JP4988694B2 - 金属イオンアフィニティークロマトグラフィーによるペプチドの精製 - Google Patents
金属イオンアフィニティークロマトグラフィーによるペプチドの精製 Download PDFInfo
- Publication number
- JP4988694B2 JP4988694B2 JP2008321467A JP2008321467A JP4988694B2 JP 4988694 B2 JP4988694 B2 JP 4988694B2 JP 2008321467 A JP2008321467 A JP 2008321467A JP 2008321467 A JP2008321467 A JP 2008321467A JP 4988694 B2 JP4988694 B2 JP 4988694B2
- Authority
- JP
- Japan
- Prior art keywords
- gst
- protein
- buffer
- amino acid
- adsorbent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 0 CC=C(CC(COC1CCCC1)O)CC(CO[C@]1*CCC1)O Chemical compound CC=C(CC(COC1CCCC1)O)CC(CO[C@]1*CCC1)O 0.000 description 1
- DNESHXZRWPOZTB-UHFFFAOYSA-N CCc1cc(CC)c(CC)cc1C Chemical compound CCc1cc(CC)c(CC)cc1C DNESHXZRWPOZTB-UHFFFAOYSA-N 0.000 description 1
Images




















































Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J7/00—Chemical treatment or coating of shaped articles made of macromolecular substances
- C08J7/12—Chemical modification
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 and B01D15/30 - B01D15/36, e.g. affinity, ligand exchange or chiral chromatography
- B01D15/3804—Affinity chromatography
- B01D15/3828—Ligand exchange chromatography, e.g. complexation, chelation or metal interaction chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/261—Synthetic macromolecular compounds obtained by reactions only involving carbon to carbon unsaturated bonds
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/262—Synthetic macromolecular compounds obtained otherwise than by reactions only involving carbon to carbon unsaturated bonds, e.g. obtained by polycondensation
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/22—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising organic material
- B01J20/26—Synthetic macromolecular compounds
- B01J20/265—Synthetic macromolecular compounds modified or post-treated polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J20/282—Porous sorbents
- B01J20/285—Porous sorbents based on polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J20/286—Phases chemically bonded to a substrate, e.g. to silica or to polymers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/30—Processes for preparing, regenerating, or reactivating
- B01J20/32—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating
- B01J20/3231—Impregnating or coating ; Solid sorbent compositions obtained from processes involving impregnating or coating characterised by the coating or impregnating layer
- B01J20/3242—Layers with a functional group, e.g. an affinity material, a ligand, a reactant or a complexing group
- B01J20/3244—Non-macromolecular compounds
- B01J20/3265—Non-macromolecular compounds with an organic functional group containing a metal, e.g. a metal affinity ligand
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J45/00—Ion-exchange in which a complex or a chelate is formed; Use of material as complex or chelate forming ion-exchangers; Treatment of material for improving the complex or chelate forming ion-exchange properties
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/08—Linear peptides containing only normal peptide links having 12 to 20 amino acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B11/00—Preparation of cellulose ethers
- C08B11/02—Alkyl or cycloalkyl ethers
- C08B11/04—Alkyl or cycloalkyl ethers with substituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B15/00—Preparation of other cellulose derivatives or modified cellulose, e.g. complexes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/0006—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid
- C08B37/0009—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid alpha-D-Glucans, e.g. polydextrose, alternan, glycogen; (alpha-1,4)(alpha-1,6)-D-Glucans; (alpha-1,3)(alpha-1,4)-D-Glucans, e.g. isolichenan or nigeran; (alpha-1,4)-D-Glucans; (alpha-1,3)-D-Glucans, e.g. pseudonigeran; Derivatives thereof
- C08B37/0021—Dextran, i.e. (alpha-1,4)-D-glucan; Derivatives thereof, e.g. Sephadex, i.e. crosslinked dextran
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/0006—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid
- C08B37/0036—Galactans; Derivatives thereof
- C08B37/0039—Agar; Agarose, i.e. D-galactose, 3,6-anhydro-D-galactose, methylated, sulfated, e.g. from the red algae Gelidium and Gracilaria; Agaropectin; Derivatives thereof, e.g. Sepharose, i.e. crosslinked agarose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2220/00—Aspects relating to sorbent materials
- B01J2220/50—Aspects relating to the use of sorbent or filter aid materials
- B01J2220/54—Sorbents specially adapted for analytical or investigative chromatography
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Analytical Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Polymers & Plastics (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- General Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Polyethers (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Description
本発明に関連する融合タンパク質中に「タグ」として組み込むのに適したオリゴペプチド、
そのアミノ末端又はカルボキシ末端又は両方において、あるいは目的タンパク質の内部アミノ酸配列位置において、少なくとも1つのこのようなオリゴペプチドに融合している目的タンパク質を含む当該タイプの融合タンパク質、
このような融合タンパク質をコードするポリヌクレオチド構築体、例えばベクター、
このようなポリヌクレオチド構築体を含む宿主細胞、
当該タイプの融合タンパク質を産生する方法であって、融合タンパク質が発現され、その融合タンパク質が培地から回収される条件下にある増殖培地中で後者のタイプの宿主細胞を培養する方法、及び
目的タンパク質を精製する方法であって、(目的タンパク質を含む)このような融合タンパク質及び他のタンパク質(外来性タンパク質)を含有するタンパク質試料を、本発明による金属イオン含有官能化ポリマー基材と接触させる方法に関する。
各トリアザシクロアルカン環中のa、b及びcの各々は、互いに独立して、かつ他のあらゆるトリアザシクロアルカン環から独立した1−3の整数であり、
各トリアザシクロアルカン環中の各−CH2−基の1つ又は両方の水素原子は、互いに独立して、かつ他のあらゆるトリアザシクロアルカン環から独立して、ある置換基で任意に独立に置換することができ;
各トリアザシクロアルカン環中の各−NH−基の水素原子は、互いに独立して、かつ他のあらゆるトリアザシクロアルカン環から独立して、ある置換基で任意に置換することができ;
nが0であるときは、RはH又はある置換基であり;
nが1であるとき、すなわち、官能基が2つのトリアザシクロアルカン環を含むときは、Rは、2つのトリアザシクロアルカン環を連結する(すなわち、各々に共有結合する)二官能基であり、この二官能基は任意に1つ以上の金属イオン配位ドナー原子を含み;
nが2又は3のとき、すなわち、官能基がそれぞれ3つ又は4つのトリアザシクロアルカン環を含むときは、Rは、それぞれ3つ又は4つのトリアザシクロアルカン環を連結する(すなわち、各々に共有結合する)(n+1)官能基であり、この(n+1)官能基は任意に1つ以上の金属イオン配位ドナー原子を含み;
Xはリンカー/スペーサー基である。
[CH2]m− (式中、mは2、3又は4である)、
第1及び第2の官能基を有する二官能試薬の第1の官能基と第1の反応を起こすことが可能な反応性官能基を有するポリマー基材を選択するステップであって、当該第1の反応の結果、ポリマー基材と二官能試薬の共有結合が形成され、得られた共有結合試薬の第2の官能基が、続いて、N、O及びSから独立に選択される少なくとも3つの金属イオン配位ドナー原子を含む金属イオン配位環式リガンド基を含む化学種中に存在する反応性官能基と第2の反応を起こすことができ、第2の反応の結果、当該化学種と共有結合試薬の共有結合が形成されるステップと、
このポリマー基材を二官能試薬と反応させるステップと、
得られた共有結合試薬を当該化学種と反応させるステップとを含む。
A:金属イオン配位大環状リガンドの選択基準:上述したように、本発明に関連して定義される官能化ポリマー基材の極めて重要で貴重な適用例は、タンパク質の精製におけるその金属イオンを含有する態様の使用である。当該タンパク質は、融合タンパク質の形をしており、目的タンパク質がそのアミノ又はカルボキシ末端においてオリゴペプチド「タグ」、特に本発明によるヒスチジン含有オリゴペプチドに融合している。また、目的タンパク質又はポリペプチドの2つの分子、あるいはそのアミノ又はカルボキシ末端がそれぞれオリゴペプチド「タグ」、特に本発明によるヒスチジン含有オリゴペプチドに結合している2つの異なる目的タンパク質又はポリペプチドの同時融合によって、新しい融合タンパク質構造が生成され、それによってオリゴペプチド「タグ」がエンドの位置(すなわち、内部の位置)に置かれて、目的タンパク質又はポリペプチドの2つの分子を連結する。特定の理論に拘泥するものではないが、本発明に関連して本発明者らが実施した研究において得られた結果に基づいて、本発明者らは、本発明のこの側面の根底にある現象は、金属イオン/リガンド錯体とN末端、C末端又はエンド位置のオリゴペプチド「タグ」として融合タンパク質内に存在する相補オリゴペプチド配列との分子相互作用を可能にし、金属イオン/リガンド錯体及び「タグ」のペプチド配列内のその同族の結合相手の分子サイズ及び表面配向によって決まる「分子カセット」とみなすことが適切であると考える。したがって、既述されたすべてのキレート系と比較して、導入したペプチド「タグ」によって組換えタンパク質を捕捉する本発明の官能化ポリマー基材における金属イオン結合キレートリガンド系の主要な特性及び利点は、少なくとも以下の特徴の非排他的なリストに関係付けることができると考えられる。
本発明に関連するいくつかの特徴を挙げることができる。第1に、本固定化キレート大環状リガンドの配位構造は、異なる立体電子効果(steri−electronic effects)パターンを生じ、金属イオン結合定数に関して異なる親和性をもたらす。第2に、スキーム4−6から明らかなように、単核及び二核の開いた構造、非サンドイッチ及びサンドイッチ構造に対して金属イオンキレート錯体を固定化することによって、特に金属イオンの性質、キレートリガンドの化学構造、ビス(並びにトリス及びテトラキス)構造における架橋基Rの立体特性及びンホメーション特性、及び溶媒組成に応じて、分子認識の平衡が純粋な配位型相互作用から純粋なアニオン交換型相互作用に移行した、基本的に諸特性が異なる吸着剤が得られる。サンドイッチ及び非サンドイッチ構造の形成は、大環状分子の第二級アミン基を非配位基で官能化することによって制御できることに留意されたい。これらの特徴はすべて、これらのIMACシステムがオリゴペプチド「タグ」の配列特性(sequence attributes)を高い親和性で認識できるかどうかに影響する。実際、金属イオン結合中心間の分子距離、金属イオン結合中心間の相互作用配位部位表面への接近、オリゴペプチド「タグ」配列中の特異的アミノ酸残基との相互作用配位部位の表面接触性(facial accessibility)、分極率、これらの中心の溶媒和力、それぞれの結合エネルギー、したがってこれらの相互作用に対する親和性を含めて、架橋基Rの諸特性は、これらの新しいIMACシステムのいくつかの主要な側面を規定している。
5’ 1−2−3−4−5−6−7−...−[標的タンパク質] → 5’ 2−3−4−5−6−7−...−[標的タンパク質]で示される)の例がもたらされる。ここでの重要な側面は、切断によってアミノ酸配列の「違相(out−of−phase)」シフトが起こらないことを確実にすることである。これは、精製操作中のオリゴペプチド「タグ」と固定化金属イオン/リガンド錯体の相互作用に対して効果がない場合もあり得るが、融合タンパク質からのオリゴペプチド「タグ」のタンパク質分解切断を不十分にするアミノ酸残基の好ましくない配置をもたらす恐れがある。したがって、「同相の(in−phase)」切断変異体は、親オリゴペプチド「タグ」配列「ライブラリ」の特別なサブセット(T−サブセット)であり、組成又は配列上の特徴の類似性、及びIMAC結合能力又は酵素による切断特性によって、それぞれの親オリゴペプチド「タグ」に関係付けられる。
5’ 1−2−3−4−5−6−7−...−[標的タンパク質] → 5’ 1−2−3−6−7−...−[標的タンパク質]で示される)の例がもたらされる。
5’ 1−2−3−4−5−6−7−...−14−15−16−17−[標的タンパク質] → 5’ 1,2−3−14−15−16−17−−4−5−6−7−...−[標的タンパク質]で示される)の例がもたらされる。
HTNIHQDQHNHFHR (本明細書ではNT2と略記する) (配列番号2)
HAMLDRAHDHGTR (配列番号3)
SLHEHHSGDNLR (配列番号4)
THYNAVHSHNTLQ (配列番号5)
DIHHWTDHLQSSTH (配列番号6)
LYNHYSHTAHVNHL (配列番号7)
このようなペプチド(オリゴペプチド)「タグ」は、示したアミノ酸配列の1つに加えて、オリゴペプチド「タグ」のC末端又はN末端に結合した2−6又はそれ以上の追加のアミノ酸残基などの1つ以上の追加のアミノ酸残基を含むことができることを理解されたい。このような配列伸長は、伸長がオリゴペプチド「タグ」のN末端においてであり「タグ」システムを大腸菌などの原核生物発現系を用いて組換えタンパク質の産生に利用しようとするときには、例えば、2位と1位(1位のLysは、オリゴペプチド「タグ」のN末端から数えて最初のアミノ残基である)にあるMet−Lysなどのジペプチド単位であることが適切である。周辺質の分泌のない原核生物発現系の場合、オリゴペプチド「タグ」のN末端伸長の好ましい第1のアミノ酸残基は、配列Met−Xaaを含む。ここで、Xaaは、プロリン以外のあらゆるアミノ酸残基である。これらのアミノ酸配列伸長を含めることによって、原核生物宿主細胞における標的タンパク質の発現が増加する別の利点がもたらされる可能性がある。
このようなポリペプチドをコードするベクターなどのポリヌクレオチド構築体;
このようなポリヌクレオチド構築体を含む宿主細胞(例えば、大腸菌などの原核生物)を、ポリペプチドの発現及び培養培地からのそのポリペプチドの回収が可能な条件下、適切な増殖培地中で培養することによって得られるポリペプチド;
このようなポリヌクレオチド構築体を含む宿主細胞、より具体的には原核生物細胞(例えば、大腸菌株)又は真核生物細胞(例えば、Saccharomyces cerevisiae、Pichia pastoris細胞、チャイニーズハムスター卵巣(CHO)細胞、又はBHK、HEK若しくはCOSなどの他の哺乳動物細胞系);及び
ポリペプチドの発現及びそのポリペプチドの培養培地からの回収が可能な条件下、適切な増殖培地中で当該タイプの宿主細胞を培養することを含む、当該タイプのポリペプチドを産生するための方法;
さらに別の本発明の側面は、目的タンパク質を精製するための方法に関し、この方法は、
本発明による少なくとも1つのオリゴペプチド(すなわち、オリゴペプチド「タグ」)にそのアミノ末端又はカルボキシ末端において融合している目的タンパク質を含む融合タンパク質(すなわち、オリゴペプチド(すなわち、「タグ」)が既述したエンド位置にある融合タンパク質を含めて、既述したタイプの1つの融合タンパク質)であるポリペプチドと、他の(外来性)タンパク質とを含有するタンパク質試料を、ポリペプチド(融合タンパク質)が金属イオン含有官能化ポリマー基材に結合してそれと錯体を形成する条件下で、本発明による金属イオン含有官能化ポリマー基材と接触させるステップと、
その錯体を緩衝溶液で洗浄して他の(外来性)タンパク質を除去するステップと、
結合ポリペプチドを洗浄した錯体から溶出させるステップとを含む。
Richman-Atkins反応[J.E. Richman及びT.J. Atkins, Journal of the American Chemical Society, 96(1974) 2268; J.E. Richman, W.F. Oettle及びT.J. Atkins, Organic Synthesis 58(1978)86]を用いて、金属イオンに配位してするリガンド、1,4,7−トリアザシクロノナン(略称、tacn又はTACN)(Sigma−Aldrich Company,St.Louis,MO,USAより購入可能)を調製しうる。後述のtacnのトリヒドロブロミド塩は、5M 臭化水素酸からの遊離リガンドの結晶化により得た。
窒素分析は、Dairy Technical Services Ltd., Melbourne, Australiaにより行われた。銅及びニッケル分析は、Varian AA-1475原始吸光分析装置を用いて行った。拡散反射電子スペクトルは、Cary 5G 分光光度計により記録した。ESRスペクトルは、Varian E−12分光計を用い、約9.6GHz(X−バンド)において操作して、77Kにて記録し、g値が2.0036であるα,α‘−ジフェニル−β−ジピークリルヒドラジル(DPPH)を基準とした。試料は、標準的な内径3mmの試験管に入れ測定し、液体窒素フロ−式クリオスタット中で凍結させた。
キレート試薬(tacn)をマトリックス(基材)上に固定化する第一段階では、エピクロロヒドリン(又はカルボニルジイミダゾールなど他の適切な活性化化合物)を用いてマトリックスを共有結合により活性化した。このため、セファロースTMCL−6Bゲル(19g、湿重量)に10mLの2M NAOHと37.5mgの水素化ホウ素ナトリウム(98%)を混合した。この混合物を25℃で2時間インキュベートした後、12mLのエピクロロヒドリンを加え、混合物を25℃で一晩ゆっくりと攪拌した。こうして得たエポキシで活性化されたセファロースTMゲルを吸引ろ過により回収し、Milli−QTM水で完全に洗浄して残った未反応のエピクロロヒドリンを除去した。次に、水(30mL)にtacnの三臭化水素酸塩5mmolを溶解し、固体NAOHを加えてpHを11に調整し、最終容量を水で50mLにして、0.1M tacnのリガンド溶液を調製した。本溶液の一部(20mL)を、上記のように調製したエポキシで活性化した10g(吸引乾燥した重量)のセファロースTMCL−6Bに添加した。振蘯ウォーターバスを用いて、25℃でこの懸濁液を一晩混合した。得られた官能化ゲルの一部を吸引ろ過(ウォーターポンプ使用)により回収し、Milli−QTM水(5x200mL)、0.1M KNO3(pH4.0)を含有する50mM 酢酸ナトリウム緩衝液50mL、最後にMilli−QTM水(5x200mL)でよく洗浄した。im−tacn吸着剤の一部を次に20mLの20%(V/V)エタノール/水溶液に懸濁し、使用するまで4℃で保存した。
使用した0.1M dtneリガンド溶液の最終的な容量が25mLである点を除き、使用した操作は実施例1に述べたものと本質的に同様である。
0.1M dtnpリガンド溶液の最終的な容量が25mLである点を除き、操作は実施例1に述べたものと本質的に同様である。
典型的には、約5g(湿重量)のim−tacn吸着剤を、Milli−QTM水中に調製した50mM 境界域硬度の金属イオンの硝酸塩溶液50mL、すなわちCu(NO3)2、Ni(N03)2、Zn(N03)2、Co(N03)2、Mn(N03)2又はCr(NO3)3の50mM溶液と、25℃で30分間インキュベートした。次いで、前記様々な固定化金属イオン吸着剤(ゲル)を、Milli−QTM水(5x200mL)で完全に洗浄した。ゆるく結合した金属イオンを吸着剤から除去するために、0.1MのKNO3を含有する50mM 酢酸ナトリウム緩衝液(pH4.0)を、50mLの緩衝液に対して5g(湿重量)の吸着剤という比率で、固定化金属吸着剤ゲルとともに10分間インキュベートした。次いで、Milli−QTM水を用いてゲルをよく洗浄した。続いて、様々な固定化金属イオン吸着剤(ゲル)の一部を、20mLの20%(V/V)エタノール水溶液に懸濁して、使用するまで4℃で保存した。
約5g(湿重量)のim−dtne吸着剤を使用した以外、用いた操作は実施例4とまったく同様である。
約5g(湿重量)のim−dtnp吸着剤を使用した以外、用いた操作は実施例4とまったく同様である。
ヒト血清の調製
血液細胞とフィブリノゲンを取り除くため、新鮮なヒト血液サンプル(20mL)を4℃で24時間沈殿させた。血清上清をSorvallTM RT6000冷却遠心分離機(DuPont CO.、Newtown、CT、USA)を用い、5000xgと4℃で20分間遠心分離した。上清を除去し、5000xgと4℃でさらに20分間遠心分離した。上清を分取(0.2mL)し、1.5mLのチューブに入れて、−20℃で保存した。
im−Cu2+−dtne吸着剤(1mL)を、4.0cm(長さ)x0.8cm(内径)のカラムなどのBio−RadTM Econo−column(Hercules、CA、USA)に充填して、20mM 炭酸ナトリウム及び0.1M NaClを含有するpH9.5の平衡緩衝液で平衡化させた(表1参照)。平衡緩衝液で5倍に希釈したヒト血清の分取試料(200μL)を平衡化カラムにロードした。ヒト血清サンプルを5分間にわたって載せた後、カラム壁からサンプルをゲル中に洗い流すために平衡緩衝液(300μL)を加えた。さらに5分間おいた後、未結合のタンパク質を溶出させるために平衡緩衝液(5mL)をカラム・リザーバーに導入した。
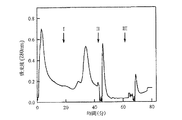
使用した平衡緩衝液が0.1M NaClを含有する20mM リン酸カリウム緩衝液、pH7.0(表2参照)である点と結合ヒト血清タンパク質を表2に示した一連の溶出緩衝液(5mL)を用いて吸着剤から溶出した点を除き、用いた操作は実施例7と実質的に同様である。結果を図3及び図4に示す。
使用した緩衝液が0.1M NaClを含有する20mM 酢酸ナトリウム緩衝液、pH4.0(表3参照)である点と結合したヒト血清タンパク質を表3に示した一連の溶出緩衝液(5mL)を用いて吸着剤から溶出した点を除き、用いた操作は実施例7と実質的に同様である。結果を図5及び図2に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cu2+−dtnpである点を除き、用いた操作は実施例7と実質的に同様である。結果を図1及び図2に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cu2+−dtnpである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3及び図4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cu2+−dtnpである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5及び図2に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−dtneである点を除き、用いた操作は実質的に実施例7と同様である。結果を図1及び図6に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−dtneである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3及び図4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−dtneである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5及び図6に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−dtnpである点を除き、用いた操作は実質的に実施例7と同様である。結果を図1及び図6に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−dtnpである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3及び図4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−dtnpである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5及び図6に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−dtneである点を除き、用いた操作は実質的に実施例7と同様である。結果を図1に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−dtneである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−dtnpである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−dtnpである点を除き、用いた操作は実質的に実施例7と同様である。結果を図1に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−dtnpである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−dtnpである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−dtneである点を除き、用いた操作は実質的に実施例7と同様である。結果を図1及び図7に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−dtneである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3及び図7に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−dtneである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5及び図7に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−dtnpである点を除き、用いた操作は実質的に実施例7と同様である。結果を図1及び図7に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−dtnpである点を除き、用いた操作は実質的に実施例8と同様である。結果を図3及び図7に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−dtnpである点を除き、用いた操作は実質的に実施例9と同様である。結果を図5及び図7に示す。
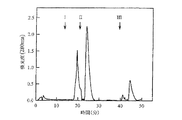
これらの分離操作を通じて、プログラム化されたFPLCシステム(Pharmacia Fine Chemicals、 Uppsala、 Sweden)を用いた。im−Cu2+−dtne/セファロースTMCL−6B吸着剤(1mL)をAmersham PharmaciaTM HR5/10カラム(充填容積 5cm×内径0.5cm)に充填した。クロマトグラフィー操作を通じて、0.5mL/minの流速を用いた。カラムは平衡緩衝液(下記参照)を用いて平衡化させた。平衡緩衝液で5倍に希釈したヒト血清溶液(200μL)を、前記充填したカラムに200μLのインジェクションループを通じてロードした。洗浄及び濃度勾配溶出操作については全て下記に述べる。ヒト血清タンパク質のクロマトグラフィー操作経過をモニターし記録するために、PharmaciaTM UV−1 単一光路モニター(280nm)、REC−482 ツーチャンネルレコーダー、及びコンピューター化されたデータ自動記録装置を使用した。全ての画分はFRAC−100 画分コレクターで1mL画分として採集し、Bio−RadTM Dyeとビシンコニン酸(BCA)法を用いて分析した。ピーク画分はSDS−PAGEにより分析した。結果を図8及び図9に示す。
平衡緩衝液:20mM 炭酸ナトリウム緩衝液/0.1M NaCl、pH9.5
洗浄緩衝液:15mLの平衡緩衝液
溶出操作:1)pH線形勾配法で10mL、20分間(20mM 炭酸ナトリウム緩衝液/100mM NaCl、pH9.5から20mM 酢酸ナトリウム緩衝液/100mM NaCl、pH4.0)
2)塩濃度線形勾配法で10mL、20分間(20mM 酢酸ナトリウム緩衝液、pH4.0中の0.1Mから1.0M NaCl)
3)イミダゾール濃度線形勾配法で10mL、20分間(20mM 酢酸ナトリウム緩衝液/1.0M NaCl、pH4.0中の0から250mMイミダゾール)
[実施例32] 固定化Cu2+−dtnp吸着剤をFPLCTMシステムとともに用いたヒト血清タンパク質の分離
im−Cu2+−dtnp/セファロースTMCL−6Bを吸着剤として用い、以下の溶出プロトコルを使用した点を除き、用いた操作は実質的に実施例31と同様である。結果を図10及び図11に示す。
平衡緩衝液:20mM 炭酸ナトリウム緩衝液/0.1M NaCl、pH9.5
洗浄緩衝液:5mLの平衡緩衝液
溶出操作:1)pH線形勾配法で5mL、10分間(20mM 炭酸ナトリウム緩衝液/0.1M NaCl、pH9.5から20mM酢酸ナトリウム緩衝液/0.1M NaCl、pH4.0)
2)2mLの20mM 酢酸ナトリウム緩衝液/0.1M NaCl、pH4.0
3)塩濃度線形勾配法で7mL、14分間(20mM 酢酸ナトリウム緩衝液、pH4.0中の0.1Mから1.0M NaCl)
4)1mLの20mM 酢酸ナトリウム緩衝液/1.0M NaCl、pH4.0
5)イミダゾール線形勾配法で5mL、10分間(20mM 酢酸ナトリウム緩衝液/1.0M NaCl、pH4.0中で0から250mM イミダゾール)
6)1mLの20mM 酢酸ナトリウム緩衝液/1.0M NaCl及び250mM イミダゾール、pH4.0
[実施例33] 固定化Cu2+−dtnp吸着剤をFPLCTMシステムとともに用いたヒト血清タンパク質の分離
im−Cu2+−dtnp/セファロースTMCL−6Bを吸着剤として使用し、以下の溶出プロトコルを使用した点を除いて、用いた操作は実質的に実施例31と同様である。結果を図12及び図13に示す。
平衡緩衝液:20mM 炭酸ナトリウム緩衝液/0.1M NaCl、pH9.5
洗浄緩衝液:10mLの平衡緩衝液
溶出緩衝液:緩衝液A:20mM 炭酸ナトリウム緩衝液/1.0M NaCl、pH9.5;
緩衝液B:20mM 酢酸ナトリウム緩衝液/1.0M NaCl、pH4.0
溶出操作:1)塩濃度線形勾配法で10mL、20分間(20mM 炭酸ナトリウム緩衝液、pH9.5中で0.1Mから1.0M NaCl)
2)1mLの緩衝液A
3)直線濃度勾配法で5mL、10分間(緩衝液Aから緩衝液B;最終緩衝液組成:緩衝液A50%及び緩衝液B50%)
4)緩衝液Aと緩衝液Bの50:50(V/V)混合物を8mL
5)直線濃度勾配法で5mL、10分間(50/50V/Vの緩衝液A+緩衝液Bから緩衝液B100%に)
6)10mLの20mM 酢酸ナトリウム緩衝液/1.0M NaCl、pH4.0
[実施例34] 組換えタンパク質GST−δATPase−His6タンパク質の結合についてのim−Cu2+−tacn吸着剤のスクリーニング
0.5mLから20mLまでのim−Cu2+−tacn/セファロースTMCL−6Bゲルを含有するBio−RadTM Econo−column[典型的なカラムサイズは4.0cm(長さ)×0.8cm(内径)から12cm(長さ)×1.5cm(内径)](Hercules、CA、USA)を、50mM リン酸ナトリウム緩衝液、300mM 塩化ナトリウム及び10%グリセロールを含む平衡緩衝液pH8.0によって平衡化させた。部分的に精製された融合タンパク質GST−δATPase−His6(E.Coli株K12 DH5alphaF’において産生される)の分取。DH5alphaF’株の起源と遺伝子型は次の通りである:F’、80dlacZΔM15、endA1、recA1、hsdR17(rk−, mk+)、supE44、thi−1、gyrA96、reiA1、Δ(lacZYA−argF)U169、deoR、λ−。[http://wheat.dw.usda.gov/qqpaqes/probes/strains.htmlを参照]。細胞発現システムから回収したE.Coliの細胞ペレットを細胞破砕して得た目的の組換えタンパク質GST−δATPase−His6を含有する粗E.Coli細胞破砕液(4.0cm×0.8cmのカラムには0.3mLで、これより大きなカラムには比例して容量を増やした)をカラムにロードした。未結合のタンパク質と弱く結合したタンパク質を溶出するためには、平衡緩衝液(4.0cm×0.8cmのカラムには5mL、これより大きなカラムには比例して容量を増やした)を使用した。タンパク質がそれ以上溶出されなくなったら、それぞれ、20mM イミダゾール、40mM イミダゾール、250mM イミダゾールを含有する大量の平衡緩衝液(4.0cm×0.8cmのカラムには2mL;これより大きなカラムには比例して容量を増やした)を順次流して結合したタンパク質を溶出させ、1mLずつ画分を回収した。未結合画分と溶出画分のタンパク質濃度は、ビシンコニン酸(BCA)試薬(Pierce Chemical Co.、Rockford、IL、USA)及びBio−RadTM Dye(BioRad、Richmond、CA、USA)法により測定し、酵素活性はGST基質1−クロロ−2,4−ジニトロベンゼン(CDNB)を用いて測定した。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−tacnである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−tacnである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−tacnである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Mn2+−tacnである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cr3+−tacnである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cu2+−DTNEである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−DTNEである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−DTNEである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−DTNEである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Mn2+−DTNEである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cr3+−DTNEである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cu2+−DTNPである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Ni2+−DTNPである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Zn2+−DTNPである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Co2+−DTNPである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Mn2+−DTNPである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
用いた吸着剤がセファロースTMCL−6B上のim−Cr3+−DTNPである点を除いて、用いた操作は実質的に実施例34と同様である。吸着剤に結合した融合タンパク質GST−δATPase−His6のパーセンテージを表4に示す。
実施例34のように得た粗E.Coli抽出液(3mL)を、300mMの塩化ナトリウムと10%グリセロールを含有する50mM リン酸ナトリウム緩衝液、pH8.0(平衡緩衝液)の存在下で平衡化させた1mLのim−Cu2+−tacn/セファロースTMCL−6Bゲルと混合した。4℃で30分間インキュベートした後、4℃で5分間、3,000×gで混合物を遠心分離にかけ、上清を取り除いた。未結合のタンパク質又は弱く結合したタンパク質を除去するため、ゲルのペレットを5mLの平衡緩衝液で2度洗浄した。次いで、洗浄したゲルをBio−RadTM Econo−column(10mL)(Hercules、CA、USA)に充填した。さらに平衡緩衝液(20mL)を使用して、未結合のタンパク質又は弱く結合したタンパク質を溶出させた。
用いた吸着剤がim−Ni2+−tacn セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Zn2+−tacn セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Cu2+−dtne セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Ni2+−dtne セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Zn2+−dtne セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Cu2+−dtnp セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Ni2+−dtnp セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
用いた吸着剤がim−Zn2+−dtnp セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例52と同様である。結果を図14−16に示す。
im−Ni2+−tacn吸着剤(1mL)を、5cmの高さになるようにPharmacia HR5/10カラム(充填容積 5cm×内径0.5cm)に充填した。クロマトグラフィー操作を通じて、0.5mL/minの流速を用いた。Bio−RadTM Econo−column(10mL)の中で、平衡緩衝液(300mM 塩化ナトリウムと10% グリセロールを含有する50mMリン酸ナトリウム緩衝液、pH8.0)を用いてゲルを平衡化させた後、2mLのインジェクションループを通じて、実施例52に記載されているように取得した2mLのE.Coli抽出液を充填したカラムにロードした。次にゲルを15mLの平衡緩衝液で溶出させた。この溶出操作では、平衡緩衝液(10mL)中のイミダゾールを20分間にわたって0mMから250mMまで増加させる線形グラジエントを用いた後、250mMイミダゾールを含有する平衡緩衝液5mLで均一濃度溶出を行った。溶出タンパク質のクロマトグラフィー曲線をモニターし記録するために、PharmaciaTM UV−1単一光路モニター(280nm)、REC−482 ツーチャンネルレコーダーとコンピューター化した自動記録装置を使用した。全ての画分(1mL)はFRACTM−100 画分コレクターで回収した。未結合画分と溶出画分のタンパク質濃度はビシンコニン酸(BCA)試薬(Pierce Chemical Co.、Rockford、IL、USA)及びBio−RadTM Dye(BioRad、Richmond、CA、USA)法により測定し、酵素活性はGST基質1−クロロ−2,4−ジニトロベンゼン(CDNB)を用いて測定した。ピーク画分はSDS−PAGEにより性質決定した。結果を図17−18に示す。
im−Cu2+−tacn/セファロースTMCL−6B吸着剤(1mL)をBio−RadTM Econo−column(10mL)に充填し、平衡緩衝液(150mM 塩化ナトリウムを含有する50mM リン酸ナトリウム緩衝液、pH8.0)で平衡化させた。特定の組換えC末端ヘキサヒスチジンタグ付きFasciola hepatica カテプシン L5(rF.h カテプシン L5)発現システムを含有し、培地で成長させた被トランスフェクトSaccharomyces cerevisiae酵母細胞から得た粗酵母抽出上清液(30mL)をim−Cu2+−tacn セファロースTMにロードした。ブレークスルー画分(未結合画分)は、後にタンパク質含量と酵素活性を決定するために回収した。次いで、10mLの平衡緩衝液でカラムを洗浄して、未結合のタンパク質又は弱く結合したタンパク質を取り除いた。次に、タンパク質不純物を溶出させるために、150mM 塩化ナトリウムを含有する50mM リン酸ナトリウムpH6.0を10mL使用して洗浄操作を行った。その後、この洗浄緩衝液にさらにそれぞれ125mM イミダゾール(溶出緩衝液A)、250mM イミダゾール(溶出緩衝液B)、及び500mM イミダゾール(溶出緩衝液C)を含有する3種類の溶液各5mLを用いて結合タンパク質を溶出させた。ブレークスルー、洗浄及び溶出操作における全ての画分を回収して、BCA及びBio−RadTM DyeアッセイならびにSDS−PAGEによりタンパク質含量を分析した。酵素活性は、Barrett, A.J.[Biochemical Journal、187(1980) 909-912]ならびにByanastati, A.、Brown, M.A.、Kembhavi, A.A.、Nicklin, M.J.H.、Sayers, C.A.、Sunter, D.C.及びBarrett, A.J.[Biochemical Journal, 211(1983) 129-138]に記載されている蛍光アッセイにより測定した。結果を図19に示す。
用いた吸着剤がim−Ni2+−tacn/セファロースTMCL−6Bである点を除いて、用いた操作は実質的に実施例62と同様である。結果を図19に示す。
im−Cu2+−tacn セファロースTMCL−6B吸着剤(0.5mL)を、2.5cmの高さまでPharmacia HR5/5カラム(充填容積 2.5cm×内径0.5cm)に充填した。組換えC末端ヘキサヒスチジンタグ付きFasciola hepatica カテプシン L5(rF.h カテプシン L5)を含む酵母上清(246mL)を、前記充填したカラムに0.2mL/分の流速でロードした。洗浄操作は、30mLの平衡緩衝液(150mM NaClを含有する50mM リン酸ナトリウム緩衝液、pH8.0)及び21mLの溶出緩衝液A(150mM NaClを含有する50mM リン酸ナトリウム、pH6.0)を使用して行った。次いで、緩衝液Aから緩衝液Bへの直線濃度勾配法(15mL、30分)でカラムからの溶出を行った後、5mLの緩衝液Bで均一濃度溶出を行った。溶出緩衝液Bは、150mM NaClと500mM イミダゾールを含有する50mM リン酸ナトリウム緩衝液、pH6.0である。全ての画分(1mL)をFRAC−100 画分コレクターで回収した。タンパク質含量は、Bio−Radアッセイにより分析した。酵素活性は実施例62のような蛍光アッセイで測定した。ピーク画分はSDS−PAGEにより性質決定した。結果を図20及び図21に示す。
AMA−1を含む被トランスフェクトSaccharomyces cerevisiae酵母細胞の粗上清を透析緩衝液(150mM NaCl及び10%グリセロールを含有する50mMリン酸ナトリウム、pH8.0)を使用し4℃で一晩透析した後、新しい透析緩衝液を使用して4℃で4時間にわたる透析を2度行った。
用いた吸着剤がim−Cu2+−dtnp セファロースTMCL−6Bである点を除き、用いた操作は実質的に実施例65と同様である。結果を図22に示す。
用いた吸着剤がim−Ni2+−tacn セファロースTMCL−6Bである点を除き、用いた操作は実質的に実施例65と同様である。結果を図22に示す。
用いた吸着剤がim−Ni2+−dtnp セファロースTMCL−6Bである点を除き、用いた操作は実質的に実施例65と同様である。結果を図22に示す。
本研究で使用した緩衝液:
平衡緩衝液:300mM 塩化カリウム、20mM イミダゾール及び0.12% TritonTM X−100を含有する50mM リン酸カリウム緩衝液、pH8.0
洗浄緩衝液:1M 塩化カリウム、40mM イミダゾール及び0.12% TritonTM X−100を含有する50mM リン酸カリウム緩衝液、pH8.0
溶出緩衝液:A.500mM 塩化カリウム、125mM イミダゾール及び0.12% TritonTM X−100を含有する50mM リン酸カリウム緩衝液、pH8.0、
B.500mM 塩化カリウム、250mM イミダゾール及び0.12% TritonTM X−100を含有する50mM リン酸カリウム緩衝液、pH8.0、
C.500mM 塩化カリウム、500mM イミダゾール及び0.12% TritonTM X−100を含有する50mM リン酸カリウム緩衝液、pH8.0
im−Cu2+−tacn セファロースTMCL−6B吸着剤を平衡緩衝液で平衡化させた後、その0.2mLを1.8mLのエッペンドルフ・チューブに分け入れた。im−Ni2+−NTAカラム(Chelating SepharoseTM Fast Flow、Amersham Pharmacia、Uppsala、Sweden)を用いて予め分離された、部分精製タンパク質溶液(500μL)及びSaccharomyces cerevisiae BJ3505株で発現させて得られた粗タンパク質混合物をエッペンドルフチューブに加え、4℃で90分間インキュベートした。混合物を1000×gで1分間遠心分離にかけた後、上清を取り除いた。前記吸着剤を1mLの平衡緩衝液で洗浄した後、続いてタンパク質不純物を取り除くために1mLの洗浄緩衝液で5回洗浄した。それぞれ、0.5mLの溶出緩衝液A、溶出緩衝液B、溶出緩衝液Cを順次4回使用して、結合タンパク質の溶出を行った。未結合画分、洗浄画分及び溶出画分はSDS−PAGE分析のために回収した。結果を図23に示す。
用いた吸着剤がim−Ni2+−tacn/セファロースTMCL−6Bである点を除き、用いた操作は実質的に実施例69と同様である。SDS−PAGEの分析結果を図23に示す。
im−Cu2+−tacn セファロースTMCL−6Bゲル(1mL)をBio−RadTM Econo−columnに充填し、平衡緩衝液(300mM 塩化カリウム、0.1mM 2−アミノエチルイソチオウロニウムブロミド、1mM グルタミン、0.2mM フェニルメチルスルホニルフルオリド、20μM ピリドキサール-L-リン酸、及び2mM β−メルカプトエタノールを含有する50mM リン酸カリウム緩衝液、pH8.0、Ni緩衝液と表記)で平衡化させた。
Mettle AE50 5桁天秤上でHMYOタンパク質を正確に秤量し、以下の平衡緩衝液中に希釈することによって、1.0mg/mLのウマ骨格筋ミオグロビン(horse skeletal muscle myoglobin、HMYO)を含有する試料を調製した:
緩衝液A:0.5M 塩化ナトリウムを含有する20mM 酢酸ナトリウム緩衝
液、pH4.0
緩衝液B:0.5M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH6.0
緩衝液C:0.5M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH7.0
緩衝液D:0.5M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH8.0
緩衝液E:0.5M 塩化ナトリウムを含有する20mM 炭酸ナトリウム緩衝
液、pH9.5
吸引乾燥したim−M2+−dtne セファロースTMCL−6Bゲル(約0.05g)(ここで、M2+はCu2+、Ni2+、Zn2+又はCo2+を表す)を0.5mLのタンパク質溶液を含むDuraporeTMメンブレンチューブ(UltrafreeTM−CL、0.1μm、 Nihon Millipore Kogyo K.K、Japan)の中で、時計回りのローテーター(RATEKTM Instruments、Mitcham、VIC、Australia)を用いてゆっくり回転させながら、25℃で90分間インキュベートした。上清を回収した後、SorvallTM RT6000冷却遠心分離機(DuPont Co.、Newtown、CT、USA)を用いて3000×gで混合物を遠心分離し、分析用逆相(RP)HPLC法を用いて、直ちにサンプルの遊離タンパク質濃度を分析した。結果を図25及び図26に示す。
im−M2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除き、用いた操作は実質的に実施例72と同様である。結果を図25及び図26に示す。
タンパク質がニワトリ卵白ゾチーム(hen egg white lysozyme、HEWL)である点を除いて、用いた操作は実質的に実施例72と同様である。結果を図27及び図28に示す。
im−Mn2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除いて、用いた操作は実質的に実施例74と同様である。結果を図27及び図28に示す。
タンパク質がウマ心臓チトクロムC(horse heart cytochrome C 、HHCC)である点を除いて、用いた操作は実質的に実施例72と同様である。結果を図29及び図30に示す。
im−M2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除いて、用いた操作は実質的に実施例76と同様である。結果を図29及び図30に示す。
タンパク質が牛乳α−ラクトアルブミン(cow milk α−lactalbumin、αLAC)である点を除いて、用いた操作は実質的に実施例72と同様である。結果を図31及び図32に示す。
im−Mn2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除いて、用いた操作は実質的に実施例78と同様である。結果を図31及び図32に示す。
以下の平衡緩衝液を使用した点を除いて、用いた操作は実質的に実施例72と同様である。
液、pH8.0
緩衝液G:0.5M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH8.0
緩衝液H:1.0M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH8.0
緩衝液I:2.0M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH8.0
緩衝液J:3.0M 塩化ナトリウムを含有する20mM リン酸カリウム緩衝
液、pH8.0
結果を図33に示す。
im−Mn2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除いて、用いた操作は実質的に実施例80と同様である。結果を図33に示す。
タンパク質がニワトリ卵白ゾチーム(HEWL)である点を除いて、用いた操作は実質的に実施例80と同様である。結果を図34に示す。
im−Mn2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除いて、用いた操作は実質的に実施例82と同様である。結果を図34に示す。
タンパク質がウマ心臓チトクロムC(HHCC)である点を除いて、用いた操作は実質的に実施例80と同様である。結果を図35に示す。
im−Mn2+−dtnp セファロースTMCL−6B吸着剤を使用した点を除いて、用いた操作は実質的に実施例84と同様である。結果を図35に示す。
PCRテンプレートの消化:
GSTのソースはpGEX3XGSTである。これを制限エンドヌクレアーゼEcoRV及びBamHIで消化した。条件:約3.45μgのプラスミド、20ユニットのBamHI、20ユニットのEcoRV、10mM Tris−HCl pH8.0、0.5mM MgCl2、100mM NaCl、1mM 2−メルカプトエタノール、総容量50μL。この混合物を37℃で2時間インキュベートした。2つのDNAフラグメントを1%アガロースゲル上で分離し、所望のフラグメントを単離した。
60pmolのフォワードプライマー、60pmolのリバースプライマー、2.5ユニットのDNAポリメラーゼ、0.5mM dNTP、10mM Tris−HCl pH8.3、1.5mM MgCl2、50mM KClを含む混合物に、GSTをコードする50ngと25ngのDNAフラグメントを加えた。反応混合物の容量は50μLである。GST cDNAはPCRによって増幅した。概略を述べると、前記反応混合物をサーモサイクラーに入れ、最初の変性ステップを95℃で5分間行い、94℃、50℃及び72℃でそれぞれ1、2及び3分間を2サイクル行った後、94℃、55℃、72℃で1、2、及び3分間を40サイクル、次いで、最後の伸長ステップを72℃で5分間実施するという条件を使用した。反応混合物を1%アガロースゲルに流し、PCR産物を単離した。
リバースプライマーの配列: 5’−TAAAAGCTTTTACAGATCCGATTAAGG−3’
PCR産物末端の修飾:
約300ngの上記単離されたPCR産物サンプルを、3ユニットのT4DNAポリメラーゼ、10ユニットのT4ポリヌクレオチドキナーゼ、1mM rATP、0.5mM dNTP、50mM Tris−HCl pH7.5、10mM MgCl2、1mM ジチオスレイトール(DTT)及び5μgのBSAを含有する100μLの反応混合物に加えた。反応は37℃で1時間にわたり行った。次に、0.5μLの0.5M EDTAを前記混合物に加え、75℃で10分間インキュベートした後、氷上で冷却した。DNAはフェノール/クロロフォルム抽出を行った後、エタノール沈殿により回収した。
様々な量の修飾PCR産物(約36ng、12ng及び4ng)を含む10μLの反応溶液を、50ngのシャトルベクターpBluescript II SK+(SmaIで切断、脱リン酸化)、2.5ユニットのT4 DNAリガーゼ、40mM Tris−HCl pH7.8、10mM MgCl2、10mM DTT及び0.5mM ATPと混合した。ライゲーション反応溶液は室温で一晩インキュベートした。
標準的な技法を用いて、塩化ルビジウムで処理したE.coli DH5α細胞を5μLの上記ライゲーション反応混合溶液で形質転換した。800μgのIPTG(Isopropyl−β−Thiogalactoside)とX−gal(5−ブロモ−4−クロロ−3−インドリル−β−D−ガラクトシド)を塗布したLB Ampプレート(トリプトン 10g/L、酵母抽出物 5g/L、NaCl 10g/L、寒天 15g/Lを添加しオートクレーブした後、アンピシリン 100mg/Lを追加)上に、様々に希釈した形質転換混合物を播種した。プレートは37℃で一晩インキュベートした。
修飾されたプラスミドDNAは、標準的な技術を用いて、上記のDH5α形質転換体から単離した。所望の挿入物を有するプラスミドを含むと考えられるクローンを同定するために、異なるクローンから得たプラスミドDNAを様々な制限エンドヌクレアーゼで消化した。多くのクローンから得たプラスミドDNAを制限エンドヌクレアーゼPvuIIで消化した。条件は、約300μgのDNA、10ユニットのPvuII、10mM Tris−HCl pH7.5、10mM MgCl2、50mM NaCl,1mM DTE、容量20μL。温度は37℃で、反応時間は3時間であった。1%アガロースゲル上でDNAフラグメントを分離することにより、正しいサイズのフラグメントを含むプラスミドの同定が可能となった。PvuII酵素によるスクリーニングで選択したプラスミドに対して、制限エンドヌクレアーゼBspHIを用いた2回目のスクリーニングを行った。条件は、約300μgのDNA、10ユニットのBspHI、50mM 酢酸カリウム、20mM Tris−酢酸、10mM 酢酸マグネシウム、1mM DTT、pH7.9、容量20μL。温度は37℃で、反応時間は2時間であった。消化されたDNAを1%アガロースゲル上に流し、正しいサイズのフラグメントを含むプラスミドを同定した。
陽性クローン(所望の挿入物を有するpBluescript)から得たプラスミドを制限エンドヌクレアーゼBspHI及びHindIIIとともにインキュベートした。条件は、約500ngのプラスミドDNA、20ユニットのBspHI、40ユニットのHindIII、50mM 酢酸カリウム、20mM Tris−酢酸、10mM 酢酸マグネシウム、1mM DTT、pH7.9、容量70μLであった。温度は37℃であり、反応時間は2.5時間であった。DNAフラグメントを2%アガロースゲル上で分離し、所望のフラグメントを単離した。
発現プラスミドpTrc 99A(Pharmacia Biotech)を制限エンドヌクレアーゼNcoI及びHindIIIとともにインキュベートした。条件は、約4.25μgのプラスミド、20ユニットのNcoI、20ユニットのHindIII、10mM Tris−HCl、pH8.0、5mM MgCl2、100mM NaCl、1mM 2−メルカプトエタノール、容量200μLであった。温度は37℃であり、反応時間は2時間であった。2つのDNAフラグメントを1%アガロースゲル上で分離し、所望のフラグメントを単離した。
様々な量の挿入プレップDNA(約25ng、8.3ng及び2.75ng)を含有し、約50ngの発現プラスミドpTrc 99A(NcoI/HindIIIで切断)、2.5ユニットのT4 DNAリガーゼ、40mM Tris−HCl pH7.8、10mM MgCl2、10mM DTT及び0.5mM ATPと混合した10μLの反応液を調製した。温度は16℃であり、反応時間は少なくとも1日であった。5μLの該反応混合溶液をE.coli DH5α細胞を形質転換するために使用した。修飾された発現プラスミドは、制限エンドヌクレアーゼEcoRV/HindIIIを使用した酵素スクリーニングを行うことよって、単離し同定した。
標準的な技術を用いて、塩化ルビジウム処理したE.coli細胞株JM105を約100ngの修飾された発現プラスミドで形質転換した。プラスミド含有細胞を選択するために、異なる希釈度の形質転換細胞をLB Ampプレート上に播種した。
フォワードプライマーの配列が5’−TAAATCATGAAACATCACCATCACCATCACCAGATGTCCCCTATACTAGGT−3’である点を除いて、操作は実質的に実施例86と同様である。
多くのクローンから得たプラスミドDNAを、制限エンドヌクレアーゼPvuIIとともにインキュベートした。条件は、約150μgのプラスミド、10ユニットのPvuII、10mM Tris−HCl、pH7.5、10mM MgCl2、50mM NaCl、1mM DTE、容量20μLであった。温度は37℃であり、反応時間は1時間であった。1%アガロースゲル上で2つのDNAフラグメントを分離して、正しいサイズのフラグメントを含むプラスミドの同定を可能とした。これらの選択したプラスミドに対して、制限エンドヌクレアーゼHindIII及びSalIを用いた2回目の酵素スクリーニングを行った。二重消化の条件は、約150μgのプラスミド、20ユニットのHindIII、20ユニットのSalI、50mM Tris−HCl、pH7.5、10mM MgCl2、100mM NaCl、1mM DTE、容量40μLであった。温度は37℃であり、反応時間は4時間であった。消化されたDNAを1%アガロースゲル上に流し、正しいサイズのフラグメントを含むプラスミドを同定した。選択したプラスミドから得た約300ngの未切断DNAを、DNA配列決定のためのテンプレートとして使用した。他の配列決定条件は実施例86と同様である。
陽性クローンから得たプラスミドDNAを制限エンドヌクレアーゼBspHI及びHindIIIとともにインキュベートした。条件は、約150ngのプラスミド、30ユニットのBspHI、60ユニットのHindIII、50mM 酢酸カリウム、20mM Tris−酢酸 pH7.9、10mM 酢酸マグネシウム、1mM DTT、容量100μL。温度は37℃であり、反応時間は22時間であった。
CT2をコードするフォワードプライマーの配列が5’−TAAATCATGAAACACCAACACCAACATCAACATCAACATCAACATCAAGTCGACCAGATGTCCCCTATACTAGGT−3’である点を除いて、操作は実質的に実施例86と同様である。
異なるクローンから得た被修飾プラスミドDNAを制限エンドヌクレアーゼPvuIIのみで消化した。
陽性クローンから得たプラスミドDNAを制限エンドヌクレアーゼBspHI及びHindIIIとともにインキュベートした。条件は、約150ngのプラスミド、30ユニットのBspHI、60ユニットのHindIII、50mM 酢酸カリウム、20mM Tris−酢酸 pH7.9、10mM 酢酸マグネシウム、1mM DTT、容量100μLであった。温度は37℃であり、反応時間は約22時間であった。
NT2をコードするフォワードプライマーの配列が5’−TAAATCATGAAACACACCAACATCCACCAGGACCAGCACAACCACTTCCACCGTGTCGACCAGATGTCCCCTATACTAGGT−3’である点を除いて、用いた操作は実質的に実施例86と同様である。
GST−NT2をコードする所望のプラスミド挿入物を含むクローンを同定した。DNA配列決定によりその配列が正しいことが証明された。
E.coli細胞株JM105を実施例86−89に記載されているように形質転換するとともに、未修飾発現ベクターpTrc 99A及びGSTの親ベクターpGEX3XGSTにより形質転換した。LB Amp培地を含むペトリ皿上で、形質転換細胞を37℃で一晩増殖させた。100μg/mLのアンピシリンを補充したLB培地(トリプトン 10g/L、酵母抽出物 5g/L、NaCl 10g/L、オートクレーブ処理した)に前記ペトリ皿から得たシングルコロニーを接種し、37℃で振蘯しながら一晩インキュベートした。100μg/mLのアンピシリンを補充した5mLの2×YT培地(トリプトン 16g/L、酵母抽出物 10g/L、NaCl 5g/L、pH7.0、オートクレーブ処理)(2×YTA)に、一晩培養した培養物の1/10希釈物を接種し、600nmでの吸光度(A600)がpTrc形質転換体の場合には0.6−1.0、pGEX3XGST形質転換体の場合には0.6−0.8になるまで、37℃で振蘯しながらインキュベートした。最終濃度が1mM(pTrc形質転換体)又は0.1m(pGEX3XGST形質転換体)になるようにIPTGを加えることによって、遺伝子発現を誘導した。誘導から5時間後(pTrc形質転換体)又は1時間後(pGEX3XGST形質転換体)に、1.5mLの培養液から細胞を採取した。300μLの氷冷した1×PBS(140mM NaCl、2.7mM KCl、10mM Na2HP04、1.8mM KH2PO4、pH 7.3)に細胞を再懸濁し、超音波で破砕した。ペレット化した後、細胞破砕液を回収し冷凍した。
大規模タンパク質発現に用いるために、各タグ化GST融合タンパク質の各クローンを選択した。100mLの培地(2×YTA)に一晩培養した培養物の1/100希釈物を接種した点を除き、実質的に実施例90に記載されているようにこれを行った。誘導後、細胞を採取するまでに、培養物をさらに6時間インキュベートした。次に、5mLの1×PBS中に細胞を再懸濁(GST−CT2を含む細胞の場合に限り、10mL中に再懸濁)し、超音波で破砕した。最終濃度が1%になるようにTriton X−100を添加した後、融合タンパク質を可溶化しやすくするために30分間インキュベートした。ペレット化して後、細胞破砕液を回収し、冷凍した。
信頼性の高い機能的GST活性のアッセイは、CDNB(1−クロロ−2,4−ジニトロベンゼン)アッセイであり、該アッセイでは、GSTはグルタチオンによるCDNBの誘導化を触媒する。この複合体は波長340nmの光学密度を測定することによりモニターできる。本アッセイは、情報冊子(GST Gene Fusion System、3rdedn.、Revision 1、Pharmacia Biotech)に概説されているとおりに行った。簡潔に述べると、各組換えGST融合タンパク質から得た10μLの細胞破砕液のサンプルを、実施例90及び91に記載されているように、96−ウェルのプレート中で、pH6.5の100mM KH2P04、1mM 1−クロロ−2,4−ジニトロベンゼン(CDNB)及び1mM 還元グルタチオンを含有する190μLの反応混合溶液に添加し、5分間にわたって、1分置きにプレートリーダー中で340nmの吸光度を測定した。本アッセイで測定したところによれば、4つの組換え融合タンパク質は何れも機能的なGST活性を示した。
Ni2+−tacn及びNi2+−NTAカラムを用いて、粗細胞破砕液(実施例91参照)を精製した。各カラムは充填容量が0.5mLで、緩衝液B(緩衝液B=10mMイミダゾールを追加した緩衝液A、pH8.4;緩衝液A=445mM NaCl、7.5mM Na2HP04及び2.5mM NaH2PO4・2H20、pH7.2)で平衡化させた。カラムに125μLの粗細胞破砕液(総容量0.5mLとなるように緩衝液Bで1/4に希釈)をロードした。次いで、溶出前に緩衝液Bでカラムを2度洗浄した。緩衝液1による溶出(緩衝液1=500mM イミダゾールを追加した緩衝液A、pH9.71)(1mL)に続いて、10M 水酸化ナトリウムでpH8.0に調整された200mMのマロン酸ナトリウム溶液(1mL)による2度目の溶出を行う、2つの溶出操作を用いた。
操作は実施例93に記載のものと同じである。
操作は実施例93に記載のものと同じである。
操作は実施例93に記載のものと同じである。
粗細胞破砕液(実施例91参照)をNi2+−tacnカラムにロードした。カラムは充填容量が0.5mLで、緩衝液C(緩衝液C=緩衝液B+50mM イミダゾール、pH9.0)で平衡化させた。緩衝液Cで1/4に希釈した125μLの粗細胞破砕液を含む0.5mLのサンプルをカラムにロードした。サンプルをカラムに通した後、カラムを緩衝液C(5mL)で洗浄した。一連の段階的(均一濃度)溶出を行った。各溶出では、様々な濃度のイミダゾールを追加した緩衝液Aを1mLずつ3回使用した。最初の溶出は、100mM イミダゾール pH9.3を追加した緩衝液Aを用いて行った。イミダゾールの濃度は、次の溶出に進むごとに50mMずつ増加させた。初回の様々な溶出条件から得られたサンプルを、分析のためにSDS−ポリアクリルアミドゲルにかけた。
操作は実施例97に記載のものと同じである。
操作は実施例97に記載のものと同じである。
操作は実施例97に記載のものと同じである。
2度目の段階的(均一濃度)溶出を行った。充填容量が0.5mLのNi2+−tacnカラムを緩衝液Bで平衡化させた。実施例90及び91に記載されているように、緩衝液D(緩衝液D=緩衝液A+12.5mMイミダゾール、pH8.5)で1/5に希釈した500μLの粗細胞破砕液を含む2.5mLのサンプルをカラムにロードした。フロースルー画分を2度カラムに再度ロードした。次いで、緩衝液B(5mL)をカラムに流した。一連の段階的(均一濃度)溶出を次に行った。各溶出では、様々な濃度のイミダゾールを追加した緩衝液Aを1mLずつ3回使用した。最初の溶出は、緩衝液A+25mM イミダゾールを用いて行った。100mMの濃度になるまで、次の溶出に進むごとに25mMずつイミダゾール濃度を増加させ、その後は50mMずつ増加させた。初回の様々な溶出条件から得られたサンプルを、分析のためにSDS−ポリアクリルアミドゲル分析にかけた。
緩衝液Dで1/5に希釈した1mLの粗細胞破砕液を含む5mLのサンプルをカラムにロードした点を除き、用いた操作は実質的に実施例101に記載のものと同様である。
緩衝液Dで1/5に希釈した300μLの粗細胞破砕液を含む1.5mLのサンプルをカラムにロードした点を除き、用いた操作は実質的に実施例101に記載のものと同様である。
緩衝液Dで1/5に希釈した100μLの粗細胞破砕液を含む0.5mLのサンプルをカラムにロードした点を除き、用いた操作は実質的に実施例101に記載のものと同様である。
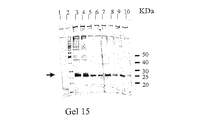
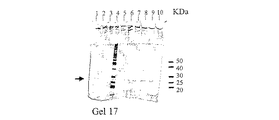
細胞破砕液(実施例90参照)を分取して、SDS ローディング緩衝液(還元状態)と混合し、100℃で1分間加熱した。次に、サンプルを12%SDS−ポリアクリルアミドゲルにロードし、適切な時間にわたって泳動させた。60μLの細胞破砕液と30μLの分子量マーカー[BenchMark Protein Ladder(Gibco BRL)及び10KDa Protein Ladder(Gibco BRL)]をゲルにロードした。pGEX3XGST形質転換体を陽性対照として用いた。N末端タグ化組換えGST融合タンパク質のGST成分は、C末端のアミノ酸が1つ切断されている。親GSTはC末端にアミノ酸が14個追加された完全長タンパク質として発現される。したがって、親GSTのC末端にはアミノ酸が15個追加されており、N末端タグ化GST融合タンパク質のGST成分より約1.65KDa大きい泳動を示す。pTrcで形質転換されたE.coli株JM105を陰性対照として用いた。バンドを可視化するため、ゲルはクーマシーブルーで染色した。
レーン1、GST−CT1 クローンNo.1;レーン2、GST−CT1 クローンNo2;レーン5、GST−CT1 クローンNo.3;レーン6、GST−CT1 クローンNo.4;レーン7、GST−CT1 クローンNo.5;レーン11、GST−CT1 クローンNo.6;レーン12、GST−CT1 クローンNo.7;レーン13、GST−CT1 クローンNo.8; レーン14、GST−CT1 クローンNo9;レーン15、GST−CT1 クローンNo.10;レーン8、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン9、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照。分子量マーカーがレーン3、4及び10に示されている。
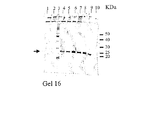
用いた操作は実質的に実施例105に記載のものと同様である。
レーン1、GST−CT2 クローンNo.1; レーン2、GST−CT2 クローンNo.2;レーン3、GST−CT2 クローンNo.3;レーン6、GST−CT2 クローンNo.4;レーン7、GST−CT2 クローンNo.5;レーン11、GST−CT2 クローンNo.6;レーン 12、GST− CT2 クローンNo.7;レーン13、GST−CT2 クローンNo.8;レーン14、GST−CT2 クローンNo.9;レーン15、GST−CT2 クローンNo.10;レーン8、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン9、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照。分子量マーカーが、レーン4、5及び10に示されている。
用いた操作は実質的に実施例105に記載のものと同様である。
レーン1、GST−NT1 クローンNo.1; レーン2、GST−NT1 クローンNo.2;レーン3、GST−NT1 クローンNo.3;レーン4、GST−NT1 クローンNo.4;レーン7、GST−NT1 クローンNo.5;レーン11、GST−NT1 クローンNo.6;レーン 12、GST− T1 クローンNo.7;レーン13、GST−NT1 クローンNo.8;レーン14、GST−NT1 クローンNo.9;レーン15、GST−NT1 クローンNo.10;レーン8、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン9、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照。分子量マーカーが、レーン5、6及び10に示されている
[実施例108] N末端タグ化組換えグルタチオン−S−トランスフェラーゼ融合タンパク質GST−NT2を小規模発現した後に得られた様々なクローンのSDS−PAGE分析
用いた操作は実質的に実施例105に記載のものと同様である。
レーン1、GST−NT2 クローンNo.1;レーン2、GST−NT2 クローンNo.2;レーン3、GST−NT2 クローンNo.3;レーン4、GST−NT2 クローンNo.4;レーン5、GST−NT2 クローンNo.5;レーン11、GST−NT2 クローンNo.6;レーン12、GST−NT2 クローンNo.7;レーン13、GST−NT2 クローンNo.8;レーン14、GST−NT2 クローンNo.9;レーン8、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン9、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照。分子量マーカーが、レーン6、7及び10に示されている。
5μLの細胞破砕液と1μLの分子量マーカー[BenchMark Protein Ladder(Gibco BRL)]をゲルにロードした点を除き、用いた操作は実質的に実施例105に記載のものと同様である。バンドを可視化するためにゲルは銀染色を行った。
レーン1、GST−CT1 クローンNo.7; レーン2、GST−CT2 クローンNo.6;レーン6、GST−NT1 クローンNo.1;レーン7、GST−NT2 クローンNo.7;レーン4、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン5、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照。分子量マーカーがレーン3に示されている
[実施例110] N末端Met−Lys−(His)6タグ化組換えグルタチオン−S−トランスフェラーゼ融合タンパク質GST−CT1のバッチ方式精製から回収された様々な画分のSDS−PAGE分析
バッチ方式の精製(実施例93参照)から回収した細胞破砕液及び画分をSDS ローディング緩衝液(還元状態)と混合し、100℃で90秒加熱した。10μLの精製画分サンプル、5μLの陽性及び陰性対照細胞破砕液、組換え融合タンパク質を含む1.25μLの細胞破砕液及び1μLの分子量マーカー[BenchMark Protein Ladder(Gibco BRL)]を12%SDS−ポリアクリルアミドゲルにロードし、適切な時間にわたり泳動させた。バンドを可視化するために、ゲルに銀染色を行った。
レーン3−5はNi2+−tacnカラムから回収したサンプルを含み、レーン6−8はNi2+−NTAカラムから回収したサンプルを含む;レーン3及び6、洗浄2;レーン4及び7、溶出1;レーン5及び8、溶出2;レーン1、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン2、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照;レーン10、粗細胞破砕液。分子量マーカーがレーン9に示されている。
実施例94で述べたようなバッチ精製から回収した細胞破砕液と画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例110に記載のものと同様である。
レーン3−5はNi2+−tacnカラムから回収したサンプルを含み、レーン6−8はNi2+−NTAカラムから回収したサンプルを含む。レーン3及び6、洗浄2;レーン4及び7、溶出1;レーン5及び8、溶出2;レーン1、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン2、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照;レーン9、粗細胞破砕液。分子量マーカーがレーン10に示されている。
実施例95で述べたようなバッチ精製から回収した細胞破砕液と画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例110に記載した操作と同様である。
レーン5−7はNi2+−tacnカラムから回収したサンプルを含み、レーン8−10はNi2+−NTAカラムから回収したサンプルを含む;レーン5及び8、洗浄2;レーン6及び9、溶出1;レーン7及び10、溶出2。レーン1、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン2、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照;レーン3、粗細胞破砕液。分子量マーカーがレーン4に示されている
[実施例113] N末端タグ化組換えグルタチオン−S−トランスフェラーゼ融合タンパク質GST−NT2のバッチ方式精製から回収した様々な画分のSDS−PAGE分析
実施例96で述べたようなバッチ精製から回収した細胞破砕液と画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例110に記載のものと同様である。
レーン5−7はNi2+−tacnカラムから回収したサンプルを含み、レーン8−10はNi2+−NTAカラムから回収したサンプルを含む。レーン5及び8、洗浄2;レーン6及び9、溶出1;レーン7及び10、溶出2;レーン1、pTrcで形質転換されたE.coli JM105から成る陰性対照;レーン2、pGEX3XGSTで形質転換されたE.coli JM105から成る陽性対照;レーン4、粗細胞破砕液。分子量マーカーがレーン3に示されている。
実施例97で述べたように、GST−CT1を含む細胞破砕液を段階的に均一濃度で溶出した初回のサンプルを、SDS ローディング緩衝液(非還元状態)と混合し、100℃で90秒加熱した。溶出画分の最初の1mLから得た10μLの分取試料と2.5μLの細胞破砕液を12.5%SDS−ポリアクリルアミドゲルにロードした。ゲルは適切な時間にわたり泳動させ、バンドを可視化するために銀染色を行った。
以下の濃度のイミダゾールを追加した緩衝液Aにより溶出を行った:レーン1、100 mM;レーン3、150 mM;レーン4、200mM;レーン5、250mM;レーン6、300mM;レーン7、350mM;レーン8、400mM;レーン9、450mM;レーン10、500mM。細胞破砕液がレーン2に示されている。
実施例98で述べたように、段階的(均一濃度)溶出から回収した細胞破砕液の分取試料と画分をゲルにロードした点を除き、用いた操作は実質的に実施例114に記載のものと同様である。
以下の濃度のイミダゾールを追加した緩衝液Aにより溶出を行った:レーン1、100mM;レーン2、150mM;レーン3、200mM;レーン4、250mM;レーン6、300mM;レーン7、350mM; レーン8、400mM;レーン9、450mM;レーン10、500mM。細胞破砕液がレーン5に示されている。
実施例99で述べたように、段階的(均一濃度)溶出から回収した細胞破砕液と画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例114に記載のものと同様である。
以下の濃度のイミダゾールを追加した緩衝液Aにより溶出を行った:レーン2、100mM;レーン3、150mM;レーン4、200mM;レーン5、250mM;レーン6、300mM;レーン7、350mM; レーン8、400mM;レーン9、450mM;レーン10、500mM;細胞破砕液がレーン1に示されている。
実施例100で述べたように、段階的(均一濃度)溶出から回収した細胞破砕液と画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例114に記載のものと同様である。
以下の濃度のイミダゾールを追加した緩衝液Aにより溶出を行った:レーン1、100mM;レーン2、150mM;レーン4、200mM;レーン5、250mM;レーン6、300mM;レーン7、350mM; レーン8、400mM;レーン9、450mM;レーン10、500 mM。細胞破砕液がレーン3に示されている。
実施例101で述べたように、GST−CT1を含む細胞破砕液の2度目の段階的均一濃度溶出によって得られたサンプルを、SDS ローディング緩衝液(非還元状態)と混合し、100℃で90秒加熱した。溶出画分の最初の1mLから得た16μLの分取試料と0.5μLの分子量マーカー[BenchMark Protein Ladder (Gibco BRL)]を12.5%SDS−ポリアクリルアミドゲルにロードした。ゲルは適切な時間にわたり泳動させ、バンドを可視化するために銀染色を行った。
以下の濃度のイミダゾールを追加した緩衝液Aを用いて溶出を行った:レーン2、25mM;レーン3、50mM;レーン4、75mM;レーン5、100mM;レーン6、150mM;レーン7、200mM;レーン8、250mM;レーン9、300mM;レーン10、350 mM。分子量マーカーがレーン1に示されている。
実施例102で述べたように、段階的(均一濃度)精製から回収した画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例118に記載のものと同様である。
以下の濃度のイミダゾールを追加した緩衝液Aを用いて溶出を行った:レーン1、25mM;レーン3、50mM;レーン4、75mM; レーン5、100mM;レーン6、150mM;レーン7、200mM;レーン 8、250mM;レーン9、300mM;レーン10、350 mM。分子量マーカーがレーン2に示されている。
実施例103で述べたように、段階的(均一濃度)精製から回収した画分の分取試料をゲルにロードした点を除き、用いた操作は実質的に実施例118に記載したものと同様である。
以下の濃度のイミダゾールを追加した緩衝液Aを用いて溶出を行った:レーン1、100mM;レーン2、150mM;レーン4、200mM;レーン5、250mM;レーン6、300mM;レーン7、350mM;レーン8、400mM;レーン9、450mM、レーン10、500mM。分子量マーカーがレーン3に示されている。
実施例104で述べたように、段階的(均一濃度)精製から回収した画分をゲルにロードした点を除き、用いた操作は実質的に実施例118に記載のものと同様である。
以下の濃度のイミダゾールを追加した緩衝液Aを用いて溶出を行った。レーン1、50mM;レーン2、75mM;レーン3、100mM;レーン5、150mM;レーン6、200mM;レーン7、250mM;レーン8、300mM;レーン9、350mM;レーン10、400mM。分子量マーカーがレーン4に示されている。
本実施例では、セファロースTMCL−4Bの表面上に事前に作製したリガンドを固定化する方法を示す。固定化した各種ビス(tacn)リガンド(Leth、Lprop、Lbut、LOX、Lmx及びLpxと表記)とともに、前記リガンドtacn(=1,4,7−トリアザシクロノナン)を下記のスキーム7に示す。これらの大環状高分子(macrocycle)およびビス(大環状高分子)のセファロースTMCL−4Bへの結合(共有結合)には、多糖類基材等のヒドロキシポリマー基材(ここでは、セファロースTMCL−4Bを例示した)の表面に1以上のアミノ基を含む分子を結合させるのに一般的な応用性がある技術を採用した。当該プロセスの第1段階では、エポキシ活性化ポリマー基材(本件の場合、エポキシ活性化セファロースTMCL−4Bゲル)を作製するために、塩基性条件下でエピクロロヒドリンによる前記基材(本件の場合、セファロースTMCL−4Bゲル)の処理を行う。次いで、前記リガンドの共有結合は、求電子性のエポキシド表面基を有するその構造中に存在する求核性第二級アミン基の反応を介して達成することができる。スキーム8(下記)には、当該ポリマー基材(例えば、セファロースTMCL−4B)の表面の模式図が示されており、リガンドは各ケースで単一のスペーサー基を介して付着している。前記リガンド内に2個以上の第二級アミン基が存在していると仮定すると、複数の固定化結合がある程度生じることがある。すなわち、少量のリガンドが、2個以上のエポキシド基と反応することによって、前記ポリマー基材表面に結合することがある。
前記リガンドは吸着剤の唯一の窒素含有成分であるので、窒素分析によってリガンドの固定化の程度を測定することができた(表5、下記)。ビス(tacn)リガンドと比較した場合、固定化tacn吸着剤に高い表面吸着率(リガンド表面密度)が観察された。ビス(tacn)リガンド内の第二級アミン基はより多く用いることができるため、求電子性の表面のエポキシド基と統計的に反応し易いと予想されるにもかかわらず、このようなことが生じる。ビス(tacn)リガンドの方が表面吸着率が低かったのは、ビス(tacn)リガンドの方がスパンが大きいために、複数のエポキシド基と反応するリガンド数を増加させ、ゲル構造内の架橋レベルを高めたためであるかもしれない。さらに、ビス(tacn)リガンドのかさが大きいために、立体的に緻密度が高いtacnリガンドと同数のエポキシド基にはアクセスできないという可能性もある。
本実施例では、ビス(tacn)リガンドを多糖類基材(本実施例では、セファロースTMCL−4B)等のヒドロキシポリマー基材の表面上に構築するための(幅広い一般性があると考えられている)方法を示す。ポリマー支持体の表面上の適切なポリ(求電子体)に結合することにより、ビス(tacn)、トリス(tacn)、テトラキス(tacn)...等のその他の誘導体やこれらの構造的類縁体を構築することができるため、前記方法は幅広い一般性を有していると考えられる。ビス(tacn)リガンドの場合、前記合成方法では、トリス(求電子体)の求電子基のうち1つを用いてアミノ化したポリマー基材表面を処理し、次に、残り2個の求電子基をtacnマクロサイクルと反応させる(スキーム9参照、下記)。前述のように、当該ポリマー基材(本件ではセファロースTMCL−4Bゲル)をまずエポキシ活性化させ(例えば、実施例122を参照、上記)、表面をアミノ化するために過剰のメチルアミンと反応させる。次に、該分子の求電子基のうちの1つとアミノ化したゲル表面との間に反応を生じさせる目的で、大過剰(XSと省略)の1,3,5−トリス(ブロモメチル)ベンゼンで前記活性化したポリマー基材を処理して、2個の求電子基を続くtacnとの反応に利用できる状態にし、固定化したビス(tacn)リガンドを構築する。反応性の高いベンジル誘導体の結合は、塩基を添加せずに行ってもよい(これは、ブロモメチル基と固定化した第二級アミン基との縮合時に遊離されるプロトンが、生成された第三級アミンにプロトンを付加し、さらなる反応からある程度の保護を与えて、第四級窒素中心を生じるという予測と一致している)。
[実施例124] 金属イオン結合に関する考証
本実施例では、新しい官能化ゲル上へのCu2+とNi2+イオンの固定化、及びその生成物の性質決定について記述する。当該ゲルを適切な金属硝酸塩の溶液と混合した後に、弱く結合したイオンを除去するために、酢酸塩緩衝液(pH5)で洗浄することによって、固定化を達成した。ゲルによるCu2+イオンの急激な取り込みは、前記ゲルの色が青に変色することで直ちに識別できるが、Ni2+の結合に関しては、適当な時間内に明るい紫−青にゲルを着色するためには、温度を上昇させながらインキュベーションすることが不可欠であることがわかった。このため、Ni2+イオンと当該固定化したリガンド間の反応は、極めて緩慢なようである。60℃で1時間のインキュベートした後における、ゲルによるCu2+及びNi2+の取り込みレベルは、対応するリガンド表面密度(表7、下記参照)に基づいて予測した最大取り込みレベルのそれぞれ約70%と40%であることが明らかとなった。Ni2+処理したゲルの場合、Cu2+イオンの溶液と混合すると前記ゲルの試料が青くなった事実によって、配位していないリガンドドナー原子が存在することが確認された。ニッケル(II)tacn錯体は速度論的に不活性であることが知られているので、Cu2+イオンによるNi2+イオンの置換が原因である可能性はない(例えば、Yang, RとZompa, L.J., Inorganic Chemistry, 15(1976) 1499;and Murphy, L.J. and Zompa, L.J., Inorganic Chemistry,18(1979) 3278を参照)。このような低い負荷の下では、固定化二核ニッケル(II)錯体種が低いレベルで形成されるにすぎないと予測されること、及び(本発明に記載された態様で)ビス(tacn)リガンドのIMACへの付与を研究する重要な理由の一つがタンパク質の二核錯体への結合を探索することであったことに鑑みれば、Ni2+添加した支持体は、対応するCu2+添加した吸着剤よりも有利ではないように思われる。さらに、実際的な面から見ると、使用する吸着剤の調製に過大な時間が費やされないように、IMAC吸着剤による金属イオンの最大取り込みは相当な速さで達成しなければならない。このため、かかる観察と考察に基づいて、容量が大きいCu2+添加吸着剤の実験に比重を置いた。
Ni2+の取り込みとは異なり、Cu2+の結合が最大に達しなかったのは、平衡の達成が遅いせいであるとは思われないので、その理由を考察することは興味深い。IDA−アガロース支持体へのCu2+結合に関しては、金属イオン対リガンドの割合がおよそ1対1であると報告されているが(例えば、Porath, J. and Belew, M. Journal of Chromatography,516(1990) 333; and Jiang, W. and Hearn, M.T.W. Analytical Biochemistry, 242(1996) 45を参照)、観察された約70%という結合の程度は、N−ドナーのみを用いて又はエーテル型のO−ドナーとともにN−ドナーを用いて固定化リガンドを取り込んだ様々な支持体について観察された程度と合致している(例えば、Gros, C.、Rabiet, F.、Denat, F.、Brandes, S.、Collet, H.、Guilard, R.、Journal of the Chemical Society, Dalton Transaction,(1996)1209; van Berkel, P.M.、Driessen,W.L.,Kodhaas,G.J.A.A.、Reedijk,J.、Sherrington,D.C. Journal of the Chemical Society,Chemical Communication(1995)147;Dudler,V.、Lindoy,L.F.、Sallin, D.、Schlaepfer, C.W., Australian Journal of Chemistry, 40(1987) 1557を例として参照)。これらの固定化された系のいくつかについて、結合が金属イオンに依存することは、3次元ポリマーネットワークに付随する立体効果の差を反映しているものと解釈されている(上記引用文中のvan Berkel,P.M.らとDudler,V.らを参照)。好ましくないコンフォメーション変化が誘発されると、このような立体障害により、幾つかの部位で、ある腫の金属イオンを結合する能力が減少するか、又は特定部位への金属イオンのアクセスが制限される可能性がある。ここで報告した吸着剤に関しては、Cu2+:リガンド比の減少は、CuL2 2+種の形成も原因の1つである可能性があり、ゲル表面上の隣接するリガンド(L)由来のtacn環の対の間に、Cu2+イオンが挟まれている(スキーム12)。しかし、比較的高い結合度が得られたということは、tacn−及びビス(tacn)−官能化吸着剤については、それぞれ、CuL2+とCu2L2+が主要種であると思われることを示唆している。リガンド表面密度から予測した最大限結合度に対するパーセントとして表したCu2+イオンの取り込みは、何れのケースでも実質的に同一であることに注目することが重要である。
吸着剤表面上に固定化したCu2+イオンの配位環境に関する情報を得るために、Cu2+添加支持体の拡散反射率とESRスペクトルを記録した。拡散反射率のスペクトルはそれぞれ、可視領域の約640nmに中心が存在する極めて広いバンドを示したが、溶液中の非固定化ビス(tacn)リガンドの銅(II)錯体について観察されたものと類似しており、四角錐又は正方体が歪曲した八面体配位環境に銅(II)が存在していることを示唆している。「遊離」錯体に対して1100nm付近に観察された低強度のバンドは、ゲルの拡散反射率のスペクトル中では分離されなかった。
真空乾燥したセファロースTMCL−4B(300g)を水(5×300mL)で徹底的に洗浄して、丸底フラスコに入れ、オーバーヘッド機械式スターラーを用いて1M NaOH(300mL)及びNaBH4(1.2g)と室温で1時間混合した。次に、エピクロロヒドリン(100mL)を加えて、更に6時間、前記懸濁液を攪拌した。生成されたエポキシ活性化ゲルを減圧濾過によって収集して、水(5×300mL)、20%EtOH/水(5×300mL)で徹底的に洗浄し、さらにもう一度水(5×300mL)で洗浄した。各リガンドの3.75mmol量の臭化水素酸塩を水(40mL)に溶解することにより、リガンドtacn及びビス(tacn)リガンドLeth、Lprop、Lbut、Lox、Lmx及びLpxの75mM 溶液を調製して、6M NaOHでpH11に調節し、その後、水を加えて50mLにした。真空乾燥したエポキシ活性化ゲル60g分に前記溶液を加えて、振盪式ウォーターバスを用いて60℃で15時間、前記懸濁液を混合した。生成された官能化ゲルを減圧濾過で収集し、水(5×200mL)、50mM 酢酸/0.1M KNO3(pH4)(2×200mL)で洗浄後、もう一度水(5×200mL)で洗浄した。ゲルは、必要なときまで、20%EtOH/水に4℃で保管した。
真空乾燥したエポキシ活性化セファロースCL−4Bゲル(80g)(前述のように調製)を24%メチルアミン水溶液中に懸濁し、この混合物を60℃で15時間、攪拌した。次に、生成されたアミノ化ゲルを減圧濾過により濾過して、水(10×200mL)、50mM 酢酸/0.1M KNO3/pH4緩衝液(4×100mL)で洗浄し、もう一度水(10×200mL)で洗浄した。アミノ化したゲルの一部(15g)を窒素分析のために取り分けた。残りを、100mLの5%トリエチルアミン/水、20%アセトニトリル/5%トリエチルアミン/水、40%アセトニトリル/5%トリエチルアミン/水、60%アセトニトリル/5%トリエチルアミン/水、80%アセトニトリル/5%トリエチルアミン/水、95%アセトニトリル/5%トリエチルアミンで3回ずつ連続して洗浄し、最後にアセトニトリル(6×100mL)で洗浄した。その後、ゲルにアセトニトリルを加えて、総量170mLの懸濁液とした。アセトニトリル(30mL)中の1,3,5−トリス(ブロモメチル)ベンゼン(10g、28mmol)の溶液を加えて、その混合物を室温で18時間、速い速度で攪拌した。次に、前記ゲルを濾過して、アセトニトリル(5×100mL)を用いて洗浄した。前記ゲルにアセトニトリルを添加して、総量130mLの懸濁液にした。次に、アセトニトリル(40mL)中の遊離塩基型tacn(6.5g、50mmol)の溶液を加えて、その混合物を室温で22時間、勢いよく攪拌した。前記ゲルを濾過して、50mLアセトニトリル、75%アセトニトリル/水、50%アセトニトリル/水、25%アセトニトリル/水、水、50mM 酢酸/0.1M KNO3/pH4緩衝液で連続して2回ずつ洗浄し、その後、再度、水で洗浄した。ゲルは20%EtOH/水に4℃で保管した。
リガンドの固定化の程度を測定するために、官能化ゲルの窒素含有量を分析した。各ゲルにつき、20%アセトン/水(50mL)中に、約20gの真空乾燥した材料を2分間懸濁した。次に、ゲルが乾燥しきらないように注意しながら、静かに減圧濾過して溶媒の大部分を除去した(この段階でゲルが乾燥すると、吸着剤が凝集する原因になることがわかっている)。このゲルを、全く同様の方法で更に20%アセトン/水で洗浄した。前記ゲルが純アセトン中に移行するまでアセトンの量を増加した(20%ずつ増加)溶液を用いて、この二重洗浄操作を繰り返した。次に、徹底的に真空乾燥する前に、このゲルを再度アセトン(50mL)で洗浄した後、24時間、高真空の下で乾燥させた。Kjeldahl法を用いて、乾燥した吸着剤の総窒素含量を分析した。
該当する金属硝酸塩(5mL)の50mM水溶液を含む10mLシンチレーションバイアルに真空乾燥した吸着剤のうち0.5gを加え、60℃で1時間、倒置して混合した。Cu2+添加に関しては、Cu2+の取り込みにおける経時的な変化を確立するために、0.5時間から20時間までの期間にわたって、一連の行程として室温でこの操作を実行した。次に、減圧濾過によりゲルを収集し、水(3×50mL)、20mM NaOAc/1M NaCl/pH5緩衝液(2×25mL)、及び水(3×50mL)で連続して洗浄し、前記溶液の添加と減圧濾過によるその除去の間に3分間の平衡時間を与えた。
室温で30分間、50mLのシンチレーションバイアル中で、50mMのCu(No3)2・3H2O(50mL)を用いて前記真空乾燥したゲルのうち4gをインキュベートすることにより、官能化吸着剤上にCu2+イオンを大量に添加(結合)させたが、取り込みの予備実験(実施例124;上記参照)により、この時間で十分なことが示されていた。次に、金属イオン添加吸着剤を減圧濾過で収集して、水(5×100mL)で洗浄し、20mM NaOAc/1M NaCl/pH5緩衝液(50mL)中に15分間懸濁した。前記ゲルを濾過し、更に一定分量(50mL)のpH5緩衝液で洗浄後、水(3×100mL)で洗浄して、3分間の洗浄平衡化の時間を維持した。
真空乾燥したCu2+添加吸着剤0.5gを20mLシンチレーションバイアルに入れて、下記の溶液(10mL)とともに、室温で所定の時間、インキュベートした。次に、さらに20mM Na2HPO4/1M NaCl/pH7緩衝液(10mL)、続いて水(3×50mL)で洗浄する前に前記ゲルを減圧濾過によって単離し、洗浄の間には3分間平衡化させた。
2)20mM Na2B4O7/1M NaCl/pH9緩衝液、10分
3)200mM イミダゾール、10分
4)200mM Na2EDTA、30分
Cu2+−及びNi2+−添加セファロースTMCL−4B吸着剤の金属分析
金属イオン添加吸着剤の試料ごとに、約0.5gの真空乾燥した材料を純アセトン中に移行させ、無金属イオンゲルに対して上述の方法を用いて高真空下で乾燥させた。次に、約20gの乾燥させた吸着剤を正確に秤量して20mLの密封シンチレーションバイアルに入れ、頻繁に倒置させながら、50℃で4時間にわたり4M HCl(4mL)中で消化した。生成した明黄褐色の溶液を8mLの容量になるように水で希釈した後、原子吸光分光法により銅の含有量を分析した。膨潤したゲル容積について吸着剤の金属含量を測定するために、50%(v/v)ゲル/水懸濁液の分取試料(1mL)を用いて分析を繰り返し、ゲルを水中に沈降させ、ゲル上の液体量がベッド容量と等しくなるように調整することによって調製した。
上記の操作に従って、高真空下で乾燥させたゲル試料を用いて、拡散反射率のスペクトルを記録した。20mM NaOAc/1M NaCl/pH5緩衝液で洗浄したゲルの真空乾燥試料を用いて、ESRスペクトルを記録した。
実施例86に記述したように、GSTをコードする配列のC末端を修飾して完全長の構築物を得るために、GST−NT1を発現するように構築されたプラスミドの部位特異的突然変異誘発(SDM)を行った。リジン残基(コドンAAA)をコードするようにGSTのSDMを行うことによって、単一塩基の変化が導入された(停止コドン(TAA)を変化させた)。N末端がタグ化された組換えGST融合タンパク質(GST−NT1)発現プラスミド中に存在するこの終止コドンを除去すると、N末端がタグ化された組換えGST融合タンパク質(GST−NT1)を発現するために使用できるプラスミドが得られ、該融合タンパク質においては、4アミノ酸Lys−Ser−Asp−LeuだけGSTのC末端がそれぞれ伸長する。C末端のリジン残基の復活により、完全長のGST配列に加えて、例えば、元の構築物を作製するために使用されたpGEX3XGSTベクターに由来する更に3つのC末端残基がコードされるので、これにより得られた発現プラスミドは、完全長グルタチオンS−トランスフェラーゼ〔GST(FL)〕と命名された。
実施例86に記載されているように、N末端がタグ化されたGST融合タンパク質GST−NT1を発現するために構築された発現プラスミドを、鋳型DNAとして使用した。望ましい塩基の変更を導入するために使用したオリゴヌクレオチドは下記のとおりであり、下線が付されている。
オリゴヌクレオチドNo.2:5‘GCTTTTACAGATCCGATTTAGGAGGATGGTCGCC3’
125ngのオリゴヌクレオチドNo.1、125ngのオリゴヌクレオチドNo.2、5μLの10X反応緩衝液、及び1μLのdNTPを含む混合物に約25ngのdsDNAテンプレートを加えた。前記反応混合物の量を二重蒸留水で50μLになるように調節した。この混合物に2.5UのPfu Turbo DNAポリメラーゼを添加し、反応混合物を鉱油で覆った。
上記の反応から得た5μLのDpnl処理DNAを50μLのXL−1−Blue Supercompetent細胞に添加し、氷上でその混合物を30分間インキュベートした。次に、この混合物に対して、42℃で45秒間、ヒートパルスを行った後、2分間、氷冷した。0.5mLのSOC培地〔5mM MgCl2及び、5mM MgS04、20mMグルコースを追加して濾過殺菌した5mLのSOB(トリプトン20g/L及び酵母エキス5g/L、2.5mM NaCl、625μM KCl、加圧滅菌処理)を添加した後に、形質転換反応を37℃1時間でインキュベートした。異なる希釈度の形質転換混合物をLB Ampプレート(寒天15g/Lにトリプトン10g/L、酵母エキス5g/L、NaCl10g/Lを添加して加圧滅菌した後、アンピシリン100mg/Lを補充した)上に播種した。前記プレートを、37℃で一晩、インキュベートした。
標準的な技術を用いて、上記XL−1 Blue形質転換体から修飾されたプラスミドDNAを単離した。単離したプラスミドDNAのDNA塩基配列決定は下記のように実行した。約200ngの修飾されたプラスミドDNA、5pmolのプライマー、6μLのターミネーターを15μLの容積に事前に混合する。前記反応による混合物を2つ準備し、一方には、
(a)pTrc フォワードプライマー: 5‘TTGACAATTAATCATCCGGC3’を入れ、
他方には、
(b)pTrc リバースプライマー:5‘ CCAGGCAAATTCTGTTTTATCAG3’を入れた。
標準技術を用いて、塩化ルビジウム処理した大腸菌細胞の菌株JM105、BL21、TOP10を、約100ngの修飾発現プラスミドでそれぞれ形質転換した。プラスミド含有細胞を選択するため、様々な希釈度の形質転換細胞をLB Ampプレート上に播種した。
GSTをコードする配列のC末端を修飾するために、実施例88に記載のGST−CT2を発現するために構築されたプラスミドの部位特異的突然変異誘発(SDM)を行った点を除いて、使用した操作は、基本的に実施例127に記述した操作と同一であった。
GSTをコードする配列のC末端を修飾するために、実施例89に記載のGST−NT2を発現するために構築されたプラスミドの部位特異的突然変異誘発(SDM)を行った点を除いて、使用した操作は、基本的に実施例127に記述した操作と同一であった。
実施例128に記述の通りに、GST(FL)−CT2発現プラスミドを用いて、大腸菌細胞株JM105を形質転換した。LB Amp培地を含むぺトリ皿上で、形質転換した細胞を37℃で一晩繁殖させた。100μg/mLアンピシリンを補充したLB培地(トリプトン10g/L、酵母エキス5g/L、NaCl 10g/L、加圧滅菌処理)に、前記ぺトリ皿の単一コロニーを播種し、一晩中振盪させながら37℃でインキュベートした。100μg/mLアンピシリン(2×YTA)を補充した500mLの2×YT培地(トリプトン16g/L、酵母エキス10g/L、NaCl 5g/L、pH7.0、加圧滅菌処理)に、一晩培養物の1/100希釈液を播種した後、600nmの吸光度(A600)が0.6−1.0になるまで、37℃で振盪させながらインキュベートした。最終濃度が1mMになるようにIPTGを加えることにより、遺伝子発現を誘導した。誘導から6−7時間後に、前記培地の細胞を採集した。25mLの氷冷1×PBS(140mM NaCl、2.7mM KCl、10mM Na2HPO4、1.8mM KH2PO4、pH7.3)中に細胞を再懸濁し、超音波処理することによって溶解した。ペレット化した後に、前記細胞破砕液を収集した。
実施例127に記載したようにGST(FL)−NT1発現プラスミドで大腸菌細胞株JM105を形質転換した点を除いて、使用した操作は、基本的に実施例130に記述した操作と同一であった。
実施例129に記載したようにGST(FL)−NT2発現プラスミドで大腸菌細胞株JM105を形質転換した点を除いて、使用した操作は、基本的に実施例130に記述した操作と同一であった。
GST(FL)−CT2発現プラスミドで大腸菌細胞株BL21を形質転換した点を除いて、使用した操作は、基本的に実施例130に記述した操作と同一であった。
GST(FL)−NT1発現プラスミドで大腸菌細胞株BL21を形質転換した点を除いて、使用した操作は、基本的に実施例131に記述した操作と同一であった。
GST(FL)−NT2発現プラスミドで大腸菌細胞株BL21を形質転換した点を除いて、使用した操作は実質的に実施例132に記述した操作と同一であった。
GST(FL)−CT2発現プラスミドで大腸菌細胞株TOP10を形質転換した点を除いて、使用した操作は実質的に実施例130に記述した操作と同一であった。
GST(FL)−NT1発現プラスミドで大腸菌細胞株TOP10を形質転換した点を除いて、使用した操作は実質的に実施例131に記述した操作と同一であった。
GST(FL)−NT2発現プラスミドで大腸菌細胞株TOP10を形質転換した点を除いて、使用した操作は実質的に実施例132に記述した操作と同一であった。
適切なクロマトグラフィーの吸着剤として、固定化したNi2+−tacn、Cu2+−tacn、Ni2+−dtne、Cu2+−dtne、Ni2+−dtnp、Cu2+−dtnp又はその他の金属イオンをキレートした大環状トリス−又はテトラキス−tacn系を用いて、基本的に実施例93に記述したバッチ法又はカラムクロマトグラフィ法によって、実施例128(上記)に記述したとおりに発現したN末端タグ化GST(FL)融合タンパク質GST(FL)−CT2を精製することができる。緩衝液B(緩衝液B=10mMイミダゾール、pH8.4を追加した緩衝液A;緩衝液A=445mM NaCl、7.5mM Na2HPO4及び2.5mM NaH2PO4.2H2O、pH7.2)を用いて各カラムをインキュベートした。ゲル1mLにつき250μLの粗細胞破砕液(緩衝液Bで4分の1に希釈)をカラムに添加した。次に、溶出前に、緩衝液Bでカラムを2回洗浄した。以下の2つの溶出操作を使用した。緩衝液1(緩衝液1=50mM イミダゾール、pH9.7を追加した緩衝液A)で溶出後に、2度目の溶出を200mM マロン酸ナトリウムで行った(これは、10M 水酸化ナトリウムでpH8.0に調節した)。あるいは、これらのGST融合タンパク質を、グルタチオンセファロースTM4B(GS4B)を用いて精製することもできる。本ケースでは、Pharmacia Biotech出版の情報冊子GST Gene Fusion System第3版改訂1に従ってバッチ精製を行った。簡潔に記述すると、実施例130に記載したように、1XPBS(140mM NaCl、2.7mM KCl、10mM Na2HPO4、1.8mM KH2PO4、pH7.3)で洗浄及び平衡化したベッド容積が0.75mLのGS4Bに、24mLの細胞破砕液を加えた。生じた混合物を室温で約1時間、繰り返し回転させながらインキュベートした。前記混合物を500Xgで5分間、遠心分離した。上清を収集して、ゲルを1XPBSで徹底的に洗浄した。融合タンパク質を溶出緩衝液(50mMトリス−HCl pH8、10mM 還元型グルタチオン)で溶出した。
実施例131に記述した通りに粗細胞破砕液を精製した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例132に記述した通りに粗細胞破砕液を精製した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例133に記述した通りに粗細胞破砕液を精製した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例134に記述した通りに粗細胞破砕液を、EDTAとともにインキュベートされなかった精製タンパク質を用いて分画した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例135に記述した通りに粗細胞破砕液を、EDTAとともにインキュベートされなかった精製タンパク質を用いて分画した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例136に記述した通りに粗細胞破砕液を、EDTAとともにインキュベートされなかった精製タンパク質を用いて分画した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例137に記述した通りに粗細胞破砕液を、EDTAとともにインキュベートされなかった精製タンパク質を用いて分画した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例138に記述した通りに粗細胞破砕液を、EDTAとともにインキュベートされなかった精製タンパク質を用いて分画した点を除いて、使用した操作は、実質的に実施例139に記述した操作と同様であった。
実施例139、142、145に記載したように、様々な大腸菌株で発現精製された精製透析N末端タグ化GST(FL)−CT2融合タンパク質の分子量を、マトリックス支援レーザー脱離イオン化飛行時間型質量分析装置(MALDI−TOF−MS)によって測定した。非還元タンパク質試料がグルタチオンセファロースTM4B(GS4B)精製手順から発生したため、予想した理論上の分子量より[M+H]+の実験値の方が高くなり、実施例136及び142、145に記載したように、グルタチオン精製時に還元グルタチオン(GSH)によってGSTタンパク質がS−チオレートされるという事実と合致していた。しかし、βメルカプトエタノールを添加して前記タンパク質試料を還元後に脱塩すると、[M+H]+の実験値は予想理論分子量と一致した。従って、MALDI−TOF MSによって分析したタンパク質試料は全て、このようにして調製した。
実施例139に記載したように、大腸菌細胞株JM105中で発現させたGST(FL)−CT2のスペクトルは、平均[M+H]+値の28,006±5.57ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
実施例142に記載したように、大腸菌細胞株BL21中で発現させたGST(FL)−CT2のスペクトルは、平均[M+H]+値28,008.10±5.29ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
実施例145に記載したように、大腸菌細胞株TOP10に発現したGST(FL)−CT2のスペクトルは、平均[M+H]+値28,003.00±3.46ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
GST(FL)−NT1の理論上の分子量が27,965.3ダルトンであるという点を除けば、使用した操作は実質的に例148に記述したものと同じであるが、
MALDI−TOF MSによる、大腸菌細胞株JM105中で発現させたGST(FL)−NT1の分子量測定
実施例140に記載したように、大腸菌細胞株JM105中で発現させたGST(FL)−NT1のスペクトルは、平均[M+H]+値27,953.00±2.65ダルトンのシングルピークを示した。実験データは無傷のタンパク質と一致した。
実施例143に記載したように、大腸菌細胞株BL21中で発現させたGST(FL)−NT1のスペクトルは、平均[M+H]+値27,951.00±4.36ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
実施例146に記載されているとおりに、大腸菌細胞株TOP10中で発現したGST(FL)−NT1のスペクトルは、平均[M+H]+値27,954.67±6.66ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
GST(FL)−NT2の理論上の分子量が、28,218.6ダルトンである点を除いて、使用した操作は実質的に実施例148に記述したものと同じである。
実施例141に記載したように、大腸菌細胞株JM105中で発現させたGST(FL)−NT2のスペクトルは、平均[M+H]+値28,208.00±8.44ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
実施例144に記載したように、大腸菌細胞株BL21で発現させたGST(FL)−NT2のスペクトルは、平均[M+H]+値28,214.33±5.86ダルトンのシングルピークを示した。実験データは、変化を受けていないタンパク質のデータと合致していた。
実施例147に記載したように、大腸菌細胞株TOP10中で発現させたGST(FL)−NT2のMALDI−MSスペクトルは下記の通り。
(b)マイナーピークは(約25%相対強度)の平均[M+H]+値は27,815.83±8.04ダルトン、
(c)マイナーピーク(約13%相対強度)の平均[M+H]+値は26,301.0±6.90ダルトンであった。
この種の理論上の分子量は27,822.11ダルトンである。
この種の理論上の分子量は26,312.55ダルトンである。
等量のDTT(20mM)を添加して室温で4−6分間、混合物をインキュベートすることによって、N末端切断酵素ジペプチジルアミノペプチターゼ1[例えば Aldrich−Sigma(St Louis、MO、USA)又はUnizyme Laboratories(Horsholm、Denmark)から入手可能なDAP−1(E.C.3.4.14.1)](10U/mL)を活性化した。消化反応混合物には、実施例139に記述の大腸菌細胞株JM105中で発現させたN末端タグ化GST(FL)−CT2タンパク質50μg及び12.5mUの活性化したDAP−1が含まれた。タンパク質が懸濁された緩衝液(20mM リン酸ナトリウム、150mM NaCl、pH6.9)を加えることによって、前記反応混合物の量を70μLに調節した。次に、前記消化反応混合物をウォーターバスに入れて37℃で5分ないし約23時間にわたって、インキュベートした。
以下の点を除き、操作は実質的に実施例151に記述したものと同じであった。実施例140に記述したように、精製N末端タグ化GST(FL)融合タンパク質GST(FL)−NT1を大腸菌細胞株JM105中で発現させ、緩衝液(20mM 酢酸ナトリウム、150mM NaCl、pH5.5)に対して更に透析を行った。透析後、標準物質としてウシ血清アルブミン(BSA)を用いたビシンコニン酸(BCA)試薬(Pierce Chemical Co.、Rockford、IL、USA)を使用して、タンパク質の濃度を測定した。前記DAP−1消化反応混合物を、5分乃至約24時間にわたってインキュベートした。
以下の点を除いて、N末端タグ化GST(FL)融合タンパク質GST(FL)−NT2のDAP−1消化の操作は、実質的に実施例151に記述した操作と同様であった。
GST(FL)−CT2をDAP−1で処理した。消化反応の条件は実施例151に記載されているとおりである。5分から約23時間のインキュベーション時間の最後に、一定分量の消化反応物を取り出し、消化反応を停止させるために、室温で5分間、グアニジンHCl(最終濃度1Mになるように加えた)とともにインキュベートした。次に、この混合物を、MALDI−TOF MSによる後の分析のために、実施例148に記載したように、更に調製した。簡潔に記述すると、反応を停止した消化反応混合物にβ−メルカプトエタノール(最終濃度50mMになるように)を加え、C18ZipTipTMを用いて脱塩する前に、−20℃で保管した。次に室温で15分間インキュベートした。
(この種の理論的分子量は、26,156.3ダルトンである)
ピーク(2b) MKHQHQHQHQHQHQVD 1QMSP―――――[GST]
(この種の理論的分子量は25,942.1ダルトンである)
ここで、1Vと1Qは、DAP−1消化後に得られる末端切断されたGST(FL)−CT2バリアントのN末端アミノ酸残基に対応する。DAP−1の特異性が顕著であることを前提にすれば、DAP−1の停止位置1SP―――――GSTまでさらに切断が起こる(ここで、1SはN末端アミノ酸残基である)。
以下の点を除き、操作は実質的に実施例154に記述したものと同様であった。
(この種の理論上の分子量は26,654.9ダルトンである)
ピーク(2b) MKHHHNSWDHDINRVD 1QMSP――――[GST]
(この種の理論上の分子量は25,942.1ダルトンである)
ピーク(2c) MKHHHNSWDHDINRVDQM 1SP―――― [GST]
(この種の理論上の分子量は25,682.80ダルトンである)
ここで、1D・・・、1Q・・・及び1S・・・は、DAP−1消化後に得られる、GST(FL)−NT1の末端切断バリアントのN末端アミノ酸残基に対応する。DAP−1の顕著な特異性を前提とすれば、DAP−1の停止位置1SP―――――[GST]まで切断が起こる。シナピン酸(分子量が224.2Da増加)及びβ−メルカプトエタノール(MW78.13)の付加物に対応する副次的ピークもスペクトル中に明瞭に観察される。
以下の点を除いて、操作は実質的に実施例154に記述したものと同様であった。
(この種の理論上の分子量は25,942.1ダルトンである)
ピーク(2b) MKHTNIHQDQHNHFHRVDQM 1SP――[GST]
この種の理論上の分子量は25,682.8Daである。
DAP−1消化前及び消化後に、実施例151、152、及び153に記載したように、様々なN末端タグ化組換えグルタチオンS−トランスフェラーゼ完全長融合タンパク質の一定分量を、SDS添加緩衝液(還元性)と混合し、100℃で90秒間加熱した。次に、試料を18%SDS−ポリアクリルアミドゲル上に添加して、適宜の時間展開させた。前記ゲル上に、0.4μLの未消化(すなわち、非DAP−1処理)タンパク質、0.6μLのDAP消化反応混合物、及び8μLの分子量マーカー[BenchMark Protein Ladder(Gibco BRL)]をゲルに添加した。バンドを可視化するために、前記ゲルを銀染色した。染色したゲルを図56に示す。
基本的に実施例92及び152に記述した操作で、DAP−1消化反応混合物を調製した。簡潔に記述すると、等量のDTT(20mM)を加え、室温で4−6分間前記混合物をインキュベートすることによって、DAP−1[EC3.4.14.1](10U/mL)を活性化した。前記消化反応混合物には、実施例140及び152に記載したように大腸菌細胞株JM105中で発現された50μgのN末端タグ化GST(FL)−NT1タンパク質及び12.5mUの活性化DAP−1が含有されていた。タンパク質を懸濁した緩衝液(20mM 酢酸ナトリウム、150mM NaCl、pH5.5)を加えることによって、前記反応混合物の量を70μLに調整した。次に、消化反応混合物をウォーターバスの中に入れ、37℃で約24時間インキュベートした。
下記の点を除いて、操作は、基本的に実施例158に記述したものと同様であった。
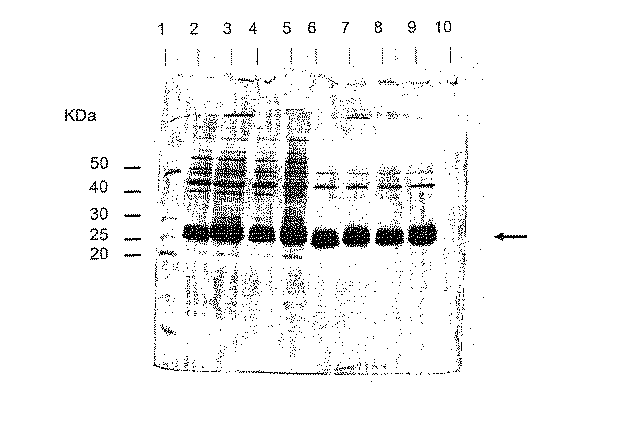
以下の様々なアフィニテイータグ(すなわち、ヘキサヒスチジン(GST−CT1)、His−Glnの交互リピート(GST−CT2)、新しいタグ1(GST−NT1)とNT2(GST−NT2))を、実施例91に記載されているようにN末端に融合させたグルタチオン S−トランスフェラーゼ(GST)の大規模な発現から得られた粗細胞破砕液(CCL)を、単一段階の均一濃度溶出操作によって精製した。粗細胞破砕液をNi2+−tacn及びCu2+−tacnの両カラムにかけた。各カラム(10mL エコノカラム、Bio−RadTM)のベッド容積は0.5mLで、緩衝液A1[緩衝液A1=緩衝液A(20mMリン酸緩衝液、500mM NaCl、pH8)+10mM イミダゾール、pH8]により平衡化した。前記カラムに0.5mL(CCL)+4.5mLの緩衝液A1を含有する5mLの試料を添加した。試料がカラムを通過した後に、カラムを緩衝液A1(5mL)で洗浄した。緩衝液A2(緩衝液A2=緩衝液A+20mM イミダゾール、pH8)を用いて,2度目の洗浄(5mL)を行った。3×1mLの溶出緩衝液(BA+500mMイミダゾール、pH8)をカラムにロードすることによって溶出を行った。
実施例160に記載したように、それぞれをNi2+−tacn及びCu2+−tacnカラムにかけた様々なN末端タグ化GST融合タンパク質の単一段階均一濃度溶出から収集した第一の溶出分画を一定分量採って、SDS添加緩衝液(還元性)と混合し、100℃で90秒間加熱した。次に、試料を15%SDS-ポリアクリルアミドゲル上に添加し、適切な時間にわたって展開した。16μLの溶出分画と2μLの分子量マーカー[Benchmark protein ladder(Gibco,BRL)]を前記ゲル上に添加した。バンドを可視化するために、前記ゲルを銀染色した。染色したゲルを図57に示す。
基本的に実施例127に記載したように生成されたプラスミド構築物を用いて、1リットルの振盪容器中でN末端タグ化融合タンパク質NT2−VD−hGH、すなわちMKHTNIHQDQHNHFHRVD−hGHを発現した。ペレットを収集して水で洗浄した。次に、250μLのリゾチーム(30mg/mL)と1.25μLのベンゾナーゼを加えることによって、溶解緩衝液(20mL 20mM Na2HPO4、500mM NaCl、pH8)中で250mLの前記ペレットを溶解した。懸濁液を5℃で1時間放置した後に、遠心分離を行った。上清を収集して80mLの溶解緩衝液で希釈した後、3mL/分の速度で15mLの固定化Ni2+−tacnカラムに導入し、続けて150mL緩衝液を加えた。次いで、2%溶出緩衝液(20mM Na2HP04、500mM NaCl、500mL イミダゾール、pH8)を含む150mLの溶解緩衝液でカラムを洗浄した後、4%溶出緩衝液を含む150mL溶解緩衝液で洗浄した。その後、75mLの総量について、4%から100%の溶出緩衝液を用いて勾配溶出を行い、続いて100%溶出緩衝液75mLを用いて溶出した。
さらに、推定精度50ダルトンのMALDI−TOF MSによって多数の試料を分析し、単離したタグ化生成物の分子量が24,444ダルトンであることを明らかにした(理論値は24,402ダルトン)。切断と2度目のNi2+−tacnカラム溶出の後には、22,156ダルトンの分子量が測定された(理論値は24,125ダルトン)。hGH標準試料の分子量は22,147ダルトンであった。
基本的に実施例127に記載したように生成されたプラスミド構築物を用いて、1リットルの振盪容器中でN末端タグ化融合タンパク質CT2−VD−hGH、すなわちMKHQHQHQHQHQHQVD−hGHを発現した。ペレットを収集して水で洗浄した。次に、実施例162に記載したように、250mLから得られたペレットを溶解緩衝液中で溶解した。懸濁液を5℃で1時間放置した後に、懸濁液の遠心分離を行った。上清を収集して、その半分の量を40mLの溶解緩衝液で希釈後、1mL/分の速度で5mLの固定化Ni2+−tacnカラムに導入した後、20mL緩衝液を流した。次に、前記カラムを2%溶出緩衝液(20mM Na2HP04、500mM NaCl、500mL イミダゾール、pH8)を含む50mLの溶解緩衝液で洗浄後、4%の溶出緩衝液を含む50mLの溶解緩衝液で洗浄した。その後、総量25mLに対して4%から100%の溶出緩衝液を用いて勾配溶出を行った後、100%の溶出緩衝液25mLを用いて溶出した。
本結果から、2つの異なるM2+−tacnカラムを連続して用いることによって、非hGH関連物質の含量が著しく減少することが実証された。
Claims (15)
- 下記からなる群から選択されるアミノ酸配列を備えたヒスチジン含有オリゴペプチド:
HHHNSWDHDINR(配列番号1)
HTNIHQDQHNHFHR(配列番号2)
HAMLDRAHDHGTR(配列番号3)
SLHEHHSGDNLR(配列番号4)
THYNAVHSHNTLQ(配列番号5)
DIHHWTDHLQSSTH(配列番号6)
LYNHYSHTAHVNHL(配列番号7)、
但し、前記アミノ酸配列が配列番号1であるときは、当該ヒスチジン含有オリゴペプチドはアミノ酸配列HHHNSWDHDINRからなる。 - 前記アミノ酸配列がアミノ酸配列Met−Xaaを含むN末端伸長部を有し、前記Xaaがプロリン残基以外のアミノ酸残基である、請求項1に記載のヒスチジン含有オリゴペプチド。
- 前記アミノ酸配列が、多くとも10アミノ酸残基のアミノ酸配列を備えたN末端伸長部を有する、請求項1に記載のヒスチジン含有オリゴペプチド。
- 請求項1乃至3の何れか1項に記載された少なくとも1つのオリゴペプチドに目的のタンパク質分子のアミノ末端又はカルボキシ末端が融合したタンパク質を備えた融合タンパク質であるポリペプチド。
- 前記オリゴペプチドのアミノ酸配列の前記アミノ末端及びカルボキシ末端が、目的のタンパク分子にそれぞれ融合する、請求項4に記載のポリペプチド。
- 酵素及び化学的切断部位からなる群から選択される切断部位に前記オリゴペプチドのアミノ酸配列が隣接して存在する、請求項5に記載のポリペプチド。
- 前記2つの対象タンパク質分子が同一タンパク質の分子である、請求項5又は6に記載のポリペプチド。
- 前記2つの対象タンパク質分子が異なる分子である、請求項5又は6に記載のポリペプチド。
- 前記ポリペプチドを発現させる条件下で、適切な増殖培地中において、請求項4乃至8の何れか1項に記載のポリペプチドをコードするポリヌクレオチド構築物を備える宿主細胞を培養すること、及び前記培地から当該ポリペプチドを回収することにより取得可能なポリペプチド。
- 前記宿主細胞がEsherichia coliの菌株である、請求項9に記載のポリペプチド。
- 請求項4乃至8の何れか1項に記載のポリペプチドをコードするポリヌクレオチド構築物。
- ベクターである、請求項11に記載のポリヌクレオチド構築物。
- 請求項11又は12に記載のポリヌクレオチド構築物を含む宿主細胞。
- Esherichia coliの菌株である、請求項13に記載の宿主細胞。
- 請求項4乃至10の何れか1項に記載のポリペプチドを生産する方法であって、前記ポリペプチドを発現させる条件下で、適切な増殖培地中において、請求項13又は14に記載されている宿主細胞を培養することと、前記培地から前記ポリペプチドを回収することを備えた方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DKPA200101681 | 2001-11-12 | ||
| DKPA200101681 | 2001-11-12 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003544084A Division JP4291152B2 (ja) | 2001-11-12 | 2002-11-12 | 金属イオンアフィニティークロマトグラフィーによるペプチドの精製 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009142279A JP2009142279A (ja) | 2009-07-02 |
| JP4988694B2 true JP4988694B2 (ja) | 2012-08-01 |
Family
ID=34778135
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003544084A Expired - Fee Related JP4291152B2 (ja) | 2001-11-12 | 2002-11-12 | 金属イオンアフィニティークロマトグラフィーによるペプチドの精製 |
| JP2008321467A Expired - Fee Related JP4988694B2 (ja) | 2001-11-12 | 2008-12-17 | 金属イオンアフィニティークロマトグラフィーによるペプチドの精製 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2003544084A Expired - Fee Related JP4291152B2 (ja) | 2001-11-12 | 2002-11-12 | 金属イオンアフィニティークロマトグラフィーによるペプチドの精製 |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US7456276B2 (ja) |
| EP (2) | EP1446426B1 (ja) |
| JP (2) | JP4291152B2 (ja) |
| CN (2) | CN101979413A (ja) |
| AT (2) | ATE509943T1 (ja) |
| AU (1) | AU2002340787B2 (ja) |
| DE (1) | DE60231055D1 (ja) |
| ES (1) | ES2322338T3 (ja) |
| WO (1) | WO2003042249A2 (ja) |
Families Citing this family (40)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1534755B1 (en) | 2002-05-10 | 2011-10-12 | Bio-Layer Pty Limited | Generation of surface coating diversity |
| AU2003903504A0 (en) * | 2003-07-08 | 2003-07-24 | Johnson, Daniel | Improvements in sensor chips |
| KR20060065643A (ko) * | 2003-07-24 | 2006-06-14 | 머크 앤드 캄파니 인코포레이티드 | 스타필로코쿠스 아우레우스에 대한 방어 면역 반응을유도하는 폴리펩타이드 |
| AU2004296412B2 (en) | 2003-12-12 | 2011-03-10 | Anteo Technologies Pty Ltd | A method for designing surfaces |
| DE602004032379D1 (de) | 2003-12-19 | 2011-06-01 | Novo Nordisk As | Prozessierung von peptiden und proteinen |
| AU2005251949B2 (en) * | 2004-06-14 | 2011-07-21 | Monash University | Peptide purification by means of hard metal ion affinity chromatography |
| WO2006002472A1 (en) * | 2004-07-02 | 2006-01-12 | Bio-Layer Pty Ltd | Use of metal complexes |
| US20100029911A1 (en) * | 2004-11-24 | 2010-02-04 | Aplagen Gmbh | Method For Solid-Phase Peptide Synthesis And Purification |
| JP2009073819A (ja) * | 2007-08-29 | 2009-04-09 | Fujifilm Corp | 生理活性物質の精製方法 |
| CN102858756A (zh) * | 2010-03-16 | 2013-01-02 | 生物辐射实验室股份有限公司 | 1,4,7-三氮杂-环壬烷的偶联物、此类偶联物的二核金属配合物、以及1,4,7-三氮杂-环壬烷和偶联物的使用方法 |
| CN102947340A (zh) * | 2010-05-10 | 2013-02-27 | 拜欧雷耶私人有限公司 | 结合系统 |
| JP5565621B2 (ja) * | 2010-07-23 | 2014-08-06 | 三菱レイヨン株式会社 | 多孔質膜用支持体の製造方法及び多孔質膜用支持体 |
| US9029166B2 (en) * | 2010-08-31 | 2015-05-12 | Symrise Ag | Method of identifying natural substances capable of complexation |
| US9062106B2 (en) | 2011-04-27 | 2015-06-23 | Abbvie Inc. | Methods for controlling the galactosylation profile of recombinantly-expressed proteins |
| WO2013082518A1 (en) * | 2011-12-02 | 2013-06-06 | Life Technologies Corporation | Increasing protein recovery in metal affinity chromatography |
| CN102600639B (zh) * | 2012-01-06 | 2014-09-10 | 东南大学 | 一种利用谷胱甘肽分离混合液中半抗原的方法及应用 |
| WO2013158279A1 (en) | 2012-04-20 | 2013-10-24 | Abbvie Inc. | Protein purification methods to reduce acidic species |
| US9067990B2 (en) | 2013-03-14 | 2015-06-30 | Abbvie, Inc. | Protein purification using displacement chromatography |
| WO2013158273A1 (en) | 2012-04-20 | 2013-10-24 | Abbvie Inc. | Methods to modulate c-terminal lysine variant distribution |
| WO2013176754A1 (en) | 2012-05-24 | 2013-11-28 | Abbvie Inc. | Novel purification of antibodies using hydrophobic interaction chromatography |
| HK1211981A1 (en) | 2012-09-02 | 2016-06-03 | Abbvie Inc. | Methods to control protein heterogeneity |
| US9512214B2 (en) | 2012-09-02 | 2016-12-06 | Abbvie, Inc. | Methods to control protein heterogeneity |
| EP2812091B1 (en) | 2012-09-17 | 2021-03-10 | W.R. Grace & CO. - CONN. | Chromatography media and devices |
| HK1207960A1 (en) | 2013-03-12 | 2016-02-19 | Abbvie Inc. | Human antibodies that bind human tnf-alpha and methods of preparing the same |
| US10023608B1 (en) | 2013-03-13 | 2018-07-17 | Amgen Inc. | Protein purification methods to remove impurities |
| US9499614B2 (en) | 2013-03-14 | 2016-11-22 | Abbvie Inc. | Methods for modulating protein glycosylation profiles of recombinant protein therapeutics using monosaccharides and oligosaccharides |
| WO2014159579A1 (en) | 2013-03-14 | 2014-10-02 | Abbvie Inc. | MUTATED ANTI-TNFα ANTIBODIES AND METHODS OF THEIR USE |
| US9017687B1 (en) | 2013-10-18 | 2015-04-28 | Abbvie, Inc. | Low acidic species compositions and methods for producing and using the same using displacement chromatography |
| JP5725462B2 (ja) * | 2013-07-26 | 2015-05-27 | 国立大学法人広島大学 | アスベスト結合タンパク質の利用 |
| US9598667B2 (en) | 2013-10-04 | 2017-03-21 | Abbvie Inc. | Use of metal ions for modulation of protein glycosylation profiles of recombinant proteins |
| US9085618B2 (en) | 2013-10-18 | 2015-07-21 | Abbvie, Inc. | Low acidic species compositions and methods for producing and using the same |
| US8946395B1 (en) | 2013-10-18 | 2015-02-03 | Abbvie Inc. | Purification of proteins using hydrophobic interaction chromatography |
| US9181337B2 (en) | 2013-10-18 | 2015-11-10 | Abbvie, Inc. | Modulated lysine variant species compositions and methods for producing and using the same |
| US20150139988A1 (en) | 2013-11-15 | 2015-05-21 | Abbvie, Inc. | Glycoengineered binding protein compositions |
| ES2887110T3 (es) | 2014-01-16 | 2021-12-21 | Grace W R & Co | Medios para cromatografía de afinidad y dispositivos para cromatografía |
| PL3137209T3 (pl) | 2014-05-02 | 2023-01-02 | W.R. Grace & Co. - Conn. | Funkcjonalizowany materiał nośnikowy i sposoby wytwarzania oraz stosowania funkcjonalizowanego materiału nośnikowego |
| KR102566292B1 (ko) | 2015-06-05 | 2023-08-10 | 더블유.알. 그레이스 앤드 캄파니-콘. | 흡착성 바이오프로세싱 정화제와 이의 제조 및 사용 방법 |
| JP7526360B2 (ja) * | 2020-08-18 | 2024-07-31 | ザルトリウス ビーアイエー セパレーションズ ディー.オー.オー. | マルチモーダル金属アフィニティー処理aavカプシド |
| CA3202317A1 (en) | 2021-01-19 | 2022-07-28 | Lg Energy Solution, Ltd. | Battery and current collector applied thereto, and battery pack and vehicle including the same |
| CN113607868B (zh) * | 2021-06-15 | 2022-03-15 | 广东省农业科学院农业生物基因研究中心 | 一种用于磷酸化蛋白质组学的在线自动化分析装置和分析方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5169936A (en) * | 1989-04-14 | 1992-12-08 | Biogen, Inc. | Protein purification on immobilized metal affinity resins effected by elution using a weak ligand |
| EP0818951A4 (en) * | 1995-04-04 | 2002-10-16 | Lilly Co Eli | Amyloid precursor protein PROTEASE |
| CA2277372C (en) * | 1998-07-13 | 2011-03-15 | Tran Quang Minh | Affinity immobilised metal resins |
| AUPP806999A0 (en) | 1999-01-08 | 1999-02-04 | Csl Limited | Process for purifying immunoglobulins |
| US20040031072A1 (en) * | 1999-05-06 | 2004-02-12 | La Rosa Thomas J. | Soy nucleic acid molecules and other molecules associated with transcription plants and uses thereof for plant improvement |
| US20090087878A9 (en) * | 1999-05-06 | 2009-04-02 | La Rosa Thomas J | Nucleic acid molecules associated with plants |
-
2002
- 2002-11-12 CN CN2009101734745A patent/CN101979413A/zh active Pending
- 2002-11-12 AT AT09151225T patent/ATE509943T1/de not_active IP Right Cessation
- 2002-11-12 AT AT02774483T patent/ATE421977T1/de not_active IP Right Cessation
- 2002-11-12 EP EP02774483A patent/EP1446426B1/en not_active Expired - Lifetime
- 2002-11-12 EP EP09151225A patent/EP2055713B1/en not_active Expired - Lifetime
- 2002-11-12 AU AU2002340787A patent/AU2002340787B2/en not_active Ceased
- 2002-11-12 ES ES02774483T patent/ES2322338T3/es not_active Expired - Lifetime
- 2002-11-12 DE DE60231055T patent/DE60231055D1/de not_active Expired - Lifetime
- 2002-11-12 CN CNA028269713A patent/CN1612896A/zh active Pending
- 2002-11-12 US US10/495,302 patent/US7456276B2/en not_active Expired - Fee Related
- 2002-11-12 WO PCT/DK2002/000758 patent/WO2003042249A2/en not_active Ceased
- 2002-11-12 JP JP2003544084A patent/JP4291152B2/ja not_active Expired - Fee Related
-
2008
- 2008-06-23 US US12/143,905 patent/US20080293917A1/en not_active Abandoned
- 2008-12-17 JP JP2008321467A patent/JP4988694B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009142279A (ja) | 2009-07-02 |
| DE60231055D1 (de) | 2009-03-19 |
| EP2055713A1 (en) | 2009-05-06 |
| CN1612896A (zh) | 2005-05-04 |
| US20060287432A1 (en) | 2006-12-21 |
| EP1446426A2 (en) | 2004-08-18 |
| US20080293917A1 (en) | 2008-11-27 |
| US7456276B2 (en) | 2008-11-25 |
| AU2002340787B2 (en) | 2009-09-17 |
| ES2322338T3 (es) | 2009-06-19 |
| ATE421977T1 (de) | 2009-02-15 |
| EP1446426B1 (en) | 2009-01-28 |
| ATE509943T1 (de) | 2011-06-15 |
| JP4291152B2 (ja) | 2009-07-08 |
| WO2003042249A2 (en) | 2003-05-22 |
| JP2005526487A (ja) | 2005-09-08 |
| WO2003042249A8 (en) | 2004-04-08 |
| WO2003042249A3 (en) | 2003-11-27 |
| EP2055713B1 (en) | 2011-05-18 |
| CN101979413A (zh) | 2011-02-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4988694B2 (ja) | 金属イオンアフィニティークロマトグラフィーによるペプチドの精製 | |
| AU2002340787A1 (en) | Peptide purification by means of metal ion affinity chromatography | |
| JP6862008B2 (ja) | 突然変異免疫グロブリン結合ポリペプチド | |
| JP5298242B2 (ja) | アフィニティークロマトグラフィー用担体およびイムノグロブリンを単離する方法 | |
| Chaga et al. | Immobilized metal ion affinity chromatography on Co2+‐carboxymethylaspartate–agarose Superflow, as demonstrated by one‐step purification of lactate dehydrogenase from chicken breast muscle | |
| EP3221338B1 (en) | Mutated immunoglobulin-binding polypeptides | |
| AU2011201741A1 (en) | Peptide purification by means of hard metal ion affinity chromatography | |
| CN109219613B (zh) | 分离方法 | |
| JP5396933B2 (ja) | 液体クロマトグラフィー用充填剤、及び生体高分子の分離精製方法 | |
| JPWO2011118599A1 (ja) | アフィニティークロマトグラフィー用充填剤およびイムノグロブリンを単離する方法 | |
| KR20100070994A (ko) | 액체 크로마토그래피용 충전 재료 및 해당 충전 재료에 의한 생체고분자의 분리·정제 방법 | |
| JP2019523215A (ja) | 分離マトリックスを保存する方法 | |
| JPH10502339A (ja) | イオン化可能原子団を有する疎水性クロマトグラフィー用樹脂 | |
| Mooney et al. | Application of an IMAC cassette for the purification of N-terminally tagged proteins | |
| JP6722187B2 (ja) | アフィニティー担体およびイムノグロブリンを単離する方法 | |
| CN109678932B (zh) | 一种IgG抗体亲和的小分子肽及其应用 | |
| Tagliavia et al. | Regeneration and Recycling of Supports for Biological Macromolecules Purification |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20110906 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20111206 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20111209 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120306 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120327 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120426 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150511 Year of fee payment: 3 |
|
| LAPS | Cancellation because of no payment of annual fees |