JP6874012B2 - グラフェン及びグラフェンナノプレートレットを含有する組成物並びにその調製方法 - Google Patents
グラフェン及びグラフェンナノプレートレットを含有する組成物並びにその調製方法 Download PDFInfo
- Publication number
- JP6874012B2 JP6874012B2 JP2018538894A JP2018538894A JP6874012B2 JP 6874012 B2 JP6874012 B2 JP 6874012B2 JP 2018538894 A JP2018538894 A JP 2018538894A JP 2018538894 A JP2018538894 A JP 2018538894A JP 6874012 B2 JP6874012 B2 JP 6874012B2
- Authority
- JP
- Japan
- Prior art keywords
- graphene
- composition
- polymer
- sonication
- solvent
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images











Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/0066—Use of inorganic compounding ingredients
- C08J9/0071—Nanosized fillers, i.e. having at least one dimension below 100 nanometers
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82B—NANOSTRUCTURES FORMED BY MANIPULATION OF INDIVIDUAL ATOMS, MOLECULES, OR LIMITED COLLECTIONS OF ATOMS OR MOLECULES AS DISCRETE UNITS; MANUFACTURE OR TREATMENT THEREOF
- B82B3/00—Manufacture or treatment of nanostructures by manipulation of individual atoms or molecules, or limited collections of atoms or molecules as discrete units
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B32/00—Carbon; Compounds thereof
- C01B32/15—Nano-sized carbon materials
- C01B32/182—Graphene
- C01B32/184—Preparation
- C01B32/19—Preparation by exfoliation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F112/00—Homopolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F112/02—Monomers containing only one unsaturated aliphatic radical
- C08F112/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F112/06—Hydrocarbons
- C08F112/08—Styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F12/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F12/02—Monomers containing only one unsaturated aliphatic radical
- C08F12/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F12/06—Hydrocarbons
- C08F12/08—Styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/44—Polymerisation in the presence of compounding ingredients, e.g. plasticisers, dyestuffs, fillers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/46—Polymerisation initiated by wave energy or particle radiation
- C08F2/56—Polymerisation initiated by wave energy or particle radiation by ultrasonic vibrations
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J9/00—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof
- C08J9/04—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent
- C08J9/12—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent
- C08J9/14—Working-up of macromolecular substances to porous or cellular articles or materials; After-treatment thereof using blowing gases generated by a previously added blowing agent by a physical blowing agent organic
- C08J9/141—Hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
- C08K3/042—Graphene or derivatives, e.g. graphene oxides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B82—NANOTECHNOLOGY
- B82Y—SPECIFIC USES OR APPLICATIONS OF NANOSTRUCTURES; MEASUREMENT OR ANALYSIS OF NANOSTRUCTURES; MANUFACTURE OR TREATMENT OF NANOSTRUCTURES
- B82Y30/00—Nanotechnology for materials or surface science, e.g. nanocomposites
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/14—Saturated hydrocarbons, e.g. butane; Unspecified hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2203/00—Foams characterized by the expanding agent
- C08J2203/18—Binary blends of expanding agents
- C08J2203/182—Binary blends of expanding agents of physical blowing agents, e.g. acetone and butane
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2325/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Derivatives of such polymers
- C08J2325/02—Homopolymers or copolymers of hydrocarbons
- C08J2325/04—Homopolymers or copolymers of styrene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2325/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring; Derivatives of such polymers
- C08J2325/02—Homopolymers or copolymers of hydrocarbons
- C08J2325/04—Homopolymers or copolymers of styrene
- C08J2325/06—Polystyrene
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Nanotechnology (AREA)
- Inorganic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Manufacturing & Machinery (AREA)
- Carbon And Carbon Compounds (AREA)
- Processes Of Treating Macromolecular Substances (AREA)
- Polymerisation Methods In General (AREA)
- Graft Or Block Polymers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Description
本発明は、溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する組成物、並びに溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する前記組成物の、黒鉛材料から出発し、超音波技術を利用する調製方法に関する。
本テキストでは、溶媒に分散したグラフェン及びグラフェンナノプレートレットを含有する組成物は、溶媒中に沈積物及び分離相の形成が観察されない、グラフェン及びグラフェンナノプレートレットの安定分散系を指す。
黒鉛材料に加えて、本特許出願で記載及び請求する組成物を調製するための出発材料として非黒鉛状炭素、例えば、カーボンブラックが存在することもある。
本発明の方法対象は、グラフェンの場合は単原子層が得られるまで、又はグラフェンナノプレートレットの場合は限定数の層が得られるまで、高剥離度を有する、グラフェン及びグラフェンナノプレートレットを含有する組成物を得られるようにする。
剥離という用語は、多層材料(例えば本発明で用いる黒鉛材料)の層が互いに分離されるプロセスを指す(「ゾル、ゲル、網目構造、及び無機-有機ハイブリッド材料の構造及び加工に関する用語の定義(Definitions of terms relating to the structure and processing of sols, gels, networks, and inorganic-organic hybrid materials)」−IUPAC勧告2007)。
・グラフェン及びグラフェンナノプレートレットを含有するポリマーコンポジット若しくはポリマーナノコンポジット又はマスターバッチ(後者は濃縮ポリマーコンポジットとしても知られる);これらの化合物は高い機械的性能、高い耐熱性、静電気防止能力、電磁絶縁特性を有するか、又は高い熱伝導度を有する;
・それから高い断熱能力、耐熱性及び高い機械的性能を有する物品及び発泡ポリマーフォームを調製できる発泡性ポリマーコンポジット又は発泡性ポリマーナノコンポジット;
・記載及び請求する組成物に由来し、記載及び請求する方法によって得られる固形のグラフェン及びグラフェンナノプレートレット。
ポリマーナノコンポジットは、相ドメインの少なくとも1つがナノメートルのオーダーの少なくとも1つの寸法を有するコンポジットと定義される。
出願人は、ポリマーがビニル芳香族型である、発泡性でもあるポリマーコンポジット、又はポリマーナノコンポジット、をも保護することをも企図し;前記コンポジットは、本テキストで記載及び請求するポリマー組成物から出発して得ることができる。
場合により発泡性である前記ポリマーコンポジット又はポリマーナノコンポジットは、高い断熱能力、耐熱性及び高い機械的性能を有するポリマー顆粒、製品又は発泡ポリマー品、発泡ポリマーフォームの調製に使用可能であり、これらも本発明の対象である。
本テキストでは、用語「GRS」は、グラフェン及びグラフェンナノプレートレットを指す。
本テキストでは、用語「グラフェン及びグラフェンナノプレートレット」は、グラフェン、若しくはグラフェンナノプレートレット、又は両方を指す。
本テキストでは、用語「溶媒」は、成分の想定される溶解特性を全く考慮せずに、グラフェン及びグラフェンナノプレートレットがその中に分散される流体相を指す。
本テキストでは、用語「液相」は、液相と、その中に分散した固体粒子(例えばグラフェンナノプレートレット)とを含む組成物をも指す。
本発明の目的のために、本テキスに示す全ての作業条件は、明示的に特定していなくても、好ましい条件であるとみなすべきである。
本発明の目的のために、用語「含む」又は「含める」は、用語「に存する」又は「から本質的に成る」をも含む。
本発明の目的のために、範囲の定義は、特に指定のない限り、極値を含める。
グラフェンの製造方法は、2つのマクロカテゴリー、すなわち、ボトムアップ法及びトップダウン法に分類することができる。
CVD(化学蒸着)、SiC上でのエピタキシャル成長、アーク放電、カーボンナノチューブのアンジッピング(unzipping)等の種々の方法は最初のカテゴリーに属する。これらの方法は、単層グラフェン、又は高純度及び高側面寸法を有する二層若しくは数層を得られるようにするが、それらは高価であり、小規模なので、ポリマーナノコンポジットの製造に適さない。例えば、US 2011/244661は、カーボンナノチューブのアンジッピングによってグラフェンのナノリボン(nmオーダーの幅を有する細片)を得る方法を記載している。この方法は、カーボンナノチューブを部分的に酸化し、それらを有機溶媒に分散させ、ナノチューブを開くために溶液を機械的撹拌に供することにある。
グラフェンの大規模な別の製造方法は、黒鉛酸化物の剥離及び還元に基づいている。後者は、黒鉛を硝酸又は硫酸の存在下でKMnO4、KClO3及びNaNO3を用いて黒鉛を酸化することによるStaudenmaier又はHummersの方法(及びその変法)を経て得られる(Staudenmaier, L. Ber. Dtsch. Chem. Ges. 1898, 31, 1481-87; Hummers, W. S., Jr., Offerman, R.E. J. Am. Chem. Soc. 1958, 80, 1339)。
黒鉛自体に関して、酸化物は、sp2混成炭素上の層の縁に位置するカルボニル基及びカルボキシル基に加えて、基平面のsp3混成炭素上にヒドロキシル基及びエポキシ基を有する。従って黒鉛酸化物は高度に親水性であり、水中で容易に剥離され、さらに、層間の距離は、元の基平面の上及び下のsp3炭素のシフト及び共有結合で結ばれた酸素原子の存在のため、黒鉛層間に存在する距離に比べて大きい(酸化物の0.6〜1nmに対して黒鉛の約0.34nm)。
この方法の主な欠点は、存在する官能基がグラフェンの共役電子構造を破壊し、欠陥及び無秩序を導入することである。官能基の空間分布及び構造欠陥に強く左右される光学特性及び電気特性が強く影響を受け、黒鉛酸化物は実際には電気絶縁性であり、熱的に不安定である。
さらに、光学特性もグラフェンとは完全に異なる。特に、黒鉛酸化物は、波長にかかわらず、赤外線吸収体であるというその特性を失い、このことが、発泡ポリマーフォームの断熱を高めるためのその用途を無効にする。
導入される位相欠陥は、孤立型(六角形セルの代わりに五角形及び七角形の存在)及び広範型(ほぼ無定形の炭素構造)として分類することができる。
黒鉛酸化物の還元は、化学的又は熱的のどちらでも伝導率(電気的及び光学的)を部分的にだけであるが、いずれの場合でも純粋グラフェンに対して数桁小さい値まで回復させ得る。実際には、酸化物の還元は完全ではなく、欠陥及び無秩序としても、かなりの酸素含量が構造内部に残る。
還元後、酸化領域内のsp2炭素の網目構造は回復されるが、最初に用いた黒鉛の結晶化度は、いずれの場合でも失われる。特に、sp3混成を有する炭素原子の存在は、このようにして生成されたグラフェンの電子工学的能力及び赤外線との相互作用を大きく制限する。還元を受けた無秩序領域は、さらに、基平面の内側にも外側にも応力及び変形をもたらす。
化学的還元は、種々の還元剤(例えば、ヒドラジン、水素化ホウ素ナトリウム等)を用いて行なわれるが、使用する化学的還元剤の危険な性質及びコストがそれらの応用を制限し得る。
熱的還元は、溶媒の使用を必要としないという利点を有するが、CO2の損失に起因する構造欠陥のため、層に非常にしわが寄ってつぶれてしまう。
WO 2012/166001は、硫酸による黒鉛の処理後、過マンガン酸カリウムによる酸化及び最終的に酸化物のアルコールによる還元を経てグラフェンを製造する方法を記載する。
類似して、CN 103408000は、大きい側面寸法及び大きい表面積を有する黒鉛酸化物の調製方法を記載する。この場合にも、天然黒鉛を硝酸及び硫酸で処理し、引き続き強い酸化剤(過酸化水素)を用いて酸化する。
上述したように、上記特許に記載の方法により得られる黒鉛酸化物は、構造欠陥の存在を特徴とする材料であり、たとえ還元されても、出発黒鉛材料の結晶構造を取り戻さない。さらに、これらの特許で請求された方法は、多くの場合腐食性で環境に優しい化学薬品(例えば強酸及び酸化剤)の使用を想定し;結果として、工業化は複雑かつ高価になる。
黒鉛の直接剥離は、適切な溶媒中の超音波処理によって液相内で達成可能である。この方法は種々の利点を有し:それは容易にスケーラブルとなる可能性があり、その結果としてそれは工業的量で、場合により官能化された、単層又は数層を有するグラフェンをおそらく製造することができる。この製造モードに一般的に存在する欠点は、以下のように要約できる:グラフェンシート又はグラフェンナノプレートレットを溶液から分離又は抽出する際の低収率及び困難さ。特に、濾過又は遠心分離による抽出は、必然的にグラフェン層のある程度の積み重ね又は再集合ということになり、その結果この方法の有効性を低下させる。さらに、超音波を用いた過度に強力な処理は、層の側面寸法の過剰な低減となる恐れがあり、アスペクト比(側面寸法と厚さの比)の低下をもたらす。
Hernandezらの上記論文“High-yield production of Graphene by liquid-phase exfoliation of graphite”において、著者らは、黒鉛から出発し、約1質量%の単層グラフェン収率で、非酸化グラフェンを得、欠陥が存在することなく、N-メチル-ピロリドン(NMP)中0.01mg/mlまでの濃度を有するグラフェンの分散系を得た。
しかしながら、用いた溶媒(NMP)は多くの欠点を有する:毒性、刺激性及び催奇形性であることに加えて、それは高い沸点(203℃)をも有し、このことは、とりわけ、溶媒の完全な除去が必要とされる電子工学分野におけるその取扱い及び適応性を制限し、そうでなくても性能に影響を及ぼす可能性がある。
WO 2013/010211は、層状材料(従って必ずしもグラフェンでない)を適切な界面活性剤中での超音波処理によって剥離する方法を記載する。
CN 103112848は、0.5〜50時間の超音波処理によって、キトサンの酸性水溶液中の黒鉛の分散系を含むグラフェンを調製する方法を記載する。
そして分散系を放置してデカントし、最低数の層を有する剥離材料を回収するために上清を遠心分離機にかける。
KR 20110077606は、10〜20分間のプロパノール中の黒鉛の超音波処理及び非剥離黒鉛粒子を排除するためのその後の4,500〜5,500rpmでの遠心分離を含む。
層間に位置する適切な物質で黒鉛を処理して、いわゆる黒鉛層間化合物(Graphite Intercalation Compound)(GIC)を生じさせる。インターカレーションに典型的に用いられる方法は、硝酸、硫酸又は両方の混合物中の黒鉛の分散を含むが、塩素酸カリウム、クロム酸、過マンガン酸カリウム、過塩素酸等の他の物質を使用することも多い。インターカレーションされた黒鉛が引き続き高温(700〜1000℃)への急速加熱を受け、その間に粒子がそれらの元の体積を80〜1,000倍まで膨張させ、黒鉛の結晶層に垂直な方向に特定のアコーディオン構造を得る。
この方法は種々の文書、例えばUS 2010/140792及びWO 2011/162727に記載されており、インターカレーションの下流で、層の分離のために、電気化学的、熱的、音響学的性質、マイクロ波及び超音波プロセスの使用を考慮する。
WO 2008/060703は、黒鉛をギ酸若しくは酢酸、水又はその組み合わせでインターカレーションし、そのインターカレーションされた化合物を引き続き少なくとも1,450℃の温度で超臨界流体及び熱的剥離にさらし、インターカレーションされた黒鉛を不活性ガスのプラズマ中に供給することによるナノ構造体(ナノチューブ、フラーレン及びグラフェンナノプレートレット)の製造方法を記載する。
インターカレーション法の欠点は、環境に優しい物質の使用によって表され;さらに、この方法は液相内にも気相内にも様々な種の硫黄化合物及び窒素化合物を生成し、これは浄化処理を必要とする。インターカレーションのさらなる欠点は、この方法によれば、剥離度の制御が困難であり、数層のみを有するグラフェンナノプレートレットの製造に利用できないことである。
KR 20130068515も、フッ素と黒鉛材料を含有するイオン液体の混合物を超音波に供し、引き続きマイクロ波に供してから再び超音波に供することによって、イオン液体を用いてグラフェンを製造する方法を記載する。
論文“Preparation of Graphene by Using an Intense Cavitation Field in a Pressurized Ultrasonic Reactor”(Stengl V.; Chem. Eur. J. 2012, 18, 14047-14054)において、著者らは、天然黒鉛から出発し、加圧超音波反応器内でのキャビテーションを利用して、高品質の非酸化グラフェンを製造する方法を提案した。水とエチレングリコールの混合物(9:1の比)に黒鉛が添加されて50分間キャビテーションを受けた。超音波反応器内で5バールの圧力にて、液体に伝達される300W/cm2超の強度を用いてキャビテーションが行なわれた。
この方法は、次のさら工程で、溶媒中のグラフェンナノプレートレットの懸濁液にモノマー又はポリマーをさらに添加し、ナノコンポジット前駆体を得ることを含み、この前駆体は、溶媒を除去するか又はモノマーを重合させることによって固体に変換することができる。
全ての場合に、これらの方法は、グラフェン又はグラフェンナノプレートレットを含むポリマーコンポジットを得ることができないか、或いはいずれの場合でも単一工程では不可能である。溶液からのグラフェン又はナノプレートレットの抽出は繊細な作業であり、グラフェン又はナノプレートレットの凝集を引き起こしやすく、結果として処理の有効性を減じる。さらに、それは溶媒、界面活性剤及び他の化学薬品の使用を必要とするので、かなり環境に影響を与える恐れがあり、作業を冗長、高価かつ複雑にする。
論文“Sonochemical Preparation of Functionalized Graphenes”(Hangxun Xu and Kenneth S. Suslick; J. Am. Chem. Soc. 2011, 133, 9148-9151)において、著者らは、粒子形態の黒鉛(天然)から出発するスチレンで官能化されたグラフェンを調製するための単一工程法を提案した。この方法は、天然黒鉛をスチレンと混合し、この混合物に高強度超音波を照射することを提供し;超音波処理は、20kHzの振動数及び50W/cm2の強度で0℃にて2時間、アルゴン流内でプローブを利用して行なわれる。この方法は、単層グラフェン又は典型的に5未満の数層を有するグラフェンへの黒鉛の剥離、及び、同時に、スチレンの重合及び生じたポリスチレン鎖によるグラフェンの官能化を達成できるようにする。
結果的に、この方法の最後には、ポリスチレン鎖がグラフェンの表面に吸収されている官能化グラフェンが得られる。この論文は、実際に、指定条件下での超音波処理中に、スチレンが重合して反応性ラジカルを形成し、一方で黒鉛の3D構造が破壊されてグラフェンの2D構造を形成すると説明している。この時点で、反応性ラジカルがグラフェンの表面に結合し、その結果、ポリスチレンで官能化されたグラフェンを形成する。また、この論文は、得られた官能化グラフェンは、通常の有機溶媒中で安定かつ溶解性であり、前記グラフェンに基づく材料の調製に使用できるとも主張する。
Suslickらの論文は、提案されたシステムの重合能力について言及していない。いずれの場合でも、当業者は、使用条件下では、相当量のポリマー、ひいてはポリマー化合物を得ることはかなり困難であると考えるであろう。
Suslickらが用いた条件は、本テキストにおいてさらに明らかになるように、本特許出願で記載及び請求する方法で用いる条件とは異なる。実際に、Suslikらの論文で用いた条件に対して、本発明の方法は、より短い超音波処理時間で行なわれ、超音波処理は、脱気環境下で典型的にずっと高い温度で、より高い圧力にて達成される。出願人は、本発明の方法は、超音波処理中に気相の非存在下で実施されるが、Suslikらによれば、超音波処理はアルゴンの存在下で達成されることをもう一度指摘したい:本特許出願で記載及び請求する方法を行なえるためには、通常は使用溶媒に溶解している少量のガスも除去するのが好ましい。
以降「Kruusら」と称する論文“Polymerization of Styrene Initiated by Ultrasonic Cavitation”(P. Kruus, D. McDonald, and T.J. Patraboy; J. Phys. Chem. 1987, 91, 3041-3047)において、著者らは、超音波処理中のスチレンの重合を研究し、反応温度(バルク温度)及び反応媒体との関連でスチレンの重合速度を評価している。Kruusらの論文では、超音波処理は60Wの出力、21W/cm2の出力密度、及び20kHzの超音波振動数で行なわれ;超音波処理はアルゴンの存在下で行なわれる。アルゴンの存在は、キャビテーションノイズを制限するためにも着色化合物の形成に関して重合に有利に働くためにも必要である。
Kruusらの論文において、著者らは、スチレンの重合速度は重合温度の低下とともに低減すると結論づけた。特に、48℃未満で、重合速度は著しく低減し、着色化合物の生成が観察される。高蒸気圧の炭化水素液体、例えば、n-ヘキサン、n-ヘプタン、トルエン又はシクロヘキサン等の添加は、重合速度を上昇させ、着色化合物の形成を抑制する。
Lawrieらの論文では、スチレンの超音波処理が記載され、86Wの出力、34W/cm2の出力密度、及び20kHzの超音波振動数にて種々のアルゴン流量の存在下で行なわれる。
この研究は、アルゴン流が存在すると、スチレンの重合が目に見え、アルゴン流が中断されると、スチレンの変換が劇的に減少することを明らかにする。さらに、アルゴン流が中断される時間帯において、著者らは、生じたポリマーの脱重合及び着色化合物の形成を観察している。
上記論文の教示に基づいて、低温は、超音波を受けるときにもスチレンの重合を非常に危うくするとの結論を下すことができる。さらに、Suslickが提供したデータ(論文及び論文に添付された関連補足情報参照)から、グラフェンに結合したポリマーの18%が、初期ポリマーに対して数十ppmオーダーであると推定することができる。従って、Suslickが得た転化率は、本テキストで記載及び請求する方法で得られる転化率とは対照的に、 極端に低いと結論を下すことができる。
さらに、上記論文の教示によれば、ガス流(例えば、アルゴン)の非存在下で超音波処理を行なうと、スチレン重合が中断され、脱重合が始まる。対照的に、本特許出願で記載及び請求する方法は、超音波処理中により高温及び気相の非存在下で行なわれる。
WO 2011/042800は、最良の断熱性を有し、不透熱性剤としてグラフェンナノプレートレットを含有する、発泡性熱可塑性ポリマーに基づく組成物を記載する。これらの発泡性化合物から得られるポリマーフォームは、黒鉛、石炭、アルミニウム粒子等の他の不透熱性剤を含有するものに比べて高い断熱を特徴とすることが分かる。
しかしながら、上記2つの特許出願はどちらもナノプレートレットの製造又は関連化合物の製造のために超音波を使用しない。
A. Ciesielski, P. Samoriの“Graphene via sonication assisted liquid-phase exfoliation”, Chem. Soc. Rev., 2014, 43, 381-398は、液相中での超音波処理を経るグラフェンの製造の種々の方法及び得られた結果を記載する。超音波処理に必要な時間が詳細に考察され、1,000時間に達し得る。高濃度で溶液を得るためには冗長な超音波処理時間が必要であり(論文に示される図6参照)、1mg/mlに等しい濃度のためには少なくとも100時間必要である。同論文が確証するように、冗長な超音波処理時間は、得られるナノ粒子の側面寸法の減少をもたらし、側面寸法は多くの用途にとって重大な意味を持つパラメーターなので、このことは望ましくない。
前述の公知技術の欠点及び制限は、本テキストで記載及び請求する組成物及び方法によって克服される。
a)それは、溶媒の総質量に対して少なくとも1質量%のビニル芳香族ポリマーを含有し、
b)それは、溶媒の総質量に対して0.001質量%〜10質量%の範囲の質量濃度のグラフェン及びグラフェンナノプレートレット(GRS)を含み;
ここで、前記ビニル芳香族ポリマーは、関連ビニル芳香族モノマー単独又は50質量%までのさらなる共重合可能モノマーとの混合物の部分又は全体重合によって、及び前記未反応モノマーの可能性のある含量と、形成されるビニル芳香族ポリマーの含量との合計が、溶媒の総質量に対して少なくとも10質量%に等しい条件で得られる。
a)黒鉛材料を、主溶媒の総質量に対して少なくとも10質量%のビニル芳香族モノマーを単独又は50質量%までのさらなる共重合可能モノマーとの混合物で含む主溶媒と接触させて、出発組成物を形成する段階;
b)前記出発組成物を、18kHz〜1000kHzの範囲内に含まれる振動数スペクトル、及び2絶対バール以上の圧力を特徴とする超音波に、前記組成物との接触時にガス状態の分離流体相がその中に存在してはいけない容器又は超音波処理チャンバー内で供する段階
を含み;
前記方法は、主溶媒中に存在する少なくとも1%のビニル芳香族モノマーを重合させることを特徴とする。
本テキストで記載及び請求する組成物が得られた時点で、引き続く主溶媒のさらなる重合において、グラフェン及びグラフェンナノプレートレットを含有するポリマーコンポジット若しくはポリマーナノコンポジット、又はマスターバッチ(濃縮ポリマーコンポジットとしても知られる)を調製することができる。
或いは、本テキストで記載及び請求する方法で前記ポリマーコンポジット若しくはポリマーナノコンポジット又はマスターバッチを直接得ることができ、同方法中に重合を完了する。
本テキストで記載及び請求する組成物から得ることができるポリマー組成物、ポリマーナノコンポジット又は濃縮物は、高剥離度並びに高度の化学的純度及び結晶純度でグラフェン及びグラフェンナノプレートレットを含有するという利点を有する。
本発明の方法対象は、有利なことに単純であり、大規模製造のために容易に拡大でき、かつ低コスト及び低環境影響を有する。
本発明の方法では、有利なことに「溶媒交換」作業、すなわち剥離のために用いる主溶媒を用途に必要な溶媒と交換することを行なう必要がない。
実際に、本発明の方法では、剥離は、ポリマーコンポジット又はポリマーナノコンポジット中に存在するポリマーがそれから重合によって得られるモノマーを含有する主溶媒中で直接達成される。
実際に、「溶媒交換」作業は、一般的に環境上の観点から課題を伴う方法の複雑化を意味することに加えて、剥離した黒鉛材料の層の部分的凝集につながる恐れがあり、従って結果として生じるポリマーナノコンポジットの品質を低下させる。
本特許出願の対象である方法は、有利なことに健康、安全及び環境(HSE)並びにナノ材料の取り扱いに関する問題を伴わない。実際に、作業中、ナノ粉末は遊離状態で形成されない。ナノ粉末は常に主溶媒と共に溶液中にあるか又は微細なナノコンポジットのポリマー成分中に直接組み込まれている。
本テキストで記載及び請求する方法は、有利には主溶媒の界面張力と無関係である。
実際に、前記方法は、黒鉛材料自体の界面張力と異なる界面張力を有する溶媒、又はいずれの場合にもより適していると考えられる溶媒に属さない溶媒、例えば、文献に記載されているものとは異なり、スチレン及びグリセロール等の溶媒を使用しても、黒鉛材料の剥離が達成されるようにし、経時的に安定な溶液の形成を可能にする。例えば、既に言及したHernandezらの論文“High-yield production of graphene by liquid-phase exfoliation of graphite”の補足情報を参照されたい。表S1は、グラフェンの剥離及び分散に最良の溶媒を示し:ベンゾイルベンゾアートに比べてアセトンはずっと有効でないように見え(約70%低い);スチレン及びグリセロールは言及さえされていない。
グリセロールに関する限り、上記WO 2011/014347は、グラフェンナノプレートレット中の黒鉛の剥離のための50の異なる溶媒を分析している。グリセロールは、高出力超音波を用いてグラフェンナノプレートレットを製造する際に効果のない溶媒の例と考えられている:生データは、90°以上の接触角を有する溶媒、例えばグリセロールは、黒鉛材料から出発する、高出力超音波処理によるグラフェンナノプレートレットの製造には効果がないことを示している。
他方で、本テキストで記載及び請求する方法では、驚くべきことに、グリセロールを用いても、時がたつにつれて安定性のあるグラフェンナノプレートレットの溶液を得ることができる。
本発明のさらなる目標及び利点は、本発明の好ましい実施形態を表す純粋に例示目的及び非限定目的で提供する下記説明及び添付図面からさらに明白に表れることになる。
以下、本特許出願の対象である本発明を詳細に説明する。
まず第一に、本発明は、溶媒と、前記溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットとを含有する組成物に関する。前記組成物は、それが溶媒の総質量に対して少なくとも1質量%のビニル芳香族ポリマーを含有し、かつ溶媒の総質量に対して0.001質量%〜10質量%の範囲のグラフェン及びグラフェンナノプレートレット(GRS)の質量濃度を有することを特徴とする。
前記ビニル芳香族ポリマーは、関連ビニル芳香族モノマー単独又は50質量%までのさらなる共重合可能モノマーとの混合物の部分重合又は全重合によって、及び場合によりさらなる未反応共重合可能モノマーと混合された前記未反応ビニル芳香族モノマーの可能性のある含量と、前記ビニル芳香族ポリマーの含量との合計が、溶媒の総質量に対して少なくとも10質量%に等しいという条件で得られる。
ビニル芳香族ポリマーの含量は、好ましくは溶媒の総質量に対して少なくとも5質量%、さらに好ましくは少なくとも10質量%である。ビニル芳香族ポリマーの含量は、好ましくは溶媒の総質量に対して70質量%以下である。
ビニル芳香族ポリマーの含量は、好ましくは溶媒の総質量に対して5質量%〜70質量%の範囲、さらに好ましくは溶媒の総質量に対して10質量%〜70質量%の範囲である。
場合によりさらなる未反応共重合可能モノマーと混合された前記未反応ビニル芳香族モノマーの可能性のある含量と、前記芳香族ビニルポリマーの含量との合計は、溶媒の総質量に対して好ましくは少なくとも20質量%、さらに好ましくは少なくとも80質量%に等しい。
記載及び請求する組成物中のグラフェン及びグラフェンナノプレートレット(GRS)の質量濃度は、溶媒の総質量に対して好ましくは0.05質量%〜5質量%、もっとさらに好ましくは0.2質量%〜2.5質量%の範囲である。
厚さは、酸化ケイ素でコーティングされたシリコンウェーハ上に沈積したGRSのいくつかのサンプル内で、間欠接触原子間力顕微鏡(AFMタッピングモード)を用いて測定される。
GRSは、好ましくは10以上、さらに好ましくは30以上、もっとさらに好ましくは50以上、もっとさらに好ましくは100以上の炭素/酸素(C/O)モル比を有し得る。比(C/O)は、GRSのX線光電子分光法(XPS)によって得られる。
記載及び請求する組成物において、グラフェン及びグラフェンナノプレートレットは、好ましくは10以上の炭素/酸素モル比及び50nm以下、かついずれの場合にも単一グラフェン層の厚さ以上の平均厚さを有する。
得られる組成物は、ASTM D5296-11に従って高速サイズ排除クロマトグラフィーで測定される50〜500キロダルトンの範囲の質量平均分子量(MW)を有し、3〜50の範囲のメルトフローインデックス(200℃、5kgでのMFI)を有する。流動性指数は、ISO 1133、第4版、2005に従って測定される。分子量分布は、高速サイズ排除クロマトグラフィーを利用し、屈折率検出器を用いてASTM D5296-11に従って測定され、GRSからの分離及び引き続く沈殿後のポリマーに関するものである。
a)黒鉛材料を、主溶媒の総質量に対して少なくとも10質量%のビニル芳香族モノマーを単独又は50質量%までのさらなる共重合可能モノマーとの混合物で含む主溶媒と接触させて、出発組成物を形成する工程;
b)前記出発組成物を、2絶対バール以上の圧力で、及び前記組成物との接触時にガス状態の分離流体相がその中に存在してはいけない容器又は超音波処理チャンバー内で18kHz〜1000kHzの範囲の振動数スペクトルを特徴とする超音波に供する工程
を含み;
前記方法は、主溶媒中に存在する少なくとも1%のビニル芳香族モノマーを重合させることを特徴とする。
a)黒鉛材料を、主溶媒の総質量に対して少なくとも10質量%のビニル芳香族モノマーを単独又は50質量%までの他の共重合可能モノマーとの混合物で含む主溶媒と接触させて、出発組成物を形成する工程;
b)前記出発組成物を、2絶対バール以上の圧力で、及び前記組成物との接触時にガス状態の分離流体相がその中に存在してはいけない容器又は超音波処理チャンバー内で、18kHz〜1000kHzの範囲の振動数スペクトルを特徴とする超音波に供する工程
を含み;
前記方法は、主溶媒中に存在する少なくとも1%のビニル芳香族モノマーを重合させることを特徴とする。
記載及び請求する方法、及び全てのその好ましい実施形態において、主溶媒中に存在する少なくとも1%、好ましくは少なくとも5%、さらに好ましくは10%〜80%、もっとさらに好ましくは15%〜50%のモノマーが重合する。
本テキストで記載及び請求する方法は、連続流モードで、又は連続流を用いて、又はバッチ流モード、又は不連続流モードのどれでも行なうことができる。
記載及び請求する方法の最後に、部分的又は全体的に重合した組成物が得られ、その中にグラフェン及びグラフェンナノプレートレットが溶媒及び好ましくは本テキストで記載及び請求する組成物に永続的に分散している。
前記組成物は、本テキストでさらに詳細に説明するように、さらなる重合工程を引き続き受けて重合を完了し、それによってポリマーコンポジット、ポリマーナノコンポジット又はマスターバッチ(濃縮ポリマーナノコンポジットとしても知られる)を形成し得る。
主溶媒に含まれる用いた重合可能モノマー、好ましくはビニル芳香族モノマーの重合を部分的又は全体的に続けることによって、溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する組成物の部分的又は完全のどちらかの重合工程が直接超音波処理中に起こる。
完全重合が超音波処理中に起こるとき、グラフェン及びグラフェンナノプレートレットの高分散を特徴とするグラフェン及びグラフェンナノプレートレットを含有するポリマーコンポジット又はポリマーナノコンポジットが直接得られる。
部分又は完全重合中に、主溶媒中に存在するモノマーの少なくとも1%、さらに好ましくは主溶媒中に存在するモノマーの少なくとも5%、もっとさらに好ましくは10%〜80%、もっとさらに好ましくは15%〜50%が重合する。
主溶媒は、超音波処理の有効性に直接影響することに加えて(実施例で記載するように)、超音波処理中に組成物の粘度及び所望の重合度を制御するために有利に使用し得る。さらに、主溶媒は、例えば、エチルベンゼンを用いた制御されない暴走する反応の誘発を防止するために使用することもできる。
溶媒は、好ましくは場合によりさらなる重合可能コモノマー及び重合中に形成される可能性のある対応ポリマーと混合したビニル芳香族モノマーである。
主溶媒は、好ましくはビニル芳香族モノマー、ビニル芳香族ポリマーとビニル芳香族モノマーを溶解できる有機化合物、ペンタン及びその混合物から選択され;主溶媒は、さらに好ましくはビニル芳香族モノマー、エチルベンゼン及びその混合物から選択され;スチレン、エチルベンゼン及びその混合物がもっとさらに好ましい。
開始剤のうち、過酸化物が好ましく;さらに好ましくは1,1-ジ(tert-ブチルペルオキシ)シクロヘキサン(Trigonox 22E50, AkzoNobel)、tert-ブチルペルオキシ2-エチルヘキシルカルボナート(Trigonox 117, AkzoNobel)、過酸化ジベンゾイル(Perkadox L-W75, AkzoNobel)、過酸化ジクミル(Perkadox BC-FF, AkzoNobel)、又は炭素原子間に弱い結合を有する物質、例えば2,3-ジメチル-2,3-ジフェニルブタン(Perkadox 30, Akzo Nobel)から選択される。さらなる化学薬品、特に開始剤は、出発組成物の調製段階又は超音波処理中に添加可能である。
本テキストで記載及び請求する最終組成物、又はポリマーコンポジット若しくはポリマーナノコンポジット中に存在するグラフェン及びグラフェンナノプレートレットは、50nm以下、さらに好ましくは20nm以下、もっとさらに好ましくは5nm以下、及びいずれの場合にも単一グラフェン層の厚さ以上の平均厚さを特徴とする。
厚さは、酸化ケイ素でコーティングされたシリコンウェーハ上に沈積したGRSのいくつかのサンプルについて、間欠接触原子間力顕微鏡(AFMタッピングモード)を用いて測定される。
比(C/O)は、GRSのX線光電子分光法(XPS)によって得られる。
これらの特徴は、誘導発泡性コンポジットの場合に得られるポリマーナノコンポジットに、良い導電性、良い機械的性能及び良い断熱能力を与える。
ポリマーコンポジット、ポリマーナノコンポジット又はマスターバッチが調製されたら、ポリマー材料の変換の分野で採用される通常の技術、例えば、押出、射出成形及び圧縮成形等によってこれらを加工して誘導ポリマーを得ることができる。
既に示したように、本発明の目的のために用いる黒鉛材料は、本質的に黒鉛炭素から成る。出発黒鉛材料の黒鉛炭素は、好ましくは合成黒鉛、天然黒鉛、膨張黒鉛、高秩序熱分解性黒鉛(highly ordered pyrolytic graphite)(HOPG)、鱗状(vein)黒鉛、又は層間(intercalated)黒鉛から選択可能である。
黒鉛材料と共に、好ましくはコークス若しくはカーボンブラック、又は黒鉛化コークスから選択される非黒鉛炭素が存在することもできる。
このために、例えば、いずれの種類の撹拌機をも使用でき、或いは静的ミキサー又は動的ミキサー、例えば、ローブポンプ又は押出機等を使用することができる。必要な時間は、組成物流体の作製に必要とされる時間である。典型的に、通常は30秒で既に十分であるが、より長い時間も同様に使用可能である。使用する混合システムと適合性の粘度を得るために室温とは異なる温度で作業を行なうのが有用なことがあり:例えば、船舶用プロペラ撹拌機を使用する場合、0.2〜5,000cPの範囲内の粘度を得るような温度でなければならない。
結果的に、前記組成物と接触するガス状態の第2の流体相が超音波処理チャンバー内に存在してはいけない。特に、大気又は他のガス、例えば、窒素若しくはアルゴン等と直接接触している、従って超音波処理を受けた出発組成物と接触するガス状態の第2の相を形成する開放容器で行なわれる超音波処理は、本発明の範囲に入らない。
処理中に超音波処理ループ内を循環する出発組成物は、好ましくは少なくとも0.1Pa・秒かつ1,000Pa・秒以下、さらに好ましくは0.1Pa・秒以上かつ100Pa・秒以下の粘度を有する。この目的で、超音波処理中に出発組成物を希釈するのに適した第2の溶媒を使用するのが有用なことがある。前記第2の溶媒は、例えば、エチルベンゼンであってよく、或いはそれは主溶媒と同一であり得る。前記第2の溶媒の添加は任意である。前記第2の溶媒は、超音波処理中に添加される。
超音波処理は、好ましくは超音波がその中で適用されるチャンバーの容積に対する超音波発生器により生じた電力として計算される少なくとも60W/cm3の特定超音波出力で行なわれる。もっとさらに好ましくは、特定超音波出力は少なくとも110W/cm3である。
超音波処理の有効持続時間は、好ましくは20分以下かつ0.1秒以上であり;それはもっとさらに好ましくは0.5秒〜5分、もっとさらに好ましくは1秒〜1分の範囲、もっとさらに好ましくは2秒〜30秒の範囲である。
有効時間は、組成物が超音波照射を効果的に受ける時間を指す。
例えば、バッチモードでは、前記時間は、超音波照射が引き起こされる時間全体に相当する。連続流モードでは、前記時間は(超音波の連続的発生の場合)、超音波がその中で発生されるチャンバー内の滞留時間に相当し、一般的に占有体積と体積流量との比として計算される。超音波が周期的に発生される場合、すなわち超音波の発生が超音波が実質的に発生しない時間と交互に起こるとき、結果として有効時間は、滞留時間に有効超音波発生時間の割合を掛けることによって計算される。
分のオーダーの超音波処理時間は、エネルギー及び経済的観点から、より有効であるのみならず、工業規模での製造のスケールアップをも容易にする。さらに、実際の超音波処理時間を短縮することによって、グラフェンナノプレートレット及びグラフェンの変化も減少し、特に、グラフェンナノプレートレット及びグラフェンの粒子の側面寸法の低減が制限され、これはアスペクト比(側面寸法と厚さの比)の低下をもたらすことになる。アスペクト比の低下は、結果として生じるポリマーコンポジットの電気的及びレオロジー的浸出閾値を高め、それは赤外線の吸収能を減じ、機械的性能を低下させるので一般的に望ましくない。
出発組成物の加圧は、超音波処理の前又はその間に起こり得る。
本特許出願で記載及び請求する最終組成物を得るためには、大気圧より高く、特に上記範囲内の圧力で作業することが不可欠である。特に、このことは、前述したように、少なくとも30日間安定した分散系を得るため、並びに単一工程で剥離及び重合を達成するために不可欠であることが判明した。
結果的に、最新技術の教示によれば、圧力上昇はキャビテーションを弱めるので、超音波処理を弱める恐れがあり、超音波処理に有利に働くように発振器(出力発振器)の強度を高くしなければならない。
対照的に、本発明の教示によれば、音波処理中に高い圧力値が不可欠である(上述したように)。結果的に、最新技術で教示されることによれば、当業者にとって、圧力値の増加は、処理自体を無効にすることになる。
超音波処理の前又はその間の加圧は、技術上周知のいずれの手順によっても行なえる。例えば、ポンピング及び/又は供給デバイス、例えばギアポンプ等を利用して超音波処理前に加圧を行なうことができる。前記ポンプは、例えば、超音波処理がその中で行なわれる回路に出発組成物がそれを用いて供給されるものと同じであり得る。別のモードによれば、さらに詳細に後述するように、加圧は、超音波を受けた流体と直接又は間接的に接触して置かれる膨張流体を用いて行なうことができる。さらなる手順によれば、圧力下で超音波処理を受ける流れを維持するように、対向流(例えば流れ方向と反対のポンピング方向を有する)の超音波処理後に、第2のポンピングデバイスを配置することができる。
直接又は間接膨張タンクの安定化手法は、バッチスキームのため又は連続スキームのため、或いは連続とバッチの両モードでの使用を想定できるプラントで使用可能であり;従ってそれは特にフレキシブルである。間接膨張タンクは、2つのチャンバーを分離する可動性又は可撓性セプタムを含有する装置を含み、2つのチャンバーの一方は安定化すべき流体、すなわちこの文脈では出発組成物を含有し、他方は安定化流体、好ましくは不活性ガスを含有する。安定化流体は、セプタムに対して圧力をかけ、セプタムは可動性又は可撓性なので、安定化すべき流体に対して同様に圧力をかける。従って2つの流体は、互いに接触することなく同圧で維持される。例えば、熱膨張、注入若しくはサンプリング、又は化学反応に起因する液体の体積の変動は、圧力の大きな変動をもたらすことになるため、液体の圧縮率よりガスの圧縮率が大きい。他方で、ガスは、圧力の如何なる実質的な変化もなしでこれらの変動を吸収することができる。結果的に、上述したような装置の使用は、圧力の安定化を可能にする。
このモードによれば、処理される組成物を気相と直接接触させて置くか、或いは同様に気相と接触している液体状態の膨張流体と直接接触させて置く。いずれの場合にも気相は、例えば、処理される組成物の温度の変動に従う該組成物の体積の変動による圧力の安定化を可能にするのに十分大きい体積を有する。気相の体積は、例えば、処理される組成物の体積の1/10以上である。
直接膨張タンクモードでは、2つの相の一方に圧力を加えると、直接接触によって両相が同圧になる。このモードは、吸収すべき体積の広い変動を可能にし、かつ何らかの形でデバイスの有効性を減じるセプタムの必然的な機械的剛性によって影響を受けないので、間接膨張タンクの使用に比べて有利である。セプタムは、化学的及び物理的にも出発組成物と適合性でなければならない。
しかしながら、超音波処理の有効性は、前記組成物中のガスの存在に大いに左右されるので、前記組成物の部分上のガスの吸収を制限することが望ましい。実際に、一方で、気泡がキャビテーションの核として作用する場合、超音波法に有利に働き、他方で、気泡は、吸収効果に起因する衝撃波の強度を減じる。液相と気相との間の直接接触は、気相の一部を液体に緩徐に溶解させ、経時的に超音波の機能性を変える。
液体・ガス接触面の好ましい低減方法は、十分小さい直径(例えば1〜8リットルの範囲の容積を有する超音波処理ループに対して3mm〜15mmで変動する直径)を有する垂直パイプ、又は対応容積が、少なくとも圧力のバランスを保つために移動しなければない液体の体積より大きいような長さを有し、かつ少なくとも10に等しい、好ましくは少なくとも20に等しい、もっとさらに好ましくは50より大きい長さ・直径比を有する等価な装置内部で液体・ガス接触を行なうことである。このようにして、体積の大きな変動を可能にしながら、接触を最小限にすることができる。
処理される組成物とガスとの間の接触領域は、超音波処理チャンバーの外側である。液体とガスとがその中で接触しているパイプは、さらに詳細に後述するように、例えば、側面ノズル又は他の分岐を利用して超音波処理ループに接続することができる。従って、前記パイプ内に含まれる全ての液体は、超音波処理ループと接続されている限り、処理される組成物がその中を循環しない領域内にある。
このモードによれば、処理される組成物を、膨張流体と直接接触させて、例えば垂直パイプ、又は一方の流体の他方の流体への拡散を最小限にするように十分小さい直径を有する等価な装置に置く。垂直パイプ又は等価な装置は、例えば、対応容積が少なくとも圧力のバランスを保つために動くべき液体の体積より大きくなるような長さを有し、かつ少なくとも10に等しい、好ましくは少なくとも20に等しい、もっとさらに好ましくは50より大きい長さ・直径比を有する。
前述したように、同様に膨張流体を直接又は間接安定化デバイスに接続することができる。この場合、直接安定化デバイスは、接触面を制限する必要がないので、小さい表面を必ずしも持つ必要がない。実際に膨張流体は直接安定化デバイスとプロセス流体との接触との間にある。
上記手順とは別に又は上記手順と組み合わせて、バッチモードでも連続モードでも、当業者に周知の他のデバイス、例えば、逃がし弁、破裂板、安全弁。及びポンピングデバイス、例えば所望逆圧を維持するためにその速度が調整されるギアポンプ等を使用することもできる。さらに詳細に後述するように、直接モード又は間接モードに従って超音波を適用することができる。これは、本発明の対象である方法を連続モードで行なってもバッチモード行なっても真実である。
適切な応力で膨張及び収縮できる活性材料を含有する目的で知られるいずれのデバイスによっても超音波を発生させ得る。これらの活性材料は、好ましくは適切な高電圧交流電界を受けた圧電セラミックス、又は超磁歪材料から選択される。活性材料の長さの周期的変動は、一般的に極端に小さく(例えば、50ミクロン以下)、従って活性材料に加えられた電界/磁界の共振周波数に近い共振周波数を有する適切に成形された金属材料に典型的に引き起こされる適切な増幅を必要とし得る。
本明細書では以後トランスデューサーと定義される前記金属材料は、一方で直接モードによれば活性材料に適用され、他方でそれは処理すべき出発組成物と直接接触する(「直接トランスデューサー」)。
他方で、間接モードによれば、トランスデューサーは一方で活性材料に適用され、他方で、それは処理すべき出発組成物がその中に含まれる容器と、接着又は溶接によって接触している(「間接トランスデューサー」)。後者の場合、音波は最初に、処理すべき組成物がその中に導入される容器の壁を通過してから、処理すべき組成物に移動する。
本発明の教示によれば、いずれの超音波発生モードも使用可能である。直接モードが好ましい。
容器内で用いるトランスデューサーの数に特に制限はない。トランスデューサーの数は組成物に適用すべき超音波出力に基づいて計算され:例えば、1リットル未満の小体積については、単一のトランスデューサーで十分なことがあるが、例えば1リットル超の大きい体積ついては、2つ以上のトランスデューサーの使用が便利であり得る。
トランスデューサーの共振周波数は、適用するトランスデューサーの全てで必ずしも同一でない。種々のトランスデューサー間にいずれの角度も形成され得る。本発明のモードによれば、複数のトランスデューサーが適用される場合、対抗が存在するように、すなわち同トランスデューサーの末端間の空間内を組成物が移動するようにトランスデューサーを適用するのが有利なことがある。
以前に特定したように、本発明の対象である方法は、連続モード、又はバッチモードで行なうことができる。
バッチモードでは、処理すべき出発組成物を、例えば前記組成物をポンプ又は重力を用いて貯蔵タンクから収集することによって、超音波処理を行なうのに適したデバイスに投入する。処理すべき出発組成物を次に加圧し、所望温度に至らしめてから超音波処理を所要時間行ない、処理された組成物を最後にデバイスから放出する。
他方で、連続モードでは、例えばポンプを用いて出発組成物を連続的に超音波処理チャンバーに投入してから、超音波処理を行なう。出発組成物は典型的に、デバイスを去る前に超音波処理チャンバー内で、再循環ポンプによって様々な回数再循環される。このようにして、出発組成物は、超音波の作用を様々な回数受け、機器は、連続サイクルで(24時間/24時間)動作することができる。これは、温度、圧力及び流速条件を経時的に一定に保つようにもし、その結果、得られる生成物の生産量及び品質の不変性を最大にする。
本発明の対象である方法を連続モードで行なうとき、超音波処理がその中で行なわれるデバイス(超音波処理チャンバー)は、超音波処理ループの全容積の最大50%、好ましくは最大15%、もっとさらに好ましくは最大2%である出発組成物が占有する容積を有する。
連続手順でもバッチ手順でも、前述した直接モード又は間接モードに従って超音波を適用することができる。直接モードを適用するのが好ましい。
超音波の発生がその上で行なわれる表面は、超音波処理チャンバーの表面の好ましくは少なくとも10%に等しく、もっとさらに好ましくは前記チャンバーの表面の少なくとも30%に等しい。
上記で定義したパルスモードは、0.0001〜1、さらに好ましくは0.0005〜0.3、もっとさらに好ましくは0.001〜0.05の範囲の、本特許出願で「デューティサイクル」と定義される、アクティブ期とアクティブ期及びパッシブ期の合計との比に従って行なわれなければならない。
実際に、例えば連続流モードを考えると、超音波処理ループ内の流体の所望どおりの小さい体積に従って、これは、それが超音波処理チャンバー内に留まる時間(アクティブ期)中に超音波の作用を受けることになる。超音波処理チャンバーを去ると、流体の前記体積はもはや超音波場の影響を受けない。超音波処理チャンバーの入口又は出口の最も近くでは、流体の前記体積がずっと低い出力(少なくとも10倍低い)を有する超音波場を受け得る。従って、超音波処理チャンバー外部の前記流体体積によって費やされる時間がパッシブ期であり、一方、超音波処理チャンバー内で費やされる時間がアクティブ期である。
超音波出力を経時的に調節する場合、超音波の周期的中断時間中に超音波処理チャンバーに流体の体積が入り得るので、アクティブ期をさらに減らすことができる。
いずれの場合にも、超音波出力の調節の有無にかかわらず、超音波処理ループ内の流体の体積は、アクティブサイクルに続いてパッシブサイクルその他があるので、わずかな時間のみ超音波の作用を受ける。
連続流モードでは、時間調節なしで(例えば経時的に一定出力で)超音波が発生される場合、デューティサイクルは、超音波処理チャンバーの容積と超音波処理ループの容積との比に相当する。実際には、溶液は超音波処理ループ内で連続的に再循環されるが、超音波処理チャンバー内に溶液が留まるわずかな時間に超音波を受けるだけである。
再び連続流モードでは、超音波の発生は経時的に調節され、デューティサイクルは、超音波処理チャンバーの容積と超音波処理ループの容積との比に、超音波の発生が活発な時間割合を掛けることによって、或いはさらに一般的には、超音波発生器に適用される平均出力の割合を掛けることによって与えられる。
出発組成物は、好ましくは20℃以上、さらに好ましくは40℃以上、もっとさらに好ましくは60℃以上、もっとさらに好ましくは90℃以上の温度で超音波処理を受ける。
また、処理は110℃以下の温度で達成され得る。出発組成物は、好ましくは200℃以下、さらに好ましくは170℃以下、もっとさらに好ましくは150℃以下、もっとさらに好ましくは130℃以下の温度で超音波処理を受ける。
出発組成物は、好ましくは20℃〜200℃、好ましくは40℃〜200℃、もっとさらに好ましくは40℃〜170℃、もっとさらに好ましくは60℃〜130℃の範囲の温度で超音波処理を受ける。
当技術分野では、超音波処理に通常用いられる温度は室温、場合によっては、室温未満及び0℃に近い温度であることが知られている。
実際には、過度に高い温度は、一般的に超音波処理法の効果を低くする。同時に、使用する温度条件は、採用するモノマーの重合において工業的に用いられる範囲外である。
他方で、記載及び請求する方法は、単一工程で黒鉛材料のGRSへの剥離、及び同時に重合可能モノマーの重合と共に、驚くべきことに有効な超音波処理を達成できることを実証する。
1つ以上の熱交換器を用いて、或いは抵抗器又はサーマルジャケット、コイル、電気抵抗器、ペルチェセルを用いて、或いは熱源と同じ超音波を利用して、温度を所要範囲内に維持することができる。温度制御は、場合により、熱交換器内で処理される組成物を連続的又は周期的に再循環させることによって、組成物がその中で超音波を受けるチャンバー(超音波処理チャンバー)の外部で行なわれる。
温度は指定範囲内で変動し得るので、時間内及び空間内で必ずしも一定でない。
例えば、超音波を室温で組成物に適用し、徐々に組成物を目標温度に至らせることができる。或いは、好ましい場合、組成物を予熱して、既に目標温度の組成物にだけ超音波を適用することができる。或いはこの場合もやはり、所定スキームに従って、経時的に及び/又は空間内で温度は変動し得る。
超音波処理チャンバーと呼ばれる、超音波処理がその中で行なわれる容積は、組成物がその中に供給され、その中で前述した温度、圧力及び音の強さ条件下で超音波場が適用され得る任意のデバイスである。
連続流モードでは、超音波処理がその中で行なわれるチャンバーは、有利には超音波処理ループの全容積の最大50%、好ましくは最大15%、もっとさらに好ましくは最大2%である、混合物が占有する容積を有する。これは、処理自体をより効果的にする。このようにして、実際には、低減した体積の流体に超音波の作用を集中させることができ、結果として短いが、より強烈な、結果的により有効な作用をもたらす。増強した有効性は、生成されるコンポジットの量当たりの電気エネルギーの消費を少なくすることもできる。
連続流モードでもバッチモードでも、超音波の発生がその上で行なわれる表面は、超音波処理がその中で行なわれるチャンバーの表面の少なくとも10%、もっとさらに好ましくは前記チャンバーの表面の少なくとも30%に等しくてよい。
この処理は、存在するガス、特に出発組成物の充填前に装置の容積中に存在するガス、及び前記組成物中に存在するガス及び/又は超音波処理中に生成されるガス(例えば、溶媒の低沸点分解生成物)の除去を可能にする。
超音波の同作用は、溶解ガスからの液体の分離を促進し;従って、バッチモードでも連続流モードでも、処理中に脱気を行なうと有利であり得る。
本発明の対象である方法は、分散及び剥離に加えて、同時に重合可能モノマーの部分的又は全体的重合を行なえるようにし、それによってポリマーコンポジットの直接合成を達成する。
最新技術によれば、ポリマーコンポジットを得るためには、生成されたグラフェン及びグラフェンナノプレートレットを予め抽出し、その後にポリマーに分散させなければならない。これは、複雑な補足単位作業及び溶媒の環境に優しくない除去プロセスを暗示するのみならず、グラフェン及びグラフェンナノプレートレットの抽出は、以前に剥離された層の部分的再凝集を必然的に伴うので、処理の有効性をも減じる。
前述したように、本特許出願の対象である方法に用いる溶媒は、少なくとも10質量%のビニル芳香族モノマーを単独又は50質量%までの他の共重合可能モノマーとの混合物で含有しなければならない。
本明細書及び特許請求の範囲で使用する場合、用語「ビニル芳香族モノマー」は、本質的に下記一般式に相当する生成物を指す。

好ましいビニル芳香族モノマーは、下記:スチレン、α-メチルスチレン、メチルスチレン、エチルスチレン、ブチルスチレン、ジメチルスチレン、モノ、ジ、トリ、テトラ及びペンタクロロスチレン、ブロモスチレン、メトキシスチレン、アセトキシスチレン等から選択され得る。好ましいビニル芳香族モノマーは、スチレン及びα-メチルスチレンである。
既に特定したように、一般式(I)を有するビニル芳香族モノマーは、単独又は50質量%までのさらなる共重合可能モノマーとの混合物で使用することができる。これらのさらなる共重合可能モノマーの例は(メタ)アクリル酸、(メタ)アクリル酸のC1-C4アルキルエステル、例えばアクリル酸メチル、メタクリル酸メチル、アクリル酸エチル、メタクリル酸エチル、アクリル酸イソプロピル、アクリル酸ブチル等、(メタ)アクリル酸のアミド及びニトリル、例えばアクリルアミド、メタクリルアミド、アクリロニトリル、メタクリロニトリル等、ブタジエン、エチレン、ジビニルベンゼン、無水マレイン酸等である。好ましい共重合可能モノマーは、アクリロニトリル、無水マレイン酸、メタクリル酸グリシジル及びメタクリル酸メチルである。
実際には、さらなる重合を進める前に、溶媒に分散したグラフェン及びグラフェンナノプレートレットを含有する最終組成物を種々の手順に従って処理することができる。
第1の手順では、前記最終組成物をデカントしたままにして非剥離黒鉛材料を沈殿させ、沈降物を形成し、結果としてグラフェン及びグラフェンナノプレートレットを含有する第1の液相を分離することができる。相のデカンティング後、分離された第1の液相(第1の上清)を、グラフェン及びグラフェンナノプレートレットを含有する第2の液相を非剥離黒鉛材料からさらに分離するのに十分な速度で遠心分離に供することができる。
第2の代替手順では、超音波処理後に、溶媒に分散したグラフェン及びグラフェンナノプレートレットを含有する最終組成物を、前もってデカントせずに、そのまま遠心分離に供して、非剥離黒鉛材料から、グラフェン及びグラフェンナノプレートレットを含有する最終液相を分離する。
デカンティング中、少なくとも2分、好ましくは20分以上、もっとさらに好ましくは少なくとも1時間に等しい時間前記最終組成物を放置して沈殿させ、結果として沈降物を形成する。
他方で、連続遠心分離機の場合、超音波処理に由来する最終組成物又はデカンティング後に分離された遠心分離しなければならない液相を連続的に遠心分離機に供給し、少なくとも第2の液相(第2の上清)を連続的に抽出する。
デカンティング及び/又は遠心分離後、溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有し、前述した手順に従って分離され、場合によりさらなる添加剤が添加された第1、第2の液相又は最終相を所望温度に至らしめて、重合を完了させ、本特許出願で記載及び請求するポリマーコンポジット、ポリマーナノコンポジット又はポリマーマスターバッチを形成する。前記化合物は、高い機械的性能、高い耐熱性、静電気防止能力、電磁絶縁特性、及び高い熱伝導度を有することが判明した。
さらなる添加剤は、難燃剤、自己消火剤、協力剤、核形成剤、潤滑剤、顔料、制酸剤及びラジカルスカベンジャーから選択される。
従って本特許出願のさらなる対象は、前述したポリマーコンポジット又はポリマーナノコンポジット又はマスターバッチ及び少なくとも1種の発泡剤を含有する発泡性ポリマーコンポジット又は発泡性ポリマーナノコンポジットに関する。
前記発泡性ポリマーコンポジット又は発泡性ポリマーナノコンポジットから出発して、高い断熱能力、耐熱性及び高い機械的性能を有する物品及び発泡ポリマーフォームを調製することができる。
重合法は、最新技術で知られる手順、例えば、懸濁重合、エマルション重合、溶液重合又は連続塊状重合に従って行なわれる。
連続塊状重合の場合、参考文献として本特許出願に組み込まれる例えばUS 6,348,549及びUS 2,593,399に記載されているように、本テキストで前述した手順に従って分離された第1又は第2の液相を溶融状態で重合することができる。場合により溶媒及び揮発性生成物の除去後に得られたポリマーを次に押し出し、カットして顆粒にする(ストランドカット)。
或いは、例えば本特許出願に参考文献として組み込まれるWO 2007/048536に記載のもののような水用リングタイプの造粒機を用いて、複数の穴がその中にあり、ヘッドで造粒されるダイプレートに溶融ポリマーを通すことによって、ポリマーコンポジット、又はポリマーナノコンポジットを得、場合により顆粒状態で濃縮される(マスターバッチ)。
このようにして得られたポリマーコンポジット、ポリマーナノコンポジット又はポリマーマスターバッチは、好ましくは最新技術で知られる任意の方法、例えば本特許出願に参考文献として組み込まれるUS 2,673,194、US 4,500,692、EP 2475709に記載されているように、例えば懸濁重合、又は連続塊状重合に従って、高断熱を有する発泡性ポリスチレン(EPS)の製造に使用可能である。
連続塊状重合中に、ポリマーナノコンポジットを他の成分、例えばポリスチレン、難燃剤及び関連協力剤、発泡剤、可能な核形成剤及び他の不透熱性剤に加えることができる。成分を例えば静的ミキサーを用いて混合し、場合により基準温度(例えば170℃)に至らしめてからダイプレートに供給し、その上にある一連の穴を通して組成物を押し出す。ダイプレートの穴から去る組成物を、例えば一連の回転ナイフを用いて造粒し、例えば水又は別の冷却流体を用いて十分に冷却して、発泡性ポリマー組成物を顆粒にカットできるようにする。例えば、前記造粒機は、水中冷却タイプのもの、又は本特許出願に参考文献として組み込まれるUS 7,320,585に記載のように、複数のスプレーノズルから出るウォータージェットタイプのものであってよい。さらに詳細な説明のためには、本特許出願に参考文献として組み込まれる特許出願WO 2003/053651、WO 2008/141766及びWO 2008/141767を参照することができる。
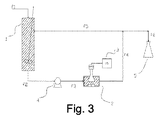
図1は、バッチ流モードの本発明の方法を説明する。黒鉛材料と、少なくとも10質量%のビニル芳香族モノマーを含む主溶媒と、可能な他のさらなる添加剤とをその中で接触して置き、機械的又は磁気的撹拌機を用いて混合する目的に適したビーカーその他の容器内で出発組成物を調製する。
出発組成物を調製したらすぐに、それをストリームf1経由で、超音波処理のために想定される圧力に耐えることができる適切なデバイス(1)に導入する。超音波発生器(3)に接続された発振器(2)によって、予測される時間出発組成物に超音波を直接適用し、その後、処理済組成物をデバイスの下部(f2)から除去する。
本方法のバッチモードでは、デバイス(1)は、デバイスの全容積が満たされるまで処理すべき出発組成物が充填されるので、ガスは存在しない。デバイスの加圧は、前述した手順の1つに従って行なわれる。例えば、ポンプを用いて組成物を供給することによって、結果として容器(1)全体を加圧し、或いは例えば容器の頂部の開口部(f3)によってデバイス(1)に接続された、本テキストで前述した直接又は間接膨張タンク加圧デバイスを適用することによって行なわれる。特に、超音波処理チャンバー及び直接膨張タンクは、前述したように液体サージ流体を用いて、単一ユニットに統合可能である。この場合、組成物の脱気を促進するように、膨張タンクは有利には、容器(1)の上部を占有することができ、一方、超音波処理チャンバーは下部を占有する。
デバイス(1)に充填された出発組成物は、連続、又はパルス超音波場を受ける。既に述べたように、バッチモードでは、時間サイクルで音波の発生を調節することによってパルスモードが得られ:発振器のトランスデューサーが、限られた時間電場/磁場によって励起された(以降、「アクティブ」期と定義される)後、活性材料の励起がないか、又は励起自体が極端に少ない(以降、「パッシブ」期と定義される)時間がある。
デバイス(1)に、任意に撹拌システムを設けることができる(図1に示さず)。前記撹拌システムは、好ましくはポンプを介した出発組成物の循環によるか、又は容器自体の内部に位置する撹拌機によって得られる。
溶液の導入前に(1)に存在するガスは、容器の頂部に作られた開口部(f3)から除去可能である。圧力制御は、供給ポンプによって、及び/又はデバイスの頂部(f3)又は下部(f2)から多少の溶液を取り除くことによって達成可能である。超音波処理が終了したら、処理済組成物を、等体積のガス又は液体で同容積を置き換えることによって即座に除去するすることができる。すなわち、処理済組成物は、同デバイス(1)内でデカンティング処理を受けることができる。デカンティング後、2つの相、すなわち上相と下相が形成され:生成物は、デバイスの側面部に適用されたノズル(図1に示さず)によって、又は浸漬管を用いて、上相の部分から除去される。デバイスの下部に層別化された残存する下相は次に処分されるか、又はその後のバッチで再利用される。
デカンティングに加えて、或いはその代わりに、濃縮処理を行なうこができ、これは溶媒の一部を蒸発させ、結果として生じた蒸気を引き続き排除し、濃縮生成物を収集することにある。この作業は、デバイス(1)内でもそれとは別に目的に適した装置でも行なうことができる。
容器(1)には、図2〜9に示すように、任意に撹拌システムを設けることができる。技術上周知のいずれの撹拌システムをも、例えばプロペラ、羽根車又はアンカー又はリボン撹拌機を使用可能であり、それらに接続したモーターで駆動し;或いは適切な外部磁場によって動く撹拌要素、例えば、アンカーを使用してよい。
ポンプが超音波処理デバイスの上流に位置する場合(図3のように)、デバイスの下流に逆止弁、又は圧力逃がし弁を適用して、出発組成物が、超音波処理中に、同デバイス(1)の圧力より高い所定圧力で確実に留まるようにすることができる。
このことは、例えば、より低い圧力又は大気圧でさえデバイス(1)をデザインすることができるので、コスト低減を可能にし、デバイス(1)の構築を単純にもする。
使用すべき交換器のタイプについては特に制限はないが、浄化作業が簡単な交換器、例えば、プロセス流体がチューブ側面を通過するチューブ・イン・チューブ交換器又はチューブ・バンドル交換器等が好ましい。また、この場合、超音波処理及び熱交換器の経路の下流で、組成物(f5)がデバイス(1)にストリームf6を経て戻る。超音波処理の最後に、処理済組成物がストリームf7によって、適切な容器(5)に回収される。
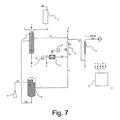
図6に示す実施形態モードは、プロセス流体(f11)と接触して置かれる発泡性流体を用いて、膨張流体がその中で気相と接触する特殊な加圧タンク又は等価システム(8)によって回路の加圧を実現する。既に述べたように、この加圧タンク又は等価システムは、垂直に配置される簡単なパイプから成る。前記膨張流体は、組成物と、ストリームf10 bisによって装入された安定化ガス(f10)との間に挿入されて、プロセス流体の液相中に存在するガスの溶解を防止する(膨張流体は動かないので、サージ流体内で溶解したガスのプロセス流体への拡散は極端に遅く、実際に無視できる)。
処理済組成物を超音波処理の最後に又は連続的にデカンターに供給することができる。連続的に供給する場合、超音波処理チャンバー(2)を去る流体の一部がストリームf7によってデカンター(10)に運ばれ、一方で残存部分は交換器(6)を通過して再び超音波処理ループに再循環される。処理済組成物(f7)は、デカンティングデバイス(10)内に留まることがあり、その結果、黒鉛材料の非剥離粒子が下部に沈殿し、一方で懸濁液中に残留するほとんど剥離された粒子は、デバイスの上部で、好ましくはデバイス(10)内部に存在する適切なセプタムを介して容易に回収され得る。
沈殿部分は容器(10)の下部から収集され、有利にはストリームf12によってローディングタンク(1)に再循環されて、超音波を再び受ける。処理済組成物は、この目的に適した容器(5)に、ストリームf13によって回収される。
デカンティングデバイス(10)から、同デバイスの上部で処理された組成物(f13)が取り除かれ、目的に適した容器(5)に回収される。黒鉛材料の非解離部分を含有する溶液が代わりにデカンティングデバイス(10)の下部から処分され、或いは、有利には、デバイス(11)に戻して再利用され、その結果、超音波プロセス(f12)を再び受ける。
タンク(11)の存在は、より長い時間連続モードでの使用を可能にし、さらに、当然に、大量の組成物を処理できるようにする。
貯蔵タンク(11)の代用として、組成物を主回路に供給できるいずれのデバイスをも使用することができる。例えば、代わりに、黒鉛材料を、少なくとも10質量%の重合可能モノマーを含む溶媒と接触状態に置くことによって、連続モードでも、混合システム又は静的ミキサーを備えたタンクを用いて、組成物の調製を超音波処理ループの上流で直接達成することができる。
この濃縮は、技術上周知のいずれの方法によっても達成可能である。例えば、最高揮発性溶媒のストリッピングをもたらすように、ガスの注入によって濃縮を達成することができる。これとは別に又はこれと組み合わせて、(12)中の組成物をより高温状態にすることによって、或いは再び、これとは別に又はこれと組み合わせて、(12)内の圧力を下げることによって、濃縮を達成することができる。
本発明によれば、不活性ガス、典型的に窒素を用いるストリッピングによって、及び圧力をより低い値、典型的に100mBarに下げることによって濃縮を行なうのが好ましい。
濃縮器(12)は、好ましくは、伝熱制御流体がその中で循環するジャケットを用いて、一定温度、例えば(10)内の組成物と同じ温度で維持される。抽出されたガス(g)は、場合により濃縮され、再利用、例えば供給原料混合容器(11)に再び供給され得る。(12)内の濃縮は、組成物中のGRSの濃度を少なくとも20%、さらに好ましくは少なくとも50%、もっとさらに好ましくは少なくとも100%高められるようにする。
以下、本発明及び出願の範囲のより良い理解のためいくつかの例を提供するが、決して本発明の範囲に対する如何なる限定をも表さない。
簡潔さのため、下記では、用語「部」は質量部を意味し、「g」又は「gr」はグラムを意味し、「l」又は「lt」はリットルを意味する。
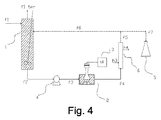
この例では、図4に示すように、連続流モードに従うグラフェン及びグラフェンナノプレートレットを含有する組成物の調製方法を説明する。
液体中の黒鉛の均質組成物を得るように、室温で15分間マグネチックアンカースターラーを用いて2部の市販のTimcal製合成黒鉛SFG6を98部のスチレン(Sigma Aldrich)に分散させる。
次に撹拌を停止し、組成物を即座に室温でポンプ(図示せず)を用いてローディングタンク(1)に投入する。循環ポンプ(4)を同時に起動する。投入中、容器(1)を完全に組成物で満たすために、超音波処理ループに最初に含まれたガスを容器(1)の上部に位置するノズルによって連続的に抽出する(f1 ter)。組成物がストリーム「f1 ter」を経て容器(1)から流出しはじめたときに組成物の投入を終わらせる。
組成物の投入が完了したらすぐに、ストリーム「f1 ter」を遮断し、供給ポンプを作動させて、容器(1)の圧力を13絶対バールの値まで上昇させる。
前述のデカンティングから得られた「生成物1A」を8,000rpmで30分間遠心分離(遠心分離機モデルSorvall Evolution RC、最大回転数25,000rpm)に供して、完全には剥離しなかった黒鉛材料を除去する。遠心分離の最後に、「生成物1B」と称する上清を収集する。これは、部分的に重合した、超音波処理プロセスの最終生成物である。
サンプルの固体、すなわち生じたポリマー並びに存在するグラフェン及びグラフェンナノプレートレットの百分率を決定するため、真空オーブンシステム(VOS)を用いる残留スチレンのストリッピングに「生成物1B」に供する。VOSは本質的に加熱デバイスであり、その中に少量の「生成物1B」(約3グラム)を230℃の温度で30分間挿入する。圧力は、10分間600mbarにしてから、さらに10分間400mbarにし、最後に試験の残り時間50mbarにする。このようにして得られたポリマーナノコンポジット生成物を「生成物1C」と呼ぶ。前記「生成物1C」においては、未反応残存モノマーは除去されており;記載方法(VOS)は、最終ポリマーナノコンポジット生成物を得るために可能なモードである。
熱重量分析(TGA)を生成物1Cについて行なって、GRSの百分率を得た。
このために、サンプルを室温から600℃まで窒素雰囲気内で、50℃/分の速度にて(この工程中に、ポリマーマトリックスが排除される)、引き続き600から850℃まで空気中で、この場合もやはり50℃/分の速度にて加熱した。この工程では、空気中の無機炭素、すなわちGRSが、存在する唯一の無機炭素材料であるので、これが燃焼する。窒素雰囲気から空気への変化後に起こる質量損失、すなわち窒素雰囲気内600℃での質量と、空気中850℃での質量との差から炭素の百分率を計算する。この値は、1.2質量%に等しいことが分かった。従って、合計に対する「生成物1B」中のGRSの百分率は0.22質量%に等しく、すなわち約2.2g/lの濃度に等しい。
残りの「生成物1A」を閉じた透明の第2の容器内で30日間静置した。如何なる目に見える沈降をも示さなかった。これは、「生成物1A」も既に濃縮された安定懸濁液であることを示唆している。
実施例1を繰り返すが、0.05部の開始剤Perkadox BC-FF(AkzoNobel製)、2部の黒鉛SFG6及び97.95部のスチレンを使用し、実施例1に記載のことと類似して、デカンティング後に「生成物2A」を得、遠心分離後に「生成物2B」を得、残留スチレンのストリッピング後に「生成物2C」を得た。「生成物2B」をTEM分析に供した。写真(図11)から、良い剥離度を観察することができる。AFMによって、GRSが10nm以下の厚さを有することをも推定した。実施例1に記載の同方法及び熱重量分析(TGA)によって「生成物2B」中のGRSの濃度を決定した。結果は2.5g/lである。固相率は22%に等しいことが分かった。
実施例1を繰り返すが、0.2部の開始剤Perkadox BC-FF、2部の黒鉛SFG6及び97.8部のスチレンを使用し、実施例1に記載のことと類似して、デカンティング後に「生成物3A」を得、遠心分離後に「生成物3B」を得、ストリッピング後に「生成物3C」を得る。実施例1及び2と同様に、TEM及びAFM分析を行ない、「生成物3B」に採用した同手順に従ってGRSの濃度を決定した。「生成物3B」のAFM分析から、GRSが10nm以下の厚さを有することを推定できるが、実施例1に記載の同方法及び熱重量分析(TGA)に従って測定された「生成物3B」中の濃度は4g/lである。固相率は25%に等しいことが分かった。この場合にも、「生成物3A」を閉じた透明容器内で静置した。結果は、安定した濃縮懸濁液であり、30日後に沈降の如何なる明白な兆候を示さなかった。
実施例1を繰り返したが、10部のポリスチレンEdistir N1782(Mw=180,000g/モル;MFI(200℃、5kg)=7.5g/10’)、2部の市販のTimcal製合成黒鉛SFG6及び88部のスチレン(Sigma Aldrich)で構成される組成物を用いた。実施例1に記載のことと類似して、デカンティング後に「生成物4A」を得、遠心分離後に「生成物4B」を得、ストリッピング後に「生成物4C」を得た。図12から明らかなように、「生成物4B」をTEM分析に供した。この場合にも、AFM分析を行なって、GRSの厚さが10nm以下であることを推定した。「生成物4B」についてのGRSの濃度は3g/lであり、実施例1に記載の同方法及び熱重量分析(TGA)を用いて得られる。
実施例1を繰り返すが、0.05部の開始剤Perkadox L-W75(AkzoNobel製)、2部の黒鉛SFG6、30部のエチルベンゼン及び67.95部のスチレンを使用する。
実施例1に記載のことと類似して、デカンティング後に「生成物5A」を得、遠心分離後に「生成物5B」を得、ストリッピング後に「生成物5C」を得た。「生成物5B」をTEM分析に供した。AFMによって、GRSの厚さが10nm以下であることをも評価した。実施例1に記載の同方法及び熱重量分析(TGA)を用いて得られた「生成物5B」について計算されたGRSの濃度は、約1.1g/lである。
この例は、ポリマーナノコンポジットの調製方法を説明する。
撹拌機を備えたガラス製オートクレーブに「生成物1B」の一部を挿入する。溶液を120℃に加熱し、この温度で真空下にて約1時間維持した。この場合もやはり実施例1に記載の方法で測定した固形生成物の百分率は35%であることが分かった。
このようにして得られた生成物を次に、オーブンの中にあるスチール製シリンダーに挿入し、150℃で3時間維持して、重合を完了させる。いずれの可能性のある残留スチレンをも排除するため、得られた生成物を次に30分間230℃の温度で真空下(50絶対mbar)に置く。このようにして得られた、「生成物1D」と称する材料を冷却し、シリンダーから抽出して粉砕する。この材料を、実施例1に記載の手順に従う熱重量分析(TGA)に供し、1%に等しい、存在するGRSの量を得た。
この材料は、ポリマー材料の変換の分野で用いられる通常の技術、例えば、押出、射出成形、圧縮成形等によって引き続き加工することができる。
実施例1を繰り返したが、溶液の(1)への投入後に、ポンプ(4)及び加圧を開始し、熱交換器(6)を用いて組成物を80℃の温度に至らしめ、それをポンプ(4)を用いて循環内で常に維持した後、超音波を開始する。実施例1に記載のことと類似して、デカンテーション後に生成物7Aを得、遠心分離後に7Bを得、ストリッピング後に7Cを得た。
実施例1に記載の同手順及び熱重量分析(TGA)を使用することで得られた、合計に対する「生成物7B」中のGRSの百分率は0.25質量%である。固相率は24%に等しいことが分かった。
実施例1を繰り返すが、図6に示すスキームに従って、加圧タンク(8)に含まれる膨張流体によって加圧を行なう。
このために、ストリーム(f10 bis)を介して加圧タンク(8)をスチレン(使用するサージ流体)で満たす。組成物の投入中(ストリームf1)、ポンプ(4)を作動させ、超音波処理ループに最初に含まれたガスを、容器(1)の上部に位置するノズルから連続的に除去し(ストリーム「e」)、専用ポンプ(図示せず)を用いて受取容器(9)を真空下で維持する。次にストリーム(f11)に対する弁を開いて膨張流体を組成物と接触状態に置く。可能性のあるいずれのガスポケットをも排除して、超音波処理ループを完全に組成物で満たすために、流れ(a)、(b)及び(d)に対する弁をも短時間開く。次にアルゴンガス(加圧ガス)をストリームf10を介して供給することによって加圧を達成し、超音波発生器を起動し、この場合もやはり実施例1に示すように、ポンプ(4)を制御する。次に実施例1と同じ熱プロファイル及び処理時間で試験を行なった。使用圧力は13絶対バールに等しかった。
この場合にも、30日後に如何なる目に見える沈降もなく安定懸濁液が得られる。実施例1に記載のことと類似して、デカンティング後に「生成物8A」を得、遠心分離後に「生成物8B」を得、ストリッピング後に「生成物8C」を得た。実施例1に記載の同手順及び熱重量分析(TGA)に従って決定した「生成物8B」中のGRSの濃度は、0.25質量%に等しいことが分かった。
TEM写真は、良い剥離度を示し、AFM分析は、10nmを超えない厚さを有するGRSの存在を示す。
実施例8を繰り返すが、8絶対バールの圧力を使用する。このようにして、実施例1に記載のことと類似して、デカンティング後に「生成物9A」を得、遠心分離後に「生成物9B」を得、ストリッピング後に「生成物9C」を得た。実施例1に記載の同方法及び熱重量分析(TGA)を用いて得た「生成物9B」中のGRSの濃度は、約0.06質量%に等しいことが分かった。
実施例8を繰り返すが、21絶対バールの圧力を使用する。このようにして、実施例1で既に述べた方法に従って「生成物10A」、「生成物10B」及び「生成物10C」を得た。前述の実施例に記載の同方法を用いて得た「生成物10B」中のGRSの濃度は、約0.15質量%に等しいことが分かった。
加圧を行なわずに、従って試験の持続時間全体にわたって大気圧を維持して実施例1を繰り返す。処理の最後に、輸送容器に溶液を抽出する。1時間後、容器内の溶液は完全に2つの相に分離することが判明し、下部に実質的に全ての黒鉛材料が沈殿した(図14、左側の容器)。
約500W/cm3の特定超音波処理出力で実施例1を繰り返す。実施例1に記載のことと類似して、デカンティング後に「生成物11A」を得、遠心分離後に「生成物11B」を得、ストリッピング後に「生成物11C」を得た。実施例1に記載の同方法及び熱重量分析(TGA)を用いて得た「生成物11B」中のGRSの濃度は、0.35質量%に等しいことが分かった。
実施例1を繰り返したが、2部の黒鉛SFG6、10部のスチレン及び88部のグリセロール(Carlo Erba)を使用した。処理後に抽出された生成物を閉じた透明の容器に入れた。30日後、このようにして得られた懸濁液は均質であることが分かった。生成物中のGRSの濃度は、0.4質量%に等しいことが分かった。
実施例1を繰り返したが、2部の黒鉛SFG6及びスチレンの代わりに98部のN-メチル-ピロリドン(Carlo Erba)を使用した。
処理の最後に、溶液を透明容器に抽出する。1時間後、容器内の溶液が完全に2つの相に分離し、実質的に全ての黒鉛材料が下部に沈殿することが判明した。
実施例1を繰り返したが、2部の黒鉛SFG6及びスチレンの代わりに98部のエチルベンゼンを使用した。
処理の最後に、溶液を透明容器に抽出する。1時間後、容器内の溶液が完全に2つの相に分離し、実質的に全ての黒鉛材料が下部に沈殿することが判明した。
液体中の黒鉛の均質な分散系を得るため、2部の市販のTimcal製合成黒鉛SFG6を室温で15分間マグネチックアンカースターラーを用いて98部のスチレン(Sigma Aldrich)に分散させる。組成物を受取デバイス(1)(図1)に室温で充填し、利用可能な全容積を満たし、同容器内に以前に含まれたガスを上部フランジに位置するノズル(f3)から除去する。組成物もストリーム(f1 bis)によって容器(1)からサイフォン(1 bis)に通して、容器(1)に存在するガスを除去する。サイフォン(1 bis)を次にアルゴンシリンダーに接続し、そのレデューサーを13絶対バールに設定する。このようにして、容器(1)を同圧で加圧し、同時に容器(1)にアルゴンが入らないようにする。
加圧を可能にすることに加えて、同接続は、全超音波処理プロセスにわたって圧力を一定に保てるようにもする(例えば、熱膨張を吸収することによって)。
発振器は、処理すべき組成物を含有する受取デバイス(1)内に直接浸漬される。受取デバイス(1)は、組成物を加熱するためにその中で油が循環するジャケットを備えている。このようにして、処理中、最初は室温で、130℃の最大温度に至るまで、55℃/時間の速度で組成物を加熱する。
処理が2時間続き、その最後に、受取デバイスの下部から直接材料を収集し、黒鉛材料が沈降するかどうかを観察するために静置する。
この場合にも、収集組成物は、1カ月後でさえ安定かつ均質であることが分かった。
この例は、GRS含有発泡性ポリスチレンの調製を説明する。
48.48部のポリスチレンEdistir N1782、1.8部のEmerald 3,000(Chemturaにより販売;ポリマー臭化難燃剤)、0.22部のPerkadox 30及び実施例6で得られた50部の「生成物1D」を2軸押出機内で混合する。次にn-ペンタン(75%)とイソペンタン(25%)の混合物を5部加える。このようにして調製した組成物を一連の静的ミキサーを用いて混合し;ギアポンプは混合物の圧力を200bargまで上昇させる。その後に熱交換器を用いて混合物を約170℃に冷却する。
次に組成物を加熱ダイプレートに送り、そこで組成物は0.5mmの直径を有する多数の穴を介して押し出され、ウォータージェットによって即座に冷却され、一連の回転ナイフによってカットされる(例えば、特許US 7,320,585に示されるように)。造粒チャンバー内の圧力は、5相対バールであり、カッティング速度は、1.2mmの平均径を有する顆粒を得るように設定される。遠心分離乾燥機により顆粒を乾燥させ、引き続き後述するようにコーティングで被覆する。
1,000部の乾燥顆粒当たり1部のステアリン酸亜鉛及び0.2部のグリセロールと共に3部のグリセリルモノステアラートを顆粒に添加すことによってコーティングを調製する。連続スクリューミキサーを用いてコーティングの添加剤を顆粒と混合する。
このようにコーティングされた生成物を100℃の温度で蒸気を用いて17g/l(かさ密度)で発泡させ、24時間寝かして最後にブロック(1040×1030×550mm)の鋳造に用いる。
ブロックをカットしてフラットシートを調製し、その上で熱伝導率の測定を行なった。測定された熱伝導率は、31mW/mKに等しかった。
サンプルブロックから得られたいくつかのシートを70℃の温度のオーブンに2日間入れてから、9×19×2cmの寸法を有する試験サンプルを得た。これらの試験サンプルを、規格DIN 4102に従う耐火試験に供した。
サンプルを、規格EN ISO 844に従う機械的圧縮強度試験にも供した。
10%圧縮歪みは、113kPaに等しいことが分かった。
この例は、黒鉛含有発泡性ポリスチレンの調製を説明する。
実施例14に記載の手順に従うが、50部の「生成物1D」の代わりに、如何なる超音波処理をも受けていない0.5部の黒鉛SFG6及び49.5部のEdistir N1782を用いて(従ってN1782は全部で97.98部に達する)発泡性ポリスチレンフォームを調製した。遠心分離乾燥機を用いて顆粒を乾燥させ、引き続き実施例14に記載どおりにコーティングで被覆した。
実施例1に記載の方法を用いる熱重量分析に顆粒を供し、0.45質量%に等しいGRS値を得た。
実施例14に示すように、熱伝導率、圧縮強度及び耐火測定用の試験サンプルを調製した。
得られた結果は、33mW/mKの熱伝導率、108kPaの10%圧縮歪みである。耐火試験は合格した。
実施例1を繰り返すが、図9のスキームを使用する。加圧タンク(8)に含まれる膨張流体を用いて加圧を達成する。超音波処理ループで用いた圧力は12bargに等しかった。
前記膨張流体はスチレンから成り、加圧化剤としてアルゴンを使用する。
貯蔵タンク(11)から超音波処理ループへの組成物の流速は、再循環ポンプ(4)の流速の1%に等しく、(11)と(1)との間に位置する追加のギアポンプ(13)を用いて制御され、速度制御は周波数変換器による。
超音波処理ループ内に含まれる流体の小部分を超音波処理ループの最高点(図9中の点d)から周期的に除去し、排水タンク(9)に回収する。
超音波処理済流体の一部を超音波処理ループから回収し、デカンター(10)に送る。超音波処理ループの圧力を12bargで一定に維持するので、(10)に送られる流体の流速は、貯蔵タンク(11)から超音波処理ループに供給される組成物の流速と実質的に等しい。
デカンター(10)の有効容積は、90分に等しいデカンター内の組成物の滞留時間を確保できるようなものである。デカンターの圧力は、超音波処理ループの圧力より0.5バール低く維持される。
デカンター(10)から濃縮器(12)に送られる上清の流速は、(11)から超音波処理ループに供給される流速の10%に等しい。残りの量は、デカンター(10)の下部から除去され、貯蔵タンク(11)に戻して再利用される。
濃縮器(12)は、100絶対mbarの圧力で維持される。圧力は、(e)に回収されたガスを熱交換器によって濃縮し、濃縮できないガスを真空ポンプで除去することによって制御される。
濃縮器(12)の下部から窒素を導入し、揮発性溶媒のストリッピングを達成する(図9に示さず)。
濃縮器を去る組成物(f15)をVOSに供し、40%に等しい固体値を得た。VOSから得られた固体生成物について上記方法に従って行なった熱重量分析は、3.2%に等しい値を与えた。これは、濃縮器を去るGRSの濃度が約1.3%に等しかったことを示す。
実施例1を繰り返すが、超音波の開始前に、溶液を30分で60℃に至らしめるために交換器(6)を使用する。次に実施例1におけるように超音波を開始する。
超音波処理の全持続時間は90分である。加熱は、最初の40分間は毎分1.5度に等しく、次の30分間は毎分約1度、最後の20分間は毎分0.5に等しく、試験の最後に160℃の温度に達するまで行なう。
超音波処理の開始から最初の50分の後に、回路内で捕捉された可能性のあるガスを排除するため、上部ベント弁を数秒間再び開く。
この場合もやはり実施例1に従って、デカンティング後に「生成物16A」を得、遠心分離後に「生成物16B」を得、ストリッピング後に「生成物16C」を得た。実施例1に記載の同方法及び熱重量分析(TGA)を用いて、「生成物16B」中のGRSの濃度を得た。結果は、5.5g/lである。固相率は30%であることが分かった。
この例は、ガスの存在下で間接モードに従う超音波処理を説明する(超音波浴)。
液体中の黒鉛の均質分散系を得るため、室温で15分間マグネチックアンカースターラーを用いて2部の市販のTimcal製合成黒鉛SFG6を98部のスチレン(Sigma Aldrich)に分散させる。分散系を超音波浴(Branson 8210モデル)内部のビーカーに導入してから大気と接触させる。次に超音波を開始する。2時間に等しい処理時間後に、第1のサンプルを収集する。さらに4時間の処理後に、第2のサンプルを収集する。両サンプルを静置する。翌々日に、両サンプル中の黒鉛材料が完全に下部に沈殿することが判明した。
膨張流体を投入せず、かつストリーム(a)、(b)及び(d)上の弁からガスポケットの抽出を行なわずに、実施例8を繰り返す。結果的に、超音波処理中に、ポンプ(4)によってガスが超音波処理チャンバー中に同伴され、そこでガスが組成物と直接接触する。処理の2時間後に、サンプルを抽出し、ビーカー内で静置する。翌々日に、ビーカー内の黒鉛材料が下部に完全に沈殿することが判明した。
この例は、超音波発生調節モードに従う超音波処理を説明する。
実施例1を繰り返すが、経時的な超音波の発生を調節する。すなわち発生される超音波出力を周期的に変える。さらに詳細には、15分の時間、一定出力で超音波を発生させ、5分の時間、超音波の発生を中断し(ゼロ出力)、これを交互に行なう。実際の持続時間は不変のままであり(72秒)、従って試験の全持続時間は9600秒である。
実施例1に記載のことと類似して、デカンティング後に「生成物17A」を得、遠心分離後に「生成物17B」を得、ストリッピング後に「生成物16C」を得た。「生成物17B」をTEM分析に供し;AFMによって、GRSが10nmを超えない厚さを有することを推定した。実施例1に記載の同方法及び熱重量分析(TGA)を用いて「生成物17B」中のGRSの濃度を得た。結果は5g/lである。
実施例1を繰り返すが、より長い全処理時間(ひいてはより長い実際の超音波処理時間)を用いる。全時間は28,800秒(8時間)に等しく;デューティサイクルは1%に等しいので、実際の超音波処理は288秒に等しい。
一定圧力で、室温(20℃)から始め、最初の30分間は毎分約2度、次の30分間は毎分約1度、次の20分間は毎分約0.5度、試験の開始から80分後に120℃の温度に達するまで、温度を上昇させて、試験を行なう。次に温度を一定に保ち、試験の残りの時間120℃に制御する。実施例1に記載のことと類似して、デカンティング後に「生成物18A」を得、遠心分離後に「生成物18B」を得、ストリッピング後に「生成物18C」を得た。「生成物18B」をTEM及びAFM分析に供し、この場合にも10nmを超えないGRSの厚さを推定することができる。実施例1に記載の同方法及び熱重量分析(TGA)を用いて「生成物18B」中のGRSの濃度を得た。結果は0.26質量%、すなわち約2.6g/lである。固相率は30%に等しいことが分かった。
本発明の実施例及び比較例の全ての生成物のサンプルを閉じた透明の容器内の調製の下流に置く。
上述した本発明の全ての実施例のサンプルは、安定懸濁液を示し、すなわち明るい光(例えば最大強度に設定したNikon Speedlight SB-80DXのフラッシュ)によるサンプルの照射でさえ、得られた画像にも、或いは視覚的にさえ、生成物の沈降は見られない。生成物は、均一に不透明かつ暗く見える。この特性は、1カ月後でさえ維持される。
対照的に、比較例のサンプルは、最初は均一の分布を示すが、1日以下の後には、生成物の強い沈降を観察することができる。
安定分散系は、黒鉛材料の良い剥離が得られたことを示す。従って実施例に由来するサンプルは、GRSにおける黒鉛材料の良い分散及び剥離を示すが、比較例のサンプルは、不十分な分散及び剥離を示す。
特に、大気圧で処理を行なうと(比較例1)、サンプルの収集の1時間後に既に黒鉛材料が沈殿したことを観察することができ、本発明が請求する方法は一般的に大気圧では有効でないことを示唆している。このことは、本発明を、対照的に大気圧で行なわれる当技術分野で知られる大半の方法と区別する。
比較例3も、生成物の急速沈降を示した。
驚くべきことに、得られた懸濁液は、経時的に安定であるのみならず、文献公知の手順よりずっと高い濃度でGRSを含有する。例えば、Hernandezらは、30分間超音波で分散系を処理することによって、NMP中約0.01g/lの濃度を得た。対照的に、実施例1で得られたGRSの濃度はずっと高い。より高い濃度が得られたが、ずっと長い超音波処理時間を使用した。例えば、Khanらは、460時間の超音波処理後に約1.2g/lの濃度を得た。100時間未満の超音波処理時間では、濃度が常に1g/l未満であることが分かった。
既に特定したように、このような長い超音波処理時間は、工業的実行のために不利であるのみならず、剥離した黒鉛材料に容易に欠陥を生じさせ得る。
比較例5及び6は、超音波処理済流体からガスを除去しなければ、或いはいずれの場合にも、処理される組成物が、覆っている分離気相(大気圧)と接触している場合、本発明の結果を得ることができないことを示す。特に、比較例5は、伝統的超音波浴で処理された同組成物は、本発明で得られる結果をもたらさないことを示す。
Claims (31)
- 溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する組成物であって、下記:
a)前記溶媒の総質量に対して少なくとも1質量%のビニル芳香族ポリマーを含有する、
b)前記溶媒の総質量に対して0.001質量%〜10質量%の範囲の質量濃度のグラフェン及びグラフェンナノプレートレット(GRS)を含む、
ことを特徴とし、
前記ビニル芳香族ポリマーは、関連ビニル芳香族モノマー単独又は50質量%までのさらなる共重合可能モノマーとの混合物の部分又は全体重合によって、及び前記未反応モノマーの可能性のある含量と、形成される前記ビニル芳香族ポリマーの含量との合計が、前記溶媒の総質量に対して少なくとも10質量%に等しいという条件で得られ、そして、少なくとも30日間、外観上明白な沈殿物又は分離相の形成もないことを特徴とする組成物。 - 前記芳香族ビニルポリマーの含量が、前記溶媒の総質量に対して少なくとも5質量%である、請求項1に記載の組成物。
- 前記未反応モノマーの可能性のある含量と前記ビニル芳香族ポリマーの含量との合計が、前記溶媒の総質量に対して少なくとも20質量%である、請求項1又は2に記載の組成物。
- 前記ビニル芳香族モノマーが、全ての前記モノマー及び前記ビニル芳香族ポリマーの質量の合計に対して少なくとも10質量%である、請求項1〜3のいずれか1項に記載の組成物。
- 前記グラフェン及びグラフェンナノプレートレットの質量濃度が、前記溶媒の総質量に対して0.05質量%と5質量%との間に含まれる、請求項1〜4のいずれか1項に記載の組成物。
- 前記グラフェン及びグラフェンナノプレートレットが、50nm以下の平均厚さを有する、請求項1〜5のいずれか1項に記載の組成物。
- 前記グラフェン及びグラフェンナノプレートレットが、10以上の炭素/酸素モル比を有する、請求項1〜6のいずれか1項に記載の組成物。
- 前記グラフェン及びグラフェンナノプレートレットが、50nm以下の平均厚さを有し、かつ10以上の炭素/酸素モル比を有する、請求項7に記載の組成物。
- 溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有し、そして、少なくとも30日間、外観上明白な沈殿物又は分離相の形成もない、少なくとも部分的に重合した組成物の、黒鉛材料から出発する調製方法であって、下記工程:
a)前記黒鉛材料を、主溶媒の総質量に対して少なくとも10質量%のビニル芳香族モノマーを単独又は50質量%までの追加の共重合可能モノマーとの混合物で含む前記主溶媒と接触させて、出発組成物を形成する工程、
b)前記出発組成物を、18kHzと1000kHzの間に含まれる振動数スペクトルを特徴とする超音波、及び2絶対バール以上の圧力に、気相が存在しない容器又は超音波処理チャンバー内で供する工程、
を含み;
前記主溶媒中に存在する少なくとも1%の前記ビニル芳香族モノマーを重合させることを特徴とする方法。 - 請求項1〜8のいずれか1項に記載の少なくとも部分的に重合した組成物の、黒鉛材料から出発する調製方法であって、下記工程:
a)前記黒鉛材料を、主溶媒の総質量に対して少なくとも10質量%のビニル芳香族モノマーを単独又は50質量%までの他の共重合可能モノマーとの混合物で含む前記主溶媒と接触させて、出発組成物を形成する工程、
b)前記出発組成物を、18kHzと1000kHzの間に含まれる振動数スペクトルを特徴とする超音波に、2絶対バール以上の圧力で、かつ気相が存在しない容器又は超音波処理チャンバー内で供する工程、
を含み;
前記主溶媒中に存在する少なくとも1%の前記ビニル芳香族モノマーを重合させることを特徴とする方法。 - 工程(a)及び(b)を同時又は順次に行う、請求項9又は10に記載の方法。
- 工程(a)及び(b)を、連続モードに従って又は不連続モードに従って行なう、請求項9〜11のいずれか1項に記載の方法。
- 請求項9〜12のいずれか1項に記載の方法であって、そのようにして調製された前記組成物を、非剥離黒鉛材料を沈殿させて、沈降物を形成し、かつ前記グラフェン及び剥離グラフェンナノプレートレット並びに前記非剥離黒鉛材料の一部を含有する第1の液相を分離するためにデカントする方法。
- デカンティング後に、前記非剥離黒鉛材料の一部から、前記グラフェン及び剥離グラフェンナノプレートレットを含有する第2の液相を分離するために、前記第1の液相を遠心分離に供する、請求項13に記載の方法。
- 溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する前記組成物を直接、遠心分離に供して、前記非剥離黒鉛材料から、前記グラフェン及び剥離グラフェンナノプレートレットを含有する最終液相を分離する、請求項9〜12のいずれか1項に記載の方法。
- デカンティング及び/又は遠心分離後に、前記第1の液相又は第2の液相の重合を完了させ、そのようにしてグラフェン及びグラフェンナノプレートレットを含有するポリマーコンポジット、ポリマーナノコンポジット又はポリマーマスターバッチを形成する、請求項13又は15に記載の方法。
- 溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する前記組成物を直接、前記超音波処理(工程(b))中に、部分的又は全体的に重合させる、請求項9〜12のいずれか1項に記載の方法。
- 前記主溶媒がビニル芳香族モノマーであり、場合によりさらなる重合可能コモノマー及び前記重合中に生じる可能性のある対応ポリマーとの混合物状態である、請求項9〜17のいずれか1項に記載の方法。
- 前記主溶媒が、ビニル芳香族モノマー、前記ビニル芳香族ポリマーを溶解できる有機化合物、ペンタン及びその混合物から選択される、請求項9〜17のいずれか1項に記載の方法。
- 前記黒鉛材料の他に、非黒鉛状炭素も存在する、請求項9〜19のいずれか1項に記載の方法。
- 前記黒鉛材料が、合成黒鉛、天然黒鉛、膨張黒鉛、高秩序熱分解性黒鉛、鱗状黒鉛、又は層間黒鉛から選択される、請求項9〜20のいずれか1項に記載の方法。
- 前記非黒鉛炭素が、コークス、カーボンブラック、又は黒鉛化コークスから選択される、請求項20に記載の方法。
- 前記超音波処理中に第2の溶媒を添加する、請求項9〜22のいずれか1項に記載の方法。
- 前記超音波処理を、超音波が適用されるチャンバーの容積に対する超音波発生器によって発生される電気出力として測定される、少なくとも60W/cm3の特定超音波出力まで行なう、請求項9〜23のいずれか1項に記載の方法。
- 前記出発組成物が超音波を受ける有効時間が、20分以下かつ0.1秒以上である、請求項9〜24のいずれか1項に記載の方法。
- 前記超音波処理中に、前記出発組成物を、8絶対バールと50絶対バールの間の圧力に至らしめる、請求項9〜25のいずれか1項に記載の方法。
- 加圧後に、前記出発組成物の圧力を、直接膨張タンク又は間接膨張タンクによって安定に保つ、請求項9〜26のいずれか1項に記載の方法。
- 前記出発組成物が前記超音波処理を受ける工程を20℃以上の温度で行なう、請求項9〜27のいずれか1項に記載の方法。
- 前記超音波を、直接モードに従うか、又は間接モードに従って伝達し、或いは前記超音波を、前記出発組成物が超音波を受けるアクティブ期と、前記組成物が超音波を受けないか又は前記アクティブ期中の伝達出力より少なくとも10倍小さい出力の超音波を受けるパッシブ期とを交互に行なうパルス状で伝達し、前記アクティブ期と、前記アクティブ期及び前記パッシブ期の合計との比が、0.0001と1の間である、請求項9〜28のいずれか1項に記載の方法。
- 溶媒に永続的に分散したグラフェン及びグラフェンナノプレートレットを含有する組成物が、前記超音波処理(工程(b))で直接完全に重合されて、前記グラフェン及びグラフェンナノプレートレットを含有する、ポリマーコンポジット、ポリマーナノコンポジット又はポリマーマスターバッチを形成する、請求項17に記載の方法。
- 少なくとも1種の発泡剤が、前記グラフェン及びグラフェンナノプレートレットを含有する、ポリマーコンポジット、ポリマーナノコンポジット又はポリマーマスターバッチに添加されて、発泡性ポリマーコンポジット又は発泡性ポリマーナノコンポジットを形成する、請求項16又は30に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IT102016000008311 | 2016-01-27 | ||
| ITUB2016A000159A ITUB20160159A1 (it) | 2016-01-27 | 2016-01-27 | Composizione contenente grafene e nano piastrine grafeniche e loro procedimento di preparazione. |
| PCT/IB2017/050420 WO2017130136A1 (en) | 2016-01-27 | 2017-01-26 | Composition containing graphene and graphene nanoplatelets and preparation process thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2019506494A JP2019506494A (ja) | 2019-03-07 |
| JP6874012B2 true JP6874012B2 (ja) | 2021-05-19 |
Family
ID=55860942
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018538894A Active JP6874012B2 (ja) | 2016-01-27 | 2017-01-26 | グラフェン及びグラフェンナノプレートレットを含有する組成物並びにその調製方法 |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US11912840B2 (ja) |
| EP (1) | EP3408303A1 (ja) |
| JP (1) | JP6874012B2 (ja) |
| CN (1) | CN108699172B (ja) |
| BR (1) | BR112018015190B1 (ja) |
| CA (1) | CA3011499C (ja) |
| IT (1) | ITUB20160159A1 (ja) |
| MX (1) | MX2018009119A (ja) |
| MY (1) | MY191677A (ja) |
| RU (1) | RU2744709C2 (ja) |
| WO (1) | WO2017130136A1 (ja) |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12406992B2 (en) * | 2018-10-08 | 2025-09-02 | Sicona Battery Technologies Pty Ltd | Dispersible edge functionalised graphene platelets |
| CA3124118A1 (en) * | 2018-12-21 | 2020-06-25 | Bio Industrial Technology, Incorporated | In situ production and functionalization of carbon materials via gas-liquid mass transfer and uses thereof |
| CN111676001A (zh) * | 2020-05-29 | 2020-09-18 | 成都西油华巍科技有限公司 | 一种钻井液用抗高温抗饱和盐润滑剂及其制备方法 |
| JP7377480B2 (ja) * | 2020-06-11 | 2023-11-10 | 博 小林 | 黒鉛粒子の集まりからグラフェンの集まりを製造し、該グラフェンをαオレフィン誘導体の微細結晶の集まりで覆い、該αオレフィン誘導体の微細結晶の集まりで覆われたグラフェンの集まりから1枚1枚のグラフェンを取り出す方法 |
| KR102727835B1 (ko) * | 2022-10-12 | 2024-11-08 | 주식회사 케이비엘러먼트 | 질소가 도핑된 그래핀 제조 방법 |
| KR102727833B1 (ko) * | 2022-10-12 | 2024-11-08 | 주식회사 케이비엘러먼트 | 안정적인 플라즈마 제트 생성이 가능한 그래핀 제조 방법 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ATE196158T1 (de) * | 1997-05-14 | 2000-09-15 | Basf Ag | Graphitpartikel enthaltende expandierbare styrolpolymerisate |
| US7745528B2 (en) * | 2006-10-06 | 2010-06-29 | The Trustees Of Princeton University | Functional graphene-rubber nanocomposites |
| ITMI20071003A1 (it) * | 2007-05-18 | 2008-11-19 | Polimeri Europa Spa | Compositi a base di polimeri vinilaromatici aventi migliorate proprieta' di isolamento termico e procedimento per la loro preparazione |
| US20090022649A1 (en) * | 2007-07-19 | 2009-01-22 | Aruna Zhamu | Method for producing ultra-thin nano-scaled graphene platelets |
| IT1396193B1 (it) * | 2009-10-07 | 2012-11-16 | Polimeri Europa Spa | Composizioni polimeriche nanocomposite termoplastiche espansibili con migliorata capacita' di isolamento termico. |
| IT1396918B1 (it) * | 2009-11-03 | 2012-12-20 | Polimeri Europa Spa | Procedimento per la preparazione di nanopiastrine grafeniche ad elevata disperdibilita' in matrici polimeriche a bassa polarita' e relative composizioni polimeriche |
| KR20130038951A (ko) * | 2010-03-25 | 2013-04-18 | 세키스이가가쿠 고교가부시키가이샤 | 수지 조성물, 합성 수지 시트, 합성 수지 성형품 및 합성 수지 적층체 |
| US9884934B2 (en) * | 2011-02-04 | 2018-02-06 | Sekisui Chemical Co., Ltd. | Method for producing exfoliated graphite-polymer composite material |
| ES2684343T3 (es) * | 2012-08-27 | 2018-10-02 | Sekisui Chemical Co., Ltd. | Material compuesto de resina y grafito en copos y método para producir el mismo |
| ITMI20130834A1 (it) * | 2013-05-22 | 2014-11-23 | Versalis Spa | Procedimento di polimerizzazione cationica per la sintesi di polimeri nano-strutturati contenenti grafene |
| US9851735B2 (en) | 2014-01-02 | 2017-12-26 | Lutron Electronics Co., Inc. | Wireless load control system |
| US9789366B1 (en) * | 2016-09-28 | 2017-10-17 | Callaway Golf Company | Graphene core for a golf ball |
-
2016
- 2016-01-27 IT ITUB2016A000159A patent/ITUB20160159A1/it unknown
-
2017
- 2017-01-26 US US16/072,383 patent/US11912840B2/en active Active
- 2017-01-26 JP JP2018538894A patent/JP6874012B2/ja active Active
- 2017-01-26 CN CN201780007978.2A patent/CN108699172B/zh active Active
- 2017-01-26 MY MYPI2018702561A patent/MY191677A/en unknown
- 2017-01-26 WO PCT/IB2017/050420 patent/WO2017130136A1/en not_active Ceased
- 2017-01-26 MX MX2018009119A patent/MX2018009119A/es unknown
- 2017-01-26 CA CA3011499A patent/CA3011499C/en active Active
- 2017-01-26 BR BR112018015190-4A patent/BR112018015190B1/pt active IP Right Grant
- 2017-01-26 RU RU2018129141A patent/RU2744709C2/ru active
- 2017-01-26 EP EP17711316.4A patent/EP3408303A1/en active Pending
-
2023
- 2023-11-28 US US18/520,592 patent/US12534586B2/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| MX2018009119A (es) | 2019-07-04 |
| WO2017130136A1 (en) | 2017-08-03 |
| US20190031852A1 (en) | 2019-01-31 |
| RU2018129141A3 (ja) | 2020-02-27 |
| CN108699172B (zh) | 2021-07-09 |
| US12534586B2 (en) | 2026-01-27 |
| RU2018129141A (ru) | 2020-02-27 |
| BR112018015190A2 (pt) | 2018-12-26 |
| EP3408303A1 (en) | 2018-12-05 |
| JP2019506494A (ja) | 2019-03-07 |
| CA3011499C (en) | 2023-08-29 |
| MY191677A (en) | 2022-07-07 |
| US11912840B2 (en) | 2024-02-27 |
| US20240101778A1 (en) | 2024-03-28 |
| CA3011499A1 (en) | 2017-08-03 |
| ITUB20160159A1 (it) | 2017-07-27 |
| CN108699172A (zh) | 2018-10-23 |
| BR112018015190B1 (pt) | 2022-11-29 |
| RU2744709C2 (ru) | 2021-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6874012B2 (ja) | グラフェン及びグラフェンナノプレートレットを含有する組成物並びにその調製方法 | |
| US9315388B2 (en) | Production of graphene materials in a cavitating fluid | |
| Dong et al. | Mechanical reinforcement from two-dimensional nanofillers: model, bulk and hybrid polymer nanocomposites | |
| RU2537311C2 (ru) | Вспениваемые термопластичные нанокомпозиционные полимерные композиции с улучшенной теплоизоляционной способностью | |
| Gao et al. | Production of graphene quantum dots by ultrasound-assisted exfoliation in supercritical CO2/H2O medium | |
| TWI633054B (zh) | 用於製造含石墨烯之奈米結構聚合物之陽離子聚合方法 | |
| CN107250236A (zh) | 树脂中的石墨剥离 | |
| Naznin et al. | Enhancement of thermal and mechanical properties of PMMA composites by incorporating mesoporous micro-silica and GO | |
| Wang et al. | The fabrication and properties of a graphite nanosheet/polystyrene composite based on graphite nanosheets treated with supercritical water | |
| Namvari et al. | Crosslinking hydroxylated reduced graphene oxide with RAFT-CTA: a nano-initiator for preparation of well-defined amino acid-based polymer nanohybrids | |
| WO2013077107A1 (ja) | 炭素質材料-ポリマー複合材料の製造方法及び炭素質材料-ポリマー複合材料 | |
| CN116789110A (zh) | 一种制备石墨烯/碳纳米管复合材料的方法 | |
| CN107337751B (zh) | 一种碳材料催化的烯烃自由基聚合及聚合方法 | |
| JP2016029002A (ja) | 薄片化黒鉛、電極材料及び薄片化黒鉛−樹脂複合材料 | |
| Yiting | Miniemulsion Polymerization using Graphene Oxide as Surfactant: In Situ Grafting of Polymer | |
| Guo et al. | Efficient production of few-layer graphene by shear exfoliation of graphite in an aqueous liquid | |
| Hordy | Direct growth of carbon nanotubes from stainless steel grids and plasma functionalization for poly (vinyl alcohol) composite production | |
| He et al. | Preparation of Nanocomposites | |
| Malikov et al. | Intercalation of Graphite through Sonochemical Suspension Copolymerization of Styrene and Methyl Methacrylate for PSMMA/Graphene nanocomposite formation | |
| HK1170518B (en) | Expandable thermoplastic nanocomposite polymeric compositions with an improved thermal insulation capacity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20181026 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191101 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20200907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20201202 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20210322 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20210421 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6874012 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |