JP2017222071A - ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイス - Google Patents
ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイス Download PDFInfo
- Publication number
- JP2017222071A JP2017222071A JP2016118473A JP2016118473A JP2017222071A JP 2017222071 A JP2017222071 A JP 2017222071A JP 2016118473 A JP2016118473 A JP 2016118473A JP 2016118473 A JP2016118473 A JP 2016118473A JP 2017222071 A JP2017222071 A JP 2017222071A
- Authority
- JP
- Japan
- Prior art keywords
- layer
- gas barrier
- group
- compound
- film
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Images



Landscapes
- Laminated Bodies (AREA)
- Electroluminescent Light Sources (AREA)
Abstract
【課題】本発明の課題は、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管したときの、ガスバリアー層と他構成層との境界面で発生する微細発泡を抑制したガスバリアーフィルムを提供することである。【解決手段】本発明のガスバリアーフィルムは、樹脂基材上に下地層(Y)と、ガスバリアー層(X)とを有し、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)の関係、及び前記金属原子(M)のモル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)との関係が、特定の関係式を満たすことを特徴とする。【選択図】図1
Description
本発明は、ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイスに関する。より詳しくは、本発明は、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管したときの、ガスバリアー層と他構成層との境界面で発生する微細発泡を抑制したガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイスに関する。
近年、ガスバリアーフィルムは、フレキシブル性を有する電子デバイス、例えば、太陽電池素子、有機エレクトロルミネッセンス(以下、ELと略記する。)素子、液晶表示素子等のフレキシブル電子デバイスへの展開が要望され、様々な分野での検討がなされている。しかし、これらフレキシブル電子デバイスにおいては、ガラス基材レベルの非常に高度のガスバリアー性が要求されている。
このようなガスバリアーフィルムにおいて、基材上にガスバリアー層を形成する方法としては、例えば、ヘキサメチルジシロキサン(以下、HMDSOと略記する。)に代表される有機ケイ素化合物を用いて、減圧下、酸素プラズマで酸化しながら基板上にガスバリアー層を成膜する化学気相成長法(CVD法:Chemical Vapor Deposition、化学蒸着法ともいう。)や、半導体レーザーを用いて金属Siを蒸発させ酸素の存在下で基板上に堆積させてガスバリアー層を形成する物理気相成長法(PVD法:Physical Vapor Deposition、例えば、真空蒸着法やスパッタ法)といった気相成長法が知られている。
樹脂基材上に、有機ケイ素化合物を用いて前記化学気相成長法、真空蒸着法やスパッタ法でガスバリアー層を形成したガスバリアーフィルムは確かに高度なガスバリアー性を実現できるが、有機EL素子の基材として用いた場合に、湾曲させたまま高温高湿などの環境に置くと、ガスバリアー層−素子境界やガスバリアー層−下層境界に微細発泡が現れることがあった。当該微細発泡が発生すると、ガスバリアー性が低下するため、例えば、有機EL素子の場合はダークスポット等の発光欠陥となりやすい。最近では、湾曲した画面を有する有機ELディスプレイも出現し、湾曲状態で高温高湿下に保管される場合もあるため、上記ダークスポットの発生は問題である。
本発明者の検討によれば、これは湾曲という物理的ストレスによる前記ガスバリアー層境界での微細クラックの発生や、ガスバリアー層自体が有するピンホールによって、基材側からしみ出す水分が、ガスバリアー層界面の当該箇所において微細発泡となるものと推察された。
特許文献1には、従来のガスバリアー層に比較して水蒸気バリアー性がより高いガスバリアーフィルムが、アルミニウム等の金属原子、酸素原子、及び硫黄原子又はリン原子を含む特定の組成比を有する金属化合物により構成されるガスバリアー層によって実現できると記載されているが、本発明者の検討によれば、上記ガスバリアーフィルムを具備した電子デバイスを、湾曲させたまま高温高湿などの環境に保管した場合に、微細クラックの発生やピンホールによる前記微細発泡の抑制については不十分であった。
また、特許文献2には、金属酸化物とリン化合物とイオン価が1以上3以下である陽イオンを特定のモル比で含むコーティング液を調製し、基材上に塗布して前駆体を形成後熱処理することで、高いガスバリアー性を有する多層構造体を形成する技術が開示されているが、上記電子デバイスを湾曲させたまま高温高湿などの環境に保管したときの、ガスバリアー層−素子境界やガスバリアー層−下層境界に現れる微細発泡についてはなんら記載されていない。
したがって、ガスバリアーフィルムを具備する電子デバイスを湾曲させたまま高温高湿などの環境に保管したときの、ガスバリアー層−素子境界やガスバリアー層−下層境界に発生する微細発泡を抑制したガスバリアーフィルムが要望されている。
本発明は、上記問題・状況に鑑みてなされたものであり、その解決課題は、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管したときの、ガスバリアー層と他構成層との境界面で発生する微細発泡を抑制したガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイスを提供することである。
本発明者は、上記課題を解決すべく、上記問題の原因等について検討する過程において、樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)を備えるガスバリアーフィルムであって、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを、特定のモル比で含有することによって、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管した場合の、ガスバリアー層(X)と他構成層との境界で発生する微細発泡を抑制できることを見出した。
すなわち、本発明に係る上記課題は、以下の手段により解決される。
1.樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)とを備えるガスバリアーフィルムであって、
前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、
前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、
前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とするガスバリアーフィルム。
前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、
前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、
前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とするガスバリアーフィルム。
関係式(1) 0.8≦NM/NP≦4.5
関係式(2) 0.001≦FZ×NZ/NM≦0.60
2.前記ガスバリアー層(X)が、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚が15〜50nmの範囲内であることを特徴とする第1項に記載のガスバリアーフィルム。
関係式(2) 0.001≦FZ×NZ/NM≦0.60
2.前記ガスバリアー層(X)が、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚が15〜50nmの範囲内であることを特徴とする第1項に記載のガスバリアーフィルム。
関係式(3) 1.6≦x≦1.9、0.15≦y≦0.40
関係式(4) 4.0≦2x+2y<4.3
3.樹脂基材上に少なくとも下地層(Y)と、これに接してガスバリアー層(X)とを形成するガスバリアーフィルムの製造方法であって、
前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、
前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、
前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすように調整することを特徴とするガスバリアーフィルムの製造方法。
関係式(4) 4.0≦2x+2y<4.3
3.樹脂基材上に少なくとも下地層(Y)と、これに接してガスバリアー層(X)とを形成するガスバリアーフィルムの製造方法であって、
前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、
前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、
前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすように調整することを特徴とするガスバリアーフィルムの製造方法。
関係式(1) 0.8≦NM/NP≦4.5
関係式(2) 0.001≦FZ×NZ/NM≦0.60
4.前記ガスバリアー層(X)を、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚を15〜50nmの範囲内に調整することを特徴とする第3項に記載のガスバリアーフィルムの製造方法。
関係式(2) 0.001≦FZ×NZ/NM≦0.60
4.前記ガスバリアー層(X)を、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚を15〜50nmの範囲内に調整することを特徴とする第3項に記載のガスバリアーフィルムの製造方法。
関係式(3) 1.6≦x≦1.9、0.15≦y≦0.40
関係式(4) 4.0≦2x+2y<4.3
5.前記ガスバリアー層(X)を、対向ローラー型のロールtoロール成膜装置を用いて、真空プラズマCVD法によって形成することを特徴とする第3項又は第4項に記載のガスバリアーフィルムの製造方法。
関係式(4) 4.0≦2x+2y<4.3
5.前記ガスバリアー層(X)を、対向ローラー型のロールtoロール成膜装置を用いて、真空プラズマCVD法によって形成することを特徴とする第3項又は第4項に記載のガスバリアーフィルムの製造方法。
6.第1項又は第2項に記載のガスバリアーフィルムを具備することを特徴とする有機エレクトロルミネッセンスデバイス。
本発明の上記手段により、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管した場合の、ガスバリアー層と他構成層との境界で発生する微細発泡を抑制したガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイスを提供することができる。
本発明の効果の発現機構ないし作用機構については、明確にはなっていないが、以下のように推察している。
有機ケイ素化合物を用いて化学気相成長法、真空蒸着法やスパッタ法でガスバリアー層を形成したガスバリアーフィルムを、有機EL素子等の電子デバイスの基材として用いた場合に、電子デバイスを湾曲させたまま高温高湿などの環境に置くと、ガスバリアー層−素子境界やガスバリアー層−下地層境界に微細発泡が発生することがあり、当該微細発泡によってガスバリアー性が低下し、有機EL素子にダークスポット等の欠陥が発生しやすい。
本発明者の検討によれば、これは電子デバイスを湾曲させた状態で高温高湿下においたときの物理的ストレスによって、前記ガスバリアー層境界で微細クラックが発生したり、ガスバリアー層自体が有するピンホールによって、基材側から微量の水分がガスバリアー層境界の当該箇所にしみ出すことで、ガスバリアー層−素子境界やガスバリアー層−下層境界において微細発泡が発生するものと推察された。
本発明者は、前記微細発泡の発生を抑制する手段を検討する中で、前記ガスバリアー層を化学気相成長法による薄膜なガスバリアー層(X)を形成し、さらに当該ガスバリアー層(X)と基材の間に、リン酸アルミニウム等の特定の金属化合物を含有する下地層を設けることによって、前記下地層の金属化合物が微量水分を吸着することによって当該微量水分のガスバリアー層への移動を遮断し、前記ガスバリアー層境界における微細発泡を抑制できることを突き止めた。
本発明の有機ケイ素化合物等を用いて形成する薄膜なガスバリアー層(X)は、柔軟性に富み緻密なガスバリアー層を形成できることから、デバイスを湾曲させた場合のクラックの発生や、ガスバリアー層自体のピンホール抑制には有利であると考えられる。しかしながら、もともと薄膜であるがため、ガスバリアー層(X)にピンホールなどの欠陥ができてしまった場合には、基材側からの水分の影響を受けやすいという欠点があった。
本発明は、前記薄膜なガスバリアー層(X)に接して、前記特定の金属化合物を含有する下地層をガスバリアー層(X)と基材の間に設けることにより、微量水分を下地層内でトラップできることから、当該薄膜なガスバリアー層(X)の柔軟性を活かすことができ、クラックの発生やピンホールを抑制し、電子デバイスを湾曲した状態で高温高湿下に保管した場合の、ガスバリアー層と他構成層との境界で発生する微細発泡を抑制できたものと推察している。
本発明のガスバリアーフィルムは、樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)とを備えるガスバリアーフィルムであって、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、前記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、前記関係式(2)で表される関係を満たすことを特徴とする。この特徴は、各請求項に係る発明に共通する又は対応する技術的特徴である。
本発明の実施態様としては、本発明の効果発現の観点から、前記ガスバリアー層(X)が、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、前記関係式(3)で表される関係及び前記関係式(4)で表される関係を満たし、かつ層厚が15〜50nmの範囲内であることが、ガスバリアー層として柔軟性に優れ、薄膜で高度なガスバリアー性を実現する観点から、好ましい。
本発明のガスバリアーフィルムの製造方法は、樹脂基材上に少なくとも下地層(Y)と、これに接してガスバリアー層(X)とを形成するガスバリアーフィルムの製造方法であって、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、前記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、前記関係式(2)で表される関係を満たすように調整することを特徴とする。
また、前記ガスバリアー層(X)を、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、前記関係式(3)で表される関係及び前記関係式(4)で表される関係を満たし、かつ層厚を15〜50nmの範囲内に調整することが、透明性、ガスバリアー性及び柔軟性(フレキシビリティ)をバランスしたガスバリアー層を形成する観点から、好ましい。
さらに、前記ガスバリアー層(X)を、対向ローラー型のロールtoロール成膜装置を用いて、真空プラズマCVD法によって形成することが、緻密でピンホールの発生を抑制したガスバリアー層を生産性よく製造でき、好ましい。
本発明のガスバリアーフィルムは、有機エレクトロルミネッセンスデバイスに具備されることが好ましく、当該デバイスを湾曲した状態で高温高湿下に保管した場合でも、前記微量水分によるダークスポット等の欠陥を防ぐことが可能である。
以下、本発明とその構成要素、及び本発明を実施するための形態・態様について詳細な説明をする。なお、本願において、「〜」は、その前後に記載される数値を下限値及び上限値として含む意味で使用する。
≪本発明のガスバリアーフィルムの概要≫
本発明のガスバリアーフィルムは、樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)とを備えるガスバリアーフィルムであって、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とし、かかる構成によって、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管した場合の、ガスバリアー層と他構成層との境界で発生する微細発泡を抑制したガスバリアーフィルムを提供する。
本発明のガスバリアーフィルムは、樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)とを備えるガスバリアーフィルムであって、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とし、かかる構成によって、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管した場合の、ガスバリアー層と他構成層との境界で発生する微細発泡を抑制したガスバリアーフィルムを提供する。
関係式(1) 0.8≦NM/NP≦4.5
関係式(2) 0.001≦FZ×NZ/NM≦0.60
本発明のガスバリアーフィルムは、基材上に前記ガスバリアー層(X)を形成させた積層体としてガスバリアー性を算出した際、JIS K 7129−1992に準拠した方法で測定された25±0.5℃、90±2%RHの環境下の水蒸気透過度が、0.01g/m2・24h以下のガスバリアーフィルムであることが好ましく、更には、JIS K 7126−2006に準拠した方法で測定された、85℃・85%RH環境下での酸素透過度が、1×10−3mL/m2・24h・atm以下、水蒸気透過度が、1×10−3g/m2・24h以下の高ガスバリアー性であるガスバリアーフィルムであることが好ましい。
関係式(2) 0.001≦FZ×NZ/NM≦0.60
本発明のガスバリアーフィルムは、基材上に前記ガスバリアー層(X)を形成させた積層体としてガスバリアー性を算出した際、JIS K 7129−1992に準拠した方法で測定された25±0.5℃、90±2%RHの環境下の水蒸気透過度が、0.01g/m2・24h以下のガスバリアーフィルムであることが好ましく、更には、JIS K 7126−2006に準拠した方法で測定された、85℃・85%RH環境下での酸素透過度が、1×10−3mL/m2・24h・atm以下、水蒸気透過度が、1×10−3g/m2・24h以下の高ガスバリアー性であるガスバリアーフィルムであることが好ましい。
また、前記ガスバリアー層(X)の平均組成を「SiOxCy」と表すとは、前記ガスバリアー層(X)を構成する化合物、又は当該化合物の平均組成をいう。
〈XPSによる組成分析〉
本発明に係るガスバリアー層(X)中に含有されるSiOxCyにおける組成x及びyは、X線光電子分光法(XPS:Xray Photoelectron Spectroscopy)によって、層厚方向における元素濃度分布を測定し平均することによって求めることができる。
本発明に係るガスバリアー層(X)中に含有されるSiOxCyにおける組成x及びyは、X線光電子分光法(XPS:Xray Photoelectron Spectroscopy)によって、層厚方向における元素濃度分布を測定し平均することによって求めることができる。
本発明におけるXPS分析は下記の条件で行ったものであるが、装置や測定条件が変わっても本発明の主旨に即した測定方法であれば問題なく適用できるものである。
本発明の主旨に即した測定方法とは、主に層厚方向の解像度であり、測定点1点あたりのエッチング深さ(下記のスパッタイオンとデプスプロファイルの条件に相当)は3nm以下であることが好ましく、2nm以下であることがより好ましく、1nm以下であることがさらに好ましい。
以下に、本発明に適用可能なXPS分析の具体的な条件の一例を示す。
・分析装置:アルバック・ファイ社製QUANTERA SXM
・X線源:単色化Al−Kα
・スパッタイオン:Ar(2keV)
・デプスプロファイル:SiO2換算スパッタ厚さで、所定の厚さ間隔で測定を繰り返し、深さ方向のデプスプロファイルを求める。この厚さ間隔は、1nmとした(深さ方向に1nmごとのデータが得られる。)。
・X線源:単色化Al−Kα
・スパッタイオン:Ar(2keV)
・デプスプロファイル:SiO2換算スパッタ厚さで、所定の厚さ間隔で測定を繰り返し、深さ方向のデプスプロファイルを求める。この厚さ間隔は、1nmとした(深さ方向に1nmごとのデータが得られる。)。
・定量:バックグラウンドをShirley法で求め、得られたピーク面積から相対感度係数法を用いて定量した。データ処理は、アルバック・ファイ社製のMultiPakを用いる。
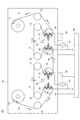
図1は、本発明のガスバリアーフィルムの構成例を示す断面図である。
図1は、樹脂基材1上に下地層(Y)2を形成し、その上にガスバリアー層(X)3を積層している本発明のガスバリアーフィルムFの最小構成を示している。
図1では、他の機能層は示していないが、帯電防止層、バックコート層、ブリードアウト防止層、ハードコート層等を適宜積層してもよい。さらに、樹脂基材1の両側に本発明に係る下地層2及びガスバリアー層3を形成した構成でもよい。
本発明では、本発明のガスバリアーフィルムFのガスバリアー層(X)3上に直接、又は平滑層等の他の機能層を介して、後述する有機EL素子を形成して、有機ELデバイスとすることが好ましい。
〔1〕樹脂基材
樹脂基材の材質は特に制限されず、様々な材質からなる基材を用いることができる。樹脂基材の材質としては、例えば、熱可塑性樹脂、熱硬化性樹脂等の樹脂等が挙げられる。これらの中でも、熱可塑性樹脂及び繊維集合体が好ましく、熱可塑性樹脂がより好ましい。樹脂基材の形態は、特に制限されず、フィルム又はシート等の層状であってもよい。樹脂基材としては、単層であってもよいし、複層であってもよい。
樹脂基材の材質は特に制限されず、様々な材質からなる基材を用いることができる。樹脂基材の材質としては、例えば、熱可塑性樹脂、熱硬化性樹脂等の樹脂等が挙げられる。これらの中でも、熱可塑性樹脂及び繊維集合体が好ましく、熱可塑性樹脂がより好ましい。樹脂基材の形態は、特に制限されず、フィルム又はシート等の層状であってもよい。樹脂基材としては、単層であってもよいし、複層であってもよい。
樹脂基材に用いられる熱可塑性樹脂としては、例えば、ポリエチレン、ポリプロピレン等のポリオレフィン系樹脂;ポリエチレンテレフタレート(PET)、ポリエチレン−2,6−ナフタレート、ポリブチレンテレフタレートやこれらの共重合体等のポリエステル系樹脂;ナイロン−6、ナイロン−66、ナイロン−12等のポリアミド系樹脂;ポリビニルアルコール、エチレン−ビニルアルコール共重合体等のヒドロキシ基含有ポリマー;ポリスチレン;ポリ(メタ)アクリル酸エステル;ポリアクリロニトリル;ポリ酢酸ビニル;ポリカーボネート;ポリアリレート;再生セルロース;セルロースエステル;ポリイミド;ポリエーテルイミド;ポリスルフォン;ポリエーテルスルフォン;ポリエーテルエーテルケトン;アイオノマー樹脂等が挙げられる。樹脂基材の材料としては、ポリエチレン、ポリプロピレン、ポリエチレンテレフタレート、ナイロン−6、及びナイロン−66からなる群より選ばれる少なくとも1種の熱可塑性樹脂が好ましい。
前記熱可塑性樹脂からなるフィルムを樹脂基材として用いる場合、樹脂基材は延伸フィルムであってもよいし無延伸フィルムであってもよい。得られるガスバリアーフィルムの加工適性(例えば、印刷やラミネートに対する適性)が優れることから、延伸フィルム、特に二軸延伸フィルムが好ましい。二軸延伸フィルムは、同時二軸延伸法、逐次二軸延伸法、及びチューブラ延伸法のいずれかの方法で製造された二軸延伸フィルムであってもよい。
前記熱可塑性樹脂からなるフィルムを樹脂基材として用いる場合、樹脂基材は延伸フィルムであってもよいし無延伸フィルムであってもよい。得られるガスバリアーフィルムの加工適性(例えば、印刷やラミネートに対する適性)が優れることから、延伸フィルム、特に二軸延伸フィルムが好ましい。二軸延伸フィルムは、同時二軸延伸法、逐次二軸延伸法、及びチューブラ延伸法のいずれかの方法で製造された二軸延伸フィルムであってもよい。
樹脂基材の厚さは5〜500μm程度が好ましく、更に好ましくは15〜250μmである。
その他、基材の種類、基材の製造方法等については、特開2013−226758号公報の段落「0125」〜「0136」に開示されている技術を適宜採用することができる。
〔2〕下地層(Y)
本発明に係る下地層(Y)は、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とする。
本発明に係る下地層(Y)は、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とする。
関係式(1) 0.8≦NM/NP≦4.5
関係式(2) 0.001≦FZ×NZ/NM≦0.60
〔2.1〕金属酸化物(A)
金属酸化物(A)を構成する金属原子(M)は、原子価が2価以上であることが好ましい。金属原子(M)としては、例えば、マグネシウム、カルシウム等の周期表第2族の金属原子;チタン、ジルコニウム等の周期表第4族の金属原子;亜鉛等の周期表第12族の金属原子;ホウ素、アルミニウム等の周期表第13族の金属原子;ケイ素等の周期表第14族の金属原子等を挙げることができる。なお、ホウ素及びケイ素は半金属原子に分類される場合があるが、本明細書ではこれらを金属原子に含めるものとする。金属原子(M)は1種類であってもよいし、2種類以上であってもよい。これらの中でも、金属酸化物(A)の生産性や得られるガスバリアーフィルムのガスバリアー性や水蒸気バリアー性がより優れることから、金属原子(M)は、アルミニウム、チタン、及びジルコニウムからなる群より選ばれる少なくとも1種であることが好ましく、アルミニウムであることがより好ましい。すなわち、金属原子(M)はアルミニウムを含むことが好ましい。
金属原子(M)に占める、アルミニウム、チタン及びジルコニウムの合計の割合は、通常60モル%以上であり、100モル%であってもよい。また、金属原子(M)に占める、アルミニウムの割合は、通常50モル%以上であり、100モル%であってもよい。金属酸化物(A)は、液相合成法、気相合成法、固体粉砕法等の方法によって製造される。
関係式(2) 0.001≦FZ×NZ/NM≦0.60
〔2.1〕金属酸化物(A)
金属酸化物(A)を構成する金属原子(M)は、原子価が2価以上であることが好ましい。金属原子(M)としては、例えば、マグネシウム、カルシウム等の周期表第2族の金属原子;チタン、ジルコニウム等の周期表第4族の金属原子;亜鉛等の周期表第12族の金属原子;ホウ素、アルミニウム等の周期表第13族の金属原子;ケイ素等の周期表第14族の金属原子等を挙げることができる。なお、ホウ素及びケイ素は半金属原子に分類される場合があるが、本明細書ではこれらを金属原子に含めるものとする。金属原子(M)は1種類であってもよいし、2種類以上であってもよい。これらの中でも、金属酸化物(A)の生産性や得られるガスバリアーフィルムのガスバリアー性や水蒸気バリアー性がより優れることから、金属原子(M)は、アルミニウム、チタン、及びジルコニウムからなる群より選ばれる少なくとも1種であることが好ましく、アルミニウムであることがより好ましい。すなわち、金属原子(M)はアルミニウムを含むことが好ましい。
金属原子(M)に占める、アルミニウム、チタン及びジルコニウムの合計の割合は、通常60モル%以上であり、100モル%であってもよい。また、金属原子(M)に占める、アルミニウムの割合は、通常50モル%以上であり、100モル%であってもよい。金属酸化物(A)は、液相合成法、気相合成法、固体粉砕法等の方法によって製造される。
金属酸化物(A)は、加水分解可能な特性基が結合した金属原子(M)を有する化合物(L)の加水分解縮合物であってもよい。該特性基の例には、後述する一般式[I]のR1が含まれる。化合物(L)の加水分解縮合物は、実質的に金属酸化物(A)とみなすことが可能である。そのため、本明細書において、「金属酸化物(A)」は「化合物(L)の加水分解縮合物」と読み替えることが可能であり、また、「化合物(L)の加水分解縮合物」を「金属酸化物(A)」と読み替えることも可能である。
<加水分解可能な特性基が結合した金属原子(M)を含有する化合物(L)>
リン化合物(B)との反応の制御が容易になり、得られるガスバリアーフィルムのガスバリアー性が優れることから、化合物(L)は、下記一般式[I]で表される化合物(L1)を少なくとも1種含むことが好ましい。
<加水分解可能な特性基が結合した金属原子(M)を含有する化合物(L)>
リン化合物(B)との反応の制御が容易になり、得られるガスバリアーフィルムのガスバリアー性が優れることから、化合物(L)は、下記一般式[I]で表される化合物(L1)を少なくとも1種含むことが好ましい。
一般式[I] M1(R1)m(R2)n―m
式中、M1は、アルミニウム、チタン、及びジルコニウムからなる群より選ばれる。R1は、ハロゲン原子(フッ素原子、塩素原子、臭素原子、ヨウ素原子)、NO3、置換基を有していてもよい炭素数1〜9のアルコキシ基、置換基を有していてもよい炭素数1〜9のアシロキシ基、置換基を有していてもよい炭素数3〜9のアルケニルオキシ基、置換基を有していてもよい炭素数5〜15のβ−ジケトナト基、又は置換基を有していてもよい炭素数1〜9のアシル基を有するジアシルメチル基である。R2は、置換基を有していてもよい炭素数1〜9のアルキル基、置換基を有していてもよい炭素数7〜10のアラルキル基、置換基を有していてもよい炭素数2〜9のアルケニル基、又は置換基を有していてもよい炭素数6〜10のアリール基である。mは1〜nの整数である。nはM1の原子価に等しい。R1が複数存在する場合、R1は互いに同一であってもよいし異なっていてもよい。R2が複数存在する場合、R2は互いに同一であってもよいし異なっていてもよい。
式中、M1は、アルミニウム、チタン、及びジルコニウムからなる群より選ばれる。R1は、ハロゲン原子(フッ素原子、塩素原子、臭素原子、ヨウ素原子)、NO3、置換基を有していてもよい炭素数1〜9のアルコキシ基、置換基を有していてもよい炭素数1〜9のアシロキシ基、置換基を有していてもよい炭素数3〜9のアルケニルオキシ基、置換基を有していてもよい炭素数5〜15のβ−ジケトナト基、又は置換基を有していてもよい炭素数1〜9のアシル基を有するジアシルメチル基である。R2は、置換基を有していてもよい炭素数1〜9のアルキル基、置換基を有していてもよい炭素数7〜10のアラルキル基、置換基を有していてもよい炭素数2〜9のアルケニル基、又は置換基を有していてもよい炭素数6〜10のアリール基である。mは1〜nの整数である。nはM1の原子価に等しい。R1が複数存在する場合、R1は互いに同一であってもよいし異なっていてもよい。R2が複数存在する場合、R2は互いに同一であってもよいし異なっていてもよい。
R1のアルコキシ基としては、例えば、メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、ベンジロキシ基、ジフェニルメトキシ基、トリチルオキシ基、4−メトキシベンジロキシ基、メトキシメトキシ基、1−エトキシエトキシ基、ベンジルオキシメトキシ基、2−トリメチルシリルエトキシ基、2−トリメチルシリルエトキシメトキシ基、フェノキシ基、4−メトキシフェノキシ基等が挙げられる。
R1のアシロキシ基としては、例えば、アセトキシ基、エチルカルボニルオキシ基、n−プロピルカルボニルオキシ基、イソプロピルカルボニルオキシ基、n−ブチルカルボニルオキシ、イソブチルカルボニルオキシ基、sec−ブチルカルボニルオキシ基、tert−ブチルカルボニルオキシ基、n−オクチルカルボニルオキシ基等が挙げられる。
R1のアルケニルオキシ基としては、例えば、アリルオキシ基、2−プロペニルオキシ基、2−ブテニルオキシ基、1−メチル−2−プロペニルオキシ基、3−ブテニルオキシ基、2−メチル−2−プロペニルオキシ基、2−ペンテニルオキシ基、3−ペンテニルオキシ基、4−ペンテニルオキシ基、1−メチル−3−ブテニルオキシ基、1,2−ジメチル−2−プロペニルオキシ基、1,1−ジメチル−2−プロペニルオキシ基、2−メチル−2−ブテニルオキシ基、3−メチル−2−ブテニルオキシ基、2−メチル−3−ブテニルオキシ基、3−メチル−3−ブテニルオキシ基、1−ビニル−2−プロペニルオキシ基、5−ヘキセニルオキシ基等が挙げられる。
R1のβ−ジケトナト基としては、例えば、2,4−ペンタンジオナト基、1,1,1−トリフルオロ−2,4−ペンタンジオナト基、1,1,1,5,5,5−ヘキサフルオロ−2,4−ペンタンジオナト基、2,2,6,6−テトラメチル−3,5−ヘプタンジオナト基、1,3−ブタンジオナト基、2−メチル−1,3−ブタンジオナト基、2−メチル−1,3−ブタンジオナト基、ベンゾイルアセトナト基等が挙げられる。
R1のジアシルメチル基のアシル基としては、例えば、ホルミル基、アセチル基、プロピオニル基(プロパノイル基)、ブチリル基(ブタノイル基)、バレリル基(ペンタノイル基)、ヘキサノイル基等の炭素数1〜6の脂肪族アシル基;ベンゾイル基、トルオイル基等の芳香族アシル基(アロイル基)等が挙げられる。
R2のアルキル基としては、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、sec−ブチル基、tert−ブチル基、n−ペンチル基、イソペンチル基、n−ヘキシル基、イソヘキシル基、3−メチルペンチル基、2−メチルペンチル基、1,2−ジメチルブチル基、シクロプロピル基、シクロペンチル基、シクロヘキシル基等が挙げられる。
R2のアラルキル基としては、例えば、ベンジル基、フェニルエチル基(フェネチル基)等が挙げられる。
R2のアルケニル基としては、例えば、ビニル基、1−プロペニル基、2−プロペニル基、イソプロペニル基、3−ブテニル基、2−ブテニル基、1−ブテニル基、1−メチル−2−プロペニル基、1−メチル−1−プロペニル基、1−エチル−1−エテニル基、2−メチル−2−プロペニル基、2−メチル−1−プロペニル基、3−メチル−2−ブテニル基、4−ペンテニル基等が挙げられる。
R2のアリール基としては、例えば、フェニル基、1−ナフチル基、2−ナフチル基等が挙げられる。
R1及びR2における置換基としては、例えば、炭素数1〜6のアルキル基;メトキシ基、エトキシ基、n−プロポキシ基、イソプロポキシ基、n−ブトキシ基、イソブトキシ基、sec−ブトキシ基、tert−ブトキシ基、n−ペンチルオキシ基、イソペンチルオキシ基、n−ヘキシルオキシ基、シクロプロピルオキシ基、シクロブチルオキシ基、シクロペンチルオキシ基、シクロヘキシルオキシ基等の炭素数1〜6のアルコキシ基;メトキシカルボニル基、エトキシカルボニル基、n−プロポキシカルボニル基、イソプロポキシカルボニル基、n−ブトキシカルボニル基、イソブトキシカルボニル基、sec−ブトキシカルボニル基、tert−ブトキシカルボニル基、n−ペンチルオキシカルボニル基、イソペンチルオキシカルボニル基、シクロプロピルオキシカルボニル基、シクロブチルオキシカルボニル基、シクロペンチルオキシカルボニル基等の炭素数1〜6のアルコキシカルボニル基;フェニル基、トリル基、ナフチル基等の芳香族炭化水素基;フッ素原子、塩素原子、臭素原子、ヨウ素原子等のハロゲン原子;炭素数1〜6のアシル基;炭素数7〜10のアラルキル基;炭素数7〜10のアラルキルオキシ基;炭素数1〜6のアルキルアミノ基;炭素数1〜6のアルキル基を有するジアルキルアミノ基が挙げられる。
R1としては、ハロゲン原子、NO3、置換基を有していてもよい炭素数1〜6のアルコキシ基、置換基を有していてもよい炭素数1〜6のアシロキシ基、置換基を有していてもよい炭素数5〜10のβ−ジケトナト基、又は置換基を有していてもよい炭素数1〜6のアシル基を有するジアシルメチル基が好ましい。
R2としては、置換基を有していてもよい炭素数1〜6のアルキル基が好ましい。M1としては、アルミニウムが好ましい。M1がアルミニウムの場合、mは、好ましくは3である。
化合物(L1)の具体例としては、例えば、硝酸アルミニウム、酢酸アルミニウム、トリス(2,4−ペンタンジオナト)アルミニウム、トリメトキシアルミニウム、トリエトキシアルミニウム、トリ−n−プロポキシアルミニウム、トリイソプロポキシアルミニウム、トリ−n−ブトキシアルミニウム、トリ−sec−ブトキシアルミニウム、トリ−tert−ブトキシアルミニウム等のアルミニウム化合物;テトラキス(2,4−ペンタンジオナト)チタン、テトラメトキシチタン、テトラエトキシチタン、テトライソプロポキシチタン、テトラ−n−ブトキシチタン、テトラキス(2−エチルヘキソキシ)チタン等のチタン化合物;テトラキス(2,4−ペンタンジオナト)ジルコニウム、テトラ−n−プロポキシジルコニウム、テトラ−n−ブトキシジルコニウム等のジルコニウム化合物が挙げられる。これらの中でも、化合物(L1)としては、トリイソプロポキシアルミニウム及びトリ−sec−ブトキシアルミニウムから選ばれる少なくとも一つの化合物が好ましい。化合物(L)は1種類を単独で使用してもよいし、2種類以上を併用してもよい。
化合物(L)において、本発明の効果が得られる限り、化合物(L)に占める化合物(L1)の割合に特に限定はない。化合物(L1)以外の化合物が化合物(L)に占める割合は、例えば、20モル%以下が好ましく、10モル%以下がより好ましく、5モル%以下がさらに好ましく、0モル%であってもよい。
化合物(L)が加水分解されることによって、化合物(L)が有する加水分解可能な特性基の少なくとも一部がヒドロキシ基に変換される。さらに、その加水分解物が縮合することによって、金属原子(M)が酸素原子(O)を介して結合された化合物が形成される。この縮合が繰り返されると、実質的に金属酸化物とみなしうる化合物が形成される。なお、このようにして形成された金属酸化物(A)の表面には、通常、ヒドロキシ基が存在する。
本明細書においては、[金属原子(M)のみに結合している酸素原子(O)のモル数]/[金属原子(M)のモル数]の比が0.8以上となる化合物を金属酸化物(A)に含めるものとする。ここで、金属原子(M)のみに結合している酸素原子(O)は、M−O−Mで表される構造における酸素原子(O)であり、M−O−Hで表される構造における酸素原子(O)のように金属原子(M)と水素原子(H)に結合している酸素原子は除外される。金属酸化物(A)における前記比は、0.9以上が好ましく、1.0以上がより好ましく、1.1以上がさらに好ましい。この比の上限は特に限定されないが、金属原子(M)の原子価をnとすると、通常、n/2で表される。
前記加水分解縮合が起こるためには、化合物(L)が加水分解可能な特性基を有していることが重要である。それらの基が結合していない場合、加水分解縮合反応が起こらない、若しくは極めて緩慢になるため、目的とする金属酸化物(A)の調製が困難になる。
化合物(L)の加水分解縮合物は、例えば、公知のゾル−ゲル法で採用される手法によって特定の原料から製造してもよい。前記原料には、化合物(L)、化合物(L)の部分加水分解物、化合物(L)の完全加水分解物、化合物(L)の部分加水分解縮合物、及び化合物(L)の完全加水分解物の一部が縮合したものからなる群より選ばれる少なくとも1種を用いることができる。
〔2.2〕リン化合物(B)
リン化合物(B)は、金属酸化物(A)と反応可能な部位を有し、典型的には、そのような部位を複数有する。リン化合物(B)としては、無機リン化合物が好ましい。リン化合物(B)としては、金属酸化物(A)と反応可能な部位(原子団又は官能基)を2〜20個有する化合物が好ましい。そのような部位には、金属酸化物(A)の表面に存在する官能基(例えば、ヒドロキシ基)と縮合反応可能な部位が含まれる。そのような部位としては、例えば、リン原子に直接結合したハロゲン原子、リン原子に直接結合した酸素原子等が挙げられる。金属酸化物(A)の表面に存在する官能基(例えば、ヒドロキシ基)は、通常、金属酸化物(A)を構成する金属原子(M)に結合している。
リン化合物(B)は、金属酸化物(A)と反応可能な部位を有し、典型的には、そのような部位を複数有する。リン化合物(B)としては、無機リン化合物が好ましい。リン化合物(B)としては、金属酸化物(A)と反応可能な部位(原子団又は官能基)を2〜20個有する化合物が好ましい。そのような部位には、金属酸化物(A)の表面に存在する官能基(例えば、ヒドロキシ基)と縮合反応可能な部位が含まれる。そのような部位としては、例えば、リン原子に直接結合したハロゲン原子、リン原子に直接結合した酸素原子等が挙げられる。金属酸化物(A)の表面に存在する官能基(例えば、ヒドロキシ基)は、通常、金属酸化物(A)を構成する金属原子(M)に結合している。
リン化合物(B)としては、例えば、リン酸、4分子以上のリン酸が縮合したポリリン酸、亜リン酸、ホスホン酸、亜ホスホン酸、ホスフィン酸、亜ホスフィン酸等のリンのオキソ酸、及びこれらの塩(例えば、リン酸ナトリウム)、並びにこれらの誘導体(例えば、ハロゲン化物(例えば、塩化ホスホリル)、脱水物(例えば、五酸化二リン))等が挙げられる。
リン化合物(B)は、1種を単独で使用してもよいし、2種以上を併用してもよい。これらのリン化合物(B)の中でも、リン酸を単独で使用するか、リン酸とそれ以外のリン化合物(B)とを併用することが好ましい。リン酸を用いることによって、後述する第1コーティング液(U)の安定性と得られる下地層のガスバリアー性及び水蒸気バリアー性が向上する。
<金属酸化物(A)とリン化合物(B)との比率>
本発明に係る下地層(Y)において、前記NMと前記NPとが、0.8≦NM/NP≦4.5の関係を満たすものであり、1.0≦NM/NP≦3.6の関係を満たすものが好ましく、1.1≦NM/NP≦3.0の関係を満たすものがより好ましい。NM/NPの値が4.5を超えると、金属酸化物(A)がリン化合物(B)に対して過剰となり、金属酸化物(A)とリン化合物(B)との結合が不充分となり、また、金属酸化物(A)の表面に存在するヒドロキシ基の量が多くなるため、ガスバリアー性とその安定性が低下する傾向がある。一方、NM/NPの値が0.8未満であると、リン化合物(B)が金属酸化物(A)に対して過剰となり、金属酸化物(A)との結合に関与しない余剰なリン化合物(B)が多くなり、また、リン化合物(B)由来のヒドロキシ基の量が多くなりやすく、やはりガスバリアー性とその安定性が低下する傾向がある。
本発明に係る下地層(Y)において、前記NMと前記NPとが、0.8≦NM/NP≦4.5の関係を満たすものであり、1.0≦NM/NP≦3.6の関係を満たすものが好ましく、1.1≦NM/NP≦3.0の関係を満たすものがより好ましい。NM/NPの値が4.5を超えると、金属酸化物(A)がリン化合物(B)に対して過剰となり、金属酸化物(A)とリン化合物(B)との結合が不充分となり、また、金属酸化物(A)の表面に存在するヒドロキシ基の量が多くなるため、ガスバリアー性とその安定性が低下する傾向がある。一方、NM/NPの値が0.8未満であると、リン化合物(B)が金属酸化物(A)に対して過剰となり、金属酸化物(A)との結合に関与しない余剰なリン化合物(B)が多くなり、また、リン化合物(B)由来のヒドロキシ基の量が多くなりやすく、やはりガスバリアー性とその安定性が低下する傾向がある。
なお、前記比は、下地層(Y)を形成するための第1コーティング液(U)における、金属酸化物(A)の量とリン化合物(B)の量との比によって調整できる。下地層(Y)におけるモル数(NM)とモル数(NP)との比は、通常、第1コーティング液(U)における比であって金属酸化物(A)を構成する金属原子(M)のモル数とリン化合物(B)を構成するリン原子のモル数との比と同じである。
<反応生成物(D)>
反応生成物(D)は、金属酸化物(A)とリン化合物(B)との反応で得られる。ここで、金属酸化物(A)とリン化合物(B)とさらに他の化合物とが反応することで生成する化合物も反応生成物(D)に含まれる。反応生成物(D)は、反応に関与していない金属酸化物(A)及び/又はリン化合物(B)を部分的に含んでいてもよい。
反応生成物(D)は、金属酸化物(A)とリン化合物(B)との反応で得られる。ここで、金属酸化物(A)とリン化合物(B)とさらに他の化合物とが反応することで生成する化合物も反応生成物(D)に含まれる。反応生成物(D)は、反応に関与していない金属酸化物(A)及び/又はリン化合物(B)を部分的に含んでいてもよい。
〔2.3〕陽イオン(Z)
陽イオン(Z)のイオン価(FZ)は、1〜3の範囲である。陽イオン(Z)は周期表第2〜7周期の元素を含む陽イオンである。陽イオン(Z)としては、例えば、リチウムイオン、ナトリウムイオン、カリウムイオン、マグネシウムイオン、カルシウムイオン、チタンイオン、ジルコニウムイオン、ランタノイドイオン(例えば、ランタンイオン)、バナジウムイオン、マンガンイオン、鉄イオン、コバルトイオン、ニッケルイオン、銅イオン、亜鉛イオン、ホウ素イオン、アルミニウムイオン、及びアンモニウムイオン等が挙げられ、中でも、リチウムイオン、ナトリウムイオン、カリウムイオン、マグネシウムイオン、カルシウムイオン、亜鉛イオンが好ましい。陽イオン(Z)は1種類であってもよいし、2種類以上を含んでいてもよい。陽イオン(Z)の働きについては、陽イオン(Z)は、金属酸化物(A)やリン化合物(B)のヒドロキシ基との相互作用によって、無機化合物粒子の肥大化を抑制し、より小さな粒子の充填によってガスバリアー層の緻密性が向上する結果、電子デバイスの性能低下を抑制しているものと推察される。そのため、より高い性能低下抑制機能が必要となる場合は、イオン結合を形成できるイオン価(FZ)が小さい陽イオンを用いることが好ましい。
陽イオン(Z)のイオン価(FZ)は、1〜3の範囲である。陽イオン(Z)は周期表第2〜7周期の元素を含む陽イオンである。陽イオン(Z)としては、例えば、リチウムイオン、ナトリウムイオン、カリウムイオン、マグネシウムイオン、カルシウムイオン、チタンイオン、ジルコニウムイオン、ランタノイドイオン(例えば、ランタンイオン)、バナジウムイオン、マンガンイオン、鉄イオン、コバルトイオン、ニッケルイオン、銅イオン、亜鉛イオン、ホウ素イオン、アルミニウムイオン、及びアンモニウムイオン等が挙げられ、中でも、リチウムイオン、ナトリウムイオン、カリウムイオン、マグネシウムイオン、カルシウムイオン、亜鉛イオンが好ましい。陽イオン(Z)は1種類であってもよいし、2種類以上を含んでいてもよい。陽イオン(Z)の働きについては、陽イオン(Z)は、金属酸化物(A)やリン化合物(B)のヒドロキシ基との相互作用によって、無機化合物粒子の肥大化を抑制し、より小さな粒子の充填によってガスバリアー層の緻密性が向上する結果、電子デバイスの性能低下を抑制しているものと推察される。そのため、より高い性能低下抑制機能が必要となる場合は、イオン結合を形成できるイオン価(FZ)が小さい陽イオンを用いることが好ましい。
なお、陽イオン(Z)が、イオン価が異なる複数種の陽イオンを含む場合、前記FZ×NZの値は、陽イオンごとに計算した値を合計することによって得られる。例えば、陽イオン(Z)が1モルのナトリウムイオン(Na+)と2モルのカルシウムイオン(Ca2+)とを含む場合、FZ×NZ=1×1+2×2=5となる。
陽イオン(Z)は、溶媒に溶解した際に陽イオン(Z)を生じるイオン性化合物(E)を第1コーティング液(U)に溶解させることで下地層(Y)に添加することができる。陽イオン(Z)のカウンターイオンとしては、例えば、水酸化物イオン、塩化物イオン、硫酸イオン、硫酸水素イオン、硝酸イオン、炭酸イオン、炭酸水素イオン等の無機陰イオン;酢酸イオン、ステアリン酸イオン、シュウ酸イオン、酒石酸イオン等の有機酸陰イオン等が挙げられる。陽イオン(Z)のイオン性化合物(E)は、溶解することによって陽イオン(Z)を生じる金属化合物(Ea)又は金属酸化物(Eb)(金属酸化物(A)を除く)であってもよい。
<金属酸化物(A)と陽イオン(Z)との比率>
本発明に係る(Y)において、前記FZとNZとNMとが、0.001≦FZ×NZ/NM≦0.60の関係を満たすものであり、0.001≦FZ×NZ/NM≦0.30の関係を満たすものが好ましく、0.01≦FZ×NZ/NM≦0.30の関係を満たすものがより好ましい。
本発明に係る(Y)において、前記FZとNZとNMとが、0.001≦FZ×NZ/NM≦0.60の関係を満たすものであり、0.001≦FZ×NZ/NM≦0.30の関係を満たすものが好ましく、0.01≦FZ×NZ/NM≦0.30の関係を満たすものがより好ましい。
<リン化合物(B)と陽イオン(Z)との比率>
本発明に係る下地層(Y)において、前記FZとNZとNPが、0.0008≦FZ×NZ/NP≦1.35の関係を満たすものが好ましく、0.001≦FZ×NZ/NP≦1.00の関係を満たすものがより好ましく、0.0012≦FZ×NZ/NP≦0.35の関係を満たすものがさらに好ましく、0.012≦FZ×NZ/NP≦0.29の関係を満たすものが特に好ましい。
本発明に係る下地層(Y)において、前記FZとNZとNPが、0.0008≦FZ×NZ/NP≦1.35の関係を満たすものが好ましく、0.001≦FZ×NZ/NP≦1.00の関係を満たすものがより好ましく、0.0012≦FZ×NZ/NP≦0.35の関係を満たすものがさらに好ましく、0.012≦FZ×NZ/NP≦0.29の関係を満たすものが特に好ましい。
〔2.4〕重合体(C)
下地層(Y)は、特定の重合体(C)をさらに含んでもよい。重合体(C)は、例えば、カルボニル基、ヒドロキシ基、カルボキシ基、カルボン酸無水物基、及びカルボキシ基の塩からなる群より選ばれる少なくとも1種の官能基を含有する重合体である。
下地層(Y)は、特定の重合体(C)をさらに含んでもよい。重合体(C)は、例えば、カルボニル基、ヒドロキシ基、カルボキシ基、カルボン酸無水物基、及びカルボキシ基の塩からなる群より選ばれる少なくとも1種の官能基を含有する重合体である。
ヒドロキシ基を有する重合体(C)の具体例としては、ポリケトン;ポリビニルアルコール、炭素数4以下のα−オレフィン単位を1〜50モル%含有する変性ポリビニルアルコール、ポリビニルアセタール(例えば、ポリビニルブチラール)等のポリビニルアルコール系重合体;セルロース、デンプン、シクロデキストリン等の多糖類;ポリ(メタ)アクリル酸ヒドロキシエチル、ポリ(メタ)アクリル酸、エチレン−アクリル酸共重合体等の(メタ)アクリル酸系重合体;エチレン−無水マレイン酸共重合体の加水分解物、スチレン−無水マレイン酸共重合体の加水分解物、イソブチレン−無水マレイン酸交互共重合体の加水分解物等のマレイン酸系重合体等が挙げられる。これらの中でも、ポリビニルアルコール系重合体が好ましく、具体的には、ポリビニルアルコール及び炭素数4以下のα−オレフィン単位を1〜15モル%含有する変性ポリビニルアルコールが好ましい。
ポリビニルアルコール系重合体のケン化度としては、特に限定されないが、75.0〜99.85モル%が好ましく、80.0〜99.5モル%がより好ましい。ポリビニルアルコール系重合体の粘度平均重合度は、100〜4000が好ましく、300〜3000がより好ましい。また、ポリビニルアルコール系重合体の20℃での4質量%水溶液の粘度は1.0〜200mPa・sが好ましく、11〜90mPa・sがより好ましい。前記ケン化度、粘度平均重合度及び4質量%水溶液の粘度は、JIS K 6726(1994年)に従って求めた値である。
重合体(C)は、重合性基を有する単量体(例えば、酢酸ビニル、アクリル酸)の単独重合体であってもよいし、2種以上の単量体の共重合体であってもよいし、カルボニル基、ヒドロキシ基及び/又はカルボキシ基を有する単量体と該基を有さない単量体との共重合体であってもよい。
重合体(C)の分子量に特に制限はない。より優れたガスバリアー性及び物理特性(例えば、耐傷性)を有する下地層を得るために、重合体(C)の数平均分子量は、5000以上であることが好ましく、8000以上であることがより好ましく、10000以上であることがさらに好ましい。重合体(C)の数平均分子量の上限は特に限定されず、例えば、1500000以下である。
ガスバリアー性をより向上させるために、下地層(Y)における重合体(C)の含有量は、下地層(Y)の質量を基準(100質量%)として、50質量%以下であることが好ましく、40質量%以下であることがより好ましく、30質量%以下であることがさらに好ましく、20質量%以下であってもよい。重合体(C)は、下地層(Y)中の他の成分と反応していてもよいし、反応していなくてもよい。
〔2.5〕下地層(Y)中の他の成分
下地層(Y)は、金属酸化物(A)、化合物(L)、リン化合物(B)、反応生成物(D)、陽イオン(Z)又はその化合物(E)、酸(加水分解縮合に使用する酸触媒、解膠時の酸等)、及び重合体(C)に加え、他の成分を含んでいてもよい。他の成分としては、例えば、陽イオン(Z)を含まない炭酸塩、塩酸塩、硝酸塩、炭酸水素塩、硫酸塩、硫酸水素塩、ホウ酸塩等の無機酸金属塩;陽イオン(Z)を含まない酢酸塩、ステアリン酸塩、シュウ酸塩、酒石酸塩等の有機酸金属塩;層状粘土化合物;架橋剤;重合体(C)以外の高分子化合物;可塑剤;酸化防止剤;紫外線吸収剤;難燃剤等が挙げられる。
下地層(Y)は、金属酸化物(A)、化合物(L)、リン化合物(B)、反応生成物(D)、陽イオン(Z)又はその化合物(E)、酸(加水分解縮合に使用する酸触媒、解膠時の酸等)、及び重合体(C)に加え、他の成分を含んでいてもよい。他の成分としては、例えば、陽イオン(Z)を含まない炭酸塩、塩酸塩、硝酸塩、炭酸水素塩、硫酸塩、硫酸水素塩、ホウ酸塩等の無機酸金属塩;陽イオン(Z)を含まない酢酸塩、ステアリン酸塩、シュウ酸塩、酒石酸塩等の有機酸金属塩;層状粘土化合物;架橋剤;重合体(C)以外の高分子化合物;可塑剤;酸化防止剤;紫外線吸収剤;難燃剤等が挙げられる。
下地層(Y)における前記の他の成分の含有量は、下地層(Y)の質量に対して50質量%以下であることが好ましく、20質量%以下であることがより好ましく、10質量%以下であることがさらに好ましく、5質量%以下であることが特に好ましく、0質量%(他の成分を含まない)であってもよい。
〔2.6〕下地層(Y)の物性
下地層(Y)の厚さ(下地層が2層以上の下地層(Y)を有する場合には各下地層(Y)の厚さの合計)は、0.05〜4.0μmであることが好ましく、0.1〜2.0μmであることがより好ましい。下地層(Y)を薄くすることによって、柔軟性が増すため、その力学的特性を基材自体の力学的特性に近づけることもできる。本発明に係る下地層が2層以上の層(Y)を有する場合、ガスバリアー性の観点から、下地層(Y)1層当たりの厚さは0.05μm以上であることが好ましい。下地層(Y)の厚さは、下地層(Y)の形成に用いられる後述する第1コーティング液(U)の濃度や、その塗工方法によって制御することができる。
下地層(Y)の厚さ(下地層が2層以上の下地層(Y)を有する場合には各下地層(Y)の厚さの合計)は、0.05〜4.0μmであることが好ましく、0.1〜2.0μmであることがより好ましい。下地層(Y)を薄くすることによって、柔軟性が増すため、その力学的特性を基材自体の力学的特性に近づけることもできる。本発明に係る下地層が2層以上の層(Y)を有する場合、ガスバリアー性の観点から、下地層(Y)1層当たりの厚さは0.05μm以上であることが好ましい。下地層(Y)の厚さは、下地層(Y)の形成に用いられる後述する第1コーティング液(U)の濃度や、その塗工方法によって制御することができる。
下地層(Y)の赤外線吸収スペクトルにおいて、800〜1400cm−1の領域における最大吸収波数は1,080〜1130cm−1の範囲にあることが好ましい。金属酸化物(A)とリン化合物(B)とが反応して反応生成物(D)となる過程において、金属酸化物(A)に由来する金属原子(M)とリン化合物(B)に由来するリン原子(P)とが酸素原子(O)を介して結合したM−O−Pで表される結合を形成する。その結果、赤外線吸収スペクトルにおいて該結合由来の特性吸収帯が生じる。本発明者らによる検討の結果、M−O−Pの結合に基づく吸収帯が1080〜1130cm−1の領域に見られる場合には、得られたガスバリアーフィルムが優れたガスバリアー性を発現することがわかった。特に、該特性吸収帯が、一般に各種の原子と酸素原子との結合に由来する吸収が見られる800〜1400cm−1の領域において最も強い吸収である場合には、得られたガスバリアーフィルムがさらに優れたガスバリアー性を発現することがわかった。
これに対し、金属アルコキシドや金属塩等の金属化合物とリン化合物(B)とをあらかじめ混合した後に加水分解縮合させた場合には、金属化合物に由来する金属原子とリン化合物(B)に由来するリン原子とがほぼ均一に混ざり合い反応した複合体が得られる。その場合、赤外線吸収スペクトルにおいて、800〜1400cm−1の領域における最大吸収波数が1080〜1130cm−1の範囲から外れるようになる。
下地層(Y)の赤外線吸収スペクトルにおいて、800〜1400cm−1の領域における最大吸収帯の半値幅は、得られるガスバリアーフィルムのガスバリアー性の観点から、200cm−1以下が好ましく、150cm−1以下がより好ましく、100cm−1以下がさらに好ましく、50cm−1以下が特に好ましい。
下地層(Y)の赤外線吸収スペクトルは、フーリエ変換赤外分光光度計を用い、減衰全反射法で測定できる。一例として、測定条件は以下のとおりである。
装置:パーキンエルマー株式会社製Spectrum One
測定モード:減衰全反射法
測定領域:800〜1400cm−1
〔2.7〕下地層(W)
本発明に係る下地層(Y)以外に、下地層(W)をさらに含んでもよい。下地層(W)は、リン原子を含有する官能基を有する重合体(G1)を含むことが好ましい。下地層(W)は、下地層(Y)に隣接して配置されることが好ましい。すなわち、下地層(W)及び下地層(Y)は、互いに接触するように配置され下地層を形成することが好ましい。また、下地層(W)は、下地層(Y)を挟んで樹脂基材と反対側(好ましくは反対側の表面)に配置されることが好ましい。
測定モード:減衰全反射法
測定領域:800〜1400cm−1
〔2.7〕下地層(W)
本発明に係る下地層(Y)以外に、下地層(W)をさらに含んでもよい。下地層(W)は、リン原子を含有する官能基を有する重合体(G1)を含むことが好ましい。下地層(W)は、下地層(Y)に隣接して配置されることが好ましい。すなわち、下地層(W)及び下地層(Y)は、互いに接触するように配置され下地層を形成することが好ましい。また、下地層(W)は、下地層(Y)を挟んで樹脂基材と反対側(好ましくは反対側の表面)に配置されることが好ましい。
下地層(W)は、ヒドロキシ基及び/又はカルボキシ基を有する重合体(G2)をさらに含んでもよい。重合体(G2)としては、重合体(C)と同じものを使用することができる。重合体(G1)について、以下に説明する。
<重合体(G1)>
リン原子を含有する官能基を有する重合体(G1)が有するリン原子を含有する官能基としては、例えば、リン酸基、亜リン酸基、ホスホン酸基、亜ホスホン酸基、ホスフィン酸基、亜ホスフィン酸基、及びこれらの塩、並びにこれらから誘導される官能基(例えば、(部分)エステル化合物、ハロゲン化物(例えば、塩化物)、脱水物)等を挙げられる。中でも、リン酸基及び/又はホスホン酸基が好ましく、ホスホン酸基がより好ましい。
リン原子を含有する官能基を有する重合体(G1)が有するリン原子を含有する官能基としては、例えば、リン酸基、亜リン酸基、ホスホン酸基、亜ホスホン酸基、ホスフィン酸基、亜ホスフィン酸基、及びこれらの塩、並びにこれらから誘導される官能基(例えば、(部分)エステル化合物、ハロゲン化物(例えば、塩化物)、脱水物)等を挙げられる。中でも、リン酸基及び/又はホスホン酸基が好ましく、ホスホン酸基がより好ましい。
重合体(G1)としては、例えば、アクリル酸6−[(2−ホスホノアセチル)オキシ]ヘキシル、メタクリル酸2−ホスホノオキシエチル、メタクリル酸ホスホノメチル、メタクリル酸11−ホスホノウンデシル、メタクリル酸1,1−ジホスホノエチル等のホスホノ(メタ)アクリル酸エステル類の重合体;ビニルホスホン酸、2−プロペン−1−ホスホン酸、4−ビニルベンジルホスホン酸、4−ビニルフェニルホスホン酸等のホスホン酸類の重合体;ビニルホスフィン酸、4−ビニルベンジルホスフィン酸等のホスフィン酸類の重合体;リン酸化デンプン等が挙げられる。重合体(G1)は、少なくとも1種の前記リン原子含有官能基を有する単量体の単独重合体であってもよいし、2種類以上の単量体の共重合体であってもよい。また、重合体(G1)として、単一の単量体からなる重合体を2種以上混合して使用してもよい。中でも、ホスホノ(メタ)アクリル酸エステル類の重合体及び/又はビニルホスホン酸類の重合体が好ましく、ビニルホスホン酸類の重合体がより好ましい。重合体(G1)は、ポリ(ビニルホスホン酸)又はポリ(2−ホスホノオキシエチルメタクリレート)であることが好ましく、ポリ(ビニルホスホン酸)であってもよい。また、重合体(G1)は、ビニルホスホン酸ハロゲン化物やビニルホスホン酸エステル等のビニルホスホン酸誘導体を単独又は共重合した後、加水分解することによっても得ることができる。
また、重合体(G1)は、少なくとも1種のリン原子含有官能基を有する単量体と他のビニル単量体との共重合体であってもよい。リン原子含有官能基を有する単量体と共重合することができる他のビニル単量体としては、例えば、(メタ)アクリル酸、(メタ)アクリル酸エステル類、アクリロニトリル、メタクリロニトリル、スチレン、核置換スチレン類、アルキルビニルエーテル類、アルキルビニルエステル類、パーフルオロアルキルビニルエーテル類、パーフルオロアルキルビニルエステル類、マレイン酸、無水マレイン酸、フマル酸、イタコン酸、マレイミド、フェニルマレイミド等が挙げられる。これらの中でも、(メタ)アクリル酸エステル類、アクリロニトリル、スチレン、マレイミド、及びフェニルマレイミドが好ましい。
より優れた耐屈曲性を有するガスバリアーフィルムを得るために、リン原子含有官能基を有する単量体に由来する構成単位が重合体(G1)の全構成単位に占める割合は、10モル%以上であることが好ましく、20モル%以上であることがより好ましく、40モル%以上であることがさらに好ましく、70モル%以上であることが特に好ましく、100モル%であってもよい。
重合体(G1)の分子量に特に制限はないが、数平均分子量が1000〜100000の範囲にあることが好ましい。数平均分子量がこの範囲にあると、下地層(W)を積層することによる耐屈曲性の改善効果と、後述する第2コーティング液(V)の粘度安定性とを、高いレベルで両立することができる。また、下地層(Y)を積層する場合、リン原子一つ当たりの重合体(G1)の分子量が100〜500の範囲にある場合に耐屈曲性の改善効果をより高めることができる。
下地層(W)は、重合体(G1)のみによって構成されてもよいし、重合体(G1)及び重合体(G2)のみによって構成されてもよいし、他の成分をさらに含んでもよい。下地層(W)に含まれる他の成分としては、例えば、炭酸塩、塩酸塩、硝酸塩、炭酸水素塩、硫酸塩、硫酸水素塩、ホウ酸塩等の無機酸金属塩;酢酸塩、ステアリン酸塩、シュウ酸塩、酒石酸塩等の有機酸金属塩;シクロペンタジエニル金属錯体(例えば、チタノセン)、シアノ金属錯体(例えば、プルシアンブルー)等の金属錯体;層状粘土化合物;架橋剤;重合体(G1)及び重合体(G2)以外の高分子化合物:可塑剤;酸化防止剤;紫外線吸収剤;難燃剤等が挙げられる。下地層(W)における前記の他の成分の含有率は、50質量%以下であることが好ましく、20質量%以下であることがより好ましく、10質量%以下であることがさらに好ましく、5質量%以下であることが特に好ましく、0質量%(他の成分を含まない)であってもよい。
下地層(W)の一層当たりの厚さは、本発明のガスバリアーフィルムの物理的ストレス(例えば、屈曲)に対する耐性がより良好になる観点から、0.003μm以上であることが好ましい。下地層(W)の厚さの上限は特に限定されないが、1.0μm以上では物理的ストレスに対する耐性の改善効果は飽和に達する。そのため、下地層(W)の合計の厚さの上限は、経済性の観点から1.0μmとすることが好ましい。下地層(W)の厚さは、下地層(W)の形成に用いられる後述する第2コーティング液(V)の濃度や、その塗工方法によって制御することができる。
〔2.8〕下地層(Y)の製造方法
本発明に係る下地層(Y)の製造方法は、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、前記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、前記関係式(2)で表される関係を満たすように調整することを特徴とし、工程〔I〕、〔II〕及び〔III〕を含むことが好ましい。
本発明に係る下地層(Y)の製造方法は、前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、前記関係式(1)で表される関係を満たし、かつ、前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、前記関係式(2)で表される関係を満たすように調整することを特徴とし、工程〔I〕、〔II〕及び〔III〕を含むことが好ましい。
工程〔I〕では、金属酸化物(A)と、リン化合物(B)と、陽イオン(Z)のイオン性化合物(E)とを混合することによって、金属酸化物(A)、リン化合物(B)、及び陽イオン(Z)を含む第1コーティング液(U)を調製する。工程〔II〕では、樹脂基材上に第1コーティング液(U)を塗工することによって、樹脂基材上に下地層(Y)の前駆体層を形成する。工程〔III〕では、当該前駆体層を110℃以上の温度で熱処理することによって、樹脂基材上に下地層(Y)を形成する。
<工程〔I〕第1コーティング液(U)の調製>
工程〔I〕では、金属酸化物(A)と、リン化合物(B)と、陽イオン(Z)のイオン性化合物(E)とを混合する。これらを混合するにあたり、溶媒を添加してもよい。第1コーティング液(U)において、イオン性化合物(E)から陽イオン(Z)が生成される。第1コーティング液(U)は、金属酸化物(A)、リン化合物(B)、及び陽イオン(Z)の他に、他の化合物を含んでもよい。
工程〔I〕では、金属酸化物(A)と、リン化合物(B)と、陽イオン(Z)のイオン性化合物(E)とを混合する。これらを混合するにあたり、溶媒を添加してもよい。第1コーティング液(U)において、イオン性化合物(E)から陽イオン(Z)が生成される。第1コーティング液(U)は、金属酸化物(A)、リン化合物(B)、及び陽イオン(Z)の他に、他の化合物を含んでもよい。
第1コーティング液(U)において、前記NMとNPとは、前記の関係式を満たすことが好ましい。また、前記NMとNZとFZとは、前記の関係式を満たすことが好ましい。さらに、前記NPとNZとFZとは、前記の関係式を満たすことが好ましい。
工程〔I〕は、以下の工程〔I−a〕〜〔I−c〕を含むことが好ましい。
工程〔I−a〕:金属酸化物(A)を含む液体を調製する工程、
工程〔I−b〕:リン化合物(B)を含む溶液を調製する工程、
工程〔I−c〕:前記工程〔I−a〕及び〔I−b〕で得られた金属酸化物(A)を含む液体とリン化合物(B)を含む溶液とを混合する工程。
工程〔I−b〕:リン化合物(B)を含む溶液を調製する工程、
工程〔I−c〕:前記工程〔I−a〕及び〔I−b〕で得られた金属酸化物(A)を含む液体とリン化合物(B)を含む溶液とを混合する工程。
工程〔I−b〕は、工程〔I−a〕より先又は後のいずれに行われてもよく、工程〔I−a〕と同時に行われてもよい。以下、各工程について、より具体的に説明する。
工程〔I−a〕では、金属酸化物(A)を含む液体を調製する。当該液体は、溶液又は分散液である。当該液体は、例えば、公知のゾル−ゲル法で採用されている手法に従い、例えば、上述した化合物(L)、水、及び必要に応じて酸触媒や有機溶媒を混合し、化合物(L)を縮合又は加水分解縮合することによって調製することができる。化合物(L)を縮合又は加水分解縮合することによって金属酸化物(A)の分散液を得た場合、必要に応じて、当該分散液に対して特定の処理(前記したような解膠や濃度制御のための溶媒の加減等)を行ってもよい。工程〔I−a〕は、化合物(L)及び化合物(L)の加水分解物からなる群より選ばれる少なくとも1種を縮合(例えば、脱水縮合)させる工程を含んでもよい。工程〔I−a〕において使用できる有機溶媒の種類に特に制限はなく、例えば、メタノール、エタノール、イソプロパノール等のアルコール類、水、及びこれらの混合溶媒が好ましい。当該液体中における金属酸化物(A)の含有量は、0.1〜30質量%の範囲にあることが好ましく、1〜20質量%の範囲にあることがより好ましく、2〜15質量%の範囲にあることがさらに好ましい。
例えば、金属酸化物(A)が酸化アルミニウムである場合、酸化アルミニウムの分散液の調製では、まず必要に応じて酸触媒でpH調整した水溶液中でアルミニウムアルコキシドを加水分解縮合することによって酸化アルミニウムのスラリーを得る。次に、そのスラリーを特定量の酸の存在下に解膠することによって、酸化アルミニウムの分散液が得られる。なお、アルミニウム以外の金属原子を含有する金属酸化物(A)の分散液も、同様の方法で製造できる。酸としては、例えば、塩酸、硫酸、硝酸、酢酸、乳酸及び酪酸が好ましく、硝酸及び酢酸がより好ましい。
工程〔I−b〕では、リン化合物(B)を含む溶液を調製する。前記溶液は、リン化合物(B)を溶媒に溶解することによって調製できる。リン化合物(B)の溶解性が低い場合には、加熱処理や超音波処理を施すことによって溶解を促進してもよい。溶媒としては、リン化合物(B)の種類に応じて適宜選択すればよいが、水を含むことが好ましい。リン化合物(B)の溶解の妨げにならない限り、溶媒は有機溶媒(例えば、メタノール)を含んでもよい。
リン化合物(B)を含む溶液中におけるリン化合物(B)の含有量は、0.1〜99質量%の範囲にあることが好ましく、45〜95質量%の範囲にあることがより好ましく、55〜90質量%の範囲にあることがさらに好ましい。
工程〔I−c〕では、金属酸化物(A)を含む液体とリン化合物(B)を含む溶液とを混合する。混合時の温度を30℃以下(例えば、20℃)に維持することによって、保存安定性に優れた第1コーティング液(U)を得ることができる。
陽イオン(Z)を含む化合物(E)は、工程〔I−a〕、工程〔I−b〕、及び工程〔I−c〕からなる群より選ばれる少なくとも一つの工程で添加してもよいし、それらのうちのいずれか一つの工程で添加してもよい。例えば、化合物(E)は、工程〔I−a〕の金属酸化物(A)を含む液体又は工程〔I−b〕のリン化合物(B)を含む溶液に添加してもよく、工程〔I−c〕における金属酸化物(A)を含む液体とリン化合物(B)を含む溶液との混合液に添加してもよい。
また、第1コーティング液(U)は、重合体(C)を含んでもよい。第1コーティング液(U)に重合体(C)を含ませる方法は、特に制限されない。例えば、重合体(C)は、金属酸化物(A)を含む液体、リン化合物(B)を含む溶液、及びこれらの混合液のいずれかに、溶液として添加・混合させてもよく、粉末又はペレットの状態で添加した後に溶解させてもよい。リン化合物(B)を含む溶液に重合体(C)を含有させることによって、金属酸化物(A)を含む液体とリン化合物(B)を含む溶液とを混合した際の金属酸化物(A)とリン化合物(B)との反応速度が遅くなり、その結果、経時安定性に優れた第1コーティング液(U)が得られる。
第1コーティング液(U)は、必要に応じて、塩酸、硝酸、酢酸、トリフルオロ酢酸、及びトリクロロ酢酸から選ばれる少なくとも1種の酸化合物(J)を含んでもよい。酸化合物(J)の含有量は、0.1〜5.0質量%の範囲にあることが好ましく、0.5〜2.0質量%の範囲にあることがより好ましい。これらの範囲では、酸化合物(J)の添加による効果が得られ、かつ酸化合物(J)の除去が容易である。金属酸化物(A)を含む液体中に酸成分が残留している場合は、その残留量を考慮して酸化合物(J)の添加量を決定すればよい。
工程〔I−c〕で得られた混合液は、そのまま第1コーティング液(U)として使用できる。この場合、通常、金属酸化物(A)を含む液体やリン化合物(B)を含む溶液に含まれる溶媒が、第1コーティング液(U)の溶媒となる。また、前記混合液に有機溶媒の添加、pHの調製、粘度の調製、添加物の添加等の処理を行って第1コーティング液(U)を調製してもよい。有機溶媒としては、例えば、リン化合物(B)を含む溶液の調製に用いられる溶媒等が挙げられる。
第1コーティング液(U)の保存安定性、及び第1コーティング液(U)の樹脂基材に対する塗工性の観点から、第1コーティング液(U)の固形分濃度は、1〜20質量%の範囲にあることが好ましく、2〜15質量%の範囲にあることがより好ましく、3〜10質量%の範囲にあることがさらに好ましい。第1コーティング液(U)の固形分濃度は、例えば、シャーレに第1コーティング液(U)を所定量加え、当該シャーレごと加熱して溶媒等の揮発分を除去し、残留した固形分の質量を、最初に加えた第1コーティング液(U)の質量で除することで算出できる。
第1コーティング液(U)は、ブルックフィールド形回転粘度計(SB型粘度計:ローターNo.3、回転速度60rpm)で測定された粘度が、塗工時の温度において3000mPa・s以下であることが好ましく、2500mPa・s以下であることがより好ましく、2000mPa・s以下であることがさらに好ましい。当該粘度が3000mPa・s以下であることによって、第1コーティング液(U)のレベリング性が向上し、外観により優れる下地層を得ることができる。また、第1コーティング液(U)の粘度としては、50mPa・s以上が好ましく、100mPa・s以上がより好ましく、200mPa・s以上がさらに好ましい。
第1コーティング液(U)において、前記NMとNPとは、0.8≦NM/NP≦4.5の関係を満たす。また、第1コーティング液(U)において、前記NMとNZとFZとは、0.001≦FZ×NZ/NM≦0.60の関係を満たす。さらに、第1コーティング液(U)において、前記FZとNZとNPが、0.0008≦FZ×NZ/NP≦1.35の関係を満たすものが好ましい。
<[工程〔II〕第1コーティング液(U)の塗工>
工程〔II〕では、樹脂基材上に第1コーティング液(U)を塗工することによって、樹脂基材上に下地層(Y)の前駆体層を形成する。第1コーティング液(U)は、樹脂基材の少なくとも一方の面の上に直接塗工してもよい。また、第1コーティング液(U)を塗工する前に、樹脂基材の表面を公知のアンカーコーティング剤で処理したり、樹脂基材の表面に公知の接着剤を塗工したりする等して、樹脂基材の表面に易接着層を形成しておいてもよい。
工程〔II〕では、樹脂基材上に第1コーティング液(U)を塗工することによって、樹脂基材上に下地層(Y)の前駆体層を形成する。第1コーティング液(U)は、樹脂基材の少なくとも一方の面の上に直接塗工してもよい。また、第1コーティング液(U)を塗工する前に、樹脂基材の表面を公知のアンカーコーティング剤で処理したり、樹脂基材の表面に公知の接着剤を塗工したりする等して、樹脂基材の表面に易接着層を形成しておいてもよい。
第1コーティング液(U)を樹脂基材上に塗工する方法は、特に限定されず、公知の方法を採用することができる。塗工方法としては、例えば、キャスト法、ディッピング法、ロールコーティング法、グラビアコート法、スクリーン印刷法、リバースコート法、スプレーコート法、キスコート法、ダイコート法、メタリングバーコート法、チャンバードクター併用コート法、カーテンコート法等が挙げられる。
通常、工程〔II〕において、第1コーティング液(U)中の溶媒を除去することによって、下地層(Y)の前駆体層が形成される。溶媒の除去方法に特に制限はなく、公知の乾燥方法を適用することができる。乾燥方法としては、例えば、熱風乾燥法、熱ロール接触法、赤外線加熱法、マイクロ波加熱法等が挙げられる。乾燥処理温度は、樹脂基材の流動開始温度よりも0〜15℃以上低いことが好ましい。第1コーティング液(U)が重合体(C)を含む場合には、乾燥処理温度は、重合体(C)の熱分解開始温度よりも15〜20℃以上低いことが好ましい。乾燥処理温度は70〜200℃の範囲にあることが好ましく、80〜180℃の範囲にあることがより好ましく、90〜160℃の範囲にあることがさらに好ましい。溶媒の除去は、常圧下又は減圧下のいずれで実施してもよい。また、後述する工程〔III〕における熱処理によって、溶媒を除去してもよい。
層状の樹脂基材の両面に下地層(Y)を積層する場合、第1コーティング液(U)を樹脂基材の一方の面に塗工した後、溶媒を除去することによって第1の層(第1の下地層(Y)の前駆体層)を形成し、次いで、第1コーティング液(U)を樹脂基材の他方の面に塗工した後、溶媒を除去することによって第2の層(第2の下地層(Y)の前駆体層)を形成してもよい。それぞれの面に塗工する第1コーティング液(U)の組成は同一であってもよいし、異なってもよい。
<[工程〔III〕下地層(Y)の前駆体層の処理>
工程〔III〕では、工程〔II〕で形成された前駆体層(下地層(Y)の前駆体層)を140℃以上の温度で熱処理することによって、下地層(Y)を形成する。この熱処理温度は第1コーティング液(U)の塗工後の乾燥処理温度よりも高いことが好ましい。
工程〔III〕では、工程〔II〕で形成された前駆体層(下地層(Y)の前駆体層)を140℃以上の温度で熱処理することによって、下地層(Y)を形成する。この熱処理温度は第1コーティング液(U)の塗工後の乾燥処理温度よりも高いことが好ましい。
工程〔III〕では、金属酸化物(A)同士がリン原子(リン化合物(B)に由来するリン原子)を介して結合される反応が進行する。別の観点では、工程〔III〕では、反応生成物(D)の生成反応が進行する。当該反応を充分に進行させるため、熱処理の温度は、140℃以上であることが好ましく、170℃以上であることがより好ましく、180℃以上であることがさらに好ましい。熱処理温度が低いと、充分な反応度を得るのにかかる時間が長くなり、生産性が低下する原因となる。熱処理の温度の好ましい上限は、樹脂基材の種類等によって異なる。例えば、ポリアミド系樹脂からなる熱可塑性樹脂フィルムを樹脂基材として用いる場合には、熱処理の温度は270℃以下であることが好ましい。また、ポリエステル系樹脂からなる熱可塑性樹脂フィルムを樹脂基材として用いる場合には、熱処理の温度は240℃以下であることが好ましい。熱処理は、空気中、窒素雰囲気下、又はアルゴン雰囲気下等で実施することができる。
熱処理の時間は0.1秒〜1時間の範囲にあることが好ましく、1秒〜15分の範囲にあることがより好ましく、5〜300秒の範囲にあることがさらに好ましい。
また、下地層(Y)の前駆体層又は下地層(Y)に紫外線を照射する工程を含んでもよい。例えば、紫外線照射は、工程〔II〕の後(例えば、塗工された第1コーティング液(U)の溶媒の除去がほぼ終了した後)で行ってもよい。
樹脂基材と下地層(Y)との間に易接着層を配置するために、第1コーティング液(U)を塗工する前に、樹脂基材の表面を公知のアンカーコーティング剤で処理したり、樹脂基材の表面に公知の接着剤を塗工したりしてもよい。
本発明に係る下地層の製造方法は、工程〔i〕及び〔ii〕をさらに含んでもよい。工程〔i〕では、リン原子を含有する重合体(G1)と溶媒とを含む第2コーティング液(V)を調製する。工程〔ii〕では、下地層(Y)に隣接して配置された下地層(W)を第2コーティング液(V)を用いて形成する。工程〔i〕の順序は特に限定されず、工程〔I〕、〔II〕又は〔III〕と並行して行ってもよく、工程〔I〕、〔II〕又は〔III〕の後に行ってもよい。工程〔ii〕は、工程〔II〕又は〔III〕の後に行うことができる。下地層(Y)又は下地層(Y)の前駆体層に第2コーティング液(V)を塗工することによって、下地層(Y)と接するように下地層(Y)に積層された下地層(W)を形成できる。重合体(G2)を含む下地層(W)を形成する場合、第2コーティング液(V)は重合体(G2)を含む。第2コーティング液(V)において、重合体(G1)と重合体(G2)との質量比は、重合体(G1):重合体(G2)が15:85〜100:0の範囲にあることが好ましく、15:85〜99:1の範囲にあることがより好ましい。当該質量比の第2コーティング液(V)を用いることによって、重合体(G1)と重合体(G2)との質量比が当該範囲にある下地層(W)を形成できる。第2コーティング液(V)は、重合体(G1)(及び必要に応じて重合体(G2))を溶媒に溶解することによって調製できる。
第2コーティング液(V)に用いられる溶媒は、含まれる重合体の種類に応じて適宜選択すればよいが、水、アルコール類、又はそれらの混合溶媒であることが好ましい。重合体の溶解の妨げにならない限り、溶媒は、テトラヒドロフラン、ジオキサン、トリオキサン、ジメトキシエタン等のエーテル;アセトン、メチルエチルケトン等のケトン;エチレングリコール、プロピレングリコール等のグリコール;メチルセロソルブ、エチルセロソルブ、n−ブチルセロソルブ等のグリコール誘導体;グリセリン;アセトニトリル;ジメチルホルムアミド等のアミド;ジメチルスルホキシド;スルホラン等を含んでもよい。
第2コーティング液(V)における固形分(重合体(G1)等)の濃度は、溶液の保存安定性や塗工性の観点から、0.01〜60質量%の範囲にあることが好ましく、0.1〜50質量%の範囲にあることがより好ましく、0.2〜40質量%の範囲にあることがさらに好ましい。固形分濃度は、第1コーティング液(U)に関して記載した方法と同様の方法によって求めることができる。
通常、工程〔ii〕において、第2コーティング液(V)中の溶媒が除去されることによって、下地層(W)が形成される。第2コーティング液(V)の溶媒の除去方法は特に限定されず、公知の乾燥方法を適用することができる。乾燥方法としては、例えば、熱風乾燥法、熱ロール接触法、赤外線加熱法、マイクロ波加熱法等を挙げることができる。乾燥温度は、樹脂基材の流動開始温度よりも0〜15℃以上低いことが好ましい。乾燥温度は70〜200℃の範囲にあることが好ましく、150〜200℃の範囲にあることがより好ましい。溶媒の除去は、常圧下又は減圧下のいずれで実施してもよい。また、工程〔ii〕を、前述の工程〔II〕と工程〔III〕との間に実施する場合は、工程〔III〕における熱処理によって溶媒を除去してもよい。
樹脂基材の両面に、下地層(Y)を介して下地層(W)を形成してもよい。その場合の一例では、第2コーティング液(V)を一方の面に塗工した後に溶媒を除去することによって第1の下地層(W)を形成する。次に、第2コーティング液(V)を他方の面に塗工した後に溶媒を除去することによって第2の下地層(W)を形成する。それぞれの面に塗工する第2コーティング液(V)の組成は同一であってもよいし、異なってもよい。
〔3〕ガスバリアー層(X)
本発明のガスバリアーフィルムは、樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)を備えることを特徴とする。言い換えれば、樹脂基材上に形成される前記下地層(Y)上に、ガスバリアー層として機能する前記ガスバリアー層(X)が積層されガスバリアーフィルムを構成する。
本発明のガスバリアーフィルムは、樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)を備えることを特徴とする。言い換えれば、樹脂基材上に形成される前記下地層(Y)上に、ガスバリアー層として機能する前記ガスバリアー層(X)が積層されガスバリアーフィルムを構成する。
本発明に係るガスバリアー層(X)は、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚が15〜50nmの範囲内であることが好ましい。この範囲とすることで、透明性、ガスバリアー性及び柔軟性(フレキシビリティ)をバランスさせることができる。
関係式(3) 1.6≦x≦1.9、0.15≦y≦0.40
関係式(4) 4.0≦2x+2y<4.3
前記酸素成分x及び炭素成分yの値の範囲は、透明性、ガスバリアー性及び柔軟性の観点から最適化したときの領域である。酸素成分xが大きくなると透明性が向上する傾向にあり、小さくしていくとガスバリアー性が向上する。また、炭素成分yが増加すると柔軟性が増加する傾向にあるが、透明性が劣化する傾向にある。したがって、上記x及びyの値の範囲内であれば、透明性、ガスバリアー性及び柔軟性のバランスがとれるものと推察される。
関係式(4) 4.0≦2x+2y<4.3
前記酸素成分x及び炭素成分yの値の範囲は、透明性、ガスバリアー性及び柔軟性の観点から最適化したときの領域である。酸素成分xが大きくなると透明性が向上する傾向にあり、小さくしていくとガスバリアー性が向上する。また、炭素成分yが増加すると柔軟性が増加する傾向にあるが、透明性が劣化する傾向にある。したがって、上記x及びyの値の範囲内であれば、透明性、ガスバリアー性及び柔軟性のバランスがとれるものと推察される。
加えて、例えば、ヘキサメチルジシロキサン(HMDSO)を原料としたCVD法によるガスバリアー層の成膜では、当該ガスバリアー層は、Si−O−Si結合か、Si−CH2−Si結合を主として有すると考えられる。全てがSi−O−Si結合か、Si−CH2−Si結合であれば、2x+2y=4となるが、実際は、少量のSi−CH3が存在するため、4<2x+2yとなるものと考えられる。したがって、2x+2yが4.3以上となる場合は、過剰にSi−CH3が存在すると考えられ、ガスバリアー性は大きく低下する。2x+2yが4未満となる場合は、いわゆる酸素欠損状態となり、透明性が劣化する。
したがって、本発明に係るガスバリアー層は、当該ガスバリアー層に含まれる化合物の平均組成をSiOxCyで表したときに、上記x及びyの値の範囲内であって、かつ2x+2yで規定されるSi−CH3の含有量を制御することによって、ガスバリアー性の向上及び透明性の向上を図ることができるものと推察される。
さらに、本発明に係るガスバリアー層(X)は、前記yとガスバリアー層(X)の厚さ(nm)との積が、4〜12の範囲内であることが、ガスバリアー性と、透明性をバランスする観点から、好ましい。
特にガスバリアー層の場合において、前記yと厚さ(nm)との積が大きくなると、ガスバリアー性が向上し、逆に波透明性が劣化する方向であり、前記yと厚さ(nm)との積が小さくなると、ガスバリアー性が劣化し、当該透明性が向上する方向であり、前記yと厚さ(nm)との積が4〜12の範囲内である場合に良好な性能バランスが得られるものと推察される。
本発明に係るガスバリアー層(X)は、化学気相成膜法(CVD法)によって形成されることが好ましい。
化学気相成膜法は、前記基材上に、目的とする薄膜の成分を含む原料ガスを供給し、基材表面又は気相での化学反応により膜を堆積する方法である。また、化学反応を活性化する目的で、プラズマなどを発生させる方法などがあり、熱CVD法、触媒化学気相成長法、光CVD法、真空プラズマCVD法など公知のCVD方式等が挙げられる。特に限定されるものではないが、成膜速度や処理面積、得られるガスバリアー層(X)は、ガスバリアー層の柔軟性(フレキシビリティ)やガスバリアー性の観点から、真空プラズマCVD法を適用することが好ましい。
例えば、ケイ素化合物を原料化合物として用い、分解ガスに酸素を用いれば、ケイ素酸化物が生成する。これはプラズマ空間内では非常に活性な荷電粒子・活性ラジカルが高密度で存在するため、プラズマ空間内では多段階の化学反応が非常に高速に促進され、プラズマ空間内に存在する元素は熱力学的に安定な化合物へと非常な短時間で変換されるためである。
前記平均組成SiOxCyにおける酸素成分x及び炭素成分yを制御するには、真空プラズマCVD法を用いることが好ましく、原料ガスの種類及び供給速度、プラズマ強度、成膜装置及び成膜速度等を制御することで達成することができる。
例えば、成膜原料と酸素の供給量とその比率、成膜時の搬送速度、成膜回数等を適宜組み合わせることにより制御することができる。
以下好ましい成膜装置であって、対向ローラー型のロールtoロール成膜装置を使用して、真空プラズマCVD法によってガスバリアー層を製造する場合を例示して説明する。
図2は、本発明に係るガスバリアー層(X)の形成に好ましく用いられる真空プラズマCVD装置の一例を示す模式図である。
図2に示すとおり、成膜装置100は、送り出しローラー10と、搬送ローラー11〜14と、第1及び第2成膜ローラー15、16と、巻取りローラー17と、ガス供給管18と、プラズマ発生用電源19と、磁場発生装置20及び21と、真空チャンバー30と、真空ポンプ40と、制御部41と、を有する。
送り出しローラー10、搬送ローラー11〜14、第1及び第2成膜ローラー15、16及び巻取りローラー17は、真空チャンバー30に収容されている。
送り出しローラー10は、あらかじめ巻き取られた状態で設置されている樹脂基材1aを搬送ローラー11に向けて送り出す。送り出しローラー10は、紙面に対して垂直方向に延在した円筒状のローラーであり、図示しない駆動モーターにより反時計回りに回転(図2の矢印を参照)することにより、送り出しローラー10に巻回された基材1aを搬送ローラー11に向けて送り出す。
搬送ローラー11〜14は、送り出しローラー10と略平行な回転軸を中心に回転可能に構成された円筒状のローラーである。搬送ローラー11は、基材1aに適当な張力を付与しつつ、基材1aを送り出しローラー10から成膜ローラー15に搬送するためのローラーである。搬送ローラー12、13は、成膜ローラー15で成膜された基材1bに適当な張力を付与しつつ、基材1bを成膜ローラー15から成膜ローラー16に搬送するためのローラーである。さらに、搬送ローラー14は、成膜ローラー16で成膜された基材1bに適当な張力を付与しつつ、基材1bを成膜ローラー16から巻取りローラー17に搬送するためのローラーである。
第1成膜ローラー15及び第2成膜ローラー16は、送り出しローラー10と略平行な回転軸を有し、互いに所定距離だけ離間して対向配置された成膜ローラー対である。成膜ローラー15は、基材1aを成膜し、成膜された基材1bに適当な張力を付与しつつ、基材1bを成膜ローラー16へ搬送する。成膜ローラー16は、基材1bを成膜し、成膜された基材1cに適当な張力を付与しつつ、基材1cを搬送ローラー14へ搬送する。
図2に示す例では、第1成膜ローラー15と第2成膜ローラー16との離間距離は、点Aと点Bとを結ぶ距離である。第1及び第2成膜ローラー15、16は、導電性材料で形成された放電電極であり、第1成膜ローラー15と第2成膜ローラー16とは、それぞれは互いに絶縁されている。なお、第1及び第2成膜ローラー15、16の材質や構成は、電極として所望の機能を達成できるように適宜選択することができる。
さらに、第1成膜ローラー15及び第2成膜ローラー16は、それぞれ独立に調温してもよい。第1成膜ローラー15及び第2成膜ローラー16の温度は、特に制限されるものではないが、例えば−30〜100℃であるが、基材1aのガラス転移温度を超えて過度に高温に設定すると、基材が熱によって変形等を生じるおそれがある。
第1及び第2成膜ローラー15、16の内部には、磁場発生装置20及び21が、各々設置されている。第1成膜ローラー15と第2成膜ローラー16とにはプラズマ発生用電源19により、プラズマ発生用の高周波電圧が印加される。それにより、第1成膜ローラー15と第2成膜ローラー16との間の成膜部Sに電場が形成され、ガス供給管18から供給される成膜ガスの放電プラズマが発生する。プラズマ発生用電源19の電源周波数は任意に設定できるが、本構成の装置としては、例えば60〜100kHzであり、印加される電力は、有効成膜幅1mに対して、例えば1〜10kWである。
巻取りローラー17は、送り出しローラー10と略平行な回転軸を有し、基材1cを巻き取り、ローラー状にして収容する。巻取りローラー17は、図示しない駆動モーターにより反時計回りに回転(図2の矢印を参照)することにより、基材1cを巻き取る。
送り出しローラー10から送り出された基材1aは、送り出しローラー10と巻取りローラー17との間で、搬送ローラー11〜14、第1及び第2成膜ローラー15、16に巻き掛けられることにより適当な張力を保ちつつ、これらの各ローラーの回転により搬送される。なお、基材1a、1b、1c(以下、基材1a、1b、1cを「基材1a〜1c」とも総称する。)の搬送方向は矢印で示されている。基材1a〜1cの搬送速度(ラインスピード)(例えば、図2の点Cにおける搬送速度)は、原料ガスの種類や真空チャンバー30内の圧力などに応じて適宜調整されうる。搬送速度は、送り出しローラー10及び巻取りローラー17の駆動モーターの回転速度を制御部41によって制御することにより調整される。搬送速度を遅くすると、形成される領域の厚さが厚くなる。
基材の搬送速度(ライン速度)は、原料ガスの種類やチャンバー内の圧力等に応じて適宜調整することができるが、0.25〜100m/minの範囲内とすることが好ましく、0.5〜20m/minの範囲内とすることがより好ましい。ライン速度が前記範囲内であれば、樹脂基材の熱に起因する皺も発生し難く、形成されるガスバリアー層の層厚も十分に制御可能となる。
また、この成膜装置を用いる場合、基材1a〜1cの搬送方向を図2の矢印で示す方向(以下、順方向と称する)とは反対方向(以下、逆方向と称する)に設定してガスバリアー性フィルムの成膜工程を実行することもできる。具体的には、制御部41は、巻取りローラー17によって基材1cが巻き取られた状態において、送り出しローラー10及び巻取りローラー17の駆動モーターの回転方向を上述の場合とは逆方向に回転するように制御する。このように制御すると、巻取りローラー17から送り出された基材1cは、送り出しローラー10と巻取りローラー17との間で、搬送ローラー11〜14、第1及び第2成膜ローラー15、16に巻き掛けられることにより適当な張力を保ちつつ、これらの各ローラーの回転により逆方向に搬送される。
成膜装置100を用いてガスバリアー層を形成する場合は、基材1aを順方向及び逆方向に搬送して成膜部Sを往復させることにより、ガスバリアー層の形成(成膜)工程を複数回繰り返すこともできる。
ガス供給管18は、真空チャンバー30内にプラズマCVDの原料ガスなどの成膜ガスを供給する。ガス供給管18は、成膜部Sの上方に第1成膜ローラー15及び第2成膜ローラー16の回転軸と同じ方向に延在する管状の形状を有しており、複数箇所に設けられた開口部から成膜部Sに成膜ガスを供給する。また、成膜装置を連結する場合(タンデム型)は、ガス供給管18から供給される成膜ガスは、成膜装置ごとに同一でもよいが、異なっていてもよい。さらに、これらのガス供給管から供給される供給ガス圧についても、同一でもよいが異なっていてもよい。
原料ガスには、ケイ素化合物を使用することができる。ケイ素化合物としては、例えば、ヘキサメチルジシロキサン(HMDSO)、1,1,3,3−テトラメチルジシロキサン、ビニルトリメチルシラン、メチルトリメチルシラン、ヘキサメチルジシラン、メチルシラン、ジメチルシラン、トリメチルシラン、ジエチルシラン、プロピルシラン、フェニルシラン、ビニルトリエトキシシラン、ビニルトリメトキシシラン、テトラメトキシシラン、ジメチルジシラザン、トリメチルジシラザン、テトラメチルジシラザン、ペンタメチルジシラザン、ヘキサメチルジシラザン等が挙げられる。これ以外にも、特開2008−056967号公報の段落「0075」に記載の化合物を使用することもできる。これらのケイ素化合物の中でも、化合物の取り扱いやすさや得られるガスバリアー性フィルムの高いガスバリアー性などの観点から、ガスバリアー層の形成においては、HMDSOを使用することが好ましい。なお、これらのケイ素化合物は、2種以上が組み合わせて使用されてもよい。また、原料ガスには、ケイ素化合物の他にモノシランが含有されてもよい。
成膜ガスとしては、原料ガスの他に反応ガスが使用されてもよい。反応ガスとしては、原料ガスと反応して酸化物、窒化物などのケイ素化合物となるガスが選択される。薄膜として酸化物を形成するための反応ガスとしては、例えば、酸素ガス、オゾンガスを使用することができる。なお、これらの反応ガスは、2種以上を組み合わせて使用してもよい。
成膜ガスとしては、原料ガスを真空チャンバー30内に供給するために、さらにキャリアガスが使用されてもよい。また、成膜ガスとして、プラズマを発生させるために、さらに放電用ガスが使用されてもよい。キャリアガス及び放電ガスとしては、例えば、アルゴンなどの希ガス、及び水素や窒素が使用される。
以下、代表例として、原料ガスとしてのヘキサメチルジシロキサン(有機ケイ素化合物:HMDSO、(CH3)6Si2O)と、反応ガスである酸素(O2)の系について説明する。
原料ガスとしてのヘキサメチルジシロキサン(HMDSO、(CH3)6Si2O)と、反応ガスである酸素(O2)とを含有する成膜ガスを、プラズマCVD法により反応させて、ケイ素−酸素系の薄膜を形成する場合、その成膜ガスにより下記反応式(1)で表される反応が起こり、二酸化ケイ素SiO2からなる薄膜が形成される。
反応式(1):(CH3)6Si2O+12O2→6CO2+9H2O+2SiO2
このような反応においては、ヘキサメチルジシロキサン1モルを完全酸化するのに必要な酸素量は12モルである。そのため、成膜初期では、成膜ガス中に、ヘキサメチルジシロキサン1モルに対し、酸素を12モル以上含有させて完全に反応させることにより、酸素原子比率が高く、均一な組成の二酸化ケイ素膜を形成することができるが、成膜中〜後期で原料のガス流量比を理論比である完全反応の原料比以下の流量に制御して、非完全反応を遂行させ、本発明に係るSiOxCyの比率を高めることができる。
このような反応においては、ヘキサメチルジシロキサン1モルを完全酸化するのに必要な酸素量は12モルである。そのため、成膜初期では、成膜ガス中に、ヘキサメチルジシロキサン1モルに対し、酸素を12モル以上含有させて完全に反応させることにより、酸素原子比率が高く、均一な組成の二酸化ケイ素膜を形成することができるが、成膜中〜後期で原料のガス流量比を理論比である完全反応の原料比以下の流量に制御して、非完全反応を遂行させ、本発明に係るSiOxCyの比率を高めることができる。
なお、実際のプラズマCVD装置のチャンバー内の反応では、原料のヘキサメチルジシロキサンと反応ガスである酸素は、ガス供給部から成膜領域へ供給されて成膜されるので、反応ガスの酸素のモル量(流量)が原料のヘキサメチルジシロキサンのモル量(流量)の12倍のモル量(流量)であったとしても、現実には完全に反応を進行させることはできず、酸素の含有量を化学量論比に比して大過剰に供給して初めて反応が完結すると考えられる。例えば、CVD法により完全酸化させて酸化ケイ素を得るために、酸素のモル量(流量)を原料のヘキサメチルジシロキサンのモル量(流量)の20倍以上程度とする場合もある。そのため、原料のヘキサメチルジシロキサンのモル量(流量)に対する酸素のモル量(流量)は、化学量論比である12倍量以下(より好ましくは、10倍以下)の量であることが好ましい。このような比でヘキサメチルジシロキサン及び酸素を含有させることにより、完全に酸化されなかったヘキサメチルジシロキサン中の炭素原子や水素原子がガスバリアー層中に取り込まれ、所望したガスバリアー層を形成することが可能となって、得られるガスバリアーフィルムに優れたガスバリアー性及び屈曲耐性を発揮させることが可能となる。なお、成膜ガス中のヘキサメチルジシロキサンのモル量(流量)に対する酸素のモル量(流量)が少なすぎると、酸化されなかった炭素原子や水素原子がガスバリアー層中に過剰に取り込まれることになる。
磁場発生装置20、21は、第1成膜ローラー15と第2成膜ローラー16との間の成膜部Sに磁場を形成する部材である。これらの磁場発生装置20、21は、第1及び第2成膜ローラー15、16の回転に追随せず、所定位置に格納されている。
真空チャンバー30は、送り出しローラー10、搬送ローラー11〜14、第1及び第2成膜ローラー15、16、及び巻取りローラー17を密封して減圧された状態を維持する。真空チャンバー30内の圧力(真空度)は、原料ガスの種類などに応じて適宜調整することができる。成膜部Sの圧力は、0.1〜50Paであることが好ましい。
真空ポンプ40は、制御部41に通信可能に接続されており、制御部41の指令に従って真空チャンバー30内の圧力を適宜調整する。
制御部41は、成膜装置100の各構成要素を制御する。制御部41は、送り出しローラー10及び巻取りローラー17の駆動モーターに接続されており、これらの駆動モーターの回転数を制御することにより、基材1aの搬送速度を調整する。また、駆動モーターの回転方向を制御することにより、基材1aの搬送方向を変更する。また、制御部41は、図示しない成膜ガスの供給機構と通信可能に接続されており、成膜ガスの各々の成分ガスの供給量を制御する。また、制御部41は、プラズマ発生用電源19と通信可能に接続されており、プラズマ発生用電源19の出力電圧及び出力周波数を制御する。さらに、制御部41は、真空ポンプ40に通信可能に接続されており、真空チャンバー30内を所定の減圧雰囲気に維持するように真空ポンプ40を制御する。
制御部41は、CPU(Central Processing Unit)、HDD(Hard Disk Drive)、RAM(Random Access Memory)、及びROM(Read Only Memory)を備える。HDDには、成膜装置100の各構成要素を制御して、ガスバリアー性フィルムの製造方法を実現する手順を記述したソフトウェアプログラムが格納されている。そして、成膜装置100の電源が投入されると、上記ソフトウェアプログラムが上記RAMにロードされ上記CPUによって逐次的に実行される。また、上記ROMには、上記CPUが上記ソフトウェアプログラムを実行する際に使用する各種データ及びパラメーターが記憶されている。
該ガスバリアー層は、単層でもよいし2層以上の積層構造であってもよい。該ガスバリアー層が2層以上の積層構造である場合、各ガスバリアー層は同じ組成であってもよいし異なる組成であってもよい。また、積層構造の場合は、図2で示した成膜装置を連結したタンデム型の成膜装置を用いることも好ましい。
ガスバリアー層(X)の厚さは、前述のとおり15〜50nmの範囲内であることが好ましい。この範囲であれば、ガスバリアー性と柔軟性との両立という利点が得られる。ガスバリアー層(X)の厚さは、透過型電子顕微鏡(Transmission Electron Microscope;TEM)観察により測定することができる。
〔4〕遷移金属を含有するガスバリアー層
本発明に係るガスバリアー層(X)上には、さらに遷移金属を含有するガスバリアー層を積層することも、ガスバリアー性向上の観点から好ましい。
本発明に係るガスバリアー層(X)上には、さらに遷移金属を含有するガスバリアー層を積層することも、ガスバリアー性向上の観点から好ましい。
前記ガスバリアー層は、遷移金属を主成分として含有する層をいい、遷移金属を主成分とするとは、当該ガスバリアー層において、50質量%以上を占める成分が遷移金属であり、当該遷移金属は好ましくは遷移金属酸化物である。
本発明に用いられる遷移金属としては特に制限されず、任意の遷移金属が単独で又は組み合わせて用いられる。ここで、遷移金属とは、長周期型周期表の第3族元素から第11族元素を指し、遷移金属としては、Sc、Ti、V、Cr、Mn、Fe、Co、Ni、Cu、Y、Zr、Nb、Mo、Tc、Ru、Pd、Ag、La、Ce、Pr、Nd、Pm、Sm、Eu、Gd、Tb、Dy、Ho、Er、Tm、Yb、Lu、Hf、Ta、W、Re、Os、Ir、Pt、及びAuなどが挙げられる。
中でも、良好なガスバリアー性が得られる遷移金属としては、Nb、Ta、V、Zr、Ti、Hf、Y、La、Ce等が挙げられる。これらの中でも、種々の検討結果から、特に第5族元素であるNb、Ta、及びVが、ガスバリアー層(X)に含有される非遷移金属であるSiに対する結合が生じやすく、遷移金属及び非遷移金属の複合組成領域を形成する。特に当該複合組成領域が酸素欠損組成である場合に、著しいガスバリアー性の向上効果が得られる。これは、Siと第5族元素(特に、Nb)との結合が特に生じやすく、前記複合組成領域において緻密なガスバリアー層を形成するためであると考えられる。さらに、光学特性の観点から、遷移金属は、透明性が良好な化合物であるNb及びTaが特に好ましい。
上記酸素欠損組成とは、当該複合組成領域の組成を、下記化学組成式(1)で表したとき、当該複合組成領域の少なくとも一部の組成が、下記関係式(5)で規定する条件を満たすこととをいう。また、当該複合組成領域における酸素欠損程度を表す酸素欠損度指標としては、当該複合組成領域における(2y+3z)/(a+bx)を算出して得られる値の最小値を用いるものとする。
化学組成式(1): (Ma)(Mb)xOyNz
関係式(5) : (2y+3z)/(a+bx)<1.0
(ただし式中、Ma:非遷移金属、Mb:遷移金属、O:酸素、N:窒素、x,y,z:化学量論係数、0.02≦x≦49、0<y、0≦z、a:M1の最大価数、b:M2の最大価数を表す。)
上記関係式(5)は、ガスバリアー層の複合組成領域が、非遷移金属(Ma)と遷移金属(Mb)との酸素欠損組成を含んでいることを表している。
関係式(5) : (2y+3z)/(a+bx)<1.0
(ただし式中、Ma:非遷移金属、Mb:遷移金属、O:酸素、N:窒素、x,y,z:化学量論係数、0.02≦x≦49、0<y、0≦z、a:M1の最大価数、b:M2の最大価数を表す。)
上記関係式(5)は、ガスバリアー層の複合組成領域が、非遷移金属(Ma)と遷移金属(Mb)との酸素欠損組成を含んでいることを表している。
前記遷移金属を含有するガスバリアー層の形成は、公知の気相成膜法を用いることができる。気相成膜法としては、特に制限されず、例えば、スパッタ法、蒸着法、イオンプレーティング法、イオンアシスト蒸着法等の物理気相成長(PVD)法、プラズマCVD(chemical vapordepositinon)法、ALD(Atomic Layer Deposition)法などの化学気相成長(CVD)法が挙げられる。中でも、機能性素子へのダメージを与えることなく成膜が可能となり、高い生産性を有することから、物理気相成長(PVD)法により形成することが好ましく、スパッタ法により形成することがより好ましい。
スパッタ法による成膜は、2極スパッタリング、マグネトロンスパッタリング、中間的な周波数領域を用いたデュアルマグネトロンスパッタリング(DMS)、イオンビームスパッタリング、ECRスパッタリングなどを単独で又は2種以上組み合わせて用いることができる。また、ターゲットの印加方式はターゲット種に応じて適宜選択され、DC(直流)スパッタリング、及びRF(高周波)スパッタリングのいずれを用いてもよい。
スパッタ法は、遷移金属の単体又はその酸化物とを含む複数のスパッタリングターゲットを用いた多元同時スパッタであってもよい。これらのスパッタリングターゲットを作製する方法や、これらのスパッタリングターゲットを用いて複合酸化物からなる薄膜を作製する方法については、例えば、特開2000−160331号公報、特開2004−068109号公報、特開2013−047361号公報などの記載が適宜参照されうる。
前記遷移金属を含有するガスバリアー層の厚さは、1〜500nmの範囲が好ましい、より好ましくは10〜300nmの範囲である。
〔5〕アンカー層、密着層
〔5.1〕アンカー層
本発明においては、樹脂基材上に少なくとも1層のアンカー層を有することが、樹脂基材と下地層(Y)との密着性を向上し、使用環境変動における機械的又は熱的ストレスによる層の損傷や欠陥を防ぐ観点から、好ましい態様である。
〔5.1〕アンカー層
本発明においては、樹脂基材上に少なくとも1層のアンカー層を有することが、樹脂基材と下地層(Y)との密着性を向上し、使用環境変動における機械的又は熱的ストレスによる層の損傷や欠陥を防ぐ観点から、好ましい態様である。
本発明に用いられるアンカー層は樹脂を含有する有機ポリマー層であることが好ましく、用いられる樹脂としては、ポリエステル樹脂、イソシアネート樹脂、ウレタン樹脂、アクリル樹脂、エチレンビニルアルコール樹脂、ビニル変性樹脂、エポキシ樹脂、変性スチレン樹脂、変性シリコーン樹脂、及びアルキルチタネート等を単独で又は2種以上組み合わせて使用することができる。
好ましくは、下記重合性化合物とシランカップリング剤と重合開始剤を含む重合性組成物を層状にした後硬化して形成することもできる。
重合性組成物を層状にする方法としては、本発明では樹脂基材上に重合性組成物を塗布して形成することができる。塗布組成物を塗布する方法としては、任意の適切な方法が採用され得る。具体的には例えば、スピンコート法、ロールコート法、フローコート法、インクジェット法、スプレーコート法、プリント法、ディップコート法、流延成膜法、バーコート法、グラビア印刷法等が挙げられる。塗布液を塗布した後は、塗膜を乾燥させることが好ましい。塗膜を乾燥することによって、塗膜中に含有される有機溶媒を除去することができる。形成方法については、従来公知である特開2014−151571号公報の段落「0058」〜「0064」、特開2011−183773号公報の段落「0052」〜「0056」等を参照して採用することができる。
上記塗布法の中では、電子デバイスは水分や親水性溶媒によって劣化する懸念があるため、一般的な溶媒塗布は好ましくなく、窒素雰囲気下、無溶媒、若しくは、親水性溶媒の含有量が少ない塗布組成物を用いたインクジェット方式を好ましく適用することができる。当該インクジェット方式は、例えば、国際公開第2014/176365号、国際公開第2015/100375号、国際公開第2015/112454号等に記載の技術内容を参照して採用することができる。具体的には、KATEEVA社製のYIELDjet(登録商標)Platformを用いてアンカー層を形成することも好ましい実施態様である。
また、重合性組成物を層状にする別の方法としては、公知のフラッシュ蒸着法といった気相成膜法を用いることができる。
例えば、重合性化合物とシランカップリング剤と重合開始剤を含む重合性組成物を、減圧雰囲気下において、加熱によって揮発させて基材、電極層又は有機機能層上に蒸着膜として形成することが好ましい。
当該蒸着膜を形成する方法は、特開2008−142941号公報、特開2004−314626号公報等に記載されているような公知の方法を用いることができる。
一例として、真空装置内に基材及びその上に形成された有機機能層を設置し、真空装置の中に設置された加熱ボートに前記重合性組成物を入れ、10Pa程度の減圧下、前記重合性組成物を200℃程度に加熱し、基材及び有機機能層を被覆しながら、所望の層厚になるように蒸着膜を形成することができる。
得られた蒸着膜に真空環境下で高圧水銀灯等を用いて紫外線を照射して、蒸着した重合性組成物を硬化させてアンカー層を形成する。
(重合性化合物)
本発明で用いられる重合性化合物は、エチレン性不飽和結合を末端又は側鎖に有する化合物、又はエポキシ又はオキセタンを末端又は側鎖に有する化合物である。これらのうち、エチレン性不飽和結合を末端又は側鎖に有する化合物が好ましい。エチレン性不飽和結合を末端又は側鎖に有する化合物の例としては、(メタ)アクリレート系化合物、アクリルアミド系化合物、スチレン系化合物、無水マレイン酸等が挙げられ、(メタ)アクリレート系化合物が好ましい。(メタ)アクリレート系化合物としては、(メタ)アクリレート、ウレタン(メタ)アクリレートやポリエステル(メタ)アクリレート、エポキシ(メタ)アクリレート等が好ましい。スチレン系化合物としては、スチレン、α−メチルスチレン、4−メチルスチレン、ジビニルベンゼン、4−ヒドロキシスチレン、4−カルボキシスチレン等が好ましい。
本発明で用いられる重合性化合物は、エチレン性不飽和結合を末端又は側鎖に有する化合物、又はエポキシ又はオキセタンを末端又は側鎖に有する化合物である。これらのうち、エチレン性不飽和結合を末端又は側鎖に有する化合物が好ましい。エチレン性不飽和結合を末端又は側鎖に有する化合物の例としては、(メタ)アクリレート系化合物、アクリルアミド系化合物、スチレン系化合物、無水マレイン酸等が挙げられ、(メタ)アクリレート系化合物が好ましい。(メタ)アクリレート系化合物としては、(メタ)アクリレート、ウレタン(メタ)アクリレートやポリエステル(メタ)アクリレート、エポキシ(メタ)アクリレート等が好ましい。スチレン系化合物としては、スチレン、α−メチルスチレン、4−メチルスチレン、ジビニルベンゼン、4−ヒドロキシスチレン、4−カルボキシスチレン等が好ましい。
(シランカップリング剤)
本発明で用いられるシランカップリング剤は、例えば、ハロゲン含有シランカップリング剤(2−クロロエチルトリメトキシシラン、2−クロロエチルトリエトキシシラン、3−クロロプロピルトリメトキシシラン、3−クロロプロピルトリエトキシシランなど)、エポキシ基含有シランカップリング剤[2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリエトキシシラン、3−(3,4−エポキシシクロヘキシル)プロピルトリメトキシシラン、2−グリシジルオキシエチルトリメトキシシラン、2−グリシジルオキシエチルトリエトキシシラン、3−グリシジルオキシプロピルトリメトキシシラン、3−グリシジルオキシプロピルトリエトキシシランなど]、アミノ基含有シランカップリング剤(2−アミノエチルトリメトキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、2−[N−(2−アミノエチル)アミノ]エチルトリメトキシシラン、3−[N−(2−アミノエチル)アミノ]プロピルトリメトキシシラン、3−(2−アミノエチル)アミノ]プロピルトリエトキシシラン、3−[N−(2−アミノエチル)アミノ]プロピル メチルジメトキシシランなど)、メルカプト基含有シランカップリング剤(2−メルカプトエチルトリメトキシシラン、3−メルカプトプロピルトリメトキシシラン、3−メルカプトプロピルトリエトキシシランなど)、ビニル基含有シランカップリング剤(ビニルトリメトキシシラン、ビニルトリエトキシシランなど)、(メタ)アクリロイル基含有シランカップリング剤(2−メタクリロイルオキシエチルトリメトキシシラン、2−メタクリロイルオキシエチルトリエトキシシラン、2−アクリロイルオキシエチルトリメトキシシラン、3−メタクリロイルオキシプロピルトリメトキシシラン、3−メタクリロイルオキシプロピルトリエトキシシラン、3−アクリロイルオキシプロピルトリメトキシシランなど)などを挙げることができる。これらの中では、(メタ)アクリロイル基を含有するシランカップリング剤((メタ)アクリロイル基含有シランカップリング剤)が好ましく用いられる。
本発明で用いられるシランカップリング剤は、例えば、ハロゲン含有シランカップリング剤(2−クロロエチルトリメトキシシラン、2−クロロエチルトリエトキシシラン、3−クロロプロピルトリメトキシシラン、3−クロロプロピルトリエトキシシランなど)、エポキシ基含有シランカップリング剤[2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリエトキシシラン、3−(3,4−エポキシシクロヘキシル)プロピルトリメトキシシラン、2−グリシジルオキシエチルトリメトキシシラン、2−グリシジルオキシエチルトリエトキシシラン、3−グリシジルオキシプロピルトリメトキシシラン、3−グリシジルオキシプロピルトリエトキシシランなど]、アミノ基含有シランカップリング剤(2−アミノエチルトリメトキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、2−[N−(2−アミノエチル)アミノ]エチルトリメトキシシラン、3−[N−(2−アミノエチル)アミノ]プロピルトリメトキシシラン、3−(2−アミノエチル)アミノ]プロピルトリエトキシシラン、3−[N−(2−アミノエチル)アミノ]プロピル メチルジメトキシシランなど)、メルカプト基含有シランカップリング剤(2−メルカプトエチルトリメトキシシラン、3−メルカプトプロピルトリメトキシシラン、3−メルカプトプロピルトリエトキシシランなど)、ビニル基含有シランカップリング剤(ビニルトリメトキシシラン、ビニルトリエトキシシランなど)、(メタ)アクリロイル基含有シランカップリング剤(2−メタクリロイルオキシエチルトリメトキシシラン、2−メタクリロイルオキシエチルトリエトキシシラン、2−アクリロイルオキシエチルトリメトキシシラン、3−メタクリロイルオキシプロピルトリメトキシシラン、3−メタクリロイルオキシプロピルトリエトキシシラン、3−アクリロイルオキシプロピルトリメトキシシランなど)などを挙げることができる。これらの中では、(メタ)アクリロイル基を含有するシランカップリング剤((メタ)アクリロイル基含有シランカップリング剤)が好ましく用いられる。
また、その他の(メタ)アクリロイル基含有シランカップリング剤としては、1,3−ビス(アクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(メタクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−アクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−メタクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザン、アクリロイルオキシメチルメチルトリシラザン、メタクリロイルオキシメチルメチルトリシラザン、アクリロイルオキシメチルメチルテトラシラザン、メタクリロイルオキシメチルメチルテトラシラザン、アクリロイルオキシメチルメチルポリシラザン、メタクリロイルオキシメチルメチルポリシラザン、3−アクリロイルオキシプロピルメチルトリシラザン、3−メタクリロイルオキシプロピルメチルトリシラザン、3−アクリロイルオキシプロピルメチルテトラシラザン、3−メタクリロイルオキシプロピルメチルテトラシラザン、3−アクリロイルオキシプロピルメチルポリシラザン、3−メタクリロイルオキシプロピルメチルポリシラザン、アクリロイルオキシメチルポリシラザン、メタクリロイルオキシメチルポリシラザン、3−アクリロイルオキシプロピルポリシラザン、3−メタクリロイルオキシプロピルポリシラザンが好ましく、更に、化合物の合成・同定が容易であるといった観点から、1,3−ビス(アクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(メタクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−アクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−メタクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザンが特に好ましい。
本発明で用いられるシランカップリング剤は、下記一般式で表される化合物が好ましく用いられるが、当該シランカップリング剤の合成方法は、特開2009−67778号公報を参照することができる。

(重合開始剤)
本発明における重合性組成物は、通常、重合開始剤を含む。重合開始剤を用いる場合、その含有量は、重合に関与する化合物の合計量の0.1モル%以上であることが好ましく、0.5〜2モル%であることがより好ましい。このような組成とすることにより、活性成分生成反応を経由する重合反応を適切に制御することができる。光重合開始剤の例としてはBASFジャパン社から市販されているイルガキュア(Irgacure)シリーズ(例えば、イルガキュア651、イルガキュア754、イルガキュア184、イルガキュア2959、イルガキュア907、イルガキュア369、イルガキュア379、イルガキュア819など)、ダロキュア(Darocure)シリーズ(例えば、ダロキュアTPO、ダロキュア1173など)、クオンタキュア(Quantacure)PDO、ランベルティ(Lamberti)社から市販されているエザキュア(Ezacure)シリーズ(例えば、エザキュアTZM、エザキュアTZT、エザキュアKTO46など)等が挙げられる。
本発明における重合性組成物は、通常、重合開始剤を含む。重合開始剤を用いる場合、その含有量は、重合に関与する化合物の合計量の0.1モル%以上であることが好ましく、0.5〜2モル%であることがより好ましい。このような組成とすることにより、活性成分生成反応を経由する重合反応を適切に制御することができる。光重合開始剤の例としてはBASFジャパン社から市販されているイルガキュア(Irgacure)シリーズ(例えば、イルガキュア651、イルガキュア754、イルガキュア184、イルガキュア2959、イルガキュア907、イルガキュア369、イルガキュア379、イルガキュア819など)、ダロキュア(Darocure)シリーズ(例えば、ダロキュアTPO、ダロキュア1173など)、クオンタキュア(Quantacure)PDO、ランベルティ(Lamberti)社から市販されているエザキュア(Ezacure)シリーズ(例えば、エザキュアTZM、エザキュアTZT、エザキュアKTO46など)等が挙げられる。
本発明では、シランカップリング剤と重合性化合物と重合開始剤を含む重合性組成物を、光(例えば、紫外線)、電子線、又は熱線にて、硬化させるが、光によって硬化させることが好ましい。特に、重合性組成物を25℃以上の温度(例えば、30〜130℃)をかけて加熱した後に、硬化させることが好ましい。このような構成とすることにより、シランカップリング剤の加水分解反応を進行させ、重合性組成物を効果的に硬化させかつ、基材や有機機能層等にダメージを与えずに成膜することができる。
照射する光は、通常、高圧水銀灯若しくは低圧水銀灯による紫外線である。照射エネルギーは0.1J/cm2以上が好ましく、0.5J/cm2以上がより好ましい。重合性化合物として、(メタ)アクリレート系化合物を採用する場合、空気中の酸素によって重合阻害を受けるため、重合時の酸素濃度若しくは酸素分圧を低くすることが好ましい。窒素置換法によって重合時の酸素濃度を低下させる場合、酸素濃度は2%以下が好ましく、0.5%以下がより好ましい。減圧法により重合時の酸素分圧を低下させる場合、全圧が1000Pa以下であることが好ましく、100Pa以下であることがより好ましい。また、100Pa以下の減圧条件下で0.5J/cm2以上のエネルギーを照射して紫外線重合を行うのが特に好ましい。
本発明に用いられるアンカー層は、平滑で、膜硬度が高いことが好ましい。アンカー層の平滑性は1μm角の平均粗さ(Ra値)として1nm未満であることが好ましく、0.5nm未満であることがより好ましい。モノマーの重合率は85%以上であることが好ましく、88%以上であることがより好ましく、90%以上であることが更に好ましく、92%以上であることが特に好ましい。ここでいう重合率とはモノマー混合物中の全ての重合性基(例えば、アクリロイル基及びメタクリロイル基)のうち、反応した重合性基の比率を意味する。重合率は赤外線吸収法によって定量することができる。
アンカー層の層厚については特に限定はないが、薄すぎると層厚の均一性を得ることが困難になるし、厚すぎると外力によりクラックを発生してガスバリアー性が低下する。かかる観点から、アンカー層の厚さは50〜2000nmが好ましく、200〜1500nmがより好ましい。
アンカー層の表面にはパーティクル等の異物、突起が無いことが要求される。このため、アンカー層の成膜はクリーンルーム内で行われることが好ましい。クリーン度はクラス10000以下が好ましく、クラス1000以下がより好ましい。
有機層の硬度は高いほうが好ましい。有機層の硬度が高いと、無機層が平滑に成膜されその結果としてガスバリアー性能が向上する。有機層の硬度はナノインデンテーション法に基づく微小硬度として表すことができる。有機層の微小硬度は100N/mm以上であることが好ましく、150N/mm以上であることがより好ましい。
〔5.2〕密着層
本発明に係るガスバリアー層(X)と、後述する有機EL素子の間に平滑層を兼ねた密着層を形成することも、ガスバリアー層(X)と素子間の密着性を向上する観点から好ましい。
本発明に係るガスバリアー層(X)と、後述する有機EL素子の間に平滑層を兼ねた密着層を形成することも、ガスバリアー層(X)と素子間の密着性を向上する観点から好ましい。
密着層としては、重合性基を有する有機ケイ素化合物を含有する密着層を形成することが好ましく、前記密着層の厚さが、5nm以下であることが好ましい。
重合性基を有する有機ケイ素化合物は、特に限定されるものではないが、シランカップリング剤であることが好ましく、例えば、ハロゲン含有シランカップリング剤(2−クロロエチルトリメトキシシラン、2−クロロエチルトリエトキシシラン、3−クロロプロピルトリメトキシシラン、3−クロロプロピルトリエトキシシランなど)、エポキシ基含有シランカップリング剤[2−(3,4−エポキシシクロヘキシル)エチルトリメトキシシラン、2−(3,4−エポキシシクロヘキシル)エチルトリエトキシシラン、3−(3,4−エポキシシクロヘキシル)プロピルトリメトキシシラン、2−グリシジルオキシエチルトリメトキシシラン、2−グリシジルオキシエチルトリエトキシシラン、3−グリシジルオキシプロピルトリメトキシシラン、3−グリシジルオキシプロピルトリエトキシシランなど]、アミノ基含有シランカップリング剤(2−アミノエチルトリメトキシシラン、3−アミノプロピルトリメトキシシラン、3−アミノプロピルトリエトキシシラン、2−[N−(2−アミノエチル)アミノ]エチルトリメトキシシラン、3−[N−(2−アミノエチル)アミノ]プロピルトリメトキシシラン、3−(2−アミノエチル)アミノ]プロピルトリエトキシシラン、3−[N−(2−アミノエチル)アミノ]プロピル メチルジメトキシシランなど)、メルカプト基含有シランカップリング剤(2−メルカプトエチルトリメトキシシラン、3−メルカプトプロピルトリメトキシシラン、3−メルカプトプロピルトリエトキシシランなど)、ビニル基含有シランカップリング剤(ビニルトリメトキシシラン、ビニルトリエトキシシランなど)、(メタ)アクリロイル基含有シランカップリング剤(2−メタクリロイルオキシエチルトリメトキシシラン、2−メタクリロイルオキシエチルトリエトキシシラン、2−アクリロイルオキシエチルトリメトキシシラン、3−メタクリロイルオキシプロピルトリメトキシシラン、3−メタクリロイルオキシプロピルトリエトキシシラン、3−アクリロイルオキシプロピルトリメトキシシランなど)などを挙げることができる。
これらの中では、(メタ)アクリロイル基を含有するシランカップリング剤((メタ)アクリロイル基含有シランカップリング剤)が好ましい。
(メタ)アクリロイル基含有シランカップリング剤としては、1,3−ビス(アクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(メタクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−アクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−メタクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザン、アクリロイルオキシメチルメチルトリシラザン、メタクリロイルオキシメチルメチルトリシラザン、アクリロイルオキシメチルメチルテトラシラザン、メタクリロイルオキシメチルメチルテトラシラザン、アクリロイルオキシメチルメチルポリシラザン、メタクリロイルオキシメチルメチルポリシラザン、3−アクリロイルオキシプロピルメチルトリシラザン、3−メタクリロイルオキシプロピルメチルトリシラザン、3−アクリロイルオキシプロピルメチルテトラシラザン、3−メタクリロイルオキシプロピルメチルテトラシラザン、3−アクリロイルオキシプロピルメチルポリシラザン、3−メタクリロイルオキシプロピルメチルポリシラザン、アクリロイルオキシメチルポリシラザン、メタクリロイルオキシメチルポリシラザン、3−アクリロイルオキシプロピルポリシラザン、3−メタクリロイルオキシプロピルポリシラザンが好ましく、更に、化合物の合成・同定が容易であるといった観点から、1,3−ビス(アクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(メタクリロイルオキシメチル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−アクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザン、1,3−ビス(γ−メタクリロイルオキシプロピル)−1,1,3,3−テトラメチルジシラザンが特に好ましい。
なお、(メタ)アクリロイル基含有シランカップリング剤の市販品としては、KBM−5103、KBM−502、KBM−503、KBE−502、KBE−503、KR−513(信越化学工業社製)などが挙げられる。これらの(メタ)アクリロイル基含有シランカップリング剤は、1種のみが単独で用いられてもよいし、2種以上が併用されてもよい。
密着層の形成は、重合性組成物を塗布して形成することができ、例えば上記(メタ)アクリロイル基含有化合物を適当な溶媒に溶解させた溶液をガスバリアー層(X)の表面に塗布し、乾燥させる方法が例示される。この際、上記溶液に適当な光重合開始剤を添加しておき、上記溶液を塗布し、乾燥させて得られた塗膜に、光照射処理を施して(メタ)アクリロイル基含有化合物の一部を重合させて重合性ポリマーとしてもよい。
塗布組成物を塗布する方法としては、任意の適切な方法が採用され得る。具体的には例えば、スピンコート法、ロールコート法、フローコート法、インクジェット法、スプレーコート法、プリント法、ディップコート法、流延成膜法、バーコート法、グラビア印刷法等が挙げられる。
また、気相成膜法によって成膜することもでき、気相成膜法は公知の方法で用いることができる。気相成膜法としては、特に制限されず、例えば、スパッタ法、蒸着法、イオンプレーティング法、イオンアシスト蒸着法等の物理気相成長(PVD)法、プラズマCVD法、ALD(Atomic Layer Deposition)法などの化学気相成長(CVD)法が挙げられる。中でも、プラズマCVD法が好ましい。
〔6〕電子デバイス
本発明のガスバリアーフィルムは、優れたガスバリアー、透明性、耐屈曲性を有する。このため、本発明のガスバリアーフィルムは、電子デバイス等のパッケージ、光電変換素子(太陽電池素子)や有機エレクトロルミネッセンス(EL)素子、液晶表示素子等の等の電子デバイスに用いられるガスバリアーフィルム及びこれを用いた電子デバイスなど、様々な用途に使用することができる。
本発明のガスバリアーフィルムは、優れたガスバリアー、透明性、耐屈曲性を有する。このため、本発明のガスバリアーフィルムは、電子デバイス等のパッケージ、光電変換素子(太陽電池素子)や有機エレクトロルミネッセンス(EL)素子、液晶表示素子等の等の電子デバイスに用いられるガスバリアーフィルム及びこれを用いた電子デバイスなど、様々な用途に使用することができる。
すなわち、本発明は、電子デバイス本体と、本発明のガスバリアーフィルム又は本発明の製造方法によって得られるガスバリアーフィルムと、を含む電子デバイスを提供する。
電子デバイスの好ましい適用例とは、液晶表示素子(LCD)、太陽電池(PV)、量子ドットフィルム、有機エレクトロルミネッセンス(EL)素子などをいうが、これらに限定されるものではない。中でも本発明のガスバリアーフィルムは優れたガスバリアー性を有するため、量子ドットフィルム及び有機エレクトロルミネッセンス(EL)素子に好適に用いることができ、特に有機エレクトロルミネッセンス(EL)素子に好適であり、本発明の有機エレクトロルミネッセンスデバイスを提供する。
〔6.1〕量子ドットフィルム
以下、量子ドットフィルム(量子ドット含有樹脂層)の主要な構成要素である量子ドット(QDともいう。)及び樹脂等について説明する。
以下、量子ドットフィルム(量子ドット含有樹脂層)の主要な構成要素である量子ドット(QDともいう。)及び樹脂等について説明する。
〈量子ドット〉
一般に、ナノメートルサイズの半導体物質で量子閉じ込め(quantum confinement)効果を示す半導体ナノ粒子は、「量子ドット」とも称されている。このような量子ドットは、半導体原子が数百個から数千個集まった10数nm程度以内の小さな塊であるが、励起源から光を吸収してエネルギー励起状態に達すると、量子ドットのエネルギーバンドギャップに相当するエネルギーを放出する。
一般に、ナノメートルサイズの半導体物質で量子閉じ込め(quantum confinement)効果を示す半導体ナノ粒子は、「量子ドット」とも称されている。このような量子ドットは、半導体原子が数百個から数千個集まった10数nm程度以内の小さな塊であるが、励起源から光を吸収してエネルギー励起状態に達すると、量子ドットのエネルギーバンドギャップに相当するエネルギーを放出する。
したがって、量子ドットは、量子サイズ効果によりユニークな光学特性を有することが知られている。具体的には、(1)粒子のサイズを制御することにより、様々な波長、色を発光させることができる、(2)吸収帯が広く、単一波長の励起光で様々なサイズの微粒子を発光させることができる、(3)蛍光スペクトルが良好な対称形である、(4)有機色素に比べて耐久性、耐退色性に優れる、といった特徴を有する。
QD含有樹脂層が含有する量子ドットは公知のものであってもよく、当業者に既知の任意の方法を使用して生成することができる。例えば、好適なQD及び好適なQDを形成するための方法には、米国特許第6225198号明細書、米国特許出願公開第2002/0066401号明細書、米国特許第6207229号明細書、同第6322901号明細書、同第6949206号明細書、同第7572393号明細書、同第7267865号明細書、同第7374807号明細書、米国特許出願第11/299299号、及び米国特許第6861155号明細書に記載のものが挙げられる。
QDは、任意の好適な材料、好適には無機材料及びより好適には無機導体又は半導体材料から生成される。好適な半導体材料には、II−VI族、III−V族、IV−VI族及びIV族の半導体を含む、任意の種類の半導体が含まれる。
好適な半導体材料には、Si、Ge、Sn、Se、Te、B、C(ダイアモンドを含む。)、P、BN、BP、BAs、AlN、AlP、AlAs、AlSb、GaN、GaP、GaAs、GaSb、InN、InP、InAs、InSb、AlN、AlP、AlAs、AlSb、GaN、GaP、GaAs、GaSb、ZnO、ZnS、ZnSe、ZnTe、CdS、CdSe、CdSeZn、CdTe、HgS、HgSe、HgTe、BeS、BeSe、BeTe、MgS、MgSe、GeS、GeSe、GeTe、SnS、SnSe、SnTe、PbO、PbS、PbSe、PbTe、CuF、CuCl、CuBr、CuI、Si3N4、Ge3N4、Al2O3、(Al、Ga、In)2(S、Se、Te)3、Al2CO、及び二つ以上のこのような半導体の適切な組合せが含まれるが、これらに限定されない。
本発明においては、次のようなコア/シェル型の量子ドット、例えば、CdSe/ZnS、InP/ZnS、PbSe/PbS、CdSe/CdS、CdTe/CdS、CdTe/ZnS等も好ましく使用できる。
〈樹脂〉
QD含有樹脂層には、QDを保持するバインダーとして樹脂を用いることができる。例えば、ポリカーボネート系、ポリアリレート系、ポリスルホン(ポリエーテルスルホンも含む。)系、ポリエチレンテレフタレート、ポリエチレンナフタレート等のポリエステル系、ポリエチレン系、ポリプロピレン系、セロ−ファン系、セルロースジアセテート、セルローストリアセテート、セルロースアセテートプロピオネート、セルロースアセテートブチレート等のセルロースエステル系、ポリ塩化ビニリデン系、ポリビニルアルコール系、エチレンビニルアルコール系、シンジオタクティックポリスチレン系、ノルボルネン系、ポリメチルペンテン系、ポリエーテルケトン系、ポリエーテルケトンイミド系、ポリアミド樹脂、フッ素樹脂、ナイロン系、ポリメチルメタクリレート等のアクリル系樹脂等を挙げることができる。
QD含有樹脂層には、QDを保持するバインダーとして樹脂を用いることができる。例えば、ポリカーボネート系、ポリアリレート系、ポリスルホン(ポリエーテルスルホンも含む。)系、ポリエチレンテレフタレート、ポリエチレンナフタレート等のポリエステル系、ポリエチレン系、ポリプロピレン系、セロ−ファン系、セルロースジアセテート、セルローストリアセテート、セルロースアセテートプロピオネート、セルロースアセテートブチレート等のセルロースエステル系、ポリ塩化ビニリデン系、ポリビニルアルコール系、エチレンビニルアルコール系、シンジオタクティックポリスチレン系、ノルボルネン系、ポリメチルペンテン系、ポリエーテルケトン系、ポリエーテルケトンイミド系、ポリアミド樹脂、フッ素樹脂、ナイロン系、ポリメチルメタクリレート等のアクリル系樹脂等を挙げることができる。
QD含有樹脂層は、厚さが50〜200μmの範囲内であることが好ましい。
なお、QD含有樹脂層におけるQDの含有量は、使用する化合物によって最適量は異なるが、一般的には15〜60体積%の範囲内であることが好ましい。
〔6.2〕有機EL素子
本発明に係る有機EL素子は、例えば、本発明のガスバリアー層上に、直接又は前記密着層等を介して、陽極、第1有機機能層群、発光層、第2有機機能層群、陰極が積層されて構成されていることが好ましい。第1有機機能層群は、例えば、正孔注入層、正孔輸送層、電子阻止層等から構成され、第2有機機能層群は、例えば、正孔阻止層、電気輸送層、電子注入層等から構成されている。第1有機機能層群及び第2有機機能層群はそれぞれ1層のみで構成されていても良いし、第1有機機能層群及び第2有機機能層群はそれぞれ設けられていなくても良い。
本発明に係る有機EL素子は、例えば、本発明のガスバリアー層上に、直接又は前記密着層等を介して、陽極、第1有機機能層群、発光層、第2有機機能層群、陰極が積層されて構成されていることが好ましい。第1有機機能層群は、例えば、正孔注入層、正孔輸送層、電子阻止層等から構成され、第2有機機能層群は、例えば、正孔阻止層、電気輸送層、電子注入層等から構成されている。第1有機機能層群及び第2有機機能層群はそれぞれ1層のみで構成されていても良いし、第1有機機能層群及び第2有機機能層群はそれぞれ設けられていなくても良い。
以下に、本発明に係る有機EL素子に用いることができる代表的な素子構成として、以下の構成を挙げることができるが、これらに限定されるものではない。
(1)陽極/発光層/陰極
(2)陽極/発光層/電子輸送層/陰極
(3)陽極/正孔輸送層/発光層/陰極
(4)陽極/正孔輸送層/発光層/電子輸送層/陰極
(5)陽極/正孔輸送層/発光層/電子輸送層/電子注入層/陰極
(6)陽極/正孔注入層/正孔輸送層/発光層/電子輸送層/陰極
(7)陽極/正孔注入層/正孔輸送層/(電子阻止層/)発光層/(正孔阻止層/)電子輸送層/電子注入層/陰極
上記の中で(7)の構成が好ましく用いられるが、これに限定されるものではない。
(1)陽極/発光層/陰極
(2)陽極/発光層/電子輸送層/陰極
(3)陽極/正孔輸送層/発光層/陰極
(4)陽極/正孔輸送層/発光層/電子輸送層/陰極
(5)陽極/正孔輸送層/発光層/電子輸送層/電子注入層/陰極
(6)陽極/正孔注入層/正孔輸送層/発光層/電子輸送層/陰極
(7)陽極/正孔注入層/正孔輸送層/(電子阻止層/)発光層/(正孔阻止層/)電子輸送層/電子注入層/陰極
上記の中で(7)の構成が好ましく用いられるが、これに限定されるものではない。
本発明のガスバリアーフィルムを具備する有機EL素子を図で説明する。
図4は、本発明のガスバリアーフィルムを具備する有機EL素子の断面図である。
上記の代表的な素子200の構成において、樹脂基材211、下地層(Y)212、ガスバリアー層(X)213が積層されたガスバリアーフィルム210上に、第1電極215が形成され、次いで陽極と陰極を除く層である発光性を有する有機機能層216、及び第2電極217が形成される。また、本発明に係る有機EL素子では、第1電極215又は第2電極217が、それぞれ陽極又は陰極を構成する。樹脂基材211及び下地層(Y)212間には前記アンカー層(不図示)を設けることも好ましい。
第2電極上には、有機EL素子全体を封止する封止部材221及び封止層222を形成する。
〈有機機能層〉
上記構成において、発光層は、単層又は複数層で構成される。発光層が複数の場合は、各発光層の間に非発光性の中間層を設けてもよい。
上記構成において、発光層は、単層又は複数層で構成される。発光層が複数の場合は、各発光層の間に非発光性の中間層を設けてもよい。
また、必要に応じて、発光層と陰極との間に正孔阻止層(正孔障壁層)や電子注入層(陰極バッファー層)等を設けてもよく、また、発光層と陽極との間に電子阻止層(電子障壁層)や正孔注入層(陽極バッファー層)等を設けてもよい。
電子輸送層は、電子を輸送する機能を有する層である。電子輸送層には、広い意味で電子注入層及び正孔阻止層も含まれる。また、電子輸送層は、複数層で構成されていてもよい。
正孔輸送層は、正孔を輸送する機能を有する層である。正孔輸送層には、広い意味で正孔注入層及び電子阻止層も含まれる。また、正孔輸送層は、複数層で構成されていてもよい。
〈タンデム構造〉
また、前記有機機能層は、少なくとも1層の発光層を含む有機機能層を複数積層した、いわゆるタンデム構造の素子であってもよい。
また、前記有機機能層は、少なくとも1層の発光層を含む有機機能層を複数積層した、いわゆるタンデム構造の素子であってもよい。
タンデム構造の代表的な素子構成としては、例えば以下の構成を挙げることができる。
(1)陽極/第1有機機能層/中間機能層/第2有機機能層/陰極
(2)陽極/第1有機機能層/中間機能層/第2有機機能層/中間機能層/第3有機機能層/陰極
ここで、上記第1有機機能層、第2有機機能層及び第3有機機能層は全て同じであっても、異なっていてもよい。また、二つの有機機能層が同じであり、残る一つが異なっていてもよい。
(1)陽極/第1有機機能層/中間機能層/第2有機機能層/陰極
(2)陽極/第1有機機能層/中間機能層/第2有機機能層/中間機能層/第3有機機能層/陰極
ここで、上記第1有機機能層、第2有機機能層及び第3有機機能層は全て同じであっても、異なっていてもよい。また、二つの有機機能層が同じであり、残る一つが異なっていてもよい。
また、各有機機能層は直接積層されていても、中間機能層を介して積層されていてもよい。中間機能層は、例えば、中間電極、中間導電層、電荷発生層、電子引抜層、接続層又は中間絶縁層等から構成され、陽極側の隣接層に電子を、陰極側の隣接層に正孔を供給する機能を持った層であれば、公知の材料構成を用いることができる。
中間機能層に用いられる材料としては、例えば、ITO(インジウム・スズ酸化物)、IZO(インジウム・亜鉛酸化物)、ZnO2、TiN、ZrN、HfN、TiOX、VOX、CuI、InN、GaN、CuAlO2、CuGaO2、SrCu2O2、LaB6、RuO2、Al等の導電性無機化合物層や、Au/Bi2O3等の2層膜や、SnO2/Ag/SnO2、ZnO/Ag/ZnO、Bi2O3/Au/Bi2O3、TiO2/TiN/TiO2、TiO2/ZrN/TiO2等の多層膜、またC60等のフラーレン類、オリゴチオフェン等の導電性有機物層、金属フタロシアニン類、無金属フタロシアニン類、金属ポルフィリン類、無金属ポルフィリン類等の導電性有機化合物層等が挙げられるが、本発明はこれらに限定されない。
タンデム型の有機機能層の具体例としては、例えば、米国特許第6337492号明細書、米国特許第7420203号明細書、米国特許第7473923号明細書、米国特許第6872472号明細書、米国特許第6107734号明細書、米国特許第6337492号明細書、国際公開第2005/009087号、特開2006−228712号公報、特開2006−24791号公報、特開2006−49393号公報、特開2006−49394号公報、特開2006−49396号公報、特開2011−96679号公報、特開2005−340187号公報、特許第4711424号、特許第3496681号、特許第3884564号、特許第4213169号、特開2010−192719号公報、特開2009−076929号公報、特開2008−078414号公報、特開2007−059848号公報、特開2003−272860号公報、特開2003−045676号公報、国際公開第2005/094130号等に記載の素子構成や構成材料等が挙げられるが、これらに限定されない。
〈発光層〉
本発明に係る有機EL素子に用いる発光層は、電極又は隣接層から注入される電子と正孔とが再結合し、励起子を経由して発光する場を提供する層である。発光層において、発光する部分は発光層の層内であっても、発光層と隣接層との界面であってもよい。
本発明に係る有機EL素子に用いる発光層は、電極又は隣接層から注入される電子と正孔とが再結合し、励起子を経由して発光する場を提供する層である。発光層において、発光する部分は発光層の層内であっても、発光層と隣接層との界面であってもよい。
発光層の厚さの総和は、特に制限されず、形成する膜の均質性、発光時に必要とされる電圧及び駆動電流に対する発光色の安定性等の観点から決められる。発光層の厚さの総和は、例えば、2nm〜5μmの範囲に調整することが好ましく、より好ましくは2〜500nmの範囲に調整され、更に好ましくは5〜200nmの範囲に調整される。また、発光層の個々の層厚としては、2nm〜1μmの範囲に調整することが好ましく、より好ましくは2〜200nmの範囲に調整され、更に好ましくは3〜150nmの範囲に調整される。
発光層は、発光ドーパント(発光性ドーパント化合物、ドーパント化合物、単にドーパントともいう。)と、ホスト化合物(マトリックス材料、発光ホスト化合物、単にホストともいう。)とを含有することが好ましい。
〈発光ドーパント〉
発光層に用いられる発光ドーパントとしては、蛍光発光性ドーパント(蛍光ドーパント、蛍光性化合物ともいう。)及びリン光発光性ドーパント(リン光ドーパント、リン光性化合物ともいう。)が好ましく用いられる。これらのうち、少なくとも1層の発光層がリン光発光ドーパントを含有することが好ましい。
発光層に用いられる発光ドーパントとしては、蛍光発光性ドーパント(蛍光ドーパント、蛍光性化合物ともいう。)及びリン光発光性ドーパント(リン光ドーパント、リン光性化合物ともいう。)が好ましく用いられる。これらのうち、少なくとも1層の発光層がリン光発光ドーパントを含有することが好ましい。
発光層中の発光ドーパントの濃度については、使用される特定のドーパント及びデバイスの必要条件に基づいて、任意に決定することができる。光ドーパントの濃度は、発光層の層厚方向に対し、均一な濃度で含有されていてもよく、また任意の濃度分布を有していてもよい。
また、発光層は、複数種の発光ドーパントが含まれていてもよい。例えば、構造の異なるドーパント同士の組み合わせや、蛍光発光性ドーパントとリン光発光性ドーパントとを組み合わせて用いてもよい。これにより、任意の発光色を得ることができる。
本発明に係る有機EL素子が発光する色は、「新編色彩科学ハンドブック」(日本色彩学会編、東京大学出版会、1985)の108頁の図4.16において、分光放射輝度計CS−2000(コニカミノルタ(株)製)で測定した結果をCIE色度座標に当てはめたときの色で決定される。
本発明に係る有機EL素子は、1層又は複数層の発光層が、発光色の異なる複数の発光ドーパントを含有し、白色発光を示すことも好ましい。白色を示す発光ドーパントの組み合わせについては特に限定はないが、例えば青と橙や、青と緑と赤の組み合わせ等が挙げられる。
本発明に係る有機EL素子における白色としては、2度視野角正面輝度を前述の方法により測定した際に、1000cd/m2でのCIE1931表色系における色度がx=0.39±0.09、y=0.38±0.08の領域内にあることが好ましい。
〈リン光発光性ドーパント〉
リン光発光性ドーパントは、励起三重項からの発光が観測される化合物であり、具体的には、室温(25℃)にてリン光発光する化合物であり、25℃においてリン光量子収率が0.01以上の化合物である。発光層に用いるリン光発光性ドーパントにおいて、好ましいリン光量子収率は0.1以上である。
リン光発光性ドーパントは、励起三重項からの発光が観測される化合物であり、具体的には、室温(25℃)にてリン光発光する化合物であり、25℃においてリン光量子収率が0.01以上の化合物である。発光層に用いるリン光発光性ドーパントにおいて、好ましいリン光量子収率は0.1以上である。
上記リン光量子収率は、第4版実験化学講座7の分光IIの398頁(1992年版、丸善)に記載の方法により測定できる。溶液中でのリン光量子収率は種々の溶媒を用いて測定できる。発光層に用いるリン光発光性ドーパントは、任意の溶媒のいずれかにおいて上記リン光量子収率(0.01以上)が達成されればよい。
リン光発光性ドーパントの発光は、原理として2種挙げられる。
一つは、キャリアが輸送されるホスト化合物上で、キャリアの再結合によるホスト化合物の励起状態が生成される。このエネルギーをリン光発光性ドーパントに移動させることでリン光発光性ドーパントからの発光を得るというエネルギー移動型である。もう一つは、リン光発光性ドーパントがキャリアトラップとなり、リン光発光性ドーパント上でキャリアの再結合が起こり、リン光発光性ドーパントからの発光が得られるというキャリアトラップ型である。いずれの場合においても、リン光発光性ドーパントの励起状態のエネルギーは、ホスト化合物の励起状態のエネルギーよりも低いことが条件となる。
リン光発光性ドーパントは、本発明に係る有機EL素子の発光層に使用される公知の材料から適宜選択して用いることができる。
公知のリン光発光性ドーパントの具体例としては、以下の文献に記載されている化合物等が挙げられる。
Nature,395,151(1998)、Appl.Phys.Lett.,78,1622(2001)、Adv.Mater.,19,739(2007)、Chem.Mater.,17,3532(2005)、Adv.Mater.,17,1059(2005)、国際公開第2009/100991号、国際公開第2008/101842号、国際公開第2003/040257号、米国特許出願公開第2006/0202194号明細書、米国特許出願公開第2007/0087321号明細書、米国特許出願公開第2005/0244673号明細書、Inorg.Chem.,40,1704(2001)、Chem.Mater.,16,2480(2004)、Adv.Mater.,16,2003(2004)、Angew.Chem.lnt.Ed.,2006,45,7800、Appl.Phys.Lett.,86,153505(2005)、Chem.Lett.,34,592(2005)、Chem.Commun.,2906(2005)、Inorg.Chem.,42,1248(2003)、国際公開第2009/050290号、国際公開第2002/015645号、国際公開第2009/000673号、米国特許出願公開第2002/0034656号明細書、米国特許第7332232号明細書、米国特許出願公開第2009/0108737号明細書、米国特許出願公開第2009/0039776号明細書、米国特許第6921915号明細書、米国特許第6687266号明細書、米国特許出願公開第2007/0190359号明細書、米国特許出願公開第2006/0008670号明細書、米国特許出願公開第2009/0165846号明細書、米国特許出願公開第2008/0015355号明細書、米国特許第7250226号明細書、米国特許第7396598号明細書、米国特許出願公開第2006/0263635号明細書、米国特許出願公開第2003/0138657号明細書、米国特許出願公開第2003/0152802号明細書、米国特許第7090928号明細書、Angew.Chem.lnt.Ed.,47,1(2008)、Chem.Mater.,18,5119(2006)、Inorg.Chem.,46,4308(2007)、Organometallics,23,3745(2004)、Appl.Phys.Lett.,74,1361(1999)、国際公開第2002/002714号、国際公開第2006/009024号、国際公開第2006/056418号、国際公開第2005/019373号、国際公開第2005/123873号、国際公開第2005/123873号、国際公開第2007/004380号、国際公開第2006/082742号、米国特許出願公開第2006/0251923号明細書、米国特許出願公開第2005/0260441号明細書、米国特許第7393599号明細書、米国特許第7534505号明細書、米国特許第7445855号明細書、米国特許出願公開第2007/0190359号明細書、米国特許出願公開第2008/0297033号明細書、米国特許第7338722号明細書、米国特許出願公開第2002/0134984号明細書、米国特許第7279704号明細書、国際公開第2005/076380号、国際公開第2010/032663号、国際公開第2008/140115号、国際公開第2007/052431号、国際公開第2011/134013号、国際公開第2011/157339号、国際公開第2010/086089号、国際公開第2009/113646号、国際公開第2012/020327号、国際公開第2011/051404号、国際公開第2011/004639号、国際公開第2011/073149号、特開2012−069737号公報、特開2012−195554号公報、特開2009−114086号公報、特開2003−81988号公報、特開2002−302671号公報、特開2002−363552号公報等である。
中でも、好ましいリン光発光性ドーパントとしては、Irを中心金属に有する有機金属錯体が挙げられる。さらに好ましくは、金属−炭素結合、金属−窒素結合、金属−酸素結合、金属−硫黄結合の少なくとも一つの配位様式を含む錯体が好ましい。
〈蛍光発光性ドーパント〉
蛍光発光性ドーパントは、励起一重項からの発光が可能な化合物であり、励起一重項からの発光が観測される限り特に限定されない。
蛍光発光性ドーパントは、励起一重項からの発光が可能な化合物であり、励起一重項からの発光が観測される限り特に限定されない。
蛍光発光性ドーパントとしては、例えば、アントラセン誘導体、ピレン誘導体、クリセン誘導体、フルオランテン誘導体、ペリレン誘導体、フルオレン誘導体、アリールアセチレン誘導体、スチリルアリーレン誘導体、スチリルアミン誘導体、アリールアミン誘導体、ホウ素錯体、クマリン誘導体、ピラン誘導体、シアニン誘導体、クロコニウム誘導体、スクアリウム誘導体、オキソベンツアントラセン誘導体、フルオレセイン誘導体、ローダミン誘導体、ピリリウム誘導体、ペリレン誘導体、ポリチオフェン誘導体及び希土類錯体系化合物等が挙げられる。
また、蛍光発光性ドーパントとして、遅延蛍光を利用した発光ドーパント等を用いてもよい。
遅延蛍光を利用した発光ドーパントの具体例としては、例えば、国際公開第2011/156793号、特開2011−213643号公報、特開2010−93181号公報等に記載の化合物が挙げられる。
〈ホスト化合物〉
ホスト化合物は、発光層において主に電荷の注入及び輸送を担う化合物であり、有機EL素子においてそれ自体の発光は実質的に観測されない。
ホスト化合物は、発光層において主に電荷の注入及び輸送を担う化合物であり、有機EL素子においてそれ自体の発光は実質的に観測されない。
好ましくは室温(25℃)においてリン光発光のリン光量子収率が、0.1未満の化合物であり、さらに好ましくは、リン光量子収率が0.01未満の化合物である。また、発光層に含有される化合物の内で、その層中での質量比が20%以上であることが好ましい。
また、ホスト化合物の励起状態エネルギーは、同一層内に含有される発光ドーパントの励起状態エネルギーよりも高いことが好ましい。
ホスト化合物は、単独で用いてもよく、又は複数種併用して用いてもよい。ホスト化合物を複数種用いることで、電荷の移動を調整することが可能であり、有機EL素子の高効率化が可能となる。
発光層に用いるホスト化合物としては、特に制限はなく、従来の有機EL素子で用いられる化合物を用いることができる。例えば、低分子化合物や、繰り返し単位を有する高分子化合物でもよく、又はビニル基やエポキシ基のような反応性基を有する化合物でもよい。
公知のホスト化合物としては、正孔輸送能又は電子輸送能を有しつつ、発光の長波長化を防ぎ、さらに、有機EL素子を高温駆動時や素子駆動中の発熱に対する安定性の観点から、高いガラス転移温度(Tg)を有することが好ましい。ホスト化合物としては、Tgが90℃以上であることが好ましく、より好ましくは120℃以上である。
ここで、ガラス転移点(Tg)とは、DSC(Differential Scanning Colorimetry:示差走査熱量法)を用いて、JIS K 7121−2012に準拠した方法により求められる値である。
本発明に係る有機EL素子に用いられる、公知のホスト化合物の具体例としては、以下の文献に記載の化合物等が挙げられるが、本発明はこれらに限定されない。
特開2001−257076号公報、同2002−308855号公報、同2001−313179号公報、同2002−319491号公報、同2001−357977号公報、同2002−334786号公報、同2002−8860号公報、同2002−334787号公報、同2002−15871号公報、同2002−334788号公報、同2002−43056号公報、同2002−334789号公報、同2002−75645号公報、同2002−338579号公報、同2002−105445号公報、同2002−343568号公報、同2002−141173号公報、同2002−352957号公報、同2002−203683号公報、同2002−363227号公報、同2002−231453号公報、同2003−3165号公報、同2002−234888号公報、同2003−27048号公報、同2002−255934号公報、同2002−260861号公報、同2002−280183号公報、同2002−299060号公報、同2002−302516号公報、同2002−305083号公報、同2002−305084号公報、同2002−308837号公報、米国特許出願公開第2003/0175553号明細書、米国特許出願公開第2006/0280965号明細書、米国特許出願公開第2005/0112407号明細書、米国特許出願公開第2009/0017330号明細書、米国特許出願公開第2009/0030202号明細書、米国特許出願公開第2005/0238919号明細書、国際公開第2001/039234号、国際公開第2009/021126号、国際公開第2008/056746号、国際公開第2004/093207号、国際公開第2005/089025号、国際公開第2007/063796号、国際公開第2007/063754号、国際公開第2004/107822号、国際公開第2005/030900号、国際公開第2006/114966号、国際公開第2009/086028号、国際公開第2009/003898号、国際公開第2012/023947号、特開2008−074939号公報、特開2007−254297号公報、EP2034538号明細書等である。
〈電子輸送層〉
本発明に係る有機EL素子における電子輸送とは、電子を輸送する機能を有する材料により行われ、陰極より注入された電子を発光層に伝達する機能を有する。
本発明に係る有機EL素子における電子輸送とは、電子を輸送する機能を有する材料により行われ、陰極より注入された電子を発光層に伝達する機能を有する。
電子輸送材料は単独で用いてもよく、また複数種を併用して用いてもよい。
電子輸送層の総層厚については特に制限はないが、通常は2nm〜5μmの範囲であり、より好ましくは2〜500nmであり、さらに好ましくは5〜200nmである。
また、本発明に係る有機EL素子においては、発光層で生じた光を電極から取り出す際、発光層から直接取り出される光と、光を取り出す電極と対極に位置する電極で反射されてから取り出される光とが、干渉を起こすことが知られている。光が陰極で反射される場合は、電子輸送層の総層厚を数nm〜数μmの間で適宜調整することにより、この干渉効果を効率的に利用することが可能である。
一方で、電子輸送層の層厚を厚くすると電圧が上昇しやすくなるため、特に層厚が厚い場合においては、電子輸送層の電子移動度は10−5cm2/Vs以上であることが好ましい。
電子輸送層に用いられる材料(以下、電子輸送材料という。)としては、電子の注入性若しくは輸送性又は正孔の障壁性のいずれかを有していればよく、従来公知の化合物の中から任意のものを選択して用いることができる。
例えば、含窒素芳香族複素環誘導体、芳香族炭化水素環誘導体、ジベンゾフラン誘導体、ジベンゾチオフェン誘導体及びシロール誘導体等が挙げられる。
上記含窒素芳香族複素環誘導体としては、カルバゾール誘導体、アザカルバゾール誘導体(カルバゾール環を構成する炭素原子の一つ以上が窒素原子に置換)、ピリジン誘導体、ピリミジン誘導体、ピラジン誘導体、ピリダジン誘導体、トリアジン誘導体、キノリン誘導体、キノキサリン誘導体、フェナントロリン誘導体、アザトリフェニレン誘導体、オキサゾール誘導体、チアゾール誘導体、オキサジアゾール誘導体、チアジアゾール誘導体、トリアゾール誘導体、ベンズイミダゾール誘導体、ベンズオキサゾール誘導体及びベンズチアゾール誘導体等が挙げられる。
芳香族炭化水素環誘導体としては、ナフタレン誘導体、アントラセン誘導体及びトリフェニレン等が挙げられる。
また、配位子にキノリノール骨格やジベンゾキノリノール骨格を有する金属錯体、例えば、トリス(8−キノリノール)アルミニウム(Alq3)、トリス(5,7−ジクロロ−8−キノリノール)アルミニウム、トリス(5,7−ジブロモ−8−キノリノール)アルミニウム、トリス(2−メチル−8−キノリノール)アルミニウム、トリス(5−メチル−8−キノリノール)アルミニウム、ビス(8−キノリノール)亜鉛(Znq)等及びこれらの金属錯体の中心金属がIn、Mg、Cu、Ca、Sn、Ga又はPbに置き替わった金属錯体も、電子輸送材料として用いることができる。
その他、メタルフリー若しくはメタルフタロシアニン又はそれらの末端がアルキル基やスルホン酸基等で置換されているものも、電子輸送材料として好ましく用いることができる。また、発光層の材料として例示したジスチリルピラジン誘導体も、電子輸送材料として用いることができるし、正孔注入層、正孔輸送層と同様にn型−Si、n型−SiC等の無機半導体も電子輸送材料として用いることができる。
また、これらの材料を高分子鎖に導入した、又はこれらの材料を高分子の主鎖とした高分子材料を用いることもできる。
本発明に係る有機EL素子では、ゲスト材料として電子輸送層にドープ材をドープして、n性の高い(電子リッチ)電子輸送層を形成してもよい。ドープ材としては、金属錯体及びハロゲン化金属等の金属化合物や、その他のn型ドーパントが挙げられる。
このような構成の電子輸送層の具体例としては、例えば、特開平4−297076号公報、同10−270172号公報、特開2000−196140号公報、同2001−102175号公報、J.Appl.Phys.,95,5773(2004)等の文献に記載されたものが挙げられる。
本発明に係る有機EL素子に用いられる、公知の好ましい電子輸送材料の具体例としては、以下の文献に記載の化合物等が挙げられるが、これらに限定されない。
米国特許第6528187号明細書、米国特許第7230107号明細書、米国特許出願公開第2005/0025993号明細書、米国特許出願公開第2004/0036077号明細書、米国特許出願公開第2009/0115316号明細書、米国特許出願公開第2009/0101870号明細書、米国特許出願公開第2009/0179554号明細書、国際公開第2003/060956号明細書、国際公開第2008/132085号、Appl.Phys.Lett.,75,4(1999)、Appl.Phys.Lett.,79,449(2001)、Appl.Phys.Lett.,81,162(2002)、Appl.Phys.Lett.,81,162(2002)、Appl.Phys.Lett.,79,156(2001)、米国特許第7964293号明細書、国際公開第2004/080975号、国際公開第2004/063159号、国際公開第2005/085387号、国際公開第2006/067931号、国際公開第2007/086552号、国際公開第2008/114690号、国際公開第2009/069442号、国際公開第2009/066779号、国際公開第2009/054253号、国際公開第2011/086935号、国際公開第2010/150593号、国際公開第2010/047707号、EP2311826号、特開2010−251675号公報、特開2009−209133号公報、特開2009−124114号公報、特開2008−277810号公報、特開2006−156445号公報、特開2005−340122号公報、特開2003−45662号公報、特開2003−31367号公報、特開2003−282270号公報、国際公開第2012/115034号等が挙げられる。
より好ましい電子輸送材料としては、ピリジン誘導体、ピリミジン誘導体、ピラジン誘導体、トリアジン誘導体、ジベンゾフラン誘導体、ジベンゾチオフェン誘導体、カルバゾール誘導体、アザカルバゾール誘導体、ベンズイミダゾール誘導体が挙げられる。
〈正孔阻止層〉
正孔阻止層は、広い意味では電子輸送層の機能を有する層である。好ましくは、電子を輸送する機能を有しつつ、正孔を輸送する能力が小さい材料を含有する。電子を輸送しつつ正孔を阻止することで、電子と正孔の再結合確率を向上させることができる。
正孔阻止層は、広い意味では電子輸送層の機能を有する層である。好ましくは、電子を輸送する機能を有しつつ、正孔を輸送する能力が小さい材料を含有する。電子を輸送しつつ正孔を阻止することで、電子と正孔の再結合確率を向上させることができる。
また、上述の電子輸送層の構成を、必要に応じて正孔阻止層として用いることができる。
本発明に係る有機EL素子に設ける正孔阻止層は、発光層の陰極側に隣接して設けられることが好ましい。
本発明に係る有機EL素子において、正孔阻止層の厚さは、好ましくは3〜100nmの範囲であり、さらに好ましくは5〜30nmの範囲である。
正孔阻止層に用いられる材料としては、上述の電子輸送層に用いられる材料が好ましく用いられ、また、上述のホスト化合物として用いられる材料も正孔阻止層に好ましく用いられる。
〈電子注入層〉
電子注入層(「陰極バッファー層」ともいう。)は、駆動電圧低下や発光輝度向上のために陰極と発光層との間に設けられる層である。電子注入層の一例は、「有機EL素子とその工業化最前線(1998年11月30日エヌ・ティー・エス社発行)」の第2編第2章「電極材料」(123〜166頁)に記載されている。
電子注入層(「陰極バッファー層」ともいう。)は、駆動電圧低下や発光輝度向上のために陰極と発光層との間に設けられる層である。電子注入層の一例は、「有機EL素子とその工業化最前線(1998年11月30日エヌ・ティー・エス社発行)」の第2編第2章「電極材料」(123〜166頁)に記載されている。
本発明に係る有機EL素子において、電子注入層は必要に応じて設けられ、上述のように陰極と発光層との間又は陰極と電子輸送層との間に設けられる。
電子注入層は、ごく薄い膜からなる層であることが好ましく、素材にもよるがその層厚は0.1〜5nmの範囲が好ましい。また構成材料が断続的に存在する不均一な膜からなる層であってもよい。
電子注入層は、特開平6−325871号公報、同9−17574号公報、同10−74586号公報等にもその詳細が記載されている。電子注入層に好ましく用いられる材料の具体例としては、ストロンチウムやアルミニウム等に代表される金属、フッ化リチウム、フッ化ナトリウム、フッ化カリウム等に代表されるアルカリ金属化合物、フッ化マグネシウム、フッ化カルシウム等に代表されるアルカリ土類金属化合物、酸化アルミニウムに代表される金属酸化物、リチウム8−ヒドロキシキノレート(Liq)等に代表される金属錯体等が挙げられる。また、上述の電子輸送材料を用いることも可能である。
また、上記の電子注入層に用いられる材料は単独で用いてもよく、複数種を併用して用いてもよい。
〈正孔輸送層〉
正孔輸送層は、正孔を輸送する機能を有する材料からなる。正孔輸送層は、陽極より注入された正孔を発光層に伝達する機能を有する層である。
正孔輸送層は、正孔を輸送する機能を有する材料からなる。正孔輸送層は、陽極より注入された正孔を発光層に伝達する機能を有する層である。
本発明に係る有機EL素子において、正孔輸送層の総層厚に特に制限はないが、通常は5nm〜5μmの範囲であり、より好ましくは2〜500nmであり、さらに好ましくは5〜200nmである。
正孔輸送層に用いられる材料(以下、正孔輸送材料という。)は、正孔の注入性又は輸送性、電子の障壁性のいずれかを有していればよい。正孔輸送材料は、従来公知の化合物の中から任意のものを選択して用いることができる。正孔輸送材料は単独で用いてもよく、また複数種を併用して用いてもよい。
正孔輸送材料は、例えば、ポルフィリン誘導体、フタロシアニン誘導体、オキサゾール誘導体、オキサジアゾール誘導体、トリアゾール誘導体、イミダゾール誘導体、ピラゾリン誘導体、ピラゾロン誘導体、フェニレンジアミン誘導体、ヒドラゾン誘導体、スチルベン誘導体、ポリアリールアルカン誘導体、トリアリールアミン誘導体、カルバゾール誘導体、インドロカルバゾール誘導体、イソインドール誘導体、アントラセンやナフタレン等のアセン系誘導体、フルオレン誘導体、フルオレノン誘導体、ポリビニルカルバゾール、芳香族アミンを主鎖若しくは側鎖に導入した高分子材料又はオリゴマー、ポリシラン、導電性ポリマー又はオリゴマー(例えば、PEDOT:PSS、アニリン系共重合体、ポリアニリン、ポリチオフェン等)等が挙げられる。
トリアリールアミン誘導体としては、α−NPDに代表されるベンジジン型や、MTDATAに代表されるスターバースト型、トリアリールアミン連結コア部にフルオレンやアントラセンを有する化合物等が挙げられる。
また、特表2003−519432号公報や特開2006−135145号公報等に記載されているヘキサアザトリフェニレン誘導体も正孔輸送材料として用いることができる。
さらに、不純物をドープしたp性の高い正孔輸送層を用いることもできる。例えば、特開平4−297076号公報、特開2000−196140号公報、同2001−102175号公報の各公報、J.Appl.Phys.,95,5773(2004)等に記載された構成を正孔輸送層に適用することもできる。
また、特開平11−251067号公報、J.Huang et.al.著文献(Applied Physics Letters,80(2002),p.139)に記載されているような、いわゆるp型正孔輸送材料やp型−Si、p型−SiC等の無機化合物を用いることもできる。さらにIr(ppy)3に代表されるような中心金属にIrやPtを有するオルトメタル化有機金属錯体も好ましく用いられる。
正孔輸送材料としては、上記のものを使用することができるが、トリアリールアミン誘導体、カルバゾール誘導体、インドロカルバゾール誘導体、アザトリフェニレン誘導体、有機金属錯体、芳香族アミンを主鎖若しくは側鎖に導入した高分子材料又はオリゴマー等が好ましく用いられる。
本発明に係る有機EL素子に用いられる正孔輸送材料の具体例としては、上記で挙げた文献の他、以下の文献に記載の化合物等が挙げられるが、これらに限定されない。
Appl.Phys.Lett.,69,2160(1996)、J.Lumin.,72−74,985(1997)、Appl.Phys.Lett.,78,673(2001)、Appl.Phys.Lett.,90,183503(2007)、Appl.Phys.Lett.,90,183503(2007)、Appl.Phys.Lett.,51,913(1987)、Synth.Met.,87,171(1997)、Synth.Met.,91,209(1997)、Synth.Met.,111,421(2000)、SID Symposium Digest,37,923(2006)、J.Mater.Chem.3,319(1993)、Adv.Mater.,6,677(1994)、Chem.Mater.,15,3148(2003)、米国特許出願公開第2003/0162053号明細書、米国特許出願公開第2002/0158242号明細書、米国特許出願公開第2006/0240279号明細書、米国特許出願公開第2008/0220265号明細書、米国特許第5061569号明細書、国際公開第2007/002683号、国際公開第2009/018009号、EP650955、米国特許出願公開第2008/0124572号明細書、米国特許出願公開第2007/0278938号明細書、米国特許出願公開第2008/0106190号明細書、米国特許出願公開第2008/0018221号明細書、国際公開第2012/115034号、特表2003−519432号公報、特開2006−135145号公報、米国特許出願番号13/585981号等が挙げられる。
〈電子阻止層〉
電子阻止層は、広い意味では正孔輸送層の機能を有する層である。好ましくは、正孔を輸送する機能を有しつつ電子を輸送する能力が小さい材料からなる。電子阻止層は、正孔を輸送しつつ電子を阻止することで、電子と正孔の再結合確率を向上させることができる。
電子阻止層は、広い意味では正孔輸送層の機能を有する層である。好ましくは、正孔を輸送する機能を有しつつ電子を輸送する能力が小さい材料からなる。電子阻止層は、正孔を輸送しつつ電子を阻止することで、電子と正孔の再結合確率を向上させることができる。
また、上述の正孔輸送層の構成を必要に応じて、本発明に係る有機EL素子の電子阻止層として用いることができる。有機EL素子に設ける電子阻止層は、発光層の陽極側に隣接して設けられることが好ましい。
電子阻止層の厚さとしては、好ましくは3〜100nmの範囲であり、更に好ましくは5〜30nmの範囲である。
電子阻止層に用いられる材料としては、上述の正孔輸送層に用いられる材料が好ましく用いることができる。また、上述のホスト化合物として用いられる材料も、電子阻止層として好ましく用いることができる。
〈正孔注入層〉
正孔注入層(「陽極バッファー層」ともいう。)は、駆動電圧低下や発光輝度向上のために陽極と発光層との間に設けられる層である。正孔注入層の一例は、「有機EL素子とその工業化最前線(1998年11月30日エヌ・ティー・エス社発行)」の第2編第2章「電極材料」(123〜166頁)に記載されている。
正孔注入層(「陽極バッファー層」ともいう。)は、駆動電圧低下や発光輝度向上のために陽極と発光層との間に設けられる層である。正孔注入層の一例は、「有機EL素子とその工業化最前線(1998年11月30日エヌ・ティー・エス社発行)」の第2編第2章「電極材料」(123〜166頁)に記載されている。
正孔注入層は必要に応じて設けられ、上述のように陽極と発光層との間、又は陽極と正孔輸送層との間に設けられる。
正孔注入層は、特開平9−45479号公報、同9−260062号公報、同8−288069号公報等にもその詳細が記載されている。
正孔注入層に用いられる材料は、例えば上述の正孔輸送層に用いられる材料等が挙げられる。中でも、銅フタロシアニンに代表されるフタロシアニン誘導体、特表2003−519432号公報や特開2006−135145号公報等に記載されているようなヘキサアザトリフェニレン誘導体、酸化バナジウムに代表される金属酸化物、アモルファスカーボン、ポリアニリン(エメラルディン)やポリチオフェン等の導電性高分子、トリス(2−フェニルピリジン)イリジウム錯体等に代表されるオルトメタル化錯体、トリアリールアミン誘導体等が好ましい。
上述の正孔注入層に用いられる材料は単独で用いてもよく、また複数種を併用して用いてもよい。
〈添加剤〉
本発明に係る有機EL素子を構成する有機機能層は、更に他の添加剤を含んでもよい。
本発明に係る有機EL素子を構成する有機機能層は、更に他の添加剤を含んでもよい。
添加剤としては、例えば臭素、ヨウ素及び塩素等のハロゲン元素やハロゲン化化合物、Pd、Ca、Na等のアルカリ金属やアルカリ土類金属、遷移金属の化合物や錯体、塩等が挙げられる。
添加剤の含有量は、任意に決定することができるが、含有される層の全質量%に対して1000ppm以下であることが好ましく、より好ましくは500ppm以下であり、さらに好ましくは50ppm以下である。
ただし、電子や正孔の輸送性を向上させる目的や、励起子のエネルギー移動を有利にするための目的などによってはこの範囲内ではない。
〈有機機能層の形成方法〉
本発明に係る有機EL素子の有機機能層(正孔注入層、正孔輸送層、発光層、正孔阻止層、電子輸送層、電子注入層等)の形成方法について説明する。
本発明に係る有機EL素子の有機機能層(正孔注入層、正孔輸送層、発光層、正孔阻止層、電子輸送層、電子注入層等)の形成方法について説明する。
有機機能層の形成方法は、特に制限はなく、従来公知の、例えば、真空蒸着法、湿式法(ウェットプロセス)等により形成することができる。
湿式法としては、スピンコート法、キャスト法、インクジェット法、印刷法、ダイコート法、ブレードコート法、ロールコート法、スプレーコート法、カーテンコート法、LB法(ラングミュア−ブロジェット法)等がある。均質な薄膜が得られやすく、かつ高生産性の点から、ダイコート法、ロールコート法、インクジェット法、スプレーコート法等のロールtoロール方式に適性の高い方法が好ましい。
湿式法において、有機機能層の材料を溶解又は分散する液媒体としては、例えば、メチルエチルケトン、シクロヘキサノン等のケトン類、酢酸エチル等の脂肪酸エステル類、ジクロロベンゼン等のハロゲン化炭化水素類、トルエン、キシレン、メシチレン、シクロヘキシルベンゼン等の芳香族炭化水素類、シクロヘキサン、デカリン、ドデカン等の脂肪族炭化水素類、DMF、DMSO等の有機溶媒を用いることができる。
また、超音波、高剪断力分散やメディア分散等の分散方法により分散することができる。
有機機能層を構成する各層の形成に蒸着法を採用する場合、その蒸着条件は使用する化合物の種類等により異なるが、一般にボート加熱温度50〜450℃、真空度10−6〜10−2Pa、蒸着速度0.01〜50nm/秒、基板温度−50〜300℃、層厚0.1nm〜5μm、好ましくは5〜200nmの範囲で適宜選ぶことが望ましい。
本発明に係る有機EL素子の形成は、1回の真空引きで一貫して有機機能層から陰極まで作製するのが好ましいが、途中で取り出して異なる成膜法を施しても構わない。その際は作業を乾燥不活性ガス雰囲気下で行うことが好ましい。
また、層毎に異なる形成方法を適用してもよい。
〈第1電極〉
第1電極215は、仕事関数の大きい(4eV以上、好ましくは4.3eV以上)金属、合金、電気伝導性化合物及びこれらの混合物からなる電極物質が用いられる。このような電極物質の具体例としては、AuやAg等の金属及びこれらの合金、CuI、ITO(インジウム・スズ酸化物)、SnO2、ZnO等の導電性透明材料が挙げられる。また、IDIXO(In2O3−ZnO)等の非晶質で透明導電膜を作製可能な材料を用いてもよい。
第1電極215は、仕事関数の大きい(4eV以上、好ましくは4.3eV以上)金属、合金、電気伝導性化合物及びこれらの混合物からなる電極物質が用いられる。このような電極物質の具体例としては、AuやAg等の金属及びこれらの合金、CuI、ITO(インジウム・スズ酸化物)、SnO2、ZnO等の導電性透明材料が挙げられる。また、IDIXO(In2O3−ZnO)等の非晶質で透明導電膜を作製可能な材料を用いてもよい。
第1電極はこれらの電極物質を蒸着やスパッタリング等の方法により薄膜を形成し、フォトリソグラフィー法で所望の形状のパターンを形成する。又は、パターン精度を余り必要としない(100μm以上程度)場合は、上記電極物質を蒸着法又はスパッタリング法で形成する際に、所望の形状のマスクを介してパターン形成してもよい。
有機導電性化合物のように塗布可能な物質を用いる場合には、印刷方式、コーティング方式等の湿式成膜法を用いることもできる。
第1電極側から発光光を取り出す場合には、透過率を10%より大きくすることが望ましい。また、第1電極としてのシート抵抗は数百Ω/sq.以下が好ましい。また、第1電極の厚さは、材料にもよるが、通常10nm〜1μm、好ましくは10〜200nmの範囲で選ばれる。
特に、第1電極は、銀を主成分として構成された層であって、銀又は銀を主成分とした合金を用いて構成されることが好ましい。このような第1電極の形成方法としては、塗布法、インクジェット法、コーティング法、ディップ法等のウェットプロセスを用いる方法や、蒸着法(抵抗加熱、EB法等)、スパッタ法、CVD法等のドライプロセスを用いる方法等が挙げられる。中でも蒸着法が好ましく適用される。
第1電極を構成する銀(Ag)を主成分とする合金は、一例として銀マグネシウム(AgMg)、銀銅(AgCu)、銀パラジウム(AgPd)、銀パラジウム銅(AgPdCu)、銀インジウム(AgIn)等が挙げられる。
以上のような第1電極は、銀又は銀を主成分とした合金の層が、必要に応じて複数の層に分けて積層された構成であってもよい。
さらに、この第1電極は、厚さが4〜15nmの範囲にあることが好ましい。厚さ15nm以下では、層の吸収成分及び反射成分が低く抑えられ、第1電極の光透過率が維持されるため好ましい。また、厚さが4nm以上であることにより、層の導電性も確保される。
なお、第1電極として銀を主成分として構成された層を形成する場合には、Pd等を含む他の導電層や、窒素化合物、硫黄化合物等の有機機能層を、第1電極の下地層として形成してもよい。下地層を形成することにより、銀を主成分として構成された層の成膜製の向上や、第1電極の抵抗率の低下及び第1電極15の光透過性を向上させることができる。
〈第2電極〉
第2電極217としては、仕事関数の小さい(4eV以下)金属(電子注入性金属と称する。)、合金、電気伝導性化合物及びこれらの混合物からなる電極物質が用いられる。このような電極物質の具体例としては、ナトリウム、ナトリウム−カリウム合金、マグネシウム、リチウム、マグネシウム/銅混合物、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、インジウム、リチウム/アルミニウム混合物、アルミニウム、希土類金属等が挙げられる。
第2電極217としては、仕事関数の小さい(4eV以下)金属(電子注入性金属と称する。)、合金、電気伝導性化合物及びこれらの混合物からなる電極物質が用いられる。このような電極物質の具体例としては、ナトリウム、ナトリウム−カリウム合金、マグネシウム、リチウム、マグネシウム/銅混合物、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、インジウム、リチウム/アルミニウム混合物、アルミニウム、希土類金属等が挙げられる。
これらの中で、電子注入性及び酸化等に対する耐久性の点から、電子注入性金属と、この電子注入性金属よりも仕事関数の値が大きく安定な第二金属との混合物、例えば、マグネシウム/銀混合物、マグネシウム/アルミニウム混合物、マグネシウム/インジウム混合物、アルミニウム/酸化アルミニウム(Al2O3)混合物、リチウム/アルミニウム混合物、アルミニウム等が好適である。
第2電極は、上記電極物質を蒸着やスパッタリング等の方法を用いて、作製することができる。また、第2電極のシート抵抗は、数百Ω/sq.以下が好ましい。また、第2電極の厚さは通常10nm〜5μm、好ましくは50〜200nmの範囲である。
また、第2電極に上記金属を1〜20nmの膜厚で作製した後に、第1電極の説明で挙げる導電性透明材料をその上に作製することで、透明又は半透明の第2電極を作製することができる。これを応用することで、第1電極と第2電極の両方が透過性を有する素子を作製することができる。
〈封止部材〉
本発明に係る有機EL素子は、図4に示すように、有機機能層の全体を覆う封止部材221と封止層222が貼り合わされることにより、固体封止されていてもよい。
本発明に係る有機EL素子は、図4に示すように、有機機能層の全体を覆う封止部材221と封止層222が貼り合わされることにより、固体封止されていてもよい。
封止部材221は、少なくとも有機機能層を覆う状態で設けられ、第1電極及び第2電極の端子部分(図示省略)を露出させる状態で設けられている。また、封止部材221に電極を設け、有機EL素子の第1電極及び第2電極の端子部分と、この電極とを導通させるように構成されていてもよい。
封止部材221は、第2電極217と封止層222とを貼合するための樹脂封止材料から構成される。また、樹脂封止材料に加えて、無機封止材料を用いてもよい。例えば、有機機能層を無機封止材料で覆った後、樹脂封止材料により封止層と第2電極とを貼合する構成としてもよい。
樹脂封止材料は、封止層222を第2電極217側に固定するために用いられる。また、封止部材と第1電極との間に挟持された有機機能層を封止するためのシール材として用いられる。
封止層を第2電極に貼合するためには、任意の硬化型の樹脂封止材料を用いて接着することが好ましい。樹脂封止材料には、接着する封止部材との密着性の向上の観点から、好適な接着剤を適宜選択することができる。
このような樹脂封止材料としては、熱硬化性樹脂を用いることが好ましい。
熱硬化性接着剤としては、例えば、分子の末端又は側鎖にエチレン性二重結合を有する化合物と熱重合開始剤とを主成分とする樹脂等を用いることができる。より具体的には、エポキシ系樹脂、アクリル系樹脂等からなる熱硬化性接着剤を使用することができる。また、有機EL素子の製造工程で用いる貼合装置及び硬化処理装置に応じて、溶融タイプの熱硬化性接着剤を使用してもよい。
また、このような樹脂封止材料としては、光硬化性樹脂用いることも好ましい。例えば、ポリエステル(メタ)アクリレート、ポリエーテル(メタ)アクリレート、エポキシ(メタ)アクリレート、ポリウレタン(メタ)アクリレート等の各種(メタ)アクリレートを主成分とした光ラジカル重合性樹脂や、エポキシやビニルエーテル等の樹脂を主成分とした光カチオン重合性樹脂や、チオール・エン付加型樹脂等が挙げられる。これら光硬化性樹脂の中でも、硬化物の収縮率が低く、アウトガスも少なく、また長期信頼性に優れるエポキシ樹脂系の光カチオン重合性樹脂が好ましい。
また、このような樹脂封止材料としては、化学硬化型(二液混合)の樹脂を用いることができる。また、ホットメルト型のポリアミド、ポリエステル、ポリオレフィンを用いることができる。また、カチオン硬化タイプの紫外線硬化型エポキシ樹脂を用いることができる。
なお、有機EL素子を構成する有機材料は、熱処理により劣化する場合がある。このため、室温から80℃までに接着硬化できる樹脂封止材料を使用することが好ましい。
上記無機封止材料は、樹脂封止材料とともに、第1電極、有機機能層及び第2電極からなる発光ユニットを封止する部材である。このため、無機封止材料は、この有機機能層を劣化させる水分や酸素等の侵入を抑制する機能を有する材料を用いることが好ましい。
また、無機封止材料は、有機機能層に直接接する構成であるため、有機機能層との接合性に優れた材料を用いることが好ましい。
無機封止材料としては、封止性が高い無機酸化物、無機窒化物、無機炭化物等の化合物により形成されることが好ましい。
具体的には、SiOx、Al2O3、In2O3、TiOx、ITO(インジウム・スズ酸化物)、AlN、Si3N4、SiOxN、TiOxN、SiC等により形成することができる。
無機封止材料は、ゾル−ゲル法、蒸着法、CVD、ALD(Atomic Layer Deposition)、PVD、スパッタリング法等の公知な手法により形成可能である。
また、無機封止材料は、大気圧プラズマ法において、原料(原材料ともいう。)である有機金属化合物、分解ガス、分解温度、投入電力などの条件を選択することで、酸化ケイ素、酸化ケイ素を主体とした無機酸化物、又は無機酸窒化物や無機酸化ハロゲン化物等のような、無機炭化物、無機窒化物、無機硫化物及び無機ハロゲン化物等の混合物等の組成を作り分けることができる。
例えば、ケイ素化合物を原料化合物として用い、分解ガスに酸素を用いれば、ケイ素酸化物が生成する。また、シラザン等を原料化合物として用いれば、酸化窒化ケイ素が生成する。これはプラズマ空間内では非常に活性な荷電粒子・活性ラジカルが高密度で存在するため、プラズマ空間内で多段階の化学反応が非常に高速に促進され、プラズマ空間内の元素が熱力学的に安定な化合物へと非常に短時間で変換されるためである。
このような無機封止材料を形成するための原料は、ケイ素化合物であれば、常温常圧下で気体、液体、固体いずれの状態であっても構わない。気体の場合にはそのまま放電空間に導入できるが、液体、固体の場合は、加熱、バブリング、減圧、超音波照射等の手段により気化させて使用する。また、溶媒によって希釈して使用してもよく、溶媒は、メタノール、エタノール、n−ヘキサン等の有機溶媒及びこれらの混合溶媒を使用できる。なお、これらの希釈溶媒は、プラズマ放電処理中において、分子又は原子に分解されるため、影響をほとんど無視することができる。
このようなケイ素化合物としては、シラン、テトラメトキシシラン、テトラエトキシシラン、テトラn−プロポキシシラン、テトライソプロポキシシラン、テトラn−ブトキシシラン、テトラt−ブトキシシラン、ジメチルジメトキシシラン、ジメチルジエトキシシラン、ジエチルジメトキシシラン、ジフェニルジメトキシシラン、メチルトリエトキシシラン、エチルトリメトキシシラン、フェニルトリエトキシシラン、(3,3,3−トリフルオロプロピル)トリメトキシシラン、ヘキサメチルジシロキサン、ビス(ジメチルアミノ)ジメチルシラン、ビス(ジメチルアミノ)メチルビニルシラン、ビス(エチルアミノ)ジメチルシラン、N,O−ビス(トリメチルシリル)アセトアミド、ビス(トリメチルシリル)カルボジイミド、ジエチルアミノトリメチルシラン、ジメチルアミノジメチルシラン、ヘキサメチルジシラザン、ヘキサメチルシクロトリシラザン、ヘプタメチルジシラザン、ノナメチルトリシラザン、オクタメチルシクロテトラシラザン、テトラキスジメチルアミノシラン、テトライソシアナートシラン、テトラメチルジシラザン、トリス(ジメチルアミノ)シラン、トリエトキシフルオロシラン、アリルジメチルシラン、アリルトリメチルシラン、ベンジルトリメチルシラン、ビス(トリメチルシリル)アセチレン、1,4−ビストリメチルシリル−1,3−ブタジイン、ジ−t−ブチルシラン、1,3−ジシラブタン、ビス(トリメチルシリル)メタン、シクロペンタジエニルトリメチルシラン、フェニルジメチルシラン、フェニルトリメチルシラン、プロパルギルトリメチルシラン、テトラメチルシラン、トリメチルシリルアセチレン、1−(トリメチルシリル)−1−プロピン、トリス(トリメチルシリル)メタン、トリス(トリメチルシリル)シラン、ビニルトリメチルシラン、ヘキサメチルジシラン、オクタメチルシクロテトラシロキサン、テトラメチルシクロテトラシロキサン、ヘキサメチルシクロテトラシロキサン、Mシリケート51等が挙げられる。
また、これらケイ素を含む原料ガスを分解して無機封止材料を得るための分解ガスとしては、水素ガス、メタンガス、アセチレンガス、一酸化炭素ガス、二酸化炭素ガス、窒素ガス、アンモニアガス、亜酸化窒素ガス、酸化窒素ガス、二酸化窒素ガス、酸素ガス、水蒸気、フッ素ガス、フッ化水素、トリフルオロアルコール、トリフルオロトルエン、硫化水素、二酸化硫黄、二硫化炭素及び塩素ガス等が挙げられる。
上述のケイ素を含む原料ガスと分解ガスとを適宜選択することで、酸化ケイ素、又は窒化物、炭化物等を含有する無機封止材料を得ることができる。
大気圧プラズマ法においては、これらの反応性ガスに対して、主にプラズマ状態になりやすい放電ガスを混合し、プラズマ放電発生装置にガスを送りこむ。このような放電ガスとしては、窒素ガス及び/又は周期表の第18族元素、具体的には、ヘリウム、ネオン、アルゴン、クリプトン、キセノン、ラドン等が用いられる。これらの中でも特に、窒素、ヘリウム、アルゴンが好ましく用いられる。
上記放電ガスと反応性ガスを混合し、薄膜形成(混合)ガスとして大気圧プラズマ放電発生装置(プラズマ発生装置)に供給することで膜形成を行う。放電ガスと反応性ガスの割合は、得ようとする膜の性質によって異なるが、混合ガス全体に対し、放電ガスの割合を50%以上として反応性ガスを供給する。
〈封止層〉
封止層222は、有機EL素子を覆うものであって、例えば、板状(フィルム状)の封止層222が封止部材221を介して第2電極側に固定されている。
封止層222は、有機EL素子を覆うものであって、例えば、板状(フィルム状)の封止層222が封止部材221を介して第2電極側に固定されている。
板状(フィルム状)の封止部材としては、具体的には、ガラス基板、ポリマー基板が挙げられ、これらの基板材料をさらに薄型のフィルム状にして用いてもよい。ガラス基板としては、特にソーダ石灰ガラス、バリウム・ストロンチウム含有ガラス、鉛ガラス、アルミノケイ酸ガラス、ホウケイ酸ガラス、バリウムホウケイ酸ガラス、石英等を挙げることができる。また、ポリマー基板としては、ポリカーボネート、アクリル、ポリエチレンテレフタレート、ポリエーテルサルファイド及びポリサルフォン等を挙げることができる。
また、封止層としては、樹脂フィルムがラミネート(ポリマー膜)された金属箔を用いることが好ましい。樹脂フィルムがラミネートされた金属箔は、光取り出し側の基材として用いることはできないが、低コストであり、透湿性の低い封止材料である。このため、光取り出しを意図しない封止層として好適である。
なお、金属箔とは、スパッタや蒸着等で形成された金属薄膜や、導電性ペースト等の流動性電極材料から形成された導電膜と異なり、圧延等で形成された金属の箔又はフィルムを指す。
金属箔としては、金属の種類に特に限定はなく、例えば銅(Cu)箔、アルミニウム(Al)箔、金(Au)箔、黄銅箔、ニッケル(Ni)箔、チタン(Ti)箔、銅合金箔、ステンレス箔、スズ(Sn)箔、高ニッケル合金箔等が挙げられる。これらの各種の金属箔の中で特に好ましい金属箔としてはAl箔が挙げられる。
金属箔の厚さは6〜50μmが好ましい。6μm以上の場合は、金属箔に用いる材料によっては使用時にピンホールがあくことなく、必要とするガスバリアー性(透湿度、酸素透過率)を得ることができる。50μm以下であることにより、金属箔に用いる材料によるコストの増加を抑えることができ、有機EL素子が厚くなりすぎることを抑制できるため、フィルム状の封止部材を用いる利点がある。
樹脂フィルムがラミネートされた金属箔において、樹脂フィルムとしては、機能性包装材料の新展開(株式会社 東レリサーチセンター)に記載の各種材料を用いることが可能である。例えば、ポリエチレン系樹脂、ポリプロピレン系樹脂、ポリエチレンテレフタレート系樹脂、ポリアミド系樹脂、エチレン−ビニルアルコール共重合体系樹脂、エチレン−酢酸ビニル共重合体系樹脂、アクリロニトリル−ブタジエン共重合体系樹脂、セロハン系樹脂、ビニロン系樹脂、塩化ビニリデン系樹脂等を用いることができる。ポリプロピレン系樹脂及びナイロン系樹脂等の樹脂は、延伸されていてもよく、さらに塩化ビニリデン系樹脂がコートされていてもよい。また、ポリエチレン系樹脂は、低密度と高密度のいずれを用いてもよい。
封止層222は、JIS K 7126−1987に準拠した方法で測定された酸素透過度が1×10−3mL/m2・24h・atm以下、JIS K 7129−1992に準拠した方法で測定された、水蒸気透過度(25±0.5℃、相対湿度(90±2)%RH)が1×10−3g/m2・24h以下であることが好ましい。
また、以上のような封止層は、凹板状に加工して用いてもよい。この場合、上述した基板部材に対してサンドブラスト加工、化学エッチング加工等の加工が施され、凹状が形成される。
また、これに限らず、金属材料を用いてもよい。金属材料としては、ステンレス、鉄、銅、アルミニウム、マグネシウム、ニッケル、亜鉛、クロム、チタン、モリブデン、シリコン、ゲルマニウム及びタンタルからなる群から選ばれる1種以上の金属又は合金が挙げられる。このような金属材料は、薄型のフィルム状にして封止層として用いることにより、有機EL素子が設けられた発光パネル全体を薄型化できる。
中でも、素子を薄型化できるということから、封止層として薄型のフィルム状にしたガスバリアー性を有するポリマー基板(ガスバリアーフィルム)を好ましく使用することができ、本発明のガスバリアーフィルムを封止層として用いることも可能である。
フィルム状のポリマー基板は、JIS K 7126−1987に準拠した方法で測定された酸素透過度が1×10−3mL/m2・24h・atm以下、JIS K 7129−1992に準拠した方法で測定された、水蒸気透過度(25±0.5℃、相対湿度(90±2)%RH)が、1×10−3g/m2・24h以下であることが好ましい。
また、以上のような基板材料は、凹板状に加工して封止部材として用いてもよい。この場合、上述した基板部材に対してサンドブラスト加工、化学エッチング加工等の加工が施され、凹状が形成される。
〈取り出し電極〉
取り出し電極(不図示)は、第1電極と外部電源とを電気的に接続するものであって、その材料としては特に限定されるものではなく公知の素材を好適に使用できるが、例えば、3層構造からなるMAM電極(Mo/Al・Nd合金/Mo)等の金属膜を用いることができる。
取り出し電極(不図示)は、第1電極と外部電源とを電気的に接続するものであって、その材料としては特に限定されるものではなく公知の素材を好適に使用できるが、例えば、3層構造からなるMAM電極(Mo/Al・Nd合金/Mo)等の金属膜を用いることができる。
〈補助電極〉
補助電極(不図示)は、第1電極の抵抗を下げる目的で設けるものであって、第1電極の電極層に接して設けられる。補助電極を形成する材料は、金、白金、銀、銅、アルミニウム等の抵抗が低い金属が好ましい。これらの金属は光透過性が低いため、光取り出し面からの発光光の取り出しの影響のない範囲でパターン形成される。
補助電極(不図示)は、第1電極の抵抗を下げる目的で設けるものであって、第1電極の電極層に接して設けられる。補助電極を形成する材料は、金、白金、銀、銅、アルミニウム等の抵抗が低い金属が好ましい。これらの金属は光透過性が低いため、光取り出し面からの発光光の取り出しの影響のない範囲でパターン形成される。
このような補助電極の形成方法としては、蒸着法、スパッタリング法、印刷法、インクジェット法、エアロゾルジェット法等が挙げられる。補助電極の線幅は、光を取り出す開口率の観点から50μm以下であることが好ましく、補助電極の厚さは、導電性の観点から1μm以上であることが好ましい。
〈保護膜、保護板〉
有機EL素子は、封止材の上に保護膜又は保護板を更に備えていても良い。
有機EL素子は、封止材の上に保護膜又は保護板を更に備えていても良い。
保護膜又は保護板は、基板との間に有機EL素子本体部(有機機能層、各種電極及び配線)及び封止材を挟んで、有機EL素子本体部を機械的に保護するものである。特に、封止材として封止膜が用いられている場合には、有機EL素子本体部に対する機械的な保護が十分ではないため、保護膜又は保護板が設けられていることが好ましい。
保護膜又は保護板としては、ガラス板、ポリマー板、薄型のポリマーフィルム、金属板、薄型の金属フィルム、又はポリマー材料膜や金属材料膜が用いられる。このうち、軽量かつ素子の薄膜化という観点からポリマーフィルムが用いられることが好ましい。
以下、実施例を挙げて本発明を具体的に説明するが、本発明はこれらに限定されるものではない。なお、実施例において「部」又は「%」の表示を用いるが、特に断りがない限り「質量部」又は「質量%」を表す。
実施例1
≪下地層(Y)用第1コーティング液U−1〜U−5の調製≫
下地層(Y)に用いる第1コーティング液U−1〜U−5を以下の手順にて調製した。
≪下地層(Y)用第1コーティング液U−1〜U−5の調製≫
下地層(Y)に用いる第1コーティング液U−1〜U−5を以下の手順にて調製した。
<第1コーティング液U−1の調製>
蒸留水230質量部を撹拌しながら70℃に昇温した。その蒸留水に、トリイソプロポキシアルミニウム88質量部を1時間かけて滴下し、液温を徐々に95℃まで上昇させ、発生するイソプロパノールを留出させることによって加水分解縮合を行った。得られた液体に、60質量%の硝酸水溶液4.0質量部を添加し、95℃で3時間撹拌することによって加水分解縮合物の粒子の凝集体を解膠させた。その後、その液体に濃度が1.0モル%の水酸化ナトリウム水溶液2.24質量部を加え、固形分濃度が酸化アルミニウム換算で10質量%になるように濃縮した。こうして得られた液体18.66質量部に対して、蒸留水58.19質量部、メタノール19.00質量部、及び5質量%のポリビニルアルコール水溶液(株式会社クラレ製PVA124;ケン化度98.5モル%、粘度平均重合度2400、20℃での4質量%水溶液粘度60mPa・s)0.50質量部を加え、均一になるように撹拌し、金属酸化物(A)を含む液体である分散液を得た。続いて、液温を15℃に維持した状態で前記分散液を撹拌しながらリン化合物(B)を含む溶液である85質量%のリン酸水溶液3.66質量部を滴下して加え、滴下完了後からさらに30分間撹拌を続け、FZ×NZ/NM=0.0005、NM/NP=1.15の値を有する第1コーティング液U−1を得た。
蒸留水230質量部を撹拌しながら70℃に昇温した。その蒸留水に、トリイソプロポキシアルミニウム88質量部を1時間かけて滴下し、液温を徐々に95℃まで上昇させ、発生するイソプロパノールを留出させることによって加水分解縮合を行った。得られた液体に、60質量%の硝酸水溶液4.0質量部を添加し、95℃で3時間撹拌することによって加水分解縮合物の粒子の凝集体を解膠させた。その後、その液体に濃度が1.0モル%の水酸化ナトリウム水溶液2.24質量部を加え、固形分濃度が酸化アルミニウム換算で10質量%になるように濃縮した。こうして得られた液体18.66質量部に対して、蒸留水58.19質量部、メタノール19.00質量部、及び5質量%のポリビニルアルコール水溶液(株式会社クラレ製PVA124;ケン化度98.5モル%、粘度平均重合度2400、20℃での4質量%水溶液粘度60mPa・s)0.50質量部を加え、均一になるように撹拌し、金属酸化物(A)を含む液体である分散液を得た。続いて、液温を15℃に維持した状態で前記分散液を撹拌しながらリン化合物(B)を含む溶液である85質量%のリン酸水溶液3.66質量部を滴下して加え、滴下完了後からさらに30分間撹拌を続け、FZ×NZ/NM=0.0005、NM/NP=1.15の値を有する第1コーティング液U−1を得た。
<第1コーティング液U−2の調製>
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.20、NM/NP=1.15になるように調製した以外は同様にして、第1コーティング液U−2を調製した。
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.20、NM/NP=1.15になるように調製した以外は同様にして、第1コーティング液U−2を調製した。
<第1コーティング液U−3の調製>
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.62、NM/NP=1.15になるように調製した以外は同様にして、第1コーティング液U−3を調製した。
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.62、NM/NP=1.15になるように調製した以外は同様にして、第1コーティング液U−3を調製した。
<第1コーティング液U−4の調製>
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.20、NM/NP=5.50になるように調製した以外は同様にして、第1コーティング液U−4を調製した。
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.20、NM/NP=5.50になるように調製した以外は同様にして、第1コーティング液U−4を調製した。
<第1コーティング液U−5の調製>
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.20、NM/NP=0.30になるように調製した以外は同様にして、第1コーティング液U−5を調製した。
第1コーティング液U−1の調製において、1.0モル%の水酸化ナトリウム水溶液の添加量を変更し、FZ×NZ/NM=0.20、NM/NP=0.30になるように調製した以外は同様にして、第1コーティング液U−5を調製した。
≪有機EL素子101の作製≫
<ガスバリアーフィルム101の作製>
(基材の準備)
厚さ125μmの両面易接着層付きポリエチレンテレフタレート(PET)フィルム(東レ(株)製、UH13)を基材として用いた。ただし、当該基材の片面には、高屈折率の易接着層が設けられており、有機層及びガスバリアー層は当該高屈折率の易接着層側に形成するものとした。
<ガスバリアーフィルム101の作製>
(基材の準備)
厚さ125μmの両面易接着層付きポリエチレンテレフタレート(PET)フィルム(東レ(株)製、UH13)を基材として用いた。ただし、当該基材の片面には、高屈折率の易接着層が設けられており、有機層及びガスバリアー層は当該高屈折率の易接着層側に形成するものとした。
(アンカー層の形成)
重合性化合物(ダイセルサイテック社製、トリメチロールプロパントリアクリレート(TMPTA))100質量部、光重合開始剤(BASFジャパン社製、イルガキュア184)、前記化1で表される有機ケイ素化合物a1を3質量部、及びメチルエチルケトン(MEK)を含む重合性組成物を調製した。MEKの含有量は、乾燥層厚が1μmになるように調整し、光重合開始剤の含有量は、重合性組成物中に3質量%とした。調製した重合性組成物をバーコーターで基材上に塗布後、乾燥し、紫外線照射により硬化させて、アンカー層を形成した。
重合性化合物(ダイセルサイテック社製、トリメチロールプロパントリアクリレート(TMPTA))100質量部、光重合開始剤(BASFジャパン社製、イルガキュア184)、前記化1で表される有機ケイ素化合物a1を3質量部、及びメチルエチルケトン(MEK)を含む重合性組成物を調製した。MEKの含有量は、乾燥層厚が1μmになるように調整し、光重合開始剤の含有量は、重合性組成物中に3質量%とした。調製した重合性組成物をバーコーターで基材上に塗布後、乾燥し、紫外線照射により硬化させて、アンカー層を形成した。
上記有機ケイ素化合物a1は、特開2009−67778号公報に記載の方法を参酌して合成した。
(ガスバリアー層(X)の形成)
上記アンカー層を形成した樹脂基材を用いて、アンカー層上に下記方法によってガスバリアー層(X)を、下記表1記載の成膜条件B1にて層厚30nmにて形成した。
上記アンカー層を形成した樹脂基材を用いて、アンカー層上に下記方法によってガスバリアー層(X)を、下記表1記載の成膜条件B1にて層厚30nmにて形成した。
図2に記載の対向する成膜ロールからなる成膜部を有する装置を2台つなげたタイプ(図3:第1成膜部、第2成膜部を有するタンデム型CVD成膜装置。図中の符号において「’」のついた符号は、それぞれ図2の各部位と同一である。))のロールtoロール型CVD成膜装置を用いた。有効成膜幅を1000mmとし、成膜条件は、搬送速度、第1成膜部、第2成膜部それぞれの原料ガス(HMDSO)の供給量、酸素ガスの供給量、真空度、印加電力で調整した。成膜回数(装置のパス数)は1パスとした。その他の条件として、電源周波数は84kHz、成膜ロールの温度は全て10℃とした。膜厚は断面TEM観察で求めた。
<有機EL素子101の作製>
(第1透明電極の形成)
前記形成したガスバリアーフィルム101のガスバリアー層(X)上に、厚さ150nmのITO(インジウム・スズ酸化物(Indiumu Tin Oxide))を市販のスパッタリング装置を用いてスパッタ法により成膜し、フォトリソグラフィー法によりパターニングを行い、第一透明電極を形成した。なお、パターンは発光面積が50mm平方になるようなパターンとした。
(第1透明電極の形成)
前記形成したガスバリアーフィルム101のガスバリアー層(X)上に、厚さ150nmのITO(インジウム・スズ酸化物(Indiumu Tin Oxide))を市販のスパッタリング装置を用いてスパッタ法により成膜し、フォトリソグラフィー法によりパターニングを行い、第一透明電極を形成した。なお、パターンは発光面積が50mm平方になるようなパターンとした。
(有機発光層の形成)
(正孔輸送層の形成)
第一透明電極の上に、以下に示す正孔輸送層形成用塗布液を、25℃、相対湿度50%RHの環境下で、スピンコーターで塗布した後、下記の条件で乾燥及び加熱処理を行い、正孔輸送層を形成した。正孔輸送層形成用塗布液は乾燥後の厚さが50nmになるように塗布した。
(正孔輸送層の形成)
第一透明電極の上に、以下に示す正孔輸送層形成用塗布液を、25℃、相対湿度50%RHの環境下で、スピンコーターで塗布した後、下記の条件で乾燥及び加熱処理を行い、正孔輸送層を形成した。正孔輸送層形成用塗布液は乾燥後の厚さが50nmになるように塗布した。
〈正孔輸送層形成用塗布液の準備〉
ポリエチレンジオキシチオフェン・ポリスチレンスルホネート(PEDOT/PSS、ヘレオス社製 Clevios P VP AI 4083)を純水で65%、メタノール5%で希釈した溶液を正孔輸送層形成用塗布液として準備した。
ポリエチレンジオキシチオフェン・ポリスチレンスルホネート(PEDOT/PSS、ヘレオス社製 Clevios P VP AI 4083)を純水で65%、メタノール5%で希釈した溶液を正孔輸送層形成用塗布液として準備した。
〈乾燥及び加熱処理条件〉
正孔輸送層形成用塗布液を塗布した後、成膜面に向け高さ100mm、吐出風速1m/s、幅手の風速分布5%、温度100℃で溶媒を除去した後、引き続き、加熱処理装置を用い温度150℃で裏面伝熱方式の熱処理を行い、正孔輸送層を形成した。
正孔輸送層形成用塗布液を塗布した後、成膜面に向け高さ100mm、吐出風速1m/s、幅手の風速分布5%、温度100℃で溶媒を除去した後、引き続き、加熱処理装置を用い温度150℃で裏面伝熱方式の熱処理を行い、正孔輸送層を形成した。
(発光層の形成)
上記で形成した正孔輸送層上に、以下に示す白色発光層形成用塗布液を、下記の条件によりスピンコーターで塗布した後、下記の条件で乾燥及び加熱処理を行い、発光層を形成した。白色発光層形成用塗布液は乾燥後の厚さが40nmになるように塗布した。
上記で形成した正孔輸送層上に、以下に示す白色発光層形成用塗布液を、下記の条件によりスピンコーターで塗布した後、下記の条件で乾燥及び加熱処理を行い、発光層を形成した。白色発光層形成用塗布液は乾燥後の厚さが40nmになるように塗布した。
〈白色発光層形成用塗布液〉
ホスト材として下記化学式H−Aで表される化合物1.0gと、ドーパント材として下記化学式D−Aで表される化合物を100mg、ドーパント材として下記化学式D−Bで表される化合物を0.2mg、ドーパント材として下記化学式D−Cで表される化合物を0.2mg、100gのトルエンに溶解し白色発光層形成用塗布液として準備した。
ホスト材として下記化学式H−Aで表される化合物1.0gと、ドーパント材として下記化学式D−Aで表される化合物を100mg、ドーパント材として下記化学式D−Bで表される化合物を0.2mg、ドーパント材として下記化学式D−Cで表される化合物を0.2mg、100gのトルエンに溶解し白色発光層形成用塗布液として準備した。
〈塗布条件〉
塗布工程を窒素ガス濃度99%以上の雰囲気で、塗布温度を25℃とした。
塗布工程を窒素ガス濃度99%以上の雰囲気で、塗布温度を25℃とした。
〈乾燥及び加熱処理条件〉
白色発光層形成用塗布液を塗布した後、成膜面に向け高さ100mm、吐出風速1m/s、幅手の風速分布5%、温度60℃で溶媒を除去した後、引き続き、温度130℃で加熱処理を行い、発光層を形成した。
白色発光層形成用塗布液を塗布した後、成膜面に向け高さ100mm、吐出風速1m/s、幅手の風速分布5%、温度60℃で溶媒を除去した後、引き続き、温度130℃で加熱処理を行い、発光層を形成した。
(電子輸送層の形成)
上記で形成した発光層の上に、以下に示す電子輸送層形成用塗布液を下記の条件によりスピンコーターで塗布した後、下記の条件で乾燥及び加熱処理し、電子輸送層を形成した。電子輸送層形成用塗布液は、乾燥後の厚さが30nmになるように塗布した。
上記で形成した発光層の上に、以下に示す電子輸送層形成用塗布液を下記の条件によりスピンコーターで塗布した後、下記の条件で乾燥及び加熱処理し、電子輸送層を形成した。電子輸送層形成用塗布液は、乾燥後の厚さが30nmになるように塗布した。
〈塗布条件〉
塗布工程は窒素ガス濃度99%以上の雰囲気で、電子輸送層形成用塗布液の塗布温度を25℃とした。
塗布工程は窒素ガス濃度99%以上の雰囲気で、電子輸送層形成用塗布液の塗布温度を25℃とした。
〈電子輸送層形成用塗布液〉
電子輸送層は下記化学式E−Aで表される化合物を2,2,3,3−テトラフルオロ−1−プロパノール中に溶解し0.5質量%溶液とし電子輸送層形成用塗布液とした。
電子輸送層は下記化学式E−Aで表される化合物を2,2,3,3−テトラフルオロ−1−プロパノール中に溶解し0.5質量%溶液とし電子輸送層形成用塗布液とした。
〈乾燥及び加熱処理条件〉
電子輸送層形成用塗布液を塗布した後、成膜面に向け高さ100mm、吐出風速1m/s、幅手の風速分布5%、温度60℃で溶媒を除去した後、引き続き、加熱処理部で、温度200℃で加熱処理を行い、電子輸送層を形成した。
電子輸送層形成用塗布液を塗布した後、成膜面に向け高さ100mm、吐出風速1m/s、幅手の風速分布5%、温度60℃で溶媒を除去した後、引き続き、加熱処理部で、温度200℃で加熱処理を行い、電子輸送層を形成した。
(電子注入層の形成)
上記で形成した電子輸送層上に、電子注入層を形成した。まず、基板を減圧チャンバー に投入し、5×10−4Paまで減圧した。あらかじめ、真空チャンバーにタンタル製蒸着ボートに用意しておいたフッ化セシウムを加熱し、厚さ3nmの電子注入層を形成した。
上記で形成した電子輸送層上に、電子注入層を形成した。まず、基板を減圧チャンバー に投入し、5×10−4Paまで減圧した。あらかじめ、真空チャンバーにタンタル製蒸着ボートに用意しておいたフッ化セシウムを加熱し、厚さ3nmの電子注入層を形成した。
(第2電極(反射電極)の形成)
上記で形成した電子注入層の上であって、第1透明電極の取り出し電極になる部分を除く部分に、5×10−4Paの真空下で、第2電極形成材料としてアルミニウム(Al)を使用し、取り出し電極を有するように蒸着法にて、発光面積が50mm平方になるようにマスクパターン成膜し、厚さ150nmの第2電極を積層した。
上記で形成した電子注入層の上であって、第1透明電極の取り出し電極になる部分を除く部分に、5×10−4Paの真空下で、第2電極形成材料としてアルミニウム(Al)を使用し、取り出し電極を有するように蒸着法にて、発光面積が50mm平方になるようにマスクパターン成膜し、厚さ150nmの第2電極を積層した。
(裁断)
以上のように、第2電極までが形成された各積層体を、再び窒素雰囲気に移動し、規定の大きさに、紫外線レーザーを用いて裁断し、有機EL素子を作製した。
以上のように、第2電極までが形成された各積層体を、再び窒素雰囲気に移動し、規定の大きさに、紫外線レーザーを用いて裁断し、有機EL素子を作製した。
(電極リード接続)
作製した有機EL素子に、ソニーケミカル&インフォメーションデバイス株式会社製の異方性導電フィルムDP3232S9を用いて、フレキシブルプリント基板(ベースフィルム:ポリイミド12.5μm、圧延銅箔18μm、カバーレイ:ポリイミド12.5μm、表面処理NiAuメッキ)を接続した。
作製した有機EL素子に、ソニーケミカル&インフォメーションデバイス株式会社製の異方性導電フィルムDP3232S9を用いて、フレキシブルプリント基板(ベースフィルム:ポリイミド12.5μm、圧延銅箔18μm、カバーレイ:ポリイミド12.5μm、表面処理NiAuメッキ)を接続した。
圧着条件:温度170℃(別途熱伝対を用いて測定したACF温度140℃)、圧力2MPa、10秒で圧着を行った。
(封止)
透明封止基材として、下記ガスバリアー層付きPET基材を用いて、有機EL素子101を作製した。PET基材は、厚さ125μmのPETフィルム(帝人デュポンフィルム株式会社製、テイジンテトロンフィルム(登録商標)K)を用いた。
透明封止基材として、下記ガスバリアー層付きPET基材を用いて、有機EL素子101を作製した。PET基材は、厚さ125μmのPETフィルム(帝人デュポンフィルム株式会社製、テイジンテトロンフィルム(登録商標)K)を用いた。
当該ガスバリアー層付きPET基材を用いた透明封止基材の接着は、接着剤としてエポキシ系熱硬化型接着剤(巴川製紙所社製エレファンCS)を用い、酸素濃度10ppm以下、水分濃度10ppm以下のグローブボックス内で、80℃、0.04MPa荷重下、減圧(1×10−3MPa以下)吸引20秒、プレス20秒の条件で、有機EL素子に向けて、前記ガスバリアー層付きPET基材のガスバリアー層が素子側になるように真空プレスした。
その後、グローブボックス内で、110℃のホットプレート上で30分間加熱して接着層を熱硬化させた。
〈ガスバリアー層付きPET基材〉
無機前駆体化合物を含有する塗布液として、無触媒のパーヒドロポリシラザン20質量%ジブチルエーテル溶液(AZエレクトロニックマテリアルズ(株)製アクアミカ NN120−20)とアミン触媒を固形分の5質量%含有するパーヒドロポリシラザン20質量%ジブチルエーテル溶液(AZエレクトロニックマテリアルズ(株)製アクアミカ NAX120−20)とを混合して用い、アミン触媒を固形分の1質量%に調整した後、さらに、ジブチルエーテルで希釈することにより5質量%ジブチルエーテル溶液として作製した。
無機前駆体化合物を含有する塗布液として、無触媒のパーヒドロポリシラザン20質量%ジブチルエーテル溶液(AZエレクトロニックマテリアルズ(株)製アクアミカ NN120−20)とアミン触媒を固形分の5質量%含有するパーヒドロポリシラザン20質量%ジブチルエーテル溶液(AZエレクトロニックマテリアルズ(株)製アクアミカ NAX120−20)とを混合して用い、アミン触媒を固形分の1質量%に調整した後、さらに、ジブチルエーテルで希釈することにより5質量%ジブチルエーテル溶液として作製した。
PET基材に乾燥後の膜厚として300nmになるように塗布後、赤外線で基材温度80℃、乾燥時間5分、乾燥雰囲気の露点5℃の条件下で乾燥させた。
乾燥後、樹脂基材を25℃まで徐冷し、真空紫外線照射装置内で、塗布面に真空紫外線照射による改質処理を行った。真空紫外線照射装置の光源としては、172nmの真空紫外線を照射する二重管構造を有するXeエキシマランプを用いた。
〈改質処理装置〉
株式会社エム・ディ・コム製エキシマ照射装置MODEL:MECL−M−1−200、光波長172nm、ランプ封入ガス Xe
〈改質処理条件〉
エキシマ光強度 3J/cm2(172nm)
ステージ加熱温度 100℃
照射装置内の酸素濃度 1000ppm
≪有機EL素子102の作製≫
ガスバリアーフィルム101の作製において、アンカー層とガスバリアー層(X)の間に下記手順にて下地層(Y)を形成した以外は同様にしてガスバリアーフィルム102を作製し、有機EL素子102を作製した。
株式会社エム・ディ・コム製エキシマ照射装置MODEL:MECL−M−1−200、光波長172nm、ランプ封入ガス Xe
〈改質処理条件〉
エキシマ光強度 3J/cm2(172nm)
ステージ加熱温度 100℃
照射装置内の酸素濃度 1000ppm
≪有機EL素子102の作製≫
ガスバリアーフィルム101の作製において、アンカー層とガスバリアー層(X)の間に下記手順にて下地層(Y)を形成した以外は同様にしてガスバリアーフィルム102を作製し、有機EL素子102を作製した。
<下地層(Y)の形成>
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−1をコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−1をコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
樹脂基材/アンカー層/下地層(Y)を積層した樹脂フィルムの赤外線吸収スペクトルを測定した結果、800〜1400cm−1の領域における最大吸収波数は1107cm−1であり、前記領域における最大吸収帯の半値幅は37cm−1であった。
≪有機EL素子103の作製≫
ガスバリアーフィルム101の作製において、アンカー層上に下記手順にて下地層(Y)を形成し、ガスバリアー層(X)を形成しなかった以外は同様にしてガスバリアーフィルム103を作製し、有機EL素子103を作製した。
ガスバリアーフィルム101の作製において、アンカー層上に下記手順にて下地層(Y)を形成し、ガスバリアー層(X)を形成しなかった以外は同様にしてガスバリアーフィルム103を作製し、有機EL素子103を作製した。
<下地層(Y)の形成>
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−2をコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−2をコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
≪有機EL素子104〜106の作製≫
ガスバリアーフィルム101の作製において、アンカー層上に下記手順にて、第1コーティング液U−3、U−4及びU−5をそれぞれコートして下地層(Y)を形成した以外は同様にして、表2記載のガスバリアーフィルム104〜106を作製し、有機EL素子104〜106を作製した。
ガスバリアーフィルム101の作製において、アンカー層上に下記手順にて、第1コーティング液U−3、U−4及びU−5をそれぞれコートして下地層(Y)を形成した以外は同様にして、表2記載のガスバリアーフィルム104〜106を作製し、有機EL素子104〜106を作製した。
<下地層(Y)の形成>
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−3、U−4及びU−5をそれぞれコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−3、U−4及びU−5をそれぞれコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
≪有機EL素子107の作製≫
ガスバリアーフィルム101の作製において、アンカー層上に下記手順にて下地層(Y)を形成し、さらにガスバリアー層(X)を成膜条件B1にて形成した以外は同様にしてガスバリアーフィルム107を作製し、有機EL素子107を作製した。
ガスバリアーフィルム101の作製において、アンカー層上に下記手順にて下地層(Y)を形成し、さらにガスバリアー層(X)を成膜条件B1にて形成した以外は同様にしてガスバリアーフィルム107を作製し、有機EL素子107を作製した。
<下地層(Y)の形成>
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−2をコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
上記樹脂基材に形成したアンカー層上に、乾燥後の厚さが0.5μmとなるようにバーコーターを用いて第1コーティング液U−2をコートし、塗工後のフィルムを100℃で5分間乾燥することによって下地層(Y)の前駆体層を形成した。続いて、180℃で1分間熱処理することによって下地層(Y)を形成した。
≪有機EL素子108〜110の作製≫
ガスバリアーフィルム107の作製において、ガスバリアー層(X)を成膜条件B1の代わりに成膜条件B2、B3及びB4にて形成した以外は同様にして、表2に記載のガスバリアーフィルム108〜110を作製し、有機EL素子108〜110を作製した。
ガスバリアーフィルム107の作製において、ガスバリアー層(X)を成膜条件B1の代わりに成膜条件B2、B3及びB4にて形成した以外は同様にして、表2に記載のガスバリアーフィルム108〜110を作製し、有機EL素子108〜110を作製した。
≪有機EL素子111〜113の作製≫
有機EL素子107の作製において、ガスバリアーフィルム107に用いた樹脂基材(PET)の代わりに下記基材を用いた以外は同様にしてガスバリアーフィルム111〜113を作製し、有機EL素子111〜113を作製した。
有機EL素子107の作製において、ガスバリアーフィルム107に用いた樹脂基材(PET)の代わりに下記基材を用いた以外は同様にしてガスバリアーフィルム111〜113を作製し、有機EL素子111〜113を作製した。
ポリエチレンナフタレートフィルム(PEN):厚さ125μm、帝人デュポンフィルム社製テオネックスQ83
シクロオレフィンポリマーフィルム(COP):厚さ125μm、日本ゼオン(株)製ゼオノアフィルム
ポリカーボネートフィルム(PC):厚さ125μm、帝人社製ピュアエース
≪有機EL素子114〜118の作製≫
有機EL素子107の作製において、表2記載のように、樹脂基材種類、ガスバリアー層(X)の成膜条件(B5及びB6)、ガスバリアー層(X)の層厚を変化させた以外は同様にして、ガスバリアーフィルム114〜118を作製し、有機EL素子114〜118を作製した。
シクロオレフィンポリマーフィルム(COP):厚さ125μm、日本ゼオン(株)製ゼオノアフィルム
ポリカーボネートフィルム(PC):厚さ125μm、帝人社製ピュアエース
≪有機EL素子114〜118の作製≫
有機EL素子107の作製において、表2記載のように、樹脂基材種類、ガスバリアー層(X)の成膜条件(B5及びB6)、ガスバリアー層(X)の層厚を変化させた以外は同様にして、ガスバリアーフィルム114〜118を作製し、有機EL素子114〜118を作製した。
≪評価≫
上記作製した有機EL素子101〜118を用いて下記評価を実施した。
上記作製した有機EL素子101〜118を用いて下記評価を実施した。
〔湾曲高温高湿保管後のダークスポット(黒点)評価〕
上記作製した各有機EL素子を、φ(直径)50mmの半円形のロールに、当該ロールの形状になるように力を印加して曲面状に巻き付け、85℃、85%RHの環境下で100時間の加速劣化処理を施した後、加速劣化処理を施していない各有機EL素子とともに、ダークスポット評価を行った。
上記作製した各有機EL素子を、φ(直径)50mmの半円形のロールに、当該ロールの形状になるように力を印加して曲面状に巻き付け、85℃、85%RHの環境下で100時間の加速劣化処理を施した後、加速劣化処理を施していない各有機EL素子とともに、ダークスポット評価を行った。
加速劣化処理を施した有機EL素子及び加速劣化処理を施していない有機EL素子に対し、それぞれ1mA/cm2の電流を印加し、24時間連続発光させた後、100倍のマイクロスコープ((株)モリテックス製MS−804、レンズMP−ZE25−200)でパネルの一部分を拡大し、撮影を行った。撮影画像を2mm四方に切り抜き、ダークスポット(黒点)の発生面積比率を求め、下式に従って素子劣化耐性率を算出し、下記の基準に従って評価した。下記基準において3以上であれば実用上問題ないと判断することができる。
素子劣化耐性率=(加速劣化処理を施していない有機EL素子で発生した黒点の面積/加速劣化処理を施した有機EL素子で発生した黒点の面積)×100(%)
5:素子劣化耐性率が、98%以上である
4:素子劣化耐性率が、90%以上、98%未満である
3:素子劣化耐性率が、60%以上、90%未満である
2:素子劣化耐性率が、20%以上、60%未満である
1:素子劣化耐性率が、20%未満である
以上の有機EL素子の構成内容と評価結果を表2に示す。
5:素子劣化耐性率が、98%以上である
4:素子劣化耐性率が、90%以上、98%未満である
3:素子劣化耐性率が、60%以上、90%未満である
2:素子劣化耐性率が、20%以上、60%未満である
1:素子劣化耐性率が、20%未満である
以上の有機EL素子の構成内容と評価結果を表2に示す。
表2から、本発明のガスバリアーフィルム107〜118は、湾曲高温高湿保管後のダークスポット(黒点)の発生の抑制に優れており、ガスバリアーフィルムを具備する電子デバイスを湾曲した状態で高温高湿下に保管したときの、ガスバリアー層と他構成層との境界で発生する微細発泡の抑制に優れた効果を有することが分かる。
実施例2
実施例1で作製したガスバリアーフィルム107のガスバリアー層(X)を第1層として、さらに市販の酸素欠損型酸化ニオブターゲット(組成はNb12O29)を用いて、DC方式により第1層上にスパッタ成膜し、第2層のガスバリアー層を形成して、樹脂基材上にアンカー層、下地層(Y)を含めて4層構成のガスバリアーフィルム201を作製した。
実施例1で作製したガスバリアーフィルム107のガスバリアー層(X)を第1層として、さらに市販の酸素欠損型酸化ニオブターゲット(組成はNb12O29)を用いて、DC方式により第1層上にスパッタ成膜し、第2層のガスバリアー層を形成して、樹脂基材上にアンカー層、下地層(Y)を含めて4層構成のガスバリアーフィルム201を作製した。
スパッタ電源パワーは4.0W/cm2、酸素分圧を12%とし、層厚が10nmとなるように成膜時間を設定した。
形成したガスバリアー層について、XPS分析により、厚さ方向の組成分布プロファイルを測定した。なお、XPS分析条件は以下のとおりである。
〈XPS分析条件〉
・装置:アルバック・ファイ社製QUANTERA SXM
・X線源:単色化Al−Kα
・スパッタイオン:Ar(2keV)
・デプスプロファイル:SiO2換算スパッタ厚さで、所定の厚さ間隔で測定を繰り返し、深さ方向のデプスプロファイルを得た。この厚さ間隔は、1nmとした(深さ方向に1nmごとのデータが得られる)。
・装置:アルバック・ファイ社製QUANTERA SXM
・X線源:単色化Al−Kα
・スパッタイオン:Ar(2keV)
・デプスプロファイル:SiO2換算スパッタ厚さで、所定の厚さ間隔で測定を繰り返し、深さ方向のデプスプロファイルを得た。この厚さ間隔は、1nmとした(深さ方向に1nmごとのデータが得られる)。
・定量:バックグラウンドをShirley法で求め、得られたピーク面積から相対感度係数法を用いて定量した。データ処理は、アルバック・ファイ社製のMultiPakを用いた。
〈複合組成領域の厚さの測定〉
上記XPS組成分析から得られたデータから、ガスバリアー層の組成は、(Si)(Nb)xOyNzで表すことができる。第1層と第2層との界面領域で、非遷移金属であるSiと遷移金属であるNbとが共存し、かつ遷移金属Nb/Siの原子数比率の値xが、0.02≦x≦49の範囲内にある領域を「複合組成領域」とし、当該領域の有無とその厚さ(nm)を測定したところ、厚さ8nmの複合組成領域が形成された。
上記XPS組成分析から得られたデータから、ガスバリアー層の組成は、(Si)(Nb)xOyNzで表すことができる。第1層と第2層との界面領域で、非遷移金属であるSiと遷移金属であるNbとが共存し、かつ遷移金属Nb/Siの原子数比率の値xが、0.02≦x≦49の範囲内にある領域を「複合組成領域」とし、当該領域の有無とその厚さ(nm)を測定したところ、厚さ8nmの複合組成領域が形成された。
〈複合組成領域の酸素欠損指標の計算〉
上記XPS分析データを用いて、各測定点における(2y+3z)/(a+bx)の値を計算した。ここで、非遷移金属はSiであるため、a=4、また、遷移金属はNbであるため、b=5である。(2y+3z)/(a+bx)の値の最小値を求め、これを酸素欠損度指標としたところ、(2y+3z)/(a+bx)<1.0となり酸素欠損の状態であった。
上記XPS分析データを用いて、各測定点における(2y+3z)/(a+bx)の値を計算した。ここで、非遷移金属はSiであるため、a=4、また、遷移金属はNbであるため、b=5である。(2y+3z)/(a+bx)の値の最小値を求め、これを酸素欠損度指標としたところ、(2y+3z)/(a+bx)<1.0となり酸素欠損の状態であった。
作製したガスバリアーフィルム107及び201を用いて、下記方法にてガスバリアー性(水蒸気透過度)評価、及び当該ガスバリアーフィルムを基材として用いて実施例1と同様に有機EL素子を作製し、湾曲高温高湿保管後のダークスポット(黒点)発生の評価を実施した。
<ガスバリアー性>
本発明のガスバリアーフィルムの水蒸気透過度を測定する方法として、下記Ca法による測定を行った。
本発明のガスバリアーフィルムの水蒸気透過度を測定する方法として、下記Ca法による測定を行った。
(水蒸気バリアー性評価用セルの作製)
各ガスバリアーフィルム試料のガスバリアー層面に、真空蒸着装置(日本電子製真空蒸着装置JEE−400)を用い、ガスバリアーフィルム試料の蒸着させたい部分(12mm×12mmを9か所)以外をマスクし、金属カルシウムを蒸着させた。その後、真空状態のままマスクを取り去り、シート片側全面にアルミニウムをもう一つの金属蒸着源から蒸着させた。アルミニウム封止後、真空状態を解除し、速やかに乾燥窒素ガス雰囲気下で、厚さ0.2mmの石英ガラスに封止用紫外線硬化樹脂(ナガセケムテックス製)を介してアルミニウム封止側と対面させ、紫外線を照射することで、評価用セルを作製した。
各ガスバリアーフィルム試料のガスバリアー層面に、真空蒸着装置(日本電子製真空蒸着装置JEE−400)を用い、ガスバリアーフィルム試料の蒸着させたい部分(12mm×12mmを9か所)以外をマスクし、金属カルシウムを蒸着させた。その後、真空状態のままマスクを取り去り、シート片側全面にアルミニウムをもう一つの金属蒸着源から蒸着させた。アルミニウム封止後、真空状態を解除し、速やかに乾燥窒素ガス雰囲気下で、厚さ0.2mmの石英ガラスに封止用紫外線硬化樹脂(ナガセケムテックス製)を介してアルミニウム封止側と対面させ、紫外線を照射することで、評価用セルを作製した。
得られた両面を封止した試料を60℃、90%RHの高温高湿下で保存し、特開2005−283561号公報に記載の方法に基づき、金属カルシウムの腐食量からセル内に透過した水分量を計算した。
なお、ガスバリアーフィルム面以外からの水蒸気の透過がないことを確認するために、比較試料としてガスバリアーフィルム試料の代わりに、厚さ0.2mmの石英ガラス板を用いて金属カルシウムを蒸着した試料を、同様な60℃、90%RHの高温高湿下保存を行い、1000時間経過後でも金属カルシウム腐食が発生しないことを確認した。
以上により測定された各ガスバリアーフィルムの透過水分量を評価した。
(使用した装置及び材料)
蒸着装置:日本電子(株)製真空蒸着装置JEE−400
恒温恒湿度オーブン:Yamato Humidic ChamberIG47M
水分と反応して腐食する金属:カルシウム(粒状)
水蒸気不透過性の金属:アルミニウム(3〜5mmφ、粒状)
蒸着装置:日本電子(株)製真空蒸着装置JEE−400
恒温恒湿度オーブン:Yamato Humidic ChamberIG47M
水分と反応して腐食する金属:カルシウム(粒状)
水蒸気不透過性の金属:アルミニウム(3〜5mmφ、粒状)
表3の結果から、ガスバリアーフィルム107のガスバリアー性に対して、ガスバリアーフィルム201は、SiとNbによる「複合組成領域」を形成することで、顕著に高いガスバリアー性を示した。また、当該ガスバリアーフィルム201を基材として具備した有機ELデバイスは、実施例1と同様に、湾曲高温高湿保管後のダークスポット(黒点)発生の抑制に、優れた効果を再現した。
F ガスバリアーフィルム
1 樹脂基材
2 下地層(Y)
3 ガスバリアー層(X)
S 成膜部
1a 樹脂基材
1b、1c、1d、1e 成膜された基材
10 送り出しローラー
11、12、13、14 搬送ローラー
15 第1成膜ローラー
16 第2成膜ローラー
17 巻取りローラー
18 ガス供給管
19 プラズマ発生用電源
20、21 磁場発生装置
30 真空チャンバー
40 真空ポンプ
41 制御部
100 成膜装置
101 成膜装置(タンデム)
200 有機EL素子
211 樹脂基材
212 下地層(Y)
213 ガスバリアー層(X)
215 第1電極
216 発光ユニット層
217 第2電極
221 封止部材
222 封止層
1 樹脂基材
2 下地層(Y)
3 ガスバリアー層(X)
S 成膜部
1a 樹脂基材
1b、1c、1d、1e 成膜された基材
10 送り出しローラー
11、12、13、14 搬送ローラー
15 第1成膜ローラー
16 第2成膜ローラー
17 巻取りローラー
18 ガス供給管
19 プラズマ発生用電源
20、21 磁場発生装置
30 真空チャンバー
40 真空ポンプ
41 制御部
100 成膜装置
101 成膜装置(タンデム)
200 有機EL素子
211 樹脂基材
212 下地層(Y)
213 ガスバリアー層(X)
215 第1電極
216 発光ユニット層
217 第2電極
221 封止部材
222 封止層
Claims (6)
- 樹脂基材上に積層された下地層(Y)と、これに接してガスバリアー層(X)とを備えるガスバリアーフィルムであって、
前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、
前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、
前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすことを特徴とするガスバリアーフィルム。
関係式(1) 0.8≦NM/NP≦4.5
関係式(2) 0.001≦FZ×NZ/NM≦0.60 - 前記ガスバリアー層(X)が、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚が15〜50nmの範囲内であることを特徴とする請求項1に記載のガスバリアーフィルム。
関係式(3) 1.6≦x≦1.9、0.15≦y≦0.40
関係式(4) 4.0≦2x+2y<4.3 - 樹脂基材上に少なくとも下地層(Y)と、これに接してガスバリアー層(X)とを形成するガスバリアーフィルムの製造方法であって、
前記下地層(Y)が、金属酸化物(A)とリン化合物(B)とイオン価(FZ)が1〜3の範囲内である陽イオン(Z)とを含有し、
前記リン化合物(B)が、前記金属酸化物(A)と反応可能な部位を有する化合物であり、前記下地層(Y)において、前記金属酸化物(A)を構成する金属原子(M)のモル数(NM)と、前記リン化合物(B)に由来するリン原子のモル数(NP)とが、下記関係式(1)で表される関係を満たし、かつ、
前記下地層(Y)において、前記モル数(NM)と、前記陽イオン(Z)のモル数(NZ)と、前記イオン価(FZ)とが、下記関係式(2)で表される関係を満たすように調整することを特徴とするガスバリアーフィルムの製造方法。
関係式(1) 0.8≦NM/NP≦4.5
関係式(2) 0.001≦FZ×NZ/NM≦0.60 - 前記ガスバリアー層(X)を、平均組成をSiOxCy(x及びyは化学量論係数)で表したときに、下記関係式(3)で表される関係及び下記関係式(4)で表される関係を満たし、かつ層厚を15〜50nmの範囲内に調整することを特徴とする請求項3に記載のガスバリアーフィルムの製造方法。
関係式(3) 1.6≦x≦1.9、0.15≦y≦0.40
関係式(4) 4.0≦2x+2y<4.3 - 前記ガスバリアー層(X)を、対向ローラー型のロールtoロール成膜装置を用いて、真空プラズマCVD法によって形成することを特徴とする請求項3又は請求項4に記載のガスバリアーフィルムの製造方法。
- 請求項1又は請求項2に記載のガスバリアーフィルムを具備することを特徴とする有機エレクトロルミネッセンスデバイス。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016118473A JP2017222071A (ja) | 2016-06-15 | 2016-06-15 | ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイス |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016118473A JP2017222071A (ja) | 2016-06-15 | 2016-06-15 | ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイス |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2017222071A true JP2017222071A (ja) | 2017-12-21 |
Family
ID=60686159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016118473A Pending JP2017222071A (ja) | 2016-06-15 | 2016-06-15 | ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイス |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2017222071A (ja) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020262110A1 (ja) * | 2019-06-25 | 2020-12-30 | 住友化学株式会社 | 有機電子デバイスの製造方法 |
| TWI777505B (zh) * | 2020-05-21 | 2022-09-11 | 日商吳羽股份有限公司 | 組成物及其製造方法 |
| WO2025206303A1 (ja) * | 2024-03-29 | 2025-10-02 | 株式会社クラレ | 多層構造体およびその製造方法、多層構造体を含む電子デバイス用保護シート並びに電子デバイス |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003340971A (ja) * | 2002-05-24 | 2003-12-02 | Dainippon Printing Co Ltd | ガスバリア性プラスチックフィルム |
| JP2003342735A (ja) * | 2002-05-24 | 2003-12-03 | Dainippon Printing Co Ltd | ガスバリア性フィルム |
| JP2010027480A (ja) * | 2008-07-23 | 2010-02-04 | Saitama Univ | エレクトロルミネッセンス材料、その製造方法及びエレクトロルミネッセンス素子 |
| JP2010228309A (ja) * | 2009-03-27 | 2010-10-14 | Toppan Printing Co Ltd | バリア性ptp底材及びその製造方法 |
| JP2012061671A (ja) * | 2010-09-15 | 2012-03-29 | Toppan Printing Co Ltd | ガスバリア性積層フィルム |
| WO2015141225A1 (ja) * | 2014-03-18 | 2015-09-24 | 株式会社クラレ | 多層構造体およびその製造方法、それを用いた包装材および製品、電子デバイスの保護シートならびにコーティング液 |
| JP2016040120A (ja) * | 2014-03-18 | 2016-03-24 | 株式会社クラレ | 包装材およびそれを用いた製品 |
| JP2016040119A (ja) * | 2014-03-18 | 2016-03-24 | 株式会社クラレ | 電子デバイスの保護シート |
| JP2016087815A (ja) * | 2014-10-30 | 2016-05-23 | 凸版印刷株式会社 | 透明ガスバリア性フィルム |
-
2016
- 2016-06-15 JP JP2016118473A patent/JP2017222071A/ja active Pending
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003340971A (ja) * | 2002-05-24 | 2003-12-02 | Dainippon Printing Co Ltd | ガスバリア性プラスチックフィルム |
| JP2003342735A (ja) * | 2002-05-24 | 2003-12-03 | Dainippon Printing Co Ltd | ガスバリア性フィルム |
| JP2010027480A (ja) * | 2008-07-23 | 2010-02-04 | Saitama Univ | エレクトロルミネッセンス材料、その製造方法及びエレクトロルミネッセンス素子 |
| JP2010228309A (ja) * | 2009-03-27 | 2010-10-14 | Toppan Printing Co Ltd | バリア性ptp底材及びその製造方法 |
| JP2012061671A (ja) * | 2010-09-15 | 2012-03-29 | Toppan Printing Co Ltd | ガスバリア性積層フィルム |
| WO2015141225A1 (ja) * | 2014-03-18 | 2015-09-24 | 株式会社クラレ | 多層構造体およびその製造方法、それを用いた包装材および製品、電子デバイスの保護シートならびにコーティング液 |
| JP2016040120A (ja) * | 2014-03-18 | 2016-03-24 | 株式会社クラレ | 包装材およびそれを用いた製品 |
| JP2016040119A (ja) * | 2014-03-18 | 2016-03-24 | 株式会社クラレ | 電子デバイスの保護シート |
| JP2016087815A (ja) * | 2014-10-30 | 2016-05-23 | 凸版印刷株式会社 | 透明ガスバリア性フィルム |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020262110A1 (ja) * | 2019-06-25 | 2020-12-30 | 住友化学株式会社 | 有機電子デバイスの製造方法 |
| TWI777505B (zh) * | 2020-05-21 | 2022-09-11 | 日商吳羽股份有限公司 | 組成物及其製造方法 |
| WO2025206303A1 (ja) * | 2024-03-29 | 2025-10-02 | 株式会社クラレ | 多層構造体およびその製造方法、多層構造体を含む電子デバイス用保護シート並びに電子デバイス |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN105390617B (zh) | 有机电致发光元件 | |
| JP5565454B2 (ja) | ガスバリアフィルムの製造方法、有機エレクトロルミネッセンス用樹脂基材、それを用いた有機エレクトロルミネッセンス素子 | |
| KR101885053B1 (ko) | 유기 일렉트로루미네센스 소자 | |
| US20150099126A1 (en) | Gas barrier film, substrate for electronic device and electronic device | |
| US10032826B2 (en) | Light extraction substrate, method for manufacturing light extraction substrate, organic electroluminescent element, and method for manufacturing organic electroluminescent element | |
| CN105026141A (zh) | 气体阻隔膜、气体阻隔膜的制造方法、及有机电致发光元件 | |
| JPWO2016039060A1 (ja) | ガスバリア性フィルム、及び、有機エレクトロルミネッセンス素子 | |
| CN105593013A (zh) | 气体阻隔性膜的制造方法 | |
| WO2016143660A1 (ja) | 有機エレクトロルミネッセンス素子 | |
| JP2017222071A (ja) | ガスバリアーフィルム、その製造方法及び有機エレクトロルミネッセンスデバイス | |
| WO2014141821A1 (ja) | 電子デバイス及び電子デバイスの製造方法 | |
| JP2006299145A (ja) | ガスバリア性フィルム、ガスバリア性フィルムを用いた有機エレクトロルミネッセンス用樹脂基材および有機エレクトロルミネッセンス素子 | |
| JP6424513B2 (ja) | 有機エレクトロルミネッセンス素子 | |
| KR20160145141A (ko) | 유기 일렉트로 루미네센스 소자 | |
| JP6477468B2 (ja) | 有機エレクトロルミネッセンス素子 | |
| JP6773048B2 (ja) | 発光装置 | |
| WO2017158920A1 (ja) | 有機エレクトロルミネッセンス素子の製造方法 | |
| JP2016170879A (ja) | 有機エレクトロルミネッセンス素子 | |
| JPWO2014148595A1 (ja) | 有機エレクトロルミネッセンス素子及び照明装置 | |
| JP2016190442A (ja) | ガスバリアフィルム、透明導電部材、及び、有機エレクトロルミネッセンス素子 | |
| JP2016054097A (ja) | 有機エレクトロルミネッセンス素子、及び、基板 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190327 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20191213 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200204 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20200804 |