JP7202009B2 - siRNA細胞内送達のための脂質膜構造体 - Google Patents
siRNA細胞内送達のための脂質膜構造体 Download PDFInfo
- Publication number
- JP7202009B2 JP7202009B2 JP2019525575A JP2019525575A JP7202009B2 JP 7202009 B2 JP7202009 B2 JP 7202009B2 JP 2019525575 A JP2019525575 A JP 2019525575A JP 2019525575 A JP2019525575 A JP 2019525575A JP 7202009 B2 JP7202009 B2 JP 7202009B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- mmol
- lipid
- integer
- sirna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images
































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/713—Double-stranded nucleic acids or oligonucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Synthetic bilayered vehicles, e.g. liposomes or liposomes with cholesterol as the only non-phosphatidyl surfactant
- A61K9/1271—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers
- A61K9/1272—Non-conventional liposomes, e.g. PEGylated liposomes or liposomes coated or grafted with polymers comprising non-phosphatidyl surfactants as bilayer-forming substances, e.g. cationic lipids or non-phosphatidyl liposomes coated or grafted with polymers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/51—Nanocapsules; Nanoparticles
- A61K9/5107—Excipients; Inactive ingredients
- A61K9/5123—Organic compounds, e.g. fats, sugars
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C219/00—Compounds containing amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C219/02—Compounds containing amino and esterified hydroxy groups bound to the same carbon skeleton having esterified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C219/04—Compounds containing amino and esterified hydroxy groups bound to the same carbon skeleton having esterified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C219/06—Compounds containing amino and esterified hydroxy groups bound to the same carbon skeleton having esterified hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being acyclic and saturated having the hydroxy groups esterified by carboxylic acids having the esterifying carboxyl groups bound to hydrogen atoms or to acyclic carbon atoms of an acyclic saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/30—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by doubly bound oxygen or sulfur atoms or by two oxygen or sulfur atoms singly bound to the same carbon atom
- C07D211/32—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by doubly bound oxygen or sulfur atoms or by two oxygen or sulfur atoms singly bound to the same carbon atom by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/02—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms containing only hydrogen and carbon atoms in addition to the ring hetero elements
- C07D295/027—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms containing only hydrogen and carbon atoms in addition to the ring hetero elements containing only one hetero ring
- C07D295/03—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms containing only hydrogen and carbon atoms in addition to the ring hetero elements containing only one hetero ring with the ring nitrogen atoms directly attached to acyclic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/145—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
- C07D295/15—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings to an acyclic saturated chain
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/52—Constant or Fc region; Isotype
- C07K2317/53—Hinge
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/55—Fab or Fab'
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/74—Inducing cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/75—Agonist effect on antigen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Dispersion Chemistry (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Psychiatry (AREA)
- Genetics & Genomics (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Physics & Mathematics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Nanotechnology (AREA)
- Optics & Photonics (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
本願は、2017年6月15日に、日本に出願された特願2017-117708号に基づき優先権を主張し、その内容をここに援用する。
特に、siRNAなどの優れた送達効率と高い安全性を両立させ、LNPの粒子径の減少に伴うsiRNAなどの送達活性の低下の克服を可能とする新規化合物及び脂質膜構造体を提供することが本発明の課題である。


で表される脂質化合物又はその塩が提供される。
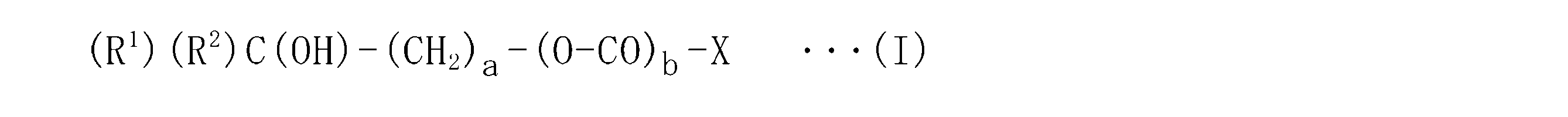
bは0又は1の整数を示す。bが0の場合には-O-CO-基が存在せず、単結合であることを意味する。

ただし、bとcが同時に0となる場合には、qが3~5の整数であり、r及びtが1であり、sが1であり、かつu+vが6~10の整数である場合を除く。

リン脂質及びリン脂質誘導体としては、例えば、ホスファチジルエタノールアミン、ホスファリジルコリン、ホスファチジルセリン、ホスファチジルイノシトール、ホスファチジルグリセロール、カルジオリピン、スフィンゴミエリン、セラミドホスホリルエタノールアミン、セラミドホスホリルグリセロール、セラミドホスホリルグリセロールホスファート、1,2-ジミリストイル-1,2-デオキシホスファチジルコリン、プラスマロゲン、ホスファチジン酸などを挙げることができ、これらは1種又は2種以上を組み合わせて用いることができる。これらリン脂質における脂肪酸残基は特に限定されないが、例えば、炭素数12~20の飽和又は不飽和の脂肪酸残基を挙げることができ、具体的には、ラウリン酸、ミリスチン酸、パルミチン酸、ステアリン酸、オレイン酸、リノール酸などの脂肪酸由来のアシル基を挙げることができる。また、卵黄レシチン、大豆レシチンなどの天然物由来のリン脂質を用いることもできる。
飽和又は不飽和の脂肪酸エステルとしては、グリセロールの1又は2個の水酸基が脂肪酸とエステル結合したグリセリン脂肪酸エステルが挙げられる。当該グリセリン脂肪酸エステル中の脂肪酸残基は、例えば、パルミチン酸、オレイン酸、ステアリン酸、アラキドン酸、ミリスチン酸などの炭素数12~20の飽和又は不飽和の脂肪酸由来のアシル基が挙げられる。具体的には、ジミリストイルグリセロール(DMG)、ジステアロイルグリセロール(DSG)等が挙げられる。
例えば、本発明の脂質膜構造体の核内移行を促進するために、例えば、脂質膜構造体を3糖以上のオリゴ糖化合物で表面修飾することもできる。3糖以上のオリゴ糖化合物の種類は特に限定されないが、例えば、3個ないし10個程度の糖ユニットが結合したオリゴ糖化合物を用いることができ、好ましくは3個ないし6個程度の糖ユニットが結合したオリゴ糖化合物を用いることができる。
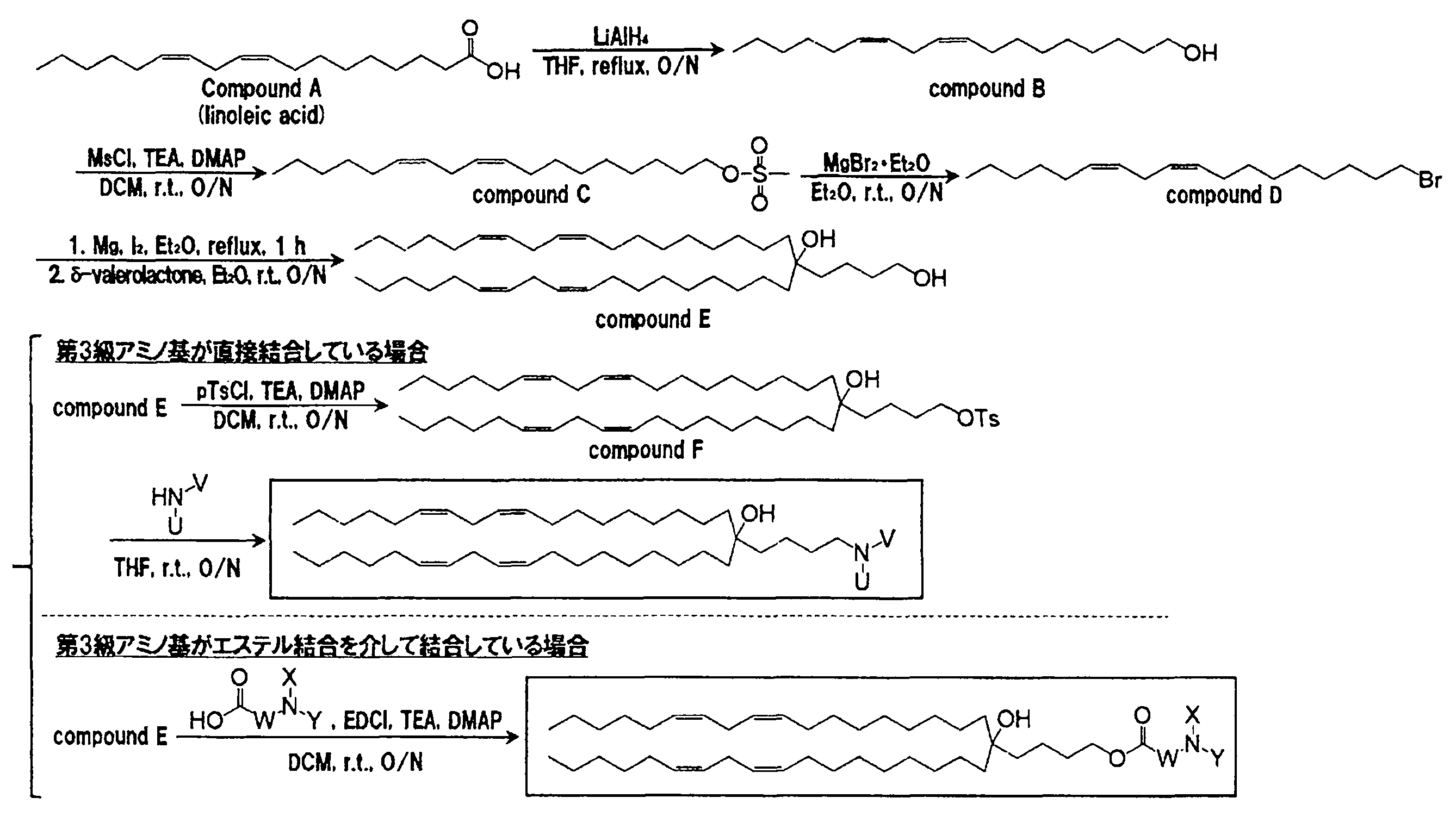
本発明の脂質化合物を以下のスキームに従って合成した。疎水性足場がYSK12(特許文献6)と同じ場合、すなわち、一般式(A)中のcが0である脂質を合成する場合には、出発としてリノール酸(Compound A)を用いた。リノール酸を水素化リチウムアルミニウムで還元後(Compound B)、水酸基をメシル化することで活性化し(Compound C)、臭化マグネシウムを作用させることで臭素化した(Compound D)。δ-Valerolactoneを基質としてグリニャール反応を行うことで2本のリノール酸由来疎水性足場を連結した(Compound E)。第3級アミノ基を炭化水素鎖に直接結合させる場合には、第1級水酸基をトシル化によって活性化し(Compound F)、求核置換反応によりアミノ基を導入した。



6-ブロモヘキサン-1-オール 20.0 g(110.5 mmol)を150 mLの1,2-ジクロロエタンに溶解し、4℃に冷却した。tert-ブチルジメチルクロロシラン(TBSCl) 18.0 g(120 mmol)を加えたのち、トリエチルアミン(TEA) 19.5 mL(140 mmol)を滴下し、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去し、ヘキサン300 mLを加えて懸濁させ、セライト濾過によって不溶物を除去することで粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して、((6-ブロモヘキシル)オキシ)(tert-ブチル)ジメチルシラン 26.0 g(88.0 mmol)を無色オイルとして得た。収率は80%であった。
4 mLのジエチルエーテルに((6-ブロモヘキシル)オキシ)(tert-ブチル)ジメチルシラン1.2 g(4.06 mmol)を溶解し、削り屑状マグネシウム2.43 g(100 mmol)を加え、続いてヨウ素1欠片加えた。室温で10分静置した後、オイルバスで40℃に加熱しながら撹拌し、21 mLのジエチルエーテルに溶解した((6-ブロモヘキシル)オキシ)(tert-ブチル)ジメチルシラン 24.8 g(83.94 mmol)を滴下した。40℃で2時間反応させた後、4℃に冷却した。続いて、δ-バレロラクトン3.67 mL(39.6 mmol)を添加し、室温で一晩反応させた。次に、4℃に冷却し、5%硫酸を滴下することで残留したマグネシウムを溶解させた。ジエチルエーテルで希釈し、有機層を水及び飽和食塩水で分液洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)ウンデカン-1,5-ジオール 14.0 g(26.3 mmol)を無色オイルとして得た。δ-バレロラクトンからの収率は66%であった。
11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)ウンデカン-1,5-ジオール 14.0 g(26.3 mmol)を50 mLのジクロロメタンに溶解し、DMAP(N,N-ジメチル-4-アミノピリジン) 321 mg(2.63 mmol)とジイソプロピルエチルアミン(DIPEA)5.50 mL(39.5mmol)を加え、4℃に冷却した。続いて、p-トルエンスルホニルクロリド(pTsCl)6.02g(31.6 mmol)を徐々に加えていった後、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去し、酢酸エチルで懸濁し、水及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)-5-ヒドロキシウンデシル 4-メチルベンゼンスルホネート 12.4 g(28.0 mmol)を無色オイルとして得た。収率は69%であった。
11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)-5-ヒドロキシウンデシル 4-メチルベンゼンスルホネート 12.4 g(18.0 mmol)に30 mLのテトラヒドロフランを加え、4℃に冷却した。続いて、ジプロピルアミン 7.38 mL(54.0 mmol)を加えた後、室温で11日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、0.5 N 水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して11-(4-(ジイソプロピルアミノ)ブチル)-2,2,3,3,19,19,20,20-オクタメチル-4,18-ジオキサ-3,19-ジシラヘニコサン-11-オール 7.27 g(11.8 mmol)を薄黄色オイルとして得た。収率は66%であった。
11-(4-(ジイソプロピルアミノ)ブチル)-2,2,3,3,19,19,20,20-オクタメチル-4,18-ジオキサ-3,19-ジシラヘニコサン-11-オール 7.27 g(11.8 mmol)に酢酸2.23 mL(39 mmol)及び26 mLの1.0 M テトラブチルアンモニウムフルオリドのテトラヒドロフラン溶液を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、逆相シリカゲルクロマトグラフィー{溶離溶媒;水(0.1%トリフルオロ酢酸):アセトニトリル0.1%トリフルオロ酢酸)(連続勾配)}に供することにより精製して7-(4-(ジイソプロピルアミノ)ブチル)トリデカン-1,7,13-トリオール 3.43 g(8.85 mmol)を薄黄色オイルとして得た。収率は75%であった。
7-(4-(ジイソプロピルアミノ)ブチル)トリデカン-1,7,13-トリオール 388 mg(1.0 mmol)を5 mLのジクロロメタンに溶解し、続いて、オレイルクロリド900 mg(3.0 mmol)を加えた後、4℃に冷却した。TEA 697 μL(5.0 mmol)を滴下し、室温で3時間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-(4-(ジイソプロピルアミノ)ブチル)-7-ヒドロキシトリデカン-1,13-ジイル ジオレアート(CL4H6)570 mg(0.622 mmol)を薄黄色オイルとして得た。収率は62%であった。

7-(4-(ジイソプロピルアミノ)ブチル)トリデカン-1,7,13-トリオール 77.5 mg(0.20 mmol)を1 mLのジクロロメタンに溶解し、続いて、ミリストイルクロリド 197 mg(0.80 mmol)を加えた後、4℃に冷却した。DIPEA 205 μL(1.2 mmol)を滴下し、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-(4-(ジイソプロピルアミノ)ブチル)-7-ヒドロキシトリデカン-1,13-ジイル ジテトラデカノエート(CL4C6)93 mg(0.115 mmol)を薄黄色オイルとして得た。収率は58%であった。

7-(4-(ジイソプロピルアミノ)ブチル)トリデカン-1,7,13-トリオール 77.5 mg(0.20 mmol)を1 mLのジクロロメタンに溶解し、続いて、パルミトイルクロリド 220 mg(0.80 mmol)を加えた後、4℃に冷却した。DIPEA 205 μL(1.2 mmol)を滴下し、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-(4-(ジイソプロピルアミノ)ブチル)-7-ヒドロキシトリデカン-1,13-ジイル ジパルミテート(CL4D6)143 mg(0.164 mmol)を薄黄色オイルとして得た。収率は82%であった。

11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)ウンデカン-1,5-ジオール 5.33 g(10.0 mmol)を50 mLのジクロロメタンに溶解し、DMAP 122 mg(1.0 mmol)と1-メチルピペリジン-4-カルボオキシ酸 塩酸塩 2.16 g(12.0 mmol)を加えた。続いて、EDCI 2.49 g(13.0 mmol)を徐々に加えていった後、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)-5-ヒドロキシウンデシル 1-メチルピペリジン-4-カルボオキシレート 5.01 g(7.61 mmol)を無色オイルとして得た。収率は76%であった。
11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)-5-ヒドロキシウンデシル 1-メチルピペリジン-4-カルボオキシレート 5.01 g(7.61 mmol)に酢酸1.43 mL(25 mmol)及び20 mLの1.0 M テトラブチルアンモニウムフルオリドのテトラヒドロフラン溶液を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、逆相シリカゲルクロマトグラフィー{溶離溶媒;水(0.1%トリフルオロ酢酸):アセトニトリル0.1%トリフルオロ酢酸)(連続勾配)}に供することにより精製して5,11-ジヒドロキシ5-(6-ヒドロキシヘキシル)ウンデシル 1-メチルピペリジン-4-カルボオキシレート 2.34 g(5.45 mmol)を薄黄色オイルとして得た。収率は72%であった。
5,11-ジヒドロキシ5-(6-ヒドロキシヘキシル)ウンデシル 1-メチルピペリジン-4-カルボオキシレート 430 mg(1.00 mmol)を10 mLのジクロロメタンに溶解した。続いて、オレイン酸 706 mg(2.50 mmol)、DMAP 24.4 mg(0.20 mmol)及びEDCI 671 mg(3.5 mmol)を加え、室温で2時間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-ヒドロキシ7-(4-((1-メチルピペリジン-4-カルボニル)オキシ)ブチル)トリデカン-1,13-ジイル ジオレアート(CL15H6)569 mg(0.594 mmol)を薄黄色オイルとして得た。収率は59%であった。

5,11-ジヒドロキシ5-(6-ヒドロキシヘキシル)ウンデシル 1-メチルピペリジン-4-カルボオキシレート 85.9 mg(0.20 mmol)を1.5 mLのジクロロメタンに溶解し、続いて、lauroyl chloride 143 mg(0.60 mmol)を加えた後、4℃に冷却した。TEA 139 μL(1.00 mmol)を滴下し、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-ヒドロキシ7-(4-((1-メチルピペリジン-4-カルボニル)オキシ)ブチル)トリデカン-1,13-ジイル ジドデカノエート(CL15B6)101. 2 mg(0.127 mmol)を薄黄色オイルとして得た。収率は64%であった。

5,11-ジヒドロキシ5-(6-ヒドロキシヘキシル)ウンデシル 1-メチルピペリジン-4-カルボオキシレート 85.9 mg(0.20 mmol)を1.5 mLのジクロロメタンに溶解し、続いて、ミリストイルクロリド 163 mg(0.60 mmol)を加えた後、4℃に冷却した。TEA 139 μL(1.00 mmol)を滴下し、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-ヒドロキシ7-(4-((1-メチルピペリジン-4-カルボニル)オキシ)ブチル)トリデカン-1,13-ジイル ジテトラデカノエート(C
L15C6)116 mg(0.136 mmol)を薄黄色固体として得た。収率は68%であった。

5,11-ジヒドロキシ5-(6-ヒドロキシヘキシル)ウンデシル 1-メチルピペリジン-4-カルボオキシレート 85.9 mg(0.20 mmol)を1.5 mLのジクロロメタンに溶解し、続いて、パルミトイルクロリド 181 mg(0.60 mmol)を加えた後、4℃に冷却した。TEA 139 μL(1.00 mmol)を滴下し、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-ヒドロキシ7-(4-((1-メチルピペリジン-4-カルボニル)オキシ)ブチル)トリデカン-1,13-ジイル ジパルミテート(CL15D6)114 mg(0.126 mmol)を薄黄色固体として得た。収率は63%であった。

5,11-ジヒドロキシ5-(6-ヒドロキシヘキシル)ウンデシル 1-メチルピペリジン-4-カルボオキシレート 85.9 mg(0.20 mmol)を1.0 mLのジクロロメタンに溶解し、続いて、stearoyl chloride 181 mg(0.80 mmol)を加えた後、4℃に冷却した。TEA 139 μL(1.00 mmol)を滴下し、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-ヒドロキシ7-(4-((1-メチルピペリジン-4-カルボニル)オキシ)ブチル)トリデカン-1,13-ジイル ジステアレート(CL15E6)141mg(0.146 mmol)を薄黄色固体として得た。収率は73%であった。

4℃に冷却したテトラヒドロフラン(THF)190 mLに、水素化リチウムアルミニウム2.73 g(72 mmol)を懸濁した。そこへリノール酸10 g(36 mmol)を滴下し、10分間撹拌した。その後、オイルバスで加熱しながら一晩還流した。これを冷却した後、1 mol/Lの水酸化ナトリウム水溶液100 mLを加えて反応を停止させた。次に、酢酸エチル100 mLを加えて希釈した後、濾過し、濾液を飽和炭酸水素ナトリウム水溶液を用いて洗浄した。続いて、有機層を回収して、そこに無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して、(9z,12z)- オクタジエン-1-オール 8.68 g(32.6 mmol)を無色オイルとして得た。収率は91%であった。
(9z,12z)- オクタジエン-1-オール 8.68 g(32.6 mmol)を100 mLのジクロロメタンに溶解した後、N,N-ジメチル-4-アミノピリジン(DMAP)366 mg(3.26 mmol)、トリエチルアミン(TEA)6.8 mL(48.9 mmol)を加えた。続いて、滴下ロートを用いて、50 mLのジクロロメタンで希釈したメタンスルホニルクロリド(MsCl)3.03 mL(39.1 mmol)を滴下し、室温で一晩撹拌した。反応液を回収して、飽和炭酸水素ナトリウム水溶液を用いて洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して、(9z, 12z)- オクタジエン-1-メタンスルホネート 10.64 g(30.9 mmol)を無色オイルとして得た。収率は95%であった。
(9z,12z)- オクタジエン-1-メタンスルホネート 10.64 gを140 mLのジエチルエーテルに溶解した後、臭化マグネシウムエチルエテレート 16.0 g(61.8 mmol)を加え、室温で一晩撹拌した。反応液を回収して、100 mLの飽和炭酸水素ナトリウム水溶液を用いて洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して、18-ブロモ-オクタデカ-(6z,9z)-ジエン 8.85 g(26.9 mmol)を無色オイルとして得た。収率は87%であった。
1.5 mLのジエチルエーテルに18-ブロモ-オクタデカ-(6z,9z)-ジエン 50 g(1.52 mmol)を溶解し、削り屑状マグネシウム609 mg(25.1 mmol)を加え、続いてヨウ素1欠片を加えた。室温で10分間静置した後、オイルバスで45℃に加熱しながら撹拌し、6 mLのジエチルエーテルに溶解した18-ブロモ-オクタデカ-(6z, 9z)-ジエン 5.0 g(15.2 mmol)を滴下した。45℃で1時間反応させた後、室温に冷却した。続いて、δ-バレロラクトン300 μL(3.23 mmol)を添加し、室温で1時間反応させた。次に、4℃に冷却し、濾過した後、濾液を飽和炭酸水素ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して、4-[(9z, 12z)-オクタジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 1.64 g(2.73 mmol)を無色オイルとして得た。δ-バレロラクトンからの収率は85%であった。
4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 301 mg(0.50 mmol)を5.0 mLのジクロロメタンに溶解し、DMAP 6.11 mg(0.05 mmol)とTEA 83.6 μL(0.60 mmol)を加え、続いてp-トルエンスルホニルクロリド(pTsCl) 95.3 mg(0.50 mmol)を加えた後、室温で一晩撹拌した。続いて、反応液にシリカゲルを加え、ロータリーエバポレーターを用いて溶媒を留去した。その後、シリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して293 mg(0.39 mmol)を無色オイルとして得た。収率は78%であった。
4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 293 mg(0.39 mmol)に10 mLの 2.0 MジメチルアミンのTHF溶液を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、100 mLのジクロロメタンを加え、100 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 155 mg(0.25 mmol)を薄黄色オイルとして得た。収率は64%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 650 mg(0.86 mmol)を4 mLのジクロロメタンに溶解し、エチルメチルアミン0.86 mL(10 mmol)を加え、40℃で3日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 432 mg(0.673 mmol)を薄黄色オイルとして得た。収率は78%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 603 mg(0.80 mmol)を4 mLのジクロロメタンに溶解し、ジエチルアミン1.04 mL(10 mmol)を加え、40℃で3日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 401 mg(0.611 mmol)を薄黄色オイルとして得た。収率は77%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 189 mg(0.25 mmol)を1.5 mLの1,2-ジクロロエタンに溶解し、ジプロピルアミン41 μL(0.3 mmol)を加え、室温で8日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 81 mg(0.118 mmol)を薄黄色オイルとして得た。収率は47%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 189 mg(0.25 mmol)を1.5 mLの1,2-ジクロロエタンに溶解し、N-ベンジルメチルアミン39 μL(0.3 mmol)を加え、室温で8日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 97.4 mg(0.138 mmol)を薄黄色オイルとして得た。収率は55%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 189 mg(0.25 mmol)を1.5 mLの1,2-ジクロロエタンに溶解し、ピペリジン30 μL(0.3 mmol)を加え、室温で8日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 85.0 mg(0.127 mmol)を薄黄色オイルとして得た。収率は51%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 227 mg(0.30 mmol)を2 mLの1,2-ジクロロエタンに溶解し、モルフォリン87.1 mg(1.0 mmol)を加え、室温で7日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLのジクロロメタンを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 60.0 mg(0.09 mmol)を薄黄色オイルとして得た。収率は30%であった。
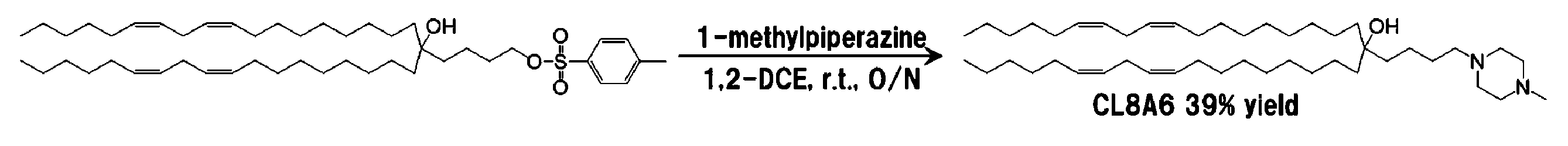
4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 227 mg(0.30 mmol)を2 mLの1,2-ジクロロエタンに溶解し、1-メチルピペラジン100.2 mg(1.0 mmol)を加え、室温で7日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLのジクロロメタンを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 79.0 mg(0.116 mmol)を薄黄色オイルとして得た。収率は39%であった。

4-[(9z, 12z)-オクタジエニル]-1-p-トルエンスルホニル-(13z, 16z)-トリコサジエン-4-オール 189 mg(0.25 mmol)を2 mLの1,2-ジクロロエタンに溶解し、1-イソプロピルピペラジン42.7 μL(0.3 mmol)を加え、室温で8日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することで精製して 116 mg(0.163 mmol)を薄黄色オイルとして得た。収率は65%であった。
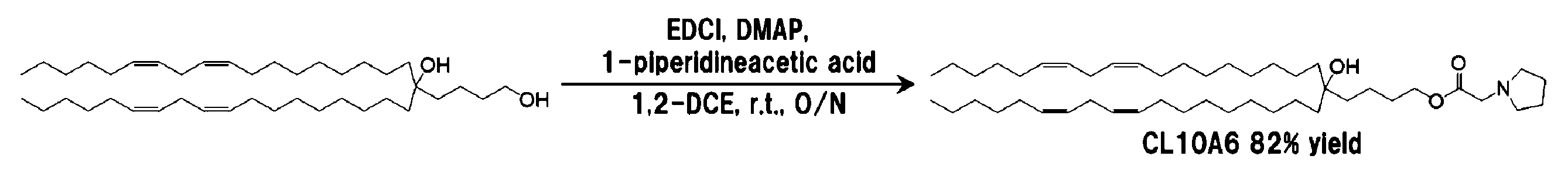
4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 120.2 mg(0.20 mmol)を1.0 mLの1,2-ジクロロエタンに溶解し、1-ピロリジン酢酸38.7 mg(0.30 mmol)を加え、続いてDMAP 6.1 mg(0.05 mmol)及び1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド塩酸塩(EDCI)57.5 mg(0.30 mmol)を加えた後、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して117 mg(0.164 mmol)を薄黄色オイルとして得た。収率は82%であった。
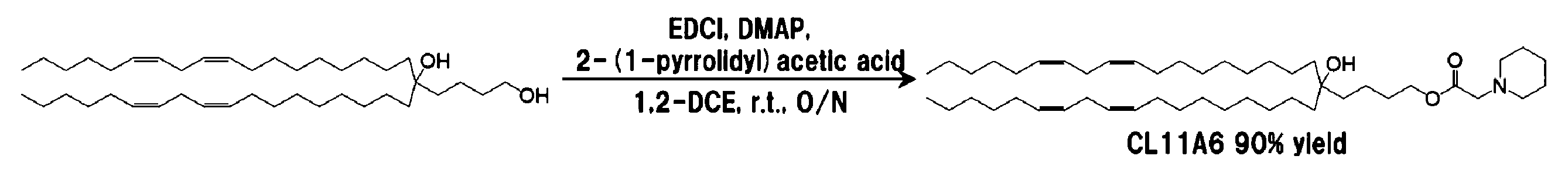
4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 120.2 mg(0.20 mmol)を1.0 mLの1,2-ジクロロエタンに溶解し、1-ピペリジン酢酸43.0 mg(0.30 mmol)を加え、続いてDMAP 6.1 mg(0.05 mmol)及びEDCI 57.5 mg(0.30 mmol)を加えた後、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して130 mg(0.179 mmol)を薄黄色オイルとして得た。収率は90%であった。

4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 601 mg(1.0 mmol)を5.0 mLのジクロロメタンに溶解し、3-(ジメチルアミノ)プロピオン酸塩酸塩153.6 mg(1.0 mmol)を加え、続いてDMAP 12.2 mg(0.1 mmol)及びEDCI 230 mg(1.2 mmol)を加えた後、室温で一晩撹拌した。50 mLのジクロロメタンを加え、50 mLの1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して351 mg(0.501 mmol)を薄黄色オイルとして得た。収率は50%であった。

4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 180 mg(0.30 mmol)を2.0 mLの1,2-ジクロロエタンに溶解し、3-(ジエチルアミノ)プロピオン酸塩酸塩72.7 mg(0.40 mmol)を加え、続いてDMAP 6.0 mg(0.05 mmol)及びEDCI 96 mg(0.50 mmol)を加えた後、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLのジクロロメタンを加え、5 mLの1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して151 mg(0.207 mmol)を薄黄色オイルとして得た。収率は69%であった。

4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 120.2 mg(0.20 mmol)を1.0 mLの1,2-ジクロロエタンに溶解し、1-ピペリジンプロピオン酸47.2 mg(0.30 mmol)を加え、続いてDMAP 6.1 mg(0.05 mmol)及びEDCI 57.5 mg(0.30 mmol)を加えた後、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去した後、5 mLの酢酸エチルを加え、5 mLの0.5 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して80.4 mg(0.109 mmol)を薄黄色オイルとして得た。収率は55%であった。

4-[(9z, 12z)-オクタデカジエニル]-(13z, 16z)-トリコサジエン-1,4-ジオール 842 mg(1.40 mmol)を10 mLの1,2-ジクロロエタンに溶解し、1-メチル-4-ピペリジンカルボン酸200 mg(1.40 mmol)を加え、続いてDMAP 17.1 mg(0.14 mmol)及びEDCI 383 mg(2.0 mmol)を加えた後、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去した後、50 mLの酢酸エチルを加え、50 mLの1 M水酸化ナトリウム水溶液で洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して877 mg(1.21 mmol)を無色オイルとして得た。収率は86%であった。

11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)-5-ヒドロキシウンデシル 4-メチルベンゼンスルホネート 8.78 g(12.78 mmol)に50 mLの2.0 MジメチルアミンのTHF溶液を加え、室温で6日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、飽和炭酸水素ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して6.20 g(11.07 mmol)を無色オイルとして得た。収率は87%であった。
11-(4-(ジメチルアミノ)ブチル)-2,2,3,3,19,19,20,20-オクタメチル-4,18-ジオキサ-3,19-ジシラヘニコサン-11-オール 6.20 g(11.07 mmol)に酢酸1.90 mL(33.21 mmol)及び24.4 mLの1.0 M テトラブチルアンモニウムフルオリドのテトラヒドロフラン溶液を加え、室温で2時間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、逆相シリカゲルクロマトグラフィー{溶離溶媒;水(0.1%トリフルオロ酢酸):アセトニトリル0.1%トリフルオロ酢酸)(連続勾配)}に供することにより精製して2.86 g(8.63 mmol)を薄黄色オイルとして得た。収率は80%であった。
7-(4-(ジメチルアミノ)ブチル)トリデカン-1,7,13-トリオール 431 mg(1.30 mmol)を5 mLのジクロロメタンに溶解し、ミリスチン酸 713 mg(3.12 mmol)及びDMAP 31.8 mg(0.26 mmol)を加え、続いてEDCI 748 mg(3.90 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-(4-(ジメチルアミノ)ブチル)-7-ヒドロキシトリデカン-1,13-ジイル ジテトラデカノエート(CL1C6)472 mg(0.627 mmol)を薄黄色オイルとして得た。収率は48%であった。

7-(4-(ジメチルアミノ)ブチル)トリデカン-1,7,13-トリオール 431 mg(1.30 mmol)を5 mLのジクロロメタンに溶解し、パルミチン酸 800 mg(3.12 mmol)及びDMAP 31.8 mg(0.26 mmol)を加え、続いてEDCI 748 mg(3.90 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-(4-(ジメチルアミノ)ブチル)-7-ヒドロキシトリデカン-1,13-ジイル ジパルミテート(CL1D6)557 mg(0.689 mmol)を薄黄色オイルとして得た。収率は53%であった。

7-(4-(ジメチルアミノ)ブチル)トリデカン-1,7,13-トリオール 1.99 g(6.0 mmol)を20 mLのジクロロメタンに溶解し、オレイン酸 4.07 g(14.4 mmol)及びDMAP 147 mg(1.20 mmol)を加え、続いてEDCI 3.45 g(18.0 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して7-(4-(ジメチルアミノ)ブチル)-7-ヒドロキシトリデカン-1,13-ジイル ジオレエート(CL1H6)2.56 g(2.98 mmol)を薄黄色オイルとして得た。収率は50%であった。

11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)ウンデカン-1,5-ジオール 12.73 g(23.9 mmol)を50 mLのジクロロメタンで溶解し、3-(ジメチルアミノ)プロパン酸塩酸塩 4.04 g(26.3 mmol)及びDMAP 293 mg(2.4 mmol)を加え、続けてEDCI 5.50 g(28.7 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、0.5 M 水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して6.66 g(10.54 mmol)を薄黄色オイルとして得た。収率は44%であった。
11-((tert-ブチルジメチルシリル)オキシ)-5-(6-((tert-ブチルジメチルシリル)オキシ)ヘキシル)-5-ヒドロキシウンデシル 3-(ジメチルアミノ)プロパノエート 6.66 g(10.54 mmol)に酢酸1.82 mL(31.6 mmol)及び21.1 mLの1.0 M テトラブチルアンモニウムフルオリドのTHF溶液を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、逆相シリカゲルクロマトグラフィー{溶離溶媒;水(0.1%トリフルオロ酢酸):アセトニトリル0.1%トリフルオロ酢酸)(連続勾配)}に供することにより精製して2.40 g(5.95 mmol)を薄黄色オイルとして得た。収率は57%であった。
5,11-ジヒドロキシ-5-(6-ヒドロキシヘキシル)ウンデシル 3-(ジメチルアミノ)プロパノエート 800 mg(2.0 mmol)を5 mLのジクロロメタンに溶解し、ミリスチン酸 1.005 g(4.4 mmol)及びDMAP 48.9 mg(0.40 mmol)を加え、続いてEDCI 959 mg(5.0 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。濾液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して737 mg(0.894 mmol)を白色固体として得た。収率は45%であった。

5,11-ジヒドロキシ-5-(6-ヒドロキシヘキシル)ウンデシル 3-(ジメチルアミノ)プロパノエート 800 mg(2.0 mmol)を5 mLのジクロロメタンに溶解し、パルミチン酸 1.128 g(4.4 mmol)及びDMAP 48.9 mg(0.40 mmol)を加え、続いてEDCI 959 mg(5.0 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。濾液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して690 mg(0.784 mmol)を白色固体として得た。収率は39%であった。

5,11-ジヒドロキシ-5-(6-ヒドロキシヘキシル)ウンデシル 3-(ジメチルアミノ)プロパノエート 800 mg(2.0 mmol)を5 mLのジクロロメタンに溶解し、オレイン酸 1.243 g(4.4 mmol)及びDMAP 48.9 mg(0.40 mmol)を加え、続いてEDCI 959 mg(5.0 mmol)を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。濾液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して874 mg(0.937 mmol)を無色オイルとして得た。収率は47%であった。

8-ブロモオクタン-1-オール 17.78 g(85.0 mmol)を100 mLの1,2-ジクロロエタンに溶解し、4℃に冷却した。TBSCl 13.86 g(92.0 mmol)を加えたのち、TEA 15.33 mL(110 mmol)を滴下し、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去し、ヘキサン300 mLを加えて懸濁させ、セライト濾過によって不溶物を除去することで粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して、14.0 g(44.3 mmol)を無色オイルとして得た。収率は51%であった。
4 mLのジエチルエーテルに((8-ブロモオクチル)オキシ)(tert-ブチル)ジメチルシラン 0.70 g(2.17 mmol)を溶解し、削り屑状マグネシウム1.26 g(52 mmol)を加え、続いてヨウ素1欠片加えた。室温で10分静置した後、オイルバスで40℃に加熱しながら撹拌し、11 mLのジエチルエーテルに溶解した((8-ブロモオクチル)オキシ)(tert-ブチル)ジメチルシラン 13.3 g(41.13 mmol)を滴下した。40℃で2時間反応させた後、4℃に冷却した。続いて、δ-バレロラクトン1.81 mL(19.5 mmol)を添加し、室温で一晩反応させた。次に、4℃に冷却し、5%硫酸を滴下することで残留したマグネシウムを溶解させた。ジエチルエーテルで希釈し、有機層を水及び飽和食塩水で分液洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して8.00 g(13.58 mmol)を無色オイルとして得た。δ-バレロラクトンからの収率は70%であった。
13-((tert-ブチルジメチルシリル)オキシ)-5-(8-((tert-ブチルジメチルシリル)オキシ)オクチル)トリデカン-1,5-ジオール 8.00 g(13.58 mmol)を30 mLの1,2-ジクロロエタンに溶解し、DMAP 183 mg(1.50 mmol)とTEA 2.79 mL(20.0 mmol)を加え、4℃に冷却した。続いて、pTsCl 2.86 g(15.0 mmol)を徐々に加えていった後、室温で2時間反応させた。ロータリーエバポレーターを用いて溶媒を留去し、酢酸エチルで懸濁し、水及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して無色オイルとして得た。
13-((tert-ブチルジメチルシリル)オキシ)-5-(8-((tert-ブチルジメチルシリル)オキシ)オクチル)-5-ヒドロキシトリデシル 4-メチルベンゼンスルホネート 10.09 g(13.58 mmol)に30 mLの1,2-ジクロロエタンを加え、4℃に冷却した。続いて、ジプロピルアミン 3.71 mL(27.2 mmol)を加えた後、室温で10日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、0.2 N 水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して5.86 g(8.72 mmol)を薄黄色オイルとして得た。収率は64%であった。
13-(4-(ジイソプロピルアミノ)ブチル)-2,2,3,3,23,23,24,24-オクタメチル-4,22-ジオキサ-3,23-ジシラペンタコサン-13-オール 4.50 g(6.70 mmol)に酢酸1.72 mL(30 mmol)及び20 mLの1.0 M テトラブチルアンモニウムフルオリドのTHF溶液を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、逆相シリカゲルクロマトグラフィー{溶離溶媒;水(0.1%トリフルオロ酢酸):アセトニトリル0.1%トリフルオロ酢酸)(連続勾配)}に供することにより精製して2.03 g(4.57 mmol)を薄黄色オイルとして得た。収率は68%であった。
9-(4-(ジイソプロピルアミノ)ブチル)ヘプタデカン-1,9,17-トリオール 222 mg(0.50 mmol)を2.5 mLの1,2-ジクロロエタンに溶解し、4℃に冷却した。続いて、オレイルクロリド451 mg(1.50 mmol)を加えた後、TEA 836 μL(6.0 mmol)を滴下し、室温で3時間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.2 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して68.4 mg(0.070 mmol)を薄黄色オイルとして得た。収率は14%であった。

10-ブロモデカン-1-オール 25.0 g(105.4 mmol)を100 mLの1,2-ジクロロエタンに溶解し、4℃に冷却した。TBSCl 17.3 g(115 mmol)を加えたのち、TEA 19.5 mL(140 mmol)を滴下し、室温で一晩撹拌した。ロータリーエバポレーターを用いて溶媒を留去し、ヘキサン300 mLを加えて懸濁させ、セライト濾過によって不溶物を除去することで粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して21.0 g(59.8 mmol)を無色オイルとして得た。収率は57%であった。
4 mLのジエチルエーテルに((10-ブロモデシル)オキシ)(tert-ブチル)ジメチルシラン 1.05 g(2.99 mmol)を溶解し、削り屑状マグネシウム1.75 g(72.0 mmol)を加え、続いてヨウ素1欠片加えた。室温で10分静置した後、オイルバスで40℃に加熱しながら撹拌し、11 mLのジエチルエーテルに溶解した((10-ブロモデシル)オキシ)(tert-ブチル)ジメチルシラン 19.95 g(56.81 mmol)を滴下した。40℃で2時間反応させた後、4℃に冷却した。続いて、δ-バレロラクトン3.67 mL(39.6 mmol)を添加し、室温で一晩反応させた。次に、4℃に冷却し、5%硫酸を滴下することで残留したマグネシウムを溶解させた。ジエチルエーテルで希釈し、有機層を水及び飽和食塩水で分液洗浄した。続いて、有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して11.95 g(18.52 mmol)を無色オイルとして得た。δ-バレロラクトンからの収率は69%であった。
15-((tert-ブチルジメチルシリル)オキシ)-5-(10-((tert-ブチルジメチルシリル)オキシ)デシル)ペンタデカン-1,5-ジオール 6.00 g(9.30 mmol)を30 mLの1,2-ジクロロエタンに溶解し、DMAP 114 mg(0.93 mmol)とTEA 3.24 mL(23.25 mmol)を加え、4℃に冷却した。続いて、pTsCl 2.13 g(11.16 mmol)を徐々に加えていった後、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去し、酢酸エチルで懸濁し、水及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ヘキサン:酢酸エチル(連続勾配)}に供することにより精製して無色オイルとして得た。
15-((tert-ブチルジメチルシリル)オキシ)-5-(10-((tert-ブチルジメチルシリル)オキシ)デシル)-5-ヒドロキシペンタデシル 4-メチルベンゼンスルホネート 1.66 g(2.08 mmol)に5 mLのTHFを加え、4℃に冷却した。続いて、ジプロピルアミン 569 μL(4.16 mmol)を加えた後、室温で21日間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、1 M水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して1.80 g(2.47 mmol)を薄黄色オイルとして得た。収率は27%であった。
15-(4-(ジイソプロピルアミノ)ブチル)-2,2,3,3,27,27,28,28-オクタメチル-4,26-ジオキサ-3,27-ジシラノナコサン-15-オール 1.80 g(2.47 mmol)に酢酸515 μL(9.0 mmol)及び6 mLの1.0 M テトラブチルアンモニウムフルオリドのTHF溶液を加え、室温で一晩反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、逆相シリカゲルクロマトグラフィー{溶離溶媒;水(0.1%トリフルオロ酢酸):アセトニトリル0.1%トリフルオロ酢酸)(連続勾配)}に供することにより精製して1.01 g(2.02 mmol)を薄黄色オイルとして得た。収率は82%であった。
11-(4-(ジイソプロピルアミノ)ブチル)ヘニコサン-1,11,21-トリオール 250 mg(0.50 mmol)を4 mLのジクロロメタンに溶解し、4℃に冷却した。続いて、オレイルクロリド602 mg(2.0 mmol)を加えた後、DMAP 12.2 mg(0.10 mmol)及びピリジン 322 μL(4.0 mmol)を滴下し、室温で3時間反応させた。ロータリーエバポレーターを用いて溶媒を留去した後、酢酸エチルで懸濁し、濾過によって不溶物を除去した。ろ液を0.5 N水酸化ナトリウム水溶液及び飽和食塩水で分液洗浄した。有機層に無水硫酸ナトリウムを加えて脱水した。これを濾過した後、ロータリーエバポレーターを用いて溶媒を留去し、粗生成物を得た。粗生成物をシリカゲルクロマトグラフィー{溶離溶媒;ジクロロメタン:メタノール(連続勾配)}に供することにより精製して42.5 mg(0.041 mmol)を薄黄色オイルとして得た。収率は8.3%であった。
疎水性足場がリノール酸由来であり、様々な親水性部位を有するpH感受性カチオン性脂質の評価を行った。LNPは特許文献6の実施例(例2)の方法に準じて、各pH感受性カチオン性脂質、cholesterol、methoxy polyethyleneglycol 2000 dimirystoylglycerol(PEG-DMG 2000)をモル比50:50:0.75-1.5とし、アルコール希釈法(図1)によって調製した。相的光散乱法によって算出される平均粒子径は80-120 nmであり、siRNA搭載率は90%以上であった。
親水性部位をCL4及びCL15の2種に固定し、疎水性足場の化学構造を変化させた際の影響を例2と同様にして評価した。TNSを用いてpKaを測定したところ、CL4では6.25-6.40、CL15では6.80-7.25と疎水性足場構造変化による影響は認められなかった(図4A及び4B)。In vitroノックダウン活性に関しては、CL15Bを除く他の誘導体で、従来の疎水性足場を有するCL15Aよりも優れていた(図5)。特にオレイン酸を疎水性足場に持つCL15HではCL15Aと比べて約3倍程度の高い活性を示した。In vivo F7ノックダウン活性に関しては、CL4、CL15共に疎水性足場C、Dの活性は低い一方で、疎水性足場Hが疎水性足場Aと同程度以上の活性を示した(図6A及び6B)。
CL4H-LNPのin vivoにおける安全性を検討した。ICRマウス(4週齢、雌性)に7 mg siRNA/kgで静脈内投与し、24時間後における血漿中alanin transaminase(ALT)及びaspartatetransaminase(AST)値及び投与前後の体重変化の測定を行った。比較対象として、開発者が以前に開発したpH感受性カチオン性脂質YSK13-C3を用い、脂質組成はYSK13-C3-LNPで最適化されたpH感受性脂質:cholesterol:PEG-DMG 2000 = 70:30:3(モル比)とした。YSK13-C3-LNPはALT及びAST共に10,000を超える強い肝毒性を示した一方、CL4H-LNPはPBS投与群と同レベルの軽微な肝毒性に留まった(図9A)。また、体重変化についてはYSK13-C3-LNP投与群では体重低下が認められた一方で、CL4H-LNPでは体重増加に転じた(図9B)。以上の結果から、CL4Hは安全性に優れる脂質化合物であることが示された。
親水性部位をCL4に固定した3種の脂質化合物(CL4H6、CL4H8、及びCL4H10)を用いて、前記疎水性足場1の化学構造を変化させた際の影響を例2と同様にして評価した。TNSを用いてpKaを測定したところ、CL4H6では6.35、CL4H8では6.10、CL4H10では5.85であり、疎水性足場1の炭素鎖の長さが長いほどpKaは低下した (図11)。In vivo F7ノックダウン活性は、ICRマウスへF7に対するsiRNAを搭載したLNPを静脈内投与してから24時間後に、血漿中F7酵素活性を測定した。この結果、3種類の誘導体のいずれも活性を有していた。なかでもCL4H6が最も優れた活性を示した(図12)。
CL4H6と、非特許文献11等に記載されている脂質化合物YSK05及びYSK13-C3について、In vivo F7ノックダウン活性を測定し、投与から24時間後のsiRNAの残存量との関係を調べた。In vivo F7ノックダウン活性は、各脂質化合物を用いて例2と同様にしてF7に対するsiRNAを搭載したLNPを調製し、各LNPを、ICRマウス(4週齢、雌性)に0.01 mg siRNA/kgで静脈内投与し、投与後30分後と24時間後に、各マウスから肝臓を回収し、肝臓中のsiRNAをqRT-PCR法により定量した。投与後30分後の肝臓におけるsiRNAは、いずれの脂質化合物を用いたLNPでも同程度であり、肝臓移行量はほぼ同等であることが確認された(図13A)。一方で、投与後24時間後の肝臓におけるsiRNA残存量には大きな差が認められた(図13B)。CL4H6-LNPを投与した肝臓のsiRNA残存量は、YSK05-LNPを投与した肝臓の17.3倍、YSK13-C3-LNPを投与した肝臓の4.8倍であった。また、各LNPを投与したマウスのF7ノックダウンにおけるED50を求め、この値と投与後24時間後の肝臓におけるsiRNA残存量は、反比例の関係にあった(図13C)。これらの結果は、CL4H6-LNPが効率的にエンドソームを脱出し、siRNAを細胞質へ送達していることを示唆していた。
CL4H6-LNPをmethoxy polyethyleneglycol 2000 distearoylglycerol(PEG-DSG 2000)でさらに修飾することによって血中の滞留時間を長期化し、さらにがん細胞におけるノックダウン活性を評価した。
具体的には、まず、例2と同様にしてCL4H6-LNP を用いてPLK1に対するsiRNAを搭載したLNPを調製した。得られたCL4H6-LNPを、pH6.0の10%EtOH水溶液にPEG-DSG 2000と共に分散させ、60℃で30分間インキュベートすることにより、CL4H6-LNP をPEG-DMG 2000でさらに修飾した。このPEG-DSG修飾CL4H6-LNPを、ICRマウス(4週齢、雌性)に0.5 mg siRNA/kgで静脈内投与し、血中PEG-DSG修飾CL4H6-LNP量を経時的に定量した。PEG-DSG修飾CL4H6-LNPのマウスへ投与した量(ID)を100%とした相対血中PEG-DSG修飾CL4H6-LNP量の経時的変化を図17に示す。
CD45に対するsiRNAを搭載したCL4H6を含むLNPを骨髄由来マクロファージに導入し、CD45遺伝子のノックダウン活性を測定した。
まず、脂質組成がカチオン性脂質(CL4H6)、cholesterol、及びPEG-DMG 2000をモル比60:40:2とし、さらにCD45に対するsiRNAを用いた以外は例2と同様にして、CD45に対するsiRNAを様々な濃度で搭載したCL4H6-LNPを調製した。
ICRマウス骨髄細胞から誘導したマクロファージの培養培地に、CD45に対するsiRNAを搭載したCL4H6-LNPを添加し、24時間培養した。比較対象として、当該マクロファージにリポフェクタミン試薬(Lipofectamine RNAiMAX、Thermo Fisher Scientific社製)を用いてCD45に対するsiRNAをトランスフェクションし、24時間培養した。各マクロファージの24時間培養後におけるCD45遺伝子の発現量を、qRT-PCR法で測定した。CD45に対するsiRNA未投与のマクロファージ(NT)におけるCD45の発現量を100%とした各マクロファージの相対CD45発現量(%)の結果を図19に示す。CD45に対するsiRNAをCL4H6-LNPに搭載してトランスフェクションしたマクロファージのほうが、リポフェクタミン試薬を用いてCD45に対するsiRNAをトランスフェクションしたマクロファージよりも10倍以上高い効率で遺伝子ノックダウンを誘導した。
CD45に対するsiRNAを搭載したLNPを、OSRC2細胞皮下移植マウスに投与し、CD45遺伝子のノックダウン活性を測定した。
まず、脂質組成がカチオン性脂質(CL4H6又はYSK05)、cholesterol、及びPEG-DSG 2000をモル比70:30:2とし、さらにCD45に対するsiRNAを用いた以外は例2と同様にして、CD45に対するsiRNAを搭載したLNPを調製した。
各LNPを、OSRC2細胞皮下移植マウスに2 mg siRNA/kg/doseで2日連続して静脈内投与した。最終投与から48時間後における腫瘍関連マクロファージのCD45遺伝子の発現量を、フローサイトメトリーで測定した。siRNA未投与の腫瘍関連マクロファージ(NT)におけるCD45発現量を100%とした場合の各腫瘍関連マクロファージの相対CD45発現量(%)の結果を図20に示す。CD45に対するsiRNAをCL4H6-LNPに搭載して投与したマウスのほうが、YSK05-LNPに搭載して投与したマウスよりも、腫瘍関連マクロファージにおけるCD45の相対発現量が低かった。すなわち、CL4H6は、腫瘍関連マクロファージにおいて優れた遺伝子ノックダウンを誘導した。
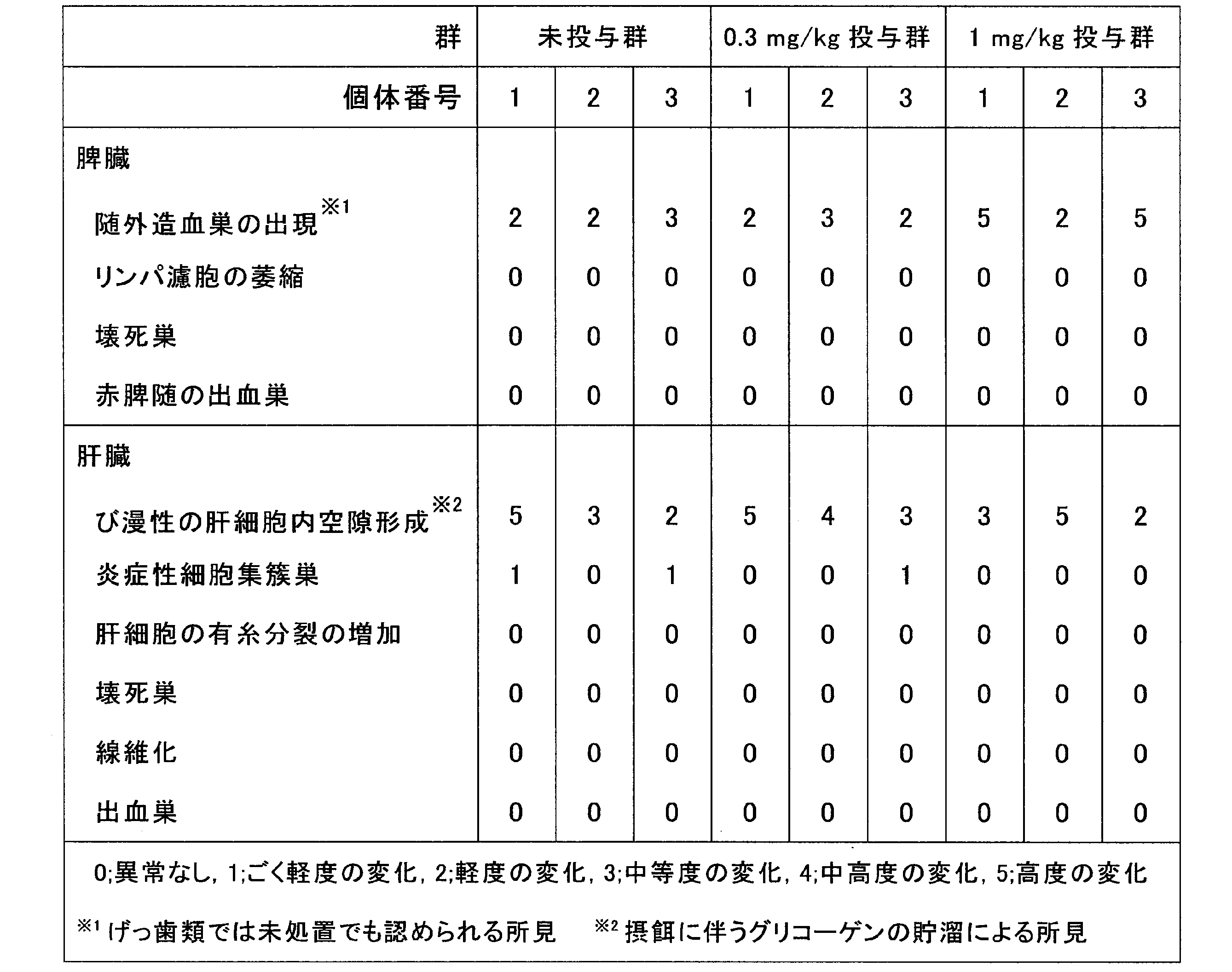
CL4H6を含むLNPをマウスに反復投与し、安全性を評価した。LNPに搭載するsiRNAとしては、マウスに対する薬理活性のないsiRNAを用いた。
まず、pH感受性カチオン性脂質としてCL4H6を用いて、例2と同様にしてヒトPLK1に対するsiRNAを搭載したLNP(CL4H6-LNP)を調製した。得られたCL4H6-LNPを、ICRマウス(4週齢、雌性)に、3又は4日ごとに、0.3 mg siRNA/kg 又は1 mg siRNA/kgで静脈内に反復投与した。具体的には、投与開始日(0日目)、投与開始日から4、7、11、14、18、21、及び23日目に、CL4H6-LNPを静脈内投与した。0.3 mg siRNA/kg は、F7に対するsiRNAを搭載したCL4H6-LNPのED50(0.0025 mg siRNA/kg)(例6参照)の120倍の投与量であり、1 mg siRNA/kg はその400倍の投与量である。


Claims (16)
- 下記の式(I):



で表される脂質化合物又はその塩。 - r及びtが0であり、q+s+uが8~18の整数である、請求項1に記載の脂質化合物又はその塩。
- r及びtが0であり、q+s+uが10~16の整数である、請求項2に記載の脂質化合物又はその塩。
- rが1であり、tが0であり、qが5~9の整数であり、s+uが5~9の整数である、請求項1に記載の脂質化合物又はその塩。
- vが5~12の整数である、請求項1ないし4のいずれか1項に記載の脂質化合物又はその塩。
- aが4であり、bが0又は1である、請求項1ないし5のいずれか1項に記載の脂質化合物又はその塩。
- bが0であり、Xが式(B)で表される基〔ただし、dは0であり、R3及びR4はそれぞれ独立にC1-4アルキル基(R3が示すC1-4アルキル基は1個のフェニル基で置換されていてもよい)を示すか、あるいはR3及びR4が互いに結合する場合には、1-ピロリジニル基、1-ピペリジニル基、1-モルホリニル基、又は1-ピペラジニル基(当該1-ピロリジニル基、1-ピペリジニル基、1-モルホリニル基、又は1-ピペラジニル基は1個のC1-4アルキル基で置換されていてよい)を形成する〕である、請求項1ないし6のいずれか1項に記載の脂質化合物又はその塩。
- bが1であり、Xが式(B)で表される基〔ただし、dは0~3の整数であり、R3及びR4はそれぞれ独立にC1-4アルキル基(R3が示すC1-4アルキル基は1個のフェニル基で置換されていてもよい)を示すか、あるいはR3及びR4が互いに結合する場合には、1-ピロリジニル基、1-ピペリジニル基、1-モルホリニル基、又は1-ピペラジニル基(当該1-ピロリジニル基、1-ピペリジニル基、1-モルホリニル基、又は1-ピペラジニル基は1又は2個の同一又は異なるC1-4アルキル基で置換されていてよい)を形成する〕である、請求項1ないし6のいずれか1項に記載の脂質化合物又はその塩。
- bが1であり、Xが5~7員非芳香族ヘテロ環基(当該基は炭素原子により(O-CO)b-に結合する)であり、当該5~7員非芳香族ヘテロ環基がピロリジニル基、ピペリジニル基、モルホリニル基、又はピペラジニル基(当該ピロリジニル基、ピペリジニル基、モルホリニル基、又はピペラジニル基は、1又は2個の同一又は異なるC1-4アルキル基で置換されていてよい)である、請求項1ないし6のいずれか1項に記載の脂質化合物又はその塩。
- 細胞内にsiRNAを送達するための脂質膜構造体の脂質成分として用いられる、請求項1ないし9のいずれか1項に記載の脂質化合物又はその塩。
- 脂質成分として請求項1ないし9のいずれか1項に記載の脂質化合物又はその塩を含む脂質膜構造体。
- リポソームである、請求項11に記載の脂質膜構造体。
- siRNAが内部に封入されている請求項11又は12に記載の脂質膜構造体。
- 細胞内の標的遺伝子をノックダウンするために用いられる、請求項13に記載の脂質膜構造体。
- 前記細胞が、免疫細胞又はがん細胞である、請求項14に記載の脂質膜構造体。
- 患者から樹状細胞を分離・採取し、in vitroで当該樹状細胞の細胞内にsiRNAを導入した後、標的遺伝子がノックダウンされた樹状細胞をその患者に投与する免疫療法において、樹状細胞における標的遺伝子をノックダウンするために用いられる、請求項15に記載の脂質膜構造体。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017117708 | 2017-06-15 | ||
| JP2017117708 | 2017-06-15 | ||
| PCT/JP2018/022940 WO2018230710A1 (ja) | 2017-06-15 | 2018-06-15 | siRNA細胞内送達のための脂質膜構造体 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2018230710A1 JPWO2018230710A1 (ja) | 2020-04-16 |
| JP7202009B2 true JP7202009B2 (ja) | 2023-01-11 |
Family
ID=64660932
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2019525575A Active JP7202009B2 (ja) | 2017-06-15 | 2018-06-15 | siRNA細胞内送達のための脂質膜構造体 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US11517528B2 (ja) |
| EP (1) | EP3640237B1 (ja) |
| JP (1) | JP7202009B2 (ja) |
| KR (1) | KR102676927B1 (ja) |
| CN (1) | CN110740985B (ja) |
| CA (1) | CA3067192A1 (ja) |
| WO (1) | WO2018230710A1 (ja) |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114127044A (zh) * | 2019-04-25 | 2022-03-01 | 英特利亚治疗股份有限公司 | 可电离的胺类脂质和脂质纳米颗粒 |
| JP7721128B2 (ja) * | 2019-05-30 | 2025-08-12 | 国立大学法人北海道大学 | 脂質ナノ粒子 |
| AU2021355199A1 (en) * | 2020-10-02 | 2023-05-25 | National University Corporation Hokkaido University | Lipid nanoparticle |
| WO2022260678A1 (en) * | 2021-06-11 | 2022-12-15 | Dnalite Therapeutics, Inc. | Compositions and methods for biological delivery vehicles |
| EP4259099A1 (en) | 2020-12-14 | 2023-10-18 | Particella, Inc. | Biological delivery systems |
| US20240050477A1 (en) * | 2021-02-17 | 2024-02-15 | National University Corporation Hokkaido University | Lipid nanoparticle |
| JP7761921B2 (ja) * | 2021-05-21 | 2025-10-29 | 国立大学法人北海道大学 | 脂質ナノ粒子 |
| CN113264842B (zh) | 2021-07-21 | 2022-03-01 | 苏州科锐迈德生物医药科技有限公司 | 一种脂质化合物及包含其的脂质载体、核酸脂质纳米粒组合物和药物制剂 |
| WO2023067561A1 (en) * | 2021-10-22 | 2023-04-27 | Seqirus Inc. | Ionizable cationic lipid for messenger rna delivery |
| JP7698851B2 (ja) * | 2021-12-16 | 2025-06-26 | Jsr株式会社 | 組成物の純化方法 |
| WO2023188830A1 (ja) | 2022-03-31 | 2023-10-05 | 国立大学法人北海道大学 | 脂質ナノ粒子 |
| JP2025081791A (ja) * | 2022-03-31 | 2025-05-28 | 日東電工株式会社 | がん送達用医薬組成物または免疫賦活用組成物 |
| KR102549868B1 (ko) | 2022-04-22 | 2023-06-30 | 주식회사 무진메디 | 재조합 프로타민을 이용한 지질 나노입자 기반 약물 전달체 및 이의 제조방법 |
| KR102475540B1 (ko) | 2022-04-26 | 2022-12-08 | 주식회사 무진메디 | 약물이 담지된 지질 나노입자 제조 방법 및 제조 장치 |
| US20250313860A1 (en) * | 2022-05-20 | 2025-10-09 | The University Of British Columbia | Expression of exogenous proteins in donor platelets treated with lipid nanoparticles |
| CN119585236A (zh) | 2022-07-19 | 2025-03-07 | 国立大学法人北海道大学 | 中性脂质及脂质纳米粒 |
| EP4559896A4 (en) | 2022-07-19 | 2025-10-22 | Univ Hokkaido Nat Univ Corp | PH-SENSITIVE CATIONIC LIPID AND LIPID NANOPARTICLE |
| JPWO2024024156A1 (ja) * | 2022-07-29 | 2024-02-01 | ||
| CN120813562A (zh) * | 2023-02-09 | 2025-10-17 | 斯奇络斯公司 | 可离子化阳离子化合物 |
| CN121013730A (zh) | 2023-03-15 | 2025-11-25 | 联合免疫股份有限公司 | 脂质颗粒 |
| JPWO2024248146A1 (ja) | 2023-05-31 | 2024-12-05 | ||
| KR102898792B1 (ko) | 2023-06-26 | 2025-12-19 | 주식회사 무진메디 | 이온화 가능한 펩타이드를 이용한 지질 나노입자 기반 약물 전달체 및 이의 제조방법 |
| TW202602853A (zh) * | 2024-03-21 | 2026-01-16 | 日商日東電工股份有限公司 | pH—敏感性陽離子脂質、包含其之脂質奈米顆粒及傳遞核酸之方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120172411A1 (en) | 2010-09-17 | 2012-07-05 | Protiva Biotherapeutics, Inc. | Novel trialkyl cationic lipids and methods of use thereof |
| JP2014500233A (ja) | 2010-09-20 | 2014-01-09 | メルク・シャープ・エンド・ドーム・コーポレイション | オリゴヌクレオチドの送達のための新規な低分子量カチオン性脂質 |
| WO2015178343A1 (ja) | 2014-05-20 | 2015-11-26 | 国立大学法人北海道大学 | siRNA細胞内送達のための脂質膜構造体 |
| JP2016084297A (ja) | 2014-10-24 | 2016-05-19 | 国立大学法人北海道大学 | 脂質膜構造体 |
| WO2016153012A1 (ja) | 2015-03-24 | 2016-09-29 | 協和発酵キリン株式会社 | 核酸含有脂質ナノ粒子 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01249717A (ja) | 1988-03-30 | 1989-10-05 | Fuji Photo Film Co Ltd | リポソームの製造方法 |
| JPH0720857B2 (ja) | 1988-08-11 | 1995-03-08 | テルモ株式会社 | リポソームおよびその製法 |
| JP3220180B2 (ja) | 1991-05-23 | 2001-10-22 | 三菱化学株式会社 | 薬剤含有タンパク質結合リポソーム |
| JP3415131B1 (ja) | 2002-06-03 | 2003-06-09 | メビオファーム株式会社 | リポソーム製剤 |
| JP4628955B2 (ja) | 2003-10-01 | 2011-02-09 | 独立行政法人科学技術振興機構 | 核移行能を有するポリアルギニン修飾リポソーム |
| JP4692983B2 (ja) | 2004-07-12 | 2011-06-01 | 独立行政法人科学技術振興機構 | リポソーム封入物質がエンドソームから脱出可能なリポソーム |
| EP1867726A4 (en) | 2005-03-24 | 2008-09-24 | Univ Hokkaido Nat Univ Corp | FOR THE EFFECTIVE DELIVERY OF A GIVEN SUBSTANCE IN THE CORE CORE CAPABLE LIPOSOME |
| WO2007102481A1 (ja) | 2006-03-07 | 2007-09-13 | National University Corporation Hokkaido University | 目的物質の核内送達用ベクター |
| JP5794541B2 (ja) | 2010-04-21 | 2015-10-14 | 国立大学法人北海道大学 | 核内移行性を有する脂質膜構造体 |
| JP6456813B2 (ja) | 2015-12-25 | 2019-01-23 | 日本特殊陶業株式会社 | スパークプラグの製造装置 |
-
2018
- 2018-06-15 KR KR1020197036750A patent/KR102676927B1/ko active Active
- 2018-06-15 CN CN201880038874.2A patent/CN110740985B/zh active Active
- 2018-06-15 CA CA3067192A patent/CA3067192A1/en active Pending
- 2018-06-15 EP EP18816591.4A patent/EP3640237B1/en active Active
- 2018-06-15 JP JP2019525575A patent/JP7202009B2/ja active Active
- 2018-06-15 WO PCT/JP2018/022940 patent/WO2018230710A1/ja not_active Ceased
- 2018-06-15 US US16/622,109 patent/US11517528B2/en active Active
-
2022
- 2022-11-17 US US17/989,260 patent/US20230132645A1/en active Pending
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20120172411A1 (en) | 2010-09-17 | 2012-07-05 | Protiva Biotherapeutics, Inc. | Novel trialkyl cationic lipids and methods of use thereof |
| JP2014500233A (ja) | 2010-09-20 | 2014-01-09 | メルク・シャープ・エンド・ドーム・コーポレイション | オリゴヌクレオチドの送達のための新規な低分子量カチオン性脂質 |
| WO2015178343A1 (ja) | 2014-05-20 | 2015-11-26 | 国立大学法人北海道大学 | siRNA細胞内送達のための脂質膜構造体 |
| JP2016084297A (ja) | 2014-10-24 | 2016-05-19 | 国立大学法人北海道大学 | 脂質膜構造体 |
| WO2016153012A1 (ja) | 2015-03-24 | 2016-09-29 | 協和発酵キリン株式会社 | 核酸含有脂質ナノ粒子 |
Non-Patent Citations (2)
| Title |
|---|
| SATO, Y. et al.,Molecular Therapy,2016年,Vol. 24, No. 4,pp. 788-795 |
| 中村 孝司、他,化学工業,化学工業社,2016年,7月号,第477~482頁 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110740985A (zh) | 2020-01-31 |
| EP3640237A1 (en) | 2020-04-22 |
| US11517528B2 (en) | 2022-12-06 |
| JPWO2018230710A1 (ja) | 2020-04-16 |
| KR102676927B1 (ko) | 2024-06-24 |
| EP3640237A4 (en) | 2021-06-09 |
| US20200129431A1 (en) | 2020-04-30 |
| KR20200018782A (ko) | 2020-02-20 |
| EP3640237B1 (en) | 2024-02-28 |
| US20230132645A1 (en) | 2023-05-04 |
| CA3067192A1 (en) | 2018-12-20 |
| EP3640237C0 (en) | 2024-02-28 |
| WO2018230710A1 (ja) | 2018-12-20 |
| CN110740985B (zh) | 2022-08-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7202009B2 (ja) | siRNA細胞内送達のための脂質膜構造体 | |
| JP6640750B2 (ja) | カチオン性脂質 | |
| EP4291242A1 (en) | Polyoxazoline-lipid conjugates and lipid nanoparticles and pharmaceutical compositions including same | |
| KR20230138496A (ko) | 핵산 전달에 적합한 지질 | |
| JP6570188B2 (ja) | siRNA細胞内送達のための脂質膜構造体 | |
| TW202337498A (zh) | 用於rna遞送之可離子化陽離子脂質 | |
| KR20230080451A (ko) | 지질 나노 입자 | |
| TW202329986A (zh) | 脂質化合物及脂質奈米顆粒組合物 | |
| JP2019151589A (ja) | 脂質ナノ粒子 | |
| TW201408324A (zh) | KRAS基因表現抑制RNAi醫藥組合物 | |
| JP6605477B2 (ja) | 核酸送達のためのカチオン性脂質 | |
| KR20260033596A (ko) | 이온화 가능한 지질 | |
| JP6774965B2 (ja) | カチオン性脂質としての化合物 | |
| WO2023191050A1 (ja) | がん送達用医薬組成物または免疫賦活用組成物 | |
| WO2018225873A1 (ja) | 核酸含有ナノ粒子 | |
| EP4563142A1 (en) | Lipid nanoparticle and pharmaceutical composition | |
| WO2026037391A1 (en) | Compositions and methods for Antigen-presenting cell-specific delivery of nucleic acids | |
| WO2018062233A1 (ja) | カチオン性脂質としての化合物 | |
| CN121909024A (zh) | 包封有核酸的配体修饰脂质纳米颗粒的制造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20191129 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20210526 |
|
| A871 | Explanation of circumstances concerning accelerated examination |
Free format text: JAPANESE INTERMEDIATE CODE: A871 Effective date: 20211209 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220301 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20220425 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20220621 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20220805 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221019 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20221019 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20221206 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20221216 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7202009 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |