JP2020002137A - テトラヒドロフラン化合物の製造方法 - Google Patents
テトラヒドロフラン化合物の製造方法 Download PDFInfo
- Publication number
- JP2020002137A JP2020002137A JP2019132088A JP2019132088A JP2020002137A JP 2020002137 A JP2020002137 A JP 2020002137A JP 2019132088 A JP2019132088 A JP 2019132088A JP 2019132088 A JP2019132088 A JP 2019132088A JP 2020002137 A JP2020002137 A JP 2020002137A
- Authority
- JP
- Japan
- Prior art keywords
- compound
- tetrahydrofuran
- raw material
- wtppm
- producing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/06—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/02—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings
- C07D307/04—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D307/06—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to ring carbon atoms
- C07D307/08—Preparation of tetrahydrofuran
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/26—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers and other compounds
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Furan Compounds (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Polyethers (AREA)
Abstract
【解決手段】2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触させて、2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留により分離するテトラヒドロフラン化合物の精製方法であって、原料テトラヒドロフラン化合物中の水分量が4900wtppm以下であることを特徴とする。
【選択図】なし
Description
いて、原料テトラヒドロフラン化合物中に含まれる2,3−ジヒドロフランを蒸留分離が
容易な高沸点化合物に変換して分離する方法に関する。また、該方法により得られたテト
ラヒドロフラン化合物を原料として用いるポリエーテルポリオールの製造方法に関する。
合物の溶剤として使用される他に、ポリテトラメチレンエーテルグリコール(以下、「P
TMG」と略記することがある。)などのポリエーテルポリオールの原料モノマーとして
も有用な化合物として知られている。
ロキシ化合物を、脱水環化反応を行うことにより環状エーテルを製造する方法が使用され
ることが多い。また、1,4−ブタンジオ−ル(以下、「1,4BG」と略記することが
ある。)とテレフタル酸とからポリブチレンテレフタレート(以下、「PBT」と略記す
ることがある。)を製造する際に副生するTHFを精製する方法も挙げられる。これらの
方法で得られたTHFには、その製法にもよるが2,3−ジヒドロフラン(以下、「2,
3DHF」と略記することがある。)などの不純物が含まれることがある。THF中に2
,3DHFが存在すると、得られるPTMGは、酸価の上昇や色相の悪化が生じるため、
工業原料としての価値が著しく低下するという問題がある。
点55℃)はTHF(沸点66℃)と沸点が近接しているため、通常の蒸留などで分離す
るには高価な設備と多量の熱量が必要とされる。そのため、該成分の除去方法として、貴
金属触媒により水素化する方法(特許文献1)、相当量の水の存在下で陽イオン交換樹脂
に接触させて2,3DHFを2−ヒドロキシテトラヒドロフラン(以下、「OTF」と略
記することがある。)に変換した後に、水素化反応を行い2,3DHFを分離除去する方
法(特許文献2)、鉱酸又はイオン交換樹脂に接触させ、2,3DHFをヒドロキシテト
ラヒドロフランやジヒドロフラン由来の縮合物に変換し、分離除去する方法(特許文献3
)等が提案されている。
価であるという点などから、安全確保に係る設備や触媒維持コストが多くかかるため、工
業的に十分有利な方法とは言えなかった。
理と2,3DHFの除去を兼ねる場合には有効である。しかし、水分の少ないTHF中に
存在する2,3DHFを除去する場合は、分離負荷の大きい水を加えることは脱水に必要
とする蒸留塔などの設備費用や、分離に係る熱量の観点から工業的に有効な手段とは言え
なかった。また水素化工程を必須とすることから上記と同様の問題もあった。
って、多量のOTFが生成することに加え、鉱酸を用いた場合はアルカリ成分による中和
工程が必要であることから、プロセスが煩雑化し工業的に有効な手段とは言えなかった。
変換したとしても、蒸留によってOTFをTHFと完全に分離することが難しいことが判
明した。また2,3DHFからOTFへの変換反応は可逆反応であることから、水分が少
ない蒸留塔の塔底部などで濃縮されたOTFは2,3DHFに再変換され、精製THF中
に含有されるという問題があることが判明した。
の2,3DHFを、容易に分離除去できる工業的に有利な精製方法を提供することにある
。また、テトラヒドロフラン化合物中の2,3DHFを、蒸留塔の塔底液中で濃縮されて
も2,3DHFに戻らない不可逆の成分へ変換することで、テトラヒドロフラン化合物の
精製負荷を低減できる工業的に有利な精製方法を提供することにある。
ステルを製造した際に水と共に副生するTHFは、安価で調達可能であるが、2,3DH
Fが多く含まれ、そのままポリエーテルポリオールの製造原料に用いると、得られるポリ
エーテルポリオールに着色が生じるという問題があることが判明した。
本発明は上記状況に鑑みてなされたもので、その目的は、色相に優れたポリエーテルポ
リオールの原料となるTHFを、精製負荷が少なく製造する方法を提供することにある。
る水分濃度や、反応器内液相中の水分濃度を制御して酸触媒と接触させれば、2,3DH
Fと水の反応により生成するOTFを抑制しつつ、2,3DHFをTHFと容易に分離が
可能な高沸点化合物に変換し得ることを見出した。また、原料となるTHFにヒドロキシ
基含有化合物を添加して酸触媒と接触させれば、2,3DHFをTHFと容易に分離が可
能な高沸点化合物に変換し得ることを見出した。さらに、かかる方法により2,3DHF
が分離除去されたTHFを原料として用いることにより、特に色相の優れたポリエーテル
ポリオールが得られることを見出した。本発明はこれらの知見に基づいて成し遂げられた
ものである。
[1]2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触
させて2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留によりテトラヒド
ロフラン化合物と高沸点化合物とを分離する精製テトラヒドロフラン化合物の製造方法で
あって、原料テトラヒドロフラン化合物中の水分量が4900wtppm以下である精製
テトラヒドロフランの製造方法。
[2]2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触
させて2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留によりテトラヒド
ロフラン化合物と高沸点化合物とを分離する精製テトラヒドロフラン化合物の製造方法で
あって、反応器内液相中の水分量が4900wtppm以下である精製テトラヒドロフラ
ンの製造方法。
[3]2,3−ジヒドロフランを含む原料テトラヒドロフラン化合物にヒドロキシ基含有
化合物を添加し、酸触媒の存在下で2,3−ジヒドロフランを高沸点化合物に変換した後
に、蒸留によりテトラヒドロフラン化合物と高沸点化合物とを分離する精製テトラヒドロ
フランの製造方法。
[4]原料テトラヒドロフラン化合物中の水分量が4900wtppm以下である[2]
又は[3]に記載の精製テトラヒドロフランの製造方法。
[5]反応器内液相中の水分濃度が4900wtppm以下である[1]、[3]、[4
]の何れか1項に記載の精製テトラヒドロフランの製造方法。
[6]高沸点化合物の分子量が100以上である[1]〜[5]の何れか1項に記載の精
製テトラヒドロフランの製造方法。
[7]高沸点化合物が2,3−ジヒドロフラン縮合物である[1]、[2]、[4]〜[
6]の何れか1項に記載の精製テトラヒドロフランの製造方法。
[8]原料テトラヒドロフラン化合物中の2,3−ジヒドロフラン濃度が10wtppm
以上5000wtppm以下である[1]〜[7]の何れか1項に記載の精製テトラヒド
ロフランの製造方法。
[9]原料テトラヒドロフラン化合物が、1,4−ブタンジオールを原料の一つとして使
用するポリエステル製造工程で副生されたものである[1]〜[8]の何れか1項に記載
の精製テトラヒドロフランの製造方法。
[10]精製テトラヒドラフラン化合物中の2−ヒドロキシテトラヒドロフラン濃度が8
00wtppm以下である[1]〜[9]の何れか1項に記載の精製テトラヒドロフラン
の製造方法。
[11]酸触媒が固体酸触媒である[1]〜[10]の何れか1項に記載の精製テトラヒ
ドロフランの製造方法。
[12]固体酸触媒が酸性陽イオン交換樹脂である[11]に記載の精製テトラヒドロフ
ランの製造方法。
[13]ヒドロキシ基含有化合物の添加量が、2,3−ジヒドロフランに対して、モル比
で0.2以上10以下である[3]〜[12]の何れか1項に記載の精製テトラヒドロフ
ランの製造方法。
[14]ヒドロキシ基含有化合物が、炭素数2〜12の脂肪族アルコールまたは脂環式構
造をもつアルコールである[3]〜[13]の何れか1項に記載の精製テトラヒドロフラ
ンの製造方法。
[15]高沸点化合物がアルコキシ化合物である[3]〜[14]の何れか1項に記載の
精製テトラヒドロフランの製造方法。
[16]ヒドロキシ基含有化合物が、2個以上のヒドロキシ基を有する化合物である[3
]〜[15]の何れか1項に記載の精製テトラヒドロフランの製造方法。
[17]2個以上のヒドロキシ基を有する化合物が、1,4−ブタンジオール又はポリテ
トラメチレンエーテルグリコールである[16]に記載の精製テトラヒドロフランの製造
方法。
[18]2個以上のヒドロキシ基を有する化合物が、ポリテトラメチレンエーテルグリコ
ールを製造する際に副生して得られた2〜6量体のポリテトラメチレンエーテルグリコー
ルである請求項[16]又は[17]に記載の精製テトラヒドロフランの製造方法。
[19]蒸留による分離に用いる蒸発缶または蒸留塔の理論段数が0段以上100段以下
である[1]〜[18]の何れか1項に記載の精製テトラヒドロフランの製造方法。
[20]蒸留による分離により回収する留出液の割合が、原料テトラヒドロフラン化合物
の供給量に対して、80wt%以上である[1]〜[19]の何れか1項に記載の精製テ
トラヒドロフランの製造方法。
[21][1]〜[20]の何れか1項に記載の製造方法によって得られた精製テトロヒ
ドロフランに対して、開環重合反応触媒の存在下で、開環重合反応を行うポリエーテルポ
リオールの製造方法。
除去することができると共に、高純度のテトラヒドロフラン化合物を効率的に得ることが
できる。
造した際に副生する2,3DHF含有テトラヒドロフラン化合物から、2,3DHFを簡
便な方法で効果的に除去することができると共に、精製後のテトラヒドロフラン化合物を
原料に用いることで着色の少ないポリエーテルポリオールを製造することができる。
施態様の一例であり、本発明はこれらの内容に特定されるものではなく、その要旨の範囲
内で種々変形して実施することができる。
ラン化合物(以下、「原料テトラヒドロフラン化合物」と略記することがある。)を酸触
媒と接触させて、2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留により
分離するテトラヒドロフラン化合物の精製方法であって、原料テトラヒドロフラン化合物
中の水分量が4900wtppm以下であることに特徴を有するテトラヒドロフラン化合
物の精製方法である。
合物を酸触媒と接触させて、2,3−ジヒドロフランを高沸点化合物に変換させた後に、
蒸留により分離するテトラヒドロフラン化合物の精製方法であって、反応器内液相中の水
分量が4900wtppm以下であることに特徴を有するテトラヒドロフラン化合物の精
製方法である。
加し、酸触媒の存在下で、2,3−ジヒドロフランを高沸点化合物に変換した後に、蒸留
により分離することに特徴を有するテトラヒドロフラン化合物の精製方法である。
方法であり、テトラヒドロフラン化合物の製造方法と同義である。
本発明において、テトラヒドロフラン化合物とは、テトラヒドロフランとその誘導体を
意味する。テトラヒドロフラン誘導体としては、例えば、テトラヒドロフランの水素原子
が、メチル基、エチル基、プロピル基等のアルキル基で置換された化合物が挙げられる。
さらに具体的には、例えば、2−メチルテトラヒドロフラン、3−メチルテトラヒドロフ
ラン、2,5−ジメチルテトラヒドロフラン等が挙げられる。
れないが、通常10質量%以上、好ましくは50質量%以上、より好ましくは80質量%
以上、更に好ましくは95質量%以上、特に好ましくは98質量%以上である。
されることから、原料テトラヒドロフラン化合物中に含まれるテトラヒドロフランの割合
が高い程、本発明の効果が得られやすい傾向にある。
のであれば特に限定されず、化学法により得られるもの、バイオ法と化学法を組み合せて
得られるもの、化学製造工程で副生するもの等の何れのものも使用することができる。
オマス資源の発酵由来の製造法によって得られた1,4BGを脱水環化することで得られ
るTHF、バイオマス資源由来のフルフラールから得られるTHFや、1,4BGを原料
にポリエステルを製造する工程(例えば、1,4BGとテレフタル酸からPBTを製造す
る工程、1,4BGとコハク酸からポリブチレンサクシネートを製造する工程等)で1,
4BGの脱水環化反応によって、水と共に副生するTHF等が挙げられる。これらの方法
は、それ自体既知の通常用いられるものである。また、テトラヒドロフラン誘導体も、既
知の方法により得られるものである。例えば、3−メチルテトラヒドロフランは、2−メ
チル−1,4−ブタンジオールの脱水環化により得られる。
合物」と略記することがある。)は、他の手法で得られたTHF化合物に比べて安価であ
るものの、テトラヒドロフラン中に2,3DHF等の不純物が多く含まれる傾向にあり、
そのままポリエーテルポリオールの製造原料に用いると得られるポリエーテルポリオール
に着色が生じるという問題があった。それ故、本発明においては、このTHFを原料とし
て上記精製方法を実施し、得られた高純度のTHFをポリエーテルポリオールの製造原料
として用いるのが特に好ましい。
であることが好ましく、より好ましくは3000wtppm以下、さらに好ましくは10
00wtppm以下である。下限は特に限定されないが、好ましくは10wtppm以上
、より好ましくは300ppm以上、さらに好ましくは500ppm以上である。
に着色が生じにくいことから、本発明の方法を用いて2,3DHFを除去する必要性は低
く、高すぎると、2,3DHFをテトラヒドロフラン化合物と容易に分離できる高沸点化
合物に変換する際に(以下、「高沸化」と略記することがある。)必要とする酸触媒の量
が多くなることや、酸触媒の寿命が短くなるため設備費用が多大となり、工業的には好ま
しくない傾向にある。
以下であり、好ましくは3000wtppm以下、さらに好ましくは1500wtppm
以下、より好ましくは500wtppm以下である。下限は特に限定されないが、10w
tppm以上であることが好ましく、より好ましくは50wtppm以上である。
900wtppm以下であることが好ましく、より好ましくは3000wtppm以下、
さらに好ましくは1500wtppm以下、特に好ましくは500wtppm以下である
。下限は特に限定されないが、10wtppm以上であることが好ましく、より好ましく
は50wtppm以上である。
除去することができず精製THF中に含まれ、ポリエーテルポリオール製造時の重合反応
を阻害する問題がある。また、原料テトラヒドロフラン化合物中の水分濃度が高い程、2
,3DHFの加水分解反応が優勢となることから反応器中にOTFが多量に生成されるこ
ととなる。原料テトラヒドロフラン化合物中の水分濃度の下限は特に限定されず、完全な
無水であっても差し支えは無いが、10wtppm未満まで脱水を行うことは、費用、労
力の面から工業的に好ましくない。
有化合物は、水を含んでいることがある。この場合、ヒドロキシ基含有化合物を添加した
後の原料THF化合物中の水分濃度は上記と同様に計算される。
6℃)と近く、蒸留による分離除去が困難である。2,3DHFはポリエーテルポリオー
ルの製造工程で容易に開環重縮合を生じ、ポリエーテルポリオールの色相を悪化させる高
沸点化合物と化す。
,3DHFをテトラヒドロフラン化合物と容易に分離できる高沸点化合物に変換し、蒸留
等の簡便な方法でテトラヒドロフラン化合物と高沸点化合物とを分離除去することが効果
的である。
含有化合物を添加して付加させることで、2,3DHFをテトラヒドロフラン化合物と容
易に分離できる高沸点化合物に変換し、蒸留等の簡便な方法でテトラヒドロフラン化合物
と高沸点化合物とを分離除去することも効果的である。
えば、アルコール化合物および2個以上のヒドロキシ基を有する化合物(以下、ジオール
成分ともいう。)が挙げられる。
つアルコール、水酸基を持つ芳香族化合物などが挙げられる。具体的には、例えば、エタ
ノール、プロパノール、ブタノール、ブテノール、ヘプタノール、ヘキサノール、ヘキセ
ノール、シクロヘキサノール、ヘプタノール、デカノール、ドデカノールなどの炭素数2
〜12の脂肪族アルコールまたは脂環式構造をもつアルコール、フェノールなどの水酸基
を持つ芳香族化合物などが挙げられる。
が好ましく、得られる高沸点化合物が蒸留によってTHFと分離しやすい、ブタノール、
ヘキサノール、ドデカノール等の炭素数4〜12の脂肪族アルコールがより好ましい。
、2個の水酸基が2個の異なる炭素に結合している脂肪族又は脂環式化合物が好ましい。
具体的には、例えば、エチレングリコール、プロパンジオール、ブチンジオール、ブテン
ジオール、ブタンジオール、ペンチンジオール、ペンテンジオール、ペンタンジオール、
ヘキシンジオール、ヘキセンジオール、ヘキサンジオール、シクロヘキサンジオール、ヘ
プチンジオール、ヘプテンジオール、ヘプタンジオール、オクチンジオール、オクテンジ
オール、オクタンジオール、ノニンジオール、ノネンジオール、ノナンジオール、デカン
ジオール、ウンデカンジオール、ドデカンジオールなどの炭素数2〜12の脂肪族又は脂
環式化合物、ポリエーテルポリオールなどが挙げられる。ポリエーテルポリオールとして
は、ポリテトラメチレンエーテルグリコール、ポリトリメチレンエーテルグリコールなど
が好ましく用いられる。
好ましく、1,4−ブタンジオール、2〜6量体のポリテトラメチレンエーテルグリコー
ルが更に好ましい。
合せで用いてもよい。
モル比で、0.2以上であることが好ましく、より好ましくは0.3以上、さらに好まし
くは0.5以上であり、また上限は、10以下であることが好ましく、より好ましくは5
以下、さらに好ましくは3以下、特に好ましくは2以下である。なお、加えるヒドロキシ
基含有化合物が、分子量の異なる化合物が混在している場合は、その平均分子量を基準と
してよい。このモル比が低すぎると、高沸化が起こりにくく処理が難しくなる。また、高
すぎると添加するヒドロキシ基含有化合物に係るコストが高くなると共に、未反応のヒド
ロキシ基含有化合物を反応液から除去する際の蒸留負荷が高くなり、熱量の観点から好ま
しくない。
機構は不明であるが、以下に仮説を示す。
HFとヒドロキシ基の付加反応や縮合反応が生じ、THFに比べて高沸点の化合物に変換
されたと考えられる。これら2,3DHFから変換される高沸点化合物の沸点は、THF
の沸点よりも高いことから、蒸留によって容易に分離することができる。
応させた場合、高沸点化合物としてアルコキシテトラヒドロフランが生じると考えられる
。例として2,3DHFを含むTHF化合物にメタノールを添加、付加した場合、得られ
る高沸点化合物としてメトキシテトラヒドロフラン(沸点112℃)が、THF化合物に
エタノールを添加、付加した場合はエトキシテトラヒドロフラン(沸点171℃)が得ら
れると考えられる。
反応させた場合、高沸点化合物としてアセタール化合物が生じると考えられる。例として
2,3DHFと1,4ブタンジオールを酸触媒存在下で反応させた場合、1,4ブタンジ
オール構造や1,4ブタンジオールの縮合重合体構造が付加されたテトラヒドロフラン誘
導体が高沸点化合物として得られると考えられる。
ことで2,3DHFの高沸点化合物への変換が抑制されて、テトラヒドロフラン化合物と
蒸留等の簡便な方法では分離が難しいOTFが副生する。OTFは多段蒸留や過剰の還流
比をかければ分離することも可能であるが、特にテトラヒドロフラン化合物の回収率が高
い蒸留では熱量の負荷が高いと共に、蒸留塔底部に濃縮されることでOTFが2,3DH
Fに戻って精製テトラヒドロフラン化合物に混入する恐れがある。そのため、原料テトラ
ヒドロフラン化合物と酸触媒が接触する際の水分濃度を低くすることや、反応器内液相中
の水分濃度を低くすることで、OTFの副生量を低減して精製負荷を下げることが可能と
なる。
混入させて使用することも可能ではあるが、該成分は低水分下で酸触媒と接触すると容易
に分解して2,3DHFと化すため、ポリエーテルポリオールの色相悪化が問題となる。
そのため、2,3DHFを含む原料テトラヒドロフラン化合物を、本発明の方法により精
製した後のテトラヒドロフラン化合物(精製テトラヒドロフラン化合物)中のOTF濃度
は、800wtppm以下であることが好ましく、より好ましくは100wtppm以下
、さらに好ましくは50wtppm以下、特に好ましくは20wtppm以下である。
フラン化合物)中の2,3DHF濃度は、通常24wtppm以下、好ましくは20wt
ppm以下、より好ましくは15wtppm以下、さらに好ましくは10wtppm以下
である。
本発明の方法においては、原料テトラヒドロフラン化合物を酸触媒と接触させて、2,
3DHFを高沸点化合物に変換させた後に、蒸留によりテトラヒドロフラン化合物と高沸
点化合物とを分離する。また、本発明の第三の発明では、原料テトラヒドロフラン化合物
に、ヒドロキシ基含有化合物を添加し、酸触媒の存在下で、2,3DHFを高沸点化合物
に変換させた後に、蒸留によりテトラヒドロフラン化合物と高沸点化合物とを分離する。
これにより、厳密な分離を行わずともOTFの含有量が少ない高純度のテトラヒドロフラ
ン化合物を得ることができる。
されないが、好ましくはOTFの沸点より高く(沸点80℃)、より好ましくは110℃
以上、更に好ましくは200℃以上、特に好ましくは250℃以上の沸点を持つ化合物で
ある。また、高分子縮合物等の分解によって沸点を測定できない又は沸点を有さない材料
についても、高沸点化合物とする。
120以上、さらに好ましくは160以上、特に好ましくは180以上である。分子量の
上限は特に限定されないが、通常1000以下である。
た2量体以上の化合物等、2,3DHFとアルコール化合物との付加反応により生成され
るアルコキシテトラヒドロフラン化合物、2,3DHFとジオール成分との付加反応によ
り生成されるアセタール化合物が挙げられる。
ないが、酸触媒接触後の溶液の中和処理等が不要な点から固体酸触媒が好ましい。
固体酸触媒としては、例えば、陽イオン交換樹脂、モンモリロナイトを主成分とする活
性白土〔例えば、水澤化学社製のガレオンアース(商品名)〕、硫酸化ジルコニア、フル
オロスルホン酸含有樹脂〔例えば、DuPont社製のNafion(商品名)〕、リン
酸、ヘテロポリ酸(リンタングステン酸、リンモリブデン酸、ケイタングステン酸)、ス
ルホン酸化合物等が好ましいものとして挙げられる。これらの中で、陽イオン交換樹脂、
活性白土がより好ましく、陽イオン交換樹脂がさらに好ましい。
換樹脂、メタクリル酸やアクリル酸などのカルボン酸基を交換基として持つ弱酸性陽イオ
ン交換樹脂などの酸性陽イオン交換樹脂が挙げられる。また、イオン交換樹脂樹脂の型式
もゲル型およびポーラス型、ハイポーラス型のいずれを選んでもよい。
3DHFの高沸化処理における反応形式として、固定床流通反応形式、懸濁床流通反応形
式、回分反応形式等の公知の反応形式を使用することができる。また用いることのできる
反応器は特に限定されず、反応槽、反応容器、反応釜、反応塔等と同じ意味内容で用いら
れるものであれば何れのものでもよい。工業的には、連続生産が可能となる固定床流通反
応形式が好ましい。
間当たりの通液量に対して、通常0.05倍以上、好ましくは0.1倍以上、より好まし
くは0.5倍以上であり、上限は、通常10倍以下、好ましくは5倍以下、より好ましく
は3倍以下である。滞留時間(反応時間)で言えば、通常3分以上、好ましくは6分以上
、より好ましくは30分以上であり、上限は、通常600分以下、好ましくは300分以
下、より好ましくは90分以下である。この値が小さすぎると十分に2,3DHFの高沸
化が行われないと共に触媒の交換頻度が高くなる。また、大きすぎると反応器に係る建設
費が高くなるため工業的に不利となる。
以上、より好ましくは5wt%以上であり、上限は、通常50wt%以下、好ましくは3
0wt%以下、より好ましくは20wt%以下である。また、滞留時間(反応時間)は、
通常3分以上、好ましくは6分以上、より好ましくは30分以上であり、上限は、通常6
00分以下、好ましくは300分以下、より好ましくは90分以下である。これらの値が
小さすぎると十分に2,3DHFの高沸化が行われないと共に触媒の交換頻度が高くなる
傾向がある。また、これらの値が大きすぎると反応器に係る建設費が高くなるため工業的
に不利な傾向となる。
0wtppm以下であることが好ましく、より好ましくは3000wtppm以下、さら
に好ましくは1500wtppm以下、特に好ましくは500wtppm以下である。下
限は特に限定されないが、10wtppm以上であることが好ましく、より好ましくは5
0wtppm以上である。
であり、好ましくは3000wtppm以下、さらに好ましくは1500wtppm以下
、特に好ましくは500wtppm以下である。下限は特に限定されないが、10wtp
pm以上であることが好ましく、より好ましくは50wtppm以上である。本発明にお
いて、「反応器内液相中の水分濃度が特定の値以下である」とは、反応中の反応器内液相
中の水分濃度を特定の値以下に維持することを意味する。
成される傾向にある。また、完全な無水であっても差し支えは無いが、反応器内の溶液中
の水分を10wtppm未満まで脱水を行うことは、費用、労力の面から工業的に不利な
傾向にある。
く、原料テトラヒドロフラン化合物中の水分量及び原料中のOTF等の脱水反応が生じる
化合物量の和から計算によって求めてもよい。
、上限は、通常120℃以下、好ましくは100℃以下、より好ましくは90℃以下であ
る。この値が低すぎると副生するOTFの分解が難しくなるため、反応液中に含まれるO
TFの濃度が高くなる傾向にある。この値が高すぎると酸触媒への熱負荷や加熱に係る熱
量の観点から好ましくない傾向にある。ここで、反応温度とは、反応器内の温度、即ち原
料テトラヒドロフラン化合物を酸触媒へ接触させて、2,3DHFを高沸点化合物への変
換反応を行う温度である。
0kPa以上であり、上限は、通常1000kPa以下、好ましくは500kPa以下で
ある。
とが好ましい。分離するために用いる設備として蒸発缶を用いてもよいし、充填塔、棚段
塔などを有した蒸留塔を用いてもよい。
、より好ましくは1段以上、さらに好ましくは4段以上であり、上限は、100段以下で
あることが好ましく、より好ましくは10段以下である。100段より大きい段数では塔
が大きくなりすぎ、設備建設のための経済性が悪化する場合がある。
1以上であり、上限は、通常10以下、好ましくは5以下、より好ましくは3以下である
。この値が小さすぎると十分な分離が行えない可能性があり、大きすぎると蒸発に要する
熱量が多大となるため経済的に悪化する場合がある。
トラヒドロフラン化合物の供給量に対して、80wt%以上であることが好ましく、より
好ましくは90wt%以上、さらに好ましくは95wt%以上である。この値が低すぎる
と蒸発缶の缶出部や蒸留塔の塔底部に含まれるTHFが多くなり、塔底部の溶液の廃棄や
別途蒸留を行う必要が生じる場合がある。塔底部の溶液の廃棄や別途蒸留プロセスを加え
る場合は、いずれも工業的に不利な傾向となる。なお、蒸留塔を用いる場合、留出液は、
通常蒸留塔の塔頂部より回収する液である。
率的に得られる方法であり、本発明の精製方法はテトラヒドロフラン化合物の製造方法と
同義である。
本発明の方法により精製されたテトラヒドロフラン化合物は、2,3DHFが効果的に
除去された高純度の化合物であり、ポリエーテルポリオールの製造原料として、特に好適
に用いることができる。
テトロヒドロフラン化合物に対して、開環重合反応触媒の存在下で、開環重合反応を行う
ことに特徴を有するものである。
より得られるものである。以下、ポリアルキレンエーテルグリコール、具体的にはポリテ
トラメチレンエーテルグリコールを代表例として、本発明のポリエーテルポリオールの製
造方法について説明する。
一般に、テトラヒドロフラン化合物等の環状エーテルは酸化されやすく過酸化物を形成
しやすい。テトラヒドロフラン化合物中の過酸化物濃度は特に制限されないが、重合時の
着色や副反応を抑制するために、通常50wtppm以下で用いられる。テトラヒドロフ
ラン化合物中の過酸化物濃度は2,6−ジ−(t−ブチル)−p−クレゾール等の酸化防
止剤を添加することでも制御することができる。
使用される場合がある。具体的には、例えば、炭素数2〜12、好ましくは2〜8の脂肪
族又は芳香族カルボン酸から誘導されるカルボン酸無水物が挙げられる。無水物の原料と
なるカルボン酸はモノカルボン酸であるのが好ましいが、ポリカルボン酸を用いてもよい
。上記カルボン酸の具体例として、酢酸、プロピオン酸、酪酸、吉草酸、カプロン酸、ヘ
プタン酸、カプリル酸、ペラルゴン酸、マレイン酸、コハク酸等の脂肪族カルボン酸;安
息香酸、フタル酸、ナフタリン酸等の芳香族カルボン酸が挙げられる。これらの中で、価
格や入手のしやすさから脂肪族カルボン酸から誘導される無水物が好ましく、反応性や製
造物の需給の観点から無水酢酸がより好ましい。
対して、通常0.03モル以上、好ましくは0.04モル以上、より好ましくは0.05
モル以上、さらに好ましくは0.06モル以上であり、上限は、通常0.30モル以下、
好ましくは0.28モル以下、より好ましくは0.26モル以下、さらに好ましくは0.
25モル以下、特に好ましくは0.22モル以下である。
物由来の着色が起こりにくくなり、製造されるポリアルキレンエーテルグリコールジエス
テルの色相が悪化するのを防ぎ、転化率の低下に伴う生産量低下を抑制することができる
。また、上記下限値以上とすることにより、十分に開環重合反応を進行させることができ
る。
媒であれば特に限定されないが、ルイス酸性を有する固体酸系触媒を用いるのが好ましい
。固体酸系触媒としては、金属酸化物からなる固体酸触媒が好適に使用される。金属とし
ては、好ましくは周期表〔IUPAC無機化学命名法改訂版(1998年)による〕の第
3族、第4族、第13族もしくは第14族に属する金属元素からなる金属酸化物、または
、これらの金属元素を含む複合酸化物が用いられる。具体的には、酸化イットリウム、チ
タニア、ジルコニア、アルミナ、シリカなどの金属酸化物;ジルコニアシリカ、ハフニア
シリカ、シリカアルミナ、チタニアシリカ、チタニアジルコニアのような複合酸化物が好
ましい。また、これらの複合酸化物にさらに他の金属元素を含有する複合酸化物を用いて
もよい。
金属の塩またはそのアルコキシドを含有する混合溶液に、必要により、酸、アルカリ又は
水を添加することにより、沈澱物あるいはゲルを固体酸触媒前駆体として形成させる方法
が挙げられる。上記沈澱物またはゲルの調製方法としては、共沈殿法、ゾル−ゲル法、混
練法、含浸法などが挙げられる。本発明においては、適当な担体上に金属塩及び/又は金
属アルコキシドを担持させ、固相状態(実質的に水を含まない状態)においてアルカリや
アミン等の塩基性物質を接触させる過程を経て固体酸触媒前駆体を得る方法が好ましい。
ルゴン等の不活性ガス雰囲気、空気あるいは希釈酸素ガス等の酸化性ガス雰囲気下で焼成
し、所望の(複合)酸化物を得ることができる。焼成温度は、通常600℃以上、好まし
くは700℃以上であり、上限は、通常1150℃以下、好ましくは1000℃以下であ
る。上記温度範囲で焼成することにより、活性、安定性に優れた固体酸触媒を得ることが
できる。
るいは連続反応であるか回分反応であるかによって異なるが、懸濁床連続反応の場合には
、通常、反応系の全化合物中の0.001〜50重量%、好ましくは0.01〜30重量
%、特に好ましくは0.1〜20重量%である。
用することにより、ポリアルキレンエーテルグリコールジエステルを得ることができる。
ポリアルキレンエーテルグリコールジエステルは加水分解反応やエステル交換反応を行う
等の公知の方法でポリアルキレンエーテルグリコールに変換することができる。例えば、
テトラヒドロフラン化合物としてTHF、カルボン酸無水物として無水酢酸を使用する場
合には、ポリテトラメチレンエーテルグリコールジメチルエステル(以下、「PTME」
と略記することがある。)を炭素数1〜4の脂肪族アルコールと混合し、エステル交換触
媒存在下でのアルコリシス反応によりエステル交換を行うことで、PTMGを得ることが
できる。
に用いられるものが使用される。また、反応方式も公知の方法であれば特に限定されない
。具体的な反応方式として、テトラヒドロフラン化合物とカルボン酸無水物、触媒をそれ
ぞれ一定量測り取り、その量を反応器に仕込んで重合させる方法(回分方式);テトラヒ
ドロフラン化合物、カルボン酸無水物及び触媒がそれぞれ反応器内で一定量存在するよう
に連続的に供給すると同時に、目的生成物であるポリアルキレンエーテルグリコールジエ
ステルを含む反応液を連続的に抜き取る方法(連続方式)が挙げられる。中でも、生産性
に優れることから、連続方式が好ましい。
以上であり、上限は、通常50℃以下、好ましくは49℃以下である。開環重合反応温度
が上記上限を超えると、ポリアルキレンエーテルグリコールジエステルに着色が生じるこ
とがある。また、上記下限未満では、収率が低下することがある。なお、本発明における
開環重合反応温度とは、反応器内の液温を意味する。
a、好ましくは常圧〜5MPaの圧力の範囲から選択される。反応時間に特に限定はない
が、ポリアルキレンエーテルグリコールジエステルの収率、経済性の観点から、通常0.
1〜20時間、好ましくは0.5〜15時間の範囲である。ここで言う反応時間とは、回
分方式においては、反応温度まで上昇した時点から反応が終了して冷却を開始するまでの
時間を意味し、連続方式においては、反応器中での重合反応液の滞留時間のことを意味す
る。
ンエーテルグリコールジエステルの取り出し及び加水分解工程、触媒の再生工程等を加え
てもよい。回分反応方式の場合、反応終了後、先ず触媒と反応液を濾過分別し、反応液よ
り、未反応原料を留去することで、重合体のみを容易に得ることができる。さらに、反応
後の触媒は、よく洗浄後、付着した有機物を燃焼することにより容易に活性を回復するこ
とができる。
の方法であれば特に限定されないが、気液接触装置にポリアルキレンエーテルグリコール
ジエステルを含む反応液を供給し、未反応原料を分離して回収する工程が含まれることが
好ましい。また、これら未反応原料の回収工程は一種又は複数を組み合わせてもよい。な
お、気液接触装置とは、ポリアルキレングリコールジエステルを含む反応液に対して不活
性ガスを接触させる工程にて使用される装置を意味する。
均分子量を下回る、ポリオキシアルキレングリコールを意味し、具体的には2〜6量体の
ポリアルキレンエーテルグリコールやそのジエステル体も含む。
程において得られたジオール成分を用いてもよい。THFを原料にポリテトラメチレンエ
ーテルグリコールを製造する場合、2〜6量体のポリテトラメチレンエーテルグリコール
ジエステルや2〜6量体のポリテトラメチレンエーテルグリコールを未反応原料の回収工
程で分離し、ジオール成分としてTHF化合物に添加する方法が好ましく用いられる。得
られた未反応原料が2〜6量体のポリテトラメチレンエーテルグリコールのジエステル体
であった場合、さらに加水分解工程を経てジオール成分に変換してから、THF化合物に
添加してもよい。
装置として例えば充填塔、スプレー塔、スクラバー、濡壁塔等;液体連続相中に気体を分
散させる形式の気液接触装置として、例えば気泡塔、段塔、気泡攪拌槽等が挙げられる。
これらは、単独で用いても、複数で用いてもよい。中でも、液体/気体の比が小さく滞留
時間が短くでき、重合物の加熱劣化を避けることができるため、気体連続相中に液体を分
散させる形式の気液接触装置が好ましい。より好ましくは、気液接触面積が大きくできる
充填塔、スプレー塔、スクラバーであり、特に制御が容易な充填塔が工業的に有利な傾向
にある。上記充填塔における充填物は、ラシヒリングやポールリングに代表される不規則
充填物でも規則充填物でもよい。
少なくとも一種が含まれることが好ましく、これらの中で、窒素がより好ましい。
るが、通常10以上100以下である。過大な体積比は、通気ガスの損失につながるので
工業的に不利な傾向となる。塔段数は、滞留時間に依存するが、5〜30段が好適である
。
kPa、処理温度は通常100〜200℃、好ましくは140〜180℃で運転される。
上記処理温度が低すぎると、残存する未反応原料の十分な分離が行えない傾向にあり、高
すぎるとカルボン酸無水物の分解が起こりやすくなり、分解生成物由来の着色がより顕在
化しやすくなる傾向がある。また、処理時間は好ましくは10〜240分、より好ましく
は15〜180分、特に好ましくは30〜120分である。処理時間が短すぎると未反応
原料が十分に分離されない傾向があり、長すぎるとカルボン酸無水物の分解が進行し、分
解生成物由来の着色が顕在化しやすい傾向となる。
ば、SUS、ハステロイ(商標名)、チタン、ガラス等が挙げられる。中でも、耐腐食性の
観点からは、SUS、ハステロイ(商標名)が好ましい。より具体的には、例えば、SUS
304、SUS316、SUS316L、SUS317、SUS317L、SUS329
J4L等が挙げられる。
エステルの品質劣化を引き起こすことなく効率よく未反応原料を回収することができる。
、ハーゼン色数米国公衆衛生協会の規格に規定されているハーゼン色数(APHA値)で
示すことができる。APHA値は特に限定されないが、通常35以下、好ましくは30以
下、より好ましくは25以下、さらに好ましくは15以下である。なお、ハーゼン色数は
、[実施例]の項において説明する方法を用いて測定した値である。
特に限定されないが、通常1.0mgKOH/g以下、好ましくは0.5mgKOH/g
以下、より好ましくは0.3mgKOH/g以下、さらに好ましくは0.1mgKOH/
g以下である。ここで、酸価は、試料1g中に含有する酸を中和するのに必要とする水酸
化カリウムのmg数である。
応やエステル交換反応を行う等の公知の方法でポリアルキレンエーテルグリコールに変換
することができる。
ものであれば特に限定されないが、通常はリチウム、ナトリウム、カリウム、セシウム、
ルビジウム等のアルカリ金属アルコキシドが用いられる。中でも、ナトリウム、カリウム
のアルコキシドが好ましい。具体的には、ナトリウムメトキシド、ナトリウムエトキシド
、ナトリウムイソプロポキシド、カリウムメトキシド、カリウムエトキシド、カリウムイ
ソプロポキシド等が挙げられる。中でも、汎用性が高く安価であることから、ナトリウム
メトキシドがより好ましい。
反応圧力は、通常0.1〜2.0MPa、好ましくは1.0〜1.5MPaあり、反応温
度は、通常60〜180℃の範囲である。
エーテルグリコールジエステルにおける値と同様である。
ることにより、酸価が低く、特に色調に優れるポリアルキレンエーテルグリコールを得る
ことができる。
ンエーテルグリコールは、弾性繊維、熱可塑性ポリエステルエラストマー、熱可塑性ポリ
ウレタンエラストマー、コーティング材などの用途に好適に用いることができる。
下の実施例に限定されるものではない。なお、以下の比較例、実施例において用いた分析
方法、反応器、触媒等は次のとおりである。
り本発明をさらに詳細に説明する。具体的には、以下の比較例1、2、実施例1〜4、6
〜8は、PBT副生THF化合物に主に含まれる2,3DHF、水分がTHF化合物の精
製工程に与える影響及びポリエーテルポリオールの着色への影響を調べるため、市販品の
THFに2,3DHF、水を添加した原料を用いたモデル実験である。また、比較例3、
実施例5は、PBT副生THF化合物(実液)を用いた実験例である。ゆえに、本発明の
技術的範囲は、その要旨を超えない限り以下の実験例により限定されるものではない。
THF中の水分分析はカールフィッシャー法を用いて行った。有機成分の分析はガスク
ロマトグラフィー(装置:島津製作所社製、型番GC−2014、カラムDB−1)によ
り行い、修正面積百分率により算出した。各成分の係数は、THFを基準として、有効炭
素数に基づいて下記の様に定めた。
THFに対する係数
2,3DHF 1.006
OTF 1.0629
なお、各成分の濃度(含有量)は、100wt%から水分濃度を差し引いた値を算出し
、その値から残る成分の割合(wtppm)をガスクロマトグラフィーの各成分の面積百
分率により計算した。
三菱化学社製の強酸性陽イオン交換樹脂(ダイヤイオンPK216LH)を、内径20
mmの円筒型ガラス反応器に30cc充填し、150cc/hrの流量で三菱化学社製の
THFを6hr通液した。本処理を終えた直後に反応器出口から得られるTHF中に含ま
れる水分は約100wtppmであった。本処理を前処理として実施した反応器を用いた
。
ポリアルキレンエーテルグリコールジエステル(具体的にはPTME)の着色の程度は
、ハーゼン色数米国公衆衛生協会の規格に規定されているハーゼン色数(APHA値)で
表した。ハーゼン色数はキシダ化学社製のAPHA色数標準液(No.500)を希釈し
て調製した標準液を使用し、JIS K0071−1(1998年)に準じて比色して求
めた。色差計は日本電色工業社製の測色色差計ZE−2000を使用し、セル厚み:10
mmの条件で測定した。
ポリアルキレンエーテルグリコールジエステル(具体的にはPTME)の数平均分子量
(Mn)は、PTMEのテトラヒドロフラン溶液を調製後、GPC装置〔東ソー社製、製
品名「HLC−8220」、カラム:TskgelSuperHZM−N(4本)〕を用
いて測定した。GPCのキャリブレーションには、英国POLYMER LABORAT
ORIES社製のPOLYTETRAHYDROFURANキャリブレーションキットを
使用した。
THFの開環重合反応触媒として、27.2%硝酸ジルコニア水溶液にCARiACT
Q15(登録商標)(富士シリシア化学社製のシリカ担体)を含浸し乾燥処理を実施し、
その後、重炭酸アンモニウム水溶液で中和・洗浄を行った後、乾燥および900℃で焼成
処理を行ったものを用いた。
三菱化学社製のTHFに、和光純薬社製の2,3DHFを添加して1000wtppm
に調製した。本原料をガラス製の300ccフラスコ反応器に135g、ダイセル社製の
無水酢酸を16.5g、触媒(開環重合反応触媒)を6g仕込み、窒素雰囲気下にて反応
温度40℃で6時間反応させた。この反応液から触媒を濾過分離して得られた重合反応液
を、撹拌子を備えたガラス製丸底フラスコに100g入れて、500cc/minの流量
で窒素をバブリングさせながら、常圧下でバス温度170℃にて2時間加熱して未反応原
料を留去し、PTMEを得た。ゲルパーミエーションクロマトグラフィー(GPC)およ
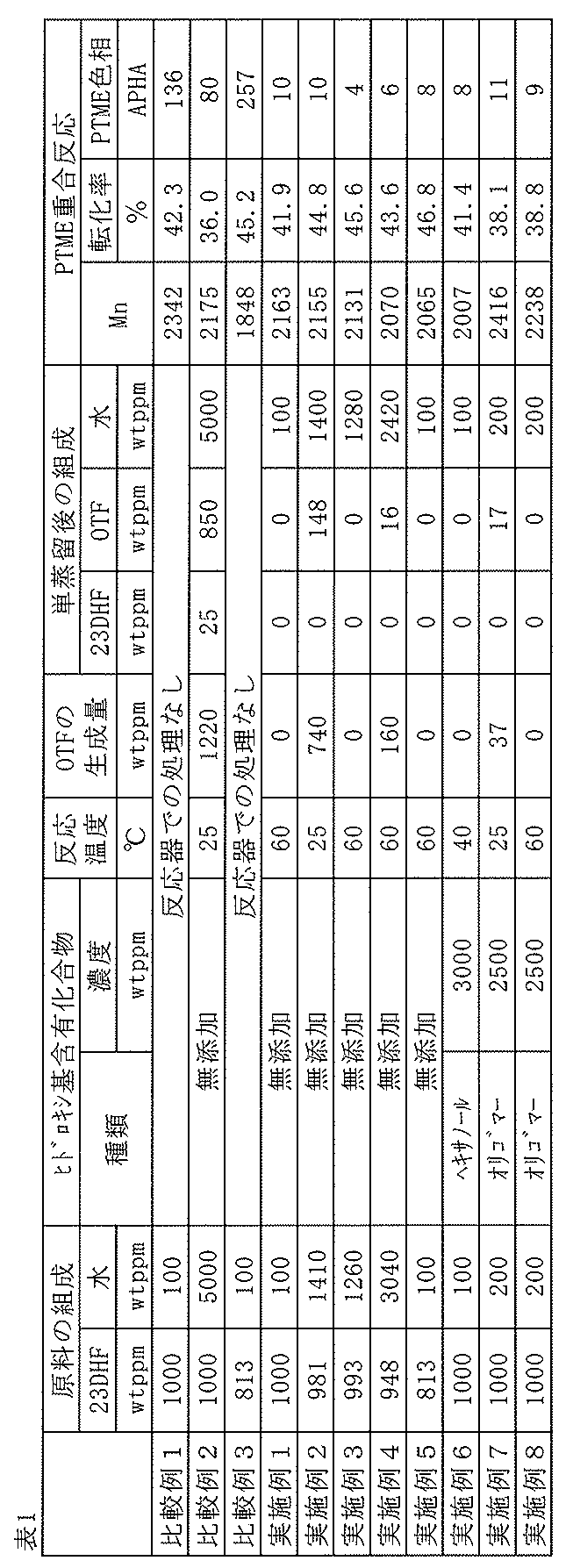
び比色分析を行い、得られたPTMEの品質を確認した。その結果を表1に示す。
三菱化学社製のTHFに、蒸留水を添加して5000wtppmに、和光純薬社製の2
,3DHFを添加して1000wtppmに調製した。本原料を前処理の終えた固定床反
応器に25℃、0.2MPaの条件で30cc/hrで流通させた。反応開始後24〜4
8hrの液を製品タンクに溜め込んだ。この製品タンクに溜め込まれた反応液中の2,3
DHFは0wtppm、OTFは1220wtppm、水は5000wtppmであった
。
窒素をバブリングさせながら、常圧下でバス温度100℃にて2時間加熱し、原料に対し
て99.8wt%のTHFを留出させた。本留出液(単蒸留後)の組成を表1に示す。
本留出液を用いて比較例1と同様に、重合反応を実施し、得られたPTMEの品質を確
認した。その結果を表1に示す。
表1に示す組成のPBT副生THF化合物を原料として、比較例1と同様の方法で重合
反応を行い、得られたPTMEの品質を確認した。その結果を表1に示す。
三菱化学社製のTHFに、和光純薬製の2,3DHFを添加して1000wtppmに
調製した。この時の水分濃度は約100wtppmであった。本原料を前処理の終えた固
定床反応器に60℃、0.2MPaの条件で30cc/hrで流通させた。反応開始後2
4〜48時間経過後の液を製品タンクに溜め込んだ。この製品タンクに溜め込まれた反応
液中の2,3DHFは0wtppm、OTFは0wtppm、水は100wtppmであ
った。
窒素をバブリングさせながら、常圧下でバス温度100℃にて2時間加熱し、原料に対し
て99.8wt%のTHFを留出させた。本留出液(単蒸留後)の組成を表1に示す。
認した。その結果を表1に示す。
三菱化学社製のTHFに、和光純薬製の2,3DHFを添加して、表1に示す組成の原
料を調製した。本原料を、実施例1と同様の方法で、前処理の終えた固定床反応器に流通
させた後に蒸留してTHFを留出させ、この留出液を用いて重合反応を実施し、得られた
PTMEの品質を確認した。表1に、固定床の反応温度、反応後のOTF生成量、本留出
液(単蒸留後)の組成、PTMEの品質を示す。
表1に示す組成のPBT副生THF化合物を原料として、実施例1と同様の方法で、前
処理の終えた固定床反応器に流通させた後に蒸留してTHFを留出させ、この留出液を用
いて重合反応を実施し、得られたPTMEの品質を確認した。表1に、固定床の反応温度
、反応後のOTF生成量、留出液(単蒸留後)の組成、PTMEの品質を示す。
三菱化学社製のTHFに、和光純薬社製の2,3DHFを添加して1000wtppm
に調製したものに、和光純薬社製ヘキサノールを添加して3000wtppm(23DH
Fに対するモル比:2.06)に調製した。この時の水分濃度は約100wtppmであ
った。本原料を前処理の終えた固定床反応器に40℃、0.2MPaの条件で30cc/
hrで流通させた。反応開始後24〜48hrの液を製品タンクに溜め込んだ。この製品
タンクに溜め込まれた反応液中の2,3DHFは0wtppm、OTFは0wtppm、
水は100wtppmであった。本反応液を、ガラス製ナシ型フラスコに1000g入れ
て、30cc/minの流量で窒素をバブリングさせながら、常圧下でバス温度100℃
にて2時間加熱して原料に対して99.5wt%のTHFを留出させた。
認した。表1に、固定床の反応温度、反応後のOTF生成量、留出液(単蒸留後)の組成
、PTMEの品質を示す。
三菱化学社製のTHFに、和光純薬社製の2,3DHFを添加して1000wtppm
に調製したものに、PTMGの2〜6量体で構成される平均分子量250のオリゴマーを
加えて2500wtppm(23DHFに対するモル比:0.70)に調製した。この時
の水分濃度は約200wtppmであった。本原料を前処理の終えた固定床反応器に25
℃、0.2MPaの条件で30cc/hrで流通させた。反応開始後24〜48hrの液
を製品タンクに溜め込んだ。この製品タンクに溜め込まれた反応液中の2,3DHF濃度
は0wtppm、OTFは37wtppm、水は200wtppmであった。本反応液を
、ガラス製ナシ型フラスコに1000g入れて、30cc/minの流量で窒素をバブリ
ングさせながら、常圧下でバス温度100℃にて2時間加熱して原料に対して99.5w
t%のTHFを留出させた。
認した。表1に、固定床の反応温度、反応後のOTF生成量、留出液(単蒸留後)の組成
、PTMEの品質を示す。
三菱化学社製のTHFに、和光純薬社製の2,3DHFを添加して1000wtppm
に調製したものに、PTMGの2〜6量体で構成される平均分子量250のオリゴマーを
加えて2500wtppm(23DHFに対するモル比:0.70)に調製した。この時
の水分濃度は約200wtppmであった。本原料を前処理の終えた固定床反応器に60
℃、0.2MPaの条件で30cc/hrで流通させた。反応開始後24〜48hrの液
を製品タンクに溜め込んだ。この製品タンクに溜め込まれた反応液中の2,3DHFは0
wtppm、OTFは0wtppm、水は200wtppmであった。本反応液を、ガラ
ス製ナシ型フラスコに1000g入れて、30cc/minの流量で窒素をバブリングさ
せながら、常圧下でバス温度100℃にて2時間加熱して原料に対して99.8wt%の
THFを留出させた。
認した。表1に、固定床の反応温度、反応後のOTF生成量、留出液(単蒸留後)の組成
、PTMEの品質を示す。
F化合物から純度の高いTHF化合物を得ることができると共に、得られた精製THF化
合物を原料として製造したPTMEの色相を著しく良くすることが可能である。また、原
料中の水分濃度を下げる、かつOTFの発生量を抑制することで、精製に係る分離負荷を
下げることができるため、設備機器費用の削減や蒸気使用量の低減が可能である。
[1]2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触
させて2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留によりテトラヒド
ロフラン化合物と高沸点化合物とを分離する精製テトラヒドロフラン化合物の製造方法で
あって、原料テトラヒドロフラン化合物中の水分量が4900wtppm以下である精製
テトラヒドロフランの製造方法。
[2]2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触
させて2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留によりテトラヒド
ロフラン化合物と高沸点化合物とを分離する精製テトラヒドロフラン化合物の製造方法で
あって、反応器内液相中の水分量が4900wtppm以下である精製テトラヒドロフラ
ンの製造方法。
[3]原料テトラヒドロフラン化合物中の水分量が4900wtppm以下である[2]
に記載の精製テトラヒドロフランの製造方法。
[4]反応器内液相中の水分濃度が4900wtppm以下である[1]に記載の精製テ
トラヒドロフランの製造方法。
[5]高沸点化合物の分子量が100以上である[1]〜[4]の何れか1項に記載の精
製テトラヒドロフランの製造方法。
[6]高沸点化合物が2,3−ジヒドロフラン縮合物である[1]〜[5]の何れか1項
に記載の精製テトラヒドロフランの製造方法。
[7]原料テトラヒドロフラン化合物中の2,3−ジヒドロフラン濃度が10wtppm
以上5000wtppm以下である[1]〜[6]の何れか1項に記載の精製テトラヒド
ロフランの製造方法。
[8]原料テトラヒドロフラン化合物が、1,4−ブタンジオールを原料の一つとして使
用するポリエステル製造工程で副生されたものである[1]〜[7]の何れか1項に記載
の精製テトラヒドロフランの製造方法。
[9]精製テトラヒドラフラン化合物中の2−ヒドロキシテトラヒドロフラン濃度が80
0wtppm以下である[1]〜[8]の何れか1項に記載の精製テトラヒドロフランの
製造方法。
[10]酸触媒が固体酸触媒である[1]〜[9]の何れか1項に記載の精製テトラヒド
ロフランの製造方法。
[11]固体酸触媒が酸性陽イオン交換樹脂である[10]に記載の精製テトラヒドロフ
ランの製造方法。
[12]蒸留による分離に用いる蒸発缶または蒸留塔の理論段数が0段以上100段以下
である[1]〜[11]の何れか1項に記載の精製テトラヒドロフランの製造方法。
[13]蒸留による分離により回収する留出液の割合が、原料テトラヒドロフラン化合物
の供給量に対して、80wt%以上である[1]〜[11]の何れか1項に記載の精製テ
トラヒドロフランの製造方法。
[14][1]〜[13]の何れか1項に記載の製造方法によって得られた精製テトロヒ
ドロフランに対して、開環重合反応触媒の存在下で、開環重合反応を行うポリエーテルポ
リオールの製造方法。
Claims (21)
- 2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触させ
て2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留によりテトラヒドロフ
ラン化合物と高沸点化合物とを分離する精製テトラヒドロフラン化合物の製造方法であっ
て、原料テトラヒドロフラン化合物中の水分量が4900wtppm以下である精製テト
ラヒドロフランの製造方法。 - 2,3−ジヒドロフランを含有する原料テトラヒドロフラン化合物を酸触媒と接触させ
て2,3−ジヒドロフランを高沸点化合物に変換させた後に、蒸留によりテトラヒドロフ
ラン化合物と高沸点化合物とを分離する精製テトラヒドロフラン化合物の製造方法であっ
て、反応器内液相中の水分量が4900wtppm以下である精製テトラヒドロフランの
製造方法。 - 2,3−ジヒドロフランを含む原料テトラヒドロフラン化合物にヒドロキシ基含有化合
物を添加し、酸触媒の存在下で2,3−ジヒドロフランを高沸点化合物に変換した後に、
蒸留によりテトラヒドロフラン化合物と高沸点化合物とを分離する精製テトラヒドロフラ
ンの製造方法。 - 原料テトラヒドロフラン化合物中の水分量が4900wtppm以下である請求項2又
は3に記載の精製テトラヒドロフランの製造方法。 - 反応器内液相中の水分濃度が4900wtppm以下である請求項1、3、4の何れか
1項に記載の精製テトラヒドロフランの製造方法。 - 高沸点化合物の分子量が100以上である請求項1〜5の何れか1項に記載の精製テト
ラヒドロフランの製造方法。 - 高沸点化合物が2,3−ジヒドロフラン縮合物である請求項1、2、4〜6の何れか1
項に記載の精製テトラヒドロフランの製造方法。 - 原料テトラヒドロフラン化合物中の2,3−ジヒドロフラン濃度が10wtppm以上
5000wtppm以下である請求項1〜7の何れか1項に記載の精製テトラヒドロフラ
ンの製造方法。 - 原料テトラヒドロフラン化合物が、1,4−ブタンジオールを原料の一つとして使用す
るポリエステル製造工程で副生されたものである請求項1〜8の何れか1項に記載の精製
テトラヒドロフランの製造方法。 - 精製テトラヒドラフラン化合物中の2−ヒドロキシテトラヒドロフラン濃度が800w
tppm以下である請求項1〜9の何れか1項に記載の精製テトラヒドロフランの製造方
法。 - 酸触媒が固体酸触媒である請求項1〜10の何れか1項に記載の精製テトラヒドロフラ
ンの製造方法。 - 固体酸触媒が酸性陽イオン交換樹脂である請求項11に記載の精製テトラヒドロフラン
の製造方法。 - ヒドロキシ基含有化合物の添加量が、2,3−ジヒドロフランに対して、モル比で0.
2以上10以下である請求項3〜12の何れか1項に記載の精製テトラヒドロフランの製
造方法。 - ヒドロキシ基含有化合物が、炭素数2〜12の脂肪族アルコールまたは脂環式構造をも
つアルコールである請求項3〜13の何れか1項に記載の精製テトラヒドロフランの製造
方法。 - 高沸点化合物がアルコキシ化合物である請求項3〜14の何れか1項に記載の精製テト
ラヒドロフランの製造方法。 - ヒドロキシ基含有化合物が、2個以上のヒドロキシ基を有する化合物である請求項3〜
15の何れか1項に記載の精製テトラヒドロフランの製造方法。 - 2個以上のヒドロキシ基を有する化合物が、1,4−ブタンジオール又はポリテトラメ
チレンエーテルグリコールである請求項16に記載の精製テトラヒドロフランの製造方法
。 - 2個以上のヒドロキシ基を有する化合物が、ポリテトラメチレンエーテルグリコールを
製造する際に副生して得られた2〜6量体のポリテトラメチレンエーテルグリコールであ
る請求項16又は17に記載の精製テトラヒドロフランの製造方法。 - 蒸留による分離に用いる蒸発缶または蒸留塔の理論段数が0段以上100段以下である
請求項1〜18の何れか1項に記載の精製テトラヒドロフランの製造方法。 - 蒸留による分離により回収する留出液の割合が、原料テトラヒドロフラン化合物の供給
量に対して、80wt%以上である請求項1〜19の何れか1項に記載の精製テトラヒド
ロフランの製造方法。 - 請求項1〜20の何れか1項に記載の製造方法によって得られた精製テトロヒドロフラ
ンに対して、開環重合反応触媒の存在下で、開環重合反応を行うポリエーテルポリオール
の製造方法。
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2014124326 | 2014-06-17 | ||
| JP2014124326 | 2014-06-17 | ||
| JP2014134714 | 2014-06-30 | ||
| JP2014134714 | 2014-06-30 | ||
| JP2014137186 | 2014-07-02 | ||
| JP2014137186 | 2014-07-02 | ||
| JP2015058286 | 2015-03-20 | ||
| JP2015058286 | 2015-03-20 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015122199A Division JP6601006B2 (ja) | 2014-06-17 | 2015-06-17 | テトラヒドロフラン化合物の製造方法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| JP2020002137A true JP2020002137A (ja) | 2020-01-09 |
Family
ID=54935550
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015122199A Active JP6601006B2 (ja) | 2014-06-17 | 2015-06-17 | テトラヒドロフラン化合物の製造方法 |
| JP2019132088A Pending JP2020002137A (ja) | 2014-06-17 | 2019-07-17 | テトラヒドロフラン化合物の製造方法 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2015122199A Active JP6601006B2 (ja) | 2014-06-17 | 2015-06-17 | テトラヒドロフラン化合物の製造方法 |
Country Status (3)
| Country | Link |
|---|---|
| JP (2) | JP6601006B2 (ja) |
| TW (1) | TW201605818A (ja) |
| WO (1) | WO2015194568A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023145554A1 (ja) | 2022-01-28 | 2023-08-03 | 保土谷化学工業株式会社 | テトラヒドロフランと植物由来2-メチルテトラヒドロフランとの共重合反応物、およびその製造方法 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5470259A (en) * | 1977-10-31 | 1979-06-05 | Basf Ag | Purification of tetrahydrofuran |
| JPS61200979A (ja) * | 1985-02-28 | 1986-09-05 | Mitsubishi Chem Ind Ltd | 粗テトラヒドロフランの精製方法 |
| JPH10237057A (ja) * | 1996-12-26 | 1998-09-08 | Mitsubishi Chem Corp | 粗テトラヒドロフランの精製方法 |
| WO1999016762A1 (en) * | 1997-09-30 | 1999-04-08 | Hodogaya Chemical Co., Ltd. | Process for purifying tetrahydrofurans used as starting material for polyether polyols |
| JP2000143652A (ja) * | 1998-11-13 | 2000-05-26 | Mitsubishi Chemicals Corp | 粗テトラヒドロフランの精製方法 |
| JP2002234883A (ja) * | 2000-12-04 | 2002-08-23 | Mitsubishi Chemicals Corp | テトラヒドロフランの精製方法 |
| JP2006503050A (ja) * | 2002-08-20 | 2006-01-26 | ビーエーエスエフ アクチェンゲゼルシャフト | テトラヒドロフランの製造方法 |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS4930353A (ja) * | 1972-07-15 | 1974-03-18 | ||
| JPS5033062B2 (ja) * | 1972-09-01 | 1975-10-27 | ||
| JPS4976861A (ja) * | 1972-12-01 | 1974-07-24 | ||
| JP2002515888A (ja) * | 1996-10-21 | 2002-05-28 | イーストマン ケミカル カンパニー | 3―アルキルテトラヒドロフランの製造方法 |
| JP2003089694A (ja) * | 2001-07-11 | 2003-03-28 | Mitsubishi Chemicals Corp | テトラヒドロフランの精製方法 |
| KR101398614B1 (ko) * | 2009-10-30 | 2014-05-23 | 가부시키가이샤 히타치세이사쿠쇼 | 테트라히드로푸란의 정제 방법 및 정제 시스템 |
| CN107011290B (zh) * | 2012-10-18 | 2019-07-26 | 三菱化学株式会社 | 四氢呋喃的制造方法 |
| JP6191213B2 (ja) * | 2013-04-19 | 2017-09-06 | 東レ株式会社 | テトラヒドロフラン含有物の精製方法 |
-
2015
- 2015-06-16 WO PCT/JP2015/067359 patent/WO2015194568A1/ja not_active Ceased
- 2015-06-17 TW TW104119536A patent/TW201605818A/zh unknown
- 2015-06-17 JP JP2015122199A patent/JP6601006B2/ja active Active
-
2019
- 2019-07-17 JP JP2019132088A patent/JP2020002137A/ja active Pending
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5470259A (en) * | 1977-10-31 | 1979-06-05 | Basf Ag | Purification of tetrahydrofuran |
| JPS61200979A (ja) * | 1985-02-28 | 1986-09-05 | Mitsubishi Chem Ind Ltd | 粗テトラヒドロフランの精製方法 |
| JPH10237057A (ja) * | 1996-12-26 | 1998-09-08 | Mitsubishi Chem Corp | 粗テトラヒドロフランの精製方法 |
| WO1999016762A1 (en) * | 1997-09-30 | 1999-04-08 | Hodogaya Chemical Co., Ltd. | Process for purifying tetrahydrofurans used as starting material for polyether polyols |
| JP2000143652A (ja) * | 1998-11-13 | 2000-05-26 | Mitsubishi Chemicals Corp | 粗テトラヒドロフランの精製方法 |
| JP2002234883A (ja) * | 2000-12-04 | 2002-08-23 | Mitsubishi Chemicals Corp | テトラヒドロフランの精製方法 |
| JP2006503050A (ja) * | 2002-08-20 | 2006-01-26 | ビーエーエスエフ アクチェンゲゼルシャフト | テトラヒドロフランの製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2016172712A (ja) | 2016-09-29 |
| TW201605818A (zh) | 2016-02-16 |
| JP6601006B2 (ja) | 2019-11-06 |
| WO2015194568A1 (ja) | 2015-12-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8247580B2 (en) | Process for preparing ε-caprolactone | |
| US20100168445A1 (en) | Process for preparing epsilon-caprolactone | |
| EP2351726B1 (en) | High-purity 1,6-hexanediol and manufacturing method thereof | |
| JP2011503021A5 (ja) | ||
| SG174394A1 (en) | Method for producing 1,6-hexanediol and caprolactone | |
| JP5939061B2 (ja) | 1,4−ブタンジオール含有組成物 | |
| US20120059174A1 (en) | Process for preparing epsilon-caprolactone and 1,6-hexanediol | |
| JP6244807B2 (ja) | テトラヒドロフランの製造方法 | |
| JP2020002137A (ja) | テトラヒドロフラン化合物の製造方法 | |
| JP5109419B2 (ja) | 1,6−ヘキサンジオールの精製方法 | |
| KR20140117590A (ko) | 개선된 가알칸올분해 프로세스 | |
| RU2571082C2 (ru) | СПОСОБ ПОЛУЧЕНИЯ ε-КАПРОЛАКТОНА И 1,6-ГЕКСАНДИОЛА | |
| CA2744126A1 (en) | Process for preparing 1,6-hexanediol | |
| JP2016017041A (ja) | テトラヒドロフランの精製方法 | |
| JP7508982B2 (ja) | ポリアルキレンエーテルグリコールジエステルの製造方法 | |
| JP6263877B2 (ja) | ポリアルキレンエーテルグリコールジエステルの製造方法 | |
| JP2012236819A (ja) | テトラヒドロフランの製造方法 | |
| CN101080433B (zh) | 聚醚多元醇化合物的制造方法 | |
| JP6264016B2 (ja) | ポリアルキレンエーテルグリコールのジエステルの製造方法及びポリアルキレンエーテルグリコールの製造方法 | |
| JP2015017238A (ja) | ポリアルキレンエーテルグリコールジエステルの製造方法 | |
| JP2014169361A (ja) | ポリアルキレンエーテルグリコールの製造方法 | |
| JP2014181327A (ja) | ポリアルキレンエーテルグリコールジエステルの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20190806 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190806 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20200611 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200630 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20210105 |