JP4536655B2 - 細胞導入剤、細胞導入方法、細胞導入剤の製造方法、細胞導入剤製造用組成物、および、細胞導入剤製造用キット - Google Patents
細胞導入剤、細胞導入方法、細胞導入剤の製造方法、細胞導入剤製造用組成物、および、細胞導入剤製造用キット Download PDFInfo
- Publication number
- JP4536655B2 JP4536655B2 JP2005505672A JP2005505672A JP4536655B2 JP 4536655 B2 JP4536655 B2 JP 4536655B2 JP 2005505672 A JP2005505672 A JP 2005505672A JP 2005505672 A JP2005505672 A JP 2005505672A JP 4536655 B2 JP4536655 B2 JP 4536655B2
- Authority
- JP
- Japan
- Prior art keywords
- cell
- cell introduction
- introduction agent
- producing
- composition
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000000203 mixture Substances 0.000 title claims description 86
- 238000004519 manufacturing process Methods 0.000 title claims description 49
- 238000000034 method Methods 0.000 title claims description 31
- 210000004027 cell Anatomy 0.000 claims description 244
- VSIIXMUUUJUKCM-UHFFFAOYSA-D pentacalcium;fluoride;triphosphate Chemical compound [F-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O VSIIXMUUUJUKCM-UHFFFAOYSA-D 0.000 claims description 124
- 229910052586 apatite Inorganic materials 0.000 claims description 122
- 239000003795 chemical substances by application Substances 0.000 claims description 115
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 claims description 92
- 239000011246 composite particle Substances 0.000 claims description 73
- 239000002245 particle Substances 0.000 claims description 71
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 claims description 58
- 229910001424 calcium ion Inorganic materials 0.000 claims description 58
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 claims description 39
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 34
- 108091033319 polynucleotide Proteins 0.000 claims description 26
- 102000040430 polynucleotide Human genes 0.000 claims description 26
- 239000002157 polynucleotide Substances 0.000 claims description 26
- 238000012546 transfer Methods 0.000 claims description 23
- 229940085991 phosphate ion Drugs 0.000 claims description 12
- 238000002156 mixing Methods 0.000 claims description 10
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 claims description 8
- 210000004102 animal cell Anatomy 0.000 claims description 4
- 229910001427 strontium ion Inorganic materials 0.000 claims description 3
- PWYYWQHXAPXYMF-UHFFFAOYSA-N strontium(2+) Chemical compound [Sr+2] PWYYWQHXAPXYMF-UHFFFAOYSA-N 0.000 claims description 2
- 108020004414 DNA Proteins 0.000 description 94
- 239000013076 target substance Substances 0.000 description 62
- 108090000623 proteins and genes Proteins 0.000 description 38
- 239000002609 medium Substances 0.000 description 37
- 230000014509 gene expression Effects 0.000 description 32
- 239000000243 solution Substances 0.000 description 29
- 238000001890 transfection Methods 0.000 description 29
- 229910052731 fluorine Inorganic materials 0.000 description 22
- 239000011737 fluorine Substances 0.000 description 21
- 229910052588 hydroxylapatite Inorganic materials 0.000 description 21
- XYJRXVWERLGGKC-UHFFFAOYSA-D pentacalcium;hydroxide;triphosphate Chemical compound [OH-].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O XYJRXVWERLGGKC-UHFFFAOYSA-D 0.000 description 21
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 19
- 239000001506 calcium phosphate Substances 0.000 description 18
- 229960001714 calcium phosphate Drugs 0.000 description 18
- 229910000389 calcium phosphate Inorganic materials 0.000 description 18
- 235000011010 calcium phosphates Nutrition 0.000 description 18
- 230000008859 change Effects 0.000 description 18
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 18
- 210000001163 endosome Anatomy 0.000 description 17
- 239000013612 plasmid Substances 0.000 description 17
- YCKRFDGAMUMZLT-UHFFFAOYSA-N Fluorine atom Chemical compound [F] YCKRFDGAMUMZLT-UHFFFAOYSA-N 0.000 description 16
- 108060001084 Luciferase Proteins 0.000 description 14
- 239000005089 Luciferase Substances 0.000 description 14
- 239000000463 material Substances 0.000 description 13
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- -1 Enedynes Chemical compound 0.000 description 10
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 10
- 239000013078 crystal Substances 0.000 description 10
- 239000003814 drug Substances 0.000 description 10
- 238000012258 culturing Methods 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 230000000694 effects Effects 0.000 description 9
- 229910052712 strontium Inorganic materials 0.000 description 9
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 8
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 8
- XDHNQDDQEHDUTM-UHFFFAOYSA-N bafliomycin A1 Natural products COC1C=CC=C(C)CC(C)C(O)C(C)C=C(C)C=C(OC)C(=O)OC1C(C)C(O)C(C)C1(O)OC(C(C)C)C(C)C(O)C1 XDHNQDDQEHDUTM-UHFFFAOYSA-N 0.000 description 8
- 239000012091 fetal bovine serum Substances 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 241000700605 Viruses Species 0.000 description 7
- 238000002441 X-ray diffraction Methods 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 239000000872 buffer Substances 0.000 description 7
- 239000011575 calcium Substances 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- 238000004090 dissolution Methods 0.000 description 7
- 230000012202 endocytosis Effects 0.000 description 7
- 239000000843 powder Substances 0.000 description 7
- 102000004169 proteins and genes Human genes 0.000 description 7
- 230000002378 acidificating effect Effects 0.000 description 6
- 230000003247 decreasing effect Effects 0.000 description 6
- 239000007788 liquid Substances 0.000 description 6
- PUZPDOWCWNUUKD-UHFFFAOYSA-M sodium fluoride Chemical compound [F-].[Na+] PUZPDOWCWNUUKD-UHFFFAOYSA-M 0.000 description 6
- CIOAGBVUUVVLOB-UHFFFAOYSA-N strontium atom Chemical compound [Sr] CIOAGBVUUVVLOB-UHFFFAOYSA-N 0.000 description 6
- XDHNQDDQEHDUTM-XJKSCTEHSA-N (3z,5e,7r,8s,9r,11e,13e,15s,16r)-16-[(2s,3r,4s)-4-[(2r,4r,5s,6r)-2,4-dihydroxy-5-methyl-6-propan-2-yloxan-2-yl]-3-hydroxypentan-2-yl]-8-hydroxy-3,15-dimethoxy-5,7,9,11-tetramethyl-1-oxacyclohexadeca-3,5,11,13-tetraen-2-one Chemical compound CO[C@H]1\C=C\C=C(C)\C[C@@H](C)[C@H](O)[C@H](C)\C=C(/C)\C=C(OC)\C(=O)O[C@@H]1[C@@H](C)[C@@H](O)[C@H](C)[C@]1(O)O[C@H](C(C)C)[C@@H](C)[C@H](O)C1 XDHNQDDQEHDUTM-XJKSCTEHSA-N 0.000 description 5
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Natural products CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 230000007423 decrease Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 238000001415 gene therapy Methods 0.000 description 5
- 210000003494 hepatocyte Anatomy 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 238000011160 research Methods 0.000 description 5
- 238000010361 transduction Methods 0.000 description 5
- 230000026683 transduction Effects 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 238000000134 MTT assay Methods 0.000 description 4
- 231100000002 MTT assay Toxicity 0.000 description 4
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 4
- 230000009368 gene silencing by RNA Effects 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 230000001293 nucleolytic effect Effects 0.000 description 4
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 4
- 238000005215 recombination Methods 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 241000699666 Mus <mouse, genus> Species 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 3
- 108700019146 Transgenes Proteins 0.000 description 3
- 108700005077 Viral Genes Proteins 0.000 description 3
- 229930192649 bafilomycin Natural products 0.000 description 3
- XDHNQDDQEHDUTM-ZGOPVUMHSA-N bafilomycin A1 Natural products CO[C@H]1C=CC=C(C)C[C@H](C)[C@H](O)[C@H](C)C=C(C)C=C(OC)C(=O)O[C@@H]1[C@@H](C)[C@@H](O)[C@H](C)[C@]1(O)O[C@H](C(C)C)[C@@H](C)[C@H](O)C1 XDHNQDDQEHDUTM-ZGOPVUMHSA-N 0.000 description 3
- 229960005069 calcium Drugs 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000003915 cell function Effects 0.000 description 3
- 230000003833 cell viability Effects 0.000 description 3
- 239000000460 chlorine Substances 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- 238000000975 co-precipitation Methods 0.000 description 3
- 210000000805 cytoplasm Anatomy 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 238000002329 infrared spectrum Methods 0.000 description 3
- 239000003112 inhibitor Substances 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 238000001638 lipofection Methods 0.000 description 3
- 210000004962 mammalian cell Anatomy 0.000 description 3
- 229910052748 manganese Inorganic materials 0.000 description 3
- 238000005259 measurement Methods 0.000 description 3
- 230000006798 recombination Effects 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000012679 serum free medium Substances 0.000 description 3
- 239000011775 sodium fluoride Substances 0.000 description 3
- 235000013024 sodium fluoride Nutrition 0.000 description 3
- 229910001631 strontium chloride Inorganic materials 0.000 description 3
- AHBGXTDRMVNFER-UHFFFAOYSA-L strontium dichloride Chemical compound [Cl-].[Cl-].[Sr+2] AHBGXTDRMVNFER-UHFFFAOYSA-L 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- VRYALKFFQXWPIH-PBXRRBTRSA-N (3r,4s,5r)-3,4,5,6-tetrahydroxyhexanal Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)CC=O VRYALKFFQXWPIH-PBXRRBTRSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- 108020005544 Antisense RNA Proteins 0.000 description 2
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 102000008186 Collagen Human genes 0.000 description 2
- 108010035532 Collagen Proteins 0.000 description 2
- 238000004566 IR spectroscopy Methods 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- 102100032709 Potassium-transporting ATPase alpha chain 2 Human genes 0.000 description 2
- 108010083204 Proton Pumps Proteins 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- PMMURAAUARKVCB-UHFFFAOYSA-N alpha-D-ara-dHexp Natural products OCC1OC(O)CC(O)C1O PMMURAAUARKVCB-UHFFFAOYSA-N 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000007853 buffer solution Substances 0.000 description 2
- 230000003139 buffering effect Effects 0.000 description 2
- 229920001436 collagen Polymers 0.000 description 2
- 239000003184 complementary RNA Substances 0.000 description 2
- 210000004748 cultured cell Anatomy 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 238000005265 energy consumption Methods 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000003068 molecular probe Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 229920000447 polyanionic polymer Polymers 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 238000003151 transfection method Methods 0.000 description 2
- 229960003048 vinblastine Drugs 0.000 description 2
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- WHTVZRBIWZFKQO-AWEZNQCLSA-N (S)-chloroquine Chemical compound ClC1=CC=C2C(N[C@@H](C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-AWEZNQCLSA-N 0.000 description 1
- 101150028074 2 gene Proteins 0.000 description 1
- AZKSAVLVSZKNRD-UHFFFAOYSA-M 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide Chemical compound [Br-].S1C(C)=C(C)N=C1[N+]1=NC(C=2C=CC=CC=2)=NN1C1=CC=CC=C1 AZKSAVLVSZKNRD-UHFFFAOYSA-M 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 241000186361 Actinobacteria <class> Species 0.000 description 1
- JQDZUSDVVHXANW-UHFFFAOYSA-N C1=CC(=C23)C4=NC5=CC=CC=C5N4C(=O)C2=CC=CC3=C1NCCN1CCOCC1 Chemical compound C1=CC(=C23)C4=NC5=CC=CC=C5N4C(=O)C2=CC=CC3=C1NCCN1CCOCC1 JQDZUSDVVHXANW-UHFFFAOYSA-N 0.000 description 1
- 101100297347 Caenorhabditis elegans pgl-3 gene Proteins 0.000 description 1
- 108010041986 DNA Vaccines Proteins 0.000 description 1
- 230000007064 DNA hydrolysis Effects 0.000 description 1
- 229940021995 DNA vaccine Drugs 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 238000001157 Fourier transform infrared spectrum Methods 0.000 description 1
- 101150066002 GFP gene Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000934888 Homo sapiens Succinate dehydrogenase cytochrome b560 subunit, mitochondrial Proteins 0.000 description 1
- 208000026350 Inborn Genetic disease Diseases 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 101710204212 Neocarzinostatin Proteins 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 102100025393 Succinate dehydrogenase cytochrome b560 subunit, mitochondrial Human genes 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 238000000333 X-ray scattering Methods 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000003698 anagen phase Effects 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000003432 anti-folate effect Effects 0.000 description 1
- 229940127074 antifolate Drugs 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 210000004507 artificial chromosome Anatomy 0.000 description 1
- 238000001636 atomic emission spectroscopy Methods 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006143 cell culture medium Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 150000001804 chlorine Chemical class 0.000 description 1
- 229960003677 chloroquine Drugs 0.000 description 1
- WHTVZRBIWZFKQO-UHFFFAOYSA-N chloroquine Natural products ClC1=CC=C2C(NC(C)CCCN(CC)CC)=CC=NC2=C1 WHTVZRBIWZFKQO-UHFFFAOYSA-N 0.000 description 1
- 230000008045 co-localization Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 229910021641 deionized water Inorganic materials 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 208000016361 genetic disease Diseases 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- GPRLSGONYQIRFK-UHFFFAOYSA-N hydron Chemical compound [H+] GPRLSGONYQIRFK-UHFFFAOYSA-N 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000013028 medium composition Substances 0.000 description 1
- 239000000155 melt Substances 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- QZGIWPZCWHMVQL-UIYAJPBUSA-N neocarzinostatin chromophore Chemical compound O1[C@H](C)[C@H](O)[C@H](O)[C@@H](NC)[C@H]1O[C@@H]1C/2=C/C#C[C@H]3O[C@@]3([C@@H]3OC(=O)OC3)C#CC\2=C[C@H]1OC(=O)C1=C(O)C=CC2=C(C)C=C(OC)C=C12 QZGIWPZCWHMVQL-UIYAJPBUSA-N 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000013630 prepared media Substances 0.000 description 1
- 229910052761 rare earth metal Inorganic materials 0.000 description 1
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 1
- 238000001878 scanning electron micrograph Methods 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000007974 sodium acetate buffer Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 230000001502 supplementing effect Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
- 229950009268 zinostatin Drugs 0.000 description 1
Images

























Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K48/00—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy
- A61K48/0008—Medicinal preparations containing genetic material which is inserted into cells of the living body to treat genetic diseases; Gene therapy characterised by an aspect of the 'non-active' part of the composition delivered, e.g. wherein such 'non-active' part is not delivered simultaneously with the 'active' part of the composition
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
[1] 目的の物質を細胞内に導入するための細胞導入剤であって、
少なくとも前記目的の物質およびリン酸カルシウム系材料から構成される複合体粒子を含有し、
pH8.0からpH6.0に変化させた場合において、pH6.0に変化させてから所定時間内に、pH8.0において存在していた前記複合体粒子の少なくとも50%が溶解する特性を有することを特徴とする細胞導入剤。
[2] 前記複合体粒子の平均粒径が、500nm以下であることを特徴とする[1]に記載の細胞導入剤。
[3] 前記リン酸カルシウム系材料が、炭酸アパタイトであることを特徴とする[1]または[2]に記載の細胞導入剤。
[4] 前記目的の物質が、マイナスに帯電している物質である[1]〜[3]のいずれか一項に記載の細胞導入剤。
[5] 前記目的の物質が、薬剤、タンパク質、およびポリヌクレオチドからなる群より選ばれる少なくとも一種の物質であることを特徴とする[1]〜[4]のいずれか一項に記載の細胞導入剤。
[6] [1]〜[5]のいずれか一項に記載の細胞導入剤を用いて、目的の物質を細胞内に導入することを特徴とする細胞導入方法。
[7] 目的の物質を細胞内に導入するために用いられ前記目的の物質および炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤の製造方法であって、
少なくともカルシウムイオン、リン酸イオン、炭酸水素イオン、および前記目的の物質を含有する組成物を調製することにより、前記複合体粒子を形成する工程を含むことを特徴とする細胞導入剤の製造方法。
[8] 前記カルシウムイオンおよび前記目的の物質を含有する第1の溶液を調製する工程、
前記リン酸イオンおよび前記炭酸水素イオンを含有する第2の溶液を調製する工程、および、
前記第1の溶液と前記第2の溶液とを混合して前記組成物を調製する工程を含むことを特徴とする[7]に記載の細胞導入剤の製造方法。
[9] 前記組成物のカルシウムイオン濃度が、0.1mM以上であることを特徴とする[7]または[8]に記載の細胞導入剤の製造方法。
[10] 前記組成物のリン酸イオン濃度が、0.1mM以上であることを特徴とする[7]〜[9]のいずれか一項に記載の細胞導入剤の製造方法。
[11] 前記組成物の炭酸水素イオン濃度が、1.0mM以上であることを特徴とする[7]〜[10]のいずれか一項に記載の細胞導入剤の製造方法。
[12] 前記組成物が、さらにフッ素イオンまたはストロンチウムイオンを含有することを特徴とする[7]〜[11]のいずれか一項に記載の細胞導入剤の製造方法。
[13] 前記組成物のpHが、pH6.0〜9.0であることを特徴とする[7]〜[12]のいずれか一項に記載の細胞導入剤の製造方法。
[14] 前記組成物を10℃以上に保持することにより、前記複合体粒子を形成することを特徴とする[7]〜[13]のいずれか一項に記載の細胞導入剤の製造方法。
[15] 目的の物質を細胞内に導入するために用いられ前記目的の物質および炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤を製造するための細胞導入剤製造用組成物であって、
少なくともカルシウムイオン、リン酸イオン、および炭酸水素イオンを含有し、
前記目的の物質を添加することにより前記細胞導入剤を製造することを特徴とする細胞導入剤製造用組成物。
[16] 目的の物質を細胞内に導入するために用いられ前記目的の物質および炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤を製造するための細胞導入剤製造用組成物であって、
少なくともリン酸イオン、および炭酸水素イオンを含有し、
前記目的の物質とカルシウムイオンを添加することにより前記細胞導入剤を製造することを特徴とする細胞導入剤製造用組成物。
[17] 目的の物質を細胞内に導入するために用いられ前記目的の物質および炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤を製造するための細胞導入剤製造用キットであって、
少なくともカルシウムイオンを含有する第1成分と、
少なくともリン酸イオンおよび炭酸水素イオンを含有する第2成分とからなり、
前記第1成分に前記目的の物質を添加した後、当該第1成分および前記第2成分を混合することにより、前記複合体粒子を含有する細胞導入剤を製造することを特徴とする細胞導入剤製造用キット。
〔I〕細胞導入剤
〔II〕細胞導入剤の製造方法
〔III〕細胞導入方法
本発明の細胞導入剤は、目的の物質を細胞内に導入するために用いられるものであり、少なくとも目的の物質およびリン酸カルシウム系材料から構成される複合体粒子を含有するものである。
次に、本発明の細胞導入剤の製造方法について説明する。本発明の細胞導入剤の製造方法は、少なくともカルシウムイオン、リン酸イオン、炭酸水素イオン、および、目的の物質の4成分を必須成分として含有する組成物を調製することにより、前記複合体粒子を形成することを特徴とする方法である。この組成物は水溶液として調製することが好ましい。
本発明の細胞導入方法は、本発明の細胞導入剤を用いて、目的の物質を細胞内に導入することを特徴とする。ここで、細胞に導入する目的物質は、細胞機能を調整することができるマイナスに帯電した物質であることが好ましい。
〔1〕細胞導入剤の調製
3〜6μ1の1M塩化カルシウム溶液を2μ1のプラスミドDNAを含む無血清炭酸水素イオン緩衝(DMEMもしくはWE)培地(pH7.5)1mlに懸濁し、37℃で30分培養し、DNA/炭酸アパタイトの複合体粒子を生じさせた。

生成した粒子を化学分析、赤外分光法、X線回折によって分析した。
上記と同じ方法で、6mMカルシウムイオンを用いDNAを加えず炭酸アパタイトを生成し、遠心分離および蒸留脱イオン水で沈殿した粒子を凍結乾燥させた。他のアパタイト粒子も上記のように精製して凍結乾燥させた。カルシウムとリンの成分はSPS1500 VR Atomic Absorption Spectrophotometerを用いて測定した。炭素とフッ素はCHNS−932(Leco,USA)とSX−elements micro analyzer,YS−10(Yanaco,日本)をそれぞれ用いて測定した。
上記のように調製したアパタイト粒子のフーリエ変換赤外分光分析はFT/IR−230(JASCO)を用いて行った。サンプルをすり鉢の中にいれ、そして約1mgを完全に300mgの分光用の臭化カリウムと混ぜ合わせた。臭化カリウムの窓板の中で0.5torr減気圧下において8000kgの加重を加えることで薄いペレットを調製した。
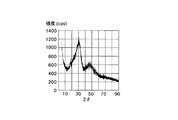
調製した粒子のX線粉体解析はM18XHF−SRA回折システムを用いて行った。
〔1〕ルシフェラーゼの発現レベル測定
実施例1〔1〕によって調製した本発明の細胞導入剤のトランスフェクション効率を調べるため、本発明の細胞導入剤を用いたルシフェラーゼの発現レベルを測定した。
細胞の生存率が高いためにトランスフェクション効率が高く現れたのではないことを明らかにするためにHeLa細胞を用いてMTTアッセイを行った。
(走査型電子顕微鏡(SEM))
遺伝子導入のプロトコールで述べた様に調製したDNA−炭酸アパタイトの混合液の液滴を炭素コートのSEMのステージに加え、乾燥させた後に、SEM(S−800、日立、日本)で観察した。
巨大粒子は小さい粒子よりも食作用によって取り込まれにくいので、本発明者らは炭酸塩の結晶の大きさを抑制したときの効果の検証を、アパタイトに吸着させたPI(propidium iodide)ラベルしたプラスミドDNAの細胞への取り込みを観察することで行った。
細胞導入におけるpHの影響を調べるため、V−ATPase(エンドソームを酸性化するのに用いられるプロトンポンプ)の特異的阻害剤であるバフィロマイシンAlを用いてトランスフェクション効率の変化を調べた。HeLa細胞をDNA/炭酸アパタイト複合体粒子および200nMバフィロマイシンAlとともに6時間培養した。5mMのEDTA含有PBSで洗ったあと、細胞を1日培養し、ルシフェラーゼの発現を検出した。結果を図4−1に示す。
アパタイトの溶解度とトランスフェクション効率の間の関係を調べるために、生成したアパタイトの液中に酸を添加した後の、溶液の濁度を溶解度の指標として測定した。
Claims (17)
- ポリヌクレオチドを細胞内に導入するために用いられ前記ポリヌクレオチドおよび炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤の製造方法であって、
少なくともカルシウムイオン、リン酸イオン、炭酸水素イオン、および前記ポリヌクレオチドを含有する組成物を調製することにより、前記複合体粒子を形成する工程を含むことを特徴とする細胞導入剤の製造方法。 - 前記カルシウムイオンおよび前記ポリヌクレオチドを含有する第1の溶液を調製する工程、
前記リン酸イオンおよび前記炭酸水素イオンを含有する第2の溶液を調製する工程、および、
前記第1の溶液と前記第2の溶液とを混合して前記組成物を調製する工程を含むことを特徴とする請求項1に記載の細胞導入剤の製造方法。 - 前記組成物のカルシウムイオン濃度が、0.1mM以上であることを特徴とする請求項1または2に記載の細胞導入剤の製造方法。
- 前記組成物のリン酸イオン濃度が、0.1mM以上であることを特徴とする請求項1〜3のいずれか一項に記載の細胞導入剤の製造方法。
- 前記組成物の炭酸水素イオン濃度が、1.0mM以上であることを特徴とする請求項1〜4のいずれか一項に記載の細胞導入剤の製造方法。
- 前記組成物が、さらにフッ素イオンまたはストロンチウムイオンを含有することを特徴とする請求項1〜5のいずれか一項に記載の細胞導入剤の製造方法。
- 前記組成物のpHが、pH6.0〜9.0であることを特徴とする請求項1〜6のいずれか一項に記載の細胞導入剤の製造方法。
- 前記組成物を10℃以上に保持することにより、前記複合体粒子を形成することを特徴とする請求項1〜7のいずれか一項に記載の細胞導入剤の製造方法。
- 前記細胞導入剤は、ポリヌクレオチドを、ヒトから単離された、またはヒト以外の動物細胞の細胞内に導入するために用いられる、請求項1〜8のいずれか一項に記載の細胞導入剤の製造方法。
- 前記複合体粒子の平均粒径が、500nm以下であることを特徴とする請求項1に記載の細胞導入剤の製造方法。
- 前記ポリヌクレオチドが、DNAおよび/またはRNAであることを特徴とする請求項1〜11のいずれか一項に記載の細胞導入剤の製造方法。
- 請求項1〜11のいずれか一項に記載の製造方法により製造される細胞導入剤を用いて、ポリヌクレオチドを、ヒトから単離された、またはヒト以外の動物細胞の細胞内に導入することを特徴とする細胞導入方法。
- 前記ポリヌクレオチドが、DNAおよび/またはRNAであることを特徴とする請求項12に記載の細胞導入方法。
- ポリヌクレオチドを細胞内に導入するために用いられ前記ポリヌクレオチドおよび炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤を製造するための細胞導入剤製造用組成物であって、
少なくともカルシウムイオン、リン酸イオン、および炭酸水素イオンを含有し、
前記ポリヌクレオチドを添加することにより前記細胞導入剤を製造することを特徴とする細胞導入剤製造用組成物。 - ポリヌクレオチドを細胞内に導入するために用いられ前記ポリヌクレオチドおよび炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤を製造するための細胞導入剤製造用組成物であって、
少なくともリン酸イオン、および炭酸水素イオンを含有し、
前記ポリヌクレオチドとカルシウムイオンを添加することにより前記細胞導入剤を製造することを特徴とする細胞導入剤製造用組成物。 - 前記ポリヌクレオチドが、DNAおよび/またはRNAであることを特徴とする請求項14または15に記載の細胞導入剤製造用組成物。
- ポリヌクレオチドを細胞内に導入するために用いられ前記ポリヌクレオチドおよび炭酸アパタイトから構成される複合体粒子を含有する細胞導入剤を製造するための細胞導入剤製造用キットであって、
少なくともカルシウムイオンを含有する第1成分と、
少なくともリン酸イオンおよび炭酸水素イオンを含有する第2成分とからなり、
前記第1成分に前記ポリヌクレオチドを添加した後、当該第1成分および前記第2成分を混合することにより、前記複合体粒子を含有する細胞導入剤を製造することを特徴とする細胞導入剤製造用キット。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US42529102P | 2002-11-12 | 2002-11-12 | |
| US46727703P | 2003-05-02 | 2003-05-02 | |
| PCT/JP2003/014376 WO2004043495A1 (en) | 2002-11-12 | 2003-11-12 | Delivery agent, method of delivering a target substance to cells, method for producing delivery agent, composition for producing delivery agent, and kit for producing delivery agent |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2006509838A JP2006509838A (ja) | 2006-03-23 |
| JP4536655B2 true JP4536655B2 (ja) | 2010-09-01 |
Family
ID=32314583
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2005505672A Expired - Lifetime JP4536655B2 (ja) | 2002-11-12 | 2003-11-12 | 細胞導入剤、細胞導入方法、細胞導入剤の製造方法、細胞導入剤製造用組成物、および、細胞導入剤製造用キット |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JP4536655B2 (ja) |
| AU (1) | AU2003279570A1 (ja) |
| WO (1) | WO2004043495A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12486504B2 (en) | 2021-01-19 | 2025-12-02 | Hirofumi Yamamoto | Drug delivery composition, method for producing same and use thereof |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2007043972A (ja) * | 2005-08-10 | 2007-02-22 | Tokyo Institute Of Technology | アパタイト粒子及びその作製方法、遺伝子−アパタイト粒子複合体及びその作製方法、並びに遺伝子導入方法及び遺伝子導入用キット |
| JP2007063194A (ja) * | 2005-08-31 | 2007-03-15 | Tokyo Institute Of Technology | 物質導入担体、物質導入方法及び物質導入用キット |
| JP2007153842A (ja) * | 2005-12-07 | 2007-06-21 | Hideki Aoki | ビタミンアパタイト複合体 |
| JP2008193977A (ja) * | 2007-02-14 | 2008-08-28 | National Institute Of Advanced Industrial & Technology | 遺伝子送達方法および基材 |
| JP5416510B2 (ja) * | 2009-08-26 | 2014-02-12 | 花王株式会社 | 歯の美白剤又は光沢剤のスクリーニング方法 |
| JP5436650B1 (ja) | 2012-11-28 | 2014-03-05 | 浩文 山本 | スーパーアパタイト超微細ナノ粒子 |
| JP6443907B2 (ja) * | 2014-02-17 | 2018-12-26 | 浩文 山本 | 造影剤の腫瘍への集積を促進するための集積促進剤 |
| EP3115454B1 (en) * | 2014-03-04 | 2020-02-12 | Hirofumi Yamamoto | Novel rna sequence having anti-tumour activity |
| JP6588765B2 (ja) * | 2015-08-07 | 2019-10-09 | 学校法人 神野学園 | Dna吸着担体及びその利用方法 |
| US10449155B2 (en) | 2015-11-09 | 2019-10-22 | Medical Corporation Ijunkai | Drug introducing agent for administration into a living body and manufacturing method |
| JP6302531B2 (ja) * | 2015-11-09 | 2018-03-28 | 医療法人 医潤会 | 生体投与を目的とした薬物導入剤および製造方法 |
| WO2018030338A1 (ja) * | 2016-08-08 | 2018-02-15 | 浩文 山本 | アジュバント組成物 |
| EP3616702B1 (en) * | 2017-04-24 | 2022-12-14 | Hirofumi Yamamoto | Prophylactic or therapeutic agent for inflammatory bowel disease |
| WO2018220665A1 (ja) * | 2017-05-29 | 2018-12-06 | 敏宏 赤池 | 細胞導入剤 |
| CN116637159A (zh) * | 2023-06-20 | 2023-08-25 | 上海章合生物科技有限公司 | 一种竹液粒子复合物及其制备方法和应用 |
| CN116870104A (zh) * | 2023-08-07 | 2023-10-13 | 上海章合生物科技有限公司 | 竹液粒子复合物在治疗皮肤炎的药物或日化品中的应用 |
| CN117180373A (zh) * | 2023-08-18 | 2023-12-08 | 上海章合生物科技有限公司 | 竹液及其粒子复合物在促进抗体分泌和增加免疫因子的药物或日化品中的应用 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5407659A (en) * | 1991-10-22 | 1995-04-18 | Mallinckrodt Medical, Inc. | Treated calcium/oxyanion-containing particles for medical diagnostic imaging |
| US5962427A (en) * | 1994-02-18 | 1999-10-05 | The Regent Of The University Of Michigan | In vivo gene transfer methods for wound healing |
| AU723740B2 (en) * | 1995-06-06 | 2000-09-07 | Howmedica Inc. | Biocompatible hydroxyapatite formulations and uses therefor |
| JP2002348234A (ja) * | 2001-05-28 | 2002-12-04 | Purotekku:Kk | 薬物封入無機物微粒子、その製造法及び薬物封入無機物微粒子製剤 |
| JP3898046B2 (ja) * | 2001-12-07 | 2007-03-28 | 独立行政法人科学技術振興機構 | ハイドロキシアパタイトの製造方法及びその利用 |
-
2003
- 2003-11-12 JP JP2005505672A patent/JP4536655B2/ja not_active Expired - Lifetime
- 2003-11-12 WO PCT/JP2003/014376 patent/WO2004043495A1/en not_active Ceased
- 2003-11-12 AU AU2003279570A patent/AU2003279570A1/en not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12486504B2 (en) | 2021-01-19 | 2025-12-02 | Hirofumi Yamamoto | Drug delivery composition, method for producing same and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2006509838A (ja) | 2006-03-23 |
| AU2003279570A1 (en) | 2004-06-03 |
| WO2004043495A1 (en) | 2004-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4536655B2 (ja) | 細胞導入剤、細胞導入方法、細胞導入剤の製造方法、細胞導入剤製造用組成物、および、細胞導入剤製造用キット | |
| Elangovan et al. | Chemically modified RNA activated matrices enhance bone regeneration | |
| ES2667644T3 (es) | UTR que aumentan la eficacia de la traducción de las moléculas de ARN | |
| ES2587963T3 (es) | ARN con una combinación de nucleótidos no modificados y modificados para la expresión de proteínas | |
| Park et al. | Chondrogenesis of human mesenchymal stem cells mediated by the combination of SOX trio SOX5, 6, and 9 genes complexed with PEI-modified PLGA nanoparticles | |
| JP4613011B2 (ja) | invivo遺伝子伝達を改善する医薬組成物 | |
| Yuan et al. | Three birds, one stone: an osteo‐microenvironment stage‐regulative scaffold for bone defect repair through modulating early osteo‐immunomodulation, middle neovascularization, and later osteogenesis | |
| JP5697123B2 (ja) | 酸性化ポリエチレンイミンを用いる細胞への核酸導入方法 | |
| Menale et al. | Mesenchymal stromal cell-seeded biomimetic scaffolds as a factory of soluble RANKL in Rankl-deficient osteopetrosis | |
| Tenkumo et al. | Gene transfection of human mesenchymal stem cells with a nano‐hydroxyapatite–collagen scaffold containing DNA‐functionalized calcium phosphate nanoparticles | |
| Jiang et al. | Bone response to the multilayer BMP‐2 gene coated porous titanium implant surface | |
| US10717980B2 (en) | MicroRNA-200 based approaches for modulating bone formation inhibition and bone regeneration | |
| CN109355310B (zh) | Ros响应的基因递送载体及其制备方法和应用 | |
| Takeshita et al. | Biomineralization of hydroxyapatite on DNA molecules in SBF: morphological features and computer simulation | |
| Wang et al. | An osteoimmunomodulatory Ca2+/Zn2+-doped scaffold promotes M2 macrophage polarization via the src-mediated chemoking signaling pathway to enhance osteoinduction | |
| US12453801B2 (en) | Nucleic acid-calcium phosphate nanoparticle complexes and application thereof in biomineralization | |
| Zhao et al. | Effective delivery of bone morphogenetic protein 2 gene using chitosan–polyethylenimine nanoparticle to promote bone formation | |
| Zhao | Zeng | |
| Yu et al. | 3-D scaffold platform for optimized non-viral transfection of multipotent stem cells | |
| Meng et al. | An injectable miRNA-activated matrix for effective bone regeneration in vivo | |
| Song et al. | MiR-148b laden titanium implant promoting osteogenic differentiation of rat bone marrow mesenchymal stem cells | |
| US20160017368A1 (en) | Inorganic coatings for the enhancement of chemical transfection | |
| WO2019010304A1 (en) | MICROPARTICLES COATED WITH MINERALS FOR GENES ADMINISTRATION IN CHRONIC WOUND THERAPY | |
| US20070077306A1 (en) | Apatite particle, method of producing the same, apatite particle-gene complex, and method of gene transfection | |
| Düzgüneş et al. | Genetic nanomedicine: gene delivery by targeted lipoplexes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20061010 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20071023 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A711 Effective date: 20071023 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100126 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100326 |
|
| RD02 | Notification of acceptance of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7422 Effective date: 20100326 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20100329 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100525 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100616 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20130625 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 4536655 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| S111 | Request for change of ownership or part of ownership |
Free format text: JAPANESE INTERMEDIATE CODE: R313113 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |