JP4288263B2 - p−ヒドロキシ−ミルナシプランの立体異性体およびその使用方法 - Google Patents
p−ヒドロキシ−ミルナシプランの立体異性体およびその使用方法 Download PDFInfo
- Publication number
- JP4288263B2 JP4288263B2 JP2005501895A JP2005501895A JP4288263B2 JP 4288263 B2 JP4288263 B2 JP 4288263B2 JP 2005501895 A JP2005501895 A JP 2005501895A JP 2005501895 A JP2005501895 A JP 2005501895A JP 4288263 B2 JP4288263 B2 JP 4288263B2
- Authority
- JP
- Japan
- Prior art keywords
- carbonyl
- compound
- treatment agent
- alkyl
- aryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 C*N(*)C(C)=O Chemical compound C*N(*)C(C)=O 0.000 description 2
Images































































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/24—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a ring other than a six-membered aromatic ring of the carbon skeleton
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/06—Anti-spasmodics, e.g. drugs for colics, esophagic dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/16—Emollients or protectives, e.g. against radiation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/07—Optical isomers
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Virology (AREA)
- Immunology (AREA)
- Pain & Pain Management (AREA)
- Dermatology (AREA)
- Pulmonology (AREA)
- Diabetes (AREA)
- Urology & Nephrology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Addiction (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Rheumatology (AREA)
- Molecular Biology (AREA)
- Endocrinology (AREA)
- Psychiatry (AREA)
- Vascular Medicine (AREA)
- Otolaryngology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Tropical Medicine & Parasitology (AREA)
Description
有効性と認容性は、精神の抑鬱および機能的体細胞性障害(Functional Somatic Disorders)を含むその他の精神障害に対する薬物療法の選択を決定する際の重要な因子である。三環性の抗抑鬱剤(TCA)から、選択的セロトニン再摂取抑制剤(SSRI)への移行は、TCAの有害な副作用の原因となる直接的な受容体相互作用失くすだけでなく、ノルエピネフリンの再摂取を阻害する能力をも含む。単一の神経伝達物質、セロトニンに対して選択性を有することから、特により重篤な鬱においては、SSRIがTCAよりも効果が低くなる傾向にあることの理由が説明され得る(Lopez-Ibor J. et al., 1996, Int. Clin.
Psychopharm., 11:41-46)。より古いTCAは、有意な行動毒性、特に、精神運動性および認識障害および鎮静に関連している。SSRIには、主としてこれらの作用はない。しかし、吐気や消化不良などの消化管障害は、これらの作用物質に共通している(Hindmarch I., 1997, Human Psychopharmacology,
12:115-119)。例えば、広く処方されてきたSSIRセルトラリン (Zoloft(登録商標)、Pfizer)については、治療の中断と関連する3つの主な副作用は、吐気、不眠および下痢であった(Physician’s Desk Reference, 57th Edition, 2003,
Thomson Medical)。
Neuropharmacology, 24:1211-1219; Palmier et al., 1989, Eur. J. Clin. Pharmacol.,
37:235-238)。現時点での臨床的な証拠は、これらの新規な作用物質は、SSRIと比較して、有効性の向上および/または作用の迅速な開始をもたらすかもしれないということを示唆している(Tran P.V. et al., 2003,
J. Clin. Psychopharmacol., 23:78-86)。ミルナシプランの最近の試験によれば、この化合物は、鬱病に関連する、および無関係の痛みを除去するのに効果的であることを示唆されている(Briley M., 2003, Curr. Opin. Investig. Drugs, 4:42-45; Cypress
Bioscience Inc., Cypress Bioscience Inc. Announces Final Results of Milnacipran Phase II Clinical Trial in Fibromyalgia, Media
Release, March 21, 2003, Available from: URL: http://www.cypressbio.com)。
24:1211-1219; Palmier et al., 1989, Eur. J. Clin. Pharmacol., 37:235-238)が、ドーパミンの摂取には影響を及ぼさない。ミルナシプランは、アルファおよびベータアドレナリン作動性、ムスカリン作動性、ヒスタミン作動性、およびドーパミン作動性の受容体に対して親和性を持たない。このことは、ミルナシプランは、アンチコリン作動性効果、鎮静効果および刺激効果を生成する能力が低いことを示唆している。ミルナシプランは、慢性投与後、ラット皮質においてベータアドレナリン受容体の数に影響を及ぼさない(Briley M. et al., Int. Clin. Psychopharmac.,
1996, 11:10-14)。ミルナシプランに関する更なる情報については、Merck Index、第12版、見出し6281に見出すことができる。
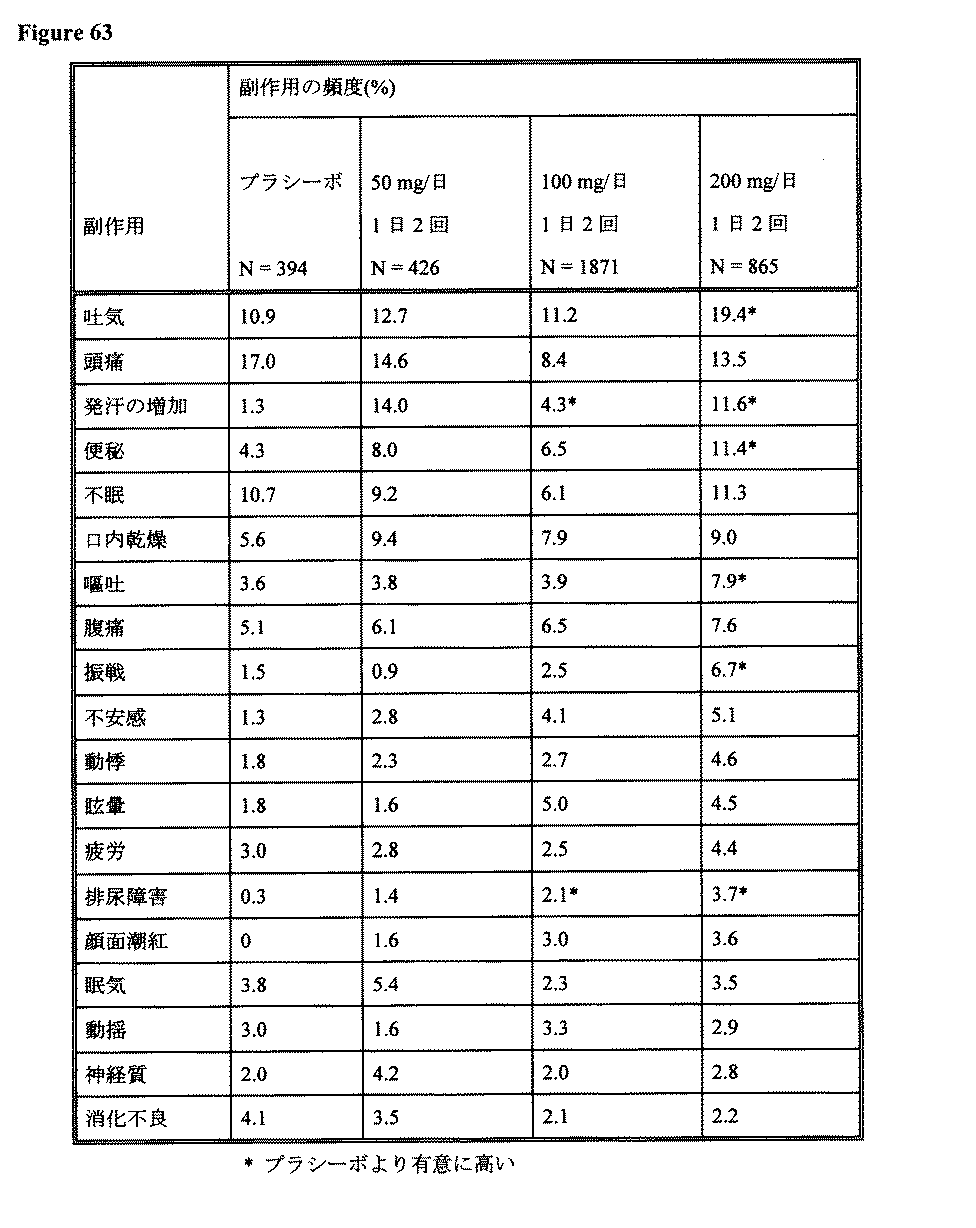
12:99-108)。二重盲検、無作為抽出で、多機関で研究を行ったところ、1日2回100mg/日のミルナシプラン投与において最も頻繁に自発的に報告された副作用は、腹痛(13%)、便秘(10%)および頭痛(9%)であった。興味深いことに、同様の研究において、ミルナシプランを200mg/日で1日2回投与した場合、副作用に関連する痛みは減少した(頭痛は8%、腹痛7%となった)が、吐気および嘔吐が著明な副作用として、患者の7%において認められた(Guelfi J.D., 1998, Int. Clin. Psychopharm.,
13:121-128)。219例の高齢の鬱病患者を対象とした二重盲検による比較研究においては、TCAイミプラミンレシピエントよりも、ミルナシプランレシピエントにおいてより頻繁に報告された唯一の副作用は吐気であった。患者に対して、1日2回8週間、ミルナシプランまたはイミプラミンのいずれかを75〜100mg/日投与した(Tignol J. et al., 1998, Acta Psychiatrica
Scandinavica, 97:157-165)。ミルナシプランを10例の患者に静脈投与したとき、5例が吐気を報告したことが観察された。吐気は、主にミルナシプラン血漿濃度がピークに達したときに報告された(Caron J. et al., 1993, Eur. Neuropsychopharmacol.,
3:493-500)。本研究は、吐気がミルナシプラン血漿濃度と直接的に関連していることを明らかに証明した。さらに、本研究においては、薬物は静脈投与されるので、吐気は中枢作用的に媒介された副作用であり得ることが示唆されている。他の研究からのデータによれば、ミルナシプランは、胃の刺激によって局所的に媒介された吐気もまた誘発し得る可能性を示唆している(血漿濃度がピークに達する前にさえ、吐気が急速に発症することが観察された)。
96:497-504)。ミルナシプラン副作用を減少させるために長期的用量増加(4週間)を行った線維筋痛症の最近の臨床試験において、患者から報告された最も一般的な用量関連性の副作用は吐気であった(Cypress Bioscience Inc., Cypress Bioscience Inc.
Announces Final Results of Milnacipran Phase II Clinical Trial in Fibromyalgia、Media
Release, March 21, 2003)。
Psychopharmacology., 5:49-56)。毎日100〜250mgのミルナシプラン投与計画が、線維筋痛症治療に関して最近報告された(米国特許第6,602,911号)。用量関連性の緊急の副作用および必要な用量に到達するために長期間滴定する必要性があることから、現在入手可能な製剤を用いた用量の範囲の上限に到達することは、非常に難しいであろう。
37, 4519-4522; Deprez et al. Eur. J. Drug Metab. Pharmacokinet. 1998,
23, 166-171参照。さらに、ミルナシプランの(−)−レブロ(levro)エナンチオマー(F2696、(−)−1R、2S−ミルナシプラン)は、はるかに力価が低い。id.参照。
便宜上、本明細書、実施例、および添付の請求の範囲において採用した特定の用語をここに集めた。
McGraw Hill Book Company, New York, (1977年版) pp. 251-259に記載されている。Hammett定数値は、通常、電子供与基(NH2ではσ[P]=−0.66)では負の値、電子吸引基(ニトロ基ではσ[P]=0.78)では正の値となる。σ[P]は、パラ置換を意味する。電子吸引基の例としては、ニトロ、アクリル、ホルミル、アルキルスルホニル、アリールスルホニル、トリフルオロメチル、シアノ、クロリドなどが挙げられる。電子供与基の例としては、アミノ、メトキシなどが挙げられる。
of Organic Chemistryの各版の第1刊に記載されている。このリストは典型的に、Standard List of Abbreviationsというタイトルの表中に示されている。前記リストに含まれている略語、および通常の技量を有する有機化学者によって用いられている略語の全てを本明細書において引用によって援用する。
Wiley: New York, 1991)。
4−メトキシベンジルシアニドとエナンチオマーとして純粋なエピクロルヒドリン(図1)(双方とも市販されており入手可能)の縮合反応によって、対応するラクトンCS1590およびCS1591を十分な収率で取得した。引き続いて、ブチルリチウムおよびジエチルアミンから生成されたリチウムジエチルアミドの存在下でラクトンを切断し、CS1608と対応するエナンチオマーをそれぞれ供給した。1級アルコールCS1608およびCS1609のアジドCS1628およびC1648への変換は、対応するメシラートのin situ生成、それに続く、アジ化ナトリウムとの求核性の置き換えによって、ポット処理において達成された。このプロトコルの後、所望のアジドが収率36〜40%で得られた。続いて、ホウ素トリブロミドの存在下で、−30℃で48時間、保護基の除去を行い、脱保護されたフェノールCS1649およびCS1658を収率66%で得た。標準的な反応条件下でのCS1649およびCS1658のアジド部分の最終的な還元によって、所望の標的化合物CS1665およびCS1710が得られた。ジオキサン中で塩酸を使用して、塩酸塩の調製を行い、続いて溶媒を除去した。
個別のエナンチオマーの別の単離手順は、エナンチオマーのラセミ混合物からの溶解によるものである。今日、陽イオン性薬物のキャピラリー電気泳動法によるキラル分離は、負に帯電したシクロデキストリン(CD)を、流れる緩衝液に付加することによって行われる。一方、陰イオン性または中性薬物の分離は、二重性CD系(中性CDおよび帯電性CDの混合物)の使用が必要である。いくつかの塩基性薬物(イダゾキサン、エファロキサン(efaroxan)、ミルナシプラン)のキラル分離は、硫酸塩d−β-CD(S−β−CD)およびヒドロキシプロピル−γ−CD(HP−γ−CD)との混合物を用いて研究されてきた。下記のパラメータ(中性CDの性質および濃度、S−β−CDの濃度)が、多くの分離ファクター(電気泳動移動度、選択性、効率性、非対称ファクター、分解能)に及ぼす影響は、第2の移動エナンチオマーの対称性を向上させるために二重CD系が塩基性薬物のキラル分離に有用であることを示している。実際、中性のCDは、移動回転による電気移動の範囲を減少させる。最終的に、0.5mg/mLのS−β-CDと5mg/mLのHP−γ−CD二重系がイダゾキサンエファロキサンおよびミルナシプランエナンチオマーを9分未満でキラル分離させた。Grard, S. et al. Electrophoresis 2000,
21, 3028-3034を参照されたい。
CS1814、CS1713、CS1714および種々の参照化合物の生物学的試験の結果を図32〜62に示す。CS1814(バイアル番号1)、CS1713(バイアル番号2)およびCS1714(バイアル番号3)を種々の放射性リガンド結合アッセイでCYP450 3A4を10μMの開始濃度での阻害について評価した。図59および60に示したように、セロトニン輸送体結合部位(バイアル番号1、Ki=6.73nM、バイアル番号2、Ki=3.88nM、バイアル番号3、Ki=8.15nM)およびノルエピネフリン輸送体結合部位(バイアル番号1、Ki=0.218μM、バイアル番号2、Ki=0.112μM、バイアル番号3、Ki=1.68μM)からの放射性リガンドのズレに関して、有意な活性(≧50%)が認められた。
いくつかの態様において、本発明の化合物は、下記式Aで表された単離された化合物である。
Rは、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、へテロアリール、アリールアルキル、ホルミル、アシル、シリル、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R1は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R2は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R3は、それぞれ個別に、H,アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R4は、1〜4回存在する、または存在しない、
R4は、もし存在すれば、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R80は、それぞれ個別に、アリール、シクロアルキル,シクロアルケニル、ヘテロアシル、またはポリシクリル部分を表し、
mは、それぞれ個別に、0〜8の間の整数であり、そして
化合物は、単一のエナンチオマーであり;または、
それらの薬学的に許容される塩もしくはプロドラッグである。
Rは、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、へテロアリール、アリールアルキル、ホルミル、アシル、シリル、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R1は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R2は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R3は、それぞれ個別に、H,アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R4は、1〜4回存在する、または存在しない、
R4は、もし存在すれば、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R80は、それぞれ個別に、アリール、シクロアルキル,シクロアルケニル、ヘテロアシル、またはポリシクリル部分を表し、
mは、それぞれ個別に、0〜8の間の整数であり、そして
化合物は、単一のエナンチオマーであり;または、
それらの薬学的に許容される塩もしくはプロドラッグである。
1−(4−メトキシ−フェニル9−3−オキサ−ビシクロ[3.1.0]ヘキサン−2−オン(CS1590)、1R、2S 1−(4−メトキシ−フェニル9−3−オキサ−ビシクロ[3.1.0]ヘキサン−2−オン(CS1591)、1S、2R 2−ヒドロキシメチル−1−(4−メトキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1608)、1R、2S 2−ヒドロキシメチル−1−(4−メトキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1609)、1S、2R 2−アジドメチル−1−(4−メトキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1628)、1R、2S 2−アジドメチル−1−(4−メトキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1648)、1S、2R 2−アジドメチル−1−(4−ヒドロキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1649)、1R、2S 2−アジドメチル−1−(4−ヒドロキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1658)、1S、2R 2−アミノメチル−1−(4−ヒドロキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1665)、1R、2S 2−アミノメチル−1−(4−ヒドロキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1710)、およびラセミ型2−アミノメチル−1−(4−ヒドロキシ−フェニル)−シクロプロパンカルボン酸ジエチルアミド(CS1814)からなる群から選択される。
最終的な脱保護段階の前に作製することができる、本発明の化合物のいくつかの保護された誘導体は、そのような薬理活性を有していないかもしれないにもかかわらず、非経口的もしくは経口的に投与され、その後体内で代謝され、薬理学的に活性のある本発明の化合物となり得るということを当該技術分野の当業者は理解するであろう。そのような誘導体は、したがって、「プロドラッグ」と記載される。さらに、本発明のいくつかの化合物は、本発明の他の化合物のプロドラッグとして機能し得る。批判的に、本発明の化合物のプロドラッグの全ては、本発明の範囲に含まれる。本明細書においてこれまでに記載したような新規な中間物質、および本発明の他の化合物を製造するためのそれらの使用もまた本発明の一部をなしている。
別の局面において、本発明は、1以上の上記薬学的に許容される単体(添加物)および/または希釈剤とともに製剤される上記化合物の治療的有効量を含む薬学的に許容される組成物を提供する。下記に詳細に示すように、本発明の薬学的組成物は、固体もしくは液体剤型で投与するために具体的に製剤することができる。下記に適用されるものを含む。(1)経口投与、例えば、飲薬(水性もしくは非水性溶液、または懸濁液)、錠剤(例えば、頬側、舌下および全身吸収を目標としたもの)、ボーラス、粉末、顆粒、舌に投与するためのパスタ剤;(2)非経口投与、例えば、皮下、筋内、静脈内もしくは硬膜外注射、例えば、無菌溶液もしくは懸濁液、または持続性放出製剤;(3)局所投与、例えば、クリーム、軟膏、または徐放パッチ、または皮膚に適用されるスプレー;(4)膣内もしくは腸内投与、例えば、ペッサリー、クリームもしくは泡剤;(5)舌下投与;(6)眼内投与;(7)経皮投与;または(8)経鼻投与。
pharmacy”(第20版、Lippincott Williams
& Wilkins, Baltimore, MD, 2000)に記載のようにして調製することができる。分散系は、典型的に2つの種類のデバイス、リザーバーおよび基質からなり、当該技術分野において公知であり、記載がある。基質デバイスは、通常、薬物を、緩やかにポリマー担体を錠剤形態に溶解させながら圧縮することによって調製することができる。基質デバイスを調製するときに用いられる物質の3つの主なタイプは、不溶性プラスチック、親水性ポリマー、および脂質化合物である。プラスチック材料としては、限定されないが、メチルアクリレート−メチルメタクリレート、ポリビニルクロリド、およびポリエチレンがある。親水性ポリマーとしては、限定されないが、メチルセルロース、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ナトリウムカルボキシメチルセルロース、およびカルボポール(carbopol)934、酸化ポリスチレンが含まれる。脂質化合物には、限定されないが、カルナバワックスおよびグリセロールトリステレート(glyceryl tristearate)などの種々のワックスが含まれる。
Freedman and CO., San Francisco, U.S.A., 1969 or "Livestock Feeds and
Feeding" O and B books, Corvallis, Ore., U.S.A., 1977)
本発明の化合物は、1以上の治療薬と組み合わせて患者に投与してもよい。補助薬または薬物を主要な薬物と混合して、単一の錠剤、丸薬、カプセル、または非経口投与用の溶液などに製剤してもよい。あるいは、主要な薬物と補助薬を別個の組成物、例えば、別個の錠剤もしくは溶液を介して投与してもよい。主要な薬物は、補助薬と同時に投与してもよく、または主要な薬物は、完全な薬物とともに断続的に投与してもよい。補助薬の用量は、通常、治療を受ける患者の健康状態、所望の治療の範囲、同時に行う治療の性質および種類、もしあれば、治療の頻度および所望の効果の性質を含む多くの要因に依存するであろう。一般的に、補助薬の用量範囲は、体重1kg当たり1日0.001〜約250mg/kgであることが多い。体重約70kgの正常の成人では、典型的に体重1kg当たり1日約0.1〜約25mg/kgが好ましい。しかしながら、この一般的な用量範囲は、治療を受ける被験体の年齢および体重、意図されている投与経路、投与される特定の薬物などにしたがって変化させる必要がある。2以上の異なる活性剤が治療と組み合わせて使用されているので、各薬物の力価およびそれらを一緒に用いることによって達成される相互作用を考慮しなければならない。しかしながら、用量の範囲の決定および特定の哺乳動物にとっての最適な用量も十分に、本開示の利点を持つ、当該技術分野の当業者の能力の範囲内である。いくつかの態様において、本発明の化合物は、鎮痛剤、抗炎症剤、解熱剤、抗抑鬱剤、抗てんかん薬、抗ヒスタミン剤、抗片頭痛薬、抗ムスカリン作動薬、抗不安剤、鎮静剤、催眠薬、抗精神病薬、気管支拡張剤、抗喘息薬、心臓血管薬、コルチコステロイド、ドーパミン作動薬、電解液、胃腸薬、筋弛緩剤、栄養剤、ビタミン剤、副交感神経作動薬、刺激剤、抗ナルコレプシー薬、および食思減退薬などの他の化合物と組み合わせて投与することができる。
対象となる化合物は、薬学的、アグロケミカルもしくは他の生物学的もしくは医学に関連する活性、または材料に関連する質のスクリーニングを行うためのコンビナトリアルライブラリーを利用する、コンビナトリアルケミストリーの方法を用いて容易に調製される。本発明の目的のためのコンビナトリアルライブラリーは、所望の特性についてスクリーニングすることができる、化学的に関連する化合物の混合物であり、前記ライブラリーは、溶液の形状でもよいし、または固体支持体に共有結合されていてもよい。多くの関連する化合物の、単一の反応における調製は、行う必要のある多くのスクリーニングプロセスを大いに減らし、単純化する。適切な生物学的、薬学的、農薬学的または物理学的特性のスクリーニングは、従来の方法によって行ってもよい。
116:2661: Kerr et al. (1993) JACS
115:252;PCT国際公開公報 WO92/10092号、WO93/09668号およびWO91/07087号;Lerner et alらのPCT国際公開公報 WO93/20242号)を参照されたい。したがって、約16〜1,000,000以上のダイバーソマーを持つ、種々のライブラリーを合成し、特別の活性もしくは特性についてスクリーニングすることができる。
94/08051号に記載の望ましい技法に適用される対象となる反応、例えば、加水分解可能または光分解可能基によるポリマービーズに結合させる、例えば、基質の部分基質の1つに位置づけるといった反応を用いて合成することができる。Stillらの技法によれば、ライブラリーは、1セットのビーズ上に合成される。各ビーズは、ビーズ上の特別のダイバーソマーを同定する1セットのタグを含んでいる。酵素阻害剤を発見するために特に好適なある態様において、該ビーズは、浸透膜の表面に分散させることができ、ダイバーソマーは、ビーズリンカーの溶解によってビーズから放出され得る。各ビーズからのダイバーソマーは、膜を超えてアッセイ帯域まで分散し、酵素アッセイと相互作用することになる。多くのコンビナトリアル方法の詳細な説明を以下に示す。
コンビナトリアルケミストリーの分野において増大している傾向は、例えば、化合物のサブフェムトモル量の特徴を調べる、および直接的にコンビナトリアルライブラリーから選択された化合物の化学的構造を決定するために用いることができるマススペクトロメトリー(MS)などの技法の開発である。例えば、ライブラリーが、不溶性支持体基質上に設けられている場合、化合物の別個のポピュレーションをまず最初に支持体から放出させ、MSによって特徴付けることができる。別の態様において、MSサンプル調製技法の一部として、そのようなMS技法をMALDIとして使用して、特に、当初から化合物を基質につなぐために、不安定な結合が用いられている場合に、基質から化合物を放出させることができる。例えば、ライブラリーから選択されたビーズは、MALDIステップにおいて、基質からダイバーソマーを放出し、MS分析するために該ダイバーソマーをイオン化するために、照射することができる。
当該方法のライブラリーは、マルチピンライブラリーフォーマットを用いることができる。簡潔に言えば、Geysenと共同研究者(Geysen et al. (1984) PNAS
81:3998-4002)は、マイクロタイタープレートフォーマットに配列されたポリアクリル酸グレートの(grated)ポリエチレンピン上でのパラレル合成によって化合物ライブラリーを作製する方法を導入した。Geysenの技法を使用して、マルチピン法を用いて、1週間に数千もの化合物を合成およびスクリーニングすることができる。そして、結合された化合物は、多くのアッセイにおいて再利用することができる。適切なリンカー部分をピンに付加して用い、純度の評価およびさらなる評価を行った後、化合物を支持体から切断してもよい(Bray et al. (1990) Tetrahedron Lett 31:5811-5814; Valerio et al. (1991) Anal Biochem 197:168-177; Bray et al. (1991) Tetrahedron Lett 32:6163-6166参照)。
さらに別の態様において、化合物の多様なライブラリーは、分割−結合−組換え法を用いて、1セットのビーズ上に設けることができる(例えば、Houghten (1985) PNAS
82:5131-5135; and 米国特許第4,631,211号;第5,440,016号;第5,480,971号参照)。簡潔に述べれば、名前が示唆しているように、縮退がライブラリーに導入される各合成ステップにおいては、ビーズをライブラリーの特定の位置に付加されるべき異なる置換基の数と等しい別個のグループに分割して入れ、別個の反応において異なる置換基を結合し、そして次の反復のために1つのプールに組み込んだ。
82:5131-5135)。バッグを適切な反応溶液に置くことによって、置換基を化合物を持つ樹脂に結合する。一方、全てに共通のステップ、例えば、樹脂洗浄や脱保護は、1つの反応容器において同時に行われる。合成が終わったとき、各バッグは、単一の化合物を含有している。
合成基質上のその位置によって化合物の同定がなされるコンビナトリアル合成のスキームを空間アドレス可能合成と呼ぶ。ある態様において、コンビナトリアルプロセスは、化学的試薬を、固体支持体の特異的な位置への添加をコントロールすることによって行うことができる(Dower et al. (1991) Annu Rep Med Chem 26:271-280; Fodor, S.P.A. (1991) Science
251:767; Pirrung et al. (1992) 米国特許第5,143,854号; Jacobs et al. (1994) Trends Biotechnol 12:19-26)。フォトリソグラフィーの空間分解能によって小型化が可能になる。この技法は、感光性保護基を用いた保護/脱保護反応によって行うことができる。
37:1233-1251に示されている。合成基質は、感光性ニトロベラトリルオキシカルボニル(NVOC)保護アミノリンカーまたは他の感光性リンカーの共有結合的付着を介して結合するために調製される。結合のための合成支持体の特異的な領域を活性化するために光が選択的に用いられる。感光保護基を光によって除去(脱保護)した結果、選択された領域が活性化される。活性化の後、それぞれ感光性保護基をアミノ末端に有している第1のアミノ酸アナログのセットを表面全体に曝露する。先行するステップにおいて、光によってアドレス指定された領域にのみ結合が生じる。反応を止め、プレートを洗浄し、基質を第2のマスクを介して再び照射し、第2の保護構築ブロックを用いた反応の異なる領域を活性化する。マスクのパターンおよび反応物の配列によって生成物およびそれらの位置を定義する。このプロセスは、フォトリソグラフィー技法を用いているので、合成され得る化合物の数は、適切な分解能によってアドレス指定され得る合成部位の数によってのみ限定される。各化合物の位置は、正確に知られている。したがって、その他の分子との相互作用を直接的に評価することができる。
さらに別の態様において、対象となる方法は、コードされたタグシステムを備えた化合物ライブラリーを使用する。近年、所与のビーズの反応ステップを独自にコードするタグ用いた化学的指標系および推測でそれが持つ構造を採用して、コンビナトリアルライブラリーからの活性化合物の同定が向上している。概念上、このアプローチは、活性が発現したペプチドに由来するファージディスプレイライブラリーを模倣しているが、活性ペプチドの構造は、対応するゲノムDNA配列から推論される。合成コンビナトリアルライブラリーの第1のコード化は、コードとしてDNAを採用した。配列可能なバイオ−オリゴマー(例えば、オリゴヌクレオチドおよびペプチド)、および更なる非配列可能タグを用いたバイナリーコード化を含む、他の形態の種々のコード化が報告されている。
コンビナトリアル合成ライブラリーをコードするオリゴヌクレオチドを用いる原則は、1992年に記載されている(Brenner et al. (1992) PNAS 89:5381-5383)。そのようなライブラリーの例は、翌年にも報告されている(Needles et al. (1993) PNAS
90:10700-10704)。各々、特異的なジヌクレオチド(それぞれTA、TC、CT、AT、TT、CAおよびAC)によってコードされているArg、Gln、Phe、Lys、Val、d−ValおよびThr(3文字アミノ酸コード)の全ての組み合わせからなる、名目上77(=823,543)個のペプチドのコンビナトリアルライブラリーを、固体支持体上で一連のペプチドおよびオリゴヌクレオチド合成を交互に行うことによって調製した。本研究において、ビーズ上のアミン結合機能性は、ビーズを、オリゴヌクレオチド合成のための保護OH基およびペプチド合成のための保護NH2基(ここでは、比率は1:20)を生成する試薬を用いて同時にプレインキュベートし、ペプチドまたはオリゴヌクレオチド合成へ特異的に分化させた。完了時、タグはそれぞれ69量体からなり、そのうち14ユニットはコードを有していた。ビーズ結合ライブラリーを、蛍光標識した抗体、および強く蛍光発光した結合抗体を含有するビーズを蛍光活性化された細胞分類によって回収した(FACS)。DNAタグは、PCRによって増幅し、配列させ、予測されるペプチドを合成した。そのような技法を行った後、当該方法で使用するために化合物ライブラリーを誘導することができ、タグのオリゴヌクレオチド配列は、特定のビーズが行う連続的なコンビナトリアル反応を同定し、したがって、ビーズ上で化合物を同定する。
試験化合物ライブラリーをコードするための別の形態は、バイナリーコードとして使用される一連の配列可能な電気泳動的な分子のタグ付けを採用する(Ohlmeyer et al. (1993) PNAS
90:10922-10926)。タグの例としては、トリメチルシリルエーテルとして、電子捕獲型ガスクロマトグラフィー(ECGC)によって、フェムトモル量未満で検出可能なハロ芳香族アルキルエーテルがあげられる。アルキル鎖の長さ、ならびに芳香族ハライド置換基の性質および位置を変化させることで、原則的に240(例えば、1012まで)の異なる分子をコードすることができる、少なくとも40のそのようなタグの合成が可能となる。最初の報告(Ohlmeyer et al., 上掲)においては、該タグは、ペプチドライブラリーの約1%の入手可能なアミン基へ、光分裂可能なo−ニトロベンジルリンカーを介して結合している。このアプローチは、ペプチド様もしくは他のアミン含有分子を調製する場合に便利である。しかしながら、本質的にあらゆるコンビナトリアルライブラリーをコードすることが可能な、より万能な系が開発された。ここで、化合物は、光分裂可能なリンカーを解して固体支持体に付着し、タグは、カテコールエーテルリンカーに、カルベンのビーズ基質への挿入を介して結合することになる(Nestler et al. (1994) J Org Chem 59:4723-4724)。この直交な付着法によって、溶液中のアッセイのためのライブラリーメンバーの選択的付着および、続く、タグセットの酸性の分解後のECGCによるデーコーディングを可能とする。
薬物開発プロセスにおいて、潜在力のある治療薬または薬物候補物質は、使用するには、安全性と有効性の両方が実証されなければならない。薬物開発プロセスにおいて、潜在力のある薬物候補物質について、安全性を実証するための毒性学的評価を行う。
Laboratory Manual(Cold Spring Harbor Press, Cold Spring Harbor, N.Y.)(1989); Nucleic
Acid Hybridization, A Practical Approach (Hames, B. D., and Higgins, S. J. eds, IRL Press,
Oxford)(1985); WO 95/21944; Chalifour, et al., Anal. Biochem. (1994) 216:
299-304; Nguyen et al., Genomics (1995) 29: 207-216; Pietu et al., Genome Res. (1996) 6: 492-503;
and Zhao et al., Gene (1995) 166: 207-213に記載されている。
候補化学物質を、飽和点またはそれに近い点で水に溶解する。この保存溶液を段階希釈して、ハエのインスタント培地を再水和する(Fisher Scientific)。特に、ある毒性アッセイは、純粋な化学物質の保存溶液で再水和したハエのインスタント培地を含む。一方、別のアッセイは、化学物質の10%溶液(水中)で再水和する。このフォーマットは、それぞれの被検化学物質について4〜5ログ用量(log dose)範囲でデータを作成するために使用することができるであろう。
CS1590およびCS1591の合成
撹拌棒、サーモメーターおよびガスアダプターを備えた200mLの三口丸底フラスコに、4−メトキシフェニルアセトニトリル(9.38g、63.76mmol)およびベンゼン(70mL)を充填した。その反応混合物を0℃に冷却させ、ナトリウムアミド(4.97g、127.5mmol)を加え、さらに2時間この温度で撹拌した。この期間の後、(R)−エピクロルヒドリン(5.9g、63.76mmol)を添加し、得られた反応混合物を一晩撹拌し、その溶媒を減圧下で還元し、残基をエタノール(50mL)および水溶性水酸化ナトリウム(1mol/L、40mL)中に溶解した。次いでその溶液を加熱し、一晩還流させ、濃縮した塩酸を添加し、pH=1に調節した。その水相を第3ブチルメチルエーテル(200mL)および酢酸エチル(200m1)を用いて抽出した。有機相を結合させ、飽和塩化ナトリウムで洗浄し、乾燥し(MgSO4)、溶媒を減圧下で還元し、粗CS1590を得た。それを、溶離剤として、酢酸エチル/ジクロロメタン(1:4)を用いたシリカゲル上で、カラムクロマトグラフィーによって精製した。所望の生成物を含有する画分を結合させ、減圧下で還元し、CS1590(5.42g、41.7%)をオフホワイトの固体として得た。
CS1608およびCS1609の合成
撹拌棒、サーモメーターおよびガスアダプターを備えた200mLの三口丸底フラスコに、n−ブチルリチウム(1.6mol/L、29.8mL、47.7mmol)を充填し、0℃に冷却し、エチルアミン(3.49g、47.7mmol)を加えた。その溶液を20分間撹拌し、−78℃に冷却させ、CS1590(6.08g、29.8mmol)のテトラヒドロフラン溶液(50mL)を添加した。その反応混合物を一晩室温に加温し、その後その反応混合物をクエンチングし、塩化アンモニウム(200ml)の飽和水溶液に入れ、次いで酢酸エチルで抽出した。有機相を分離し、乾燥し(MgSO4)、溶媒を減圧下で還元し、粗CS1608(8.19g、98%)を得た。それをさらなる精製を行わずに次のステップに用いた。
CS1628およびCS1648の合成
撹拌棒、サーモメーターおよびガスアダプターを備えた200mLの三口丸底フラスコに、CS1608(5.6g、20.19mmol)およびN,N−ジメチルホルムアミド(20mL)を充填し、0℃に冷却し、アジ化ナトリウム(5.2g、80.76mmol)、トリエチルアミン(10.2g、100.95mmol)およびメタンスルホクロリド(4.6g、60.57mmol)を添加した。その懸濁液を24時間撹拌し、水(200ml−)中に入れてクエンチングし、酢酸エチル(2×200mL)で抽出し、乾燥し(MgSO4)、その溶媒を減圧下で還元し、粗CS1628を得た。それを、溶離剤としてヘプタン/酢酸エチル(5:1)を用いたシリカゲル上で精製し、CS1628(2.2g、36%)をオフホワイトの固体として得た。
CS1649およびCS1658の合成
撹拌棒、サーモメーターおよびガスアダプターを備えた50mLの三口丸底フラスコに、CS1628(2.2g、7.27mmol)およびジクロロメタン(200mL)を充填し、−35℃に冷却し、三臭化ホウ素のジクロロメタン溶液(1.0mol/L、21.8mL、21.8mmol)を加えた。その反応混合物を48時間、−28℃で保ち、−40℃に冷却し、メタノールを添加した。得られた混合物を水(200ml)中に注ぎ、酢酸エチル(2×200mL)で抽出し、乾燥し(MgSO4)、その溶媒を減圧下で還元し、粗CS1649を得た。それを、溶離剤としてヘプタン/酢酸エチル(2:1)を用いたシリカゲル上でカラムクロマトグラフィーにより精製し、CS1649(1.39g、66.5%)をオフホワイトの固体として得た。
CS1665およびCS1710の合成
200mLの水素化ボトルに、CS1649(1.1g、3.81mmol)、メタノール(50mL−)および触媒量のPd/Cを充填した。完全な変換が観察されるまで、その反応物に1バールの水素圧をかけ、その反応混合物をセライトのパッドを介して濾過し、減圧下で溶媒を除去し、粗CS1655を得た。それを、溶離剤として、ジクロロメタン/メタノール/トリエチルアミン(10:0.5:0.25)を用いたシリカゲル上でカラムクロマトグラフィーによって精製し、CS1665(0.80g、80%)をオフホワイトの固体として得た。
CS1713およびCS1714の合成
10mLの丸底フラスコに、CS1665(0.51g、1.71mmol)およびジオキサン(5mol/L、10mL)を充填した。混合物を室温で1時間撹拌し、減圧下で溶媒を除去し、C1713(0.43g、84%)をオフホワイトの固体として得た。
CS1814の調製
マグネティック撹拌棒を備えた10mLのフラスコに、CS1665/2(120mg、0.46mmol)、CS1710/1(120mg、0.46mmol)および塩酸の入ったジオキサン(5mol/L、5ml)を充填した。その懸濁液を1時間撹拌し、減圧下で還元し、残基を再び塩酸の入ったジオキサン(5mol/L、1ml)に取り上げた。その懸濁液をさらに1時間撹拌し、減圧下で還元し、CS1814(240mg量)をオフホワイトの固体として得た。その固体をメタノール(10mL、均質溶液)に溶解し、20mLの丸底フラスコに移し、そのフラスコをさらに5mLのメタノールで洗浄し、上記溶液と混合し(合計量約15mL、均質溶液)、減圧下で還元し、CS1814(240mg量)をオフホワイトの固体として得た。その固体を高真空化で乾燥させた。この物質の50mgを取り出し、メタノール(10ml−)中に溶解させ、続いて、旋光性を測定した。後にその溶液を20mLのフラスコ(均質溶液)に移し、減圧下で溶媒を除去した。
CS1814および参照化合物の生物学的試験
CS1814および種々の参照化合物の生物学的試験の結果を図32〜40および59に示す。図59のデータは、CS1814は、ノルエピネフリン輸送体の阻害についてはIC50=0.22μM、セロトニン輸送体の阻害についてはIC50=12.7nMであることを示している。CS1814の結合定数は、ノルエピネフリン輸送体についてはKi=0.218μM、セロトニン輸送体については、Ki=6.73nMである。
Pharma Servicesにおいて実験によって取得)を用いて、ChengとPrusoffの式(Cheng, Y., Prusoff, W.H., Biochem. Pharmacol.
22:3099−3108, 1973)を使って計算したものである。示されている場合、競合する結合曲線の傾斜を定義するHill係数(nH)は、Data Analysis Toolbox(登録商標)を用いて計算した。Hill係数が1.0と有意に異なる場合、結合のズレは、単一の結合部位を持つ質量作用の法則に従わないことを示唆するかもしれない。IC50、Kiおよび/またはnHのデータが、Standard Error of the Mean (SEM)なしで示されている場合、データは、量的に不十分であり、示された値(Ki、IC50、nH)の解釈には注意が必要である。
118050 CYP450、1A2
起源:ヒト組換えSf9昆虫細胞
基質剤:5μMの3−シアノ−7−エトキシクマリン
賦形剤:0.1%DMSO
プレインキュベーション時間/温度:なし
インキュベーション時間/温度:30分/37℃
インキュベーション緩衝液:75mMのリン酸カリウム緩衝液、pH7.5
定量方法:3−シアノ−7−ヒドロキシクマリンの分光蛍光法による定量化
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えSf9昆虫細胞
基質剤:25μMの3−シアノ−7−エトキシクマリン
賦形剤:0.1%DMSO
プレインキュベーション時間/温度:なし
インキュベーション時間/温度:45分/37℃
インキュベーション緩衝液:75mMのリン酸カリウム緩衝液、pH7.5
定量方法:3−シアノ−7−ヒドロキシクマリンの分光蛍光法による定量化
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えSf9昆虫細胞
基質剤:25μMの3−シアノ−7−エトキシクマリン
賦形剤:0.1%DMSO
プレインキュベーション時間/温度:なし
インキュベーション時間/温度:45分/37℃
インキュベーション緩衝液:75mMのリン酸カリウム緩衝液、pH7.5
定量方法:3−シアノ−7−ヒドロキシクマリンの分光蛍光法による定量化
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えSf9昆虫細胞
基質剤:50μMの3−シアノ−7−エトキシクマリン
賦形剤:0.1%DMSO
プレインキュベーション時間/温度:なし
インキュベーション時間/温度:45分/37℃
インキュベーション緩衝液:75mMのリン酸カリウム緩衝液、pH7.5
定量方法:3−シアノ−7−ヒドロキシクマリンの分光蛍光法による定量化
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えSf9昆虫細胞
基質剤:50μMの7−ベンジルオキシ−4−(トリフルオロメチル)−クマリン
賦形剤:0.1%DMSO
プレインキュベーション時間/温度:なし
インキュベーション時間/温度:30分/37℃
インキュベーション緩衝液:75mMのリン酸カリウム緩衝液、pH7.5
定量方法:7−ヒドロキシ−4−(トリフルオロメチル)−クマリンの分光蛍光法による定量化
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:1nMの[3H]DPCPX
賦形剤:0.1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:20mMのHEPES、pH7.4、10mMのMgCl2、100mMのNaCl
非特異的リガンド:100μM R(−)−PIA
Kd:1.4nM*
Bmax:2.7pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えHEK−293細胞
リガンド:0.05μMの[3H]CG5−21680
賦形剤:0.1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、10mMのMgCl2、1mMのEDTA、2U/mLのアデノシンデアミナーゼ
非特異的リガンド:50μMのNECA
Kd:0.064μM*
Bmax:7pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット顎下腺
リガンド:0.25nMの[3H]プラゾシン
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、0.5mMのEDTA、pH7.4
非特異的リガンド:10μMのフェントラミン
Kd:0.17nM*
Bmax:0.18pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット肝臓
リガンド:0.25nMの[3H]プラゾシン
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、0.5mMのEDTA、pH7.4
非特異的リガンド:10μMのフェントラミン
Kd:0.31nM*
Bmax:0.18pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えHEK−293細胞
リガンド:0.6nMの[3H]プラゾシン
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸
非特異的リガンド:10μMのフェントラミン
Kd:0.58nM*
Bmax:0.17pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換え昆虫Sf9細胞
リガンド:1nMの[3H]MK−912
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:75mMのトリス塩酸、pH7.4、12.5mMのMgCl2、2mMのEDTA
非特異的リガンド:10μMのWB−4101
Kd:0.6nM*
Bmax:4.6pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO−K1細胞
リガンド:2.5nMの[3H]ラウオイシン(Rauwoiscine)
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、1mMのEDTA、12.5mMのMgCl2、pH7.4、0.2%BSA、25℃
非特異的リガンド:10μMのプラゾシン
Kd:2.1nM*
Bmax:2.1pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えRex16細胞
リガンド:0.03nMの[125I]シアノピンドロール
賦形剤:0.1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのトリス塩酸、5mMのEDTA、1.5mMのCaCl2、120mMのNaCl、1.4mMのアスコルビン酸、10mg/LのB5A,pH7.4
非特異的リガンド:100μMの5(−)−プロプラノール
Kd:0.041nM*
Bmax:0.072pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO−NBR1細胞
リガンド:0.2nMの[3H]CGP−12177
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、0.5mMのEDTA、5.0mMのMgCl2、120mMのNaCl、pH7.4
非特異的リガンド:10μMの1Cl−118551
Kd:0.44nM*
Bmax:0.437pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトHs729細胞
リガンド:2.5nMの[3H](Des−Arg10)−カリジン
賦形剤:0.1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:20mMのHEPES、125mMのN−メチル−Dグルカミン、5mMのKCl、1mMの1,10−フェナントロリン、pH7.4
非特異的リガンド:10μMの(Des−Arg9−Leu8)−ブラジキニン
Kd:0.5nM*
Bmax:0.059pmole/mgのタンパク質*
特異的結合:70%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組み換えCHO−K1細胞
リガンド:0.2nMの[3H]ブラジキニン
賦形剤:0.1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:24mMのTES−NH4OH、pH6.8、1mMの1,10−フェナントロリン、0.3%BSA
非特異的リガンド:5μMのブラジキニン
Kd:0.29nM*
Bmax:2pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳
リガンド:2nMの[3H]ジルチアゼム
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:50mMのトリス塩酸、0.1%BSA、pH7.4、25℃
非特異的リガンド:10μMのジルチアゼム
Kd:0.016μM*
Bmax:0.21pmole/mgのタンパク質*
特異的結合:73%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット大脳皮質
リガンド:0.1nMの[3H]ニトレンジピン
賦形剤:1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.7、25℃
非特異的リガンド:1μMのニトレンジピン
Kd:0.18nM*
Bmax:0.23pmole/mgのタンパク質*
特異的結合:91%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット前頭葉
リガンド:10pMの[125H]ω−コノトキシン GVIA
賦形剤:1%DMSO
インキュベーション時間/温度:30分/4℃
インキュベーション緩衝液:20mMのトリス塩酸、pH7.4、0.1%BSA
非特異的リガンド:0.1μMのω−コノトキシン GVIA
Kd:0.051nM*
Bmax:0.88pmole/mgのタンパク質*
特異的結合:96%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:1.4nMの[3H]SCH−23390
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/37℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、150mMのNaCl、1.4mMのアスコルビン酸、0.001%BSA
非特異的リガンド:10μMの(+)−ブタクラモール
Kd:1.4nM*
Bmax:0.63pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.16nMのスピペロン
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、150mMのNaCl、1.4mMのアスコルビン酸、0.001%BSA
非特異的リガンド:10μMのハロペリドール
Kd:0.08nM*
Bmax:0.48pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.7nMのスピペロン
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/37℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、150mMのNaCl、1.4mMのアスコルビン酸、0.001%BSA
非特異的リガンド:25μMのS(−)−スルピリド
Kd:0.36nM*
Bmax:1.1pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.5nMの[3H]スピペロン
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、150mMのNaCl、1.4mMのアスコルビン酸、0.001%BSA
非特異的リガンド:10μMのハロペリドール
Kd:0.27nM*
Bmax:1pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.03nMの[125I]エンドセリン−1
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/37℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、0.5mMのCaCl2、0.05%のトウィーン−20、1mg/mlのBSA
非特異的リガンド:0.1μMのエンドセリン−1
Kd:0.048nM*
Bmax:0.35pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO−K1細胞
リガンド:0.1nMの[125I]エンドセリン−1
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのHEPES、1mMのCaCl2、5mMのMgCl2、0.5%のBSA、pH7.4 (プロテアーゼなし)
非特異的リガンド:0.1μMのエンドセリン−1
Kd:0.085nM*
Bmax:4.3pmole/mgのタンパク質*
特異的結合:75%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトA431細胞
リガンド:0.05nMの[125I] 上皮成長因子(EGF)(ネズミ)
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのHEPES、138mMのNaCl、5mMのKCl、1.2mMのMgSO4、1.2mMのCaCl2、1mg/mlのBSA、pH7.7
非特異的リガンド:10nMの上皮成長因子(EGF)(ヒト)
Kd1:0.032nM*
Kd2:0.03nM*
Bmax1:1pmole/mgのタンパク質*
Bmax2:4.1pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換え昆虫Sf9細胞
リガンド:0.5nMの[3H]エストラジオール
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:10mMのトリス塩酸、pH7.5、10%グリセロール、1mMのDTT、1mg/mlのBSA
非特異的リガンド:1μMのジエチルスチルベステロール
Kd:0.2nM*
Bmax:1400pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳(マイナス、小脳)
リガンド:1nMの[3H]ムスシモール
賦形剤:1%DMSO
インキュベーション時間/温度:10分/4℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:0.1μMのムスシモール
Kd:3.8nM*
Bmax:1.8pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳(マイナス、小脳)
リガンド:1nMの[3H] フルニトラゼパム
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのNa−Kホスフェート、pH
非特異的リガンド:10μMのジアゼパム
Kd1:4.4nM*
Kd2:0.3nM*
Bmax1:1.2pmole/mgのタンパク質*
Bmax2:4.1pmole/mgのタンパク質*
特異的結合:91%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳
リガンド:0.6nMの[3H]CGP−54626
賦形剤:1%DMSO
インキュベーション時間/温度:20分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、2.5mMのCaCl2、pH7.4、25℃
非特異的リガンド:100μMのCGP−54626
Kd:2.3nM*
Bmax:1.1pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトHeLa53細胞
リガンド:6nMの[3H] デキサメタゾン
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:RPMI1640、10mMのHEPES、pH7.2
非特異的リガンド:20μMのデキサメタゾン
Kd:5nM*
Bmax:61000R/細胞*
特異的結合:75%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳(マイナス小脳)
リガンド:5nMの[3H]カイニン酸
賦形剤:1%DMSO
インキュベーション時間/温度:60分/4℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:1000μMのL−グルタメート
Kd:0.012μM*
Bmax:0.35pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット大脳皮質
リガンド:2nMの[3H]CGP−39653
賦形剤:1%DMSO
インキュベーション時間/温度:20分/4℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:1000μMのL−グルタメート
Kd:0.019μM*
Bmax:2.3pmole/mgのタンパク質*
特異的結合:70%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット大脳皮質
リガンド:0.33nMの[3H]MDL−105519
賦形剤:1%DMSO
インキュベーション時間/温度:30分/4℃
インキュベーション緩衝液:50mMのHEPES、pH7.7
非特異的リガンド:10μMのMDL−105519
Kd:6nM*
Bmax:3.7pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット大脳皮質
リガンド:4nMの[3H]TCP
賦形剤:1%DMSO
インキュベーション時間/温度:45分/25℃
インキュベーション緩衝液:10mMのトリス塩酸、pH7.7
非特異的リガンド:1μMのジゾルシピン(Dizolcipine)(MK−801)
Kd:8.4nM*
Bmax:0.78pmole/mgのタンパク質*
特異的結合:94%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO−K1細胞
リガンド:1.2nM[3H]ピリラミン
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、2mMのMgCl2、100mMのNaCl、250mMのスクロース
非特異的リガンド:1μMのピリラミン
Kd:1.1nM*
Bmax:6.7pmole/mgのタンパク質*
特異的結合:94%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO−K1細胞
リガンド:0.1nM[125I]アミノポテンチジン(Aminopotentidine)
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのKH2PO4/Na2HPO4、pH7.4
非特異的リガンド:3μMのチオチジン
Kd:0.45nM*
Bmax:6.9pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO−K1細胞
リガンド:3nM[3H]R(−)−α−メチルヒスタミン(RAMH)
賦形剤:1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、10mMのMgCl2、0.04%のBSA
非特異的リガンド:1μMのR(−)−α−メチルヒスタミン(RAMH)
Kd:2.4nM*
Bmax:4.2pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット大脳皮質
リガンド:2nMの[3H] イダゾキサン
賦形剤:1%DMSO
インキュベーション時間/温度:30分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、0.5mMのEDTA、pH7.4、25℃
非特異的リガンド:1μMのイダゾキサン
Kd:4nM*
Bmax:0.14pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:マウス3T3細胞
リガンド:10pMの[125I]インターロイキン−Iα(IL−1α)
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/37℃
インキュベーション緩衝液:RPMI1640、20mMのHEPES、0.1%アジ化ナトリウム、1%のBSA、pH7.2
非特異的リガンド:0.03μMのインターロイキン−Iα(IL−1α)
Kd:6pM*
Bmax:8.3fmole/mgのタンパク質*
特異的結合:70%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Duncan Hartley 由来モルモット肺
リガンド:0.2nMの[3H] ロイコトリエンD4(LTD4)
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、0.01%のBSA、5mMのCaCl2、5mMのMgCl2、100μg/mLのバシトラシン、1mMのベンズアミジン、0.1mMのフェニルメチルスルホニルフロリド
非特異的リガンド:0.1μMのロイコトリエンD4(LTD4)
Kd:0.2nM*
Bmax:0.24pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換え昆虫Sf9細胞
リガンド:0.29nMの[3H] メトスコポラミン
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、10mMのMgCl2、1mMのEDTA
非特異的リガンド:1μMのアトロピン
Kd:0.092nM*
Bmax:2.1pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換え昆虫Sf9細胞
リガンド:0.29nMの[3H] メトスコポラミン
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、10mMのMgCl2、1mMのEDTA
非特異的リガンド:1μMのアトロピン
Kd:0.16nM*
Bmax:4.9pmole/mgのタンパク質*
特異的結合:96%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換え昆虫Sf9細胞
リガンド:0.29nMの[3H] メトスコポラミン
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、10mMのMgCl2、1mMのEDTA
非特異的リガンド:1μMのアトロピン
Kd:0.078nM*
Bmax:3.2pmole/mgのタンパク質*
特異的結合:96%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトSK−N−MC細胞
リガンド:0.013nMの[125I]ペプチドYY
賦形剤:1%DMSO
インキュベーション時間/温度:45分/25℃
インキュベーション緩衝液:HBSS、2mg/mlのBSA、1mMのMgCl2、1mMのCaCl2
非特異的リガンド:0.1μMのニューロペプチドY(ヒト、ラット)
Kd:0.62nM*
Bmax:5800R/細胞受容体/細胞*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトKAN−TS神経芽腫細胞
リガンド:20pMの[125I]ペプチドYY
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/37℃
インキュベーション緩衝液:25mMのHEPES、2.5mMのCaCl2、1mMのMgCl2、0.1%のバシトラシン、pH7.4
非特異的リガンド:1μMのニューロペプチドY(13−36)(ブタ)
Kd:0.012nM*
Bmax:0.5pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトIMR−32細胞
リガンド:0.1nMの[125I]エピバチジン(Epibatidine)
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:300μMの(−)−ニコチン
Kd:0.22nM*
Bmax:0.46pmole/mgのタンパク質*
特異的結合:97%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.9nMのナルトリンドール
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのトリス塩酸、5mMのMgCl2、pH7.4
非特異的リガンド:10μMのナロキソン
Kd:0.49nM*
Bmax:8.6pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えHEK−293細胞
リガンド:0.6nMの[3H] シプレノルフィン
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:10μMのナロキソン
Kd:0.4nM*
Bmax:1.1pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.6nMの[3H] シプレノルフィン
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:10μMのナロキソン
Kd:0.41nM*
Bmax:3.8pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ICRマウス脳
リガンド:3nMの[3H]PDBu
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:5mMのCaCl2を含有する20mMのトリス塩酸、pH7.4、25℃
非特異的リガンド:1μMのPDBu
Kd:8.7nM*
Bmax:26pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト血小板
リガンド:0.12nMの[3H]PAF
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、100mMのKCl、5mMのEDTA、5mMのMgCl2、0.25%のBSA(w/v)
非特異的リガンド:1μMのPAF
Kd:0.13nM*
Bmax:120R/細胞*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:シリアンハムスター膵臓ベータ細胞HIT−T15
リガンド:5nMの[3H] グリベンクラミド
賦形剤:1%DMSO
インキュベーション時間/温度:2時間/25℃
インキュベーション緩衝液:50mMのMOPS、0.1mMのCaCl2、pH7.4
非特異的リガンド:1μMのグリブリド
Kd:0.64nM*
Bmax:1pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ニュージーランドアルビノラビット膀胱
リガンド:8nMの[3H]α、β−メチレン−ATP
賦形剤:1%DMSO
インキュベーション時間/温度:30分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:100μMのβ、γ−メチレン−ATP
Kd1:2.2nM*
Kd2:2.2μM*
Bmax1:2pmole/mgのタンパク質*
Bmax2:790pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳
リガンド:0.1nMの[35S]ATP−αS
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:10μMのADP−βS
Kd:0.015μM*
Bmax:16pmole/mgのタンパク質*
特異的結合:87%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:1.5nMの[3H]8−OH−DPAT
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、10mMのMgSO4、0.5mMのEDTA、0.1%のアスコルビン酸、pH7.4
非特異的リガンド:10μMのメテルゴリン
Kd:2nM*
Bmax:1.3pmole/mgのタンパク質*
特異的結合:75%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えHEK−293細胞
リガンド:0.69nMの[3H]GR−65630
賦形剤:1%DMSO
インキュベーション時間/温度:60分/25℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.5、1mMのEDTA、5mMのMgCl2
非特異的リガンド:10μMのMDL−72222
Kd:0.2nM*
Bmax:11pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒトジャーカット(jurkat)細胞
リガンド:8nMの[3H] ハロペリドール
賦形剤:1%DMSO
インキュベーション時間/温度:4時間/25℃
インキュベーション緩衝液:5mMのK2HPO4/KH2PO4緩衝液、pH7.5
非特異的リガンド:10μMのハロペリドール
Kd:5.8nM*
Bmax:0.71pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistar ラット脳
リガンド:3nMの[3H] イフェンプロジル
賦形剤:1%DMSO
インキュベーション時間/温度:60分/37℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4
非特異的リガンド:10μMのイフェンプロジル
Kd:4.8nM*
Bmax:1.3pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistar ラット脳
リガンド:2nMの[3H] サキシトキシン
賦形剤:1%DMSO
インキュベーション時間/温度:30分/4℃
インキュベーション緩衝液:(1)ホモゲナイズ緩衝液、140mMのNaCl、20mMのトリス塩酸、pH7.1、1mMのPMSF、(2)75mMのHepes/140mMのNaCl、pH7.5、(3)アッセイ緩衝液、ホモゲナイズ緩衝液:緩衝液(2)=1:4
非特異的リガンド:10μMのテトロドトキシン
Kd:1.4nM*
Bmax:3.7pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistar ラット脳
リガンド:5nMの[3H] バトラコトキシニンA 20−α−ベンゾエート
賦形剤:1%DMSO
インキュベーション時間/温度:60分/37℃
インキュベーション緩衝液:50mMのトリス塩酸、pH7.4、25℃、50mMのHEPES、130mMのコリン−Cl、5.4mMのKCl、0.8mMのMgSO4、7H2O(またはMgCl2)、5.5mMのグルコース、40μg/mlのLqTx
非特異的リガンド:100μMのベラトリジン
Kd:0.052μM*
Bmax:0.7pmole/mgのタンパク質*
特異的結合:77%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.25nMの[3H] SR−140333
賦形剤:1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:20mMのHEPES、pH7.4、1mMのMnCl2,0.01%のBSA
非特異的リガンド:2μMのL−703、606
Kd:0.3nM*
Bmax:10pmole/mgのタンパク質*
特異的結合:85%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ラット組換え大腸菌
リガンド:1.5nMの[3H] ミボレロン
賦形剤:1%DMSO
インキュベーション時間/温度:4時間/4℃
インキュベーション緩衝液:50mMのトリス塩酸(pH7.5)、0.8MのNaCl、10%のグリセロール、2mMのジチオトレイトール、1mg/mlのBSA、2%エタノール
非特異的リガンド:10μMのミボレロン
Kd:3nM*
Bmax:930pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えCHO細胞
リガンド:0.15nMの[125I]RTI−55
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:100mMのNaCl、50mMのトリス塩酸、1μMのロイペプチン、10μMのPM5F、pH7.4
非特異的リガンド:10μMのノミフェンシン
Kd:0.58nM*
Bmax:0.047pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えMDCK細胞
リガンド:0.2nMの[125I]RTI−55
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:50mMのトリス塩酸、100mMのNaCl、1μMのロイペプチン、10μMのPM5F、pH7.4
非特異的リガンド:10μMのデシプラミン
Kd:0.024nM*
Bmax:2.5pmole/mgのタンパク質*
特異的結合:75%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組換えHEK−293細胞
リガンド:0.15nMの[125I]RTI−55
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:100mMのNaCl、50mMのトリス塩酸、1μMのロイペプチン、10μMのPM5F、pH7.4
非特異的リガンド:10μMのイミプラミン
Kd:0.17nM*
Bmax:0.41pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値のf50%
起源:Wistarラット大脳皮質
リガンド:6nMの[3H]GABA
賦形剤:1%DMSO
インキュベーション時間/温度:20分/25℃
インキュベーション緩衝液:10mMのNa−HEPES、120mMのNaCl、4mMのCaアセテート、10μMのイソグバシン、10μMの(−)バクロフェン、pH7.5
非特異的リガンド:10μMのNO−711
Kd:0.3μM*
Bmax:60pmole/mgのタンパク質*
特異的結合:80%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
*は、ヒストリカル値を示す。
カタログ番号、引用文献
118050. Crespi, C. L.,
Miller, V. P. and Penman, B.W. (1997)、 Microtiter plate assays for
inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem 248(l):
188 - 190.Gentest Technical
Bulletin (Version 4.2: Revised 27 September 2000) A high throughput method for
measuring cytochrome P450 inhibition. Gentest Technical Bulletin
(Version 4-2. Revised 27 September 2000).118060. Crespi, C. L., Miller, V. P. and
Penman, B.W. (1997) Microtiter plate assays for inhibition of human,
drug-metabolizing cytochromes P450. Anal Biochem 248(1):
188-190.Gentest Technical Bulletin (Version 4.2: Revised 27 September 2000)
A high throughput method for measuring cytochrome P450 inhibition. Gentest
Technical Bulletin (Version 4-2: Revised 27 September
2000).118070. Crespi, C. L.,
Miller, V. P. and Penman, B.W. (1997) Microtiter plate assays for inhibition of
human, drug-metabolizing cytochromes P450. Anal Biochem 248(1):
188 - 190.Gentest Technical Bulletin (Version 4.2: Revised 27 September
2000)
A high throughput method for measuring cytochrome P450 inhibition. Gentest
Technical Bulletin (Version 4-2: Revised 27 September
2000).118080. Crespi, C. L.,
Miller, V. P. and Penman, B.W. (1997) Microtiter plate assays for inhibition of
human, drug-metabolizing cytochromes P450. Anal Biochem 248(1):
188 - 190.Gentest Technical Bulletin (Version 4.2: Revised 27 September 2000) A
high throughput method for measuring cytochrome P450 inhibition. Gentest
Technical Bulletin (Version 4-2: Revised 27 September 2000).118090. Crespi, C. L., Miller, V. P. and Penman, B.W. (1997)
Microtiter plate assays for inhibition of human, drug-metabolizing cytochromes
P450. Anal Biochem 248M: 188 - 190.Gentest Technical Bulletin
(Version 4.2: Revised 27 September 2000) A high throughput method for measuring
cytochrome P450 inhibition. Gentest Technical Bulletin (Version 4.2:
Revised 27 September 2000).200510.
Libert, F., Sande, J.V., Lefort, A., Czernilofsky, A., Dumont, J.E.,
Vassart, G., Ensinger, H.A. and Mendla, K.D. (1992). Cloning and functional
characterization of a human Al adenosine receptor. Biochem. Biophys. Res.
Commun. 187:919- 926.200610.
Varani, K., Gessi, S., Dalpiaz, A. and Borea, P. A. (1996) Pharmacological
and biochemical characterization of purified A2A adenosine receptors in human
platelet membranes by [3H]CG521680 binding. Br. J. Pharmacol. 117:1693-1701203100.
Michel, A.D., Loury, D.N., Whiting, R.L. (1989) Identification of a single
a1A-adrenoceptor corresponding to the a1A-subtype in rat submaxillary gland.
Br. J. Pharmacol. 98:883-889.203200. Garcia-Sainz, J.A.,
Romero-Avila, M.T., Hernandez, R.A., Macias-Silva, M., Olivares-Reyes, A.,
Gonzalez-Espinosa, C. (1992) Species heterogeneity of hepatic al-adrenoceptors:
at A-, al B-, and at C-subtypes. Biochem. Biophys. Res. Comm. 186:760-767.Michel,
A.D., Loury, D.N., and Whiting, R.L. (1989) Identification of a single al
A-adrenoceptor corresponding to the al A subtype in the rat submaxillary gland.
Br.
J. Pharmacol. 98:833-889.203400. Kenny, B. A., Chalmers, D. H., Philpott, P. C. and Naylor A.
M. (1995) Characterization of an al D-adrenoceptor mediating the contractile
response of rat aorta to noradrenaline. British Journal of Pharmacology.
115: 981 - 986203620.
UhlSn, S., Porter, A.C., Neubig, R.R. (1994) The novel alpha-2
adrenergic radioligand [3H]MK912 is alpha-2C selective among human alpha-2A,
alpha-2B and alpha-
2C adrenoceptors. J. Pharmacol. Exp. Ther. 271:1558-1565.203710. Uhlen S., Dambrova, M., Nasman, J.,
Schioth, H.B., Gu, Y., Wikberg-Matsson, A., Wikberg, J.E., (1998) Alpha 213-
and alpha 2C- adrenoceptors. comparison with MK912, RX821002, rauwolscine and
yohimbine. Eur. J. Pharmacol. 343 (1): 93-101.204010. Feve, B., Elhadri, K.,
Quignard-Boulange, A., Pairault, J. (1994) Transcriptional down-regulation by
insulin of the b3-adrenergic receptor expression in 3T3-F442A adipocytes: a
mechanism for repressing the CAMP signalling pathway. Proc. NatL Acad. Sci.
USA 91:5677-5681.204110.
McCrea, K.E. and Hill S.J. (1993) Salmeterol, a long-acting
b2-adrenoceptor agonist mediating cyclic AMP accumulation in a neuronal cell
line. Brit. J. Pharmacol. 110:619-626.204410. Galli, A., De Felice, L., Duke, B.-J.,
Moore, K., Blakely, R. (1995) Sodium dependent norepinephrine induced currents
in norepinephrine transporter transfected HEK293 cells blocked by cocaine and
antidepressants. J. Exp. Biol. 198:2197-2212.212500. Menke, J., Borkowski, J.A., Bierilo,
K.K., MacNeil, T., Derrick, A.W., Schneck, K.A., Ransom, R.W. Strader, C.D.,
Linemeyer, D.L., Hess, J.F. (1994) Expression cloning of a human B1 bradykinin
receptor. J. Biol. Chem. 269:21583-21586.212610. Eggerickx, D., Raspe, E., Bertrand, D.,
Vassart, G., Parmentier, M. (1992)
Molecular cloning, functional expression and pharmacological characterization
of a human bradykinin B2 receptor gene. Biochem Biophys Res Commun 187 (3):
1306 - 1313.214510. Schoemaker, H.
and Langer S.Z. (1985) [3H]Diltiazem binding to calcium channel antagonist
recognition sites in rat cerebral cortex. Eur. J. Pharmacol. 111:273-277.214600. Ehlert, F.J., Roeske, W.R., Itoga, E.,
and Yamamura, H.I. (1982) The binding of [3H]nitrendipine to receptors for
calcium channel antagonists in the heart, cerebral cortex and ileum of rats. Life Sci. 30:2191-2202.Gould
R.J., Murphy, K.M.M., Snyder, S.H. (1982) [3H]nitrendipine-labeled calcium
channels discriminate inorganic calcium agonists and antaggonists. Proc Natl. Acad. Sci. USA 79:3656-3650.216000. Moresco, R.M., Govoni, S., Battaini,
F., Trivulzio, S., Trabucchi, M. (1990) Omegaconotoxin binding decreases in
aged rat brain. Neurobiol. of Aging 11:433-436.219500. Dearry, A., Gingrich, J.A., Falardeau,
P., Fremeau, R.Tjr., Bates, M.D., Caron, M.G. (1990) Molecular cloning and
expression of the gene for a human D1 dopamine receptor. Nature 347:72-76.
Sunahara, R.K., Niznik, H.B., Weiner, D.M., Stormann, T.M., Brann, M.R.,
Kennedy, J.L., Gelernter, J.E., Rozmahel, R., Yang, Y., Israel, Y., Seeman, P.,
and O'Dowd, B.F. (1990) Human Dopamine D1 receptor encoded by an intronless
gene on chromosome 5. Nature 347:80-83. Zhou, Q.-Y., Grandy,
D.K., Thambi, L., Kushner, J.A., Van To[, H.H.M., Cone, R., Pribnow, D., Salon,
J. Bunzow, J.R., and Civelli, O. (1990) Cloning and expression of human and rat
D1 dopamine receptors. Nature 347:76-80.219600. Bunzo, J.R., Van To[, H.H.M., Grandy,
D.K., Albert, P., Salon, J., Christie, M., Machida, C.A., Neve, K.A., and
Civelli, O. (1988) Cloning and expression of rat D2 dopamine receptor cDNA Nature
336:783-787.Grandy, D.K., Marchionni, M.A., Makam, H., Stofko, R.E.,
Alfano, M., Frothingham, L, Fischer, J.B. Burke-Howie, K.J., Bunzow, J.R.,
Seiver, A.C., Civelli, O. (1989) Cloning of the cDNA and gene for a human D2
dopamine receptor. Proc. NatL Acad. Sci. USA 86:9762-9766. Hayes,
G., Biden, T.J., Selbie, L.A., and Shine, J. (1992) Structural subtypes of the
dopamine D2 receptor are functionally distinct: Expression of the clone D2A and
D2B subtypes in a heterologous cell line. Molec. Endocrin.
6:920-926.219800. Sokoloff, P.,
Giros, B., Martres, M.P., Bouthenet, M.L., Schwartz, J.C. (1990)
Molecular cloning and characterization of a novel dopamine receptor (D3) as a
target for neuroleptics. Nature 347:146-151.219900. Van Tot, H.H.M., Bunzow, J.R., Guan,
H.C., Sunahara, R.K., Seeman, P., Niznik, H.B., Civelli, 0. (1991) Cloning of
the gene for a human dopamine D4 receptor with high affinity for the
antipsychotic clozapine. Nature 350:610-614.Van To[, H.H.M., Wu,
C.M., Guan, H.-C., Ohara, K., Bunzow, J.R., Civelli, 0., Kennedy, J., Seeman,
P. Niznik, H.B., and Jovanovic, V. (1992) Multiple dopamine D4 receptor
variants in the human population. Nature 358:149-152.220320. Giros, B. and Caron., M.G. (1993). Molecular characterization of the
dopamine transporter. Trends. Pharmacol. Sci. 14:
43-49.220320. Gu, H., Wall, S.,
Rudnick, G. (1994) Stable expression of biogenic amine transporters reveals
differences in inhibitor sensitivity, kinetics, and ion dependence. J. Biol.
Chem. 269(10):7124-7130.224010. Pharmacological characterization of a potent nonpeptide
endothelia receptor antagonist, 97 - 139. The Journal of Pharmacology and
Experimental Therapeutics. 268: 1122 - 1127.224110. Cain, M.J., Garlick, R.K. and Sweetman,
P.M. (1991) Endothelia-1 receptor binding assay for high throughput chemical
screening. J Cardiovasc Pharmacol 17 Suppl 7:
5150-i51Chiou, W.J., Magnuson, S.R., Dixon, D., Sundy, S., Opgenorth, T.J. and
Wu-Wong, J.R. (1997) Dissociation characteristics of endothelia receptor
agonists and antagonists in cloned human type-B endothelia225500. Dittadi, R., Gion, M., Brazzale, A.,
Bruscagnin, G. (1990) Radioligand binding assay of epidermal growth factor
receptor: Causes of variability and standardization of the
assay. Clin. Chem. 36:849-854.Massague, J. (1983) Epidermal
growth factor-like transforming growth factodr: II. Interaction with epidermal
growth factor receptors in human placenta membranes and A431 cells. J. Biol.
Chem. 258:13614-13620.226010.
Obourn, J. D., Koszewski, N. J. and Notides, A. C. (1993) Hormone-and
DNA-binding mechanism of the recombinant human estrogen receptor. Biochemistry.
32: 6229 - 6236.226400.
Shank, R.P., Baldy, W.J., Matucci, L.C., Villani, F.J. Jr. (1990) Ion
and temperature effects on the binding of gamma-aminobutyrate to its receptors
and the high-affinity transport system. J. Neurochem. 54:2007-2015.226500. Enna, S.J., and Snyder, S.H. (1976)
Influences of ions, enzymes and detergents on gamma-aminobutyric acid-receptor
binding in synaptic membranes of rat brain. Mol. Pharmacol. 13:442-453.
Martinin, C., Rigacci, T., Lucacchini, A. (1983) [3H]muscimol binding site on
purified benzodiazepine receptor. J. Neurochem. 41:1183-1185.Snodgrass,
S.R. (1978) Use of [3H]muscimol for GABA receptor studies. Nature 273:392-394.226600. Damm, H.W., Mueller, W.E., Schlaefer,
U., Wollert, U. (1978) [3H]flunitrazepam: Its advantages as a ligand, for the
identification of benzodiazepine receptors in rat brain
membranes. Res. Comm. Chem. Pathol. Pharmacol. 22:597-600.Speth,
R.C., Wastek, G.J., and Yamamura, H.I. (1979) Benzodiacepam receptors:
temperature dependence of [3H]flunitrazepam binding. Life Sci. 24:351-357.228510. Facklam, M. and Bowery, N., G. (1993)
Solubiliaztion and characterization of GABAB receptor binding sites from
porcine brain synaptic membranes. Br. J. Pharmacol. 110:
1291-1296228510. Mathivet P.,
Bernasconi, R., Barry, J. D., Marescaux, C., Bittiger, H. (1992)
Binding characteristics of y-hydroxybutyric acid as a weak but selective GABAB
receptor agonist. Eur. J. Pharmacol. 321: 67-75232010. Cidlowski, J. A. and Cidlowski, N. B.
(1981) Regulation of glucocorticoid receptors by glucocorticoids in Cultured
HeLa S3 Cells. Endocrinology 109: 1975 - 1982.232700. London, E.D. and Coyle J.T. (1979)
Specific binding of [3H]kainic acid to receptor sites in rat brain. Mol.
Pharmacol. 15:492-505.232810.
Sills, M. A. Fagg, G. Pozza, M. Angst, C. Brundish, D. E. Hurt, S. D.
Wilusz, E. J. and Williams, M. (1991).
[3H]CGP 39653: a new N-methyl-D-aspartate antagonist
radioligand with low nanomolar affinity in rat brain. European Journal of
Pharmacology 192: 19-24.232910. Seifel, B.W., Sreekrishna, K., Baron, B.M. (1996) Binding of
the radiolabeled glycine antagonist [3H]MDS105,519 to homomeric NMDA-NR1a
receptors. Eur. J.
Pharmacol. 312:357-365.233000.
Goldman, M.E., Jacobson, A.E., Rice, K.C., Paul, S.M. (1985)
Differentiation of [3H] phencyclidine and (+)-[3H]SKF-10047 binding sites in
rat cerebral cortex. FEBS Lett.
190:333-336.239610. De
Backer, M. D., Gommeren, W., Moereels, H., Nobels, G., Van Gompel, P., Leysen,
J. E. and Luyten, W. H. (1993) Genomic cloning, heterologous expression and
pharmacological characterization of a human histamine H1 receptor. Biochemical
and Biophysical Research Communications. 1601 - 1608239710. Ruat, M., Traiffort, E., Bouthenet, M.
L., Schwartz, J. C., Hirschfeld, J., Buschauer, A. anad Schunack, W. (1990)
Reversible and irreversible labeling and autoradiographic localization of the
cerebral histamine H2 receptor using [1251]iodinated probes. Proceedings
of the National Academy of Sciences of the United States of America. 87(5):
1658- 1662.239810. Yanai, K.,
Ryu, J. H., Sakai, N., Takahashi, T., Iwata, R., Ido, T., Murakami, K. and
Watanabe, T.(1994) Binding characteristics of a histamine H3-receptor
antagonist, [3H]S-methylthioperamide: comparison with [3H](R)a methylhistamine
binding to rat tissues. Japanese Journal of Pharmacology. 65(2):
107 - 112. Zhu, Y., Michalovich, D., Wu, H., Tan, K. B., Dytko, G. M.,
Mannan, 1. J., Boyce, R., Alston, J. Tierney, L. A., Li, X., Herrity, N. C.,
Vawter, L., Sarau, H. M., Ames, R.S., Davenport, C. M., Hieble, J. P., Wilson,
S., Bergsma, D. J. et al. (2001) Cloning, expression, and pharmacological characterization
of a novel human histamine receptor. Molecular pharmacology. 59(3):
434 - 441, 2001.241000. Brown,
C.M., Mackinnon, A.C., McGrath, J.C., Spedding, M., Kilpatrick, A.T. (1990)
a2-Adrenoceptor subtypes and imidazoline-like binding in the rat brain. Br.
J. Pharmacol. 99:803-809.243510. Chin, J., Cameron, P.M., Rupp, E., and Schmidt, J.A. (1987)
Identification of a high affinity receptor for native interleukin-1 a and
interleukin-1 b on normal human lung
fibroblasts. J. Exp. Med. 165:70-86.250600. Bruns, R.F., Thomsen, W.J., Pugsley,
T.A. (1983) Binding of leukotrienes C4 and D4 to membranes from guinea pig
lung: regulation by ions and guanine nucleotides. Life Sci. 33:645-653.Mong,
S., Wu, H.-L, Hogabaoom, G.K., Clark, M.A., Crooke, S.T. (1984) Characterization
of the leukotriene D4 receptor in guinea pig lung. Eur. J. Pharmacol. 102:1-11.252600. Buckley, NJ., Bonner, T.I., Buckley,
C.M., Brann, M.R. (1989) Antagonist binding properties of five clonal
muscarinic receptors expressed in CHO-K1 cell. Mot. Pharmacol. 35:469-476.
Luthin, G.R. and Wolfe, B.B. (1984) Comparison of [3H]pirenzepine and
3H]quinuctidinyl-benzilate binding to muscarine cholinergic receptors in rat
brain. J. Pharmacol. Exp. Ther. 228:648-665. Watson, M., Yamamura, H.I., and Roeske,
W.R. (1983) A unique regulatory profile and regional distribution of
[3H]prienzepine binding in the rat provide evidence for distinct M1 and M2
muscarinic receptor subtypes. Life Sci. 32:3001-3011.252700. Buckley, N.J., Bonner, T.I., Buckley,
C.M., Brann, M.R. (1989) Antagonist binding properties of five clonal
muscarinic receptors expressed in CHO-Kt cell. Mot. Pharmacol. 35:469-476. Delmendo, R.E, Michel, A.D., and
Whiting, R.L. (1989) Affinity of muscarinic receptor antagonists for the three
putative muscarinic binding sites. Br. J. Pharmacol. 96:457-464.252800. Buckley, NJ., Bonner, T.I., Buckley,
C.M., Brann, M.R. (1989) Antagonist binding properties of five clonal
muscarinic receptors expressed in CHO-K1 cell. Mot. Pharmacol. 35:469-476.255510. Patacchini, R. and Maggi, C.A. (1995)
Tachykinin receptors and receptor subtypes. Arch. Int. Pharmacodyn. 329:161-184.257000. Fuhlendorff, J., Gether, U., Aakerlund,
L., Langeland-Hohansen, N., Thogersen, H., Melberg, S.G., Olsen, U.B.,
Thastrup, 0., and Schwartz, T.W. (1990) [Leu31,Pro34]neuropeptide Y: a specific
Y1 receptor agonist. Proc.
Natl. Acad. Sci. USA 87:182-186.
Sheikh, S.P., O'Hare, M.M., Tortroa, 0., Schwartz, T.W. (1989) Binding of
monoiodinated neuropeptide Y to hippocampal membranes and human neuroblastoma
cell line. J. Biol. Chem. 264:6648-6654.257110. Rose, P.M., Fernandes, P., Lynch, J.S.,
Frazier, S.T., Fisher, S.M., Kodukuta, K., Kienzle, B., and Seethala, R. (1995)
Cloning and functional expression of a cDNA encoding a human type 2
neuropeptide Y receptor. J. Biol. Chem. 270(39):22661-22664.258590. Davila-Garcia, M. 1., Musachio, J. L.,
Perry, D. C., Xiao, Y., Horti, A., London, E. D., Dannals, R. F. and Kellar, K.
J. (1997) [1251]IPH, an epibatidine analog, binds with high affinity to
neuronal nicotinic cholinergic receptors. The journal of pharmacology and
experimental therapeutics. Z821: 445 - 451.258590. Whiteaker, P., Jimenez, M., McIntosh, J. M., Collins, A. C.
and Marks, M.J. (2000)
Identification of a novel nicotinic binding site in mouse brain using
[(125)1]-epibatidine. British journal of pharmacology. 131(4) :
729 - 739.260110. Simonin, F. et
al. (1994) The human d-opioid receptor: Genomic organization, cDNA cloning,
functional expression, and distribution in human brain. Mol. Pharmacol. 46:
1015-1021.260210. Patricia,
M., et al.. (1992) Pharmacological profiles of fentanyl analogs as + and +
opiate receptors. Eur. J. Pharmacol. 213: 219-225. Simonin, F., et al. (1995) Kappa-opioid
receptor in humans: cDNA and genomic cloning, chromosomal assignment,
functional expression, pharmacology and expression pattern in the central
nervous system. PNAS U.S.A. 92 15 : 1431-1437.260410. Wang, J.B., Johnson, P.S., Persico,
A.M., Hawkins, A. L., Griffin, C. A., and Uhl, G.R. (1994) Human mu opiate
receptor: cDNA and genomic clones, pharmacologic characterization and
chromosomal assignment. FEBS
Lett. 338:217-222.264500.
Ashendel, C.L. (1985) The phorbol ester receptor: a
phospholipid-regulated protein kinase. Biochem. Biophys. Acta 822:219-242.265010. Herbert, J. M., Castro-Faria-Neto, H.
C., Barbosa-Filho, J. M., Cordeiro, R. S. B., Tibirica, E. (1997)
Pharmacological evidence for the putative existence of two different subtypes
of PAF receptors on platelets and leukocytes; studies with yangambin. J. Lipid
Mediat. Cell Signal. 17: 1-14.265600. Gaines, K.L., Hamilton, S. Boyd, A.E. 3rd (1988)
Characterizatrion of the sulfonylurea receptor on beta cell membranes. J. Biol.
Chem. 263:2589-2592.268700. Bo,
X., and Burnstock, G. (1990) High- and low-affinity binding sites for
[3H]-a,b-methylene ATP in rat urinary bladder membranes. Br. J. Pharmacol.
101:291-296.Ziganshin, A.U., Hoyle, C.H., Bo, X., Lambrecht, G.,
Mutschler, E., Baumert, H.G., Burnstock. G. (1993) PPADS selectively
antagonized P2X-purinoceptor-mediated responses in the rabbit urinary bladder.
Br. J. Pharmacol. 110:1491-1495.268810. Boyer, J. L., Cooper, C. L. and Harden T. K. (1990) [132P]3'-0-(4-Benzoyi)benzoyl
ATP as a photoaffinity label for a phospholipase C-coupled P2Y-Purinergic
receptor. J. Biol Chem. Vol. 265 No. 23: pp. 13515 - 13520.271110. Martin, G.R. and Humphrey, P.P.A.
(1994) Receptor for 5-hydroxytryptamine: current perspectives on classification
and nomenclature. Neuropharm. 33:261-273.271910. 1. Millerk, W. E. , Fletcher, P. W.,
and Teitler, M. (1992) Membrane-bound and solubilized brain 5-HT3 receptor:
improved radioligand binding assay using bovine area postrema or rat cortex and
the radioligand [3H]GR65630, [3H]BRL43694, and [3H]LY278584 Synapase, 11:58
- 66.Boess, F. G., Steward, L. J., Steele, J. A., Liu, D., Reid, J., Glencorse,
T. A. and Martin, 1. L. (1997) Analysis of the ligand binding site of the 5-HT,
receptor using site- directed mutagenesis: importance of glutamate 106. Neuropharmacology,
36: 637 - 647.274020. Gu,
H., Wall, S., Rudnick, G. (1994) Stable expression of biogenic amine
transporters reveals differences in inhibitor sensitivity, kinetics, and ion
dependence. J. Biol. Chem. 269(10):7124-7130.278110. Ganapathy, M. E., Prasad, P. D., Huang,
W., Seth, P. , Leibach, F. H. and Ganapathy, V. (1999) Molecular and
ligand-binding characterization of the s-receptor in the Jurkat human T
lymphocyte cell line. Pharmacol
Exp. Ther 289: 251 - 260.278200. Hashimoto, K., and London, E.D. (1993) Further
characterization of [3H]Ifenprodil binding to sigma receptors in rat brain. Eur.
J. Pharmacol. 236:159-163.279450. Doucette, G.J. Logan, M. M., Ramsdell, J. S. and Van Dolah,
F. M. (1997)
Development and preliminary validation of a microtiter plate-based receptor
binding assay for paralytic shellfish poisoning toxins. Toxicon, 35 (5):
625 - 636.279510. Catterall, W.A.,
Morrow, C.S., Daly, J.W., Brown, G.B. (1981) Binding of batrachotoxin A
20-alpha-benzoate to a receptor site associated with sodium channels in
synaptic nerve ending particles. J. Biol. Chem. 256:8922-8927.285010. Chang, C. and Liao,
S. (1987) Topographic recognition of cyclic hydrocarbons and related compounds
by receptors for androgens, estrogens, and glucocorticoids. J. Steroid
Biochem. 27(1-3): 123 - 131.
Traish, A. M., Muller R. E. and Wotiz, H. H. (1986) Binding of 7a,
17a-dimethyl-19-nortestosterone (mibolerone) to androgen and progesterone
receptors in human and
animal tessues. Endocrinology 118(4): 1327 - 1333.
CS1814、CS1713およびCS1714の生物学的試験
CS1814、CS1713、CS1714および種々の参照化合物の生物学的試験の結果を図41〜58および60〜62に示す。方法は、信頼性および再生性を最大化するために、科学文献から採用した。参照は、各アッセイのインテグラルパートとして行い、得られた結果の有効性を確認した。アッセイは、下記に示した条件下で行った。各アッセイの文献の符号については、下記にまとめている。本明細書においてそれを引用によって援用する。
Pharma Servicesにおいて実験によって取得)を用いて、ChengとPrusoffの式(Cheng, Y., Prusoff, W.H., Biochem. Pharmacol.
22:3099−3108, 1973)を使って計算したものである。示されている場合、競合する結合曲線の傾斜を定義するHill係数(nH)は、Data Analysis Toolbox(登録商標)を用いて計算した。Hill係数が1.0と有意に異なる場合、結合のズレは、単一の結合部位を持つ質量作用の法則に従わないことを示唆するかもしれない。IC50、Kiおよび/またはnHのデータが、Standard Error of the Mean (SEM)なしで示されている場合、データは、量的に不十分であり、示された値(Ki、IC50、nH)の解釈には注意が必要である。
118090 CYP450、3A4
起源:ヒト組換えSf9昆虫細胞
基質剤:50μMの7−ベンジルオキシ−4−(トリフルオロメチル)−クマリン
賦形剤:0.1%DMSO
プレインキュベーション時間/温度:なし
インキュベーション時間/温度:30分/37℃
インキュベーション緩衝液:75mMのリン酸カリウム緩衝液、pH7.5
定量方法:7−ヒドロキシ−4−(トリフルオロメチル)−クマリンの分光蛍光法による定量化
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組み換えCHO−K1細胞
リガンド:0.2nMの[3H]ブラジキニン
賦形剤:0.1%DMSO
インキュベーション時間/温度:90分/25℃
インキュベーション緩衝液:24mMのTES−NH4OH、pH6.8、1mMの1,10−フェナントロリン、0.3%BSA
非特異的リガンド:5μMのブラジキニン
KD:0.29nM*
BMAX:2pmole/mgのタンパク質*
特異的結合:90%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:Wistarラット脳
リガンド:2nMの[3H]ジルチアゼム
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:50mMのトリス塩酸、0.1%BSA、pH7.4、25℃
非特異的リガンド:10μMのジルチアゼム
KD:0.016μM*
BMAX:0.21pmole/mgのタンパク質*
特異的結合:73%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組み換えMDCK細胞
リガンド:0.2nMの[125I]RTI−55
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:50mMのトリス塩酸、100mMのNaCl、1μMのロイペプチン、10μMのPMSF、pH7.4
非特異的リガンド:10μMのデシプラミン
KD:0.024μM*
BMAX:2.5pmole/mgのタンパク質*
特異的結合:75%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値の50%以上
起源:ヒト組み換えHEK−293細胞
リガンド:0.15nMの[125I]RTI−55
賦形剤:1%DMSO
インキュベーション時間/温度:3時間/4℃
インキュベーション緩衝液:100mMのNaCl、50mMのトリス塩酸、1μMのロイペプチン、10μMのPMSF、pH7.4
非特異的リガンド:10μMのイミプラミン
KD:0.17nM*
BMAX:0.41pmole/mgのタンパク質*
特異的結合:95%*
定量方法:放射リガンド結合
有意性評価基準:刺激または阻害の最大値のf50%
標的:ヒトMDCK細胞イヌ腎臓
賦形剤:0.4%DMSO
インキュベーション時間/温度:30分/25℃
インキュベーション緩衝液:5mMのトリス塩酸、7.5mMのHEPES、120mMのNaCl、5.4mMのKCl、1.2mMのCaCl2、1.2mMのMgSO4、5mMのグルコース、1mMのアスコルビン酸、pH7.1
定量方法:Alamar Blue の分光蛍光法による定量化
有意性評価基準−Ag:該当なし
有意性評価基準−Ant:蛍光強度の、賦形剤対照に対する50%以上の減少
標的:ヒトHEK−293細胞ヒト胎児性腎臓
賦形剤:0.4%DMSO
インキュベーション時間/温度:30分/25℃
インキュベーション緩衝液:5mMのトリス塩酸、7.5mMのHEPES、120mMのNaCl、5.4mMのKCl、1.2mMのCaCl2、1.2mMのMgSO4、5mMのグルコース、1mMのアスコルビン酸、pH7.1
定量方法:Alamar Blue の分光蛍光法による定量化
有意性評価基準−Ag:該当なし
有意性評価基準−Ant:蛍光強度の、賦形剤対照に対する50%以上の減少
標的:ヒトMDCK細胞イヌ腎臓
賦形剤:0.4%DMSO
インキュベーション時間/温度:10分/25℃
インキュベーション緩衝液:5mMのトリス塩酸、7.5mMのHEPES、120mMのNaCl、5.4mMのKCl、1.2mMのCaCl2、1.2mMのMgSO4、5mMのグルコース、1mMのアスコルビン酸、pH7.1
定量方法:[3H]ノルエピネフリンの定量化
有意性評価基準−Ag:該当なし
有意性評価基準−Ant:デシプラミン反応に対する、[3H]ノルエピネフリン摂取の50%以上の阻害
標的:ヒトHEK−293細胞ヒト胎児性腎臓
賦形剤:0.4%DMSO
インキュベーション時間/温度:10分/25℃
インキュベーション緩衝液:5mMのトリス塩酸、7.5mMのHEPES、120mMのNaCl、5.4mMのKCl、1.2mMのCaCl2、1.2mMのMgSO4、5mMのグルコース、1mMのアスコルビン酸、pH7.1
定量方法:[3H]セロトニン摂取の定量
有意性評価基準−Ag:該当なし
有意性評価基準−Ant:フルキセチン(fluxetine)反応に対する[3H]セロトニン摂取の50%以上の阻害
(カタログ番号、引用)
118090.
Crespi, C. L., Miller, V. P and Penman, B.W. (1997) Microtiter plate
assays for inhibition of human, drug-metabolizing cytochromes P450. Anal Biochem 248(1):
188-190.
Gentest Technical Bulletin (Version 4.2: Revised 27 September 2000) A high
throughput method for measuring cytochrome P450 inhibition. Gentest Technical Bulletin
(Version 4.2) Revised 27 September
2000).
204410. Galli,
A., DeFelice, L., Duke, B.-J., Moore, K. Blakely, R. (1995) Sodium dependent
norepinephrine induced currents in norephinephrine transporter transfected
HEK293 cells blocked by cocaine and antidepressants. J. Exp. Biol. 198:2197-2212.
212610.
Eggerickx, D., Raspe, E. Bertrand, D., Vassart, G., Parmentier, M.
(1992) Molecular cloning, functional expression and pharmacological
characterization of a human bradykinin B2 receptor gene. Biochem Biophys Res
Commun 187 (3): 1306 - 1313.
214510.
Schoemaker, H. and Langer, S.Z. (1985) [3H]Diltiazem binding to calcium
channel antagonist recognition sites in rat cerebral cortex. Eur. J. Pharmacol. 111:273-277.
274020. Gu,
H., Wall, S., Rudnick, G. (1994) Stable expression of biogenic amine
transporters reveals differences in inhibitor sensitivity, kinetics, and ion
dependence. J. Biol. Chem. 269(10):7124-7130.
302000. Galli,
A., DeFelice, L. Duke, B.-J., and Blakely, R. (1995) Sodium dependent
norephinephrine-induced currents in
norepinephrine-transporter-transfected
HEK-293 cells blocked by cocaine and antidepressants. J. Exp. Biol. 198:2197-2212.
302100. Page,
B., Page, M. and Noel, C. (1993) A new fluorometric assay for cytotoxicity
measurements in vitro. Ing. I
3:473-476, 1993.
364000. Gu,
H., Wall, S., Rudnick, G. (1994) Stable expression of biogenic amine
transporter reveals differences in inhibitor sensitivity, kinetics, and ion
dependence. J. Biol. Chem. 269(1):7124-7130.
364100. Page,
B., Page, M. and Noel, C. (1993) A new fluorometric assay for cytotoxicity measurements
in vitro. Int. J. Oncology 3:473-476,
1993.
1. 米国特許第4,478,836号
2. 米国特許第5,034,541号
3. 米国特許第5,621,142号
4. Moret, C. et al. Neuropharmacology 1985,
24, 1211-1219.
5. Bonnaud, B. et al. J. Med. Chem. 1987, 30, 318-325.
6. Shuto, S. et al. J. Med. Chem. 1995, 38, 2964-2968.
7. Viazzo, P. et al. Tetrahedron Lett. 1996, 37, 4519-4522.
8. Shuto, S. et al. Tetrahedron Lett. 1996, 37, 641-644.
9. Shuto, S. et al. J. Med. Chem. 1996, 39, 4844-4852.
10. Shuto, S. et al. J. Med. Chem. 1998, 41, 3507-3514.
11. Deprez, D.
et al. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 166-171.
12. Puozzo, C.
et al. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 273-279.
13. Puozzo C.
et al. Eur. J. Drug Metab. Pharmacokinet. 1998, 23, 280-286.
14. Shuto, S. et al. Jpn. J. Pharmacol. 2001, 85, 207-213.
15. Doyle, M.P. et al. Adv. Synth. Catal. 2001, 343, 299-302.
16. Kazuta, Y. et al. Bioorg. Med. Chem. 2002, 10, 1777-1791.
17. Labat, L. et al. J. Chromatogr. B 2002, 773, 17-23.
18. Grard, S. et al. Electrophoresis 2000, 21, 3028-3034.
本明細書において引用されている、すべての特許および文献を本明細書に引用によって援用する。
当業者なら、ルーチンの実験法を用いるだけで、ここに記載された本発明の特定の実施形態に対して多くの均等物を認識または確認することになろう。そのような均等物は、以下の請求項に包含されることが意図される。
Claims (47)
- 式A(化1)で表される、単離された化合物。
Rは、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、へテロアリール、アリールアルキル、ホルミル、アシル、シリル、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R1は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R2は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R3は、それぞれ個別に、H,アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R4は、1〜4回存在する、または存在しない、
R4は、もし存在すれば、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R80は、それぞれ個別に、アリール、シクロアルキル,シクロアルケニル、ヘテロ環、またはポリシクリル部分を表し、
mは、それぞれ個別に、0〜8の間の整数であり、そして
化合物は、単一のエナンチオマーであり;または、
それらの薬学的に許容される塩もしくはプロドラッグである。 - 式B(化2)で表される、単離された化合物。
Rは、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、へテロアリール、アリールアルキル、ホルミル、アシル、シリル、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R1は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R2は、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R3は、それぞれ個別に、H,アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、または−(CH2)m−R80を表し、
R4は、1〜4回存在する、または存在しない、
R4は、もし存在すれば、それぞれ個別に、H、アルキル、シクロアルキル、アルケニル、アリール、ヘテロアリール、アリールアルキル、シアノ、ハロゲン、ヒドロキシル、アルコキシル、アリールオキシ、アリールアルキルオキシ、アミノ、アルキルアミノ、アリールアミノ、アリールアルキルアミノ(arylakylamino)、スルフヒドリル、アルキルチオ、アリールチオ、アリールアルキルチオ(arylakylthio)、ニトロ、アジド、アルキルセレノ、ホルミル、アシル、カルボキシル、シリル、シリルオキシ、(アルキルオキシ)カルボニル、(アリールオキシ)カルボニル、(アリールアルキルオキシ)カルボニル、(アルキルアミノ)カルボニル、(アリールアミノ)カルボニル、(アリールアルキルアミノ)カルボニル、アルキルスルホニル、アリールスルホニル、または−(CH2)m−R80を表し、
R80は、それぞれ個別に、アリール、シクロアルキル,シクロアルケニル、ヘテロ環、またはポリシクリル部分を表し、
mは、それぞれ個別に、0〜8の間の整数であり、そして
化合物は、単一のエナンチオマーであり;または、
それらの薬学的に許容される塩もしくはプロドラッグである。 - XがOを表す、請求項1または2に記載の化合物。
- RがHを表す、請求項1または2に記載の化合物。
- R1がHを表す、請求項1または2に記載の化合物。
- R2がHを表す、請求項1または2に記載の化合物。
- R3がアルキルを表す、請求項1または2に記載の化合物。
- R4が存在しない、請求項1または2に記載の化合物。
- XがOを表し、RがHを表す、請求項1または2に記載の化合物。
- XがOを表し、RがHを表し、R1がHを表す、請求項1または2に記載の化合物。
- XがOを表し、RがHを表し、R1がHを表し、R2がHを表す、請求項1または2に記載の化合物。
- XがOを表し、RがHを表し、R1がHを表し、R2がHを表し、R3がアルキルを表す、請求項1または2に記載の化合物。
- XがOを表し、RがHを表し、R1がHを表し、R2がHを表し、R3がアルキルを表し、R4が存在しない、請求項1または2に記載の化合物。
- XがOを表し、RがHを表し、R1がHを表し、R2がHを表し、R3がエチルを表し、R4が存在しない、請求項1または2に記載の化合物。
- 請求項1または2に記載の化合物、および薬学的に許容される添加剤を含む、製剤。
- 請求項1または2に記載の化合物、ならびに鎮痛剤、抗炎症剤、解熱剤、抗抑鬱剤、抗てんかん薬、抗ヒスタミン剤、抗片頭痛薬、抗ムスカリン作動薬、抗不安剤、鎮静剤、催眠薬、抗精神病薬、気管支拡張剤、抗喘息薬、心臓血管薬、コルチコステロイド、ドーパミン作動薬、電解液、胃腸薬、筋弛緩剤、栄養剤、ビタミン剤、副交感神経作動薬、刺激剤、抗ナルコレプシー薬、および食思減退薬からなる群から選択される化合物を含む、製剤。
- 請求項1または2に記載の化合物、ならびにアセクロフェナク、アセトアミノフェン、アドメキセチン(adomexetine)、アルモトリプタン、アルプラゾラム、アマンタジン、アムシノニド、アミノシクロプロパン、アミトリプチリン、アモロジピン(amolodipine)、アモキサピン、アンフェタミン、アリピラゾール、アスピリン、アトモキセチン、アザセロトニン(azasetron)、アザタジン、ベクロメタソン、ベナクチジン、ベノキサプロフェン、ベルモプロフェン、ベタメタゾン、ビシファジン(bicifadine)、ブロモクリプチン、ブデソニド、ブプレノルフィン、ブプロピオン、ブスピロン、ブトルファノール、ブトリプチリン、カフェイン、カルバマゼピン、カルビドーパ、カリソプロドール、セレコシブ、クロルジアゼポキシド、クロルプロマジン、サリチル酸コリン、シタロプラム、クロミプラミン、クロナゼパム、クロニジン、クロニタゼン、クロラゼペート、クロチアゼパム、クロキサゾラム、クロザピン、コデイン、コルチコステロン、コルチゾン、シクロペンザプリン、シプロヘプタジン、デメキシプチリン(demexiptiline)、デシプラミン、デソモルフィン(desomorphine)、デキサメタゾン、デキサナビノール、デキストロアンフェタミンスルフェート、デキストロモラミド、デキストロプロポキシフェン、デゾシン、ジアゼパム、ジベンゼピン、ジクロフェナクナトリウム、ジフルニサール、ジヒドロコデイン、ジヒドロエルゴタミン、ジヒドロモルフィン、ジメタクリン、ジバルプロキセス、ジザトリプタン(dizatriptan)、ドラセトロン、ドネペジル、ドチエピン、ドキセピン、デュロキセチン、エルゴタミン、エスシタロプラム、エスタゾラム、エトスクシミド、エトドラク、フェモキセチン、フェナメート、フェノプロフェン、フェンタニル、フルジアゼパム、フルオキセチン、フルフェナジン、フルラゼパム、フルビプロフェン、フルタゾラム、フルボキサミン、フロバトリプタン、ガバペンチン、ガランタミン、ゲピロン、ギンコビルボア(ginko bilboa)、グラニセトロン、ハロペリドール、フペルジンA,ヒドロコドン、ヒドロコルチゾン、ヒドロモルホン、ヒドロキシジン、イブプロフェン、イミプラミン、インディプロン、インドメタシン、インドプロフェン、イプリンドール、イプサピロン、ケタセリン、ケトプロフェン、ケトロラク、レソピトロン(lesopitron)、レボドーパ、リパーゼ、ロフェプラミン、ロラゼパム、ロクサピン、マプロチリン、マチンドール、メフェナム酸、メラトニン、メリトラセン、メマンチン、メペリジン、メプロバメート、メサラミン、メタプラミン、メタキサロン、メタドン、メタドンメタンフェタミン、メトカルバモール、メチルドーパ、メチルフェニデート、メチルサリチラート、メチセルジド、メトクロプラミド、ミアンセリン、ミフェプリストン、ミルナシプラン、ミナプリン、ミルタザピン、モクロベミド、モダフィニル、モリンドン、モルフィン、モルフィンヒドロクロリド、ナブメトン、ナドロール、ナプロキセン、ナラトリプタン、ネファゾドン、ニューロンチン、ノミフェンシン、ノルトリプチリン、オランザピン、オルサラジン、オンダンセトロン、オピプラモール、オルフェナドリン、オキサフロザン(oxaflozane)、オキサプラジン(oxaprazin)、オキサゼパム、オキシトリプタン、オキシコドン、オキシモルフォン、パンクレリパーゼ、パレコシブ(parecoxib)、パロキセチン、ペモリン、ペンタゾシン、ペプシン、ペルフェナジン、フェナセチン、フェンジメトラジン、フェンメトラジン、フェニルブタゾン、フェニトイン、ホスファチジルセリン、ピモジド、ピルリンドール、ピロキシカム、ピゾチフェン、ピゾチリン、パラミペキソール(pramipexole)、プレドニソロン、プレドニソン、プレガバリン、プロパノロール、プロピゼピン(propizepine)、プロポキシフェン、プロトリプチリン、クアゼパム、キヌプラミン(quinupramine)、レボキシチン、レセルピン、リスペリドン(risperidone)、リタンセリン、リバスチグミン、リザトリプタン、ロフェコシブ、ロピニロール、ロチゴチン、サルサレート、セルトラリン、シブトラミン、シルデナフィル、スルファサラジン、スリンダク、スマトリプタン、タクリン、テマゼパム、テトラベノジン(tetrabenozine)、チアジド、チオリダジン、チオチキセン、チアプリド、チアシピロン(tiasipirone)、チザニジン、トフェナシン、トルメチン、トロキサトン、トピラメート、トラマドール、トラゾドン、トリアゾラム、トリフルオロペラジン、トリメトベンズアミド、トリミブラミン、トロピセトロン、バルデコシブ(valdecoxib)、バルプロ酸、ベンラファキシン、ビロキサジン、ビタミンE、ジメルジン、ジプラシドン、ゾルミトリプタン、ゾルピデム、およびゾピクロンからなる群から選択される化合物を含む、製剤。
- 請求項1または2に記載の化合物の治療的有効量を含む、鬱病を患う哺乳動物の処置剤。
- 請求項1または2に記載の化合物の治療的有効量を含む、線維筋痛症候群を患う哺乳動物の処置剤。
- 請求項1または2に記載の化合物の治療的有効量を含む、機能的体細胞性障害、鬱病、線維筋痛症候群、慢性疲労症候群、痛み、注意欠陥多動性障害、および内臓痛症候群(VPS)、過敏性腸症候群(IBS)、非心臓性胸痛(NCCP)、機能性消化不良、間質性膀胱炎、本態性外陰痛(essential vulvodynia)、尿道症候群、睾丸痛、抑鬱性障害、主要な抑鬱性障害、胸腺機能不全, 非定型の鬱病、不安障害、全身性不安障害、恐怖症, 強迫性障害、パニック障害、心的外傷後ストレス障害、月経前不快性障害、顎関節障害、非定型の顔面痛、片頭痛、ならびに緊張性頭痛を含む精神的障害を患う哺乳動物の処置剤。
- 前記哺乳動物が霊長類、ウマ、イヌ、ネコである、請求項18、19または20に記載の処置剤。
- 前記哺乳動物がヒトである、請求項18、19または20に記載の処置剤。
- 前記処置剤が、経口投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、静脈投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、舌下投与剤である、請求項18、19または20に記載の処置剤。
- 前記処理剤が、眼からの投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、経皮投与剤である、請求項18、19または20に記載の処置剤。
- 前記処理剤が、直腸投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、膣投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、局所投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、筋内投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、皮下投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、頬側投与剤である、請求項18、19または20に記載の処置剤。
- 前記処置剤が、経鼻投与剤である、請求項18、19または20に記載の処置剤。
- 選択的セロトニン再摂取抑制剤および請求項1または2に記載の化合物を含む組成物。
- 選択的ノルエピネフリン再摂取抑制剤、および請求項1または2に記載の化合物を含む組成物。
- 選択的セロトニン再摂取抑制剤、選択的ノルエピネフリン再摂取抑制剤、および請求項1または2に記載の化合物を含む組成物。
- 選択的ノルエピネフリン再摂取抑制剤が、ミルナシプランである、請求項36または37に記載の組成物。
- 前記化合物が、下記式で表される化合物の塩酸塩である、請求項35〜37のいずれか1項に記載の組成物。
- 前記化合物が、下記式で表される化合物の塩酸塩である、請求項35〜37のいずれか1項に記載の組成物。
- 前記組成物が、下記式で表される化合物の塩酸塩をさらに含む、請求項35〜40のいずれか1項に記載の組成物。
- 選択的セロトニン再摂取抑制剤および下記式で表される化合物の塩酸塩を含む組成物。
- 選択的ノルエピネフリン再摂取抑制剤および下記式で表される化合物の塩酸塩を含む組成物。
- 選択的セロトニン再摂取抑制剤、選択的ノルエピネフリン再摂取抑制剤、および下記式で表される化合物の塩酸塩を含む組成物。
- 選択的セロトニン再摂取抑制剤および下記式で表される化合物の塩酸塩を含む組成物。
- 選択的ノルエピネフリン再摂取抑制剤および下記式で表される化合物の塩酸塩を含む組成物。
- 選択的セロトニン再摂取抑制剤、選択的ノルエピネフリン再摂取抑制剤および下記式で表される化合物の塩酸塩を含む組成物。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US42164002P | 2002-10-25 | 2002-10-25 | |
| US42306202P | 2002-11-01 | 2002-11-01 | |
| US44514203P | 2003-02-05 | 2003-02-05 | |
| PCT/US2003/033681 WO2004039320A2 (en) | 2002-10-25 | 2003-10-22 | STEREOISOMERS OF p-HYDROXY-MILNACIPRAN, AND METHODS OF USE THEREOF |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2006503920A JP2006503920A (ja) | 2006-02-02 |
| JP2006503920A5 JP2006503920A5 (ja) | 2009-04-09 |
| JP4288263B2 true JP4288263B2 (ja) | 2009-07-01 |
Family
ID=32234198
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005501895A Expired - Fee Related JP4288263B2 (ja) | 2002-10-25 | 2003-10-22 | p−ヒドロキシ−ミルナシプランの立体異性体およびその使用方法 |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7038085B2 (ja) |
| EP (1) | EP1578719A4 (ja) |
| JP (1) | JP4288263B2 (ja) |
| AU (1) | AU2003284342B2 (ja) |
| CA (1) | CA2503381A1 (ja) |
| MX (1) | MXPA05004381A (ja) |
| WO (1) | WO2004039320A2 (ja) |
Families Citing this family (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10041479A1 (de) * | 2000-08-24 | 2002-03-14 | Sanol Arznei Schwarz Gmbh | Neue pharmazeutische Zusammensetzung zur Verabreichung von N-0923 |
| DE10041478A1 (de) | 2000-08-24 | 2002-03-14 | Sanol Arznei Schwarz Gmbh | Neue pharmazeutische Zusammensetzung |
| US20040048779A1 (en) * | 2002-05-06 | 2004-03-11 | Erwin Schollmayer | Use of rotigotine for treating the restless leg syndrome |
| US7704527B2 (en) | 2002-10-25 | 2010-04-27 | Collegium Pharmaceutical, Inc. | Modified release compositions of milnacipran |
| ES2239196T3 (es) * | 2002-12-02 | 2005-09-16 | Schwarz Pharma Ag | Suministro iontoforetico de rotigotina para el tratamiento de la enfermedad de parkinson. |
| AU2003230189A1 (en) * | 2003-01-29 | 2004-08-23 | Nitin Bhalachandra Dharmadhikari | Oral controlled release pharmaceutical composition containing metaxalone as active agent |
| EP1603585A2 (en) | 2003-03-14 | 2005-12-14 | Bristol-Myers Squibb Company | Polynucleotide encoding a novel human g-protein coupled receptor variant of hm74, hgprbmy74 |
| NZ527142A (en) | 2003-07-23 | 2006-03-31 | Douglas Pharmaceuticals Ltd | A stable suspension formulation |
| DE10334188B4 (de) * | 2003-07-26 | 2007-07-05 | Schwarz Pharma Ag | Verwendung von Rotigotin zur Behandlung von Depressionen |
| DE10334187A1 (de) * | 2003-07-26 | 2005-03-03 | Schwarz Pharma Ag | Substituierte 2-Aminotetraline zur Behandlung von Depressionen |
| AR047759A1 (es) * | 2003-09-26 | 2006-02-22 | Vertex Pharma | Derivados de fenil - piperazina como moduladores de receptores muscarnicos |
| EP1547592A1 (en) * | 2003-12-23 | 2005-06-29 | Schwarz Pharma Ag | Intranasal formulation of rotigotine |
| DE102004011392A1 (de) * | 2004-01-13 | 2005-08-04 | Grünenthal GmbH | Schwache bis mittelstarke Opioide oder Kombinationen dieser Opioide mit Antidepressiva für die Behandlung von Depressionen, Angststörungen und Schmerzen |
| US20070259865A1 (en) * | 2004-07-14 | 2007-11-08 | Astellas Pharma Inc. | Agent for Promoting the Recovery from Dysfunction After the Onset of Central Neurological Disease |
| WO2006036686A2 (en) * | 2004-09-24 | 2006-04-06 | University Of Maryland, Baltimore | Method of treating organophosphorous poisoning |
| US9132135B2 (en) * | 2004-09-24 | 2015-09-15 | University Of Maryland, Baltimore | Method of treating organophosphorous poisoning |
| EP1688161A1 (en) * | 2004-11-02 | 2006-08-09 | Switch Biotech Aktiengesellschaft | Use of pirlindole for the treatment of diseases which are characterized by proliferation of t-lymphocytes and/or hyperproliferation of keratinocytes in particular atopic dermatitis and psoriasis |
| US20060223875A1 (en) * | 2005-03-08 | 2006-10-05 | Phil Skolnick | Methods and compositions for production, formulation and use of 1-aryl-3-azabicyclo[3.1.0]hexanes |
| WO2007098356A1 (en) * | 2006-02-16 | 2007-08-30 | Neurocrine Biosciences, Inc. | Congeners of milnacipran as monoamine re-uptake |
| WO2007127421A2 (en) * | 2006-04-28 | 2007-11-08 | Merck & Co., Inc. | Process for the synthesis of (+) and (-)-1-aryl-3-azabicyclo[3.1.0]hexanes |
| JP2009536667A (ja) | 2006-05-09 | 2009-10-15 | ブレインセルス,インコーポレイティド | 5ht受容体介在性の神経新生 |
| TWI392670B (zh) * | 2006-06-22 | 2013-04-11 | Ucb Pharma Gmbh | 經取代的2-胺基萘滿之於製造用於預防、減緩及/或治療各種類型疼痛之藥物上的用途 |
| AU2007272501A1 (en) * | 2006-07-12 | 2008-01-17 | Elan Pharma International Limited | Nanoparticulate formulations of modafinil |
| GB0618879D0 (en) | 2006-09-26 | 2006-11-01 | Zysis Ltd | Pharmaceutical compositions |
| EP1987815A1 (en) * | 2007-05-04 | 2008-11-05 | Schwarz Pharma Ag | Oronasopharyngeally deliverable pharmaceutical compositions of dopamine agonists for the prevention and/or treatment of restless limb disorders |
| NZ584363A (en) | 2007-11-28 | 2012-11-30 | Ucb Pharma Gmbh | Drug formulations of Rotigotine which comprise the thermodynamically stable polymorphic form 2 |
| SG157299A1 (en) | 2008-05-09 | 2009-12-29 | Agency Science Tech & Res | Diagnosis and treatment of kawasaki disease |
| FR2944011B1 (fr) | 2009-04-03 | 2011-05-20 | Pf Medicament | Derives d'aminocyclobutane ou d'aminocyclobutene, leur procede de preparation et leur utilisation a titre de medicaments. |
| US20100298397A1 (en) * | 2009-05-19 | 2010-11-25 | Singh Nikhilesh N | Method of treatment of obsessive compulsive disorder with ondansetron |
| SMT201700578T1 (it) * | 2009-11-20 | 2018-01-11 | Tonix Pharma Holdings Ltd | Metodo e composizioni per trattare i sintomi associati al disturbo da stress post-traumatico usando ciclobenzaprina |
| US20110319389A1 (en) | 2010-06-24 | 2011-12-29 | Tonix Pharmaceuticals, Inc. | Methods and compositions for treating fatigue associated with disordered sleep using very low dose cyclobenzaprine |
| ES2705027T3 (es) | 2010-08-19 | 2019-03-21 | Buck Institute For Age Res | Métodos de tratamiento del deterioro cognitivo leve (DCL) y trastornos relacionados |
| US20120136416A1 (en) * | 2010-11-19 | 2012-05-31 | President And Fellows Of Harvard College | Caged Neuropeptides: modulation and measurement of synapse and signaling activity; and methods for drug evaluation, pharmaceuticals preparation and treatment |
| US11998516B2 (en) | 2011-03-07 | 2024-06-04 | Tonix Pharma Holdings Limited | Methods and compositions for treating depression using cyclobenzaprine |
| WO2012145234A2 (en) * | 2011-04-21 | 2012-10-26 | Emory University | Cyclopropyl derivatives and methods of use |
| CN103030567A (zh) * | 2012-07-25 | 2013-04-10 | 浙江工业大学 | 一种普萘洛尔药物对映体拆分方法 |
| SMT202400324T1 (it) | 2013-03-15 | 2024-11-15 | Tonix Pharma Ltd | Formulazioni eutettiche di ciclobenzaprina cloridrato e mannitolo |
| WO2014203140A1 (en) | 2013-06-22 | 2014-12-24 | Wockhardt Limited | Solid oral pharmaceutical compositions comprising fixed dose combination of acetaminophen, dicyclomine and dextropropoxyphene or salts thereof |
| CA3042642A1 (en) | 2013-08-12 | 2015-02-19 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded immediate release abuse deterrent pill |
| US10172797B2 (en) | 2013-12-17 | 2019-01-08 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| US9492444B2 (en) | 2013-12-17 | 2016-11-15 | Pharmaceutical Manufacturing Research Services, Inc. | Extruded extended release abuse deterrent pill |
| CA2955229C (en) | 2014-07-17 | 2020-03-10 | Pharmaceutical Manufacturing Research Services, Inc. | Immediate release abuse deterrent liquid fill dosage form |
| MY186047A (en) | 2014-09-18 | 2021-06-17 | Tonix Pharma Holdings Ltd | Eutectic formulations of cyclobenzaprine hydrochloride |
| AU2015336065A1 (en) | 2014-10-20 | 2017-05-04 | Pharmaceutical Manufacturing Research Services, Inc. | Extended release abuse deterrent liquid fill dosage form |
| MX2020006140A (es) | 2017-12-11 | 2020-08-13 | Tonix Pharma Holdings Ltd | Tratamiento con ciclobenzaprina para la agitacion, psicosis y deterioro cognitivo en la demencia y enfermedades neurodegenerativas. |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US478836A (en) * | 1892-07-12 | Eyeglass-guard | ||
| FR2508035A1 (fr) | 1981-06-23 | 1982-12-24 | Fabre Sa Pierre | Derives d'aryl-1-aminomethyl-2 cyclopropanes carboxamides (z), leur preparation et leur application en tant que medicaments utiles dans le traitement des troubles du systeme nerveux central |
| JP2780115B2 (ja) | 1989-07-24 | 1998-07-30 | 旭化成工業株式会社 | 虚血性脳細胞障害に対する細胞保護剤 |
| WO1995022521A1 (en) * | 1994-02-22 | 1995-08-24 | Asahi Kasei Kogyo Kabushiki Kaisha | Aminoalkylcyclopropane derivative |
| US6602911B2 (en) * | 2001-11-05 | 2003-08-05 | Cypress Bioscience, Inc. | Methods of treating fibromyalgia |
-
2003
- 2003-10-22 JP JP2005501895A patent/JP4288263B2/ja not_active Expired - Fee Related
- 2003-10-22 AU AU2003284342A patent/AU2003284342B2/en not_active Ceased
- 2003-10-22 EP EP03776524A patent/EP1578719A4/en not_active Withdrawn
- 2003-10-22 US US10/691,465 patent/US7038085B2/en not_active Expired - Lifetime
- 2003-10-22 CA CA002503381A patent/CA2503381A1/en not_active Abandoned
- 2003-10-22 MX MXPA05004381A patent/MXPA05004381A/es active IP Right Grant
- 2003-10-22 WO PCT/US2003/033681 patent/WO2004039320A2/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| AU2003284342B2 (en) | 2009-03-05 |
| US7038085B2 (en) | 2006-05-02 |
| AU2003284342A1 (en) | 2004-05-25 |
| MXPA05004381A (es) | 2006-02-10 |
| EP1578719A4 (en) | 2006-07-05 |
| CA2503381A1 (en) | 2004-05-13 |
| US20040142904A1 (en) | 2004-07-22 |
| JP2006503920A (ja) | 2006-02-02 |
| WO2004039320A2 (en) | 2004-05-13 |
| EP1578719A2 (en) | 2005-09-28 |
| WO2004039320A3 (en) | 2004-06-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4288263B2 (ja) | p−ヒドロキシ−ミルナシプランの立体異性体およびその使用方法 | |
| US7671087B2 (en) | Amines that inhibit a mammalian anandamide transporter, and methods of use thereof | |
| JP2002517457A (ja) | β2−アドレナリン作動性レセプターアゴニスト | |
| Brust et al. | Molecular imaging of σ1 receptors in vivo: current status and perspectives | |
| WO2002022579A2 (en) | Antipsychotic sulfonamide-heterocycles, and methods of use thereof | |
| CN1156454A (zh) | N-取代的3-氮杂双环[3,2,0]庚烷衍生物及其制备方法和应用 | |
| US7361666B2 (en) | Heterocyclic analgesic compounds and methods of use thereof | |
| WO2002046156A2 (en) | 4,4-disubstituted piperidines for use as dopamine, serotonin and norepinephrine ligands | |
| US7115664B2 (en) | Peptidomimetic ligands for cellular receptors and ion channels | |
| US6982271B1 (en) | Tropane analogs | |
| EP1998771B1 (en) | Use of tetrahydropyridines in the treatment of central nervous system disorders | |
| US7030122B2 (en) | 1,4-disubstituted piperazine ligands for neurotransmitter receptors | |
| US20030176314A1 (en) | Compounds for the treatment of pain | |
| WO2003026657A1 (en) | Compounds for the treatment of pain | |
| US20080234247A1 (en) | Heterocyclic analgesic compounds and methods of use thereof | |
| WO2002083071A2 (en) | Heterocyclic analgesic compounds and methods of use thereof | |
| JP2007509168A (ja) | ドーパミン、ノルエピネフリンおよびセロトニンの輸送体に選択的なヘテロ環式化合物およびその治療的使用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20080930 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20081226 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090115 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090129 |
|
| A524 | Written submission of copy of amendment under article 19 pct |
Free format text: JAPANESE INTERMEDIATE CODE: A524 Effective date: 20090129 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20090310 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20090330 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20120403 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| LAPS | Cancellation because of no payment of annual fees |