別途定義しない限り、本開示に関連して使用される科学用語および技術用語は、当業者により一般に理解されている意味を有するものとする。
さらに、文脈により別途必要とされない限り、単数形の用語は複数形を含むものとし、複数形の用語は単数形を含むものとする。
一般に、本明細書で説明される細胞および組織の培養、分子生物学、ならびにタンパク質およびオリゴヌクレオチド化学またはポリヌクレオチド化学、ならびにハイブリダイゼーションに関連して用いられる命名法およびこれらの技術は、当技術分野で公知でありかつ一般に使用されるものである。
組換えDNA、オリゴヌクレオチド合成および組織培養および形質転換(例えば、エレクトロポレーション、リポフェクション)について標準的技術が使用sれる。酵素反応および精製技術は、製造業者の仕様書に従って実施するか、または当技術分野で一般的に達成されているようにもしくは本明細書で説明したように実施する。本開示の実行は、反対のことを具体的に示さない限り、当技術分野の技術内のウイルス学、免疫学、微生物学、分子生物学および組換えDNA技術の従来の方法を用い、これらの多くが例示を目的として下記で説明される。そのような技術は文献で十分に説明されている。例えば、Sambrook,et al.Molecular Cloning:A Laboratory Manual(2nd Edition,1989);Maniatis et al.Molecular Cloning:A Laboratory Manual(1982);DNA Cloning:A Practical Approach,vol.I&II(D.Glover,ed.);Oligonucleotide Synthesis(N.Gait,ed.,1984);Nucleic Acid Hybridization(B.Hames&S.Higgins,eds.,1985);Transcription and Translation(B.Hames&S.Higgins,eds.,1984);Animal Cell Culture(R.Freshney,ed.,1986);Perbal,A Practical Guide to Molecular Cloning(1984)を参照されたい。
本明細書で説明する分析化学、合成有機化学、ならびに医薬化学および製薬化学に関連して用いられる命名法ならびにそれらの実験室手順および技術は、当技術分野で公知でありかつ一般に使用されるものである。化学合成、化学分析、薬剤の調製、製剤および送達ならびに患者の処置について標準的技術が使用される。
用語「核酸」は、本明細書で使用される場合、少なくとも1個の核酸塩基(例えば、DNA中に見出される天然に存在するプリン塩基またはピリミジン塩基(例えば、アデニン「A」、グアニン「G」、チミン「T」およびシトシン「C」)またはRNA中に見出される天然に存在するプリン塩基またはピリミジン塩基(例えば、A、G、ウラシル「U」およびC))を含むDNA、RNA、miRNAまたはその誘導体もしくは模倣物の少なくとも1個の分子または鎖を概して指す。用語「核酸」は、用語「オリゴヌクレオチド」を包含する。
「RNA」は、本明細書では、機能性RNA(例えば、限定されないがmRNA、tRNA、rRNA、触媒RNA、siRNA、miRNA、アンチセンスRNA、lncRNAおよびpiRNA)を指す。
当業者に理解されるように、一本鎖の描写は、相補鎖の配列も規定する。そのため、核酸は、描写した一本鎖の相補鎖も包含する。そのため、用語核酸は、相補DNAを包含する。当業者にも認識されるように、核酸の多くのバリアントを所与の核酸と同じ目的のために使用することができる。そのため、核酸は、実質的に同一の核酸およびその相補体も包含する。当業者にも理解されるように、プライマー等の一本鎖核酸は、ハイブリダイゼーション条件(好ましくはストリンジェントなハイブリダイゼーション条件)下で標的配列にハイブリダイズすることができる。そのため、核酸は、ハイブリダイゼーション条件下で標的配列にハイブリダイズするプライマーも包含する。
用語「オリゴヌクレオチド」は、約3~約200個の核酸塩基の長さの少なくとも1種の分子を指す。
これらの定義は少なくとも1種の一本鎖分子を指すが、いくつかの実施形態では、この少なくとも1種の一本鎖分子に部分的に、実質的にまたは完全に相補的な少なくとも1種の追加の鎖も包含する。従って、いくつかの実施形態では、前記定義は二本鎖分子を指す。
そのため、一実施形態では、核酸は、少なくとも1種の二本鎖分子であって、この分子の鎖を含む特定の配列の1種または複数の相補鎖または「相補体」を含む二本鎖分子を指す。
「遺伝子」は、本明細書で使用される場合、転写調節配列および/もしくは翻訳調節配列、ならびに/またはコード領域、ならびに/または非翻訳配列(例えば、イントロン、5’-非翻訳配列および3’-非翻訳配列)を含むゲノム遺伝子であり得る。遺伝子のコード領域は、アミノ酸配列または機能性RNA(例えば、tRNA、rRNA、触媒RNA、siRNA、miRNA、アンチセンスRNA、lncRNAおよびpiRNA)をコードするヌクレオチド配列であり得る。遺伝子は、コード領域(例えば、エクソンおよびmiRNA)であって、任意選択で、このコード領域に連結された5’-非翻訳配列または3’-非翻訳配列を含むコード領域に対応するmRNAまたはcDNAでもあり得る。遺伝子は、コード領域および/またはこのコード領域に連結された5’-非翻訳配列もしくは3’-非翻訳配列の全てまたは一部を含む、in vitroで生成された増幅核酸分子でもあり得る。
用語「ストリンジェントな条件」または「高ストリンジェンシー条件」は、本明細書で使用される場合、規定のレベルの相補性を有する核酸間に結合対を生じさせるのに適しているが、前記規定のレベルよりも下方の相補性を示す核酸間での結合対の形成に適していない条件に対応する。ストリンジェントな条件は、ハイブリダイゼーション条件および洗浄条件の両方の組み合わせでありかつ配列依存性である。この条件は、当業者に既知の方法に従って変更し得る(Tijssen,1993,Laboratory Techniques in Biochemistry and Molecular Biology-Hybridization with Nucleic Acid Probes,Part I,Chapter 2“Overview of principles of hybridization and the strategy of nucleic acid probe assays”,Elsevier,New York)。一般に、高ストリンジェンシー条件は、熱融点(Tm)と比べて約5℃低いように選択され、好ましくは、完全に塩基対形成された二本鎖のTmに近い温度であるように選択される(Andersen,Nucleic acid Hybridization,Springer,1999,p.54)。ハイブリダイゼーション手順は当技術分野で公知であり、例えば、Ausubel,F.M.,Brent,R.,Kingston,R.E.,Moore,D.D.,Seidman,J.G.,Smith,J.A.,Struhl,K.eds.(1998)Current protocols in molecular biology.V.B.Chanda,series ed.New York:John Wiley&Sonsで説明されている。
高ストリンジェンシー条件は、概して、約50℃~約68℃でハイブリダイズさせることであって、前記温度は、概して、例えば5×SSC/5×Denhardt’s溶液/1.0%SDS中で標的配列とハイブリダイズさせる核酸の最高融解温度TMに対応する、ハイブリダイズさせること、および約60℃~約68℃において0.2×SSC/0.1%SDSで洗浄することを含む。
例えば、本発明に関連して、オリゴヌクレオチドに含まれるプライマー配列は、概して、本明細書において下記で定義される溶解組成物および逆転写組成物をさらに含むコンパートメント(特に液滴)中または複数のコンパートメント(特に複数の液滴)中で約50℃~約68℃において相補核酸(例えば、相補的なRNA配列、cDNA配列またはDNA配列)とハイブリダイズする。
用語「抗体」は、免疫グロブリン分子および免疫グロブリン分子の免疫学的に活性な部分(即ち、抗原に免疫特異的に結合する抗原結合部位を含む分子)を指す。このように、用語抗体は、抗体分子全体だけでなく抗体断片および抗体のバリアント(例えば、ヒト化抗体等の誘導体)も包含する。特定の従来の抗体では、2本の重鎖がジスルフィド結合により互いに連結され、各重鎖はジスルフィド結合により軽鎖に連結されている。2種の軽鎖、即ちラムダ(λ)およびカッパ(κ)が存在する。抗体分子の機能活性を決定する5種の主要な重鎖クラス(またはアイソタイプ):IgM、IgD、IgG、IgAおよびIgEが存在する。各鎖は、別々の配列ドメインを含む。軽鎖は、2種のドメイン、即ち可変ドメイン(VL)および定常ドメイン(CL)を含む。重鎖は、4種のドメイン、即ち可変ドメイン(VH)ならびに3種の定常ドメイン(CH1、CH2およびCH3、まとめてCHと称される)を含む。軽鎖の可変領域(VL)および重鎖の可変領域(VH)は、両方とも抗原に対する結合認識および特異性を決定する。軽鎖の定常領域ドメイン(CL)および重鎖の定常領域ドメイン(CH)は、重要な生物学的特性(例えば、抗体鎖会合、分泌、経胎盤性移動性、補体結合およびFc受容体(FcR)への結合)を付与する。Fv断片は、免疫グロブリンのFab断片のN末端部分であり、1本の軽鎖および1本の重鎖の可変部分からなる。抗体の特異性は、抗体結合部位と抗原決定基との間の構造的相補性に存在する。抗体結合部位は、超可変領域または相補性決定領域(CDR)に主に由来する残基で構成されている。場合により、非超可変領域またはフレームワーク領域(FR)に由来する残基が全体的なドメイン構造に影響を及ぼし、そのため結合部位に影響を及ぼす。相補性決定領域(CDR)は、天然免疫グロブリン結合部位の天然Fv領域の結合親和性および特異性を一緒に規定するアミノ酸配列を指す。免疫グロブリンの軽鎖および重鎖は、それぞれ3種のCDRを有し、それぞれL-CDR1、L-CDR2、L-CDR3およびH-CDR1、H-CDR2、H-CDR3と呼ばれる。従って、抗原結合部位は6種のCDRを含み、重鎖および軽鎖のV領域の各々からのCDRセットを含む。フレームワーク領域(FR)は、CDR間に挿入されているアミノ酸配列を指し、即ち、Kabatら(Sequences of Proteins of Immunological Interest(National Institutes of Health,Bethesda,Md.,1991)により定義されているように、単一種において異なる免疫グロブリン間で比較的保存されている免疫グロブリンの軽鎖可変領域および重鎖可変領域の一部を指す。
「抗体断片」は、インタクトな抗体の一部を含み、好ましくは、インタクトな抗体の抗原結合領域または可変領域を含む。抗体断片の例として、Fab断片、Fab’断片、F(ab’)2断片およびFv断片;二重特異性抗体;線状抗体(米国特許第5,641,870号明細書;Zapata et al.,Protein Eng.8(10):1057-1062[1995]を参照されたい);一本鎖抗体分子;ならびに抗体断片から形成された多特異的抗体が挙げられる。
用語抗体は、一本鎖抗体(例えば、ラクダ抗体またはナノボディまたはVHH)をさらに示す。
用語「T細胞レセプター」は、本明細書では、T細胞(即ちTリンパ球)の表面上に存在する抗原認識分子を指す。この定義は、当技術分野で既知である用語の理解を明白に含み、例えば、インバリアントなCD3鎖との複合体として細胞膜で発現される高度に可変のアルファ鎖もしくはベータ鎖のジスルフィド結合ヘテロ二量体を含むか、またはこのヘテロ二量体からなるレセプター、あるいはT細胞のサブセット上でCD3との複合体として細胞膜で発現される可変のガンマ鎖およびデルタ鎖を含むか、またはこれらの鎖からなるレセプターを含む。
「抗体遺伝子」および「T細胞レセプター遺伝子」は、T細胞およびB細胞の成熟の初期段階で発生するリンパ球中でのみ起こる、V(D)J組換えと呼ばれる固有の遺伝子組換え機構を経る。この機構は、体細胞組換えをさらに含み、B細胞およびT細胞のそれぞれで見出される抗体/免疫グロブリン(Ig)およびT細胞レセプター(TCR)の多様なレパートリーをもたらす。
単一細胞の核酸を捕捉しかつバーコードを付与する方法
「捕捉する」は、本明細書では、コンパートメント(特に液滴)中または複数のコンパートメント(特に複数の液滴)中で単一細胞の核酸をプライマーにハイブリダイズさせることを指す。
「バーコードを付与する」は、本明細書では、前記バーコードが付与された核酸を、別の遺伝子配列(即ち、別の固有のバーコード配列)が付加されている核酸から区別することを可能にする遺伝子配列(本明細書において下記でさらに定義されるいわゆるバーコード配列)を核酸に付加することを指す。
本発明者らは、単一細胞のmRNAの一段階逆転写であって、単一細胞が3nL未満の体積で捕捉され、好ましくはサブナノリットル体積の液滴で捕捉され、バーコードが付与されたcDNAが、処理量が増加し(倍数5~10倍;バーコードが付与されたプライマーと共に100,000個の細胞が1時間で封入される)かつコストが低減されて生成される、一段階逆転写を開発している。本発明者らは、細胞のサイズおよび形状とは無関係に、3nL未満の効率的なcDNA合成用マイクロ反応器に至る実験条件を確立した。
従って、本発明は、単一細胞の核酸を捕捉しかつバーコードを付与する方法であって、
a)複数のコンパートメント内に含まれる複数の細胞を提供することであって、これらのコンパートメントの少なくともいくつかは、単一細胞、逆転写酵素、および少なくとも1つのタイプのオリゴヌクレオチドを含み、この少なくとも1つのタイプのオリゴヌクレオチドは、バーコード配列およびプライマー配列を含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、これらの複数のコンパートメントのコンパートメントは、これらの複数のコンパートメントの他のコンパートメントに含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含む、提供すること、
b)コンパートメント内の細胞の少なくともいくつかを溶解させて、この細胞から核酸を放出させること、
c)放出された核酸の少なくともいくつかをコンパートメントの少なくともいくつかの中の前記オリゴヌクレオチドにハイブリダイズさせること、
d)コンパートメントの少なくともいくつかの中のプライマー配列を使用して、前記オリゴヌクレオチドにハイブリダイズされた放出された核酸を逆転写すること
を含み、
複数のコンパートメントは、3nL未満の体積を有し、
好ましくは、マイクロ流体液滴中の各タイプのオリゴヌクレオチドの濃度は、少なくとも100nMである、方法に言及する。
「コンパートメント」(「容器」とも称される)は、本明細書では、例えばプレート、ウェル、チューブ、チャネル、ナノウェル、ナノドロップ、ナノチューブまたはナノチャネルを指す。
本発明に関連して、好ましい実施形態では、コンパートメントは、液滴であり、より好ましくはマイクロ流体液滴である。
従って、好ましい実施形態では、複数のコンパートメントは、複数の液滴であり、より好ましくは複数のマイクロ流体液滴である。
いくつかの実施形態では、少なくとも1つのタイプのオリゴヌクレオチドを、この少なくとも1つのタイプのオリゴヌクレオチドを最初に粒子(a particle)に結合させ、次いでこの粒子をコンパートメントに組み込んだ後、この粒子から放出させることにより、コンパートメントに導入する。
本発明の特定の実施形態では、少なくとも1つのタイプのオリゴヌクレオチドを粒子に最初に結合させることにより、1つのタイプのオリゴヌクレオチドのみの各コンパートメントへの導入が容易になる。
上記によれば、一実施形態では、工程a)は、前記複数のコンパートメント内に含まれる複数の粒子を提供することをさらに含み、これらのコンパートメントの少なくともいくつかは、1つの粒子(a particle)をさらに含む。
関連する実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、前記粒子に結合されている。
従って、好ましい実施形態では、本発明は、単一細胞の核酸を捕捉しかつバーコードを付与する方法であって、
a)複数のコンパートメント内に含まれる複数の細胞および複数の粒子を提供することであって、これらのコンパートメントの少なくともいくつかは、単一細胞、逆転写酵素、および少なくとも1つのタイプのオリゴヌクレオチドを含む粒子(a particle)を含み、この少なくとも1つのタイプのオリゴヌクレオチドは、任意選択で粒子に結合されており、この少なくとも1つのタイプのオリゴヌクレオチドは、バーコード配列およびプライマー配列を含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、複数のコンパートメントのコンパートメントは、複数のコンパートメントの他のコンパートメントに含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含む、提供すること、
b)コンパートメント内の細胞の少なくともいくつかを溶解させて、この細胞から核酸を放出させること、
c)放出された核酸の少なくともいくつかをコンパートメントの少なくともいくつかの中の前記オリゴヌクレオチドにハイブリダイズさせること、
d)コンパートメントの少なくともいくつかの中のプライマー配列を使用して、前記オリゴヌクレオチドにハイブリダイズされた放出された核酸を逆転写すること
を含み、
複数のコンパートメントは、3nL未満の体積を有し、
好ましくは、粒子(a particle)を含むコンパートメント中の各タイプのオリゴヌクレオチドの濃度は、少なくとも100nMである、方法に言及する。
本発明に関連して、「粒子」は、マイクロ粒子を指す。
一実施形態では、この粒子は、ヒドロゲル粒子、ポリマー粒子または磁性粒子である。
この粒子は、不規則的なまたは規則的な形状を有することができる。例えば、この粒子は、球形、楕円形または立方体であり得る。
一実施形態では、粒子および細胞は、同時にまたは任意の適切な順序で連続してコンパートメントに導入され得、またはコンパートメントに封入され得る。
好ましい一実施形態では、粒子および細胞は、同時にまたは任意の適切な順序で連続して液滴に封入され得る。
本発明に関連して、いくつかの実施形態では、粒子はヒドロゲル粒子である。
「ヒドロゲル粒子」は、例えば「Assay and other reactions involving droplets」という名称の国際特許出願国際公開第2008/109176号パンフレットで説明されている。ヒドロゲルの例として、アガロース、ポリ(エチレングリコール)ジアクリレートまたはアクリルアミドベースのゲル、例えばビスアクリルアミド、ポリアクリルアミド、ストレプトアビジンアクリルアミド、ポリ-N-イソプロピルアクリルアミドもしくはポリN-イソプロピルポリアクリルアミドまたはこれらの混合物が挙げられるが、これらに限定されない。一実施形態では、このヒドロゲル粒子は、アクリルアミド、ビスアクリルアミドおよびストレプトアビジンアクリルアミドを含む。
例えば、モノマーの水溶液をコンパートメント(例えば、液滴)に分散し、次いで重合させて例えばゲルを形成することができる。別の例は、カルシウムイオンの添加によりゲル化され得るアルギン酸等のヒドロゲルである。場合により、例えばLinkらにより2006年2月23日に出願され、2007年1月4日に米国特許出願公開第2007/000342号明細書として公開された「Electronic Control of Fluidic Species」という名称の米国特許出願第11/360,845号明細書、またはAhnらにより2007年1月24日に出願された「Fluidic Droplet Coalescence」という名称の米国特許出願第11/698,298号明細書(それぞれは、その全体が参照により本明細書に組み込まれる)で論じられているように、例えば、水相との同時流により、油相を通した同時流により、または2個の異なる液滴の合体により、ゲル化開始剤(アクリルアミドの場合には過硫酸アルミニウムおよびTEMED、またはアルギン酸塩の場合にはCa2+)をコンパートメント(例えば、液滴)に添加することができる。
別のセットの実施形態では、この粒子は、1種または複数のポリマーを含むことができ、そのため、本明細書では「ポリマー粒子」と称される。例示的なポリマーとして、ポリスチレン(PS)、ポリカプロラクトン(PCL)、ポリイソプレン(PIP)、ポリ(乳酸)、ポリエチレン、ポリプロピレン、ポリアクリロニトリル、ポリイミド、ポリアミド、ならびに/またはこれらのおよび/もしくは他のポリマーの混合物および/もしくはコポリマーが挙げられるが、これらに限定されない。
加えて、いくつかの実施形態では、この粒子は磁性であり得、そのため「磁性粒子」と称され、粒子の磁気操作を可能にすることができる。例えば、この粒子は、鉄または他の磁性材料を含むことができる。この粒子は、この粒子が、付着した他の分子(例えば、タンパク質、核酸または小分子)を有することができるように官能化もされ得る。そのため、本発明のいくつかの実施形態は、例えば核酸、タンパク質、小分子または本明細書で説明したもの等の他の種のライブラリを規定する粒子のセットを対象とする。いくつかの実施形態では、この粒子は蛍光性であり得る。
一実施形態では、この粒子はストレプトアビジンを含む。ストレプトアビジンは、本明細書において上記で定義された粒子の表面に共役され得る。
一実施形態では、ヒドロゲル粒子は、1pL~1000pLのサイズを有し、例えば、1pL~500pL、1pL~400pL、1pL~400pL、1pL~300pL、例えば5pL~300pL、5pL~250pL、5pL~200pL、10pL~250pL、10pL~200pLのサイズを有し、好ましくは10pL~200pLのサイズを有する。
当業者に理解されるようにかつセクション「複数のマイクロ流体液滴を調製する方法」でさらに説明されるように、1個のコンパートメント(例えば、液滴)に封入された細胞の数は、確率分布(例えば、ポアソン分布)に従い、例えば、本発明の複数のマイクロ流体液滴を調製する方法で使用される第1の流体中の細胞の濃度、第2の流体中の粒子の濃度、主チャネルおよび副チャネルの配置、第1の流体、第2の流体および担体流体の注入パラメータに左右される。
従って、本発明の方法の工程a)では、水性組成物に含まれる複数の細胞を複数のコンパートメントに導入し、具体的には複数のマイクロ流体液滴に封入し、1個のコンパートメントに導入される(具体的には1個の液滴に封入される)細胞の数は、「複数のマイクロ流体液滴を調製する方法」で使用されるパラメータに応じてポアソン分布に従う。このパラメータを、例えば1個または0個の細胞が入ったコンパートメントを得るのに適合させることができ、そのため、コンパートメントがいくつかの細胞を含むことが回避される。
本発明者らが示すように、細胞を導入するためにまたは封入するために(好ましくは封入するために)使用されるパラメータは、単一細胞を含むコンパートメントの少なくともいくつかを得るのに適合させることができる。少なくともいくつかのコンパートメントは、本発明に関連して、本明細書において下記でさらに定義されるように、逆転写酵素および少なくとも1つのタイプのオリゴヌクレオチドをさらに含む。
従って、一実施形態では、工程a)の少なくともいくつかのコンパートメント(好ましくは工程a)の液滴)は、1秒当たり1~2000個のコンパートメントの速度で生成され、例えば1秒当たり1~1000個のコンパートメント、1秒当たり1~800個のコンパートメント、1秒当たり1~700個のコンパートメント、1秒当たり1~600個のコンパートメント、1秒当たり1~500個のコンパートメント、1秒当たり1~400個のコンパートメント、1秒当たり1~300個のコンパートメント、1秒当たり1~200個のコンパートメント、1秒当たり1~100個のコンパートメント、1秒当たり1~80個のコンパートメント、1秒当たり1~70個のコンパートメント、1秒当たり1~50個のコンパートメント、例えば1秒当たり10~300個のコンパートメント、1秒当たり50~300個のコンパートメント、1秒当たり100~300個のコンパートメント、1秒当たり150~300個のコンパートメント、1秒当たり150~250個のコンパートメント、1秒当たり175~250個のコンパートメント、典型的には1秒当たり1~1000個のコンパートメント、好ましくは1秒当たり175~250個のコンパートメントの速度で生成される。
さらに、当業者に理解されるように、様々なパラメータに基づいて、1個のコンパートメント当たり0個、1個または2個の粒子を含むようにコンパートメントを生成し得、そのため、コンパートメントが3個以上の粒子を含むことが回避される。特定の一実施形態では、1個のコンパートメント当たり0個または1個の粒子を含むようにコンパートメントが生成され、そのため、コンパートメントが複数の粒子を含むことが回避される。
特定の例では、当業者に理解されるように、様々なパラメータ(例えば、粒子濃度および流速等)に基づいて、1個の液滴当たり0個、1個または2個の粒子を含むように液滴を生成し得、そのため、液滴が3個以上の粒子を含むことが回避される。特定の一実施形態では、1個の液滴当たり0個または1個の粒子を含むように液滴が生成され、そのため、液滴が複数の粒子を含むことが回避される。
従って、一実施形態では、粒子は、約2個以下の粒子/コンパートメントでコンパートメントに導入もしくは封入され、好ましくは、さらなる実施形態では、粒子は、約1個以下の粒子/コンパートメントでコンパートメントに導入もしくは封入され、または粒子は、好ましくは1個の粒子/コンパートメントでコンパートメントに導入もしくは封入され、または粒子は、平均して1個の粒子/コンパートメントでコンパートメントに導入もしくは封入される。
一実施形態では、特にコンパートメントとして液滴に言及する場合、粒子は、本明細書において上記で定義された粒子の数で液滴に好ましくは封入される。上記に従って、液滴に言及する場合、工程a)で生成される液滴のサイズは、確率分布(例えば、ポアソン分布)に従うことも当業者に理解されるであろう。マイクロ流体液滴であって、この液滴の少なくともいくつかは、特許請求の範囲の通りである、マイクロ流体液滴を生成するために工程a)で使用されるパラメータを調節して、特定の体積を有する複数のマイクロ液滴流体を得ることができることがさらに理解されるであろう。
「液滴」は、概して、体積の尺度を指し、本発明に関連して、第2の流体で取り囲まれている第1の流体の分離部分をさらに指す。液滴は、必ずしも球形ではなく、さらに例えば外部環境に応じて他の形状を仮定し得ることに留意されたい。
「複数のコンパートメントは3nL未満の体積を有する」は、複数のコンパートメント中の各コンパートメントが3nL未満の体積を有することを意味する。
本発明に関連して、「コンパートメント」または「複数のコンパートメント」(好ましくは「液滴」または「複数の液滴」)は、3nL未満の体積を有する。一実施形態では、前記複数のマイクロ流体液滴は、2.5nL未満、2nL未満、1.5nL未満、1nL未満、0.5nL未満、例えば0.1nL~3nL、0.5nL~3nL、1nL~3nL、典型的には0.1nL、0.5nL、1nL、1.2nL、1.4nL、1.6nL、1.8nL、2.0nL、2.2nL、2.4nL、2.6nL、2.8nL、3nLの体積を有する。
好ましい一実施形態では、複数のコンパートメントは、1nL以下の体積を有する。
先行技術の開示とは対照的に、本発明者らは、本発明の方法を使用して、単一細胞の全トランスクリプトームを効率的に捕捉しかつバーコードを付与し得ること(実施例10で実証されている)、あるいは本明細書において上記で定義されたオリゴヌクレオチドの1つのタイプの濃度が100nm以上である場合、3nL未満の体積で遺伝子特異的なトランスクリプトームを特異的に捕捉しかつバーコードを付与し得ることを実証した。
「トランスクリプトーム」は、概して、1個の細胞中または細胞の集団中の全てのメッセンジャーRNA分子のセットまたは全てのメッセンジャーRNA分子の一部を指す。従って、「細胞のトランスクリプトーム」または「単一細胞のトランスクリプトーム」は、本明細書では、1個の細胞中の全てのメッセンジャーRNA分子のセットまたは全てのメッセンジャーRNA分子の一部を指す。
そのため、当業者に理解されるように、遺伝子特異的なトランスクリプトームは、1種の遺伝子に由来する全てのメッセンジャーRNA分子のセットを指す。
当業者に知られているように、様々な遺伝子産物(いわゆるアイソフォーム)が1種の遺伝子でコードされ得る。従って、遺伝子特異的なトランスクリプトームは、1種の特定の遺伝子の少なくとも1種の特定のアイソフォームのメッセンジャーRNA分子(例えば、1種の特定の遺伝子の1種、2種、3種または4種の特定のアイソフォームのメッセンジャーRNA分子)または1種の特定の遺伝子の全てのアイソフォームのメッセンジャーRNA分子をさらに指すことができる。
従って、本発明に関連して、一実施形態では、核酸は、cDNAまたはRNAであり、好ましくはRNAである。
一実施形態では、RNAは、mRNA、tRNA、rRNA、触媒RNA、siRNA、miRNA、アンチセンスRNA、lncRNAおよびpIRNAからなる群から選択され、好ましくはmRNAである。
さらなる実施形態では、mRNAは、ポリA配列(ポリAテールとも呼ばれる)を含む。
さらに、本発明に関連して、特定の一実施形態では、「放出された核酸の少なくともいくつか」は、少なくとも1個の核酸を指し、好ましくは、少なくとも2個、少なくとも3個、少なくとも4個、少なくとも5個の核酸またはより多くを指す。特定の1つでは、工程c)およびd)の核酸は、1個、2個、3個、4個、5個、6個、7個、8個、9個または10個の核酸を指す。
本発明の方法を使用して、単一細胞の1個の特定の核酸から全ての核酸までを転写することができる。
従って、本発明に関連して、さらなる実施形態では、工程c)およびd)の「放出された核酸の少なくともいくつか」は、1~100000個の核酸を指し、例えば1~80000個、1~60000個、1~40000個、例えば1個、1000個、2000個、4000個、6000個、8000個、10000個、12000個、14000個、16000個、20000個、25000個、30000個、45000個、50000個の核酸を指す。従って、一実施形態では、核酸は、本明細書において全ての遺伝子のRNAを指す。
上述したように、本発明者らにより実証されたように、本発明の方法は、遺伝子特異的なトランスクリプトームを捕捉および転写することも指す。
従って、一実施形態では、核酸は、本明細書においてa)特定の遺伝子のRNAを指す。当業者に理解されるように、前記遺伝子は、目的の任意の遺伝子であり得る。
一実施形態では、前記特定の遺伝子は、抗体重鎖可変遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子、抗体軽鎖定常遺伝子、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子、およびデルタT細胞レセプター遺伝子、およびガンマT細胞レセプター遺伝子からなる群から選択される。
一実施形態では、工程c)およびd)で言及される核酸は、遺伝子特異的核酸であり、この遺伝子は、抗体重鎖可変遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子、抗体軽鎖定常遺伝子、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子、およびデルタT細胞レセプター遺伝子、およびガンマT細胞レセプター遺伝子からなるリストから選択され得る。
特定の一実施形態では、工程c)およびd)で言及される核酸は、少なくとも2種の遺伝子に特異的な核酸であり、この少なくとも2種の遺伝子は、抗体重鎖可変遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子、抗体軽鎖定常遺伝子、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子、およびデルタT細胞レセプター遺伝子、およびガンマT細胞レセプター遺伝子からなるリストから選択される。
一実施形態では、工程c)およびd)で言及される核酸は、遺伝子特異的核酸であり、この遺伝子は、抗体重鎖可変異遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子、抗体軽鎖定常遺伝子、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子、およびデルタT細胞レセプター遺伝子、およびガンマT細胞レセプター遺伝子からなるリストから選択され得、好ましくは、抗体重鎖可変遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子および抗体軽鎖定常遺伝子からなるリストから選択され得る。
さらに特定の実施形態では、工程c)およびd)で言及される核酸は、少なくとも3種の遺伝子に特異的な核酸であり、この少なくとも3種の遺伝子は、抗体重鎖可変遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子、抗体軽鎖定常遺伝子、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子およびデルタT細胞レセプター遺伝子からなるリストから選択され、好ましくは、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子、およびデルタT細胞レセプター遺伝子、およびガンマT細胞レセプター遺伝子からなるリストから選択される。
細胞のトランスクリプトームが捕捉される場合、本明細書において下記でさらに定義されるように、ポリTプライマー配列等のプライマー配列(前記プライマー配列は全てのmRNAに特異的である)を有するオリゴヌクレオチドが使用されるが、遺伝子特異的トランスクリプトームが捕捉されてバーコードが付与される場合、本明細書において下記でさらに定義されるように、遺伝子特異的プライマー配列を含むオリゴヌクレオチドが使用されることが当業者に理解されるであろう。
「少なくとも1つのタイプのオリゴヌクレオチド」は、本発明に関連して使用される場合、バーコード配列およびプライマー配列を含むオリゴヌクレオチド(本明細書において上記で定義されている)を指し、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定する。一実施形態では、この少なくとも1つのタイプのオリゴヌクレオチドは、5’から3’へとバーコード配列およびプライマー配列を含む。
文言「少なくとも1つのタイプのオリゴヌクレオチド」中の「少なくとも1つ」は、少なくとも2つ、少なくとも3つ、少なくとも4つ、少なくとも5つ、少なくとも6つ、少なくとも7つ、少なくとも8つ、少なくとも9つ、少なくとも10のタイプまたはより多くのオリゴヌクレオチドを指す。1個のコンパートメント中に存在するオリゴヌクレオチドの異なるタイプの数は、捕捉されてバーコードが付与されるRNAの遺伝子の数に左右される。
上述したように、1つのタイプのオリゴヌクレオチドは、このオリゴヌクレオチドのプライマー配列により、別のタイプのオリゴヌクレオチドと区別される。
全トランスクリプトームは、1つのプライマー配列ポリdTプライマーを使用して転写され得る。オリゴヌクレオチドのタイプの数は、捕捉されてバーコードが付与されるトランスクリプトームの特定の遺伝子の数に少なくとも対応することが理解されるであろう。
従って、さらなる一実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、1~100のタイプのオリゴヌクレオチドを指し、1~80、1~60、1~40、1~30、1~20、1~10のタイプのオリゴヌクレオチドを指し、好ましくは1つ、2つ、3つ、4つ、5つ、6つ、7つ、8つ、9つのタイプのオリゴヌクレオチドを指す。
従って、一実施形態では、1個のコンパートメントに含まれる粒子に結合された様々なタイプのオリゴヌクレオチドは、同一のバーコード配列を含む。
一実施形態では、コンパートメントが液滴である場合、粒子は前記液滴に封入される。
一実施形態では、1個のコンパートメントに封入された粒子に結合された様々なタイプのオリゴヌクレオチドは、同一のバーコード配列を含む。
本発明の方法で定義されたように、好ましくは、コンパートメント中の各タイプのオリゴヌクレオチドの濃度は、少なくとも100nMである。
従って、いくつかの実施形態では、コンパートメント中の各タイプのオリゴヌクレオチドの濃度は、少なくとも100nMであり、好ましくは100nM超である。
いくつかの実施形態では、コンパートメント中の各タイプのオリゴヌクレオチドの濃度は、少なくとも150nMであり、少なくとも200nMであり、少なくとも300nMであり、少なくとも400nMであり、少なくとも500nMであり、少なくとも600nMであり、少なくとも700nMであり、少なくとも800nMであり、少なくとも900nMであり、および少なくとも1μMであり、例えば100nM~5μMであり、100nM~4μMであり、100nM~3μMであり、100nM~2μMであり、100nM~1μMであり、好ましくは100nM~500nMである。
一例では、プライマー配列は、ポリTプライマー配列であり、およびオリゴヌクレオチドの濃度は、100nM~3300nM(3.3μMに対応する)である。
さらなる例では、プライマー配列は、遺伝子特異的プライマー配列であり、およびオリゴヌクレオチドの濃度は、100nMまたは1000nM(1μMに対応する)である。
例えば、本発明者らは、1個の細胞当たりのオリゴヌクレオチド密度/プライマー濃度が、コンパート体積に関連して効率的な捕捉およびバーコード添付を可能にする(1個のコンパートメント当たり1個の粒子という仮定である)ことを実証する。
「バーコード配列」または「バーコード」と単に呼ばれるものは、本明細書では、配列により別の核酸配列から区別され得る固有の核酸配列を指し、そのため、核酸配列を一意的に標識することが可能になり、その結果、別のバーコード配列を担持する別の核酸から区別され得る。
一実施形態では、バーコード配列は、例えば核酸が一緒にプールされた後であっても、単一細胞により放出された核酸を他の細胞から放出された核酸から一意的に識別する。
いくつかの実施形態では、バーコード配列を使用して、例えば異なる細胞または他の供給源から生じる数十、数百またはさらに数千の核酸を区別することができる。
一実施形態では、バーコード配列は、任意の適切な長さであり得る。このバーコード配列は、好ましくは、バーコード配列を他のバーコード配列から区別するのに十分な長さである。一実施形態では、バーコード配列は、5個、6個、7個、8個、9個、10個、11個、12個、13個、14個、15個、16個、17個、18個、19個、20個、21個、22個、23個、24個、25個、30個、35個、40個、45個、50個、55個、60個、65個、70個、72個、74個、76個、78個、80個、85個、90個またはより多くのヌクレオチドの長さを有し、例えば、50~85個、60~80個、70~80個のヌクレオチドの長さを有する。
一実施形態では、バーコード配列は、複数のバーコード配列からなり、これらのバーコード配列は異なる。
関連する実施形態では、異なるバーコード配列を潜在的なバーコード配列の「プール」から得ることができる。バーコード配列が複数のバーコード配列からなる場合、これらのバーコード配列を潜在的なバーコード配列の同一のまたは異なるプールから得ることができる。配列のプールを、例えば無作為に、または例えばバーコード配列の読み取りでエラーを検出し得るようにかつ場合により訂正し得るように特定の距離(例えば、ハミング距離)だけ離れることにより配列がエラー検出および/もしくは訂正を可能にするように、任意の適切な技術を使用して選択することができる。このプールは、任意の数の潜在的バーコード配列を有し得、例えば、少なくとも100個、少なくとも300個、少なくとも500個、少なくとも1,000個、少なくとも3,000個、少なくとも5,000個、少なくとも10,000個、少なくとも30,000個、少なくとも50,000個、少なくとも100,000個、少なくとも300,000個、少なくとも500,000個、または少なくとも1,000,000個のバーコード配列を有し得る。
1つの「プール」または複数のプールから得られた異なるバーコード配列を連結する方法は当業者に既知であり、リガーゼの使用および/またはアニーリングもしくはプライマー伸長法の使用が挙げられるが、これらに限定されない。
リガーゼの非限定的例としてDNAリガーゼが挙げられ、例えば、DNAリガーゼI、DNAリガーゼII、DNAリガーゼIII、DNAリガーゼIV、T4 DNAリガーゼ、T7 DNAリガーゼ、T3 DNAリガーゼ、大腸菌(E.coli)DNAリガーゼ、Taq DNAリガーゼまたは同類のものが挙げられる。多くのそのようなリガーゼは、商業的に購入することができる。
一実施形態では、バーコード配列は、二本鎖または一本鎖の核酸である。
「プライマー配列」は、概して、10~50個のヌクレオチドの長さの短い一本鎖核酸であり、捕捉されて典型的にはPCRで増幅されるかまたは典型的にはRTで逆転写される目的の核酸と完全にまたはほぼ完全に一致するように設計されている。プライマー配列は、このプライマー配列がハイブリダイズする核酸に「特異的」であり、即ち、このプライマー配列は、ストリンジェンシーハイブリダイゼーション条件下で好ましくはハイブリダイズし、より好ましくは高ストリンジェンシーハイブリダイゼーション条件下でハイブリダイズするか、またはこのプライマー配列がハイブリダイズする核酸(標的配列とも称される)に相補的であるかもしくはほぼ相補的である。
典型的には、プライマー配列は、核酸合成の開始点として機能し、核酸ポリメラーゼ等のポリメラーゼ酵素がプライマー配列を伸長させて相補鎖を複製することを可能にする。プライマー配列は、標的核酸に相補的であり得、かつハイブリダイズし得る。いくつかの実施形態では、プライマー配列は、合成プライマー配列である。いくつかの実施形態では、プライマー配列は、天然には存在しないプライマー配列である。プライマー配列は、概して、10~50個のヌクレオチドの長さを有する。例えば、プライマー配列は、10~40個、10~30個、10~20個、25~50個、15~40個、15~30個、20~50個、20~40個または20~30個のヌクレオチドの長さを有することができる。いくつかの実施形態では、プライマー配列は、18~24個のヌクレオチドの長さを有する。
一実施形態では、プライマー配列は、本発明に関連して使用されるオリゴヌクレオチドの3’側に位置する(即ち、このプライマーはバーコード配列と比べて3’位置である)。
一実施形態では、プライマー配列は、ポリT配列、ランダムDNA配列および遺伝子特異的配列からなる群から選択される。
「ポリT配列」は、本明細書で言及される場合、10~50個、10~40個、10~30個、10~20個、25~50個、15~40個、15~30個、20~50個、20~40個または20~30個のチミン「T」を含む配列である。このポリT配列は、mRNA中に存在するポリAテールとハイブリダイズする。
一部の実施形態では、ランダムDNA配列は任意の適切な長さであり得、例えば、6~50個、6~50個、6~40個、6~30個、6~20個、10~50個、10~40個、25~50個、15~40個、15~30個、20~50個、20~40個または20~30個のヌクレオチドであり得る。
特定の一実施形態では、プライマー配列は、遺伝子特異的配列であり、およびこの遺伝子は、抗体重鎖可変遺伝子、抗体重鎖定常遺伝子、抗体軽鎖可変遺伝子、抗体軽鎖定常遺伝子、アルファT細胞レセプター遺伝子、ベータT細胞レセプター遺伝子、デルタT細胞レセプター遺伝子からなる群から選択される。
文言「抗体重鎖可変遺伝子」、「抗体重鎖定常遺伝子」、「抗体軽鎖可変遺伝子」および「抗体軽鎖定常遺伝子」中の用語「抗体」は、本明細書において上記で定義された通りである。
文言「アルファT細胞レセプター遺伝子」、「ベータT細胞レセプター遺伝子」および「デルタT細胞レセプター遺伝子」または「ガンマT細胞レセプター遺伝子」中の用語「T細胞レセプター」は、本明細書において上記で定義された通りである。
単語「遺伝子」は、本明細書において上記で定義された通りである。
特定の一実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、少なくとも2つのタイプのオリゴヌクレオチドであり、この2つのタイプのオリゴヌクレオチドでは、少なくとも1つのタイプのオリゴヌクレオチドが抗体重鎖可変遺伝子に特異的なプライマー配列を含み、他の少なくとも1つのタイプのオリゴヌクレオチドが抗体軽鎖可変遺伝子に特異的なプライマー配列を含む。
別の特定の実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、少なくとも2つのタイプのオリゴヌクレオチドであり、この少なくとも2つのタイプのオリゴヌクレオチドでは、少なくとも1つのタイプのオリゴヌクレオチドがアルファT細胞レセプター遺伝子、またはベータT細胞レセプター遺伝子、またはガンマT細胞レセプターに特異的なプライマー配列を含み、他の少なくとも1つのタイプのオリゴヌクレオチドがデルタT細胞レセプター遺伝子に特異的なプライマー配列を含む。
別の特定の実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、少なくとも3つのタイプのオリゴヌクレオチドであり、この少なくとも3つのタイプのオリゴヌクレオチドでは、少なくとも1つのタイプのオリゴヌクレオチドがアルファT細胞レセプター遺伝子に特異的なプライマー配列を含み、第2のタイプのオリゴヌクレオチドがベータT細胞レセプター遺伝子に特異的なプライマー配列を含み、第3のタイプのオリゴヌクレオチドがデルタT細胞レセプター遺伝子に特異的なプライマー配列を含む。
一実施形態では、オリゴヌクレオチドは、プロモーター配列および/またはスペーサー配列をさらに含む。
プロモーター配列の例として、T7プロモーター、T3プロモーターまたはSP6プロモーターが挙げられるが、これらに限定されない。
オリゴヌクレオチドを粒子に一時的に結合させることにより多量のオリゴヌクレオチドを有する粒子の生成が可能になることが当業者に理解されるであろう。さらに、少なくとも1つのタイプのオリゴヌクレオチドを粒子に最初に結合させることにより、各コンパートメント(特に各液滴)へのオリゴヌクレオチドの導入が容易になり、これらの少なくとも1つのタイプのオリゴヌクレオチドは、同一のバーコード配列を有する。
従って、一実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、粒子に共有結合または非共有結合されている。
「非共有結合されている」は、本明細書では、例えばストレプトアビジン-ビオチン結合を指す。アビジンビオチン結合またはヒスタグおよびニッケル結合等の他の非共有結合が当業者に既知である。
「共有結合されている」は、本明細書では、例えばアミノ結合またはアクリダイトホスホラミダイト結合を指す。
「ストレプトアビジン」は、概して、細菌ストレプトマイセス・アビジニイ(Streptomyces avidinii)から精製された52.8kDaのタンパク質を指す。ストレプトアビジンホモ四量体は、約10-14モル/Lのオーダーの解離定数(Kd)でビオチンに対して非常に高い親和性を有し、ビオチンのストレプトアビジンへの結合は、自然界において既知の最も強力な非共有相互作用の1つである。
好ましい実施形態では、この非共有結合は、ストレプトアビジン-ビオチン結合である。
ストレプトアビジン-ビオチン結合は、当業者に既知である。従って、一実施形態では、本明細書で定義された粒子は、ストレプトアビジンを含む。従って、同一の実施形態では、本明細書で定義された少なくとも1つのタイプのオリゴヌクレオチドは、ビオチンを含む。換言すると、少なくとも1つのタイプのオリゴヌクレオチドは、ビオチンで官能化されている。
少なくとも1つのタイプのオリゴヌクレオチドを粒子に連結するために使用される結合のタイプとは無関係に、少なくとも1つのタイプのオリゴヌクレオチドは、少なくとも1つのリンカー配列をさらに含むことができる。
従って、さらなる実施形態では、「少なくとも1つのタイプのオリゴヌクレオチド」または単に「オリゴヌクレオチド」は、少なくとも1つのリンカー配列をさらに含み、前記リンカー配列は、好ましくは5’末端で含まれている。従って、一実施形態では、少なくとも1つのタイプのオリゴヌクレオチドは、5’から3’へとリンカー配列、バーコード配列およびプライマー配列を含む。
一実施形態では、「リンカー配列」は、それによりオリゴヌクレオチドが粒子に任意選択的に結合される配列である。
「本明細書において任意選択的に結合される」は、粒子に結合された少なくとも1つのタイプのオリゴヌクレオチドがコンパートメントまたは複数のコンパートメントに充填されると、少なくとも1つのタイプのオリゴヌクレオチドが放出され得、そのため、このコンパートメントは、粒子と、前記粒子に結合されていない少なくとも1つのタイプのオリゴヌクレオチドとを含む可能性を指す。
好ましくは、リンカー配列は、開裂可能なリンカー配列であり、例えば、酵素的等の適切な刺激および/または光開裂の適用で開裂され得るリンカー配列である。
「開裂可能なリンカー」として、TEV、トリプシン、トロンビン、カテプシンB、カテスピンD、カテプシンK、カスパーゼルマトリックスメタロプロテイナーゼ(caspase lumatrix metalloproteinase)配列、ホスホジエステル、リン脂質、エステル、-ガラクトース、ジアルキルジアルコキシシラン、シアノエチル基、スルホン、エチレングリコールジスクシネート、2-N-アシルニトロベンゼンスルホンアミド、a-チオフェニルエステル、不飽和ビニルスルフィド、活性化後のスルホンアミド、マロンジアルデヒド(MDA)-インドール誘導体、レブリノイルエステル、ヒドラゾン、アシルヒドラゾン、アルキルチオエステル、ジスルフィド架橋、アゾ化合物、2-ニトロベンジル誘導体、フェナシルエステル、8-キノリニルベンゼンスルホネート、クマリン、ホスホトリエステル、ビス-アリールヒドラゾン、ビマン ビ-チオプロピオン酸誘導体、パラメトキシベンジル誘導体、tert-ブチルカルバメート類似体、ジアルキルジアルコキシシランまたはジアリールジアルコキシシラン、オルトエステル、アセタール、アコニチル、ヒドラゾン、b-チオプロピオネート、ホスホルアミデート、イミン、トリチル、ビニルエーテル、ポリケタール、アルキル2-(ジフェニルホスフィノ)ベンゾエート誘導体、アリルエステル、8-ヒドロキシキノリンエステル、ピコリン酸エステル、隣接ジオールおよびセレン化合物を挙げることができるが、これらに限定されない(例えば、Leriche G,Chisholm L,Wagner Aを参照されたい。
開裂可能なリンカーは、当業者に公知であり、化学生物学でさらに説明されており、例えば、Leriche H.et al.(Bioorg Med Chern.15;20(2):571-82.2012)でさらに説明されている。開裂の条件および試薬として、酵素、求核/塩基性試薬、還元剤、光照射、求電子/酸性試薬、有機金属試薬および金属試薬、ならびに酸化剤が挙げられるが、これらに限定されない。
上述したように、一実施形態では、本発明の方法は、粒子に結合されたオリゴヌクレオチドの少なくともいくつかを、細胞を溶解させる前または溶解させた後に前記粒子から放出させる工程をさらに含む。
細胞を溶解させた後および前記オリゴヌクレオチドにハイブリダイズされた放出された核酸を逆転写する前に、または細胞を溶解させた後および前記オリゴヌクレオチドにハイブリダイズされた放出された核酸を逆転写した後に、オリゴヌクレオチドの少なくともいくつかを放出させる工程がさらに起こり得る。
オリゴヌクレオチドの少なくともいくつかを放出させるために選択された時点に応じて、用語「オリゴヌクレオチドの少なくともいくつか」は、例えば、上記で定義されたように、細胞により放出された核酸またはDNA/RNA二本鎖にハイブリダイズされたオリゴヌクレオチドの少なくともいくつかを指す可能性があることを当業者は理解するであろう。
一実施形態では、オリゴヌクレオチドの少なくともいくつかを、任意の手段(例えば、酵素、求核/塩基性試薬、還元剤、光照射、求電子/酸性試薬、有機金属試薬および金属試薬、ならびに酸化剤)を使用して放出させることができる。
一実施形態では、オリゴヌクレオチドの少なくともいくつかを、酵素開裂および/または光開裂を使用して放出させることができる。例えば、エンドヌクレアーゼを使用して、リンカー配列または任意の他の配列を開裂してオリゴヌクレオチドの少なくともいくつかを粒子から放出させることができる。
さらなる実施形態では、オリゴヌクレオチドを放出させることは、ストレプトアビジンビオチン等の結合を破壊することを指す。ストレプトアビジンビオチン結合を破壊する方法は当業者に既知であり、ストレプトアビジンの酵素消化および/またはストレプトアビジンの変性が挙げられる。
一実施形態では、ストレプトアビジンの酵素消化によりオリゴヌクレオチドを放出させる。
本発明に関連して、「細胞」は、生物学で使用される通常の意味が与えられ、例えば、細胞は、生物(例えば、単細胞生物)の機能的に独立した単位として、または全体として生物の原因に対する特定の機能を実行することに特化されている多細胞生物(例えば、植物および哺乳動物)中の副単位として存在し得る自律的な自己複製単位を指す。しかしながら、「細胞」は、有糸分裂刺激が適用される場合、典型的には依然として細胞分裂が可能である静止細胞をさらに指すことができる。
一実施形態では、細胞は、原核細胞または真核細胞を指し、好ましくは真核細胞を指す。
「真核細胞」を原核細胞から区別する決定的な特徴は、遺伝物質を含む膜結合細胞小器官(特に核)を有しかつ核膜で覆われていることである。
本発明に関連して、「真核細胞」は、哺乳動物細胞、植物細胞および真菌細胞からなる群から選択され、好ましくは哺乳動物細胞からなる群から選択される。
「哺乳動物」は、本明細書ではあらゆる哺乳動物を指し、ヒト、飼育動物および家畜、動物園、スポーツまたはペットの動物、例えばイヌ、ネコ、ウシ、ウマ、ヒツジ、ブタ、ヤギ、ウサギ等が挙げられる。好ましくは、哺乳動物はヒトである。
従って、一実施形態では、細胞は、哺乳動物細胞、操作された哺乳動物細胞もしくは細胞株、または哺乳動物の免疫細胞である。
一実施形態では、哺乳動物細胞は免疫細胞である。
一実施形態では、免疫細胞は、B細胞、T細胞またはハイブリドーマであり得るが、これらに限定されず、好ましくはB細胞であり得る。
一実施形態では、細胞または複数の細胞は、本明細書では、様々なタイプの細胞、または様々な条件に曝露された同一のタイプもしくは起源の細胞を指す。
特定の一実施形態では、細胞は非哺乳動物細胞である。
さらに特定の一実施形態では、非哺乳動物細胞は、酵母細胞、トリ細胞またはサメ細胞である。
本発明の方法の工程d)は、コンパートメントの少なくともいくつかの中のプライマー配列を使用して、前記オリゴヌクレオチドにハイブリダイズされた放出された核酸を逆転写することを指す。逆転写は、コンパートメントの少なくともいくつかに含まれる逆転写酵素(RT)を使用して実施される。
本発明に関連して、「逆転写酵素(RT)」は、逆転写と呼ばれるプロセスにおいてRNAテンプレートから相補DNA(cDNA)を生成するために使用される酵素である。
一実施形態では、逆転写酵素は、Superscriptase I、Superscriptase II、Superscriptase III、Superscriptase IV、Murine Leukemia RT、SmartScribe RTまたはMultiScribe RTからなる群から選択される。
一実施形態では、逆転写酵素は、1~50U/μlの濃度であり、好ましくは5~25U/μLであり、例えば12.5U/μlである。
「逆転写」または「RT反応」は、全細胞RNAまたはポリ(A)RNA、逆転写酵素、プライマー、dNTPおよびRNアーゼ阻害剤を使用することにより一本鎖RNAが一本鎖相補RNA(cDNA)に逆転写されるプロセスである。逆転写の生成物は、一本鎖cDNAであって、この一本鎖cDNAのテンプレートRNAにハイブリダイズされた一本鎖cDNAを含むRNA/DNA二本鎖であることが当業者に理解されるであろう。さらに理解されるように、前記RNA/DNA二本鎖は、逆転写に使用されるプライマー配列を含むオリゴヌクレオチドにさらに連結されている。
従って、本発明の方法の工程d)で核酸を逆転写した後、コンパートメントまたは複数のコンパートメントは、cDNAをさらに含むことが当業者に理解されるであろう。
従って、一実施形態では、コンパートメントの少なくともいくつかは、単一細胞の溶解物からの核酸の逆転写により生成される単一細胞のcDNAをさらに含む。
一実施形態では、前記cDNAは一本鎖相補DNAを指す。
さらなる実施形態では、前記cDNAはRNA/DNA二本鎖に含まれる。
一実施形態では、RNA/DNA二本鎖は、逆転写され、かつ少なくとも1つのタイプのオリゴヌクレオチドのプライマー配列にハイブリダイズされたRNAを指す。
当業者に理解されるように、一実施形態では、RNA/DNA二本鎖は、工程c)で核酸(好ましくはmRNA)がハイブリダイズされ、かつ工程d)で逆転写に使用されたプライマー配列を含むオリゴヌクレオチドに連結されている。
一実施形態では、コンパートメントまたは複数のコンパートメントは、逆転写酵素組成物を含む。
一実施形態では、逆転写酵素組成物は、プロテアーゼ阻害剤、dNTPおよび/またはDTTを含み、好ましくはプロテアーゼ阻害剤、dNTPおよびDTTを含む。
一実施形態では、DTTは、1mM~10mMの濃度であり、好ましくは5mMの濃度である。
一実施形態では、このプロテアーゼ阻害剤は、複数のプロテアーゼ阻害剤を含む。
一実施形態では、このプロテアーゼ阻害剤は、ロイペプチンヘミ硫酸塩、ペプスタチンA、AEBSF、アプロチニン、ベスタチン塩酸塩、E-64およびPMSFからなるリストから選択される。
例えば、プロテアーゼ阻害剤は、ロイペプチンヘミ硫酸塩、ペプスタチンA、AEBSF、アプロチニン、ベスタチン塩酸塩、E-64およびPMSFの1つまたは複数を含むことができる。
本明細書で使用される場合、用語「dNTP」は、デオキシヌクレオシド三リン酸を指し、例えば、デオキシアデノシン-5’-三リン酸(dATP、「A」)、デオキシシチジン-5’-三リン酸(dCTP、「C」)、デオキシグアノシン-5’-三リン酸(dGTP、「G」)、デオキシチミジン-5’-三リン酸(dTTP、「T」)、またはデオキシウリジン-5’-三リン酸(dUTP、「U」)を指す。用語「dNTP」は、A、C、G、TもしくはUの塩基対の模倣が可能な改変塩基および塩基類似体または縮重様式での塩基対形成が可能な改変塩基および塩基類似体(例えば、ヌクレオチド類似体と呼ばれる、AもしくはG、CもしくはT、AもしくはC、GもしくはT、GもしくはC、またはAもしくはTと対形成する塩基)を含むデオキシヌクレオシド三リン酸を指すことも意図されている。本明細書において下記でさらに説明するように、前記ヌクレオチド類似体を例えば精製に使用することができる。
一実施形態では、dNTPは、0.01mM~10mMの濃度であり、好ましくは0.1~1mMの濃度であり、より好ましくは0.5mMの濃度である。
一実施形態では、逆転写酵素組成物はRNアーゼ阻害剤をさらに含む。
本発明に関連して、コンパートメントまたは複数のコンパートメントは水性組成物を含む。
本発明に関連して、「水性組成物」は、本発明の方法で使用される細胞に概して適合されており、本明細書において下記で定義される緩衝溶液を概して含む。
本発明の方法の工程b)は、コンパートメント内の細胞の少なくともいくつかを溶解させることを指す。
本発明に関連して、前記「細胞溶解」は、酵素的手段、物理的手段および/もしくは化学的手段またはこれらの任意の組み合わせにより達成され得、特に酵素的手段、物理的手段および/または化学的手段により達成され得る。他の細胞破壊方法を使用することもできる。
従って、一実施形態では、酵素的細胞溶解、物理的細胞溶解および/または化学的細胞溶解を使用して、工程a)でコンパートメントまたは複数のコンパートメントを溶解させる。
細胞を除去する「酵素的方法」は、当技術分野で十分に確立されている。酵素は一般に市販されており、ほとんどの場合、もともと生物学的起源から単離された。一般に使用される酵素として、リゾチーム、リゾスタフィン、ザイモラーゼ(zymolase)、ムタノリシン、グリカナーゼ、プロテアーゼおよびマンノースが挙げられる。
当業者に既知であるように、「化学的細胞溶解」は、脂質-脂質、脂質-タンパク質およびタンパク質-タンパク質の相互作用を破壊することにより、細胞を取り囲む脂質バリアを破壊する化学物質(例えば、界面活性剤)を使用して達成される。細胞溶解に理想的な界面活性剤は、細胞のタイプおよび供給源に左右される。非イオン性界面活性剤および両イオン性界面活性剤は、より穏やかな界面活性剤である。この目的のために、Triton Xシリーズの非イオン性界面活性剤および3-[(3-コラミドプロピル)ジメチルアミノ]-l-プロパンスルホネート(CHAPS)、両性イオン界面活性剤が一般に使用される。対照的に、イオン性界面活性剤は強い可溶化剤であり、タンパク質を変性させる傾向があり、それによりタンパク質の活性および機能を破壊する。当技術分野では、細胞を破壊するために、タンパク質に結合して変性させるイオン性界面活性剤であるSDSが広く使用される。
「物理的細胞溶解」は、超音波処理、氷ショックまたはエレクトロポレーションの使用を指す。
一例では、コンパートメント内の細胞を氷で溶解させる。
好ましい一実施形態では、工程a)の細胞溶解は、本発明に関連して、コンパートメント(特に液滴)を破壊しない。
上記によれば、一実施形態では、コンパートメントまたは複数のコンパートメントは、溶解組成物を含む。
一実施形態では、この溶解組成物は、リゾチーム、リゾスタフィン、ザイモラーゼ、ムタノリシン、グリカナーゼ、プロテアーゼおよびマンノースからなる群から選択される酵素を含む。
本発明に関連して、好ましい一実施形態では、溶解組成物は、塩化マグネシウム、界面活性剤、緩衝溶液およびRNアーゼ阻害剤を含む。
一実施形態では、塩化マグネシウムを1mM~20mMの濃度で使用する。
一実施形態では、この界面活性剤は、Triton-X-100、NP-40、Nonidet P40およびTween-20およびIGEPAL CA 630からなる群から選択される。
一実施形態では、この界面活性剤は、0.1%~10%の濃度である。
この緩衝溶液の非限定的な例として、トリス-HCl、ヘペス-KOH、ピペス-NaOH、マイレン酸、リン酸、クエン酸、リンゴ酸、ギ酸、乳酸、コハク酸、酢酸、ピバル(トリメチル酢)酸、ピリジン、ピペラジン、ピコリン酸、L-ヒスチジン、MES、ビス-トリス、ビス-トリスプロパン、ADA、ACES、MOPSO、PIPES、イミダゾール、MOPS、BES、TES、HEPES、DIPSO、TAPSO、TEA(トリエタノールアミン)、N-エチルモルホリン、POPSO、EPPS、HEPPS、HEPPSO、トリス、トリシン、グリシルグリシン、ビシン、TAPS、モルホリン、N-メチルジエタノールアミン、AMPD(2-アミノ-2-メチル-1,3-プロパンジオール)、ジエタノールアミン、AMPSO、ホウ酸、CHES、グリシン、CAPSO、エタノールアミン、AMP(2-アミノ-2-メチル-1-プロパノール)、ピペラジン、CAPS、1,3-ジアミノプロパン、CABS、またはピペリジンを挙げることができる(www.reachdevices.com/Protein/BiologicalBuffers.htmlも参照されたい)。RNアーゼ阻害剤の非限定的な例として、RNアーゼOUT、IN、SuperIN Rnアーゼ、ならびに広範囲のRNアーゼ(例えば、A、B、C、1およびT1)を標的とする阻害剤を挙げることができる。
一例では、この溶解組成物は、概して、0.2%Triton、3mM MgCl2、50mMトリス-HCl pH7.4である。
本発明の方法の工程c)は、コンパートメントの少なくともいくつかにおいて、放出された核酸の少なくともいくつかを前記オリゴヌクレオチドにハイブリダイズさせることを指す。
工程c)のハイブリダイゼーションは、本明細書では、オリゴヌクレオチド中に存在するプライマー配列が、放出された核酸の相補的核酸配列にアニールする現象を指し、従って、当業者に既知であるように、使用する温度は、使用するプライマー配列および/またはRT酵素に左右される。
一例では、工程c)およびd)を、典型的には例えば550rpmでのコンパートメントの混合中に例えば55℃で1時間にわたりまたは50℃で2時間にわたりコンパートメントをインキュベートすることにより実施する。
一実施形態では、工程d)の逆転写により生成されたcDNAを回収し、典型的にはその後の増幅および配列決定ライブラリの調製にさらに使用する。
従って、一実施形態では、本発明の方法は、コンパートメントの少なくともいくつかにおいて逆転写により生成された単一細胞のcDNAを回収することを含む。
「回収すること」は、本明細書では、コンパートメントの少なくともいくつかでの逆転写により生成されたcDNAを前記複数のコンパートメントから単離することを指す。
一実施形態では、回収することは、本明細書では、逆転写により生成されたcDNAを含むコンパートメントを収集すること、または前記cDNAを含む前記コンパートメントに含まれる水性組成物を収集し、この水性溶液に含まれるcDNAを分離することを指す。
特定の一実施形態では、回収することは、本明細書では、逆転写により生成されたcDNAを含むマイクロ流体液滴を収集すること、このマイクロ流体液滴を破壊すること、および水性組成物に含まれるcDNAを前記マイクロ流体液滴の油相から分離することを指す。
核酸(特にcDNA)をマイクロ流体液滴から単離する方法は、当業者に既知であり、例えば、マイクロ流体液滴を収集すること、および典型的にはペルフルオロオクタノール(V/Vエマルジョン)を使用してマイクロ流体液滴を破壊することを含む。次いで、前工程で得られたエマルジョンを、水相および油相が分離されるまでインキュベートすることである。一例では、水相を例えば4℃において10000gで10分にわたり概して遠心分離し、cDNAを含む上清を回収する。
一実施形態では、この方法は、組み込まれていないオリゴヌクレオチドを除去する工程をさらに含み、好ましくは、組み込まれていないオリゴヌクレオチドをコンパートメントの水性組成物から除去する工程をさらに含む。好ましい一実施形態では、組み込まれていないオリゴヌクレオチド組成物をコンパートメントの少なくともいくつかから除去する工程を、本明細書において上記で定義された逆転写により生成されたcDNAを回収する工程後に行う。
好ましくは、組み込まれていないオリゴヌクレオチドを除去する工程は、本明細書において下記で定義される増幅工程および/または配列決定工程に先行する。
組み込まれていないオリゴヌクレオチドを除去する工程は、組み込まれていないバーコード配列を除去することを包含することが当業者に理解されるであろう。
一実施形態では、組み込まれていないオリゴヌクレオチドを除去することは、コンパート面の少なくともいくつかの水性組成物と精製基体とを接触させることであって、この精製基体は、組み込まれていないオリゴヌクレオチドを除去する、接触させることを含む。一実施形態では、この精製基体は、ビーズまたは粒子を含み、このビーズおよび粒子は、任意選択でカラムを形成する。さらなる例では、組み込まれていないオリゴヌクレオチドを、例えばアクリルアミドゲルを使用するサイズ選択により除去する。
一実施形態では、組み込まれていないオリゴヌクレオチドを除去する工程は、コンパートメントの少なくともいくつかの水性組成物とエキソヌクレアーゼとを接触させて、コンパートメントの少なくともいくつかの水性組成物内の組み込まれていないオリゴヌクレオチドを分解することを含む。
この工程の特定の実施形態では、エキソヌクレアーゼは、cDNAを含む水性組成物由来の一本鎖核酸配列を分解する。
工程d)で得られたcDNAは、RNA/DNA複合体の形態で概して存在し、そのため、前記エキソヌクレアーゼから保護されることが当業者に理解されるであろう。
一実施形態では、このcDNAは、cDNA配列またはcDNA分子の精製を容易にする1種または複数のヌクレオチド類似体(本明細書において上記で定義されている)を含む。
当業者に理解されるように、特定の実施形態では、精製済みcDNAは、組み込まれていないオリゴヌクレオチドを含まない。従って、特定の実施形態では、精製済みcDNAは、組み込まれていないバーコード配列を含まない。
一実施形態では、このcDNAをRNアーゼAおよび/またはRNアーゼHでさらに処理する。
「RNアーゼA」は、C残基およびU残基で一本鎖RNAを特異的に分解するエンドリボヌクレアーゼである。
一実施形態では、RNアーゼAは、10~1000μg/μLの濃度であり、好ましくは50~200μg/μLの濃度であり、例えば100μg/μLの濃度である。
「RNアーゼH」は、加水分解機構によるRNAの開裂を触媒する非配列特異的エンドヌクレアーゼのファミリーである。RNアーゼHのリボヌクレアーゼ活性によりDNA/RNA二本鎖基質中のRNAの3’-O-P結合が開裂され、3’-ヒドロキシル終端生成物および5’-リン酸終端生成物が生成される。
一実施形態では、RNアーゼHは、10~1000μg/μLの濃度であり、好ましくは50~200μg/μLの濃度であり、例えば100μg/μLの濃度である。
一実施形態では、このcDNAをプロテイナーゼKでさらに処理する。
「プロテイナーゼK」は、広域スペクトルのセリンプロテアーゼであり、疎水性アミノ酸の次にタンパク質を優先的に消化する。
一実施形態では、プロテイナーゼKは、0.1~5mg/mLの濃度であり、好ましくは0.1~1mg/mLの濃度であり、例えば0.8mg/mLの濃度である。
一実施形態では、本方法は、本発明の方法の工程d)で得られたcDNAを増幅させる工程をさらに含む。一実施形態では、前記増幅工程を、組み込まれていないオリゴヌクレオチドを除去した後に実施する。一実施形態では、前記増幅工程を、本明細書において下記で定義される配列決定工程前に実施する。
一実施形態では、この増幅させる工程は、多重反応、分離型ポリメラーゼ連鎖反応(PCR)または線形増幅で実施される。
一実施形態では、この線形増幅は、in vitroでの転写である。
一実施形態では、工程d)で生成されたcDNAを、qPCR(例えば、一重qPCR反応および/または多重qPCR反応)を使用して定量する。
さらなる実施形態では、本方法は、工程d)で得られたcDNAを配列決定する工程をさらに含む。
本発明に関連して、一実施形態では、cDNAを配列決定する工程は、本明細書では、cDNAを配列決定ライブラリに最初に接触させること、およびそれぞれcDNAに対応する配列決定ライブラリからの目的の配列を増幅させることを指す。
一実施形態では、cDNAを配列決定する工程は、配列決定ライブラリに対して次世代配列決定(NGS)プロトコルを実施することを含むことができる。
特定の実施形態では、このNGSプロトコルは、試薬キットの1つのフローセル当たり4pM~20pMの量の配列決定ライブラリをロードすることを含む。
一実施形態では、このNGS配列決定プロトコルは、この量の配列決定ライブラリまたは試薬キットのフローセルに5~60%PhiXを添加する工程をさらに含む。
複数のマイクロ流体液滴
本発明は、複数のコンパートメントであって、これらのコンパートメントの少なくともいくつかは、(i)単一細胞または核酸を含む単一細胞溶解物と、(ii)少なくとも1つのタイプのオリゴヌクレオチドと、(iii)逆転写酵素とを含み、少なくとも1つのタイプのオリゴヌクレオチドは、バーコード配列およびプライマー配列を含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、複数のコンパートメントのコンパートメントは、複数のコンパートメントの他のコンパートメントに含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含み、
複数のコンパートメントは、3nL未満の体積を有し、
好ましくは、コンパートメント中の各タイプのオリゴヌクレオチドの濃度は、少なくとも100nMである、複数のコンパートメントにさらに言及する。
本明細書において上記で説明したように、いくつかの実施形態では、少なくとも1つのオリゴヌクレオチドを粒子(例えば、ヒドロゲルまたはポリマー粒子または磁性粒子)に最初に結合させることにより、少なくとも1つのタイプのオリゴヌクレオチドをコンパートメントに導入する。
従って、一実施形態では、コンパートメントの少なくともいくつかは、(iv)粒子をさらに含み、ii)の少なくとも1つのタイプのオリゴヌクレオチドは、iV)の粒子に好ましくは結合されている。
従って、特定の一実施形態では、本発明は、複数のコンパートメントであって、これらのコンパートメントの少なくともいくつかは、(i)単一細胞または核酸を含む単一細胞溶解物と、(ii)少なくとも1つのタイプのオリゴヌクレオチドと、(iii)逆転写酵素と、(iv)粒子(a particle)とを含み、ii)の少なくとも1つのタイプのオリゴヌクレオチドは、前記粒子(iv)に任意選択で結合されており、少なくとも1つのタイプのオリゴヌクレオチドは、バーコード配列およびプライマー配列を含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、複数のコンパートメントのコンパートメントは、複数のコンパートメントの他のコンパートメントに含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含み、
複数のコンパートメントは、3nL未満の体積を有し、
好ましくは、コンパートメント中の各タイプのオリゴヌクレオチドの濃度は、少なくとも100nMである、複数のコンパートメントにさらに言及する。
一実施形態では、iii)の少なくとも1つのタイプのオリゴヌクレオチドは、iv)の粒子に非共有結合または共有結合され得る。非共有結合および共有結合は、本明細書において上記で定義された通りである。
特定の一実施形態では、複数のコンパートメントは、1nL以下の体積を有する。
一実施形態では、コンパートメントの少なくともいくつかは、v)単一細胞溶解物からの核酸の逆転写によって生成された単一細胞のcDNAをさらに含む。
用語「細胞」、「核酸」、「オリゴヌクレオチド」または「各タイプのオリゴヌクレオチド」、「逆転写酵素」、「バーコード配列」、「プライマー配列」、「コンパートメントの体積」、「各タイプのオリゴヌクレオチドの濃度」、「粒子」、「結合された」は、本明細書において上記セクション「単一細胞の核酸を捕捉しかつバーコードを付与する方法」で定義された通りである。
前のセクション「単一細胞の核酸を捕捉しかつバーコードを付与する方法」で説明されている特徴は、複数のコンパートメントに関する本説明に完全に適用可能である。
複数のマイクロ流体液滴を調製する方法
本発明は、複数のマイクロ流体液滴を調製する方法であって、
- 第1の流体源を提供する工程であって、第1の流体は細胞の懸濁液を含む、工程、
- 第2の流体源を提供する工程であって、第2の流体は、少なくとも1つのタイプのオリゴヌクレオチドを含み、この少なくとも1つのタイプのオリゴヌクレオチドは、1つのバーコード配列とプライマー配列とを含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、複数のマイクロ流体液滴の液滴は、複数のマイクロ流体液滴の他の液滴に含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含む、工程、
- 担体流体を提供する工程であって、この担体流体は、第1の流体および第2の流体と非混和性である、工程、
- この担体流体をチップの主チャネルに注入する工程、
- このチップの少なくとも副チャネルに第2の流体および第1の流体を注入することにより、担体流体中に液滴の流れを生じさせる工程であって、この副チャネルは、主チャネル中で開口しており、それぞれの生成された液滴は、第1の流体および第2の流体の混合物を含む、工程
を含み、
第1の流体中の細胞の濃度、第2の流体中のオリゴヌクレオチドの濃度、主チャネルおよび副チャネルの配置、第1の流体、第2の流体および担体流体の注入パラメータは、各液滴が単一細胞のみを含み、かつ3nL未満の体積を示すように適合されており、好ましくは、各液滴中の各タイプのオリゴヌクレオチドの濃度が少なくとも100nMであるように適合されている、方法にさらに言及する。
一実施形態では、第2の流体は、複数の粒子をさらに含み、各粒子は、この粒子に結合された少なくとも1つのタイプのオリゴヌクレオチドを含み、用語「結合された」は、本明細書において上記で定義された通りである。
同一の実施形態では、第1の流体中の細胞の濃度、第2の流体中の粒子の濃度、各粒子に結合されたオリゴヌクレオチドの数、主チャネルおよび副チャネルの配置、第1の流体、第2の流体および担体流体の注入パラメータは、各液滴が単一細胞および粒子(a particle)のみを含み、かつ3nL未満の体積を示すように適合されており、好ましくは、各液滴中の各タイプのオリゴヌクレオチドの濃度が少なくとも100nMであるように適合されている。
一実施形態では、第2の流体は、逆転写酵素をさらに含み、この逆転写酵素は、本明細書において上記で定義された通りである。
一実施形態では、第2の流体は、溶解組成物をさらに含む。
一実施形態では、第2の流体は、本明細書において上記で定義された逆転写組成物をさらに含む。
代替実施形態では、第2の流体は、逆転写組成物を含まず、かつ逆転写酵素を含まず、同一の実施形態では、複数のマイクロ流体液滴を調製する方法は、第3の流体源を提供することであって、第3の流体は逆転写酵素を含む、提供することをさらに含む。
関連する実施形態では、第3の流体は、逆転写酵素組成物をさらに含む。
従って、さらなる実施形態では、本発明は、複数のマイクロ流体液滴を調製する方法であって、
- 第1の流体源を提供する工程であって、第1の流体は、細胞の懸濁液を含む、工程、
- 第2の流体源を提供する工程であって、第2の流体は、少なくとも1つのタイプのオリゴヌクレオチドを含み、この少なくとも1つのタイプのオリゴヌクレオチドは、1つのバーコード配列とプライマー配列とを含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、複数のマイクロ流体液滴の液滴は、複数のマイクロ流体液滴の他の液滴に含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含む、工程、
- 第3の流体源を提供する工程であって、第3の流体は、逆転写酵素を含む、工程、
- 担体流体を提供する工程であって、この担体流体は、第1の流体、第2の流体および第3の流体と非混和性である、工程、
- この担体流体をチップの主チャネルに注入する工程、
- このチップの少なくとも副チャネルに第1の流体、第2の流体および第3の流体を注入することにより、担体流体中に液滴の流れを生じさせる工程であって、副チャネルは、主チャネル中で開口しており、それぞれの生成された液滴は、第1の流体、第2の流体および第3の流体の混合物を含む、工程
を含み、
第1の流体中の細胞の濃度、第2の流体中のオリゴヌクレオチドの濃度、主チャネルおよび副チャネルの配置、第1の流体、第2の流体、第3の流体および担体流体の注入パラメータは、各液滴が単一細胞および粒子のみを含み、かつ3nL未満の体積を示すように適合されており、好ましくは、各液滴中の各タイプのオリゴヌクレオチドの濃度が少なくとも100nMであるように適合されている、方法に言及する。
同一の実施形態では、第2の流体は、複数の粒子をさらに含み得、各粒子は、この粒子に結合された少なくとも1つのタイプのオリゴヌクレオチドを含み、用語「結合された」は、本明細書において上記で定義された通りである。
同一の実施形態では、第1の流体中の細胞の濃度、第2の流体中の粒子の濃度、各粒子に結合されたオリゴヌクレオチドの数、主チャネルおよび副チャネルの配置、第1の流体、第2の流体および第3の流体および担体流体の注入パラメータは、各液滴が単一細胞および粒子(a particle)のみを含み、かつ3nL未満の体積を示すように適合されており、好ましくは、各液滴中の各タイプのオリゴヌクレオチドの濃度が少なくとも100nMであるように適合されている。
関連する実施形態では、第2の流体または第3の流体は、溶解組成物をさらに含むことができる。
さらに関連する実施形態では、第3の流体は、逆転写酵素組成物をさらに含む。
さらなる実施形態では、本発明は、複数のマイクロ流体液滴を調製する方法であって、
- 第1の流体源を提供する工程であって、第1の流体は、細胞の懸濁液を含む、工程、
- 第2の流体源を提供する工程であって、第2の流体は、少なくとも1つのタイプのオリゴヌクレオチドを含み、この少なくとも1つのタイプのオリゴヌクレオチドは、1つのバーコード配列とプライマー配列とを含み、それぞれの異なるプライマー配列は、異なるオリゴヌクレオチドタイプを規定し、複数のマイクロ流体液滴の液滴は、複数のマイクロ流体液滴の他の液滴に含まれるバーコード配列から識別可能な1つまたは複数のバーコード配列を含む、工程、
- 第3の流体源を提供する工程であって、第3の流体は、逆転写酵素を含む、工程、
- 第4の流体源を提供する工程であって、第4の流体は、溶解組成物を含む、工程、
- 担体流体を提供する工程であって、この担体流体は、第1の流体、第2の流体、第3の流体および第4の流体と非混和性である、工程、
- この担体流体をチップの主チャネルに注入する工程、
- このチップの少なくとも副チャネルに第1の流体、第2の流体、第3の流体および第4の流体を注入することにより、担体流体中に液滴の流れを生じさせる工程であって、副チャネルは、主チャネル中で開口しており、それぞれの生成された液滴は、第1の流体、第2の流体、第3の流体および第4の流体の混合物を含む、工程
を含み、
第1の流体中の細胞の濃度、オリゴヌクレオチドの濃度、主チャネルおよび副チャネルの配置、第1の流体、第2の流体、第3の流体、第4の流体および担体流体の注入パラメータは、各液滴が単一細胞および粒子(a particle)のみを含み、かつ3nL未満の体積を示すように適合されており、好ましくは、各液滴中の各タイプのオリゴヌクレオチドの濃度が少なくとも100nMであるように適合されている、方法に言及する。
関連する実施形態では、第3の流体は、逆転写酵素組成物をさらに含む。
一実施形態では、第2の流体は、複数の粒子をさらに含み、各粒子は、この粒子に結合された少なくとも1つのタイプのオリゴヌクレオチドを含み、用語「結合された」は、本明細書において上記で定義された通りである。
同一の実施形態では、第1の流体中の細胞の濃度、第2の流体中の粒子の濃度、各粒子に結合されたオリゴヌクレオチドの数、主チャネルおよび副チャネルの配置、第1の流体、第2の流体、第3の流体、第4の流体および担体流体の注入パラメータは、各液滴が単一細胞および粒子(a particle)のみを含み、かつ3nL未満の体積を示すように適合されており、好ましくは、各液滴中の各タイプのオリゴヌクレオチドの濃度が少なくとも100nMであるように適合されている。
本明細書において上記で説明されている方法では、一実施形態において、粒子および細胞を同時にまたは任意の適切な順序で連続して液滴に封入することができる。
「溶解組成物」は、本明細書において上記セクション「単一細胞の核酸を捕捉しかつバーコードを付与する方法」で定義されたのと同じ成分を含む。しかしながら、当業者に理解されるように、第2の流体の溶解組成物中に存在する成分の濃度は、セット方法により得られる複数のマイクロ流体液滴中に存在する成分の濃度と比較して増加するであろう。換言すると、複数のマイクロ流体液滴中に存在する溶解組成物の成分の濃度は、第2の流体中に存在する成分の濃度と比較して減少するであろう。
例えば、第2の流体中に存在する溶解組成物の成分の濃度は、最終マイクロ流体液滴中または複数のマイクロ液体流体中に存在する濃度と比べて3~7倍高く、好ましくは3~5倍高く、例えば4倍高い。
複数のマイクロ液体液滴を調製する方法に関連して、「逆転写酵素組成物」は、本明細書において上記で定義されたのと同じ成分を含む。しかしながら、当業者にさらに理解されるように、第2または第3の流体の逆転写酵素組成物に含まれる成分の濃度は、前記方法により得られる複数のマイクロ液滴流体中に存在する逆転写酵素組成物に含まれる成分の濃度と比較して増加するであろう。
例えば、第2または第3の流体中に存在する逆転写酵素組成物の成分の濃度は、複数のマイクロ流体液滴(本明細書において上記で定義された通りである)中に存在する濃度と比べて2~7倍高く、好ましくは2~5倍高く、例えば2.6倍高い場合がある。
一実施形態では、第1および第2の流体源は、接合の形態で構築されている。
さらなる実施形態では、第1、第2および第3の流体源は、接合の形態で構築されている。
さらなる実施形態では、第1、第2、第3および第4の流体源は、接合の形態で構築されている。
この接合は、例えば、T接合、Y接合、チャネル内チャネル接合(例えば、同軸配置である、または内部チャネルと、この内部チャネルの少なくとも一部を取り囲む外部チャネルとを含む)、交差(または「X」)接合、流れ収束接合、または液滴の生成に適した他の接合であり得る。例えば、Linkらにより2004年4月9日に出願され、2004年10月28日に国際公開第2004/091763号パンフレットとして公開された「Formation and Control of Fluidic Species」という名称の国際特許出願第PCT/米国特許出願公開第2004/010903号明細書、またはStoneらにより2003年6月30日に出願され、2004年1月8日に国際公開第2004/002627号パンフレットとして公開された「Method and Apparatus for Fluid Dispersion」という名称の国際特許出願第PCT/米国特許出願公開第2003/020542号明細書を参照されたい。
いくつかの実施形態では、この接合は、実質的に単分散の液滴を生成するように構成され得、かつ配置され得る。
一実施形態では、複数のマイクロ液体流体は、約2個以下の粒子/液滴が存在するように調製され、好ましくは複数のマイクロ流体液滴は、約1個以下の粒子/液滴が存在するように調製され、または1個の粒子/液滴でマイクロ流体液滴を優先的に調製し、または平均して1個の粒子/液滴でマイクロ流体液滴を調製される。
粒子/液滴の量を充填割合と称することもできる。例えば、平均充填割合は、約1個未満の粒子/液滴であり得、約0.9個未満の粒子/液滴であり得、約0.8個未満の粒子/液滴であり得、約0.7個未満の粒子/液滴であり得、約0.6個未満の粒子/液滴であり得、約0.5個未満の粒子/液滴であり得、約0.4個未満の粒子/液滴であり得、約0.3個未満の粒子/液滴であり得、約0.2個未満の粒子/液滴であり得、約0.1個未満の粒子/液滴であり得、約0.05個未満の粒子/液滴であり得、約0.03個未満の粒子/液滴であり得、約0.02個未満の粒子/液滴であり得、または約0.01個未満の粒子/液滴であり得る。場合により、より低い粒子充填割合を選択して、2個以上の粒子を有する液滴が生成される確率を最小化することができる。そのため、例えば、液滴の少なくとも約50%、少なくとも約60%、少なくとも約70%、少なくとも約80%、少なくとも約90%、少なくとも約95%、少なくとも約97%、少なくとも約98%または少なくとも約99%は、粒子を含まないか、または1個のみの粒子を含む場合がある。
同様に、いくつかの実施形態では、複数のマイクロ液体流体は、平均して各液体が1個未満の細胞を有するように調製される。同一の実施形態では、細胞/液滴の量を充填割合と称することができる。
例えば、平均充填割合は、約1個未満の細胞/液滴であり得、約0.9個未満の細胞/液滴であり得、約0.8個未満の細胞/液滴であり得、約0.7個未満の細胞/液滴であり得、約0.6個未満の細胞/液滴であり得、約0.5個未満の細胞/液滴であり得、約0.4個未満の細胞/液滴であり得、約0.3個未満の細胞/液滴であり得、約0.2個未満の細胞/液滴であり得、約0.1個未満の細胞/液滴であり得、約0.05個未満の細胞/液滴であり得、約0.03個未満の細胞/液滴であり得、約0.02個未満の細胞/液滴であり得、または約0.01個未満の細胞/液滴であり得る。場合により、より低い細胞充填割合を選択して、2個以上の細胞を有する液滴が生成される確率を最小化することができる。そのため、例えば、液滴の少なくとも約50%、少なくとも約60%、少なくとも約70%、少なくとも約80%、少なくとも約90%、少なくとも約95%、少なくとも約97%、少なくとも約98%または少なくとも約99%は、細胞を含まないか、または1個のみの細胞を含む場合がある。加えて、液滴内の粒子充填の平均割合および細胞充填の平均割合は同一であっても異なっていてもよいことに留意すべきである。
場合により、1秒当たり比較的多数の液滴が調製され得、例えば、1秒当たり少なくとも約10個、1秒当たり少なくとも約30個、1秒当たり少なくとも約50個、1秒当たり少なくとも約100個、1秒当たり少なくとも約300個、1秒当たり少なくとも約500個、1秒当たり少なくとも約1,000個、1秒当たり少なくとも約3,000個、1秒当たり少なくとも約5,000個、1秒当たり少なくとも約10,000個、1秒当たり少なくとも約30,000個、1秒当たり少なくとも約50,000個、1秒当たり少なくとも約100,000個の液滴等が調製され得る。具体的には、1秒当たり1~2000個の液滴が調製され得、例えば、1秒当たり1~800個の液滴、1秒当たり1~700個の液滴、1秒当たり1~600個の液滴、1秒当たり1~500個の液滴、1秒当たり1~400個の液滴、1秒当たり1~300個の液滴、1秒当たり1~200個の液滴、1秒当たり1~100個の液滴、1秒当たり1~80個の液滴、1秒当たり1~70個の液滴、1秒当たり1~50個の液滴、例えば、1秒当たり10~300個のマイクロ流体液滴、1秒当たり50~300個の液滴、1秒当たり100~300個の液滴、1秒当たり150~300個の液滴、1秒当たり150~250個の液滴、1秒当たり175~250個のマイクロ流体液滴、典型的には1秒当たり1~1000個の液滴、好ましくは1秒当たり175~250個のマイクロ流体液滴が調製され得る。
場合により、既に論じたように、液滴のいくつかまたは全ては、例えば液滴(例えば、好ましくは本明細書において上記で定義された1つのバーコード配列を含む)の少なくともいくつかの中に存在するオリゴヌクレオチドのタイプに基づいて識別可能であり得る。場合により、少なくとも約50%、少なくとも約60%、少なくとも70%、少なくとも約である。
複数の液滴の体積は、本明細書において上記セクション「単一細胞の核酸を捕捉しかつバーコードを付与する方法」で定義された通りである。
一実施形態では、本明細書における上記の方法に従って複数のマイクロ流体液滴を調製した後、この方法は、本明細書において上記で定義された単一細胞の核酸を捕捉しかつバーコードを付与する方法の工程b)~d)、および本明細書において上記で定義された単一細胞の核酸を捕捉しかつバーコードを付与する方法の他のさらなる工程をさらに含むことができる。
本発明は、先の出願米国特許出願第62/264,414号明細書で説明されている項目にさらに関し、この明細書の内容は参照により本明細書に組み込まれる。
従って、本発明は、少なくとも1個の細胞から少なくとも2種の遺伝子配列を同定する方法であって、
(a)少なくとも、第1の細胞と、複数の第1の固有のDNAバーコードに連結された第1の担体と、逆転写酵素とを、第1の細胞が第1の水性組成物内で少なくとも2種のRNA分子を放出するような条件下、第1の固有のDNAバーコードが少なくとも2種のRNA分子の各々にハイブリダイズして少なくとも2種のDNA/RNA二本鎖を形成するような条件下、および第1の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物が第1の水性組成物内で生成されるような条件下で封入して第1の水性組成物を形成することであって、第1の水性組成物の体積は、5ナノリットル未満である、形成すること、
(b)組み込まれてないDNAバーコード配列を除去すること、および
(c)少なくとも2種のcDNA転写産物の各々を配列決定すること
を含み、
それにより、少なくとも1個の細胞から少なくとも2種の遺伝子配列を同定する、方法にさらに言及する。
さらなる項目によれば、本発明は、少なくとも1個の細胞から少なくとも2種の遺伝子配列を同定する方法であって、
(a)少なくとも、第1の細胞と、複数の第1の固有のDNAバーコードに連結された第1の担体とを、第1の細胞が第1の水性組成物内で少なくとも2種のRNA分子を放出するような条件下、および第1の固有のDNAバーコードが少なくとも2種のRNA分子の各々にハイブリダイズして少なくとも2種のDNA/RNA二本鎖を形成するような条件下で封入して第1の水性組成物を形成すること、
(b)第1の水性組成物と逆転写酵素とを、第1の固有のバーコードを含む少なくとも2種のcDNA転写産物が第1の水性組成物内で生成されるような条件下で接触させることであって、第1の水性組成物の体積は、5ナノリットル未満である、接触させること、
(c)組み込まれていないDNAバーコード配列を除去すること、および
(d)少なくとも2種のcDNA転写産物の各々を配列決定すること
を含み、
それにより、少なくとも1個の細胞から少なくとも2種の遺伝子配列を同定する、方法に言及する。
一項目によれば、本発明の方法の方法は、少なくとも2種のcDNA転写産物の各々を配列決定する前に少なくとも2種のcDNA転写産物の各々を増幅させる工程をさらに含む。
一項目によれば、この増幅させる工程は、多重反応、分離型ポリメラーゼ連鎖反応(PCR)または線形増幅で実施される。
一項目によれば、この線形増幅は、in vitroでの転写である。
さらなる項目によれば、本発明の方法は、少なくとも2種のcDNA転写産物の各々を増幅させる前に第1の水性組成物内で第1の担体から少なくとも2種のDNA/RNA二本鎖の各々を放出させる工程をさらに含む。
別の項目によれば、本発明の方法は、少なくとも2種のcDNA転写産物の各々を増幅させた後に第1の水性組成物内で第1の担体から少なくとも2種のDNA/RNA二本鎖の各々を放出させる工程をさらに含む。
一項目によれば、本発明の方法は、第1の水性組成物と逆転写酵素とを接触させる前に第1の水性組成物内で第1の担体から少なくとも2種のDNA/RNA二本鎖の各々を放出させる工程をさらに含む。
さらなる項目によれば、第1の水性組成物は、第1の溶解組成物をさらに含む。
さらなる項目によれば、第1の水性組成物は、第1の逆転写酵素組成物をさらに含む。
さらなる項目によれば、第1の細胞は免疫細胞である。
さらなる項目によれば、この免疫細胞は、B細胞、T細胞またはハイブリドーマであり、好ましくはB細胞である。
別の項目によれば、第1の細胞は哺乳動物細胞である。
別の項目によれば、第1の細胞は非哺乳動物細胞である。
一項目によれば、この非哺乳動物細胞は酵母細胞である。
別の項目によれば、この非哺乳動物細胞はトリ細胞である。
別の項目によれば、この非哺乳動物細胞はサメ細胞である。
さらなる項目によれば、少なくとも2種のRNA分子は、第1の標的配列を含む第1のRNA分子と、第2の標的配列を含む第2のRNA分子とを含む。
特定の項目によれば、第1の細胞は免疫細胞であり、少なくとも2種のRNA分子は、重鎖可変領域(VH)をコードする配列を含む第1のRNA分子と、軽鎖可変領域(VL)をコードする配列を含む第2のRNA分子とを含み、少なくとも2種のDNA/RNA二本鎖は、重鎖可変領域(VH)をコードする配列を含む第1のDNA/RNA二本鎖と、軽鎖可変領域(VL)をコードする配列を含む第2のDNA/RNA二本鎖とを含み、第1の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物は、VHおよび第1の固有のDNAバーコード配列をコードする配列を含む第1のcDNA転写産物と、軽鎖可変領域(VL)および第1の固有のDNAバーコード配列をコードする配列を含む第2のcDNA転写産物とを含む。
特定の項目によれば、第1の細胞は哺乳動物細胞であり、少なくとも2種のRNA分子は、T細胞レセプターα鎖(TCR-α)をコードする配列を含む第1のRNA分子と、T細胞レセプターβ鎖(TCR-β)をコードする配列を含む第2のRNA分子とを含み、少なくとも2種のDNA/RNA二本鎖は、TCR-αをコードする配列を含む第1のDNA/RNA二本鎖と、TCR-βをコードする配列を含む第2のDNA/RNA二本鎖とを含み、第1の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物は、TCR-αおよび第1の固有のバーコード配列をコードする配列を含む第1のcDNA転写産物と、TCR-βおよび第1の固有のDNAバーコード配列をコードする配列を含む第2のcDNA転写産物とを含む。
一項目によれば、少なくとも2種のRNA分子は、ポリA配列を含む第1のRNA分子と、ポリA配列を含む第2のRNA分子とを含む。
さらなる項目によれば、第1の固有のDNAバーコード配列は、5’から3’へと第1の固有のバーコード配列(このバーコード配列は一本鎖または二本鎖である)および逆転写用の第1のプライミング配列(このプライミング配列は一本鎖である)を含むDNA配列を含む。
さらなる項目によれば、第1の固有のDNAバーコード配列は、第1の固有のバーコード配列および第1のプライミング配列に対して5’にリンカー配列をさらに含む。
さらなる項目によれば、この方法は、
(a)少なくとも、第2の細胞と、複数の第2の固有のDNAバーコードに連結された第2の担体と、逆転写酵素とを、第2の細胞が第2の水性組成物内で少なくとも2種のRNA分子を放出するような条件下、第2の固有のDNAバーコードが少なくとも2種のRNA分子の各々にハイブリダイズして少なくとも2種のDNA/RNA二本鎖を形成するような条件下、および第2の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物が第2の水性組成物内で生成されるような条件下で封入して第2の水性組成物を形成することであって、第2の水性組成物の体積は、5ナノリットル未満である、形成すること、
(b)組み込まれていないDNAバーコード配列を除去すること、および
(c)少なくとも2種のcDNA配列の各々を配列決定すること
を含む。
別の項目によれば、この方法は、
(a)少なくとも、第2の細胞と、複数の第2の固有のDNAバーコードに連結された第2の担体とを、第2の細胞が第2の水性組成物内で少なくとも2種のRNA分子を放出するような条件下、および第2の固有のDNAバーコードが少なくとも2種のRNA分子の各々にハイブリダイズして少なくとも2種のDNA/RNA二本鎖を形成するような条件下で封入して第2の水性組成物を形成すること、
(b)第2の水性組成物と逆転写酵素とを、第2の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物が第2の水性組成物内で生成されるような条件下で接触させることであって、第2の水性組成物の体積は、5ナノリットル未満である、接触させること、
(b)組み込まれてないDNAバーコード配列を除去すること、および
(c)少なくとも2種のcDNA転写産物の各々を配列決定すること
を含む。
一項目によれば、本発明の方法は、少なくとも2種のcDNA転写産物の各々を配列決定する前に少なくとも2種のcDNA転写産物の各々を増幅させる工程をさらに含む。
一項目によれば、前記増幅させる工程は、多重反応、分離型ポリメラーゼ連鎖反応(PCR)または線形増幅で実施される。
さらなる項目によれば、この線形増幅は、in vitroでの転写である。
別の項目によれば、本発明の方法は、少なくとも2種のcDNA転写産物の各々を増幅させる前に第2の水性組成物内で第2の担体から少なくとも2種のDNA/RNA二本鎖の各々を放出させる工程をさらに含む。
別の項目によれば、本発明の方法は、少なくとも2種のcDNA転写産物の各々を増幅させた後に第2の水性組成物内で第2の担体から少なくとも2種のDNA/RNA二本鎖の各々を放出させる工程をさらに含む。
さらなる項目によれば、本発明の方法は、第2の水性組成物と逆転写酵素とを接触させる前に第2の水性組成物内で第2の担体から少なくとも2種のDNA/RNA二本鎖の各々を放出させる工程をさらに含む。
一項目によれば、第2の水性組成物は、第2の溶解組成物をさらに含む。
別の項目によれば、第2の水性組成物は、第2の逆転写酵素組成物をさらに含む。
さらなる項目によれば、第2の細胞は免疫細胞である。
さらなる項目によれば、この免疫細胞は、B細胞、T細胞またはハイブリドーマであり、好ましくはB細胞である。
別の項目によれば、第2の細胞は哺乳動物細胞である。
別の項目によれば、第2の細胞は、本明細書において上記の項目の1つに従って定義された非哺乳動物細胞である。
特定の一項目によれば、第2の細胞は免疫細胞であり、少なくとも2種のRNA分子は、重鎖可変領域(VH)をコードする配列を含む第1のRNA分子と、軽鎖可変領域(VL)をコードする配列を含む第2のRNA分子とを含み、少なくとも2種のDNA/RNA二本鎖は、重鎖可変領域(VH)をコードする配列を含む第1のDNA/RNA二本鎖と、軽鎖可変領域(VL)をコードする配列を含む第2のDNA/RNA二本鎖とを含み、第2の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物は、VHおよび第2の固有のDNAバーコード配列をコードする配列を含む第2のcDNA転写産物と、軽鎖可変領域(VL)および第2の固有のDNAバーコード配列をコードする配列を含む第2のcDNA転写産物とを含む。
さらに特定の一項目によれば、第2の細胞は哺乳動物細胞であり、少なくとも2種のRNA分子は、T細胞レセプターα鎖(TCR-α)をコードする配列を含む第1のRNA分子と、T細胞レセプターβ鎖(TCR-β)をコードする配列を含む第2のRNA分子とを含み、少なくとも2種のDNA/RNA二本鎖は、TCR-αをコードする配列を含む第1のDNA/RNA二本鎖と、TCR-βをコードする配列を含む第2のDNA/RNA二本鎖とを含み、第1の固有のDNAバーコードを含む少なくとも2種のcDNA転写産物は、TCR-αおよび第1の固有のバーコード配列をコードする配列を含む第1のcDNA転写産物と、TCR-βおよび第1の固有のDNAバーコード配列をコードする配列を含む第2のcDNA転写産物とを含む。
さらなる一項目によれば、第2の固有のDNAバーコード配列は、5’から3’へと第2の固有のバーコード配列(このバーコード配列は一本鎖または二本鎖である)および逆転写用の第2のプライミング配列(このプライミング配列は一本鎖である)を含むDNA配列を含む。
一項目によれば、第1の溶解組成物および第2の溶解組成物は同一である。
一項目によれば、第1の固有のバーコード配列および第2の固有のDNAバーコード配列は異なる。
一項目によれば、第1の水性組成物の体積は、約10~5000ピコリットル(端点を含む)である。例えば、第1の水性組成物の体積は、0.01、0.05、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0ナノリットルまたはその間の任意の量であり得る。
特定の一項目によれば、第1の水性組成物の体積は、3nL未満の体積を有する。より具体的な項目によれば、第1の水性組成物の体積は、2.5nL未満、2nL未満、1.5nL未満、1nL未満、0.5nL未満、例えば0.1nL~3nL、0.5nL~3nL、1nL~3nL、典型的には0.1nL、0.5nL、1nL、1.2nL、1.4nL、1.6nL、1.8nL、2.0nL、2.2nL、2.4nL、2.6nL、2.8nL、3nLの体積を有する。
一項目によれば、第2の水性組成物の体積は、約10~5000ピコリットル(端点を含む)である。例えば、本開示の第2の水性組成物の体積は、0.01、0.05、0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.5、2.0、2.5、3.0、3.5、4.0、4.5、5.0ナノリットルまたはその間の任意の量であり得る。
特定の一項目によれば、第2の水性組成物の体積は、3nL未満の体積を有する。より具体的な項目によれば、第2の水性組成物の体積は、2.5nL未満、2nL未満、1.5nL未満、1nL未満、0.5nL未満、例えば0.1nL~3nL、0.5nL~3nL、1nL~3nL、典型的には0.1nL、0.5nL、1nL、1.2nL、1.4nL、1.6nL、1.8nL、2.0nL、2.2nL、2.4nL、2.6nL、2.8nL、3nLの体積を有する。
一項目によれば、第1の水性組成物中の第1の固有のDNAバーコード配列の濃度は、少なくとも100nMであり、好ましくは100nM超である。
一項目によれば、第2の水性組成物中の第2の固有のDNAバーコード配列の濃度は、少なくとも100nMであり、好ましくは100nM超である。
一項目によれば、第1の溶解組成物および/または第2の溶解組成物は、塩化マグネシウム、界面活性剤、緩衝溶液およびRNアーゼ阻害剤を含む。
一項目によれば、塩化マグネシウムは、1mM~20mM(端点を含む)の濃度である。
一項目によれば、この界面活性剤は、Triton-X-100、Nonidet P40およびTween-20からなる群から選択される。
一項目によれば、この界面活性剤は、0.1%~10%(端点を含む)の濃度である。
一項目によれば、緩衝溶液は、トリス-HCl、ヘペス-KOHおよびピペス-NaOHからなる群から選択される。
さらなる一項目によれば、このRNアーゼ阻害剤は、RNアーゼOUT、INおよびSuperIN RNアーゼからなる群から選択される。
一項目によれば、第1の逆転写酵素組成物および/または第2の逆転写酵素組成物は、プロテアーゼ阻害剤、dNTPおよびDTTを含む。
一項目によれば、このプロテアーゼ阻害剤は、複数のプロテアーゼ阻害剤を含む。
一項目によれば、このプロテアーゼ阻害剤は、ロイペプチンヘミ硫酸塩、ペプスタチンA、AEBSF、アプロチニン、ベスタチン塩酸塩、E-64およびPMSFの1つまたは複数を含む。
一項目によれば、逆転写酵素は、Superscriptase I、Superscriptase II、Superscriptase III、Superscriptase IV、Moloney Leukemia RT、SmartScribe RTまたはMultiScribe RTである。
一項目によれば、組み込まれていないDNAバーコード配列を除去することは、第1の水性組成物と精製基体とを接触させることであって、この精製基体は、組み込まれていないDNAバーコード配列を除去する、接触させることを含む。
別の項目によれば、組み込まれていないDNAバーコード配列を除去することは、第2の水性組成物と精製基体とを接触させることであって、この精製基体は、組み込まれていないDNAバーコード配列を除去する、接触させることを含む。
一項目によれば、この精製基体は、ビーズまたは粒子を含む。
一項目によれば、このビーズまたは粒子は、カラムを形成する。
一項目によれば、少なくとも2種のcDNA転写産物の各々を配列決定することは、配列決定ライブラリに対して次世代配列決定(NGS)プロトコルを実施することを含む。
一項目によれば、このNGSプロトコルは、試薬キットの1つのフローセル当たり8pM~14pMの量の配列決定ライブラリをロードすることを含む。
一項目によれば、このNGS配列決定プロトコルは、この量の配列決定ライブラリまたは試薬キットのフローセルに10~25%PhiXを添加する工程をさらに含む。
一項目によれば、本発明の方法は、第1のcDNA転写産物の配列および第2のcDNA転写産物の配列を発現ベクターに挿入することをさらに含む。
用語「ベクター」は、これらの項目に関連して使用される場合、シャトルベクターおよび発現ベクターを含む。典型的には、このプラスミド構築物は、細菌中でのプラスミドの複製および選択それぞれのために、複製起点(例えば、複製のColE1起点)および選択可能マーカー(例えば、アンピシリン耐性またはテトラサイクリン耐性)も含む。「発現ベクター」は、細菌中または真核細胞中での本発明の抗体断片等の抗体の発現に必須の制御配列または調節要素を含むベクターを指す。
一項目によれば、本発明の方法は、第1のcDNA転写産物および第2のcDNA転写産物を配列決定ライブラリに接触させる工程、および第1のcDNA転写産物および第2のcDNA転写産物それぞれに対応する少なくとも2種の目的の配列を配列決定ライブラリから増幅させる工程をさらに含む。
一項目によれば、本発明の方法は、第1のcDNA転写産物に対応する目的の第1の配列および第2のcDNA転写産物に対応する目的の配列を発現ベクターに挿入することをさらに含む。
一項目によれば、本発明の方法は、宿主細胞中で発現ベクターを発現させ、それにより組換えポリペプチドを産生することをさらに含む。
一項目によれば、この宿主細胞は哺乳動物細胞である。
一項目によれば、この哺乳動物細胞は、293細胞、293T細胞、HeLa細胞、CHO細胞またはU2OS細胞である。
一項目によれば、第1の水性組成物は液滴である。
一項目によれば、第1の水性組成物は、第1の容器またはこの容器の任意のコンパートメントに含まれている。
一項目によれば、第1の容器またはこの容器の任意のコンパートメントは、プレート、ウェル、チューブ、チャネル、ナノウェル、ナノチューブまたはナノチャネルである。
一項目によれば、第2の水性組成物は液滴である。
一項目によれば、第2の水性組成物は、第2の容器またはこの容器の任意のコンパートメントに含まれている。
一項目によれば、第2の容器またはこの容器の任意のコンパートメントは、プレート、ウェル、チューブ、チャネル、ナノウェル、ナノチューブまたはナノチャネルである。
一項目によれば、第1の容器は、3~5ナノリットル(端点を含む)の最大体積を有する。
一項目によれば、第1の容器および/または第2の容器は、3~5ナノリットル(端点を含む)の最大体積を有する。
一項目によれば、第1の担体は、固体担体または液体担体である。
一項目によれば、この固体担体は、ビーズ、ヒドロゲルビーズ、ポリスチレンビーズ、磁性粒子または表面である。
一項目によれば、この液体担体は、エマルジョン、溶液、緩衝液、水性溶液、非水性溶液である。
一項目によれば、第2の担体は、固体担体または液体担体である。
一項目によれば、この固体担体は、ビーズ、ヒドロゲルビーズ、ポリスチレンビーズ、磁性粒子または表面である。
一項目によれば、この液体担体は、エマルジョン、溶液、緩衝液、水性溶液、非水性溶液である。
本出願全体を通して、用語「および/または」は、接続する事例の1つまたは複数が起こり得ることを包含すると解釈されるべき文法上の接続詞である。例えば、語句「配列はエラー検出および/または訂正を可能にする」における文言「エラー検出および/または訂正」は、この配列がエラー検出を可能にし得、かつこの配列がエラー訂正を可能にし得ること、またはこの配列がエラー検出を可能にし得るかもしくはこの配列がエラー訂正を可能にし得ることを示す。
本出願および添付した特許請求の範囲で使用される場合、単数形「1つの(a)」、「1つの(an)」および「その(the)」は、内容が別途明確に示さない限り、複数の参照(例えば、言及された複数の対象)を含む。
本出願全体を通して、用語「含む」は、全ての具体的に言及された特徴および任意の追加の特定されていない特徴を包含すると解釈されるべきである。本明細書で使用される場合、用語「含む」の使用は、具体的に言及された特徴以外の特徴が存在しない(即ち「からなる」)実施形態も開示する。
説明全体において、1つのセクションで説明された特徴は、本明細書の他のセクションに完全に適用可能である。例えば、セクション「単一細胞の核酸を捕捉しかつバーコードを付与する方法」で示された「プライマー」に関する説明は、「複数のマイクロ流体液滴」と呼ばれるセクションおよび「複数のマイクロ流体液滴を調製する方法」と呼ばれるセクションに完全に適用可能であり、または例えば、セクション「単一細胞の核酸を捕捉しかつバーコードを付与する方法」で示された「少なくとも1つのタイプのオリゴヌクレオチド」に関する説明は、「複数のマイクロ流体液滴」と呼ばれるセクションおよび「複数のマイクロ流体液滴を調製する方法」と呼ばれるセクションに完全に適用可能である。
ここで、下記の実施例を参照して本発明をより詳細に説明する。本明細書で引用されている全ての文献および特許文献は、参照により本明細書に組み込まれる。上記の説明で本発明を詳細に例証しかつ説明しているが、実施例は、実例または代表例であって限定的でないと見なされるべきである。
配列表
配列番号1は、核酸配列
CAACGTGACTGGAGTTCAGACGTGTGCT CTTCCGATCTCTGCTTCAGCGAAGGGTTTC
からなる、アンチセンスプライマーTop_SBS12-ATP5G3のヌクレオチド配列を示す。
配列番号2は、核酸配列
CAACGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTAC AGACACGACAAGAGCGA’
からなる、アンチセンスプライマーTop_SBS12-RPS29のヌクレオチド配列を示す。
配列番号3は、核酸配列
CAACGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGAT CCATGGACCATGTTGCT
からなる、アンチセンスプライマーTop_SBS12-ZNF780Aのヌクレオチド配列を示す。
配列番号4は、核酸配列ACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGGTGCTGCAACAGTAGGAからなる、センスプライマーSBS3-ATP5G3のヌクレオチド配列を示す。
配列番号5は、核酸配列ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTACCTCGTT GCACTGCTGAからなる、センスプライマーSBS3-RPS29のヌクレオチド配列を示す。
配列番号6は、核酸配列ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGAGCGTTACTGCTGCACAからなる、センスプライマーSBS3-ZNF780Aのヌクレオチド配列を示す。
配列番号7は、核酸配列CAACTGCAACTCTGGATCCAGCTCからなる、マウスATP5G3用のアンチセンスプライマーのヌクレオチド配列を示す。
配列番号8は、下記の核酸配列ACACTCTTTCCCTACACGACGCTCTTCCGAT CTTCGCCTGTCACCTAGATCCAからなる、マウスATP5G3用のセンスプライマーのヌクレオチド配列を示す。
配列番号9は、核酸配列GGCCAGTGGATAGACAGATGGGGGからなる、VH用のRTプライマーVH_1のヌクレオチド配列を示す。
配列番号10は、核酸配列GGCCAGTGGATAGACCGATGGGGCからなる、VH用のRTプライマーVH_2のヌクレオチド配列を示す。
配列番号11は、核酸配列GGCCAGTGGATAGACTGATGGGGGからなる、VH用のRTプライマーVH_3のヌクレオチド配列を示す。
配列番号12は、核酸配列GTCACCGCAGCCAGGGACCAAGGGからなる、VH用のRTプライマーVH_4のヌクレオチド配列を示す。
配列番号13は、核酸配列GCGTTTCATTTCCAGCTTGGからなる、VH用のRTプライマーVLk_1のヌクレオチド配列を示す。
配列番号14は、核酸配列GCGTTTGATTTCCAGCTTGGからなる、VH用のRTプライマーVLk_2のヌクレオチド配列を示す。
配列番号15は、核酸配列GCGTTTTATTTCCAATTTTGからなる、VH用のRTプライマーVLk_3のヌクレオチド配列を示す。
配列番号16は、核酸配列GAATTTAATACGACTCACTATAGGGAGAからなる、T7アンチセンスPCR1プライマーのヌクレオチド配列を示す。
配列番号17は、核酸配列TAACTGCAGGTGTCCACTCCからなる、センスプライマーMmLH_AG1のヌクレオチド配列を示す。
配列番号18は、核酸配列CAGCTACAGGTGTCCACTCCからなる、センスプライマーMmLH_AG2のヌクレオチド配列を示す。
配列番号19は、核酸配列TTTATCAAGGTGTGCATTGTからなる、センスプライマーMmLH_AG3のヌクレオチド配列を示す。
配列番号20は、核酸配列GAACTGCAGGCGTCCACTCTからなる、センスプライマーMmLH_AG4のヌクレオチド配列を示す。
配列番号21は、核酸配列TAACTGCAGGTGTTCACTCCからなる、センスプライマーMmLH_AG5のヌクレオチド配列を示す。
配列番号22は、核酸配列TCCCAAGCTGTGTCCTATCCからなる、センスプライマーMmLH_AG6のヌクレオチド配列を示す。
配列番号23は、核酸配列TTCCAAGCTGTGTCCTGTCCからなる、センスプライマーMmLH_AG7のヌクレオチド配列を示す。
配列番号24は、核酸配列CTTTTAAAGGTATTCACTGTからなる、センスプライマーMmLH_AG8のヌクレオチド配列を示す。
配列番号25は、核酸配列TTTTAAAAGGGGTCCAGTGTからなる、センスプライマーMmLH_AG9のヌクレオチド配列を示す。
配列番号26は、核酸配列TTTTAAAAGGTGTCCAGTGTからなる、センスプライマーMmLH_AG10のヌクレオチド配列を示す。
配列番号27は、核酸配列TTTTAAATGGTATCCAGTGTからなる、センスプライマーMmLH_AG11のヌクレオチド配列を示す。
配列番号28は、核酸配列CTGCCCAAAGTGCCCAAGCAからなる、センスプライマーMmLH_AG12のヌクレオチド配列を示す。
配列番号29は、核酸配列CTGCCCAAAGTATCCAAGCAからなる、センスプライマーMmLH_AG13のヌクレオチド配列を示す。
配列番号30は、核酸配列ATGRASTTSKGGYTMARCTKGRTTTからなる、センスプライマーMmLHaのヌクレオチド配列を示す。
配列番号31は、核酸配列ATGRAATGSASCTGGGTYWTYCTCTTからなる、センスプライマーMmLHbのヌクレオチド配列を示す。
配列番号32は、核酸配列ATGGACTCCAGGCTCAATTTAGTTTTCCTからなる、センスプライマーMmLHc1のヌクレオチド配列を示す。
配列番号33は、核酸配列ATGGCTGTCYTRGBGCTGYTCYTCTGからなる、センスプライマーMmLHc2のヌクレオチド配列を示す。
配列番号34は、核酸配列ATGGVTTGGSTGTGGAMCTTGCYATTCCTからなる、センスプライマーMmLHc3のヌクレオチド配列を示す。
配列番号35は、核酸配列ATGAAATGCAGCTGGRTYATSTTCTTからなる、センスプライマーMmLHd1のヌクレオチド配列を示す。
配列番号36は、核酸配列ATGGRCAGRCTTACWTYYTCATTCCTからなる、センスプライマーMmLHd2のヌクレオチド配列を示す。
配列番号37は、核酸配列ATGATGGTGTTAAGTCTTCTGTACCTからなる、センスプライマーMmLHd3のヌクレオチド配列を示す。
配列番号38は、核酸配列ATGGGATGGAGCTRTATCATSYTCTTからなる、センスプライマーMmLHe1のヌクレオチド配列を示す。
配列番号39は、核酸配列ATGAAGWTGTGGBTRAACTGGRTからなる、センスプライマーMmLHe2のヌクレオチド配列を示す。
配列番号40は、核酸配列ATGGRATGGASCKKIRTCTTTMTCTからなる、センスプライマーMmLHe3のヌクレオチド配列を示す。
配列番号41は、核酸配列ATGAACTTYGGGYTSAGMTTGRTTTからなる、センスプライマーMmLHf1のヌクレオチド配列を示す。
配列番号42は、核酸配列ATGTACTTGGGACTGAGCTGTGTATからなる、センスプライマーMmLHf2のヌクレオチド配列を示す。
配列番号43は、核酸配列ATGAGAGTGCTGATTCTTTTGTGからなる、センスプライマーMmLHf3のヌクレオチド配列を示す。
配列番号44は、核酸配列ATGGATTTTGGGCTGATTTTTTTTATTGからなる、センスプライマーMmLHf4のヌクレオチド配列を示す。
配列番号45は、核酸配列ATGRAGWCACAKWCYCAGGTCTTTからなる、VLセンスプライマーMmLKaのヌクレオチド配列を示す。
配列番号46は、核酸配列ATGGAGACAGACACACTCCTGCTATからなる、VLセンスプライマーMmLKbのヌクレオチド配列を示す。
配列番号47は、核酸配列ATGGAGWCAGACACACTSCTGYTATGGGTからなる、VLセンスプライマーMmLKcのヌクレオチド配列を示す。
配列番号48は、核酸配列ATGAGGRCCCCTGCTCAGWTTYTTGGIWTCTTからなる、VLセンスプライマーMmLKd1のヌクレオチド配列を示す。
配列番号49は、核酸配列ATGGGCWTCAAGATGRAGTCACAKWYYCWGGからなる、VLセンスプライマーMmLKd2のヌクレオチド配列を示す。
配列番号50は、核酸配列ATGAGTGTGCYCACTCAGGTCCTGGSGTTからなる、VLセンスプライマーMmLKe1のヌクレオチド配列を示す。
配列番号51は、核酸配列ATGTTGGGAYCGKTTTYAMMCTTTTCAATTGからなる、VLセンスプライマーMmLKe2のヌクレオチド配列を示す。
配列番号52は、核酸配列ATGGAAGCCCCAGCTCAGCTTCTCTTCCからなる、VLセンスプライマーMmLKe3のヌクレオチド配列を示す。
配列番号53は、核酸配列ATGAGIMMKTCIMTTCAITTCYTGGGからなる、VLセンスプライマーMmLKf1のヌクレオチド配列を示す。
配列番号54は、核酸配列ATGAKGTHCYCIGCTCAGYTYCTIRGからなる、VLセンスプライマーMmLKf2のヌクレオチド配列を示す。
配列番号55は、核酸配列ATGGTRTCCWCASCTCAGTTCCTTGからなる、VLセンスプライマーMmLKf3のヌクレオチド配列を示す。
配列番号56は、核酸配列ATGTATATATGTTTGTTGTCTATTTCTからなる、VLセンスプライマーMmLKf4のヌクレオチド配列を示す。
配列番号57は、核酸配列ATGAAGTTGCCTGTTAGGCTGTTGGTGCTからなる、VLセンスプライマーMmLKg1のヌクレオチド配列を示す。
配列番号58は、核酸配列ATGGATTTWCARGTGCAGATTWTCAGCTTからなる、VLセンスプライマーMmLKg2のヌクレオチド配列を示す。
配列番号59は、核酸配列ATGGTYCTYATVTCCTTGCTGTTCTGGからなる、VLセンスプライマーMmLKg3のヌクレオチド配列を示す。
配列番号60は、核酸配列ATGGTYCTYATVTTRCTGCTGCTATGGからなる、VLセンスプライマーMmLKg4のヌクレオチド配列を示す。
配列番号61は、核酸配列attgctcaggttctttctccからなる、VLセンスプライマーLL_AG_1のヌクレオチド配列を示す。
配列番号62は、核酸配列cagtcataatgtccagaggaからなる、VLセンスプライマーLL_AG_2のヌクレオチド配列を示す。
配列番号63は、核酸配列cagtcataatgtccaggggaからなる、VLセンスプライマーLL_AG_3のヌクレオチド配列を示す。
配列番号64は、核酸配列cagtcatactattcagaggaからなる、VLセンスプライマーLL_AG_4のヌクレオチド配列を示す。
配列番号65は、核酸配列cagtcatagtgtctaatggaからなる、VLセンスプライマーLL_AG_5のヌクレオチド配列を示す。
配列番号66は、核酸配列cagtcatattgaccaatggaからなる、VLセンスプライマーLL_AG_6のヌクレオチド配列を示す。
配列番号67は、核酸配列cagtcatattgtccagtggaからなる、VLセンスプライマーLL_AG_7のヌクレオチド配列を示す。
配列番号68は、核酸配列cggtatctggtacctgtggaからなる、VLセンスプライマーLL_AG_8のヌクレオチド配列を示す。
配列番号69は、核酸配列cgagtccagcctcaagcagtからなる、VLセンスプライマーLL_AG_9のヌクレオチド配列を示す。
配列番号70は、核酸配列gaatcacaggcataatatgtからなる、VLセンスプライマーLL_AG_10のヌクレオチド配列を示す。
配列番号71は、核酸配列gaatcccaggcatgatatgtからなる、VLセンスプライマーLL_AG_11のヌクレオチド配列を示す。
配列番号72は、核酸配列gcatgtctggtgcctgtgcaからなる、VLセンスプライマーLL_AG_12のヌクレオチド配列を示す。
配列番号73は、核酸配列ggaccacggtctcagctgtcからなる、VLセンスプライマーLL_AG_13のヌクレオチド配列を示す。
配列番号74は、核酸配列ggacttcagcctccagatgtからなる、VLセンスプライマーLL_AG_14のヌクレオチド配列を示す。
配列番号75は、核酸配列ggatatcaggtgcccagtgtからなる、VLセンスプライマーLL_AG_15のヌクレオチド配列を示す。
配列番号76は、核酸配列ggatccctggagccactgggからなる、VLセンスプライマーLL_AG_16のヌクレオチド配列を示す。
配列番号77は、核酸配列ggatccctggatccactgcaからなる、VLセンスプライマーLL_AG_17のヌクレオチド配列を示す。
配列番号78は、核酸配列ggatctctggagtcagtgggからなる、VLセンスプライマーLL_AG_18のヌクレオチド配列を示す。
配列場合79は、核酸配列ggattcaggaaaccaacggtからなる、VLセンスプライマーLL_AG_19のヌクレオチド配列を示す。
配列番号80は、核酸配列ggattccagcctccagaggtからなる、VLセンスプライマーLL_AG_20のヌクレオチド配列を示す。
配列番号81は、核酸配列ggattcctgcttccagcagtからなる、VLセンスプライマーLL_AG_21のヌクレオチド配列を示す。
配列番号82は、核酸配列ggattcgggaaaccaacggtからなる、VLセンスプライマーLL_AG_22のヌクレオチド配列を示す。
配列番号83は、核酸配列ggatttcagcctccacaggtからなる、VLセンスプライマーLL_AG_23のヌクレオチド配列を示す。
配列番号84は、核酸配列ggctccaaggcatgagctgtからなる、VLセンスプライマーLL_AG_24のヌクレオチド配列を示す。
配列番号85は、核酸配列ggcttcatggtgctcagtgtからなる、VLセンスプライマーLL_AG_25のヌクレオチド配列を示す。
配列番号86は、核酸配列ggcttacagacgcaggatgtからなる、VLセンスプライマーLL_AG_26のヌクレオチド配列を示す。
配列番号87は、核酸配列ggcttacagatgccagatgtからなる、VLセンスプライマーLL_AG_27のヌクレオチド配列を示す。
配列番号88は、核酸配列gggtatctggtacctgtgggからなる、VLセンスプライマーLL_AG_28のヌクレオチド配列を示す。
配列番号89は、核酸配列gggtatctggtgcctgtgcaからなる、VLセンスプライマーLL_AG_29のヌクレオチド配列を示す。
配列番号90は、核酸配列gggttccaggttccactggtからなる、VLセンスプライマーLL_AG_30のヌクレオチド配列を示す。
配列番号91は、核酸配列ggttatatggtgctgatgggからなる、VLセンスプライマーLL_AG_31のヌクレオチド配列を示す。
配列番号92は、核酸配列ggttcccaggtgccagatgtからなる、VLセンスプライマーLL_AG_32のヌクレオチド配列を示す。
配列番号93は、核酸配列ggttgtctggtgttgaaggaからなる、VLセンスプライマーLL_AG_33のヌクレオチド配列を示す。
配列番号94は、核酸配列ggtttccaggtatcagatgtからなる、VLセンスプライマーLL_AG_34のヌクレオチド配列を示す。
配列番号95は、核酸配列ggtttccaggtgcaagatgtからなる、VLセンスプライマーLL_AG_35のヌクレオチド配列を示す。
配列番号96は、核酸配列ggtttgcaggtggtaaatgtからなる、VLセンスプライマーLL_AG_36のヌクレオチド配列を示す。
配列番号97は、核酸配列ggtttttaggtgccagatgtからなる、VLセンスプライマーLL_AG_37のヌクレオチド配列を示す。
配列番号98は、核酸配列gtgtcacagtgtcaaagggaからなる、VLセンスプライマーLL_AG_38のヌクレオチド配列を示す。
配列番号99は、核酸配列gtgtctctgattctagggcaからなる、VLセンスプライマーLL_AG_39のヌクレオチド配列を示す。
配列番号100は、核酸配列gtgtgtctggtgctcatgggからなる、VLセンスプライマーLL_AG_40のヌクレオチド配列を示す。
配列番号101は、核酸配列gttttcaaggtaccagatatからなる、VLセンスプライマーLL_AG_41のヌクレオチド配列を示す。
配列番号102は、核酸配列gttttcaaggtaccagatgtからなる、VLセンスプライマーLL_AG_42のヌクレオチド配列を示す。
配列番号103は、核酸配列
CAAGCAGAAGACGGCATACGAGATインデックスGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT
からなる、イルミナインデックスアンチセンス(Illumina Index Antisense)プライマーのヌクレオチド配列を示し、インデックスは標準イルミナインデックスを表し、この標準イルミナインデックスは6~7塩基長である。
配列番号104は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATGTGAAGCTTCAGGAGTC
からなる、VHセンスプライマーVHs1のヌクレオチド配列を示す。
配列場合105は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGGTGCAGCTGAAGGAGTC
からなる、VHセンスプライマーVHs2のヌクレオチド配列を示す。
配列番号106は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGGTGCAGCTGAAGCAGTC
からなる、VHセンスプライマーVHs3のヌクレオチド配列を示す。
配列番号107は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGGTTACTCTGAAAGAGTC
からなる、VHセンスプライマーVHs4のヌクレオチド配列を示す。
配列番号108は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGAGGTCCAGCTGCAACAATCT
からなる、VHセンスプライマーVHs5のヌクレオチド配列を示す。
配列番号109は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGAGGTCCAGCTGCAGCAGTC
からなる、VHセンスプライマーVHs6のヌクレオチド配列を示す。
配列番号110は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGGTCCAACTGCAGCAGCCT
からなる、VHセンスプライマーVHs7のヌクレオチド配列を示す。
配列番号111は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGAGGTGAAGCTGGTGGAGTC
からなる、VHセンスプライマーVHs8のヌクレオチド配列を示す。
配列番号112は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGAGGTGAAGCTGGTGGAATC
からなる、VHセンスプライマーVHs9のヌクレオチド配列を示す。
配列番号113は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATGTGAACTTGGAAGTGTC
からなる、VHセンスプライマーVHs10のヌクレオチド配列を示す。
配列番号114は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGAGGTCCAGCTGCAACAGTC
からなる、VHセンスプライマーVHs11のヌクレオチド配列を示す。
配列番号115は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGAGGTGCAGCTGGAGGAGTC
からなる、VHセンスプライマーVHs12のヌクレオチド配列を示す。
配列番号116は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATATTGTGATGACGCAGGCT
からなる、VLセンスプライマーVKs1のヌクレオチド配列を示す。
配列番号117は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATATTGTGATAACCCAG
からなる、VLセンスプライマーVKs2のヌクレオチド配列を示す。
配列番号118は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGACATTGTGCTGACCCAATCT
からなる、VLセンスプライマーVKs3のヌクレオチド配列を示す。
配列番号119は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGACATTGTGATGACCCAGTCT
からなる、VLセンスプライマーVKs4のヌクレオチド配列を示す。
配列番号120は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATATTGTGCTAACTCAGTCT
からなる、VLセンスプライマーVKs5のヌクレオチド配列を示す。
配列番号121は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATATCCAGATGACACAG ACT
からなる、VLセンスプライマーVKs6のヌクレオチド配列を示す。
配列番号122は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGACATCCAGCTGACTCAGTCT
からなる、VLセンスプライマーVKs7のヌクレオチド配列を示す。
配列番号123は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCAAATTGTTCTCACCCAGTCT
からなる、VLセンスプライマーVKs8のヌクレオチド配列を示す。
配列番号124は、核酸配列
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTGATGTTTTGATGACCCAAACT
からなる、VLセンスプライマーVKs9のヌクレオチド配列を示す。
配列番号125は、核酸配列GATGGTGGGAAGATGGATACからなる、プライマーRT_VLkのヌクレオチド配列を示す。
配列番号126は、核酸配列AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTからなる、PCR3で使用されるセンスプライマーのヌクレオチド配列を示す。
配列番号127は、核酸配列CAAGCAGAAGACGGCATACGAGAT
からなる、PCR3で使用されるアンチセンスプライマーのヌクレオチド配列を示す。
配列番号128は、CDR3のヌクレオチド配列CARDWSRSWYLAPNGPDLDYWを示す。
配列番号129は、CDR3のヌクレオチド配列CCSYAGGSTLVFを示す。
配列番号130は、CDR3のヌクレオチド配列CARGGKSDDGNFRYFDHWを示す。
配列番号131は、CDR3のヌクレオチド配列CQQRSSWPPGWTFを示す。
配列番号132は、CDR3のヌクレオチド配列CAKSFGFGGVIVIGGYFLHWを示す。
配列番号133は、CDR3のヌクレオチド配列CQQYDNLPLTFを示す。
配列番号134は、CDR3のヌクレオチド配列CARHKTTSGWYSPLDYWを示す。
配列番号135は、CDR3のヌクレオチド配列CQQYSGSVWTFを示す。
配列番号136は、CDR3のヌクレオチド配列CARGVKAAGRTPNWFGPWを示す。
配列番号137は、CDR3のヌクレオチド配列CQSYDSSLSGHVVFを示す。
配列番号138は、CDR3のヌクレオチド配列CAREVSADILTGYYDYWを示す。
配列番号139は、CDR3のヌクレオチド配列CQHYDNLPPTFを示す。
本開示がより効率的に理解され得るように、下記に実施例を記載する。この実施例は説明を目的とするのみであり、いかなる方法でも本発明を限定すると解釈されるべきではないことを理解すべきである。この実施例全体にわたり、別途注記した場合を除いて、分子クローニング反応および他の標準的な組換えDNA技術を、市販の試薬を使用してManiatis et al.,Molecular Cloning-A Laboratory Manual,2nd ed.,Cold Spring Harbor Press(1989)で説明されている方法に従って実行した。
実施例1:バルク対液滴およびRTの効率測定
細胞から始まるRT反応の最適化をバルク反応で実施している。この実験は、液滴条件(特に1個の細胞当たりの反応緩衝液の体積、1個の細胞当たりの試薬の量)を模倣する。バルク条件が液滴条件を模倣することを確認するために、バルク中でおよび液滴中でRT反応を並行して実施した。RT反応の効率を評価するために、この結果を、同一の細胞集団から抽出した精製済みRNAと比較した。
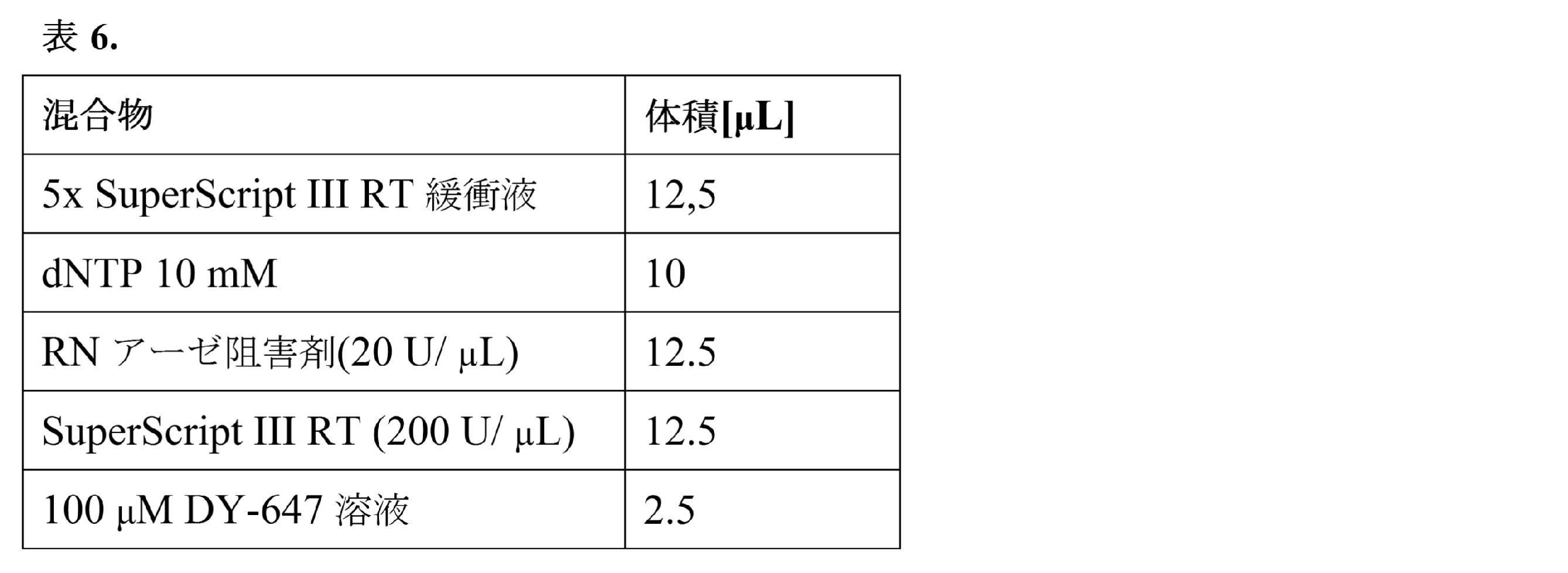
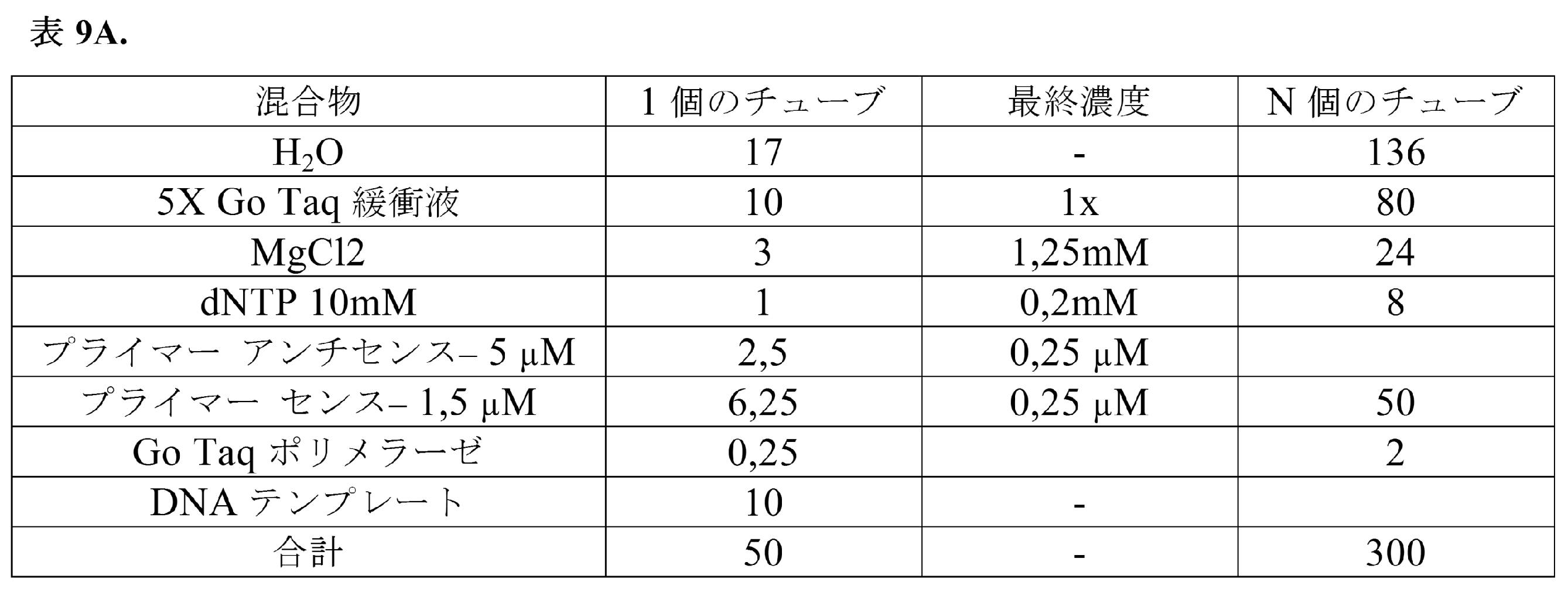
比較はqPCRデータに基づく。液滴実験のCp値(二次導関数最大算出法により決定された交点値)をバルク実験および精製済みRNA試料のcDNA合成と比較した。混合物(溶解緩衝液4× 50μL;RT緩衝液5× 17.6μL;H2O 22.4μL;ポリdTオリゴ 100μM 2μL;プロテアーゼ阻害剤4μL;DY-647 100μM 4μL)を含む溶解緩衝液と、RT混合物緩衝液(一本鎖反応緩衝液5× 22.5μL;0.1M DTT 10μL;10mM各dNTP 10μL;RNアーゼ阻害剤2.5μL;Superscript III RT酵素2.5μL;DY-647 100μM溶液2.5μL)と、PBSで予め洗浄し、ペレット化し、かつDY-647 100μM溶液1μL+細胞懸濁溶液(Percoll 300μL;NaCl 1.5M 33.3μL;Pluronic10% 10μL;ヘペス1M 25μL;血清低IgG5% 50μL;培地DMEM581.7μL)49μLに再懸濁した200,000個のヒトRamos細胞とを同時に流すことにより、液滴ベースの実験を実施した。溶解緩衝液1×は、50mM トリスHCl pH7.5、75mM NaCl、3mM MgCl2、0.2%Triton X-100である。Fluigentポンプを備えたフィードバックモジュールを使用して、下記の流速が得られることを確実にする(表2)。
100pLの液滴を生成し(CVは<5%の幅)、HFE-7500を充填したチューブ中において氷上でエマルジョンを回収した。エマルジョンの製造が終了すると、HFE-7500が入ったシリンジを使用して、リザーバーのより長いチューブに取り付けた。出口に接続された針に油を充填し、このシリンジを塞いで気泡の形成を防止した。チップからチューブを直ちに切断し、プランジャーを緩やかに引き出して、チューブ中に依然として存在するエマルジョンを完全に吸引する。回収チューブ内での気泡を避ける。エマルジョン含有チューブを60分にわたり55℃でサーモミキサー中に置いた。RTを15分にわたり70℃で不活性化し、次いで4℃で1分間不活性化した。ペルフルオロオクタノール(エマルジョンの体積/体積)を使用してエマルジョンを回収して破壊し、油相および水相が分離するまでインキュベートした。4℃において10,000gで10分にわたり水相を遠心分離し、上清を回収する。37℃で15分にわたりRNアーゼA 2.5μLと共にインキュベートし、37℃で一晩プロテイナーゼK 4μLと共にインキュベートすることにより、RNAおよびタンパク質を消化する。
1個の細胞当たり100pLを模倣する反応体積(20μLの最終反応)で、200,000個のヒトRamos細胞(上記のようにPBSで予め洗浄した)または200,000個のヒトRamos細胞から抽出(Machery Nagel抽出キット)したRNAを混合することにより、バルク実験を実施した。この混合物を表3に示す。
混合した細胞およびRNAを氷上で10分にわたりインキュベートし、次いでRTを55℃で1時間にわたり実施した。RTを15分にわたり70℃で不活性化した。cDNAを上記のように処理した。
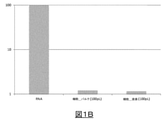
全てのcDNAを、(0.8×比で)RNAClean XP Beckmanを使用して精製し、H2O 20μLで溶出させた。cDNAを3回希釈し、Ramos細胞中において様々なレベルで発現されることが分かっている3組のハウスキーピング遺伝子用のqPCR反応(Roche LightCycler 480マスターミックス)のテンプレートとして使用した。純粋なRNA条件からのCp値と細胞反応からのCp値とを比較し(図1A)、次いでRT効率を評価するために使用した(RNA試料を100%効率と見なした:図1B)。
まとめると、この実験は、同一の体積/細胞を模倣する液滴およびバルクをベースとする実験が、試験した3種の遺伝子に関して同等の結果を与えることを示す(図1A)。次に、標準条件を使用すると、RT効率の平均は、試験した3種の遺伝子に関して約2%である(図1B)。この値は事前の報告と一致する。
ヒト遺伝子のqPCR用のアンチセンスプライマー配列
Top_SBS12-ATP5G3:
/5Phos/CAACGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTGCTTCAGCGAAGGGTTTC(配列番号1)
Top_SBS12-RPS29:
/5Phos/CAACGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTACAGACACGACAAGAGCGA(配列番号2)
Top_SBS12-ZNF780A:
/5Phos/CAACGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGATCCATGGACCATGTTGCT(配列番号3)
ヒト遺伝子のqPCR用のセンスプライマー配列
SBS3-ATP5G3:ACACTCTTTCCCTACACGACGCTCTTCCGATCTCAGGTGCTGCAACAGTAGGA(配列番号4)
SBS3-RPS29:ACACTCTTTCCCTACACGACGCTCTTCCGATCTTTACCTCGTTGCACTGCTGA(配列番号5)
SBS3-ZNF780A:ACACTCTTTCCCTACACGACGCTCTTCCGATCTAGAGCGTTACTGCTGCACA(配列番号6)
実施例2:RT効率での液滴サイズの影響
実施例1に従って試薬/体積を増加させることにより、RT効率での1個の細胞当たりの緩衝液体積の影響を評価した。Cp値を精製済みRNA条件と比較した(図2)。100pL体積では1%RT効率が達成されており、この効率は報告されたものと比べて高く、1nL反応/細胞での>10%効率まで増加している。
実施例3:RT効率でのRT酵素、溶解緩衝液の量の影響
サブナノリットル体積でRT効率をさらに改善するために、RT濃度、溶解緩衝液およびRT反応の試薬の増加の影響を評価した。本発明者らは、(供給業者により説明された通りの)標準cDNA合成において、精製済みRNA条件をベースとして、これらの変更のRT効率への影響を比較した。
図3Aは、液滴ベースの実験でのRT濃度の増加の影響を示す。RT酵素を5倍に増加させることにより、cDNA合成が平均して10倍増加した(3つの異なる遺伝子のCp値に基づく)。最大の影響が低発現遺伝子に関して達成されており、これは、RT合成効率の本当の効果を立証する。
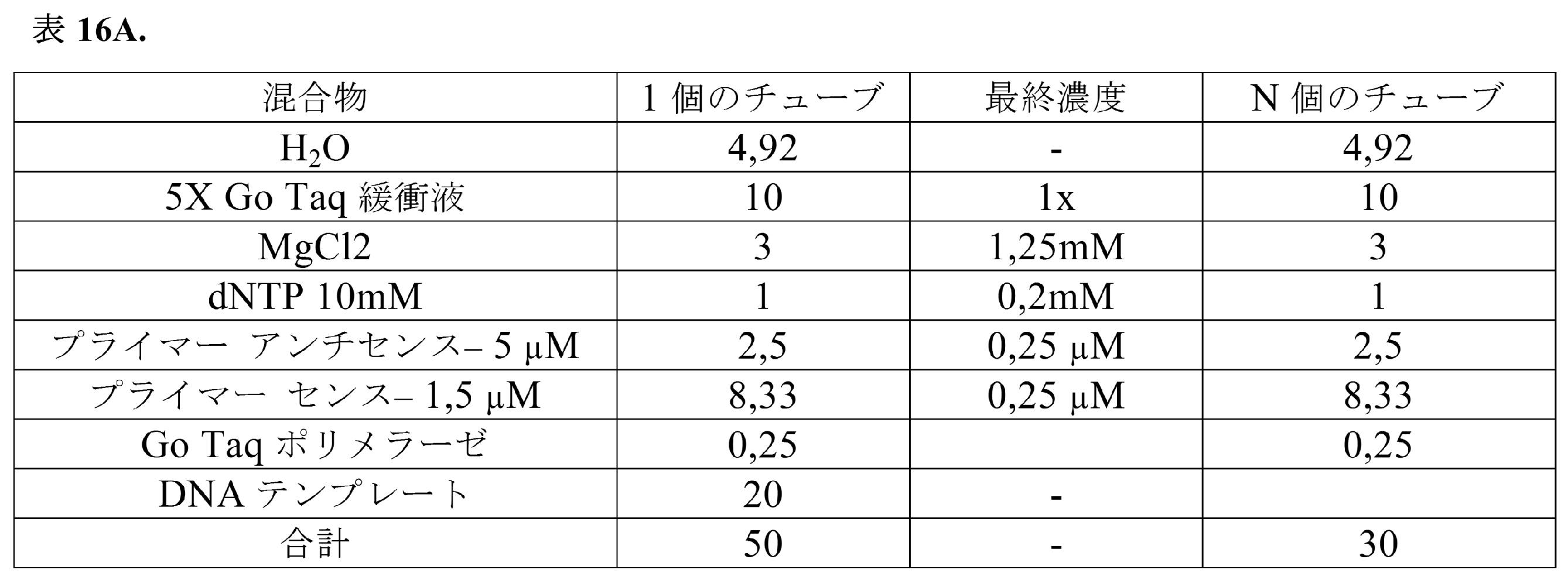
細胞のマイクロ流体封入を、RTプライマーおよびRT混合物と一緒に実施例1で説明したように実施した。混合物(溶解緩衝液4× 50μL;RT緩衝液5× 17,6μL;H2O 22.4μL;ポリdTオリゴ 100μM 2μL;プロテアーゼ阻害剤4μL;DY-647 100μM 4μL)を含む溶解緩衝液と、RT混合物緩衝液(一本鎖反応緩衝液5× 22.5μL;0.1M DTT 10μL;10mM各dNTP 10μL;RNアーゼ阻害剤2.5μL;Superscript III RT酵素2.5μL;DY-647 100μM溶液2.5μL)と、PBSで予め洗浄し、ペレット化し、かつDY-647 100μM溶液1μL+細胞懸濁溶液(Percoll 300μL;NaCl 1.5M 33.3μL;Pluronic10% 10μL;ヘペス1M 25μL;血清低IgG5% 50μL;培地DMEM581.7μL)49μLに再懸濁した200,000個のヒトRamos細胞とを同時に流すことにより、液滴ベースの実験を実施した。溶解緩衝液1×は、50mMトリスHCl pH7.5、75mM NaCl、3mM MgCl2、0.2%Triton X-100である。
RT酵素の濃度が上記の実施例と比べて5×高いエマルジョンでは、本発明者らは、混合物(溶解緩衝液4× 50μL;RT緩衝液5× 27.6μL;H2O 12.4μL;ポリdTオリゴ100μM 2μL;プロテアーゼ阻害剤4μL;DY-647 100μM 4μL)を含む溶解緩衝液と、RT混合物緩衝液(一本鎖反応緩衝液5× 12.5μL;0.1M DTT 10μL;10mM各dNTP 10μL;RNアーゼ阻害剤2.5μL;Superscript III RT酵素12.5μL;DY-647 100μM溶液2.5μL)とを同時に流し、次いで上記のようにかつ実施例1のように細胞を封入した。cDNA精製およびqPCRを実施例1のように実行した。図3Bは、効率的なcDNA合成への溶解緩衝液の影響を示す。MgCl2(効率的なRT酵素活性に必須である)を補完した低張溶解緩衝液を低張水溶液と比較した。MgCl2を補完した低張溶解緩衝液は、1個の細胞当たりサブナノリットル反応体積での効率的なcDNA合成に必須である。実施例1で説明したように、溶解条件の影響を、バルク反応においてかつ1個の細胞当たり100pLを模倣する体積で細胞から始めて分析した。1個の細胞当たり100pLを模倣する反応体積(20μLの最終反応)で、200,000個のヒトRamos細胞(実施例1のようにPBSで予め洗浄した)を混合することにより、バルク実験を実施した。この混合物を下記の表4で説明する。
逆転写酵素(RT)反応混合物中の細胞を最初に氷上で10分にわたりインキュベートして、細胞を溶解させた。次いで、この反応物を(750rpmで振盪しつつ)55℃において1時間にわたりインキュベートすることにより、RTを始動させた。RT酵素を15分にわたり70℃で不活性化させた。RT反応物を10,000g 4℃で10分にわたり遠心分離することにより、細胞破片をペレット化した。懸濁液中のcDNAを上清で回収し、(750rpmで振盪しつつ)37℃において15分にわたりRNアーゼA 2.5μLにより、および(750rpmで振盪しつつ)37℃においてONにわたりプロテイナーゼK 4μLにより、順次処理した。0.8×ビーズ/溶液比でRNAクリーンビーズ(Beckman)を使用してcDNAを精製し、DNアーゼおよびRNアーゼフリー水20μLで溶出させた。精製済みcDNAを使用して、qPCRによりcDNA合成効率を評価した。
図3Cは、cDNA合成の効率におけるRT量/溶解条件の変化の影響を再現する。全体的なRT効率が約40倍改善された(1.2%から49%超へ;精製済みRNAを100%に設定した)。
図3Dは、RT効率における液滴体積の増加ならびにRT条件の変化(RT酵素およびRNアーゼ阻害剤の増加量ならびに溶解緩衝液の変化)の両方のさらなる影響を示す。実施例5で説明するように、マウスハイブリドーマ細胞を100pLまたは500pLのいずれかの液滴に封入して、並行液滴ベースの単一細胞RNAseq実験を実施した。捕捉されたATP5G3ハウスキーピング遺伝子にバーコードが付与されたcDNAのRT効率分析は、100pLと比較して、500pLの液滴ベースのRNAseqが、生成されたcDNAの量の2倍の増加を示した。この結果は、他の遺伝子に関してヒト細胞によりおよびマウスハイブリドーマ系において一貫して繰り返されている。
マウスATP5G3遺伝子のqPCR用のアンチセンスプライマー配列
/5Phos/CAAC TGCAACTCTGGATCCAGCTC(配列番号7)
マウスATP5G3遺伝子のqPCR用のセンスプライマー配列
ACACTCTTTCCCTACACGACGCTCTTCCGATCTTCGCCTGTCACCTAGATCCA(配列番号8)
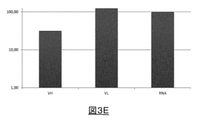
精製済みRNA条件と比較して100%のRT効率に達するために、RT反応条件をさらに最適化した(細胞の封入の最中のPercollの除去、広域スペクトルRNアーゼ阻害剤の選択、cDNAの回収および精製の最適化)。免疫グロブリンを発現するマウスハイブリドーマ細胞を500pLの液滴に封入し、この液滴中で液滴ベースの単一細胞VH/VL mRNAの捕捉およびcDNA合成を実施した。並行して、同一の細胞集団からRNAを抽出して精製し、VHおよびVLのmRNAの逆転写を、供給業者の推奨で説明されているように、チューブベースのアッセイ(tube based assay)で実施した。リアルタイム定量PCRによりVH/VL cDNA合成効率を評価した。図3Eは、精製済みRNAと比較した液滴ベースのアプローチのRT効率の%を示し、この%は、軽鎖免疫グロブリン可変遺伝子の場合には100%超であり、重鎖免疫グロブリン可変遺伝子の場合には約30%である。VHの場合のより低い値は、バーコードが付与された試料へのより低いPCR効率に起因する可能性がある。
細胞を細胞洗浄緩衝液(CWB;DMEM F12培地、0.1%Pluronic、25mMヘペス、5%低IgG血清)で洗浄し、100μM DY-647溶液2.6μLを含むCWB緩衝液47.4μL中においてマイクロ流体設計で同時に流した。単一細胞のバーコードならびに液滴中でのVHおよびVLのmRNAの捕捉およびcDNA合成のためのプライマーを担持するヒドロゲルビーズライブラリを1×BW緩衝液で10回洗浄し、4℃において2分にわたり2500gで回転させた。二本鎖バーコードを2分にわたり22℃において変性溶液(H2O 700μL+1M NaOH 300μL)1mL中で変性させた。このビーズをBW緩衝液で3回洗浄し、FITCビオチン100μM 10μLで標識し、10分にわたり回転台上で室温においてインキュベートした。BW緩衝液での3回の洗浄後、このビーズを70℃で2分にわたり加熱し、上清を除去した。最後に、このビーズを溶解/Pi/DTT/色素含有緩衝液(溶解緩衝液10× 80μL;プロテアーゼ阻害剤8μL、Dye 647 100μM 8μLおよび1M DTT 4μL)200μLで洗浄した。このビーズを4℃において2分にわたり2500gで回転させ、上清を除去して緩衝液/HgB混合物50μLを残した。
溶解緩衝液中に単一細胞のバーコードおよびプライマーを担持するヒドロゲルビーズと、RT混合物緩衝液(一本鎖反応緩衝液5× 52μL;0.1M DTT 10μL;10mM各dNTP 13μL;SUPERin RNアーゼ阻害剤16.25μL;Superscript III RT酵素16.25μL;DY-647 100μM溶液2.5μL)と、15,000個のマウスハイブリドーマ細胞とを同時に流すことにより、液滴ベースの実験を実施した。
RTプライマーおよびRT混合物との細胞の封入を表5に示すように実施した。
実施例4:ハイブリドーマ混合物
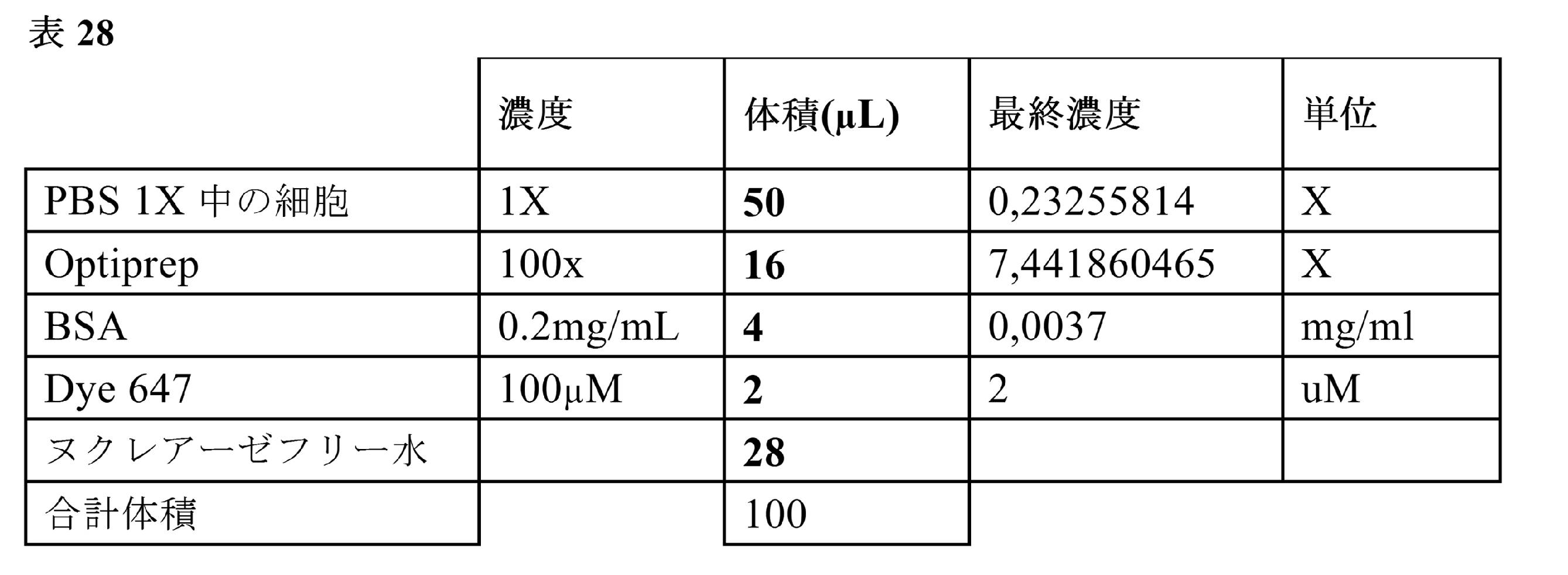
サブナノリットルの液滴中でのVH/VL遺伝子の対形成の正確さを評価するために、既知の抗体配列を発現するマウスハイブリドーマ細胞株を等しい割合で混合した。これらの細胞をPBSで洗浄し、細胞懸濁緩衝液(Percoll300μL;NaCl 1.5M 33.3μL;Pluronic10% 10μL;ヘペス1M 25μL;血清低IgG5% 50μL;培地DMEM581.7μL)47.5μLおよび100μM DY-647 2.5μLに再懸濁させた。
細胞を、単一細胞のバーコードならびに液滴中でのVHおよびVLのmRNAの捕捉ならびにcDNA合成のためのプライマーを担持するヒドロゲルビーズライブラリと同時に封入した。
RTプライマー配列:
VH_1:GGCCAGTGGATAGACAGATGGGGG(配列番号9)
VH_2:GGCCAGTGGATAGACCGATGGGGC(配列番号10)
VH_3:GGCCAGTGGATAGACTGATGGGGG(配列番号11)
VH_4:GTCACCGCAGCCAGGGACCAAGGG(配列番号12)
VLk_1:GCGTTTCATTTCCAGCTTGG(配列番号13)
VLk_2:GCGTTTGATTTCCAGCTTGG(配列番号14)
VLk_3:GCGTTTTATTTCCAATTTTG(配列番号15)
ヒドロゲルビーズを1×BW緩衝液で10回洗浄し、4℃において2分にわたり2500gで回転させた。二本鎖バーコードを2分にわたり22℃において変性溶液(H2O 700μL+1M NaOH 300μL)1mL中で変性させた。このビーズをBW緩衝液で3回洗浄し、FITCビオチン20μM 10μLで標識し、1時間にわたり回転台上で室温においてインキュベートした。4×溶解緩衝液(0.8% TritonX100、12mM MgCl2、200mM トリス-HCl pH7.4)での3回の洗浄後、このビーズを70℃で2分にわたり加熱し、上清を除去した。このビーズを溶解緩衝液4× 50μL、5×一本鎖RT緩衝液27.6μL、プロテアーゼ阻害剤4μL、DY-647 100μM 4μL、H2O 4.4μLおよび0.1M DTT 10μLに可溶化した。
ビーズおよび細胞を、表6で説明するRT混合物と同時に流した。
Fluigentポンプを備えたフィードバックモジュールを使用して、表7に示す下記の流速が得られたことを確実にする。
100~110pLの液滴を確実に生成する。エマルジョンを生成すると、オリゴ光開裂が200mW/cm2で30秒にわたり進行した。このエマルジョンを60分にわたり55℃でインキュベートし、RTを15分にわたり70℃で不活性化した。チューブを4℃で1分にわたり冷却した。このエマルジョンを、シリンジプランジャーを押すことにより新たな1.5mL DNA LoBindチューブに移し、30秒間ボルテックスしてビーズからのオリゴ拡散を可能にした。このエマルジョンを効率的に破壊してcDNAを回収するために、シリンジで、液滴を少しも取ることなく可能な限り多くの油を除去した。パーフルオロオクタノール(エマルジョンの体積/体積)を添加し、時に優しくボルテックス(5秒)しつつおよび優しく回転させつつ(5秒)、室温で5~10分にわたり混合して相を分離した。
2回目の光開裂が200mW/cm2で60秒にわたり進行した。このエマルジョンを30秒にわたりボルテックスし、室温で5分にわたりインキュベートしてビーズからのオリゴ拡散を可能にした。
油相をシリンジで除去した。このエマルジョンを30秒間ボルテックスし、室温で5分にわたりインキュベートしてビーズからのオリゴ拡散を可能にした。cDNAを、RNアーゼA 2.5μLでエッペンドルフに処理し、37℃で15分にわたりDNアーゼ/RNアーゼフリーDNA LoBindチューブに入れた。このcDNA溶液をプロテイナーゼK 4μLで処理し、50℃で1時間にわたりまたは37℃で一晩にわたり1.5mLのDNアーゼ/RNアーゼフリーDNA LoBindチューブに入れた。
cDNAおよびヒドロゲルビーズを含む水相を30μMフィルタ(Pierceスピンカラム)にアプライし、H2Oで洗浄した。ろ過したcDNAの25%を1×比のRNA Clean UPビーズを使用して精製し、DNアーゼおよびRNアーゼフリー水40μLに溶出させた。VH/VL PCR反応を下記のように実施した。
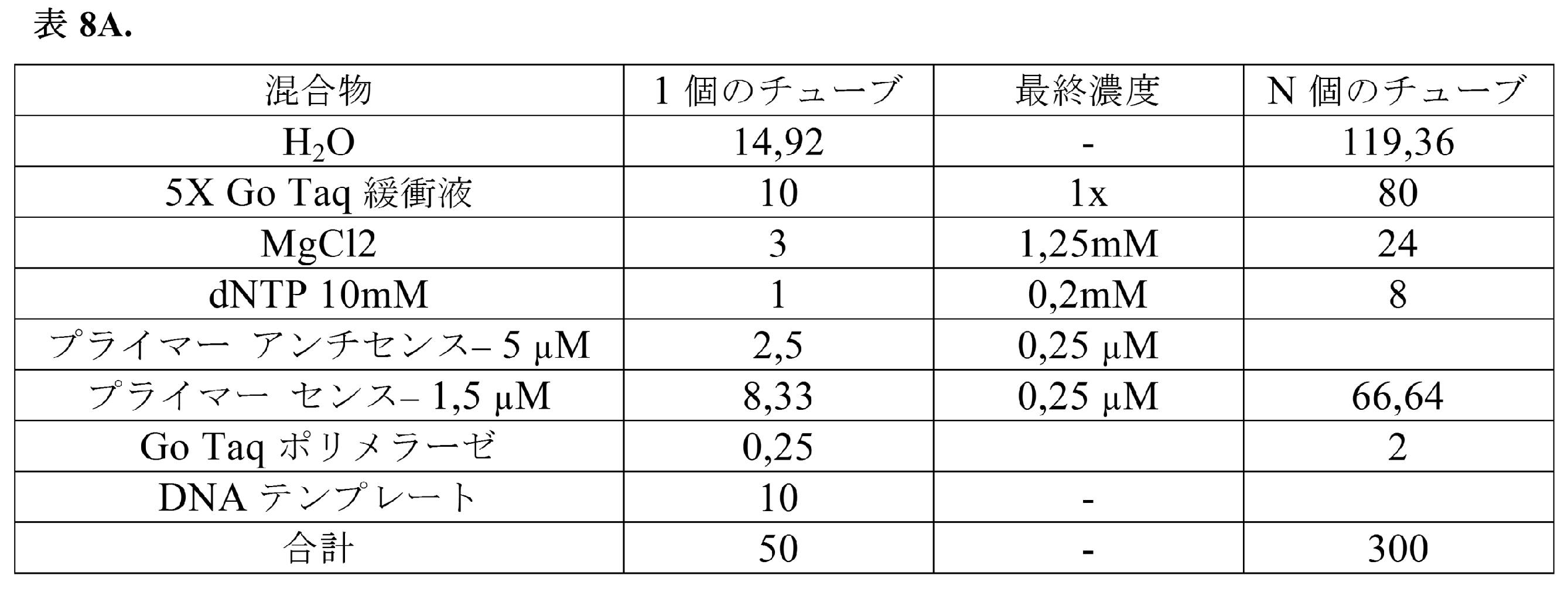
表8Aおよび表8Bで説明されているように、4*10μLの精製済みcDNAを使用してPCR1を実施した。
このPCRを、1×の比でAMPure XPビーズを使用して精製し、水20μLで溶出させた。このPCRの10%を2%アガロースゲルに流した。精製済みPCR1の50%を表9および表9Bで下記に示すようにPCR2にかけた。
このPCRを、1×の比でAMPure XPビーズを使用して精製し、水20μLで溶出させた。このPCRの10%を2%アガロースゲルに流した。このゲルを切断し、バンドを秤量した。バンドを、Gel Extractionキット(QIAGEN(商標))を使用して精製し、水10μLで溶出させた。
PCRプライマーセット
1回目のPCR反応
T7アンチセンスPCR1プライマー配列
GAATTTAATACGACTCACTATAGGGAGA(配列番号16)
2回目のネステッドPCR反応
イルミナインデックスアンチセンスプライマー
CAAGCAGAAGACGGCATACGAGATインデックスGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT(配列番号103)
増幅されて品質管理されたVHおよびVL配列決定ライブラリを多重化させ(それぞれ50%)、2*300 PE読み取りを使用してMiSeqで配列決定した。
図4Aは、バーコードが付与された単一細胞VH/VLのネステッドPCR増幅を示す。
図4Bは、VHおよびVLの読み取り数に基づいてプロットされた同一バーコード(同一細胞に由来する)を共有する対の配列読み取りを示す。
図4Cは、各鎖の読み取り閾値の関数として正確なハイブリドーマVL/VH対形成率を示す。
図4Dは、(両方の鎖の)読み取り閾値の関数としてハイブリドーマ対の対形成率の数を示す。
実施例5:ハイブリドーマの希釈
サブナノリットルの液滴でのVH/VL遺伝子の捕捉効率を評価するために、既知の抗体配列を発現する3種のマウスハイブリドーマ細胞株を60%-30%-10%の割合で混合した。合計で10,000個の細胞(および240,000個のヒト非Ig発現細胞)をPBSで洗浄し、細胞懸濁緩衝液(Percoll300μL;NaCl 1.5M 33.3μL;Pluronic10% 10μL;ヘペス1M 25μL;血清低IgG5% 50μL;培地DMEM581.7μL)97.4μLおよび100μM DY-647 2.6μLに再懸濁させた。
細胞を、単一細胞のバーコードならびに液滴中でのVHおよびVLのmRNAの捕捉ならびにcDNA合成のためのプライマーを担持するヒドロゲルビーズライブラリと同時に封入した。
RTプライマー配列:
VH_1:GGCCAGTGGATAGACAGATGGGGG(配列番号9)
VH_2:GGCCAGTGGATAGACCGATGGGGC(配列番号10)
VH_3:GGCCAGTGGATAGACTGATGGGGG(配列番号11)
VH_4:GTCACCGCAGCCAGGGACCAAGGG(配列番号12)
RT_VLk:GATGGTGGGAAGATGGATAC(配列番号125)
ヒドロゲルビーズを1×BW緩衝液で10回洗浄し、4℃において2分にわたり2500gで回転させた。二本鎖バーコードを2分にわたり22℃において変性溶液(H2O 700μL+1M NaOH 300μL)1mL中で変性させた。このビーズをBW緩衝液で3回洗浄し、FITCビオチン20μM 10μLで標識し、10分にわたり回転台上で室温においてインキュベートした。BW緩衝液での3回の洗浄後、このビーズを70℃で2分にわたり加熱し、上清を除去した。このビーズを溶解*緩衝液(10×溶解緩衝液{2%Triton、30mM MgCl2、500mM トリス-HCl pH7.4}80μL、プロテアーゼ阻害剤8μL、DY-647 8μL、DTT 1M 4μL)200μLで洗浄した。このビーズを溶解*緩衝液50μL中で維持した。
RT混合物を表14に示すように調製した。
Fluigentポンプを備えたフィードバックモジュールを使用して、表14に示す流速を用いて細胞、ヒドロゲルビーズおよびRT混合物を500pLの液滴に同時に封入した。
RT反応、エマルジョンの破壊、ヒドロゲルビーズを含む水相およびcDNAを実施例4で説明したように回収/処理した。
回収したcDNAの半分を、1×ビーズ/水相体積でRNAClean Beckmanビーズを使用して連続して2回精製した。表15Aおよび表16Bで説明するように、溶出したcDNAをPCR(実施例4で列挙したPCRプライマー)によるVH/VLの増幅に使用した。
VHおよびVLのPCR1産物を、それぞれ0.8×比および1×比でAMPure Beckmanビーズを使用して2回精製した。表17Aおよび表17Bに示すようにPCR1産物の半分をPCR2反応に使用した。
増幅産物をアガロースゲルでサイズ選択し、ゲル精製し、品質管理し、かつMiSeq 2*300 PE読み取りで配列決定した。
図5Aは、液滴体積(100pLまたは500pL)およびcDNA精製法(1回の溶出対2回の溶出(1.1または1.2と命名し、2回の精製(2.1と称する)と比較する))によって決まる、バーコードが付与された単一細胞VH/VLのネステッドPCR増幅を示す。
図5Bは、500pLの液滴および改善されたcDNA精製(2回の精製)での、VHおよびVLの読み取り数に基づいてプロットした同一バーコード(同一細胞に由来する)を共有する対の配列読み取りを示す。
図5Cは、500pLの液滴および改善されたcDNA精製(2回の精製)での、(両方の鎖の)読み取り閾値の関数としてハイブリドーマ対の対形成率の数を示す。
図5Dは、(両方の鎖の)読み取り閾値、液滴体積(100pLまたは500pL)およびcDNA精製法(1回の溶出対2回の溶出(1.1または1.2と命名し、2回の精製(2.1と称する)と比較する))の関数としてハイブリドーマ対の対形成率の正確な対形成率を示す。
図5Fは、液滴体積(100pLまたは500pL)およびcDNA精製法(1回の溶出対2回の溶出(1.1または1.2と命名し、2回の精製(2.1と称する)と比較する))によって決まる、1本の鎖当たり10回の読み取りの閾値での回収したハイブリドーマ対分布を示す。cDNA精製が改善された(2回の精製)500pLの液滴体積により、最初の60%-30%-10%のハイブリドーマ比がもたらされる。
実施例6:マウス一次B細胞のスクリーニング
破傷風トキソイド特異的抗体を分泌する一次マウス脾臓B細胞を選別し、細胞洗浄緩衝液(DMEM/F12細胞培養培地915μL;血清低IgG 50μL;ヘペス1M 25μL;PluronicF68 10% 10μL)500μLで2回洗浄した。最後に、CWB 97.4μLおよびDY-647 2.6μLに再懸濁した。
ヒドロゲルビーズを担持する単一細胞を実施例5のように調製した。
細胞、ヒドロゲルビーズおよびRT混合物の500pLの液滴への同時封入を実施例5で説明したように実施した。エマルジョンを実施例5で説明したように破壊した。
ヒドロゲルビーズおよびcDNAを含む水相を2つの等体積に分割した。この水相の半分を(37℃で15分にわたり)RNアーゼA 1μLおよび(37℃で一晩)プロテイナーゼK 4μLで処理し、次いでこの物質の半分を精製した。この水相の半分を4℃において10分にわたり10,000gで遠心分離した。回収した上清を上記のように処理し、次いでこの物質の半分を精製した。
cDNAを、1×ビーズ:水相体積比を使用してRNACleanビーズで2回精製し、表18Aおよび表18Bに示すように1回目のVH/VL PCR増幅に使用した。
VHおよびVLの増幅産物をそれぞれ0.8×および1×のAMPureビーズ比で2回精製した。図18Aおよび図18Bに示すように、精製した物質の半分を2回目のネステッドPCR反応に使用した。
次いで、VH/VL増幅産物を、PCR精製キット(QIAGEN)を使用して精製した。10分の1を3回目のPCR反応に使用し、表19Aおよび表19Bに示すように実施した。
増幅産物をアガロースゲルでサイズ選択し、ゲル精製し、品質管理し、かつMiSeq 2*300 PE読み取りで配列決定した。遠心分離したcDNA配列決定ライブラリの情報と遠心分離していないcDNA配列決定ライブラリの情報とを組み合わせることにより、分析を行った。
1回目および2回目のPCR反応で使用したセンスVH PCRプライマーおよびセンスVL PCRプライマーは、文献(Rohatgi,et al.,2008 Dec 31;339(2):205-19)で説明されているものであった。
PCR1およびPCR2に使用したアンチセンスプライマーは、実施例4で説明した通りであった。
PCR3で使用したセンスプライマー:
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT(配列番号126)。
PCR3で使用したアンチセンスプライマー:CAAGCAGAAGACGGCATACGAGAT(配列番号127)。
図6Aは、(VH鎖およびVL鎖の両方の)読み取り閾値の関数として一次B細胞のVH/VL対形成の数を示す。
図6Bは、1本の鎖当たりの読み取り閾値に基づいて、重鎖および軽鎖の異なる相補性決定領域(CDR)の数を示す。
図6Cは、VHおよびVLの読み取りの数に基づいてプロットした同一バーコード(同一細胞に由来する)を共有する対の配列読み取りを示す。
図6Dは、(1本の鎖当たり40回の読み取りの閾値での)対の配列における重可変遺伝子の使用を示す。
図6Eは、(1本の鎖当たり40回の読み取りの閾値での)対の配列におけるカッパ可変遺伝子の使用を示す。
図6Fは、(1本の鎖当たり40回の読み取りの閾値での)対の配列における重J遺伝子ファミリーの使用を示す。
図6Gは、(1本の鎖当たり40回の読み取りの閾値での)対の配列におけるカッパJ遺伝子ファミリーの使用を示す。
実施例7:遺伝子特異的プライマー濃度の影響の評価
RT反応における遺伝子特異的プライマー濃度の影響を、細胞から始まるマウス抗体のVHおよびVLに関するバルク反応で評価している(図7を参照されたい)。このバルク反応は、液滴条件(特に1個の細胞当たりの反応緩衝液の体積、1個の細胞当たりの試薬の量)を模倣することを目的としていた。
従って、40000個のマウス9E10ハイブリドーマ細胞(細胞洗浄緩衝液(DMEM F12培地、0.1%Pluronic、25mMヘペス、5%低IgG血清)で予め洗浄した)を、1個の細胞当たり500pL(20μLの最終反応)を模倣する反応体積で使用した。この混合物は下記の表21で示す通りであった。
次いで、VH遺伝子およびVL遺伝子に特異的なプライマーの様々な量を、表22に示す濃度で添加した。
ペレット化した細胞を氷上で10分にわたりインキュベートして混合物にし、溶解を生じさせた。この混合物を55℃で1時間にわたりインキュベートし、550rpmで混合した。次いで、RTを550rpmで15分にわたり70℃において不活性化した。次いで、cDNAをRNアーゼAおよびPKで処理して、微量のPCR阻害剤を除去した。本発明者らは、H2O qsp 95μLを添加し、RNAアーゼA(10mg/mLストック濃度、100ug/μL最終濃度)1μLを添加し、37℃で15分にわたりDNアーゼ/RNアーゼフリーDNA LoBindチューブに入れた。次いで、本発明者らは、プロテイナーゼK(20mg/mLストック濃度、0.8mg/mL最終濃度)4μLを添加し、このチューブを50℃で1時間にわたり置いた。本発明者らは、70℃で15分にわたりPKを不活性化した。次いで、cDNAを、1×比(1体積のビーズ溶液対1体積のcDNA)でRNACLeanupビーズを使用して精製し、続いてBeckman RNA CleanupビーズSOPを使用して精製した。本発明者らは、2分にわたりDNアーゼ/RNアーゼフリーH2O 40μLでcDNAを溶出させ、溶出したcDNAを回収した。本発明者らは、H2O 40μLでビーズを2回溶出させた。これら2回の溶出をプールし、本発明者らは、H2O 40μLで1回の溶出のみを行うことを除いて前と全く同じように1×RNACleanupビーズ(Beckman)を使用して2回目の精製工程を始める。条件毎に生成されたcDNAの量を、多重qPCR反応に基づく絶対的定量(absolute quantification)qPCRを使用して測定した。
遺伝子特異的プライマー濃度の影響を、細胞から始まるヒト抗体のVHおよびVLに関する液滴でのRT反応で評価した(図8Aおよび図8Bを参照されたい)。PBMCから精製され、かつ5日にわたり活性化された健康なヒトドナー由来の5000個のヒト活性化スイッチドメモリ(switched memory)B細胞を、重/軽_カッパ/軽_ラムダ鎖を捕捉するための各遺伝子特異的プライマーの100nMまたは10nMのいずれかを含む500pLの液滴に封入した。各試薬(細胞、溶解およびRTの試薬)をマイクロ流体チップ中で同時に流して500pLの液滴を生成した。溶解/Pi/DTT/色素含有溶液を下記の表23で示すように調製した。
この混合物50μLを封入に使用し、チップへの吸引まで氷上に置いた。2.6×RT混合調製物を表24で示すように調製した。
細胞を洗浄し、下記の試薬を含む細胞懸濁緩衝液(CSB)に再懸濁した。
PluronicF68 10% 10μl
ヘペス1M 25μl
血清低IgG 50μl
培地DMEM/F12 915μl
生成した液滴を氷上で回収し、溶解を生じさせた。エマルジョンを55℃で1時間にわたりインキュベートし、550rpmで混合した。次いで、RTを550rpmで15分にわたり70℃において不活性化した。次いで、cDNAをRNアーゼAおよびPKで処理して、微量のPCR阻害剤を除去した。H2O qsp 95μLを添加し、RNAアーゼA(10mg/mLストック濃度、100μg/μL最終濃度)1μLを添加し、37℃で15分にわたりDNアーゼ/RNアーゼフリーDNA LoBindチューブに入れた。次いで、プロテイナーゼK(20mg/mLストック濃度、0.8mg/mL最終濃度)4μLを添加し、このチューブを50℃で1時間にわたり置いた。70℃で15分にわたりPKを不活性化した。次いで、cDNAを、0.8×比(1体積のビーズ溶液対1体積のcDNA)でRNACLeanupビーズを使用して精製し、続いてBeckman RNA CleanupビーズSOPを使用して精製した。2分にわたりDNアーゼ/RNアーゼフリーH2O 40μLでcDNAを溶出させ、溶出したcDNAを回収した。
条件毎に生成されたcDNAの量を、多重qPCR反応に基づく絶対的定量qPCRを使用して測定した。
RT反応における遺伝子特異的プライマー濃度の影響を、細胞から始まる非抗体遺伝子に関するバルク反応で評価した(図9を参照されたい)。本発明者らは、液滴条件(特に1個の細胞当たりの反応緩衝液の体積、1個の細胞当たりの試薬の量)を模倣することを目的とした。
本発明者らは、1個の細胞当たり500pLを模倣する反応体積(20μL最終反応)で、40000個のヒトJurkat細胞(細胞洗浄緩衝液(DMEM F12培地、0.1%Pluronic、25mMヘペス、5%低IgG血清)で予め洗浄した)を使用した。この混合物は表25で示した通りであった。
ペレット化した細胞を氷上で10分にわたりインキュベートして混合物にし、溶解を生じさせた。この混合物を55℃で1時間にわたりインキュベートし、550rpmで混合した。次いで、RTを550rpmで15分にわたり70℃において不活性化した。次いで、cDNAをRNAseq AおよびPKで処理して、微量のPCR阻害剤を除去した。H2O qsp 95uLμLを添加し、RNAアーゼA(10mg/mLストック濃度、100ug/uLμμ最終濃度)1μLを添加し、37℃で15分にわたりDNアーゼ/RNアーゼフリーDNA LoBindチューブに入れた。次いで、プロテイナーゼK(20mg/mLストック濃度、0.8mg/mL最終濃度)4μLを添加し、このチューブを50℃で1時間にわたり置いた。70℃で15分にわたりPKを不活性化した。次いで、cDNAを、1×比(1体積のビーズ溶液対1体積のcDNA)でRNACLeanupビーズを使用して精製し、続いてBeckman RNA CleanupビーズSOPを使用して精製した。2分にわたりDNアーゼ/RNアーゼフリーH2O 40μLでcDNAを溶出させ、溶出したcDNAを回収した。H2O 40μLでビーズを2回溶出させた。これら2回の溶出を一緒にプールした。条件毎に生成されたcDNAの量を、一重qPCR反応に基づく相対的定量(relative quantification)qPCRを使用して測定した。
実施例8:RT反応におけるポリdTプライマー濃度の影響の評価
細胞から始まるヒト全トランスクリプトーム捕捉のためのRT反応におけるポリdTプライマー濃度の影響の評価を液滴反応で実施している。95%のヒトB細胞Ramos細胞と5%のヒトT細胞Leukemia Jurkat細胞とで構成された20,000個の細胞の混合物を溶解緩衝液、RT試薬、および3.3uMまたは100nMまたは33nMのいずれかのポリdTプリマーと共に500pLの液滴に封入した。全ての試薬をマイクロ流体チップ中で同時に流して500pLの液滴を生成した。
RT試薬を含む混合物を下記の表26で示すように調製した。
次いで、ポリdT RTプライマーを表27で示すように様々な濃度で添加した。
細胞を、表28に示す濃度を含む混合物に再懸濁した。
最終混合物は、表29に示す成分および濃度で構成されている。
生成した液滴を氷上で回収し、溶解を生じさせた。エマルジョンを50℃で2時間にわたりインキュベートし、次いでRTを15分にわたり70℃で不活性化した。次いで、エマルジョンを氷で冷却し、80%(体積/体積)のHFE-7500および20%(体積/体積)のパーフルオロオクタノールで構成された溶液1体積を添加して液滴を破壊した。次いで、cDNAを4℃において14,000gで15分間遠心分離し、上清を回収した。過剰のプライマーおよびプライマー二量体を消化するために、cDNAを37℃で30分にわたりExonuclease I(NEB、参照#M0293;20,000ユニット/ml)1μL、Hinf1(NEB、#R0155;10,000ユニット/ml)1μLと共にインキュベートした。80℃で10分にわたりExonuclease I酵素およびHinf1酵素を不活性化した。次いで、cDNAを、Beckman Agencourt AMPure SOPを使用して精製した後、1.2×比(1.2体積のビーズ対1体積のcDNA)でAgencourt AMPureビーズを使用して精製した。cDNAを2分にわたりDNアーゼ/RNアーゼフリー水17μLで溶出させ、溶出したcDNAを回収した。条件毎に生成されたcDNAの量を、一重qPCR反応に基づく相対的定量qPCRを使用して測定した。
実施例9:遺伝子特異的逆転写への遺伝子特異的プライマー濃度の影響
マウス免疫グロブリンの可変重鎖(VH)および可変軽鎖(VL)の逆転写の捕捉効率(1uMの各遺伝子特異的プライマーに対して正規化されている)。9E10個のハイブリドーマ細胞を、液滴条件を模倣するRT反応におけるバルクで処理した。この実験は、液滴条件(特に1個の細胞当たりの反応緩衝液の体積、1個の細胞当たりの試薬の量)を模倣する。各条件に関して、本発明者らは、1uM~10nMの各VHおよびVL遺伝子特異的プライマーを使用した。比較はqPCRデータに基づく。Cp値(二次導関数最大算出法により決定された交点値)を各プライマー条件と比較した。(n=2)。エラーバー=標準偏差(図7)。
ヒト免疫グロブリンの可変重鎖(VH)および可変軽カッパ鎖(VLk)およびラムダ鎖(VLl)の逆転写の捕捉効率(100nMの各遺伝子特異的プライマーに対して正規化されている)。ヒトスイッチド活性化メモリB細胞(活性化後5日)を溶解試薬およびRT試薬と共に500pLの液滴に封入し、この混合物に、100nMまたは10nMのいずれかの各遺伝子特異的プライマーを添加した。(左側)比較は、各免疫グロブリン可変鎖のqPCRデータに基づく。Cp値(二次導関数最大算出法により決定された交点値)を各プライマー条件と比較した。(右側)2段階のネステッドPCR反応は、Sybr Goldで染色した2%アガロースゲルにロードした各VHおよびVLのアンプリコンを増幅させた。(n=2)。エラーバー=標準偏差(図8Aおよび図8B)。
ヒトハウスキーピング遺伝子RPS29およびTRAT1(高発現遺伝子から低発現遺伝子)の捕捉効率(1uMの各遺伝子特異的プライマーに対して正規化されている)。ヒトT細胞Leukemia(Jurkat細胞)を、液滴条件を模倣するおよび1uM、100nM、10nM、1nMのいずれかの各遺伝子特異的プライマーでのRT反応におけるバルクで処理した。比較は各特異的遺伝子のqPCRデータに基づく。(各qPCRに関してn=2,3 cDNA希釈)エラーバー=標準偏差。最も高く発現された遺伝子はプライマー濃度の影響を受けにくいが、最も低く発現された遺伝子の捕捉効率およびRTは10nMのRTプライマー濃度で影響を受ける(図9)。
実施例10:全トランスクリプトーム捕捉および逆転写へのポリdTプライマー濃度の影響
ヒトハウスキーピング遺伝子RPS29およびTRAT1(高発現遺伝子から低発現遺伝子)の捕捉効率。ヒトT細胞Leukemia(Jurkat)細胞を、溶解試薬およびRT試薬と共に500pLの液滴に封入し、この混合物に3.3μMまたは100または33nMのいずれかのポリdTプライマーを添加した。比較は各特異的遺伝子のqPCRデータに基づく。(n=1)。n.d.=不検出。最も高く発現された遺伝子および最も低く発現された遺伝子の両方がプライマー濃度の影響を受け、特に低ポリdTプライマー濃度で影響を受ける(図10)。