JP5897576B2 - レンチウイルスベクターの半−安定産生 - Google Patents
レンチウイルスベクターの半−安定産生 Download PDFInfo
- Publication number
- JP5897576B2 JP5897576B2 JP2013526469A JP2013526469A JP5897576B2 JP 5897576 B2 JP5897576 B2 JP 5897576B2 JP 2013526469 A JP2013526469 A JP 2013526469A JP 2013526469 A JP2013526469 A JP 2013526469A JP 5897576 B2 JP5897576 B2 JP 5897576B2
- Authority
- JP
- Japan
- Prior art keywords
- semi
- gene
- stable
- cell line
- packaging cell
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 239000013598 vector Substances 0.000 title claims description 54
- 238000004519 manufacturing process Methods 0.000 title description 46
- 210000004027 cell Anatomy 0.000 claims description 148
- 238000004806 packaging method and process Methods 0.000 claims description 74
- 108090000623 proteins and genes Proteins 0.000 claims description 60
- 230000014509 gene expression Effects 0.000 claims description 43
- 230000010354 integration Effects 0.000 claims description 40
- 238000000034 method Methods 0.000 claims description 27
- 239000003550 marker Substances 0.000 claims description 24
- 102000004169 proteins and genes Human genes 0.000 claims description 24
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 claims description 22
- 238000012546 transfer Methods 0.000 claims description 20
- 108700004025 env Genes Proteins 0.000 claims description 17
- 241000701447 unidentified baculovirus Species 0.000 claims description 17
- 101150030339 env gene Proteins 0.000 claims description 15
- 102100034349 Integrase Human genes 0.000 claims description 13
- 108020004684 Internal Ribosome Entry Sites Proteins 0.000 claims description 13
- 108700004029 pol Genes Proteins 0.000 claims description 13
- 101710091045 Envelope protein Proteins 0.000 claims description 12
- 101710188315 Protein X Proteins 0.000 claims description 12
- 108700004030 rev Genes Proteins 0.000 claims description 12
- 108700004026 gag Genes Proteins 0.000 claims description 10
- 230000001105 regulatory effect Effects 0.000 claims description 9
- 229930193140 Neomycin Natural products 0.000 claims description 7
- 241000713880 Spleen focus-forming virus Species 0.000 claims description 7
- 108010084455 Zeocin Proteins 0.000 claims description 7
- 229930027917 kanamycin Natural products 0.000 claims description 7
- 229960000318 kanamycin Drugs 0.000 claims description 7
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 claims description 7
- 229930182823 kanamycin A Natural products 0.000 claims description 7
- 229960004927 neomycin Drugs 0.000 claims description 7
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 claims description 7
- 239000013613 expression plasmid Substances 0.000 claims description 6
- 241000713666 Lentivirus Species 0.000 claims description 4
- 210000005260 human cell Anatomy 0.000 claims description 4
- 101100118916 Gibbon ape leukemia virus env gene Proteins 0.000 claims description 3
- 101001001300 Human cytomegalovirus (strain Towne) 65 kDa phosphoprotein Proteins 0.000 claims description 3
- 239000003242 anti bacterial agent Substances 0.000 claims description 3
- 238000012258 culturing Methods 0.000 claims description 3
- 101150098213 rev gene Proteins 0.000 claims description 3
- 108091006104 gene-regulatory proteins Proteins 0.000 claims description 2
- 102000034356 gene-regulatory proteins Human genes 0.000 claims description 2
- 230000003115 biocidal effect Effects 0.000 claims 2
- 101710177291 Gag polyprotein Proteins 0.000 claims 1
- 101710125418 Major capsid protein Proteins 0.000 claims 1
- 101710150344 Protein Rev Proteins 0.000 claims 1
- 108010089520 pol Gene Products Proteins 0.000 claims 1
- 108020004414 DNA Proteins 0.000 description 39
- 208000002267 Anti-neutrophil cytoplasmic antibody-associated vasculitis Diseases 0.000 description 37
- 239000013612 plasmid Substances 0.000 description 34
- 238000003146 transient transfection Methods 0.000 description 14
- 239000013607 AAV vector Substances 0.000 description 11
- 208000031886 HIV Infections Diseases 0.000 description 10
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 10
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 10
- 239000000523 sample Substances 0.000 description 10
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 9
- 238000001890 transfection Methods 0.000 description 9
- 210000000349 chromosome Anatomy 0.000 description 8
- 239000000543 intermediate Substances 0.000 description 8
- 238000010586 diagram Methods 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 108010027225 gag-pol Fusion Proteins Proteins 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 230000006798 recombination Effects 0.000 description 7
- 238000005215 recombination Methods 0.000 description 7
- 230000001052 transient effect Effects 0.000 description 7
- 108700010908 HIV-1 proteins Proteins 0.000 description 6
- 238000002105 Southern blotting Methods 0.000 description 6
- 238000010367 cloning Methods 0.000 description 6
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 241000700605 Viruses Species 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 101150047047 gag-pol gene Proteins 0.000 description 5
- 230000030279 gene silencing Effects 0.000 description 5
- 238000001415 gene therapy Methods 0.000 description 5
- 230000002068 genetic effect Effects 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- 101100524324 Adeno-associated virus 2 (isolate Srivastava/1982) Rep78 gene Proteins 0.000 description 4
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 4
- 108010067390 Viral Proteins Proteins 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 238000001378 electrochemiluminescence detection Methods 0.000 description 4
- 238000001476 gene delivery Methods 0.000 description 4
- 230000006801 homologous recombination Effects 0.000 description 4
- 238000002744 homologous recombination Methods 0.000 description 4
- 238000007901 in situ hybridization Methods 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 238000010899 nucleation Methods 0.000 description 4
- 238000013518 transcription Methods 0.000 description 4
- 230000035897 transcription Effects 0.000 description 4
- 238000010361 transduction Methods 0.000 description 4
- 230000026683 transduction Effects 0.000 description 4
- 230000010474 transient expression Effects 0.000 description 4
- 230000003612 virological effect Effects 0.000 description 4
- 208000030507 AIDS Diseases 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 238000010222 PCR analysis Methods 0.000 description 3
- 108700005077 Viral Genes Proteins 0.000 description 3
- 230000003321 amplification Effects 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 210000005220 cytoplasmic tail Anatomy 0.000 description 3
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 3
- 108010030074 endodeoxyribonuclease MluI Proteins 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 238000010353 genetic engineering Methods 0.000 description 3
- 238000009396 hybridization Methods 0.000 description 3
- 230000007774 longterm Effects 0.000 description 3
- 238000003199 nucleic acid amplification method Methods 0.000 description 3
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 3
- 239000002953 phosphate buffered saline Substances 0.000 description 3
- 108091008146 restriction endonucleases Proteins 0.000 description 3
- 230000001177 retroviral effect Effects 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000013605 shuttle vector Substances 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 241001430294 unidentified retrovirus Species 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- 108091093088 Amplicon Proteins 0.000 description 2
- 235000005881 Calendula officinalis Nutrition 0.000 description 2
- 240000001432 Calendula officinalis Species 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- 238000002965 ELISA Methods 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 102100021519 Hemoglobin subunit beta Human genes 0.000 description 2
- 108091005904 Hemoglobin subunit beta Proteins 0.000 description 2
- 229920000209 Hexadimethrine bromide Polymers 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 102100034864 Homeobox protein Hox-D9 Human genes 0.000 description 2
- 101001037162 Homo sapiens Homeobox protein Hox-D1 Proteins 0.000 description 2
- 101000962573 Homo sapiens Homeobox protein Hox-D10 Proteins 0.000 description 2
- 101000962591 Homo sapiens Homeobox protein Hox-D11 Proteins 0.000 description 2
- 101001037158 Homo sapiens Homeobox protein Hox-D3 Proteins 0.000 description 2
- 101001041136 Homo sapiens Homeobox protein Hox-D4 Proteins 0.000 description 2
- 101001019776 Homo sapiens Homeobox protein Hox-D8 Proteins 0.000 description 2
- 101001019766 Homo sapiens Homeobox protein Hox-D9 Proteins 0.000 description 2
- 101000716729 Homo sapiens Kit ligand Proteins 0.000 description 2
- 108010048209 Human Immunodeficiency Virus Proteins Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- 241000713869 Moloney murine leukemia virus Species 0.000 description 2
- 241000714177 Murine leukemia virus Species 0.000 description 2
- 101710182846 Polyhedrin Proteins 0.000 description 2
- 101710149951 Protein Tat Proteins 0.000 description 2
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 108700019146 Transgenes Proteins 0.000 description 2
- 108091093126 WHP Posttrascriptional Response Element Proteins 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 208000022806 beta-thalassemia major Diseases 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000001185 bone marrow Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 238000010370 cell cloning Methods 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000012761 co-transfection Methods 0.000 description 2
- 238000010924 continuous production Methods 0.000 description 2
- 230000029087 digestion Effects 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 210000004700 fetal blood Anatomy 0.000 description 2
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 2
- 238000012239 gene modification Methods 0.000 description 2
- 238000012226 gene silencing method Methods 0.000 description 2
- 230000005017 genetic modification Effects 0.000 description 2
- 235000013617 genetically modified food Nutrition 0.000 description 2
- 238000011194 good manufacturing practice Methods 0.000 description 2
- 102000055151 human KITLG Human genes 0.000 description 2
- 210000003917 human chromosome Anatomy 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 210000003041 ligament Anatomy 0.000 description 2
- 238000013507 mapping Methods 0.000 description 2
- 230000031864 metaphase Effects 0.000 description 2
- 210000005087 mononuclear cell Anatomy 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 230000003595 spectral effect Effects 0.000 description 2
- 230000010473 stable expression Effects 0.000 description 2
- 239000006228 supernatant Substances 0.000 description 2
- 238000004448 titration Methods 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 238000010200 validation analysis Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- SNBCLPGEMZEWLU-QXFUBDJGSA-N 2-chloro-n-[[(2r,3s,5r)-3-hydroxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methyl]acetamide Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CNC(=O)CCl)[C@@H](O)C1 SNBCLPGEMZEWLU-QXFUBDJGSA-N 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- 101100111800 Caenorhabditis elegans best-24 gene Proteins 0.000 description 1
- 101710205625 Capsid protein p24 Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 241001227713 Chiron Species 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 238000007399 DNA isolation Methods 0.000 description 1
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 1
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 1
- 241000702421 Dependoparvovirus Species 0.000 description 1
- 238000008157 ELISA kit Methods 0.000 description 1
- UPEZCKBFRMILAV-JNEQICEOSA-N Ecdysone Natural products O=C1[C@H]2[C@@](C)([C@@H]3C([C@@]4(O)[C@@](C)([C@H]([C@H]([C@@H](O)CCC(O)(C)C)C)CC4)CC3)=C1)C[C@H](O)[C@H](O)C2 UPEZCKBFRMILAV-JNEQICEOSA-N 0.000 description 1
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 1
- 101710121417 Envelope glycoprotein Proteins 0.000 description 1
- 241000701959 Escherichia virus Lambda Species 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 241001663880 Gammaretrovirus Species 0.000 description 1
- 208000031448 Genomic Instability Diseases 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 101001002657 Homo sapiens Interleukin-2 Proteins 0.000 description 1
- 101000799461 Homo sapiens Thrombopoietin Proteins 0.000 description 1
- 108010061833 Integrases Proteins 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- 241000283923 Marmota monax Species 0.000 description 1
- 208000024556 Mendelian disease Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000011755 Phosphoglycerate Kinase Human genes 0.000 description 1
- 101710177166 Phosphoprotein Proteins 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 239000012980 RPMI-1640 medium Substances 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 108091027981 Response element Proteins 0.000 description 1
- 238000012300 Sequence Analysis Methods 0.000 description 1
- 101710149279 Small delta antigen Proteins 0.000 description 1
- 241000256251 Spodoptera frugiperda Species 0.000 description 1
- 206010043391 Thalassaemia beta Diseases 0.000 description 1
- 101001099217 Thermotoga maritima (strain ATCC 43589 / DSM 3109 / JCM 10099 / NBRC 100826 / MSB8) Triosephosphate isomerase Proteins 0.000 description 1
- 208000026487 Triploidy Diseases 0.000 description 1
- 102100022563 Tubulin polymerization-promoting protein Human genes 0.000 description 1
- 241000711975 Vesicular stomatitis virus Species 0.000 description 1
- 108700022715 Viral Proteases Proteins 0.000 description 1
- 230000001464 adherent effect Effects 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- UPEZCKBFRMILAV-UHFFFAOYSA-N alpha-Ecdysone Natural products C1C(O)C(O)CC2(C)C(CCC3(C(C(C(O)CCC(C)(C)O)C)CCC33O)C)C3=CC(=O)C21 UPEZCKBFRMILAV-UHFFFAOYSA-N 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000036436 anti-hiv Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 208000005980 beta thalassemia Diseases 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 210000002230 centromere Anatomy 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 230000008045 co-localization Effects 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229960000633 dextran sulfate Drugs 0.000 description 1
- 229960003722 doxycycline Drugs 0.000 description 1
- XQTWDDCIUJNLTR-CVHRZJFOSA-N doxycycline monohydrate Chemical compound O.O=C1C2=C(O)C=CC=C2[C@H](C)[C@@H]2C1=C(O)[C@]1(O)C(=O)C(C(N)=O)=C(O)[C@@H](N(C)C)[C@@H]1[C@H]2O XQTWDDCIUJNLTR-CVHRZJFOSA-N 0.000 description 1
- UPEZCKBFRMILAV-JMZLNJERSA-N ecdysone Chemical compound C1[C@@H](O)[C@@H](O)C[C@]2(C)[C@@H](CC[C@@]3([C@@H]([C@@H]([C@H](O)CCC(C)(C)O)C)CC[C@]33O)C)C3=CC(=O)[C@@H]21 UPEZCKBFRMILAV-JMZLNJERSA-N 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 1
- 101150098622 gag gene Proteins 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 238000012500 good manufacturing practice method Methods 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 238000005286 illumination Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 238000011031 large-scale manufacturing process Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 235000004213 low-fat Nutrition 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 208000017830 lymphoblastoma Diseases 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 238000007857 nested PCR Methods 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 229940029358 orthoclone okt3 Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 238000009520 phase I clinical trial Methods 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 230000001124 posttranscriptional effect Effects 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 239000013608 rAAV vector Substances 0.000 description 1
- 101150066583 rep gene Proteins 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 108010056030 retronectin Proteins 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 238000013341 scale-up Methods 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000012163 sequencing technique Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- YEENEYXBHNNNGV-XEHWZWQGSA-M sodium;3-acetamido-5-[acetyl(methyl)amino]-2,4,6-triiodobenzoate;(2r,3r,4s,5s,6r)-2-[(2r,3s,4s,5r)-3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound [Na+].CC(=O)N(C)C1=C(I)C(NC(C)=O)=C(I)C(C([O-])=O)=C1I.O[C@H]1[C@H](O)[C@@H](CO)O[C@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 YEENEYXBHNNNGV-XEHWZWQGSA-M 0.000 description 1
- 208000003265 stomatitis Diseases 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 108700004027 tat Genes Proteins 0.000 description 1
- ABZLKHKQJHEPAX-UHFFFAOYSA-N tetramethylrhodamine Chemical compound C=12C=CC(N(C)C)=CC2=[O+]C2=CC(N(C)C)=CC=C2C=1C1=CC=CC=C1C([O-])=O ABZLKHKQJHEPAX-UHFFFAOYSA-N 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 208000005925 vesicular stomatitis Diseases 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
Images




Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
- C12N15/867—Retroviral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
- C12N15/866—Baculoviral vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/85—Vectors or expression systems specially adapted for eukaryotic hosts for animal cells
- C12N15/86—Viral vectors
- C12N15/867—Retroviral vectors
- C12N15/8673—Special methods for packaging systems
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
- C12N7/02—Recovery or purification
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/14011—Baculoviridae
- C12N2710/14111—Nucleopolyhedrovirus, e.g. autographa californica nucleopolyhedrovirus
- C12N2710/14141—Use of virus, viral particle or viral elements as a vector
- C12N2710/14144—Chimeric viral vector comprising heterologous viral elements for production of another viral vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2740/00—Reverse transcribing RNA viruses
- C12N2740/00011—Details
- C12N2740/10011—Retroviridae
- C12N2740/16011—Human Immunodeficiency Virus, HIV
- C12N2740/16051—Methods of production or purification of viral material
- C12N2740/16052—Methods of production or purification of viral material relating to complementing cells and packaging systems for producing virus or viral particles
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2750/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssDNA viruses
- C12N2750/00011—Details
- C12N2750/14011—Parvoviridae
- C12N2750/14111—Dependovirus, e.g. adenoassociated viruses
- C12N2750/14141—Use of virus, viral particle or viral elements as a vector
- C12N2750/14143—Use of virus, viral particle or viral elements as a vector viral genome or elements thereof as genetic vector
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/40—Systems of functionally co-operating vectors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2800/00—Nucleic acids vectors
- C12N2800/50—Vectors for producing vectors
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Chemical & Material Sciences (AREA)
- Zoology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Plant Pathology (AREA)
- Physics & Mathematics (AREA)
- Virology (AREA)
- Molecular Biology (AREA)
- Biophysics (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Description
本発明の第1の態様によれば、
i. 第1の発現カセットがレンチウイルスのgagおよびpol遺伝子をコードしかつ第2の発現カセットがレンチウイルスのrevおよび選択マーカーをコードする2つの発現カセットを含むAAV ITRに隣接した組込みカセットを含有するバキュロウイルス主鎖を含むハイブリッドベクター;および
ii. プロモーターの調節下にあるAAV Repオープンリードフレーム(ORF)を含有する発現プラスミド
から成るレンチウイルスの構造および調節タンパク質を安定発現する系が提供される。
i. レンチウイルスのgag、polおよびrevを安定して発現する細胞から成る半-安定レンチウイルスのパッケージング細胞株を培養するステップであって、かかる細胞は、安定してゲノム中に組込まれたAAV ITRに隣接した2つの発現カセットを含む組込みカセットの少なくとも1つのコピーを含有し、第1の発現カセットはレンチウイルスのgagおよびpol遺伝子をコードしかつ第2の発現カセットはレンチウイルスのrevおよび選択マーカーをコードすることを特徴とする前記ステップ;
ii.半-安定パッケージング細胞株にenv遺伝子を挿入するステップ;
iii.半-安定パッケージング細胞株に導入ベクターを挿入するステップ
を含むものである前記方法が提供される。
本発明の好ましい特徴および実施形態の詳細な説明を非限定の例を用いて説明しよう。
本発明はHIV-1-に基づくパッケージング細胞株を作製する新しい戦略を提供する。LV用の産生系の最適化は、LV技法に基づく遺伝子治療薬開発のために解決を要する重要な課題である。この技法を用いる臨床試験数が増加するにも関わらず、LVはかかる試験においてなお一過性トランスフェクションプロトコルを用いて作製されている。この方法では、LVの作製は未だに非常に高価でありかつ多数の患者向けには不満足である。この理由で、LV用の安定パッケージング細胞株を開発する多くの努力がなされてきた。安定なレンチウイルスパッケージング細胞株の開発における重要な課題の1つは宿主細胞を遺伝子操作するための正しいビヒクルの選定である。多くの場合、宿主細胞はプラスミドを用いて遺伝子操作で作られてきたが、かかる場合、ゲノム不安定性および遺伝子サイレンシング現象も観察されている。他の2例ではgag/polおよびrev遺伝子を安定して組み込むために、レトロウイルスベクターが使われている。今まで開発された安定パッケージング細胞株はいずれも臨床試験に用いられていない。
本発明の半-安定パッケージング細胞株は、HIV-1 gag-polおよびrev遺伝子を発現する組換えバキュロ-AAVパッケージング構築物の少なくとも1つのコピーを運ぶ宿主細胞から成る。バキュロ-AAVパッケージングベクターのゲノム組込みは、ITR-介在型AAVベクター組込みを得る目的でAAV repタンパク質の一過性発現により得ている。好ましくは、2つの発現カセットはテール・ツー・テール配向であり、それぞれが構成的プロモーターおよびポリAにより駆動され;、好ましくは、プロモーターはCMV、CMV IE、PGK、SV40、eF1α、SFFVおよびRSVから選択される。より好ましくは、構成的プロモーターはCMV IEプロモーターである。
i. 前記の半-安定パッケージング細胞株を培養するステップ、
ii. 半-安定パッケージング細胞株にenv遺伝子を挿入するステップ、
iii. 半-安定パッケージング細胞株に導入ベクターを挿入するステップ
を含む前記方法を提供する。
プラスミド
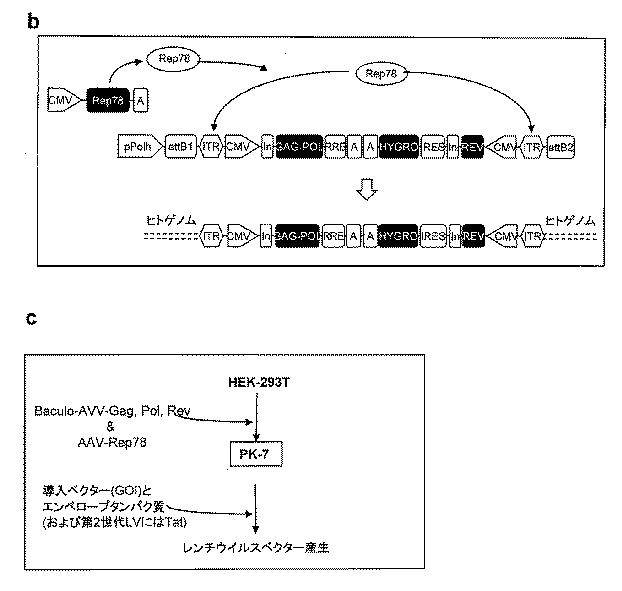
野生型HIV-1 gag、polおよびrev遺伝子を、pCG719-pKLgagpol(以後、簡略化のためにCMV-GPRと呼ぶ)(図1a、模式図9)およびpCG720-pKrev(以後、CMV-Revと呼ぶ)(図1a、模式図5)プラスミドからそれぞれMluI/NarIおよびMluI/NotI消化により切除した[25]。そのウイルス遺伝子を2つの異なる発現カセット(各カセットはCMV IEプロモーターにより駆動され、polyA配列を運ぶ)に入れて、Gateway(登録商標)pENTRTM4シャトルベクター(Invitrogen、Co.、Carlsbad、CA)中にテール・ツー・テール配向で挿入した。第1のカセットはgagとpol遺伝子を発現するが、第2のカセットはrev遺伝子と選択マーカーハイグロマイシン耐性(hygro)遺伝子を発現し;hygroはIRESの下流にクローニングしてバイシストロン(bi-cistronic)翻訳を可能にした。2つの発現ユニットを感染性AAVゲノムを運ぶ組換えpSUB201プラスミドのXbaI部位中に導入した[17]。得られる5'ITR-CMV-gagpol-polyA-polyA-hygro-IRES-rev-CMV-ITR3'カセットを次いで切除し、Gateway(登録商標)pENTRTM4シャトルベクター(Invitrogen, Co., Carlsbad, CA)中に挿入した。組換えハイブリッドバキュロ-AAVパッケージングベクター(バキュロ-AAV-GPR)(図1a、模式図1)を、2つのカセットを含有するpENTRTM4シャトルエントリーベクターとBaculoDirect直鎖DNA(BaculoDirectTMバキュロウイルス発現系, Invitrogen, Co.)との間のバクテリオファージλ部位-特異的組換え系を用いて得た。相同組換え中に、BaculoDNAのポリヘドリン遺伝子はその際、GPR二重カセットで置換えられた。pABCMV-rep78発現プラスミド(CMV-AAV-rep78)は、Recchia et al., 2004[18]に記載の発現ベクターpABS.43のCMV IEプロモーター下のAAV-rep78 ORFをクローニングすることにより得た(図1a、模式図4)。pMD.Gプラスミド(CMV-VSV-G)[19]は小胞性口内炎エンベロープ糖タンパク質(VSV-G)をコードする(図1a、模式図6)。第3世代導入ベクター、pCCLsin.PPT.hPGK.eGFP.WPRE.Amp(SIN-eGFP)[20]は構成的プロモーターhPGK下のeGFP遺伝子(図 1a、模式図2)を発現する。抗-HIV-1 Chim3トランスジーンを発現する第2世代PΔN-Chim3導入ベクターはPorcellini ら, 2009 & 2010 [21,22]に記載されている(図1a、模式図3)。pCMV-RD114-TR(CMV-RD114-TR)(図1a、模式図7)プラスミドは、ネコ内因性レトロウイルスRD114エンベロープの細胞外および膜貫通ドメインならびにA-MLVenv 4070Aの細胞質テール(TR)から作られたキメラRD114-TRエンベロープをコードする[23]。第2世代パッケージングpCMV-ΔR8.74(CMV-GPRT)構築物(図1a、模式図8)はHIV-1gag、pol、revおよびtat遺伝子をコードする[19]。
Spodoptera frugiperda(Sf9)昆虫細胞(Invitrogen, Co.)は10%FCS(EuroClone Ltd、UK)およびペニシリン-ストレプトマイシンとグルタミン(PSG)の組み合わせを補充したTC-100培地(Invitrogen, Co.)の懸濁液中で27℃にてCO2の非存在のもとで増殖した。ヒト胎児腎293T(HEK293T)細胞およびその誘導体クローン(PK-7)は10%FCSおよびPSGを補充したDulbecco改変Eagle培地(DMEM)で増殖した。CEM A3.01およびSupT1 Tリンパ芽球腫細胞は10%FCSおよびPSGを補充したRPMI 1640中で増殖した。CD34+造血幹細胞(HSC)および新生児白血球は臍帯血(UCB)からFicoll-Hypaque 勾配遠心分離(Lymphoprep, Nycomed Pharma AS, Oslo, Norway)で精製した。勾配分離の後、回収したUCB単核細胞環から、CD34マイクロビーズキットおよびMiniMACS分離カラム(Miltenyi Biotec, Sunnyvale, CA)を用いてポジティブ選択により、CD34+ HSCを単離した。CD34+細胞純度(>92%)はFACS分析(FACS Calibur BD Bioscience, San Jose, CA)およびFlowJoソフトウエア(Tree Star, Inc., Ashland, OR)により、抗-CD34-PEAb(BD PharmingenTM, San Diego, CA)を用いて確立した。CD34+細胞はヒト幹細胞因子(h-SCF)100ng/ml(R&D Systems, Minneapolis, MN)、h-Flt3L 100 ng/ml(Peprotech, Rocky Hill, NJ)、h-IL-620ng/ml(R&D Systems)およびヒトトロンボポエチン(h-Tpo)20ng/ml(Peprotech)を含有する20%血清Iscove改変Dulbecco培地(IMDM)で24時間、前刺激し、形質導入の間、同じ培地中で維持した。新生児白血球は48時間、RPMI中の可溶性抗-ヒトCD3(30ng/ml)(Orthoclone OKT3, Janssen-Cilag, UK)および組換えヒトIL-2(rhIL-2)50U/ml(Chiron, Emeryville, CA)で刺激し、次いで、10%FCS、PSG、およびrhIL-2を補充したRPMI中に保った。
組換えハイブリッドバキュロ-AAV-GPR DNAゲノムを運ぶバキュロウイルスをGateway(登録商標)適合化バキュロウイルスDNAシステム(Invitrogen、Co.)を用いてBaculoDirect法に従い作製した。組換えバキュロウイルス力価をプラークアッセイにより評価すると、Sf9細胞中の3継代のウイルス増幅後に1×1011 pfu/mlに対応した。1.5×106 HEK293T細胞を4μgのAAV-rep78発現プラスミドでトランスフェクトし、24時間後に組換えバキュロ-AAV-GPRに1,000のMOIにて感染することによりPK-7クローンを得た。細胞をハイグロマイシン無しで4日間維持し、次いで5×105細胞を、ハイグロマイシン(100μg/ml)の連続希釈濃度の存在する22枚の10cmディッシュに播いた。22枚のディッシュをELISAによるp24gag産生についてスクリーニングした。細胞を3.7×104細胞/ディッシュで播いたわずか1枚のディッシュが上清中に十分なp24gagを放出した。このディッシュは40コロニーを含有し、それらを全て拾い上げてスクリーニングした。そのうちの、p24gag産生に対してポジティブの評価を得た3コロニーをさらに特徴付けた。
HEK293T細胞から産生される偽型LVは、次のプラスミド:パッケージング構築物CMV-GPR(第3世代)[またはCMV-GPRT(第2世代)]、VSV-GまたはRD114-TRエンベロープ構築物、および第3世代SIN-eGFP[24]または第2世代PΔN-Chim3導入ベクター[21]の一過性共トランスフェクションにより得た。パッケージング:エンベロープ:導入ベクターの比は、特に断らない限り、6.5:3.5:10μg DNAであった。PK-7クローンからのLVはenv発現プラスミドと導入ベクターを共トランスフェクトすることにより作製した。一過性トランスフェクションは標準Ca++-リン酸法またはFugene6(登録商標)系を用いて製造業者の取扱説明書(Roche Diagnostics Corporation, Indianapolis, IN)に従って実施し、類似の結果を得た。上清をトランスフェクション後48時間に収穫し、0.45μmフィルターを通して濾過した。力価を、SupT1、CEMA3.01、一次活性化末梢血液単核細胞(PBMC)および臍帯血由来のCD34+ HSCについて、実験の型に応じて計算した。概要を説明すると、SupT1と一次活性化末梢血液単核細胞細胞はポリブレン(8μg/ml)(Sigma-Aldrich, StLouis, MO)の存在のもとで2サイクルのスピノキュレーション(1,240×g、1時間)により形質導入し、一晩静置して相を分離し;CD34+ HSCは24時間レトロネクチンをコーティングしたプレート(Takara Bio、Otsu、Japan)上でポリブレン無しで形質導入した。形質導入効率はeGFP発現(SIN-eGFP)のフローサイトメトリー分析(FACS Calibur BD Bioscience, San Jose, CA)またはPorcellini et al., 2009および2010[21,22]に記載のFlowJoソフトウエア(Tree Star, Inc., Ashland, OR)を用いるΔLNFGR発現(PΔN-Chim3)によってモニターした。5〜20%ポジティブ細胞の範囲の形質導入値だけを用いて、次式:
TU=[細胞数×(%GFP/100)]/vol sup(mlで)に従って各LV調製物の力価を計算した。
ゲノムDNA(gDNA)をQIAamp Miniキット(QIAGEN GmbH、Germany)により製造業者の取扱説明書に従って単離した。バキュロウイルスDNAをウイルス粒子からQIAamp DNAマイクロキット(QIAGEN)により抽出した。示した制限酵素を用いて一晩消化後、10μgのgDNAを0.8%アガロースゲルに流し、サザンキャピラリートランスファーによりナイロン膜上にブロットし(Duralon, Stratagene, TX, USA)、次いでPerfectHyb PLUSハイブリダイゼーションバッファー中の1×106 dpm/mlの32P-ランダムプライムド標識した600-bp CMVまたは11-kb GPRカセットとハイブリダイズさせた。
AAV-Rep78プラスミドの残りの組込みのスクリーニングに対するPCR分析を300ngのゲノムDNAについて、プライマーのセット:AAV-Rep78 フォワード: 5'-CGG GCT GCT GGC CCA CCA GG-3'; AAV-Rep78 リバース: 5'-ATG CCG GGG TTT TAC GAG ATT GTG-3'を次のPCR条件:98℃にて7分間、30サイクルの94℃にて30秒間、66℃にて30秒間、および72℃にて1.5分間で用いることにより、実施した。
培養上清中の物理的LV産生をAlliance HIV-1 p24抗原ELISAキット(Perkin Elmer Life and Analytical Sciences, Inc. Waltham, MA)により製造業者取扱説明書に従い、1pg p24gagが1×104 物理的粒子に対応すると仮定して測定した。
PK-7細胞から誘導した全細胞および核抽出物ならびに単離された細胞を含まないVLPまたはLV由来のウイルスタンパク質を先に記載のように調製した[21,22]。タンパク質をSDS-PAGEによりサイズ分画し、Hybond ECLニトロセルロース膜(GE Healthcare、Life Sciences、UK Ltd、UK)にエレクトロブロットした。膜を5%低脂肪ドライミルクでブロックし、次いで適当な一次Abとインキュベートした。AIDS患者から得た抗-HIV-1血清を1:2,000希釈で用い;HIV-1revMoAb(Rev-6, sc-69730)(S. Cruz Biotechnology, Inc., S. Cruz, CA)を1:200希釈で用いた。Ab結合を強化化学発光系ECL(ECL, Amersham)により、製造業者の取扱説明書に従って可視化した。
組込まれたGPRベクターのコピー数を定量TaqMan PCRにより、ABI Prism 7,900 FAST 計器(Applied Biosystems, Foster City, CA)を用いて確立し、SDS 2.3ソフトウエア (Applied Biosystems)により分析した。ゲノムDNA(gDNA)を、次のプライマーとgag遺伝子から誘導したプローブを用いて増幅した:フォワード:5'-ACA TCA AGC AGC CAT GCA AAT-3';リバース:5'-ATC TGG CCT GGT GCA ATA GG-3';プローブ:FAM 5'-CAT CAA TGA GGA AGC TGC AGA ATG GGA タグ A-3' TAMRA。PCR条件は次の通りであった:2分間50℃にておよび5分間95℃にて、次いで40サイクルの15秒間95℃にておよび15秒間60℃にて(0.1℃/サイクルの増分で)。
ゲノムDNAをPK-7細胞からQIAamp DNA Miniキット(QIAGEN)により製造業者の取扱説明書に従って抽出し、そしてBglIIおよびBamHIを用いて37℃にて一晩消化した。5'-GATC-3'粘着末端と適合しうるアダプター76-bpオリゴヌクレオチドリンカーのライゲーションを標準条件で実施した。LM-PCRは次のネスト化プライマーのカップルを用いて行った:ITRフォワード:16s:5'-GTA GCA TGG CGG GTT AAT CA-3'、および17s/ロングネスト化:5'-TTA ACT ACA AGG AAC CCC タグ TGA TGG-3';リンカーリバースプライマー:リンカー-1:5'-GTA ATA CGA CTC ACT ATA GGG C-3'およびリンカー-2ネスト化:5'-AGG GCT CCG CTT AAG GGA C-3'。リンカー配列は5'-GAT CGT CCC TTA AGC GGA GCC CTA タグ TGA GTC GTA TTA CCA GGG AAT TCG CCT CGG GAT ATC ACT CAG CAT AAT G-3'に対応する。2ラウンドのLM-PCRをAmpliTaq Gold DNA ポリメラーゼ (Applied Biosystems)を用いて行い、それぞれ30サイクルから成った(95℃にて30秒間、52℃にて30秒間、72℃にて2分間)。PCRアンプリコンをTOPO(登録商標)クローニングキット(Invitrogen、Co.)を用いてクローニングし、そしておよそ100〜200bpのインサートを運ぶプラスミドコロニーを選択し、配列決定を行った。配列相同性はBLASTサーチ、NCBIにより同定した。
分裂中期染色体はPK-7細胞をコルヒチン(10μg/ml)(Sigma#C9754)で37℃にて2時間処理することによって得た。リン酸バッファー生理食塩水(PBS)で洗浄した後、細胞を低浸透圧溶液(75mM KCl)中に室温にて6分間保持し、メタノール/酢酸(3:1)で4回洗浄して固定し、次いで清浄なガラススライド上に延ばした。細胞遺伝子サンプルを70%ホルムアミド溶液中で2分間72℃にて変性し、冷温の70%、85%、および95%エタノールの連続洗浄により脱水し次いで空気乾燥した。特異的プローブを次の通り調製した:GPRカセットを含有する13-kbプラスミドDNAはランダムプライムドDNA標識キット(Roche Applied Science、Indianapolis、IN)を用いてSpectrumOrangeTM-dUTP (Vysis、Inc.、Downers Grove、IL)で標識したが、ヒトhox4遺伝子を含有する対照30-kbコスミドDNAはFISHBrightTM核酸標識キット(Kreatech Biotechnology、Amsterdam、The Netherlands)を用いて標識した。ハイブリダイゼーションは、5ng/μlの各プローブを含む250μlの50%ホルムアミド、2×SSC、および10%硫酸デキストランおよび50ng/μlのヒトC0T-1 DNAハイブリダイゼーションバッファー(Invitrogen)をインキュベートすることにより実施した。サンプルを変性したプローブで10分間75℃にてコートし、Parafilm(登録商標)Mで覆い、そして一晩37℃にて加湿室でインキュベートした。サンプルを1回、0.4×SSC中で、pH=7、72℃にて2分間;1回、4×0.0025% Tween-20を含有するSSC中で、pH=7、RTにて30秒間;ならびに2回、PBS 1×RTにて1分間洗浄した。スライドを0.02μg/μlの49,6-ジアミジノ-2-フェニルインドール(DAPI)(Sigma)で対比染色した。可視化と写真撮影はNikon 80i正立顕微鏡(Nikon Instruments S.p.A.、Italy)で、(FITC)およびスペクトルオレンジ色(スペクトルオレンジ色)フィルター照明を用いて行った。画像はGenikonソフトウエア(Nikon)で処理した。
第2または第3世代LVの連続産生用のRD-MolPackパッケージング細胞株を得るために、いくつかのHEK293T由来の中間体クローンを得た。最初のものは、PK-7と名付けられ、組換えハイブリッドバキュロ-AAVベクター(rhバキュロ-AAV-GPR)(図1a、模式図1)を使ってHIV-1 gag、polおよびrev遺伝子を安定して組み込むことにより得た。この送達系は、一過性で与えられるAAV-rep78タンパク質のインテグラーゼ活性を利用し、AAV ITR-隣接組込みカセットを切除してヒト染色体中に組み込む(図1b)。rh-バキュロ-AAVベクターはBaculoDirect Linear DNAとITR-隣接GPRカセットを含有するGateway(登録商標)pENTRTM4エントリープラスミドの間の相同組換えにより作製した(図1a、模式図1)。Sf9昆虫細胞における3サイクル(p3)の組換えバキュロウイルス増幅の後、ハイブリッドバキュロ-AAV DNAの力価と潜在的組換え事象をプラークアッセイおよびウイルスのゲノムDNAサザンブロットによりそれぞれチェックした。p3における力価は6×1010pfu/mlに対応し、サザンブロット分析は単一の鋭いバンドを現し、ウイルス増幅プロセス中に組換え事象が無いことを示した(図3)。
8クローンより優れているPK-7クローンをさらなる遺伝子操作の対象に選定した(表1)。
PK-7クローンからの半-安定LV産生がHEK293T細胞からの一過性LV産生のそれと全体に比較し得るかどうかをよく確立するために、両方の細胞型を、同じ量の必要なプラスミドによりトランスフェクトし、異なる標的細胞に対するトランスフェクションのパーセントおよびそれらのそれぞれのLVの効力を測定した(表3)。この条件において、HEK293T細胞での11回の実験のトランスフェクションのパーセントの平均値は90.54±3.6 SEMであり、そしてPK-7細胞での12回の実験のそれは91±5.3 SEMであり、PK-7細胞がその高いレベルのトランスフェクション能力を維持することを示した。そこで、LV力価を、本発明者らの標準参照細胞型としてのSupT1細胞、靭帯血液由来のCD34+ HSC、および抗-CD3/IL-2活性化靭帯血単核細胞(表3でT lymph.と記した)(表 3)において計算した。
Claims (19)
- i. 第1の発現カセットがレンチウイルスのgag遺伝子およびpol遺伝子をそして第2の発現カセットがレンチウイルスのrev遺伝子および抗生物質耐性遺伝子である選択マーカーをコードする2つの発現カセットを含むAAV ITRに隣接した組込みカセットを含有するバキュロウイルスの主鎖を含むハイブリッドベクター;および
ii. プロモーターの調節下にあるAAV repタンパク質オープンリーディングフレーム(ORF)を含有する発現プラスミド
から成る、レンチウイルスの構造および調節タンパク質を安定して発現する系。 - ハイブリッドベクターの2つの発現カセットがテール・ツー・テール配向でありかつそれぞれが構成的プロモーターおよびポリAにより駆動される、請求項1に記載の系。
- プロモーターがCMV、CMV IE、PGK、SV40、eF1α、SFFVおよびRSVから選択される、請求項1または2に記載の系。
- プロモーターがCMV IEプロモーターである、請求項1〜3のいずれか1項に記載の系。
- 選択マーカーがハイグロマイシン、カナマイシン、ネオマイシン、ゼオマイシン耐性遺伝子から選択される、請求項1〜4のいずれか1項に記載の系。
- 選択マーカーがハイグロマイシン耐性遺伝子である、請求項5に記載の系。
- 選択マーカーが内部リボソーム侵入部位(IRES)下流にクローニングされる、請求項1〜6のいずれか1項に記載の系。
- repタンパク質がrep78である、請求項1〜7のいずれか1項に記載の系。
- 第1の発現カセットがレンチウイルスのgag遺伝子およびpol遺伝子をコードしそして第2の発現カセットがレンチウイルスのrev遺伝子および抗生物質耐性遺伝子である選択マーカーをコードする2つの発現カセットを含む、AAV ITRに隣接した組込みカセットの少なくとも1つのコピーを安定してそのゲノム中に組み込んで含有することで特徴付けられる、安定してレンチウイルスのgag、polおよびrevタンパク質を発現する細胞から成る半-安定なレンチウイルスのパッケージング細胞株。
- 細胞がHEK293、HEK293-T、HEK293-SF、TE671、HT1080またはHeLaから選択されるヒト細胞株である、請求項9に記載の半-安定レンチウイルスのパッケージング細胞株。
- 2つの発現カセットがテール・ツー・テール配向でありかつそれぞれが構成的プロモーターおよびポリAにより駆動される、請求項9または10のいずれか1項に記載の半-安定レンチウイルスのパッケージング細胞株。
- プロモーターがCMV、CMV IE、PGK、SV40、eF1α、SFFVおよびRSVから選択される、請求項9または11のいずれか1項に記載の半-安定レンチウイルスのパッケージング細胞株。
- プロモーターがCMV IEプロモーターである、請求項12に記載の半-安定レンチウイルスのパッケージング細胞株。
- 選択マーカーがハイグロマイシン、カナマイシン、ネオマイシン、ゼオマイシン耐性遺伝子から選択される、請求項9〜13のいずれか1項に記載の半-安定レンチウイルスのパッケージング細胞株。
- 選択マーカーがハイグロマイシン耐性遺伝子である、請求項14に記載の半-安定レンチウイルスのパッケージング細胞株。
- 選択マーカーが内部リボソーム侵入部位(IRES)下流にクローニングされている、請求項9〜15のいずれか1項に記載の半-安定レンチウイルスのパッケージング細胞株。
- i. 請求項9〜16いずれか1項に記載の半-安定パッケージング細胞株を培養するステップ、
ii. 半-安定パッケージング細胞株にenv遺伝子を挿入するステップ、および
iii. 半-安定パッケージング細胞株に導入ベクターを挿入するステップ
を含むレンチウイルスベクター産生する方法。 - env遺伝子がVSV-G env遺伝子、MLV 4070 env遺伝子、RD114 env遺伝子、キメラエンベロープタンパク質RD114-TRをコードする遺伝子、キメラエンベロープタンパク質RD114-proをコードする遺伝子、バキュロウイルスGP64 env遺伝子またはGALV env遺伝子から選択される、請求項17に記載の方法。
- env遺伝子が、キメラエンベロープタンパク質RD114-TRをコードする遺伝子である、請求項17または18に記載の方法。
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10175088 | 2010-09-02 | ||
| EP10175088.3 | 2010-09-02 | ||
| PCT/EP2011/065089 WO2012028680A1 (en) | 2010-09-02 | 2011-09-01 | Semi-stable production of lentiviral vectors |
| EP11749849.3A EP2480677B1 (en) | 2010-09-02 | 2011-09-01 | Semi-stable production of lentiviral vectors |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2013537800A JP2013537800A (ja) | 2013-10-07 |
| JP5897576B2 true JP5897576B2 (ja) | 2016-03-30 |
Family
ID=49582836
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2013526469A Expired - Fee Related JP5897576B2 (ja) | 2010-09-02 | 2011-09-01 | レンチウイルスベクターの半−安定産生 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US9133481B2 (ja) |
| EP (1) | EP2480677B1 (ja) |
| JP (1) | JP5897576B2 (ja) |
| KR (1) | KR101891626B1 (ja) |
| CN (1) | CN103080322B (ja) |
| AU (1) | AU2011298332B2 (ja) |
| CA (1) | CA2809447C (ja) |
| WO (1) | WO2012028680A1 (ja) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI2480678T1 (sl) | 2010-09-02 | 2014-06-30 | Molmed Spa | Stabilno proizvajanje lentivirusnih vektorjev |
| CN103842501B (zh) | 2011-10-05 | 2017-12-08 | 莫尔梅德股份有限公司 | 病毒载体纯化系统 |
| US11185555B2 (en) | 2016-04-11 | 2021-11-30 | Noah James Harrison | Method to kill pathogenic microbes in a patient |
| GB201715052D0 (en) | 2017-09-19 | 2017-11-01 | Oxford Genetics Ltd | Vectors |
| EP3696272A1 (en) * | 2017-12-22 | 2020-08-19 | Oxford BioMedica (UK) Limited | Retroviral vector |
| JP2023535632A (ja) | 2020-07-27 | 2023-08-18 | アンジャリウム バイオサイエンシズ エージー | Dna分子の組成物、その作製方法、及びその使用方法 |
| CN116731974B (zh) * | 2023-05-23 | 2024-11-29 | 杭州荣谷生物科技有限公司 | 病毒的制备方法和应用 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SI2480678T1 (sl) | 2010-09-02 | 2014-06-30 | Molmed Spa | Stabilno proizvajanje lentivirusnih vektorjev |
-
2011
- 2011-09-01 JP JP2013526469A patent/JP5897576B2/ja not_active Expired - Fee Related
- 2011-09-01 AU AU2011298332A patent/AU2011298332B2/en not_active Ceased
- 2011-09-01 CN CN201180042482.1A patent/CN103080322B/zh not_active Expired - Fee Related
- 2011-09-01 US US13/819,989 patent/US9133481B2/en active Active
- 2011-09-01 KR KR1020137008386A patent/KR101891626B1/ko not_active Expired - Fee Related
- 2011-09-01 CA CA2809447A patent/CA2809447C/en not_active Expired - Fee Related
- 2011-09-01 EP EP11749849.3A patent/EP2480677B1/en active Active
- 2011-09-01 WO PCT/EP2011/065089 patent/WO2012028680A1/en not_active Ceased
Also Published As
| Publication number | Publication date |
|---|---|
| KR20130101526A (ko) | 2013-09-13 |
| AU2011298332A1 (en) | 2013-03-21 |
| CA2809447A1 (en) | 2012-03-08 |
| JP2013537800A (ja) | 2013-10-07 |
| EP2480677B1 (en) | 2015-03-18 |
| CA2809447C (en) | 2019-12-03 |
| AU2011298332B2 (en) | 2015-05-07 |
| KR101891626B1 (ko) | 2018-09-28 |
| US9133481B2 (en) | 2015-09-15 |
| US20130164840A1 (en) | 2013-06-27 |
| CN103080322A (zh) | 2013-05-01 |
| EP2480677A1 (en) | 2012-08-01 |
| CN103080322B (zh) | 2015-01-14 |
| WO2012028680A1 (en) | 2012-03-08 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5897577B2 (ja) | レンチウイルスベクターの安定な産生 | |
| JP5897576B2 (ja) | レンチウイルスベクターの半−安定産生 | |
| JP2022513359A (ja) | ウイルスベクターを調製するための組成物および方法 | |
| HK1184822B (en) | Semi-stable production of lentiviral vectors | |
| HK1187953B (en) | Stable production of lentiviral vectors | |
| van Heuvel | Optimization of retroviral packaging cells for scale-up of vector production | |
| Phillips | Viral Vectors for HIV Gene Therapy | |
| Heuvel | Optimization of retroviral packaging cells for scale-up of vector production |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20140828 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20151013 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20160104 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20160125 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20160223 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20160302 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 5897576 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |