本発明の一つ以上の実施形態の詳細が、以下の付随の記述に記載される。本発明の実行または試験においては、本明細書に記載のものに類似するか同等の任意の方法または材料を使用できるが、好ましい方法および材料を以下に記載する。本発明の他の特徴、目的および利点は、記載から明らかになる。明細書においては、文脈が逆を明示しない限り、単数形には複数も含まれる。本明細書において用いられる全ての技術的および科学的用語は、特に断りがない限り、本発明が属する技術の当業者により一般に理解されるのと同じ意味を有する。矛盾が生じる場合には、本明細書が優先する。
SELEX(商標)方法
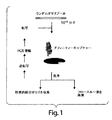
アプタマーを生成する好ましい方法は、概して図1に示され、in vitro選択と呼もばれる、“Systematic Evolution of Ligands by Exponential Enrichment(試験管内人工進化法)”(「SELEX(商標)」)と称されるプロセスによる。SELEX(商標)プロセスは、標的分子に対する高特異性結合をもつ核酸分子のin vitro進化のための方法であり、例えば、1990年6月11日出願の、現在は放棄されている米国特許出願第07/536428号、“Nucleic Acid Ligands”という題の米国特許第5,475,096号、および“Nucleic Acid Ligands”という題の米国特許第5,270,163号(WO91/19813も参照)に記載されている。SELEX(商標)は、選択および増幅の反復的サイクルを実行することにより、任意の所望のレベルの標的結合親和性をもつ、本明細書において「核酸リガンド」とも称されるアプタマーを得るために使用できる。
SELEX(商標)プロセスは、核酸が様々な二次元および三次元構造を形成するための十分な能力と、モノマーかポリマーかを問わず事実上任意の化学化合物とのリガンドとして働く(すなわち特異的結合対を形成する)ために利用可能な十分な化学的可転性をそれらのモノマーの中に有するという、固有の洞察に基づく。任意のサイズまたは組成の分子が、標的となりうる。
SELEX(商標)プロセスは、標的を結合する能力に基づく。したがって、SELEX(商標)手順から得られたアプタマーは、標的に結合する特性を有する。しかし、単なる標的結合からは、アプタマー結合作用により標的に及ぼされうる機能効果に関する情報が、仮にあるとしても提供されない。
標的分子の特性の変更には、アプタマーが標的の特性の変化をもたらすために標的上の一定の位置で結合することが必要である。理論的には、SELEX(商標)法により、各アプタマーが標的の異なる部位に結合する、多数のアプタマーの同定がもたらされうる。実行においては、アプタマー―標的結合相互作用は、相互作用のための安定的で利用可能な構造的インターフェイスを提供する、標的上の一つまたは比較的少数の好ましい結合部位で生じることが多い。さらに、SELEX(商標)法が生理的標的分子に実行される場合には、技術者は通常、標的に対するアプタマーの位置を制御できない。したがって、標的上のアプタマー結合部位の位置が、所望の効果につながりうる、あるいは標的分子に影響し得ない、複数の潜在的結合部位の一つまたはその付近である可能性も、そうでない可能性もある。
アプタマーが、その標的結合能力によって、効果をもつことが認められる場合でも、その効果の存在を予測し、または効果がどのようなものになるかを予知する方法はない。SELEX(商標)実験の実施においては、技術者は、アプタマーが、標的に対するアプタマーを得ることができる範囲で、標的を結合する特性を有するということしか確実には分からない。同定されるアプタマーの一部が標的に結合する以上の効果も有することを期待してSELEX(商標)を行うことはできるが、それは不確実である。
SELEX(商標)プロセスは、開始点としてランダム化配列を含む一本鎖オリゴヌクレオチドの大きなライブラリまたはプールに依存する。オリゴヌクレオチドは、修飾または非修飾DNA、RNAまたはDNA/RNAハイブリッドでありうる。いくつかの例においては、プールは、100%縮重または部分的縮重オリゴヌクレオチドを含む。他の例においては、プールは、ランダム化された配列に導入された少なくとも一つの固定配列および/または保存配列を含む、縮重または部分的縮重オリゴヌクレオチドを含む。他の例においては、プールは、オリゴヌクレオチドプールの全ての分子により共有される配列を含みうる少なくとも一つの固定配列および/または保存配列を、5’および/または3’末端に含む、縮重または部分的縮重オリゴヌクレオチドを含む。固定配列は、後述のCpGモチーフ、PCRプライマのハイブリダイゼーション部位、RNAポリメラーゼ(例えばT3、T4、T7、およびSP6)のプロモータ配列、制限部位、またはポリA領域またはポリT領域等のホモポリマー配列、触媒核、アフィニティーカラムへの選択的結合の部位、転写を促進するリーダー配列、および目的のオリゴヌクレオチドのクローニングおよび/または配列決定を容易にする他の配列等、予め選択された目的のために導入された、プールのオリゴヌクレオチドに共通の配列である。
プールのオリゴヌクレオチドは、効率的な増幅に必要な固定配列ならびに縮重配列部分を含むのが好ましい。典型的には、開始プールのオリゴヌクレオチドは、30〜40のランダムなヌクレオチドの内部領域に隣接する固定5’および3’末端配列を含む。縮重ヌクレオチドは、ランダムに切断された細胞核酸から、化学合成およびサイズ選択を含む多数の方法により作成できる。選択/増幅の反復の前または間に、突然変異誘発によりテスト核酸において配列変異を導入または増加することもできる。
オリゴヌクレオチドの縮重配列部分は、任意の長さであり得、リボヌクレオチドおよび/またはデオキシリボヌクレオチドを含み得、修飾または非天然ヌクレオチドまたはヌクレオチド類似体を含みうる。例えば、米国特許第5,958,691号;第5,660,985号;第5,958,691号;第5,698,687号;第5,817,635号;第5,672,695号、および国際公開第92/07065号を参照。縮重オリゴヌクレオチドは、当該分野で周知のオリゴヌクレオチド固相合成技術を使用してホスホジエステル結合ヌクレオチドから合成されうる。例えば、Froehler等、Nucl.Acid Res.14:5399―5467(1986)およびFroehler等、Tet.Lett.27:5575―5578(1986)を参照。トリエステル合成法等の液相法を用いて、ランダムオリゴヌクレオチドを合成することもできる。例えば、Sood等、Nucl.Acid Res.4:2557(1977)およびHirose等、Tet.Lett.,28:2449(1978)を参照。自動DNA合成装置で行われる典型的な合成は、ほとんどのSELEX(商標)実験に十分な数である、1016〜1017の個々の分子を生じる。配列設計における十分に大きな縮重配列の領域により、各合成分子が固有の配列を表わす可能性が増加する。
オリゴヌクレオチドの開始ライブラリは、DNA合成装置での自動化された化学合成により生成されうる。縮重配列を合成するために、四つ全てのヌクレオチドの混合物を、合成プロセスの間に各ヌクレオチド添加ステップにおいて加え、ヌクレオチドの確率的導入を可能にする。前述の通り、一実施形態においては、ランダムオリゴヌクレオチドは完全縮重配列を含むが、他の実施形態では、縮重オリゴヌクレオチドは非ランダムまたは部分的にランダムな配列の連なりを含みうる。各添加ステップで四つのヌクレオチドを異なるモル比で加えることにより、部分的にランダムな配列が生成されうる。
RNAライブラリが開始ライブラリとして用いられる場合には、典型的に、DNAライブラリを合成し、選択的PCR増幅してから、T7RNAポリメラーゼまたは修飾T7 RNAポリメラーゼを用いてDNAライブラリをin vitroで転写し、転写されたライブラリを精製することにより、それが生成される。そして、RNAまたはDNAライブラリが、結合に好ましい条件下で標的と混合され、同じ一般的選択スキームを用いて結合、分離および増幅の段階的な反復が行われ、結合能および選択性の事実上任意の所望の基準が達成される。より詳しくは、SELEX(商標)法は、核酸の開始プールを含む混合物から開始し、(a)結合に好ましい条件下で混合物を標的と接触させるステップと;(b)標的分子に特異的に結合した核酸から、未結合核酸を分離するステップと;(c)核酸―標的複合体を選択的に解離させるステップと;(d)リガンドが濃縮された核酸の混合物を得るために、核酸―標的複合体から解離された核酸を増幅するステップと;(e)標的分子に対する高特異性、高親和性核酸リガンドを得るために、結合、分離、解離、および増幅のステップを、必要な回数のサイクルで繰り返すステップを含む。RNAアプタマーが選択される場合においては、SELEX(商標)法は、(i)ステップ(d)の増幅の前に、核酸―標的複合体から解離された核酸を逆転写するステップと;(ii)プロセスを再開する前に、ステップ(d)からの増幅された核酸を転写するステップをさらに含む。
多数の可能性のある配列および構造を含む核酸混合物の中においては、所与の標的に対する様々な結合親和性がある。標的に対する親和性が高い(解離定数が低い)ものは、最も標的と結合しやすい。分離、解離および増幅の後、第二核酸混合物が生成され、結合親和性がより高い候補が濃縮される。得られた核酸混合物が一つまたは数個の配列のみから主に構成されるまで、追加的な選択のラウンドにより次第に最適なリガンドが選ばれる。そして、これらがクローニングされ、配列決定され、リガンドまたはアプタマーとして1)標的結合親和性;および/または2)標的機能に影響する能力につき個々に試験されうる。
所望の目標が達成されるまで、選択および増幅のサイクルが繰り返される。最も一般的な場合においては、サイクルの反復により結合強度に有意な改善が達成されなくなるまで、選択/増幅が継続される。方法は、典型的には、約1014の異なる核酸種のサンプリングに使用されるが、約1018もの数の異なる核酸種のサンプリングにも使用されうる。一般に、5〜20サイクルの手順において、核酸アプタマー分子が選択される。一実施形態においては、最初の選択ステージにおいてのみ不均質性が導入され、複製プロセスの全体にわたって生じることはない。
SELEX(商標)法の一実施形態においては、選択プロセスが、選択された標的に最も強く結合する核酸リガンドを単離することにおいて非常に効率的であるために、選択および増幅のサイクルが一度で足りる。そのような効率的な選択は、例えば、カラムに結合した標的と結びつく核酸の能力が、カラムが最も親和性の高い核酸リガンドの分離および単離を十分に許容できるような態様で働く、クロマトグラフィタイプのプロセスにおいて生じうる。
多くの場合には、単一の核酸リガンドが同定されるまでSELEX(商標)プロセスの反復的ステップを行うことが、必ずしも望ましいとは限らない。標的特異的核酸リガンド溶液は、多数の保存配列、および標的に対する核酸リガンドの親和性に大きく影響せずに置換または加えられうる多数の配列を有する、核酸構造またはモチーフのファミリーを含みうる。SELEX(商標)プロセスを完了前に終了することにより、核酸リガンド溶液ファミリーの多数のメンバーの配列を決定することが可能である。
種々の核酸の一次構造、二次構造および三次構造が存在することが知られている。非ワトソン―クリック型の相互作用に関与することが最も一般的に示されている構造またはモチーフは、ヘアピンループ、対称および非対称のバルジ、シュードノットおよびこれらの無数の組み合わせと呼ばれる。このようなモチーフの公知の場合のほぼ全てが、それらがわずか30ヌクレオチド以内の核酸配列において形成されうることを示唆する。この理由から、連続したランダム化セグメントを用いたSELEX(商標)手順が、約20〜50、いくつかの実施形態においては約30〜40の間のヌクレオチドのランダム化セグメントを含む核酸配列で開始されることが好ましいことが多い。一実施例においては、5’―固定:ランダム:3’―固定配列は、約30〜約40ヌクレオチドのランダム配列を含む。
多くの特定の目的を達成するために、基本的なSELEX(商標)法が改変されている。例えば、米国特許第5,707,796号は、曲がったDNA等の特定の構造特性を有する核酸分子を選択するための、ゲル電気泳動と合わせたSELEX(商標)プロセスの利用を記載する。米国特許第5,763,177号は、標的分子に結合および/または光架橋および/またはこれを光不活性化しうる光反応性基を含む核酸リガンドを選択するための、SELEX(商標)ベースの方法を記載する。米国特許第5,567,588号および第5,861,254号は、標的分子に対して高い親和性および低い親和性を持つオリゴヌクレオチドの非常に効率的な分離を達成する、SELEX(商標)ベースの方法を記載する。米国特許第5,496,938号は、SELEX(商標)プロセスが実施された後に、改善された核酸リガンドを得るための方法を記載する。米国特許第5,705,337号は、リガンドを標的に共有結合するための方法を記載する。
SELEX(商標)法を用いて、標的分子上の一つ以上の部位に結合する核酸リガンドを得ること、および、標的上の特定の部位に結合する非核酸種を含む核酸リガンドを得ることもできる。SELEX(商標)法は、核酸結合タンパク質およびその生物学的機能の一部として核酸に結合することが知られていないタンパク質、ならびに補因子および他の小分子等の、巨大分子および小分子を含む任意の想定可能な標的に結合する核酸リガンドを単離および同定するための手段を提供する。例えば、米国特許第5,580,737号は、カフェインとその密接に関連するアナログ、テオフィリンに対して高い親和性で結合し得る、SELEX(商標)法により同定される核酸配列を開示する。
Counter−SELEX(商標)プロセスは、一つ以上の非標的分子への交差反応性をもつ核酸リガンド配列を排除することにより、標的分子に対する核酸リガンドの特異性を改善する方法である。Counter−SELEX(商標)プロセスには、(a)核酸の候補混合物を調製するステップと;(b)候補混合物を標的に接触させるステップであり、候補混合物と比較して標的に対する親和性が増加している核酸が、候補混合物の残部から分離され得る、ステップと;(c)候補混合物の残部から親和性が増加した核酸を分離するステップと;(d)標的から親和性が増加した核酸を選択的に解離するステップと;(e)非標的核酸分子(単数または複数)に対する特異的親和性をもつ核酸リガンドが除去されるように、親和性の増加した核酸を一つ以上の非標的分子と接触させる、ステップと;(f)標的分子への結合につき比較的高い親和性および特異性を有する核酸配列が濃縮された核酸の混合物を得るために、標的分子だけに対して特異的親和性をもつ核酸を増幅する、ステップが含まれる。SELEX(商標)法について上述したように、所望の目標が達成されるまで、選択および増幅のサイクルが必要に応じて繰り返される。
治療剤およびワクチンとしての核酸の使用において直面する一つの潜在的な問題は、そのホスホジエステル形態におけるオリゴヌクレオチドが、所望の効果が現れる前に、エンドヌクレアーゼおよびエキソヌクレアーゼ等の細胞内酵素および細胞外酵素により体液中で急速に分解されうることである。したがって、SELEX(商標)法は、リガンドに、in vivo安定性の改善または送達特性の改善等の特性の改善を与える修飾ヌクレオチドを含む高親和性核酸リガンドの同定を含む。このような修飾の例には、糖位および/またはリン酸位および/または塩基位での化学的置換が含まれる。修飾ヌクレオチドを含むSELEX(商標)―同定核酸が、例えば、リボースの2’位、ピリミジンの5位、およびプリンの8位で化学的に修飾されたヌクレオチド誘導体を含むオリゴヌクレオチドを記載する米国特許第5,660,985号、様々な2’修飾ピリミジンを含むオリゴヌクレオチドを記載する米国特許第5,756,703号、および、2’―アミノ(2’―NH2)、2’―フルオロ(2’―F)、および/または2’―O―メチル(2’―OMe)置換基で修飾された一つ以上のヌクレオチドを含む、高特異的核酸リガンドを記載する米国特許第5,580,737号に記載されている。
本発明において意図される核酸リガンドの修飾には、核酸リガンド塩基または全体としての核酸リガンドに追加的電荷、極性、疎水性、水素結合、静電相互作用、および流動性を導入する他の化学基を提供するものが含まれるが、これらに限定されない。ヌクレアーゼに対して抵抗性のオリゴヌクレオチド集団を生成するための修飾には、一つ以上の置換ヌクレオチド間結合、改変された糖、改変された塩基、またはそれらの組み合わせも含まれうる。このような修飾には、2’―位糖修飾、5―位ピリミジン修飾、8―位プリン修飾、環外アミンでの修飾、4―チオウリジンの置換、5―ブロモもしくは5―ヨードウラシルの置換;骨格修飾、ホスホロチオエートまたはアルキルホスフェート修飾、メチル化、および、イソ塩基のイソシチジンおよびイソグアノシン等の異常な塩基対の組み合わせが含まれるが、これに限られない。修飾には、キャッピング等の3’修飾および5’修飾も含まれうる。修飾には、キャッピング、例えばエキソヌクレアーゼ抵抗性を増加させるための3’―3’―dTキャップの付加等の、3’修飾および5’修飾も含まれうる(例えば、各々参照により全体として本明細書に組み込まれる、米国特許第5,674,685号;第5,668,264号;第6,207,816号;および第6,229,002号を参照。)
一実施形態においては、P(O)O基がP(O)S(「チオエート」)、P(S)S(「ジチオエート」)、P(O)NR2(「アミデート」)、P(O)R、P(O)OR’、COまたはCH2(「ホルムアセタール」)、または3’―アミン(―NH―CH2―CH2―)により置換されたオリゴヌクレオチドが提供され、各RまたはR’は、独立してHであるか置換もしくは非置換アルキルである。―O―、―N―、または―S―結合を介して、隣接するヌクレオチドに結合基が結合され得る。オリゴヌクレオチドにおける全ての結合が同一である必要はない。
さらなる実施形態においては、オリゴヌクレオチドは修飾糖基を含み、例えば一つ以上のヒドロキシル基が、ハロゲン、脂肪族基で置換されるか、エーテルまたはアミンとして官能化される。一実施形態においては、フラノース残基の2’位が、O―メチル、O―アルキル、O―アリル、S―アルキル、S―アリル、またはハロ基のいずれかにより置換される。2’―修飾糖の合成方法が、例えばSproat等、Nucl.AcidRes.19:733―738(1991);Cotton等、Nucl.Acid Res.19:2629―2635(1991);およびHobbs等、Biochemistry12:5138―5145(1973)に記載される。他の修飾は、当技術分野の技術者に公知である。このような修飾は、SELEX(商標)プロセス前修飾またはSELEX(商標)プロセス後修飾(先に同定された未修飾リガンドの修飾)であるか、SELEX(商標)プロセスへの組み込みによりなされうる。
SELEX(商標)プロセス前修飾またはSELEX(商標)プロセスへの組み込みによりなされるものは、そのSELEX(商標)標的に対する特異性と、安定性の改善、例えばin vivo安定性の両方を有する核酸リガンドを生じる。核酸リガンドに対するSELEX(商標)プロセス後修飾((例えばSELEX(商標)プロセス前修飾によりヌクレオチドが導入された、先に同定されたリガンドの切断、欠失、置換または追加的なヌクレオチド修飾)は、核酸リガンドの結合能に悪影響を与えることなく、安定性、例えばin vivo安定性の改善をもたらしうる。選択的に、修飾ヌクレオチドがSELEX(商標)プロセス前修飾により導入されたアプタマーが、SELEX(商標)プロセス後修飾(すなわちSELEXの後のSELEX(商標)後修飾プロセス)により、さらに修飾されうる。
SELEX(商標)法は、米国特許第5,637,459号および第5,683,867号に記載されるように、選択されたオリゴヌクレオチドを他の選択されたオリゴヌクレオチドおよび非オリゴヌクレオチドの機能単位と組み合わせるステップを含む。SELEX(商標)法は、例えば米国特許第6,011,020号、第6,051,698号、および国際公開第98/18480号に記載されるように、選択された核酸リガンドを、診断用または治療用複合体において親油性または非免疫原性の高分子量化合物と組み合わせるステップをさらに含む。これらの特許および特許出願は、広範囲の形および他の特性の組み合わせを、オリゴヌクレオチドの効率的増幅および複製特性、および他の分子の所望の特性とともに教示する。
SELEX(商標)法による、小さなフレキシブルペプチドに対する核酸リガンドの同定も調査されている。小ペプチドは、フレキシブルな構造を有し、通常は複数のコンホーマの平衡状態で溶液中に存在し、そのため、フレキシブルペプチドを結合する際のコンホメーションのエントロピーの損失により結合親和性が制限されうると当初は考えられた。しかし、溶液中における小ペプチドに対する核酸リガンドの同定の実現可能性が、米国特許第5,648,214号において示された。この特許においては、11アミノ酸ペプチドのサブスタンスPに対する高親和性RNA核酸リガンドが同定された。
SELEX(商標)プロセスの一部として、標的と結合するように選択された配列は、その後、所望の結合親和性を有する最小の配列を決定するために、選択的に最小化される。選択された配列および/または最小化された配列は、例えば結合親和性を増加し、あるいは配列中のどの位置が結合活性に必須であるか決定するために、配列のランダムまたは定方向突然変異誘発を行うことにより選択的に修飾される。
修飾ヌクレオチドSELEX(商標)法
アプタマーが、治療薬および/または特定の種類の診断薬としての使用に適するためには、合成が安価であり、in vivoで安全および安定していることが好ましい。野生型RNAおよびDNAアプタマーは通常、ヌクレアーゼにより分解されやすいため、in vivoで安定してない。2’―位における修飾基の導入により、ヌクレアーゼ分解に対する抵抗が大きく高められうる。
さらに、アプタマーが選択されるオリゴヌクレオチドライブラリへの化学修飾の導入により、オリゴヌクレオチドライブラリの構造的および化学的多様性が増加し、特に望ましい特性を有する高親和性および高特異的アプタマーが得られる標的数が、潜在的に増加する。例えば、多くの用途においては、完全2’―OMeアプタマーを得ることが望ましいが、そのようなアプタマーを、標的の機能を所与の用途において十分な程度に調節、例えば阻害するアプタマーをもたらす単一のプールの組成物、例えば最初のヌクレオチド以外の全てが2’―OMeであるオリゴヌクレオチドから得ることは必ずしも可能でないだろう。この場合には、dCmD転写組成物から得られるライブラリなど、一つ以外の全てのヌクレオチドが2’―OMeであるオリゴヌクレオチドを含むプール等の他のプール組成物からアプタマーを選択することが望ましいだろう。それはまた望ましい、および、別の実施形態においては、本発明は、2’―OMe修飾ならびに核酸塩基の位置に側鎖修飾を有するオリゴヌクレオチドを含むプール組成物からアプタマーを選択する方法を提供する。
2’―フルオロおよび2’―アミノ基がオリゴヌクレオチドプールに良好に導入され、そこからその後アプタマーが選択されている。しかし、これらの修飾は、得られるアプタマーの合成費用を大きく増加させ、また、修飾オリゴヌクレオチドの分解およびその後のDNA合成の基質としてのヌクレオチドの使用により、修飾ヌクレオチドがホストDNAに再利用される可能性があるために、一部の場合には安全性の問題をもたらしうる。
本明細書に提供される、2’―O―メチル(「2’―OMe」)ヌクレオチドを含むアプタマーは、これらの欠点の多くを克服する。2’―OMeヌクレオチドを含むオリゴヌクレオチドは、ヌクレアーゼ抵抗性であり、合成が安価である。2’―OMeヌクレオチドは、生物系に遍在するが、天然のポリメラーゼは2’―OMe NTPsを生理学的条件下で基質として受け入れず、したがって、2’―OMeヌクレオチドのホストDNAへの再利用に関する安全性の問題がない。2’―修飾アプタマーを生成するために使用されるSELEX(商標)法が、例えば、2002年12月3日に出願の米国特許仮出願第60/430,761号、2003年7月15日に出願の米国特許仮出願第60/487,474号、2003年11月4日に出願の米国特許仮出願第60/517,039号、2003年12月3日に出願の米国特許出願第10/729,581号、2004年6月21日に出願の、“Method for in vitro Selection of 2’―O―methyl Substituted Nucleic Acids”と題した米国特許出願第10/873,856号、2005年6月30日に出願の、Materials and Methods for the Generation of Fully 2’―Modified Nucleic Acid Transcriptsと題した米国特許仮出願第60/696,292号、および2006年6月30日に出願の、Materials and Methods for the Generation of Fully 2’―Modified Nucleic Acid Transcriptsと題した米国特許出願第11/480,188号に記載されており、これらの各々は参照により全体として本明細書に組み込まれる。
本発明は、アプタマー標的に結合してその機能を調節するアプタマーであり、オリゴヌクレオチドを非修飾オリゴヌクレオチドよりも酵素的および化学的分解、ならびに熱的および物理的分解に対してより安定させるために修飾されたヌクレオチド(例えば2’―位に修飾を有するヌクレオチド)を含む、アプタマーを含む。本発明には、アプタマー標的に結合してその機能を調節するアプタマーであり、2’位、例えば2’―OMe修飾、および核酸塩基の位置、例えばピリミジンの5位の側鎖修飾で修飾されたヌクレオチドを含む、アプタマーも含まれる。2’―OMeを含むアプタマーのいくつかの例が文献にあるが(例えばRuckman等、J.Biol.Chem,1998 273,20556―20567―695)、これらは、CおよびU残基が2’―フルオロ(2’―F)置換され、AおよびG残基が2’―OHである修飾された転写産物のライブラリのin vitro選択により生成された。機能的配列が同定されると、各AおよびG残基の2’―OMe置換に対する寛容性が試験され、2’―OMe残基としての2’―OMe置換に寛容な全てのAおよびG残基を有するアプタマーが再合成された。この二段階様式で生成されるアプタマーのほとんどのAおよびG残基は、2’―OMe残基による置換に寛容であるが、平均的に約20%は許容しない。したがって、この方法を用いて生成されるアプタマーは、2〜4の2’―OH残基を含む傾向があり、安定性および合成費用が結果的に損なわれる。本発明の方法は、SELEX(商標)法(および/または本明細書に説明されるものを含むその任意の変化形および改善形態)によりアプタマーが選択および濃縮されるオリゴヌクレオチドプールにおいて使用される、安定化されたオリゴヌクレオチドを生成する、転写反応物への修飾ヌクレオチドの導入により、アプタマーオリゴヌクレオチドを2’―OMe修飾ヌクレオチドにより再合成することにより、選択されたアプタマーオリゴヌクレオチドを安定化する必要性をなくす。
一実施形態においては、本発明は、ATP、GTP、CTP、TTP、およびUTPヌクレオチドの2’―OH、2’―F、2’―デオキシ、および2’―OMe修飾の組み合わせを含むアプタマーを提供する。別の実施形態においては、本発明は、ATP、GTP、CTP、TTP、およびUTPヌクレオチドの2’―OH、2’―F、2’―デオキシ、2’―OMe、2’―NH2、およびT―メトキシエチル修飾の組み合わせを含むアプタマーを提供する。好ましい実施形態においては、本発明は、全てまたは実質的に全て2’―OMe修飾されたATP、GTP、CTP、TTP、および/またはUTPヌクレオチドを含むアプタマーを提供する。別の好ましい実施形態においては、本発明は、全てのヌクレオチド三リン酸基、ATP、GTP、CTP、TTPおよび/またはUTPが、一つを除いて2’―OMe修飾されたアプタマーを提供する。例えば、2’―OMe ATP、GTP、TTPを含むが、CTPがデオキシであるアプタマーである。本発明は、任意の先行する2’修飾の組み合わせとの組み合わせにおける、側鎖による核酸塩基修飾を含むアプタマーをさらに提供する。
別の好ましい実施形態においては、本発明は、開始ヌクレオチドが2’修飾である必要がないことを除いて、三つの2’―OMe修飾ヌクレオチド三リン酸基および一つのデオキシまたは2’―OHヌクレオチド三リン酸基を含むアプタマーを提供する。特定の実施形態では、アプタマーはデオキシシチジンヌクレオチド三リン酸を含み、残りのヌクレオチド三リン酸は、開始ヌクレオチドが2’修飾である必要がないことを除いて、2’―OMe修飾される。別の実施形態では、アプタマーはデオキシチミジンヌクレオチド三リン酸を含み、残りのヌクレオチド三リン酸は、開始ヌクレオチドが2’修飾である必要がないことを除いて、2’―OMe修飾されている。別の実施形態では、アプタマーは2’―OHウリジンヌクレオチド三リン酸を含み、残りのヌクレオチド三リン酸は、開始ヌクレオチドが2’修飾である必要がないことを除いて、2’―OMe修飾されている。別の実施形態では、アプタマーはデオキシグアノシンヌクレオチド三リン酸を含み、残りのヌクレオチドは、開始ヌクレオチドが2’修飾である必要がないことを除いて、2’―OMeが修飾される。別の実施形態では、アプタマーはデオキシアデノシンヌクレオチド三リン酸を含み、残りのヌクレオチドは、開始ヌクレオチドが2’修飾である必要がないことを除いて、2’―OMe修飾されている。
修飾ポリメラーゼ
本発明の2’修飾アプタマーは、野生型ポリメラーゼ、例えば野生型T7ポリメラーゼより高い、フラノース2’位に大きな置換基を有する修飾ヌクレオチドの導入率を有する修飾ポリメラーゼ、例えば変異T7ポリメラーゼを用いてつくられる。例えば、639位のチロシン残基がフェニルアラニンに変えられている(Y639F)変異T7ポリメラーゼは、2’デオキシ、2’アミノ―、および2’フルオロ―ヌクレオチド三リン酸(NTPs)を容易に基質として利用し、様々な用途のために修飾RNAsを合成するために広く使われている。しかし、この変異T7ポリメラーゼは、2’―OMeまたは2’―アジド(2’―N3)置換基等の大きな2’―置換基を伴うNTPsを容易に利用(すなわち導入)できないことが報告されている。大きな2’置換基の導入のために、Y639F変異に加えて、784位のヒスチジンがアラニン残基に変えられた変異T7ポリメラーゼが、記載されており(Y639F/H784A)、修飾ピリミジンNTPsを導入するために、限られた状況において使われている。Padilla,R.およびSousa,R.,Nucleic Acids Res.,2002,30(24):138を参照。639位のチロシン残基がフェニルアラニンに変えられ、784位のヒスチジン残基がアラニンに変えられ、378位のリジン残基がアルギニンに変えられている変異T7RNAポリメラーゼ(Y639F/H784A/K378R)が、修飾プリンおよびピリミジンNTPs、例えば2’―OMe NTPsを導入するために、限られた状況において使用されているが、転写のための2’―OH GTPのスパイクを含む。Burmeister等、(2005)Chemistry and Biology,12:25―33を参照。転写のための2’―OH GTPスパイクの包含により、完全に2’―OMeではなく2’―OH GTPの存在に依存しうるアプタマーが生じうる。
784位のヒスチジンがアラニン残基に変えられた(H784A)変異T7ポリメラーゼも、説明されている。Padilla等、Nucleic Acids Research,2002,30:138。Y639F/H784A変異およびH784A変異T7ポリメラーゼの両方において、アラニン等のより小さなアミノ酸残基への変更により、より大きなヌクレオチド基質、例えば2’―OMe置換ヌクレオチドの導入が可能になる。Chelliserry,K.およびEllington,A.D.,(2004)Nature Biotech,9:1155―60を参照。大きな2’修飾基質をより容易に導入する、T7RNAポリメラーゼの活性部位に変異を伴う追加的なT7RNAポリメラーゼ、例えば639位のチロシン残基がロイシンに変えられた(Y639L)変異T7RNAポリメラーゼが、記載されている。しかし、そのような変異により与えられる基質特異性の増加のために活性が犠牲になり、転写収率が低下することが多い。Padilla RおよびSousa,R.,(1999)Nucleic Acids Res.,27(6):1561を参照。T7RNAポリメラーゼ変異P266Lが、プロモータクリアランスを促進することが記載されている(Guillerez等(2005)Proc.Nat.Acad.Sci.USA,102(17)5958)。ポリメラーゼは、プロモータに結合された開始コンホメーションから、結合されていない伸長コンホメーションへと変化する。以上の変異ポリメラーゼのいずれも、完全2’―OMe転写産物をもたらすことが報告されていない。
本発明は、オリゴヌクレオチドの転写収率を増加させる材料および方法を提供する。一実施形態においては、本発明は、オリゴヌクレオチドに修飾ヌクレオチドを酵素的に導入するために、修飾T7RNAポリメラーゼ(T7RNAポリメラーゼの修飾は、野生型T7RNAポリメラーゼ、配列番号33に対するもの)を使用するための、方法および条件を提供する。好ましい実施形態においては、本発明の転写方法で使用される変異T7RNAポリメラーゼは、2’―OH GTPの存在を要しない。好ましい実施形態においては、修飾ポリメラーゼは、639位のチロシン残基がロイシン残基に変えられ、784位のヒスチジン残基がアラニン残基に変えられた(Y639L/H784A)、変異T7RNAポリメラーゼである。別の好ましい実施形態においては、修飾ポリメラーゼは、639位のチロシン残基がロイシン残基に変えられ、784位のヒスチジン残基がアラニン残基に変えられ、378位のリジン残基がアルギニン残基に変えられた(Y639L/H784A/K378R)、変異T7RNAポリメラーゼである。別の実施形態においては、本発明の方法に用いられる修飾ポリメラーゼは、639位のチロシン残基がロイシン残基に変えられ(Y639L)、さらに別の実施形態においては、変異T7RNAポリメラーゼは、639位のチロシン残基がロイシン残基に変えられ、378位のリジン残基がアルギニン残基に変えられ(Y639L/K378R)ている。いかなる理論にも拘束されるものではないが、K378R変異は、ポリメラーゼの活性部位の近くでなく、したがってサイレント変異であると考えられる。別の実施形態においては、本発明の方法に用いられる修飾ポリメラーゼは、266位のプロリン残基がロイシンに変えられ、639位のチロシン残基がロイシンに変えられ、784位のヒスチジン残基がアラニン残基に変えられた(P266L/Y639L/H784A)変異T7RNAポリメラーゼであり、さらに別の実施形態においては、変異T7RNAポリメラーゼは、266位のプロリン残基がロイシンに変えられ、639位のチロシン残基がロイシン残基に変えられ、784位のヒスチジン残基がアラニン残基に変えられ、378位のリジン残基がアルギニン残基に変えられている(P266L/Y639L/H784A/K378R)。
変異T7RNAポリメラーゼのアミノ酸配列が、以下に示される。
2’修飾(例えば2’―OMeまたは2’―OMeおよび核酸塩基側鎖修飾)RNA転写物のプールを生成するために、変異ポリメラーゼが2’―修飾NTPsを許容する条件下における、Y639F、Y639F/K378R、Y639F/H784A、Y639F/H784A/K378R、Y639L/H784A、Y639L/H784A/K378R、Y639L、Y639L/K378R、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378R変異T7RNAポリメラーゼが使用されうる。好ましいポリメラーゼは、Y639L/H784A変異T7RNAポリメラーゼである。別の好ましいポリメラーゼは、Y639L/H784A/K378R変異T7RNAポリメラーゼである。本発明における別の好ましいポリメラーゼは、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378R変異T7RNAポリメラーゼである。他のT7RNAポリメラーゼ、特に大きな2’―置換基に対する高い寛容性をもつものも、本発明の方法で使用できる。本明細書に開示される条件を用いたテンプレート指向性重合において使用される場合には、Y639L/H784A、Y639L/H784A/K378R、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378R変異T7RNAポリメラーゼが、Y639F、Y639F/K378R、Y639F/H784A、Y639F/H784A/K378R、Y639L、またはY639L/K378R変異T7RNAポリメラーゼを用いて達成されるより高い転写収率で、2’―OMe GTPを含み、核酸塩基の位置で側鎖修飾もされた2’―OMe NTPSも含む、全て2’―OMeのNTPsの導入のために使用されうる。Y639L/H784A、Y639L/H784A/K378R、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378R変異T7RNAポリメラーゼは、2’―修飾、例えば2’―OMeを含むオリゴヌクレオチドの高収率を達成するために、2’―OH GTPを伴って使用されうるがその必要はない。
好ましい実施形態においては、本発明のY639L/H784AまたはY639L/H784A/K378R変異T7RNAポリメラーゼが、より高い完全2’―OMe転写収率を促進するために、MNA転写混合物とともに使用される。いくつかの実施形態においては、MNA転写混合物の少なくとも一つのオリゴヌクレオチドは、核酸塩基の位置における側鎖変更も含む。別の好ましい実施形態においては、本発明のY639L/H784AまたはY639L/H784A/K378R変異T7RNAポリメラーゼが、転写産物の最初のヌクレオチド以外の全てのグアノシン三リン酸が2’―OMeである、より高い転写収率を促進するために、dCmD、dTmV、rTmV、rUmVまたはdUmV転写混合物とともに使用される。いくつかの実施形態においては、本発明のY639L/H784AまたはY639L/H784A/K378R変異T7RNAポリメラーゼが、dCmD、dTmV、rTmV、rUmVまたはdUmV転写混合物とともに使用され、転写産物の最初のヌクレオチド以外の全てのグアノシン三リン酸が2’―OMeである、より高い転写収率を促進するために、混合物中の少なくとも一つヌクレオチドタイプがその核酸塩基の位置の側鎖修飾も含み、一つのヌクレオチドタイプが核酸塩基の側鎖修飾も含む。いくつかの実施形態においては、Y639L/H784AまたはY639L/H784A/K378R変異T7RNAポリメラーゼが、rRmY、dRmY、rGmH、fGmH、dGmH、dAmB、rRdY、dRdYまたはrN転写混合物とともに使用されうる。いくつかの実施形態においては、Y639L/H784A、Y639L/H784A/K378R、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378R変異T7RNAポリメラーゼのいずれか一つが、dCmD、dTmV、rUmV、dUmV、dGmH、rGmH、交互混合物、fGmH、rAmBまたはdAmB転写反応混合物において使用されうる。特定の実施形態においては、Y639L/H784A、Y639L/H784A/K378R、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378R変異T7RNAポリメラーゼのいずれか一つが、dCmD、dTmV、rUmVまたはdUmV転写反応混合物において使用されうる。
本明細書で使用されるところでは、2’―OMe A、G、CおよびU三リン酸(2’―OMe ATP、2’―OMe UTP、2’―OMe CTP、および2’―OMe GTP)だけを含む転写混合物が、MNA混合物と呼ばれ、そこから選択されるアプタマーはMNAアプタマーと呼ばれ、2’―O―メチルヌクレオチドだけを含む。2’―OMe CおよびU、ならびに2’―OH AおよびGを含む転写混合物は「rRmY」混合物と呼ばれ、そこから選択されるアプタマーは「rRmY」と呼ばれる。デオキシAおよびG、ならびに2’―OMe UおよびCを含む転写混合物は「dRmY」混合物と呼ばれ、そこから選択されたアプタマーは「dRmY」アプタマーと呼ばれる。2’―OMe A、CおよびU、ならびに2’―OH Gを含む転写混合物は「rGmH」混合物と呼ばれ、そこから選択されるアプタマーは「rGmH」アプタマーと呼ばれる。2’―OMe A、C、UおよびG、ならびに2’―OMe A、UおよびC、ならびに2’―F Gを交互に含む転写混合物は「交互混合物」と呼ばれ、そこから選択されるアプタマーは「交互混合物」アプタマーと呼ばれる。2’―OMe A、U、およびC、ならびに2’―F Gを含む転写混合物は「fGmH」混合物と呼ばれ、そこから選択されるアプタマーは「fGmH」アプタマーと呼ばれる。2’―OMe A、U、およびC、ならびにデオキシGを含む転写混合物は「dGmH」混合物と呼ばれ、そこから選択されるアプタマーは「dGmH」アプタマーと呼ばれる。デオキシA、ならびに2’―OMe C、GおよびUを含む転写混合物は「dAmB」混合物と呼ばれ、そこから選択されるアプタマーは「dAmB」アプタマーと呼ばれる。2’―OH Aならびに2’―OMe C、GおよびUを含む転写混合物は「rAmB」混合物と呼ばれ、そこから選択されるアプタマーは「rAmB」アプタマーと呼ばれる。2’―OHアデノシン三リン酸およびグアノシン三リン酸およびデオキシシチジン三リン酸およびチミジン三リン酸を含む転写混合物はrRdY混合物と呼ばれ、そこから選択されるアプタマーは「rRdY」アプタマーと呼ばれる。2’―OMe A、UまたはT、およびG、ならびにデオキシCを含む転写混合物は「dCmD」混合物と呼ばれ、そこから選択されるアプタマーは「dCmD」アプタマーと呼ばれる。2’―OMe A、G、およびC、ならびにデオキシTを含む転写混合物は「dTmV」混合物と呼ばれ、そこから選択されるアプタマーは「dTmV」アプタマーと呼ばれる。2’―OMe A、C、およびG、ならびに2’―OH Uを含む転写混合物は「rUmV」混合物と呼ばれ、そこから選択されるアプタマーは「rUmV」アプタマーと呼ばれる。2’―OMe A、C、およびG、ならびに2’―デオキシUを含む転写混合物は「dUmV」混合物と呼ばれ、そこから選択されるアプタマーは「dUmV」アプタマーと呼ばれる。全2’―OHのヌクレオチドを含む転写混合物は「rN」混合物と呼ばれ、そこから選択されるアプタマーは「rN」、「rRrY」またはRNAアプタマーと呼ばれ、全デオキシヌクレオチドを含む転写混合物は「dN」混合物と呼ばれ、そこから選択されるアプタマーは「dN」または「dRdY」またはDNAアプタマーと呼ばれる。
2’―修飾オリゴヌクレオチドは、完全に修飾ヌクレオチドから、または修飾ヌクレオチドのサブセットから合成できる。全てのヌクレオチドが修飾され、全てが同じ修飾を含みうる。全てのヌクレオチドが修飾されるが、異なる修飾を含みうる。例えば、同じ塩基を含む全てのヌクレオチドが、一つのタイプの修飾を有し、他の塩基を含むヌクレオチドは、異なるタイプの修飾を有しうる。全てのプリンヌクレオチドは、一つのタイプの修飾を有し(または修飾されず)、全てのピリミジンヌクレオチドは別の、異なるタイプの修飾を有しうる(または修飾されない)。この様式で、例えばリボヌクレオチド(2’―OH)、デオキシリボヌクレオチド(2’―デオキシ)、2’―F、および2’―OMeヌクレオチドを含め、任意の修飾の組み合わせを用いて、転写産物または転写産物のプールが生成される。さらに、修飾オリゴヌクレオチドは、ヌクレオチド間結合(例えばホスホロチオエート)および糖(例えば2’―OMe)および塩基(例えばイノシン)での修飾等、同時に二つ以上の修飾をもつヌクレオチドを含みうる。別の実施形態においては、同時に二つ以上の修飾をもつヌクレオチドは、例えば2’―OMe修飾および側鎖修飾を核酸塩基の位置で含む。
転写条件
2’―修飾SELEX(商標)法の転写条件に、多数の因子が重要であると決定されている。2’修飾、特に2’―OMe修飾ヌクレオチドを含む転写混合物による転写条件のために記載される因子は、2’修飾および側鎖核酸塩基修飾ヌクレオチド三リン酸を含む転写混合物による本発明の転写方法と関連しても用いられる。本発明に用いられる側鎖修飾ヌクレオチド三リン酸が、説明されている。例えば、各々が参照により全体として本明細書に組み込まれる、Vaught JD,Dewey T,Eaton BE.J Am Chem Soc.2004;126:11231―11237;Nieuwlandt D,West M,Cheng X,Kirshenheuter G,Eaton BE.Chembiochem.2003,4,651―654;Ito T,Ueno Y,Komatsu Y,Matsuda A.Nucleic Acids Res.2003;31,2514―2523;Dewey,TM,Mundt,AA,Crouch,GJ,Zyzniewyski,MC,Eaton,BE.J Am Chem Soc 1995,117,8474―8475;Jager S,Rasched G,Kornreich―Leshem H,Engeser M,Thum O,Famulok M.J Am Chem Soc.2005,127,15071―15082;Tarasow TM,Tarasow SL,Eaton BE.Nature.1997,389,54―57;Liu D,Gugliotti LA,Wu T,Dolska M,Tkachenko AG,Shipton MK,Eaton BE,Feldheim DL.Langmuir.2006,22,5862―5866;Gugliotti LA,Feldheim DL,Eaton BE.J Am Chem Soc.2005,127,17814―17818;Gugliotti LA,Feldheim DL,Eaton BE.Science.2004,304,850―852;Kuwahara M,Hanawa K,Ohsawa K,Kitagata R,Ozaki H,Sawai H.Bioorg Med Chem.2006,14,2518―2526;Saitoh H,Nakamura A,Kuwahara M,Ozaki H,Sawai H.Nucleic Acids Res Suppl.2002,2,215―216;Kuwahara M,Ohbayashi T,Hanawa K,Shoji A,Ozaki AN,Ozaki H,Sawai H.Nucleic Acids Res Suppl.2002,2,83―84.Mehedi Masud M,Ozaki―Nakamura A,Kuwahara M,Ozaki H,Sawai H Chembiochem.2003,4,584―548;Masud MM,Ozaki―Nakamura A,Satou F,Ohbayashi T,Ozaki H,Sawai H.Nucleic Acids Res Suppl.2001;(l):21―2;Obayashi T,Masud MM,Ozaki AN,Ozaki H,Kuwahara M,Sawai H.Bioorg Med Chem Lett.2002,12,1167―70;Sawai H,Ozaki―Nakamura A,Mine M,Ozaki H..Bioconjug Chem.2002,2,309―316;Ohbayashi T,Kuwahara M,Hasegawa M,Kasamatsu T,Tamura T,Sawai H Org Biomol Chem.2005,3,2463―2468;Shoji A,Hasegawa T,Kuwahara M,Ozaki H,Sawai H.Bioorg Med Chem Lett.2006 Oct 28;[Epub ahead of print]Kuwahara M,Nagashima J,Hasegawa M,Tamura T,Kitagata R,Hanawa K,Hososhima S,Kasamatsu T,Ozaki H,Sawai H.Nucleic Acids Res.2006,34,5383―5394;Ito Y.Nucleic Acids Symp Ser.997,37,259―260;Schoetzau T,Langner J,Moyroud E,Roehl I,Vonhoff S,Klussmann S.Bioconjug Chem.2003,5,919―926;Mehedi Masud M,Ozaki―Nakamura A,Kuwahara M,Ozaki H,Sawai H.Chembiochem.2003,4,584―588;およびEbara Y,Kaihatsu K,Ueji S.Nucleic Acids Symp Ser.1999,42,175―176を参照。
例えば、特定のリーダー配列/変異ポリメラーゼの組み合わせが使用されるいくつかの条件下で、修飾転写産物の収率の増加が観察できる。リーダー配列は、DNA転写テンプレートの5’末端の固定配列の3’末端に導入されうる配列である。リーダー配列は典型的に、6〜15ヌクレオチド長であり、所定のヌクレオチド組成により構成され、例えば全てプリンであるか、プリンおよびピリミジンヌクレオチドの特定の混合物でありうる。
特にY639L/H784A、Y639L/H784A/K378R、P266L/Y639L/H784AまたはP266L/Y639L/H784A/K378Rと組み合わせて、本発明の変異ポリメラーゼおよび転写条件とともに使用されうるテンプレートの例は、ARC2118(配列番号7)、ARC2119(配列番号31)、および以下の実施例に示されるとおりである。これらの変異ポリメラーゼとともに使用されるテンプレートのリーダー配列は、選択的である。
さらに、2’―OH GTPの存在は、修飾ヌクレオチドを導入した転写産物を得る上で歴史的に重要な因子となっている。転写は、二相に分割されうる:第一相は開始であり、その間に、NTPがGTP(または別の置換グアノシン)の3’―末端に加えられて、ジヌクレオチドが得られ、これが約10〜12ヌクレオチド延長される;第二相は伸長であり、その間に、転写が最初の約10〜12ヌクレオチドの添加を越えて進む。Y639F/K378R変異またはY639F/H784A/K378R変異T7RNAポリメラーゼを含む転写混合物に加えられた少量の2’―OH GTPおよび過量の2’OMe GTPが、ポリメラーゼによる2’―OH GTPを用いた転写の開始を可能にするために十分であることが以前に分かった(また、2’―OH GTPを伴わない場合よりも高い、2’―OMeを含む転写産物の転写収率をもたらした)が、転写が伸長相に入ると、2’―OMeおよび2’―OH GTPの間の選別の減少、および2’―OH GTPに対する過量の2’OMe GTPにより、主に2’―OMe GTPの導入が許容される。
本発明は、GTPが開始ヌクレオチドを除いて2’―OMeである転写産物をもたらす、高収率の2’―OMe転写のために転写混合物において2’―OH GTPを必要としない変異T7RNAポリメラーゼ、例えばY639L/H784A、Y639L/H784A/K378R、P266L/Y639L/H7S4AまたはP266L/Y639L/H784A/K378Rを提供する。一実施形態において、高収率は、平均して投入転写テンプレートにつき少なくとも一つの転写産物である。
転写産物への2’―OMe置換ヌクレオチドの導入における別の因子は、転写混合物における二価マグネシウムおよびマンガン(Mn2+)の使用である。塩化マグネシウムおよび塩化マンガンの濃度の異なる組み合わせが、2―’O―メチル化転写産物の収率に影響することがわかっており、塩化マグネシウムおよび塩化マンガンの最良の濃度は、二価金属イオンと複合体を形成する転写反応混合物中のNTPsの濃度に依存する。全て2―’O―メチル化された転写産物(すなわち全2’―OMe ATP、2’―OMe UTP、2’―OMe CTP、および2’―OMe GTPヌクレオチド)の最も高い収率を得るためには、各NTPが0.5mMの濃度で存在するときには、約5mMの濃度の塩化マグネシウムおよび約1.5mMの濃度の塩化マンガンが好ましい。各NTPの濃度が1.0mMであるときには、約6.5mMの濃度の塩化マグネシウムおよび2.0mMの濃度の塩化マンガンが好ましい。各NTPが1.5mMの濃度で存在するときには、約8mMの濃度の塩化マグネシウムおよび3.0mMの濃度の塩化マンガンの濃度が好ましい。各NTPの濃度が2.0mMであるときには、約9.5mMの濃度の塩化マグネシウムおよび3.0mMの濃度の塩化マンガンが好ましい。いずれのケースにおいても、これらの濃度からの最大二倍の逸脱は、なお有意な量の修飾転写産物を与える。
2’―OH GMP、グアノシン、または2’―OH糖位以外の位置で置換された他の2’―OHグアノシンを転写に入れることも、2’―OH GTPを含まない転写混合物に重要である。この効果は、開始ヌクレオチドに対するポリメラーゼの特異性から生じる。その結果、この様式で生成される任意の転写産物の5’―末端ヌクレオチドは、2’―OHグアノシン(2’―OH G)である可能性が高い。GMP(またはグアノシン)の好ましい濃度は0.5mMであり、より好ましくは1mMである。PEG、好ましくはPEG―8000を転写反応物に含むことが、修飾ヌクレオチドの導入を最大化するのに有用であることも分かっている。
転写産物への2’―OMe ATP(100%)、2’―OMe UTP(100%)、2’―OMe CTP(100%)および2’―OMe GTP(100%)(「MNA」)の最大限の導入のために、以下の条件が使用されうる:HEPESバッファー200mM、DTT40mM、スペルミジン2mM、PEG―8000 10%(w/v)、TritonX―100 0.01%(w/v)、MgCl2 8mM、MnCl2 3.0mM、2’―OMe NTP(各)1.5mM、2’―OH GMP 1mM、pH 7.5、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nM、無機ピロホスファターゼ0.025単位/ml、およびDNAテンプレート。いくつかの実施形態においては、DNAテンプレートは、好ましくは約5〜500nMの濃度で存在しうる。
転写産物への2’―OMe ATP(100%)、2’―OMe UTP(100%)または2’―OMe TTP(100%)、2’―OMe GTP(100%)および2’―デオキシCTP(100%)(「dCmD」)の最大限の導入のために、以下の条件が使用されうる:HEPESバッファー200mM、DTT40mM、スペルミジン2mM、PEG―8000 10%(w/v)、TritonX―100 0.01%(w/v)、MgCl2 8mM、MnCl2 3.0mM、2’―OMe NTP(ATP、UTPまたはTTP、GTP)1.5mM、2―デオキシCTP 1.5mM、2’―OH GMP 1mM、pH7.5、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nM、無機ピロホスファターゼ0.025単位/ml、およびDNAテンプレート。いくつかの実施形態においては、DNAテンプレートは、好ましくは約5〜500nMの濃度で存在しうる。
転写産物への2’―OMe ATP(100%)、2’―OMe CTP(100%)、2’―OMe GTP(100%)および2’―デオキシTTP(100%)(「dTmV」)の最大限の導入のために、以下の条件が使用されうる:HEPESバッファー200mM、DTT40mM、スペルミジン2mM、PEG―8000 10%(w/v)、TritonX―100 0.01%(w/v)、MgCl2 8mM、MnCl2 2.5mM、2’―OMe NTP(ATP、CTP、GTP)1.5mM、2―デオキシTTP 1.5mM、2’―OH GMP 1mM、pH7.5、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nM、無機ピロホスファターゼ5単位/ml、およびDNAテンプレート。いくつかの実施形態においては、DNAテンプレートは、好ましくは約5〜500nMの濃度で存在しうる。
転写産物への2’―OMe ATP(100%)、2’―OMe CTP(100%)、2’―OMe GTP(100%)および2’―デオキシUTP(100%)(「dUmV」)の最大限の導入のために、以下の条件が使用されうる:HEPESバッファー200mM、DTT40mM、スペルミジン2mM、PEG―8000 10%(w/v)、TritonX―100 0.01%(w/v)、MgCl2 8mM、MnCl2 3.0mM、2’―OMe NTP(ATP、CTP、GTP)1.5mM、2―デオキシUTP 1.5mM、2’―OH GMP 1mM、pH7.5、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nM、無機ピロホスファターゼ0.025単位/ml、およびDNAテンプレート。いくつかの実施形態においては、DNAテンプレートは、好ましくは約5〜500nMの濃度で存在しうる。
転写産物への2’―OMe ATP(100%)、2’―OMe CTP(100%)、2’―OMe GTP(100%)および2’―OH UTP(100%)(「rUmV」)の最大限の導入のために、以下の条件が使用されうる:HEPESバッファー200mM、DTT40mM、スペルミジン2mM、PEG―8000 10%(w/v)、TritonX―100 0.01%(w/v)、MgCl2 8mM、MnCl2 3.0mM、2’―OMe NTP(ATP、CTP、GTP)1.5mM、2’―OH UTP 1.5mM、2’―OH GMP 1mM、pH7.5、Y639L/H784A/K378R変異T7RNAテンプレート200nM、無機ピロホスファターゼ0.025単位/ml、およびDNAテンプレート。いくつかの実施形態においては、DNAテンプレートは、好ましくは約5〜500nMの濃度で存在しうる。
選択的に、上述の転写条件で使用されるDNAテンプレートは、両方のテンプレートが同一の条件下で転写されたときに、そのようなリーダー配列を含まないテンプレートに対して転写収率を増加させる、全てプリンのリーダー配列を含む。別の実施形態においては、リーダー配列は、両方が同一の条件下で転写されたときに、そのようなリーダー配列を含まないテンプレートに対して転写収率を増加させる、プリンおよびピリミジンの混合物である。本明細書で使用されるところの、一単位の無機ピロホスファターゼは、pH7.2および25℃で一分あたり1.0モルの無機オルトリン酸塩を遊離させる酵素の量として定義される。反応は、約1〜24時間行われうる。
各場合においてその後、転写産物が、アプタマーを同定および/または所与の標的に対する結合特異性を有する保存配列を決定するために、SELEX(商標)プロセスへのインプットに使用されうる。得られる配列はすでに安定化され、SELEX(商標)後修飾プロセスからこのステップが除去され、結果的により安定化されたアプタマーが得られる。
後述するように、2’置換ヌクレオチドを導入した有用な収率の転写産物は、上述の条件以外の条件下で得ることもできる。例えば、上述の転写条件に対する変更には、以下が含まれる。
HEPESバッファーの濃度は、0〜1Mの範囲でありうる。本発明は、例えばトリス―ヒドロキシメチル―アミノメタンを含む、5〜10の間のpKaを有する他のバッファーの使用も予定する。
DTTの濃度は、0〜400mMの範囲でありうる。本発明の方法は、例えばメルカプトエタノールを含む他の還元剤の使用も提供する。
スペルミジンおよび/またはスペルミンの濃度は、0〜20mMの範囲でありうる。
PEG―8000の濃度は、0〜50%(w/v)の範囲でありうる。本発明の方法は、例えば他の分子量のPEGまたは他のポリアルキレングリコールを含む他の親水性ポリマーの使用も提供する。
TritonX―100の濃度は、0〜0.1%(w/v)の範囲でありうる。本発明の方法は、例えば他のTriton―X界面活性剤を含む他の界面活性剤を含む他の非イオン性界面活性剤の使用も提供する。
MgCl2の濃度は、0.5mM〜50mMの範囲でありうる。MnCl2の濃度は、0.15mM〜15mMの範囲でありうる。MgCl2およびMnCl2の両方が記載の範囲内になければならず、好ましい実施形態では、MgCl2:MnCl2が約10対約3の比率で存在し、好ましくは、比率は約3〜5:1、より好ましくは比率は約3〜4:1である。
2’―OMe NTPの濃度(各NTP)は、5μM〜5mMの範囲でありうる。
デオキシNTPの濃度(各NTP)は、5μM〜5mMの範囲でありうる。
2’―OH NTPの濃度(各NTP)は、5μM〜5mMの範囲でありうる。
2’―OH GTPの濃度(2’―OMe GTPが反応混合物中5μM〜5mMの範囲である場合)は、0μM〜300μMの範囲でありうる。好ましい実施形態においては、2’―OH GTPの非存在下(0μM)で転写が生じる。
2’―糖の位置以外の位置で置換された2’―OH GMP、グアノシンまたは他の2’―OH Gの濃度は、0〜5mMの範囲でありうる。2’―OH GTPが反応物に含まれない場合は、2’―OH GMPが必要であり、5μM〜5mMの範囲でありうる。
DNAテンプレートの濃度は、5nM〜5μMの範囲でありうる。
変異ポリメラーゼの濃度は、2nM〜20μMの範囲でありうる。
無機ピロホスファターゼは、0〜100単位/mlの範囲でありうる。
pHは、pH6〜pH9の範囲でありうる。本発明の方法は、修飾ヌクレオチドを導入した大半のポリメラーゼの活性のpHの範囲内で実施されうる。
転写反応は、約一時間〜数週間、好ましくは約1〜約100時間行われうる。
さらに、本発明の方法は、例えばEDTA、EGTA、およびDTTを含めて、転写反応条件におけるキレート剤の選択的使用を提供する。
Aptamer Medicinal Chemistry(アプタマー医薬品化学)
所望の標的に結合するアプタマーが同定されたら、同定されたアプタマー配列の結合および/または機能的特性をさらに増加させるために、いくつかの技術が選択的に実行されうる。SELEX(商標)プロセス(例えば2’―修飾SELEX(商標))により同定される所望の標的に結合するアプタマー、例えばMNA、dCmD、dTmV、rUmV、dUmVアプタマーが、所望の結合および/または機能的特性を有する最小のアプタマー配列(本明細書において「最小化コンストラクト」とも称される)を得るために、選択的に切断されうる。これを達成する一つの方法は、最小化コンストラクトのデザインを知るための、フォールディングプログラムおよび配列分析(例えば保存モチーフおよび/または共変動を探すための、選択から得られるクローン配列のアライニング)の使用による。最小化コンストラクトのデザインを知るために、アプタマー配列の5’および3’境界を決定するために、生化学的プロービング実験も行われうる。その後、最小化コンストラクトが化学合成され、それらが得られた非最小化配列と比較して、結合および機能的特性につき試験されうる。一連の5’、3’および/または内部欠損を含むアプタマー配列の変異体も、直接化学合成され、それらが得られた非最小化アプタマー配列と比較して、結合および/または機能的特性につき試験されうる。
さらに、ドープ再選択を用いて、MNA、dCmD、dTmV、rUmVまたはdUmVアプタマー等の単一の活性アプタマー配列(すなわち、(2’―修飾SELEX(商標)プロセスを含むSELEX(商標)プロセスにより同定される、所望の標的に結合するアプタマー)または単一の最小化アプタマー配列の範囲内の配列要件を調査できる。目的の単一の配列に基づいて設計されている合成の縮重プールを用いて、ドープ再選択が行われる。縮重のレベルは通常、野生型ヌクレオチド、すなわち目的の単一の配列から70%〜85%変動する。一般に、中立変異を伴う配列がドープ再選択プロセスにより同定されるが、場合によっては、配列変化が親和性の改善をもたらしうる。そして、ドープ再選択を用いて同定されるクローンからの合成配列情報を用いて、最小結合モチーフを同定し、医薬品化学研究をたすけうる。
MNA、dCmD、dTmV、rUmVまたはdUmVアプタマー等、(2’―修飾SELEXプロセスおよびドープ再選択含む)SELEX(商標)プロセスを用いて同定されるアプタマー配列および/または最小化アプタマー配列は、配列のランダムまたは定方向突然変異を行って結合親和性および/または機能的特性を高めるため、あるいは配列のどの位置が結合活性および/または機能的特性に必須であるかを決定するために、アプタマー医薬品化学を用いてSELEX(商標)後選択により選択的に修飾されてもよい。
アプタマー医薬品化学は、一連の変異アプタマーが化学合成されるアプタマー改善技術である。これらの一連の変異体は典型的に、単一の置換基の導入により親アプタマーと異なり、この置換基の位置により互いに異なる。そして、これらの変異体が互いと親に対して比較される。特性の改善は十分に大きくなりうるため、特定の治療基準を達成するのに、単一の置換基の包含だけで足りうる。
あるいは、単一の変異体組から収集された情報を用いて、二つ以上の置換基が同時に導入されたさらなる変異体組を設計できる。一つの設計戦略においては、全ての単一置換基の変異体がランク付けされ、トップ4が選択され、これら4つの単一置換基変異体の全ての可能な二重(6)、三重(4)および四重(1)の組み合わせが合成および検定される。第二の設計戦略においては、最良の単一置換基変異体が新しい親と考えられ、この最高ランクの単一置換基変異体を含む全ての可能な二重置換基変異体が合成および検定される。他の戦略が用いられてもよく、さらに改良された変異体の同定を続けながら置換基の数が段階的に増加するように、これらの戦略が繰り返し行われうる。
アプタマー医薬品化学は特に、全体的でなく局所的な置換基の導入を調査する方法として用いることができる。アプタマーが転写により生成されるライブラリの中に発見されるため、SELEX(商標)プロセスの間に導入される任意の置換基は、全体的に導入されなければならない。例えば、ヌクレオチド間にホスホロチオエート結合を導入することが望ましい場合には、全てのA(または全てのG、C、T、U等)に導入することしかできない(全体的に置換される)。ホスホロチオエートがいくつかのA(またはいくつかのG、C、T、U等)(局地的に置換)に必要であるが他のAでは寛容性がないアプタマーは、このプロセスにより容易に発見できない。
アプタマー医薬品化学プロセスにより利用されうる置換基の種類は、それらを固相合成試薬として生成し、オリゴマー合成スキームに導入する能力によってのみ制限される。プロセスは、もちろんヌクレオチドだけに限られない。アプタマー医薬品化学スキームには、立体的大きさ、疎水性、親水性、親油性、疎油性、正電荷、負電荷、中性電荷、双性イオン、分極率、ヌクレアーゼ抵抗性、構造的剛性、構造的柔軟性、タンパク質結合特性、質量等を導入する置換基が含まれうる。アプタマー医薬品化学スキームには、塩基―修飾、糖―修飾またはホスホジエステル結合―修飾が含まれうる。
治療用アプタマーの文脈で有益となりうる置換基の種類を考える際には、以下の一つ以上のカテゴリに入る置換を導入することが望ましい:
(1)すでに体内に存在する置換基、例えば2’―デオキシ、2’―リボ、2’―O―メチルプリンまたはピリミジンまたは5―メチルシトシン。
(2)既に承認された治療薬の一部である置換基、例えばホスホロチオエート―結合オリゴヌクレオチド。
(3)上の二つのカテゴリのうち一つに加水分解または分解する置換基、例えばメチルホスホネート―結合オリゴヌクレオチド。
本発明のアプタマーは、本明細書に記載されるアプタマー医薬品化学により開発されるアプタマーを含む。
本発明のアプタマーの標的結合親和性は、アプタマーと標的(例えばタンパク質)の間の一連の結合反応により評価でき、トレース32P―標識アプタマーが緩衝培地において標的の希釈系とともにインキュベートされ、その後真空濾過マニホルドを用いてニトロセルロース濾過により分析される。本明細書においてドットブロット結合アッセイと称されるこの方法は、(上から下に)ニトロセルロース、ナイロンフィルタ、およびゲルブロット紙からなる三層濾過媒体を使用する。標的に結合したRNAがニトロセルロースフィルタに捕らえられ、標的に結合していないRNAがナイロンフィルタに捕らえられる。ゲルブロット紙は、他のフィルタの支持媒体として含まれる。濾過の後に、濾過層が分離され、乾燥させられて蛍光スクリーンに曝露され、リン光画像化システムを使用して定量される。定量結果を用いてアプタマー結合曲線を生成でき、そこから解離定数(KD)を計算できる。好ましい実施形態においては、結合反応を実行するために用いられる緩衝培地は、1X DulbeccoのPBS(Ca++およびMg++)と0.1mg/mL BSAである。
一般に、標的の機能的活性を調節するアプタマーの能力、すなわちアプタマーの機能的活性は、標的の生物学的機能に応じたin vitroおよびin vivoモデルを使用して評価できる。いくつかの実施形態においては、本発明のアプタマーは、標的の公知の生物学的機能を阻害でき、他の実施形態では、本発明のアプタマーは、標的の公知の生物学的機能を刺激しうる。アプタマー標的の公知の機能を測定するために設計されたin vitroおよびin vivoモデルを使用して、本発明のアプタマーの機能的活性を評価できる。
本発明のアプタマーは、当業者により用いられる任意の数の技術により、診断目的にルーチン的に採用されうる。診断への利用には、in vivoおよびin vitroの両方の診断用途が含まれうる。診断薬は、利用者が特定の場所または濃度での所与の標的の存在を同定することを可能にするだけで足りる。単に標的と結合対を形成する能力が、診断目的でプラスのシグナルを誘発するために十分でありうる。当業者は、そのようなリガンドの存在を追跡するために標識タグを導入するために、公知技術の方法により任意のアプタマーを適応させることもできる。そのようなタグは、多数の診断法において用いられうる。
免疫賦活性モチーフを有するアプタマー
脊椎動物の免疫系による細菌DNAの認識は、特定の配列コンテキストにおける非メチル化CGジヌクレオチドの認識に基づく(「CpGモチーフ」)。このようなモチーフを認識する一つのレセプターは、異なる微生物成分を認識することにより先天性の免疫反応に関与するToll―likeレセプター(〜10メンバー)のファミリーのメンバー、Toll―likeレセプター9(「TLR9」)である。TLR9は、非メチル化オリゴデオキシヌクレオチド(「ODN」)CpG配列を配列特異的様式で結合する。CpGのモチーフの認識が防御機構を誘発し、先天性および最終的に後天性の免疫反応につながる。例えばマウスにおけるTLR9の活性化は、抗原提示細胞の活性化、MHCクラスIおよびII分子のアップレギュレーション、および重要な共刺激分子およびIL―12およびIL―23を含むサイトカインの発現を誘導する。この活性化は、TH1サイトカインIFN―ガンマの強いアップレギュレーションを含めて、直接および間接的にBおよびT細胞反応を増強する。CpG配列に対する反応は、集合的に、感染症からの保護、ワクチンに対する免疫反応の改善、喘息に対する有効な反応、および抗体依存―細胞媒介細胞傷害の改善につながる。したがって、CpG ODNsは、感染症からの保護を提供し、免疫アジュバントまたは癌治療薬(単独療法またはmAbや他の療法と組み合わせて)として機能し、喘息およびアレルギー反応を減少させうる。
本発明のアプタマー、例えばMNA、dCmD、dTmV、rUmVまたはdUmVアプタマーは、一つ以上のCpGまたは他の免疫賦活性配列を含みうる。このようなアプタマーは、例えば本明細書に説明されるSELEX(商標)プロセスを用いて、様々な戦略により同定または生成されうる。一般に、戦略は二つの群に分けられうる。群一においては、戦略は、標的のための結合部位ならびにCpGモチーフまたは他の免疫賦活性配列の両方を含むアプタマーを同定または生成することを目指し、標的(以下「非CpG標的」)は、CpGモチーフまたは他の免疫賦活性配列を認識することが知られており、CpGモチーフに結合するときに免疫反応を刺激することが知られているもの以外の標的である。この群の第一戦略は、CpGモチーフが固定領域またはその一部としてプールの各メンバーに導入されているオリゴヌクレオチドプールを用いて(例えば、いくつかの実施形態においては、プールメンバーのランダム化領域が、CpGモチーフが導入された固定領域を含む)、SELEX(商標)を行って特異的非CpG標的に対するアプタマーを得るステップと、CpGモチーフを含むアプタマーを同定するステップを含む。この群の第二戦略は、SELEX(商標)を行って特異的非CpG標的、好ましくは標的に対するアプタマーを得、選択の後CpGモチーフを5’および/または3’末端に加えるかCpGモチーフをアプタマーの領域、好ましくは非必須領域に組み込むステップを含む。この群の第三の戦略は、SELEX(商標)を行って特異的非CpG標的に対するアプタマーを得るステップであり、プールの合成の間に、プールの各メンバーのランダム化領域のCpGモチーフが濃縮されるように、一つ以上のヌクレオチド添加ステップにおいて様々なヌクレオチドのモル比が偏らせられる、ステップと、CpGモチーフを含むアプタマーを同定するステップを含む。この群の第四の戦略は、SELEX(商標)を行って特異的非CpG標的に対するアプタマーを得るステップと、CpGモチーフを含むアプタマーを同定するステップを含む。この群の第五の戦略は、SELEX(商標)を行って特異的非CpG標的に対するアプタマーを得るステップと、結合時に免疫反応を刺激するがCpGモチーフを含まないアプタマーを同定するステップを含む。
群二においては、戦略は、CpGモチーフのレセプター(例えばTLR9または他のtoll―likeレセプター)により結合され、結合時に免疫反応を刺激する、CpGモチーフおよび/または他の配列を含むアプタマーの同定または生成を目指す。この群の第一戦略は、CpGモチーフがプールの各メンバーに、固定領域またはその一部として導入されているオリゴヌクレオチドプールを用いて(例えばいくつかの実施形態においては、プールメンバーのランダム化領域は、CpGモチーフが導入された固定領域を含む)、SELEX(商標)を行ってCpGモチーフまたは他の免疫賦活性配列に結合し、結合時に免疫反応を刺激することが知られている標的に対するアプタマーを得るステップと、CpGモチーフを含むアプタマーを同定するステップを含む。この群の第二戦略は、SELEX(商標)を行って、CpGモチーフまたは他の免疫賦活性配列に結合し、結合時に免疫反応を刺激することが知られている標的に対するアプタマーを得てから、CpGモチーフを5’および/または3’末端に加えるかCpGモチーフをアプタマーの領域、好ましくは非必須領域に組み込むステップを含む。この群の第三の戦略は、SELEX(商標)を行って、CpGモチーフまたは他の免疫賦活性配列に結合し、結合時に免疫反応を刺激することが分かっている標的に対するアプタマーを得るステップであり、プールの合成の間に、プールの各メンバーのランダム化された領域がCpGモチーフにおいて濃縮されるように、一つ以上のヌクレオチド添加ステップにおいて様々なヌクレオチドのモル比が偏らせられるステップと、CpGモチーフを含むアプタマーを同定するステップを含む。この群の第四の戦略は、SELEX(商標)を行って、CpGモチーフまたは他の免疫賦活性配列に結合し、結合時に免疫反応を刺激することが知られている標的に対するアプタマーを得るステップと、CpGモチーフを含むアプタマーを同定するステップを含む。この群の第五の戦略は、SELEX(商標)を行って、CpGモチーフまたは他の免疫賦活性配列に結合することが知られている標的に対するアプタマーを得るステップと、結合時に免疫反応を刺激するがCpGモチーフを含まないアプタマーを同定するステップを含む。
各々が異なるイベントのカスケードにおける認識、サイトカインおよび他の分子の放出、一定の細胞型の活性化により生じる、多様なCpGモチーフのクラスが同定されている。例えば、参照により本明細書に組み込まれる、CpG Motifs in Bacterial DNA and Their Immune Effects,Annu.Rev.Immunol.2002,20:709―760を参照。追加的な免疫賦活性モチーフは、参照により各々が本明細書に組み込まれる以下の米国特許において開示される:米国特許第6,207,646号;第6,239,116号;第6,429,199号;第6,214,806号;第6,653,292;第6,426,434号;第6,514,948号および第6,498,148号。これらのCpGまたは他の免疫賦活性モチーフが、アプタマーに導入されうる。アプタマーの選択は、治療される病気または疾患に依存する。好ましい免疫賦活性モチーフは以下の通りであり(左から右に5’から3’で示される)、「r」がプリンを表わし、「y」はピリミジンを表わし、「X」は任意のヌクレオチドを表わす:
,rCGy;rrCGyy,XCGX,XXCGXX,およびX
1がGまたはAであり、X
2がCでなく、Y
1がGでなく、Y
2が好ましくはTであるX
1X
2CGY
1Y
2。
CpGモチーフに結合し、結合時に免疫反応を刺激することが知られている標的以外の特異的標的(「非CpG標的」)に結合するアプタマーにCpGモチーフが導入される場合においては、CpGは、アプタマーの非必須領域に位置するのが好ましい。アプタマーの非必須領域は、部位特異的変異誘発、欠失分析および/または置換分析により同定されうる。しかし、非CpG標的に結合するアプタマーの能力を大きく妨げない任意の位置が使用されうる。CpGモチーフは、アプタマー配列に埋め込まれるのに加えて、5’および3’末端のいずれかまたは両方に加えられるなど、アプタマーに付着されうる。非CpG標的に結合するアプタマーの能力が大きく妨げられない限り、任意の付着の位置または手段が使用されうる。
本明細書で使用されるところの、「免疫反応の刺激」は、(1)特異的反応(例えばTh1反応の誘発)または一定の分子の産生の誘発、または(2)特異的反応(例えばTh2反応の阻害または抑制)または一定の分子の阻害または抑制を意味しうる。
アプタマー治療薬の薬物動態および体内分布の調節
アプタマーを含む全てのオリゴヌクレオチドベースの治療薬の薬物動態学的特性が、所望の薬学的用途に適合するように調整されることが重要である。細胞外標的に対するアプタマーには(アンチセンスおよびRNAiベースの治療薬の場合のように)細胞内送達と関連した問題がないが、このようなアプタマーはなお、標的器官および組織に分布され、所望の薬剤投与計画と整合した一定期間体内にとどまることが可能でなければならない。
したがって、本発明は、アプタマー組成物の薬物動態、および、特にアプタマー薬物動態を調整する能力に影響を及ぼすための、材料および方法を提供する。アプタマー薬物動態の調整可能性(すなわち調節の可能性)は、アプタマーに対する修飾部分(例えばPEGポリマー)の結合および/または修飾ヌクレオチド(例えば2’―フルオロまたは2’―O―メチル)の導入により核酸の化学組成を変更することにより達成される。アプタマー薬物動態の調整可能性は、既存の治療的応用の改善、あるいは新しい治療用途の開発において用いられる。例えば、迅速な薬物クリアランスまたはターンオフが求められうる抗腫瘍または救急処置の場合等、いくつかの治療用途においては、循環中のアプタマーの滞留時間を減少させることが望ましい。あるいは、他の治療用途、例えば治療薬の全身循環が求められる維持療法においては、循環中のアプタマーの滞留時間を増加させることが望まれうる。
さらに、アプタマー薬物動態の調整可能性は、対象におけるアプタマー治療薬の生体分布を改変するために用いられる。例えば、いくつかの治療用途においては、特定のタイプの組織または特定の臓器(または一連の臓器)を標的とする努力においてアプタマー治療薬の生体分布を変更することが望まれうる。これらの用途においては、アプタマー治療薬は、優先的に特定の組織または臓器(単数または複数)に蓄積する。他の治療用途においては、アプタマー治療薬が患部組織に優先的に蓄積するように、所与の病気、細胞傷害または他の異常な症状と関連した細胞マーカまたは症状を示す組織を標的とすることが望まれうる。例えば、2004年3月5日に出願の、“Controlled Modulation of the Pharmacokinetics and Biodistribution of Aptamer Therapeutics”と題した米国特許仮出願第60/550790号、および2005年3月7日に出願の、“Controlled Modulation of the Pharmacokinetics and Biodistribution of Aptamer Therapeutics”と題した米国特許出願第10/―――,―――号に記載されるように、アプタマー治療薬のPEG化(例えば20kDaのPEGポリマーによるPEG化)が、PEG化されたアプタマー治療薬が炎症組織に優先的に蓄積するように、炎症組織の標的化に使用される。
アプタマー治療薬(例えばアプタマー結合体または修飾ヌクレオチド等、化学的に変更されたアプタマー)の薬物動態学的および生体分布プロフィールを決定するために、様々なパラメータがモニタされる。このようなパラメータには、例えば半減期(t1/2)、血漿クリアランス(Cl)、分布体積(Vss)、濃度―時間曲線下面積(AUC)、血清または血しょう中濃度の最大値(Cmax)の観察、およびアプタマー組成物の平均滞留時間(MRT)が含まれる。本明細書において使用されるところの「AUC」という用語は、アプタマー投与後の時間に対するアプタマー治療薬の血しょう中濃度のプロット下の面積を指す。AUC値を用いて、所与のアプタマー治療薬の生物学的利用能(すなわちアプタマー投与後の循環中の投与アプタマー治療薬の割合)および/または総クリアランス(Cl)(すなわちアプタマー治療薬が循環から除去される速度)が推定される。分布体積は、アプタマー治療薬の血しょう中濃度を体内に存在するアプタマーの量に関連させる。Vssが大きいほど、アプタマーが血漿外でより多く見られる(すなわち血管外遊出が多い)。
本発明は、アプタマー、例えばMNA、dCmD、dTmV、rUmVまたはdUmVアプタマーを小分子、ペプチドまたはポリマー末端基等の調節部分に結合することにより、または修飾ヌクレオチドをアプタマーに導入することにより、in vivoで、安定化されたアプタマー組成物、例えばMNA、dCmD、dTmV、rUmVまたはdUmVアプタマーの薬物動態および生体分布を、制御された様式で調節する材料および方法を提供する。本明細書に記載されるように、修飾部分の結合および/またはヌクレオチド(単数または複数)化学組成の変更は、循環におけるアプタマー滞留時間および組織への分布の基本的側面を変更する。
ヌクレアーゼによるクリアランスに加えて、オリゴヌクレオチド治療薬は、腎臓の濾過により除去される。そのようなものとして、静脈内投与されるヌクレアーゼ耐性オリゴヌクレオチドは、濾過を遮断できない限り、典型的に<10分のin vivo半減期をもつ。これは、血流から組織への迅速分布を促進すること、またはオリゴヌクレオチドの見掛けの分子量を糸球体の有効なカットオフサイズより上に増加させることにより達成できる。後述されるPEGポリマーに対する小さな治療薬の結合(PEG化)は、循環中のアプタマーの滞留時間を劇的に延長し、これにより投与頻度を減少させ、血管標的に対する有効性を高めうる。
アプタマーは、高分子量ポリマー、例えばPEG;ペプチド、例えばTat(HIV Tatタンパク質の13―アミノ酸フラグメント(Vives等(1997),J.Biol.Chem.272(25):16010―7))、Ant(ショウジョウバエアンテナペディアホメオチックタンパク質の第三のらせんに由来する16―アミノ酸配列(Pietersz等(2001),Vaccine 19(11―12):1397―405))およびArg7(ポリアルギニン(Arg7)で構成される短い正に帯電する細胞浸透ペプチド(Rothbard等(2000),Nat.Med.6(11):1253―7;Rothbard,J等(2002)、J.Med.Chem.45(17):3612―8));および小分子、例えばコレステロール等の脂肪親和性化合物等、様々な修飾部分に結合されうる。本明細書に説明される様々な結合体の中で、アプタマーのin vivo特性は、PEG基との錯体形成により最も大きく変更される。例えば、20kDaのPEGポリマーとの、混合2’Fおよび2’―OMe修飾アプタマー治療薬の錯体化は、腎臓濾過を妨げ、健常および炎症組織へのアプタマー分布を促進する。さらに、20kDaのPEGポリマー―アプタマー結合体は、アプタマーの腎臓濾過を防止する上で、40kDaのPEGポリマーとほぼ同じくらい有効である。PEG化の一つの効果はアプタマークリアランスにあるが、20kDa部分の存在により生じる全身的曝露の延長により、組織、特に高灌流臓器および炎症部位の組織へのアプタマーの分布も促進される。アプタマー―20kDaPEGポリマー結合体は、PEG化されたアプタマーが炎症組織に優先的に蓄積するように、アプタマー分布を炎症部位に導く。いくつかの場合には、20kDaのPEG化アプタマー結合体は、例えば腎臓細胞等の細胞の内部にアクセスできる。
修飾ヌクレオチドを用いて、アプタマーの血漿クリアランスを調節することもできる。例えば、in vivoおよびin vitroで高いヌクレアーゼ安定性をもつために現世代のアプタマーに典型的である、2’―Fおよび2’―OMe安定化化学を導入する非結合アプタマーは、非修飾アプタマーと比較して、血漿からの迅速な減少(すなわち迅速な血漿クリアランス)、および組織、主に腎臓への迅速な分布を示す。
PEG誘導体化核酸
上述の通り、高分子量の非免疫原性ポリマーによる核酸の誘導体化は、核酸の薬物動態学的および薬力学的特性を変更し、それらをより有効な治療剤とする可能性を有する。活性の好ましい変化には、ヌクレアーゼによる分解に対する抵抗の増加、腎臓による濾過の減少、免疫系に対する曝露の減少、および治療薬の体内分布の変更を含みうる。
本発明のアプタマー組成物は、ポリアルキレングリコール(「PAG」)部分により誘導体化されうる。PAG―誘導体化核酸の例は、参照により全体として本明細書に組み込まれる、2003年11月21日に出願の米国特許出願第10/718,833号に見られる。本発明で使用される典型的なポリマーには、ポリエチレンオキシド(「PEO」)としても知られるポリエチレングリコール(「PEG」)およびポリプロピレングリコール(ポリイソプロピレングリコールを含む)が含まれる。さらに、異なるアルキレンオキシド(例えばエチレンオキシドおよびプロピレンオキシド)のランダムまたはブロックコポリマーが、多くの用途において使用されうる。その最も一般的な形において、PEG等のポリアルキレングリコールは、各末端でヒドロキシル基で終了する直鎖状ポリマーである:HO―CH2CH2O―(CH2CH2O)n―CH2CH2―OH。このポリマー、アルファ―、オメガ―ジヒドロキシルポリエチレングリコールは、HO―PEG―OHとしても表わすことができ、―PEG―記号は以下の構造単位を表すと理解され:―CH2CH2O―(CH2CH2O)n―CH2CH2―、nが典型的に約4〜約10,000の範囲である。
示されるように、PEG分子は二機能であり、「PEGジオール」と呼ばれることもある。PEG分子の終末部は、化合物の反応部位における他の化合物に対するPEGの付着のために、活性化または機能的部分に転換されうる、比較的非反応性のヒドロキシ部分、―OH基である。このような活性化されたPEGジオールは、本明細書において二活性化されたPEGsと称される。例えば、PEGジオールの末端部分は、アミノ部分との選択反応のために、比較的非反応性のヒドロキシ部分、―OHを、N―ヒドロキシスクシンイミドからのスクシンイミジル活性エステル部分と置換することにより、活性カルボネートエステルとして官能化されている。
多くの用途においては、PEG分子が単官能性(または単活性化)であるように、一端のPEG分子を本質的に非反応性の部分でキャップすることが望ましい。一般に活性化PEGsのための複数の反応位置をもつタンパク質治療薬の場合には、二官能性の活性化PEGsは広範な架橋に至り、機能性が低い凝集物を生成する。単活性化PEGsを生成するために、PEGジオール分子の末端上の一つのヒドロキシ部分が、典型的に非反応性メトキシ末端部分、―OCH3と置換される。PEG分子の他方のキャップされていない末端は、典型的に、表面またはタンパク質等の分子上の反応部位における付着のために活性化されうる反応性末端部分に転換される。
PAGsは、水および多くの有機溶媒中における溶解性、無毒性、および非免疫原性の性質を典型的に有するポリマーである。PAGsの一つの使用は、ポリマーを不溶性分子に共有結合させて、得られるPAG―分子「結合体」を可溶性にすることである。例えば、水不溶性薬物パクリタキセルは、PEGに連結されると水溶性になることが示されている。Greenwald等、J.Org.Chem.,60:331―336(1995)。PAG結合体は、溶解性および安定性を高めるためだけでなく、分子の血液循環半減期を延長するためにも用いられることが多い。
本発明のポリアルキル化化合物は、典型的には5〜80kDaのサイズであるが、任意のサイズを使用でき、その選択はアプタマーおよび用途に依存する。本発明の他のPAG化合物は、10〜80kDaのサイズである。本発明のさらに他のPAG化合物は、10〜60kDaのサイズである。例えば、PAGポリマーは、少なくとも10、20、30、40、50、60、または80kDaのサイズでありうる。このようなポリマーは直鎖状または分枝ポリマーでありうる。いくつかの実施形態においては、ポリマーは、PEGである。
生物学的に発現されたタンパク質治療薬に対して、核酸治療薬は典型的に、活性化されたモノマーヌクレオチドから化学合成される。同じ反復的モノマー合成を用いてPEGを導入することにより、PEG―核酸結合体が調製されうる。例えば、ホスホラミダイト形態への変換により活性化されたPEGsが、固相オリゴヌクレオチド合成に導入されうる。あるいは、オリゴヌクレオチド合成は、反応性PEG付着部位の部位特異的導入により完了されうる。最も一般的には、これは、5’―末端における遊離一級アミンの添加により達成されている(固相合成の最後のカップリングステップにおいて修飾因子ホスホラミダイトを用いて導入される)。このアプローチを用いて、反応性PEG(例えば、反応してアミンと結合を形成するように活性化されたもの)が精製オリゴヌクレオチドと組み合わせられ、溶液中で共役反応が行われる。
治療薬の生体分布を変更するPEG結合の能力は、結合体の見掛け上のサイズ(例えば流体力学的半径に関して測定される)を含む、多数のファクターに関連する。より大きな結合体(>10kDa)は、腎臓を介した濾過をより効果的に遮断し、その結果小さな高分子(例えばペプチド、アンチセンスオリゴヌクレオチド)の血中半減期を増加させることが知られている。濾過を遮断するPEG結合体の能力は、約50kDaまでのPEGのサイズとともに増加することが分かっている(半減期が腎臓による除去よりマクロファージ媒介性代謝により定められるため、さらなる増加による有益な効果は最小限である)。
高分子量のPEGs(>10kDa)の産生は困難で、非効率的で高価でありうる。高分子量PEG―核酸結合体の合成へのルートとして、過去の研究は、より高分子量の活性化PEGsの生成に集中している。このような分子を生成する一方法は、活性化された基を担持する中心コアに二つ以上のPEGsが付着する、分枝した活性化PEGの形成を伴う。これらの高分子量のPEG分子の末端部、すなわち比較的非反応性のヒドロキシル(―OH)部分は、他の化合物に対する化合物上の反応部位における一つ以上のPEGsの付着のために、活性化または機能的部分に転換されうる。分枝活性化PEGsは、二つ以上の末端を有し、二つ以上の末端が活性化されている場合においては、このような活性化された高分子量PEG分子は、本明細書において多活性化PEGsと呼ばれる。いくつかの場合には、分岐PEG分子の全ての末端が活性化されるわけではない。分岐PEG分子の任意の二つの末端が活性化される場合においては、このようなPEG分子は、二活性化PEGsと呼ばれる。分岐PEG分子の一つの末端だけが活性化されるいくつかの場合においては、このようなPEG分子は単活性化と呼ばれる。このアプローチの例として、反応のためにその後活性化される、リシンコアに対する二つのモノメトキシPEGsの付着により調製される、活性化PEGが説明されている(Harris等、Nature,vol.2:214―221,2003)。
本発明は、多重PEG化核酸を含む高分子量PEG―核酸(好ましくは、アプタマー)結合体の合成に、別の費用効果的ルートを提供する。本発明は、PEG―連結多量体オリゴヌクレオチド、例えば二量体化アプタマーも含む。本発明は、PEG安定化部分が、例えばアプタマーの異なる部分を隔てるリンカーである、例えば高分子量アプタマー組成物の線形配置が、例えば、nが1以上である核酸―PEG―核酸(―PEG―核酸)nであるように、PEGが単一のアプタマー配列中に結合されている、高分子量組成物にも関する。
本発明の高分子量組成物は、少なくとも10kDaの分子量を有するものを含む。組成物は典型的に、10〜80kDaのサイズの分子量を有する。本発明の高分子量組成物は、少なくとも10、20、30、40、50、60、または80kDaのサイズである。
安定化部分は、本発明の高分子量アプタマー組成物の薬物動態学的および薬力学的特性を改善する分子または分子の部分である。いくつかの場合においては、安定化部分は、二つ以上のアプタマーまたはアプタマードメインを近づけるか、本発明の高分子量アプタマー組成物の全体的な回転自由度の減少を提供する、分子または分子の部分である。安定化部分は、ポリアルキレングリコール、線形または分枝、ホモポリマーまたはヘテロポリマーでありうるようなポリエチレングリコールでありうる。他の安定化部分には、ペプチド核酸(PNA)等のポリマーが含まれる。オリゴヌクレオチドも安定化部分になりうる;このようなオリゴヌクレオチドには、修飾ヌクレオチド、および/またはホスホロチオエート等の修飾結合を含みうる。安定化部分は、アプタマー組成物の一体的部分、すなわちアプタマーに共有結合された部分でありうる。
本発明の組成物には、二つ以上の核酸部分が少なくとも一つのポリアルキレングリコール部分に共有結合される、高分子量アプタマー組成物を含む。ポリアルキレングリコール部分は、安定化部分として働く。ポリアルキレングリコールが核酸部分を一つの分子において結合するように、ポリアルキレングリコールがアプタマーのいずれかの末端で共有結合された組成物においては、ポリアルキレングリコール部分は連結部分であると言われる。このような組成物においては、共有結合分子の一次構造には、線形配置の核酸―PAG―核酸が含まれる。一つの例は、核酸―PEG―核酸の一次構造を有する組成物である。別の例は、線形配置:核酸―PEG―核酸―PEG―核酸である。
核酸―PEG―核酸結合体を生成するために、核酸が、単一の反応部位を持つように(例えば単活性化)最初に合成される。好ましい実施形態においては、この反応部位は、オリゴヌクレオチドの固相合成の最終ステップにおける修飾因子ホスホラミダイトの添加により5’―末端に導入されるアミノ基である。修飾オリゴヌクレオチドの脱保護および精製の後、それは、活性化PEGの自発的加水分解を最小化する溶液において高濃度で再構築される。好ましい実施形態においては、オリゴヌクレオチドの濃度は1mMであり、再構築された溶液はpH8.3の200mM NaHCO3―バッファー、を含む。結合体の合成は、高度に精製された二官能性PEGのゆっくりとした段階的添加により開始される。好ましい実施形態においては、PEGジオールが、スクシンイミジルプロピオン酸による誘導体化により、両末端で活性化(二活性化)される。反応の後、完全―、部分―、および非―結合種を分離するために、PEG―核酸結合体が、ゲル電気泳動または液体クロマトグラフィにより精製される。(例えばランダムまたはブロックコポリマーとして)連結された複数のPAG分子または小さなPAG鎖が連結されて、様々な長さ(または分子量)が達成されうる。様々な長さのPAG鎖の間に、非PAGリンカーが使用されうる。
2’―O―メチル、2’―フルオロおよび他の修飾ヌクレオチド修飾は、ヌクレアーゼに対してアプタマーを安定化し、そのin vivo半減期を増加させる。3’―3’―dTキャップも、エキソヌクレアーゼ抵抗を増加させる。例えば、参照により各々が全体として本明細書に組み込まれる、米国特許第5,674,685号;第5,668,264号;第6,207,816号;および第6,229,002号を参照。
反応性核酸のPAG―誘導体化
高分子量PAG―核酸―PAG結合体は、二つ以上の反応部位を含む核酸と単官能性活性化PEGの反応により調製されうる。一実施形態においては、核酸は二反応性または二活性化され、二つの反応部位:従来のホスホラミダイト合成によりオリゴヌクレオチドに導入された5’―アミノ基および3’―アミノ基、例えば図2に示されるような3’―5’―ジ―PEG化を含む。代替的実施形態では、例えばピリミジンの5―位、プリンの8―位またはリボースの2’―位を一級アミンの付着のための部位として用いて、内部の位置に反応部位が導入されうる。このような実施形態においては、核酸がいくつかの活性化または反応性部位を有することができ、多重活性化されていると言われる。合成および精製の後、修飾オリゴヌクレオチドは、自発的加水分解を最小化しながら、オリゴヌクレオチド反応部位との選択反応を促進する条件下で、単活性化PEGと組み合わせられる。好ましい実施形態においては、モノメトキシ―PEGがスクシンイミジルプロピオン酸で活性化され、pH8.3で共役反応が行われる。二置換PEGの合成を誘発するために、オリゴヌクレオチドに対して化学量論的過剰のPEGが提供される。反応の後、完全、部分的、および非結合種を分離するために、PEG―核酸結合体がゲル電気泳動または液体クロマトグラフィにより精製される。
連結ドメインは、それに付着した一つ以上のポリアルキレングリコール部分も有しうる。このようなPAGsは、様々な長さであり得、組成物の所望の分子量を達成するために、適切な組み合わせにおいて用いられうる。
特定のリンカーの効果は、その化学組成および長さの両方により影響されうる。長すぎる、短すぎる、または標的と好適でない立体化学的および/またはイオン相互作用を形成するリンカーは、アプタマーと標的の間の複合体形成を妨げる。核酸の間の距離をつなぐのに必要であるより長いリンカーは、リガンドの有効濃度を減ずることにより、結合安定性を減じうる。したがって、標的に対するアプタマーの親和性を最大化するために、リンカー組成物および長さを最適化することが必要である場合が多い。
本明細書にあげられる全ての刊行物および特許文献は、そのような刊行物および文献の各々が特に個々に参照により本明細書に組み込まれるものと示されるのと同様に、参照により本明細書に組み込まれる。刊行物および特許文献の引用は、いずれかが関連する従来技術であるとの承認を意図するものでなく、内容または同の日付に関するいかなる承認も構成しない。以上本発明を記述してきたが、当業者には当然のことながら、本発明は様々な実施形態において実施でき、以上の記載および以下の実施例は説明を目的としたものであり、後続の請求の範囲を制限するものではない。
(実施例1)ポリメラーゼ発現および精製
本発明の方法に用いられる変異T7RNAポリメラーゼは、以下のように調製されうる。T7RNAポリメラーゼ(図3Aおよび3Bにそれぞれ示され、Bull,J.J等、J.Mol.Evol.,57(3),241―248(2003)に記載される核酸およびアミノ酸配列を変異させて、LA変異体(Y639L/H784A)、LAR変異体(Y639L/H784A/K378R)、LLA変異体(P266L/Y639L/H784A)またはLLAR変異体P266L/Y639L/H784A/K378R)を生じうる。T7RNAポリメラーゼが、発現ベクター(T7RNAポリメラーゼ発現ベクターの例が、参照により全体として本明細書組み込まれるに米国特許出願第5,869,320号に記載される)に含まれ、または変異誘発後に発現ベクターに挿入されうる。変異T7RNAポリメラーゼは、タンパク質精製の間の容易性のためにHis―tagを選択的に含むために、操作されうる。
639位のロイシンの変異を含む相補的オリゴヌクレオチド配列
が、合成されうる。P266L変異のための相補的オリゴヌクレオチド配列
が、合成されうる。H784A変異のための相補的オリゴヌクレオチド配列
が、合成されうる。K378R変異のための相補的オリゴヌクレオチド配列
が、合成されうる。部位特異的変異誘発が、QuikChange(登録商標)Site―Directed Mutagenesis Kit(Stratagene、カリフォルニア州ラホーヤ)を用いて、製造者の指示に従って実行されて、上述の変異の組み合わせをもつ変異ポリメラーゼをコードする核酸配列(図4)を生じうる。本発明の変異ポリメラーゼをコードする得られた核酸配列が、発現および精製のための標準的な技術を用いて、所望の発現ベクターに挿入されうる。
発現および精製
変異T7ポリメラーゼ核酸配列を含む発現ベクターは、BL21(DE3)コンピテント細胞(Stratagene、カリフォルニア州)に形質転換され、20分間氷上でインキュベートされる。チューブを2分間42℃に入れることにより、熱ショックが行われる。チューブを1分間氷に置いた後、1mlのLブロス(「LB」)が加えられ、37℃のシェーカで45分間インキュベートされる。100ulの培養液が、LB+Amp寒天プレートにプレートされ、一晩37℃でインキュベートされる。
一晩培養されたプレートからの単一のコロニーが、100mlのLB―Amp+(150ug/ml)に37℃で一晩植えつけられる。2日目に、2リットルの予熱されたLB+Ampを含む二つの4リットルフラスコに、50mlの一晩培養物が植えつけられ、OD600が0.6〜0.8の間に達するまで37℃で成長させられる。200ulの1M IPTGが各2Lの細胞培養に100uMの最終濃度で加えられ、37℃でさらに3時間成長させられる。10分間の5000rpmでのスピニングにより、細胞がペレット化される。細胞は、200mlの溶解バッファーの中に再懸濁され(溶解バッファー:50mM Tris―Cl、pH8.0、100mM NaCl、5%グリセロール、1mMイミダゾール、ベータメルカプトエタノール(「BME」)5mM)、6つの50ml円錐チューブに分割される。各チューブにつき、細胞がパワーレベル8、3×30’’で超音波破壊され、60分間11,000rpmで細菌細片が沈降され、上清が0.22uMフィルタで濾過される。イミダゾールが、10mMの最終濃度でフィルタに加えられる。
濾過液は、サンプルポンプで5mlのNi―NTAカラム(GE Healthcare Bio―Sciences、ニュージャージー州)にロードされる。カラムが、20mMのイミダゾールを含む10カラム体積(CV)のバッファーAで洗浄される(バッファーA:50mM Tris―Cl、pH8.0、100mM NaCl、5%グリセロール、10mMイミダゾール、BME10mM)。その後、カラムは、バッファーA中40mM〜70mMのイミダゾールの直線濃度勾配を伴う10CVのバッファーで洗浄される。タンパク質が、6CVのバッファーBにより溶出される(バッファーB:50mM Tris―Cl、pH8.0、100mM NaCl、5%グリセロール、250mMイミダゾール、BME10mM)。収集画分を4―12%のSDS―PAGE上で5μlの試料と照合した後、全ての目的の画分が合わせられ、1Lの透析バッファー中(透析バッファー:50mM Tris―Cl、pH7.9、100mM NaCl、50%グリセロール、0.1mM EDTA、0.1%TritonX―100、BME20mM)で一晩透析される(透析管:Spectrum Spectra/por Molecular多孔膜(VWR)MWCO:12―14000)。透析バッファーは12時間後に交換され、透析はさらに4時間行われる。T7RNAポリメラーゼの濃度が、Bradford,M.M.(1976)Anal.Biochem.72,248に記載されるBradfordアッセイを用いて測定される。
(実施例2)100%2’―O―メチルヌクレオチドを導入する転写
実施例2A:2’―OH GTPを伴わない2’―O―メチル転写
2’―OH GTPの滴定を用いて、2’―OH GTPの濃度に対するY639L/H784A/K378R変異ポリメラーゼの感受性をテストするために、実験が行われた。
Y639L/H784A/K378R変異T7RNAポリメラーゼとともに使用されたときに高い転写収率を示した二つのライブラリ、ARC2118およびARC2119を用いて、2’―OH GTPの濃度に対するY639L/H784A/K378R変異T7RNAポリメラーゼの転写の感受性がテストされた。ARC2118の配列が以下の実施例2Cに与えられ、ARC2119の配列がすぐ下に、5’から3’方向に与えられる。
1X転写バッファー(HEPES200mM、DTT40mM、スペルミジン2mM、TritonX―100 0.01%w/v)、〜200nMテンプレート、2’―OMe ATP、CTP、UTP、およびGTP各1mM、MgCl
26.5mM、MnCl
22.0mM、PEG―8000w/v 10%、GMP1mM、無機ピロホスファターゼ5単位/mL、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nMと、2’―OH GTP(0〜160uM)の滴定を用いて、一晩37℃で転写が行われた。
各条件下の転写産物が、200uLの反応混合物を用いてPAGE―ゲル分析によりアッセイされ、ImageQuant5.2版ソフトウェア(Molecular Dynamics)を用いて、PAGE―ゲル分析のUV―シャドウイングから、各条件の転写産物が定量された。図5は、PAGE―ゲル分析の定量結果をまとめ、バックグラウンドに対する各条件による転写収率の増加倍率を示す。図5にみられるように、2’―OH GTPを含まない全ての条件下でY639L/H784A/K378Rにより転写されるARC2118およびARC2119、および2’―OH GTPの非存在下での収率は、反応混合物に2’―OH GTPが含まれる場合の転写収率と同等だった。これらの結果は、転写のために2’―OH GTPを必要とするY639F/H784A/K378R変異T7RNAポリメラーゼ(データは示されない)と対照的に、Y639L/H784A/K378R変異T7RNAポリメラーゼが、転写収率増加のために2’―OH GTPの存在を必要としないことを示す。
その後、Y639L/H784A/K378R変異T7RNAポリメラーゼで使用される最適な転写条件を決定するために実験が行われた。2’―OMe NTP、マグネシウムおよびマンガン濃度の変更の転写収率への影響をテストするために、ARC2119が、Y639L/H784A/K378R変異T7RNAポリメラーゼとともに用いられた。
1X転写バッファー(HEPES200mM、DTT40mM、スペルミジン2mM、TritonX―100 0.01%)、〜200nMテンプレート、2’―OMe ATP、CTP、UTP、およびGTP(それぞれ0.5mM、1mM、1.5mM、および2mM)、MgCl2(5mM、6.5mM、8mM、および9.5mM)、MnCl2(1.5mM、2mM、2.5mM、3mM)、PEG―8000w/v 10%、GMP1mM、無機ピロホスファターゼ5単位/mL、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nMを用いて、一晩37℃で転写が行われた。
各条件下の転写産物が、200uLの反応混合物を用いてPAGE―ゲル分析によりアッセイされ、各条件の転写産物がImageQuant5.2版ソフトウェア(Molecular Dynamics)を用いて、PAGE―ゲル分析のUV―シャドウイングから定量された。図6は、PAGE―ゲル分析の定量結果をまとめ、バックグラウンドに対する各条件による転写収率の増加倍率を示す。2’―OMe NTPsのコストおよびこの実験の結果に基づき、各1.5mMの2’―OMe NTP(および8mM MgCl2、2.5mM MnCl2)が、ARC2119および本発明のY639L/H784A/K378R変異T7RNAポリメラーゼのための好ましい条件として採用された。
実施例2B:Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたMNA転写の忠実度およびバイアス:
Y639L/H784A/K378R変異T7RNAポリメラーゼを用い、2’―OH GTPを用いないMNA転写の忠実度およびバイアスを評価するために、追加的な実験が行われた。忠実度をテストするために、単一のクローン配列がPCRにより増幅され、Y639L/H784A/K378Rポリメラーゼを用いた2’―OH GTPを伴わないMNA転写をプログラムするために使用され、PAGEにより精製され、RQl DNアーゼを用いて残りのDNAテンプレートが消化され(そしてDNAテンプレートの非存在がPCRによりアッセイされ)、転写物が逆転写され(Thermoscript、Invitrogen、カリフォルニア州カールズバッド)、PCRにより増幅された。このPCR産物が配列決定された後、欠失、挿入および置換の数が計算された。この実験で配列決定された1300の塩基のうちに、欠失および挿入は観察されず、三つの置換が観察された(図7を参照)。これらの数は、30―ヌクレオチド縮重領域内にコードされる配列情報がSELEX(商標)の次のラウンドに忠実に伝達されるチャンスが、93%であることを示唆し、この数は非常に高いため、野生型RNAにつき測定される数を上回る。
ヌクレオチドのバイアスをテストするために、ライブラリARC2118が、以下の条件下:HEPES200mM、DTT40mM、スペルミジン2mM、TritonX―100 0.01%w/v、〜200nMテンプレート、2’―OMe ATP、CTP、UTP、およびGTP各1mM(2’―OH GTPなし)、MgCl2(6.5mM)、MnCl2(2mM)、PEG―8000w/v 10%、GMP1mM、無機ピロホスファターゼ5単位/mL、Y639L/H784A/K378R変異T7RNAポリメラーゼ200nMで、37℃で一晩転写され、PAGEにより精製され、残りのDNAテンプレートがRQ1 DNアーゼを用いて消化され(その後DNAテンプレートの非存在がPCRによりアッセイされ)、転写物が逆転写され、クローニングおよび配列決定の前にPCRを用いて増幅された。増幅ライブラリからの48のクローンおよび開始ライブラリからの48のクローンが、配列決定された。バイアスが生じたかを見るために、縮重領域におけるヌクレオチド頻度の値が検査された。図8により示されるように、転写後のヌクレオチド組成の割合は、各ヌクレオチド(A、T、CおよびG)の割合がほぼ等しい開始ライブラリのヌクレオチド組成の割合と非常に類似しており、Y639L/H784A/K378R変異T7RNAポリメラーゼを転写に用いてヌクレオチドバイアスが生じないことが示された。
実施例2C:P266L/Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたMNA転写
二本鎖転写テンプレートを生成するために、ポリメラーゼ連鎖反応をプログラムするために以下のDNAテンプレートおよびプライマが用いられた。Nは、ATGCの各々である確率がほぼ等しい縮重位置を示し、全ての配列が3’から5’方向にリストされる:PCRテンプレート(ARC2118)
その後、得られた二本鎖転写テンプレートを用いて、以下のように各サンプルの200uL転写混合物がプログラムされた:HEPES(200mM)、DTT(40mM)、スペルミジン(2mM)、TritonX―100(0.01%w/v)、MgCl
2(8mM)、MnCl
2(2.5mM)、PEG―8000(10%w/v)、各1.5mMの2’―OMe NTP、GMP 1mM、100―200nM転写テンプレート、無機ピロホスファターゼ(1単位)、pH7.5、T7変異ポリメラーゼP266L/Y639L/H784A/K378Rが下記のように希釈された。転写混合物は、37℃で一晩インキュベートされた(16h)。
インキュベーション後、混合物は、イソプロパノールにより沈殿され、得られたペレットが溶解され、変性PAGE(12.5%アクリルアミド)を用いて60分間25Wで定量された。サンプルがUVシャドウにより260nmで視覚化および定量された。転写収率が下表1に与えられる。
(実施例3)2’―O―メチルATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチドを導入する転写
in vitro転写を用いて2’―O―メチルヌクレオチドおよび2’―デオキシヌクレオチドの両者を導入するY639L/H784A/K378R変異ポリメラーゼの能力をテストするために実験が行われた。
DNAテンプレートARC4206およびARC4212、およびその各々のプライマ(下記に5’から3’方向に配列が与えられる)を用いて、一つの2’―デオキシCTPヌクレオチドと、2’―O―メチルATP、GTP、およびUTPである他の三つのヌクレオチドを導入するY639L/H784A/K378R変異T7RNAポリメラーゼの転写能力がテストされた。200mM HEPES、40mM DTT、2.0mM スペルミジン、0.01%w/v TritonX―100、10%PEG―8000、8mM MgCl2、2.5mM MnCl2、1.5mM dCTP、1.5mM mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼおよび〜9μg/mL変異T7ポリメラーゼ(Y639L/H784A/K378R)および0.3μMテンプレートDNAを用いて、37℃で一晩転写が行われて、2’―OMe ATP、GTP、およびUTPヌクレオチドおよび2’―デオキシCTPヌクレオチドからなるARC4206およびARC4212プールが生成された(以下に「dCmD」)。同時に、50mM HEPES、10mM DTT、0.5mM スペルミジン、0.0025% TritonX―100、10%PEG―8000、8mM MgCl2、2.5mM MnCl2、1.5mM mCTP、1.5mM mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼおよび〜9μg/mL変異T7ポリメラーゼ(Y639L/H784A/K378R)および0.3μMテンプレートDNAを用いて37℃で一晩転写が行われて、2’―OMeプリンおよびピリミジンヌクレオチドからなるARC4206およびARC4212プールが生成された(以下に「MNA」)。各条件組下の転写産物が、400uLの反応混合物を用いてPAGE―ゲル分析によりアッセイされ、各条件の転写産物がPAGE―ゲルのUV―シャドウイングから視覚化された。表2は、300mM NaOAc、20mM EDTA中37℃での一晩のPAGE―ゲルからの溶出、エタノール沈殿、その後のUV分光法による定量化後に単離されたプールに基づいて、転写収率の定量結果をまとめる。
Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたMNAおよびdCmD転写の忠実度を評価するために、追加的な実験が行われた。忠実度をテストするために、上で生成されたARC4206およびARC4212MNAおよびdCmDプールが、逆転写され(Thermoscript、Invitrogen、カリフォルニア州カールズバッド)、PCRにより増幅され、Y639L/H784A/K378Rポリメラーゼを用いた追加的なMNAおよびdCmD転写をプログラムするために使用された。プールはPAGEにより精製され、転写物がPAGE―ゲルのUV―シャドウイングから視覚化された。表3は、300mM NaOAc、20mM EDTA中での65℃での4.5時間のPAGE―ゲルからの溶出、エタノール沈殿、その後のUV分光法による定量後に単離されたプールに基づく、転写収率の定量結果をまとめる。
テンプレートは、その各々のDNAプライマにより増幅された:
(実施例4)Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたアプタマー選択
2’―OMeプリンおよびピリミジンヌクレオチド(以下に「MNA」)からなるプールを用いて、ヒトAng2(以下に「h―Ang2」)に対するアプタマーを同定するために、選択が実行された。選択戦略は、h―Ang2に特異的な高親和性アプタマーをもたらした。
ヒトAng2は、R&D Systems,Inc.(ミネソタ州ミネアポリス)から購入された。上の実施例1に記載したように、T7RNAポリメラーゼ(Y639L/H784A/K378R)が発現され、精製された。2’―OMeプリンおよびピリミジンヌクレオチドは、TriLink BioTechnologies(カリフォルニア州サンディエゴ)から購入された。
Ang2アプタマーの選択
プール調製
配列
のDNAテンプレートが、ABI EXPEDITE(商標)(Applied Biosystems、カリフォルニア州フォスターシティ)DNA合成装置を用いて合成され、標準法により脱保護された。テンプレートは、プライマ
により増幅されてから、T7RNAポリメラーゼ(Y639L/H784A/K378R)によるin vitro転写のためのテンプレートとして使用された。200mM Hepes、40mM DTT、2mMスペルミジン、0.01%w/v TritonX―100、10%PEG―8000、8mM MgCl
2、2.5mM、MnCl
2、1.5mM mCTP、1.5mM mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼ、および〜9μg/mL T7ポリメラーゼ(Y639L/H784A/K378R)および0.5μMテンプレートDNAを用いて転写が行われて、ARC2118mRmYプールが生成された。
選択
100μLの最終的体積の選択バッファー(1X DulbeccoのPBS(DPBS))において100pmolのタンパク質と330pmol(2×1014分子)のMNA ARC2118プールを1時間室温でインキュベートすることにより、選択が開始された。RNA―タンパク質複合体および未結合RNA分子が、0.45ミクロンニトロセルローススピンカラム(Schleicher and Schuell、ニューハンプシャー州キーン)を用いて分離された。カラムはKOHで前処置され(1mL 0.5M KOH中にカラムフィルタ浸漬、15分RT;スピン。1mL dH2O中にフィルタ浸漬 5分RT;スピン)、1mL 1X PBSで二回洗浄されてから、プール:Ang2複合体を含む溶液がカラムに加えられ、2分間1500xgで遠心分離された。フィルタは、非特異的結合物を除去するために、500μL DPBSにより二回洗浄された。2×100μL溶離バッファー(7M尿素、100mM酢酸ナトリウム、3mMEDTA、95℃に予熱)の添加によりRNAが溶出されてから、エタノールにより沈殿させられた。プライマ配列番号27を用いて、製造者の指示に従って、ThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により、RNAが逆転写された。配列番号26および配列番号27を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)によるPCRにより、cDNAが増幅された。プール調製のために上述のようにテンプレートが転写され、変性ポリアクリルアミドゲルで精製された。
ラウンド2は、ラウンド1と同じ方法により実行された。ラウンド3―12は、疎水性プレートに固定されたh―Ang2により行われた。各選択ラウンドは、100μLの1X DPBS中で室温で1時間Nunc Maxisorp疎水性プレートの表面に20pmolのh―Ang2を固定することにより開始された。プレートは、120μL DPBSで5×洗浄された後、ブロッキングバッファー(1X DPBS、および0.1mg/mL BSA)で1時間インキュベートされた。そして、上清が除去され、ウェルが120μL 1X DPBSで5回洗浄された。プールRNAは、1時間室温で空のウェルでインキュベートされてから、100μLのブロッキングバッファーで先にブロックされたウェルで1時間インキュベートされた。ラウンド3から先は、標的が固定されたウェルが、ポジティブ選択ステップの前に100μLブロッキングバッファー(1X PBS、0.1mg/mL tRNA、0.1mg/mL ssDNAおよび0.1mg/mL BSA)中で、1時間室温でブロックされた。全ての場合において、固定h―Ang2に結合したプールRNAは、逆転写(「RT」)混合物(3’プライマ、配列番号27、およびThermoscript RT、Invitrogen、カリフォルニア州カールズバッド)の添加により、選択プレートにおいて直接逆転写された後、1時間65℃でインキュベートされた。ラウンド1につき記載されるように、得られたcDNAがPCR(Taqポリメラーゼ、New England Biolabs、マサチューセッツ州ベヴァリー)および転写のテンプレートとして使用された。各ラウンドの条件が、表4にある。
MNAアプタマー結合分析
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース
32P―エンド標識したプールRNAが、h―Ang2と組み合わされ、DPBSバッファー中で30μLの最終的体積で、30分間室温でインキュベートされた。混合物がドットブロット装置に適用され(Minifold―1Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAはニトロセルロースフィルタに捕らえられ;非タンパク結合RNAはナイロンフィルタに捕らえられる。ラウンド9でh―Ang2結合の濃縮の開始が見られた。ラウンド9、10および12のプールテンプレートが、TOPO TAクローニングキット(Invitrogen、カリフォルニア州カールズバッド)を使用して製造者の指示に従ってクローニングされ、化学合成のために26のユニーククローンが選択され、解離定数(K
D)が決定された。簡単にいうと、合成RNAsがγ―
32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca
2+およびMg
2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてK
D値が決定された。式:RNA結合画分=振幅
*(((AptConc+[h―Ang2]+K
D)−SQRT((AptConc+[h―Ang2]+K
D)
2−4(AptConc
*[h―Ang2])))/(2
*AptConc))+バックグラウンドにデータをあてはめてK
Dsが推定された。結果は、下表5にレポートされる。
26のユニーク配列の中で、8つが類似のモチーフを共有し、類似の結合および阻害活性を有した。これらの配列は、ファミリーIとして同定される。ファミリーIIは、類似の結合および阻害活性を有した、モチーフを共有する二つの配列を含む。
MNAアプタマー機能の分析
Elisaアッセイ
いくつかアプタマーが、Tie2レセプターに対するAng2結合を妨げる能力を測定するために準備されたELISAアッセイにおいてテストされた。Tie2レセプターを捕捉するために、100μLのPBS(pH7.4)中の150ngのTie2―Fc(R&D systems 313―TI―100―CF、ミネソタ州ミネアポリス)が、96ウェルMaxisorbプレート(NUNC#446612、ニューヨーク州ロチェスター)に置かれ、4℃で一晩インキュベートされた。捕捉の間、様々な濃度の合成RNA50μLが、50μLの3.6nM Ang2(200ng/mL)(R&D systems、623―AN―025/CF、ミネソタ州ミネアポリス)(0.2% BSAを伴うPBS中)と、0.1% BSAを伴うPBS中1.8nM(100ng/mL)の最終Ang2濃度で混合され、1時間室温でインキュベートされた。一晩のインキュベーション後に捕捉溶液が除去され、200μLのTBST(25mM Tris―HCl pH7.5、150mM NaClおよび0.01% Tween20)によりプレートが三回洗浄された。そして、プレートが、5%脱脂粉ミルクを含む200μL TBSTにより室温で30分間ブロックされた。ブロッキングの後、プレートが200μLのTBSTで再び室温で三回洗浄され、合成RNA:Ang2混合物がプレートに加えられ、1時間室温でインキュベートされた。そして、プレートが200μLのTBSTで三回洗浄され、100μLのビオチン化ヤギ抗Ang2抗体(1:1000;R&D Systems BAF623、ミネソタ州ミネアポリス)が加えられ、室温で1時間インキュベートされた。200μLのTBSTにより三回洗浄したあと、100μLのHRP連結ストレプトアビジン(1:200;R&D systems#DY998、ミネソタ州ミネアポリス)が加えられ、0.5時間室温でインキュベートされた。そして、プレートが再び200μLのTBSTにより三回洗浄され、100μLのTMP溶液(Pierce、#34028)が加えられ、暗所で、室温で5分間インキュベートされた。2N H2SO4を含む100μLの溶液が加えられて反応が停止され、プレートがSpectroMaxで、450nmで読み取られた。結果は、下表5の最終的欄に与えられる。
FACSアッセイ
ヒト臍静脈内皮細胞(「HUVEC」)(ATCC)およびK293細胞、ヒトTie2レセプターを過剰発現する細胞系統を用いて、細胞膜上のTie2レセプターに対するAng2の結合を阻害する特異的MNA Ang2アプタマーのIC50が決定された。簡単にいうと、組換え哺乳類発現ベクターpCDNA3.1―Tie2が293細胞(ATCC、ヴァージニア州マナサス)にトランスフェクションされ、G418(Invitrogen、カリフォルニア州カールズバッド)による選択の後、安定したクローンが得られた。フローサイトメトリは、HUVECおよびK293細胞の両方のTie2タンパク質の発現を示した。Ang2滴定アッセイにより、HUVECおよびK293細胞に対する細胞アプタマー阻害アッセイのAng2(R&D Systems、ミネソタ州ミネアポリス)の量がさらに決定され、それぞれ1および0.1μg/mLであった。
フローサイトメトリ結合アッセイにおいては、HUVECおよびK293細胞(2×105細胞/ウェル)が、V底96ウェルプレートにペレット化され、その後MNAアプタマー/Ang2溶液中に再懸濁され、2時間インキュベートされた。アプタマー/Ang2溶液は、FACバッファー(PBS中1%BSA、0.2%アジ化ナトリウム)中のAng2と異なる用量のアプタマー(100nM、33.3nM、11.1nM、3.7nM、1.2nM、0.411nM、0.137nM、および0.0456nM)を氷上で30分間プレインキュベーションすることにより調製された。FACsバッファーによる三度の洗浄のあと、細胞はビオチン化抗ヒトAng2抗体(5μg/mL;R&D Systems、ミネソタ州ミネアポリス)で30分間インキュベートされた後、ストレプトアビジンPE(1:10;BD Biosciences、カリフォルニア州サンノゼ)によるもう30分間のインキュベーションが行われた。FACScan(BD Biosciences、カリフォルニア州サンノゼ)を用いて、FACS分析が完了された。結果は、下表5にレポートされる。
(実施例5)Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたアプタマー選択
2’―OMeプリンおよびピリミジンヌクレオチドからなるプール(以下に「mRmY」)を用いて、ヒトIgE(以下に「h―IgE」)に対するアプタマーを同定するために、選択が行われた。選択戦略は、h―IgEに特異的な高親和性アプタマーをもたらした。
ヒトIgEは、Athens Research & Technology(Cat.#16―16―090705 ジョージア州アセンズ)から購入された。実施例1に上述したように、T7RNAポリメラーゼ(Y639L/H784A/K378R)が発現され、精製された。2’―OMeプリンおよびピリミジンヌクレオチドは、TriLink BioTechnologies(カリフォルニア州サンディエゴ)から購入された。
IgEアプタマーの選択
プール調製
配列
のDNAテンプレートが、ABI EXPEDITE(商標)(Applied Biosystems、カリフォルニア州フォスターシティ)DNA合成装置用いて合成され、標準法により脱保護された。テンプレートは、プライマ
により増幅され、T7RNAポリメラーゼ(Y639L/H784A/K378R)によるin vitro転写のテンプレートとして使用された。200mM HEPES、40mM DTT、2.0mM スペルミジン、0.01%w/v TritonX―100、10% PEG―8000、8mM MgCl
2、2.5mM MnCl
2、1.5mM mCTP、1.5mM mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼ、および〜9μg/mL変異T7ポリメラーゼ(Y639L/H784A/K378R)および0.3μMテンプレートDNAを用いて転写が行われ、ARC2118 MNAプールが生成された。
選択
330pmol(2×1014分子)のMNA ARC2118プールを、BSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合した24pmolのタンパク質とともに100μLの最終体積の選択バッファー(1X DulbeccoのPBS(DPBS))中で、室温で1時間インキュベートすることにより選択が開始されれた。ウェルは、非特異的結合物を除去するために、120μL DPBSにより四回洗浄された。RNAが溶出され、プライマ配列番号27を用いて、ThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により、製造者の指示に従って逆転写された。配列番号26および配列番号27を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)を使用したPCRによりcDNAが増幅された。プール調製で上述したようにテンプレートが転写され、変性ポリアクリルアミドゲルで精製された。
全てのラウンドは、疎水性プレートに固定されたh―IgEにより行われた。各選択ラウンドが、Nunc Maxisorp疎水性プレートの表面に24pmolのh―IgEを100μLの1X DPBS中で、室温で1時間固定することにより開始された。プレートが120μL DPBSにより四回洗浄されてから、ブロッキングバッファー(1X DPBS、および0.1mg/mL BSA)で1時間インキュベートされた。そして、上清が除去され、120μL 1X DPBSによりウェルが四回洗浄された。ラウンド2から開始して、プールRNAは空のウェルで、室温で1時間インキュベートされてから、100μLのブロッキングバッファーで先にブロックされたウェルで1時間インキュベートされた。ラウンド2から先は、非―特異的競合物が、正の選択ステップに加えられた(0.1mg/mL tRNA、および0.1mg/mL ssDNA)。全ての場合において、固定されたh―IgEに結合したプールRNAは、逆転写(「RT」)混合物(3’プライマ、配列番号27、およびThermoscript RT、Invitrogen、カリフォルニア州カールズバッド)の添加後65℃で1時間インキュベートすることにより、直接選択プレートにおいて逆転写された。得られたcDNAは、ラウンド1につき記載されるように、PCR(Taqポリメラーゼ、New England Biolabs、マサチューセッツ州ベヴァリー)および転写のテンプレートとして使用された。各ラウンドの条件が、表6にある。
MNAアプタマー結合分析
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース
32P―エンド標識したプールRNAが、h―IgEと組み合わされ、DPBSバッファー中で30μLの最終体積で、30分間室温でインキュベートされた。混合物がドットブロット装置に適用され(Minifold―1 Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAは、ニトロセルロースフィルタに捕らえられ;非タンパク結合RNAは、ナイロンフィルタに捕らえられる。ラウンド8でh―IgE結合の濃縮の開始が見られた。ラウンド5、8および12のプールテンプレートが、TOPO TAクローニングキット(Invitrogen、カリフォルニア州カールズバッド)を使用して製造者の指示に従ってクローニングされた。配列決定データから、ラウンド8のプールが全配列の59%を含む単一の主要クローンに収束したことが明らかになった。この主要クローンと三つの可能性のあるミニマーが化学合成のために選択され、解離定数(K
D)が決定された。簡単にいうと、合成RNAsがγ―
32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてKD値が決定された。式:RNA結合画分=振幅
*(((AptConc+[h―IgE]+KD)−SQRT((AptConc+[h―IgE]+KD)2−4(AptConc
*[h―IgE])))/(2
*AptConc))+バックグラウンドにデータをあてはめてKDsが推定された。主要クローンは、約800pMのK
Dを有した。最善の結合のミニマーの、サルIgE(m―IgE)に対する結合もテストされたが、サルIgEタンパク質に対する交差反応結合は示されなかった。この交差反応性の欠如は、ELISAによっても確認された。3’末端に逆方向dTを伴うミニマーが、医薬品化学プロセスの親分子として用いられた。
医薬品化学
IgE特異的MNAミニマーの一つの化学組成(図9)が、化合物の血漿安定性を維持しながら親和性および能力を改善するために変更された。プロセスは、最小化IgEアプタマーの一連の誘導体の設計、合成および評価を含み、一連の各誘導体は、いずれの残基が置換に耐えるかを決定するため、各所定のヌクレオチドで単一の修飾を含んだ。第一修飾組は、各ユニーク2’―OMeヌクレオチドへのデオキシヌクレオチドの置換であった。別個の修飾ラウンドにおいては、各誘導体が異なるヌクレオチド間結合位置で単一のホスホロチオエート修飾を含む一連の誘導体が合成された。これらの修飾の最初の段階において生成されたデータを用いて、最小化アプタマーの構造活性相関(SAR)が確立された。後の修飾段階においては、最初のSARデータに基づいて設計された複合置換組でアプタマーが合成され、テストされた。複合置換パネルから、2’―OMeから2’―デオキシへの置換がその組成に導入された、39ヌクレオチドの長さのアプタマーが同定された。さらに、2’―OMeから2’―デオキシへの一つの置換およびリン酸塩からホスホロチオエートへの四つの置換がその組成に導入された、39ヌクレオチドの長さの、得られた最小化修飾アプタマーが同定された。図10に示すように、このデオキシ/ホスホロチオエート修飾アプタマーは、最小化非修飾親アプタマーならびに二つの2’―OMeからデオキシへの置換を有する最小化親アプタマーの両方と比較して、結合親和性の増加を示す。
血清安定性
最小化非修飾親アプタマーおよびデオキシ/ホスホロチオエート修飾アプタマーが、ヒト、ラットおよびサル血清における安定性を決定するためにアッセイされた。各アプタマーが、90%血清中5μMの最終濃度になるように、1mlのプール血清に加えられた。アプタマーは振盪を伴って37℃でインキュベートされ、0、0.5、1、4、24、48、72、および98時間でタイムポイントがとられた。各タイムポイントで、インキュベート試料からの90μlのストックが、10μlの0.5M EDTAに加えられ、BIACORE 2000システムを用いた後の安定性分析のために−20℃で冷凍された。
全てのバイオセンサ結合測定が、研究等級のCM5バイオセンサチップを備えたBIACORE 2000(BIACORE Inc.、ニュージャージー州ピスカタウェイ)を使用して、25℃で行われた。精製された組換えヒトIgE(Athens Research & Technology、ジョージア州アセンズ)が、アミノ―カップリング化学を用いてバイオセンサ表面に固定された。これを達成するために、二つのフローセルの表面が、まず0.1M NHS(Nヒドロキシスクシンイミド)および0.4M EDC(3―(N,Nジメチルアミン)プロピル―N―エチルカルボジイミド)の1:1混合物で、5μl/分の流速で7分間活性化された。表面活性化の後、活性化された表面に対する共有結合の確立を許容するために、一つのフローセルに50μg/mlのIgEが10μl/分で20分間注入された。次に、1MのエタノールアミンヒドロクロリドpH8.5が、残余エステルを不活性化するために、5μl/分で7分間注入された。ブランクとして使用されるフローセルには、タンパク質注入を伴わずに残余エステルを不活性化するために、1MのエタノールアミンヒドロクロリドpH8.5が7分間注入された。
全てのタイムポイントが分析される前に、調製されたチップに一連のアプタマースタンダードが通されて、標準曲線が生成された。標準曲線を確立するために、アプタマーが、4%ヒト血清および50mM EDTAが添加されたHBS―Pバッファー(10mM HEPES pH7.4、150mM NaCl、0.005% Surfactant20)に、連続的に希釈された(200nMから12.5nMへ)。全ての希釈サンプルがBiacore 2000に注入されて20μl/分で5分間結合され、3分間置かれた。チップを再生させるために、1N NaClが30μl/分で60秒間注入された。結合段階の最後のRUピーク反応がアプタマー濃度に対してプロットされ、Four―Parameterロジスティック関数を用いて標準曲線が生成された。ヒト、ラット、およびサル血清における活性アプタマー濃度を測定するために、タイムポイントサンプルが、Biacore 2000への注入の直前に最終血清濃度が4%となるように、HBS―P中22.5倍に希釈された。上で生成された標準曲線を用いてRU反応単位から濃度に変換することにより、各血清インキュベーションピリオドの機能的アプタマー濃度が計算された。追加的な品質制御措置として、二つのアプタマースタンダードが実験の最後に独立してテストされて、BIACOREで測定された濃度のスタンダードからの逸脱が20%未満であることが確認された。最小化非修飾親アプタマーおよびデオキシ/ホスホロチオエート修飾アプタマーは、ヒト、ラット、およびサル血清中でいずれも98時間で90%以上活性であることが決定された。
(実施例6)Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたアプタマー選択
2’―OMe ATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチド(以下に「dCmD」)からなるプールを用いて、ヒトフォンビルブラント因子(以下に「h―vWF」)に対するアプタマーを同定するために、選択が行われた。選択戦略から、h―vWFに特異的な高親和性アプタマーが生じた。
ヒトvWFは、EMD Biosciences Inc.(Cat.#681300、カリフォルニア州ラホーヤ)から購入された。ヒトvWF Alドメインが発現および精製された。変異ポリメラーゼ(Y639L/H784A/K378R)が、上述の実施例1に記載したように発現および精製された。2’―OMe ATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチドは、TriLink BioTechnologies(カリフォルニア州サンディエゴ)から購入された。
vWFアプタマーの選択
プール調製
配列
を伴うテンプレートが、ABI EXPEDITE(商標)(Applied Biosystems、カリフォルニア州フォスターシティ)DNA合成装置を使用して合成され、標準法により脱保護された。テンプレートは、プライマ
により増幅されてから、T7RNAポリメラーゼ(Y639L/H784A/K378R)によるin vitro転写のためのテンプレートとして使用された。200mM HEPES、40mM DTT、2mMスペルミジン、0.01%w/v TritonX―100、10%PEG―8000、8mM MgCl
2、2.5mM MnCl
2、1.5mM dCTP、1.5mM mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼ、および〜9μg/mL変異T7ポリメラーゼ(Y639L/H784A/K378R)および0.3μMテンプレートDNAを用いて転写が行われて、ARC4218dCmDプールが生成された。
選択
100μLの最終体積の選択バッファー(1X DulbeccoのPBS(DPBS)において、HSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合された20pmolのタンパク質とともに330pmol(2×1014分子)のdCmD ARC4218プールを1時間室温でインキュベートすることにより、選択が開始された。非特異的結合物を除去するために、200μL DPBSによりウェルが五回洗浄された。RNAが溶出され、プライマARC4208(配列番号30)を用いて、製造者の指示に従ってThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により逆転写された。プライマARC4219(配列番号29)およびARC4208(配列番号30)を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)によるPCRによりcDNAが増幅された。プール調製につき上述したようにテンプレートが転写された。エタノール沈殿によりテンプレートが精製され、その後Micro Bio―spinカラム(Cat.#732―6250、Bio−Rad Laboratories、カリフォルニア州ハーキュリーズ)を用いた脱塩ステップが行われた。
全てのラウンドが、疎水性プレートに固定されたh―vWFにより行われた。10pmolのh―vWFおよび10pmolのヒトvWF Alドメインを、Nunc Maxisorp疎水性プレートの表面に100μLの1X DPBS中で、室温で30分間固定することにより、最初の三つの選択ラウンドが開始された。ラウンド4で開始して、20pmolのh―vWFが疎水性プレートに2ラウンド固定された後、20pmolのヒトvWF Alドメインが疎水性プレートに一ラウンド固定された。プレートは、200μL DPBSにより三回洗浄されてから、ブロッキングバッファー(1X DPBSおよび5%HSA)で30分間インキュベートされた。それから上清が除去され、200μL DPBSによりウェルが三回洗浄された。ラウンド2から始まり、プールRNAが室温で30分間空のウェルでインキュベートされてから、100μLブロッキングバッファーにより先にブロックされたウェルで30分間インキュベートされた。ラウンド2から先は、正の選択ステップに非特異的競争物が加えられた(0.1mg/mL tRNA)。いずれの場合も、固定標的に結合されたプールRNAが、逆転写(「RT」)混合物(3’プライマ、配列番号30、およびThermoscript RT、Invitrogen、カリフォルニア州カールズバッド)の添加およびその後の65℃での1時間のインキュベートにより、直接選択プレートにおいて逆転写された。ラウンド1につき記載されるように、得られたcDNAが、PCR(Taqポリメラーゼ、New England Biolabs、マサチューセッツ州ベヴァリー)および転写のテンプレートとして用いられた。各ラウンドの条件が、表7にある。
dCmDアプタマー結合分析
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース
32P―エンド標識したプールRNAが、h―vWFと組み合わされ、DPBSバッファー中で30μLの最終体積で、室温で30分間インキュベートされた。混合物がドットブロット装置に適用され(Minifold―1 Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAは、ニトロセルロースフィルタに捕らえられ;非タンパク結合RNAは、ナイロンフィルタに捕らえられる。ラウンド5でh―vWF結合の濃縮の開始が見られた。ラウンド5およびラウンド6プールの解離定数(K
D)が決定された。簡単にいうと、プールRNAsがγ―
32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてK
D値が決定された。式:RNA結合画分=振幅
*(((AptConc+[h―vWF]+KD)−SQRT((AptConc+[h―vWF]+KD)2−4(AptConc
*[h―vWF])))/(2
*AptConc))+バックグラウンドにデータをあてはめてK
Dsが推定された。ラウンド6プールは、約2nMのK
Dを有した。プールは、Alドメインに対する結合についてもテストされ、約4nMのK
Dを有した。
(実施例7)Y639L/H784A/K378R変異T7RNAポリメラーゼを用いたアプタマー選択
2’―OMe ATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチド(以下に「dCmD」)からなるプールを用いて、ヒトIgE(以下に「h―IgE」)に対するアプタマーを同定するために選択が行われた。選択戦略から、h―IgEに特異的な高親和性アプタマーが生じた。
ヒトIgEは、Athens Research & Technology(Cat.#16―16―090705 ジョージア州アセンズ)から購入された。実施例1に上述したように、T7RNAポリメラーゼ(Y639L/H784A/K378R)が発現され、精製された。2’―OMe ATP、GTP、およびUTPヌクレオチド、および2’―デオキシCTPヌクレオチドは、TriLink BioTechnologies(カリフォルニア州サンディエゴ)から購入された。
IgEアプタマーの選択
プール調製
配列
のDNAテンプレートが、ABI EXPEDITE(商標)(Applied Biosystems、カリフォルニア州フォスターシティ)DNA合成装置用いて合成され、標準法により脱保護された。テンプレートは、プライマ
により増幅されてから、T7RNAポリメラーゼ(Y639L/H784A/K378R)によるin vitro転写のテンプレートとして使用された。200mM HEPES、40mM DTT、2.0mMスペルミジン、0.01%w/v TritonX―100、10%PEG―8000、8mM MgCl
2、2.5mM MnCl
2、1.5mM dCTP、1.5mM mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼ、および〜9μg/mL変異T7ポリメラーゼ(Y639L/H784A/K378R)および0.3μMテンプレートDNAを用いて転写が行われ、ARC4218dCmDプールが生成された。
選択
330pmol(2×1014分子)のdCmD ARC4218プールを、HSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合した20pmolのタンパク質とともに100μLの最終体積の選択バッファー(1X DulbeccoのPBS(DPBS))中で、室温で1時間インキュベートすることにより選択が開始されれた。ウェルは、非特異的結合物を除去するために、200μL DPBSにより五回洗浄された。RNAが溶出され、プライマARC4208(配列番号30)を用いて、ThermoScript RT―PCR(商標)システム(Invitrogen、カリフォルニア州カールズバッド)により、製造者の指示に従って逆転写された。プライマARC4219(配列番号29)およびARC4208(配列番号30)を用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)を用いたPCRにより、cDNAが増幅された。プール調製につき上述したように、テンプレートが転写された。エタノール沈殿によりテンプレートが精製された後、Micro Bio―spinカラム(Cat.#732―6250、Bio―Rad Laboratories、カリフォルニア州ハーキュリーズ)を用いて脱塩ステップが行われた。
全てのラウンドが、疎水性プレートに固定されたh―IgEにより行われた。各選択ラウンドが、Nunc Maxisorp疎水性プレートの表面に20pmolのh―IgEを100μLの1X DPBS中で、室温で30分間固定することにより開始された。プレートは、200μL DPBSにより三回洗浄されてから、ブロッキングバッファー(1X DPBS、および5% HSA)で30分間インキュベートされた。そして上清が除去され、200μL DPBSによりウェルが三回洗浄された。ラウンド2から開始して、プールRNAが空のウェルにおいて室温で30分間インキュベートされてから、100μLのブロッキングバッファーで先にブロックされたウェルで30分間インキュベートされた。ラウンド2から先は、非―特異的競合物が正の選択ステップに加えられた(0.1mg/mL tRNA)。全ての場合において、固定されたh―IgEに結合したプールRNAは、逆転写(「RT」)混合物(3’プライマ、配列番号30、およびThermoscript RT、Invitrogen、カリフォルニア州カールズバッド)の添加と、その後の65℃での1時間のインキュベーションにより、直接選択プレートにおいて逆転写された。得られたcDNAが、ラウンド1につき記載されるように、PCR(Taqポリメラーゼ、New England Biolabs、マサチューセッツ州ベヴァリー)および転写のテンプレートとして使用された。各ラウンドの条件が、表8にある。
dCmDアプタマー結合分析
プールのタンパク質結合親和性をモニタするために、選択の全体にわたりドットブロット結合アッセイが行われた。トレース
32P―エンド標識したプールRNAが、h―IgEと組み合わされ、DPBSバッファー中で30μLの最終体積で、室温で30分間インキュベートされた。混合物がドットブロット装置に適用され(Minifold―1 Dot Blot、Acrylic、Schleicher and Schuell、ニューハンプシャー州キーン)、(上から下へ)ニトロセルロース、ナイロン、およびゲルブロット膜とアセンブルされた。タンパク質に結合されたRNAはニトロセルロースフィルタに捕らえられ;非タンパク結合RNAはナイロンフィルタに捕らえられる。ラウンド5でh―IgE結合の濃縮の開始が見られた。ラウンド5および6プールの解離定数(K
D)が決定された。簡単にいうと、プールRNAsがγ―
32P ATPで5’末端標識され、ドットブロットアッセイおよび1X DPBS(w/Ca2+およびMg2+)(Gibco、Catalog#14040、Invitrogen、カリフォルニア州カールズバッド)のバッファー条件を用いてK
D値が決定された。式:RNA結合画分=振幅
*(((AptConc+[h―IgE]+KD)−SQRT((AptConc+[h―IgE]+KD)2―4(AptConc
*[h―IgE])))/(2
*AptConc))+バックグラウンドにデータをあてはめてKDsが推定された。ラウンド6プールは、約80nMのK
Dを有した。
(実施例8)2’―O―メチルATP、CTP、GTPおよび2’―O―メチル―5―インドリルメチレンUTPヌクレオチドを導入する転写
Y639L/H784A/K378R変異ポリメラーゼが、in vitro転写を用いて2’―O―メチルA、C、Gおよび2’―O―メチル―5インドリルメチレンUを転写産物に導入するために用いられる。修飾ウリジンが、以下にヌクレオシドとして示される:
200mM HEPES、40mM DTT、2mMスペルミジン、0.01%w/v TritonX―100、10% PEG―8000、8mM MgCl
2、2.5mM MnCl
2、1.5mM mCTP、1.5mM 5―インドリル―mUTP、1.5mM mGTP、1.5mM mATP、1mM GMP、0.01単位/μL無機ピロホスファターゼ、および〜9μg/mL変異T7ポリメラーゼ(Y639L/H784A/K378R)および0.3μMテンプレートDNAを用いて、37℃で一晩転写が行われて、5’―末端に単一の2’―ヒドロキシGMPを伴う2’―OMe A、2’―OMe G、2’―OMe Cおよび2’―OMe―5―インドリルメチレンUからなる転写産物が生成される(以下に「MNA―I」)。各条件下の転写産物が、400uLの反応混合物を用いてPAGE―ゲル分析によりアッセイされ、各条件での転写産物が、PAGE―ゲルのUV―シャドウイングから視覚化される。
(実施例9)Y639L/H784A/K378R変異T7RNAポリメラーゼおよび2’―O―メチル―5―インドリルメチレンウリジンを用いたSELEX
治療目的のタンパク質標的に対するSELEXが、複数の縮重位置および上の実施例7に記載のMNA―I転写条件を含む転写テンプレートを用いて実行される。
最終体積100μLの選択バッファー(1XDulbeccoのPBS(DPBS))において、HSAブロックされた疎水性プレート(Maxisorpプレート、Nunc、ニューヨーク州ロチェスター)に結合した20pmolのタンパク質とともに330pmol(2×1014分子)のMNA―I転写ライブラリを1時間室温でインキュベートすることにより、選択ステップが開始される。非特異的結合物を除去するために、200μL DPBSによりウェルが五回洗浄される。製造者の指示に従ってThermoScript RT―PCR(商標)(Invitrogen、カリフォルニア州カールズバッド)を用いて、選択ウェル中でRNAが逆転写される。得られたcDNAが、T7RNAポリメラーゼプロモータをコードする5’プライマを用いて、製造者の指示に従って、Taqポリメラーゼ(New England Biolabs、マサチューセッツ州ベヴァリー)によるPCRにより増幅される。得られた転写テンプレートが、MNA―I条件下で上述のように転写される。エタノール沈殿により、得られた転写産物が精製され、その後Micro Bio―spinカラム(Cat.#732―6250、Bio−Rad Laboratories、カリフォルニア州ハーキュリーズ)を用いた脱塩ステップが行われる。
10ラウンドのSELEXの後、得られた転写産物ライブラリが逆転写され、PCRにより増幅され、クローニングおよび配列決定される。個々のクローンがMNA―I条件下で転写され、標的タンパク質を結合および阻害する能力につきアッセイされる。
(実施例10)2’―OMe TTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTPヌクレオチドを導入する転写(mTmV組成物)
200nMの40ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OMe TTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG 8000、2.5単位/ml無機ピロホスファターゼ、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―OMe TTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
(実施例11)デオキシTTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTPヌクレオチドを導入する転写(dTmV組成物)
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―デオキシTTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、2.5単位/ml無機ピロホスファターゼ、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―デオキシTTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
(実施例12)2’―OH TTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTPヌクレオチドを導入する転写(rTmV組成物)
200nMの40ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OH TTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―OH TTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
(実施例13)デオキシUTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTPヌクレオチドを導入する転写(dUmV組成物)
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―デオキシUTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―デオキシUTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
(実施例14)2’―OH UTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTPヌクレオチドを導入する転写(rUmV組成物)
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OH UTP、2’―OMe ATP、2’―OMe CTP、2’―OMe GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―OH UTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
(実施例15)2’―OH GTP、デオキシTTP、2’―OMe ATP、2’―OMe CTPヌクレオチドを導入する転写(rGdTmM組成物)
200nMの二本鎖転写テンプレートARC3205、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―デオキシTTP、2’―OMe ATP、2’―OMe CTP、2’―OH GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動したバンドが観察された。2’―デオキシTTPを伴わずにインキュベートされた陰性対照転写は、観察可能なバンドを生成しなかった。
(実施例16)2’―OMe QTP、2’―OH GTP、2’―OMe ATP、2’―OMe CTPヌクレオチドを導入する転写(mQrGmM組成物)
200nMの40ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OMe QTP、2’―OMe ATP、2’―OMe CTP、2’―OH GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5およびK378R/Y639F/H784A変異T7ポリメラーゼにより、400ul転写が準備された。2’―OMe QTP(mQ)が下に示される:
溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。隣接するウェルにロードされた対応するMNA転写産物と共移動した明らかなバンドが観察された。2’―OMe QTPを伴わずにインキュベートされた陰性対照転写物は、観察可能なバンドを生成しなかった。
(実施例17)修飾および野生型T7ポリメラーゼを用いて2’―OMe UTP、2’―OMe ATP、2’―OMe GTP、2’―OMe CTPまたは2’―OH NTPsを導入する転写(rGdTmM組成物)
50nMの30ヌクレオチドの内側の連続縮重領域を伴う二本鎖転写テンプレート、200mM HEPES、40mM DTT、2mMスペルミジン、0.01%TritonX―100、各1.5mMの2’―OMe UTP、2’―OMe ATP、2’―OMe CTPおよび2’―OMe GTP、または各1.5mMの2’―OH UTP、2’―OH ATP、2’―OH CTPおよび2’―OH GTP、8mM MgCl2、2.5mM MnCl2、1mM GMP、10%PEG8000、無機ピロホスファターゼ2.5単位/ml、pH7.5および以下に示される変異または野生型T7ポリメラーゼ(WTがK378R、FがK378R/Y639F、LがK378R/Y639L、FAがK378R/Y639F/H784A、LAがK378R/Y639L/H784A)により、400ulの転写が準備された。溶液が37℃で一晩インキュベートされ、沈殿され、変性ローディングバッファーに溶解され、変性PAGEを用いて分析された後、UVシャドウイングを用いて蛍光板上で可視化された。図11に示されるように、異なる転写の収率が観察された。
説明および実施例により本発明を記載してきたが、当業者には当然のことながら、本発明は様々な実施形態において実施することができ、以上の説明および実施例は説明を目的としたものであり、以下の請求の範囲を制限するためのものではない。