JP5048984B2 - 光導波路用縮合生成物、硬化物及び光導波路デバイス - Google Patents
光導波路用縮合生成物、硬化物及び光導波路デバイス Download PDFInfo
- Publication number
- JP5048984B2 JP5048984B2 JP2006261464A JP2006261464A JP5048984B2 JP 5048984 B2 JP5048984 B2 JP 5048984B2 JP 2006261464 A JP2006261464 A JP 2006261464A JP 2006261464 A JP2006261464 A JP 2006261464A JP 5048984 B2 JP5048984 B2 JP 5048984B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- group
- general formula
- optical waveguide
- condensation product
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Landscapes
- Optical Integrated Circuits (AREA)
- Silicon Polymers (AREA)
Description
(A)一般式(1):
R1 2Si(OR2)2 (1)
〔式中、R1は、少なくとも1個の芳香族基を有する炭素原子数が6〜20個の基を示し、R2は、H(Hは重水素Dであってよい)を示す。〕
で示されるシランジオール化合物と、
(B)一般式(2):
R3Si(OR4)3 (2)
〔式中、R3は、少なくとも1個のC=C二重結合を有する有機基を示し、R4は、CnH2n+1(n=1又は2の数である)を示す。〕
で示される変性シラン化合物と
(C)一般式(3):
R5R6Si(OR7)2 (3)
〔式中、R5は、少なくとも1個のC=C二重結合を有する有機基を示し、R6は、CnH2n+1(n=1又は2の数である)を示し、R7は、CnH2n+1(n=1又は2の数である)を示す。〕
で示される変性シラン化合物と
(D)一般式(4):
R8Si(OR9)3 (4)
〔式中、R8は、CF3(CF2)n(CH2)2−基(n=0〜9の数である)又はC6X5−基(X=H又はFである)を示し、R9は、CnH2n+1(n=1又は2の数である)を示す。〕
で示される変性シラン化合物のうち、少なくとも化合物(A)、化合物(C)及び化合物(D)を、
0≦b×3+c×2+d×3−a×2≦0.4
〔式中、a乃至dはそれぞれ化合物(A)乃至(D)の総量に対する各化合物(A)乃至(D)のモル比率を示す。〕
の関係を満たす範囲内で縮合させることにより得られ、上記一般式(2)中のR 3 と一般式(3)中のR 5 のうち少なくとも一方が、CH 2 =CH−C 6 X 4 −(CH 2 ) n −基(X=H又はFであり、n=0〜2の数である)であることを特徴とする。
上記一般式(4)中のR8が、CF3(CF2)n(CH2)2−基(n=5〜7の数である)であることを特徴とする。
さらに、一般式(2)中のR 3 と一般式(3)中のR 5 のうち少なくとも一方が、CH 2 =CH−C 6 X 4 −(CH 2 ) n −基(X=H又はFであり、n=0〜2の数である)であることによって、光導波路用縮合生成物は、芳香族環を含むためCH基が少なくなって近赤外線領域での光吸収を抑制して、材料損失を低減することができ、また、−C 6 X 4 −基におけるXの一部又は全てをフッ素化することにより、更なる材料損失の低減が可能となる。
本発明では、上記一般式(1)で示されるシランジオール化合物(A)と、上記一般式(2)で示される変性シラン化合物(B)と、上記一般式(3)で示される変性シラン化合物(C)と、上記一般式(4)で示される変性シラン化合物(D)とのうち、少なくとも化合物(A)、化合物(C)及び化合物(D)を、
0≦b×3+c×2+d×3−a×2≦0.4
〔式中、a乃至dはそれぞれ化合物(A)乃至(D)の総量に対する各化合物(A)乃至(D)のモル比率を示す。〕
の関係を満たす範囲内で縮合させて、縮合生成物を得る。
0≦b×3+c×2+e×2−a×2≦0.4
〔式中、a,b,c及びeはそれぞれ化合物(A),(B),(C)及び(E)の総量に対する各化合物(A),(B),(C)及び(E)のモル比率を示す。〕
の関係を満たす範囲内で縮合させて、縮合生成物を得る。
0≦b×3+c×2+d×3+e×2−a×2≦0.4
〔式中、a乃至eはそれぞれ化合物(A)乃至(E)の総量に対する各化合物(A)乃至(E)のモル比率を示す。〕
の関係を満たす範囲内で縮合させて、縮合生成物を得る。
CF3CH2CH2−Si(OCH3)3、
CF3CH2CH2−Si(OCH2CH3)3、
CF3(CF2)3CH2CH2−Si(OCH3)3、
CF3(CF2)3CH2CH2−Si(OCH2CH3)3、
CF3(CF2)5CH2CH2−Si(OCH3)3、
CF3(CF2)5CH2CH2−Si(OCH2CH3)3、
CF3(CF2)7CH2CH2−Si(OCH3)3、
CF3(CF2)7CH2CH2−Si(OCH2CH3)3、
CF3(CF2)9CH2CH2−Si(OCH3)3、
CF3(CF2)9CH2CH2−Si(OCH2CH3)3、
C6F5Si(OCH3)3、
C6F5Si(OCH2CH3)3、
C6F5Si(OCH3)3、
C6F5Si(OCH2CH3)3、
C6F5CH2CH2Si(OCH3)3、
C6F5CH2CH2Si(OCH2CH3)3
を挙げることができる。
CF3CH2CH2−SiCH3(OCH3)2、
CF3CH2CH2−SiCH2CH3(OCH2CH3)2、
CF3(CF2)3CH2CH2−SiCH3(OCH3)2、
CF3(CF2)3CH2CH2−SiCH2CH3(OCH2CH3)2、
CF3(CF2)5CH2CH2−SiCH3(OCH3)2、
CF3(CF2)5CH2CH2−SiCH2CH3(OCH2CH3)2、
CF3(CF2)7CH2CH2−SiCH3(OCH3)2、
CF3(CF2)7CH2CH2−SiCH2CH3(OCH2CH3)2、
CF3(CF2)9CH2CH2−SiCH3(OCH3)2、
CF3(CF2)9CH2CH2−SiCH2CH3(OCH2CH3)2、
C6F5SiCH3(OCH3)2、
C6F5SiCH2CH3(OCH2CH3)2、
C6F5SiCH3(OCH3)2、
C6F5SiCH2CH3(OCH2CH3)2、
C6F5CH2CH2SiCH3(OCH3)2、
C6F5CH2CH2SiCH2CH3(OCH2CH3)2
を挙げることができる。
シランジオール化合物(A)としてジフェニルシランジオールを、変性シラン化合物(B)としてスチリルエチルトリメトキシシランを、変性シラン化合物(C)としてスチリルエチルジメトキシメチルシランを、変性シラン化合物(D)としてトリデカフルオロオクチルトリメトキシシランをそれぞれ用いた。
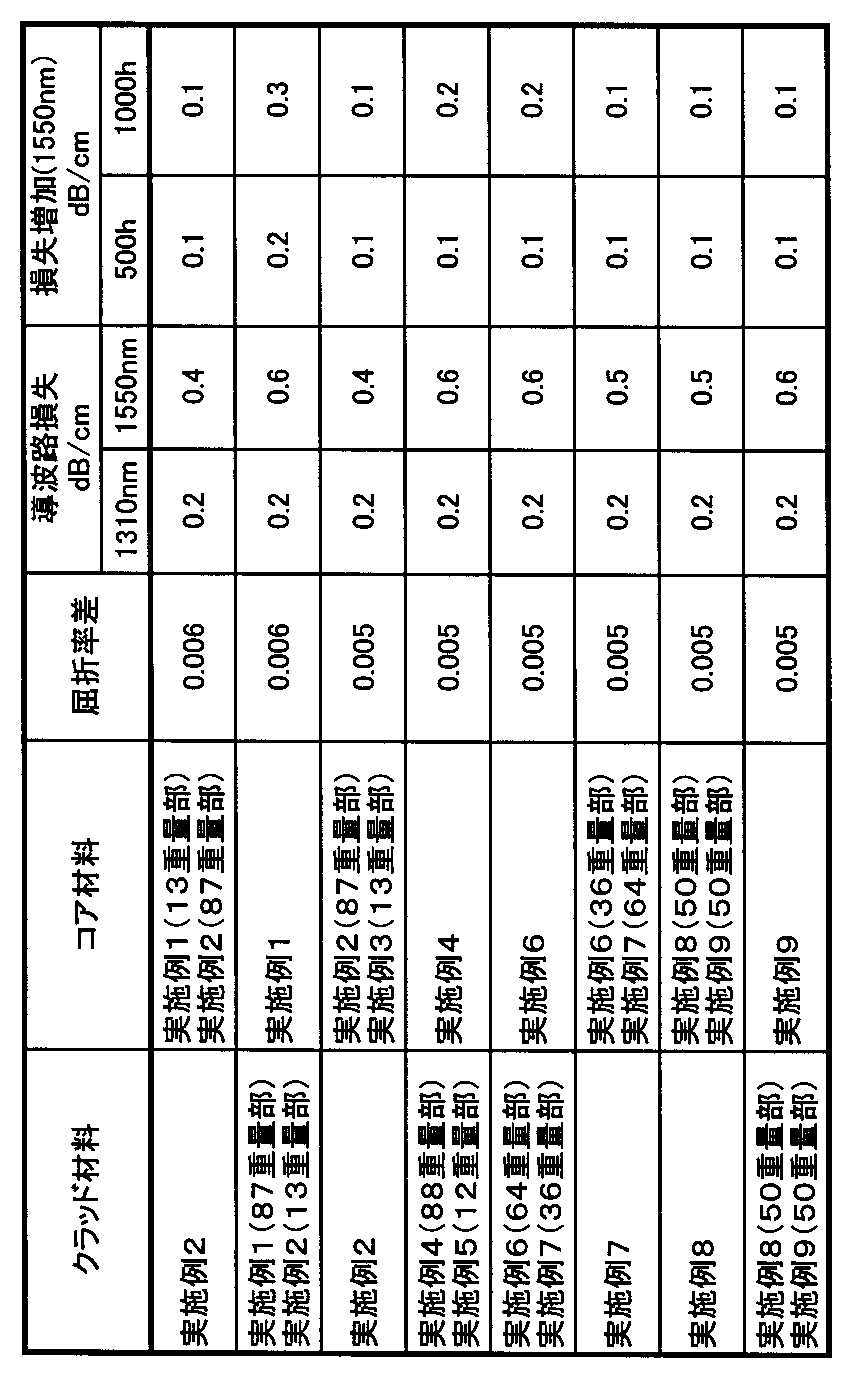
各実施例及び比較例で得られた合成材料を容器に入れ、この容器を傾けた場合の合成材料の流動の様子を目視で観察することで、この合成材料を高粘度、中粘度、低粘度に分類した。その結果、表1に示すように、実施例1〜9ではいずれも粘度が低いのに対し、比較例1、比較例2はどちらも粘度が高かった。
前記合成材料の1310nmと1550nmの近赤外光に対する材料損失を求めることにより、通信波長における特性を調べた。
(屈折率の測定)
各実施例及び比較例で得られた合成材料に、光重合開始剤として2−ベンジル−2−ジメチルアミノ−1−(4−モルフォリノフェニル)−ブタノン−1(チバスペシャルティケミカルズ株式会社製、イルガキュア369)を前記合成材料に対して1重量%の割合で添加したもの80重量部に対し、プロピレングリコールモノメチルエーテルアセテート(PGMEA)を20重量部添加して溶解させた後、これを目開き0.2μmのフィルタで濾過した。
(1)クラッド材料の調製
表2のクラッド材料の欄に示される一種単独の合成材料又は所定割合で二種混合した合成材料に、この合成材料80重量部に対して20重量部のPGMEAを添加して溶解させ、更に光重合開始剤としてイルガキュア369(チバガイギー社製)を合成材料に対して1重量%添加した。これを攪拌した後、目開き0.2μmのフィルタで濾過し、クラッド材料を調製した。
表2のコア材料の欄に示される一種単独の合成材料又は所定割合で二種混合した合成材料に、この合成材料75重量部に対して25重量部のPGMEAを添加して溶解させ、更に光重合開始剤としてイルガキュア369を合成材料に対して1重量%添加した。これを攪拌した後、目開き0.2μmのフィルタで濾過した。これにより、上記「屈折率の測定」と同一手法で測定される屈折率が、クラッド材料よりも表2の屈折率差に示す値だけ大きいコア材料を調製した。
シリコンウェハー上にクラッド材料を塗布後、スピンコーターにて1000rpmで回転させ、100℃で3分間加熱した。その後、窒素雰囲気下、20mW/cm2の強度で5分間紫外線硬化させた後、窒素雰囲気下、200℃で1時間ポストベークし、アンダークラッド層を得た。
アンダークラッド層上にコア材料を塗布後、スピンコーターにて2500rpmで回転させ、100℃で3分間加熱した。その後、マスクを通して、窒素雰囲気下、20mW/cm2の強度で6秒間紫外線硬化させ、メチルイソブチルケトン(MIBK)によって現像した。その後、100℃で3分間加熱し、更に、窒素雰囲気下、20mW/cm2の強度で5分間紫外線硬化させ、7μm×7μmのコア層を得た。
コア層の上にクラッド材料を塗布後、スピンコーターにて1000rpmで回転させ、100℃で3分間加熱した。その後、窒素雰囲気下、20mW/cm2の強度で5分間紫外線硬化させ、オーバークラッド層を得た。その後、窒素雰囲気下、200℃で1時間加熱した。
作製した各直線導波路について、カットバック法(長さの異なる複数の直線導波路の損失を測定し、傾きから単位長さあたりの損失を算出する方法)によって求めた導波路損失を、それぞれ表2に示した。
上記のようにして作製した各直線導波路について、温度85℃、湿度85%の雰囲気下中に500時間、1000時間放置後の導波路損失を測定し、初期(放置開始前)の導波路損失と比較した波長1550nmにおける損失増加をもって「吸湿による損失増加」とした(表2)。尚、波長1310nmに関しては、損失増加は見られなかった。
Claims (4)
- (A)一般式(1):
R1 2Si(OR2)2 (1)
〔式中、R1は、少なくとも1個の芳香族基を有する炭素原子数が6〜20個の基を示し、R2は、H(Hは重水素Dであってよい)を示す。〕
で示されるシランジオール化合物と、
(B)一般式(2):
R3Si(OR4)3 (2)
〔式中、R3は、少なくとも1個のC=C二重結合を有する有機基を示し、R4は、CnH2n+1(n=1又は2の数である)を示す。〕
で示される変性シラン化合物と
(C)一般式(3):
R5R6Si(OR7)2 (3)
〔式中、R5は、少なくとも1個のC=C二重結合を有する有機基を示し、R6は、CnH2n+1(n=1又は2の数である)を示し、R7は、CnH2n+1(n=1又は2の数である)を示す。〕
で示される変性シラン化合物と
(D)一般式(4):
R8Si(OR9)3 (4)
〔式中、R8は、CF3(CF2)n(CH2)2−基(n=0〜9の数である)又はC6X5−基(X=H又はFである)を示し、R9は、CnH2n+1(n=1又は2の数である)を示す。〕
で示される変性シラン化合物のうち、少なくとも化合物(A)、化合物(C)及び化合物(D)を、
0≦b×3+c×2+d×3−a×2≦0.4
〔式中、a乃至dはそれぞれ化合物(A)乃至(D)の総量に対する各化合物(A)乃至(D)のモル比率を示す。〕
の関係を満たす範囲内で縮合させることにより得られ、上記一般式(2)中のR 3 と一般式(3)中のR 5 のうち少なくとも一方が、CH 2 =CH−C 6 X 4 −(CH 2 ) n −基(X=H又はFであり、n=0〜2の数である)であることを特徴とする光導波路用縮合生成物。 - 上記一般式(4)中のR8が、CF3(CF2)n(CH2)2−基(n=5〜7の数である)であることを特徴とする請求項1に記載の光導波路用縮合生成物。
- 請求項1又は2に記載の光導波路用縮合生成物を熱硬化又は光硬化させて成ることを特徴とする硬化物。
- コア及びクラッドを含んでなる光導波路デバイスであって、前記コア及びクラッドが、それぞれ請求項3に記載の硬化物にて形成されていることを特徴とする光導波路デバイス。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006261464A JP5048984B2 (ja) | 2006-09-26 | 2006-09-26 | 光導波路用縮合生成物、硬化物及び光導波路デバイス |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006261464A JP5048984B2 (ja) | 2006-09-26 | 2006-09-26 | 光導波路用縮合生成物、硬化物及び光導波路デバイス |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2008081561A JP2008081561A (ja) | 2008-04-10 |
| JP5048984B2 true JP5048984B2 (ja) | 2012-10-17 |
Family
ID=39352748
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006261464A Active JP5048984B2 (ja) | 2006-09-26 | 2006-09-26 | 光導波路用縮合生成物、硬化物及び光導波路デバイス |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP5048984B2 (ja) |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4126521B2 (ja) * | 2000-08-04 | 2008-07-30 | 信越化学工業株式会社 | 被膜形成用組成物用フロロオルガノポリシロキサン樹脂の製造方法 |
| US6818721B2 (en) * | 2002-12-02 | 2004-11-16 | Rpo Pty Ltd. | Process for producing polysiloxanes and use of the same |
| KR100614976B1 (ko) * | 2004-04-12 | 2006-08-25 | 한국과학기술원 | 광소자 또는 디스플레이에 이용되는 무기/유기혼성올리고머, 나노혼성고분자 및 그 제조방법 |
-
2006
- 2006-09-26 JP JP2006261464A patent/JP5048984B2/ja active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2008081561A (ja) | 2008-04-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7844153B2 (en) | Active energy ray-curable organopolysiloxane resin composition, optical transmission component, and manufacturing method thereof | |
| US6832036B2 (en) | Siloxane optical waveguides | |
| EP2714811B1 (en) | Epoxy-functional radiation-curable composition containing an epoxy-functional siloxane oligomer | |
| KR101152262B1 (ko) | 활성 에너지선 경화성 오가노폴리실록산 수지 조성물, 광전송 부재, 및 광 전송 부재의 제조방법 | |
| JP5027148B2 (ja) | ケイ酸縮合生成物及びそれを使用してなる光導波路デバイス | |
| JP2007530682A (ja) | 光学材料用化合物及び製造方法 | |
| Kim et al. | Synthesis of fluorinated hybrid material for UV embossing of a large core optical waveguide structure | |
| JP3034236B2 (ja) | 光通信素子 | |
| Su et al. | Siloxane materials for optical applications | |
| JP4146277B2 (ja) | ポリマー光導波路の製造方法及びポリマー光導波路 | |
| JP5048984B2 (ja) | 光導波路用縮合生成物、硬化物及び光導波路デバイス | |
| JP4593348B2 (ja) | ポリシラン組成物並びに光導波路及びその製造方法 | |
| JP3110814B2 (ja) | シロキサン系ポリマおよび光学材料 | |
| AU2003242399B2 (en) | Hindered Siloxanes | |
| JP7754370B2 (ja) | 硬化性樹脂組成物、硬化物および光導波路 | |
| KR101190530B1 (ko) | 활성 에너지선 경화성 오가노폴리실록산 수지 조성물, 광전송 부재 및 이의 제조방법 | |
| JP2024123795A (ja) | 光学材料用組成物、光学材料、光導波路およびそれらを用いた光配線素子 | |
| JP2020134799A (ja) | ポリマー光導波路 | |
| WO2024116861A1 (ja) | 硬化性樹脂、硬化性樹脂組成物、及び、硬化物 | |
| WO2025225393A1 (ja) | 硬化性樹脂組成物、硬化性樹脂および硬化物 | |
| JP2025175174A (ja) | 硬化性樹脂組成物、硬化性樹脂および硬化物 | |
| JP2001040215A (ja) | 光学材料 | |
| JP2002309094A (ja) | ポリマ材料及びポリマ膜 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090519 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20100819 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20100915 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111122 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20120112 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120123 |
|
| A02 | Decision of refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A02 Effective date: 20120214 |
|
| A521 | Written amendment |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120514 |
|
| A911 | Transfer of reconsideration by examiner before appeal (zenchi) |
Free format text: JAPANESE INTERMEDIATE CODE: A911 Effective date: 20120524 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120626 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20120720 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20150727 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5048984 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |