JP5458231B2 - フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 - Google Patents
フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 Download PDFInfo
- Publication number
- JP5458231B2 JP5458231B2 JP2008254836A JP2008254836A JP5458231B2 JP 5458231 B2 JP5458231 B2 JP 5458231B2 JP 2008254836 A JP2008254836 A JP 2008254836A JP 2008254836 A JP2008254836 A JP 2008254836A JP 5458231 B2 JP5458231 B2 JP 5458231B2
- Authority
- JP
- Japan
- Prior art keywords
- fluorapatite
- powder
- slurry
- hydroxyapatite
- hydrogen fluoride
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images

Classifications
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B25/00—Phosphorus; Compounds thereof
- C01B25/16—Oxyacids of phosphorus; Salts thereof
- C01B25/26—Phosphates
- C01B25/32—Phosphates of magnesium, calcium, strontium, or barium
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/02—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising inorganic material
- B01J20/04—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising inorganic material comprising compounds of alkali metals, alkaline earth metals or magnesium
- B01J20/048—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof comprising inorganic material comprising compounds of alkali metals, alkaline earth metals or magnesium containing phosphorus, e.g. phosphates, apatites, hydroxyapatites
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28002—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their physical properties
- B01J20/28004—Sorbent size or size distribution, e.g. particle size
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/28—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties
- B01J20/28002—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof characterised by their form or physical properties characterised by their physical properties
- B01J20/28011—Other properties, e.g. density, crush strength
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J20/00—Solid sorbent compositions or filter aid compositions; Sorbents for chromatography; Processes for preparing, regenerating or reactivating thereof
- B01J20/281—Sorbents specially adapted for preparative, analytical or investigative chromatography
- B01J20/282—Porous sorbents
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2220/00—Aspects relating to sorbent materials
- B01J2220/50—Aspects relating to the use of sorbent or filter aid materials
- B01J2220/52—Sorbents specially adapted for preparative chromatography
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J2220/00—Aspects relating to sorbent materials
- B01J2220/50—Aspects relating to the use of sorbent or filter aid materials
- B01J2220/56—Use in the form of a bed
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/29—Coated or structually defined flake, particle, cell, strand, strand portion, rod, filament, macroscopic fiber or mass thereof
- Y10T428/2982—Particulate matter [e.g., sphere, flake, etc.]
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Analytical Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Inorganic Chemistry (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Treatment Of Liquids With Adsorbents In General (AREA)
Description
(1) カルシウム源と、リン酸源と、フッ素源とを原材料として、湿式法を用いて合成されるフッ素アパタイトを含有するスラリーに超音波を付与する第1の工程と、
前記スラリーを乾燥することにより、前記フッ素アパタイトを造粒して主としてフッ素アパタイトで構成されるフッ素アパタイト粉体を得る第2の工程とを有することを特徴とするフッ素アパタイト粉体の製造方法。
これにより、優れた粒子強度を有するフッ素アパタイト粉体を製造することができる。
これにより、優れた粒子強度を有するフッ素アパタイト粉体を製造することができる。
これにより、フッ素アパタイトを確実にスラリー中に合成することができる。
これにより、得られたフッ素アパタイト粉体は優れた粒子強度を有するものとなる。
当該フッ素アパタイト粉体を、平均粒径40±5μmの大きさに分級し、圧縮粒子強度を測定したとき、前記圧縮粒子強度が5.4MPa以上であることを特徴とするフッ素アパタイト粉体。
本発明のフッ素アパタイト粉体の製造方法は、カルシウム源と、リン酸源と、フッ素源とを原材料として、湿式法を用いて合成されるフッ素アパタイトを含有するスラリーに超音波を付与する第1の工程と、このフッ素アパタイトを含有するスラリーを乾燥することにより、主としてフッ素アパタイトで構成されるフッ素アパタイト粉体を得る第2の工程とを有する。
以下では、まず、この本発明のフッ素アパタイト粉体の第1製造方法について説明する。
まず、ハイドロキシアパタイトを含有するスラリーを調製する。
ハイドロキシアパタイトを含有するスラリーは、各種合成方法を用いて得ることができるが、カルシウム源とリン酸源との少なくとも一方を溶液として用いる湿式合成法によって得るのが好ましい。このような方法を用いると、微細なハイドロキシアパタイト(ハイドロキシアパタイトの一次粒子)がスラリー中に形成されるとともに、このハイドロキシアパタイトが均一に分散されたスラリー(ハイドロキシアパタイトスラリー)を得ることができる。
一方、第1の製造方法では、ハイドロキシアパタイトを含むスラリーとは別に、フッ素源として、フッ化水素を含有するフッ化水素含有液を調製する。
次に、前記工程[S1]で調製されたハイドロキシアパタイトスラリーと前記工程[S2]で調製されたフッ化水素含有液とを混合することにより、フッ化水素含有液を含むハイドロキシアパタイトスラリー中において、ハイドロキシアパタイトとフッ化水素とを反応させる湿式法により、フッ素アパタイトを得る。
Ca10(PO4)6(OH)2−2xF2X ・・・ (I)
[ただし、式(I)中、xは0<x≦1である。]
次に、フッ素アパタイトを含有するフッ素アパタイトスラリーを乾燥することにより、フッ素アパタイトを造粒させて、主としてフッ素アパタイトで構成されるフッ素アパタイト粉体(乾燥粒子)を得る。
以上のような工程を経て、フッ素アパタイト粉体を製造することができる。
次に、本発明のフッ素アパタイト粉体の第2製造方法について説明する。
[S1’−1] カルシウム源含有液調製工程
まず、カルシウムを含むカルシウム源(カルシウム系化合物)を含有するカルシウム源含有液を調製する。
次に、フッ素源として、フッ化水素を含有するフッ化水素含有液を調製する。
次に、リン酸源を含有するリン酸源含有液を調製する。
リン酸源としては、前記第1製造方法で挙げたのと同様のものを用いることができる。
次に、前記工程[S1’−2]および[S1’−3]でそれぞれ調製した、フッ化水素含有液およびリン酸源含有液を混合して第2の混合液を得る。
次に、前記工程[S1’−1]で調製されたカルシウム源含有液と、前記工程[S1’−4]で得られた第2の混合液とを混合することにより第1の混合液を得、この第1の混合液中において、カルシウム源(カルシウム系化合物)とフッ化水素とリン酸源とを反応させることにより、フッ素アパタイト(フッ素アパタイトの一次粒子)を得る。
次に、フッ素アパタイトを含有するフッ素アパタイトスラリーを乾燥することにより、フッ素アパタイトを造粒させて、主としてフッ素アパタイトで構成されるフッ素アパタイト粉体(乾燥粒子)を得る。
なお、この第2の製造方法では、工程[S1’]および工程[S2’]により、本発明のフッ素アパタイト粉体の製造方法における第1の工程が構成され、工程[S3’]により、第2の工程が構成される。
1.フッ素アパタイトの製造
(実施例1)
まず、水酸化カルシウム(カルシウム源)を純水に懸濁させ、その中へ、リン酸(リン酸源)水溶液を滴下していき、かつ十分に撹拌した。これにより、10wt%のハイドロキシアパタイトを含むスラリー50Lを得た。
まず、カルシウム源として水酸化カルシウム(カルシウム源)を3.11kg用意し、この水酸化カルシウムを9kgの純水に懸濁させることにより10wt%水酸化カルシウム懸濁液を調製した。
フッ素アパタイトを含むスラリーへの超音波の付与を省略した以外は、前記実施例1と同様にして、中心粒径約40μmの乾燥粒子を得た。
フッ素アパタイトを含むスラリーへの超音波の付与を省略した以外は、前記実施例2と同様にして、中心粒径約40μmの乾燥粒子を得た。
まず、水酸化カルシウム(カルシウム源)を純水に懸濁させ、その中へ、リン酸(リン酸源)水溶液を滴下していき、かつ十分に撹拌した。これにより、10wt%のハイドロキシアパタイトを含むスラリー50Lを得た。
ハイドロキシアパタイトを含むスラリーへの超音波の付与を省略した以外は、前記参考例1と同様にして、中心粒径約40μmの乾燥粒子を得た。
2−1.アパタイト凝集体の粒度分布の評価
実施例1、2および参考例1で、フッ素アパタイト(またはハイドロキシアパタイト)を含むスラリーに対する超音波付与の途中で採取した、超音波付与後1時間後、2時間後および3時間後のスラリーについて、それぞれ、粒度分布測定装置(マイクロトラック社製、「FRA」)を用いて、スラリー中に含まれるフッ素アパタイト(またはハイドロキシアパタイト)の凝集体の50%粒径を求めた。
その測定結果を表1に示す。
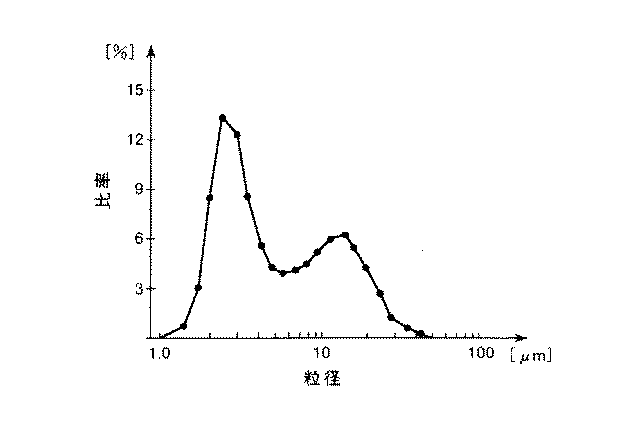
実施例1、2、比較例1、2および参考例1、2の乾燥粒子(フッ素アパタイト粉体またはハイドロキシアパタイト粉体)について、それぞれ、微小圧縮試験機(島津製作所社製、「型番MCT−W200−J」)を用いて、その粒子強度を求めた。
その測定結果を表2に示す。
Claims (7)
- カルシウム源と、リン酸源と、フッ素源とを原材料として、湿式法を用いて合成されるフッ素アパタイトを含有するスラリーに超音波を付与する第1の工程と、
前記スラリーを乾燥することにより、前記フッ素アパタイトを造粒して主としてフッ素アパタイトで構成されるフッ素アパタイト粉体を得る第2の工程とを有することを特徴とするフッ素アパタイト粉体の製造方法。 - 前記第1の工程において、前記スラリーに対する超音波の付与は、前記フッ素アパタイトの合成の後に行う請求項1に記載のフッ素アパタイト粉体の製造方法。
- 前記第1の工程において、前記フッ素アパタイトを含有するスラリーは、前記カルシウム源を含有するスラリーに、前記リン酸源と前記フッ素源との混合物を滴下して反応させることにより、合成されたハイドロキシアパタイトが有する水酸基の少なくとも一部がフッ素原子で置換されることにより得られる請求項1または2に記載のフッ素アパタイト粉体の製造方法。
- 請求項1ないし3のいずれかに記載のフッ素アパタイト粉体の製造方法を用いて製造されたことを特徴とするフッ素アパタイト粉体。
- ハイドロキシアパタイトが有する水酸基の一部が、フッ素原子で置換されてなるフッ素アパタイトを含有するスラリーを乾燥することにより得られた、前記フッ素アパタイトが造粒した主としてフッ素アパタイトで構成されるフッ素アパタイト粉体であって、
当該フッ素アパタイト粉体を、平均粒径40±5μmの大きさに分級し、圧縮粒子強度を測定したとき、前記圧縮粒子強度が5.4MPa以上であることを特徴とするフッ素アパタイト粉体。 - 請求項4または5のフッ素アパタイト粉体を吸着剤として、または、当該フッ素アパタイト粉体を焼成して得られた焼結粒子を吸着剤として、備える吸着装置。
- 請求項4または5のフッ素アパタイト粉体を吸着剤として、または、当該フッ素アパタイト粉体を焼成して得られた焼結粒子を吸着剤として、備え、複数のタンパク質を含有する液体中の前記複数のタンパク質を吸着し、それぞれの前記タンパク質と前記フッ素アパタイト粉体との吸着性の差に基づいて、各前記タンパク質を分離する吸着装置。
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008254836A JP5458231B2 (ja) | 2008-09-30 | 2008-09-30 | フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 |
| US12/568,753 US10407306B2 (en) | 2008-09-30 | 2009-09-29 | Method of producing fluoroapatite powder, fluoroapatite powder, and adsorption apparatus |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008254836A JP5458231B2 (ja) | 2008-09-30 | 2008-09-30 | フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2010083713A JP2010083713A (ja) | 2010-04-15 |
| JP5458231B2 true JP5458231B2 (ja) | 2014-04-02 |
Family
ID=42248044
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2008254836A Active JP5458231B2 (ja) | 2008-09-30 | 2008-09-30 | フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 |
Country Status (2)
| Country | Link |
|---|---|
| US (1) | US10407306B2 (ja) |
| JP (1) | JP5458231B2 (ja) |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5248904B2 (ja) * | 2008-04-18 | 2013-07-31 | Hoya株式会社 | 被覆粒子、被覆粒子の製造方法および吸着装置 |
| JP5458231B2 (ja) * | 2008-09-30 | 2014-04-02 | HOYA Technosurgical株式会社 | フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 |
| JP5509493B2 (ja) * | 2009-06-02 | 2014-06-04 | HOYA Technosurgical株式会社 | 粉体の製造方法、粉体、吸着装置 |
| JP5509494B2 (ja) * | 2009-06-02 | 2014-06-04 | HOYA Technosurgical株式会社 | 粉体の製造方法、粉体、吸着装置 |
| JP6141256B2 (ja) * | 2012-03-29 | 2017-06-07 | Hoya株式会社 | フッ素アパタイト、吸着装置および分離方法 |
| CN102961259A (zh) * | 2012-12-12 | 2013-03-13 | 郑荣荣 | 一种用于牙科修复的硬质涂层材料及其制备方法 |
| JP6068216B2 (ja) * | 2013-03-21 | 2017-01-25 | 三菱製紙株式会社 | フッ素アパタイトの製造方法およびフッ素アパタイト微粒子の製造方法 |
| JP7787598B2 (ja) | 2021-01-28 | 2025-12-17 | ナノ デンティカ エービー | フッ素アパタイト結晶を生成するための方法 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6306297B1 (en) * | 1968-07-08 | 2001-10-23 | Asahi Kogaku Kogyo Kabushiki Kaisha | Packing material for liquid chromatography and process for producing the same |
| US4157378A (en) * | 1973-01-11 | 1979-06-05 | Colgate Palmolive Company | Process for preparing fluorapatite |
| JPH01264915A (ja) * | 1988-04-15 | 1989-10-23 | Asahi Optical Co Ltd | フッ素アパタイトの製造方法 |
| JPH01308942A (ja) * | 1988-06-08 | 1989-12-13 | Mitsubishi Electric Corp | 固体表面付着微粒子計数装置 |
| US5344640A (en) * | 1991-10-22 | 1994-09-06 | Mallinckrodt Medical, Inc. | Preparation of apatite particles for medical diagnostic imaging |
| JP4172937B2 (ja) | 2002-01-11 | 2008-10-29 | Hoya株式会社 | 粉体の製造方法 |
| FR2848856B1 (fr) * | 2002-12-24 | 2007-05-25 | Cadorel Catherine | Materiau a usage medical ou veterinaire, son procede d'obtention et ses applications |
| JP2004330113A (ja) * | 2003-05-08 | 2004-11-25 | Pentax Corp | 吸着剤の製造方法および吸着剤 |
| JP5458229B2 (ja) * | 2007-08-03 | 2014-04-02 | HOYA Technosurgical株式会社 | フッ素アパタイトの製造方法、フッ素アパタイトおよび吸着装置 |
| JP5458230B2 (ja) * | 2007-08-30 | 2014-04-02 | HOYA Technosurgical株式会社 | フッ素アパタイトの製造方法、フッ素アパタイトおよび吸着装置 |
| JP5301804B2 (ja) * | 2007-10-01 | 2013-09-25 | Hoya株式会社 | フッ素アパタイトの乾燥粒子および吸着装置 |
| JP5463537B2 (ja) * | 2008-02-22 | 2014-04-09 | HOYA Technosurgical株式会社 | 分離方法 |
| JP5458231B2 (ja) * | 2008-09-30 | 2014-04-02 | HOYA Technosurgical株式会社 | フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 |
-
2008
- 2008-09-30 JP JP2008254836A patent/JP5458231B2/ja active Active
-
2009
- 2009-09-29 US US12/568,753 patent/US10407306B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| JP2010083713A (ja) | 2010-04-15 |
| US10407306B2 (en) | 2019-09-10 |
| US20100255306A1 (en) | 2010-10-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5458231B2 (ja) | フッ素アパタイト粉体の製造方法、フッ素アパタイト粉体および吸着装置 | |
| JP6752724B2 (ja) | 球状多孔質ヒドロキシアパタイト吸着剤及びその方法 | |
| JP5458230B2 (ja) | フッ素アパタイトの製造方法、フッ素アパタイトおよび吸着装置 | |
| JP5301804B2 (ja) | フッ素アパタイトの乾燥粒子および吸着装置 | |
| CN101035741B (zh) | 陶瓷颗粒群的制造方法 | |
| JP5458229B2 (ja) | フッ素アパタイトの製造方法、フッ素アパタイトおよび吸着装置 | |
| JP2016193798A (ja) | リン酸カルシウム焼結体粒子の製造方法 | |
| JP6025795B2 (ja) | 結晶性シリコチタネートの製造方法 | |
| JP5544813B2 (ja) | 球状ヒドロキシアパタイトの製造方法 | |
| JP2004330113A (ja) | 吸着剤の製造方法および吸着剤 | |
| JP5509494B2 (ja) | 粉体の製造方法、粉体、吸着装置 | |
| KR102387660B1 (ko) | 리튬화합물 제조시 발생하는 슬러지를 이용한 고순도 수산화아파타이트 분말 제조 방법 및 이에 따른 수산화아파타이트 분말 | |
| JP5509493B2 (ja) | 粉体の製造方法、粉体、吸着装置 | |
| CN108584896A (zh) | 掺镁羟基磷灰石及其制备方法 | |
| JP6356567B2 (ja) | 結晶性シリコチタネートの製造方法 | |
| JPH0832552B2 (ja) | ハイドロキシアパタイト微細単結晶及びその製造方法 | |
| JP2004284890A (ja) | 水酸アパタイトの製造方法 | |
| JP2003054943A (ja) | 板状炭酸カルシウム球状複合体およびその製造方法 | |
| JP7824477B1 (ja) | 金属含有化合物粒子の製造方法及び金属含有化合物粒子 | |
| JP6230362B2 (ja) | 粉体の製造方法 | |
| JP2021181400A (ja) | リン酸カルシウム焼結体粒子の製造方法 | |
| JP2003146656A (ja) | 板状炭酸カルシウム系の球状複合体の製造方法 | |
| JP2004224620A (ja) | 湿式粉砕法により得られるリン酸カルシウム前駆体からのリン酸カルシウム微粉末の製造方法 | |
| JP2007125478A (ja) | リン成分の捕捉剤及びその製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20110407 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20121212 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20130521 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20130719 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20130924 |
|
| A711 | Notification of change in applicant |
Free format text: JAPANESE INTERMEDIATE CODE: A712 Effective date: 20131021 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20131021 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20131219 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5458231 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |