化合物(19)のカルボン酸遊離体と化合物(20)のアミン遊離体を不活性溶媒中、脱水縮合剤及び塩基の存在下反応させた後、不活性溶媒中、シュウ酸と塩を形成させて化合物(21)を得ることができる。
化合物(20)は、特許文献4に記載の方法で合成することができる。
化合物(19)のカルボン酸遊離体及び化合物(20)のアミン遊離体を得る時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトンまたはこれらの混合溶媒等を使用することができる。
酸の水溶液としては、例えば、塩酸、硫酸水溶液、クエン酸水溶液等を使用することができる。
塩基の水溶液としては、例えば、水酸化ナトリウム水溶液、水酸化カリウム水溶液、水酸化リチウム水溶液、炭酸ナトリウム水溶液、炭酸カリウム水溶液等を使用することができる。
脱水縮合反応時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシドまたはこれらの混合溶媒等を使用することができる。
脱水縮合剤としては、例えば、N,N’−ジシクロヘキシルカルボジイミド、N,N’−ジイソプロピルカルボジイミド、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩及び1−ヒドロキシベンゾトリアゾール一水和物、1,1’−カルボニルジイミダゾール、1−プロパンホスホン酸無水物等を使用することができる。
脱水縮合反応時の塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン等を使用することができる。
シュウ酸塩(21)を合成する時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリルまたはこれらの混合溶媒等を使用することができる。
化合物(19)のカルボン酸遊離体及び化合物(20)のアミン遊離体を得る時の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜50℃の範囲であり、より好ましくは15〜35℃の範囲である。
脱水縮合反応の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは10〜40℃の範囲である。
化合物(20)の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.6モル当量の範囲である。
脱水縮合剤の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.5モル当量の範囲である。
塩基の使用量は、原料の化合物(19)に対して1〜5モル当量の範囲で使用することができ、好ましくは1〜3モル当量の範囲であり、より好ましくは2〜2.5モル当量の範囲である。
シュウ酸の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(19)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(21)は、結晶化による精製品のほか、リスラリー、再結晶等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
化合物(19)を不活性溶媒中、酸の水溶液にて処理して化合物(19)のカルボン酸遊離体を得ることができる。化合物(20)を不活性溶媒中、塩基の水溶液にて処理して化合物(20)のアミン遊離体を得ることができる。化合物(19)のカルボン酸遊離体と化合物(20)のアミン遊離体を不活性溶媒中、脱水縮合剤及び塩基の存在下反応させた後、不活性溶媒中、シュウ酸と塩を形成させて化合物(21)を得ることができる。
化合物(20)は、特許文献4に記載の方法で合成することができる。
化合物(19)のカルボン酸遊離体及び化合物(20)のアミン遊離体を得る時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトンまたはこれらの混合溶媒等を使用することができる。
酸の水溶液としては、例えば、塩酸、硫酸水溶液、クエン酸水溶液等を使用することができる。
塩基の水溶液としては、例えば、水酸化ナトリウム水溶液、水酸化カリウム水溶液、水酸化リチウム水溶液、炭酸ナトリウム水溶液、炭酸カリウム水溶液等を使用することができる。
脱水縮合反応時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトン、アセトニトリル、N,N−ジメチルホルムアミド、N,N−ジメチルアセトアミド、N−メチルピロリドン、ジメチルスルホキシドまたはこれらの混合溶媒等を使用することができる。
脱水縮合剤としては、例えば、N,N’−ジシクロヘキシルカルボジイミド、N,N’−ジイソプロピルカルボジイミド、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩、1−(3−ジメチルアミノプロピル)−3−エチルカルボジイミド塩酸塩及び1−ヒドロキシベンゾトリアゾール一水和物、1,1’−カルボニルジイミダゾール、1−プロパンホスホン酸無水物等を使用することができる。
脱水縮合反応時の塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン等を使用することができる。
シュウ酸塩(21)を合成する時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、アセトン、2−ブタノン、アセトニトリルまたはこれらの混合溶媒等を使用することができる。
化合物(19)のカルボン酸遊離体及び化合物(20)のアミン遊離体を得る時の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜50℃の範囲であり、より好ましくは15〜35℃の範囲である。
脱水縮合反応の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜100℃の範囲であり、より好ましくは10〜40℃の範囲である。
化合物(20)の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.6モル当量の範囲である。
脱水縮合剤の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜2モル当量の範囲であり、より好ましくは1〜1.5モル当量の範囲である。
塩基の使用量は、原料の化合物(19)に対して1〜5モル当量の範囲で使用することができ、好ましくは1〜3モル当量の範囲であり、より好ましくは2〜2.5モル当量の範囲である。
シュウ酸の使用量は、原料の化合物(19)に対して1〜3モル当量の範囲で使用することができ、好ましくは1〜1.5モル当量の範囲であり、より好ましくは1〜1.1モル当量の範囲である。
溶媒の使用量は、原料の化合物(19)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(21)は、結晶化による精製品のほか、リスラリー、再結晶等の方法による精製品として得ることができ、また、未精製品として得ることもできる。
工程(iii):
化合物(21)を不活性溶媒中、塩基の水溶液にて処理して化合物(21)のアミン遊離体を得ることができる。化合物(21)のアミン遊離体を溶媒中、塩基の存在下反応させることにより、化合物(XI)を得ることができる。
化合物(21)のアミン遊離体を得る時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトンまたはこれらの混合溶媒等を使用することができる。
塩基の水溶液としては、例えば、水酸化ナトリウム水溶液、水酸化カリウム水溶液、水酸化リチウム水溶液、炭酸ナトリウム水溶液、炭酸カリウム水溶液等を使用することができる。
化合物(XI)を得る時の溶媒としては、例えば、メタノール、エタノール等のアルコール系溶媒またはこれらアルコール系溶媒と水の混合溶媒等を使用することができる。
塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、ナトリウムメトキシド等を使用することができる。
脱アセチル化反応の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜80℃の範囲であり、より好ましくは10〜40℃の範囲である。
塩基の使用量は、原料の化合物(21)に対して0.1〜4モル当量の範囲で使用することができ、好ましくは0.5〜2モル当量の範囲であり、より好ましくは1〜1.6モル当量の範囲である。
溶媒の使用量は、原料の化合物(21)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(XI)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。
特許文献2に記載のように、化合物(XI)は非晶質の化合物であるが、化合物(XI)をエタノールまたはエタノールと混和する性質を有する有機溶媒とエタノールの混合液に溶解させた溶液からの結晶化により、化合物(XI)のエタノール溶媒和物を結晶として得ることができる。
化合物(21)を不活性溶媒中、塩基の水溶液にて処理して化合物(21)のアミン遊離体を得ることができる。化合物(21)のアミン遊離体を溶媒中、塩基の存在下反応させることにより、化合物(XI)を得ることができる。
化合物(21)のアミン遊離体を得る時の不活性溶媒としては、例えば、ジクロロメタン、クロロホルム、1,2−ジクロロエタン、クロロベンゼン等のハロゲン系溶媒、トルエン、キシレン、ベンゼン等の炭化水素系溶媒、テトラヒドロフラン、2−メチルテトラヒドロフラン、テトラヒドロピラン、ジエチルエーテル、ジイソプロピルエーテル、tert−ブチルメチルエーテル、シクロペンチルメチルエーテル、1,2−ジメトキシエタン、ジエトキシメタン等のエーテル系溶媒、酢酸エチル、酢酸イソプロピル、メチルイソブチルケトンまたはこれらの混合溶媒等を使用することができる。
塩基の水溶液としては、例えば、水酸化ナトリウム水溶液、水酸化カリウム水溶液、水酸化リチウム水溶液、炭酸ナトリウム水溶液、炭酸カリウム水溶液等を使用することができる。
化合物(XI)を得る時の溶媒としては、例えば、メタノール、エタノール等のアルコール系溶媒またはこれらアルコール系溶媒と水の混合溶媒等を使用することができる。
塩基としては、例えば、トリエチルアミン、トリ−n−ブチルアミン、N,N−ジイソプロピルエチルアミン、N−メチルモルホリン、炭酸カリウム、炭酸ナトリウム、炭酸セシウム、水酸化ナトリウム、水酸化カリウム、水酸化リチウム、ナトリウムメトキシド等を使用することができる。
脱アセチル化反応の温度は、通常、0℃から使用する溶媒の沸点まで可能であるが、好ましくは0〜80℃の範囲であり、より好ましくは10〜40℃の範囲である。
塩基の使用量は、原料の化合物(21)に対して0.1〜4モル当量の範囲で使用することができ、好ましくは0.5〜2モル当量の範囲であり、より好ましくは1〜1.6モル当量の範囲である。
溶媒の使用量は、原料の化合物(21)に対して1〜100質量倍の範囲で使用することができ、好ましくは1〜30質量倍の範囲であり、より好ましくは1〜10質量倍の範囲である。
化合物(XI)は、クロマトグラフィー等の方法による精製品、または未精製品として得ることができる。
特許文献2に記載のように、化合物(XI)は非晶質の化合物であるが、化合物(XI)をエタノールまたはエタノールと混和する性質を有する有機溶媒とエタノールの混合液に溶解させた溶液からの結晶化により、化合物(XI)のエタノール溶媒和物を結晶として得ることができる。
以下に実施例を示し、本発明をさらに詳しく具体的に説明するが、本発明はこれらの記載により限定的に解釈されるものではない。また、本発明の範囲を逸脱しない範囲で変化させてもよい。
下記実施例における収率は、反応条件及び後処理・単離条件により影響を受けているものがあり、最適化された反応条件及び後処理・単離条件を選択することによってさらに高い収率にすることが可能である。
下記実施例における収率は、反応条件及び後処理・単離条件により影響を受けているものがあり、最適化された反応条件及び後処理・単離条件を選択することによってさらに高い収率にすることが可能である。
本実施例に記載した機器分析データは、以下の測定機器にて測定した。
融点:融点測定器 B-545(BUCHI)
核磁気共鳴分光分析(NMR):
JNM-ECA600(JEOL RESONANCE);1H 600 MHz、13C 150 MHz
JNM-ECA500(JEOL RESONANCE);1H 500 MHz、13C 125 MHz
質量分析(MS):
GCT(micromass);イオン化法 EI
LCMS-IT-TOF(島津製作所)、LCMS-2010EV(島津製作所);イオン化法 ESI/APCI
CHN元素分析:vario MICRO cube(elementar)
イオンクロマト分析:XS-100(三菱化学)
赤外分光分析(IR):Spectrum One(Perkin Elmer)
融点:融点測定器 B-545(BUCHI)
核磁気共鳴分光分析(NMR):
JNM-ECA600(JEOL RESONANCE);1H 600 MHz、13C 150 MHz
JNM-ECA500(JEOL RESONANCE);1H 500 MHz、13C 125 MHz
質量分析(MS):
GCT(micromass);イオン化法 EI
LCMS-IT-TOF(島津製作所)、LCMS-2010EV(島津製作所);イオン化法 ESI/APCI
CHN元素分析:vario MICRO cube(elementar)
イオンクロマト分析:XS-100(三菱化学)
赤外分光分析(IR):Spectrum One(Perkin Elmer)
本明細書中で用いられている各略語を次に示す。
NMR:核磁気共鳴(nuclear magentic resonance)
DMSO−d6:ジメチルスルホキシド−d6(dimethylsulfoxide−d6)
s : シングレット(singlet)
d : ダブレット(doublet)
t : トリプレット(triplet)
q : クァルテット(quartet)
m : マルチプレット(multiplet)
dd: ダブレット オブ ダブレッツ(doublet of doublets)
td: トリプレット オブ ダブレッツ(triplet of doublets)
ddd: ダブレット オブ ダブレット オブ ダブレッツ(doublet of doublet of doublets)
J : スピン結合定数(coupling constant)
Hz : ヘルツ(Hertz)
MS:質量分析(mass spectrometry)
HRMS:高分解能質量分析(high resolution mass spectrometry)
EI:電子イオン化法(electron ionization)
ESI :エレクトロスプレーイオン化法(electrospray ionization)
APCI:大気圧化学イオン化法(atomospheric pressure chemical ionization)
mp:融点(melting point)
v/v :体積/体積
wt% :重量パーセント濃度
NMR:核磁気共鳴(nuclear magentic resonance)
DMSO−d6:ジメチルスルホキシド−d6(dimethylsulfoxide−d6)
s : シングレット(singlet)
d : ダブレット(doublet)
t : トリプレット(triplet)
q : クァルテット(quartet)
m : マルチプレット(multiplet)
dd: ダブレット オブ ダブレッツ(doublet of doublets)
td: トリプレット オブ ダブレッツ(triplet of doublets)
ddd: ダブレット オブ ダブレット オブ ダブレッツ(doublet of doublet of doublets)
J : スピン結合定数(coupling constant)
Hz : ヘルツ(Hertz)
MS:質量分析(mass spectrometry)
HRMS:高分解能質量分析(high resolution mass spectrometry)
EI:電子イオン化法(electron ionization)
ESI :エレクトロスプレーイオン化法(electrospray ionization)
APCI:大気圧化学イオン化法(atomospheric pressure chemical ionization)
mp:融点(melting point)
v/v :体積/体積
wt% :重量パーセント濃度
化合物の命名には、ACD/Name 2015 (Advanced Chemistry Development Inc.)等のソフトを使用している場合がある。
本明細書では、室温とは、特に断りが無い限り、20〜30℃を指す。
式(I)で表される化合物は、下記実施例1〜9に示す方法によって得ることができた。
実施例1
2−(2−ブロモ−5−メトキシフェニル)プロパン−2−オール(2)の合成
実施例1
2−(2−ブロモ−5−メトキシフェニル)プロパン−2−オール(2)の合成
mp : 99℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.74 ( 6H, s ), 2.69 ( 1H, s, exchangeable with D2O ), 3.80 ( 3H, s ), 6.65 ( 1H, dd, J = 8.7 and 3.2 Hz ), 7.26 ( 1H, d, J = 3.2 Hz ), 7.46 (1H, d, J = 8.7 Hz ).
MS (EI) : m/z 246 [(M+2)+], 244 (M+), 226 (base).
実施例2
1−ブロモ−4−メトキシ−2−(プロパン−2−イル)ベンゼン(3)の合成
1H NMR ( 600 MHz, CDCl3 ) : δ 1.23 ( 6H, d, J = 6.9 Hz ), 3.31 ( 1H, septet, J = 6.9 Hz ), 3.79 ( 3H, s ), 6.61 ( 1H, dd, J = 8.7 and 3.2 Hz ), 6.83 ( 1H, d, J = 3.2 Hz ), 7.41 (1H, d, J = 8.7 Hz ).
MS (EI) : m/z 230 [(M+2)+], 228 (M+), 213 (base).
実施例3
(4−ブロモフェニル)[4−メトキシ−2−(プロパン−2−イル)フェニル]メタノール(6)の合成
窒素雰囲気下、マグネシウム0.099 g及びよう素0.011 gの混合物に1-ブロモ-4-メトキシ-2-(プロパン-2-イル)ベンゼン(3)の粗生成物1.25 gのテトラヒドロフラン(8 mL)溶液を加え、2.5時間加熱還流した。氷水浴にて冷却した反応液に4-ブロモベンズアルデヒド(5)0.756 gのテトラヒドロフラン(2 mL)溶液を3〜18℃にて2分間かけて滴下した。2℃にて1時間撹拌後、氷水浴をはずして室温まで昇温(〜22℃)しながら1.5時間撹拌した。氷水浴にて冷却した反応液に20 wt%塩化アンモニウム水溶液60 mL、引き続きトルエン20 mLを加えて有機層と水層に分液した。有機層を20 wt%塩化アンモニウム水溶液20 mL、引き続き10 wt%塩化ナトリウム水溶液20 mLにて洗浄後、有機層と水層に分液した。有機層を無水硫酸マグネシウムにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して黄色油状物の(4-ブロモフェニル)[4-メトキシ-2-(プロパン-2-イル)フェニル]メタノール(6)の粗生成物1.36 gを得た。
1H NMR ( 600 MHz, CDCl3 ) : δ 1.11 ( 3H, d, J = 6.8 Hz ), 1.18 ( 3H, d, J = 6.8 Hz ), 2.04 ( 1H, d, J = 3.7 Hz, exchangeable with D2O ), 3.21 ( 1H, septet, J = 6.8 Hz ), 3.81 ( 3H, s ), 6.05 ( 1H, d, J = 3.7 Hz ), 6.72 ( 1H, dd, J = 8.7 and 2.9 Hz ), 6.85 ( 1H, d, J = 2.9 Hz ), 7.18-7.23 ( 3H, m ), 7.43-7.46 ( 2H, m ).
MS (EI) : m/z 336 [(M+2)+] , 334 (M+), 222 (base).
実施例4
1−[(4−ブロモフェニル)メチル]−4−メトキシ−2−(プロパン−2−イル)ベンゼン(7)の合成
窒素雰囲気下、(4-ブロモフェニル)[4-メトキシ-2-(プロパン-2-イル)フェニル]メタノール(6)の粗生成物1.36 g、トルエン20 mL及びtert-ブチルジメチルシラン0.953 gの混合物にトリフルオロメタンスルホン酸トリメチルシリルエステル1.2 mLを1〜7℃にて3分間かけて滴下した後、1℃にて1時間撹拌した。反応液に15 wt%炭酸ナトリウム水溶液20 mLを1〜9℃にて8分間かけて滴下後、22℃まで昇温しながら70分間撹拌した。有機層と水層に分液し、有機層を10 wt%塩化ナトリウム水溶液20 mLにて洗浄した。分液した有機層を減圧濃縮し、黄色油状物の1-[(4-ブロモフェニル)メチル]-4-メトキシ-2-(プロパン-2-イル)ベンゼン(7)の粗生成物1.26 gを得た。
1H NMR ( 600 MHz, CDCl3 ) : δ 1.11 ( 6H, d, J = 6.8 Hz ), 3.01 ( 1H, septet, J = 6.8 Hz ), 3.80 ( 3H, s ), 3.93 ( 2H, s ), 6.69 ( 1H, dd, J = 8.3 and 2.5 Hz ), 6.85 ( 1H, d, J = 2.5 Hz ), 6.94-6.98 ( 2H, m ), 7.00 (1H, d, J = 8.3 Hz ), 7.34-7.38 ( 2H, m ).
MS (EI) : m/z 320 [(M+2)+], 318 (M+), 275 (base).
実施例5
1−[(4−ブロモフェニル)メチル]−5−ヨード−4−メトキシ−2−(プロパン−2−イル)ベンゼン(8)の合成
窒素雰囲気下、1-[(4-ブロモフェニル)メチル]-4-メトキシ-2-(プロパン-2-イル)ベンゼン(7)の粗生成物1.26 gの酢酸(10 mL)溶液によう素酸カリウム水溶液(KIO3 0.281 g / H2O 5 mL)及びよう素0.821 gを加えて49〜51℃にて17時間撹拌した。反応液に酢酸10 mLを加えて52℃にて6.5時間撹拌した。反応液を炭酸カリウム水溶液(K2CO3 30.1 g / H2O 60 mL)に13〜31℃にて24分間かけて滴下後、トルエン40 mLを加えて1時間撹拌した。水10 mLを加えた後、有機層と水層に分液した。有機層をチオ硫酸ナトリウム水溶液(Na2S2O3 8.01 g / H2O 40 mL)、引き続き水40 mLにて順次洗浄した。有機層を減圧濃縮して得られた黄色油状物1.51 gにメタノール6 mLを加え、室温にて61.5時間撹拌した後、氷水浴にて冷却しながら3.5時間撹拌した。析出した固体をろ過、冷メタノール3 mLにて洗浄後、減圧乾燥して白色固体の1-[(4-ブロモフェニル)メチル]-5-ヨード-4-メトキシ-2-(プロパン-2-イル)ベンゼン(8)0.417 g[化合物(2)からの通算換算収率23.0%]を得た。
mp : 93℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.11 ( 6H, d, J = 6.8 Hz ), 3.00 ( 1H, septet, J = 6.8 Hz ), 3.88 ( 5H, s ), 6.74 ( 1H, s ), 6.93-6.97 ( 2H, m ), 7.36-7.40 ( 2H, m ), 7.49 ( 1H, s ).
MS (EI) : m/z 446 [(M+2)+], 444 (M+, base).
実施例6
1−C−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−2,3,4,6−テトラキス−O−(トリメチルシリル)−D−グルコピラノース(11)
アルゴン雰囲気下、1-[(4-ブロモフェニル)メチル]-5-ヨード-4-メトキシ-2-(プロパン-2-イル)ベンゼン(8)2.00 gにテトラヒドロフラン8.0 mLを加えて溶解し、塩化イソプロピルマグネシウム−塩化リチウム錯体のテトラヒドロフラン溶液(1.3 mol / L)3.30 mLを−21〜−18℃にて10分間かけて滴下し、−21℃にて1時間15分間撹拌した。反応液に塩化イソプロピルマグネシウム−塩化リチウム錯体のテトラヒドロフラン溶液(1.3 mol / L)0.15 mLを−21〜−20℃にて1分間かけて滴下し、−21℃にて42分間撹拌した。反応液に塩化イソプロピルマグネシウム−塩化リチウム錯体のテトラヒドロフラン溶液(1.3 mol / L)0.20 mLを−21℃にて2分間かけて滴下し、−21℃にて50分間撹拌した。反応液に2,3,4,6-テトラキス-O-(トリメチルシリル)-D-グルコノ-1,5-ラクトン(10)(97.9 wt%)2.36 gのテトラヒドロフラン(8.0 mL)溶液を−21〜−18℃にて10分間かけて滴下し、−10℃に昇温して1時間撹拌後、0℃に昇温して4時間撹拌した。反応液を飽和塩化アンモニウム水溶液30.0 mL及び水30.0 mLの混合液に加えた後、酢酸エチル100 mLを加えて有機層と水層に分液した。有機層を飽和塩化ナトリウム水溶液30.0 mLにて洗浄後、無水硫酸マグネシウムにて乾燥した。不溶物をろ別し、ろ液を減圧濃縮して淡黄色油状物の1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(トリメチルシリル)-D-グルコピラノース(11)の粗生成物3.80 gを得た。
MS (ESI/APCI dual source) : m/z 809 {[(M+2)+Na]+}, 807 [(M+Na)+].
実施例7
メチル 1−C−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−α−D−グルコピラノシド(12)の合成
1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(トリメチルシリル)-D-グルコピラノース(11)の粗生成物3.80 gにメタンスルホン酸0.117 gのメタノール(10.0 mL)溶液を加え、アルゴン雰囲気下、24〜25℃にて3時間撹拌した。氷水浴にて冷却した反応液に飽和炭酸水素ナトリウム水溶液10.0 mLを4〜16℃にて2分間かけて滴下後、水15.0 mL及び酢酸エチル70.0 mLを加え、有機層と水層に分液した。水層を酢酸エチル30.0 mLにて抽出した有機層を先の有機層と併せて飽和塩化ナトリウム水溶液30.0 mLにて洗浄した。有機層を無水硫酸マグネシウムにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して淡褐色固体のメチル 1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(12)の粗生成物1.95 gを得た。化合物(12)の粗生成物1.95 gに酢酸エチル4.00 mL及びトルエン12.0 mLを加えて溶解させた溶液に25℃にて化合物(12)の種晶0.5 mgを添加し、25℃にて30分間撹拌した。氷水浴にて冷却し、0〜16℃にて19時間、引き続き0℃にて4時間撹拌した。析出した固体をろ過、トルエン−酢酸エチル混合溶媒(3/1, v/v)6.0 mLにて洗浄後、減圧乾燥して白色固体のメチル 1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(12)0.697 g[化合物(8)からの通算収率30.3%]を得た。
mp : 160℃.
1H NMR ( 600 MHz, DMSO-d6 ) : δ 1.01 ( 3H, d, J = 6.7 Hz ), 1.09 ( 3H, d, J = 6.7 Hz ), 2.99 ( 1H, septet, J = 6.7 Hz ), 3.08 ( 3H, s ), 3.17 ( 1H, td, J = 9.7 and 5.4 Hz ), 3.28-3.33 ( 1H, m ), 3.37 ( 1H, ddd, J = 9.7, 6.0 and 1.9 Hz ), 3.46-3.54 ( 2H, m ), 3.69-3.74 ( 1H, m ), 3.76 ( 3H, s ), 3.89, 3.99 ( 2H, AB quartet, J = 15.9 Hz ), 4.21 ( 1H, d, J = 7.4 Hz, exchangeable with D2O ), 4.27 ( 1H, t, J = 6.0 Hz, exchangeable with D2O ), 4.65 ( 1H, d, J = 5.0 Hz, exchangeable with D2O ), 4.91 ( 1H, d, J = 5.4 Hz, exchangeable with D2O ), 6.90 ( 1H, s ), 7.02-7.07 ( 2H, m ), 7.16 ( 1H, s ), 7.42-7.47 ( 2H, m ).
13C NMR ( 125 MHz, DMSO-d6 ) : δ 23.5, 28.7, 36.9, 48.6, 56.3, 61.3, 70.1, 74.3, 74.4, 75.7, 79.2, 101.5, 110.6, 118.7, 124.4, 127.9, 130.4, 131.0, 131.7, 141.2, 147.7, 156.5.
MS (ESI/APCI dual source) : ポジティブモード;m/z 535 {[(M+2)+Na]+}, 533 [(M+Na)+], ネガティブモード;m/z 511 {[(M+2)-H]−}, 509 [(M-H)−].
実施例8
メチル 2,3,4,6−テトラ−O−アセチル−1−C−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−α−D−グルコピラノシド(13)の合成
アルゴン雰囲気下、メチル 1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(12)1.00 gをピリジン5.00 mLに溶解した溶液に4-ジメチルアミノピリジン(DMAP)0.024 g及び無水酢酸1.21 gを加え、21〜38℃にて4.5時間撹拌した。冷却した反応液に水5.00 mLを1〜9℃にて8分間かけて滴下し、1℃にて20分間撹拌した。酢酸エチル60.0 mL及び10 wt%塩酸30.0 mLを加え、有機層と水層に分液した。有機層を飽和炭酸水素ナトリウム水溶液30.0 mL及び飽和塩化ナトリウム水溶液20.0 mLにて順次洗浄し、無水硫酸マグネシウムにて乾燥した。不溶物をろ別し、ろ液を減圧濃縮後、減圧乾燥して白色アモルファス固体のメチル 2,3,4,6-テトラ-O-アセチル-1-C-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-α-D-グルコピラノシド(13)の粗生成物1.30 gを得た。
1H NMR ( 500 MHz, CDCl3 ) : δ 1.08 ( 3H, d, J = 6.8 Hz ), 1.14 ( 3H, d, J = 6.8 Hz ), 1.77 ( 3H, s ), 1.97 ( 3H, s ), 2.06 ( 3H, s ), 2.08 ( 3H, s ), 2.98 ( 1H, septet, J = 6.8 Hz ), 3.30 ( 3H, s ), 3.88 ( 3H, s ), 3.92 ( 2H, s ), 4.02 ( 1H, ddd, J = 10.3, 4.7 and 2.4 Hz ), 4.20 ( 1H, dd, J = 12.3 and 2.4 Hz ), 4.30 ( 1H, dd, J = 12.3 and 4.7 Hz ), 5.25 ( 1H, dd, J = 10.3 and 9.8 Hz ), 5.40 ( 1H, d, J = 9.8 Hz ), 5.57 ( 1H, t, J = 9.8 Hz ), 6.82 ( 1H, s ), 6.86-6.91 ( 2H, m ), 7.09 ( 1H, s ), 7.33-7.37 ( 2H, m ).
13C NMR ( 125 MHz, CDCl3 ) : δ 20.3, 20.6, 20.67, 20.74, 23.4, 23.8, 29.4, 37.4, 49.9, 56.0, 62.4, 68.9, 69.0, 71.5, 72.6, 101.1, 110.0, 119.6, 120.6, 128.0, 129.9, 131.30, 131.33, 140.6, 149.7, 156.9, 169.0, 169.7, 170.0, 170.6.
MS (ESI/APCI dual source) : ポジティブモード;m/z 703 {[(M+2)+Na]+}, 701 [(M+Na)+].
実施例9
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(I)の合成
mp : 127℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.04 ( 3H, d, J = 6.8 Hz ), 1.09 ( 3H, d, J = 6.8 Hz ), 1.76 ( 3H, s ), 2.00 ( 3H, s ), 2.05 ( 3H, s ), 2.06 ( 3H, s ), 2.99 ( 1H, septet, J = 6.8 Hz ), 3.80-3.86 ( 1H, m ), 3.84 ( 3H, s ), 3.89, 3.93 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.4 and 2.5 Hz ), 4.25 ( 1H, dd, J = 12.4 and 4.5 Hz ), 4.88 ( 1H, d, J = 9.1 Hz ), 5.21 ( 1H, t, J = 9.7 Hz ), 5.31-5.40 ( 2H, m ), 6.77 ( 1H, s ), 6.90-6.95 ( 2H, m ), 7.11 ( 1H, s ), 7.33-7.38 ( 2H, m ).
MS (ESI/APCI dual source) : ポジティブモード;m/z 673 {[(M+2)+Na]+}, 671 [(M+Na)+].
Anal.Calcd for C31H37BrO10 : C, 57.32 ; H, 5.74 ; Br, 12.30. Found : C, 57.32 ; H, 5.72 ; Br, 12.26. IR (KBr) cm-1 : 2966, 1747, 1252, 1224, 1042.
また、式(I)で表される化合物は、下記実施例10〜12に示す方法によっても得ることができた。
実施例10
(1S)−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−2,3,4,6−テトラキス−O−(2,2−ジメチルプロパノイル)−D−グルシトール(16)の合成
実施例10
(1S)−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−2,3,4,6−テトラキス−O−(2,2−ジメチルプロパノイル)−D−グルシトール(16)の合成
アルゴン雰囲気下、臭化亜鉛1.42 gにトルエン10.0 mLを加えた懸濁液にn-ブチルリチウム(2.65 M ヘキサン溶液)4.50 mLを23〜24℃にて2分間かけて滴下し、24〜25℃にて2時間撹拌した。この懸濁液に1-[(4-ブロモフェニル)メチル]-5-ヨード-4-メトキシ-2-(プロパン-2-イル)ベンゼン(8)5.00 gのトルエン20.0 mL溶液を1〜4℃にて20分間かけて滴下した。0℃にて1時間撹拌後、ジブチルエーテル4.00 mLを0〜4℃にて添加して26℃まで昇温しながら、1時間45分間撹拌した。この懸濁液に2,3,4,6-テトラキス-O-(2,2-ジメチルプロパノイル)-α-D-グルコピラノシルブロミド(15)6.54 gのトルエン15.0 mL溶液を25〜26℃にて3分間かけて滴下後、95〜98℃にて4時間撹拌した。反応混合物を26℃まで冷却後、20 wt%塩化アンモニウム水溶液50.0 gを加えて24℃にて15分間撹拌した。トルエン10.0 mLを加えて有機層と水層に分液した。有機層を水25.3 gにて洗浄後、減圧濃縮して褐色油状物の(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(2,2-ジメチルプロパノイル)-D-グルシトール(16)の粗生成物11.0 gを得た。
1H NMR ( 500 MHz, 50℃, acetone-d6 ):δ 0.86 ( 9H, s ), 0.96 ( 3H, d, J = 6.9 Hz ), 1.12 ( 9H, s ), 1.14 ( 3H, d, J = 6.9 Hz ), 1.18 ( 9H, s ), 1.19 ( 9H, s ), 3.07 ( 1H, septet, J = 6.9 Hz ), 3.86 ( 3H, s ), 3.92, 4.01 ( 2H, AB quartet, J = 15.9 Hz ), 4.04 ( 1H, ddd, J = 9.5, 3.8 and 1.9 Hz ), 4.12 ( 1H, dd, J = 12.4 and 3.8 Hz ), 4.23 ( 1H, dd, J = 12.4 and 1.9 Hz ), 5.07 ( 1H, d, J = 9.5 Hz ), 5.32 ( 1H, t, J = 9.5 Hz ), 5.40 ( 1H, t, J = 9.5 Hz ), 5.43 ( 1H, t, J = 9.5 Hz ), 6.90 ( 1H, s ), 7.06-7.11 ( 2H, m ), 7.26 ( 1H, s ), 7.37-7.42 ( 2H, m ).
13C NMR ( 125 MHz, 50℃, acetone-d6 ):δ 23.99, 24.02, 27.3, 27.59, 27.62, 27.8, 30.3, 38.6, 39.3, 39.45, 39.54, 39.6, 56.3, 62.9, 69.5, 73.1, 74.3, 75.2, 77.6, 109.5, 120.1, 123.4, 129.9, 131.7, 132.2, 132.4, 142.3, 150.3, 158.1, 176.9, 177.1, 177.7, 178.1.
MS (ESI/APCI dual source):ポジティブモード;m/z 841 {[(M+2)+Na]+}, 839 [(M+Na)+].
HRMS (ESI/APCI dual source) : ポジティブモード;m/z calcd for C43H61BrNaO10 [(M+Na)+] 839.3340, found :839.3321.
IR (KBr) cm-1 : 2972, 2874, 1744, 1482, 1282, 1174, 1141.
実施例11
(1S)−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(17)の合成
アルゴン雰囲気下、(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-2,3,4,6-テトラキス-O-(2,2-ジメチルプロパノイル)-D-グルシトール(16)の粗生成物11.0 g、メタノール25.0 mL及び28 wt%ナトリウムメトキシド/メタノール溶液2.17 gの混合物を2時間加熱還流した。室温まで冷却した反応液にメタノール4.00 mL及びヘプタン25.0 mLを加えて撹拌し、上層と下層に分液した。下層にメタノール2.00 mL及びヘプタン25.0 mLを加えて撹拌し、上層と下層に分液した。得られた下層にメタノール1.00 mL、硫酸(97%)1.14 g及び水1.14 gを加えて1.5時間加熱還流後、硫酸(97%)1.15 gを加えて1.5時間加熱還流した。反応液に28 wt%ナトリウムメトキシド/メタノール溶液6.50 gを2〜10℃にて加えて室温まで昇温した。反応混合物にセルロースパウダー3.08 gを添加後、ろ過して固形物をメタノール50.0 mLにて洗浄した。ろ液を減圧濃縮後、酢酸エチル25.0 mLを添加して減圧濃縮する操作を3回実施し、黒色油状物の(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(17)の粗生成物11.5 gを得た。
mp : 157℃.
1H NMR ( 500 MHz, DMSO-d6 ) : δ 1.02 ( 3H, d, J = 6.8 Hz ), 1.06 ( 3H, d, J = 6.8 Hz ), 2.98 ( 1H, septet, J = 6.8 Hz ), 3.09-3.20 ( 2H, m ), 3.27 ( 1H, td, J = 8.5 and 4.7 Hz ), 3.35-3.44 ( 2H, m ), 3.66 ( 1H, ddd, J = 11.8, 5.7 and 1.8 Hz ), 3.76 ( 3H, s ), 3.91, 3.94 ( 2H, AB quartet, J = 15.9 Hz ), 4.37 ( 1H, t, J = 5.7 Hz, exchangeable with D2O ), 4.44 ( 1H, d, J = 9.9 Hz ), 4.61 ( 1H, d, J = 5.7 Hz, exchangeable with D2O ), 4.87 ( 1H, d, J = 5.0 Hz, exchangeable with D2O ), 4.89 ( 1H, d, J = 4.7 Hz, exchangeable with D2O ), 6.84 ( 1H, s ), 7.03-7.09 ( 2H, m ), 7.13 ( 1H, s ), 7.41-7.46 ( 2H, m ).
13C NMR ( 125 MHz, DMSO-d6 ) :δ 23.5, 23.6, 28.8, 37.0, 55.7, 61.5, 70.6, 73.6, 74.2, 78.7, 81.5, 108.5, 118.6, 125.7, 128.1, 130.4, 130.5, 131.0, 141.2, 147.2, 156.7.
MS (ESI/APCI dual source):ポジティブモード;m/z 500 {[(M+2)+NH4]+}, 498 [(M+NH4)+].
MS (ESI/APCI dual source):ネガティブモード;m/z 527 {[(M+2)+HCO2]-}, 525 [(M+HCO2)-].
Anal.Calcd for C23H29BrO6 : C, 57.39 ; H, 6.07 ; Br, 16.60. Found : C, 57.14 ; H, 6.00 ; Br, 16.47.
IR (KBr) cm-1 : 3402, 2961, 2929, 1616, 1508, 1488, 1089, 1072, 1048, 1012.
実施例12
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−ブロモフェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(I)の合成
アルゴン雰囲気下、(1S)-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(17)の粗生成物5.23 gとピリジン10.0 mLの混合物に無水酢酸5.79 gを加えて23〜26℃にて71時間45分間撹拌した。反応液に水1.00 mLを加えて22〜25℃にて30分間撹拌後、トルエン25.0 mL及び10 wt%塩酸20.0 gを加えて有機層と水層に分液した。有機層に10 wt%塩酸15.1 gを加えて撹拌後、不溶物をろ過してトルエン5.00 mLにて洗浄した。ろ液を有機層と水層に分液し、有機層を2 wt%炭酸水素ナトリウム水溶液15.1 g、引き続き、水10.0 mLにて洗浄した。有機層にトルエン3.00 mL及びNH-シリカゲル(Chromatorex NH、富士シリシア化学株式会社)2.50 gを加えて22〜23℃にて16時間撹拌した。不溶物をろ別後、トルエン10.0 mLにて洗浄し、ろ液を減圧濃縮した。得られた残渣にエタノール5.00 mLを加えて減圧濃縮する操作を2回実施した。濃縮残渣にエタノール3.50 mLを加えて60℃にて加熱溶解後、ヘプタン10.5 mLを57〜59℃にて6分間かけて滴下し、徐冷しながら撹拌した。44℃にて化合物(I)の種晶0.6 mgを添加後、室温(〜21℃)にて19時間撹拌した。析出した固体をろ過、洗浄(ヘプタン:エタノール=3:1(v/v)、6.50 mL)後、50℃にて1時間減圧乾燥して白色粉末の(1S) -2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(I)2.16 g得た[化合物(8)からの通算換算収率64.9%]。
mp : 126℃.
1H NMR ( 600 MHz, CDCl3 ) : δ 1.04 ( 3H, d, J = 6.9 Hz ), 1.09 ( 3H, d, J = 6.9 Hz ), 1.76 ( 3H, s ), 2.00 ( 3H, s ), 2.05 ( 3H, s ), 2.06 ( 3H, s ), 2.99 ( 1H, septet, J = 6.9 Hz ), 3.80-3.86 ( 1H, m ), 3.84 ( 3H, s ), 3.89, 3.93 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.4 and 2.1 Hz ), 4.25 ( 1H, dd, J = 12.4 and 4.5 Hz ), 4.88 ( 1H, d, J = 9.5 Hz ), 5.22 ( 1H, t, J = 9.7 Hz ), 5.30-5.40 ( 2H, m ), 6.77 ( 1H, s ), 6.90-6.95 ( 2H, m ), 7.11 ( 1H, s ), 7.33-7.38 ( 2H, m ).
13C NMR ( 150 MHz, CDCl3 ) :δ 20.4, 20.7, 20.8, 23.4, 23.8, 29.4, 37.7, 55.7, 62.4, 68.8, 71.8, 73.9, 74.7, 76.1, 108.4, 119.6, 121.5, 128.9, 130.1, 130.6, 131.3, 140.5, 149.3, 156.7, 167.0, 169.6, 170.4, 170.8.
MS (ESI/APCI dual source) : ポジティブモード;m/z 673 {[(M+2)+Na]+}, 671 [(M+Na)+].
Anal.Calcd for C31H37BrO10 : C, 57.32 ; H, 5.74 ; Br, 12.30. Found : C, 57.37 ; H, 5.72 ; Br, 12.28.
IR (KBr) cm-1 : 2966, 1746, 1251, 1224, 1042.
式(XI)で表される化合物は、下記実施例13〜15に示す方法によって得ることができた。
実施例13
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−[5−({4−[(1E)−3−カルボキシ−3−メチルブタ−1−エン−1−イル]フェニル}メチル)−2−メトキシ−4−(プロパン−2−イル)フェニル]−D−グルシトール=ヘキサン−1−アミン塩(19)の合成
実施例13
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−[5−({4−[(1E)−3−カルボキシ−3−メチルブタ−1−エン−1−イル]フェニル}メチル)−2−メトキシ−4−(プロパン−2−イル)フェニル]−D−グルシトール=ヘキサン−1−アミン塩(19)の合成
アルゴン雰囲気下、(1S) -2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-ブロモフェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(I)3.00 g、2,2-ジメチルブタ-3-エン酸(18)0.804 g、トリ−o−トリルホスフィン0.285 g、トリエチルアミン1.88 g、アセトニトリル9.00 mL及び酢酸パラジウム0.112 gの混合物を8.5時間加熱還流した。室温まで冷却後、一晩静置した。反応混合物にアセトニトリル3.00 mLを加えて38℃にて30分間加熱撹拌後、セルロースパウダーパッドにてろ過し、アセトニトリル9.00 mLにて洗浄した。ろ液を減圧濃縮して得た残渣に酢酸エチル20.0mL及び10 wt%塩酸10.0 gを加えて有機層と水層に分液した。有機層を10 wt%塩化ナトリウム水溶液10.0 g、引き続き、水10.0 mLにて順次洗浄した。有機層を減圧濃縮後、酢酸エチル6.00 mLを加えて減圧濃縮する操作を2回実施し、残渣4.15 gを得た。この残渣に酢酸エチル12.0 g及びヘキシルアミン0.495 gを加えて加熱溶解し、ヘプタン31.5 mLを50〜55℃にて8分間かけて滴下した。51℃にて化合物(19)の種晶0.8 mgを添加後、室温(〜23℃)にて21時間45分間撹拌した。析出した固体をろ過、洗浄(ヘプタン:酢酸エチル=3:1(v/v)、12.0 mL)後、室温にて2時間減圧乾燥して白色粉末の(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-[5-({4-[(1E)-3-カルボキシ-3-メチルブタ-1-エン-1-イル]フェニル}メチル)-2-メトキシ-4-(プロパン-2-イル)フェニル]-D-グルシトール=ヘキサン-1-アミン塩(19)3.01 g(収率83.2%)を得た。
mp : 146〜147℃.
1H NMR ( 500 MHz, CDCl3 ) : δ 0.82 ( 3H, t, J = 7.3 Hz ), 1.02 ( 3H, d, J = 6.9 Hz ), 1.07 ( 3H, d, J = 6.9 Hz ), 1.09-1.16 ( 4H, m ), 1.16-1.24 ( 2H, m ), 1.27 ( 6H, s ), 1.43-1.54 ( 2H, m ), 1.76 ( 3H, s ), 2.00 ( 3H, s ), 2.047 ( 3H, s ), 2.052 ( 3H, s ), 2.59-2.66 ( 2H, m ), 3.01 ( 1H, septet, J = 6.9 Hz ), 3.80-3.85 ( 1H, m ), 3.84 ( 3H, s ), 3.91, 3.93 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.3 and 2.1 Hz ), 4.25 ( 1H, dd, J = 12.3 and 4.6 Hz ), 4.85 ( 1H, d, J = 9.5 Hz ), 5.22 ( 1H, t, J = 9.5 Hz ), 5.33 ( 1H, t, J = 9.5 Hz ), 5.41 ( 1H, t, J = 9.5 Hz ), 6.29 ( 1H, d, JAB = 16.3 Hz ), 6.44 ( 1H, d, JAB = 16.3 Hz ), 6.50 ( 3H, br s, exchangeable with D2O ), 6.76 ( 1H, s ), 6.92-6.97 ( 2H, m ), 7.12 ( 1H, s ), 7.20-7.25 ( 2H, m ).
13C NMR ( 150 MHz, CDCl3 ) :δ 14.0, 20.4, 20.7, 20.8, 22.4, 23.5, 23.8, 25.9, 26.2, 28.8, 29.3, 31.3, 38.1, 39.9, 45.5, 55.7, 62.4, 68.8, 71.7, 74.8, 76.1, 108.4, 121.3, 125.8, 126.0, 128.4, 129.5, 130.7, 135.3, 136.7, 140.2, 149.4, 156.6, 169.0, 169.6, 170.4, 170.8, 183.0.
MS (ESI/APCI dual source) : ポジティブモード;m/z 784 [(M+H)+], 705 {[(M−C6H15N)+Na]+}. MS (ESI/APCI dual source) : ネガティブモード;m/z 681 {[(M−C6H15N)−H]−}.
Anal.Calcd for C37H46O12・C6H15N : C, 65.88 ; H, 7.84 ; N, 1.79. Found : C, 65.83 ; H, 7.81 ; N, 1.80.
IR (KBr) cm-1 : 2960, 2933, 2870, 1747, 1254, 1224, 1042.
実施例14
(1S)−2,3,4,6−テトラ−O−アセチル−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール一シュウ酸塩(21)の合成
(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-[5-({4-[(1E)-3-カルボキシ-3-メチルブタ-1-エン-1-イル]フェニル}メチル)-2-メトキシ-4-(プロパン-2-イル)フェニル]-D-グルシトール=ヘキサン-1-アミン塩(19)25.0 g及び酢酸エチル117 mLの混合物に氷冷下、1M塩酸133 mLを加えた。室温まで昇温後、有機層と水層に分液した。有機層を1M塩酸65.0 mL、引き続き、10 wt%塩化ナトリウム水溶液62.5 gにて順次洗浄した。有機層を無水硫酸マグネシウム6.25 gにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して黄色油状物の化合物(19)のカルボン酸遊離体の粗生成物29.4 gを得た。
N-[2-(ジメチルアミノ)エチル]-2-メチルアラニンアミド二塩酸塩(20)12.1 gに25 wt%水酸化ナトリウム水溶液27.6 g、水5.00 g、酢酸エチル170 g及び塩化ナトリウム5.00 gを順次加えた。有機層と水層に分液し、水層を酢酸エチル162 gにて抽出した。分液した水層を再度酢酸エチル162 gにて抽出し、全ての有機層を合わせた。有機層を無水硫酸マグネシウム6.25 gにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して無色油状物の化合物(20)のアミン遊離体の粗生成物10.6 gを得た。
化合物(19)のカルボン酸遊離体の粗生成物29.4 gのN,N-ジメチルホルムアミド50.0mL溶液、化合物(20)のアミン遊離体の粗生成物10.6 gのN,N-ジメチルホルムアミド50.0mL溶液、N,N-ジメチルホルムアミド127 mL、1-ヒドロキシベンゾトリアゾール一水和物(HOBt・H2O)6.45 g及びトリエチルアミン10.7 mLの混合物に1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩(EDC・HCl)7.70 gを加え、室温にて12.5時間撹拌した。反応液を氷冷した水100 mLに加えた後、トルエン300 mLを加えた。有機層と水層に分液し、有機層を5 wt%塩化ナトリウム水溶液112 mLで2回、5 wt%塩化アンモニウム水溶液56.3 mLで1回、5 wt%塩化ナトリウム水溶液112 mLで1回順次洗浄した。有機層を無水硫酸マグネシウム7.50 gにて乾燥後、不溶物をろ別し、ろ液を減圧濃縮して淡黄色液41.15 gを得た。得られた濃縮液にトルエン58.85 gを加えて(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトールの粗生成物トルエン溶液100 gを得た。
得られたトルエン溶液40.0 gを減圧濃縮し、黄色液17.2 gを得た。酢酸エチル90.0 mLを加え、37℃に加熱した。化合物(21)の種晶0.05 g、0.5 Mシュウ酸/酢酸エチル溶液25.4 mL及び酢酸エチル30.0 mLを順次加え、1.5時間同温度にて撹拌した。室温まで放冷撹拌した後、析出した固体をろ過、洗浄(酢酸エチル50.0 mL)後、40℃にて1時間減圧乾燥して白色粉末の(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール一シュウ酸塩(21)10.7 g[化合物(19)からの換算収率90.5%]を得た。
mp : 184℃.
1H NMR ( 500 MHz, CDCl3 ):δ 1.05 ( 3H, d, J = 6.8 Hz ), 1.11 ( 3H, d, J = 6.8 Hz ), 1.35 ( 6H, s ), 1.43 ( 6H, s ), 1.79 ( 3H, s ), 2.00 ( 3H, s ), 2.05 ( 3H, s ), 2.06 ( 3H, s ), 2.87 ( 6H, s ), 3.04 ( 1H, septet, J = 6.8 Hz ), 3.22 ( 2H, t, J = 5.3 Hz ), 3.55-3.65 ( 2H, m ), 3.79-3.86 ( 1H, m ), 3.85 ( 3H, s ), 3.93, 3.99 ( 2H, AB quartet, J = 16.1 Hz ), 4.13 ( 1H, dd, J = 12.4 and 2.4 Hz ), 4.24 ( 1H, dd, J = 12.4 and 4.5 Hz ), 4.89 ( 1H, d, J = 9.0 Hz ), 5.21 ( 1H, t, J = 9.0 Hz ), 5.33 ( 1H, t, J = 9.0 Hz ), 5.37 ( 1H, t, J = 9.0 Hz ), 6.40 ( 1H, s, exchangeable with D2O ), 6.42 ( 1H, d, J = 16.1 Hz ), 6.49 ( 1H, d, J = 16.1 Hz ), 6.77 ( 1H, s ), 6.98-7.03 ( 2H, m ), 7.13 ( 1H, s ), 7.30-7.35 ( 2H, m ), 8.31 ( 1H, t, J = 5.8 Hz, exchangeable with D2O ).
13C NMR ( 125 MHz, CDCl3 ):δ 20.4, 20.65, 20.68, 20.8, 23.4, 23.9, 24.86, 24.90, 25.06, 25.09, 29.4, 35.0, 37.9, 44.4, 45.0, 55.8, 56.8, 58.8, 62.5, 68.8, 71.9, 73.9, 74.7, 76.1, 108.3, 121.5, 126.5, 128.6, 129.3, 129.6, 130.5, 133.0, 134.2, 141.2, 149.4, 156.6, 163.1, 169.4, 169.6, 170.4, 170.8, 175.5, 177.2.
MS (ESI/APCI dual source) : ポジティブモード;m/z 838 {[(M−C2H2O4)+H]+}.
Anal.Calcd for C45H63N3O12・C2H2O4 : C, 60.83 ; H, 7.06 ; N, 4.53. Found : C, 60.60 ; H, 7.02 ; N, 4.50.
IR (KBr) cm-1 : 3358, 2962, 1756, 1745, 1657, 1505, 1253, 1233.
実施例15
(1S)−1,5−アンヒドロ−1−{5−[(4−{(1E)−4−[(1−{[2−(ジメチルアミノ)エチル]アミノ}−2−メチル−1−オキソプロパン−2−イル)アミノ]−3,3−ジメチル−4−オキソブタ−1−エン−1−イル}フェニル)メチル]−2−メトキシ−4−(プロパン−2−イル)フェニル}−D−グルシトール(XI)のエタノール溶媒和物の合成
(1S)-2,3,4,6-テトラ-O-アセチル-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール一シュウ酸塩(21)10.0 g及びトルエン100 mLの混合物に5 wt%炭酸水素ナトリウム水溶液250 g、水50.0 g及びトルエン30.0 mLを加えた。有機層と水層に分液し、有機層を5 wt%塩化ナトリウム水溶液95.0 mLにて洗浄後、減圧濃縮して黄色油状物12.4 gを得た。得られた濃縮物にメタノール100 mL、水2.00 m及びトリエチルアミン2.00 gを加え、室温にて50時間撹拌後、88.5時間静置した。反応液を減圧濃縮し、得られた濃縮物にエタノール50.0 mLを加えて減圧濃縮する操作を3回繰り返し、淡黄色油状物の(1S)-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(XI)13.8 gを得た。エタノール16.2 gを加えて56℃に加熱し、ヘプタン10.0 gを加えた。化合物(XI)のエタノール溶媒和物の種晶0.0025 gを加えた後、6時間かけて25℃にし、同温度にて15時間撹拌した。さらに2時間かけて2℃にし、同温度にて2時間撹拌した。析出した固体をろ過、洗浄(ヘプタン:エタノール=63:37(v/v)、60.0 mL)後、40℃にて2時間減圧乾燥して白色粉末の(1S)-1,5-アンヒドロ-1-{5-[(4-{(1E)-4-[(1-{[2-(ジメチルアミノ)エチル]アミノ}-2-メチル-1-オキソプロパン-2-イル)アミノ]-3,3-ジメチル-4-オキソブタ-1-エン-1-イル}フェニル)メチル]-2-メトキシ-4-(プロパン-2-イル)フェニル}-D-グルシトール(XI)のエタノール溶媒和物6.08 g(収率79.4%)を得た。
mp : 105〜106℃.
1H NMR ( 600 MHz, CD3OD ):δ 1.075 ( 3H, d, J = 6.9 Hz ), 1.084 ( 3H, d, J = 6.9 Hz), 1.17 ( 2.4H, t, J = 7.2 Hz ), 1.35 ( 6H, s ), 1.45 ( 6H, s ), 2.22 ( 6H, s ), 2.39 ( 2H, t, J = 6.8 Hz ), 3.09 ( 1H, septet, J = 6.9 Hz ), 3.27 ( 2H, t, J = 6.8 Hz ), 3.36-3.41 ( 2H, m ), 3.45-3.51 ( 1H, m ), 3.57 ( 1H, t, J = 9.2 Hz ), 3.60 ( 1.6H, q, J = 7.2 Hz ), 3.63-3.68 ( 1H, m ), 3.83 ( 3H, s ), 3.85 ( 1H, dd, J = 11.8 and 1.0 Hz ), 3.98 ( 2H, s ), 4.65 ( 1H, d, J = 9.2 Hz ), 6.39 ( 1H, d, JAB = 16.1 Hz ), 6.52 ( 1H, d, JAB = 16.1 Hz ), 6.88 ( 1H, s ), 7.06-7.09 ( 2H, m ), 7.23 ( 1H, s ), 7.30-7.33 ( 2H, m ).
13C NMR ( 150 MHz, CD3OD ):δ 18.5, 24.1, 24.2, 25.2, 25.7, 30.7, 38.4, 39.2, 45.6, 46.0, 56.5, 58.1, 58.5, 59.1, 63.4, 72.3, 75.9, 76.8, 80.3, 82.5, 109.8, 126.3, 127.5, 129.9, 130.4, 130.8, 132.1, 134.8, 136.2, 142.8, 149.7, 158.7, 177.2, 178.3.
MS (ESI/APCI dual source) : ポジティブモード;m/z 670 [(M+H)+].
Anal.Calcd for C37H55N3O8・0.8C2H6O・0.2H2O : C, 65.27 ; H, 8.54 ; N, 5.92. Found : C, 65.13 ; H, 8.46 ; N, 5.85.
IR (KBr) cm-1 : 3357, 2964, 1673, 1634, 1505, 1041.
本発明により、SGLT1阻害活性を有する医薬として有用な1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の大量生産に適した製造法を提供することが可能となった。
Claims (2)
- 式(XI)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(vi)前記式(VII)で表される化合物を式(XII)で表される化合物に変換する工程と、
(vii)前記式(XII)で表される化合物を式(XIII)で表される化合物に変換する工程と、
(viii)前記式(XIII)で表される化合物を式(I)で表される化合物に変換する工程と、
(ix)前記式(I)で表される化合物を式(XIV)で表される化合物に変換する工程と、
(x)前記式(XIV)で表される化合物を式(XV)で表される化合物に変換する工程と、
(xi)前記式(XV)で表される化合物を前記式(XI)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。 - 式(I)で表される1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法であって、
(i)式(II)で表される化合物を式(III)で表される化合物に変換する工程と、
(ii)前記式(III)で表される化合物を式(IV)で表される化合物に変換する工程と、
(iii)前記式(IV)で表される化合物を式(V)で表される化合物に変換する工程と、
(iv)前記式(V)で表される化合物を式(VI)で表される化合物に変換する工程と、
(v)前記式(VI)で表される化合物を式(VII)で表される化合物に変換する工程と、
(vi)前記式(VII)で表される化合物を式(VIII)で表される化合物に変換する工程と、
(vii)前記式(VIII)で表される化合物を式(IX)で表される化合物に変換する工程と、
(viii)前記式(IX)で表される化合物を式(X)で表される化合物に変換する工程と、
(ix)前記式(X)で表される化合物を前記式(I)で表される化合物に変換する工程
を含むことを特徴とする1,5−アンヒドロ−1−置換フェニル−D−グルシトール化合物の製造方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015239306 | 2015-12-08 | ||
| JP2015239306 | 2015-12-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017105748A true JP2017105748A (ja) | 2017-06-15 |
Family
ID=59059057
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016119447A Pending JP2017105748A (ja) | 2015-12-08 | 2016-06-16 | 1,5−アンヒドロ−1−置換フェニル−d−グルシトール化合物の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017105748A (ja) |
-
2016
- 2016-06-16 JP JP2016119447A patent/JP2017105748A/ja active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1984898A (zh) | 生产薁衍生物的方法和合成该薁衍生物的中间体 | |
| JP2021530505A (ja) | フェニルピペリジニルインドール誘導体を調製する化学的プロセス | |
| CN110950765A (zh) | 一种硫酸特布他林的制备方法 | |
| KR20170131508A (ko) | 레디파스비르 및 이의 유도체의 제조방법 및 레디파스비르를 제조하기 위한 중간체 화합물 | |
| CN107245064B (zh) | 索非布韦中间体的制备方法 | |
| CN104327027B (zh) | 一类新型c‑芳基葡萄糖苷sglt2抑制剂 | |
| JP5646706B2 (ja) | C−グリコシド誘導体の製造方法 | |
| TW201912656A (zh) | 舒更葡糖鈉之製備方法及其晶型 | |
| CN111646964A (zh) | 一种碱催化的合成2h-吡喃-2-酮衍生物新方法 | |
| CN110698467A (zh) | 恩格列净的合成方法 | |
| CN112585124A (zh) | 用于合成sglt抑制剂的中间体的制备方法 | |
| JP2018505184A5 (ja) | ||
| KR20120016053A (ko) | 알킬아민 유도체의 제조방법 | |
| CN110746476A (zh) | 一种5-氮杂胞嘧啶核苷化合物及其制备方法 | |
| CN105198829B (zh) | 一种可比司他中间体的制备方法及其中间体和用途 | |
| JP2017105748A (ja) | 1,5−アンヒドロ−1−置換フェニル−d−グルシトール化合物の製造方法 | |
| CN113387797B (zh) | 一种艾乐替尼关键中间体的制备方法 | |
| US20050159611A1 (en) | Process for producing shogaols and intermediates for the synthesis thereof | |
| CN106795080B (zh) | 用于制备卤代三氟苯乙酮的方法 | |
| CN1086187C (zh) | 二环己基并18冠6的制备方法 | |
| CN105143171B (zh) | 3,4,5-三咖啡酰奎尼酸的制造方法 | |
| Hosono et al. | Enantiospecific synthesis of [(2′ S, 3′ S)-bis (hydroxymethyl) azetidin-1-yl] pyrimidine nucleosides as potential antiviral agents | |
| CN112592306A (zh) | 吡咯啉酮类化合物及其合成方法 | |
| RU2315747C2 (ru) | Способ получения ацетиленового соединения | |
| RU2838750C2 (ru) | Двухстадийный синтез селексипага с применением природосберегающих "зеленых" технологий |