JP2017145247A - C−4”位置換マクロライド誘導体を含有する感染症の予防および/又は治療剤 - Google Patents
C−4”位置換マクロライド誘導体を含有する感染症の予防および/又は治療剤 Download PDFInfo
- Publication number
- JP2017145247A JP2017145247A JP2017026792A JP2017026792A JP2017145247A JP 2017145247 A JP2017145247 A JP 2017145247A JP 2017026792 A JP2017026792 A JP 2017026792A JP 2017026792 A JP2017026792 A JP 2017026792A JP 2017145247 A JP2017145247 A JP 2017145247A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- acid
- compound
- therapeutic agent
- added
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- OFISWRJORIMAGZ-UHFFFAOYSA-N CC(C)CN(CCNC)C(C)C Chemical compound CC(C)CN(CCNC)C(C)C OFISWRJORIMAGZ-UHFFFAOYSA-N 0.000 description 1
- DBVADBHSJCWFKI-UHFFFAOYSA-N CC(C)N(CCCl)C(C)C Chemical compound CC(C)N(CCCl)C(C)C DBVADBHSJCWFKI-UHFFFAOYSA-N 0.000 description 1
- QCQZFSUBYDWVBG-UHFFFAOYSA-N CCNC(CN)=O Chemical compound CCNC(CN)=O QCQZFSUBYDWVBG-UHFFFAOYSA-N 0.000 description 1
- UGAMNTGMICNTPS-UHFFFAOYSA-N CCNC(CNC(OCc1ccccc1)=O)=O Chemical compound CCNC(CNC(OCc1ccccc1)=O)=O UGAMNTGMICNTPS-UHFFFAOYSA-N 0.000 description 1
Images


Landscapes
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
【課題】従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌、例えば耐性肺炎球菌、連鎖球菌、及びマイコプラズマなどに対しても優れた抗菌活性を示す有用な新規な化合物を有効成分として含有する感染症の予防又は治療薬の提供。
【解決手段】式[1]で表される化合物若しくは薬学的に許容されるその塩、又はそれらの水和物若しくはそれらの溶媒和物を有効成分として含有する感染症の予防又は治療薬。
【選択図】なし
【解決手段】式[1]で表される化合物若しくは薬学的に許容されるその塩、又はそれらの水和物若しくはそれらの溶媒和物を有効成分として含有する感染症の予防又は治療薬。
【選択図】なし
Description
本発明は、エリスロマイシン類似骨格を有する新規抗生物質に関する。より具体的には、本発明は、クラジノースの4”位に窒素原子を有する置換基で置換されたメチル基を有するマクロライド化合物を有効成分として含有する感染症の予防及び/又は治療のために用いる医薬に関するものである。
エリスロマイシンAはグラム陽性菌、マイコプラズマなどに起因する感染症の治療薬として広く使用されている抗生物質である。しかし、エリスロマイシンは胃酸で分解されるため、体内動態が一定しないという欠点があった。そこで酸に対する安定性を増した誘導体が検討され、その結果、クラリスロマイシン、アジスロマイシン(特許文献1及び2)、ロキシスロマイシンなどの体内動態の安定したマクロライド剤が開発されてきた。外来の呼吸器感染症を治療領域とするこれらマクロライド剤は、特に臨床分離頻度の高い肺炎球菌、連鎖球菌並びにインフルエンザ菌に対し強い抗菌活性を有する必要がある。さらに、市中肺炎からマクロライド耐性の肺炎球菌が高頻度に分離されていることから耐性肺炎球菌に有効であることも重要となっている。
近年、広範な研究の結果、エリスロマイシン耐性肺炎球菌、エリスロマイシン耐性連鎖球菌のいずれに対しても有効なマクロライドとしてAgouridasらは1995年にHMR3647(テリスロマイシン,特許文献3)を、Orらは1998年にABT−773(セスロマイシン,特許文献4)を相次いで見出した。その後、さらに薬効増強が図られた2−フルオロケトライド(特許文献5)が報告されている。
一方、クラジノースの4”位に窒素原子を有する置換基で置換されたメチル基を有するマクロライド化合物に関しては、ラクトン環内に窒素原子をもつという構造的な特徴を有しているアザライドタイプの化合物がほとんどである(特許文献6)。
更に、エリスロマイシン耐性肺炎球菌、エリスロマイシン耐性連鎖球菌のいずれに対しても有効なマクロライドとして、クラジノースの4”位に窒素原子を有する置換基で置換されたメチル基を有するマクロライド化合物については、出願人らも報告している(特許文献7、8及び9)。特にその中でも特許文献7、8に記載された実施例15が好ましい化合物である。
本発明の課題は、従来のエリスロマイシン感受性菌のみならず、エリスロマイシン耐性菌(例えば耐性肺炎球菌、耐性連鎖球菌、及びマイコプラズマ)に対しても有効な化合物を提供することにある。
そこで、本発明者らは新たなマクロライド化合物の研究を鋭意行った結果、下記に示す化合物が優れた抗菌活性を有することを見出し、本発明を完成した。
すなわち、本発明により、
(1)式[1]:
(1)式[1]:
また、上記発明の好ましい態様として、
(2)固形製剤である請求項1に記載の感染症の予防又は治療薬、
(3)剤形が錠剤、丸剤、カプセル剤、顆粒剤、散剤、又は粉剤から選択される上記(2)に記載の感染症の予防又は治療薬、及び
(4)乳糖、ショ糖、ブドウ糖、麦芽糖、果糖、マンニトール、デンプン、粉末セルロース、結晶セルロース、カルメロース、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチルセルロースからなる群から選ばれる1種以上を含む上記(3)に記載の感染症の予防又は治療薬が本発明により提供される。
(2)固形製剤である請求項1に記載の感染症の予防又は治療薬、
(3)剤形が錠剤、丸剤、カプセル剤、顆粒剤、散剤、又は粉剤から選択される上記(2)に記載の感染症の予防又は治療薬、及び
(4)乳糖、ショ糖、ブドウ糖、麦芽糖、果糖、マンニトール、デンプン、粉末セルロース、結晶セルロース、カルメロース、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチルセルロースからなる群から選ばれる1種以上を含む上記(3)に記載の感染症の予防又は治療薬が本発明により提供される。
さらに、(5)式[1]で表される化合物若しくはその薬学的に許容される塩、又はそれらの水和物若しくはそれらの溶媒和物の1日あたり投与量が1〜10000mgである、上記(1)乃至(4)に記載の感染症の予防又は治療薬が本発明により提供される。
本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は微生物、好ましくはグラム陽性菌又はグラム陰性菌などの好気性又は嫌気性細菌類、マイコプラズマやクラミジアなどに対して幅広い抗菌活性を有しており、特に従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌(例えば耐性肺炎球菌、耐性連鎖球菌、及びマイコプラズマ)などに対しても優れた抗菌活性を示すという特徴がある。
本発明において、「その薬学的に許容される塩」とは、酸付加塩又は塩基付加塩のいずれでもよく、酸付加塩としては、例えば酢酸、プロピオン酸、酪酸、ギ酸、トリフルオロ酢酸、マレイン酸、酒石酸、クエン酸、ステアリン酸、コハク酸、エチルコハク酸、ラクトビオン酸、グルコン酸、グルコヘプトン酸、安息香酸、メタンスルホン酸、エタンスルホン酸、2−ヒドロキシエタンスルホン酸、ベンゼンスルホン酸、パラトルエンスルホン酸、ラウリル硫酸、リンゴ酸、アスパラギン酸、グルタミン酸、アジピン酸、システイン、N−アセチルシステイン、塩酸、臭化水素酸、リン酸、硫酸、ヨウ化水素酸、ニコチン酸、シュウ酸、ピクリン酸、チオシアン酸、ウンデカン酸、アクリル酸ポリマー、カルボキシビニルポリマーなどの酸との塩を挙げることができ、塩基付加塩としては、例えばナトリウム塩、カリウム塩、カルシウム塩などの無機塩基との塩、モルホリン、ピペリジンなどの有機アミン、アミノ酸との塩を挙げることができるが、これらに限定されることはない。
本発明において、「抗菌剤」とはグラム陽性細菌、グラム陰性細菌やマイコプラズマといった細菌に作用してその生育を抑制又は殺菌する能力を持つ物質を意味する。菌の繁殖を抑えたり、一部の菌を殺してその数を減少させたりするようなものでもよい。グラム陽性細菌としては、例えば、ブドウ球菌属(黄色ブドウ球菌、表皮ブドウ球菌など)、連鎖球菌属(化膿連鎖球菌、B群連鎖球菌、肺炎球菌など)、腸球菌属(エンテロコッカス・フェカーリス、エンテロコッカス・フェシウムなど)が挙げられる。グラム陰性菌としては、例えば、シュードモナス属(緑膿菌など)、大腸菌属(大腸菌など)、クレブシエラ属(肺炎桿菌、クレブシエラ・オキシトカなど)、ヘモフィルス属(インフルエンザ菌、パラインフルエンザ菌など)、ボルデテラ属(百日咳菌、気管支敗血症菌など)、セラチア属(セラチア・マルセッセンスなど)、プロテウス属(プロテウス・ミラビリスなど)エンテロバクター属(エンテロバクター・クロアカなど)、カンピロバクター属(カンピロバクター・ジェジュニなど)、シトロバクター属、ビブリオ属(腸炎ビブリオ、コレラ菌など)、モルガネラ属(モルガネラ・モルガニなど)、サルモネラ属(チフス菌、パラチフス菌など)、シゲラ属(赤痢菌など)、アシネトバクター属(アシネトバクター・バウマニー、アシネトバクター・カルコアセチカスなど)、レジオネラ属(レジオネラ・ニューモフィラなど)、バクテロイデス属(バクテロイデス・フラジリスなど)、ナイセリア属(淋菌、髄膜炎菌など)、モラキセラ属(モラキセラ・カタラーリスなど)、クラミジア属(クラミジア・トラコマティス、クラミジア・シッタシーなど)及びヘリコバクター属(ヘリコバクター・ピロリなど)が挙げられる。マイコプラズマとしては、M. galli septicum、M. genitalium、M. hominis、M. hyopneumoniae、M. laboratorium、M. mycoides、M. ovipneumoniae、M. pneumonia が挙げられる。
本発明の化合物は、特に従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌(例えば耐性肺炎球菌、耐性連鎖球菌、及びマイコプラズマ)などに対しても優れた抗菌活性を示すという特徴がある。
上記式[1]で表される化合物には光学異性体が存在しうるが、式[1]で表される化合物には、それら光学異性体、及び光学異性体の混合物が含まれる。また、式[1]で表される化合物、若しくはその薬学的に許容される塩、又はそれらの各種水和物、若しくは溶媒和物も本発明の範囲に含まれる。
本発明における「溶媒和物」の「溶媒」とは特に示さない限り、例えば極性溶媒(例えば、メタノール、エタノール、1−プロパノール、2−プロパノール、ブタノール等のアルコール系の溶媒、酢酸エチル等)、不活性溶媒(例えば、クロロホルム若しくは塩化メチレン等のハロゲン化炭化水素系溶媒、ジエチルエーテル、テトラヒドロフラン若しくはジオキサン等のエーテル系溶媒、ジメチルホルムアミド、ジメチルアセトアミド等のアミド系溶媒、ジメチルスルホキシド、アセトニトリル等の非プロトン性溶媒、トルエン等の芳香族炭化水素類、又はシクロヘキサン等の炭化水素類等)、更に2−ブタノン、ヘキサン、イソプロピルエーテル、アセトン、ジクロロメタン等、又はここに例示した溶媒の混合溶媒を意味するが、これらに限定されることはない。
上記式[1]で表される本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は、優れた安全性を示す。安全性は、種々の試験によって評価されるが、たとえば、細胞毒性試験、hERG試験、シトクロムP450(CYP)活性阻害試験などで評価することができる。
上記式[1]で表される本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は、優れた代謝安定性を示す。代謝安定性は、種々の試験によって評価されるが、たとえば、ヒト肝ミクロソーム代謝安定性試験などで評価することができる。
本発明の化合物は、一つ又は二つ以上の医薬的に許容される担体、賦形剤又は希釈剤と組み合せて医薬的製剤とすることができる。上記式[1]で表される本発明の化合物若しくはその塩、又はその水和物若しくはその溶媒和物は、一般的な医薬製剤として調製される。例えば、製剤上許容しうる担体(賦形剤、結合剤、崩壊剤、矯味剤、乳化剤、希釈剤、溶解補助剤など)と混合、溶解及び/又は分散して医薬組成物とする。この医薬組成物は、錠剤、丸剤、散剤、顆粒剤、カプセル剤、液剤、乳剤、懸濁剤、注射剤、座剤、吸入剤、経皮吸収剤などの製剤として経口または非経口に適した形態で投与される。 経口投与製剤には固形製剤と液状製剤がある。本発明における固形製剤とは、製剤の全体又は集合体を構成する各要素が少なくとも一定の形を有する形態の製剤である。具体的には、例えば錠剤、丸剤、カプセル剤、顆粒剤、散剤又は粉剤が挙げられる。本発明において、内容物が液体のカプセル剤は、全体もしくは複数のカプセルの集合体を構成する一つのカプセルが一定の形を有する場合は、固形製剤に含まれる。また、用時溶解又は懸濁して服用するドライシロップ剤も、保存時に製剤全体又は粉末もしくは顆粒の個々の粒子が一定の形を有する場合、固形製剤に含まれる。それに対して、本発明における液状製剤とは、保存時から投与時まで液体の溶媒又は分散媒に溶解又は分散され、一定の形を有しないため液体として取り扱われる形態の製剤をいう。これら製剤を製造するには賦形剤、希釈剤、結合剤、崩壊剤、滑沢剤、抗酸化剤、安定化剤、保存剤、溶剤、可溶化剤、等張化剤などを添加することができる。医薬的に許容される賦形剤又は希釈剤としては、例えば、乳糖、ショ糖、ブドウ糖、麦芽糖、果糖、マンニトール、キシリトール、ソルビトール、エリスリトール、デンプン、スターチ、カルボキシメチルスターチナトリウム、粉末セルロース、結晶セルロース、カルメロース、結晶セルロース・カルメロースナトリウム、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、リン酸水素カルシウム、リン酸水素ナトリウム、リン酸水素カリウム、リン酸二水素カリウム、炭酸カルシウム、軽質無水ケイ酸、酸化チタン、メタケイ酸アルミン酸マグネシウムなどが挙げられる。結合剤としては、例えば、ヒドロキシプロプルセルロース、ヒプロメロース、デンプン、スターチ、アルファー化デンプン、部分アルファー化デンプン、ポリビニルピロリドンなどが挙げられる。崩壊剤としては、例えば、粉末セルロース、結晶セルロース、カルメロース、カルメロースカリウム、カルメロースカルシウム、カルメロースナトリウム、結晶セルロース・カルメロースナトリウム、クロスカルメロースナトリウム、低置換度ヒドロキシプロピルセルロース、デンプン、部分アルファー化デンプン、カルボキシメチルスターチナトリウム、ポビドン、クロスポビドンなどが挙げられる。滑沢剤としては、例えば、ステアリン酸、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸ポリオキシル、タルク、硬化油、ショ糖脂肪酸エステル、セタノール、ミツロウ、サラシミツロウなどが挙げられる。抗酸化剤としては、例えば、ジブチルヒドロキシトルエン(BHT)、没食子酸プロピル、ブチルヒドロキシアニソール(BHA)、トコフェロール、クエン酸、エデト酸塩などが挙げられる。溶剤としては、例えば水、生理食塩水、エタノールなどが、可溶化剤としては、例えば、ポリオキシエチレン硬化ヒマシ油、ポリソルベート類、ラウリル硫酸ナトリウム、マクロゴール類、ショ糖脂肪酸エステルなど、等張化剤可溶化剤としては、塩化ナトリウム、クエン酸、クエン酸ナトリウム、グリセリン、ソルビトール、ブドウ糖、プロピレングリコール、マクロゴール類、ホウ酸、ホウ砂、リン酸、リン酸水素塩類などが挙げられる。
本発明の化合物の投与量は、動物実験の結果に基づき、単回および反復投与したときに、一定量を超えないように定められる。試験例に開示した動物実験のデータに基づけば、成人患者に対して1日の投与量として1〜10000mg、好ましくは5〜1000mgを1日1回又は数回に分けて経口又は非経口で投与することが想定される。さらに、適量と投与回数は、投与方法、年齢、体重、性別、感受性、患者または被処置動物の症状の程度など、種々の要素を勘案し、専門医等によって決定されうる。また、本発明の化合物は、他の薬剤との組み合わせで使用することも可能である。
以下に、参考例、実施例及び試験例により本発明をさらに詳細に説明する。本発明の化合物の合成法は以下の方法に限定されず、各工程の順序を入れ替える、官能基の保護・脱保護を経る、等の当業者に周知の方法を用いて合成することもできる。
以下の参考例、実施例記載の各機器データは以下の測定機器で測定した。
NMRスペクトル:日本電子社JNM-ECA600(600MHz)、日本電子社JNM-ECA500(500MHz)
MSスペクトル:島津社LCMS−2010EVあるいはmicromass社 Platform LC
以下の参考例、実施例において、高速液体クロマトグラフィーマススペクトル(LCMS)は以下の条件により測定した。
測定機械:Agilent 2900およびAgilent 6150
カラム:Waters Acquity CSH C18,1.7μm,φ2.1x50mm
溶媒:A液;0.1%ギ酸含有水、B液;0.1%ギ酸含有アセトニトリル
(条件1)
グラジエント:0分(A液/B液=80/20)、1.2−1.4分(A液/B液=1/99)
流速:0.8mL/分、検出法:UV、ELSD
(条件2)
グラジエント:0分(A液/B液=95/5)、1.20分(A液/B液=50/50)、1.0mL/分、1.38分(A液/B液=3/97)
流速:0.8mL/分、検出法:UV、ELSD
イオン化法:ESI
参考例、実施例中の略号を以下に示す。
ESI:エレクトロスプレーイオン化法
MS:マススペクトル
CDCl3:重クロロホルム
NMR:核磁気共鳴
s:シングレット
br:幅広いピーク
d:ダブレット
m:マルチプレット
t:トリプレット
q:カルテット
NMRスペクトル:日本電子社JNM-ECA600(600MHz)、日本電子社JNM-ECA500(500MHz)
MSスペクトル:島津社LCMS−2010EVあるいはmicromass社 Platform LC
以下の参考例、実施例において、高速液体クロマトグラフィーマススペクトル(LCMS)は以下の条件により測定した。
測定機械:Agilent 2900およびAgilent 6150
カラム:Waters Acquity CSH C18,1.7μm,φ2.1x50mm
溶媒:A液;0.1%ギ酸含有水、B液;0.1%ギ酸含有アセトニトリル
(条件1)
グラジエント:0分(A液/B液=80/20)、1.2−1.4分(A液/B液=1/99)
流速:0.8mL/分、検出法:UV、ELSD
(条件2)
グラジエント:0分(A液/B液=95/5)、1.20分(A液/B液=50/50)、1.0mL/分、1.38分(A液/B液=3/97)
流速:0.8mL/分、検出法:UV、ELSD
イオン化法:ESI
参考例、実施例中の略号を以下に示す。
ESI:エレクトロスプレーイオン化法
MS:マススペクトル
CDCl3:重クロロホルム
NMR:核磁気共鳴
s:シングレット
br:幅広いピーク
d:ダブレット
m:マルチプレット
t:トリプレット
q:カルテット
参考例1 N,N-ジイソプロピル-N-メチルエタン-1,2-ジアミンの合成
<スキームA>
<スキームA>
8.9mol/Lメチルアミンのメタノール溶液135mLに氷冷下、ジイソプロピルアミノエチルクロリド塩酸塩24.0gのメタノール72mL溶液を滴下し、室温にて20分間撹拌した。反応液を減圧濃縮して得た残渣をクロロホルムに溶解し、氷冷下2 mol/L水酸化ナトリウム水溶液を加えた。反応液をクロロホルムにて2回抽出し、有機層を減圧下濃縮した後に、得られた残渣をアミノシリカゲルカラムクロマトグラフィー(ヘキサン:クロロホルム=5:1からクロロホルムのみ)にて精製し、標記化合物19.4gを得た。
MS(ESI) m/z= 159 [M+H]+
1H-NMR (400 MHz, CDCl3) δ(ppm) : 0.99 (d, J=1.71 Hz, 6 H) 1.00 (d, J=1.71 Hz, 6 H) 2.43 (s, 3 H) 2.54 -2.57 (m, 4 H) 2.96 -3.03 (m, 2 H)
MS(ESI) m/z= 159 [M+H]+
1H-NMR (400 MHz, CDCl3) δ(ppm) : 0.99 (d, J=1.71 Hz, 6 H) 1.00 (d, J=1.71 Hz, 6 H) 2.43 (s, 3 H) 2.54 -2.57 (m, 4 H) 2.96 -3.03 (m, 2 H)
参考例2 2-アミノ-N-エチルアセトアミドの合成
<スキームB>
<スキームB>
(1)N-(ベンジルオキシカルボニル)グリシン209gのクロロホルム1.0L溶液に 70%エチルアミン水溶液108mLを加え、氷冷下1-エチル-3-(3-ジメチルアミノプロピル)カルボジイミド塩酸塩249gを加えた後、室温にて終夜攪拌した。反応液に飽和重曹水を加え、クロロホルムにて抽出した。有機層を減圧下濃縮した後に、得られた残渣を酢酸エチル400mLに懸濁し、ヘキサン200mLを加え攪拌し、生じた個体をろ取しアミド体150gを得た。
<スキームC>
(2)上記の参考例2−(1)にて得られたアミド体150gのメタノール630mL溶液に 10%パラジウム炭素 15gを加え、水素雰囲気下、室温にて6日間攪拌した。反応液をろ過した後、濾液を減圧下濃縮し、標記化合物64.4gを得た。
MS(ESI) m/z= 103 [M+H]+
1H-NMR (400 MHz, CDCl3) δ(ppm) : 1.17 (t, J=7.2 Hz, 3 H) 1.38 (brs, 2 H) 3.29 - 3.37 (m, 4 H) 7.20 (brs, 1 H)
MS(ESI) m/z= 103 [M+H]+
1H-NMR (400 MHz, CDCl3) δ(ppm) : 1.17 (t, J=7.2 Hz, 3 H) 1.38 (brs, 2 H) 3.29 - 3.37 (m, 4 H) 7.20 (brs, 1 H)
参考例3 式[2]で示される化合物の製造
式[2]:
式[2]:
<スキームD>
(1)クラリスロマイシン200gをアセトン1.5Lに溶解し、無水酢酸30.3mLを滴下して、室温にて終夜攪拌した。反応液を減圧濃縮して得られた残渣に酢酸エチル、ヘキサン、水酸化ナトリウム水溶液を加えた後、飽和重曹水を加えてpH=9に調整した。析出した固体をグラスフィルターにて濾取、蒸留水で洗浄した後、減圧下乾燥してアセチル体202gを得た。
MS(ESI) m/z= 790.6 [M+H]+
MS(ESI) m/z= 790.6 [M+H]+
<スキームE>
(2)上記の参考例3−(1)で得られたアセチル体202gをクロロホルム1.8Lに溶解し、ピリジン210mLを加えた後氷冷し、トリホスゲン77.4gのクロロホルム0.8L溶液を40分間かけて滴下した。反応液を室温まで昇温した後、3時間攪拌した。反応液にピリジン158mLを加えて、氷冷下、トリホスゲン57.9gのクロロホルム溶液を滴下して、室温にて15分間攪拌した。反応液に蒸留水、飽和重曹水を加えてクロロホルムにて抽出し、有機層を無水硫酸マグネシウムで乾燥して濾過した。濾液を減圧濃縮して得られた残渣に酢酸エチルとヘキサンの1:1混合溶媒を加えて攪拌し、更にヘキサンを加え室温にて終夜攪拌した。生じた固体を濾取し、酢酸エチルとヘキサンの1:2混合溶媒で洗浄した後、減圧下乾燥してカーボネート体220gを得た。
MS(ESI) m/z= 816.5 [M+H]+
MS(ESI) m/z= 816.5 [M+H]+
<スキームF>
(3)N-クロロコハク酸イミド99.7gをクロロホルム1Lに溶解し、-25℃に冷却した。反応液にジメチルスルフィド210mLのクロロホルム0.2L溶液を20分間かけて滴下して、15分間攪拌した後、上記(2)で得られたカーボネート体のクロロホルム1L溶液を30分間かけて滴下して、15分間攪拌した。反応液にトリエチルアミン136mLのクロロホルム0.2L溶液を加えて、30分間攪拌した。反応液に飽和重曹水を加えて室温まで昇温し、クロロホルムにて分液した。有機層を無水硫酸マグネシウムで乾燥して濾過した後、濾液を減圧濃縮して得られた残渣に酢酸エチルとヘキサンの1:5の混合溶媒を加え室温にて終夜攪拌した。生じた固体を濾取し、酢酸エチルとヘキサンの1:2混合溶媒で洗浄してケトン体109gを得た。濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1からアセトン:ヘキサン:トリエチルアミン = 10:10:0.2)にて精製した後、上記と同様の方法にて結晶化してケトン体59.5gを得た。
MS(ESI) m/z= 814.5 [M+H]+
MS(ESI) m/z= 814.5 [M+H]+
<スキームG>
(4)トリメチルスルホキソニウムヨージド210g をジメチルスルホキシドとテトラヒドロフランの5:1混合溶媒1.2Lに溶解し、70%水素化ナトリウム32.6gを少量ずつ加えて、室温にて1.5時間攪拌した。氷冷下、上記(3)で得られたケトン体155gのテトラヒドロフラン0.8L溶液を滴下して、室温にて30分間攪拌した。反応液を氷冷し、蒸留水を加え、酢酸エチルを加えて分液し、得られた有機層を蒸留水で洗浄した。水層を酢酸エチルにて抽出し、有機層を蒸留水で洗浄した。集めた有機層を無水硫酸マグネシウムで乾燥して濾過した。濾液を減圧濃縮してエポキシ体146gを得た。
MS(ESI) m/z= 784.5 [M+H]+
1H-NMR (600 MHz, CDCl3) δ(ppm) : 0.90 (t, J=7.57 Hz, 3 H) 0.97 (d, J=7.34 Hz, 3 H) 1.04 (d, J=6.88 Hz, 3 H) 1.07 (s, 3 H) 1.14 (d, J=6.88 Hz, 3 H) 1.18 (d, J=5.96 Hz, 3 H) 1.21 - 1.36 (m, 7 H) 1.42 (s, 3 H) 1.47 - 1.55 (m, 1 H) 1.67 - 1.73 (m, 1 H) 1.83 - 1.98 (m, 5 H) 2.02 (d, J=1.83 Hz, 6 H) 2.18 - 2.29 (m, 1 H) 2.25 (s, 6 H) 2.58 - 2.69 (m, 1 H) 2.63 (d, J=4.13 Hz, 1 H) 2.80 - 2.89 (m, 1 H) 2.94 (d, J=4.13 Hz, 1 H) 3.12 - 3.26 (m, 1 H) 3.17 (s, 3 H) 3.34 (s, 3 H) 3.43 - 3.51 (m, 1 H) 3.66 (d, J=6.42 Hz, 1 H) 3.94 (brs, 1 H) 4.57 (d, J=7.34 Hz, 1 H) 4.73 (dd, J=10.55, 7.34 Hz, 1 H) 4.80 (q, J=6.42 Hz, 1 H) 4.98 - 5.06 (m, 2 H) 6.50 (s, 1 H)
MS(ESI) m/z= 784.5 [M+H]+
1H-NMR (600 MHz, CDCl3) δ(ppm) : 0.90 (t, J=7.57 Hz, 3 H) 0.97 (d, J=7.34 Hz, 3 H) 1.04 (d, J=6.88 Hz, 3 H) 1.07 (s, 3 H) 1.14 (d, J=6.88 Hz, 3 H) 1.18 (d, J=5.96 Hz, 3 H) 1.21 - 1.36 (m, 7 H) 1.42 (s, 3 H) 1.47 - 1.55 (m, 1 H) 1.67 - 1.73 (m, 1 H) 1.83 - 1.98 (m, 5 H) 2.02 (d, J=1.83 Hz, 6 H) 2.18 - 2.29 (m, 1 H) 2.25 (s, 6 H) 2.58 - 2.69 (m, 1 H) 2.63 (d, J=4.13 Hz, 1 H) 2.80 - 2.89 (m, 1 H) 2.94 (d, J=4.13 Hz, 1 H) 3.12 - 3.26 (m, 1 H) 3.17 (s, 3 H) 3.34 (s, 3 H) 3.43 - 3.51 (m, 1 H) 3.66 (d, J=6.42 Hz, 1 H) 3.94 (brs, 1 H) 4.57 (d, J=7.34 Hz, 1 H) 4.73 (dd, J=10.55, 7.34 Hz, 1 H) 4.80 (q, J=6.42 Hz, 1 H) 4.98 - 5.06 (m, 2 H) 6.50 (s, 1 H)
<スキームH>
(5)上記参考例3−(4)で得られたエポキシ体138gをテトラヒドロフランとジメチルホルムアミドの1:1混合溶媒1.4Lに溶解し、1,1’-カルボニルジイミダゾール85.6gを加えた。氷冷下、70%水素化ナトリウム18.1gを40分間かけて加えて、室温にて0.5時間攪拌した。反応液を氷冷し、蒸留水を加え、酢酸エチルにて抽出し、有機層を蒸留水で2回洗浄した。水層を酢酸エチルにて抽出し、有機層を蒸留水で2回洗浄した。集めた有機層を無水硫酸マグネシウムで乾燥して濾過した。濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサンからヘキサン:酢酸エチル=1:1からアセトン:ヘキサン:トリエチルアミン=10:10:0.2) にて精製した。得られた精製物に酢酸エチル、ヘキサン(1:1)を加えて、室温にて終夜攪拌した。生じた固体を濾取し、酢酸エチルとヘキサンの1:4混合溶媒にて洗浄し、式[2]で示される化合物87.1gを得た。
MS(ESI) m/z= 878.6 [M+H]+
1H-NMR (600 MHz, CDCl3) δ(ppm) : 0.85 - 1.41 (m, 25 H) 1.64 - 1.78 (m, 3 H) 1.79 (s, 3 H) 1.90 (dd, J=14.67, 5.04 Hz, 4 H) 1.86 (s, 3 H) 2.04 (s, 3 H) 2.19 - 2.28 (m, 1 H) 2.25 (s, 6 H) 2.60 - 2.68 (m, 1 H) 2.65 (d, J=4.13 Hz, 1 H) 2.86 - 2.97 (m, 1 H) 2.95 (d, J=4.13 Hz, 1 H) 3.15 (s, 3 H) 3.22 - 3.29 (m, 1 H) 3.35 (s, 3 H) 3.38 - 3.47 (m, 1 H) 3.66 (d, J=6.42 Hz, 1 H) 3.79 - 3.88 (m, 1 H) 4.56 (d, J=6.88 Hz, 1 H) 4.72 (dd, J=10.32, 7.57 Hz, 1 H) 4.79 (q, J=6.27 Hz, 1 H) 5.01 - 5.09 (m, 1 H) 5.83 (dd, J=10.55, 2.75 Hz, 1 H) 6.66 (s, 1 H) 7.07 (s, 1 H) 7.34 - 7.38 (m, 1 H) 8.08 (s, 1 H)
MS(ESI) m/z= 878.6 [M+H]+
1H-NMR (600 MHz, CDCl3) δ(ppm) : 0.85 - 1.41 (m, 25 H) 1.64 - 1.78 (m, 3 H) 1.79 (s, 3 H) 1.90 (dd, J=14.67, 5.04 Hz, 4 H) 1.86 (s, 3 H) 2.04 (s, 3 H) 2.19 - 2.28 (m, 1 H) 2.25 (s, 6 H) 2.60 - 2.68 (m, 1 H) 2.65 (d, J=4.13 Hz, 1 H) 2.86 - 2.97 (m, 1 H) 2.95 (d, J=4.13 Hz, 1 H) 3.15 (s, 3 H) 3.22 - 3.29 (m, 1 H) 3.35 (s, 3 H) 3.38 - 3.47 (m, 1 H) 3.66 (d, J=6.42 Hz, 1 H) 3.79 - 3.88 (m, 1 H) 4.56 (d, J=6.88 Hz, 1 H) 4.72 (dd, J=10.32, 7.57 Hz, 1 H) 4.79 (q, J=6.27 Hz, 1 H) 5.01 - 5.09 (m, 1 H) 5.83 (dd, J=10.55, 2.75 Hz, 1 H) 6.66 (s, 1 H) 7.07 (s, 1 H) 7.34 - 7.38 (m, 1 H) 8.08 (s, 1 H)
参考例4 式[3]で示される化合物の製造
式[3]:
式[3]:
<スキームI>
(1)参考例3で得られた式[2]で示される化合物360mgをアセトニトリル1.5mLに溶解し、1,8-ジアザビシクロ[5,4,0]-7-ウンデセン280μl、3-メタンスルホニルプロピルアミン塩酸塩273mgを加えて、室温にて1日間攪拌した。反応液に酢酸エチル、飽和塩化アンモニウム水溶液を加えて分液した。有機層を無水硫酸マグネシウムで乾燥して濾過し、濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルムからクロロホルム:メタノール:28%アンモニア水=25:1:0.1から15:1:0.1)にて精製してカーバメート体117mgを得た。
<スキームJ>
(2)上記の参考例4−(1)で得られたカーバメート体115mgをエタノール1mLに溶解し、N,N-ジエチル-N'-メチルエタン-1,2-ジアミン195μlを加えて、封管中100℃にて1日間攪拌した。反応液に酢酸エチル、飽和塩化アンモニウム水溶液を加えて分液した。有機層を無水硫酸マグネシウムで乾燥して濾過し、濾液を減圧濃縮して得られた残渣をシリカゲルカラムクロマトグラフィー(クロロホルムからクロロホルム:メタノール:28%アンモニア水=12:1:0.1)、分取用薄層クロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=20:1:0.1)にて精製し、式[3]で示される化合物62.7mgを得た。
なお、式[3]で示される化合物は、特許文献7、8に好ましい化合物として記載された実施例15である。
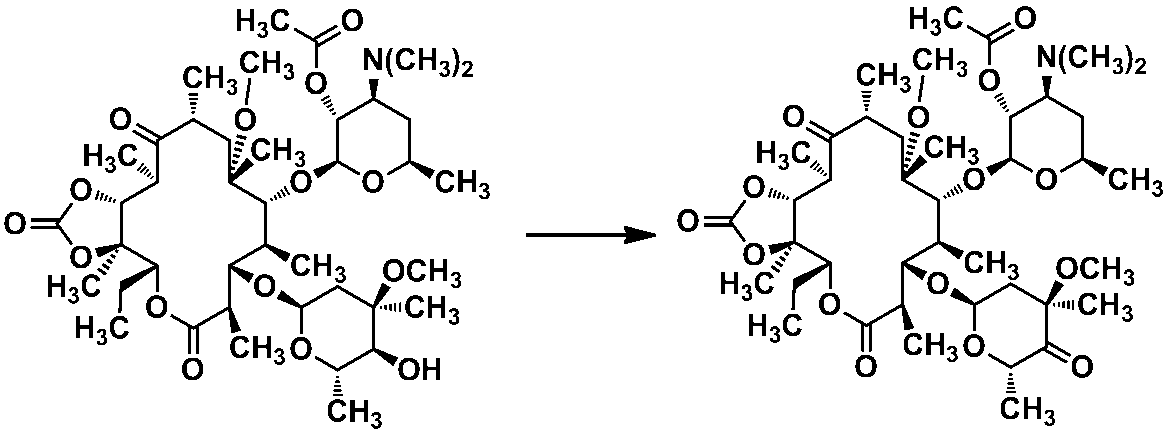
実施例1 式[1]で示される化合物の製造
式[1]:
式[1]:
<スキームK>
(1)参考例3で得られた式[2]で示される化合物277gをアセトニトリル315mLに溶解 し、参考例2で得られた化合物64.4gおよび1,8-ジアザビシクロ[5,4,0]-7-ウンデセン 191mLを加え、室温にて1.5時間攪拌した。反応液に水500mLを加え、酢酸エチル400mLにて抽出した。有機層を飽和食塩水で洗浄、硫酸マグネシウムにて乾燥濾過後、減圧下濃縮した。残渣を酢酸エチル 300mLとヘキサン 300mLより再結晶し、カーバメート体 83.5gを得た。濾液を減圧下濃縮後、再結晶(酢酸エチル 200mL, ヘキサン 200mL)することでカーバメート体34.4gを得た。さらに濾液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5)で精製した後に、酢酸エチル 100mLとヘキサン 100mLより再結晶し、カーバメート体16.3gを得た。濾液と前記カラムで得られた一部のフラクションを併せて、アミノシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=20:80 から 酢酸エチルのみ)で精製した後に、酢酸エチル 200mLとヘキサン 200mLより再結晶することでカーバメート体55.3gを得た。これらカーバメート体を合わせて189.5g得た。
MS(ESI) m/z= 912.6 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.85 - 0.90 (m, 6 H) 0.95 (d, J=7.55 Hz, 3 H) 0.99 - 1.29 (m, 19 H) 1.33 (s, 3 H) 1.44 (s, 3 H) 1.50 - 1.65 (m, 3 H) 1.68 - 1.73 (m, 1 H) 1.84 - 2.00 (m, 3 H) 2.05 (s, 3 H) 2.21 (dd, J=14.75, 3.09 Hz, 1 H) 2.26 (s, 6 H) 2.53 - 2.60 (m, 1 H) 2.62 (d, J=4.12 Hz, 1 H) 2.63 - 2.70 (m, 1 H) 2.86 - 2.91 (m, 1 H) 2.93 (s, 3 H) 2.94 (d, J=4.12 Hz, 1 H) 3.06 (q, J=6.86 Hz, 1 H) 3.27 - 3.36 (m, 2 H) 3.34 (s, 3 H) 3.41 - 3.50 (m, 1 H) 3.68 (d, J=6.17 Hz, 1 H) 3.73 (d, J=10.29 Hz, 1 H) 3.76 (s, 1 H) 4.20 (d, J=16.81 Hz, 1 H) 4.49 (d, J=16.81 Hz, 1 H) 4.63 (d, J=7.55 Hz, 1 H) 4.68 - 4.77 (m, 2 H) 5.04 (dd, J=4.63, 3.26 Hz, 1 H) 5.25 (dd, J=10.63, 2.40 Hz, 1 H) 6.17 (t, J=5.66 Hz, 1 H)
MS(ESI) m/z= 912.6 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.85 - 0.90 (m, 6 H) 0.95 (d, J=7.55 Hz, 3 H) 0.99 - 1.29 (m, 19 H) 1.33 (s, 3 H) 1.44 (s, 3 H) 1.50 - 1.65 (m, 3 H) 1.68 - 1.73 (m, 1 H) 1.84 - 2.00 (m, 3 H) 2.05 (s, 3 H) 2.21 (dd, J=14.75, 3.09 Hz, 1 H) 2.26 (s, 6 H) 2.53 - 2.60 (m, 1 H) 2.62 (d, J=4.12 Hz, 1 H) 2.63 - 2.70 (m, 1 H) 2.86 - 2.91 (m, 1 H) 2.93 (s, 3 H) 2.94 (d, J=4.12 Hz, 1 H) 3.06 (q, J=6.86 Hz, 1 H) 3.27 - 3.36 (m, 2 H) 3.34 (s, 3 H) 3.41 - 3.50 (m, 1 H) 3.68 (d, J=6.17 Hz, 1 H) 3.73 (d, J=10.29 Hz, 1 H) 3.76 (s, 1 H) 4.20 (d, J=16.81 Hz, 1 H) 4.49 (d, J=16.81 Hz, 1 H) 4.63 (d, J=7.55 Hz, 1 H) 4.68 - 4.77 (m, 2 H) 5.04 (dd, J=4.63, 3.26 Hz, 1 H) 5.25 (dd, J=10.63, 2.40 Hz, 1 H) 6.17 (t, J=5.66 Hz, 1 H)
<スキームL>
(2)上記の実施例1−(1)で得られたカーバメート体189gをメタノール 410mLに溶解し、4時間加熱還流した後、室温にて一昼夜攪拌した。反応液を減圧下濃縮した。残渣に酢酸エチル50mLおよびヘキサン300mLを加え、30分間攪拌し、生じた固体を濾取し、脱アセチル体41.2gを得た。濾液をアミノシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=20:80 から 100:0)およびシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5)で3回精製した。得られた粗精製物に酢酸エチル50mLおよびヘキサン 600mLを加え、30分間攪拌し、生じた固体を濾取し、脱アセチル体62.8gを得た。濾液をさらにシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5) で精製し、同様に酢酸エチル 20mLとヘキサン 50mLより再結晶し、脱アセチル体2.99gを得た。
MS(ESI) m/z= 870.6 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.88 (t, J=7.38 Hz, 3 H) 1.00 - 1.08 (m, 9 H) 1.09 - 1.27 (m, 1 H) 1.10 - 1.15 (m, 9 H) 1.18 (d, J=6.17 Hz, 3 H) 1.24 (d, J=7.20 Hz, 3 H) 1.36 (s, 3 H) 1.43 (s, 3 H) 1.58 (ddd, J=14.24, 10.46, 7.20 Hz, 1 H) 1.62 - 1.78 (m, 3 H) 1.88 (dd, J=14.92, 4.97 Hz, 1 H) 1.91 - 2.00 (m, 2 H) 2.23 (dd, J=14.75, 2.74 Hz, 1 H) 2.28 (s, 6 H) 2.42 - 2.50 (m, 1 H) 2.59 (dd, J=7.03, 4.29 Hz, 1 H) 2.62 (d, J=4.12 Hz, 1 H) 2.89 - 2.96 (m, 1 H) 2.93 (d, J=4.12 Hz, 1 H) 2.95 (s, 3 H) 3.08 (q, J=6.86 Hz, 1 H) 3.18 (dd, J=10.29, 7.20 Hz, 1 H) 3.27 - 3.39 (m, 2 H) 3.32 (s, 3 H) 3.42 - 3.50 (m, 1 H) 3.71 (d, J=6.52 Hz, 1 H) 3.76 (d, J=9.95 Hz, 1 H) 3.77 (s, 1 H) 4.21 (d, J=16.81 Hz, 1 H) 4.50 (d, J=10.63 Hz, 1 H) 4.52 (s, 1 H) 4.76 (q, J=6.52 Hz, 1 H) 5.04 (dd, J=4.80, 2.74 Hz, 1 H) 5.21 (dd, J=10.63, 2.40 Hz, 1 H) 6.25 (t, J=5.66 Hz, 1 H)
MS(ESI) m/z= 870.6 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.88 (t, J=7.38 Hz, 3 H) 1.00 - 1.08 (m, 9 H) 1.09 - 1.27 (m, 1 H) 1.10 - 1.15 (m, 9 H) 1.18 (d, J=6.17 Hz, 3 H) 1.24 (d, J=7.20 Hz, 3 H) 1.36 (s, 3 H) 1.43 (s, 3 H) 1.58 (ddd, J=14.24, 10.46, 7.20 Hz, 1 H) 1.62 - 1.78 (m, 3 H) 1.88 (dd, J=14.92, 4.97 Hz, 1 H) 1.91 - 2.00 (m, 2 H) 2.23 (dd, J=14.75, 2.74 Hz, 1 H) 2.28 (s, 6 H) 2.42 - 2.50 (m, 1 H) 2.59 (dd, J=7.03, 4.29 Hz, 1 H) 2.62 (d, J=4.12 Hz, 1 H) 2.89 - 2.96 (m, 1 H) 2.93 (d, J=4.12 Hz, 1 H) 2.95 (s, 3 H) 3.08 (q, J=6.86 Hz, 1 H) 3.18 (dd, J=10.29, 7.20 Hz, 1 H) 3.27 - 3.39 (m, 2 H) 3.32 (s, 3 H) 3.42 - 3.50 (m, 1 H) 3.71 (d, J=6.52 Hz, 1 H) 3.76 (d, J=9.95 Hz, 1 H) 3.77 (s, 1 H) 4.21 (d, J=16.81 Hz, 1 H) 4.50 (d, J=10.63 Hz, 1 H) 4.52 (s, 1 H) 4.76 (q, J=6.52 Hz, 1 H) 5.04 (dd, J=4.80, 2.74 Hz, 1 H) 5.21 (dd, J=10.63, 2.40 Hz, 1 H) 6.25 (t, J=5.66 Hz, 1 H)
<スキームM>
(3)上記の実施例1−(2)で得られた脱アセチル体104gをエタノール 120mLに溶解し、参考例1で得られた化合物56.5gを加え、2時間加熱還流した。反応液を減圧下濃縮した。残渣を酢酸エチルに溶解させ、飽和炭酸水素ナトリウム水溶液にて3回洗浄した後、水を加え分液した。水層を酢酸エチルで再度抽出し、水で洗浄した。あわせた有機層を飽和食塩水で洗浄、硫酸マグネシウムにて乾燥ろ過後、減圧下濃縮した。残渣を酢酸エチル 100mLとヘキサン 600mLより再結晶し、式[1]で示される化合物41.4gを得た。更に、濾液を減圧下濃縮し、残渣をシリカゲルカラムクロマトグラフィー(クロロホルム:メタノール:28%アンモニア水=99:1:0.1から85:15:1.5)で精製した後に酢酸エチル 100mLと ヘキサン 500mLより再結晶し、式[1]で示される化合物62.1gを得た。このようにして得られた式[1]で示される化合物を合せ、合計で103.5gを得た。
MS(ESI) m/z= 1028.8 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.87 (t, J=7.20 Hz, 3 H) 1.00 (m, J=10.60, 6.50 Hz, 15 H) 1.06 - 1.26 (m, 22 H) 1.38 (s, 3 H) 1.42 (s, 3 H) 1.52 - 1.79 (m, 4 H) 1.84 - 2.07 (m, 5 H) 2.29 (s, 6 H) 2.35 (s, 3 H) 2.39 - 2.55 (m, 5 H) 2.57 - 2.64 (m, 1 H) 2.83 (d, J=14.75 Hz, 1 H) 2.89 (dd, J=9.26, 7.20 Hz, 1 H) 2.94 (s, 3 H) 2.95-3.03 (m, 2 H) 3.08 (q, J=7.09 Hz, 1 H) 3.17 (dd, J=10.12, 7.38 Hz, 1 H) 3.22 - 3.32 (m, 1 H) 3.28 (s, 3 H) 3.34 - 3.48 (m, 3 H) 3.64 (d, J=7.55 Hz, 1 H) 3.73 (d, J=9.61 Hz, 1 H) 3.78 (s, 1 H) 4.08 (q, J=6.40 Hz, 1 H) 4.21 (d, J=17.15 Hz, 1 H) 4.40 (d, J=7.20 Hz, 1 H) 4.57 (d, J=16.81 Hz, 1 H) 4.95 (brs, 1 H) 4.99 (d, J=4.80 Hz, 1 H) 5.11 (dd, J=10.63, 2.06 Hz, 1 H) 6.39 (t, J=5.66 Hz, 1 H)
本発明化合物の作用は以下の薬理試験により確認された。
MS(ESI) m/z= 1028.8 [M+H]+
1H-NMR (499 MHz, CDCl3) δ(ppm) : 0.87 (t, J=7.20 Hz, 3 H) 1.00 (m, J=10.60, 6.50 Hz, 15 H) 1.06 - 1.26 (m, 22 H) 1.38 (s, 3 H) 1.42 (s, 3 H) 1.52 - 1.79 (m, 4 H) 1.84 - 2.07 (m, 5 H) 2.29 (s, 6 H) 2.35 (s, 3 H) 2.39 - 2.55 (m, 5 H) 2.57 - 2.64 (m, 1 H) 2.83 (d, J=14.75 Hz, 1 H) 2.89 (dd, J=9.26, 7.20 Hz, 1 H) 2.94 (s, 3 H) 2.95-3.03 (m, 2 H) 3.08 (q, J=7.09 Hz, 1 H) 3.17 (dd, J=10.12, 7.38 Hz, 1 H) 3.22 - 3.32 (m, 1 H) 3.28 (s, 3 H) 3.34 - 3.48 (m, 3 H) 3.64 (d, J=7.55 Hz, 1 H) 3.73 (d, J=9.61 Hz, 1 H) 3.78 (s, 1 H) 4.08 (q, J=6.40 Hz, 1 H) 4.21 (d, J=17.15 Hz, 1 H) 4.40 (d, J=7.20 Hz, 1 H) 4.57 (d, J=16.81 Hz, 1 H) 4.95 (brs, 1 H) 4.99 (d, J=4.80 Hz, 1 H) 5.11 (dd, J=10.63, 2.06 Hz, 1 H) 6.39 (t, J=5.66 Hz, 1 H)
本発明化合物の作用は以下の薬理試験により確認された。
試験例1 インビトロ抗菌活性
本発明品、実施例1の式[1]で表される化合物の各種試験菌に対するインビトロ抗菌力は、微量液体希釈法(CLSI法)に準じて測定した。また、参考例4の式[3]で表される化合物も同様に測定した。使用した試験菌を表1に示した。菌体番号A、B、C、D、E、F、G、H、I、J、K及びLの試験菌に対するMIC値(微生物生育最小阻止濃度μg/ml)を表2に示した。
本発明品、実施例1の式[1]で表される化合物の各種試験菌に対するインビトロ抗菌力は、微量液体希釈法(CLSI法)に準じて測定した。また、参考例4の式[3]で表される化合物も同様に測定した。使用した試験菌を表1に示した。菌体番号A、B、C、D、E、F、G、H、I、J、K及びLの試験菌に対するMIC値(微生物生育最小阻止濃度μg/ml)を表2に示した。
試験例2 インフルエンザ菌感受性試験
インフルエンザ菌(Haemophilus influenzae)39種類の臨床分離株を用い、試験例1と同様の手法を用いて、薬剤感受性について評価を実施した。表3に結果を示した。
インフルエンザ菌(Haemophilus influenzae)39種類の臨床分離株を用い、試験例1と同様の手法を用いて、薬剤感受性について評価を実施した。表3に結果を示した。
試験例3 インフルエンザ菌感染動物における治療効果試験
薬理効果の評価は下記に示す方法を用いた。
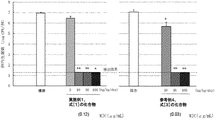
細菌として、Haemophilus influenzae ATCC43095株(菌体番号A)を用いた。チョコレート寒天培地で1晩培養した菌体を掻き取り、ヘモフィルス感受性試験培地またはフィルズエンリッチメント添加ブレインハートインフュージョン培地に懸濁後、1晩培養した。これをヘモフィルス感受性試験培地またはフィルズエンリッチメント添加ブレインハートインフュージョン培地で希釈し、接種菌液とした。マウス(ICR系、雄性、4週齢)に接種菌液0.05mLを気道内接種して感染させた。接種菌量は2.25x10^6CFU/マウスまたは9.00x10^5CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(100および200mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群6例、平均値±標準誤差)を図1に示した。
薬理効果の評価は下記に示す方法を用いた。
細菌として、Haemophilus influenzae ATCC43095株(菌体番号A)を用いた。チョコレート寒天培地で1晩培養した菌体を掻き取り、ヘモフィルス感受性試験培地またはフィルズエンリッチメント添加ブレインハートインフュージョン培地に懸濁後、1晩培養した。これをヘモフィルス感受性試験培地またはフィルズエンリッチメント添加ブレインハートインフュージョン培地で希釈し、接種菌液とした。マウス(ICR系、雄性、4週齢)に接種菌液0.05mLを気道内接種して感染させた。接種菌量は2.25x10^6CFU/マウスまたは9.00x10^5CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(100および200mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群6例、平均値±標準誤差)を図1に示した。
なお、図1についての備考は以下の通りである。媒体との有意差:Steel検定 *:p<0.05、**:p<0.01 実施例1、式[1]の化合物MIC値は4μg/mL、参考例4、式[3]の化合物MIC値は4μg/mL
以下において、試験結果としての肺内生菌数は、肺内生菌数(CFU/肺)の常用対数(常用対数は以下logと記載する)として表すこととする。
媒体投与群の肺内生菌数は5.88±0.14[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 100および200mg/kg投与群の肺内生菌数は、それぞれ3.54±0.49[log(CFU/肺)]および2.83±0.53[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(100および200mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。結果を、肺内生菌数(CFU/肺)の常用対数(常用対数は以下logと記載する)として表すこととすると、接種3日後の肺内生菌数は、媒体投与群で5.67±0.32[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 100および200mg/kg投与群の肺内生菌数は、それぞれ4.37±0.27[log(CFU/肺)]および2.53±0.23[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。以上より、実施例1の式[1]で表される化合物は、当該菌株に対して参考例4の式[3]で表される化合物と同程度の治療効果を示した。
媒体投与群の肺内生菌数は5.88±0.14[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 100および200mg/kg投与群の肺内生菌数は、それぞれ3.54±0.49[log(CFU/肺)]および2.83±0.53[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(100および200mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。結果を、肺内生菌数(CFU/肺)の常用対数(常用対数は以下logと記載する)として表すこととすると、接種3日後の肺内生菌数は、媒体投与群で5.67±0.32[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 100および200mg/kg投与群の肺内生菌数は、それぞれ4.37±0.27[log(CFU/肺)]および2.53±0.23[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。以上より、実施例1の式[1]で表される化合物は、当該菌株に対して参考例4の式[3]で表される化合物と同程度の治療効果を示した。
試験例4 エリスロマイシン耐性(erm(B)遺伝子保有)肺炎球菌感染動物における治療効果試験
薬理効果の評価は下記に示す方法を用いた。
薬理効果の評価は下記に示す方法を用いた。
細菌として、 Streptococcus pneumoniae 1101 株(臨床分離株)を用いた。使用菌株の凍結保存液を30vol%非働化ウマ血清添加トッドヒューイット液体培地に添加し、濁度(OD600)が約0.3となるまで培養した。これを30vol%非働化ウマ血清添加トッドヒューイット液体培地で希釈し、接種菌液とした。マウス(CBA/JN系、雄性、5週齢)に接種菌液0.05mLを経鼻接種して感染させた。接種菌量は7.50x10^4CFU/マウスまたは1.65x10^5CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群5〜6例、平均値±標準誤差)を図2に示した。
なお、図2についての備考は以下の通りである。媒体との有意差:Steel検定 *:p<0.05、**:p<0.01 実施例1、式[1]の化合物MIC値は0.25μg/mL、参考例4、式[3]の化合物MIC値は0.12μg/mL
媒体投与群の肺内生菌数は5.83±0.08[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 30および100mg/kg投与群の肺内生菌数は、それぞれ4.14±0.19[log(CFU/肺)]および2.28±0.24[log(CFU/肺)]であり、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(10、30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した結果、接種3日後の肺内生菌数は、媒体投与群で5.91±0.18[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 10、30および100mg/kg投与群の肺内生菌数は、それぞれ5.86±0.12[log(CFU/肺)]、5.22±0.16[log(CFU/肺)]および3.65±0.36[log(CFU/肺)]であり、参考例4の式[3]で表される化合物 100mg/kg投与群で媒体投与群と比較して有意に減少した。以上より、実施例1の式[1]で表される化合物は参考例4の式[3]で表される化合物より優れた治療効果を示した。
試験例5 エリスロマイシン耐性(mef(A)遺伝子保有)肺炎球菌感染動物における治療効果試験
薬理効果の評価は下記に示す方法を用いた。
薬理効果の評価は下記に示す方法を用いた。
細菌として、 Streptococcus pneumoniae 1028 株(臨床分離株)を用いた。使用菌株の凍結保存液を30vol%非働化ウマ血清添加トッドヒューイット液体培地に添加し、濁度(OD600)が約0.3となるまで培養した。これを30vol%非働化ウマ血清添加トッドヒューイット液体培地で希釈し、接種菌液とした。マウス(CBA/JN系、雄性、5週齢)に接種菌液0.05mLを経鼻接種して感染させた。接種菌量は3.45x10^4CFU/マウスまたは3.90x10^4CFU/マウスであった。接種翌日から1日1回2日間、実施例1の式[1]で表される化合物(3、10、30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した。接種3日後の肺内生菌数(1群5〜6例、平均値±標準誤差)を図3に示した。
なお、図3についての備考は以下の通りである。媒体との有意差:Steel検定 *:p<0.05、**:p<0.01 実施例1、式[1]の化合物MIC値は0.12μg/mL、参考例4、式[3]の化合物MIC値は0.03μg/mL
媒体投与群の肺内生菌数は6.94±0.07[log(CFU/肺)]であった。実施例1の式[1]で表される化合物 3、10、30および100mg/kg投与群の肺内生菌数は、それぞれ6.45±0.18[log(CFU/肺)]、1.30±0.00[log(CFU/肺)]、1.30±0.00[log(CFU/肺)]および1.30±0.00[log(CFU/肺)]であり、実施例1の式[1]で表される化合物 10、30および100mg/kg投与群で肺内生菌数は全例検出限界値以下を示し、媒体投与群と比較して有意に減少した。同様に、参考例4の式[3]で表される化合物(10、30および100mg/kg)または媒体(0.1mol/Lラクトビオン酸溶液および0.5w/v%炭酸水素ナトリウム溶液の等量混液)を経口投与した結果、接種3日後の肺内生菌数は、媒体投与群で7.03±0.22[log(CFU/肺)]であった。参考例4の式[3]で表される化合物 10、30および100mg/kg投与群の肺内生菌数は、それぞれ5.67±0.38[log(CFU/肺)]、1.30±0.00[log(CFU/肺)]および1.30±0.00[log(CFU/肺)]であり、参考例4の式[3]で表される化合物 10、30および100mg/kg投与群で媒体投与群と比較して有意に減少した。ただし全例で検出限界値以下を示したのは参考例4の式[3]で表される化合物 30および100mg/kg投与群のみであった。以上より、実施例1の式[1]で表される化合物は参考例4の式[3]で表される化合物より優れた治療効果を示した。
本発明の化合物又はその薬学的に許容される塩は、グラム陽性細菌、グラム陰性細菌やマイコプラズマに強い抗菌活性を有し、特に従来のマクロライド系抗生物質では十分な抗菌活性が得られなかったエリスロマイシン耐性菌(例えば耐性肺炎球菌、連鎖球菌、及びマイコプラズマ)などに対しても優れた抗菌活性を有し、医薬品として利用することができる。
Claims (5)
- 式[1]:
- 固形製剤である請求項1に記載の感染症の予防又は治療薬。
- 剤形が錠剤、丸剤、カプセル剤、顆粒剤、散剤、または粉剤から選択される請求項2に記載の感染症の予防又は治療薬。
- 乳糖、ショ糖、ブドウ糖、麦芽糖、果糖、マンニトール、デンプン、粉末セルロース、結晶セルロース、カルメロース、低置換度ヒドロキシプロピルセルロース、ヒドロキシプロピルセルロース及びヒドロキシプロピルメチルセルロースからなる群から選ばれる1種以上を含む請求項3に記載の感染症の予防又は治療薬。
- 式[1]で表される化合物若しくはその薬学的に許容される塩、又はそれらの水和物若しくはそれらの溶媒和物である有効成分の投与量が1〜10000mg/日である、請求項1から4に記載の感染症の予防又は治療薬。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016027731 | 2016-02-17 | ||
| JP2016027731 | 2016-02-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017145247A true JP2017145247A (ja) | 2017-08-24 |
Family
ID=59680621
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2017026792A Pending JP2017145247A (ja) | 2016-02-17 | 2017-02-16 | C−4”位置換マクロライド誘導体を含有する感染症の予防および/又は治療剤 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017145247A (ja) |
-
2017
- 2017-02-16 JP JP2017026792A patent/JP2017145247A/ja active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20150006082A (ko) | 거대고리 다형체, 이런 다형체를 포함하는 조성물, 및 이의 사용 및 제조방법 | |
| CN103965273B (zh) | 一种大环内酯类化合物 | |
| CN101117336A (zh) | 7-(4-肟基-3-氨基-1-哌啶基)喹啉羧酸衍生物及其制备方法 | |
| JP5874871B1 (ja) | C−4”位置換マクロライド化合物 | |
| EP3719020B1 (en) | Crystal form of beta-lactamase inhibitor and preparation method therefor | |
| JPWO2015056799A1 (ja) | ヒドロキサム酸誘導体 | |
| WO2015056800A1 (ja) | ヒドロキサム酸誘導体 | |
| JP2017145247A (ja) | C−4”位置換マクロライド誘導体を含有する感染症の予防および/又は治療剤 | |
| CN116514775B (zh) | 一种徳拉沙星葡甲胺盐新晶型及其制备方法 | |
| CN101558078B (zh) | 具有抗炎活性的大环内酯化合物 | |
| WO2017142002A1 (ja) | C-4"位置換マクロライド化合物フリー体及び塩の結晶形並びにそれらの製造方法 | |
| JP2019064921A (ja) | C−4”位置換マクロライド化合物の結晶形及びそれらの製造方法 | |
| JP2019064922A (ja) | C−4”位置換マクロライド化合物の塩、それらの結晶形及びそれらの製造方法 | |
| CN115466246B (zh) | 吡咯酰哌啶胺类化合物及其用途 | |
| JP2007527434A (ja) | 非晶質タクロリマス及びその調製 | |
| CN104803911A (zh) | 一类截短侧耳素化合物、其药物组合物、合成方法与用途 | |
| WO2018067663A2 (en) | Ketolides having antibacterial activity | |
| WO2015056798A1 (ja) | ヒドロキサム酸誘導体 | |
| JP2016199499A (ja) | (2s)−2−メチルアミノ−n−ヒドロキシ−n’,2−ジメチルプロパンジアミドを有する化合物の結晶形及びそれらの製造方法 | |
| HK1233650A1 (en) | C-4''-substituted macrolide compound | |
| JP2017105714A (ja) | 多剤排出ポンプ阻害剤 | |
| JP2023505389A (ja) | マクロライド化合物及びその慢性呼吸器疾患の治療用途 | |
| JPWO2004078771A1 (ja) | 2−フルオロ−6−o−置換ケトライド誘導体 | |
| HK40076742A (en) | Novel salt of terphenyl compound | |
| JP2021178793A (ja) | 超多剤耐性グラム陰性菌に有効な新規アミノ配糖体抗菌剤 |