JP2020002108A - 安息香酸化合物の製造方法 - Google Patents
安息香酸化合物の製造方法 Download PDFInfo
- Publication number
- JP2020002108A JP2020002108A JP2018125301A JP2018125301A JP2020002108A JP 2020002108 A JP2020002108 A JP 2020002108A JP 2018125301 A JP2018125301 A JP 2018125301A JP 2018125301 A JP2018125301 A JP 2018125301A JP 2020002108 A JP2020002108 A JP 2020002108A
- Authority
- JP
- Japan
- Prior art keywords
- formula
- compound represented
- sulfate
- reaction
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
- 0 Cc1ccc(*)cc1NC(N)=N Chemical compound Cc1ccc(*)cc1NC(N)=N 0.000 description 6
Landscapes
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
【課題】3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸(C1〜C4アルキル)エステル硫酸塩、及びこのエステルの簡便かつ高収率な製造方法の提供。【解決手段】3−アミノ−4−メチル安息香酸(C1〜C4アルキル)エステルとシアナミドとから、3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸(C1〜C4アルキル)エステル硫酸塩(式2の硫酸塩)を製造する方法を提供。(式中、R1はC1〜C4アルキル基を示す)。更に、この化合物を利用したニロチニブの製造中間体である4−メチル−3−{[4−(ピリジン−3−イル)ピリミジン−2−イル]アミノ}−安息香酸の簡便かつ高収率な製造方法を提供。【選択図】なし
Description
本発明は、安息香酸化合物の製造方法に関する。さらに詳しくは、4−メチル−3−{[4−(ピリジン−3−イル)ピリミジン−2−イル]アミノ}−安息香酸及びその中間体の製造方法に関する。
4−メチル−3−{[4−(ピリジン−3−イル)ピリミジン−2−イル]アミノ}−安息香酸(化合物5)は、慢性骨髄性白血病治療薬であるニロチニブの製造中間体として有用であることが知られている(特許文献1参照)。
この中間体(化合物5)は、3−アミノ−4−メチル安息香酸エチルエステル(化合物1a)とシアナミドとを塩酸存在下で反応させ、次いで硝酸アンモニウムを作用させて3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸エチルエステル(化合物2a)の硝酸塩を得て、これに3−ジメチルアミノ−1−(ピリジン−3−イル)−2−プロペン−1−オン(化合物3a)を反応させて4−メチル−3−{[4−(ピリジン−3−イル)ピリミジン−2−イル]アミノ}−安息香酸エチルエステル(化合物4a)とし、これを加水分解することにより合成されている(特許文献1、非特許文献1)。
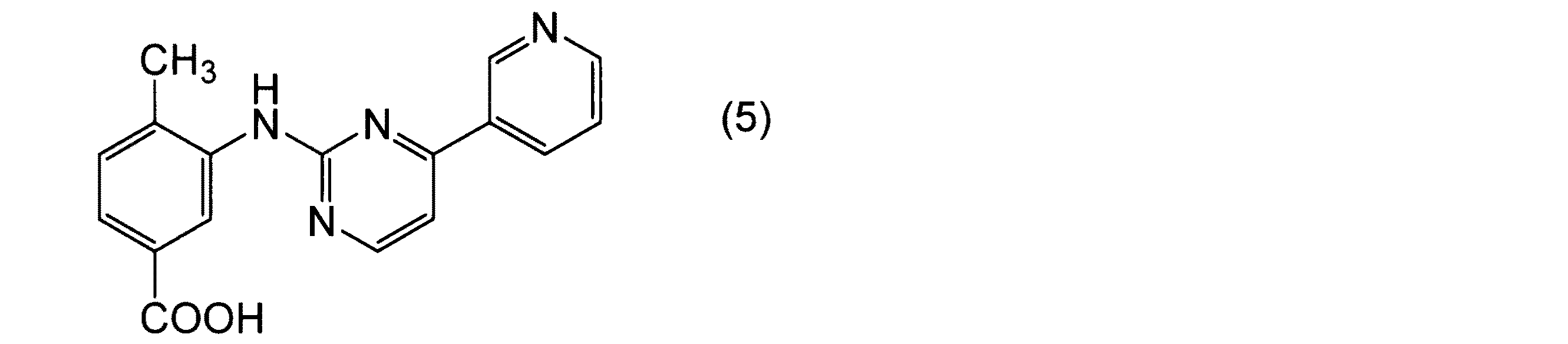
Chemical Biology & Drug Design (2014), 83(5), 592
上記の製法では、3-アミノ-4-メチル安息香酸エチルエステル(化合物1a)とシアナミドとを塩酸存在下で反応させて得られる3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸エチルエステル(化合物2a)の塩酸塩は、水溶解性が高く結晶として取得することができないため、いったん化合物2aの硝酸塩として結晶化して取得する方法が採用されている。しかし、化合物1aから化合物2aの塩酸塩を経てその硝酸塩を得る一連の工程は煩雑であり、その工程の収率は必ずしも満足のいくものではない。
そこで、本発明は、3-アミノ-4-メチル安息香酸(C1〜C4アルキル)エステルとシアナミドとから、3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸(C1〜C4アルキル)エステルを、簡便かつ高収率で製造する方法を提供することを課題とする。
また、本発明は、この方法を利用してニロチニブの製造中間体である4−メチル−3−{[4−(ピリジン−3−イル)ピリミジン−2−イル]アミノ}−安息香酸を簡便かつ高収率で製造する方法を提供することを課題とする。
本発明は、以下の態様を包含する。
[1] 式(2):
[1] 式(2):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物の硫酸塩。
[2] 硫酸塩が1/2硫酸塩である、[1]に記載の硫酸塩。
[3] R1がメチル基である、[1]又は[2]に記載の硫酸塩。
[4] 式(2):
で表される化合物の硫酸塩。
[2] 硫酸塩が1/2硫酸塩である、[1]に記載の硫酸塩。
[3] R1がメチル基である、[1]又は[2]に記載の硫酸塩。
[4] 式(2):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物の硫酸塩の製造方法であって、硫酸存在下に、式(1):
で表される化合物の硫酸塩の製造方法であって、硫酸存在下に、式(1):
(式中、R1は前記に同じ。)
で表される化合物、及びシアナミドを反応させる工程を含む、製造方法。
[5] 硫酸塩が1/2硫酸塩である、[4]に記載の製造方法。
[6] R1がメチル基である、[4]又は[5]に記載の製造方法。
[7] 式(4):
で表される化合物、及びシアナミドを反応させる工程を含む、製造方法。
[5] 硫酸塩が1/2硫酸塩である、[4]に記載の製造方法。
[6] R1がメチル基である、[4]又は[5]に記載の製造方法。
[7] 式(4):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物の製造方法であって、アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2):
で表される化合物の製造方法であって、アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2):
(式中、R1は前記に同じ。)
で表される化合物の硫酸塩、及び式(3):
で表される化合物の硫酸塩、及び式(3):
(式中、R2はC1〜C4アルキル基を示す。)
で表される化合物を反応させる工程を含む、製造方法。
[8] 硫酸塩が1/2硫酸塩である、[7]に記載の製造方法。
[9] アルコールがC1〜C4アルコール(好ましくは1−プロパノール)である、[7]又は[8]に記載の製造方法。
[10] アルカリ金属の水酸化物が水酸化リチウム又は水酸化カリウムである、[7]〜[9]のいずれかに記載の製造方法。
[11] 式(5):
で表される化合物を反応させる工程を含む、製造方法。
[8] 硫酸塩が1/2硫酸塩である、[7]に記載の製造方法。
[9] アルコールがC1〜C4アルコール(好ましくは1−プロパノール)である、[7]又は[8]に記載の製造方法。
[10] アルカリ金属の水酸化物が水酸化リチウム又は水酸化カリウムである、[7]〜[9]のいずれかに記載の製造方法。
[11] 式(5):
で表される化合物の製造方法であって、
(I)アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2):
(I)アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2):
(式中、R1は前記に同じ。)
で表される化合物の硫酸塩、及び式(3):
で表される化合物の硫酸塩、及び式(3):
(式中、R2はC1〜C4アルキル基を示す。)
で表される化合物を反応させて、式(4):
で表される化合物を反応させて、式(4):
(式中、R1は前記に同じ。)
で表される化合物を製造する工程、並びに
(II)得られた式(4)で表される化合物を加水分解する工程、
を含む、製造方法。
[12] 工程(II)の加水分解工程が、工程(I)で得られた式(4)で表される化合物を含む反応混合物に水及びアルカリ金属の水酸化物を加えて加水分解するものである、[11]に記載の製造方法。
[13] 式(5)で表される化合物の製造方法であって、
(I)硫酸存在下、式(1)で表される化合物及びシアナミドを反応させて、式(2)で表される化合物の硫酸塩を得る工程、
(II)アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、得られた式(2)で表される化合物の硫酸塩、及び式(3)で表される化合物を反応させて、式(4)で表される化合物を得る工程、並びに
(III)得られた式(4)で表される化合物を加水分解する工程、
を含む、製造方法。
で表される化合物を製造する工程、並びに
(II)得られた式(4)で表される化合物を加水分解する工程、
を含む、製造方法。
[12] 工程(II)の加水分解工程が、工程(I)で得られた式(4)で表される化合物を含む反応混合物に水及びアルカリ金属の水酸化物を加えて加水分解するものである、[11]に記載の製造方法。
[13] 式(5)で表される化合物の製造方法であって、
(I)硫酸存在下、式(1)で表される化合物及びシアナミドを反応させて、式(2)で表される化合物の硫酸塩を得る工程、
(II)アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、得られた式(2)で表される化合物の硫酸塩、及び式(3)で表される化合物を反応させて、式(4)で表される化合物を得る工程、並びに
(III)得られた式(4)で表される化合物を加水分解する工程、
を含む、製造方法。
本発明の式(2)で表される化合物の硫酸塩の製造方法によれば、硫酸の存在下、式(1)で表される化合物及びシアナミドを反応させて、式(2)で表される化合物の硫酸塩を結晶として高収率且つ高純度で得ることができる。また、従来の製造方法における、式(2)で表される化合物の塩酸塩を硝酸塩に変換して結晶化させるという煩わしい工程を省略できるため、簡便且つ効率的な製造方法である。
よって、本発明の製造方法は、式(2)で表される化合物を工業的規模で大量に製造する場合に非常に有利な方法である。また、この製造方法を用いることにより、式(4)で表される化合物、式(5)で表される化合物、及びニロチニブを効率的かつ高収率で製造することができる。
本発明における典型的な形態を説明する。式(1)で表される化合物から、式(2)で表される化合物の硫酸塩を経て、式(5)で表される化合物を製造する反応式を以下に示す。
(式中、R1はC1〜C4アルキル基を示す。R2はC1〜C4アルキル基を示す。)
R1で示されるC1〜C4アルキル基としては、鎖状又は分岐状のいずれであってもよく、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、tert−ブチル基が挙げられ、好ましくはメチル基、エチル基であり、より好ましくはメチル基である。
R2で示されるC1〜C4アルキル基としては、鎖状又は分岐状のいずれであってもよく、例えば、メチル基、エチル基、n−プロピル基、イソプロピル基、n−ブチル基、イソブチル基、tert−ブチル基が挙げられ、好ましくはメチル基、エチル基であり、より好ましくはメチル基である。
(1)→(2)の硫酸塩:
硫酸存在下に、式(1)で表される化合物とシアナミドとを反応させて、式(2)で表される化合物の硫酸塩を製造する。
硫酸存在下に、式(1)で表される化合物とシアナミドとを反応させて、式(2)で表される化合物の硫酸塩を製造する。
本反応で用いる硫酸は、例えば、濃硫酸、含溶媒の硫酸、含水の硫酸等を用いてもよい。硫酸の濃度は、通常50%(質量パーセント)以上であり、65〜90%が好ましい。硫酸の使用量は、式(1)で表される化合物1モルに対して、0.5モル以上であればよく、通常0.5〜2モルであり、好ましくは1〜1.5モルであり、より好ましくは1.1〜1.4モルである。
本反応で用いるシアナミドは、結晶のものや水溶液の形態のものが使用される。50%(質量パーセント)のシアナミドの水溶液は購入可能である。シアナミドの使用量は、式(1)で表される化合物1モルに対して、1モル以上であればよく、通常1〜3モルであり、好ましくは1.5〜2.5モルであり、より好ましくは1.7〜2.3モルである。
本反応は、通常、アルコールを溶媒として用いて実施される。アルコールとしては、例えば、メタノール、エタノール、1−プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、tert−ブタノール等が挙げられ、中でもエタノール、1−プロパノールが好ましく、エタノールが特に好ましい。溶媒の使用量は、式(1)で表される化合物100重量部に対して、100重量部以上であればよく、通常100〜1000重量部であり、好ましくは200〜800重量部であり、より好ましくは300〜700重量部である。
本反応は、式(1)で表される化合物とシアナミドと硫酸との混合の順序に特に限定はない。例えば、溶媒と式(1)で表される化合物とシアナミドとの混合物に硫酸を添加してもよいし、溶媒と硫酸と式(1)で表される化合物との混合物にシアナミドを添加してもよい。そのうち、溶媒と式(1)の化合物とシアナミドとの混合物に硫酸を添加することが好ましい。
硫酸を添加する場合、その添加時の反応液の温度は、通常0℃〜溶媒の沸点の範囲内であり、好ましくは60℃〜80℃である。また、硫酸の添加時間は、通常2〜20時間であり、好ましくは6〜12時間である。硫酸を滴下した後の反応時間は通常1〜4時間である。
次いで、本反応で得られた混合物を冷却する。冷却温度は、反応液の温度が通常−10℃〜20℃であり、収率及び操作性の観点から−5℃〜5℃が好ましい。冷却の速度は1時間に1℃〜20℃の範囲内、好ましくは1時間に5℃〜20℃の範囲で行うことができる。
冷却後の混合物には、液体中、式(2)で表される化合物の硫酸塩が析出しており、この固体(結晶)を取得し乾燥することにより、式(2)で表される化合物の硫酸塩を得る。
析出した式(2)で表される化合物の硫酸塩は、公知の固液分離方法に従って取得することができる。具体的には、濾過、デカンテーションなどの固液分離操作が挙げられる。得られた式(2)で表される化合物の硫酸塩の結晶は、必要により、溶媒による洗浄を行ってもよい。洗浄に用いる溶媒としては、特に限定されないが、反応溶媒と同じ溶媒(好ましくはエタノール)が用いられる。分離された式(2)で表される化合物の硫酸塩は、常圧または減圧下で乾燥することができる。式(2)で表される化合物としては、R1がメチル基又はエチル基である化合物が好ましく、R1がメチル基である化合物、即ち、3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸のメチルエステルがより好ましい。
本反応によれば、式(2)で表される化合物の硫酸塩を効率的且つ高収率で得ることができ、上記のように冷却及び濾過等の単純な操作を行うだけで、高純度の式(2)で表される化合物の硫酸塩を得ることができる。本反応では、通常、上記反応により得られる式(2)で表される化合物が、その生成と同時に共存する硫酸により結晶性の良い硫酸塩(特に、1/2硫酸塩)となり直ちに反応液から析出するため、反応条件により分解する等の悪影響を受けにくく安定的に高純度が保持されていると考えられる。式(2)で表される化合物の硫酸塩の純度は、通常、99%以上、好ましくは99.5%以上である。
(2)+(3)→(4):
次いで、アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2)で表される化合物の硫酸塩及び式(3)で表される化合物を反応させて、式(4)で表される化合物を製造する。本反応は、ピリミジン環を形成する縮合反応である。
次いで、アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2)で表される化合物の硫酸塩及び式(3)で表される化合物を反応させて、式(4)で表される化合物を製造する。本反応は、ピリミジン環を形成する縮合反応である。
アルコールを含む溶媒におけるアルコールとしては、通常C1〜C6のアルコール、好ましくはC1〜C4のアルコールが挙げられる。具体的には、例えば、メタノール、エタノール、1−プロパノール、イソプロパノール、1−ブタノール、2−ブタノール、tert−ブタノール等が挙げられ、好ましくは1−プロパノール、1−ブタノールであり、より好ましくは1−プロパノールである。
アルコールを含む溶媒は、アルコール以外の他の溶媒であって、本反応に悪影響を及ぼさないものを含んでいてもよい。アルコールのみからなる溶媒が好ましい。
アルカリ金属の水酸化物としては、例えば、水酸化ナトリウム、水酸化リチウム、水酸化カリウムが挙げられ、水酸化リチウム又は水酸化カリウムが好ましい。アルカリ金属の水酸化物は、式(2)で表される化合物の硫酸塩の硫酸を中和するために必要な当量に加えて、ピリミジン環を形成する本縮合反応の触媒として作用するために必要な量が存在すればよい。アルカリ金属の水酸化物の使用量は、式(2)で表される化合物1モルに対して、通常1.03〜1.2モルである。
式(3)で表される化合物の使用量は、式(2)で表される化合物の硫酸塩1モルに対して、通常1〜2モルであり、好ましくは1〜1.5モルであり、より好ましくは1.05〜1.2モルである。式(3)で表される化合物は、公知の方法、例えば、国際公開第2011/39782号に記載の方法に従い製造することができる。
本反応の反応温度(反応液の温度)は特に限定はなく、通常溶媒の還流下で行うことができる。耐圧反応器を用いた場合は、反応圧力は、0.1〜0.5MPa(ゲージ圧)、反応温度は150℃以下で行うことができる。反応時間は通常5〜30時間である。本反応により、式(4)で表されるエステル化合物が得られる。
本反応で得られた式(4)で表される化合物を含む反応混合物をそのまま次の加水分解工程に供することができる。或いは、当該反応混合物から式(4)で表される化合物を単離又は精製して次の工程に供することもできる。反応の簡便性の観点から前者が好ましい。
式(4)で表される化合物のエステル部位(−COOR1)の一部が、本縮合反応に用いた溶媒中のアルコールとエステル交換反応して混合エステル化合物を与える場合がある。しかし、次の工程で当該混合エステル化合物は支障なく加水分解されて、式(5)で表されるカルボン酸化合物を与える。
(4)→(5):
式(4)で表される化合物を加水分解して、式(5)で表されるカルボン酸化合物を得る。加水分解反応は、通常、水を含む溶媒中、アルカリ金属の水酸化物の存在下で実施することができる。
式(4)で表される化合物を加水分解して、式(5)で表されるカルボン酸化合物を得る。加水分解反応は、通常、水を含む溶媒中、アルカリ金属の水酸化物の存在下で実施することができる。
アルカリ金属の水酸化物としては、上記式(4)で表される化合物の製造工程で用いたものを使用することができる。特に、水酸化カリウム又は水酸化リチウムが好ましい。アルカリ金属の水酸化物の使用量は、式(4)で表される化合物1モルに対して、通常1モル以上であり、好ましくは1〜3モルであり、より好ましくは1.01〜1.25モルである。
本加水分解反応の反応温度(反応液の温度)は、通常0℃〜溶媒の沸点の範囲内、好ましくは50℃〜70℃である。反応時間は通常1〜3時間である。
反応後は、反応混合物に酸を添加して中和し、式(5)で表されるカルボン酸化合物を析出させる。添加する酸としては、例えば塩酸、硫酸、硝酸等の無機酸;酢酸、ギ酸、プロピオン酸、シュウ酸、クエン酸等の有機酸が挙げられる。詳細なpH調整を必要としないため酢酸を用いるのが好ましい。
酸の使用量は、式(2)で表される化合物1モルに対して、通常、2〜4モルであり、好ましくは3〜4モルである。
前記工程で得られた式(4)で表される化合物を含む反応混合物を用いて本加水分解工程に供する場合、当該反応混合物に水及びアルカリ金属の水酸化物を加えて反応させることができる。
析出した式(5)で表されるカルボン酸化合物は、公知の固液分離方法に従って取得することができる。具体的には、濾過、デカンテーションなどの固液分離操作が挙げられる。分離した式(5)で表されるカルボン酸化合物の結晶は、必要であれば、溶媒で洗浄してもよい。洗浄に用いる溶媒としては、例えば、メタノール、エタノール、イソプロピルアルコール等のアルコール溶媒、水、それらの混合溶媒等が挙げられる。特に、メタノール及び/又は水が好ましく用いられる。分離した結晶を乾燥することにより、式(5)で表されるカルボン酸化合物を得る。乾燥は、常圧又は減圧下で、通常、70℃以下、好ましくは、30〜60℃で行うことができる。
上記式(5)で表されるカルボン酸化合物は、公知の方法を用いてニロチニブに導くことができる。
以下、実施例により本発明をさらに詳細に説明するが、本発明はこれらの例に限定されるものではない。
以下の実施例において得られた3−[(アミノイミノメチル)アミノ]−4−メチル安息香酸メチルエステル(化合物(2b))の1/2硫酸塩の純度は、下記の条件に従って分析を行った。
<高速液体クロマトグラフィー(HPLC)分析条件>
・測定機器:島津製作所製LC20A
・カラム:InertSustain C18 4.6×150mm, 3μm
・移動相:A液 炭酸アンモニウム水(1.57g/1L)
B液 メタノール
・グラジエント条件:
・測定機器:島津製作所製LC20A
・カラム:InertSustain C18 4.6×150mm, 3μm
・移動相:A液 炭酸アンモニウム水(1.57g/1L)
B液 メタノール
・グラジエント条件:
[実施例1](結晶シアナミド、濃硫酸を用いた方法)
化合物(1b) 10.00g(0.06mol)、シアナミド 5.08g(0.12mol)、エタノール 50mLを仕込み、70℃まで昇温した。濃硫酸 7.12g(0.07mol)を6hかけて滴下後、還流下、2h撹拌した。反応終了後、反応マスを5℃まで冷却し、保温後、濾過、エタノール 40mLで洗浄した。得られた結晶を減圧乾燥し、化合物(2b)の1/2硫酸塩 14.53g(収率 93.7%)を得た。LC面積百分率 100%。
[実施例2](50%シアナミド水溶液、濃硫酸を用いた方法)
化合物(1b) 10.00g(0.12mol)を用い、50%シアナミド水溶液 10.16g(0.24mol)を用いた以外は同様の操作を行った。化合物(2b)の1/2硫酸塩の得量14.10g(収率 90.9%)。LC面積百分率 99.82%。
化合物(1b) 10.00g(0.12mol)を用い、50%シアナミド水溶液 10.16g(0.24mol)を用いた以外は同様の操作を行った。化合物(2b)の1/2硫酸塩の得量14.10g(収率 90.9%)。LC面積百分率 99.82%。
[実施例3](50%シアナミド水溶液、78%硫酸を用いた方法)
化合物(1b) 20.00g(0.12mol)、50%シアナミド水溶液 20.36g(0.24mol)、エタノール 100mLを仕込み、70℃まで昇温した。78%硫酸 18.27kg (0.14mol)を8hかけて滴下後、2h撹拌した。50℃まで冷却し、反応終点を確認した後、反応マスを約5時間かけて0℃まで冷却し、保温後、濾過、エタノール 36Lで洗浄した。得られた結晶を減圧乾燥し、化合物(2b)の1/2硫酸塩 28.16g(収率 90.8%)を得た。LC面積百分率 99.87%。
化合物(1b) 20.00g(0.12mol)、50%シアナミド水溶液 20.36g(0.24mol)、エタノール 100mLを仕込み、70℃まで昇温した。78%硫酸 18.27kg (0.14mol)を8hかけて滴下後、2h撹拌した。50℃まで冷却し、反応終点を確認した後、反応マスを約5時間かけて0℃まで冷却し、保温後、濾過、エタノール 36Lで洗浄した。得られた結晶を減圧乾燥し、化合物(2b)の1/2硫酸塩 28.16g(収率 90.8%)を得た。LC面積百分率 99.87%。
[実施例4](KOH使用)
1-プロパノール25mL中、化合物(2b)の1/2硫酸塩(3-[(アミノイミノメチル)アミノ]-4-メチル安息香酸メチルエステル・1/2硫酸塩)5.00g(19.5mmol)、化合物(3b) 4.13g(23.4mmol)、水酸化カリウム 1.15g(20.5mmol)を還流下、17h反応した。反応マスは冷却後、メチルエステル(化合物(4b))、プロピルエステル、カルボン酸の混合物としてそのまま加水分解反応へと進めた。縮合反応マスに水10mLを流入し、30%水酸化カリウム水溶液 7.30g(39.2mmol)を加え、60℃で1h反応した。反応終了マスに酢酸 2.34g(30.2mmol)を滴下し、60℃で1h以上撹拌後、更に酢酸 2.34g(30.2mmol)を1h以上かけて滴下した。20℃まで冷却し、保温した。得られたスラリーを濾過後、水 180mL、メタノール120mLで洗浄した。得られた結晶を減圧乾燥し、化合物(5) 5.63g(収率 94.2%)を得た。LC面積百分率 99.92%。
[実施例5](LiOH使用)
1-プロパノール263mL中、化合物(2b)の1/2硫酸塩 75.00g(0.29mol)、化合物(3b) 61.90g(0.35mol)、水酸化リチウム1水和物 12.90g(0.31mol)を還流下、15h反応した。反応マスは冷却後、メチルエステル(化合物(4b))、プロピルエステル、カルボン酸の混合物としてそのまま加水分解反応へと進めた。縮合反応マスに水225mLを流入し、水酸化リチウム1水和物18.42g(0.44mol)を加え、60℃で3h反応した。反応終了マスに32%酢酸水 49.57g(0.26mol)と1-プロパノール38mLを滴下し、60℃で30min以上撹拌後、更に32%酢酸水 143.2g(0.75mol)を6h以上かけて滴下した。20℃まで冷却し、保温した。得られたスラリーを濾過後、水 450mL、メタノール300mLで洗浄した。得られた結晶を減圧乾燥し、化合物(5) 81.69g(収率 91.1%)を得た。LC面積百分率 99.94%。
1-プロパノール263mL中、化合物(2b)の1/2硫酸塩 75.00g(0.29mol)、化合物(3b) 61.90g(0.35mol)、水酸化リチウム1水和物 12.90g(0.31mol)を還流下、15h反応した。反応マスは冷却後、メチルエステル(化合物(4b))、プロピルエステル、カルボン酸の混合物としてそのまま加水分解反応へと進めた。縮合反応マスに水225mLを流入し、水酸化リチウム1水和物18.42g(0.44mol)を加え、60℃で3h反応した。反応終了マスに32%酢酸水 49.57g(0.26mol)と1-プロパノール38mLを滴下し、60℃で30min以上撹拌後、更に32%酢酸水 143.2g(0.75mol)を6h以上かけて滴下した。20℃まで冷却し、保温した。得られたスラリーを濾過後、水 450mL、メタノール300mLで洗浄した。得られた結晶を減圧乾燥し、化合物(5) 81.69g(収率 91.1%)を得た。LC面積百分率 99.94%。
[実施例6](加圧反応による反応時間短縮)
1-プロパノール50mL、化合物(2b)の1/2硫酸塩 10.00g(39.0mmol)、化合物(3b) 8.25g(46.8mmol)、水酸化リチウム1水和物 1.72g(41.0mmol)を耐圧反応器に仕込み、110℃まで昇温したところ、反応は7hで終了した。以降、実施例5と同様の操作を行った。化合物(5) 10.42g(収率 87.2%)を得た。LC面積百分率 99.81%。
1-プロパノール50mL、化合物(2b)の1/2硫酸塩 10.00g(39.0mmol)、化合物(3b) 8.25g(46.8mmol)、水酸化リチウム1水和物 1.72g(41.0mmol)を耐圧反応器に仕込み、110℃まで昇温したところ、反応は7hで終了した。以降、実施例5と同様の操作を行った。化合物(5) 10.42g(収率 87.2%)を得た。LC面積百分率 99.81%。
本発明の製造方法を用いることにより、ニロチニブの製造中間体を簡便かつ高収率で製造することができる。
Claims (13)
- 式(2):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物の硫酸塩。 - 硫酸塩が1/2硫酸塩である、請求項1に記載の硫酸塩。
- R1がメチル基である、請求項1又は2に記載の硫酸塩。
- 式(2):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物の硫酸塩の製造方法であって、硫酸存在下に、式(1):
(式中、R1は前記に同じ。)
で表される化合物、及びシアナミドを反応させる工程を含む、製造方法。 - 硫酸塩が1/2硫酸塩である、請求項4に記載の製造方法。
- R1がメチル基である、請求項4又は5に記載の製造方法。
- 式(4):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物の製造方法であって、アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2):
(式中、R1は前記に同じ。)
で表される化合物の硫酸塩、及び式(3):
(式中、R2はC1〜C4アルキル基を示す。)
で表される化合物を反応させる工程を含む、製造方法。 - 硫酸塩が1/2硫酸塩である、請求項7に記載の製造方法。
- アルコールがC1〜C4アルコールである請求項7又は8に記載の製造方法。
- アルカリ金属の水酸化物が水酸化リチウム又は水酸化カリウムである、請求項7〜9のいずれかに記載の製造方法。
- 式(5):
で表される化合物の製造方法であって、
(I)アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、式(2):
(式中、R1は前記に同じ。)
で表される化合物の硫酸塩、及び式(3):
(式中、R2はC1〜C4アルキル基を示す。)
で表される化合物を反応させて、式(4):
(式中、R1は前記に同じ。)
で表される化合物を製造する工程、並びに
(II)得られた式(4)で表される化合物を加水分解する工程、
を含む、製造方法。 - 工程(II)の加水分解工程が、工程(I)で得られた式(4)で表される化合物を含む反応混合物に水及びアルカリ金属の水酸化物を加えて加水分解するものである、請求項11に記載の製造方法。
- 式(5):
で表される化合物の製造方法であって、
(I)硫酸存在下、式(1):
(式中、R1はC1〜C4アルキル基を示す。)
で表される化合物及びシアナミドを反応させて、式(2):
(式中、R1は前記に同じ。)
で表される化合物の硫酸塩を得る工程、
(II)アルコールを含む溶媒中、アルカリ金属の水酸化物の存在下、得られた式(2)で表される化合物の硫酸塩、及び式(3):
(式中、R2はC1〜C4アルキル基を示す。)
で表される化合物を反応させて、式(4):
(式中、R1は前記に同じ。)
で表される化合物を得る工程、並びに
(III)得られた式(4)で表される化合物を加水分解する工程、
を含む、製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018125301A JP2020002108A (ja) | 2018-06-29 | 2018-06-29 | 安息香酸化合物の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2018125301A JP2020002108A (ja) | 2018-06-29 | 2018-06-29 | 安息香酸化合物の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2020002108A true JP2020002108A (ja) | 2020-01-09 |
Family
ID=69098607
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018125301A Pending JP2020002108A (ja) | 2018-06-29 | 2018-06-29 | 安息香酸化合物の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP2020002108A (ja) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112745300A (zh) * | 2021-01-21 | 2021-05-04 | 杭州浙中医药科技有限公司 | 一种制备n-(5-羧基-2-甲基苯基)-4-(3-吡啶)-2-嘧啶胺的方法 |
| CN113968801A (zh) * | 2020-07-25 | 2022-01-25 | 扬子江药业集团南京海陵药业有限公司 | 尼洛替尼中间体及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS504038A (ja) * | 1972-12-05 | 1975-01-16 | ||
| JP2005533827A (ja) * | 2002-07-05 | 2005-11-10 | ノバルティス アクチエンゲゼルシャフト | チロシンキナーゼの阻害剤 |
| CN103694145A (zh) * | 2013-11-28 | 2014-04-02 | 山东鑫泉医药有限公司 | (2-甲基-5-硝基苯基)胍硫酸盐的合成方法 |
-
2018
- 2018-06-29 JP JP2018125301A patent/JP2020002108A/ja active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS504038A (ja) * | 1972-12-05 | 1975-01-16 | ||
| JP2005533827A (ja) * | 2002-07-05 | 2005-11-10 | ノバルティス アクチエンゲゼルシャフト | チロシンキナーゼの阻害剤 |
| CN103694145A (zh) * | 2013-11-28 | 2014-04-02 | 山东鑫泉医药有限公司 | (2-甲基-5-硝基苯基)胍硫酸盐的合成方法 |
Non-Patent Citations (1)
| Title |
|---|
| JOURNAL OF CHEMICAL RESEARCH, vol. Vol.39(4), JPN6022005544, 2015, pages 223 - 225, ISSN: 0004705703 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113968801A (zh) * | 2020-07-25 | 2022-01-25 | 扬子江药业集团南京海陵药业有限公司 | 尼洛替尼中间体及其制备方法 |
| CN112745300A (zh) * | 2021-01-21 | 2021-05-04 | 杭州浙中医药科技有限公司 | 一种制备n-(5-羧基-2-甲基苯基)-4-(3-吡啶)-2-嘧啶胺的方法 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN101959840A (zh) | 制备2-烷氧基亚甲基-4,4-二氟-3-氧丁酸烷基酯的方法 | |
| JP2013501010A (ja) | 1−(2−ハロビフェニル−4−イル)−シクロプロパンカルボン酸の誘導体の調製方法 | |
| JP2020002108A (ja) | 安息香酸化合物の製造方法 | |
| CN104016877B (zh) | 一种苯基乙酰胺类化合物及在制备米拉贝隆中的应用 | |
| EA016937B1 (ru) | Соли карбоновой и серной кислот сложных эфиров 3-(2,2,2-триметилгидразиниум) пропионата и их использование для синтеза дигидрата 3-(2,2,2-триметилгидразиниум) пропионата | |
| JPWO2011001976A1 (ja) | スレオ−3−(3,4−ジヒドロキシフェニル)−l−セリンの製造法 | |
| EA015790B1 (ru) | Способ получения 4'-[[4-метил-6-(1-метил-1н-бензимидазол-2-ил)-2-пропил-1н-бензимидазол-1-ил]метил]бифенил-2-карбоновой кислоты (телмисартана) | |
| US20120123128A1 (en) | Process for production of optically active nipecotamide | |
| CA2722818C (en) | Preparation of 1,7´-dimethyl-2´-propyl-2,5´-bi-1h-benzimidazole | |
| US9650337B2 (en) | Method of synthesising 4-piperidin-4-yl-benzene-1,3-diol and the salts of same and novel compound tert-butyl 4-(2,4-dihydroxy-phenyl)-4-hydroxy-piperidine-1-carboxylate | |
| KR20180123851A (ko) | 신규 결정형 레날리도마이드 및 이의 제조방법 | |
| CN102050750A (zh) | 制备2-氨基丁酸的新方法 | |
| WO2017050092A1 (zh) | 一种制备奥当卡替中间体的方法 | |
| KR101513561B1 (ko) | 펙소페나딘 염산염의 신규 제조방법 | |
| WO2011128699A2 (en) | Novel process | |
| KR100749162B1 (ko) | 아세클로페낙 아미노산염의 제조방법 | |
| CN118666855A (zh) | 一种制备苯并环酮类似物的流动化学方法 | |
| JPH10330313A (ja) | 安息香酸誘導体の製造方法 | |
| JP4572618B2 (ja) | 4−テトラヒドロピラニルグリシンの製法 | |
| JP4587139B2 (ja) | アミノアルコキシカルボスチリル誘導体の製造方法。 | |
| JP6219516B2 (ja) | 2−アミノ−6−メチルニコチン酸の製造方法 | |
| KR101213467B1 (ko) | 로자탄 대사체 이엑스피-3174 이수화물의 신규한 제조 방법 | |
| CN114249690A (zh) | 一种罗沙司他的合成方法 | |
| CN119409627A (zh) | 一种Boc-Cys(S-Py)-OH的制备方法 | |
| KR100929414B1 (ko) | 트로스피움 클로라이드의 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210511 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20220209 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220215 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20220809 |