JP2017186482A - 重合体、タンパク質付着防止剤および医療用デバイス - Google Patents
重合体、タンパク質付着防止剤および医療用デバイス Download PDFInfo
- Publication number
- JP2017186482A JP2017186482A JP2016078146A JP2016078146A JP2017186482A JP 2017186482 A JP2017186482 A JP 2017186482A JP 2016078146 A JP2016078146 A JP 2016078146A JP 2016078146 A JP2016078146 A JP 2016078146A JP 2017186482 A JP2017186482 A JP 2017186482A
- Authority
- JP
- Japan
- Prior art keywords
- polymer
- coating layer
- compound
- protein
- medical device
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images


Landscapes
- Apparatus Associated With Microorganisms And Enzymes (AREA)
- Materials For Medical Uses (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
【課題】タンパク質が付着しにくく、耐水性および耐久性に優れた被覆層を形成できる重合体およびタンパク質付着防止剤、ならびに該タンパク質付着防止剤を用いた被覆層を備える医療用デバイスを提供することを目的とする。【解決手段】側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位を有する、ガラス転移温度が20℃以上の重合体。基材2と、基材2上に形成された被覆層3とを備え、被覆層3が、前記重合体からなるタンパク質付着防止剤から形成されてなる層である、医療用デバイス1。【選択図】図2
Description
本発明は、重合体、タンパク質付着防止剤および医療用デバイスに関する。
合成高分子材料は、細胞培養容器、カテーテル、人工臓器等の医療用デバイスを形成する医用高分子材料として広く用いられている。医用高分子材料に用いられる代表的な合成高分子材料としては、例えば、ポリ塩化ビニル、ポリスチレン、シリコーン樹脂、ポリメタクリル酸エステル、含フッ素樹脂等の疎水性高分子や、ポリビニルアルコール、ポリ(メタクリル酸2−ヒドロキシエチル)、ポリアクリルアミド等の親水性高分子が挙げられる。しかし、これらの合成高分子材料ではタンパク質や血球等の生体成分がデバイス表面に吸着しやすい。生体成分がデバイス表面に吸着して変性すると、血栓形成、炎症反応等の生体への悪影響や、デバイスの劣化等の問題が引き起こされる。該問題は医用高分子材料において根本的かつ緊急に解決しなければならない重要な課題である。
デバイスにおいてタンパク質の吸着を抑制する方法としては、生体膜類似構造を有する2−メタクリロイルオキシエチルホスホリルコリン(MPC)や、ポリエチレングリコール(PEG)を含む合成高分子材料からなる被覆層をデバイス表面に形成することが提案されている(例えば非特許文献1)。しかし、MPCやPEGは水溶性のため、それらだけでは使用中に被覆層が溶解するという課題がある。
そこで、MPCをブチルメタクリレート等の疎水性単量体と共重合させる(特許文献1)、ヒドロキシ基とリン脂質類似構造を有するプレポリマーとジイソシアネート化合物を反応させてウレタン結合にて架橋する(特許文献2)、エポキシ基含有単量体を共重合して得られる親水性重合体を、エポキシ基の反応により材料表面に固定化する(特許文献3)等によって耐水性を高めることが提案されている。
高分子論文集 Vol.35、pp.423−427、1978
しかし、ブチルメタクリレート等の単量体を共重合させたり、重合後にジイソシアネート化合物を反応させたりして耐水性を高める方法では、重合体の組成のばらつきにより再現性が得られにくく、耐水性が不充分になることがある。また、該重合体を用いて形成した被覆層は、水と接触した際にデバイス表面から剥がれて充分な耐久性が得られないことがある。
本発明は、タンパク質が吸着しにくく、耐水性および耐久性に優れた被覆層を形成できる重合体およびタンパク質付着防止剤、ならびに該タンパク質付着防止剤を用いた被覆層を備える医療用デバイスを提供することを目的とする。
本発明は、以下の構成を有する。
[1]側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位を有する、ガラス転移温度が20℃以上の重合体。
[2]前記マレイミド誘導体が、下式(1)で表される化合物である、[1]の重合体。
[1]側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位を有する、ガラス転移温度が20℃以上の重合体。
[2]前記マレイミド誘導体が、下式(1)で表される化合物である、[1]の重合体。
(ただし、前記式(1)中、Xは、炭素数1〜10のアルキレン基、−(CH2)d−COO−(CH2)e−(ただし、d,eはそれぞれ独立に0から10の整数である。)または−C6H4−(CH2)c−(ただし、cは0〜10の整数である。)であり、aは2〜50の整数であり、R1は水素原子または1価の炭化水素基である。)
[3]下式(2)で表される化合物に由来する単位をさらに有する、[1]または[2]の重合体。
[3]下式(2)で表される化合物に由来する単位をさらに有する、[1]または[2]の重合体。
(ただし、前記式(2)中、R2は、水素原子、塩素原子またはメチル基であり、bは1〜10の整数であり、R3は炭素数1〜5のアルキル基である。R3の3つのアルキル基は、互いに同じであってもよく、異なっていてもよい。)
[4]前記[1]〜[3]のいずれかの重合体からなるタンパク質付着防止剤。
[5]表面の少なくとも一部に[1]〜[3]のいずれかに記載の重合体を含む被覆層を有する医療用デバイス。
[6]細胞培養容器である、[5]の医療用デバイス。
[4]前記[1]〜[3]のいずれかの重合体からなるタンパク質付着防止剤。
[5]表面の少なくとも一部に[1]〜[3]のいずれかに記載の重合体を含む被覆層を有する医療用デバイス。
[6]細胞培養容器である、[5]の医療用デバイス。
本発明の重合体を用いれば、タンパク質が付着しにくく、耐水性および耐久性に優れた被覆層を形成できる。
本発明のタンパク質付着防止剤を用いれば、タンパク質が付着しにくく、耐水性および耐久性に優れた被覆層を形成できる。
本発明の医療用デバイスは、タンパク質が付着しにくく、耐水性および耐久性に優れた被覆層を備えている。
本発明のタンパク質付着防止剤を用いれば、タンパク質が付着しにくく、耐水性および耐久性に優れた被覆層を形成できる。
本発明の医療用デバイスは、タンパク質が付着しにくく、耐水性および耐久性に優れた被覆層を備えている。
以下の用語の定義は、本明細書および特許請求の範囲にわたって適用される。
重合体の「ガラス転移温度(Tg)」とは、示差走査熱量測定(DSC)法で測定したゴム状態からガラス状態へ変化する際の中間点ガラス転移温度を意味する。
重合体の「数平均分子量(Mn)」および「質量平均分子量(Mw)」とは、ゲルパーミエーションクロマトグラフィ(GPC)法によってポリスチレン換算で求めた値を意味する。
「単位」とは、重合体中に存在して重合体を構成する、単量体に由来する部分を意味する。炭素−炭素不飽和二重結合を有する単量体の付加重合により生じる、該単量体に由来する単位は、該不飽和二重結合が開裂して生じた2価の単位である。また、ある単位の構造を重合体形成後に化学的に変換したものも単位という。
マレイミド誘導体の「側鎖」とは、マレイミドにおける窒素原子と結合した水素原子が置換されて形成された鎖を意味する。
「医療用デバイス」とは、治療、診断、解剖学または生物学的な検査等の医療用として用いられるデバイスであり、人体等の生体内に挿入あるいは接触させる、または生体から取り出した媒体(血液等)と接触させる如何なるデバイスも含むものとする。
重合体の「ガラス転移温度(Tg)」とは、示差走査熱量測定(DSC)法で測定したゴム状態からガラス状態へ変化する際の中間点ガラス転移温度を意味する。
重合体の「数平均分子量(Mn)」および「質量平均分子量(Mw)」とは、ゲルパーミエーションクロマトグラフィ(GPC)法によってポリスチレン換算で求めた値を意味する。
「単位」とは、重合体中に存在して重合体を構成する、単量体に由来する部分を意味する。炭素−炭素不飽和二重結合を有する単量体の付加重合により生じる、該単量体に由来する単位は、該不飽和二重結合が開裂して生じた2価の単位である。また、ある単位の構造を重合体形成後に化学的に変換したものも単位という。
マレイミド誘導体の「側鎖」とは、マレイミドにおける窒素原子と結合した水素原子が置換されて形成された鎖を意味する。
「医療用デバイス」とは、治療、診断、解剖学または生物学的な検査等の医療用として用いられるデバイスであり、人体等の生体内に挿入あるいは接触させる、または生体から取り出した媒体(血液等)と接触させる如何なるデバイスも含むものとする。
本明細書においては、式(1)で表される化合物を化合物(1)と記す。他の式で表される化合物も同様に記す。
[重合体]
本発明の重合体は、側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位(以下、単位(a1)ともいう。)を有する、ガラス転移温度が20℃以上の重合体である。本発明の重合体は、単位(a1)に加えて、後述する化合物(2)に由来する単位(以下、単位(a2)ともいう。)、単位(a1)および単位(a2)以外の他の単位(以下、単位(a3)ともいう。)をさらに有してもよい。
本発明の重合体は、側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位(以下、単位(a1)ともいう。)を有する、ガラス転移温度が20℃以上の重合体である。本発明の重合体は、単位(a1)に加えて、後述する化合物(2)に由来する単位(以下、単位(a2)ともいう。)、単位(a1)および単位(a2)以外の他の単位(以下、単位(a3)ともいう。)をさらに有してもよい。
(単位(a1))
単位(a1)は、側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位である。
マレイミド誘導体の側鎖は、ポリオキシエチレン基を有するものであればよく、ポリオキシエチレン基以外の部分の構造は特に限定されない。マレイミド誘導体の側鎖は、ポリオキシエチレン基が炭素数1〜10のアルキレン基、−(CH2)d−COO−(CH2)e−(ただし、d,eはそれぞれ独立に0から10の整数である。)または−C6H4−(CH2)c−(ただし、cは0〜10の整数である。)を介してマレイミドの窒素原子と結合した鎖であることが好ましい。マレイミド誘導体の側鎖の末端構造は、特に限定されず、ヒドロキシ基であってもよく、アルコキシ基であってもよい。
単位(a1)は、側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位である。
マレイミド誘導体の側鎖は、ポリオキシエチレン基を有するものであればよく、ポリオキシエチレン基以外の部分の構造は特に限定されない。マレイミド誘導体の側鎖は、ポリオキシエチレン基が炭素数1〜10のアルキレン基、−(CH2)d−COO−(CH2)e−(ただし、d,eはそれぞれ独立に0から10の整数である。)または−C6H4−(CH2)c−(ただし、cは0〜10の整数である。)を介してマレイミドの窒素原子と結合した鎖であることが好ましい。マレイミド誘導体の側鎖の末端構造は、特に限定されず、ヒドロキシ基であってもよく、アルコキシ基であってもよい。
ポリオキシエチレン基におけるオキシエチレン基の繰り返し数は、2〜50が好ましく、2〜25がより好ましい。オキシエチレン基の繰り返し数が下限値以上であれば、細胞をより吸着しにくい。オキシエチレン基の繰り返し数が上限値以下であれば、純度の高いマレイミド誘導体を製造しやすい。
マレイミド誘導体としては、タンパク質を吸着しにくく、優れた耐水性を発現しやすい点から、下記の化合物(1)が好ましい。
ただし、前記式(1)中、Xは、炭素数1〜10のアルキレン基、−(CH2)d−COO−(CH2)e−(d,eは0から10の整数)または−C6H4−(CH2)c−(ただし、cは0〜10の整数である。)であり、aは2〜50の整数であり、R1は水素原子または1価の炭化水素基である。
Xにおけるアルキレン基は、直鎖状であってもよく、分岐鎖状であってもよい。Xにおけるアルキレン基の炭素数は、タンパク質を吸着しにくいという観点から、1〜10が好ましく、1〜5がより好ましい。
Xにおける−C6H4−(CH2)c−中のフェニレン基は、o、mまたはp−フェニレン基のいずれでもよい。cは、タンパク質を吸着しにくいという点から0〜10の整数が好ましく、0〜5の整数がより好ましい。
Xとしては、重合体の耐熱性に優れる点では、−C6H4−(CH2)c−が好ましい。
Xとしては、重合体の耐熱性に優れる点では、−C6H4−(CH2)c−が好ましい。
aは、2〜50の整数が好ましく、2〜25の整数がより好ましい。aが下限値以上であれば、タンパク質の付着をより防止できる。aが上限値以下であれば、材料の純度が高く、均一な化合物の製造がしやすい。
R1における1価の炭化水素基としては、例えば、炭素数1〜10のアルキル基、アリール基(フェニル基、ナフチル基等)、芳香族アルキル基(ベンジル基等)等が挙げられる。R1におけるアルキル基は、直鎖状であってもよく、分岐鎖状であってもよい。1価の炭化水素基としては、タンパク質をより吸着しにくい点から、炭素数1〜10のアルキル基が好ましく、メチル基またはエチル基がより好ましい。
R1としては、タンパク質をより吸着しにくい点から、水素原子、メチル基またはエチル基が好ましい。
R1としては、タンパク質をより吸着しにくい点から、水素原子、メチル基またはエチル基が好ましい。
本発明の重合体が有する単位(a1)は、1種のみであってもよく、2種以上であってもよい。
本発明の重合体の全単位に対する単位(a1)の割合は、10〜100モル%が好ましく、20〜100モル%がより好ましく、30〜100モル%がさらに好ましい。単位(a1)の割合が下限値以上であれば、タンパク質が吸着しにくく、優れた耐水性を発現できる。単位(a1)の割合が上限値以下であれば、相対的に単位(a2)や単位(a3)の割合を高めることができ、デバイスの基材等への密着性を高めることが容易になる。
本発明の重合体の全単位に対する単位(a1)の割合は、10〜100モル%が好ましく、20〜100モル%がより好ましく、30〜100モル%がさらに好ましい。単位(a1)の割合が下限値以上であれば、タンパク質が吸着しにくく、優れた耐水性を発現できる。単位(a1)の割合が上限値以下であれば、相対的に単位(a2)や単位(a3)の割合を高めることができ、デバイスの基材等への密着性を高めることが容易になる。
(単位(a2))
単位(a2)は、下記の化合物(2)に由来する単位である。本発明の重合体は、デバイスの基材等への密着性が向上する点から、単位(a2)を有することが好ましい。
単位(a2)は、下記の化合物(2)に由来する単位である。本発明の重合体は、デバイスの基材等への密着性が向上する点から、単位(a2)を有することが好ましい。
ただし、式(2)中、R2は、水素原子、塩素原子またはメチル基であり、bは1〜10の整数であり、R3は炭素数1〜5のアルキル基である。R3の3つのアルキル基は、互いに同じであってもよく、異なっていてもよい。
R2は、重合しやすい点から、水素原子またはメチル基が好ましい。
bは、入手容易性の点から、1〜10の整数が好ましく、1〜5の整数がより好ましい。
bは、入手容易性の点から、1〜10の整数が好ましく、1〜5の整数がより好ましい。
R3のアルキル基は、直鎖状であってもよく、分岐鎖状であってもよい。R3のアルキル基としては、反応時間の観点から、炭素数1〜5のアルキル基が好ましく、炭素数1〜3のアルキル基がより好ましく、メチル基が特に好ましい。
本発明の重合体が単位(a2)を有する場合、単位(a2)は、1種のみであってもよく、2種以上であってもよい。
本発明の重合体が単位(a2)を有する場合、重合体の全単位に対する単位(a2)の割合は、0〜50モル%が好ましく、0〜30モル%がより好ましく、0〜20モル%がさらに好ましい。単位(a2)の割合が下限値以上であれば、デバイスの基材等への密着性がより優れたものとなり、優れた耐久性を有する被覆層を形成しやすい。単位(a2)の割合が上限値以下であれば、相対的に単位(a1)の割合を高めることができ、タンパク質の付着をより防止できるとともに、優れた耐水性が得られやすい。
本発明の重合体が単位(a2)を有する場合、重合体の全単位に対する単位(a2)の割合は、0〜50モル%が好ましく、0〜30モル%がより好ましく、0〜20モル%がさらに好ましい。単位(a2)の割合が下限値以上であれば、デバイスの基材等への密着性がより優れたものとなり、優れた耐久性を有する被覆層を形成しやすい。単位(a2)の割合が上限値以下であれば、相対的に単位(a1)の割合を高めることができ、タンパク質の付着をより防止できるとともに、優れた耐水性が得られやすい。
(単位(a3))
単位(a3)を形成する単量体としては、特に限定されず、以下に示す化合物が挙げられる。
例えば、CH2=CH−COO−(CH2CH2O)n−CH3(nは2〜50の整数である。)、CH2=CH−COO−(CH2CH2O)n−CH2CH3(nは2〜50の整数である。)、CH2=C(CH3)−COO−(CH2CH2O)n−CH3(nは2〜50の整数である。)、CH2=C(CH3)−COO−(CH2CH2O)n−CH2CH3(nは2〜50の整数である。)、C(CH3)=CH−COO−(CH2)3−CH3等が挙げられる。
単位(a3)を形成する単量体としては、特に限定されず、以下に示す化合物が挙げられる。
例えば、CH2=CH−COO−(CH2CH2O)n−CH3(nは2〜50の整数である。)、CH2=CH−COO−(CH2CH2O)n−CH2CH3(nは2〜50の整数である。)、CH2=C(CH3)−COO−(CH2CH2O)n−CH3(nは2〜50の整数である。)、CH2=C(CH3)−COO−(CH2CH2O)n−CH2CH3(nは2〜50の整数である。)、C(CH3)=CH−COO−(CH2)3−CH3等が挙げられる。
本発明の重合体に耐水性を付与したい場合には、単位(a3)を形成する単量体として以下の化合物を用いることができる。
CH2=CH−COO−(CH2)4−H、
CH2=CH−COO−(CH2)6−H、
CH2=CH−COO−(CH2)8−H、
CH2=CH−COO−(CH2)16−H、
CH2=CH−COO−CH2CH(C2H5)CH2CH2CH2CH3等。
なかでもCH2=CH−COO−(CH2)4−H、CH2=CH−COO−(CH2)8−H、CH2=CH−COO−(CH2)16−Hが好ましく、CH2=CH−COO−(CH2)8−H、CH2=CH−COO−(CH2)16−Hが特に好ましい。
CH2=CH−COO−(CH2)4−H、
CH2=CH−COO−(CH2)6−H、
CH2=CH−COO−(CH2)8−H、
CH2=CH−COO−(CH2)16−H、
CH2=CH−COO−CH2CH(C2H5)CH2CH2CH2CH3等。
なかでもCH2=CH−COO−(CH2)4−H、CH2=CH−COO−(CH2)8−H、CH2=CH−COO−(CH2)16−Hが好ましく、CH2=CH−COO−(CH2)8−H、CH2=CH−COO−(CH2)16−Hが特に好ましい。
そして、特に耐水性を付与したい場合は、単位(a3)を形成する単量体として以下に示す含フッ素化合物を用いることができる。
CH2=CH−COO−(CH2)2(CF2)3CF3、
CH2=CH−COO−(CH2)2(CF2)5CF3、
CH2=C(CH3)COO−(CH2)2(CF2)3CF3、
CH2=C(CH3)COO−(CH2)2(CF2)5CF3、
CH2=CH−COO−(CH2)3(CF2)3CF3、
CH2=CH−COO−(CH2)3(CF2)5CF3、
CH2=CH−COO−CH2CH(OH)CH2(CF2)3CF3、
CH2=CH−COO−CH2CH(OH)CH2(CF2)5CF3、
CH2=C(CH3)−COO−(CH2)3(CF2)3CF3、
CH2=C(CH3)−COO−(CH2)3(CF2)5CF3、
CH2=C(CH3)−COO−CH2CH(OH)CH2(CF2)3CF3、
CH2=C(CH3)−COO−CH2CH(OH)CH2(CF2)5CF3、
CH2=CHC6H4(CF2)3CF3、
CH2=CHC6H4(CF2)5CF3、
CH2=CH−COO−CH2CH2N(CH3)SO2(CF2)3CF3、
CH2=CH−COO−CH2CH2N(CH3)SO2(CF2)5CF3、
CH2=C(CH3)−COO−CH2CH2N(CH3)SO2(CF2)3CF3、
CH2=C(CH3)−COO−CH2CH2N(CH3)SO2(CF2)5CF3、
CH2=CH−COO−CH2CH2N(C2H5)SO2(CF2)3CF3、
CH2=CH−COO−CH2CH2N(C2H5)SO2(CF2)5CF3、
CH2=C(CH3)−COO−CH2CH2N(C2H5)SO2(CF2)3CF3、
CH2=C(CH3)−COO−CH2CH2N(C2H5)SO2(CF2)5CF3、
CH2=CH−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)3CF3、
CH2=CH−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)5CF3、
CH2=C(CH3)−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)3CF3、
CH2=C(CH3)−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)5CF3、
CH2=CHCONHCH2C4F9、
CH2=CHCONHCH2C5F11、
CH2=CHCONHCH2C6F13、
CH2=CHCONHCH2CH2OCOC4F9、
CH2=CHCONHCH2CH2OCOC5F11、
CH2=CHCONHCH2CH2OCOC6F13、
CH2=CH−COO−CH(CF3)2
CH2=C(CH3)−COO−CH(CF3)2等。
CH2=CH−COO−(CH2)2(CF2)3CF3、
CH2=CH−COO−(CH2)2(CF2)5CF3、
CH2=C(CH3)COO−(CH2)2(CF2)3CF3、
CH2=C(CH3)COO−(CH2)2(CF2)5CF3、
CH2=CH−COO−(CH2)3(CF2)3CF3、
CH2=CH−COO−(CH2)3(CF2)5CF3、
CH2=CH−COO−CH2CH(OH)CH2(CF2)3CF3、
CH2=CH−COO−CH2CH(OH)CH2(CF2)5CF3、
CH2=C(CH3)−COO−(CH2)3(CF2)3CF3、
CH2=C(CH3)−COO−(CH2)3(CF2)5CF3、
CH2=C(CH3)−COO−CH2CH(OH)CH2(CF2)3CF3、
CH2=C(CH3)−COO−CH2CH(OH)CH2(CF2)5CF3、
CH2=CHC6H4(CF2)3CF3、
CH2=CHC6H4(CF2)5CF3、
CH2=CH−COO−CH2CH2N(CH3)SO2(CF2)3CF3、
CH2=CH−COO−CH2CH2N(CH3)SO2(CF2)5CF3、
CH2=C(CH3)−COO−CH2CH2N(CH3)SO2(CF2)3CF3、
CH2=C(CH3)−COO−CH2CH2N(CH3)SO2(CF2)5CF3、
CH2=CH−COO−CH2CH2N(C2H5)SO2(CF2)3CF3、
CH2=CH−COO−CH2CH2N(C2H5)SO2(CF2)5CF3、
CH2=C(CH3)−COO−CH2CH2N(C2H5)SO2(CF2)3CF3、
CH2=C(CH3)−COO−CH2CH2N(C2H5)SO2(CF2)5CF3、
CH2=CH−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)3CF3、
CH2=CH−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)5CF3、
CH2=C(CH3)−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)3CF3、
CH2=C(CH3)−COO−(CH2)2N(CH2CH2CH3)SO2(CF2)5CF3、
CH2=CHCONHCH2C4F9、
CH2=CHCONHCH2C5F11、
CH2=CHCONHCH2C6F13、
CH2=CHCONHCH2CH2OCOC4F9、
CH2=CHCONHCH2CH2OCOC5F11、
CH2=CHCONHCH2CH2OCOC6F13、
CH2=CH−COO−CH(CF3)2
CH2=C(CH3)−COO−CH(CF3)2等。
また、上記以外には、単位(a3)を形成する単量体として以下の化合物が挙げられる。例えば、 N,N−ジメチルアミノエチル(メタ)アクリレート、N,N−ジエチルアミノエチル(メタ)アクリレート、N−(メタ)アクリロイルモルホリン、N−(メタ)アクリロイルペピリジン、N,N−ジメチルアミノオキシドエチル(メタ)アクリレート、N,N−ジエチルアミノオキシドエチル(メタ)アクリレート。また、2−イソシアネートエチル(メタ)アクリレート、2−イソシアネートエチル(メタ)アクリレートの3,5−ジメチルピラゾール付加体、3−イソシアネートプロピル(メタ)アクリレート、4−イソシアネートブチル(メタ)アクリレート、トリアリルイソシアヌレート、グリシジル(メタ)アクリレート、ポリオキシアルキレングリコールモノグリシジルエーテル(メタ)アクリレート。
本発明の重合体が単位(a3)を有する場合、単位(a3)は、1種のみであってもよく、2種以上であってもよい。
本発明の重合体の全単位に対する単位(a3)の割合は、50モル%以下が好ましく、30モル%以下がより好ましい。単位(a3)の割合が上限値以下であれば、タンパク質の付着をより防止できるとともに、優れた耐水性が得られやすい。
本発明の重合体の全単位に対する単位(a3)の割合は、50モル%以下が好ましく、30モル%以下がより好ましい。単位(a3)の割合が上限値以下であれば、タンパク質の付着をより防止できるとともに、優れた耐水性が得られやすい。
本発明の重合体は、タンパク質の付着をより防止することができ、耐水性および耐久性を兼ね備えた被覆層を形成しやすい点から、単位(a1)のみを有する重合体、または単位(a1)と単位(a2)とからなる重合体が好ましい。
(物性)
本発明の重合体のガラス転移温度(Tg)は、20℃以上である。本発明の重合体のTgが20℃以上であることで、水と接触した状態でもデバイス表面等から剥がれにくい優れた耐久性を有する被覆層を形成できる。本発明の重合体のガラス転移温度は、20〜250℃が好ましく、40〜250℃がより好ましい。重合体のガラス転移温度が下限値以上であれば、優れた耐久性を有する被覆層を形成しやすい。重合体のガラス転移温度が上限値以下であれば、汎用溶媒に溶かすことが可能である。
本発明の重合体のガラス転移温度(Tg)は、20℃以上である。本発明の重合体のTgが20℃以上であることで、水と接触した状態でもデバイス表面等から剥がれにくい優れた耐久性を有する被覆層を形成できる。本発明の重合体のガラス転移温度は、20〜250℃が好ましく、40〜250℃がより好ましい。重合体のガラス転移温度が下限値以上であれば、優れた耐久性を有する被覆層を形成しやすい。重合体のガラス転移温度が上限値以下であれば、汎用溶媒に溶かすことが可能である。
重合体のガラス転移温度は、重合体中の単位(a1)の割合、重合体の分子量を調節することにより調節できる。単位(a1)の割合が高いほど重合体のガラス転移温度が高くなる傾向がある。また、重合体の分子量が高いほど重合体のガラス転移温度が高くなる傾向がある。
本発明の重合体の数平均分子量(Mn)は、3000〜1000000が好ましく、5000〜500000が特に好ましい。重合体の数平均分子量が前記下限値以上であれば、耐久性に優れる。重合体の数平均分子量が前記上限値以下であれば、加工性に優れる。
本発明の重合体の質量平均分子量(Mw)は、3000〜1000000が好ましく、5000〜500000が特に好ましい。重合体の質量平均分子量が前記下限値以上であれば、耐久性に優れる。重合体の質量平均分子量が前記上限値以下であれば、加工性に優れる。
重合体の分子量分布(Mw/Mn)は、1.0〜5.0が好ましく、1.0〜3.0が特に好ましい。重合体の分子量分布が前記範囲内であれば、耐水性に優れ、かつタンパク質が吸着しにくい。
(製造方法)
本発明の重合体は、公知の方法を用いて、重合溶媒中でマレイミド誘導体、化合物(2)等の単量体の重合反応を行うことにより得られる。
重合溶媒としては、特に限定されず、例えば、ケトン類(アセトン、メチルエチルケトン、メチルイソブチルケトン等)、アルコール類(メタノール、2−プロピルアルコール等)、エステル類(酢酸エチル、酢酸ブチル等)、エーテル類(ジイソプロピルエーテル、テトラヒドロフラン、ジオキサン等)、グリコールエーテル類(エチレングリコール、プロピレングリコール、またはジプロピレングリコールのエチルエーテルまたはメチルエーテル等)およびその誘導体、脂肪族炭化水素類、芳香族炭化水素類、ハロゲン化炭化水素類(パークロロエチレン、トリクロロ−1,1,1−エタン、トリクロロトリフルオロエタン、ジクロロペンタフルオロプロパン等)、ジメチルホルムアミド、N−メチル−2−ピロリドン、ブチロアセトン、ジメチルスルホキシド(DMSO)等が挙げられる。
本発明の重合体は、公知の方法を用いて、重合溶媒中でマレイミド誘導体、化合物(2)等の単量体の重合反応を行うことにより得られる。
重合溶媒としては、特に限定されず、例えば、ケトン類(アセトン、メチルエチルケトン、メチルイソブチルケトン等)、アルコール類(メタノール、2−プロピルアルコール等)、エステル類(酢酸エチル、酢酸ブチル等)、エーテル類(ジイソプロピルエーテル、テトラヒドロフラン、ジオキサン等)、グリコールエーテル類(エチレングリコール、プロピレングリコール、またはジプロピレングリコールのエチルエーテルまたはメチルエーテル等)およびその誘導体、脂肪族炭化水素類、芳香族炭化水素類、ハロゲン化炭化水素類(パークロロエチレン、トリクロロ−1,1,1−エタン、トリクロロトリフルオロエタン、ジクロロペンタフルオロプロパン等)、ジメチルホルムアミド、N−メチル−2−ピロリドン、ブチロアセトン、ジメチルスルホキシド(DMSO)等が挙げられる。
重合反応における反応液中のすべての単量体の合計濃度は、5〜60質量%が好ましく、10〜40質量%が特に好ましい。
重合反応においては、重合開始剤を用いることが好ましい。重合開始剤としては、過酸化物(ベンジルパーオキシド、ラウリルパーオキシド、スクシニルパーオキシド、tert−ブチルパーピバレート等)、アゾ化合物等が挙げられる。
重合開始剤としては、2,2’−アゾイソブチロニトリル、2,2’−アゾビス−2−メチルブチロニトリル、ジメチル−2,2’−アゾビスイソブチレート、2,2’−アゾビス[2−(2−イミダゾリン−2イル)プロパン]、2,2’−アゾビス(4−メトキシ−2、4−ジメチルバレロニトリル)、1、1’−アゾビス(2シクロヘキサン−1−カルボニトリル)、2,2’−アゾビス(2、4−ジメチルバレロニトリル)、1、1’−アゾビス(1−アセトキシ−1−フェニルエタン)、ジメチルアゾビスイソブチレート、4,4’−アゾビス(4−シアノ吉草酸)が好ましく、2,2’−アゾイソブチロニトリル、2,2’−アゾビス[2−(2−イミダゾリン−2イル)プロパン]、4,4’−アゾビス(4−シアノ吉草酸)が特に好ましい。
重合開始剤の使用量は、単量体の合計量100質量部に対して0.1〜1.5質量部が好ましい。
重合開始剤としては、2,2’−アゾイソブチロニトリル、2,2’−アゾビス−2−メチルブチロニトリル、ジメチル−2,2’−アゾビスイソブチレート、2,2’−アゾビス[2−(2−イミダゾリン−2イル)プロパン]、2,2’−アゾビス(4−メトキシ−2、4−ジメチルバレロニトリル)、1、1’−アゾビス(2シクロヘキサン−1−カルボニトリル)、2,2’−アゾビス(2、4−ジメチルバレロニトリル)、1、1’−アゾビス(1−アセトキシ−1−フェニルエタン)、ジメチルアゾビスイソブチレート、4,4’−アゾビス(4−シアノ吉草酸)が好ましく、2,2’−アゾイソブチロニトリル、2,2’−アゾビス[2−(2−イミダゾリン−2イル)プロパン]、4,4’−アゾビス(4−シアノ吉草酸)が特に好ましい。
重合開始剤の使用量は、単量体の合計量100質量部に対して0.1〜1.5質量部が好ましい。
重合体の重合度(分子量)を調節するために、重合反応において連鎖移動剤を用いてもよい。連鎖移動剤を用いることにより重合溶媒中の単量体の濃度の合計を高められる効果もある。
連鎖移動剤としては、アルキルメルカプタン(tert−ドデシルメルカプタン、n−ドデシルメルカプタン、ステアリルメルカプタン等)、アミノエタンチオール、メルカプトエタノール、3−メルカプトプロピオン酸、2−メルカプトプロピオン酸、チオリンゴ酸、チオグリコール酸、3,3’−ジチオ−ジプロピオン酸、チオグリコール酸2−エチルヘキシル、チオグリコール酸n−ブチル、チオグリコール酸メトキシブチル、チオグリコール酸エチル、2,4−ジフェニル−4−メチル−1−ペンテン、四塩化炭素等が挙げられる。
連鎖移動剤の使用量は、単量体の合計量100質量部に対して0〜2質量部が好ましい。
連鎖移動剤としては、アルキルメルカプタン(tert−ドデシルメルカプタン、n−ドデシルメルカプタン、ステアリルメルカプタン等)、アミノエタンチオール、メルカプトエタノール、3−メルカプトプロピオン酸、2−メルカプトプロピオン酸、チオリンゴ酸、チオグリコール酸、3,3’−ジチオ−ジプロピオン酸、チオグリコール酸2−エチルヘキシル、チオグリコール酸n−ブチル、チオグリコール酸メトキシブチル、チオグリコール酸エチル、2,4−ジフェニル−4−メチル−1−ペンテン、四塩化炭素等が挙げられる。
連鎖移動剤の使用量は、単量体の合計量100質量部に対して0〜2質量部が好ましい。
重合反応における反応温度は、室温から反応液の沸点までの範囲が好ましい。重合開始剤を効率良く使う観点からは重合開始剤の半減期温度以上が好ましく、30〜90℃がより好ましい。
(作用効果)
以上説明した本発明の重合体は、前記したマレイミド誘導体に由来する単位(a1)を有しているため、耐水性に優れている。また、単位(a1)が側鎖にポリオキシエチレン基を有しているため、タンパク質が付着しにくい。また、本発明の重合体はガラス転移温度が20℃以上であるため、デバイス表面等に被覆層を形成した際、該被覆層が水と接触した状態でも動きにくく、デバイス表面等から剥がれにくいため優れた耐久性が得られる。本発明の重合体は、単位(a1)のみからなる単独重合体であっても、タンパク質が付着しにくく、耐水性および耐久性を兼ね備えた被覆層を形成できる。
以上説明した本発明の重合体は、前記したマレイミド誘導体に由来する単位(a1)を有しているため、耐水性に優れている。また、単位(a1)が側鎖にポリオキシエチレン基を有しているため、タンパク質が付着しにくい。また、本発明の重合体はガラス転移温度が20℃以上であるため、デバイス表面等に被覆層を形成した際、該被覆層が水と接触した状態でも動きにくく、デバイス表面等から剥がれにくいため優れた耐久性が得られる。本発明の重合体は、単位(a1)のみからなる単独重合体であっても、タンパク質が付着しにくく、耐水性および耐久性を兼ね備えた被覆層を形成できる。
また、本発明の重合体が、単位(a1)として、式(1)におけるXがフェニレン基や−C6H4−(CH2)c−である化合物に由来する単位を有する場合には、滅菌処理にも充分に耐え得る優れた耐熱性を有する被覆層を形成できる。また、この場合には蒸着によるドライコーティングにより被覆層を形成することもできる。
[タンパク質付着防止剤]
本発明のタンパク質付着防止剤は、本発明の重合体からなるタンパク質付着防止剤である。
本発明のタンパク質付着防止剤は、本発明の重合体からなるタンパク質付着防止剤である。
(細胞付着防止剤)
本発明のタンパク質付着防止剤の用途としては、医療用デバイスが特に有効である。
本発明のタンパク質付着防止剤は、例えば、細胞の接着を防止するため、すなわち細胞付着防止剤として用いることができる。なお、「細胞」とは、生体を構成する最も基本的な単位であり、細胞膜の内部に細胞質と各種の細胞小器官をもつものを意味する。DNAを内包する核は、細胞内部に含まれても含まれなくてもよい。
本発明のタンパク質付着防止剤の用途としては、医療用デバイスが特に有効である。
本発明のタンパク質付着防止剤は、例えば、細胞の接着を防止するため、すなわち細胞付着防止剤として用いることができる。なお、「細胞」とは、生体を構成する最も基本的な単位であり、細胞膜の内部に細胞質と各種の細胞小器官をもつものを意味する。DNAを内包する核は、細胞内部に含まれても含まれなくてもよい。
動物由来の細胞には、生殖細胞(精子、卵子等)、生体を構成する体細胞、幹細胞、前駆細胞、生体から分離された癌細胞、生体から分離され不死化能を獲得して体外で安定して維持される細胞(細胞株)、生体から分離され人為的に遺伝子改変された細胞、生体から分離され人為的に核が交換された細胞等が含まれる。
生体を構成する体細胞には、線維芽細胞、骨髄細胞、Bリンパ球、Tリンパ球、好中球、赤血球、血小板、マクロファージ、単球、骨細胞、骨髄細胞、周皮細胞、樹枝状細胞、ケラチノサイト、脂肪細胞、間葉細胞、上皮細胞、表皮細胞、内皮細胞、血管内皮細胞、肝実質細胞、軟骨細胞、卵丘細胞、神経系細胞、グリア細胞、ニューロン、オリゴデンドロサイト、マイクログリア、星状膠細胞、心臓細胞、食道細胞、筋肉細胞(例えば、平滑筋細胞、骨格筋細胞)、膵臓ベータ細胞、メラニン細胞、造血前駆細胞、単核細胞等が含まれる。体細胞には、皮膚、腎臓、脾臓、副腎、肝臓、肺、卵巣、膵臓、子宮、胃、結腸、小腸、大腸、膀胱、前立腺、精巣、胸腺、筋肉、結合組織、骨、軟骨、血管組織、血液、心臓、眼、脳、神経組織等の任意の組織から採取される細胞が含まれる。
幹細胞とは、自分自身を複製する能力と他の複数系統の細胞に分化する能力を兼ね備えた細胞であり、胚性幹細胞(ES細胞)、胚性腫瘍細胞、胚性生殖幹細胞、人工多能性幹細胞(iPS細胞)、神経幹細胞、造血幹細胞、間葉系幹細胞、肝幹細胞、膵幹細胞、筋幹細胞、生殖幹細胞、腸幹細胞、癌幹細胞、毛包幹細胞等が含まれる。
前駆細胞とは、前記幹細胞から特定の体細胞または生殖細胞に分化する途中の段階にある細胞である。
癌細胞とは、体細胞から派生して無限の増殖能を獲得した細胞である。
細胞株とは、生体外での人為的な操作により無限の増殖能を獲得した細胞であり、HCT116、Huh7、HEK293(ヒト胎児腎細胞)、HeLa(ヒト子宮頸癌細胞株)、HepG2(ヒト肝癌細胞株)、UT7/TPO(ヒト白血病細胞株)、CHO(チャイニーズハムスター卵巣細胞株)、MDCK、MDBK、BHK、C−33A、HT−29、AE−1、3D9、Ns0/1、Jurkat、NIH3T3、PC12、S2、Sf9、Sf21、High Five、Vero等が含まれる。
生体を構成する体細胞には、線維芽細胞、骨髄細胞、Bリンパ球、Tリンパ球、好中球、赤血球、血小板、マクロファージ、単球、骨細胞、骨髄細胞、周皮細胞、樹枝状細胞、ケラチノサイト、脂肪細胞、間葉細胞、上皮細胞、表皮細胞、内皮細胞、血管内皮細胞、肝実質細胞、軟骨細胞、卵丘細胞、神経系細胞、グリア細胞、ニューロン、オリゴデンドロサイト、マイクログリア、星状膠細胞、心臓細胞、食道細胞、筋肉細胞(例えば、平滑筋細胞、骨格筋細胞)、膵臓ベータ細胞、メラニン細胞、造血前駆細胞、単核細胞等が含まれる。体細胞には、皮膚、腎臓、脾臓、副腎、肝臓、肺、卵巣、膵臓、子宮、胃、結腸、小腸、大腸、膀胱、前立腺、精巣、胸腺、筋肉、結合組織、骨、軟骨、血管組織、血液、心臓、眼、脳、神経組織等の任意の組織から採取される細胞が含まれる。
幹細胞とは、自分自身を複製する能力と他の複数系統の細胞に分化する能力を兼ね備えた細胞であり、胚性幹細胞(ES細胞)、胚性腫瘍細胞、胚性生殖幹細胞、人工多能性幹細胞(iPS細胞)、神経幹細胞、造血幹細胞、間葉系幹細胞、肝幹細胞、膵幹細胞、筋幹細胞、生殖幹細胞、腸幹細胞、癌幹細胞、毛包幹細胞等が含まれる。
前駆細胞とは、前記幹細胞から特定の体細胞または生殖細胞に分化する途中の段階にある細胞である。
癌細胞とは、体細胞から派生して無限の増殖能を獲得した細胞である。
細胞株とは、生体外での人為的な操作により無限の増殖能を獲得した細胞であり、HCT116、Huh7、HEK293(ヒト胎児腎細胞)、HeLa(ヒト子宮頸癌細胞株)、HepG2(ヒト肝癌細胞株)、UT7/TPO(ヒト白血病細胞株)、CHO(チャイニーズハムスター卵巣細胞株)、MDCK、MDBK、BHK、C−33A、HT−29、AE−1、3D9、Ns0/1、Jurkat、NIH3T3、PC12、S2、Sf9、Sf21、High Five、Vero等が含まれる。
(細胞付着防止剤以外の用途)
なお、本発明のタンパク質付着防止剤は、船舶、橋梁、海上タンク、港湾施設、海底基地、海底油田掘削設備等の海洋構造物に対して用いてもよい。該海洋構造物に対して本発明のタンパク質付着防止剤を用いることで、該海洋構造物にタンパク質が吸着することが抑制される。その結果、貝類(フジツボ等)、海藻類(アオノリ、アオサ等)等の水生生物が接着することが抑制される。
なお、本発明のタンパク質付着防止剤は、船舶、橋梁、海上タンク、港湾施設、海底基地、海底油田掘削設備等の海洋構造物に対して用いてもよい。該海洋構造物に対して本発明のタンパク質付着防止剤を用いることで、該海洋構造物にタンパク質が吸着することが抑制される。その結果、貝類(フジツボ等)、海藻類(アオノリ、アオサ等)等の水生生物が接着することが抑制される。
[医療用デバイス]
本発明の医療用デバイスは、表面の少なくとも一部に本発明の重合体を含む被覆層を有する。これにより医療用デバイス表面に細胞が付着するのを防止できる。
本発明の医療用デバイスは、表面の少なくとも一部に本発明の重合体を含む被覆層を有する。これにより医療用デバイス表面に細胞が付着するのを防止できる。
医療用デバイスの具体例としては、例えば、医薬品、医薬部外品、医療用器具等が挙げられる。医療用器具としては、特に限定されず、細胞培養容器、細胞培養シート、細胞捕捉フィルター、バイアル、プラスチックコートバイアル、シリンジ、プラスチックコートシリンジ、アンプル、プラスチックコートアンプル、カートリッジ、ボトル、プラスチックコートボトル、パウチ、ポンプ、噴霧器、栓、プランジャー、キャップ、蓋、針、ステント、カテーテル、インプラント、コンタクトレンズ、マイクロ流路チップ、ドラッグデリバリーシステム材、人工血管、人工臓器、血液透析膜、ガードワイヤー、血液フィルター、血液保存パック、内視鏡、バイオチップ、糖鎖合成機器、成形補助材、包装材等が挙げられる。なかでも、細胞培養容器、細胞培養シート、細胞捕捉フィルター等に好ましく用いられる。
本発明の医療用デバイスの具体例としては、例えば、図1および図2に例示した医療用デバイス1が挙げられる。医療用デバイス1は、細胞培養容器の一つであるシャーレである。
医療用デバイス1は、基材2と、基材2上に形成された被覆層3、とを備える。基材2は、平面視形状が円形状の底面部4と、底面部4の周縁から全周にわたって立ち上がる側面部5とを備え、上方が開放された容器形状になっている。被覆層3は、基材2における内面、すなわち底面部4の上面と側面部5の内面に、本発明の重合体によって形成されている。
医療用デバイス1は、基材2と、基材2上に形成された被覆層3、とを備える。基材2は、平面視形状が円形状の底面部4と、底面部4の周縁から全周にわたって立ち上がる側面部5とを備え、上方が開放された容器形状になっている。被覆層3は、基材2における内面、すなわち底面部4の上面と側面部5の内面に、本発明の重合体によって形成されている。
本発明の医療用デバイスにおけるデバイスを構成する材料は、特に限定されず、ポリエチレンテレフタラート、ポリスチレン、ポリカーボネート、ポリプロピレン、テトラフルオロエチレン−エチレン共重合体(ETFE)等の樹脂や、ガラスが挙げられる。一般的に、材料コスト、加工コストの観点からは樹脂が好ましい。一方、高精度な分析に用いる等の用途については、材料自体の透明性が高く、蛍光が少なく、化学的に安定で、剛性に優れるガラスが望ましい。
被覆層における本発明の重合体の含有割合は95質量%以上が好ましく、99質量%以上がより好ましい。被覆層としては、本発明の重合体のみから形成される層、または本発明の重合体と架橋剤とから形成される層が挙げられる。
被覆層の厚さは、1nm〜1mmが好ましく、5nm〜800μmが特に好ましい。被覆層の厚さが前記下限値以上であれば、タンパク質が吸着しにくい。被覆層の厚さが前記上限値以下であれば、被覆層がデバイスを構成する基材の表面に密着しやすい。
被覆層の厚さは、1nm〜1mmが好ましく、5nm〜800μmが特に好ましい。被覆層の厚さが前記下限値以上であれば、タンパク質が吸着しにくい。被覆層の厚さが前記上限値以下であれば、被覆層がデバイスを構成する基材の表面に密着しやすい。
被覆層と基材との密着性を向上させるために、基材と被覆層の間に接着層を設けてもよい。接着層を形成する接着剤としては、被覆層と基材の双方に対して充分な接着力を発揮するものを適宜使用でき、例えば、シアノアクリレート系接着剤、シリコーン変性アクリル接着剤、エポキシ変性シリコーン接着剤等が挙げられる。
具体例としては、例えば、基材を形成する材料としてポリスチレンを使用する場合、シアノアクリレート系接着剤を使用する。この場合、接着剤層の基材側では、シアノアクリレート系接着剤中のシアノアクリレートモノマーが空気中または基材の表面の水分と反応して硬化する。被覆層中には本発明の重合体由来のポリオキシエチレン基が存在するため、被覆層中およびその周辺部に水分が存在する。そのため、接着剤層の被覆層側でもシアノアクリレートモノマーがそれらの水分と反応して硬化する。
(医療用デバイスの製造方法)
本発明の医療用デバイスの製造方法は、特に限定されず、例えば、下記の塗布工程と、乾燥工程とを含む方法が挙げられる。
塗布工程:基材上に、本発明の重合体と溶媒とを含む塗布液を塗布する工程。
乾燥工程:前記塗布液に由来する溶媒を除去し、前記基材上に被覆層が形成された医療用デバイスを得る工程。
本発明の医療用デバイスの製造方法は、特に限定されず、例えば、下記の塗布工程と、乾燥工程とを含む方法が挙げられる。
塗布工程:基材上に、本発明の重合体と溶媒とを含む塗布液を塗布する工程。
乾燥工程:前記塗布液に由来する溶媒を除去し、前記基材上に被覆層が形成された医療用デバイスを得る工程。
<塗布工程>
本発明の重合体と溶媒とを含む塗布液を基材上に湿式塗布する。塗布液の塗布方法としては、公知の方法を用いることができ、例えば、刷毛、ローラー、ディッピング、スプレー、ロールコーター、ダイコーター、アプリケーター、スピンコーター等の塗装装置を用いて行う方法が挙げられる。
本発明の重合体と溶媒とを含む塗布液を基材上に湿式塗布する。塗布液の塗布方法としては、公知の方法を用いることができ、例えば、刷毛、ローラー、ディッピング、スプレー、ロールコーター、ダイコーター、アプリケーター、スピンコーター等の塗装装置を用いて行う方法が挙げられる。
塗布液に用いる溶媒としては、非含フッ素溶媒、含フッ素溶媒が挙げられる。非含フッ素溶媒としては、アルコール系溶媒、含ハロゲン系溶媒等が挙げられる。例えば、エタノール、メタノール、アセトン、クロロホルム、アサヒクリンAK225(旭硝子社製)、AC6000(旭硝子社製)等が挙げられる。溶媒としては、デバイス等を溶解しない種類を選択することが好ましい。デバイスとしてポリスチレンを使用する場合、エタノール、メタノール、アサヒクリンAK225(旭硝子社製)、AC6000(旭硝子社製)が好ましい。
塗布液中の重合体の濃度は、0.0001〜10質量%が好ましく、0.0005〜5質量%が特に好ましい。重合体の濃度が前記範囲であれば、均一に塗布することができ、均一な被覆層が形成できる。
塗布液を塗布する際には、塗布液に重合体および溶媒以外の成分、例えばレベリング剤、架橋剤等を含ませてもよい。架橋剤を塗布液に配合し、被覆層中の架橋度合いを調整することで、タンパク質を付着させにくい効果がより長期にわたって持続する、より優れた耐久性を有する被覆層を形成できる。具体的には、本発明の重合体における単位(a1)の側鎖の末端がヒドロキシ基の場合は、該ヒドロキシ基と反応する架橋剤を添加することで、優れた耐久性を有する被覆層を形成できる。塗布液に架橋剤を含ませる場合は、被覆層は重合体と架橋剤とから形成される層となる。塗布液に架橋剤を含ませない場合は、被覆層は重合体のみから形成される層となる。
架橋剤としては、例えば、多官能イソシアネート化合物が挙げられる。多官能イソシアネート化合物としては、ヘキサメチレンジイソシアネート(HDI)、HDI系ポリイソシアネート、イソホロンジイソシアネート(IPDI)等が挙げられる。HDI系ポリイソシアネートには、2液型用としてビウレットタイプ、イソシアヌレートタイプ、アダクトタイプ、2官能型が挙げられ、硬化開始温度に閾値があるブロック型も挙げられる。HDI系ポリイソシアネートは、市販品を使用することができ、デュラネート(旭化成社製)等が挙げられる。
使用する多官能イソシアネート化合物は、反応温度、デバイスの材質によって適宜選択できる。例えばデバイスとしてポリスチレンを使用する場合、アサヒクリンAK225(旭硝子社製)、AC6000(旭硝子社製)等に溶解でき、かつポリスチレンの熱変形温度である80℃以下でも硬化反応が進行するビウレットタイプ、イソシアヌレートタイプが好ましい。
使用する多官能イソシアネート化合物は、反応温度、デバイスの材質によって適宜選択できる。例えばデバイスとしてポリスチレンを使用する場合、アサヒクリンAK225(旭硝子社製)、AC6000(旭硝子社製)等に溶解でき、かつポリスチレンの熱変形温度である80℃以下でも硬化反応が進行するビウレットタイプ、イソシアヌレートタイプが好ましい。
被覆層中の架橋度合いは、重合体中のヒドロキシ基量と添加する架橋剤の量、反応率によって決まり、本発明の効果を損なわない範囲で適宜調節できる。
架橋剤の使用量は、重合体の100質量部に対して、0.01〜10質量部が好ましく、0.1〜1質量部が特に好ましい。架橋剤の使用量が前記範囲の下限値以上であれば、耐久性に優れた被覆層を形成しやすい。架橋剤の使用量が前記範囲の上限値以下であれば、タンパク質が付着しにくい被覆層を形成しやすい。
架橋剤の使用量は、重合体の100質量部に対して、0.01〜10質量部が好ましく、0.1〜1質量部が特に好ましい。架橋剤の使用量が前記範囲の下限値以上であれば、耐久性に優れた被覆層を形成しやすい。架橋剤の使用量が前記範囲の上限値以下であれば、タンパク質が付着しにくい被覆層を形成しやすい。
<乾燥工程>
基材上に塗布した塗布液に由来する溶媒を除去する方法としては、特に限定されず、例えば、風乾、加熱による乾燥等の公知の乾燥方法を用いることができる。
乾燥温度は、30〜200℃が好ましい。
基材上に塗布した塗布液に由来する溶媒を除去する方法としては、特に限定されず、例えば、風乾、加熱による乾燥等の公知の乾燥方法を用いることができる。
乾燥温度は、30〜200℃が好ましい。
以上のように、本発明の重合体と溶媒とを含む塗布液を基材上に湿式塗布することで、重合体から形成されてなる被覆層を容易に形成することができる。なお、本発明の医療用デバイスの製造方法は、前記した方法には限定されず、本発明の重合体が常温(20〜25℃)で液体の場合には、該重合体をそのまま基材上に塗布して被覆層を形成してもよい。この場合には、基材の表面との密着性を向上させるために、重合体を加熱してもよい。本発明の重合体を用いた蒸着により被覆層を形成してもよい。
以上説明した本発明の医療用デバイスは、本発明の重合体から形成されてなる被覆層を表面に有するため、タンパク質が吸着しにくく、また耐水性に優れ被覆成分が溶出しにくく、耐久性にも優れている。
以下、実施例によって本発明を詳細に説明するが、本発明は以下の記載によっては限定されない。
[ガラス転移温度(Tg)]
重合体のガラス転移温度は、DSC(TAインスツメント社製)により、10℃/分の速度で−30℃から200℃まで昇降温させて測定した。降温時の2サイクル目のゴム状態からガラス状態へ変化する温度をガラス転移温度とした。
[ガラス転移温度(Tg)]
重合体のガラス転移温度は、DSC(TAインスツメント社製)により、10℃/分の速度で−30℃から200℃まで昇降温させて測定した。降温時の2サイクル目のゴム状態からガラス状態へ変化する温度をガラス転移温度とした。
[分子量]
重合体の数平均分子量(Mn)、質量平均分子量(Mw)および分子量分布(質量平均分子量(Mw)/数平均分子量(Mn))は、テトラヒドロフラン(THF)を溶媒とするGPC装置(HLC8220、東ソー社製)を用いて測定した。
重合体の数平均分子量(Mn)、質量平均分子量(Mw)および分子量分布(質量平均分子量(Mw)/数平均分子量(Mn))は、テトラヒドロフラン(THF)を溶媒とするGPC装置(HLC8220、東ソー社製)を用いて測定した。
[耐水性]
各例で使用した重合体の10mgと、水の1gとをサンプル管に秤取し、室温で1時間撹拌した後に、目視にて耐水性を確認した。評価は以下の基準で行った。
<評価基準>
○:重合体が分散せずに残存していた。
△:重合体が水中に分散していた。
×:重合体が完全に溶解し、残存していなかった。
各例で使用した重合体の10mgと、水の1gとをサンプル管に秤取し、室温で1時間撹拌した後に、目視にて耐水性を確認した。評価は以下の基準で行った。
<評価基準>
○:重合体が分散せずに残存していた。
△:重合体が水中に分散していた。
×:重合体が完全に溶解し、残存していなかった。
[タンパク質非吸着性]
<タンパク質非吸着性試験>
(1)発色液、およびタンパク質溶液の準備
発色液は、ペルオキシダーゼ発色液(3,3’,5,5’−テトラメチルベンジジン(TMBZ)、KPL社製)50mLとTMB Peroxidase Substrate(KPL社製)50mLとを混合したものを使用した。タンパク質溶液として、タンパク質(POD−goat anti mouse IgG、Biorad社製)を、リン酸緩衝溶液(D−PBS、Sigma社製)で16,000倍に希釈したものを使用した。
(2)タンパク質吸着
各ウェル表面に被覆層を形成した24ウェルマイクロプレートにおける3ウェルに、タンパク質溶液の2mLを分注し(1ウェル毎に2mLを使用)、室温で1時間放置した。ブランクとして、タンパク質溶液を96ウェルマイクロプレートにおける3ウェルに、2μL分注(1ウェル毎に2μLを使用)した。
(3)ウェル洗浄
次いで、24ウェルマイクロプレートを、界面活性剤(Tween20、和光純薬社製)を0.05質量%含ませたリン酸緩衝溶液(D−PBS、Sigma社製)の4mLで4回洗浄した(1ウェル毎に4mLを使用)。
(4)発色液分注
次いで、洗浄を終えた24ウェルマイクロプレートに、発色液の2mLを分注し(1ウェル毎に2mLを使用)、7分間発色反応を行った。2N硫酸の1mLを加えることで(1ウェル毎に1mLを使用)発色反応を停止させた。
ブランクは、96ウェルマイクロプレートに、発色液の100μLを分注し(1ウェル毎に100μLを使用)、7分間発色反応を行い、2N硫酸の50μLを加えることで(1ウェル毎に50μLを使用)発色反応を停止させた。
(5)吸光度測定準備
次いで、24ウェルマイクロプレートの各ウェルから150μLの液を取り、96ウェルマイクロプレートに移した。
(6)吸光度測定およびタンパク質吸着率Q
吸光度は、MTP−810Lab(コロナ電気社製)により、450nmの吸光度を測定した。ここで、ブランクの吸光度(N=3)の平均値をA0とした。24ウェルマイクロプレートの3ウェルから96ウェルマイクロプレートに移動させた液のそれぞれについて吸光度A1を測定した。
各吸光度A1についてタンパク質吸着率Q1を下式によりそれぞれ求め、それらの平均値をタンパク質吸着率Q(N=3)とした。
Q1=A1/{A0×(100/ブランクのタンパク質溶液の分注量)}×100
=A1/{A0×(100/2μL)}×100 [%]
<タンパク質非吸着性試験>
(1)発色液、およびタンパク質溶液の準備
発色液は、ペルオキシダーゼ発色液(3,3’,5,5’−テトラメチルベンジジン(TMBZ)、KPL社製)50mLとTMB Peroxidase Substrate(KPL社製)50mLとを混合したものを使用した。タンパク質溶液として、タンパク質(POD−goat anti mouse IgG、Biorad社製)を、リン酸緩衝溶液(D−PBS、Sigma社製)で16,000倍に希釈したものを使用した。
(2)タンパク質吸着
各ウェル表面に被覆層を形成した24ウェルマイクロプレートにおける3ウェルに、タンパク質溶液の2mLを分注し(1ウェル毎に2mLを使用)、室温で1時間放置した。ブランクとして、タンパク質溶液を96ウェルマイクロプレートにおける3ウェルに、2μL分注(1ウェル毎に2μLを使用)した。
(3)ウェル洗浄
次いで、24ウェルマイクロプレートを、界面活性剤(Tween20、和光純薬社製)を0.05質量%含ませたリン酸緩衝溶液(D−PBS、Sigma社製)の4mLで4回洗浄した(1ウェル毎に4mLを使用)。
(4)発色液分注
次いで、洗浄を終えた24ウェルマイクロプレートに、発色液の2mLを分注し(1ウェル毎に2mLを使用)、7分間発色反応を行った。2N硫酸の1mLを加えることで(1ウェル毎に1mLを使用)発色反応を停止させた。
ブランクは、96ウェルマイクロプレートに、発色液の100μLを分注し(1ウェル毎に100μLを使用)、7分間発色反応を行い、2N硫酸の50μLを加えることで(1ウェル毎に50μLを使用)発色反応を停止させた。
(5)吸光度測定準備
次いで、24ウェルマイクロプレートの各ウェルから150μLの液を取り、96ウェルマイクロプレートに移した。
(6)吸光度測定およびタンパク質吸着率Q
吸光度は、MTP−810Lab(コロナ電気社製)により、450nmの吸光度を測定した。ここで、ブランクの吸光度(N=3)の平均値をA0とした。24ウェルマイクロプレートの3ウェルから96ウェルマイクロプレートに移動させた液のそれぞれについて吸光度A1を測定した。
各吸光度A1についてタンパク質吸着率Q1を下式によりそれぞれ求め、それらの平均値をタンパク質吸着率Q(N=3)とした。
Q1=A1/{A0×(100/ブランクのタンパク質溶液の分注量)}×100
=A1/{A0×(100/2μL)}×100 [%]
[被覆層の耐久性]
(1)マイクロプレートのウェル表面に形成した被覆層の耐久性
ウェル表面に被覆層を形成した24ウェルのマイクロプレートを37℃の水に1週間浸漬させた後、60℃で2時間加熱して乾燥させた。浸漬の前後でタンパク質非吸着性試験を行ってタンパク質吸着率Qを測定し、以下の式によりタンパク質吸着率Qの上昇率を算出した。
タンパク質吸着率Qの上昇率(%)=(37℃の水に1週間浸漬させた後のタンパク質吸着率(%)÷初期のタンパク質吸着率(%)−1)×100
(1)マイクロプレートのウェル表面に形成した被覆層の耐久性
ウェル表面に被覆層を形成した24ウェルのマイクロプレートを37℃の水に1週間浸漬させた後、60℃で2時間加熱して乾燥させた。浸漬の前後でタンパク質非吸着性試験を行ってタンパク質吸着率Qを測定し、以下の式によりタンパク質吸着率Qの上昇率を算出した。
タンパク質吸着率Qの上昇率(%)=(37℃の水に1週間浸漬させた後のタンパク質吸着率(%)÷初期のタンパク質吸着率(%)−1)×100
(2)ガラスシャーレの表面に形成した被覆層の耐久性
表面に被覆層を形成したガラスシャーレに水を6mL入れ、40℃のオーブン内で24時間静置させた。次いで、水を除去した後、該ガラスシャーレをオーブンにより100℃で1時間加熱して乾燥させた。静置前と乾燥後でタンパク質非吸着性試験を行ってタンパク質吸着率Qを測定し、以下の式によりタンパク質吸着率Qの上昇率を算出した。
タンパク質吸着率Qの上昇率(%)=(水を入れて40℃で24時間静置させた後のタンパク質吸着率(%)÷初期のタンパク質吸着率(%)−1)×100
表面に被覆層を形成したガラスシャーレに水を6mL入れ、40℃のオーブン内で24時間静置させた。次いで、水を除去した後、該ガラスシャーレをオーブンにより100℃で1時間加熱して乾燥させた。静置前と乾燥後でタンパク質非吸着性試験を行ってタンパク質吸着率Qを測定し、以下の式によりタンパク質吸着率Qの上昇率を算出した。
タンパク質吸着率Qの上昇率(%)=(水を入れて40℃で24時間静置させた後のタンパク質吸着率(%)÷初期のタンパク質吸着率(%)−1)×100
[合成例1]
以下に示す合成スキームにより化合物Bを合成した。具体的には、300mL3つ口フラスコに、トリエチレングリコールモノメチルエーテルの21.5g(131mmol)、パラトルエンスルホン酸クロリドの25.0g(131mmol)およびジクロロメタンの100mLを投入した。得られた混合物に、トリエチルアミン(TEA)の18.6g(183mmol)およびジクロロメタンの50mLの混合物を0℃、窒素雰囲気下で滴下し、室温で16時間撹拌した。得られた反応混合物を500mL分液ロートに移し、1N塩酸水溶液で1回、飽和食塩水で2回有機相を洗浄した。得られた有機相を硫酸ナトリウムで乾燥させて濃縮した後、酢酸エチル:ヘキサン(体積比)=5:5の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物Bの無色透明液体を得た。化合物Bの収量は37.3g、収率は89.3%であった。
以下に示す合成スキームにより化合物Bを合成した。具体的には、300mL3つ口フラスコに、トリエチレングリコールモノメチルエーテルの21.5g(131mmol)、パラトルエンスルホン酸クロリドの25.0g(131mmol)およびジクロロメタンの100mLを投入した。得られた混合物に、トリエチルアミン(TEA)の18.6g(183mmol)およびジクロロメタンの50mLの混合物を0℃、窒素雰囲気下で滴下し、室温で16時間撹拌した。得られた反応混合物を500mL分液ロートに移し、1N塩酸水溶液で1回、飽和食塩水で2回有機相を洗浄した。得られた有機相を硫酸ナトリウムで乾燥させて濃縮した後、酢酸エチル:ヘキサン(体積比)=5:5の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物Bの無色透明液体を得た。化合物Bの収量は37.3g、収率は89.3%であった。
[合成例2]
以下に示す合成スキームにより化合物Cを合成した。具体的には、1L2つ口フラスコに、マレイミドの29.1g(300mmol)、フランの61.3g(900mmol)、ジブチルヒドロキシトルエンの22mg(0.1mmol)およびトルエンの600mLを投入した。得られた混合物を60℃、窒素雰囲気下で24時間撹拌し、反応液を氷冷した後、析出した結晶をろ過し、冷トルエンで洗浄して、化合物Cの白色粉末を得た。化合物Cの収量は43.5g、収率は87.6%であった。
以下に示す合成スキームにより化合物Cを合成した。具体的には、1L2つ口フラスコに、マレイミドの29.1g(300mmol)、フランの61.3g(900mmol)、ジブチルヒドロキシトルエンの22mg(0.1mmol)およびトルエンの600mLを投入した。得られた混合物を60℃、窒素雰囲気下で24時間撹拌し、反応液を氷冷した後、析出した結晶をろ過し、冷トルエンで洗浄して、化合物Cの白色粉末を得た。化合物Cの収量は43.5g、収率は87.6%であった。
[合成例3]
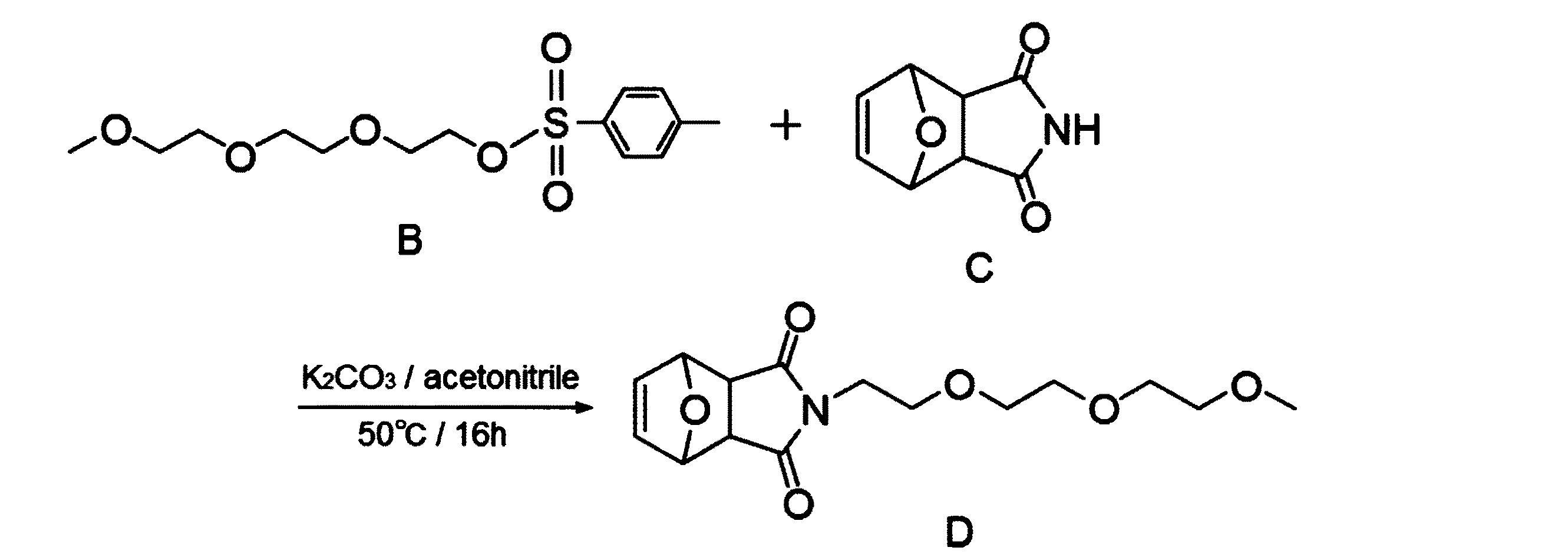
以下に示す合成スキームにより化合物Dを合成した。具体的には、300mL2つ口フラスコに、化合物Bの9.55g(30mmol)、化合物Cの4.95g(30mmol)、炭酸カリウム20.7g(150mmol)およびアセトニトリル200mLを投入した。得られた混合物を50℃、窒素雰囲気下で16時間撹拌し、炭酸カリウムをろ過で除去した。得られた反応混合物を濃縮した後、酢酸エチル:メタノール(体積比)=20:1の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物Dの無色透明液体を得た。化合物Dの収量は3.83g、収率は41.0%であった。
以下に示す合成スキームにより化合物Dを合成した。具体的には、300mL2つ口フラスコに、化合物Bの9.55g(30mmol)、化合物Cの4.95g(30mmol)、炭酸カリウム20.7g(150mmol)およびアセトニトリル200mLを投入した。得られた混合物を50℃、窒素雰囲気下で16時間撹拌し、炭酸カリウムをろ過で除去した。得られた反応混合物を濃縮した後、酢酸エチル:メタノール(体積比)=20:1の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物Dの無色透明液体を得た。化合物Dの収量は3.83g、収率は41.0%であった。
[合成例4]
以下に示す合成スキームにより、側鎖にポリオキシエチレン基を有するマレイミド誘導体である化合物(1−1)を合成した。具体的には、100mLフラスコに、化合物Dの3.5g(11.2mmol)、ジブチルヒドロキシトルエンの2.2mg(0.01mmol)、およびトルエンの50mLを投入し、4時間還流撹拌した。得られた反応混合物を濃縮した後、酢酸エチルを展開溶媒としたシリカゲルカラムクロマトグラフィーによって精製し、化合物(1−1)の無色透明液体を得た。化合物(1−1)の収量は1.87g、収率は68.4%であった。
以下に示す合成スキームにより、側鎖にポリオキシエチレン基を有するマレイミド誘導体である化合物(1−1)を合成した。具体的には、100mLフラスコに、化合物Dの3.5g(11.2mmol)、ジブチルヒドロキシトルエンの2.2mg(0.01mmol)、およびトルエンの50mLを投入し、4時間還流撹拌した。得られた反応混合物を濃縮した後、酢酸エチルを展開溶媒としたシリカゲルカラムクロマトグラフィーによって精製し、化合物(1−1)の無色透明液体を得た。化合物(1−1)の収量は1.87g、収率は68.4%であった。
[合成例5]
以下に示す合成スキームにより化合物Eを合成した。具体的には、1L3つ口フラスコに、カリウム−t−ブトキシド(t−BuOK)の13.5g(120mmol)およびテトラヒドロフラン(THF)の200mLを投入した。得られた混合物にp−アミノフェノールの10.9g(100mmol)およびテトラヒドロフランの300mLの混合物を0℃、窒素雰囲気下で滴下し、60℃で30分撹拌した。得られた反応液に化合物Bの31.8g(100mmol)およびテトラヒドロフラン100mLの混合物を60℃、窒素雰囲気下で滴下し、2時間還流撹拌した。得られた反応混合物を濃縮した後、クロロホルム500mLを加え、1L分液ロートに移し、1N塩酸水溶液で1回、飽和食塩水で2回有機相を洗浄した。得られた有機相を硫酸ナトリウムで乾燥させて濃縮した後、酢酸エチル:ヘキサン(体積比)=7:3の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物Eの無色透明液体を得た。化合物Eの収量は13.8g、収率は54%であった。
以下に示す合成スキームにより化合物Eを合成した。具体的には、1L3つ口フラスコに、カリウム−t−ブトキシド(t−BuOK)の13.5g(120mmol)およびテトラヒドロフラン(THF)の200mLを投入した。得られた混合物にp−アミノフェノールの10.9g(100mmol)およびテトラヒドロフランの300mLの混合物を0℃、窒素雰囲気下で滴下し、60℃で30分撹拌した。得られた反応液に化合物Bの31.8g(100mmol)およびテトラヒドロフラン100mLの混合物を60℃、窒素雰囲気下で滴下し、2時間還流撹拌した。得られた反応混合物を濃縮した後、クロロホルム500mLを加え、1L分液ロートに移し、1N塩酸水溶液で1回、飽和食塩水で2回有機相を洗浄した。得られた有機相を硫酸ナトリウムで乾燥させて濃縮した後、酢酸エチル:ヘキサン(体積比)=7:3の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物Eの無色透明液体を得た。化合物Eの収量は13.8g、収率は54%であった。
[合成例6]
以下に示す合成スキームにより化合物Fを合成した。具体的には、300mL3つ口フラスコに、化合物Eの12.8g(50mmol)およびアセトンの100mLを投入した。得られた混合物に無水マレイン酸の4.9g(50mmol)およびアセトンの50mLの混合物を室温、窒素雰囲気下で滴下し、室温で2時間撹拌した。得られた反応混合物を濃縮した後、トルエン:ヘキサン(体積比)=3:2の混合液で再結晶し、精製して化合物Fの黄色粉末を得た。化合物Fの収量は9.14g、収率は51.8%であった。
以下に示す合成スキームにより化合物Fを合成した。具体的には、300mL3つ口フラスコに、化合物Eの12.8g(50mmol)およびアセトンの100mLを投入した。得られた混合物に無水マレイン酸の4.9g(50mmol)およびアセトンの50mLの混合物を室温、窒素雰囲気下で滴下し、室温で2時間撹拌した。得られた反応混合物を濃縮した後、トルエン:ヘキサン(体積比)=3:2の混合液で再結晶し、精製して化合物Fの黄色粉末を得た。化合物Fの収量は9.14g、収率は51.8%であった。
1H−NMR(300MHz、溶媒:CDCl3、基準:TMS)により化合物Fの同定を行った。化合物FのNMRスペクトルは以下のとおりである。
σ=9.78(1H、COOH)、7.54(2H、Ar−H)、6.79(2H、Ar−H)、6.65(1H、CHCONH)、6.36(1H、CHCOOH)、4.02−3.53(12H、OCH2CH2O)、3.32(3H、OCH3)。
σ=9.78(1H、COOH)、7.54(2H、Ar−H)、6.79(2H、Ar−H)、6.65(1H、CHCONH)、6.36(1H、CHCOOH)、4.02−3.53(12H、OCH2CH2O)、3.32(3H、OCH3)。
[合成例7]
以下に示す合成スキームにより、側鎖にポリオキシエチレン基を有するマレイミド誘導体である化合物(1−2)を合成した。具体的には、300mLフラスコに、化合物Fの8.84g(25mmol)、ジブチルヒドロキシトルエンの8.8mg(0.04mmol)、濃硫酸の112mg(1.1mmol)、ジメチルホルムアミド(DMF)の0.4mLおよびトルエンの180mLを投入した。得られた混合物を3時間還流撹拌し、トルエンと共沸した水を適時抜き出した。得られた反応混合物を1L分液ロートに移し、クロロホルムの300mLを加え、飽和食塩水で3回有機相を洗浄した。得られた有機相を硫酸ナトリウムで乾燥させて濃縮した後、酢酸エチル:ヘキサン(体積比)=9:1の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物(1−2)の黄色粉末を得た。化合物(1−2)の収量は6.14g、収率は73.2%であった。
以下に示す合成スキームにより、側鎖にポリオキシエチレン基を有するマレイミド誘導体である化合物(1−2)を合成した。具体的には、300mLフラスコに、化合物Fの8.84g(25mmol)、ジブチルヒドロキシトルエンの8.8mg(0.04mmol)、濃硫酸の112mg(1.1mmol)、ジメチルホルムアミド(DMF)の0.4mLおよびトルエンの180mLを投入した。得られた混合物を3時間還流撹拌し、トルエンと共沸した水を適時抜き出した。得られた反応混合物を1L分液ロートに移し、クロロホルムの300mLを加え、飽和食塩水で3回有機相を洗浄した。得られた有機相を硫酸ナトリウムで乾燥させて濃縮した後、酢酸エチル:ヘキサン(体積比)=9:1の展開溶媒を用いたシリカゲルカラムクロマトグラフィーによって精製し、化合物(1−2)の黄色粉末を得た。化合物(1−2)の収量は6.14g、収率は73.2%であった。
1H−NMR(300MHz、溶媒:CDCl3、基準:TMS)により化合物(1−2)の同定を行った。化合物(1−2)のNMRスペクトルは以下のとおりである。
σ=7.22(2H、Ar−H)、6.99(2H、Ar−H)、6.84(2H、O=CCHCHC=O)、4.17−3.55(12H、OCH2CH2O)、3.38(3H、OCH3)。
σ=7.22(2H、Ar−H)、6.99(2H、Ar−H)、6.84(2H、O=CCHCHC=O)、4.17−3.55(12H、OCH2CH2O)、3.38(3H、OCH3)。
[実施例1]
100mLフラスコに、単量体として化合物(1−1)の0.70g(2.88mmol)、重合開始剤としてアゾビスイソブチロニトリル(AIBN)の7mg(0.043mmol)、および重合溶媒としてジクロロエタンの2.8gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−1)を得た。重合体(A−1)の収量は0.60g、収率は85.7%であった。
得られた重合体(A−1)をその濃度が0.05質量%となるようにエタノールに溶解させ、塗布液を調製した。ポリスチレン製の24ウェルのマイクロプレートに該塗布液を2.2mL分注し、3日間放置して溶媒を揮発させ、ウェル表面に被覆層を形成した。また、ガラス製シャーレに該塗布液を2.2mL注入し、3日間放置して溶媒を揮発させ、シャーレ表面に被覆層を形成した。
100mLフラスコに、単量体として化合物(1−1)の0.70g(2.88mmol)、重合開始剤としてアゾビスイソブチロニトリル(AIBN)の7mg(0.043mmol)、および重合溶媒としてジクロロエタンの2.8gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−1)を得た。重合体(A−1)の収量は0.60g、収率は85.7%であった。
得られた重合体(A−1)をその濃度が0.05質量%となるようにエタノールに溶解させ、塗布液を調製した。ポリスチレン製の24ウェルのマイクロプレートに該塗布液を2.2mL分注し、3日間放置して溶媒を揮発させ、ウェル表面に被覆層を形成した。また、ガラス製シャーレに該塗布液を2.2mL注入し、3日間放置して溶媒を揮発させ、シャーレ表面に被覆層を形成した。
[実施例2]
単量体として化合物(1−1)の2.19g(9.0mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の0.25g(1.0mmol)とを300mLの3つ口フラスコに秤取し、さらに重合開始剤としてAIBNの0.244gと、重合溶媒としてトルエンの13.4gとを加えた。化合物(1−1)と化合物(2−1)の仕込みモル比は化合物(1−1)/化合物(2−1)=90/10とした。反応液中の単量体の合計濃度を20質量%、開始剤濃度を1質量%とした。フラスコ内を充分にアルゴン置換した後に密封し、16時間75℃に加温することにより重合反応を行った。反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−2)を得た。重合体(A−2)の収量は2.05g、収率は84.2%であった。
重合体(A−1)の代わりに重合体(A−2)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
単量体として化合物(1−1)の2.19g(9.0mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の0.25g(1.0mmol)とを300mLの3つ口フラスコに秤取し、さらに重合開始剤としてAIBNの0.244gと、重合溶媒としてトルエンの13.4gとを加えた。化合物(1−1)と化合物(2−1)の仕込みモル比は化合物(1−1)/化合物(2−1)=90/10とした。反応液中の単量体の合計濃度を20質量%、開始剤濃度を1質量%とした。フラスコ内を充分にアルゴン置換した後に密封し、16時間75℃に加温することにより重合反応を行った。反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−2)を得た。重合体(A−2)の収量は2.05g、収率は84.2%であった。
重合体(A−1)の代わりに重合体(A−2)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
[実施例3]
100mL2口フラスコに、単量体として化合物(1−2)の2.0g(6.0mmol)、重合開始剤としてAIBNの20mg(0.12mmol)、および重合溶媒としてジクロロエタン8.0gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−3)を得た。重合体(A−3)の収量は1.64g、収率は82.0%であった。
重合体(A−1)の代わりに重合体(A−3)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
100mL2口フラスコに、単量体として化合物(1−2)の2.0g(6.0mmol)、重合開始剤としてAIBNの20mg(0.12mmol)、および重合溶媒としてジクロロエタン8.0gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−3)を得た。重合体(A−3)の収量は1.64g、収率は82.0%であった。
重合体(A−1)の代わりに重合体(A−3)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
[実施例4]
単量体として化合物(1−2)の1.59g(4.75mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の62mg(0.25mmol)とを100mL2口フラスコに秤取し、さらに重合開始剤としてAIBNの16.5mg(0.10mmol)と、重合溶媒としてジクロロエタンの6.6gとを加えた。化合物(1−2)と化合物(2−1)の仕込みモル比は化合物(1−2)/化合物(2−1)=90/10とした。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−4)を得た。重合体(A−4)の収量は0.93g、収率は58.5%であった。
重合体(A−1)の代わりに重合体(A−4)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
単量体として化合物(1−2)の1.59g(4.75mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の62mg(0.25mmol)とを100mL2口フラスコに秤取し、さらに重合開始剤としてAIBNの16.5mg(0.10mmol)と、重合溶媒としてジクロロエタンの6.6gとを加えた。化合物(1−2)と化合物(2−1)の仕込みモル比は化合物(1−2)/化合物(2−1)=90/10とした。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、反応液を氷冷した後、ヘキサンに滴下することにより重合体を沈殿させた。得られた重合体を充分にヘキサンで洗浄した後、減圧乾燥して白色粉末状の重合体(A−4)を得た。重合体(A−4)の収量は0.93g、収率は58.5%であった。
重合体(A−1)の代わりに重合体(A−4)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
[比較例1]
100mLフラスコに、単量体として2−(2−(2−エトキシエトキシ)エトキシ)エチルアクリレート(PEGA)の0.70g(2.88mmol)、重合開始剤としてAIBNの7mg(0.043mmol)、および重合溶媒としてジクロロエタンの2.8gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、減圧乾燥して、重合体(G−1)の粘調性液体を得た。重合体(G−1)の収量は0.69g、収率は98.7%であった。
重合体(A−1)の代わりに重合体(G−1)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
100mLフラスコに、単量体として2−(2−(2−エトキシエトキシ)エトキシ)エチルアクリレート(PEGA)の0.70g(2.88mmol)、重合開始剤としてAIBNの7mg(0.043mmol)、および重合溶媒としてジクロロエタンの2.8gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、減圧乾燥して、重合体(G−1)の粘調性液体を得た。重合体(G−1)の収量は0.69g、収率は98.7%であった。
重合体(A−1)の代わりに重合体(G−1)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
[比較例2]
単量体としてPEGAの2.09g(9.0mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の0.25g(1.0mmol)とを300mLの3つ口フラスコに秤取し、さらに重合開始剤としてAIBNの0.234gと、重合溶媒としてトルエンの12.9gとを加えた。PEGAと化合物(2−1)の仕込みモル比をPEGA/化合物(2−1)=90/10とした。また、反応液中の単量体の合計濃度を20質量%、開始剤濃度を1質量%とした。フラスコ内を充分にアルゴン置換した後に密封し、16時間75℃に加温することにより重合反応を行った。反応液を氷冷した後、減圧乾燥して、重合体(G−2)の粘調性液体を得た。重合体(G−2)の収量は2.30g、収率は98.5%であった。
重合体(A−1)の代わりに重合体(G−2)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
単量体としてPEGAの2.09g(9.0mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の0.25g(1.0mmol)とを300mLの3つ口フラスコに秤取し、さらに重合開始剤としてAIBNの0.234gと、重合溶媒としてトルエンの12.9gとを加えた。PEGAと化合物(2−1)の仕込みモル比をPEGA/化合物(2−1)=90/10とした。また、反応液中の単量体の合計濃度を20質量%、開始剤濃度を1質量%とした。フラスコ内を充分にアルゴン置換した後に密封し、16時間75℃に加温することにより重合反応を行った。反応液を氷冷した後、減圧乾燥して、重合体(G−2)の粘調性液体を得た。重合体(G−2)の収量は2.30g、収率は98.5%であった。
重合体(A−1)の代わりに重合体(G−2)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
[比較例3]
100mLフラスコに、単量体として2−(2−(2−エトキシエトキシ)エトキシ)エチルメタアクリレート(PEGMA)の0.70g(2.88mmol)、重合開始剤としてAIBNの7mg(0.043mmol)、および重合溶媒としてエタノール2.8gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、減圧乾燥して、重合体(G−3)の粘調性液体を得た。重合体(G−3)の収量は0.69g、収率は99.1%であった。
重合体(A−1)の代わりに重合体(G−3)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
100mLフラスコに、単量体として2−(2−(2−エトキシエトキシ)エトキシ)エチルメタアクリレート(PEGMA)の0.70g(2.88mmol)、重合開始剤としてAIBNの7mg(0.043mmol)、および重合溶媒としてエタノール2.8gを投入した。反応液中の単量体の濃度を20質量%、開始剤濃度を1質量%とした。得られた混合物を75℃、窒素雰囲気下で16時間撹拌し、減圧乾燥して、重合体(G−3)の粘調性液体を得た。重合体(G−3)の収量は0.69g、収率は99.1%であった。
重合体(A−1)の代わりに重合体(G−3)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
[比較例4]
単量体としてPEGMAの2.19g(9.0mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の0.25g(1.0mmol)とを300mLの3つ口フラスコに秤取し、さらに重合開始剤としてAIBNの0.244gと、重合溶媒としてトルエンの13.4gを加えた。PEGMAと化合物(2−1)の仕込みモル比をPEGMA/化合物(2−1)=90/10とした。また、反応液中の単量体の合計濃度を20質量%、開始剤濃度を1質量%とした。フラスコ内を充分にアルゴン置換した後に密封し、16時間75℃に加温することにより重合反応を行った。反応液を氷冷した後、減圧乾燥して、重合体(G−4)の粘調性液体を得た。重合体(G−4)の収量は2.42g、収率は99.0%であった。
重合体(A−1)の代わりに重合体(G−4)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
単量体としてPEGMAの2.19g(9.0mmol)と、化合物(2−1)(トリメトキシシリルプロピルメタクリレート、商品名「KBM−503」、信越シリコーン社製)の0.25g(1.0mmol)とを300mLの3つ口フラスコに秤取し、さらに重合開始剤としてAIBNの0.244gと、重合溶媒としてトルエンの13.4gを加えた。PEGMAと化合物(2−1)の仕込みモル比をPEGMA/化合物(2−1)=90/10とした。また、反応液中の単量体の合計濃度を20質量%、開始剤濃度を1質量%とした。フラスコ内を充分にアルゴン置換した後に密封し、16時間75℃に加温することにより重合反応を行った。反応液を氷冷した後、減圧乾燥して、重合体(G−4)の粘調性液体を得た。重合体(G−4)の収量は2.42g、収率は99.0%であった。
重合体(A−1)の代わりに重合体(G−4)を用いた以外は、実施例1と同様にしてウェル表面およびシャーレ表面にそれぞれ被覆層を形成した。
実施例および比較例における重合体のMn、Mw、Mw/Mn、Tgの測定結果、および評価結果を表1に示す。耐水性における「×〜△」は、評価が「×」と「△」の間であること、すなわち重合体の一部が溶解して残部が分散していることを意味する。
表1に示すように、本発明の重合体を用いて形成した実施例1〜4の被覆層は、耐水性に優れ、またタンパク質が付着しにくく、耐久性にも優れていた。一方、単位(a1)を有さず、ガラス転移温度が20℃未満の重合体を用いて形成した比較例1〜4の被覆層は、耐水性および耐久性が実施例1〜4の被覆層に比べて劣っていた。
1 医療用デバイス、2 基材、3 被覆層、4 底面部、5 側面部。
Claims (6)
- 側鎖にポリオキシエチレン基を有するマレイミド誘導体に由来する単位を有する、ガラス転移温度が20℃以上の重合体。
- 前記マレイミド誘導体が、下式(1)で表される化合物である、請求項1に記載の重合体。
- 下式(2)で表される化合物に由来する単位をさらに有する、請求項1または2に記載の重合体。
- 請求項1〜3のいずれか一項に記載の重合体からなるタンパク質付着防止剤。
- 表面の少なくとも一部に請求項1〜3のいずれか一項に記載の重合体を含む被覆層を有する医療用デバイス。
- 細胞培養容器である、請求項5に記載の医療用デバイス。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016078146A JP2017186482A (ja) | 2016-04-08 | 2016-04-08 | 重合体、タンパク質付着防止剤および医療用デバイス |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016078146A JP2017186482A (ja) | 2016-04-08 | 2016-04-08 | 重合体、タンパク質付着防止剤および医療用デバイス |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017186482A true JP2017186482A (ja) | 2017-10-12 |
Family
ID=60046152
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016078146A Pending JP2017186482A (ja) | 2016-04-08 | 2016-04-08 | 重合体、タンパク質付着防止剤および医療用デバイス |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017186482A (ja) |
-
2016
- 2016-04-08 JP JP2016078146A patent/JP2017186482A/ja active Pending
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6680208B2 (ja) | タンパク質付着防止剤 | |
| JP6759958B2 (ja) | 温度応答性基材、その製造方法及びその評価方法 | |
| JP6617704B2 (ja) | タンパク質付着防止剤 | |
| JPWO2017204306A1 (ja) | タンパク質付着防止剤、硬化物、硬化物の製造方法、および物品 | |
| TW201634500A (zh) | 具有抑制生物物質附著能力之離子複合材料及其製造方法 | |
| CN109563463A (zh) | 具有薄膜台阶覆盖性的涂覆膜、具备该膜的结构基体 | |
| CN109196089A (zh) | 细胞培养容器 | |
| EP3719113A1 (en) | Cell culture container capable of long-term culture, and method for manufacturing same | |
| US20180296988A1 (en) | Cell-trapping filter | |
| JP2016026520A (ja) | タンパク質付着防止用化合物、塗布液および医療用デバイス | |
| JP2017186291A (ja) | N−フェニルマレイミド誘導体およびその製造方法、n−フェニルマレイミド誘導体の前駆体、重合体、タンパク質付着防止剤、ならびに医療用デバイス | |
| US12291701B2 (en) | Cell culture container having minute volume | |
| JP2017186482A (ja) | 重合体、タンパク質付着防止剤および医療用デバイス | |
| WO2020066685A1 (ja) | タンパク質付着抑制用共重合体、共重合体の製造方法、樹脂改質剤、成形材料、共重合体含有組成物、塗膜および物品 | |
| US11396568B2 (en) | Curable composition, film, cured product, and medical member | |
| JP7401876B2 (ja) | 含フッ素重合体、膜及び医療用具 | |
| US20220135806A1 (en) | Method for producing polymer compatible with biomaterials | |
| Shaik et al. | Interpenetrating photopolymers for intraocular lens application | |
| JP2025136486A (ja) | 共重合体、医療用コーティング剤及び医療機器 | |
| JP2018053093A (ja) | 吸着制御表面を有する高分子基材及びその製造方法 | |
| JP2021165321A (ja) | 含フッ素重合体、膜及び医療用具 | |
| WO2020122193A1 (ja) | 医療用具、医療用具の製造方法、および塗布液 |