JP6607606B2 - トリアジン置換インドロカルバゾール誘導体、それからなる有機電子デバイス形成用アルコール不溶性塗膜、及びそれを用いた有機電子デバイス - Google Patents
トリアジン置換インドロカルバゾール誘導体、それからなる有機電子デバイス形成用アルコール不溶性塗膜、及びそれを用いた有機電子デバイス Download PDFInfo
- Publication number
- JP6607606B2 JP6607606B2 JP2016025894A JP2016025894A JP6607606B2 JP 6607606 B2 JP6607606 B2 JP 6607606B2 JP 2016025894 A JP2016025894 A JP 2016025894A JP 2016025894 A JP2016025894 A JP 2016025894A JP 6607606 B2 JP6607606 B2 JP 6607606B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- mmol
- triazine
- electronic device
- layer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- QVFWRFBJSIUPAD-UHFFFAOYSA-N Brc(cc1)cc(c2c3)c1[n](Cc1ccccc1)c2ccc3Br Chemical compound Brc(cc1)cc(c2c3)c1[n](Cc1ccccc1)c2ccc3Br QVFWRFBJSIUPAD-UHFFFAOYSA-N 0.000 description 1
- FODGUJRIRWUAAG-UHFFFAOYSA-N C(c1ccccc1)[n](c(c(c1c2)c3)ccc3-[n]3c4ccccc4c4c3cccc4)c1ccc2-[n]1c2ccccc2c2c1cccc2 Chemical compound C(c1ccccc1)[n](c(c(c1c2)c3)ccc3-[n]3c4ccccc4c4c3cccc4)c1ccc2-[n]1c2ccccc2c2c1cccc2 FODGUJRIRWUAAG-UHFFFAOYSA-N 0.000 description 1
- UJOBWOGCFQCDNV-UHFFFAOYSA-N c1ccc2[nH]c3ccccc3c2c1 Chemical compound c1ccc2[nH]c3ccccc3c2c1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 1
Images



















Landscapes
- Electroluminescent Light Sources (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Description

本発明のトリアジン置換インドロカルバゾール誘導体は、下記構造式(1)〜(4)のいずれかで表されることを特徴とする。
本発明の有機電子デバイスは、上記トリアジン置換インドロカルバゾール誘導体、又は上記アルコール不溶性塗膜を用いて得られることを特徴とする。
本発明の有機電子デバイスの製造方法は、上記トリアジン置換インドロカルバゾール誘導体を塗布成膜した層上に、塗布成膜により層を形成する工程を含むことを特徴とする。
[トリアジン置換インドロカルバゾール誘導体]
本発明のトリアジン置換インドロカルバゾール誘導体は、下記構造式(1)〜(4)のいずれかで表されることを特徴とする。以下、構造式(1)〜(4)で表されるトリアジン置換インドロカルバゾール誘導体を、それぞれ化合物(1)〜(4)と記載する。
本発明の有機電子デバイスは、上記トリアジン置換インドロカルバゾール誘導体よりなる。
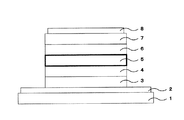
ここで、図7に有機電子デバイスの一形態である有機EL素子の典型的な層構造を示す。
上記有機EL素子は、典型的には、基板1上に陽極2として、例えば、ITO等を成膜し、その上に正孔注入層3、正孔輸送層4、発光層5、電子輸送層6、電子注入層7及び陰極8がこの順に積層されてなる。
本発明では、基板1上に、陽極2、上記した層、及び陰極8が積層されてなる有機EL素子の少なくとも一層に上記トリアジン置換インドロカルバゾール誘導体を含有させることにより、優れた有機電子デバイスを与える。具体的には、発光層5、正孔輸送層4又は電子輸送層6に上記トリアジン置換インドロカルバゾール誘導体を含有させることが好ましく、発光層5のホスト材料として、りん光発光ドーパントとともに、上記トリアジン置換インドロカルバゾール誘導体を含有させることがより好ましい。
正孔注入層3には、陽極2から発光した光を透過させるため、ポリ(エチレンジオキシチオフェン):ポリスチレンスルホン酸(PEDOT/PSS)、りんモリブデン酸、ポリアニリン、及びその他の電荷輸送性材料が用いられる。
合成物の同定に使用した機器及び測定条件は以下のとおりである。
(1)質量分析(MS)装置
日本電子(株)製JMS−K9[卓上GCQMS]及びWaters(株)製Zspray(SQ検出器2)
(2)1H核磁気共鳴(NMR)装置
日本電子(株)製(400MHz)JNM−EX270FT−NMR型
残膜率及び光学特性評価に用いた機器及び測定条件は以下のとおりである。
紫外・可視(UV−vis)分光光度計
(株)島津製作所製 UV−3150
測定条件;スキャンスピード 中速、測定範囲 200〜800nm サンプリングピッチ 0.5nm、スリット幅 0.5nm
光学特性評価に用いた機器及び測定条件は以下のとおりである。
(1)光電子収量分光(PYS)装置
住友重機械工業(株)製イオン化ポテンシャル測定装置
イオン化ポテンシャル測定装置を用いて、真空中でイオン化ポテンシャル(Ip)の測定を行った。
(2)発光量子収率(PLQY)測定装置
浜松ホトニクス(株)製 絶対PL量子収率測定装置
有機EL素子の評価に用いた機器は以下のとおりである。
EL(エレクトロルミネッセンス)測定装置
浜松ホトニクス(株)製 PHOTONIC MULTI−CHANNEL ANALYZER PMA−1
[化合物cの合成]
撹拌子を備えた三口フラスコに3,6−ジブロモ−9−フェニルカルバゾール(化合物a)2.50g(6.23mmol)、9−フェニルカルバゾール−3−ボロン酸(化合物b)1.79g(6.23mmol)、2M炭酸カリウム(K2CO3)水溶液41.6ml(炭酸カリウム83.2mmol)、及びテトラヒドロフラン(THF)125mlを加え1時間窒素バブリングを行った。そこにテトラキス(トリフェニルホスフィン)パラジウム(Pd(PPh3)4)350mg(0.31mmol)を加え、終夜加熱撹拌を行った。
反応溶液を室温に戻し、溶媒を留去した。残渣にジクロロメタンを加え再溶解した後、飽和食塩水にて洗浄を行った。有機層に無水硫酸マグネシウムを加え乾燥後、ろ別した。ろ液を濃縮後、シリカゲルカラムクロマトグラフィー(ジクロロメタン:ヘキサン=1:4)を行い、目的物である化合物cを収量1.32g(0.24mmol)、収率38%で得た。
撹拌子を備えた耐圧管に化合物c 500mg(0.89mmol)、11,12−ジヒドロインドロ[2,3−a]カルバゾール(化合物1)226mg(0.89mmol)、炭酸カリウム730mg(5.30mmol)、ヨウ化銅(CuI)600mg(3.17mg)、キノリン9mlを加え、減圧後、密閉を行い、バス温190℃にて72時間撹拌を行った。反応液を室温に戻し、ジクロロメタンを加え再溶解した後、飽和食塩水にて洗浄を行った。有機層に無水硫酸マグネシウムを加え乾燥後、ろ別した。ろ液を濃縮後、シリカゲルカラムクロマトグラフィー(ジクロロメタン:ヘキサン=3:7)を行い、化合物dを311mg(0.42mmol)、収率47%で得た。
撹拌子を備えた三口フラスコに水素化ナトリウム18mg(0.75mmol)、化合物d 111mg(0.15mmol)、N,N−ジメチルホルムアミド(DMF)3.0mlを加え窒素雰囲気下、室温にて1時間撹拌を行った。そこにN,N−ジメチルホルムアミド2.0mlに2−クロロ−4,6−ジフェニル−1,3,5−トリアジン(化合物2)201mg(0.75mmol)を溶解させ滴下した。滴下後、終夜加熱還流にて撹拌を行った。その後、トルエン及び飽和食塩水にて抽出、洗浄を行った。硫酸マグネシウムにて乾燥を行い、乾燥剤をろ別した。ろ液を濃縮後、シリカゲルカラムクロマトグラフィー(ジクロロメタン:ヘキサン=3:7)を行い、化合物(1)を収量91mg(0.094mmol)、収率64%で得た。
目的物の同定はMS及び1H−NMRにて行った。
EIMS:m/z=969.36[M+]
1H−NMR測定の結果を図8に示す。
[化合物gの合成]
撹拌子を備えた三口フラスコに3−ブロモカルバゾール(化合物e)984mg(4.00mmol)、9−フェニルカルバゾール−3−ボロン酸(化合物f)1.26g(4.40mmol)、2Mリン酸三カリウム(K3PO4)水溶液20ml(リン酸三カリウム40.0mmol)、キシレン40ml、エタノール20mlを加え1時間窒素バブリングを行った。そこにトリス(ジベンシジリデンアセトン)ジパラジウム(0)(Pd2(dba)3)256mg(0.28mmol)、2−ジシクロヘキシルホスフィノ−2’,6’−ジメトキシビフェニル(SPhos)230mg(0.56mmol)を加え終夜加熱撹拌を行った。溶媒を除去した後、トルエン及び飽和食塩水にて抽出、洗浄を行った。その後、硫酸マグネシウムにて乾燥を行い、乾燥剤をろ別した。ろ液を濃縮後、シリカゲルカラムクロマトグラフィー(ジクロロメタン:ヘキサン=1:1)にて精製を行った。目的物gを937mg(2.30mmol)、収率57%で得た。
撹拌子を備えた試験管型フラスコに化合物g594mg(0.69mmol)、4−ブロモヨードベンゼン(化合物h)195mg(0.69mmol)、リン酸三カリウム292mg(1.38mmol)、ヨウ化銅(I)3.8mg(0.02mmol)を加え30分窒素フローを行った。トランス−1,2−シクロヘキサンジアミン4.56mg(0.04mmol)の脱気1,4−ジオキサン17.5ml溶液を別途調製し、そのうち3.5mlを系内に加え、終夜加熱撹拌を行った。反応溶液を室温に戻し、溶媒を留去した。残渣にトルエンを加え再溶解した後、飽和食塩水にて洗浄を行った。有機層に無水硫酸マグネシウムを加え乾燥後、ろ別した。ろ液を濃縮し、シリカゲルカラムクロマトグラフィー(ジクロロメタン:ヘキサン=3:7)にて二回精製を行った。化合物iを収量114mg(0.20mmol)、収率29%で得た。
撹拌子を備えた25mL三口フラスコに化合物i 662mg(1.17mmol)、インドロカルバゾール(化合物1)300mg(1.17mmol)、炭酸カリウム972mg(7.04mmol)、脱水キシレン25mlを加え1時間窒素バブリングを行った。そこに酢酸パラジウム(II)10.2mg(0.045mmol)、トリ−tert−ブチルホスフィン39μl(0.234mmol)を加え、バス温135℃にて終夜加熱撹拌を行った。反応液を室温に戻し、溶媒を留去した。そこにトルエンを加え再溶解した後、飽和食塩水にて洗浄を行った。有機層に無水硫酸マグネシウムを加え乾燥後、ろ別し、ろ液を濃縮した。シリカゲルカラムクロマトグラフィー(ジクロロメタン:ヘキサン=3:7)にて精製を行った。目的物jを766mgg(1.04mmol)、収率89%で得た。
撹拌子を備えた三口フラスコに水素化ナトリウム(NaH)24mg(0.54mmol)、化合物j333mg(0.45mmol)を加えた後、系内の脱気を行い、その後、窒素雰囲気にした。そこに、N,N−ジメチルホルムアミド10mlを加え窒素雰囲気下、室温にて1時間撹拌を行った。その後、N,N−ジメチルホルムアミド6.0mlに2−クロロ−4,6−ジフェニル−1,3,5−トリアジン(化合物2)603mg(2.25mmol)を溶解させ滴下した。滴下後、終夜加熱還流にて撹拌を行った。反応液を室温に戻し、溶媒を留去した。そこにトルエンを加え再溶解した後、飽和食塩水にて洗浄を行った。有機層に無水硫酸マグネシウムを加え乾燥後、ろ別し、ろ液を濃縮した。シリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:19)にて精製し、不純物を除去した。次に、アルミナ薄層クロマトグラフィー(TLC)(トルエン:ヘキサン=1:1→1:0)にて精製を行い、化合物(2)を360mg(0.37mmol)、収率68%で得た。
目的物の同定はMS及び1H−NMRにて行った。
EIMS:m/z=969.36[M+]
1H−NMR測定の結果を図9に示す。
[化合物kの合成]
撹拌子を備えた50mL三口フラスコに3,6−ジブロモ−9−フェニルカルバゾール(化合物a)2.00g(4.99mmol)、カルバゾール2.50g(15.0mmol)、リン酸三カリウム2.33g(10.98mmol)、ヨウ化銅(I)190mg(0.100mmol)、trans−1,2−シクロヘキサンジアミン114mg(0.100mmol)、脱水1,4−ジオキサン50mlを加え、窒素雰囲気下終夜還流撹拌を行った。反応終了後、セライトろ過を行い、不溶部を除去した。ろ液を濃縮し、シリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=3:1→7:3)にて精製を行い、化合物kを収量1.04g(2.13mmol)、収率43%で得た。
撹拌子を備えた三口フラスコに化合物k 244mg(1.72mmol)、インドロカルバゾール(化合物1)411mg(1.72mmol)、炭酸カリウム1.43g(10.35mmol)、脱水キシレン35mlを加え1時間窒素バブリングを行った。そこに酢酸パラジウム(II)15mg(0.067mmol)、トリ−tert−ブチルホスフィン60μlを加えバス温135℃を保持し、終夜加熱撹拌を行った。反応終了後不溶部を除去し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=7:3)にて精製を行い、目的物である化合物lを収量817mg(1.23mmol)、収率72%で得た。
撹拌子を備えた三口フラスコに化合物lを440mg(0.66mmol)、水素化ナトリウムを44mg(1.01mmol)加え脱気した。脱気した三口フラスコに脱水N,N−ジメチルホルムアミド3mlを加え窒素雰囲気下1時間撹拌した。別途、2−クロロ−4,6−ジフェニル−1,3,5−トリアジン(化合物2)1.77g(6.6mmol)を脱気後、脱気した脱水N,N−ジメチルホルムアミド9mlを加え加熱溶解し、これを先の反応溶液に加え、バス温70℃を保持し、終夜加熱攪拌した。反応混合物を室温に戻した後、水を加え反応を終了した。ジクロロメタンで抽出した後、無水硫酸マグネシウムで乾燥し、ろ別し、溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=3:1)を行い、化合物(3)を収量255.7mg(0.286mmol)、収率43%で得た(原料45%回収)。
目的物の同定はMS及び1H−NMRにて行った。
EIMS:m/z=893.33[M+]
1H−NMR測定の結果を図10に示す。
[化合物nの合成]
撹拌子を備えた100mL三口フラスコにカルバゾール1.61g(9.63mmol)、9−ベンジル−3,6−ジブロモカルバゾール1.00g(2.41mmol)、炭酸カリウム5.32g(38.5mmol)、脱水キシレン50mlを加え1時間窒素バブリングを行った。そこに酢酸パラジウム(II)27mg(0.12mmol)、トリ−tert−ブチルホスフィン113μl(0.48mmol)を加え、バス温135℃にて終夜加熱撹拌を行った。反応終了後、セライトろ過にて不溶部を除去し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=7:3)及びシリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=2:1)にて精製を行い、化合物nを収量913mg(1.55mmol)、収率64%で得た。
撹拌子を備えた200mL三角フラスコに化合物n 799.2mg(1.36mmol)、ジメチルスルホキシド42.4mlを加え、大気下室温で撹拌した。そこにカリウムtert−ブトキシド(1MTHF溶液)13.6ml(13.6mmol)を加え、さらに撹拌を行った。1時間後反応液にイオン交換水59.2mlを加え反応を終了させた。反応混合物を吸引ろ過、減圧乾燥し、白色固体の粗生成物623mgを定量的に得た。
撹拌子を備えた25mL三口フラスコに、化合物o 623mg(1.25mmol)、4−ブロモヨードベンゼン(化合物h)530mg(1.87mmol)、リン酸三カリウム530mg(2.50mmol)、ヨウ化銅(I)7.3mg(0.0383mmol)を加え脱気した。別途trans−1,2−シクロヘキサンジアミン6.9mg(0.060mmol)を脱水1,4−ジオキサン25mlに溶解し、そのうち6.3mlを加え、バス温115℃にて終夜加熱撹拌を行った。反応終了後、セライトろ過を行い、不溶部を除去した。ろ液を濃縮し、シリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=3:1→1:1)にて精製を行い、目的物である化合物pを収量596mg(0.914mmol)、収率73%で得た(原料回収41.8mg(0.0840mmol、6.7%))。
撹拌子を備えた50mL三口フラスコに化合物p1.09g(1.67mmol)、インドロカルバゾール(化合物1)428mg(1.67mmol)、炭酸カリウム1.38g(9.98mmol)、脱水キシレン33mlを加え1時間窒素バブリングを行った。そこに酢酸パラジウム(II)14.6mg(0.065mmol)、トリ−tert−ブチルホスフィン59μl(0.25mmol)を加え、バス温135℃にて終夜加熱撹拌を行った。反応終了後、不溶部をろ別し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=2:1)及び、メタノールにて再沈殿を行い、目的物である化合物rを収量1.30g(1.57mmol)、収率94%で得た。
撹拌子を備えた100mL三口フラスコに化合物r 1.29g(1.56mmol)、水素化ナトリウム103mg(2.36mmol)を加え脱気した。脱気した脱水N,N−ジメチルホルムアミド31.4mlを加え窒素雰囲気下、1時間撹拌した。そこに別途、2−クロロ−4,6−ジフェニル−1,3,5−トリアジン(化合物2)2.10g(7.84mmol)の脱水N,N−ジメチルホルムアミド20.9ml溶液を加え、バス温70℃で終夜加熱攪拌した。反応混合物を室温に戻した後、水を加え反応を終了した。反応混合物を酢酸エチルで抽出、無水硫酸マグネシウムで乾燥、ろ別し溶媒を留去した後、アルミナカラムクロマトグラフィー(トルエン→トルエン:酢酸エチル=1000:1)及びシリカゲルカラムクロマトグラフィー(ヘキサン:ジクロロメタン=3:1→3:2)を2回行い、さらにメタノールにて再沈殿を行い、化合物(4)を収量1.27g(1.20mmol)、収率77%で得た。
目的物の同定はMS及び1H−NMRにて行った。
EIMS:m/z=1058.38[M+]
1H−NMR測定の結果を図11に示す。
以下に示す構造式を有する既存の化合物A−1及びA−2を用いた。
実施例1〜4で得られた化合物(1)〜(4)について、粉末状態での溶解度試験を行った。サンプル管に各試料1mgを秤量し、表1に示す溶媒を加えた。溶媒を加えた後、超音波及び加熱を行った。
結果を表1に示す。
化合物(1)〜(4)がアルコール溶媒に対して耐溶媒性を持っていることがわかったため、化合物(1)〜(4)及び化合物A−2について、薄膜にした場合の耐溶剤性を調べた。
石英基板上に各試料をTHFに溶解させて成膜した後、UV−vis吸収スペクトルを測定し、その後、メタノールにてリンスを行った。リンス前後でのUV−vis吸収スペクトルの変化から、残膜率を算出した。なお、メタノールは、有機EL素子の上層の塗布によく用いられる溶媒である。
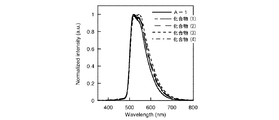
結果を図1及び2に示す。
一方、図2に示すように、化合物A−2はリンス後において、スペクトルがほとんど観測できなかった。つまり、化合物A−2はリンス処理により溶解してしまうことがわかった。
化合物(1)〜(4)を成膜して作製した試料について、光学特性評価を行った。バンドギャップ(Eg)は、UV−vis吸収スペクトルの吸収端から、イオン化ポテンシャル(Ip)はイオン化ポテンシャル測定装置(PYS;photoelectron yield spectroscopy)から、電子親和力(Ea)はバンドギャップ(Eg)及びイオン化ポテンシャル(Ip)から算出した。
さらに、化合物(1)〜(4)をホスト材料とし、ドーパントとして緑色りん光材料としてIr(ppy)3を8wt%の濃度で添加した薄膜を作製し、発光量子収率(PLQY)の測定を行った。
なお、化合物(1)〜(4)のガラス転位温度(Tg)に関しては、示差走査熱量測定(DSC)にて測定を行ったが、得られなかった。
結果を表2に示す。
下記に示す素子構造からなる塗布型有機EL素子を作製した。層構成を図3に示す。図3中、括弧内の数字は厚み(nm)を表し、ZnONPsはZnOナノ粒子を意味する。
ITO/PEDOT:PSS(30)/TFB(20)/発光層(EML)(70)/ZnO(10)/PEIE(20)/Al(100)
ITO基板上に、正孔注入材料であるPEDOT:PSSの水分散液を、大気下、6500rpmで30秒間成膜した後、200℃で10分間焼成した。次いで、グローブボックスにて、PEDOT:PSS層上に、正孔輸送材料であるTFBのp−キシレン溶液を5000rpmで30秒間成膜し、200℃で1時間焼成した。TFB層上に、発光層(EML)形成材料のTHF溶液を2500rpm(化合物A−1は3000rpm)で30秒間成膜後、130℃で10分間焼成した。さらに、発光層上に、電子注入材料であるZnOナノ粒子のメタノール分散液を2000rpmで30秒間成膜後、100℃で10分間焼成し、さらにZnO層上に、電子注入材料であるPEIEのメタノール溶液を2000rpmで30秒間成膜後、100℃で10分間焼成した。PEIE層上に、Alを1000Å/秒から2500Å/秒に蒸着速度を変化させながら真空蒸着にて成膜した。塗布成膜ではすべてスピンコーターを使用した。
ここで、発光層(EML)形成材料は以下の4種類である。いずれもIr(ppy)3が8wt%の濃度でドープされた材料である。
発光層(EML)形成材料 化合物A−1:8wt% Ir(ppy)3
化合物(1):8wt% Ir(ppy)3
化合物(2):8wt% Ir(ppy)3
化合物(3):8wt% Ir(ppy)3
化合物(4):8wt% Ir(ppy)3
PEDOT:PSS、TFB、PEIE、及びIr(ppy)3の構造式を以下に示す。
結果として、電極以外を塗布成膜した塗布型有機EL素子の作製に成功した。また、図4より、化合物(1)〜(4)は、化合物A−1と同等の効率を示し、大幅に低電圧化に成功した。低電圧化された理由は、化合物(1)〜(4)はバイポーラ性を有しているため、効率良く電荷が注入されたためである。
次に、下記に示す素子構造からなる蒸着型有機EL素子を作製した。層構成を図5に示す。括弧内の数字は厚み(nm)を表す。
ITO/PMA(10)/TFB(20)/EML(30)/BAlq2(10)/Alq3(40)/Liq(1)/Al(100)
ITO基板上に、正孔注入材料として、グローブボックスにてアセトニトリルに溶解したりんモリブデン酸(PMA)を4000rpmで30秒間成膜した後、150℃で10分間焼成した。PMA層上に、正孔輸送材料として、p−キシレンに溶解したTFBを5000rpmで30秒間成膜し、表面を拭き取った後、200℃で1時間焼成した。TFB層上に、発光層(EML)形成材料のTHF溶液を4000rpmで30秒間成膜し、表面を拭き取った後、130℃で10分間焼成した。電子輸送材料であるBAlq2及びAlq3と、電子注入材料であるLiqと、陰極Alは、発光層(EML)上にすべて真空蒸着にて成膜した。塗布成膜ではすべてスピンコーターを使用した。
ここで、発光層(EML)形成材料は以下の4種類である。いずれもIr(ppy)3が8wt%の濃度でドープされた材料である。
発光層(EML)形成材料 化合物A−1:8wt% Ir(ppy)3
化合物A−2:8wt% Ir(ppy)3
化合物(1):8wt% Ir(ppy)3
化合物(2):8wt% Ir(ppy)3
化合物(3):8wt% Ir(ppy)3
化合物(4):8wt% Ir(ppy)3
Alq3、BAlq2、及びLiqの構造式を以下に示す。
結果として、化合物A−1が若干高電圧化したが、各化合物とも同様の挙動が得られた。
2 陽極
3 正孔注入層
4 正孔輸送層
5 発光層
6 電子輸送層
7 電子注入層
8 陰極
Claims (4)
- 下記構造式(1)〜(4)のいずれかで表されるトリアジン置換インドロカルバゾール誘導体。
- 請求項1に記載のトリアジン置換インドロカルバゾール誘導体からなる有機電子デバイス形成用アルコール不溶性塗膜。
- 請求項1に記載のトリアジン置換インドロカルバゾール誘導体、又は請求項2に記載のアルコール不溶性塗膜を用いた有機電子デバイス。
- 請求項1に記載のトリアジン置換インドロカルバゾール誘導体を塗布成膜した層上に、塗布成膜により層を形成する工程を含む、有機電子デバイスの製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016025894A JP6607606B2 (ja) | 2016-02-15 | 2016-02-15 | トリアジン置換インドロカルバゾール誘導体、それからなる有機電子デバイス形成用アルコール不溶性塗膜、及びそれを用いた有機電子デバイス |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2016025894A JP6607606B2 (ja) | 2016-02-15 | 2016-02-15 | トリアジン置換インドロカルバゾール誘導体、それからなる有機電子デバイス形成用アルコール不溶性塗膜、及びそれを用いた有機電子デバイス |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2017145198A JP2017145198A (ja) | 2017-08-24 |
| JP6607606B2 true JP6607606B2 (ja) | 2019-11-20 |
Family
ID=59681934
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2016025894A Expired - Fee Related JP6607606B2 (ja) | 2016-02-15 | 2016-02-15 | トリアジン置換インドロカルバゾール誘導体、それからなる有機電子デバイス形成用アルコール不溶性塗膜、及びそれを用いた有機電子デバイス |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP6607606B2 (ja) |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018123783A1 (ja) * | 2016-12-27 | 2018-07-05 | 新日鉄住金化学株式会社 | 有機電界発光素子用材料及び有機電界発光素子 |
| JP7112708B2 (ja) * | 2018-02-14 | 2022-08-04 | 国立大学法人山形大学 | 有機el素子 |
| CN108912154B (zh) * | 2018-08-03 | 2021-04-16 | 大连理工大学 | 一种用于制备双极性电致发光材料的基材 |
| CN108752372B (zh) * | 2018-08-03 | 2021-01-22 | 大连理工大学 | 一种用于制备有机电致发光材料的化合物 |
| CN115669281A (zh) * | 2020-05-29 | 2023-01-31 | 默克专利有限公司 | 有机电致发光器件 |
| CN112250685B (zh) * | 2020-11-25 | 2022-04-19 | 中钢集团南京新材料研究院有限公司 | 一种吲哚并[2,3-a]咔唑的制备方法 |
| CN113421980B (zh) * | 2021-06-02 | 2023-02-24 | 陕西莱特迈思光电材料有限公司 | 有机电致发光器件及包含其的电子装置 |
| JPWO2022255427A1 (ja) | 2021-06-04 | 2022-12-08 | ||
| TW202402758A (zh) * | 2022-05-25 | 2024-01-16 | 日商日鐵化學材料股份有限公司 | 攝像用的光電轉換元件用材料及使用其的攝像用光電轉換元件 |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8062769B2 (en) * | 2006-11-09 | 2011-11-22 | Nippon Steel Chemical Co., Ltd. | Indolocarbazole compound for use in organic electroluminescent device and organic electroluminescent device |
| KR101233380B1 (ko) * | 2009-10-21 | 2013-02-15 | 제일모직주식회사 | 신규한 유기광전소자용 화합물 및 이를 포함하는 유기광전소자 |
| KR101986570B1 (ko) * | 2011-12-15 | 2019-06-07 | 닛테츠 케미컬 앤드 머티리얼 가부시키가이샤 | 유기 전계 발광 소자 |
| KR101638072B1 (ko) * | 2014-03-14 | 2016-07-08 | (주)피엔에이치테크 | 새로운 유기전계발광소자용 화합물 및 그를 포함하는 유기전계발광소자 |
-
2016
- 2016-02-15 JP JP2016025894A patent/JP6607606B2/ja not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017145198A (ja) | 2017-08-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6607606B2 (ja) | トリアジン置換インドロカルバゾール誘導体、それからなる有機電子デバイス形成用アルコール不溶性塗膜、及びそれを用いた有機電子デバイス | |
| CN110520407B (zh) | 化合物、包含其的涂覆组合物、使用其的有机发光二极管和用于制备其的方法 | |
| JP7504535B2 (ja) | 新規な化合物、それを含むコーティング組成物、それを用いた有機発光素子およびその製造方法 | |
| JP5453249B2 (ja) | 発光用途のための電荷輸送材料 | |
| US8227096B2 (en) | Arylamine compound and organic electroluminescence device | |
| JP2019535679A (ja) | 新規なヘテロ環式化合物およびこれを利用した有機発光素子 | |
| KR101309502B1 (ko) | 탄화수소 화합물, 전하 수송 재료, 전하 수송 재료 조성물및 유기 전계 발광 소자 | |
| KR102416120B1 (ko) | 화합물, 이를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| JP5649029B2 (ja) | 発光性組成物、有機電界発光素子、及びベンゾジフラン誘導体 | |
| JP5028934B2 (ja) | 炭化水素化合物、電荷輸送材料、電荷輸送材料組成物および有機電界発光素子 | |
| KR102096145B1 (ko) | 공중합체, 이를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| KR102922671B1 (ko) | 신규한 화합물, 이를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| JP6833252B2 (ja) | 有機発光素子およびその製造方法 | |
| JP5584971B2 (ja) | 高分子化合物の製造方法 | |
| WO2018084426A1 (ko) | 신규한 해테로 고리 화합물 및 이를 이용한 유기발광 소자 | |
| KR102352352B1 (ko) | 중합체 제조방법, 이 중합체를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| KR102929255B1 (ko) | 화합물, 이를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| JP5250967B2 (ja) | 有機化合物、電荷輸送材料、電荷輸送材料用組成物および有機電界発光素子 | |
| KR102920657B1 (ko) | 신규한 화합물, 이를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| KR102706857B1 (ko) | 신규한 고분자 및 이를 포함하는 유기 발광 소자 | |
| JP2012119471A (ja) | 有機電界発光素子用組成物、有機電界発光素子、有機el表示装置及び有機el照明 | |
| KR20200043756A (ko) | 신규한 화합물 및 이를 이용한 유기발광 소자 | |
| KR20200042808A (ko) | 신규한 화합물 및 이를 이용한 유기 발광 소자 | |
| KR102310375B1 (ko) | 공중합체, 이를 포함하는 코팅 조성물, 이를 이용한 유기 발광 소자 및 이의 제조방법 | |
| JP2018127433A (ja) | 新規ベンゾチエノベンゾチオフェン誘導体、それを用いた正孔輸送材料及び有機el素子 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20190115 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20190925 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20190926 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20191017 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6607606 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| LAPS | Cancellation because of no payment of annual fees |