JP5154797B2 - ポリピリジニウムの製法 - Google Patents
ポリピリジニウムの製法 Download PDFInfo
- Publication number
- JP5154797B2 JP5154797B2 JP2006511291A JP2006511291A JP5154797B2 JP 5154797 B2 JP5154797 B2 JP 5154797B2 JP 2006511291 A JP2006511291 A JP 2006511291A JP 2006511291 A JP2006511291 A JP 2006511291A JP 5154797 B2 JP5154797 B2 JP 5154797B2
- Authority
- JP
- Japan
- Prior art keywords
- derivative
- group
- ion
- polymerization
- sodium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Images









Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
- C08G73/06—Polycondensates having nitrogen-containing heterocyclic rings in the main chain of the macromolecule
- C08G73/0622—Polycondensates containing six-membered rings, not condensed with other rings, with nitrogen atoms as the only ring hetero atoms
- C08G73/0627—Polycondensates containing six-membered rings, not condensed with other rings, with nitrogen atoms as the only ring hetero atoms with only one nitrogen atom in the ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G73/00—Macromolecular compounds obtained by reactions forming a linkage containing nitrogen with or without oxygen or carbon in the main chain of the macromolecule, not provided for in groups C08G12/00 - C08G71/00
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Polyoxymethylene Polymers And Polymers With Carbon-To-Carbon Bonds (AREA)
- Macromolecular Compounds Obtained By Forming Nitrogen-Containing Linkages In General (AREA)
- Polyethers (AREA)
Description
また、テトラフルオロホウ酸ナトリウム等の無機化合物の存在下の有機溶媒中で1,4−ピリジニウム塩を重合させてポリ(1,4−ピリジニウム)塩を得る方法も開示されているが(特許文献1)、分子量を制御することができていない。

R’’は炭化水素基又は複素環基を表す。この炭化水素基とはアルキル基、二重結合を有する直鎖炭化水素基、シクロアルキル基、アリール基又はアラルキル基をいう。
nは少なくとも1の整数、好ましくは1〜3、最も好ましくは1を表す。
R’は水素原子、アルキル基、アルコキシ基、ハロゲン原子、ニトロ基若しくはエステル基の置換基又はピリジン環と縮合環を形成する芳香環、好ましくは水素原子を表す。ピリジン環と縮合環を形成する芳香環としては、ベンゼン環又はナフタレン環、好ましくはベンゼン環が挙げられる。ピリジン環が1のベンゼン環と縮合環を形成する場合には該縮合環はキノリン環となり、ピリジン環が2のベンゼン環と縮合環を形成する場合には該縮合環はアクリジン環となる。これらの芳香環は更に1又は複数の上記置換基を有していてもよい。
mは1〜4の整数を表す。但し、上記縮合環がキノリン環の場合は1、アクリジンの場合には2である。
複素環基は好ましくはN、O若しくはSを有するヘテロ環基であり、ヘテロ環基としては、フリル基、チエニル基、ピロリル基、イミダゾリル基、ピラゾリル基、チアゾリル基、イソチアゾリル基、オキサゾリル基、イソキサゾリル基、トリアゾリル基、オキサジアゾリル基、チアジアゾリル基、テトラゾリル基、ピリジル基、ピリミジニル基、ピリダジニル基、ピラジニル基等の単環へテロ環又はこれら同士若しくはこれらにベンゼン環やナフタレン環等の芳香環が縮環した多環ヘテロ環が挙げられるが、これらの中で含窒素ヘテロ環基が好ましい。重合開始剤において、この複素環基の炭素原子がピリジン環の窒素原子と結合していることを要する。
これらアリール基及及び複素環基は更に1又は複数の置換基を有していてもよい。この置換基として、アルキル基、アルコキシ基、ハロゲン原子、水酸基、アミノ基、ニトロ基、シリル基(−SR'''3:R'''は同一でも異なってもよく水素原子、水酸基、アルキル基、ハロゲン原子、アルコキシ基等を表す。)等が挙げられる。
Yは、この反応に用いた有機溶媒に溶解するアニオンを表す。このようなアニオンとしては、ハロゲン化物イオン、過塩素酸イオン、四フッ化ホウ酸イオン、ヘキサフルオロリン酸イオン、テトラフェニルホウ酸イオン等が挙げられる。
反応液中の重合開始剤の濃度は、通常0.0001mol/L〜5.0mol/Lであるが、仕込み比(開始剤に対するモノマーの存在比)に応じて濃度を設定することが好ましい。
反応液中のモノマーの濃度は、仕込み比(開始剤に対するモノマーの存在比)に応じて濃度を設定するが、例えば、0.01mol/L〜5.0mol/Lである。
但し、これらの濃度の上限は用いる溶媒における溶解度により制限される。
反応温度は通常0〜70℃程度、好ましくは10〜70℃程度である。
また、重合速度は重合開始剤とモノマーの濃度の積に比例するため、両者の仕込み濃度に依存し、さらに、重合温度によっても大きく変化する。従って、反応時間は数分から数日かかる場合もある。
この重合停止剤は、重合に用いたモノマーよりも塩基性の強い求核試薬が対象となる。このような重合停止剤として、例えば、アミノ基、アルコキシ基若しくはアルキル基などの電子供与基で置換したピリジン、キノリン若しくはアクリジン又はこれらの誘導体、トリフェニルホスフィン又はその誘導体、又はアルキル基若しくはアリル基を置換したアミン誘導体などが挙げられる。また、これらの官能基が分子構造の一部に含まれていれば停止剤として機能し、固体表面に固定化した分子に含まれていても構わない。
反応液中の重合停止剤の濃度は、通常0.01mol/L〜5mol/Lである。
合成例1 (モノマーの合成)
4-クロロピリジン塩酸塩(関東化学、5.0 g, 33.0 mmol)を純水 10 mLに溶かし,氷浴下、5.0 wt%炭酸水素ナトリウム水溶液を用いて中和した。生成した4-クロロピリジンをエーテルで抽出(50 mL×3回)を行い、硫酸マグネシウムで乾燥した。このエーテル溶液を氷浴下で減圧蒸留を行い、無色透明の液体4-クロロピリジン(4-ClPy)を3.44 g(収率 91 %)得た。1H-NMR(DMSO-d6)とFT-IR(KBrペレット)によって同定した。1H-NMRを図2に、FT-IRを図3に示す。
4-tert-ブチルベンジルブロミド(和光純薬、5.20 g, 23.0 mmol)に合成例1で得た4-ClPy(0.26 g, 2.3 mmol)を徐々に滴下し、室温で5時間撹拌した。析出した黄色固体を濾別し、エーテルで洗浄したのち、エタノールから再結晶を行い、N-(4´-tert-ブチルベンジル)-4-クロロピリジニウム(t-BBPy)を得た。収量0.76 g(収率 97 %)。1H-NMR(DMSO-d6)とFT-IR(KBrペレット)によって同定した。1H-NMRを図4に、FT-IRを図5に示す。
重合体の片方の末端構造は開始剤t-BBPyに由来するので、末端定量法を用いて平均分子量を求めることができる。具体的には、1H-NMRの1.3 ppm付近に現れるt-ブチル基のプロトンと6〜10 ppmに現れる重合体ポリピリジニウムのプロトン(モノマー及び重合開始剤のプロトンと区別できる)に注目して、両者の積分値の比より平均分子量を求める。その結果、図7に示すように、モノマーの転化率の増加に伴って、平均分子量の増加が見られた。
重合開始剤(t-BBPy)を用いずに実施例1と同様に反応を行ったところ、図7に示すように測定精度内で重合反応は進行しなかった。
溶解促進剤(TBABF4)を用いずに実施例1と同様に反応を行った。黄褐色固体が加熱直後より析出し、重合反応は不均一系で進行した。析出した黄褐色固体を濾別し、エーテルで洗浄して乾燥した。1H-NMRからは、ハロゲン化物イオンを対アニオンとするポリカチオンのために低い溶解性と開始剤のない熱重合による広い分子量分布をもつために、平均分子量を求めることはできなかった。
比較のため、4-クロロピリジンの重合を無溶媒で重合させた。生成物をBと呼ぶ。
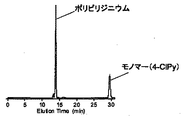
生成した重合物のGPC溶出曲線を測定した(使用機器:日本分光社製GPCポンプ PU-2089、昭和電工製屈折率検出器RI-101、日本分光製UV検出器MD-201、使用カラム:昭和電工製Asahipak GF-310HQx2本)。その結果を図10に示す。
生成物Aとして重合度(重合時間)の異なる2種類の生成物を得たが、それぞれ1H NMRより平均重合度を計算すると、59量体(図10実線)及び34量体(図10点線)であった。
NMRで求めた平均分子量の増加に伴って、GPC曲線の高分子量側(溶出時間の短い領域)が増えていることがわかる。
また、従来型の生成物BのGPC曲線がブロードであり分子量分布が広いのに対し、生成物Aは、GPC曲線が幅がシャープであり分子量分布が狭いことが分かる。
Claims (8)
- 有機溶媒中で重合開始剤及び下記一般式(化1)
- 前記重合開始剤が、下記一般式(化2)
- Y−がハロゲン化物イオン、過塩素酸イオン、四フッ化ホウ酸イオン、ヘキサフルオロリン酸イオン又はテトラフェニルホウ酸イオンであり、Xが塩素原子又は臭素原子であり、前記疎水性アニオンが過塩素酸イオン、四フッ化ホウ酸イオン、ヘキサフルオロリン酸イオン、酒石酸イオン、クエン酸イオン、ニコチン酸イオン又はビナフチル基をもつリン酸イオンである請求項2に記載の製法。
- 前記溶解促進剤が、過塩素酸テトラブチルアンモニウム、四フッ化ホウ酸テトラブチルアンモニウム、六フッ化リンテトラブチルアンモニウム、過塩素酸ナトリウム、四フッ化ホウ酸ナトリウム、六フッ化リンナトリウム、過塩素酸テトラエチルアンモニウム、四フッ化ホウ酸テトラエチルアンモニウム、六フッ化リンテトラエチルアンモニウム、テトラフェニルホウ酸ナトリウム、p-トルエンスルホン酸ナトリウム、アルキルスルホン酸ナトリウム(炭素数6〜24)、アルキルリン酸ナトリウム(炭素数6〜24)又はリン脂質(炭素数6〜24)である請求項1〜3のいずれか一項に記載の製法。
- 前記溶解促進剤がテトラフルオロホウ酸テトラブチルアンモニウムである請求項4に記載の製法。
- 前記重合開始剤がN-(4´-tert-ブチルベンジル)-4-クロロピリジニウムであり、前記モノマーが4-クロロピリジンである請求項5に記載の製法。
- 更に反応物を重合停止剤と反応させることを含み、該重合停止剤が、アミノ基、アルコキシ基若しくはアルキル基で置換したピリジン、キノリン若しくはアクリジン又はこれらの誘導体、トリフェニルホスフィン又はその誘導体、又はアルキル基若しくはアリル基を置換したアミン誘導体である請求項1〜6のいずれか一項に記載の製法。
- 反応液に前記重合停止剤を加えることにより、反応物を重合停止剤と反応させる請求項7に記載の製法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006511291A JP5154797B2 (ja) | 2004-03-23 | 2005-03-23 | ポリピリジニウムの製法 |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004084518 | 2004-03-23 | ||
| JP2004084518 | 2004-03-23 | ||
| JP2006511291A JP5154797B2 (ja) | 2004-03-23 | 2005-03-23 | ポリピリジニウムの製法 |
| PCT/JP2005/005180 WO2005090443A1 (ja) | 2004-03-23 | 2005-03-23 | ポリピリジニウムの製法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2005090443A1 JPWO2005090443A1 (ja) | 2008-05-08 |
| JP5154797B2 true JP5154797B2 (ja) | 2013-02-27 |
Family
ID=34993651
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2006511291A Expired - Fee Related JP5154797B2 (ja) | 2004-03-23 | 2005-03-23 | ポリピリジニウムの製法 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7985829B2 (ja) |
| EP (1) | EP1754738B1 (ja) |
| JP (1) | JP5154797B2 (ja) |
| KR (1) | KR100890296B1 (ja) |
| CN (1) | CN1934167B (ja) |
| WO (1) | WO2005090443A1 (ja) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8822887B2 (en) | 2010-10-27 | 2014-09-02 | Shaw Arrow Development, LLC | Multi-mode heater for a diesel emission fluid tank |
| USD729722S1 (en) | 2014-05-28 | 2015-05-19 | Shaw Development LLC | Diesel emissions fluid tank floor |
| USD729141S1 (en) | 2014-05-28 | 2015-05-12 | Shaw Development LLC | Diesel emissions fluid tank |
| CN108102092B (zh) * | 2018-01-09 | 2020-07-14 | 苏州大学 | 阳离子有机聚合物及其制备方法和应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5936662A (ja) * | 1982-08-25 | 1984-02-28 | Agency Of Ind Science & Technol | 電導性有機物 |
| JPS61271324A (ja) * | 1985-05-27 | 1986-12-01 | Showa Denko Kk | ポリピリジニウムクロライド共重合体の製造方法 |
| JPS61272234A (ja) * | 1985-05-28 | 1986-12-02 | Showa Denko Kk | ポリピリジニウムフルオライド |
| JPH04293931A (ja) * | 1990-12-14 | 1992-10-19 | Hoechst Ag | ポリ(1,4−ピリジニウム)塩及びその使用方法 |
-
2005
- 2005-03-23 EP EP05721280A patent/EP1754738B1/en not_active Expired - Lifetime
- 2005-03-23 WO PCT/JP2005/005180 patent/WO2005090443A1/ja not_active Ceased
- 2005-03-23 US US10/593,176 patent/US7985829B2/en not_active Expired - Fee Related
- 2005-03-23 CN CN2005800095388A patent/CN1934167B/zh not_active Expired - Fee Related
- 2005-03-23 KR KR1020067021252A patent/KR100890296B1/ko not_active Expired - Fee Related
- 2005-03-23 JP JP2006511291A patent/JP5154797B2/ja not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5936662A (ja) * | 1982-08-25 | 1984-02-28 | Agency Of Ind Science & Technol | 電導性有機物 |
| JPS61271324A (ja) * | 1985-05-27 | 1986-12-01 | Showa Denko Kk | ポリピリジニウムクロライド共重合体の製造方法 |
| JPS61272234A (ja) * | 1985-05-28 | 1986-12-02 | Showa Denko Kk | ポリピリジニウムフルオライド |
| JPH04293931A (ja) * | 1990-12-14 | 1992-10-19 | Hoechst Ag | ポリ(1,4−ピリジニウム)塩及びその使用方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1754738A1 (en) | 2007-02-21 |
| KR20060130734A (ko) | 2006-12-19 |
| WO2005090443A1 (ja) | 2005-09-29 |
| US20080132676A1 (en) | 2008-06-05 |
| US7985829B2 (en) | 2011-07-26 |
| CN1934167B (zh) | 2011-06-15 |
| JPWO2005090443A1 (ja) | 2008-05-08 |
| EP1754738A4 (en) | 2007-04-25 |
| CN1934167A (zh) | 2007-03-21 |
| KR100890296B1 (ko) | 2009-03-26 |
| EP1754738B1 (en) | 2012-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Marcilla et al. | Synthesis of novel polycations using the chemistry of ionic liquids | |
| Price Jr et al. | Supramolecular pseudorotaxane polymers from biscryptands and bisparaquats | |
| Peng et al. | Synthesis and characterization of photorefractive polymers containing transition metal complexes as photosensitizer | |
| Vetrichelvan et al. | Carbazole-containing conjugated copolymers as colorimetric/fluorimetric sensor for iodide anion | |
| JP6311355B2 (ja) | 導電性高分子水溶液、及び導電性高分子膜 | |
| Hmissa et al. | Autocatalytic synthesis of bifluoride ionic liquids by SuFEx click chemistry | |
| Shi et al. | Synthesis of Well‐Defined Figure‐of‐Eight‐Shaped Polymers by a Combination of ATRP and Click Chemistry | |
| JP2002003478A (ja) | N−アルコキシアルキルイミダゾリウム塩、該イミダゾリウム塩からなるイオン性液体ならびにイオン性ゲル | |
| Ikeda | Glycidyl Triazolyl Polymers: Poly (ethylene glycol) Derivatives Functionalized by Azide–Alkyne Cycloaddition Reaction | |
| Su et al. | Barbier-type nitro/nitroso addition polymerization as a versatile approach for molecular design of polyarylamines through C–N bond formation | |
| De Jesús‐Téllez et al. | Kinetic investigations of quaternization reactions of poly [2‐(dimethylamino) ethyl methacrylate] with diverse alkyl halides | |
| JP5154797B2 (ja) | ポリピリジニウムの製法 | |
| Wu et al. | Changing the shape of chromophores from “H-type” to “star-type”: increasing the macroscopic NLO effects by a large degree | |
| Hsiao et al. | Synthesis and properties of new aromatic polyamides with redox‐active 2, 4‐dimethoxytriphenylamine moieties | |
| Willot et al. | The Use of Cyclopenta [2, 1‐b; 3, 4‐b′] dithiophene Analogues for the Development of Low‐Bandgap Materials | |
| Lee et al. | Rotaxane‐type hyperbranched polymers from a crown ether host and paraquat guests containing blocking groups | |
| CN105254787B (zh) | 含腙聚苯乙炔及其制备方法 | |
| Hur et al. | Novel amphiphilic homopolymers containing meta‐and para‐pyridine moieties with living characteristics and their self‐assembly | |
| Weng et al. | Design, synthesis, and self‐assembly manipulating of polymerized ionic liquids contained imidazolium based on “Jacketing” effect | |
| Park et al. | Influence of ionic liquids as solvents for the chemical synthesis of poly (3-octylthiophene) with FeCl 3 | |
| Zhou et al. | Synthesis and characterization of tris (2, 2′‐bipyridine) ruthenium‐cored star‐shaped polymers via RAFT polymerization | |
| He et al. | Electroactive polymer with oligoanilines in the main chain and azo chromophores in the side chain: synthesis, characterization and dielectric properties | |
| Jahed et al. | Synthesis and electrochemical studies of new styrenic poly (ionic liquid) s based on the 1-methyl-1, 2, 3-benzotriazolium cation | |
| Mincheva et al. | Optimized water‐based ATRP of an anionic monomer: Comprehension and properties characterization | |
| Karagollu et al. | Synthesis, Characterization, and Ion Sensing Application of Pyrene‐Containing Chemical Probes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20080317 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20080325 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20111221 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120208 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20120723 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20120921 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20121203 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20121206 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20151214 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5154797 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| S533 | Written request for registration of change of name |
Free format text: JAPANESE INTERMEDIATE CODE: R313533 |
|
| R350 | Written notification of registration of transfer |
Free format text: JAPANESE INTERMEDIATE CODE: R350 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |