JP5113065B2 - N−置換3,4−アルキレンジオキシピロール、エステル置換ジヒドロキシピロールおよびこれらのピロールの合成法 - Google Patents
N−置換3,4−アルキレンジオキシピロール、エステル置換ジヒドロキシピロールおよびこれらのピロールの合成法 Download PDFInfo
- Publication number
- JP5113065B2 JP5113065B2 JP2008534790A JP2008534790A JP5113065B2 JP 5113065 B2 JP5113065 B2 JP 5113065B2 JP 2008534790 A JP2008534790 A JP 2008534790A JP 2008534790 A JP2008534790 A JP 2008534790A JP 5113065 B2 JP5113065 B2 JP 5113065B2
- Authority
- JP
- Japan
- Prior art keywords
- substituted
- methyl
- ethyl
- alkylenedioxypyrrole
- aryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- ZBWXTIWHVVWTKU-UHFFFAOYSA-N 1h-pyrrole-2,3-diol Chemical class OC=1C=CNC=1O ZBWXTIWHVVWTKU-UHFFFAOYSA-N 0.000 title claims abstract description 27
- 238000000034 method Methods 0.000 title claims description 33
- 230000002194 synthesizing effect Effects 0.000 title description 2
- 150000003233 pyrroles Chemical class 0.000 title 1
- 238000006243 chemical reaction Methods 0.000 claims abstract description 23
- 125000000217 alkyl group Chemical group 0.000 claims description 50
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 47
- QTBSBXVTEAMEQO-UHFFFAOYSA-N acetic acid Substances CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 46
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 46
- 125000003118 aryl group Chemical group 0.000 claims description 43
- SNOOUWRIMMFWNE-UHFFFAOYSA-M sodium;6-[(3,4,5-trimethoxybenzoyl)amino]hexanoate Chemical compound [Na+].COC1=CC(C(=O)NCCCCCC([O-])=O)=CC(OC)=C1OC SNOOUWRIMMFWNE-UHFFFAOYSA-M 0.000 claims description 41
- 229910052739 hydrogen Inorganic materials 0.000 claims description 38
- 239000001257 hydrogen Substances 0.000 claims description 36
- -1 2-ethylhexyl Chemical group 0.000 claims description 32
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 27
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 24
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 16
- 229910052757 nitrogen Inorganic materials 0.000 claims description 13
- 230000008569 process Effects 0.000 claims description 13
- 238000009833 condensation Methods 0.000 claims description 12
- 230000005494 condensation Effects 0.000 claims description 12
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 9
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 claims description 8
- 230000009467 reduction Effects 0.000 claims description 7
- 238000007363 ring formation reaction Methods 0.000 claims description 6
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 claims description 4
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 claims description 4
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- LOTBYPQQWICYBB-UHFFFAOYSA-N methyl n-hexyl-n-[2-(hexylamino)ethyl]carbamate Chemical compound CCCCCCNCCN(C(=O)OC)CCCCCC LOTBYPQQWICYBB-UHFFFAOYSA-N 0.000 claims description 4
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 claims description 4
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 claims description 4
- 238000006114 decarboxylation reaction Methods 0.000 claims description 3
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 3
- 238000006266 etherification reaction Methods 0.000 claims description 3
- 238000006386 neutralization reaction Methods 0.000 claims description 3
- 230000009466 transformation Effects 0.000 claims description 3
- KYWZDAWYRTUKGJ-UHFFFAOYSA-N 2-ethylhexyl 4-methylbenzenesulfonate Chemical compound CCCCC(CC)COS(=O)(=O)C1=CC=C(C)C=C1 KYWZDAWYRTUKGJ-UHFFFAOYSA-N 0.000 claims description 2
- NZWIYPLSXWYKLH-UHFFFAOYSA-N 3-(bromomethyl)heptane Chemical compound CCCCC(CC)CBr NZWIYPLSXWYKLH-UHFFFAOYSA-N 0.000 claims description 2
- 229910010082 LiAlH Inorganic materials 0.000 claims description 2
- LOMVENUNSWAXEN-UHFFFAOYSA-N Methyl oxalate Chemical compound COC(=O)C(=O)OC LOMVENUNSWAXEN-UHFFFAOYSA-N 0.000 claims description 2
- 238000006751 Mitsunobu reaction Methods 0.000 claims description 2
- 125000002947 alkylene group Chemical group 0.000 claims description 2
- 230000001588 bifunctional effect Effects 0.000 claims description 2
- 229910052799 carbon Inorganic materials 0.000 claims description 2
- 238000010438 heat treatment Methods 0.000 claims description 2
- 238000007127 saponification reaction Methods 0.000 claims description 2
- 150000002431 hydrogen Chemical class 0.000 claims 16
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims 4
- 150000001350 alkyl halides Chemical class 0.000 claims 4
- KAESVJOAVNADME-UHFFFAOYSA-N 1H-pyrrole Natural products C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 claims 2
- 150000008052 alkyl sulfonates Chemical class 0.000 claims 2
- 239000003638 chemical reducing agent Substances 0.000 claims 2
- 230000005595 deprotonation Effects 0.000 claims 2
- 238000010537 deprotonation reaction Methods 0.000 claims 2
- 238000000197 pyrolysis Methods 0.000 claims 2
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 claims 1
- 150000008051 alkyl sulfates Chemical class 0.000 claims 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 claims 1
- 239000000178 monomer Substances 0.000 abstract description 37
- 125000004185 ester group Chemical group 0.000 abstract description 13
- 239000000543 intermediate Substances 0.000 abstract description 12
- 238000002360 preparation method Methods 0.000 abstract description 7
- 229920000547 conjugated polymer Polymers 0.000 abstract description 5
- 238000010189 synthetic method Methods 0.000 abstract description 3
- 229920000642 polymer Polymers 0.000 description 43
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 39
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 36
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 33
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 33
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 27
- 229960000583 acetic acid Drugs 0.000 description 26
- 239000000203 mixture Substances 0.000 description 24
- 230000003647 oxidation Effects 0.000 description 17
- 238000007254 oxidation reaction Methods 0.000 description 17
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 16
- 239000000741 silica gel Substances 0.000 description 16
- 229910002027 silica gel Inorganic materials 0.000 description 16
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 15
- 239000008367 deionised water Substances 0.000 description 15
- 229910021641 deionized water Inorganic materials 0.000 description 15
- 238000006116 polymerization reaction Methods 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 150000002148 esters Chemical class 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- 239000012300 argon atmosphere Substances 0.000 description 10
- 125000004494 ethyl ester group Chemical group 0.000 description 10
- 239000000047 product Substances 0.000 description 10
- 239000007787 solid Substances 0.000 description 10
- 239000000243 solution Substances 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000011541 reaction mixture Substances 0.000 description 9
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- 230000002829 reductive effect Effects 0.000 description 8
- 230000015572 biosynthetic process Effects 0.000 description 7
- 238000004770 highest occupied molecular orbital Methods 0.000 description 7
- 150000004702 methyl esters Chemical class 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 150000001875 compounds Chemical class 0.000 description 6
- 238000001318 differential pulse voltammogram Methods 0.000 description 6
- 239000003921 oil Substances 0.000 description 6
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 5
- 150000001412 amines Chemical class 0.000 description 5
- 239000012267 brine Substances 0.000 description 5
- 238000006482 condensation reaction Methods 0.000 description 5
- 238000000151 deposition Methods 0.000 description 5
- 239000010408 film Substances 0.000 description 5
- 239000002480 mineral oil Substances 0.000 description 5
- 235000010446 mineral oil Nutrition 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 229920006254 polymer film Polymers 0.000 description 5
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 5
- 125000003047 N-acetyl group Chemical group 0.000 description 4
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 4
- 150000001408 amides Chemical class 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000004020 conductor Substances 0.000 description 4
- 230000008021 deposition Effects 0.000 description 4
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 description 4
- 150000002170 ethers Chemical class 0.000 description 4
- CXOBWJYMBBMTBX-UHFFFAOYSA-N ethyl 2-[bis(2-ethoxy-2-oxoethyl)amino]acetate Chemical compound CCOC(=O)CN(CC(=O)OCC)CC(=O)OCC CXOBWJYMBBMTBX-UHFFFAOYSA-N 0.000 description 4
- 238000003818 flash chromatography Methods 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 239000000463 material Substances 0.000 description 4
- 239000002244 precipitate Substances 0.000 description 4
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- 150000005691 triesters Chemical class 0.000 description 4
- 0 *C(C[n](c(C(O*)=O)c1[N+]([O-])=O)c(C(O*)=O)c1O)=O Chemical compound *C(C[n](c(C(O*)=O)c1[N+]([O-])=O)c(C(O*)=O)c1O)=O 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 125000003158 alcohol group Chemical group 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 239000012141 concentrate Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 239000008151 electrolyte solution Substances 0.000 description 3
- 125000001033 ether group Chemical group 0.000 description 3
- 238000007306 functionalization reaction Methods 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 230000007935 neutral effect Effects 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 238000003828 vacuum filtration Methods 0.000 description 3
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 2
- MUVUZVAQPQNBMU-UHFFFAOYSA-N 7-(carboxymethyl)-3,4-dihydro-2h-[1,4]dioxepino[2,3-c]pyrrole-6,8-dicarboxylic acid Chemical compound O1CCCOC2=C(C(O)=O)N(CC(=O)O)C(C(O)=O)=C21 MUVUZVAQPQNBMU-UHFFFAOYSA-N 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 150000005690 diesters Chemical class 0.000 description 2
- 238000009792 diffusion process Methods 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- 230000000694 effects Effects 0.000 description 2
- 229920001746 electroactive polymer Polymers 0.000 description 2
- 238000004070 electrodeposition Methods 0.000 description 2
- PQJJJMRNHATNKG-UHFFFAOYSA-N ethyl bromoacetate Chemical compound CCOC(=O)CBr PQJJJMRNHATNKG-UHFFFAOYSA-N 0.000 description 2
- 238000002474 experimental method Methods 0.000 description 2
- 230000005669 field effect Effects 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- MGFYIUFZLHCRTH-UHFFFAOYSA-N nitrilotriacetic acid Chemical compound OC(=O)CN(CC(O)=O)CC(O)=O MGFYIUFZLHCRTH-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000005809 transesterification reaction Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- ZXHQLEQLZPJIFG-UHFFFAOYSA-N 1-ethoxyhexane Chemical compound CCCCCCOCC ZXHQLEQLZPJIFG-UHFFFAOYSA-N 0.000 description 1
- MKUDTVSYIHJZFF-UHFFFAOYSA-N 7-(2-methoxyethyl)-3,4-dihydro-2h-[1,4]dioxepino[2,3-c]pyrrole Chemical compound O1CCCOC2=CN(CCOC)C=C21 MKUDTVSYIHJZFF-UHFFFAOYSA-N 0.000 description 1
- 239000004135 Bone phosphate Substances 0.000 description 1
- QBMYSWSPXOWANL-UHFFFAOYSA-N C1COC2=CNC=C2OC1CCOCC3=CC=CC=C3 Chemical compound C1COC2=CNC=C2OC1CCOCC3=CC=CC=C3 QBMYSWSPXOWANL-UHFFFAOYSA-N 0.000 description 1
- KWDMFBNFBKZQSO-UHFFFAOYSA-N CCCCC(CC)COCCC1CCOC2=CNC=C2O1 Chemical compound CCCCC(CC)COCCC1CCOC2=CNC=C2O1 KWDMFBNFBKZQSO-UHFFFAOYSA-N 0.000 description 1
- XFUOZQYFMQSLKH-UHFFFAOYSA-N CCOC(CN1C(C(O)=O)=C2OCCCOC2=C1C(O)=O)=O Chemical compound CCOC(CN1C(C(O)=O)=C2OCCCOC2=C1C(O)=O)=O XFUOZQYFMQSLKH-UHFFFAOYSA-N 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910013684 LiClO 4 Inorganic materials 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- UCXUOVPWMMNOLN-UHFFFAOYSA-N O=C(C[n]1cc2OCCCOc2c1)OCc1ccccc1 Chemical compound O=C(C[n]1cc2OCCCOc2c1)OCc1ccccc1 UCXUOVPWMMNOLN-UHFFFAOYSA-N 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- HEMHJVSKTPXQMS-DYCDLGHISA-M Sodium hydroxide-d Chemical compound [Na+].[2H][O-] HEMHJVSKTPXQMS-DYCDLGHISA-M 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 239000011260 aqueous acid Substances 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000003637 basic solution Substances 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000002484 cyclic voltammetry Methods 0.000 description 1
- 238000006264 debenzylation reaction Methods 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- CSAKSSPMVJONQE-UHFFFAOYSA-N diethyl 1-(2-ethoxy-2-oxoethyl)-3,4-dihydroxypyrrole-2,5-dicarboxylate Chemical compound CCOC(=O)CN1C(C(=O)OCC)=C(O)C(O)=C1C(=O)OCC CSAKSSPMVJONQE-UHFFFAOYSA-N 0.000 description 1
- 150000002009 diols Chemical class 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 229920001971 elastomer Polymers 0.000 description 1
- 238000002848 electrochemical method Methods 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 229940097789 heavy mineral oil Drugs 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 229920006158 high molecular weight polymer Polymers 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- ARRNBPCNZJXHRJ-UHFFFAOYSA-M hydron;tetrabutylazanium;phosphate Chemical compound OP(O)([O-])=O.CCCC[N+](CCCC)(CCCC)CCCC ARRNBPCNZJXHRJ-UHFFFAOYSA-M 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000037427 ion transport Effects 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 238000005691 oxidative coupling reaction Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002798 polar solvent Substances 0.000 description 1
- 238000005498 polishing Methods 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- RUOJZAUFBMNUDX-UHFFFAOYSA-N propylene carbonate Chemical compound CC1COC(=O)O1 RUOJZAUFBMNUDX-UHFFFAOYSA-N 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000010926 purge Methods 0.000 description 1
- 150000005839 radical cations Chemical class 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- DZCAZXAJPZCSCU-UHFFFAOYSA-K sodium nitrilotriacetate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CC([O-])=O DZCAZXAJPZCSCU-UHFFFAOYSA-K 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 239000003115 supporting electrolyte Substances 0.000 description 1
- 239000010409 thin film Substances 0.000 description 1
- UVZICZIVKIMRNE-UHFFFAOYSA-N thiodiacetic acid Chemical class OC(=O)CSCC(O)=O UVZICZIVKIMRNE-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- HRXKRNGNAMMEHJ-UHFFFAOYSA-K trisodium citrate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O HRXKRNGNAMMEHJ-UHFFFAOYSA-K 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 238000001075 voltammogram Methods 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/36—Oxygen or sulfur atoms
- C07D207/40—2,5-Pyrrolidine-diones
- C07D207/416—2,5-Pyrrolidine-diones with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to other ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/30—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members
- C07D207/34—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/36—Oxygen or sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyrrole Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
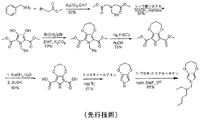
本発明はエステル置換ジヒドロキシピロール、N-置換3,4-アルキレンジオキシピロール、ならびにエステル置換ジヒドロキシピロールおよびN-置換3,4-アルキレンジオキシピロールの合成法を目的とする。
ポリ(3,4-アルキレンジオキシピロール)のファミリーは多様な生成物の作成のために有用なポリマーであることが公知である。このポリマーのファミリーのために有用なモノマーにはN-アルキル化3,4-アルキレンジオキシピロールが含まれる。そのような生成物にはエレクトロクロミック窓、鏡およびディスプレー、電子ペーパー、静電防止導体、透明導体、電界効果トランジスター、スーパーキャパシタ、電池、光起電力装置、ならびにそれらの高いバンドギャップ、低い酸化電位、生物活性、および官能基化に対する柔軟性による他の電子成分が含まれる。N-アルキル化3,4-アルキレンジオキシピロールモノマーへの現在の合成ルートは、合成経路が難しくて非効率的であり、その単離は典型的にクロマトグラフィを必要とするが、これはコストを大幅に引き上げ、その工程による生成物の処理量を著しく制限するため、高価である。
下記構造の新規N-置換3,4-アルキレンジオキシピロールのファミリーのための組成物:

下記構造
N(CH2C(O)OR)3
(式中、3つのR基は独立にメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアリールである)の窒素トリエステルを提供する段階;
窒素トリエステルをシュウ酸ジメチルまたはジエチルと縮合して、下記構造
エステル置換ジヒドロキシピロールを、下記構造
W(CHR1)m(CR2R3)n(CHR4)p-m-nW
(式中、WはCl、Br、I、スルフェート、またはスルホネートであり、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり;pは2から6であり、mは1からp-1であり、nはOからp-2である)の二官能性アルキレンと環化して、下記構造
エステル置換アルキレンジオキシピロールをけん化および中和して、下記構造
該酸置換3,4-アルキレンジオキシピロールを脱カルボキシル化して、下記構造
3,4-アルキレンジオキシピロール-酢酸を単一の反応または一連の反応により、下記構造
エステル置換ジヒドロキシピロールの調製法は窒素トリエステル、N(CH2C(O)OR)3を提供すること、および縮合反応を用いて窒素トリエステルをエステル置換ジヒドロキシピロールに変換することから開始する。縮合反応は好ましくは改変ヒンスベルク縮合反応である。ヒンスベルク縮合とは、エタノール中、ナトリウムエトキシドによるシュウ酸ジエチルとチオジグリコール酸ジエチルとの縮合を含む、論文に発表された反応(Hinsberg Chem. Ber. 1910, 43, 901)を言う。伝統的に、かつ本明細書において用いられるとおり、チオジグリコール酸の誘導体(硫黄が別のヘテロ原子で置換されている)およびシュウ酸ジエチルから3,4-ジヒドロキシ複素環を生じる任意の縮合はヒンスベルク縮合と呼ばれている。エステル置換ジヒドロキシピロールを環化して、エステル置換ジヒドロキシピロールのジヒドロキシからの酸素を含む第二の環を形成し、エステル置換アルキレンジオキシピロールとすることができる。エステル置換アルキレンジオキシピロールをけん化した後に中和し、酸置換アルキレンジオキシピロールを脱カルボキシル化して、3,4-アルキレンジオキシピロールN-酢酸を生成する。続いて、3,4-アルキレンジオキシピロールN-酢酸を、重合して共役ポリマーとすることができる様々なN-置換3,4-アルキレンジオキシピロールから変換することができる。
のジヒドロキシピロールを生成することができる。ジヒドロキシピロールは、水性酸でプロトン化型を生じた後、減圧ろ過する、単純な処理により高収率で単離することができる。さらなる精製は単純な水洗浄および固体の乾燥により達成することができる。
X(CHR1)m(CR2R3)n(CHR4)p-m-nX
(式中、XはCl、Br、もしくはI、スルフェート、またはスルホネートであり、pは2から6であり、mは1からp-1であり、かつnは0からp-2であり、かつR1、R2、R3およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールである)
の二置換アルキル鎖とのウィリアムソンエーテル化により実施することができる。または、環化をジヒドロキシピロールと下記構造
HO(CHR1)m(CR2R3)n(CHR4)p-m-nOH
(式中、pは2から6であり、nは1からp-1であり、かつmは0からp-2であり、かつR1、R2、R3およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールである)
のジオールとの間の光延反応を介して実施してもよい。環化生成物は、下記構造
により示される。この生成物は水洗浄の後、メタノール、エタノール、アセトン、または酢酸エチルなどの溶媒中での再結晶により単離することができる。
により示される。このようにして高収率で生成した3,4-アルキレンジオキシピロール-酢酸は再結晶により容易に精製される。
以下の合成法、中間体、モノマーのN-置換3,4-アルキレンジオキシピロール、およびこれらのモノマーからのポリマーの例は、例示のために示すにすぎず、いかなる様式でも本発明の範囲を限定するものではないことが理解されるべきである。
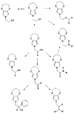
撹拌子を含み、アルゴン雰囲気下、還流冷却器を装備した1000mL丸底フラスコに、ニトリロ三酢酸(50.00g、262mmol)、エタノール(200プルーフ、500mL)、および濃硫酸(15mL)を加えた。混合物を3時間還流し、室温まで冷却し、減圧濃縮した。濃縮物をただちにジクロロメタン(DCM)(300mL)に溶解し、少量の飽和炭酸水素ナトリウムでガスの発生が止まるまで洗浄した。溶媒を除去し、得られた濃縮液を蒸留(0.1mmHgで沸点135℃)して、澄明油状物を得た(56.61g、79%)。
撹拌子を含み、アルゴン雰囲気下、還流冷却器を装備した1000mL丸底フラスコに、エタノール(200プルーフ、500mL)および金属ナトリウム(21.31g、927mmol)を加えた。ナトリウムが完全に溶解した時点で、ニトリロ三酢酸トリエチル(56.20g、204mmol)およびシュウ酸ジエチル(29.44g、204mmol)の混合物を加えた。混合物を終夜還流し、そこで混合物は澄明なゲル状溶液となった。室温まで冷却した後、混合物を脱イオン水(1200mL)に加え、氷浴中で冷却し、氷酢酸(200mL)で酸性化した。得られた白色沈澱を減圧ろ過により単離し、脱イオン水で数回洗浄して、白色固体(59.75g、89%)を得た。融点121.3〜121.8℃;
撹拌子を含み、アルゴン雰囲気下の500mL丸底フラスコに、1-(2-エトキシ-2-オキソエチル)-3,4-ジヒドロキシ-1H-ピロール-2,5-ジカルボン酸ジエチル(48.00g、146mmol)、1,3-ジブロモプロパン(29.48g、146mmol)、無水炭酸カリウム(50.36g、364mmol)、および無水ジメチルホルムアミド(DMF)(250mL)を加えた。反応混合物を105℃に加熱し、約30分後に黄緑色となった。反応混合物を12時間撹拌し、その間に混合物は黄色となり、室温まで冷却した。混合物を脱イオン水(500mL)に加え、固体を減圧ろ過により回収し、脱イオン水で数回洗浄した。固体を熱メタノールから再結晶し、黄色固体(44.90g、80%)を得た。融点139.7〜139.9℃;
100mL丸底フラスコに、7-(2-エトキシ-2-オキソエチル)-2,3,4,7-テトラヒドロ-[1,4]ジオキセピノ[2,3-c]ピロール-6,8-ジカルボン酸ジエチル(17.56g、47.5mmol)、脱イオン水(45mL)、アセトン(25mL)、および水酸化カリウム(13.34g、238mmol)を加えた。反応混合物にアルゴンを20分間通気し、次いで2.5時間還流し、濃褐色溶液を得た。有機揮発物質を減圧下で除去し、残った水層を氷浴中で冷却した。混合物に濃硫酸をpH6になるまで加え、そこで白色固体沈澱が生じた。この沈澱を赤色水溶液からろ過し、脱イオン水で数回洗浄し、乾燥後重量は13.55gであった(100%)。構造を確認するために、NaOD/D2O溶液に溶解することにより完全に脱プロトン化した試料でNMR分析を実施した。
撹拌子を含み、アルゴン雰囲気下の250mL丸底フラスコに、重質鉱油(100mL)を加えた。溶液を80〜100℃に加熱し、式中減圧/アルゴンパージを3回繰り返して酸素を除去し、その後160℃まで加熱した。持続的アルゴンブランケットを維持しながら、7-(カルボキシメチル)-2,3,4,7-テトラヒドロ-[1,4]ジオキセピノ[2,3-c]ピロール-6,8-ジカルボン酸(13.55g、47.5mmol)を少量ずつ加えた。得られたスラリーをさらに10分間撹拌し、次いで室温まで冷却した。ヘキサン(250mL)をフラスコに加え、デカントした。残った固体をメタノール(250mL)に溶解し、溶液をろ過して痕跡量の固体を除去した。溶媒を減圧下で除去した後、黄褐色固体(7.94g、85%)を単離した。
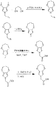
撹拌子を含み、アルゴン雰囲気下の50mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロール-7(2H)-イル)酢酸(1.00g、5.07mmol)、無水DMF(25mL)、ヨウ化メチル(0.86g、6.08mmol)、および無水K2CO3(1.05g、7.61mmol)を加えた。反応混合物を60℃に加熱し、17時間撹拌し、その後室温まで冷却し、脱イオン水(250mL)に加えた。得られた混合物を酢酸エチル(2×100mL)で抽出し、減圧濃縮し、シリカゲルパッドを通過させ(酢酸エチル)、減圧濃縮して、白色固体(0.60g、56%)を得た。TLC Rf=0.24(シリカゲル、酢酸エチル);
磁気撹拌子、窒素注入アダプター、滴下漏斗およびラバーセプタムを備えた250mL丸底フラスコに、3,4-プロピレンジオキシピロール(0.60g、4.31mmol)を加えた。次いで、無水ジメチルホルムアミド(DMF)(40mL)をフラスコに加え、混合物を0℃まで冷却した。NaH(鉱油中60%、0.19g、4.74mmol)を溶液に加えた。淡黄色スラリーを室温で2時間撹拌した後、ブロモ酢酸エチル(0.56mL、4.74mmol)を加えた。混合物を室温で終夜撹拌し、次いで脱イオン水(200mL)を加えた。粗製化合物をEt2O(3×100mL)で抽出し、合わせた有機抽出物をNa2SO4で乾燥した。溶媒を減圧下で除去し、帯赤色油状残渣を得た。化合物をシリカゲルのカラムクロマトグラフィ(ヘキサン:酢酸エチル=3:1)で精製して、無色油状物(0.63g、65%)を得た。TLC Rf=0.78(シリカゲル、酢酸エチル);
撹拌子を含み、アルゴン雰囲気下の50mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロール-7(2H)-イル)酢酸(1.00g、5.07mmol)、無水DMF(25mL)、臭化ベンジル(0.91g、5.32mmol)、および無水K2CO3(1.05g、7.61mmol)を加えた。反応混合物を60℃に加熱し、17時間撹拌し、その後室温まで冷却し、脱イオン水(250mL)に加えた。得られた沈澱を減圧ろ過により単離し、脱イオン水で洗浄し、風乾して、白色粉末(0.70g、48%)を得た。TLC Rf=0.81(シリカゲル、ヘキサン:Et2O=3:2);融点71.8〜72.7℃;
撹拌子を含み、アルゴン雰囲気下の100mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロール-7(2H)-イル)酢酸(2.00g、10.1mmol)、無水DMF(50mL)、1-ブロモ-2-エチルヘキサン(2.06g、10.6mmol)、および無水K2CO3(2.10g、15.2mmol)を加えた。反応混合物を60℃に加熱し、15時間撹拌した。室温まで冷却した後、混合物を脱イオン水(250mL)に加え、Et2O(2×100mL)で抽出した。合わせた有機層を脱イオン水(100mL)、食塩水(50mL)で洗浄し、無水Na2SO4で乾燥した。溶媒を減圧下で除去し、濃縮物をシリカゲルのフラッシュクロマトグラフィ(TEAで非活性化、Et2O:ヘキサン=7:4)で精製して、澄明油状物(2.65g、84%)を得た。TLC Rf=0.23(シリカゲル、Et2O:ヘキサン=3:2);
撹拌子を含み、アルゴン雰囲気下の乾燥した250mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロル-7(2H)-イル)酢酸(1.50g、7.61mmol)、および無水THF(100mL)を加えた。混合物をCO2/2-プロパノール浴中で冷却し、LiAlH4粉末(1.29g、34.0mmol)を注意深く加えた。気体の発生が停止した時点で、反応混合物を室温に戻し、さらに3時間撹拌した。反応をまずメタノールと、次いで氷により停止し、アルミナ凝集物があれば最少量のH2SO4を加えて不活化した。混合物をEt2O(2×100mL)で抽出し、合わせた有機層を飽和NH4Clと次いで食塩水で洗浄した。溶液をNa2SO4で乾燥し、減圧濃縮し、溶離剤として酢酸エチルを用い、シリカゲルパッド(トリエチルアミンで不活化)を通してろ過した。酢酸エチルを減圧下で除去した後、純粋な化合物を淡黄色油状物で単離した(0.70g、50%)。TLC Rf=0.67(シリカゲル、酢酸エチル);
撹拌子を含み、アルゴン雰囲気下の50mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロル-7(2H)-イル)エタノール(0.50g、2.73mmol)、ヨウ化メチル(0.47g、3.28mmol)、および無水DMF(25mL)を加えた。混合物を氷浴中で冷却し、次いで水素化ナトリウム(鉱油中60%分散液、0.22g、5.46mmol)を加えた。反応混合物を15時間撹拌し、その間に氷浴を室温に戻した。次いで、混合物を脱イオン水(100mL)に加え、Et2O(2×50mL)で抽出した。有機層を合わせ、食塩水で洗浄し、Na2SO4で乾燥し、減圧濃縮した。残渣をシリカゲルのフラッシュクロマトグラフィ(TEAで不活化、ヘキサン:Et2O=5:6)で精製して、澄明油状物(0.42g、78%)を得た。TLC Rf=0.24(シリカゲル、ヘキサン:Et2O=1:1);
撹拌子を含み、アルゴン雰囲気下の50mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロル-7(2H)-イル)エタノール(0.50g、2.73mmol)、臭化ベンジル(0.56g、3.28mmol)、および無水DMF(25mL)を加えた。混合物を氷浴中で冷却し、次いで水素化ナトリウム(鉱油中60%分散液、0.22g、5.46mmol)を加えた。反応混合物を15時間撹拌し、その間に氷浴を室温に戻した。次いで、混合物を脱イオン水(100mL)に加え、Et2O(2×50mL)で抽出した。有機層を合わせ、食塩水で洗浄し、Na2SO4で乾燥し、減圧濃縮した。残渣をシリカゲルのフラッシュクロマトグラフィ(ヘキサン:Et2O=2:1)で精製して、澄明油状物(0.52g、64%)を得た。TLC Rf=0.28(シリカゲル、ヘキサン:Et2O=1:1);
撹拌子を含み、アルゴン雰囲気下の50mL丸底フラスコに、2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロル-7(2H)-イル)エタノール(0.41g、2.24mmol)、2-エチルヘキシルトシレート(0.77g、2.69mmol)、および無水DMF(25mL)を加えた。混合物を氷浴中で冷却し、次いで水素化ナトリウム(鉱油中60%分散液、0.18g、4.48mmol)を加えた。反応混合物を15時間撹拌し、その間に氷浴を室温に戻した。次いで、混合物を脱イオン水(100mL)に加え、Et2O(2×50mL)で抽出した。有機層を合わせ、食塩水で洗浄し、Na2SO4で乾燥し、減圧濃縮した。残渣をシリカゲルのフラッシュクロマトグラフィ(TEAで不活化、ヘキサン:Et2O=5:1)で精製して、澄明油状物(0.42g、64%)を得た。TLC Rf=0.46(シリカゲル、ヘキサン:Et2O=1:1);
2-(2,3-ジヒドロ-[1,4]ジオキセピノ[2,3-c]ピロル-7(2H)-イル)酢酸から前述のとおりに合成したモノマーは、そのポリマー型では異なる電気化学的および光学的性質を有する材料を生じる。ポリマーは重合度4以上で所望の性質を示し始める。高分子量のポリマーは、重合度が1,000に近いものでも、これらのモノマーの多くで生成することができる。モノマーを重合させ、所望の性質を得る能力が、その合成および単離を上で記載している、ポリマーの側鎖となるメチル、エチル、ベンジル、および2-エチルヘキシル置換基を有するエステルモノマーから調製したポリマーについて示される。メチルおよびエチル基は全体の極性、酸化的カップリングの立体障害および共役ポリマーのπスタッキング相互作用の妨害、ならびにポリマーの溶解性に対しては最小限の寄与しかしないが、ベンジル基は分子に幾分かの立体障害を与え、πスタッキングのための手だてを与える可能性がある。ラセミの2-エチルヘキシル基はモノマーおよびポリマーの溶解性を高めうる疎水性部分であり、ポリマーのπスタッキング相互作用を妨害すると考えられる。
Claims (19)
- 下記構造のN-置換3,4-アルキレンジオキシピロール:
式中、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり;pは3から6であり、mは1からp-1であり、nは0からp-2であり;ZはHまたはC(O)ORであり、式中Rは水素、メチル、エチル、C3からC8の直鎖または分枝鎖アルキルであり;かつXはC(O)R5、CH2YR6、またはCR7=CR8R9であり、式中R5は水素、メチル、エチル、C3からC8の直鎖もしくは分枝鎖アルキル、アリール、OR10、またはNR11R12であり、式中R10、R11、およびR12は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリールまたはアルキルアリールであり、式中YはO、OC(O)、NR13、またはNR14C(O)であり、かつここでR6、R7、R8、R9、R13、およびR14は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールである。 - Rがエチルを含む、請求項1記載のN-置換3,4-アルキレンジオキシピロール。
- Rが水素またはエチルを含み、かつR1、R2、R3、およびR4が水素を含む、請求項1記載のN-置換3,4-アルキレンジオキシピロール。
- Zが水素を含み、かつR1、R2、R3、およびR4が水素を含む、請求項1記載のN-置換3,4-アルキレンジオキシピロール。
- XがC(O)R5を含み、R5がOR10を含み、かつR10が水素、メチル、エチル、ベンジル、または2-エチルヘキシルを含む、請求項1〜4のいずれか一項記載のN-置換3,4-アルキレンジオキシピロール。
- XがCH2YR6を含み、YがOであり、かつR6が水素、メチル、ベンジル、または2-エチルヘキシルを含む、請求項1〜4のいずれか一項記載のN-置換3,4-アルキレンジオキシピロール。
- 以下の段階を含む、N-置換3,4-アルキレンジオキシピロールの調製法:
下記構造
N(CH2C(O)OR)3
式中、3つのR基は独立にメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアリールである
の窒素トリエステルを提供する段階;
該窒素トリエステルをシュウ酸ジメチルまたはジエチルと縮合させて、下記構造
式中、3つのR基は独立にメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアリールである
のエステル置換ジヒドロキシピロールを得る段階;
該エステル置換ジヒドロキシピロールを、下記構造
W(CHR1)m(CR2R3)n(CHR4)p-m-nW
式中、WはCl、Br、I、スルフェート、またはスルホネートであり、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり;pは2から6であり、mは1からp-1であり、nはOからp-2である
の二官能性アルキレンと環化させて(annulating)、下記構造
式中、3つのR基は独立にメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアリールであり、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり、かつpは2から6であり、mは1からp-1であり、nはOからp-2である
のエステル置換アルキレンジオキシピロールを形成させる段階;
該エステル置換アルキレンジオキシピロールをけん化および中和して、下記構造
式中、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり、かつpは2から6であり、mは1からp-1であり、nはOからp-2である
の酸置換3,4-アルキレンジオキシピロールを形成させる段階;
該酸置換3,4-アルキレンジオキシピロールを脱カルボキシル化して、下記構造
式中、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり、かつpは2から6であり、mは1からp-1であり、nはOからp-2である
の3,4-アルキレンジオキシピロール-酢酸を形成させる段階;ならびに
単一の反応または一連の反応により、該3,4-アルキレンジオキシピロール-酢酸を、下記構造
式中、R1、R2、R3、およびR4は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールであり;pは2から6であり、mは1からp-1であり、nはOからp-2であり;かつXはC(O)R5、CH2YR6、またはCR7=CR8R9であり、式中R5は水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、OR10、またはNR11R12であり、式中R10は独立にメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリールまたはアルキルアリールであり、R11およびR12は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリールまたはアルキルアリールであり、式中YはO、OC(O)、NR13、またはNR14C(O)であり、かつここでR6、R7、R8、R9、R13、およびR14は独立に水素、メチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、アリール、またはアルキルアリールである
のN-置換3,4-アルキレンジオキシピロールに変換する段階。 - 縮合の段階がヒンスベルク縮合を含む、請求項7記載の方法。
- 環化の段階がウィリアムソンエーテル化を含み、かつWがCl、Br、I、スルフェート、またはスルホネートを含む、請求項7記載の方法。
- 環化の段階が光延反応を含み、かつWがOHを含む、請求項7記載の方法。
- 脱カルボキシル化の段階が熱分解を含み、かつZがC(O)ORを含み、RがHを含む、請求項7記載の方法。
- 熱分解が約140℃〜約200℃の温度への加熱を含む、請求項11記載の方法。
- R1、R2、R3、およびR4が水素を含む、請求項7記載の方法。
- XがC(O)R5を含み、R5がOR10を含み、式中R10がメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアルキルアリールを含む、前記N-置換3,4-アルキレンジオキシピロールを得るために、変換の段階が、塩基による前記3,4-アルキレンジオキシピロール-酢酸の脱プロトン化、および、ハロゲン化アルキル、硫酸アルキル、またはスルホン酸アルキルとの縮合を含む、請求項7記載の方法。
- XがC(O)R5を含み、R5がOR10を含み、式中R10がメチル、ベンジル、または2-エチルヘキシルを含む、前記N-置換3,4-アルキレンジオキシピロールを得るために、前記塩基がK2CO3を含み、かつ前記ハロゲン化アルキルがヨウ化メチル、臭化ベンジル、または1-ブロモ-2-エチルヘキサンを含む、請求項14記載の方法。
- XがCH2YR6を含み、YがOを含み、R6がメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアルキルアリールを含む、前記N-置換3,4-アルキレンジオキシピロールを得るために、変換の段階が、還元剤の添加による前記3,4-アルキレンジオキシピロール-酢酸の還元を含む、請求項7記載の方法。
- 前記還元剤がLiAlH4を含む、請求項16記載の方法。
- XがCH2YR6を含み、YがOを含み、R6がメチル、エチル、C3からC20の直鎖もしくは分枝鎖アルキル、またはアルキルアリールを含む、前記N-置換3,4-アルキレンジオキシピロールを得るために、塩基によるアルコールの脱プロトン化およびハロゲン化アルキルまたはスルホン酸アルキルとの縮合を介した前記X基の縮合をさらに含む、請求項16記載の方法。
- XがCH2YR6を含み、YがOを含み、R6がメチル、2-エチルヘキシル、またはベンジルを含む、前記N-置換3,4-アルキレンジオキシピロールを得るために、前記塩基がNaHを含み、かつ前記ハロゲン化アルキルまたはスルホン酸アルキルがヨウ化メチル、臭化ベンジル、または2-エチルヘキシルトシレートを含む、請求項18記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US72417805P | 2005-10-06 | 2005-10-06 | |
| US60/724,178 | 2005-10-06 | ||
| PCT/US2006/039958 WO2007041724A1 (en) | 2005-10-06 | 2006-10-06 | N-substituted 3,4-alkylenedioxypyrroles, ester substituted dihydroxypyrroles and methods for synthesis of these pyrroles |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2009511505A JP2009511505A (ja) | 2009-03-19 |
| JP5113065B2 true JP5113065B2 (ja) | 2013-01-09 |
Family
ID=37667260
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008534790A Expired - Fee Related JP5113065B2 (ja) | 2005-10-06 | 2006-10-06 | N−置換3,4−アルキレンジオキシピロール、エステル置換ジヒドロキシピロールおよびこれらのピロールの合成法 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7799932B2 (ja) |
| EP (1) | EP1931630B1 (ja) |
| JP (1) | JP5113065B2 (ja) |
| KR (1) | KR101298458B1 (ja) |
| AT (1) | ATE467615T1 (ja) |
| DE (1) | DE602006014285D1 (ja) |
| DK (1) | DK1931630T3 (ja) |
| ES (1) | ES2343818T3 (ja) |
| WO (1) | WO2007041724A1 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007041724A1 (en) | 2005-10-06 | 2007-04-12 | University Of Florida Research Foundation, Inc. | N-substituted 3,4-alkylenedioxypyrroles, ester substituted dihydroxypyrroles and methods for synthesis of these pyrroles |
| CA2805638A1 (en) | 2010-08-02 | 2012-02-09 | University Of Florida Research Foundation, Inc. | Processing method for water soluble polymeric materials |
| WO2012058416A2 (en) | 2010-10-28 | 2012-05-03 | University Of Florida Research Foundation, Inc. | Cathodically coloring yellow soluble electrochromic and light emitting polymers |
| WO2013122982A1 (en) * | 2012-02-13 | 2013-08-22 | University Of Florida Research Foundation, Inc. | Synthesis of dioxyheterocycle-based polymers |
| TWI468440B (zh) | 2012-11-27 | 2015-01-11 | Ind Tech Res Inst | 透明電致變色聚醯亞胺與其形成方法與電致變色元件 |
| WO2016081787A2 (en) | 2014-11-19 | 2016-05-26 | Biltmore Technologies, Inc. | Controlled color and opacity-changing coating system |
| WO2018039230A1 (en) | 2016-08-22 | 2018-03-01 | Georgia Tech Research Corporation | High-gap yellow and orange electrochromic polymers |
| US10207974B1 (en) | 2017-12-05 | 2019-02-19 | Chevron Phillips Chemical Company Lp | Synthesis of gamma dicarbonyl and pyrrole compounds |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4565730B2 (ja) * | 2000-10-23 | 2010-10-20 | 日本カーリット株式会社 | 固体コンデンサ及びその製造方法 |
| US6492082B1 (en) * | 2000-11-28 | 2002-12-10 | Xerox Corporation | Toner compositions comprising polypyrroles |
| US6791738B2 (en) * | 2001-11-21 | 2004-09-14 | University Of Florida | Electrochromic polymers and polymer electrochromic devices |
| WO2003055889A1 (en) | 2001-12-28 | 2003-07-10 | Agfa-Gevaert | Process for preparing a heteroaromatic compound substituted with one or more ether groups |
| WO2007041724A1 (en) | 2005-10-06 | 2007-04-12 | University Of Florida Research Foundation, Inc. | N-substituted 3,4-alkylenedioxypyrroles, ester substituted dihydroxypyrroles and methods for synthesis of these pyrroles |
-
2006
- 2006-10-06 WO PCT/US2006/039958 patent/WO2007041724A1/en not_active Ceased
- 2006-10-06 DK DK06816817.8T patent/DK1931630T3/da active
- 2006-10-06 JP JP2008534790A patent/JP5113065B2/ja not_active Expired - Fee Related
- 2006-10-06 US US11/990,042 patent/US7799932B2/en active Active
- 2006-10-06 EP EP06816817A patent/EP1931630B1/en not_active Not-in-force
- 2006-10-06 KR KR1020087010880A patent/KR101298458B1/ko not_active Expired - Fee Related
- 2006-10-06 ES ES06816817T patent/ES2343818T3/es active Active
- 2006-10-06 DE DE602006014285T patent/DE602006014285D1/de active Active
- 2006-10-06 AT AT06816817T patent/ATE467615T1/de not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| US7799932B2 (en) | 2010-09-21 |
| DE602006014285D1 (de) | 2010-06-24 |
| JP2009511505A (ja) | 2009-03-19 |
| KR101298458B1 (ko) | 2013-08-23 |
| KR20080059287A (ko) | 2008-06-26 |
| EP1931630A1 (en) | 2008-06-18 |
| EP1931630B1 (en) | 2010-05-12 |
| WO2007041724A1 (en) | 2007-04-12 |
| DK1931630T3 (da) | 2010-08-16 |
| ES2343818T3 (es) | 2010-08-10 |
| US20090149661A1 (en) | 2009-06-11 |
| ATE467615T1 (de) | 2010-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US5677417A (en) | Tetraaroxyperylene-3,4,9,10-tetracarboxylic polyimides | |
| CN109803955B (zh) | 用于制备4-铵-2,2,6,6-四烷基哌啶基盐的方法 | |
| Merz et al. | 3, 4-Dialkoxypyrroles and 2, 3, 7, 8, 12, 13, 17, 18-Octaalkoxyporphyrins | |
| JP2020196727A (ja) | 非常に優れたアルカリ安定性を有するイミダゾール及びイミダゾリウムカチオン | |
| JP5113065B2 (ja) | N−置換3,4−アルキレンジオキシピロール、エステル置換ジヒドロキシピロールおよびこれらのピロールの合成法 | |
| Koyama et al. | Polycondensations of hydroxycarboxylic acids derived from optically active aminoalcohols and acid anhydrides—syntheses of functional poly (ester‐amide) s | |
| EP0647223A1 (en) | Ion-sensitive bipyridine complexes | |
| KR20210099010A (ko) | 세포독성 벤조디아제핀 유도체의 제조 방법 | |
| Navarro et al. | Characterization of PEDOT film functionalized with a series of automated synthesis ferrocenyl-containing oligonucleotides | |
| Pleus et al. | Design of chiral poly (pyrroles) | |
| US4528389A (en) | Pentacoordinate silyl enolates and their use as polymerization initiators | |
| JP6226363B2 (ja) | ビス−ボロンジピロメテン系色素 | |
| WO2021072159A1 (en) | Anodic oxidation of 5-aminouracil | |
| SU554815A3 (ru) | Способ получени производных симмтриазоло-(4,3-а)-хинолина или их солей | |
| CN106518865B (zh) | 一种1-烯基中氮茚衍生物的制备方法 | |
| JP3475017B2 (ja) | 高純度可溶性アニリン系導電性ポリマー及びその製造方法 | |
| KR102734118B1 (ko) | 인덴 유도체의 제조 방법, 이를 이용한 메탈로센 촉매 제조방법 및 올레핀 중합체 제조방법 | |
| JPH10251401A (ja) | 鎖状炭酸エステル基を有するアルキレンオキシドオリゴマーおよびその製造方法 | |
| US4577003A (en) | Pentacoordinate silyl enolates and their use as polymerization initiators | |
| US3313850A (en) | Unsaturated acid amides containing sulphonic groups and method of preparation | |
| KR102476098B1 (ko) | 결정성 테트라하이드로-베타-카볼린의 제조방법 | |
| KR102605275B1 (ko) | 아미드 칠량체 및 이의 제조 방법 | |
| KR102605281B1 (ko) | 아미드 오량체 및 이의 제조 방법 | |
| WO1992004352A1 (fr) | Procede de production de diacetal | |
| US3246010A (en) | 1, 5-diaryl-2-pyrrole propanols and ethers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20090929 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120521 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20120522 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120821 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20120913 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121011 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151019 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5113065 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |