JP2017507174A - 抗菌剤 - Google Patents
抗菌剤 Download PDFInfo
- Publication number
- JP2017507174A JP2017507174A JP2016568088A JP2016568088A JP2017507174A JP 2017507174 A JP2017507174 A JP 2017507174A JP 2016568088 A JP2016568088 A JP 2016568088A JP 2016568088 A JP2016568088 A JP 2016568088A JP 2017507174 A JP2017507174 A JP 2017507174A
- Authority
- JP
- Japan
- Prior art keywords
- optionally substituted
- group
- optionally
- alkyl
- lys
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
- 0 *CCCC[C@](*)C(N)=O Chemical compound *CCCC[C@](*)C(N)=O 0.000 description 3
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1019—Tetrapeptides with the first amino acid being basic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06086—Dipeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0815—Tripeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K9/00—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof
- C07K9/006—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof the peptide sequence being part of a ring structure
- C07K9/008—Peptides having up to 20 amino acids, containing saccharide radicals and having a fully defined sequence; Derivatives thereof the peptide sequence being part of a ring structure directly attached to a hetero atom of the saccharide radical, e.g. actaplanin, avoparcin, ristomycin, vancomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Pharmacology & Pharmacy (AREA)
- Communicable Diseases (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
細菌感染により起こる疾患は、人間及び他の哺乳動物においてかなりの罹患率及び死亡率を有する。グラム陽性細菌は、堅い細胞壁に囲まれた典型的な脂質二重層細胞膜を有する。細胞壁は、交互D−及びL−アミノ酸を含むペプチドにより架橋されたN−アセチルグルコサミンとN−アセチルムラミン酸のポリマーであるペプチドグリカンで構成されている。
−(L−[W])nBX(I)
(式中、各Lは、独立に、柔軟なリンカー基であり、各Wは、独立に、ペプチド膜結合要素であり、nは1以上の整数であり、Xはペプチド又は非ペプチド性の膜結合又は挿入要素である)
の存在物により修飾されているポリペプチド誘導体を記載している。
V−L−W−X(II)
(式中、Vは、細菌中のペプチドグリカン生合成を阻害するグリコペプチド部分であり;
Lは、連結基であり;
Wは、ペプチド膜会合要素であり;且つ
Xは、水素又は膜挿入要素である)
を記載している。
本発明者らは、ジスルフィド結合を含む、例えば、上記構造IIにあるグリコペプチドと膜結合要素の間の連結を含む従来技術の構造が、生理的条件下で他のチオールとのジスルフィド交換を受け、化合物の分解とその結果の活性低下を起こし得ることを発見した。さらに、従来技術に記載される長いペプチド配列は、商業的な抗生物質に求められる規模での合成が困難であると同時に、生理的条件下でタンパク質分解を受けやすい。
X−W−L−V(III)
(式中、
Xは、WのN−末端に結合した親油性基であり、炭素原子がベースとなっており、且つ以下のパラメーター;
− 存在する場合、任意の芳香環の炭素原子を含む、3〜60個の炭素原子を有すること;
− 直鎖又は分岐鎖であり、且つ後者の場合、1〜6個の分岐点を有すること;
− 飽和又は不飽和であり、後者の場合、1〜8個の二重又は三重結合を含むこと;
− S、O、又はNから独立に選択され、酸性置換基中に含まれない最大6個のヘテロ原子を(存在する場合、芳香環における、存在する場合、ヘテロ原子に加えて)任意選択で有すること;
− 1個以上、例えば、2、3、4、5、又は6個の芳香環であって、縮合していてよく、且つそのそれぞれが、存在する場合、N、O、又はSから独立に選択される1、2、又は3個のヘテロ原子を含み得る、1個以上、例えば、2、3、4、5、又は6個の芳香環を任意選択で含むこと;且つ
− ヒドロキシ、アミノ、メチル、メチルアミノ、及びハロから選択される1〜6個の置換基を任意選択で有すること
を有し;
Wは、Wが、硫黄含有側鎖を有する任意のアミノ酸でなく、又はそれを含まないという条件で、塩基性アミノ酸又は2〜10個のアミノ酸からなる塩基性ペプチドであり;
Lは、式−NH−(CR1R2)m−Z−(CR3R4)n−NH−
(式中、
Zは、酸素又は−NH−、−CONH−、−NHCO−、−(OCH2CH2)p−、C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C1〜C12ヘテロアルキル、C1〜C10ヘテロアルケニル、C3〜C12シクロアルキル、C1〜C12複素環、C6〜C18アリール、若しくはC1〜C18ヘテロアリールからなる群から選択される任意選択で置換されている部分であり;且つ
R1、R2、R3、及びR4は、H、ハロゲン、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC1〜C10ヘテロアルケニル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC1〜C12複素環、任意選択で置換されているC6〜C18アリール、任意選択で置換されているC1〜C18ヘテロアリール、任意選択で置換されているカルボキシ、任意選択で置換されているカルボキサミドからなる群からそれぞれ独立に選択され;且つ
mは、0、1、2、及び3からなる群から選択される整数であり;且つ
nは、0、1、2、及び3からなる群から選択される整数であり;
但し、mとnとの両方が0ではないことを条件とし;且つ
pは、1〜10からなる群から選択される整数である)
の連結基であるか、又は
Lは、式:
(式中、c及びdは、0、1、及び2からなる群から選択される整数であり;且つ
点線は、V及びWへの結合点を示す)
の1つから選択され;且つ
Vは、細菌中のペプチドグリカン生合成を阻害するグリコペプチド部分である)
の化合物、又はその薬学的に許容できる塩若しくはプロドラッグを提供する。
本発明がさらに説明される前に、説明される実施形態は当然変わり得るため、本発明が、説明される特定の実施形態に限定されないことを理解されたい。本発明の範囲は添付される特許請求の範囲によってのみ限定されるため、本明細書で使用される用語は、特定の実施形態を説明する目的のみであり、限定的でないものとすることも理解されたい。
本発明の化合物は、式(III)のグリコペプチド抗生物質である。化合物は、以下に示される通り良好な抗菌活性並びに先に示された従来技術の式(II)の化合物よりも、グルタチオン存在下でのより良好な安定性及びインビボでのより良好な安定性を示す。
本発明の化合物において、Xは、WのN−末端に結合した親油性基であり、炭素原子がベースとなっており、且つ以下のパラメーター:
− 存在する場合、脂環式環又は芳香環の原子を含む、3〜60個の原子を有すること;
− 直鎖又は分岐鎖であり、且つ後者の場合、1個以上、例えば、2、3、4、5、又は6個の分岐点を含むこと;
− 飽和又は不飽和であり、後者の場合、1〜8、例えば、1、2、3、4、5、6、7、又は8個の二重又は三重結合を含むこと;
− S、O、又はNから独立に選択され、酸性置換基中に含まれない最大6個、例えば、1、2、3、4、5、又は6個のヘテロ原子を(存在する場合、芳香環における、存在する場合、ヘテロ原子に加えて)任意選択で有すること;
− 1個以上、例えば、2、3、4、5、又は6個の芳香環であって、縮合していてよく、且つそのそれぞれが、存在する場合、N、O、又はSから独立に選択される1、2、又は3個のヘテロ原子を含み得る、1個以上、例えば、2、3、4、5、又は6個の芳香環を任意選択で含むこと;且つ
− ヒドロキシ、アミノ、メチル、メチルアミノ、及びハロから独立に選択される1〜6個(1、2、3、4、5又は6個など)置換基を任意選択で有すること
を有する。
(式中、
各R25及びR26は、以下のパラメーター:
− 存在する場合、任意の脂環式環又は芳香環の炭素原子を含む、3〜30個の炭素原子を有すること;
− 直鎖又は分岐鎖であり、且つ後者の場合、1〜3個の分岐点を含むこと;
− 飽和又は不飽和であり、後者の場合、1〜4個の二重又は三重結合を含むこと;
− S、O、又はNから独立に選択される1、2、又は3個のヘテロ原子を(存在する場合、芳香環における、存在する場合、ヘテロ原子に加えて)任意選択で有すること;
− 1個以上、例えば、2又は3個の芳香環であって、縮合していてよく、且つそのそれぞれが、存在する場合、N、O、又はSから独立に選択される1、2、又は3個のヘテロ原子を含むことができる、1個以上、例えば、2又は3個の芳香環を任意選択で含むこと;且つ
− ヒドロキシ、アミノ、メチル、メチルアミノ、及びハロから選択される1〜3個の置換基を任意選択で有すること
を有する、親油性基であり;
tは、0、1、2、3、4、及び5からなる群から選択される整数であり;且つ
uは、0、1、2、3、4、及び5からなる群から選択される整数であり;
但し、t又はuの一方が0である場合、t又はuの他方が0でないことを条件とする)
のものである。
− 直鎖又は分岐鎖であり、且つ脂環式環又は芳香環であって、基中の炭素原子の総数は任意のそのような環の炭素原子を含む、脂環式環又は芳香環を含むことができ;
− 飽和又は不飽和であり、後者の場合、1〜4個の二重又は三重結合を含み;
− O又はNから独立に選択される1又は2個のヘテロ原子を(存在する場合、芳香環における、存在する場合、ヘテロ原子に加えて)任意選択で有し;
− 1又は2個の芳香環であって、そのいずれか若しくは両方が1個の窒素ヘテロ原子を含み得る、1又は2個の芳香環を任意選択で含み;且つ
− ヒドロキシル、アミノ、メチル、メチルアミノ、及びハロから選択される1〜3個の置換基を任意選択で有する。
nC10CO−;nC13CO−;4−PhO−PhCO−;[(4−PhO−PhCO)Lys(4−PhO−PhCO)−;(nC10CO)Lys(COnC10)−;nC10CO−Gly−;nC11CO−;nC12CO−;(4−PhO−PhCO)−Gly−;nC7CO−;nC8CO−;nC9Co−;(2−Bu−nC7CO)−;[(2−Bu−nC7CO−)Lys(2−Bu−nC7CO)]−;(9Z−9,10−デイヒドロ(deyhdro)−nC13CO)−;C6PhCO−;C7PhCO−;C5OPhCO−;(C5OPhCO−)Lys(C5OPhCO)−;C7OPhCO−;C9OPhCO−;PhOC3CO−;[PhOC3CO−Lys(PhOC3CO)]−;PhC5CO−;[PhC5CO−Lys(PhC5CO)]−;PhC8CO−;PhC9CO−;PhC11CO−;(4−Ph−PhC1CO)−;[(4−Ph−PhC1CO)−Lys(4−Ph−PhC1CO)]−;[4−(4−F−PhO)−PhCO]−;{[4−(4−Cl−PhO)−PhCO−Lys[4−(4−F−PhO)−PhCO]}−;[4−(4−Cl−PhCO)−PhCO]−;{[4−(4−Cl−PhO)−PhCO−Lys[4−(4−Cl−PhO)−PhCO]}−;(4−BnO−PhCO)−;[(4−BnO−PhCO)−Lys(4−BnO−PhCO)]−;(nC9−pip−4−CO)−;[nC7CO−Lys(COnC7)]−;[(C9OPhCO)−Lys(C9OPhCO)]−;[(PhOC3CO)−Lys(PhOC3CO)]−;{[4−(4−F−PhO)−PhCO−Lys[4−(4−F−PhO)−PhCO]}−;[PhC11CO−Lys(PhC11CO)]−;nC10CH2−;4−PhO−PhCH2−;CH3Ph−SO2−;[nC12CO−Lys(nC12−CO)]−;[nC13CO−Lys(nC13−CO)]−;(2−Bu−C7CO)−Lys(2−Bu−C7CO)−;(nC9CH2)2−;nC13CH2−;PhCH2−;(nC11−Pip−4−CO)−;(nC7−Pip−4−CO)−;(1−nC9−Pro)−;(nC9−Pip−2−CO)−;(4−NH2−PhCO)−;(4−MeNH−PhCO)−;(4−nC7NH−PhCO)−;(nC4CH2−)2;nC8CH2−;(2−NH2−nC9CO)−、3,5−Me2C7CO−;3−OH−C9CO−;(4−Cl−PhCO)−;cHexCH2CO−、及び4−nC5−cHexCO−。
本発明の化合物において、Wは、Wが、硫黄含有側鎖を有する任意のアミノ酸でなく、又はそれを含まないという条件で、塩基性アミノ酸又は2〜10個のアミノ酸からなる塩基性ペプチドである。
(式中、
Yは、式−(CR20R21)g−の基であり;
R18aは、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、任意選択で置換されているC1〜C18ヘテロアリール、−C(=NR22)−NR23R24、及びOR22からなる群から選択され、
R18bは、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から選択されるか、又は
R18a及びR18bは、それらが結合している窒素原子と共に、任意選択で置換されている複素環部分を形成するか、又は
R18a及びR18bの一方が、R20又はR21のいずれか及びそれらが結合している原子と共に、任意選択で置換されている複素環部分を形成し;
R19は、H及び任意選択で置換されているC1〜C12アルキルからなる群から選択され;
R20及びR21は、H、ハロゲン、OH、C1〜C12アルキル、C6〜C18アリール、C1〜C12ハロアルキル、C1〜C12ヒドロキシアルキル、C1〜C12アルキルオキシ、及びC1〜C12ハロアルキルオキシからなる群からそれぞれ独立に選択されるか、又は
R20及びR21は、それらが結合している炭素と共に、任意選択で置換されているC3〜C12シクロアルキル、又は任意選択で置換されているC1〜C12ヘテロシクロアルキル基を形成するか、又は
R20及びR21の一方が、R18aとR18bとの一方及びそれらが結合している原子と共に、任意選択で置換されている複素環部分を形成し;
各R22、R23、及びR24は、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から独立に選択されるか、又は
R22、R23、及びR24のいずれか2個は、それらが結合している原子と共に、任意選択で置換されている環式基を形成し;
gは、1、2、3、4、及び5からなる群から選択される整数であり;
rは、1、2、3、4、5、6、7、8、9、及び10からなる群から選択される整数である)
の1個の残基又は2〜10個の連続する残基から、より好ましくは1個の残基又は2〜5個の連続する残基からなる。
本発明の化合物において、Lは、式:
−NH−(CR1R2)m−Z−(CR3R4)n−NH−
(式中、
Zは、酸素又は−NH−、−CONH−、−NHCO−、−(OCH2CH2)p−、C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C1〜C12ヘテロアルキル、C1〜C10ヘテロアルケニル、C3〜C12シクロアルキル、C1〜C12複素環、C6〜C18アリール、若しくはC1〜C18ヘテロアリールからなる群から選択される任意選択で置換されている部分であり;且つ
R1、R2、R3、及びR4は、H、ハロゲン、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC1〜C10ヘテロアルケニル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC1〜C12複素環、任意選択で置換されているC6〜C18アリール、任意選択で置換されているC1〜C18ヘテロアリール、任意選択で置換されているカルボキシ、任意選択で置換されているカルボキサミドからなる群からそれぞれ独立に選択され;且つ
mは、0、1、2、及び3からなる群から選択される整数であり;且つ
nは、0、1、2、及び3からなる群から選択される整数であり;
但し、mとnとの両方が0ではなく;且つ
pは、1〜10からなる群から選択される整数である)
の連結基であるか、又は;
Lは、式:
(式中、c及びdは、0、1、及び2からなる群から選択される整数であり;且つ
点線は、V及びWへの結合点を示す)
の1つから選択される。
R1が、−(CO)OH、−(CO)OMe、−(CO)NH2、−(CO)NHNH2、−(CO)NHMe、−(CO)NHEt、−(CO)N(Me)2、−(CO)NHBn、若しくは−(CO)R5、又は任意選択で置換されているC1〜C12複素環、若しくは任意選択で置換されているC1〜C18ヘテロアリール部分であり;且つ
R5が、任意選択で置換されているC1〜C12複素環又は任意選択で置換されているC1〜C18ヘテロアリール部分であり;且つ
Z及びnが先に定義されている通りである。
qが、0、1、2、3、4、及び5からなる群から選択される整数であり;且つ
R1が、−(CO)OH、−(CO)OMe、−(CO)NH2、−(CO)NHNH2、−(CO)NHMe、−(CO)NHEt、−(CO)N(Me)2、−(CO)NHBn、若しくは−(CO)R5、又は任意選択で置換されているC1〜C12複素環、若しくは任意選択で置換されているC1〜C18ヘテロアリール部分である。
(R)−又は(S)−NHCH(COOH)(CH2)4NH−;
(R)−又は(S)−NHCH(CONHMe)(CH2)4NHCOCH2NH−;
(R)−又は(S)−NHCH(COOH)(CH2)4NHCOCH2NH−;
(R)−又は(S)−NHCH2CO−NH(CH2)2NH−;
(R)−又は(S)−NHCH(CONH2)−CH2−(1,3−トリアゾール)−(CH2)3−NH−;
(R)−又は(S)−NHCH(CONH2)(CH2)4NH−;
(R)−又は(S)−NHCH(CONHMe)(CH2)4NH−;
(R)−又は(S)−NHCH(COOMe)(CH2)4NH−;
(R)−又は(S)−NHCH(CONHMe)(CH2)3NH−;
(R)−又は(S)−NHCH(CONHMe)(CH2)2NH−;
(R)−又は(S)−NHCH(CONH2)(CH2)3NH−;
(R)−又は(S)−NHCH(CONH2)(CH2)2NH−;
(R)−又は(S)−NHCH(COOH)(CH2)3NH−;
(R)−又は(S)−NHCH(COOH)(CH2)2NH−;
(R)−又は(S)−NHCH2CO−NH(CH2)3NH−;
(R)−又は(S)−NHCH(CONHnC14)(CH2)4NH−;
(R)−又は(S)−NHCH(CONHEt)(CH2)4NH−;
(R)−又は(S)−NHCH(CONHBn)(CH2)4NH−;
−NH(CH2)2NH−;
−NH(CH2)3NH−;
−NH(CH2)4NH−;
−NH(CH2)5NH−;
−NH(CH2)6NH−;
−NH(CH2)2NH(CH2)2NH−;
−NH(CH2)2O(CH2)2NH−;
−NH(CH2)2O(CH2)2O(CH2)2NH−;
−NHCH2(1,3−Ph)CH2NH−;
−NHCH2(1,4−Ph)CH2NH−;
−NH(1,4−cHex)NH−;
−NHCH2(1,4−cHex)CH2NH−;
−1−ピペリジン−4−CH2NH−;
−1−ピペリジン−2−CH2NH−;及び
−1−ピペリジン−2−NH−。
(R)−又は(S)−NHCH(CONHMe)(CH2)4NH−;
(R)−又は(S)−NHCH(CONH2)(CH2)4NH−;
(R)−又は(S)−NHCH(COOH)(CH2)4NH−;
−NH(CH2)2NH−;及び
−NH(CH2)3NH−。
本発明の化合物において、Vは、細菌中のペプチドグリカン合成を阻害するグリコペプチド部分である。
(式中、
X、W、及びLは先に定義されている通りであり;
R7は、水素、炭水化物、又はアミノ炭水化物であり;
R8は、水素、OH、又は−O−マンノースであり;
R9は、−NH2、−NHCH3、又は−N(CH3)2であり;
R10は、−CH2CH(CH3)2、[p−OH,m−Cl]フェニル、p−ラムノース−フェニル、[p−ラムノース−ガラクトース]フェニル、[p−ガラクトース−ガラクトース]フェニル、[p−CH3O−ラムノース]フェニルであるか、又は[p−OH,m−(O−{m−OH,m−R11}−フェニル)]フェニルを介してR11に結合して環式環系を形成し;
R11は、−CH2−(CO)NH2、ベンジル、[p−OH]フェニル、[p−OH,m−Cl]フェニル;[p−OH,m−Cl]フェニルであるか、又は[m−OH,m−(O−{o−OH、m−R10}フェニル)]−フェニルを介してR10に結合して環式環系を形成し;
R12は、水素、又はマンノースであり;
R13は、水素、OH、又はCH2NHCH2PO3H2であり;
R14は、水素、ベータ−D−グルコピラノース、ベータ−D−グルコサミン、2−O−(アルファ−L−バンコサミニル)−ベータ−D−グルコピラノース、2−O−(アルファ−L−4−エピ−バンコサミニル)−ベータ−D−グルコピラノース、(アルファ−アクチノサミニル)−ベータ−D−グルコピラノース、(アルファ−リストサミニル)−ベータ−D−グルコピラノース、若しくは(アルファ−アコサミニル)−ベータ−D−グルコピラノース;又は前記グルコサミン若しくはグルコピラノース基のいずれか1つであって、その一級アミンでR17により任意選択で置換されている前記グルコサミン若しくはグルコピラノース基のいずれか1つであり、R17は、先に定義されている通りであり;且つ
R15及びR16は、独立に、水素又はクロロである)
のものである。
Xが式R27CO−のものであり、式中、R27が3〜15個の炭素原子を有する親油性基であり、前記親油性基が:
− 直鎖又は分岐鎖であり、且つ脂環式環又は芳香環であって、基中の炭素原子の総数は任意のそのような環の炭素原子を含む、脂環式環又は芳香環を含むことができ;
− 飽和又は不飽和であり、後者の場合、1〜4個の二重又は三重結合を含み;
− O又はNから独立に選択される1又は2個のヘテロ原子を(存在する場合、芳香環における、存在する場合、ヘテロ原子に加えて)任意選択で有し;
− 1又は2個の芳香環であって、そのいずれか若しくは両方が1個の窒素ヘテロ原子を含み得る、1又は2個の芳香環を任意選択で含み;且つ
− ヒドロキシル、アミノ、メチル、メチルアミノ、及びハロから選択される1〜3個の置換基を任意選択で有し;
Wが、アミノ酸であるか、又は任意選択で置換されているD−若しくはL−リジン、オルニチン、2,4−ジアミノ酪酸、2,3−ジアミノプロピオン酸、及びアルギニンから選択される2〜5個のタンパク質構成若しくは非タンパク質構成アミノ酸からなるペプチドであり;
Lが式−NH−(CH2)m−Z−(CH2)n−NH−のものであり;式中、Z、m、及びnが、先に定義されている通りであり;且つ
Vが、バンコマイシン、バンコマイシンアグリコン、バンコマイシンデスバンコサミン、デスメチルバンコマイシン、クロロエレモマイシン、テイコプラナイン−A2−2、リストセチンA、エレモマイシン、バルヒマイシン、アクチノイジンA、コンプレスタチン、クロロペプチン1、キスタマイシンA、アボパルシン、テラバンシン、A40926、及びオリタバンシン、並びに一級アミンでR17により任意選択で置換されているそのいずれか1つから選択され、R17は、水素、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC1〜C10ヘテロアルケニル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から選択される有機側鎖部分である、化合物である。
Xが、式CjH(2j+1)CO−のアルカン酸であり、式中、jは、7、8、9、10、11、12、又は13から選択され;
Wが、塩基性アミノ酸又は2若しくは3個のアミノ酸残基からなる塩基性ペプチドであり、Wとしての又はW内の各アミノ酸は、L−リジン、D−リジン、オルニチン、2,4−ジアミノ酪酸、及び2,3−ジアミノプロピオン酸からなる群から選択され;
Lが、
(R)−又は(S)−NHCH(CONHMe)(CH2)4NH−;
(R)−又は(S)−NHCH(CONH2)(CH2)4NH−;
(R)−又は(S)−NHCH(COOH)(CH2)4NH−;
−NH(CH2)2NH−;又は
−NH(CH2)3NH−;
からなる群から選択され;且つ
Vが、バンコマイシン、バンコマイシンアグリコン、バンコマイシンデスバンコサミン、及びデスメチルバンコマイシンからなる群から選択される、化合物である。
Ra、Rb、Rc、及びRdは、H、C1〜C12アルキル、C1〜C12ハロアルキル、C2〜C12アルケニル、C2〜C12アルキニル、C1〜C10ヘテロアルキル、C3〜C12シクロアルキル、C3〜C12シクロアルケニル、C1〜C12ヘテロシクロアルキル、C1〜C12ヘテロシクロアルケニル、C6〜C18アリール、C1〜C18ヘテロアリール、及びアシルからなる群からそれぞれ独立に選択されるか、又はRa、Rb、Rc、及びRdのいずれか2個以上が、それらが結合している原子と共に、3〜12個の環原子を有する複素環式環系を形成する。
本発明のさらなる態様は、式(III)の化合物及び薬学的に許容できる担体を含む医薬組成物である。
本発明の化合物を合成できる経路は当技術分野で周知である。一般的に、化合物は、保護された要素Wを要素Lにカップリングし、次いでそれに要素Xを取り付けることにより合成できる。最後に、X−W−LをVとカップリングして、保護基を除く。生じた化合物を、例えば、L基において修飾して、さらなる化合物を得ることができる。代替合成経路において、LをVにカッププリングし、XをWにカップリングし、最終工程として、X−WをL−Vにカップリングする。
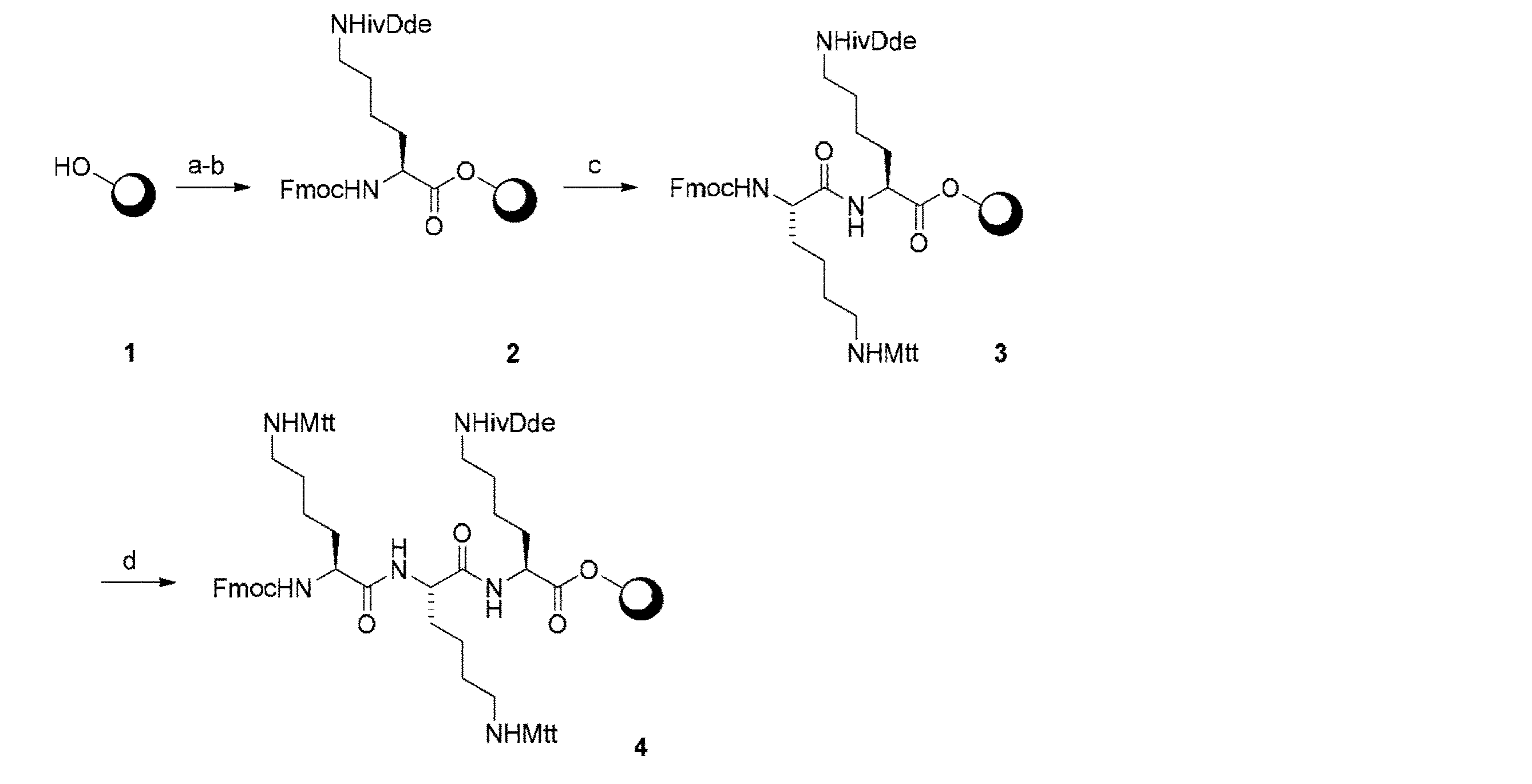
Hypogel HMBA樹脂(1.0g、ローディング0.81mmol/g、0.81mmol)を、乾燥DMF(×3)で洗浄した。Fmoc−L−Lys(ivDde)−OH(2.33g、MW 574.7、4.05mmol、5当量)の乾燥DMF溶液(10ml)に、ヒドロキシベンゾトリアゾール(HOBt、547mg、MW 135、4.05mmol、5当量)を加え、それに続いて1,3−ジイソプロピルジイミド(DIC)(627μl、d 0.815、MW 126.2、4.05mmol、5当量)を、次いで4−ジメチルアミノピリジン(DMAP、30mg、MW 122.17、0.3当量)を加えた。生じた溶液を樹脂に加え、樹脂を室温で一晩振とうした。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。樹脂をアセチル残基でキャップするために、樹脂を、グローブボックス中で、乾燥DMF(×3)及びDIPEA(848μl)で洗浄し、次いで無水酢酸(307μl、d 1.08、MW 102.09)を加え、1時間振とうし、次いで液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。
樹脂(1.45g、0.56mmol/g)を、20%ピペリジンのDMF溶液(14.5ml)により処理し、室温で1時間振とうした。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。
樹脂(1.27g、0.64mmol/g)を、DMF(×3)で洗浄した。Fmoc−L−Lys(Mtt)−OH(995mg、MW 624.8、1.59mmol、2.0当量)のDMF(9.5ml)溶液に、HBTUのDMF溶液(3.2ml、0.5M、1.59mmol)を加え、それに続いてDIPEA(1107μl、d 0.742、MW 129.25、6.35mmol、7.8当量)を加えた。溶液を10分間静置し、次いで洗浄した樹脂に加えた。樹脂を、室温で3時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。
樹脂(100mg、0.043mmol)をDMF(×3)で洗浄した。酸のDMF溶液(1ml)に、HBTUのDMF溶液(0.2ml、0.5M)、DIPEA(34.9μl、d 0.742、MW 129.25、最終濃度0.5M)を加えた。溶液を10分間静置し、次いでDMFで洗浄した樹脂に加えた。樹脂を室温で一晩振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。
樹脂(100mg、0.43mmol/g)をDMF(×3)で洗浄した。樹脂に、DMF(2ml)中のドデカンスルホニルクロリド(17.3mg、MW 268.84、1.5当量)、DIPEA(8.9μl、d 0.742、MW 129.25、1.2当量)を加え、室温で一晩振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。
樹脂(1.89g、0.43mmol/g)をDMF(×3)で洗浄した。Fmoc−L−Lys(Fmoc)−OH(1400mg、MW 624.8、1.59mmol、2.9当量)のDMF(14.2ml)溶液に、HBTUのDMF溶液(4.73ml、0.5M、2.37mmol、2.9当量)を加え、それに続いてDIPEA(1650□l、d 0.742、MW 129.25、6.35mmol、11.7当量)を加えた。溶液を10分間静置し、次いで洗浄した樹脂に加えた。樹脂を、室温で3時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄し、真空中で乾燥させた。次いで、Fmoc基を、手順(ii)に従って除去し、遊離アミンを、手順(iv−a)を利用して誘導体化した。
樹脂を、無水メタノール(2.5ml/樹脂100mg)、無水DMF(2.5ml/樹脂100mg)、及び無水DIPEA(0.5ml/樹脂100mg)により処理し、50℃の油浴中で一晩加熱した。樹脂から液体を排出し、DMF(×3)及びMeOH(×3)で洗浄し、溶媒を減圧下で除去した。
樹脂を、THF(×3)で、次いでDIPEA(×2)で洗浄し、次いで、THF中のメチルアミン2M、2ml/樹脂100mg)により処理し、室温で一晩振とうした。減圧を利用して樹脂から液体を排出し、次いで、THF(×2)、DCM(×2)、及びACN(×2)で洗浄した。N2ガスを利用して溶媒を発散させ、試料をLCMSにより品質管理のために分析した。
ペプチド(40mg)を、2%抱水ヒドラジンのDMF溶液(2.0ml)に溶解させた。生じた溶液を室温で30分間撹拌し、溶媒を減圧下で除去した。いくつかの試料をLCMSにより品質管理のために分析した。
ペプチド7(50μmol)の乾燥DMF(0.86ml)溶液を、バンコマイシン塩酸塩(89mg、FW 1485.71、59.9μmol、1.2当量)の乾燥DMF(0.86ml)溶液により処理した。この溶液に、HBTUの乾燥DMF溶液(120μl、0.5M、60μmol)、1.2当量)を加え、それに続いてDIPEA(36μl、d 0.742、FW 129.25、0.207μmol、4.1当量)を加えた。生じた溶液を室温で一晩撹拌した。試料をLCMSにより分析し、カップリングの完了を確実にして、必要な場合追加のカップリング試薬を加えた。溶媒を真空中で除去した。
バンコマイシン結合化合物(50mg)を、2%TFAと5%TESのDCM溶液(2ml)により処理し、30分間静置した。溶媒を減圧下で除去し、試料をLCMSにより分析して、完全な脱保護を確実にした。必要な場合、プロセスを繰り返した。最終化合物をH2O/ACN(1:1、1.0ml)に溶解させ、0.45μmシリンジフィルターに通して濾過し、LCMSにより分析した。
C−末端にカルボン酸を有する化合物を得るために、化合物をジオキサン/水(1:1)及びLiOHの水溶液(10当量)0.1ml、0.5M、50μmol)により処理し、室温で一晩撹拌した。試料を凍結乾燥し、H2O/ACN(1:1、1.0ml)に溶解させ、0.45μmシリンジフィルターに通して濾過し、LCMSにより分析した。
実施例1に記載した標準的な手順を、MCC000310の合成に適用した。
MCC000635の調製のために、実施例2からの共通な中間体トリペプチド4を、標準的な条件下でFmoc−L−Lys(Mtt)−OHにより処理して、ペプチド10を与えた。ペプチド10をミリスチン酸により処理すると、アルキルテールペプチド11を与え、それを次のバンコマイシンとの液相カップリングのために樹脂から切断した。
精製されたメチルエステルMCC000635(20mg)を、ジオキサン:H2O(50:50)中で、過剰のLiOH(10当量)により処理し(スキーム6)、反応の進行を、LCMSを利用してモニターした。エステルは、希薄条件中で、5℃で24時間の期間をかけて手際よく加水分解され、MCC000223を与えた。HPLC精製により、MCC223が良好な収率で生じた(10mg)。
MCC223を調製する固相経路は、HMPB樹脂(スキーム6)を利用した。酸感受性樹脂は、最終工程として使うのに魅力的であり、同時にMtt基を除去し、最終生成物を樹脂から切断する。Alloc保護基を、ivDde基の代りに使用する。固相経路は、MCC223を、8工程で、11%というかなりの全体収率(純度95%超)でHPLC精製の後に与えた。
Rink Amide AM樹脂(ローディング0.94mmol/g、300mg)を、DMF中20%ピペリジン(6.0ml)により処理し、室温で30分間振とうして、Fmoc保護基を除去した。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
Fmoc−L−Lys(Boc)−OH(703mg)をDMF(3.0ml、0.5M)に溶解させた。HBTUのDMF溶液(3.0ml、0.5M)を加え、それに続いてDIPEA(523μl)を加えた。溶液を室温で10分間静置し、次いで、先にDMFで3回洗浄した樹脂に加えた。次いで、樹脂を室温で1時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
樹脂をDMF中20%ピペリジン(6.0ml)により処理し、室温で30分間振とうした。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
Fmoc−L−Lys(ivDde)−OH(423mg)のDMF溶液(4..5ml、0.167M)を調製した。これに、HBTUのDMF溶液(1.5ml、0.5M)を加え、それに続いてDIPEA(261μl)を加えた。溶液を室温で10分間静置し、次いで、先にDMFで3回洗浄した樹脂に加えた。次いで、樹脂を室温で2時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。この工程を繰り返した。
ウンデカン酸(279mg)のDMF(3.0ml)溶液を、HBTUのDMF溶液(3.0ml、0.5M)及びDIPEA(523μl)と混合した。溶液を室温で10分間静置し、次いで、先にDMFで3回洗浄した樹脂に加えた。樹脂を室温で1時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。樹脂を真空中で乾燥させた。5mgの樹脂を切断して、反応の程度を確認した。樹脂を1.0mlアセトニトリルに溶解させ、0.45μmシリンジフィルターに通して濾過し、LCMSにより分析した。LCMS分析:RT 9.088分、ELSDにより80%、(M+H)+ 982.5、(M+2+)2+ 491.9、(M+3H)3+ 328.3。
20%TFA、5%トリエチルシランのDCM溶液(6.0ml)を調製し、樹脂(300mg)に30分間加えた。樹脂を濾過により除去し、DCM(×3)及びアセトニトリル(×3)で洗浄した。溶媒を真空中で除去した。樹脂を、1:1のACN/H2Oに溶解させ、凍結乾燥した。
グローブボックス中で、ペプチド(40mg、40.7μmol)を乾燥DMF(0.7ml、58.1mM)に溶解させた。乾燥DMSO(0.7ml、69.7mM)中のバンコマイシン.HCl(72mg、FW1485.71、48.8μmol、1.2当量)を、溶解するまで加熱し、溶液は透明であった。溶液を室温に冷却し、ペプチドに加えた。HBTU(95mg、FW 379.3)の乾燥DMF溶液(0.5ml、0.5M)の98μlの溶液と、それに続いてDIPEA(29μl、4.1当量)を加え、一晩撹拌した。LCMS分析:Rt 7.189分、ELSDにより25%、(M+2H)2+ 1207.4、(M+3H)3+ 805.3、(M+4H)4+ 604.4。
バンコマイシン誘導体を、1:1のDMF/DMSO中の2%ヒドラジン一水和物1.0mlにより処理し、室温で一晩撹拌した。0.8mlの1:1のACN/H2Oに20μlの溶液を溶解させ、30分後、2時間後、及び一晩の後LCMSにより分析することにより、反応の程度を確認した。LCMS分析:所望の生成物Rt 5.388分、ELSDにより50%、(M+2H)2+ 1000、(M+3H)3+ 667.8、(M+4H)4+ 501;1つのivDdeのみ除去されたものRt 6.376分、ELSDにより20%、(M+2H)2+ 1103、(M+3H)3+ 736.5、(M+4H)4+ 552.6。次いで、溶媒を、高真空下35℃で一晩除去した。
粗生成物を水/ACNに溶解させ、島津分取HPLCにより精製した。精製された生成物を凍結乾燥すると、白色粉末、19mgを与え、切断したペプチドの量に基づいて19%であった。
1,3−ジアミノプロパン又は1,2−ジアミノエタンのDMF溶液(2.0M、樹脂100mgあたり2.0ml)を、ガラスバイアル中で樹脂に加えた。バイアルを固く締め、50℃で一晩加熱した。樹脂及び溶液を固相反応管に移した。濾液を回収し、樹脂を、DMF(×3)、メタノール(×3)、及びアセトニトリル(×3)で洗浄した。洗液を濾液と合わせ、溶媒を減圧下で除去すると、粗製のリポペプチドを、C末端アミノ−アルキルアミドとして与えた。粗製のリポペプチドを、さらに精製せずに、バンコマイシンとの液相カップリング反応に使用した。
1.5gのHMBA Hypogel樹脂(ローディング:0.81mmol/g)をDMF(3×)で洗浄した。Fmoc−L−Lys(Mtt)−OH(3.8g、MW 624.8g/mol、5当量)の溶液を、15mLのDMF中に調製した。ヒドロキシベンゾトリアゾール(HOBt)(0.82g、MW 135、5当量)、N,N−ジイソプロピルカルボジイミド(DIC)(941μL、MW 126.2g/mol、5当量)、及び4−ジメチルアミノピリジン(DMAP)(44.5mg、MW 122.17)を、アミノ酸溶液に加えた。溶液を、事前洗浄した樹脂に加え、一晩室温で振とうした。樹脂から液体を排出し、DMF(3×)、MeOH(3×)、及びDCM(3×)で洗浄し、真空中で乾燥させた。
250mgの樹脂を、DMF(3×)で事前洗浄し、2.5mLのDMF中20%ピペリジン(樹脂100mgあたり1.0mL)により処理し、室温で1時間振とうした。樹脂から液体を排出し、DMF(3×)、MeOH(3×)、及びDCM(3×)で洗浄し、真空中で乾燥させた。
Fmoc−L−Lys(Mtt)−OH(1539mg、MW 624.8g/mol、2当量)の14.75mLのDMF中の溶液を調製した。HBTUの乾燥DMF溶液(0.5M、4.92mL)及びDIPEA(1713μL、MW 129.25)を、アミノ酸溶液に加えた。溶液を室温で10分間静置し、次いで、事前洗浄した樹脂に加え、一晩室温で振とうした。樹脂から液体を排出し、DMF(3×)、MeOH(3×)、及びDCM(3×)で洗浄し、真空中で乾燥させた。
250mgの樹脂を、DMF(3×)により事前洗浄し、2.5mLのDMF中20%ピペリジン(樹脂100mgあたり1.0mL)により処理し、室温で1時間振とうした。樹脂から液体を排出し、DMF(3×)、MeOH(3×)、及びDCM(3×)で洗浄し、真空中で乾燥させた。
ウンデカン酸(105mg、MW 186.29g/mol 5.1当量)の1.13mLのDMF中の溶液及びHBTUの乾燥DMF中の溶液(1.13mL、0.5M、5.1当量)を調製した。HBTU溶液をウンデカン酸溶液に加え、それに続いてDIPEA(196μL、MW 129.25g/mol、d 0.742、10.2当量)を加えた。溶液を室温で10分間静置し、次いで事前洗浄した樹脂(225mg)に加え、室温で一晩振とうした。樹脂から液体を排出し、DMF(3×)、MeOH(3×)、及びDCM(3×)で洗浄し、真空中で乾燥させた。
25mgの樹脂を、0.75mLの1,3−ジアミノプロパン及び0.15mLのDIPEAにより処理した。反応混合物を、一晩室温で撹拌したままにした。切断されたペプチドを、DCM(3×)、MeOH(3×)、及びACN(3×)を使用した樹脂洗液と共に回収した。溶媒を蒸発させ、真空中で乾燥させた。
バンコマイシン.HClの溶液(195.76mg、MW 1485.7g/mol、0.125M、1.2当量)を、1.05mLの乾燥DMSO中に調製した。バンコマイシンを完全に溶解させるには、穏やかな加熱が必要である。観察された色はピンクから薄茶色に変化した。乾燥DMF中のHBTU(0.26mL、MW 379.3g/mol、0.5M 1.2当量)をバンコマイシン溶液に加え、それに続いてDIPEA(78.42μL、MW 129.25g/mol、4.1当量)を加えた。溶液をペプチドに加え、室温で一晩撹拌したままにした。バンコマイシン誘導体を、高真空下で蒸発乾固させた。
2%TFA、5%トリエチルシランの乾燥DCM溶液20mLを、バンコマイシン誘導体(200mg)に加えた。溶液を室温で30分間撹拌したままにした。溶媒を蒸発させ、真空中で乾燥させ、最終生成物MCC000344を分取HPLCにより精製した。
Rink Amide AM樹脂(ローディング0.81mmol/g、300mg)を、DMF中20%ピペリジン(6.0ml)により処理し、室温で30分間振とうし、Fmoc保護基を除去した。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
Fmoc−L−プロパルギルグリシンをDMF(4.5ml、0.5M)に溶解させた。HBTUのDMF溶液(1.5ml、0.5M)を加え、それに続いてDIPEA(0.5M最終濃度)を加えた。溶液を室温で10分間静置し、次いで先にDMFで3回洗浄した樹脂に加えた。次いで、樹脂を室温で一晩振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
樹脂を、DMF中20%ピペリジン(6.0ml)により処理し、室温で30分間振とうした。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
Fmoc−L−Lys(Boc)−OH(703mg)のDMF溶液(3.0ml、0.5M)を調製した。これに、HBTUのDMF溶液(3.0ml、0.5M)を加え、それに続いてDIPEA(523μl)を加えた。溶液を室温で10分間静置し、次いで先にDMFで3回洗浄した樹脂に加えた。次いで、樹脂を室温で1時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
樹脂をDMF中20%ピペリジン(6.0ml)により処理し、室温で30分間振とうした。樹脂から液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。
ウンデカン酸(279mg)のDMF(3.0ml)溶液を、HBTUのDMF溶液(3.0ml、0.5M)及びDIPEA(523μl)と混合した。溶液を室温で10分間静置し、次いで、先にDMFで3回洗浄した樹脂に加えた。樹脂を室温で1時間振とうし、液体を排出し、DMF(×3)、MeOH(×3)、及びDCM(×3)で洗浄した。樹脂を真空中で乾燥させた。
20%TFA、5%トリエチルシランのDCM溶液(0.5ml)を調製し、樹脂に加え、30分間静置した。樹脂を、濾過により除き、DCM(×3)及びアセトニトリル(×3)で洗浄した。溶媒を真空中で除去した。残渣を、1:1のACN/H2Oに溶解させ、凍結乾燥した。1mgの試料を1.0mlのアセトニトリルに溶解させ、0.45μmシリンジフィルターに通して濾過し、LCMSにより分析した:−Rt 6.38分、ELSDにより90%、(M+H)+ 537.3、(M+2H)2+ 269.2。
バンコマイシンアジド誘導体の液相カップリング(「クリック」反応):
バンコマイシン−アジドアナログ(1.5mg、1μmol)をH2Oに溶解させた。この溶液に、CuSO4.5H2O(0.5mg、2μmol)及びアスコルビン酸ナトリウム(1mg、5μmol)を加えた。生じた溶液を、ペプチド(0.5mg、1μmol)のDMF(250μl)溶液に加え、次いで、マイクロ波中で80℃で10分間加熱した。0.125mlの溶液を、0.375mlの1:1 ACN/H2Oと混合し、0.45μmシリンジフィルターに通して濾過し、LCMSにより分析した:Rt 5.522分、ELSDにより70%、(M+2H)2+ 1034.3、(M+3H)3+ 690、(M+4H)4+ 517.7。粗生成物を水/ACNに溶解させ、島津分取HPLを利用して、凍結乾燥により精製すると、白色粉末、1.3mgが生じ、切断したペプチドの量に基づき16%であった。LCMS:Rt 5.57 95%(ELSD)[M+2H]2+ 1033.8、[M+3H]3+ 690.2、[M+4H]4+ 517.8。
化合物を、上述の経路又はその変形体の1つ以上により合成した。合成の分野の通常の技量を有する化学者は、使用される試薬の変更により(アミド結合形成のための代りのカップリング試薬の取り換えなど)、又は保護基の装着、種類、及び除去の順序を変えることにより、又は成分が組み立てられる順序を変えることにより、述べられた手順を変更しても、所望の生成物が製造されることを認識するであろう。
化合物の抗菌活性を、スタフィロコッカス・アウレウス(Staphylococcus aureus)(MRSA ATCC 43300、GISA NRS17、VISA NRS1、MRSA臨床分離株、ダプトマイシン耐性臨床分離株)、ストレプトコッカス・ニューモニエ(Streptococcus pneumoniae)(MDR ATCC 700677)、エンテロコッカス・フェカリス(Enterococcus faecalis)(VanA臨床分離株)、及びエンテロコッカス・フェシウム(Enterococcus faecium)(MDR Van A ATCC 51559)を含むいくつかの細菌株に対して実施した。
化合物を、標準的な抗生物質と共に、96ウェルノンバインディングサーフェイスプレート(NBS, Corning)のウェルにわたって連続的に2倍希釈した。標準は64μg/ml〜0.03μg/mlであり、化合物は8μg/ml〜0.003μg/mlであり、最終体積はウェルあたり50μLであった。グラム陽性細菌を、ミュラー(Muller)ヒントンブロス(MHB)(Bacto laboratories、カタログ番号211443)中で、37℃で一晩培養した。次いで、各培養物の試料を新しいMHBブロスで40倍に希釈し、37℃で2〜3時間インキュベートした。生じた対数中期培養物を、5×10e5 CFU/mLの最終濃度に希釈し、次いで、50μLを、化合物含有96ウェルプレートの各ウェルに加えた。全プレートを覆い、37℃で24時間インキュベートした。MICは、目に見える増殖を全く示さない最低濃度であった。
最小殺菌濃度(MBC)を決定するために、MIC値を決定した後に、30μlのレサズリン(0.01%)を、96ウェルプレートの各ウェルに加えた。次いで、化合物を37℃でさらに18〜24時間インキュベートした。青く発色したウェルは死んだ微生物を示し、ピンクの発色のあるウェルは生きている微生物を示す。MBC値は、青い発色のウェルの最低濃度により決定した。
MICを24時間のインキュベーションで目視で決定し、MICを、インキュベーション後に増殖が全く見られない最低濃度と定義した。MICとMBCの両方を、目視検査のみで決定した。
上記で測定された抗菌活性を、3つの代表的な細菌株に対して以下の表にまとめる。
ヒト血漿安定性アッセイを、Di, L.;Kerns, E. H.;Hong, Y.;Chen, H. Development and application of high throughput plasma stability assay for drug discovery(International Journal of Pharmaceutics 2005, 297, 110-119、改変あり)に記載の方法に従って実施した。
化学薬品:DMSO、アセトニトリル、及びリン酸緩衝食塩水(PBS pH 7.4、等張性)。
血漿:ヒト血漿(貯蔵された正常なヒト血漿ヘパリン抗凝固剤50mL(IPLA−2 N−04ロットIR09−1001、元々はInnovative Research製であり、BioCoreにより輸入された)。
消耗品:エッペンドルフチューブ(ユーカトロピン及びバンコマイシン)、低結合エッペンドルフチューブ(化合物用)、及びAgilentガラスHPLCバイアル及びインサート。
装置:37℃振とう器、ボルテクサー、遠心分離機、及びLC−MS。
ストック溶液:
全化合物は100%の水中(20μMに等しい400μLアッセイ体積中の40μL)
ユーカトロピン:100%DMSO中300μg/mL(23μMに等しい400μLアッセイ体積中の10μL)。
緩衝液のみの対照試料の調製:
化合物:40μLのバンコマイシン誘導体溶液を、それぞれエッペンドルフチューブ中の160μLのPBS(pH7.4)に加えた。600μLの冷アセトニトリルを加え、チューブをボルテックスにかけ、LC−MS分析用のガラスバイアルに移した。
ユーカトロピン:200μLの血漿と190μLの緩衝剤をボルテックスにかけ、37℃で10分間振とうした。次いで、10μLのユーカトロピン溶液を加え、チューブをボルテックスにかけた。50μLのアリコートを直ちにエッペンドルフチューブに移し、150μLの冷アセトニトリルを加え、チューブを4℃の冷蔵庫に10分間移した。次いで、残りの血漿溶液をインキュベーターに移し、37℃で振とうした。チューブを冷蔵庫から取り出し、3000rpmで15分間遠心分離し、100μLの上清をLC−MS分析用のガラスバイアルインサートに移した。50μLのアリコートを、上記の通り、1、2、3、6、及び24時間で処理し、5μLを、分析のためにトリプル四重極MSシステムに注入した。
バンコマイシン誘導体:200μLの血漿と160μLの緩衝剤をボルテックスにかけ、37℃で10分間振とうした。次いで、40μLの化合物を加え、試料を上記の通り処理した。
生理学的濃度のグルタチオンの存在下での化合物の安定性を以下のプロトコルに従って評価した:20μLの1mMの試験化合物のストック溶液を、プラスチックHPLCインサート中の180μLのグルタチオン(還元型)PBS溶液に加え、溶液に、100μMの化合物及び5mMか0.5mMのいずれかのグルタチオンの最終濃度を与えた。試料をHPLCサンプリングラックに置き、1時間に1回の間隔で10時間までサンプリングした。注入の直前に試料を調製するように注意を払った。次いで、化合物の減少に関して、UV面積を時間に対してプロットした。0時間での総バンコマイシン誘導体に対して、パーセンテージをプロットした。結果を図1に示す。
インビボ実験アッセイ:体重が25〜35gの7〜9週齢の雄のCD1マウス(SLAC Laboratory Animal Co. Ltd., Shanghai, China)を、試験に使用する前におよそ3日間順化させた。動物を、National Research CouncilのGuide for the Care and Use of Laboratory Amimalsに従って、順化及びインライフ試験の間群飼する。温度及び相対湿度を毎日モニターしながら、動物室の環境を制御する(目標条件:温度18〜26℃、相対湿度30〜70%、12時間人工光及び12時間暗がり)。製剤投与の前およそ16時間、動物に餌を与えず、次いで投薬の4時間後にCertified Rodent Diet(カタログ番号M−01F、Shanghai SLAC Laboratory Animal Co. Ltd.)に自由にアクセスさせた。水を、動物に自由に与える前に、オートクレーブにかける。化合物の製剤を、投薬日の朝に調製した。IV群の製剤を、動物に投薬する前に、0.22μmのフィルターで濾過した。用量製剤調製の後に、二連の50μLアリコートを、用量確認のために各用量製剤から除いた。
IVサンプリング時点(投薬後の時間):0、0.083、0.25、0.5、1、2、4、8、24。
SCサンプリング時点(投薬後の時間):0、0.25、0.5、1、2、4、8、24。
投薬製剤確認:製剤のアリコートを、二連で各用量製剤の中心の位置で収集した。LC−UV法を、6つの較正標準からなる較正曲線により開発した。用量製剤試料中の試験化合物の濃度を、LC−UV法により決定した。分析単位の受入基準:6つの較正標準のうち少なくとも5つが、正常値の20%以内でなくてはならない。
概要:成体(8週齢)の雌のCD1マウスを、感染の4日前及び1日前の2回のシクロホスファミドの注射により、好中球減少にさせた。105cfu MRSA(Strain ATCC 43300)の接種物を、全マウスの左右両方の大腿部に筋肉内注射した。感染開始の2時間後に、食塩水、バンコマイシン、又は本発明の化合物を、腰部に皮下注射した。さらに2時間後、50μLの血液試料を、尾部の出血から得て、抗生物質化合物の存在に関して分析した。感染後24時間で、マウスを安楽死させ、追加の血液試料を化合物分析のために回収した。次いで、大腿部を取り除き、秤量し、一定体積の食塩水中でホモジナイズした。ホモジネート溶液を濾過し、希釈し、寒天プレートの上に播種し、それを一晩37℃でインキュベートした。コロニー数を利用して、大腿部ホモジネート中のcfu、cfu/大腿部、及びcfu/大腿部のgに到達した。
体重が22±2gである10匹の雄の特定病原体不在ICRマウスの群を使用した。20μLのBHIに懸濁させたLD90〜100の投与量(2.96×106CFU/マウス)のストレプトコッカス・ニューモニエ(Streptococcus pneumoniae)(ATCC 6301)の気管内接種により、急性肺炎を誘発させた。ビヒクル(10ml/kg)、バンコマイシン、及び25mg/kgの試験物質を、感染2時間後にそれぞれ皮下に投与した。死亡率を、接種後10日間毎日記録した。ビヒクル対照群に対する50パーセント以上の(≧50%)生存率の増加は、有意な抗感染活性を示す。結果を図4に表す。
Claims (24)
- 式(III):
X−W−L−V (III)
[式中、
Xは、WのN−末端に結合した親油性基であり、炭素原子がベースとなっており、且つ以下のパラメーターを有し;
− 3〜60個の炭素原子であって、任意の脂環式環又は芳香環が存在する場合にはそれらの炭素原子を含めて3〜60個の炭素原子を有すること;
− 直鎖又は分岐鎖であって、且つ後者の場合、1〜3個の分岐点を含むこと;
− 飽和又は不飽和であって、後者の場合、1〜8個の二重又は三重結合を含むこと;
− 任意選択的に、(ヘテロ原子が存在する場合であって、これらに加えて、芳香環が存在する場合には当該芳香環中に)酸性置換基中に含まれない最大6個のヘテロ原子であって、S、O、又はNから独立して選択されるヘテロ原子を有すること;
− 任意選択的に、1個以上の芳香環であって、縮合していてよく、且つそのそれぞれが、存在する場合、N、O、又はSから独立に選択される1、2、又は3個のヘテロ原子を含み得る、1個以上の芳香環を含むこと;且つ
− 任意選択的に、ヒドロキシ、アミノ、メチル、メチルアミノ、及びハロから選択される1〜6個の置換基を有すること;
Wは、塩基性アミノ酸又は2〜10個のアミノ酸からなる塩基性ペプチドであって、但し、Wは、硫黄含有側鎖を有する任意のアミノ酸でなく、又はそれを含まず;
Lは、式−NH−(CR1R2)m−Z−(CR3R4)n−NH−
(式中、
Zは、酸素又は−NH−、−CONH−、−NHCO−、−(OCH2CH2)p−、C1〜C12アルキル、C2〜C12アルケニル、C2〜C12アルキニル、C1〜C12ヘテロアルキル、C1〜C10ヘテロアルケニル、C3〜C12シクロアルキル、C1〜C12複素環、C6〜C18アリール、若しくはC1〜C18ヘテロアリールからなる群から選択される任意選択で置換されている部分であり;且つ
R1、R2、R3、及びR4は、H、ハロゲン、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC1〜C10ヘテロアルケニル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC1〜C12複素環、任意選択で置換されているC6〜C18アリール、任意選択で置換されているC1〜C18ヘテロアリール、任意選択で置換されているカルボキシ、任意選択で置換されているカルボキサミドからなる群からそれぞれ独立に選択され;且つ
mは、0、1、2、及び3からなる群から選択される整数であり;且つ
nは、0、1、2、及び3からなる群から選択される整数であり;
但し、mとnとの両方が0ではなく;且つ
pは、1〜10からなる群から選択される整数である)
の連結基であるか、あるいは
Lは、式:
(式中、c及びdは、0、1、及び2からなる群から選択される整数であり;且つ
点線は、V及びWへの結合点を示す)
の1つから選択され;且つ
Vは、細菌中のペプチドグリカン生合成を阻害するグリコペプチド部分である]
の抗菌性化合物、又はその薬学的に許容できる塩若しくはプロドラッグ。 - Xが式R27CO−のものであり、式中、R27が3〜15個の炭素原子を有する親油性基であり、前記親油性基が:
− 直鎖又は分岐鎖であり、且つ脂環式環又は芳香環であって、前記基中の炭素原子の総数が任意の前記環の炭素原子を含む、脂環式環又は芳香環を含むことができ;
− 飽和又は不飽和であり、後者の場合、1〜4個の二重又は三重結合を含み;
− 任意選択的に、(ヘテロ原子が存在する場合であって、これらに加えて、芳香環が存在する場合には当該芳香環中に)1又は2個のヘテロ原子であって、O又はNから独立に選択されるヘテロ原子を有し;
− 任意選択に、1又は2個の芳香環であって、そのいずれか若しくは両方が1個の窒素ヘテロ原子を含み得る、芳香環を含み;且つ
− 任意選択的に、ヒドロキシル、アミノ、メチル、メチルアミノ、及びハロから選択される1〜3個の置換基を有する、請求項1に記載の化合物。 - Xが、式(IV):
(式中、
各R25及びR26が、以下のパラメーター:
− 3〜30個の炭素原子であって、任意の脂環式環又は芳香環が存在する場合にはそれらの炭素原子を含めて3〜30個の炭素原子を有すること;
− 直鎖又は分岐鎖であり、且つ後者の場合、1〜3個の分岐点を含むこと;
− 飽和又は不飽和であり、後者の場合、1〜4個の二重又は三重結合を含むこと;
− 任意選択的に、(ヘテロ原子が存在する場合であって、これらに加えて、芳香環が存在する場合には当該芳香環中に)1、2、又は3個のヘテロ原子であって、O、S、又はNから独立に選択されるヘテロ原子を有すること;
− 任意選択的に1個以上、例えば、2又は3個の芳香環であって、縮合していてよく、且つそのそれぞれが、存在する場合、N、O、又はSから独立に選択される1、2、又は3個のヘテロ原子を含むことができる、芳香環を含むこと;且つ
− 任意選択的に、ヒドロキシ、アミノ、メチル、メチルアミノ、及びハロから選択される1〜3個の置換基を有すること
を有する、炭素原子に基づく親油性鎖であり;
tが、0、1、2、3、4、及び5からなる群から選択される整数であり;且つ
uが、0、1、2、3、4、及び5からなる群から選択される整数であり;
但し、t又はuの一方が0である場合、t又はuの他方は0ではない)
のものである、請求項1に記載の化合物。 - −L−が式−NH−(CH2)m−Z−(CH2)n−NH−のものであり;式中、Z、m、及びnが請求項1に定義されている通りである、請求項1〜3のいずれか一項に記載の化合物。
- Zが、C1〜C12アルキル、NH、O、C1〜C12複素環、C6〜C18アリール、及びC3〜C12シクロアルキルからなる群から選択される、請求項4に記載の化合物。
- −L−が、式−NH−(CH2)2−NH−、−NH−(CH2)3−NH−、−NH−(CH2)4−NH−、−NH−(CH2)2−NH−(CH2)2−NH−、−NH−(CH2)2−O−(CH2)2−NH−、−NH−(CH2)3−O−(CH2)3−NH−、−NH−(1,4−Ph)−CH2−NH−、−NH−(1,3−Ph)−CH2−NH−、−NH−(1,4−cHex)−CH2−NH−、又は−NH−CH2−(1,4−cHex)−CH2−NH−のものである、請求項1〜5のいずれか一項に記載の化合物。
- −L−が、式−NH−CH(R1)−Z−(CH2)n−NH−のものであり、式中:
R1が、−(CO)OH、−(CO)OMe、−(CO)NH2、−(CO)NHNH2、−(CO)NHMe、−(CO)NHEt、−(CO)N(Me)2、−(CO)NHBn、若しくは−(CO)R5、又は任意選択で置換されているC1〜C12複素環、若しくは任意選択で置換されているC1〜C18ヘテロアリール部分であり;且つ
R5が、任意選択で置換されているC1〜C12複素環又は任意選択で置換されているC1〜C18ヘテロアリール部分であり;且つ
Z及びnが、請求項1に定義されている通りである、請求項1に記載の化合物。 - −L−が式−NH−CH(R1)−(CH2)q−NH−のものであり、式中:
qが、0、1、2、3、4、及び5からなる群から選択される整数であり;且つ
R1が、−(CO)OH、−(CO)OMe、−(CO)NH2、−(CO)NHNH2、−(CO)NHMe、−(CO)NHEt、−(CO)N(Me)2、−(CO)NHBn、若しくは−(CO)R5、又は任意選択で置換されているC1〜C12複素環、若しくは任意選択で置換されているC1〜C18ヘテロアリール部分である、請求項1又は7に記載の化合物。 - Vが、バンコマイシン、バンコマイシンアグリコン、バンコマイシンデスバンコサミン、デスメチルバンコマイシン、クロロエレモマイシン、テイコプラナイン−A2−2、リストセチンA、エレモマイシン、バルヒマイシン、アクチノイジンA、コンプレスタチン、クロロペプチン1、キスタマイシンA、アボパルシン、テラバンシン、A40926、及びオリタバンシン、並びに一級アミンでR17により任意選択で置換されているそのいずれか1つから選択され、R17は、水素、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC1〜C10ヘテロアルケニル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から選択される有機側鎖部分である、請求項1〜8のいずれか一項に記載の化合物。
- X−W−L−が、グリコペプチド遊離カルボキシル基へのアミド連結によりVに結合している、請求項9に記載の化合物。
- X−W−L−Vが、式(VI):
(式中、
X、W、及びLは、請求項1に定義されている通りであり;
R7は、水素、炭水化物、又は4−エピ−バンコサミニル、アクチノサミニル、若しくはリストサミニルを含むがこれらに限定されないアミノ炭水化物であり;
R8は、水素、OH、又は−O−マンノースであり;
R9は、−NH2、−NHCH3、又は−N(CH3)2であり;
R10は、−CH2CH(CH3)2、[p−OH,m−Cl]フェニル、p−ラムノース−フェニル、[p−ラムノース−ガラクトース]フェニル、[p−ガラクトース−ガラクトース]フェニル、[p−CH3O−ラムノース]フェニルであるか、又は[p−OH,m−(O−{m−OH,m−R11}フェニル)]フェニルを介してR11に結合して環式環系を形成し;
R11は、−CH2−(CO)NH2、ベンジル、[p−OH]フェニル、[p−OH,m−Cl]フェニル;[p−OH,m−Cl]フェニルであるか、又は[m−OH,m−(O−{o−OH,m−R10}フェニル)]−フェニルを介してR10に結合して環式環系を形成し;
R12は、水素、又はマンノースであり;
R13は、水素、OH、又はCH2NHCH2PO3H2であり;
R14は、水素、ベータ−D−グルコピラノース、ベータ−D−グルコサミン、2−O−(アルファ−L−バンコサミニル)−ベータ−D−グルコピラノース、2−O−(アルファ−L−4−エピ−バンコサミニル)−ベータ−D−グルコピラノース、(アルファ−アクチノサミニル)−ベータ−D−グルコピラノース、(アルファ−リストサミニル)−ベータ−D−グルコピラノース、若しくは(アルファ−アコサミニル)−ベータ−D−グルコピラノース;又は前記グルコサミン若しくはグルコピラノース基のいずれか1つであって、その一級アミンでR17により任意選択で置換されている前記グルコサミン若しくはグルコピラノース基のいずれか1つであり、R17は、請求項9に定義されている通りであり;且つ
R15及びR16は、独立に、水素又はクロロである)
のものである、請求項1〜8のいずれか一項に記載の化合物。 - Wが、塩基性アミノ酸又は2〜5個のアミノ酸を含む塩基性ペプチドであり、但し、Wは硫黄含有側鎖を有する任意のアミノ酸でなく、又はそれを含まない、請求項1〜11のいずれか一項に記載の化合物。
- Wが、式(V):
(式中、
Yは、式−(CR20R21)g−の基であり;
R18aは、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、任意選択で置換されているC1〜C18ヘテロアリール、−C(=NR22)−NR23R24、及びOR22からなる群から選択され、
R18bは、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から選択されるか、又は
R18a及びR18bは、それらが結合している窒素原子と共に、任意選択で置換されている複素環部分を形成するか、又は
R18a及びR18bの一方が、R20又はR21のいずれか及びそれらが結合している原子と共に、任意選択で置換されている複素環部分を形成し;
R19は、H及び任意選択で置換されているC1〜C12アルキルからなる群から選択され;
R20及びR21は、H、ハロゲン、OH、C1〜C12アルキル、C6〜C18アリール、C1〜C12ハロアルキル、C1〜C12ヒドロキシアルキル、C1〜C12アルキルオキシ、及びC1〜C12ハロアルキルオキシからなる群からそれぞれ独立に選択されるか、又は
R20及びR21は、それらが結合している炭素と共に、任意選択で置換されているC3〜C12シクロアルキル、又は任意選択で置換されているC1〜C12ヘテロシクロアルキル基を形成するか、又は
R20及びR21の一方が、R18aとR18bとの一方及びそれらが結合している原子と共に、任意選択で置換されている複素環部分を形成し;
各R22、R23、及びR24は、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から独立に選択されるか、又は
R22、R23、及びR24のいずれか2個が、それらが結合している原子と共に、任意選択で置換されている環式基を形成し;
gは、1、2、3、4、及び5からなる群から選択される整数であり;
rは、1、2、3、4、5、6、7、8、9、及び10からなる群から選択される整数である)
の1個の残基又は2〜10個の連続する残基からなる、請求項12に記載の化合物。 - Yが(CH2)gである、請求項13に記載の化合物。
- Wが、式(V):
(式中、
Yは式−(CR20R21)g−の基であり;
R18aは、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、任意選択で置換されているC1〜C18ヘテロアリール、−C(=NR22)−NR23R24、及びOR22からなる群から選択され、
R18bは、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC2〜C12アルケニル、任意選択で置換されているC2〜C12アルキニル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC2〜C12ヘテロシクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から選択されるか、又は
R18a及びR18bは、それらが結合している窒素原子と共に、任意選択で置換されている複素環部分を形成するか、又は
R18a及びR18bの一方が、R20又はR21のいずれか及びそれらが結合している原子と共に、任意選択で置換されている複素環部分を形成し;
R19は、H及び任意選択で置換されているC1〜C12アルキルからなる群から選択され;
R20及びR21は、H、ハロゲン、OH、C1〜C12アルキル、C6〜C18アリール、C1〜C12ハロアルキル、C1〜C12ヒドロキシアルキル、C1〜C12アルキルオキシ、及びC1〜C12ハロアルキルオキシからなる群からそれぞれ独立に選択されるか、又は
R20及びR21は、それらが結合する炭素と共に、任意選択で置換されているC3〜C12シクロアルキル、又は任意選択で置換されているC1〜C12ヘテロシクロアルキル基を形成するか、又は
R20及びR21の一方が、R18aとR18bとの一方及びそれらが結合している原子と共に、任意選択で置換されている複素環部分を形成し;
各R22、R23、及びR24は、H、任意選択で置換されているC1〜C12アルキル、任意選択で置換されているC1〜C12ヘテロアルキル、任意選択で置換されているC3〜C12シクロアルキル、任意選択で置換されているC6〜C18アリール、及び任意選択で置換されているC1〜C18ヘテロアリールからなる群から独立に選択されるか、又は
R22、R23、及びR24のいずれか2個が、それらが結合している原子と共に、任意選択で置換されている環式基を形成し;
gは、1、2、3、4、及び5からなる群から選択される整数であり;
rは、1、2、3、4、5、6、7、8、9、及び10からなる群から選択される整数である)
の1個の残基又は2〜5個の連続する残基からなる、請求項12に記載の化合物。 - Yが(CH2)gである、請求項15に記載の化合物。
- 前記残基又は連続する残基が、任意選択で置換されているD−若しくはL−リジン、オルニチン、2,4−ジアミノ酪酸、2,3−ジアミノプロピオン酸、及びアルギニンから選択される、請求項12〜16のいずれか一項に記載の化合物。
- Wが、−Lys−、−Lys−Lys−、−Lys−Lys−Lys−、−Orn−、−Orn−Orn−、−Orn−Orn−Orn−、−Lys−Orn−、−Orn−Lys、−Dab−、−Dab−Dab−、−Lys−Dab−、−Dab−Lys−、−Dab−Orn−、−Orn−Dab−、−Dap−、−Dap−Dap−、−Dap−Lys−、−Lys−Dap、−Dap−Orn、−Orn−Dap、−Dap−Dab−、及び−Dab−Dap−からなる群から選択され、前記アミノ酸のいずれも−L−配置又は−D−配置であり得る、請求項1〜17のいずれか一項に記載の化合物。
- 各任意選択の置換基が、ハロゲン、=O、=S、−CN、−NO2、−CF3、−OCF3、アルキル、アルケニル、アルキニル、ハロアルキル、ハロアルケニル、ハロアルキニル、ヘテロアルキル、シクロアルキル、シクロアルケニル、ヘテロシクロアルキル、ヘテロシクロアルケニル、アリール、ヘテロアリール、シクロアルキルアルキル、ヘテロシクロアルキルアルキル、ヘテロアリールアルキル、アリールアルキル、シクロアルキルアルケニル、ヘテロシクロアルキルアルケニル、アリールアルケニル、ヘテロアリールアルケニル、シクロアルキルヘテロアルキル、ヘテロシクロアルキルヘテロアルキル、アリールヘテロアルキル、ヘテロアリールヘテロアルキル、ヒドロキシ、ヒドロキシアルキル、アルキルオキシ、アルキルオキシアルキル、アルキルオキシシクロアルキル、アルキルオキシヘテロシクロアルキル、アルキルオキシアリール、アルキルオキシヘテロアリール、アルキルオキシカルボニル、アルキルアミノカルボニル、アルケニルオキシ、アルキニルオキシ、シクロアルキルオキシ、シクロアルケニルオキシ、ヘテロシクロアルキルオキシ、ヘテロシクロアルケニルオキシ、アリールオキシ、フェノキシ、ベンジルオキシ、ヘテロアリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アシルアミノ、アミノアルキル、アリールアミノ、スルホニルアミノ、スルフィニルアミノ、スルホニル、アルキルスルホニル、アリールスルホニル、アミノスルホニル、スルフィニル、アルキルスルフィニル、アリールスルフィニル、アミノスルフィニルアミノアルキル、−C(=O)OH、−C(=O)Ra、C(=O)ORa、C(=O)NRaRb、C(=NOH)Ra、C(=NRa)NRbRc、NRaRb、NRaC(=O)Rb、NRaC(=O)ORb、NRaC(=O)NRbRc、NRaC(=NRb)NRcRd、NRaSO2Rb、−SRa、SO2NRaRb、−ORa、OC(=O)NRaRb、OC(=O)Ra、及びアシルからなる群から独立に選択され、
Ra、Rb、Rc、及びRdは、H、C1〜C12アルキル、C1〜C12ハロアルキル、C2〜C12アルケニル、C2〜C12アルキニル、C1〜C10ヘテロアルキル、C3〜C12シクロアルキル、C3〜C12シクロアルケニル、C1〜C12ヘテロシクロアルキル、C1〜C12ヘテロシクロアルケニル、C6〜C18アリール、C1〜C18ヘテロアリール、及びアシルからなる群からそれぞれ独立に選択されるか、又はRa、Rb、Rc、及びRdのいずれか2個以上が、それらが結合している原子と共に、3〜12個の環原子を有する複素環式環系を形成する、請求項1〜18のいずれか一項に記載の化合物。 - 表1に開示される化合物から選択される、請求項1に記載の化合物。
- 請求項1〜20のいずれか一項に記載の任意の化合物の薬学的に許容できる塩。
- 請求項1〜20のいずれか一項に記載の化合物及び薬学的に許容できる担体、希釈剤、又は賦形剤を含む組成物。
- ヒト又は動物の体の治療の方法に使用するための、請求項1〜20のいずれか一項に記載の化合物又は請求項22に記載の組成物。
- 対象の細菌感染を治療する方法であって、対象に、有効量の請求項1〜20のいずれか一項に記載の抗菌剤又は有効量の請求項22に記載の組成物を投与することを含む、方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1402267.7A GB201402267D0 (en) | 2014-02-10 | 2014-02-10 | Antibacterial agents |
| GB1402267.7 | 2014-02-10 | ||
| PCT/AU2015/000071 WO2015117196A1 (en) | 2014-02-10 | 2015-02-10 | Antibacterial agents |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017507174A true JP2017507174A (ja) | 2017-03-16 |
| JP6602318B2 JP6602318B2 (ja) | 2019-11-06 |
Family
ID=50390736
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016568088A Active JP6602318B2 (ja) | 2014-02-10 | 2015-02-10 | 抗菌剤 |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US10947271B2 (ja) |
| EP (1) | EP3105244B1 (ja) |
| JP (1) | JP6602318B2 (ja) |
| CN (1) | CN107027309B (ja) |
| AU (1) | AU2015213478B2 (ja) |
| BR (1) | BR112016018442A2 (ja) |
| CA (1) | CA2939145C (ja) |
| GB (1) | GB201402267D0 (ja) |
| MX (1) | MX2016010385A (ja) |
| RU (1) | RU2016134602A (ja) |
| WO (1) | WO2015117196A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11174288B2 (en) | 2016-12-06 | 2021-11-16 | Northeastern University | Heparin-binding cationic peptide self-assembling peptide amphiphiles useful against drug-resistant bacteria |
| CN110312518A (zh) * | 2016-12-09 | 2019-10-08 | 昆士兰大学 | 糖蛋白抗生物素构建体 |
| WO2018102890A1 (en) * | 2016-12-09 | 2018-06-14 | The University Of Queensland | Visualization constructs |
| IL270685B (en) * | 2017-05-22 | 2022-09-01 | Insmed Inc | Cleavable derivatives of lipoglycopeptide and their uses |
| CN108060207B (zh) * | 2017-12-29 | 2021-10-15 | 河南科技大学 | 一种基于微量天然化合物的最小杀菌浓度测定方法 |
| CN109946398B (zh) * | 2019-03-28 | 2022-05-24 | 丽珠集团新北江制药股份有限公司 | 一种检测达巴万星及其杂质的方法 |
| CN116515102B (zh) * | 2022-01-21 | 2025-04-18 | 苏州万维生命科学技术有限公司 | 星型β-抗菌糖肽、其制备方法及应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001501601A (ja) * | 1996-09-12 | 2001-02-06 | メルク エンド カンパニー インコーポレーテッド | 前立腺ガンの治療において有用な共役体 |
| JP2002503708A (ja) * | 1998-02-23 | 2002-02-05 | シンソーブ・バイオテック・インコーポレーテッド | 抗生物質と毒素結合性オリゴ糖組成物を用いた細菌性赤痢の治療のための組成物と方法 |
| WO2004022101A1 (en) * | 2002-09-05 | 2004-03-18 | Adprotech Limited | Modified therapeutic agents |
| JP2004513132A (ja) * | 2000-11-03 | 2004-04-30 | ケンブリッジ・ユニバーシティ・テクニカル・サービシーズ・リミテッド | 糖ペプチド及びペプチド性膜関連成分のコンジュゲートを含んでなる抗菌薬 |
| JP2009529530A (ja) * | 2006-03-09 | 2009-08-20 | インフラザイム・ファーマシューティカルズ・リミテッド | 新規の抗生物質組成物 |
| JP2012506419A (ja) * | 2008-10-23 | 2012-03-15 | ステバ、バイオテック、ナムローゼ、フェンノートシャップ | Rgd含有ペプチド模倣体及びその使用 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5840684A (en) | 1994-01-28 | 1998-11-24 | Eli Lilly And Company | Glycopeptide antibiotic derivatives |
| GB9614871D0 (en) | 1996-07-15 | 1996-09-04 | Smithkline Beecham Plc | Compounds |
| US6017511A (en) | 1997-05-30 | 2000-01-25 | Resolution Pharmaceuticals Inc. | Glycopeptide-chelator conjugates |
| JP4381531B2 (ja) * | 1999-12-08 | 2009-12-09 | 塩野義製薬株式会社 | グリコペプチド誘導体 |
| AU2001259298A1 (en) | 2000-05-02 | 2001-11-12 | Advanced Medicine, Inc. | Polyacid glycopeptide derivatives |
| WO2002005837A1 (en) | 2000-07-17 | 2002-01-24 | Intrabiotics Pharmaceuticals, Inc. | Antimicrobial sulfonamide derivatives of lipopeptide antibiotics |
| WO2004044222A2 (en) | 2002-11-12 | 2004-05-27 | Enzon Pharmaceuticals, Inc. | Polymeric prodrugs of vancomycin |
| WO2007047608A2 (en) | 2005-10-14 | 2007-04-26 | Epix Pharmaceuticals, Inc. | Fibrin targeted therapeutics |
| WO2011019839A2 (en) | 2009-08-12 | 2011-02-17 | The Medicines Company | Glycopeptide and lipoglycopeptide antibiotics with improved solubility |
-
2014
- 2014-02-10 GB GBGB1402267.7A patent/GB201402267D0/en not_active Ceased
-
2015
- 2015-02-10 CA CA2939145A patent/CA2939145C/en active Active
- 2015-02-10 JP JP2016568088A patent/JP6602318B2/ja active Active
- 2015-02-10 MX MX2016010385A patent/MX2016010385A/es unknown
- 2015-02-10 EP EP15746637.6A patent/EP3105244B1/en active Active
- 2015-02-10 CN CN201580018660.5A patent/CN107027309B/zh not_active Expired - Fee Related
- 2015-02-10 US US15/117,704 patent/US10947271B2/en active Active
- 2015-02-10 WO PCT/AU2015/000071 patent/WO2015117196A1/en not_active Ceased
- 2015-02-10 BR BR112016018442A patent/BR112016018442A2/pt not_active Application Discontinuation
- 2015-02-10 RU RU2016134602A patent/RU2016134602A/ru not_active Application Discontinuation
- 2015-02-10 AU AU2015213478A patent/AU2015213478B2/en not_active Ceased
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001501601A (ja) * | 1996-09-12 | 2001-02-06 | メルク エンド カンパニー インコーポレーテッド | 前立腺ガンの治療において有用な共役体 |
| JP2002503708A (ja) * | 1998-02-23 | 2002-02-05 | シンソーブ・バイオテック・インコーポレーテッド | 抗生物質と毒素結合性オリゴ糖組成物を用いた細菌性赤痢の治療のための組成物と方法 |
| JP2004513132A (ja) * | 2000-11-03 | 2004-04-30 | ケンブリッジ・ユニバーシティ・テクニカル・サービシーズ・リミテッド | 糖ペプチド及びペプチド性膜関連成分のコンジュゲートを含んでなる抗菌薬 |
| WO2004022101A1 (en) * | 2002-09-05 | 2004-03-18 | Adprotech Limited | Modified therapeutic agents |
| JP2009529530A (ja) * | 2006-03-09 | 2009-08-20 | インフラザイム・ファーマシューティカルズ・リミテッド | 新規の抗生物質組成物 |
| JP2012506419A (ja) * | 2008-10-23 | 2012-03-15 | ステバ、バイオテック、ナムローゼ、フェンノートシャップ | Rgd含有ペプチド模倣体及びその使用 |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2016010385A (es) | 2017-05-12 |
| EP3105244B1 (en) | 2020-01-01 |
| EP3105244A4 (en) | 2017-11-08 |
| RU2016134602A (ru) | 2018-03-15 |
| CA2939145C (en) | 2024-05-07 |
| EP3105244A1 (en) | 2016-12-21 |
| WO2015117196A1 (en) | 2015-08-13 |
| AU2015213478B2 (en) | 2019-08-08 |
| US10947271B2 (en) | 2021-03-16 |
| AU2015213478A2 (en) | 2016-09-15 |
| CN107027309A (zh) | 2017-08-08 |
| CN107027309B (zh) | 2021-10-01 |
| GB201402267D0 (en) | 2014-03-26 |
| US20170204138A1 (en) | 2017-07-20 |
| JP6602318B2 (ja) | 2019-11-06 |
| CA2939145A1 (en) | 2015-08-13 |
| AU2015213478A1 (en) | 2016-09-15 |
| BR112016018442A2 (pt) | 2018-05-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6602318B2 (ja) | 抗菌剤 | |
| TWI665218B (zh) | 多黏菌素化合物、包含其之醫藥組成物及其用途 | |
| JP4209673B2 (ja) | 糖ペプチド及びペプチド性膜関連成分のコンジュゲートを含んでなる抗菌薬 | |
| CN114269771B (zh) | Romo1来源抗微生物肽及其变体 | |
| US11046730B2 (en) | Antimicrobial compositions | |
| TW200936153A (en) | Short fatty acid tail polymyxin derivatives and uses thereof | |
| KR20120104986A (ko) | 의료용 펩티드 | |
| CA2979163A1 (en) | 3-substituted 1,3,4-oxadiazole and thiadiazole compounds as immunomodulators | |
| KR100893426B1 (ko) | 항균 활성을 갖는 플레우로무틸린 유도체 | |
| AU2002212482A1 (en) | Antibacterial agents comprising conjugates of glycopeptides and peptidic membrane-associating elements | |
| EA010294B1 (ru) | Композиции на основе производных липопептидных антибиотиков и способы их применения | |
| ES2334547B1 (es) | Compuestos peptidicos antibacterianos. | |
| EA009294B1 (ru) | Антагонисты в1 рецептора брадикинина (варианты), фармацевтическая композиция, лекарственное средство, способ лечения заболеваний (варианты) | |
| JP5646051B2 (ja) | 癌細胞に特異的毒性を有する新規抗癌剤 | |
| Paritala et al. | Glycopeptides: insights towards resistance, clinical pharmacokinetics and pharmacodynamics | |
| CA2855753C (en) | Cationic antibacterial composition | |
| CN114650835A (zh) | 高效杀灭耐药病害菌的药物及在抑制耐药病害菌中的应用 | |
| US20230181678A1 (en) | Novel antibacterial peptide or peptide analog and use thereof | |
| ZA200300374B (en) | Novispirins: Antimicrobial peptides. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20180131 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20181221 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190322 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190402 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190621 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20190730 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190904 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190925 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20191008 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6602318 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |