JP2017156706A - トナー - Google Patents
トナー Download PDFInfo
- Publication number
- JP2017156706A JP2017156706A JP2016042555A JP2016042555A JP2017156706A JP 2017156706 A JP2017156706 A JP 2017156706A JP 2016042555 A JP2016042555 A JP 2016042555A JP 2016042555 A JP2016042555 A JP 2016042555A JP 2017156706 A JP2017156706 A JP 2017156706A
- Authority
- JP
- Japan
- Prior art keywords
- charge control
- control agent
- toner
- resin composition
- polyester resin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Landscapes
- Developing Agents For Electrophotography (AREA)
Abstract
【課題】 かぶり抑制、ドット再現性、現像性に優れたトナーを提供する。【解決手段】 以下のトナーを用いることによって、上記の課題は達成される。R1−O−またはR2−COO−で表わされる部分構造を有するポリエステル樹脂と、炭素数が12〜102の脂肪族炭化水素とを含有するポリエステル系樹脂組成物、およびヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下である荷電制御剤、を含有するトナー。【選択図】 なし
Description
本発明は、電子写真法の如き画像形成方法に使用されるトナーに関する。
電子写真画像形成装置には、高速化、省エネルギー化、小型化が求められている一方で、画質面で求められる水準も高くなってきている。また、紙情報の見た目の印象も当然重要ではあるが、画像そのものに機能を持たせることも多くなってきており、そのような観点でも画質向上は重要な命題となってきている。例えば、スマートフォンの普及で二次元バーコードの利用機会が増えている。二次元バーコードも面積の小型化が進んでおり、読み取り不良を防ぐために画質の向上は必須である。
また、紙情報の管理の点からセキュリティの意識も向上してきている。代表的な例として、紙情報が「原本」なのか「コピー」なのかを識別するために、あらかじめ「原本」に複写機では読み取れない小ドットを描画しておく地紋印字の需要も高まってきている。そのためには小ドットを適切に印字することが重要である。
一方、電子写真画像形成装置は熱帯地域での需要も伸びてきており、同時に上述したような画質への要求も次第に高まってきている。
このような地域では、日中に高温高湿であっても、夜間に気温が下がった場合は、飽和していた水蒸気が結露するなど、過酷な環境にさらされやすい。特にトナーは一般的に現像器内や容器内など空気の入れ替わりにくい空間で使用されるため、結露の影響を大きく受けて、トナー性能に大きな影響を及ぼす可能性がある。
このように従来では想定しにくかった厳しい状況においても、画質の低下を抑制できるトナーが求められている。
特許文献1では、荷電制御剤の結晶構造を制御することで、湿度を制御しないヒートサイクル条件下で放置しても、良好な現像性と耐静電オフセット性が得られるトナーが提案されている。
特許文献2では、特定の荷電制御剤と、脂肪族化合物を反応させたポリエステル樹脂を用いることで、トナーの飛び散りや選択現像性を改良できる提案がなされている。
特許文献3では、ポリエステル樹脂に脂肪族化合物を反応させることで、低温定着性と耐高温オフセット性を改良できる提案がなされている。
これらの技術によれば、トナーの定着性や画像弊害に対しては一定の効果が確認される。しかしながら、これらの技術も使用環境によって、例えば、結露の影響を大きく受けるような状況においては、かぶりやドット再現性に大きな影響を受ける可能性があった。
本発明の目的は、高湿下でヒートサイクルを繰り返す条件下でも、かぶりやドット再現性の低下を抑えることができるトナーを提供することである。
本発明は、ポリエステル系樹脂組成物、および荷電制御剤を含有するトナー粒子を有するトナーであって、
該ポリエステル系樹脂組成物は、
i)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、炭素数が12以上102以下の脂肪族炭化水素とを含有し、
ii)該ポリエステル系樹脂組成物の質量を基準として、該部分構造と炭素数が12以上102以下の該脂肪族炭化水素とを合計して、2.5質量%以上10.0質量%以下含有しており、
iii)示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、30℃以上100℃以下にピークトップを有する吸熱ピークを有し、該吸熱ピークの吸熱量が0.10J/g以上1.90J/g以下であり、
該荷電制御剤は、該荷電制御剤0.01gをヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下である、
ことを特徴とするトナーに関する。
該ポリエステル系樹脂組成物は、
i)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、炭素数が12以上102以下の脂肪族炭化水素とを含有し、
ii)該ポリエステル系樹脂組成物の質量を基準として、該部分構造と炭素数が12以上102以下の該脂肪族炭化水素とを合計して、2.5質量%以上10.0質量%以下含有しており、
iii)示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、30℃以上100℃以下にピークトップを有する吸熱ピークを有し、該吸熱ピークの吸熱量が0.10J/g以上1.90J/g以下であり、
該荷電制御剤は、該荷電制御剤0.01gをヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下である、
ことを特徴とするトナーに関する。
本発明によれば、高湿下でのヒートサイクルを受けても、かぶり、ドット再現性に優れたトナーを得ることができる。
これまでもトナー中の荷電制御剤の分散性を制御する試みが行われてきており、トナーを一定の温湿度下で保管した後の評価や、湿度を制御しないヒートサイクル下で保管した場合の性能は確認されてきた。しかし、上述したように電子写真画像形成装置は熱帯地域での需要も伸びてきている。このような地域では、日中に高温高湿であっても、夜間に気温が下がった場合は、飽和していた水蒸気が結露するなど、過酷な環境にさらされやすい。特にトナーは一般的に現像器内や容器内など空気の入れ替わりにくい空間で使用されるため、結露の影響を大きく受ける可能性がある。そして、従来のトナーでは、高湿下でヒートサイクルを受けると、トナーの帯電特性が大きく変化してしまい、その結果、かぶりが発生しやすくなり、さらにドット再現性が低下しやすくなることがわかった。
本発明者らは、高湿下でのヒートサイクルを受けたトナーにおいて、かぶりの発生や、ドット再現性の低下を抑えるために、荷電制御剤の分散性をさらに制御する検討を行った。
かぶりの発生やドット再現性の低下は、トナーの帯電分布がブロードになった場合によく見られる現象である。トナー製造直後で既にトナー中の荷電制御剤の分散が不十分であると、トナー一粒毎に帯電性能が変化しやすくなり、結果的にブロードな帯電分布のトナーになりやすい。
一方で、トナー製造直後にトナー中の荷電制御剤の分散が良い帯電分布がシャープなトナーでも、高湿下でのヒートサイクルを受けると、一般的に水分はポリエステル系樹脂組成物を可塑化するために、トナー中の低粘度成分等が徐々に動きやすくなる。そのため荷電制御剤の分布も長い時間をかけて徐々に偏りが生じやすい。ヒートサイクルは、可塑化したポリエステル系樹脂組成物を硬化させ、再び可塑化することを繰り返すので、トナー中の低粘度成分等や荷電制御剤の移動を加速させると考えられる。高湿下でのヒートサイクルは、トナーの帯電分布のブロード化を促進していると考えられる。
このように高湿下でのヒートサイクルを受けた結果、荷電制御剤の分散が低下した状態になってしまい、ブロードな帯電分布のトナーになりやすく、かぶりが発生し、ドット再現性が低下しやすくなる。
これらの結果を受けて本発明者らは、高湿下でのヒートサイクルで、荷電制御剤の移動を抑えられれば、かぶりの発生やドット再現性の低下を抑えることができるのではないかと考えた。
その方法として、荷電制御剤と親和性が近い成分を結着樹脂中に固定化すれば、荷電制御剤がその部位に留まりやすくなり、高湿下でのヒートサイクルでも荷電制御剤の移動を抑えることができると考えた。
具体的には、ポリエステル系樹脂組成物、および荷電制御剤を含有するトナー粒子を有するトナーであって、
該ポリエステル系樹脂組成物は、
i)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101である脂肪族炭化水素から水素原子が1つ脱離して形成される基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、炭素数が12〜102である脂肪族炭化水素とを含有し、
ii)該ポリエステル系樹脂組成物の質量を基準として、該部分構造と炭素数が12以上102以下である該脂肪族炭化水素とを合計して、2.5質量%以上10.0質量%以下含有しており、
iii)示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、30℃以上100℃以下に吸熱ピークを有し、該吸熱ピークの吸熱量が0.10J/g以上1.90J/g以下であり、
該荷電制御剤は、該荷電制御剤0.01gをヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下であるトナー、
を用いることによって、高湿下でのヒートサイクルでも、荷電制御剤の分散性を維持し、かぶりの発生やドット再現性の低下を抑えることを見出した。
該ポリエステル系樹脂組成物は、
i)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101である脂肪族炭化水素から水素原子が1つ脱離して形成される基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、炭素数が12〜102である脂肪族炭化水素とを含有し、
ii)該ポリエステル系樹脂組成物の質量を基準として、該部分構造と炭素数が12以上102以下である該脂肪族炭化水素とを合計して、2.5質量%以上10.0質量%以下含有しており、
iii)示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、30℃以上100℃以下に吸熱ピークを有し、該吸熱ピークの吸熱量が0.10J/g以上1.90J/g以下であり、
該荷電制御剤は、該荷電制御剤0.01gをヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下であるトナー、
を用いることによって、高湿下でのヒートサイクルでも、荷電制御剤の分散性を維持し、かぶりの発生やドット再現性の低下を抑えることを見出した。
[ポリエステル系樹脂組成物と荷電制御剤との関係]
ポリエステル系樹脂組成物と荷電制御剤との関係について、記載する。
ポリエステル系樹脂組成物と荷電制御剤との関係について、記載する。
本発明で用いられるポリエステル系樹脂組成物は、
(a)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101の脂肪族炭化水素から水素原子が1つ脱離して形成される官能基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、
(b)炭素数が12〜102の脂肪族炭化水素と、
を含有する。
(a)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101の脂肪族炭化水素から水素原子が1つ脱離して形成される官能基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、
(b)炭素数が12〜102の脂肪族炭化水素と、
を含有する。
上記(a)で規定されるポリエステル樹脂とは、炭素数が12〜102の脂肪族モノアルコール、または炭素数が12〜102の脂肪族モノカルボン酸を、反応させたポリエステル樹脂である。
また、ポリエステル系樹脂組成物において、上記(a)で規定されるポリエステル樹脂が有する部分構造と、上記(b)で規定される脂肪族炭化水素との合計量は、該ポリエステル系樹脂組成物の質量を基準として、2.5質量%以上10.0質量%以下である。
更に、該ポリエステル系樹脂組成物は、示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、30℃以上100℃以下に吸熱ピークを有し、且つ該吸熱ピークの吸熱量が0.10J/g以上1.90J/g以下である。この吸熱ピークは上記(b)で規定される脂肪族炭化水素に由来するものである。
吸熱ピークの吸熱量を上記範囲にするためには、脂肪族炭化水素の量を調整すればよい。具体的には、例えば、以下のようにして調整できる。まず、上記(a)で規定されるポリエステル樹脂の製造において用いられる脂肪族モノアルコール、脂肪族モノカルボン酸を得る際に、アルコール或いは酸で変性されていない脂肪族炭化水素を残存させる。そして、変性物と未変性物とを含有する組成物を用いて、ポリエステルとの反応を行うことによって、ポリエステル系樹脂組成物中の脂肪族炭化水素の量を調整することができる。
上記部分構造において、R1の炭素数が11以下の場合、及びR2の炭素数が10以下の場合、含有量に係る規定を満たすためには、ポリエステル系樹脂組成物中の部分構造の数を多くする必要がある。この場合には、荷電制御剤の動きを制御することが困難となる。R1の炭素数が103以上の場合、及びR2の炭素数が102以上の場合は、ポリエステル系樹脂組成物中の部分構造の数が少なくなるため、荷電制御剤の偏在が顕著となる。尚、脂肪族炭化水素と荷電制御剤との相互作用に関しては、荷電制御剤の項にて詳述する。
該ポリエステル系樹脂組成物の該部分構造と炭素数12〜102の該脂肪族炭化水素との合計が、2.5質量%より少ない場合、トナー中に形成される部分構造が少ない。そのため、荷電制御剤が安定して留まる場所が少なく、高湿下でヒートサイクルを経た場合の変動が大きくなってしまう。一方、合計が10.0質量%より多い場合、ポリエステル系樹脂組成物中の部分構造の数が多くなり、部分構造の近傍にとどまる荷電制御剤の割合が高くなってしまう。その結果、荷電制御剤の分散性が低下して、かぶりが生じやすくなり、ドット再現性が低下しやすい。
該ポリエステル系樹脂組成物の示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、吸熱ピークの吸熱量が1.90J/gより大きい場合は、部分構造を形成していない脂肪族炭化水素が多いことを示している。そのため、トナー中を移動しやすい脂肪族炭化水素が多くなり、高湿下でヒートサイクルを経た場合の変動が大きくなってしまう。その結果、かぶりが発生しやすくなり、ドット再現性が低下しやすい。吸熱ピークの吸熱量が0.10J/g未満の場合は、荷電制御剤の分散が劣るようになりやすくなるために、摩擦帯電性が不十分となりやすい。
ポリエステル系樹脂組成物の吸熱ピークが30℃未満の場合、含有される脂肪族炭化水素の移動性が高まり、高湿下でのヒートサイクルでは、部分構造の部位に荷電制御剤を留めておくことが難しくなる。その結果、荷電制御剤の分散性が劣るようになり、かぶりが生じやすくなり、ドット再現性が低下しやすい。一方、吸熱ピークが100℃より大きい場合、脂肪族炭化水素の移動性が低くなり、高湿下でのヒートサイクルでは荷電制御剤が安定した状態で部分構造の部位に留まってしまい、荷電制御剤の分散性が低下する。そのため、かぶりが発生しやすくなり、ドット再現性が低下しやすい。
次いで、荷電制御剤について記載する。
ヘキサンは脂肪族炭化水素に分類され、ポリエステル系樹脂組成物に含まれる該部分構造及び炭素数が12〜102の該脂肪族炭化水素と極性的に非常に近い関係にある。そこで、本発明者らは、ヘキサンと荷電制御剤の親和性を制御することで、高湿下でのヒートサイクルでも、荷電制御剤の分散性を維持し、かぶりの発生やドット再現性の低下を抑えられると考えた。
上記の考えに従い、検討した結果、荷電制御剤0.01gをヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下であることが重要であるとの知見を得た。
上記の透過率に係る試験において、静置して30分後の透過率が10%以下であるということは、ヘキサン中では荷電制御剤が沈降しにくいことを示している。つまり、上記のような荷電制御剤は、ヘキサンとの親和性が高く、即ち、上記(a)で規定されるポリエステル樹脂が有する部分構造や脂肪族炭化水素とも親和性が高いということを示している。
ポリエステル系樹脂組成物中の部分構造は、ポリエステル樹脂の分子末端として存在しているため、フリーで存在している場合と比べて、トナー中を移動しにくい。また、ポリエステル系樹脂組成物中の部分構造と親和性が高い荷電制御剤は、ポリエステル系樹脂組成物中の部分構造の部位に集まる。結果、高湿下でのヒートサイクルを受けた場合であっても、部分構造部位に集まった荷電制御剤は、その周辺に留まることができ、分散性を維持することができると考えられる。その結果、高湿下でのヒートサイクルでも、トナーの帯電分布をシャープに保つことができるため、かぶりやドット再現性を改善することができる。
一方、ポリエステル系樹脂組成物中に含有される脂肪族炭化水素の周囲にも荷電制御剤は集まりやすい。脂肪族炭化水素は、トナー中を移動しやすい成分であると考えられる。ポリエステル樹脂組成物中の部分構造の部位に集まっているだけであると、トナー全体に荷電制御剤が広がりにくいが、この移動しやすい成分が存在し、それと荷電制御剤が共に挙動することによって、トナー粒子全体に荷電制御剤が分散する。その結果、より均一な帯電性を有するトナーが得られるようになる。
そして、部分構造と脂肪族炭化水素の合計量が2.5〜10.0質量%であって、且つ脂肪族炭化水素の含有量が、吸熱量の規定で表される程度である場合に、留まる成分と分散する成分のバランスが好適になる。
即ち、トナー粒子中において、移動しにくい成分の周辺に留まる荷電制御剤と、移動しやすい成分と共に挙動し、トナー粒子中に分散する荷電制御剤の両方が、適度な割合で存在することによって、本願発明の効果が得られているものと考えられる。
尚、ヘキサン分散液を静置して30分後の透過率は、荷電制御剤の構造の影響を大きく受けるため、透過率を10%以下にするためには、例えば、荷電制御剤の構造を制御すればよい。
荷電制御剤は、一次粒子の個数平均粒径(D1)が0.80μm以上2.00μm以下であることが好ましい。一次粒子の個数平均粒径(D1)が上記の範囲内であれば、ポリエステル系樹脂組成物の部分構造の部位に留まりやすくなる。そのため、高湿下のヒートサイクル後のかぶりの抑制やドット再現性がより良化する。さらに高湿下のヒートサイクル後の高温高湿下の画出しでも良好な現像性が得られる。
荷電制御剤は、下記式[1]で表される化合物であることが好ましく、高湿下のヒートサイクル後の高温高湿下の画出しにおいて、初期現像性が良化する。このメカニズムについての詳細は次のように推測している。式[1]の化合物はイオン結合性の化合物であり、結晶構造を取りやすくなる。そのため、ポリエステル系樹脂組成物の部分構造の部位に規則的に配列することができ、その結果、帯電の立ち上がりがスムーズになり高温高湿下の初期現像性が良化すると考えている。
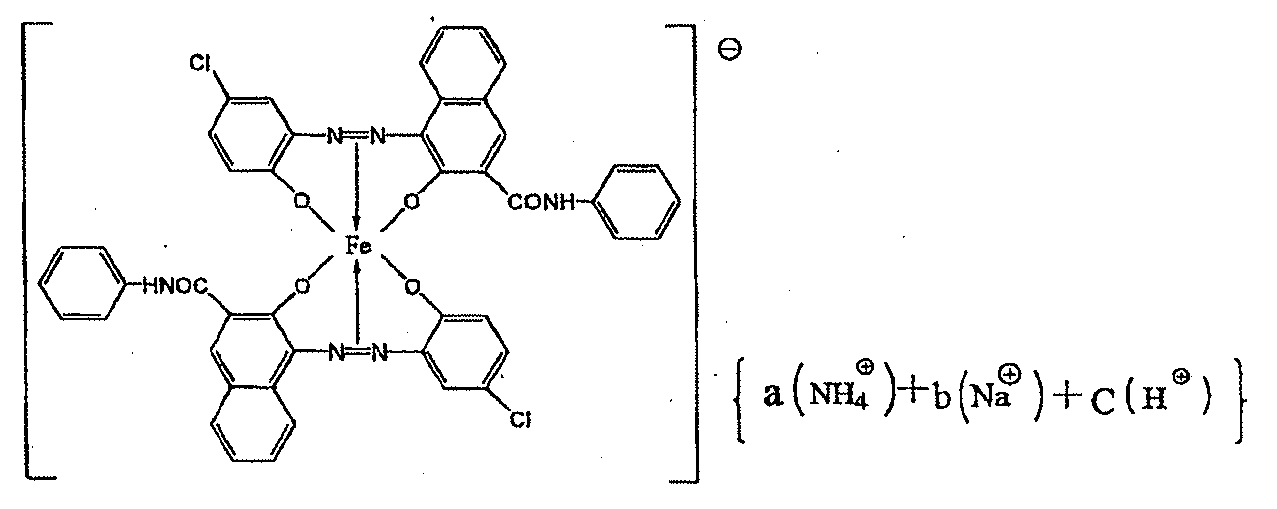
(式中、A1、A2及びA3は、それぞれ独立して、水素原子、ニトロ基又はハロゲン原子を示す。B1は水素原子又はアルキル基を示す。Mは、Fe原子、Cr原子、又はAl原子を示し、X+は、水素イオン、アルカリ金属イオン、アンモニウムイオン、アルキルアンモニウムイオン又はこれらの混合イオンを示す。)
また、荷電制御剤は、下記式[2]で表される化合物であることが更に好ましい。高湿下のヒートサイクル後の高温高湿下の画出しにおいて、初期現像性がさらに良化する。式[2]は、電気陰性度の高い置換基を複数有しているために、非常に帯電の立ち上がりが優れるためと考えている。
また、荷電制御剤は、下記式[2]で表される化合物であることが更に好ましい。高湿下のヒートサイクル後の高温高湿下の画出しにおいて、初期現像性がさらに良化する。式[2]は、電気陰性度の高い置換基を複数有しているために、非常に帯電の立ち上がりが優れるためと考えている。
(式中、X+は、水素イオン、アルカリ金属イオン、アンモニウムイオン、アルキルアンモニウムイオン又はこれらの混合イオンを示す。)
荷電制御剤をトナー粒子に内添する場合、添加量としては、結着樹脂100質量部に対して、0.1〜10質量部が好ましく、0.2〜5質量部がより好ましい。また、トナー粒子に外添する場合は、トナー粒子100質量部に対して、0.01〜5質量部が好ましく、0.01〜2質量部がより好ましい。
荷電制御剤をトナー粒子に内添する場合、添加量としては、結着樹脂100質量部に対して、0.1〜10質量部が好ましく、0.2〜5質量部がより好ましい。また、トナー粒子に外添する場合は、トナー粒子100質量部に対して、0.01〜5質量部が好ましく、0.01〜2質量部がより好ましい。
[ポリエステル系樹脂組成物に係るその他の記載]
脂肪族炭化水素を、炭素数が12〜102の脂肪族アルコールまたは脂肪族カルボン酸で変性した場合、通常、変性率は100%にならない。そのため、アルコールまたは酸で変性された脂肪族炭化水素と未変性の脂肪族炭化水素の混合物(以下、“変性・未変性混合物”と称することもある。)が得られる。この混合物をポリエステル樹脂の合成に用いることによって、脂肪族炭化水素から水素原子1つ脱離して形成される基が末端に結合したポリエステル樹脂と、脂肪族炭化水素とを含有するポリエステル系樹脂組成物を得ることができる。
脂肪族炭化水素を、炭素数が12〜102の脂肪族アルコールまたは脂肪族カルボン酸で変性した場合、通常、変性率は100%にならない。そのため、アルコールまたは酸で変性された脂肪族炭化水素と未変性の脂肪族炭化水素の混合物(以下、“変性・未変性混合物”と称することもある。)が得られる。この混合物をポリエステル樹脂の合成に用いることによって、脂肪族炭化水素から水素原子1つ脱離して形成される基が末端に結合したポリエステル樹脂と、脂肪族炭化水素とを含有するポリエステル系樹脂組成物を得ることができる。
ポリエステル系樹脂組成物の吸熱ピーク温度や吸熱量を本発明の規定を満たすようにするためには、反応条件の最適化による変性率の制御を行ったり、変性反応後に精製作業を行い、未変性の脂肪族炭化水素成分を除去したりすればよい。脂肪族炭化水素成分の変性率の好ましい範囲は、85%以上であり、より好ましくは90%以上である。
また、変性された脂肪族炭化水素と未変性の脂肪族炭化水素の混合物について、アルコール変性である場合には、水酸基価80〜110mgKOH/gが好ましく、より好ましくは90〜105mgKOH/gである。酸変性である場合には、酸価90〜150mgKOH/gが好ましく、より好ましくは95〜140mgKOH/gである。上記範囲に制御することで、ポリエステル樹脂成分と変性部位との反応性が高まり、その結果、DSCの吸熱ピーク面積を効率的に低減することが可能となるため好ましい。
また、本発明のポリエステル系樹脂組成物の製造方法としては特に制限は無いが、下記の製造方法であることが好ましい。
本発明で用いられるポリエステル系樹脂組成分は、変性・未変性混合物の存在下でポリエステルの重合反応を行う製造方法が好ましい。すなわち、変性・未変性混合物をポリエステル合成反応の初期から投入することで、効率的且つ均一にポリエステル系樹脂組成物中へ変性・未変性混合物を取り込むことができるため好ましい。
変性・未変性混合物は、炭素数が12〜102の脂肪族モノアルコールを主成分として含有することが、反応率の観点から好ましい。
ポリエステル系樹脂組成分が、R1−O−を部分構造として有するポリエステル樹脂を含有し、そして、R1が炭素数12〜102の脂肪族炭化水素の2級炭素から水素原子が1つ脱離して形成される基であることが好ましい。この場合、1つの部分構造に2本の炭化水素の分子鎖が存在することになる。2本の炭化水素の分子鎖が存在することで、さらに荷電制御剤が部分構造の部位に留まりやすくなる。そのため、高湿下でのヒートサイクルでも、荷電制御剤の分散性を維持し、かぶりの悪化やドット再現性の低下を抑えることができる。このような構成とするためには、炭素数が12〜102の脂肪族2級モノアルコールを主成分として含有する変性・未変性混合物を用いればよい。
またポリエステル系樹脂組成物に含有されるポリエステル樹脂が、ポリエステル部位とビニル重合体部位が化学的に結合したハイブリッド樹脂であることが好ましい。この場合、脂肪族モノアルコールまたは脂肪族カルボン酸は、ハイブリッド樹脂のポリエステル部位の末端に反応したものであることが好ましい。
ハイブリッド樹脂にすることで、環境に依らず帯電性が安定になり、高湿下でのヒートサイクルでも、荷電制御剤の分散性を維持し、かぶりの発生やドット再現性の低下を抑えることができるため好ましい。
ポリエステル系樹脂組成物に用いられるポリエステル樹脂或いは上記ハイブリッド樹脂のポリエステル部位を構成するためのモノマーとしては以下の化合物が挙げられる。
アルコール成分としては、以下のものが挙げられる。エチレングリコール、プロピレングリコール、1,3−ブタンジオール、1,4−ブタンジオール、2,3−ブタンジオール、ジエチレングリコール、トリエチレングリコール、1,5−ペンタンジオール、1,6−ヘキサンジオール、ネオペンチルグリコール、2−エチル−1,3−ヘキサンジオール、水素化ビスフェールA、下記(3)式で表されるビスフェノール誘導体及び下記(4)式で示されるジオール類。
(式中、Rはエチレンまたはプロピレン基を示し、x及びyはそれぞれ1以上の整数であり、かつ、x+yの平均値は2〜10である。)
上記(3)式で表されるビスフェノール誘導体を用いる場合、本発明においては、エチレンオキサイド付加体(EO)とプロピレンオキサイド(PO)付加体の比EO/POが40/60〜60/40が好ましい。EO:POの比を該範囲に制御することで、長鎖アルキル成分が樹脂中へ依り均一に分散し、保存安定性が良好になる為好ましい。
酸成分としては、以下のものが挙げられる。フタル酸、テレフタル酸、イソフタル酸、無水フタル酸の如きベンゼンジカルボン酸類またはその無水物;こはく酸、アジピン酸、セバシン酸、アゼライン酸の如きアルキルジカルボン酸類またはその無水物、またさらに炭素数6〜18のアルキル基またはアルケニル基で置換されたこはく酸もしくはその無水物;フマル酸、マレイン酸、シトラコン酸、イタコン酸の如き不飽和ジカルボン酸またはその無水物。
また、ポリエステル系樹脂組成物に用いられるポリエステル樹脂或いは上記ハイブリッド樹脂のポリエステル部位は、三価以上の多価カルボン酸またはその無水物及び/または三価以上の多価アルコールを用いても良い。三価以上の多価カルボン酸またはその無水物としては、以下のものが挙げられる。1,2,4−ベンゼントリカルボン酸、1,2,4−シクロヘキサントリカルボン酸、1,2,4−ナフタレントリカルボン酸、ピロメリット酸及びこれらの酸無水物または低級アルキルエステル。三価以上の多価アルコールとしては、以下のものが挙げられる。1,2,3−プロパントリオール、トリメチロールプロパン、ヘキサントリオール、ペンタエリスリトール。本発明のポリエステル系樹脂組成物においては、環境変動による安定性も高い芳香族系アルコールが特に好ましく、例えば1,2,4−ベンゼントリカルボン酸及びその無水物が挙げられる。
ポリエステル系樹脂組成物に、前記ハイブリッド樹脂を用いる場合、ハイブリッド樹脂のビニル重合体部位を構成するためのビニル系モノマーとしては、公知のものを用いればよく、例えば、次の化合物が挙げられる。
スチレン及びその誘導体;不飽和モノオレフィン類;不飽和ポリエン類;ハロゲン化ビニル類;ビニルエステル類;アクリル酸エステル類;メタクリル酸エステル類;不飽和二塩基酸;不飽和二塩基酸無水物;不飽和二塩基酸エステル。
また、本発明のポリエステル系樹脂組成物に、前記ハイブリッド樹脂を用いる場合、ハイブリッド樹脂のビニル重合体部位は、ビニル基を2個以上有する架橋剤で架橋された架橋構造を有してもよい。この場合に用いられる架橋剤としては、公知のものを用いることができ、例えば、以下のものが挙げられる。芳香族ジビニル化合物(ジビニルベンゼン、ジビニルナフタレン);ジアクリレート化合物類。これらの架橋剤は、他のモノマー成分100質量部に対して、0.01〜10.00質量部、さらに好ましくは0.03〜5.00質量部用いることができる。
ビニル系重合体部位の重合に用いられる重合開始剤としては、公知のものを用いればよく、例えば、アゾ系開始剤、過酸化物系開始剤が挙げられる。
ポリエステル系樹脂組成物に前記したハイブリッド樹脂を用いる場合には、ビニル系樹脂及び/またはポリエステル樹脂成分中に、両樹脂成分と反応し得るモノマー成分を含むことが好ましい。ポリエステル樹脂成分を構成するモノマーのうちビニル系樹脂と反応し得るものとしては、例えば、フマル酸、マレイン酸、シトラコン酸、イタコン酸の如き不飽和ジカルボン酸またはその無水物が挙げられる。ビニル系樹脂成分を構成するモノマーのうちポリエステル樹脂成分と反応し得るものとしては、カルボキシル基またはヒドロキシ基を有するものや、アクリル酸もしくはメタクリル酸エステル類が挙げられる。
また、上記のようなポリエステル系樹脂組成物を単独で使用してもよいが、軟化点の異なる2種類の高軟化点樹脂(H)と低軟化点樹脂(L)とを任意の範囲で混合して使用しても良い。高軟化点樹脂(H)は、軟化点が100〜170℃であることが好ましい。また、低軟化点樹脂(L)は軟化点が70℃以上100℃未満であることが好ましい。
異なる2種類の樹脂を結着樹脂として用いる場合、少なくとも結着樹脂中の50質量%以上が、ポリエステル系樹脂組成物である必要がある。
ポリエステル系樹脂組成物を結着樹脂として単独で使用する場合、軟化点Tmは90〜170℃が好ましい。さらに好ましくは100〜130℃である。Tmが上記の範囲内であれば、耐高温オフセット性と低温定着性のバランスが良好となる。
また、ポリエステル系樹脂組成物のガラス転移温度(Tg)は、保存安定性の観点から45.0℃以上であることが好ましく、50.0℃以上であることが特に好ましい。また、低温定着性の観点から、Tgは75.0℃以下であることが好ましく、65.0℃以下であることが特に好ましい。
また、ポリエステル系樹脂組成物の酸価は15.0〜30.0mgKOH/g、より好ましくは、20.0〜30.0mgKOH/gである。酸価を上記範囲に制御することで、本発明の効果が得られやすい。
本発明のトナーは、高湿下でのヒートサイクル放置後のトナーの全個数基準のポジ成分率が4.0個数%以下であることが好ましい。トナーが高湿下でヒートサイクルを受けても荷電制御剤の分散性が保持されてポジ成分の増加を抑えることができれば、かぶりが発生しにくくなる。
[その他の成分に関して]
本発明のトナーは、磁性材料を含有させ磁性トナーとしても使用しうる。この場合、磁性材料は着色剤の役割を兼ねることもできる。
本発明のトナーは、磁性材料を含有させ磁性トナーとしても使用しうる。この場合、磁性材料は着色剤の役割を兼ねることもできる。
磁性材料としては、マグネタイト、ヘマタイト、フェライトのような酸化鉄、鉄、コバルト、ニッケルのような金属あるいはこれらの金属とアルミニウム、コバルト、銅、鉛、マグネシウム、スズ、亜鉛、アンチモン、ビスマス、カルシウム、マンガン、チタン、タングステン、バナジウムのような金属の合金およびその混合物が挙げられる。これらの磁性材料は平均粒子径が2μm以下、好ましくは0.05〜0.5μmのものが好ましい。トナー中に含有させる量としては、樹脂成分100質量部に対し20〜200質量部が良い。
本発明において用いることができる着色剤について記載する。
黒色着色剤としてカーボンブラック、グラフト化カーボンを用いることができる。また、イエロー/マゼンタ/シアン着色剤を用い黒色に調色して用いてもよい。
イエロー着色剤としては、縮合アゾ化合物,イソインドリノン化合物,アンスラキノン化合物,アゾ金属錯体,メチン化合物,アリルアミド化合物に代表される化合物が挙げられる。
マゼンタ着色剤としては、縮合アゾ化合物,ジケトピロロピロール化合物,アントラキノン,キナクリドン化合物,塩基染料レーキ化合物,ナフトール化合物,ベンズイミダゾロン化合物,チオインジゴ化合物,ペリレン化合物等が挙げられる。
シアン着色剤としては、銅フタロシアニン化合物及びその誘導体,アントラキノン化合物,塩基染料レーキ化合物等が挙げられる。
本発明の着色剤は、色相角、彩度、明度、耐候性、OHP透明性、トナー中への分散性の点から選択される。該着色剤の添加量は、結着樹脂100質量部に対し1〜20質量部が好ましい。
本発明に係るトナーは、定着時の離型性を付与するために、ワックスを含有してもよい。ワックスとしては、公知のものを用いることができる。
また、本発明に係るトナーは、流動性や帯電性を向上させるために、外添剤を添加してもよい。
外添剤としては、公知のものを使用することができ、例えば、以下のものを用いることができる。フッ化ビニリデン微粉末、ポリテトラフウルオロエチレン微粉末の如きフッ素系樹脂粉末;湿式製法シリカ、乾式製法シリカの如き微粉末シリカ、微粉末酸化チタン、微粉末アルミナ、ケイ素と他の金属との複合酸化物;酸化亜鉛、酸化スズの如き酸化物;チタン酸ストロンチウムやチタン酸バリウム、チタン酸カルシウム、ジルコン酸ストロンチウムやジルコン酸カルシウムの如き複酸化物;炭酸カルシウム及び、炭酸マグネシウムの如き炭酸塩化合物等。また、これらの外添剤は、シラン化合物、チタンカップリング剤、シリコーンオイルにより表面処理を施してもよい。
トナー粒子100質量部に対して、外添剤を総量で、0.01〜3質量部使用することが好ましい。
[トナーの製造方法]
本発明に係るトナー粒子の製造方法は特に限定されず、例えば粉砕法や、乳化重合法、懸濁重合法及び溶解懸濁法などのいわゆる重合法を用いることができる。
本発明に係るトナー粒子の製造方法は特に限定されず、例えば粉砕法や、乳化重合法、懸濁重合法及び溶解懸濁法などのいわゆる重合法を用いることができる。
粉砕法では、まず、トナー粒子を構成する結着樹脂、着色剤、ワックス、電荷制御剤等を、ヘンシェルミキサ、ボールミル等の混合機により充分に混合する。次いで、得られた混合物を二軸混練押出機、加熱ロール、ニーダー、エクストルーダー等の熱混練機を用いて溶融混練し、冷却固化後、粉砕及び分級を行う。これによって、本発明に係るトナー粒子を得られる。
さらに必要に応じ所望の外添剤をヘンシェルミキサの如き混合機により充分混合し、本発明に係るトナーを得ることができる。
次に、本発明に係る各物性の測定方法に関して記載する。
<変性・未変性混合物における変性率、酸またはアルコールで変性された脂肪族炭化水素、未変性の脂肪族炭化水素の炭素数の測定方法>
変性・未変性混合物の炭素数は、ガスクロマトグラフィー質量分析装置(GC・MS)により、以下のようにして測定する。変性・未変性混合物10mgを精秤しサンプルビンに入れる。このサンプルビンに精秤した10gのヘキサンを加えてフタをした後、ホットプレートで温度150℃に加温して混合する。その後、脂肪族炭化水素が析出してこないようすばやくガスクロマトグラフィーの注入口へこのサンプルを注入して分析を行い、横軸を炭素数、縦軸をシグナルの強度とする炭素数分布のチャートを得る。次いで、得られたチャートにおいて、検出された全ピークをマススペクトルにより、変性物と未変性物とに帰属させる。
変性・未変性混合物の炭素数は、ガスクロマトグラフィー質量分析装置(GC・MS)により、以下のようにして測定する。変性・未変性混合物10mgを精秤しサンプルビンに入れる。このサンプルビンに精秤した10gのヘキサンを加えてフタをした後、ホットプレートで温度150℃に加温して混合する。その後、脂肪族炭化水素が析出してこないようすばやくガスクロマトグラフィーの注入口へこのサンプルを注入して分析を行い、横軸を炭素数、縦軸をシグナルの強度とする炭素数分布のチャートを得る。次いで、得られたチャートにおいて、検出された全ピークをマススペクトルにより、変性物と未変性物とに帰属させる。
炭素数分布のチャートにおいて、変性物、未変性物に帰属されたピークについて、成分ごとの総面積を算出する。その各成分のピーク総面積を検出した全ピークの総面積で割ることにより各成分の割合を算出し、変性物の割合を変性率とした。また、本発明における炭素数とは、上記炭素数分布チャートにおいてピークトップとなる炭素数のことを示す。
測定装置及び測定条件は、下記の通りである。
GC:Agilent Technologies 7890A GC
MS: Agilent Technologies 5975C
カラム:ULTRA ALLOY−1 P/N:UA1−30m−0.5F (フロンティア・ラボ社製)
キャリアーガス:He
オーブン:(1)温度100℃で5分ホールド、(2)30℃/分で温度360℃まで昇温、(3)温度360℃で60分ホールド
注入口:温度300℃
初期圧力:10.523 psi
スプリット比:50:1
カラム流量:1mL/min
MS質量範囲:46−800 イオン化:EI イオン源:250℃ 四重極温度:150℃
GC:Agilent Technologies 7890A GC
MS: Agilent Technologies 5975C
カラム:ULTRA ALLOY−1 P/N:UA1−30m−0.5F (フロンティア・ラボ社製)
キャリアーガス:He
オーブン:(1)温度100℃で5分ホールド、(2)30℃/分で温度360℃まで昇温、(3)温度360℃で60分ホールド
注入口:温度300℃
初期圧力:10.523 psi
スプリット比:50:1
カラム流量:1mL/min
MS質量範囲:46−800 イオン化:EI イオン源:250℃ 四重極温度:150℃
<吸熱ピークの温度及び吸熱量(ΔH)の測定方法>
示差走査熱量測定(DSC)におけるポリエステル系樹脂組成物の吸熱ピーク温度及び吸熱量は、示差走査型熱量分析装置「Q2000」(TA Instruments社製)を用いて、ASTM D3418−82に準じて測定される。装置検出部の温度補正はインジウムと合亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。
示差走査熱量測定(DSC)におけるポリエステル系樹脂組成物の吸熱ピーク温度及び吸熱量は、示差走査型熱量分析装置「Q2000」(TA Instruments社製)を用いて、ASTM D3418−82に準じて測定される。装置検出部の温度補正はインジウムと合亜鉛の融点を用い、熱量の補正についてはインジウムの融解熱を用いる。
具体的には、測定試料約2mgを精密に秤量し、これをアルミパン中に入れ、リファレンスとして空のアルミパンを用いて、測定温度範囲10〜200℃の間で、昇温速度10℃/minで常温常湿下測定を行う。尚、測定においては、一度200℃まで昇温させ、続いて10℃まで降温し、その後に再度昇温を行う。この昇温過程で得られるDSC曲線において、温度10〜200℃の範囲における最大の吸熱ピークのピークトップの温度を求める。また、吸熱ピークの吸熱量(ΔH)は、上記吸熱ピークの積分値である。
<試料の水酸基価の測定方法>
(装置及び器具)
メスシリンダー(100mL)
全量ピペット(5mL)
平底フラスコ(200mL)
グリセリン浴
(装置及び器具)
メスシリンダー(100mL)
全量ピペット(5mL)
平底フラスコ(200mL)
グリセリン浴
(試薬)
・アセチル化試薬(無水酢酸25gを全量フラスコ100mLに取り、ピリジンを加えて全量を100mLにし、十分に振り混ぜる。)
・フェノールフタレイン溶液
・0.5kmol/m3水酸化カリウムエタノール溶液
・アセチル化試薬(無水酢酸25gを全量フラスコ100mLに取り、ピリジンを加えて全量を100mLにし、十分に振り混ぜる。)
・フェノールフタレイン溶液
・0.5kmol/m3水酸化カリウムエタノール溶液
(測定法)
(a)試料を0.5〜6.0g平底フラスコに精秤して、これにアセチル化試薬5mLを全量ピペットを用いて加える。
(b)フラスコの口に小さな漏斗を置き、温度95〜100℃のグリセリン浴中に底部約1cmを浸して加熱する。フラスコの首がグリセリン浴の熱を受けて温度が上がるのを防ぐために、中に丸い穴をあけた厚紙の円板をフラスコの首の付け根にかぶせる。
(c)1時間後フラスコをグリセリン浴から取り出し、放冷後漏斗から水1mLを加えて振り動かして無水酢酸を分解する。
(d)更に、分解を完全にするため、再びフラスコをグリセリン浴中で10分間加熱し、放冷後エタノール(95vol%)5mLで漏斗及びフラスコの壁を洗う。
(e)フェノールフタレイン溶液数滴を指示薬として加え、0.5kmol/m3水酸化カリウムエタノール溶液で滴定し、指示薬の薄い紅色が約30秒間続いたときを終点とする。
(f)空試験は、試料を入れないで(a)〜(e)を行う。
(g)試料が溶解しにくい場合は、少量のピリジンを追加するか、キシレン又はトルエンを加えて溶解する。
(a)試料を0.5〜6.0g平底フラスコに精秤して、これにアセチル化試薬5mLを全量ピペットを用いて加える。
(b)フラスコの口に小さな漏斗を置き、温度95〜100℃のグリセリン浴中に底部約1cmを浸して加熱する。フラスコの首がグリセリン浴の熱を受けて温度が上がるのを防ぐために、中に丸い穴をあけた厚紙の円板をフラスコの首の付け根にかぶせる。
(c)1時間後フラスコをグリセリン浴から取り出し、放冷後漏斗から水1mLを加えて振り動かして無水酢酸を分解する。
(d)更に、分解を完全にするため、再びフラスコをグリセリン浴中で10分間加熱し、放冷後エタノール(95vol%)5mLで漏斗及びフラスコの壁を洗う。
(e)フェノールフタレイン溶液数滴を指示薬として加え、0.5kmol/m3水酸化カリウムエタノール溶液で滴定し、指示薬の薄い紅色が約30秒間続いたときを終点とする。
(f)空試験は、試料を入れないで(a)〜(e)を行う。
(g)試料が溶解しにくい場合は、少量のピリジンを追加するか、キシレン又はトルエンを加えて溶解する。
(計算)
得られた結果から下記式(1)によって試料の水酸基価を求める。
A=[{(B−C)×28.05×f}/S]+D ・・・ (1)
A:水酸基価(mgKOH/g)
B:空試験に用いた0.5kmol/m3水酸化カリウムエタノール溶液の量(mL)
C:滴定に用いた0.5kmol/m3水酸化カリウムエタノール溶液の量(mL)
f:0.5kmol/m3水酸化カリウムエタノール溶液のファクター
S:試料の質量(g)
D:試料の酸価(mgKOH/g)
28.05:水酸化カリウムの式量56.11×1/2
得られた結果から下記式(1)によって試料の水酸基価を求める。
A=[{(B−C)×28.05×f}/S]+D ・・・ (1)
A:水酸基価(mgKOH/g)
B:空試験に用いた0.5kmol/m3水酸化カリウムエタノール溶液の量(mL)
C:滴定に用いた0.5kmol/m3水酸化カリウムエタノール溶液の量(mL)
f:0.5kmol/m3水酸化カリウムエタノール溶液のファクター
S:試料の質量(g)
D:試料の酸価(mgKOH/g)
28.05:水酸化カリウムの式量56.11×1/2
<試料の酸価の測定方法>
(装置及び器具)
三角フラスコ(300mL)
ビュレット(25mL)
水浴又は熱板
(装置及び器具)
三角フラスコ(300mL)
ビュレット(25mL)
水浴又は熱板
(試薬)
・0.1kmol/m3塩酸
・0.1kmol/m3水酸化カリウムエタノール溶液(標定は、0.1kmol/m3塩酸25mLを全ピペットを用いて三角フラスコに取り、フェノールフタレイン溶液を加え、0.1kmol/m3水酸化カリウムエタノール溶液で滴定し、中和に要した量からファクターを求める。)
・フェノールフタレイン溶液(ジエチルエーテルとエタノール(99.5vol%)を体積比で1:1又は2:1で混合したもの。これらは、使用直前にフェノールフタレイン溶液を指示薬として数滴加え、0.1kmol/m3水酸化カリウムエタノール溶液で中和する。)
・0.1kmol/m3塩酸
・0.1kmol/m3水酸化カリウムエタノール溶液(標定は、0.1kmol/m3塩酸25mLを全ピペットを用いて三角フラスコに取り、フェノールフタレイン溶液を加え、0.1kmol/m3水酸化カリウムエタノール溶液で滴定し、中和に要した量からファクターを求める。)
・フェノールフタレイン溶液(ジエチルエーテルとエタノール(99.5vol%)を体積比で1:1又は2:1で混合したもの。これらは、使用直前にフェノールフタレイン溶液を指示薬として数滴加え、0.1kmol/m3水酸化カリウムエタノール溶液で中和する。)
(測定法)
(a)試料1〜20gを三角フラスコに精秤する。
(b)溶剤100mL及び指示薬としてフェノールフタレイン溶液を数滴加え、水浴上で試料が完全に溶けるまで十分に振り混ぜる。
(c)0.1kmol/m3水酸化カリウムエタノール溶液で滴定し、指示薬の薄い紅色が30秒間続いたときを終点とする。
(a)試料1〜20gを三角フラスコに精秤する。
(b)溶剤100mL及び指示薬としてフェノールフタレイン溶液を数滴加え、水浴上で試料が完全に溶けるまで十分に振り混ぜる。
(c)0.1kmol/m3水酸化カリウムエタノール溶液で滴定し、指示薬の薄い紅色が30秒間続いたときを終点とする。
(計算)
得られた結果から下記式(2)により試料の酸価を算出する。
A=5.611×B×f/S ・・・ (2)
A:酸価(mgKOH/g)
B:滴定に用いた0.1kmol/m3水酸化カリウムエタノール溶液の量(mL)
f:0.1kmol/m3水酸化カリウムエタノール溶液のファクター
S:試料の質量(g)
5.611:水酸化カリウムの式量56.11×1/10
得られた結果から下記式(2)により試料の酸価を算出する。
A=5.611×B×f/S ・・・ (2)
A:酸価(mgKOH/g)
B:滴定に用いた0.1kmol/m3水酸化カリウムエタノール溶液の量(mL)
f:0.1kmol/m3水酸化カリウムエタノール溶液のファクター
S:試料の質量(g)
5.611:水酸化カリウムの式量56.11×1/10
<Tgの測定方法>
ポリエステル系樹脂組成物のガラス転移温度(Tg)は、示差走査熱量測定(DSC)における吸熱ピーク温度及び吸熱ピーク量は、示差走査型熱量分析装置「Q2000」(TA Instruments社製)を用いて、ASTM D3418−82に準じて測定される。
ポリエステル系樹脂組成物のガラス転移温度(Tg)は、示差走査熱量測定(DSC)における吸熱ピーク温度及び吸熱ピーク量は、示差走査型熱量分析装置「Q2000」(TA Instruments社製)を用いて、ASTM D3418−82に準じて測定される。
測定試料として、ポリエステル系樹脂組成物約2mgを精密に秤量したものを用いる。これをアルミパン中に入れ、リファレンスとして空のアルミパンを用いる。測定温度範囲を30℃以上200℃以下とし、一旦、昇温速度10℃/minで30℃から200℃まで昇温した後、降温速度10℃/minで200℃から30℃まで降温し、再度、昇温速度10℃/minで200℃まで昇温させる。2回目の昇温過程で得られるDSC曲線において、比熱変化が出る前と出た後のベースラインの中間点の線と示差熱曲線との交点を、ガラス転移温度Tgとする。
<軟化点の測定方法>
ポリエステル系樹脂組成物の軟化点は、定荷重押し出し方式の細管式レオメータ「流動特性評価装置 フローテスターCFT−500D」(島津製作所社製)を用い、装置付属のマニュアルに従って行う。本装置では、測定試料の上部からピストンによって一定荷重を加えつつ、シリンダに充填した測定試料を昇温させて溶融し、シリンダ底部のダイから溶融された測定試料を押し出し、この際のピストン降下量と温度との関係を示す流動曲線を得ることができる。
ポリエステル系樹脂組成物の軟化点は、定荷重押し出し方式の細管式レオメータ「流動特性評価装置 フローテスターCFT−500D」(島津製作所社製)を用い、装置付属のマニュアルに従って行う。本装置では、測定試料の上部からピストンによって一定荷重を加えつつ、シリンダに充填した測定試料を昇温させて溶融し、シリンダ底部のダイから溶融された測定試料を押し出し、この際のピストン降下量と温度との関係を示す流動曲線を得ることができる。
本発明においては、「流動特性評価装置 フローテスターCFT−500D」に付属のマニュアルに記載の「1/2法における溶融温度」を軟化点とする。尚、1/2法における溶融温度とは、次のようにして算出されたものである。まず、流出が終了した時点におけるピストンの降下量Smaxと、流出が開始した時点におけるピストンの降下量Sminとの差の1/2を求める(これをXとする。X=(Smax−Smin)/2)。そして、流動曲線においてピストンの降下量がXとSminの和となるときの流動曲線の温度が、1/2法における溶融温度Tmである。
測定試料は、約1.0gのサンプルを、25℃の環境下で、錠剤成型圧縮機(例えば、NT−100H、エヌピーエーシステム社製)を用いて約10MPaで、約60秒間圧縮成型し、直径約8mmの円柱状としたものを用いる。
CFT−500Dの測定条件は、以下の通りである。
試験モード:昇温法
開始温度:50℃
到達温度:200℃
測定間隔:1.0℃
昇温速度:4.0℃/min
ピストン断面積:1.000cm2
試験荷重(ピストン荷重):10.0kgf(0.9807MPa)
予熱時間:300秒
ダイの穴の直径:1.0mm
ダイの長さ:1.0mm
試験モード:昇温法
開始温度:50℃
到達温度:200℃
測定間隔:1.0℃
昇温速度:4.0℃/min
ピストン断面積:1.000cm2
試験荷重(ピストン荷重):10.0kgf(0.9807MPa)
予熱時間:300秒
ダイの穴の直径:1.0mm
ダイの長さ:1.0mm
<荷電制御剤のヘキサン分散液の透過率の測定方法>
ガラス製の100mL平底ビーカーにヘキサン10mLを入れる。この中に荷電制御剤0.01gを添加する。
ガラス製の100mL平底ビーカーにヘキサン10mLを入れる。この中に荷電制御剤0.01gを添加する。
その後、超音波分散器を用いて分散を5分行う。なお、超音波分散器としては発振周波数50kHzの発振器2個を位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispension System Tetora150」(日科機バイオス社製)を使用する。
透過率は分光光度計(島津製作所 UV−3100PC)で測定する。分散液を分光光度計用セル(ジーエルサイエンス株式会社製、石英セル、型式:S10−UV−10E)に入れて透過率の測定を開始し、30分後の透過率のデータを採用する。このとき対照セルにはヘキサンを入れておく。
なお、透過率の測定条件は以下の通りとする。
(測定条件)
測定モード:タイムコース
測定値:透過率
波長:550nm
スリット幅:3.0nm
測定時間:60分
サンプリングピッチ:0.1
データ数:601
測定モード:タイムコース
測定値:透過率
波長:550nm
スリット幅:3.0nm
測定時間:60分
サンプリングピッチ:0.1
データ数:601
<トナーの摩擦帯電量の測定>
トナーの摩擦帯電量の測定は、E−SPART Analyzer MODEL EST−III ver.9.03(ホソカワミクロン社製)を用いて行う。
トナーの摩擦帯電量の測定は、E−SPART Analyzer MODEL EST−III ver.9.03(ホソカワミクロン社製)を用いて行う。
E−SPART Analyzer(ホソカワミクロン社製)に付属されている二成分フィーダー(磁石を内蔵する回転盤を有する現像剤保持台)に、トナーと、キャリア(F81−2535、パウダーテック株式会社製)とをトナー濃度が5質量%になるように混合し、更にターブラミキサーで1分間混合したサンプルを保持させる。次いで、2成分フィーダーに磁力で保持されたサンプルに、エアーノズルから窒素ガスを噴射してトナーのみを吹き飛ばし、二成分フィーダー下部にある試料導入管を通してトナーのみをE−SPART Analyzer測定部に吸引導入する。測定部に吸引導入されたトナーは、粒子径d(μm)に応じた帯電量q/d(femto−C/μm)が計測される。そして、付属のソフトによりこのデータをもとに全粒径の平均摩擦帯電量q/mを求め、全個数基準に対するポジ成分の個数の割合をポジ成分率として計算した。
なお、E−SPART Analyzerの測定条件は以下の通りとする。
窒素ガスブロー圧力 :20kPa
窒素ガスブロー時間 :1秒
窒素ガスブローのインターバル:4秒
印加電圧 :100V
カウント個数 :3000個
窒素ガスブロー圧力 :20kPa
窒素ガスブロー時間 :1秒
窒素ガスブローのインターバル:4秒
印加電圧 :100V
カウント個数 :3000個
<荷電制御剤の一次粒子の個数平均粒径(D1)の測定方法>
荷電制御剤の一次粒子の個数平均粒径(D1)は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)で測定した。
荷電制御剤の一次粒子の個数平均粒径(D1)は、フロー式粒子像分析装置「FPIA−3000」(シスメックス社製)で測定した。
具体的な測定方法は、以下の通りである。まず、ガラス製の容器中に予め不純固形物などを除去したイオン交換水約20mLを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で約3質量倍に希釈した希釈液を約0.2mL加える。更に測定試料を約0.05g加え、超音波分散器を用いて2分間分散処理を行い、測定用の分散液とする。その際、分散液の温度が10℃以上40℃以下となる様に適宜冷却する。超音波分散器としては、発振周波数50kHz、電気的出力150Wの卓上型の超音波洗浄器分散器(例えば「VS−150」(ヴェルヴォクリーア社製))を用い、水槽内には所定量のイオン交換水を入れ、この水槽中に前記コンタミノンNを約2mL添加する。
測定には、高倍率撮像ユニット(対物レンズ(20倍))を搭載した前記フロー式粒子像分析装置を用い、シース液にはパーティクルシース「PSE−900A」(シスメックス社製)を使用した。前記手順に従い調製した分散液を前記フロー式粒子像分析装置に導入し、HPF測定モードで、トータルカウントモードにて3000個の荷電制御剤の粒子を計測した。個数基準の円相当径の平均値を一次粒子の個数平均粒径(D1)とした。
<トナーの重量平均粒径(D4)の測定方法>
トナーの重量平均粒径(D4)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
トナーの重量平均粒径(D4)は、以下のようにして算出する。測定装置としては、100μmのアパーチャーチューブを備えた細孔電気抵抗法による精密粒度分布測定装置「コールター・カウンター Multisizer 3」(登録商標、ベックマン・コールター社製)を用いる。測定条件の設定及び測定データの解析は、付属の専用ソフト「ベックマン・コールター Multisizer 3 Version3.51」(ベックマン・コールター社製)を用いる。尚、測定は実効測定チャンネル数2万5千チャンネルで行う。
測定に使用する電解水溶液は、特級塩化ナトリウムをイオン交換水に溶解して濃度が約1質量%となるようにしたもの、例えば、「ISOTON II」(ベックマン・コールター社製)が使用できる。
尚、測定、解析を行う前に、以下のように前記専用ソフトの設定を行った。
前記専用ソフトの「標準測定方法(SOM)を変更」画面において、コントロールモードの総カウント数を50000粒子に設定し、測定回数を1回、Kd値は「標準粒子10.0μm」(ベックマン・コールター社製)を用いて得られた値を設定する。「閾値/ノイズレベルの測定ボタン」を押すことで、閾値とノイズレベルを自動設定する。また、カレントを1600μAに、ゲインを2に、電解液をISOTON IIに設定し、「測定後のアパーチャーチューブのフラッシュ」にチェックを入れる。
前記専用ソフトの「パルスから粒径への変換設定」画面において、ビン間隔を対数粒径に、粒径ビンを256粒径ビンに、粒径範囲を2μmから60μmまでに設定する。
具体的な測定法は以下の通りである。
(1)Multisizer 3専用のガラス製250mL丸底ビーカーに前記電解水溶液約200mLを入れ、サンプルスタンドにセットし、スターラーロッドの撹拌を反時計回りで24回転/秒にて行う。そして、専用ソフトの「アパーチャーのフラッシュ」機能により、アパーチャーチューブ内の汚れと気泡を除去しておく。
(2)ガラス製の100mL平底ビーカーに前記電解水溶液約30mLを入れる。この中に分散剤として「コンタミノンN」(非イオン界面活性剤、陰イオン界面活性剤、有機ビルダーからなるpH7の精密測定器洗浄用中性洗剤の10質量%水溶液、和光純薬工業社製)をイオン交換水で約3質量倍に希釈した希釈液を約0.3mL加える。
(3)発振周波数50kHzの発振器2個を位相を180度ずらした状態で内蔵し、電気的出力120Wの超音波分散器「Ultrasonic Dispension System Tetora150」(日科機バイオス社製)を準備する。超音波分散器の水槽内に約3.3Lのイオン交換水を入れ、この水槽中にコンタミノンNを約2mL添加する。
(4)前記(2)のビーカーを前記超音波分散器のビーカー固定穴にセットし、超音波分散器を作動させる。そして、ビーカー内の電解水溶液の液面の共振状態が最大となるようにビーカーの高さ位置を調整する。
(5)前記(4)のビーカー内の電解水溶液に超音波を照射した状態で、トナー約10mgを少量ずつ前記電解水溶液に添加し、分散させる。そして、さらに60秒間超音波分散処理を継続する。尚、超音波分散にあたっては、水槽の水温が10℃以上40℃以下となる様に適宜調節する。
(6)サンプルスタンド内に設置した前記(1)の丸底ビーカーに、ピペットを用いてトナーを分散した前記(5)の電解質水溶液を滴下し、測定濃度が約5%となるように調整する。そして、測定粒子数が50000個になるまで測定を行う。
(7)測定データを装置付属の前記専用ソフトにて解析を行い、重量平均粒径(D4)を算出する。尚、前記専用ソフトでグラフ/体積%と設定したときの、「分析/体積統計値(算術平均)」画面の「平均径」が重量平均粒径(D4)である。
以下に実施例及び比較例を挙げて本発明を更に詳細に説明するが、本発明は何らこれに制約されるものではない。
<荷電制御剤(C−1)の製造例>
水76.5部及び35%塩酸15.2部の混合溶液中に、4−クロロ−2−アミノフェノールの10部を加え、冷却下で撹拌した。氷冷し、溶液の温度が0℃〜5℃になるように維持し、水24.6部に溶解させた亜硝酸ナトリウム13.6部を塩酸水溶液に滴下し、2時間撹拌しジアゾ化した。これにスルファミン酸で過剰の亜硝酸を消失させた後、濾過を行ってジアゾ溶液とした。
水76.5部及び35%塩酸15.2部の混合溶液中に、4−クロロ−2−アミノフェノールの10部を加え、冷却下で撹拌した。氷冷し、溶液の温度が0℃〜5℃になるように維持し、水24.6部に溶解させた亜硝酸ナトリウム13.6部を塩酸水溶液に滴下し、2時間撹拌しジアゾ化した。これにスルファミン酸で過剰の亜硝酸を消失させた後、濾過を行ってジアゾ溶液とした。
次に、3−メチル−1−(3,4−ジクロロフェニル)−5−ピラゾロンの12.0部を水87部、25%水酸化ナトリウム12.1部、炭酸ナトリウム4.9部、及びn−ブタノール104.6部の混合溶液に添加し溶解させた。そこに上記ジアゾ溶液を加え、温度20℃〜22℃で4時間攪拌し、カップリング反応を行った。その後、水92.8部、25%水酸化ナトリウム水溶液43.5部を加え攪拌洗浄し、下層の水層を分液除去した。
次に、水42.2部、サリチル酸5.9部、ブタノール24.6部、及び15%炭酸ナトリウム48.5部を上記反応液に添加し攪拌した。さらに、38%塩化第二鉄水溶液15.1部と15%炭酸ナトリウム18.0部を加え、酢酸で反応液のpHを4.5に調整した。液温を温度30℃に昇温した後、8時間攪拌し錯体化反応を行った。攪拌停止後、静置して下部水層を分液した。更に水189.9部を加え攪拌洗浄し、下部水層を分液した。濾過後、水253部でケーキを洗浄した。温度60℃で24時間真空乾燥の後、モノアゾ金属錯体化合物である荷電制御剤(C−1)を得た。
赤外吸収スペクトル、可視部吸収スペクトル、元素分析(C,H,N)、原子吸光分析、マススペクトルより、荷電制御剤(C−1)の構造を同定したところ、下記の構造であることを確認した。荷電制御剤(C−1)の一次粒子の個数平均粒径(D1)は、1.41μm、ヘキサン分散試験で静置して30分後の透過率は、6.7%であった。
<荷電制御剤(C−2)〜(C−4)の製造例>
荷電制御剤(C−1)の製造例において、錯体化反応の時間を変更することで、荷電制御剤(C−2)〜(C−4)を得た。荷電制御剤荷電制御剤(C−2)〜(C−4)の構造を同定したところ、荷電制御剤(C−1)と同じ構造であることを確認した。荷電制御剤(C−2)〜(C−4)の物性を表1に示す。
荷電制御剤(C−1)の製造例において、錯体化反応の時間を変更することで、荷電制御剤(C−2)〜(C−4)を得た。荷電制御剤荷電制御剤(C−2)〜(C−4)の構造を同定したところ、荷電制御剤(C−1)と同じ構造であることを確認した。荷電制御剤(C−2)〜(C−4)の物性を表1に示す。
<荷電制御剤(C−5)の製造例>
荷電制御剤(C−1)の製造例において、3−メチル−1−(3,4−ジクロロフェニル)−5−ピラゾロンを3−メチル−1−フェニル−5−ピラゾロンに変更した。それ以外は、荷電制御剤(C−1)の製造例と同様にして、荷電制御剤(C−5)を得た。
荷電制御剤(C−1)の製造例において、3−メチル−1−(3,4−ジクロロフェニル)−5−ピラゾロンを3−メチル−1−フェニル−5−ピラゾロンに変更した。それ以外は、荷電制御剤(C−1)の製造例と同様にして、荷電制御剤(C−5)を得た。
荷電制御剤荷電制御剤(C−5)の構造を同定したところ、以下の構造であることを確認した。荷電制御剤(C−5)の物性を表1に示す。
<荷電制御剤(C−6)>
荷電制御剤(C−6)には、オリエント化学社製のS−34を使用した。荷電制御剤(C−6)の構造を以下に、物性を表1に示す。
荷電制御剤(C−6)には、オリエント化学社製のS−34を使用した。荷電制御剤(C−6)の構造を以下に、物性を表1に示す。
<荷電制御剤(C−7)>
荷電制御剤(C−7)には、保土谷化学社製のT−77を使用した。荷電制御剤(C−7)の構造を以下にしめす。尚、a+b+cは1である。また、物性を表1に示す。
荷電制御剤(C−7)には、保土谷化学社製のT−77を使用した。荷電制御剤(C−7)の構造を以下にしめす。尚、a+b+cは1である。また、物性を表1に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−1)の製造例>
脂肪族炭化水素(ピーク炭素数35)1200gをガラス製の円筒型反応容器に入れ、硼酸38.5gを温度140℃で添加した。次いで直ちに空気50容量%と窒素50容量%の酸素濃度約10容量%の混合ガスを毎分20リットルの割合で吹き込み、200℃で3.0時間反応させた。その後、反応液に温水を加え、95℃で2時間加水分解を行い、静置後上層の変性品を含む反応物を得た。変性品20質量部をn−ヘキサン100質量部に加え、未変性成分を溶解除去して、混合物(A−1)を得た。得られた混合物(A−1)の物性を表2に示す。
脂肪族炭化水素(ピーク炭素数35)1200gをガラス製の円筒型反応容器に入れ、硼酸38.5gを温度140℃で添加した。次いで直ちに空気50容量%と窒素50容量%の酸素濃度約10容量%の混合ガスを毎分20リットルの割合で吹き込み、200℃で3.0時間反応させた。その後、反応液に温水を加え、95℃で2時間加水分解を行い、静置後上層の変性品を含む反応物を得た。変性品20質量部をn−ヘキサン100質量部に加え、未変性成分を溶解除去して、混合物(A−1)を得た。得られた混合物(A−1)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−2)の製造例>
直鎖脂肪族1級モノアルコール(ピーク炭素数50)20質量部をn−ヘキサン100質量部に加え、未変性成分を溶解除去させた混合物(A−2)を得た。得られた混合物(A−2)の物性を表2に示す。
直鎖脂肪族1級モノアルコール(ピーク炭素数50)20質量部をn−ヘキサン100質量部に加え、未変性成分を溶解除去させた混合物(A−2)を得た。得られた混合物(A−2)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−3)の製造例>
脂肪族炭化水素(ピーク炭素数22)を用いた以外は、混合物(A−1)の製造例と同様にして、混合物(A−3)を得た。得られた混合物(A−3)の物性を表2に示す。
脂肪族炭化水素(ピーク炭素数22)を用いた以外は、混合物(A−1)の製造例と同様にして、混合物(A−3)を得た。得られた混合物(A−3)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−4)の製造例>
平均炭素数が50の脂肪族炭化水素(ピーク炭素数50)を用いた以外は、混合物(A−1)の製造例と同様にして、混合物(A−4)を得た。得られた混合物(A−4)の物性を表2に示す。
平均炭素数が50の脂肪族炭化水素(ピーク炭素数50)を用いた以外は、混合物(A−1)の製造例と同様にして、混合物(A−4)を得た。得られた混合物(A−4)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−5)の製造例>
n−ヘキサンによる未変性成分を溶解除去する条件を変更した以外は、混合物(A−1)の製造例と同様にして、混合物(A−5)を得た。得られた混合物(A−5)の物性を表2に示す。
n−ヘキサンによる未変性成分を溶解除去する条件を変更した以外は、混合物(A−1)の製造例と同様にして、混合物(A−5)を得た。得られた混合物(A−5)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−6)の製造例>
直鎖脂肪族1級モノアルコール(ピーク炭素数33)を用いた以外は、混合物(A−2)の製造例と同様にして、混合物(A−6)を得た。得られた混合物(A−6)の物性を表2に示す。
直鎖脂肪族1級モノアルコール(ピーク炭素数33)を用いた以外は、混合物(A−2)の製造例と同様にして、混合物(A−6)を得た。得られた混合物(A−6)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−7)の製造例>
直鎖脂肪族1級モノカルボン酸(ピーク炭素数32)20質量部をn−ヘキサン100質量部に加え、未変性成分を溶解除去して混合物(A−7)を得た。得られた混合物(A−7)の物性を表2に示す。
直鎖脂肪族1級モノカルボン酸(ピーク炭素数32)20質量部をn−ヘキサン100質量部に加え、未変性成分を溶解除去して混合物(A−7)を得た。得られた混合物(A−7)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−8)の製造例>
n−ヘキサンで未変性成分を溶解除去しなかった以外は、混合物(A−2)の製造例と同様にして、混合物(A−8)を得た。得られた混合物(A−8)の物性を表2に示す。
n−ヘキサンで未変性成分を溶解除去しなかった以外は、混合物(A−2)の製造例と同様にして、混合物(A−8)を得た。得られた混合物(A−8)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−9)の製造例>
脂肪族炭化水素(ピーク炭素数105)を用いた以外は、混合物(A−1)の製造例と同様にして、混合物(A−9)を得た。得られた混合物(A−9)の物性を表2に示す。
脂肪族炭化水素(ピーク炭素数105)を用いた以外は、混合物(A−1)の製造例と同様にして、混合物(A−9)を得た。得られた混合物(A−9)の物性を表2に示す。
<変性・未変性の脂肪族炭化水素を含有する混合物(A−10)の製造例>
n−ヘキサンで未変性成分を溶解除去しなかった以外は、混合物(A−1)の製造例と同様にして、混合物(A−10)を得た。得られた混合物(A−10)の物性を表2に示す。
n−ヘキサンで未変性成分を溶解除去しなかった以外は、混合物(A−1)の製造例と同様にして、混合物(A−10)を得た。得られた混合物(A−10)の物性を表2に示す。
<ポリエステル系樹脂組成物(B−1)の製造例>
・ビスフェールAエチレンオキサイド付加物(2.0モル付加物) 50.0mol部
・ビスフェールAプロピレンオキサイド付加物(2.3モル付加物)50.0mol部
・テレフタル酸 60.0mol部
・無水トリメリット酸 20.0mol部
・アクリル酸 10.0mol部
上記ポリエステルモノマーと、最終的に得られる樹脂組成物中における含有量が5.0質量%となる量の混合物(A−1)を添加した(尚、混合物(A−1)の添加量は、ポリエステル合成時に生成する水の量を考慮して、目的とする割合に対して少なめに添加すればよい。)。その混合物60質量部を4口フラスコに仕込み、減圧装置、水分離装置、窒素ガス導入装置、温度測定装置及び攪拌装置を装着して窒素雰囲気下にて160℃で攪拌した。そこに、ビニル重合体部位を構成するビニル系重合モノマー(スチレン:60.0mol部、アクリル酸−2−エチルヘキシル:40.0mol部)40質量部と重合開始剤としてベンゾイルパーオキサイド2.0mol部を混合したものを滴下ロートから4時間かけて滴下した。その後、160℃で5時間反応した後、230℃に昇温してテトライソブチルチタネートを0.05質量%添加し、所望の粘度となるように反応時間を調節した。
・ビスフェールAエチレンオキサイド付加物(2.0モル付加物) 50.0mol部
・ビスフェールAプロピレンオキサイド付加物(2.3モル付加物)50.0mol部
・テレフタル酸 60.0mol部
・無水トリメリット酸 20.0mol部
・アクリル酸 10.0mol部
上記ポリエステルモノマーと、最終的に得られる樹脂組成物中における含有量が5.0質量%となる量の混合物(A−1)を添加した(尚、混合物(A−1)の添加量は、ポリエステル合成時に生成する水の量を考慮して、目的とする割合に対して少なめに添加すればよい。)。その混合物60質量部を4口フラスコに仕込み、減圧装置、水分離装置、窒素ガス導入装置、温度測定装置及び攪拌装置を装着して窒素雰囲気下にて160℃で攪拌した。そこに、ビニル重合体部位を構成するビニル系重合モノマー(スチレン:60.0mol部、アクリル酸−2−エチルヘキシル:40.0mol部)40質量部と重合開始剤としてベンゾイルパーオキサイド2.0mol部を混合したものを滴下ロートから4時間かけて滴下した。その後、160℃で5時間反応した後、230℃に昇温してテトライソブチルチタネートを0.05質量%添加し、所望の粘度となるように反応時間を調節した。
反応終了後、容器から取り出し、冷却、粉砕して、ハイブリッド樹脂であるポリエステル系樹脂組成物(B−1)を得た。得られたポリエステル系樹脂組成物(B−1)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−2)の製造例>
・ビスフェールAプロピレンオキサイド付加物(2.2モル付加物)60.0mol部
・ビスフェールAエチレンオキサイド付加物(2.2モル付加物) 40.0mol部
・テレフタル酸 77.0mol部
上記ポリエステルモノマーと、最終的に得られる樹脂組成物中における含有量が、5.0質量%となる量の混合物(A−1)と、0.2質量%となる量のジブチル錫オキシドとを5リットルオートクレーブに仕込んだ。そして、該オートクレーブに、還流冷却器、水分分離装置、N2ガス導入管、温度計及び撹拌装置を付けた。オートクレーブ内にN2ガスを導入しながら230℃で重縮合反応を行った。所望の軟化点になるように反応時間を調整し、反応終了後容器から取り出し、冷却、粉砕してポリエステル系樹脂組成物(B−2)を得た。ポリエステル系樹脂組成物(B−2)の物性を表3に示す。
・ビスフェールAプロピレンオキサイド付加物(2.2モル付加物)60.0mol部
・ビスフェールAエチレンオキサイド付加物(2.2モル付加物) 40.0mol部
・テレフタル酸 77.0mol部
上記ポリエステルモノマーと、最終的に得られる樹脂組成物中における含有量が、5.0質量%となる量の混合物(A−1)と、0.2質量%となる量のジブチル錫オキシドとを5リットルオートクレーブに仕込んだ。そして、該オートクレーブに、還流冷却器、水分分離装置、N2ガス導入管、温度計及び撹拌装置を付けた。オートクレーブ内にN2ガスを導入しながら230℃で重縮合反応を行った。所望の軟化点になるように反応時間を調整し、反応終了後容器から取り出し、冷却、粉砕してポリエステル系樹脂組成物(B−2)を得た。ポリエステル系樹脂組成物(B−2)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−3)の製造例>
・ビスフェノールAエチレンオキサイド(2.2モル付加物): 45.0mol部
・ビスフェノールAプロピレンオキサイド(2.2モル付加物): 40.0mol部
・エチレングリコール: 15.0mol部
・テレフタル酸: 100.0mol部
上記ポリエステルモノマーと、最終的に得られる樹脂組成物中における含有量が5.0質量%となる量の混合物(A−2)と、0.05質量%となる量のチタンテトラブトキシドを5リットルオートクレーブに仕込んだ。そして、該オートクレーブに、還流冷却器、水分分離装置、N2ガス導入管、温度計及び撹拌装置を付けた。オートクレーブ内にN2ガスを導入しながら230℃で重縮合反応を行った。所望の軟化点になるように反応時間を調整し、反応終了後容器から取り出し、冷却、粉砕してポリエステル系樹脂組成物(B−3)を得た。ポリエステル系樹脂組成物(B−3)の物性を表3に示す。
・ビスフェノールAエチレンオキサイド(2.2モル付加物): 45.0mol部
・ビスフェノールAプロピレンオキサイド(2.2モル付加物): 40.0mol部
・エチレングリコール: 15.0mol部
・テレフタル酸: 100.0mol部
上記ポリエステルモノマーと、最終的に得られる樹脂組成物中における含有量が5.0質量%となる量の混合物(A−2)と、0.05質量%となる量のチタンテトラブトキシドを5リットルオートクレーブに仕込んだ。そして、該オートクレーブに、還流冷却器、水分分離装置、N2ガス導入管、温度計及び撹拌装置を付けた。オートクレーブ内にN2ガスを導入しながら230℃で重縮合反応を行った。所望の軟化点になるように反応時間を調整し、反応終了後容器から取り出し、冷却、粉砕してポリエステル系樹脂組成物(B−3)を得た。ポリエステル系樹脂組成物(B−3)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−4)の製造例>
・ビスフェノールAエチレンオキサイド(2.2モル付加物): 100.0mol部
・テレフタル酸: 65.0mol部
・無水トリメリット酸: 25.0mol部
・アクリル酸: 10.0mol部
上記ポリエステルモノマーの混合物60質量部と、最終的に得られる樹脂組成物中における含有量が4.8質量%となる量の混合物(A−2)とを4口フラスコに入れた。そこに、減圧装置、水分離装置、窒素ガス導入装置、温度測定装置および撹拌装置を付け、N2雰囲気下にて160℃で撹拌した。そこにビニル重合体部位を構成するビニル系重合モノマー(スチレン90.0モル部および2−エチルヘキシルアクリレート10.0モル部)40質量部および重合開始剤としてのベンゾイルパーオキサイド1質量部を滴下ロートから4時間かけて滴下した。そして、160℃で5時間反応を行った。その後、230℃に昇温して、上記ポリエステルユニットを構成するためのモノマーの総量に対して0.2質量部のチタンテトラブトキシドを添加し、得られる樹脂の軟化点が所定の値になるまで重合反応を行った。反応終了後、反応生成物を容器から取り出し、冷却し、粉砕して、ハイブリッド樹脂であるポリエステル系樹脂組成物(B−4)を得た。ポリエステル系樹脂組成物(B−4)の物性を表3に示す。
・ビスフェノールAエチレンオキサイド(2.2モル付加物): 100.0mol部
・テレフタル酸: 65.0mol部
・無水トリメリット酸: 25.0mol部
・アクリル酸: 10.0mol部
上記ポリエステルモノマーの混合物60質量部と、最終的に得られる樹脂組成物中における含有量が4.8質量%となる量の混合物(A−2)とを4口フラスコに入れた。そこに、減圧装置、水分離装置、窒素ガス導入装置、温度測定装置および撹拌装置を付け、N2雰囲気下にて160℃で撹拌した。そこにビニル重合体部位を構成するビニル系重合モノマー(スチレン90.0モル部および2−エチルヘキシルアクリレート10.0モル部)40質量部および重合開始剤としてのベンゾイルパーオキサイド1質量部を滴下ロートから4時間かけて滴下した。そして、160℃で5時間反応を行った。その後、230℃に昇温して、上記ポリエステルユニットを構成するためのモノマーの総量に対して0.2質量部のチタンテトラブトキシドを添加し、得られる樹脂の軟化点が所定の値になるまで重合反応を行った。反応終了後、反応生成物を容器から取り出し、冷却し、粉砕して、ハイブリッド樹脂であるポリエステル系樹脂組成物(B−4)を得た。ポリエステル系樹脂組成物(B−4)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−5)〜(B−11)の製造例>
混合物の種類と含有割合を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−1)の製造例と同様にして、ポリエステル系樹脂組成物(B−5)〜(B−11)を得た。得られたポリエステル系樹脂組成物(B−5)〜(B−11)の物性を表3に示す。
混合物の種類と含有割合を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−1)の製造例と同様にして、ポリエステル系樹脂組成物(B−5)〜(B−11)を得た。得られたポリエステル系樹脂組成物(B−5)〜(B−11)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−12)の製造例>
混合物の種類を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−3)の製造例と同様にして、ポリエステル系樹脂組成物(B−12)を得た。得られたポリエステル系樹脂組成物(B−12)の物性を表3に示す。
混合物の種類を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−3)の製造例と同様にして、ポリエステル系樹脂組成物(B−12)を得た。得られたポリエステル系樹脂組成物(B−12)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−13)の製造例>
混合物の種類を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−4)の製造例と同様にして、ポリエステル系樹脂組成物(B−13)を得た。得られたポリエステル系樹脂組成物(B−13)の物性を表3に示す。
混合物の種類を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−4)の製造例と同様にして、ポリエステル系樹脂組成物(B−13)を得た。得られたポリエステル系樹脂組成物(B−13)の物性を表3に示す。
<ポリエステル系樹脂組成物(B−14)〜(B−17)の製造例>
混合物の種類と添加量を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−1)の製造例と同様にして、ポリエステル系樹脂組成物(B−17)〜(B−17)を得た。得られたポリエステル系樹脂組成物(B−14)〜(B−17)の物性を表3に示す。
混合物の種類と添加量を表3に示すように変更した以外は、ポリエステル系樹脂組成物(B−1)の製造例と同様にして、ポリエステル系樹脂組成物(B−17)〜(B−17)を得た。得られたポリエステル系樹脂組成物(B−14)〜(B−17)の物性を表3に示す。
いずれのポリエステル系樹脂組成物においても、
(i)使用した“変性・未変性の脂肪族炭化水素を含有する混合物”に含有される変性物の炭素数が、ポリエステル樹脂の有する部分構造の炭素数であり、
(ii)使用した“変性・未変性の脂肪族炭化水素を含有する混合物”に含有される未変性物の炭素数が、脂肪族炭化水素の炭素数であった。
(i)使用した“変性・未変性の脂肪族炭化水素を含有する混合物”に含有される変性物の炭素数が、ポリエステル樹脂の有する部分構造の炭素数であり、
(ii)使用した“変性・未変性の脂肪族炭化水素を含有する混合物”に含有される未変性物の炭素数が、脂肪族炭化水素の炭素数であった。
<実施例1>
・ポリエステル系樹脂組成物(B−1): 100質量部
・磁性粒子(平均粒径0.12μm、八面体形状): 45質量部
・フィッシャートロプッシュワックス(サゾール社製C105、融点:105℃):
2質量部
・荷電制御剤(C−1): 2質量部
上記材料を三井ヘンシェルミキサ(三井三池化工機社製)で前混合した後、2軸押出機(商品名:PCM−30、池貝鉄工所社製)を用いて、吐出口における溶融物温度が130℃になるように、温度を設定し、溶融混練した。
・ポリエステル系樹脂組成物(B−1): 100質量部
・磁性粒子(平均粒径0.12μm、八面体形状): 45質量部
・フィッシャートロプッシュワックス(サゾール社製C105、融点:105℃):
2質量部
・荷電制御剤(C−1): 2質量部
上記材料を三井ヘンシェルミキサ(三井三池化工機社製)で前混合した後、2軸押出機(商品名:PCM−30、池貝鉄工所社製)を用いて、吐出口における溶融物温度が130℃になるように、温度を設定し、溶融混練した。
得られた混練物を冷却し、ハンマーミルで粗粉砕した後、粉砕機(商品名:ターボミルT250、ターボ工業社製)を用いて微粉砕した。得られた微粉砕粉末をコアンダ効果を利用した多分割分級機を用いて分級して、重量平均粒径(D4)6.8μmのトナー粒子1を得た。
このトナー粒子1の100質量部に対して、負帯電性疎水性シリカ1.2質量部を、三井ヘンシェルミキサ(三井三池化工機社製)を用いて外添混合し、トナー1を得た。このトナーを以下の項目について評価した。
[評価]
得られたトナー1を以下の放置条件1〜3でエージングした。
・放置条件1
25℃,60%RHで24時間放置
・放置条件2
以下のヒートサイクル環境下での放置(湿度は25%Hで一定)
<1>25℃で1時間保持
<2>11時間かけて45℃まで直線的に温度を上げる
<3>45℃で1時間保持
<4>11時間かけて25℃まで直線的に温度を下げる
上記<1>乃至<4>までを1サイクルとして、計20サイクル行った。
・放置条件3
高湿下で以下のヒートサイクル環境下での放置(湿度は95%Hで一定)
<1>25℃で1時間保持
<2>11時間かけて45℃まで直線的に温度を上げる
<3>45℃で1時間保持
<4>11時間かけて25℃まで直線的に温度を下げる
上記<1>乃至<4>までを1サイクルとして、計20サイクル行った。
得られたトナー1を以下の放置条件1〜3でエージングした。
・放置条件1
25℃,60%RHで24時間放置
・放置条件2
以下のヒートサイクル環境下での放置(湿度は25%Hで一定)
<1>25℃で1時間保持
<2>11時間かけて45℃まで直線的に温度を上げる
<3>45℃で1時間保持
<4>11時間かけて25℃まで直線的に温度を下げる
上記<1>乃至<4>までを1サイクルとして、計20サイクル行った。
・放置条件3
高湿下で以下のヒートサイクル環境下での放置(湿度は95%Hで一定)
<1>25℃で1時間保持
<2>11時間かけて45℃まで直線的に温度を上げる
<3>45℃で1時間保持
<4>11時間かけて25℃まで直線的に温度を下げる
上記<1>乃至<4>までを1サイクルとして、計20サイクル行った。
放置条件1〜3で放置したトナー1に関して評価を行った。
評価には、市販の磁性一成分現像方式のプリンターHP LaserJet Enterprise600 M603dn(ヒューレットパッカード社製:プロセススピード350mm/s)を用いた。尚、現像スリーブをスリーブ径が14mmのものに付け替え、スリーブ表面の磁束密度を80mTに変更したものを用いた。評価結果を表5に示す。
<かぶりの評価>
印字率2%となる横線パターンを1枚/1ジョブとして、ジョブとジョブの間にマシンがいったん停止してから次のジョブが始まるように設定したモードで、計12000枚の画出し試験を実施し、1枚目及び12000枚後のかぶり濃度を測定した。
印字率2%となる横線パターンを1枚/1ジョブとして、ジョブとジョブの間にマシンがいったん停止してから次のジョブが始まるように設定したモードで、計12000枚の画出し試験を実施し、1枚目及び12000枚後のかぶり濃度を測定した。
尚、リフレクトメーター(東京電色社製)を用いて、定着画像の白地部分の反射率(%)と転写材の反射率(%)とを測定し、その差をかぶり濃度(%)として算出した。
かぶりの評価はトナーの帯電分布がブロードになりやすく、かぶりに対してより厳しいと想定される低温低湿下(15℃,10%RH)で行った。各条件で放置したトナーを低温低湿下(15℃,10%RH)で24時間エージングした後、評価をスタートした。
<ドット再現性の評価>
ドット再現性の評価は、常温常湿環境下(25℃,60%RH)で実施した。印字率2%となる横線パターンを2枚/1ジョブとして、ジョブとジョブの間にマシンがいったん停止してから次のジョブが始まるように設定したモードで、計1000枚の画出しを行った。続いてXerox4200(ゼロックス社製、75g/m2紙)上に孤立ドットを印字したハーフトーン画像(画像濃度が0.5乃至0.6に設定)を出力し、光学顕微鏡により孤立1ドット100個中の欠損数を測定した。各条件で放置したトナーを常温常湿環境下(25℃,60%RH)で24時間エージングした後、評価をスタートした。
A:孤立ドット100個中の欠損が5個以下
B:孤立1ドット100個中の欠損が6個以上10個以下
C:孤立1ドット100個中の欠損が11個以上20個以下
D:孤立1ドット100個中の欠損が21個以上
ドット再現性の評価は、常温常湿環境下(25℃,60%RH)で実施した。印字率2%となる横線パターンを2枚/1ジョブとして、ジョブとジョブの間にマシンがいったん停止してから次のジョブが始まるように設定したモードで、計1000枚の画出しを行った。続いてXerox4200(ゼロックス社製、75g/m2紙)上に孤立ドットを印字したハーフトーン画像(画像濃度が0.5乃至0.6に設定)を出力し、光学顕微鏡により孤立1ドット100個中の欠損数を測定した。各条件で放置したトナーを常温常湿環境下(25℃,60%RH)で24時間エージングした後、評価をスタートした。
A:孤立ドット100個中の欠損が5個以下
B:孤立1ドット100個中の欠損が6個以上10個以下
C:孤立1ドット100個中の欠損が11個以上20個以下
D:孤立1ドット100個中の欠損が21個以上
<現像性の評価>
現像性の評価は高温高湿環境下(32℃,80%RH)で実施した。初期、及び印字率1%となる横線パターンを連続で計1000枚の画出し後に、5mm丸のベタ画像を出力し、画像濃度を測定した。画像濃度は、反射濃度計であるマクベス濃度計(マクベス社製)でSPIフィルターを使用して、5mm丸のベタ画像の反射濃度を測定することにより測定した。数値が大きい方が良いことを示す。
現像性の評価は高温高湿環境下(32℃,80%RH)で実施した。初期、及び印字率1%となる横線パターンを連続で計1000枚の画出し後に、5mm丸のベタ画像を出力し、画像濃度を測定した。画像濃度は、反射濃度計であるマクベス濃度計(マクベス社製)でSPIフィルターを使用して、5mm丸のベタ画像の反射濃度を測定することにより測定した。数値が大きい方が良いことを示す。
高温高湿環境下(32℃,80%RH)で評価を実施した理由は、トナーの帯電性が低下しやすく、現像性を評価するのに厳しい条件だからである。各条件で放置したトナーを高温高湿環境下(32℃,80%RH)で24時間エージングした後、評価をスタートした。
<実施例2〜15、比較例1〜6>
ポリエステル系樹脂組成物、荷電制御剤の種類と量を表4に記載のように変更した以外は、実施例1と同様にして、トナー2〜15、比較トナー1〜6を得た。また、実施例1と同様の評価を行い、その評価結果を表5に示す。
ポリエステル系樹脂組成物、荷電制御剤の種類と量を表4に記載のように変更した以外は、実施例1と同様にして、トナー2〜15、比較トナー1〜6を得た。また、実施例1と同様の評価を行い、その評価結果を表5に示す。
Claims (5)
- ポリエステル系樹脂組成物、および荷電制御剤を含有するトナー粒子を有するトナーであって、
該ポリエステル系樹脂組成物は、
i)R1−O−またはR2−COO−
[式中、R1は、炭素数が12〜102の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表し、R2は、炭素数が11〜101の脂肪族炭化水素から水素原子が1つ脱離して形成される基を表す。]
で表わされる部分構造を有するポリエステル樹脂と、炭素数が12〜102の脂肪族炭化水素とを含有し、
ii)該ポリエステル系樹脂組成物の質量を基準として、該部分構造と炭素数が12〜102の該脂肪族炭化水素とを合計して、2.5質量%以上10.0質量%以下含有しており、
iii)示差走査熱量測定(DSC)にて得られる温度−吸熱量曲線において、30℃以上100℃以下にピークトップを有する吸熱ピークを有し、該吸熱ピークの吸熱量が0.10J/g以上1.90J/g以下であり、
該荷電制御剤は、該荷電制御剤0.01gをヘキサン10mLに分散させた分散液について波長550nmの光の透過率の経時変化を測定した試験において、静置して30分後の透過率が10%以下である、
ことを特徴とするトナー。 - 該荷電制御剤の一次粒子の個数平均粒径(D1)が0.80μm以上2.00μm以下である、請求項1に記載のトナー。
- 該荷電制御剤は、下記式[1]で表される化合物である、請求項1または2に記載のトナー。
(式中、A1、A2及びA3は、それぞれ独立して、水素原子、ニトロ基又はハロゲン原子を示す。B1は水素原子又はアルキル基を示す。Mは、Fe原子、Cr原子、又はAl原子を示し、X+は、水素イオン、アルカリ金属イオン、アンモニウムイオン、アルキルアンモニウムイオン又はこれらの混合イオンを示す。) - 該荷電制御剤は、下記式[2]で表される化合物である、請求項1または2に記載のトナー。
(式中、X+は、水素イオン、アルカリ金属イオン、アンモニウムイオン、アルキルアンモニウムイオン又はこれらの混合イオンを示す。) - 該ポリエステル系樹脂組成物のR1−O−を形成するR1が、炭素数が12〜102の脂肪族炭化水素の2級炭素から水素原子が1つ脱離して形成される官能基である、請求項1乃至4のいずれか1項に記載のトナー。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016042555A JP2017156706A (ja) | 2016-03-04 | 2016-03-04 | トナー |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016042555A JP2017156706A (ja) | 2016-03-04 | 2016-03-04 | トナー |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017156706A true JP2017156706A (ja) | 2017-09-07 |
Family
ID=59809816
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016042555A Pending JP2017156706A (ja) | 2016-03-04 | 2016-03-04 | トナー |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017156706A (ja) |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000181125A (ja) * | 1998-12-15 | 2000-06-30 | Mitsui Chemicals Inc | 電子写真トナー |
| JP2015045850A (ja) * | 2013-08-01 | 2015-03-12 | キヤノン株式会社 | トナー |
| JP2015194737A (ja) * | 2014-03-20 | 2015-11-05 | キヤノン株式会社 | トナーおよび二成分現像剤 |
| JP2015215603A (ja) * | 2014-04-24 | 2015-12-03 | キヤノン株式会社 | 磁性トナー |
| JP2016103004A (ja) * | 2014-11-18 | 2016-06-02 | キヤノン株式会社 | トナー |
-
2016
- 2016-03-04 JP JP2016042555A patent/JP2017156706A/ja active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000181125A (ja) * | 1998-12-15 | 2000-06-30 | Mitsui Chemicals Inc | 電子写真トナー |
| JP2015045850A (ja) * | 2013-08-01 | 2015-03-12 | キヤノン株式会社 | トナー |
| JP2015194737A (ja) * | 2014-03-20 | 2015-11-05 | キヤノン株式会社 | トナーおよび二成分現像剤 |
| JP2015215603A (ja) * | 2014-04-24 | 2015-12-03 | キヤノン株式会社 | 磁性トナー |
| JP2016103004A (ja) * | 2014-11-18 | 2016-06-02 | キヤノン株式会社 | トナー |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7433869B2 (ja) | トナー | |
| JP6808538B2 (ja) | トナー | |
| JP6921678B2 (ja) | トナー製造方法及び重合体 | |
| US9417540B2 (en) | Toner and two-component developer | |
| CN1595302B (zh) | 彩色调色剂 | |
| JP2021140031A (ja) | トナー及びトナーの製造方法 | |
| KR20050014713A (ko) | 토너 및 화상 형성 방법 | |
| JP2012145934A (ja) | トナー | |
| JP5495685B2 (ja) | トナー | |
| JP5865011B2 (ja) | トナーおよび二成分系現像剤 | |
| JP6195374B2 (ja) | トナー | |
| JP5517505B2 (ja) | 磁性トナー及びその製造方法 | |
| JP6541356B2 (ja) | トナー | |
| JP2017015912A (ja) | トナー及びトナーの製造方法 | |
| JP5430168B2 (ja) | トナー | |
| JP5455746B2 (ja) | トナー粒子の製造方法及びイエロートナー | |
| JP2017173555A (ja) | 静電潜像現像用トナー及び静電潜像現像用トナーの作製方法 | |
| JP6821442B2 (ja) | トナー | |
| JP2011033916A (ja) | トナー | |
| JP6671965B2 (ja) | トナー | |
| JP6727819B2 (ja) | トナー | |
| JP2017156706A (ja) | トナー | |
| JP5414339B2 (ja) | トナー及び該トナーの製造方法 | |
| JP4468232B2 (ja) | トナー | |
| JP2017076099A (ja) | トナー |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190301 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200117 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200128 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20200804 |