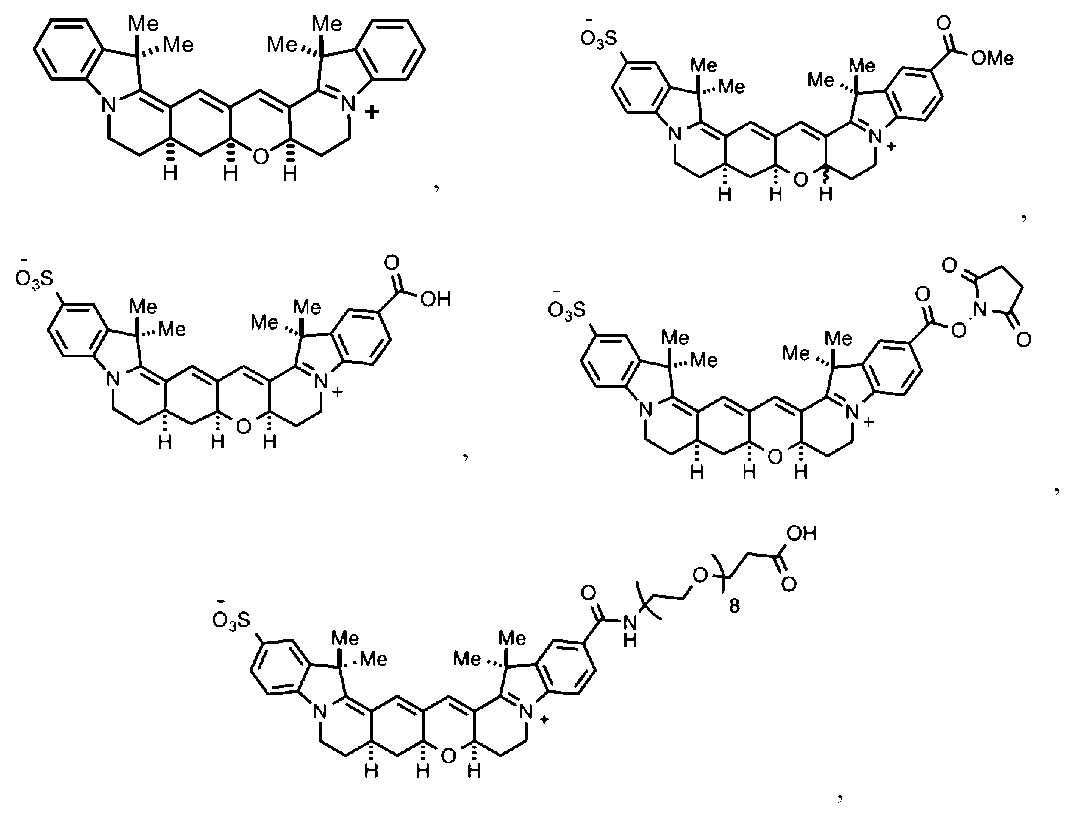
本開示は、配座が束縛されたシアニンフルオロフォア、ならびに配座が束縛されたシアニンフルオロフォアを作製および使用する方法の実施形態に関する。有利には、配座が束縛されたシアニンフルオロフォアは、遠赤および近赤外範囲の発光極大を有し、かつ/または配座が束縛されていない対応するシアニンフルオロフォアと比較して、赤方偏移している吸収および/もしくは発光極大を有する。
I.定義および略語
以下の用語および略語の説明は、本開示をより的確に説明し、本開示を実施する際に当業者を導くために提供される。本明細書で使用される場合、文脈によって別段明示されない限り、「含む(comprising)」は、「含む(including)」を意味し、単数形「a」または「an」または「the」は、複数の参照物を含む。用語「または」は、文脈によって別段明らかに示されない限り、記載された代替要素の単一要素、または2つもしくはそれよりも多い要素の組合せを指す。
別段説明されない限り、本明細書で使用されるすべての技術用語および科学用語は、本開示が属する分野の当業者に一般に理解される意味と同じ意味を有する。本明細書に記載されるものに類似のまたはそれと等価な方法および材料を、本開示の実施または試験において使用することができるが、適切な方法および材料を以下に記載する。材料、方法、および例は、単に例示的なものであり、限定することを意図しない。本開示の他の特色は、以下の詳細な説明および特許請求の範囲から明らかである。
別段指定されない限り、本明細書または特許請求の範囲で使用される場合、構成成分の量、分子量、パーセンテージ、温度、時間などを表すすべての数は、用語「約」によって修飾されると理解されるべきである。したがって、暗示的にであれ明確にであれ、別段指定されない限り、記載される数値パラメーターは近似値であり、その近似値は、求められる所望の特性および/または標準試験条件/方法の下での検出限界に応じて変わり得る。実施形態を論じられている従来技術と直接的かつ明確に区別する場合、これらの実施形態における数値は、用語「約」が記載されない限り近似ではない。
化学における一般用語の定義は、John Wiley & Sons,Inc.によって刊行されたRichard J.Lewis,Sr.(ed.),Hawley's Condensed Chemical Dictionary,1997(ISBN 0-471-29205-2)に見出すことができる。分子生物学における一般用語の定義は、Oxford University Pressによって刊行されたBenjamin Lewin,Genes VII,2000(ISBN 019879276X);Blackwell Publishersによって刊行されたKendrew et al.(eds.),The Encyclopedia of Molecular Biology,1994(ISBN 0632021829);およびWiley,John & Sons,Inc.によって刊行されたRobert A.Meyers(ed.),Molecular Biology and Biotechnology:a Comprehensive Desk Reference,1995(ISBN 0471186341)、ならびに他の類似の参考文献に見出すことができる。
アルキル:飽和炭素鎖を有する炭化水素基。その鎖は、分岐、非分岐、または環式(シクロアルキル)であってよい。低級アルキルという用語は、鎖が1~10個の炭素原子を含むことを意味する。別段特定されない限り、アルキルという用語は、置換および非置換アルキルを包含する。
アミノ:構造-N(R)R’(ここで、RおよびR’は、独立に、水素、ハロアルキル、脂肪族、ヘテロ脂肪族、アリール(例えば、必要に応じて置換されているフェニルもしくはベンジル)、ヘテロアリール、アルキルスルファノ、または他の官能基である)を有する基。「第一級アミノ」基は、-NH2である。「一置換アミノ」は、上記の通り置換されているラジカル-N(H)Rを意味し、それには、例えば、メチルアミノ、(1-メチルエチル)アミノ、フェニルアミノなどが含まれる。「二置換アミノ」は、上記の通り置換されているラジカル-N(R)R’を意味し、それには、例えば、ジメチルアミノ、メチルエチルアミノ、ジ(1-メチルエチル)アミノなどが含まれる。アミノという用語はまた、荷電三置換アミノ基、例えば、-N(R)(R’)R’’+(ここで、R、R’、およびR’’は、独立に、水素、ハロアルキル、脂肪族、ヘテロ脂肪族、アリール(例えば、必要に応じて置換されているフェニルもしくはベンジル)、ヘテロアリール、アルキルスルファノ、または他の官能基である)を包含する。
抗体:免疫グロブリン遺伝子または免疫グロブリン遺伝子の断片によって実質的にコードされた1つまたは複数のポリペプチドを含むタンパク質(またはタンパク質複合体)。認識されている免疫グロブリン遺伝子には、カッパ、ラムダ、アルファ、ガンマ、デルタ、イプシロンおよびミュー定常領域遺伝子、ならびに無数の免疫グロブリン可変領域遺伝子が含まれる。軽鎖は、カッパまたはラムダのいずれかに分類される。重鎖は、ガンマ、ミュー、アルファ、デルタ、またはイプシロンとして分類され、それによって、それぞれ免疫グロブリンクラスIgG、IgM、IgA、IgDおよびIgEが定義される。鳥類および爬虫類では、IgY抗体は、哺乳動物IgGと同等である。
免疫グロブリン(抗体)の基本構造単位は、一般に四量体である。各四量体は、ポリペプチド鎖の2つの同一の対から構成され、各対は、1つの「軽」鎖(約25kDa)および1つの「重」鎖(約50~70kDa)を有する。各鎖のN末端は、主に抗原認識に関与する約100~110個またはそれよりも多いアミノ酸の可変領域を画定する。用語「可変軽鎖」(VL)および「可変重鎖」(VH)は、それぞれ、これらの軽鎖および重鎖を指す。
IgY抗体の構造は、哺乳動物IgGの構造に類似しており、2つの重鎖(「ニュー」鎖;およそ67~70kDa)および2つの軽鎖(22~30kDa)を有する。IgY分子の分子量は、約180kDaであるが、しばしば、ゲル上では約3%炭水化物の存在に起因してスメアとして泳動する。IgY抗体の重鎖(H)は、4つの定常ドメインおよび抗原結合部位を含有する1つの可変ドメインから構成される。
本明細書で使用される場合、用語「抗体」は、インタクトな免疫グロブリンおよびいくつかの十分に特徴付けられた断片を含む。例えば、標的タンパク質(またはタンパク質もしくは融合タンパク質内のエピトープ)に結合するFab、Fv、および単鎖Fv(SCFv)も、そのタンパク質(またはエピトープ)のための特異的結合剤となる。これらの抗体断片は、以下の通り定義される。(1)Fab、抗体全体を酵素パパインで消化して、インタクトな軽鎖および1つの重鎖の一部を得ることによって生成された、抗体分子の一価の抗原結合性断片を含有する断片、(2)Fab’、抗体全体をペプシンで処理し、その後還元して、インタクトな軽鎖および重鎖の一部を生産することによって得られた抗体分子の断片(抗体分子1つ当たり2つのFab’断片が得られる)、(3)(Fab’)2、抗体全体を酵素ペプシンで処理し、その後の還元を行わないことによって得られた抗体の断片、(4)F(ab’)2、2つのジスルフィド結合によって一緒に保持された2つのFab’断片の二量体、(5)Fv、2つの鎖として発現された、軽鎖の可変領域および重鎖の可変領域を含有する遺伝子操作された断片、ならびに(6)単鎖抗体、遺伝的に融合した一本鎖分子として適したポリペプチドリンカーによって連結された、軽鎖の可変領域、重鎖の可変領域を含有する遺伝子操作された分子。これらの断片を作製する方法は、慣用的である(例えば、Harlow and Lane,Using Antibodies:A Laboratory Manual,CSHL,New York,1999を参照されたい)。本明細書で使用される場合、用語「抗体」は、部位特異的コンジュゲーションを容易にするための1つまたは複数の非天然(すなわち、天然に存在しない)アミノ酸(例えば、p-アセチル-フェニルアラニン)を含む抗体を含む。
本開示の方法において使用するための抗体は、モノクローナルであってもポリクローナルであってもよく、例えば、標的抗原などの標的に特異的に結合する。単なる例として、モノクローナル抗体は、Kohler and Milstein(Nature 256:495-97,1975)の古典的方法またはその誘導的方法に従って、マウスハイブリドーマから調製することができる。モノクローナル抗体生成についての詳細な手順は、Harlow and Lane,Using Antibodies:A Laboratory Manual,CSHL,New York,1999に記載されている。
抗原:動物内での抗体生成またはT細胞応答を刺激することができる化合物、組成物、または物質であり、動物内に注射または吸収される組成物を含む。抗原は、異種免疫原によって誘導されるものを含む、特異的液性免疫または細胞性免疫の生成物と反応する。本明細書で使用される場合、「標的抗原」は、標的化剤によって認識され、結合される抗原(抗原のエピトープを含む)である。「特異的結合」は、排他的結合を必要としない。一部の実施形態では、抗原は、細胞または組織抽出物から得られる。一部の実施形態では、標的抗原は、腫瘍細胞上の抗原である。抗原は、全長タンパク質である必要はない。使用が企図される抗原には、タンパク質の任意の免疫原性断片、例えば、抗体が特異的に結合することができる少なくとも1つのエピトープを有する任意の抗原が含まれる。
アリール:別段特定されない限り、単一の環(例えば、フェニル)または少なくとも1つの環が芳香族である複数の縮合した環(例えば、キノリン、インドール、ベンゾジオキソールなど)を有する、6~15個の炭素原子の一価の芳香族炭素環式基(ただし、結合点は、アリール基の芳香族部分の原子を介しており、結合点の芳香族部分は、その芳香環内に炭素だけを含有している)。任意の芳香環部分がヘテロ原子を含有している場合、その基は、ヘテロアリールであり、アリールではない。アリール基は、単環式、二環式、三環式または四環式である。別段特定されない限り、アリールという用語は、置換および非置換アリールを包含する。
生物学的試料:本明細書で使用される場合、「生物学的試料」は、対象(例えば、ヒトまたは動物対象)あるいは他の種類の生物、例えば植物、細菌または昆虫から得られた試料を指す。対象由来の生物学的試料には、それに限定されるものではないが、細胞、組織、血清、血液、血漿、尿、唾液、脳脊髄液(CSF)または他の体液が含まれる。本明細書に開示される方法の特定の例では、生物学的試料は、組織試料である。
配座が束縛された:用語「配座が束縛された」は、本明細書で使用される場合、中心のコンジュゲートされたポリメチン架橋領域の可撓性を分子が喪失するように修飾されているシアニン化合物を指す。用語「固定された(rigidized)」は、「配座が束縛された」と同義である。
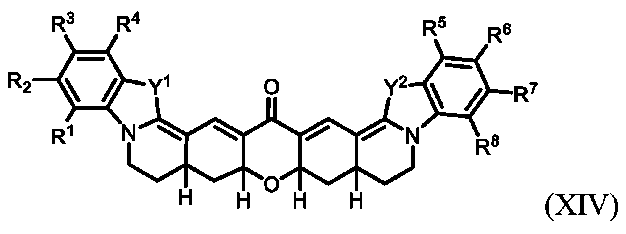
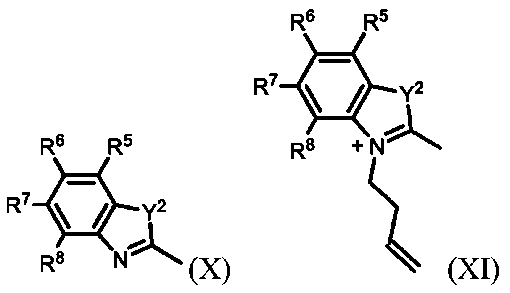
コンジュゲート可能な部分:分子を別の分子に、例えば薬物または標的化剤、例えば抗体にコンジュゲート(すなわち、カップリングまたは結合)することができる分子の一部。
薬物:本明細書で使用される場合、用語「薬物」は、対象に投与された場合に生理学的効果を有し、疾患の処置、緩和、治癒、防止もしくは診断における使用を意図されるか、または身体的もしくは精神的健全性をその他の方法で増強するために使用される物質を指す。用語「小分子薬物」は、分子量<1,000ダルトンを有する薬物を指す。
抗がん薬は、悪性腫瘍を処置するために使用される薬物である。例示的な抗がん薬として、それに限定されるものではないが、アビラテロン、アクチノマイシンD、アルトレタミン、アミホスチン、アナストロゾール、アスパラギナーゼ、ベキサロテン、ビカルタミド、ブレオマイシン、ブセレリン、ブスルファン、カルボプラチン、カルムスチン、クロラムブシル シスプラチン、クラドリビン、クロドロネート、コンブレタスタチンA4、シクロホスファミド、シプロテロン、シタラビン、ダカルバジン、ダウノルビシン、デガレリクス、ジエチルスチルベストロール、ドセタキセル、ドキソルビシン、ズオカルマイシンDM、エピルビシン、エチニルエストラジオール、エトポシド、エキセメスタン、5-フルオロウラシル、フルダラビン、フルタミド、フォリン酸、フルベストラント、ゲムシタビン、ゴセレリン、イバンドロン酸、イダルビシン、イフォスファミド、イリノテカン、ランレオチド、レナリドミド、レトロゾール、リュープロレリン、メドロキシプロゲステロン、メゲストロール、メルファラン、メスナ、メトトレキセート、オクトレオチド、パミドロネート、ペメトレキセド、マイトマイシン(mitocmycin)、ミトタン、ミトキサントロン、オキサリプラチン、パクリタキセル、ペントスタチン(pentastatin)、ピポブロマン(pipbroman)、プリカマイシン、プロカルバジン、ラルチトレキセド、スチルベストロール、ストレプトゾシン、タモキシフェン、テモゾロミド、テニポシド、トポテカン、トリプトレリン、ビンブラスチン、ビンクリスチン、ビノレルビン、およびゾレドロン(zolendronic)酸が挙げられる。
エピトープ:抗原決定基。エピトープは、抗原性である、すなわち特異的免疫応答を惹起する、分子上の特定の化学基、または連続的もしくは非連続的ペプチド配列である。抗体は、抗体の三次元構造およびマッチする(または同族の)エピトープに基づいて、特定の抗原性エピトープに結合する。
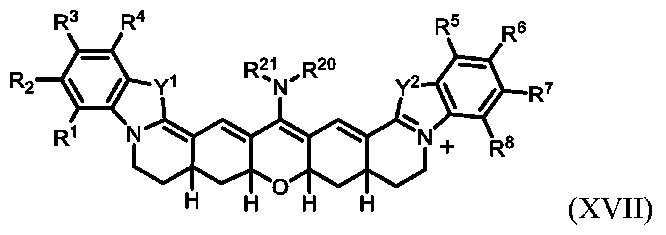
ヘテロ脂肪族:少なくとも1つのヘテロ原子を有する、すなわち1つまたは複数の炭素原子が少なくとも1つの孤立電子対を有する原子、典型的に窒素、酸素、リン、ケイ素、または硫黄で置き換えられている、脂肪族化合物または基。ヘテロ脂肪族化合物または基は、置換または非置換、分岐または非分岐、環式または非環式であってよく、それには、「複素環」、「ヘテロシクリル」、「ヘテロシクロ脂肪族」、または「複素環式」基が含まれる。
ヘテロアリール:少なくとも1つのヘテロ原子を有する、すなわち環中の1つまたは複数の炭素原子が少なくとも1つの孤立電子対を有する原子、典型的に窒素、酸素、リン、ケイ素、または硫黄で置き換えられている、芳香族化合物または基。別段特定されない限り、ヘテロアリールという用語は、置換および非置換ヘテロアリールを包含する。
リンカー:2つの部分の間に位置する分子または原子団。本明細書で使用される場合、用語「リンカー」は、シアニンフルオロフォアと標的化剤もしくは反応基との間に位置する原子団、またはシアニンフルオロフォアと薬物との間に位置する原子団を指す。
近赤外(近IR、NIR):650~2500nmの範囲内の波長。別段特定されない限り、用語「近赤外」および「NIR」は、本明細書で使用される場合、650~900nmの範囲内の波長を指す。
薬学的に許容される担体:本開示において有用な薬学的に許容される担体(ビヒクル)は、従来のものである。Remington:The Science and Practice of Pharmacy,The University of the Sciences in Philadelphia,Editor,Lippincott,Williams,& Wilkins,Philadelphia,PA,21st Edition(2005)は、本明細書に開示される1つまたは複数の配座が束縛されたシアニンフルオロフォアの薬学的送達に適した組成物および製剤を記載している。
一般に、担体の性質は、用いられる特定の投与形式に応じて変わる。例えば、非経口製剤は、通常、ビヒクルとして薬学的および生理的に許容される流体、例えば水、生理食塩水、平衡塩溶液、ブドウ糖水溶液、グリセロールなどを含む、注射可能な流体を含む。一部の例では、薬学的に許容される担体は、対象への投与(例えば、非経口、筋肉内、または皮下注射による)に適するように無菌にされ得る。生物学的に中性の担体に加えて、投与される医薬組成物は、少量の非毒性補助物質、例えば湿潤剤または乳化剤、防腐剤、およびpH緩衝剤など、例えば酢酸ナトリウムまたはソルビタンモノラウレートを含有することができる。
薬学的に許容される塩:開示される配座が束縛されたシアニンフルオロフォアの生物学的に適合可能な塩であり、その塩は、当技術分野で周知の様々な有機および無機対イオンから誘導され、その単なる例として、ナトリウム、カリウム、カルシウム、マグネシウム、アンモニウム、テトラアルキルアンモニウムなどが挙げられ、分子が塩基性官能基を含有する場合には、有機または無機酸の塩、例えば塩酸塩、臭化水素酸塩、酒石酸塩、メシル酸塩、酢酸塩、マレイン酸塩、シュウ酸塩などが挙げられる。薬学的に許容される酸付加塩は、遊離塩基の生物学的有効性を保持すると同時に、生物学的にも、または他の点でも望ましくないことがない酸パートナー、例えば無機酸、例えば塩酸、臭化水素酸、硫酸、硝酸、リン酸など、ならびに有機酸、例えば酢酸、トリフルオロ酢酸、プロピオン酸、グリコール酸、ピルビン酸、シュウ酸、マレイン酸、マロン酸、コハク酸、フマル酸、酒石酸、クエン酸、安息香酸、ケイ皮酸、マンデル酸、メタンスルホン酸、エタンスルホン酸、p-トルエンスルホン酸、サリチル酸などによって形成される塩である。薬学的に許容される塩基付加塩には、無機塩基から誘導された塩、例えばナトリウム、カリウム、リチウム、アンモニウム、カルシウム、マグネシウム、鉄、亜鉛、銅、マンガン、アルミニウムなどの塩が含まれる。例示的な塩は、アンモニウム、カリウム、ナトリウム、カルシウム、およびマグネシウム塩である。薬学的に許容される非毒性有機塩基から誘導された塩には、それに限定されるものではないが、第一級、第二級および第三級アミン、天然に存在する置換アミンを含む置換アミン、環状アミンおよび塩基性イオン交換樹脂、例えばイソプロピルアミン、トリメチルアミン、ジエチルアミン、トリエチルアミン、トリプロピルアミン、エタノールアミン、2-ジメチルアミノエタノール、2-ジエチルアミノエタノール、ジシクロヘキシルアミン、リシン、アルギニン、ヒスチジン、カフェイン、プロカイン、ヒドラバミン、コリン、ベタイン、エチレンジアミン、グルコサミン、メチルグルカミン、テオブロミン、プリン、ピペラジン、ピペリジン、N-エチルピペリジン、ポリアミン樹脂などの塩が含まれる。例示的な有機塩基は、イソプロピルアミン、ジエチルアミン、エタノールアミン、トリメチルアミン、ジシクロヘキシルアミン、コリン、およびカフェインである(例えば、参照によって本明細書に組み込まれるS.M.Berge,et al.,“Pharmaceutical Salts,”J.Pharm.Sci.,1977;66:1-19を参照されたい)。
保護基:有機化合物を合成する場合、しばしば特定の官能基は、必要とされる試薬または化学的環境を耐え抜くことはできない。これらの基は、保護されなければならない。保護基(protecting group)または保護用の基(protective group)は、その後の化学反応における化学選択性を得るために、官能基の化学修飾によって分子に導入される。様々な例示的な保護基または保護用の基は、参照によって本明細書に組み込まれるGreene's Protective Groups in Organic Synthesis,Peter G.M.Wuts and Theodora W.Greene(October 30,2006)に開示されている。
特異的結合パートナー:関与する分子の三次元構造に応じて決まる、特異的な非共有結合性の相互作用によって相互作用する一対の分子のメンバー。特異的結合パートナーの例示的な対には、抗原/抗体、ハプテン/抗体、受容体/リガンド、核酸鎖/相補的核酸鎖、基質/酵素、阻害剤/酵素、炭水化物/レクチン、ビオチン/アビジン(例えば、ビオチン/ストレプトアビジン)、およびウイルス/細胞受容体が含まれる。
置換基:反応の結果として分子内の別の分子を置き換える原子または原子団。用語「置換基」は、典型的に、親炭化水素鎖または親炭化水素環上の1つの水素原子、または置換基が二重結合によって結合する場合には2つの水素原子を置き換える原子または原子団を指す。用語「置換基」はまた、分子との複数の結合点を有する原子団を包含することができ、例えば、置換基は、親炭化水素鎖または親炭化水素環上の2つまたはそれよりも多い水素原子を置き換える。このような場合、置換基は、別段特定されない限り、親炭化水素鎖または親炭化水素環に対して任意の空間的配向で結合することができる。例示的な置換基として、例えば、アルキル、アルケニル、アルキニル、アルコキシ、アルキルアミノ、アルキルチオ、アシル、アルデヒド、アミド、アミノ、アミノアルキル、アリール、アリールアルキル、アリールアミノ、カーボネート、カルボキシル、シアノ、シクロアルキル、ジアルキルアミノ、ハロ、ハロ脂肪族(例えば、ハロアルキル)、ハロアルコキシ、ヘテロ脂肪族、ヘテロアリール、ヘテロシクロ脂肪族、ヒドロキシル、イソシアノ、イソチオシアノ、オキソ、スルホンアミド、スルフヒドリル、チオ、およびチオアルコキシ基が挙げられる。
置換されている:1つまたは複数の置換基がカップリングしている基本化合物、例えばアリールまたは脂肪族化合物またはそのラジカル(各置換基は、典型的に、基本化合物上の水素原子を置き換える)。単なる例として、それに限定されるものではないが、置換されているアリール化合物は、アリールベースの閉環にカップリングされた脂肪族基、例えばトルエンを有することができる。やはり単なる例として、それに限定されるものではないが、長鎖炭化水素は、それに結合したヒドロキシル基を有することができる。
標的:標的化剤を含む開示される配座が束縛されたシアニンフルオロフォアが特異的に結合できる、所期の分子。標的の例として、組織試料中に存在するタンパク質および核酸配列が含まれる。標的領域は、標的分子が位置するか、または潜在的に位置する可能性がある領域である。
標的化剤:標的部位、例えば対象の体内の標的化された位置、例えば特定の器官、小器官、生理学的系、組織、または病理部位、例えば腫瘍、感染領域もしくは組織傷害領域への、優先的なまたは標的化された送達を促進する薬剤。標的化剤は、様々な機序、例えば標的部位内での選択的濃縮によって、または特異的結合パートナーとの結合によって機能する。適切な標的化剤には、それに限定されるものではないが、タンパク質、ポリペプチド、ペプチド、糖タンパク質および他のグリコシル化分子、オリゴヌクレオチド、リン脂質、リポタンパク質、アルカロイド、ならびにステロイドが含まれる。例示的な標的化剤として、抗体、抗体断片、アフィボディ、アプタマー、アルブミン、サイトカイン、リンホカイン、増殖因子、成長因子、ホルモン、酵素、免疫モジュレーター、受容体タンパク質、アンチセンスオリゴヌクレオチド、アビジン、ナノ粒子などが挙げられる。標的化剤の中で特に有用なものは、抗体、核酸配列、および受容体リガンドであるが、特異的結合パートナーの任意の対を、この目的で容易に用いることができる。
処置する/処置:本明細書で使用される場合、用語「処置する」および「処置」は、状態、すなわち障害または疾患と関連する少なくとも1つの徴候または症候を阻害または低減することを意味する。腫瘍に関して、処置は、腫瘍成長の阻害および/または腫瘍体積の低減を意味することができる。処置は、例えば腫瘍の一部もしくはすべての臨床症候の重症度の低減、腫瘍の進行の緩徐(例えば、腫瘍を有する対象を延命することによって)、腫瘍再発回数の低減、対象の全体的健康もしくは福祉の改善、または特定の障害もしくは疾患に特異的な当技術分野で周知の他のパラメーターをもたらすことができる。
先の実施形態のいずれかまたはすべてでは、式I、IA~ID、II、またはIIIによる化合物は、コンジュゲート可能な部分、標的化剤または薬物を含む少なくとも1つの基を含むことができる。コンジュゲート可能な部分または標的化剤を含む例示的な基には、それに限定されるものではないが、-(CH2)nC(O)Re、-(CH2)nN(H)Re、-(CH2)nN(H)C(O)Re、-(CH2)nC(O)N(H)Re、-(CH2)nC(O)SRe、-C(O)Re、-C(O)N(H)Re、-C(O)N(H)(CH2CH2O)m(CH2)nC(O)Re -N(H)C(O)Re、-N(H)Re、または-SReが含まれ、ここで、mは、≧1の整数であり、nは、≧1の整数であり、Reは、コンジュゲート可能な部分または標的化剤である。
例示的な標的化剤には、それに限定されるものではないが、抗体、リガンド、ペプチド、核酸鎖などが含まれる。一部の例では、標的化剤は、F-アクチンに結合する二環式ヘプタペプチドである、ファロイジンである。ある特定の例では、標的化剤は、抗体である。例示的な抗体には、標的分子を認識し、結合することができる抗体、例えば疾患、感染、または環境曝露と関連するバイオマーカーが含まれる。バイオマーカーには、それに限定されるものではないが、タンパク質、ペプチド、脂質、代謝産物、および核酸が含まれる。一部の実施形態では、抗体は、腫瘍細胞の内部もしくはその表面にだけ見出されるタンパク質などの腫瘍バイオマーカー、または1つもしくは複数のがんと関連する細胞表面受容体を認識し、結合することができる。例えば、パニツムマブは、ヒト上皮増殖因子受容体1(HER1)を認識し、結合する、ヒトモノクローナル抗体であり、HER1は、数々の腫瘍型において過剰発現し、一部の炎症性疾患とも関連する。トラスツズマブおよびペルツズマブは、一部の乳がんに過剰発現するHER2/neu受容体に結合するモノクローナル抗体である。ブレンツキシマブは、古典的ホジキンリンパ腫および全身性退形成大細胞型リンパ腫に発現する細胞膜タンパク質CD30を標的にするモノクローナル抗体である。
開示される配座が束縛されたシアニンフルオロフォアの実施形態は、対応する制限されていないペンタメチンおよびヘプタメチンシアニンと比較して、量子収率の改善および/または蛍光寿命の延長を示す。一部の実施形態では、量子収率および/または蛍光寿命は、対応する制限されていないシアニンフルオロフォアの量子収率および/または蛍光寿命を、少なくとも1.1倍、少なくとも1.5倍、少なくとも2倍、少なくとも3倍、少なくとも4倍、または少なくとも5倍上回る。量子収率および/または蛍光寿命は、対応する制限されていないシアニンフルオロフォアの量子収率および/または蛍光寿命を1.1~10倍、例えば1.5~8倍、1.5~5倍、または2~5倍上回っていてもよい。有利には、吸収および発光の最大波長は、対応する制限されていないシアニンフルオロフォアと比較して赤方偏移することができる。ある特定の実施形態では、λmaxおよび/またはλemは、対応する制限されていないシアニンフルオロフォアのλmaxおよび/またはλem値と比較して、少なくとも10nm、少なくとも20nm、または少なくとも30nm、例えば10~50nm、10~40nm、10~30nm、または10~20nm、赤方偏移する。さらに、開示される配座が束縛されたシアニンフルオロフォアの一部の実施形態は、既存の遠赤および/または近IRシアニンと比較して優れた効率で、水素化物の還元から回収される。ある特定の実施形態では、これらの特性により、PALM様のSMLMが、多量のチオール(脱酸素化緩衝剤)に頼ることなく、優れた光子カウントを提供することが可能になる。
III.医薬組成物
本開示はまた、本明細書で開示されるような、少なくとも1つの配座が束縛されたシアニンフルオロフォアを含む医薬組成物を含む。医薬組成物の一部の実施形態は、薬学的に許容される担体および少なくとも1つの配座が束縛されたシアニンフルオロフォアを含む。有用な薬学的に許容される担体および賦形剤は、当技術分野で公知である。
1つまたは複数の配座が束縛されたシアニンフルオロフォアを含む医薬組成物は、例えば、投与形式および/または画像化される位置に応じて、様々な方式で製剤化することができる。非経口製剤は、薬学的かつ生理的に許容される流体ビヒクル、例えば水、生理食塩水、他の平衡塩溶液、ブドウ糖水溶液、グリセロールなどである注射可能な流体を含むことができる。賦形剤には、例えば非イオン性可溶化剤、例えばCremophor(登録商標)、またはタンパク質、例えばヒト血清アルブミンもしくは血漿調製物が含まれ得る。所望に応じて、投与される医薬組成物は、非毒性補助物質、例えば湿潤剤または乳化剤、防腐剤、およびpH緩衝剤など、例えば、酢酸ナトリウムまたはソルビタンモノラウレートを含有することもできる。
医薬組成物の形態は、選択される投与形式によって決定される。開示される医薬組成物の実施形態は、例えば、局所、眼、経口、口腔内頬側、全身、経鼻、注射、経皮、直腸、膣内などを含む実質的に任意の投与形式に適した形態、または吸入もしくは吹送による投与に適した形態をとることができる。一般に、開示される医薬組成物の実施形態は、注射、全身、または経口により投与される。
有用な注射可能な調製物には、水性または油性ビヒクル中の活性化合物(単数または複数)の無菌の懸濁液、溶液またはエマルジョンが含まれる。組成物は、製剤化剤、例えば懸濁化剤、安定化剤および/または分散化剤を含有することもできる。注射のための製剤は、単位剤形で、例えば、アンプルまたは多回用量容器で提示することができ、添加された防腐剤を含有することができる。組成物は、油性または水性ビヒクル中の懸濁液、溶液またはエマルジョンなどの形態をとることができ、製剤化剤、例えば懸濁化剤、安定化剤および/または分散化剤を含有することができる。例えば、非経口投与を、ボーラス注射または持続注入によって行うことができる。あるいは、配座が束縛されたシアニンフルオロフォアは、使用前に適切なビヒクル、例えば滅菌水を用いて再構成するための、粉末形態であってもよい。
全身製剤には、注射、例えば皮下、静脈内、筋肉内、髄腔内または腹腔内注射による投与のために設計された製剤、および経皮、経粘膜、経口または肺への投与のために設計された製剤が含まれる。
経口製剤は、液体(例えば、シロップ、液剤または懸濁液)、あるいは固体(例えば、散剤、錠剤、またはカプセル)であり得る。経口製剤は、内皮バリアを横断するための標的リガンドとカップリングさせることができる。一部の配座が束縛されたシアニンフルオロフォア製剤は、例えば二糖と一緒に噴霧乾燥によって乾燥させて、配座が束縛されたシアニンフルオロフォアの粉末を形成することができる。固体組成物は、従来の手段によって、薬学的に許容される賦形剤、例えば結合剤(例えば、アルファ化トウモロコシデンプン、ポリビニルピロリドンまたはヒドロキシプロピルメチルセルロース)、充填剤(例えば、ラクトース、マンニトール、微結晶性セルロースまたはリン酸水素カルシウム)、滑沢剤(例えば、ステアリン酸マグネシウム、タルクまたはシリカ)、崩壊剤(例えば、バレイショデンプンまたはデンプングリコール酸ナトリウム)、あるいは湿潤剤(例えば、ラウリル硫酸ナトリウム)を用いて調製される。錠剤は、当技術分野で周知の方法によって、例えば、糖、フィルムまたは腸溶コーティングを用いてコーティングすることができる。このような剤形を調製する実際の方法は、当業者に公知であり、または明らかになろう。
経口投与のための液体調製物は、例えば、エリキシル、溶液、シロップまたは懸濁液の形態をとることができる。このような液体調製物は、従来の手段によって、薬学的に許容される添加剤、例えば懸濁化剤(例えば、ソルビトールシロップ、セルロース誘導体または水素化食用脂)、乳化剤(例えば、レシチンまたはアカシア)、非水性ビヒクル(例えば、アーモンド油、油性エステル、エチルアルコール、Cremophor(登録商標)または分画植物油)、および防腐剤(例えば、p-ヒドロキシ安息香酸メチルもしくはp-ヒドロキシ安息香酸プロピル、またはソルビン酸)を用いて調製することができる。調製物は、適宜、緩衝塩、防腐剤、香味剤、着色剤および甘味剤を含有することもできる。経口投与のための調製物は、周知の通り、フルオロフォアを制御放出するために適切に製剤化することができる。
直腸および膣内投与経路では、配座が束縛されたシアニンフルオロフォア(単数または複数)は、溶液(停留浣腸のため)坐剤、または従来の坐剤基剤、例えばカカオバターもしくは他のグリセリドを含有する軟膏として製剤化することができる。
経鼻投与、または吸入もしくは吹送による投与では、配座が束縛されたシアニンフルオロフォア(単数または複数)は、エアロゾルスプレーまたはミストの形態で、加圧パックまたはネブライザーから、適切な噴射剤、例えばジクロロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、フルオロカーボン、二酸化炭素または他の適切なガスを使用して好都合に送達することができる。加圧エアロゾルの場合、投与量単位は、定量を送達するための弁を提供することによって決定することができる。
本明細書に記載される配座が束縛されたシアニンフルオロフォアを含む医薬組成物のある特定の実施形態は、正確な投与量の個々の投与に適した単位剤形に製剤化することができる。医薬組成物は、所望に応じて、配座が束縛されたシアニンフルオロフォアを含有する1つまたは複数の単位剤形を含有することができるパックまたはディスペンサーデバイスで提示することができる。パックは、例えば、金属またはプラスチック箔、例えばブリスターパックを含むことができる。パックまたはディスペンサーデバイスは、投与のための使用説明書を伴うことができる。
投与される配座が束縛されたシアニンフルオロフォアの量は、処置を受ける対象、標的(例えば、腫瘍のサイズ、位置、および特徴)、および投与様式に少なくとも部分的に応じて決まり、医薬組成物および/または造影剤投与の分野の当業者に公知の通りに決定することができる。これらの範囲内で、投与される製剤は、対象に投与した後に適切な手段によって配座が束縛されたシアニンフルオロフォアを可視化することができるのに有効な量の、本明細書に開示される配座が束縛されたシアニンフルオロフォアを含有する。ある特定の実施形態では、配座が束縛されたシアニンフルオロフォアは、分子に結合した薬物を含み、投与される製剤は、処置を受ける対象に治療上有効な用量の薬物を提供するのに有効な量の、配座が束縛されたシアニンフルオロフォアに結合した薬物を含有する。
一部の実施形態では、医薬組成物は、配座が束縛されたシアニンフルオロフォア以外の第2の薬剤を含む。第2の薬剤は、例えば、抗腫瘍剤または血管新生阻害剤であり得る。
IV.合成
配座が束縛されたトリメチンシアニン色素はこれまでに、還流条件下でマイルドな酸の溶液(例えば、酢酸)中で、またはより穏やかな条件下でより強い鉱酸溶液中でトリメチンシアニン色素を処理し、その結果、「固定された」カルボシアニン色素をその溶液から沈殿させることによって合成されている(例えば、国際公開第99/31181号を参照されたい)。しかし、この合成戦略は、遠赤および近IRシアニン、例えばペンタメチンおよびヘプタメチンシアニンに拡大することができない。今までに、式Iによる配座が束縛されたシアニンフルオロフォアを作製するための合成戦略は、公知ではなかった。
図2は、非置換の配座が束縛されたペンタメチンシアニンフルオロフォアの一実施形態を調製するための例示的な合成スキームを示す。前駆体1は、以下により詳細に記載される通り、N-アルキル化インドレニンから調製される。ジクロロメタン中でグラブス第2世代触媒およびアクロレインジメチルアセタールを使用する交差メタセシスによって、精製後に化合物3が得られた。化合物3は、加熱した(例えば、70℃)酸性化クロロホルム溶液中で四環化を受けて、化合物4を単一ジアステレオマーとして提供する。あるいは、冷却した(例えば、-78℃)三臭化ホウ素のジクロロメタン溶液中の化合物3の反応によって、化合物4および別の化合物が得られ、その別の化合物を、1:3のメタノール:0.3M HClの加熱(例えば、60℃)溶液中で、化合物4に変換することができる。
図3は、配座が束縛されたペンタメチンシアニンフルオロフォアのコンジュゲート可能なバリアントを作製するための例示的な合成スキームを示す。ホベイダ(Hoyveda)-グラブス第2世代触媒を使用する交差メタセシスは、化合物5および化合物2の間で室温において効率的に進行して、化合物6を提供し、それを大規模に精製することなく使用することができる。化合物6の四環化は、冷却した(例えば、-78℃)三臭化ホウ素のジクロロメタン溶液中で進行して、化合物7を含む混合物を提供する。1:3のメタノール:0.3M HClの加熱した(例えば、60℃)溶液中で平衡化することによって、メチルエステル6が得られ、それをけん化し、精製した後、化合物7が得られる。バイオコンジュゲーションに適した化合物を調製するために、化合物7を、アミドカップリングシーケンスを介してカルボン酸8に変換することができる。図3の例では、ファロイジンを、N-ヒドロキシスクシンイミド-エステル作製およびアミド結合形成によって分子にコンジュゲートさせて、ファロイジンコンジュゲート9を提供した。
一部の実施形態では、対称性の配座が束縛されたシアニンフルオロフォアを生成することが望ましい場合がある。このような実施形態では、式IVおよびXによる化合物は同じであり、それにより、式VおよびXIによる同じ化合物が生成される。これらの実施形態では、式IV/Xによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式V/XIによる化合物を生成する単一ステップを実施することができる。
反応のための例示的な溶媒として、それに限定されるものではないが、テトラヒドロフラン(THF)、N,N-ジメチルホルムアミド(DMF)、ジクロロメタン(DCM)、水、およびそれらの組合せが挙げられる。
有効な条件には、室温(20~26℃)から100℃の範囲の反応温度における、数分から数時間の範囲の時間にわたる反応が含まれ得る。ある特定の例では、反応温度は、室温から90℃の範囲であり、時間は10分から18時間の範囲である。一例では、溶液は、マイクロ波照射を反応温度で照射される。様々な実施形態では、溶液は、穏やかに撹拌されるか、激しく撹拌されるか、または撹拌されない。反応は、密封容器中、不活性雰囲気(例えば、アルゴン、窒素)下で進行することができる。反応の完了は、それに限定されるものではないが、視覚的な変色またはLC/MSを含む任意の適切な手段によってモニタリングすることができる。式XVIIIによる化合物は、適切な手段によって回収され、必要に応じて精製される。一部の実施形態では、化合物は、抽出、沈殿、蒸発、イオン交換、クロマトグラフィー(例えば、シリカゲルクロマトグラフィー、HPLC)、およびそれらの組合せによって回収され、かつ/または精製される。
開示される式Iによる化合物の実施形態は、生細胞の位置決定および追跡適用に有用となり得る。さらに、開示される化合物の一部の実施形態は、活性酸素種(ROS)を感知するのに有用となり得る。ある特定の実施形態では、スルホネートまたは他の極性官能基を含むことにより、抗体の標識を容易にすることができる。さらに、開示される化合物の一部の実施形態は、細胞透過性であってもよく、これは生細胞研究にとって利点である。研究上の、診断上の、およびセラノスティック(theranostic)な使用は、本開示の範囲内である。
一部の実施形態では、可視化は、可視、遠赤、または近赤外範囲の波長と選択された強度を有する所定量の光の標的化された適用により、試料または対象の標的化部分に照射すること(ここで、所定量の光は、化合物の蛍光を生じさせるのに十分である)、ならびに化合物によって放出された任意の蛍光を検出することを含む。有利には、光は、式Iによる化合物の最大吸収波長のまたはそれに近い波長を有する。例えば、試料は、600nm~2500nm、例えば600~900nm、または600~700nmの範囲内の波長を有する光を照射され得る。一部の実施形態では、光源は、レーザーである。適切な光強度は、標的部位および適用方法に応じて、1mW/cm2~1000mW/cm2、例えば1~750mW/cm2または300~700mW/cm2の範囲であり得る。近赤外光源は、Thorlabs(Newton、NJ)、Laser Components、USA(Hudson、NH)、ProPhotonix(Salem、NH)などを含む商業的供給源から得ることができる。一部の実施形態では、遠赤またはNIR光の有効量は、10~250J、例えば10~200J、10~150J、または10~100Jである。
一部の実施形態では、可視化には、蛍光透視法、単一分子局在性顕微鏡法(SMLM)、光活性化局在性顕微鏡法(PALM)、確率的光学的再構築顕微鏡法(STORM)、直接確率的光学的再構築顕微鏡法(dSTORM)、二方向画像化(BP)、経時的ラジアル開口ベース強度推定(TRABI)、蛍光共鳴エネルギー移動(FRET)、およびそれらの組合せなどの技術が含まれ得る。
一部の実施形態では、有効量の式Iによる化合物またはこの化合物を含む医薬組成物は、対象内の標的(例えば、腫瘍)に結合したフルオロフォアを可視化することによって検出および/または評価することができる状態を有すると疑われる対象に投与される。有利には、式Iによる化合物は、対象内の標的に結合することができる標的化剤を含むことができる。投与は、任意の適切な方法、例えば、静脈内、動脈内、筋肉内、腫瘍内もしくは皮下注射、または経口、鼻腔内もしくは舌下投与によって実施される。その後、投与された化合物は、対象の標的領域への、遠赤または近赤外範囲の波長と選択された強度を有する所定量の光の標的化された適用によって照射され、ここで、所定量の光は、式Iによる化合物を励起するのに十分である。標的領域(例えば、腫瘍近くの領域)に照射する場合、遠赤またはNIR光の有効量は、1~250J/cm2、例えば1~250J/cm2、例えば5~250J/cm2、10~250J/cm2、10~200J/cm2、10~150J/cm2、10~100J/cm2、または30~100J/cm2であり得る。対象の標的化部分における化合物からの任意の蛍光を検出し、それによって、対象はその状態を有すると診断される。
ある特定のセラノスティックな実施形態では、その状態は、腫瘍であり、対象の標的化部分は、その腫瘍部位を含む。投与された化合物は、腫瘍を、化合物の蛍光を誘導するのに十分な波長および強度を有する光に曝露することによって可視化される。照射は、対象の標的化領域に光を外部から適用することによって実施することができる。遠赤またはNIR光は、数センチメートルの深さまで組織に経皮的に透過することができる。他の実施形態では、照射は、光の内部適用によって、例えば内視鏡、光ファイバーカテーテル、または埋込式蛍光デバイスを使用することによって実施することができる。内部適用は、腫瘍などの標的組織が、外部から光を適用するのに適していない深さに位置する場合に使用することができる。例えば内視鏡は、光を、肺、胃、または膀胱内に送達するために使用することができる。一部の例では、腫瘍部位は、腫瘍を光に曝露する前に、外科的切開に曝露される。腫瘍は、蛍光領域を指針として使用して切除することができる。一実施形態では、治療有効量の化合物またはこの化合物を含む医薬組成物を対象に投与する前に、腫瘍の少なくとも一部が対象から切除される。独立な実施形態では、治療有効量の化合物またはこの化合物を含む医薬組成物は、腫瘍またはその一部を外科手術によって切除する前に、対象に投与される。
光を適用するための表面領域は、一般に、標的組織、例えば腫瘍もしくは腫瘍の一部、または標的組織外部の皮膚領域を含むように選択される。外部光の標的化された適用がin vivo生物学的試料にとって望ましい場合、表面領域は、適切な光アプリケーター、例えばマイクロレンズ、フレネルレンズ、またはディフューザー配置を使用することによって制御することができる。内部光の適用の標的化では、望ましい内視鏡または光ファイバーカテーテルの直径を選択することができる。一部の適用では、光散乱溶液を充填した留置カテーテルを、標的組織近くの内部に置くことができ、そのカテーテルに光ファイバー光源を挿入することができる(例えば、Madsen et al.,Lasers in Surgery and Medicine 2001,29,406-412を参照されたい)。
別の実施形態では、in vitroまたはex vivo評価を実施して、式Iによる化合物が、この化合物によって可視化され得る状態を有するか、または有すると疑われる対象から得られた組織試料に有効に結合するかどうかを決定することができる。化合物は、その状態の指標となるか、またはその状態と関連する標的分子に結合するか、または会合することができると考えられる標的化剤を含む。非限定的な一例では、標的化剤は、標的受容体に結合することができる受容体リガンドまたは抗体である。化合物は、組織試料と合わせられ、その後、試料は、有効量の近IR光を照射される。一実施形態では、組織試料は、過剰の非結合化合物を除去するために洗浄され、組織試料の蛍光が評価される。蛍光は、化合物が組織試料に結合したことを示す。
ある特定の実施形態では、式Iによる化合物は、細胞内の形状および/または構造を可視化するために利用することができる。式Iによる化合物は、細胞、例えば固定または透過性細胞内の望ましい構成成分に結合することができる標的化剤を含むことができる。例えば、化合物は、F-アクチンに結合する標的化剤として、ファロイジンを含むことができる。F-アクチンの可視化は、細胞の全体的な形状および構造を示すのに有用である。
独立な実施形態では、式Iによる化合物は、試料中のスーパーオキシドおよび水酸化物ラジカルを含む活性酸素種(ROS)の存在を検出し、かつ/または測定するために利用することができる。ROSは、がんおよびアテローム性動脈硬化症などの様々な炎症性疾患に関与している。ROSの検出は、in vitro、ex vivo、またはin vivoで実施することができる。ある特定の実施形態では、化合物は、試料を化合物と接触させる前に還元することができる。試料中に存在するROSは、化合物を酸化して、フルオロフォアを再構築することができ、そのフルオロフォアは任意の適切な方法によって検出される。蛍光は、試料中のROSの存在を示す。蛍光の強度は、試料中のROSの濃度と相関し得る。
VI.実施例
一般材料および方法
商業的に得たすべての試薬を、受け取ったまま使用した。2,3,3-トリメチルインドリン(S1)、マロンアルデヒドビス(フェニルイミン)一塩酸塩、グラブス第2世代触媒およびホベイダ-グラブス第2世代触媒は、Sigma-Aldrich(St.Louis、MO)から購入した。アクロレインジメチルアセタール(2)は、TCI America(Portland、OR)から購入した。4-ヒドラジニル安息香酸(S5)は、Oakwood Chemical(Estill、SC)に発注した。2-(2-ブロモエチル)-1,3-ジオキソランは、Acros Organics(Geel、Belgium)から購入した。NH2-PEG8-COOHは、Thermo Scientific(Waltham、MA)から購入した。アミノファロイジントシレートは、Enzo,Inc.(Farmingdale、NY)から購入した。化合物S2、S4、およびS8は、公知の手順に従って合成した(Hanessian et al.,J.Am.Chem.Soc.2004,126(19),6064-71;Hall et al.,Nucleic Acids Res.2012,40(14),e108;Park et al.,Bioconjugate Chem.2012,23(3),350-362)。順相(60Å、20~40μm、RediSep(登録商標)Rf Gold(登録商標)シリカまたは60Å、35~70μm、RediSep(登録商標)Rfシリカ)および逆相(100Å、20~40ミクロン粒径、RediSep(登録商標)Rf Gold(登録商標)逆相C18またはC18Aq)フラッシュカラムクロマトグラフィーを、CombiFlash(登録商標)Rf 200i(Teledyne Isco,Inc.、Lincoln、NE)で実施した。逆相分取HPLCを、Agilent 1260 Infinity II LCシステムを使用し、Waters,Co(Milford、MA)から得たSunFire Prep C18カラム(100Å、5μm、10×150mm)を利用して実施した。高分解能LC/MS分析を、Thermo-Fisher LTQ-Orbitrap-XLハイブリッド質量分析計システムで、Ion MAX APIエレクトロスプレーイオン源を負イオンモードで用いて実施した。分析LC/MSを、Shimadzu LCMS-2020シングル四重極を使用し、Phenomenex,Inc(Torrance、CA)から得たKinetex C18カラム(100Å、2.6μm、2.1×50mm)を利用して実施した。泳動では、0~90%MeCN/0.1%ギ酸水溶液の勾配を流速0.2mL/分で4.5分にわたって用いた。1H NMRおよび13C NMRスペクトルを、Bruker分光計(400もしくは500MHz、または100もしくは125MHz)で記録し、重水素化溶媒シグナルと比較して記録する。1H NMRスペクトルのデータを、以下の通り記録する。化学シフト(δppm)、多重度、カップリング定数(Hz)、および積分。13C NMRスペクトルのデータを、化学シフトに関して記録する。吸光度曲線は、Shimadzu UV-2550分光光度計をUVProbe 2.32ソフトウェアによって操作して得た。モル吸光係数(ε)は、PBS(50mM、pH7.4)またはメタノール(MeOH)中、ベールの法則を使用して吸光度対濃度のプロットから決定した。測定を、室温で維持した10mm経路長の石英キュベット(Hellma 111-QS)で実施した。記録値は平均である(n≧3)。蛍光トレースを、PTI QuantaMaster定常状態蛍光光度計をFelixGX 4.2.2ソフトウェアによって操作し、4nmの励起および発光スリット幅、ならびに0.1s積分速度で記録した。Quantaurus-QY分光計(浜松、モデルC11374)を使用して、絶対的蛍光量子収率(ΦF)を決定した(Suzuki et al.,Phys.Chem.Chem.Phys.11 2009,9850-9860)。この機器は、試料によって吸収された光子および放出された光子を測定するために積分球を使用する。測定は、MeOHまたはPBS(50mM、pH7.4)中250nMの濃度で実施し、自己吸収補正を、機器のソフトウェアを使用して実施した。記録値は平均である(n≧5)。データ分析および曲線フィッティングを、MS Excel 2011およびGraphPad Prism 7を使用して実施した。光強度測定を、S120VC標準Siフォトダイオード出力センサー(200~1100nm、50nW~50mW)を備えたThorlabs PM200光出力およびエネルギーメーターで実施した。略語については、JOC Standard Abbreviations and Acronyms(http://pubs.acs.org/paragonplus/submission/joceah/-joceah_abbreviations.pdf)を参照されたい。
単一分子画像化を、他所(van de Linde et al.,Nature Protocols 2011,6:991)に記載されている通り、高度斜光を用いる特注の対物型TIRFセットアップで実施した。ノーズピースステージ(IX2-NPS、Olympus)および60×油浸対物レンズ(NA 1.45 PlanApo、Olympus)を備えた倒立顕微鏡法(IX71、Olympus)を使用した。TRABI-二方向画像化を、他所(Franke et al.,Nature Methods 2017 14:41)に記載されている通り実施した。手短には、50/50ビームスプリッタ(Cairn Research)および焦点面が300nm離れている2つのEMCCDカメラ(Ixon 897およびIxon Ultra 897、Andor)を備えた、2倍拡大率の2チャネル画像スプリッタ(TwinCam、Cairn Research)を使用した。カメラを、パルスジェネレータ(DG535、Stanford Research Systems)によって同期した。3D較正実験を、LVPZTサーボコントローラー(E-662、Physik Instrumente)で駆動されるピエゾスキャナ(Pifoc、Physik Instrumente)を用いて対物レンズを移動させることによって実施した。TRABI-二方向画像化由来のZ座標は、屈折率ミスマッチについて倍率0.71(緩衝液の屈折率n
b=1.34および基質(ガラス)n
s=1.52、開口数NA=1.45)によって補正した。
(実施例1)
配座が束縛されたペンタメチンシアニンフルオロフォアの例示的な合成
(S3):2,3,3-トリメチルインドリン(S1、2.0g、12.5mmol、1.0当量)のCH
2Cl
2(50mL、0.25M)中の溶液に、0℃でS2(3.0g、15mmol、1.2当量)を添加した。溶液を室温に加温し、1時間静置した。反応体積をおよそ10mLに低減し、次に順相クロマトグラフィー(24gのシリカカラム、0~15%MeOH/CH
2Cl
2)によって精製して、S3(2.5g、6.9mmol、55%)を濃紫色の固体として得た。
1H NMR (400 MHz, CDCl
3) δ 7.71 - 7.65 (m, 1H), 7.62 - 7.54 (m, 3H), 5.82 (ddt, J = 17.3, 10.1, 7.3 Hz, 1H), 5.07 (ddd, J = 10.1, 1.5, 0.9 Hz, 1H), 4.90 (dq, J = 17.0, 1.4 Hz, 1H), 4.65 (t, J = 6.7 Hz, 2H), 2.82 (s, 3H), 2.74 (q, J = 6.9 Hz, 2H), 1.57 (s, 6H).
13C NMR (101 MHz, CDCl
3) δ 196.7, 141.6, 140.7, 132.2, 130.1, 129.5, 123.2, 120.3, 115.4, 54.6, 47.6, 32.2, 23.2, 14.9. C
15H
20N (M
+)のHRMS(ESI)計算値 214.1590、実測値 214.1588.
(1):S3(1.0g、5.0mmol、1.0当量)、S4(1.3g、5.0mmol、1.0当量)、マロンアルデヒドビス(フェニルイミン)一塩酸塩(1.3g、5.0mmol、1.0当量)、およびEt
3N(3.6mL、25mmol、5当量)のCH
2Cl
2(50mL)中の溶液に、Ac
2O(1.4mL、15mmol、3当量)を添加した。溶液を室温で2時間静置した。次に、その濃青色の溶液に、NaI水溶液(50mL、0.4M)を添加した。混合物を18時間激しく撹拌し、CH
2Cl
2(2×100mL)で分離し、NaHSO
4で乾燥させ、減圧下で濃縮した。得られた青色の残留物を、順相カラムクロマトグラフィー(40gのシリカカラム、25~75%酢酸エチル/CH
2Cl
2)によって精製して、1(2番目に溶出するピーク)を青色の固体(1.20g、1.9mmol、38%)として得た。
1H NMR (400 MHz, CD
3OD) δ 8.27 (td, J = 13.1, 3.0 Hz, 2H), 7.51 (d, J = 7.4 Hz, 2H), 7.43 (t, J = 7.7 Hz, 2H), 7.32 (d, J = 8.0 Hz, 2H), 7.28 (t, J = 7.5 Hz, 2H), 6.65 (t, J = 12.4 Hz, 1H), 6.34 (dd, J = 13.7, 6.1 Hz, 2H), 5.92 (ddt, J = 17.2, 10.1, 7.1 Hz, 1H), 5.14 - 5.03 (m, 2H), 5.01 (t, J = 3.9 Hz, 1H), 4.25 (dt, J = 14.6, 7.0 Hz, 4H), 4.02 - 3.81 (m, 4H), 2.62 (q, J = 7.0 Hz, 2H), 2.21 (td, J = 7.1, 3.9 Hz, 2H), 1.74 (s, 12H).
13C NMR (101 MHz, CD
3OD) δ 173.6, 173.2, 154.1, 154.0, 142.1, 142.1, 141.2, 141.2, 133.7, 128.3, 128.2, 125.2, 124.8, 124.8, 120.0, 122.0, 117.4, 110.9, 110.6, 103.5, 103.1, 101.6, 64.7, 49.2, 49.2, 42.7, 38.6, 31.6, 30.2, 26.7, 26.3.C
34H
41N
2O
2(M
+)のHRMS(ESI)計算値 509.3163、実測値 509.3157。
(3):1(80mg、0.13mmol、1.0当量)およびグラブス第2世代触媒(33mg、0.039mmol、0.3当量)のCH
2Cl
2(16mL、0.01M)中の濃青色の溶液に、アクロレインジメチルアセタール(2、149μL、1.3mmol、10当量)を、真空で脱気した後、アルゴン下で添加した。この反応物を40℃においてアルゴンおよび静的真空(およそ1時間当たり1回再適用した)下で5.5時間還流させた。室温に冷却した後、飽和NaCl水溶液(20mL)を添加し、18時間激しく撹拌した。次に、二相混合物を分離し、CH
2Cl
2(3×20mL)で抽出し、Na
2SO
4で乾燥させ、減圧下で蒸発させた。濃青色の残留物を、順相クロマトグラフィー(12gのシリカカラム、0~20%MeOH/CH
2Cl
2)によって精製して、3(2番目に溶出するピーク、37mg、0.060mmol、48%)を得た。不純生成物を含有していた最初に溶出するピークを合わせ、飽和NaCl水溶液(20mL)と共に再び一晩撹拌した。CH
2Cl
2中での有機抽出および順相精製を繰り返して、3(11mg、0.018mmol、14%)を得た。両方のカラムから精製した生成物を合わせて、3(48mg、0.078mmol、62%)を青色の固体として得た。
1H NMR (500 MHz, CD
3OD) δ 8.27 (td, J = 13.1, 5.9 Hz, 2H), 7.53 - 7.49 (m, 2H), 7.46 - 7.40 (m, 2H), 7.36 - 7.25 (m, 4H), 6.64 (t, J = 12.4 Hz, 1H), 6.33 (d, J = 13.7 Hz, 2H), 5.91 (dt, J = 14.9, 7.2 Hz, 1H), 5.44 (dd, J = 15.6, 5.1 Hz, 1H), 5.00 (t, J = 3.8 Hz, 1H), 4.59 (d, J = 5.1 Hz, 1H), 4.26 (dt, J = 10.8, 7.0 Hz, 4H), 3.92 (dt, J = 51.0, 7.0 Hz, 4H), 3.16 (s, 6H), 2.65 (q, J = 6.7 Hz, 2H), 2.21 (q, J = 7.0 Hz, 2H), 1.74 (d, J = 3.2 Hz, 12H).
13C NMR (101 MHz, CD
3OD) δ 173.5, 173.4, 154.1, 142.2, 142.1, 141.2, 141.2, 130.4, 130.0, 128.3, 128.3, 125.3, 124.8, 124.8, 122.0, 122.0, 110.9, 110.7, 103.5, 103.1, 103.0, 101.6, 64.7, 51.9, 49.2, 49.2, 42.5, 38.6, 30.2, 30.0, 26.6, 26.3.C
37H
47N
2O
4(M
+)のHRMS(ESI)計算値 583.3530、実測値 583.3536。
(4):封止バイアル中、H2SO4水溶液(3.5M、1.3mL)を、3(37mg、0.059mmol)のCHCl3(5.0mL)中の濃青色の溶液に添加した。この二相混合物(1:4の20%H2SO4/CHCl3)を70℃で3時間撹拌し、その時点でLC/MS分析によって出発材料の完全な消費が明らかになった。得られた溶液を飽和NaHCO3水溶液(20mL)でクエンチし、CH2Cl2(4×10mL)で抽出し、Na2SO4で乾燥させた。溶媒を減圧下で蒸発させ、青緑色の残留物を、順相クロマトグラフィー(12gのシリカ金カラム、0~15%MeOH/CH2Cl2)で精製した。得られた生成物を1:1のNaI水溶液(1.0M、15mL)およびCH2Cl2(15mL)に溶解させ、室温で5時間激しく撹拌した。二相混合物を分離し、CH2Cl2(3×10mLのCH2Cl2)で抽出し、真空中で乾燥させた。得られた残留物を、シリカを充填したピペットから溶出し(10%MeOH/90%CH2Cl2)、減圧下で蒸発させて、4(8.64mg、0.014mmol、24%)を青色の固体として得た。
化合物4は、NMR分析によって帰属したsyn-syn環接合点立体化学を有する単一ジアステレオマーであった(図4)。算出は、Spartan’14(Wavefunction,Inc.、Irvine、CA)を使用して実施した。分子力学および半経験的モデルを除き、Spartanで使用した算出方法は、Shao et al.(Phys.Chem.Chem.Phys.2016,8:3172)により実証されている。配座分布を、MMFFレベルで実施した。先のステップにおける3つの最低エネルギーの配座異性体(最低エネルギーの20kJ/mol以内)を、B3LYP/6-31G(d)、EtOH溶媒を使用して幾何構造最適化に供した。(1つをsyn-synについて、2つをsyn-antiについて、3つをanti-synについて、3つをanti-antiについて)。得られた最小値を図4に示す。環接合点(位置12、14、および15)に位置する3つすべてのメチンプロトンは、syn,synジアステレオマーと合致する、高磁場軸方向のα-窒素プロトン(3.88(tq、J=13.3、4.5Hz、2H)-位置10および17)との1D-NOESY相互作用を示した。さらに、これらのプロトンはいずれも、低磁場エカトリアルα-窒素プロトン(4.40~4.24(m,2H)-位置10および17)に対してNOEを示さなかったが、それは任意の他のジアステレオマー構造のうちの少なくとも1つのメチンプロトンについても予測される。また、syn-synジアステレオマーの形成と合致して、α-酸素プロトン(位置14および15)は、ほぼ同じカップリング定数を示したが、このことは、これらのプロトンが類似の環系にあることを示している。
(4):アルゴン下で脱気し、-78℃に冷却した(ドライアイスおよびアセトン浴)、3(11.0mg、0.018mmol、1当量)の無水CH
2Cl
2(1.8mL、0.01M)中の溶液に、BBr
3(1.0M CH
2Cl
2溶液0.21mL、0.21mmol、12当量)をゆっくり添加した。反応物の色は、BBr
3添加を行うと、濃青色から赤褐色に急速に遷移した。アルゴン下で-78℃において2時間撹拌した後、NaHCO
3水溶液(0.5M、5mL)を添加して反応をクエンチすると、急速に青色に戻った。室温に加温したら、追加のCH
2Cl
2(10mL)を提供し、二相混合物を45分間激しく撹拌した。次に、薄緑色/黄色の水層をCH
2Cl
2(3×5mL)で抽出し、合わせた濃青色の有機溶液をNa
2SO
4で乾燥させ、減圧下で蒸発させた。この青色の残留物は、4および第2の化合物のジアステレオマー混合物を含有していた(約1:1~約2:1の変動比で得られた)。分離不可能な混合物が、単一の[M]
+イオンシグナル、ジヒドロピラン領域の複雑なNMRシグナル、および単一の遠赤UV-vis吸光度最大値を示した。以下の通り混合物を平衡化して、均質な化合物4を得た。混合物をMeOH(0.9mL)およびHCl水溶液(0.3M、2.7mL)に溶解させ、60℃で20分間撹拌した。室温に冷却した後、NaHCO
3水溶液(0.5M、5mL)およびCH
2Cl
2(10mL)を添加して、二相混合物を形成し、それを30分間激しく撹拌した。薄緑色/黄色の水層をCH
2Cl
2(10×5mL)で抽出し、合わせた有機溶液をNa
2SO
4で乾燥させ、減圧下で濃縮した。青色の残留物を、CH
2Cl
2(15mL)およびNaI水溶液(0.5M、10mL)に溶解させ、室温で13時間激しく撹拌した。二相混合物を分離し、CH
2Cl
2(2×10mL)で抽出し、Na
2SO
4で乾燥させた。真空中で乾燥させた後、残留物を順相クロマトグラフィー(4gのシリカ金カラム、0~10%MeOH/CH
2Cl
2)によって精製して、4の青色の固体を単一ジアステレオマー(5.7mg、0.0095mmol、53%)として得た。
1H NMR (400 MHz, CD
3OD) δ 7.93 (s, 1H), 7.86 (s, 1H), 7.57 - 7.36 (m, 4H), 7.37 - 7.16 (m, 4H), 4.67 (dd, J = 11.5, 5.0 Hz, 1H), 4.61 (dd, J = 11.4, 5.0 Hz, 1H), 4.40 - 4.24 (m, 2H), 3.88 (tq, J = 13.3, 4.5 Hz, 2H), 2.84 (tt, J = 12.3, 4.1 Hz, 1H), 2.58 (dt, J = 11.7, 4.5 Hz, 1H), 2.49 (dt, J = 11.7, 4.5 Hz, 1H), 2.41 (dt, J = 13.3, 4.2 Hz, 1H), 2.01 (qd, J = 12.6, 5.4 Hz, 1H), 1.82 (dd, J = 12.6, 5.0 Hz, 1H), 1.78 - 1.74 (m, 12H), 1.48 (q, J = 11.8 Hz, 1H).
13C NMR (126 MHz, CD
3OD) δ 169.2, 165.8, 143.3, 142.0, 142.0, 141.5, 141.1, 140.3, 128.7, 128.3, 128.3, 125.3, 124.5, 121.9, 121.9, 114.0, 111.4, 110.2, 109.6, 72.2, 70.2, 49.1, 48.3, 42.9, 40.7, 35.1, 31.1, 27.2, 26.7, 26.7, 26.6, 26.1, 26.0.C
33H
35N
2O(M
+)のHRMS(ESI)計算値 475.2737、実測値 475.2744。計算的分析は、anti-環接合点を含有する最低エネルギーのジアステレオマーが、ポリエンのC5’に隣接している化合物4に対するジアステレオマーであったことを示している(図5)。化合物3をMeOH:HCl平衡化条件に供するだけで、ごく微量の化合物4が提供されたことが分かった。
(S6):以下を3つの別個の封止バイアルに入れた。4-ヒドラジニル-安息香酸(S5、610mg、4.0mmol、1.0当量)、3-メチル-2-ブタノン(430μL、4.0mmol、1.0当量)、KHSO
4(1.5g、12mmol、3.0当量)およびMeOH(15mL)。各バイアルをマイクロ波によって160℃に2時間加熱した。3つのバイアルを合わせた内容物を、飽和NaHCO
3水溶液(100mL)とCH
2Cl
2(2×100mL)との間に分離し、乾燥させ(NaHSO
4)、減圧下で濃縮した。得られた残留物を、順相クロマトグラフィー(40gのシリカカラム、30~100%酢酸エチル/ヘキサン)によって精製して、S6(1.4g、6.3mmol、53%)を黄褐色の油状物として提供した。
1H NMR (400 MHz, CDCl
3) δ 8.06 (dd, J = 8.1, 1.7 Hz, 1H), 7.99 (d, J = 1.6 Hz, 1H), 7.58 (d, J = 8.1 Hz, 1H), 3.95 (s, 3H), 2.34 (s, 3H), 1.36 (s, 6H).
13C (126 MHz, CDCl
3) δ 191.7, 167.3, 157.7, 145.7, 130.1, 126.9, 122.7, 119.6, 53.9, 52.1, 22.9, 15.7.C
13H
16NO
2(MH
+)のHRMS(ESI)計算値 218.1176、実測値 218.1173。
(S7):密封容器中、S6(6.0g、28mmol、1.0当量)を、MeCN(80mL)中で2-(2-ブロモエチル)-1,3-ジオキソラン(5.0mL、41mmol、1.5当量)、NaHCO
3(4.6g、55mmol、2.0当量)、およびNaI(6.2g、41mmol、1.5当量)と合わせた。反応物を95℃で14.5時間撹拌すると、溶液の色が橙色から褐色に遷移した。粗製生成物をセライトで濾過し、減圧下で濃縮し、残留物を逆相カラムクロマトグラフィー(150gのC18金カラム、0~80%MeCN/H
2O)によって精製して、S7(2.8g、8.9mmol、32%)を濃緑色の油状物として得た。
1H NMR (400 MHz, CDCl
3) δ 7.90 (dd, J = 8.3, 1.8 Hz, 1H), 7.75 (d, J = 1.7 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 4.93 (t, J = 4.5 Hz, 1H), 4.09 (d, J = 2.3 Hz, 1H), 4.04 - 3.84 (m, 8H), 3.72 (t, J = 7.2 Hz, 2H), 2.03 (td, J = 7.5, 4.5 Hz, 2H), 1.36 (s, 6H).
13C NMR (126 MHz, CDCl
3) δ 167.6, 160.7, 149.8, 137.6, 131.1, 123.3, 120.2, 104.7, 102.5, 76.4, 65.1, 51.79, 43.81, 37.39, 30.23, 30.01.C
18H
23NO
4(MH
+)のHRMS(ESI)計算値 318.1700、実測値 318.1695。
(S9):MeCN(50mL、0.38M)中、S8(4.6g、19mmol、1.0当量)の不均質混合物に、新しく調製したニートな3-ブテニルトリフレート(S2、5.1g、25mmol、1.3当量)を室温でゆっくり添加した。15分後、得られた濃色の混合物を、飽和NaHCO
3水溶液(50mL)で処理した。得られた混合物を、セライトを介して濾過し、その得られた溶液の体積を、ロータリーエバポレーターで低減した(約50mL)。この溶液を、逆相クロマトグラフィー(150gのC18Aq金カラム、0~15%MeCN/H
2O)で精製して、クリーンなS9(2.2g、7.4mmol、40%)を桃色の固体として得た。
1H NMR (400 MHz, DMSO-d
6) δ 7.35 (dd, J = 8.0, 1.7 Hz, 1H), 7.33 (d, J = 1.5 Hz, 1H), 6.57 (d, J = 8.1 Hz, 1H), 5.83 (ddt, J = 17.1, 10.2, 7.0 Hz, 1H), 5.06 (dq, J = 17.2, 1.4 Hz, 1H), 4.98 (dd, J = 10.1, 2.1 Hz, 1H), 3.94 (d, J = 1.8 Hz, 1H), 3.90 (d, J = 1.8 Hz, 1H), 3.60 (t, J = 7.1 Hz, 2H), 2.31 (q, J = 6.9 Hz, 2H), 1.26 (s, 6H).
13C NMR (126 MHz, DMSO-d
6) δ 160.9, 145.9, 139.5, 136.3, 136.1, 125.9, 120.0, 117.3, 104.4, 75.2, 44.0, 41.2, 30.5, 30.2.C
15H
19NO
3S(MH
+)のHRMS(ESI)計算値 294.1158、実測値 294.1155。
(9):S9(2.4g、7.7mmol、1.5当量)、S7(1.5g、5.1mmol、1当量)、マロンアルデヒドビス(フェニルイミン)一塩酸塩(1.6g、6.1mmol、1.2当量)、およびEt
3N(3.6mL、26mmol、5当量)のMeOH(26mL)中の溶液に、無水酢酸(1.0mL、10mmol、2当量)を添加した。室温で撹拌しながら、追加の無水酢酸(1.0mL、10mmol、2当量)を、反応の最初の1.5時間にわたって、およそ30分ごとに添加して、合計3.9mL(41mmol、8当量)にした。反応中、溶液の色は、赤色から緑色、紫色、最後に濃青色に遷移した。合計4時間の反応時間が経過した後、LC/MS分析によって、S7が完全に消費され、3種類の可能なシアニン生成物の、およそ2:1:1比の非対称性の所望の生成物と望ましくない対称性シアニンの混合物が明らかになった。濃青色の溶液を、2:1のジエチルエーテル/ヘキサン(180mL)中で沈殿させ、遠心分離し(6800rpm、8分)、上清を除去した。この磨砕を3回実施し、真空中で乾燥させた後、濃青色の残留物を、逆相クロマトグラフィー(150gの金C18カラム、0~80%MeCN/H
2O)を用いて精製して、5(0.72g、1.1mmol、22%)を濃青色の固体として得た。
1H NMR (400 MHz, CD
3OD) δ 8.35 (dt, J = 17.0, 13.0 Hz, 2H), 8.14 - 8.06 (m, 2H), 7.97 - 7.89 (m, 2H), 7.44 (d, J = 8.3 Hz, 1H), 7.34 (d, J = 8.3 Hz, 1H), 6.72 (t, J = 12.4 Hz, 1H), 6.48 (d, J = 13.9 Hz, 1H), 6.34 (d, J = 13.4 Hz, 1H), 5.90 (ddt, J = 17.2, 10.2, 7.1 Hz, 1H), 5.10 - 5.02 (m, 2H), 4.99 (t, J = 3.8 Hz, 1H), 4.30 (t, J = 6.9 Hz, 2H), 4.26 (t, J = 7.0 Hz, 2H), 3.98 - 3.84 (m, 7H), 2.64 (q, J = 7.1 Hz, 2H), 2.21 (td, J = 7.0, 3.8 Hz, 2H), 1.77 (d, J = 5.6 Hz, 12H).
13C NMR (126 MHz, CD
3OD) δ 175.8, 172.6, 166.6, 155.8, 154.2, 146.4, 143.2, 142.7, 141.4, 141.1, 133.4, 130.5, 126.7, 125.8, 123.0, 120.0, 117.8, 111.1, 110.0, 105.5, 103.6, 101.6, 64.7, 51.3, 49.7, 48.6, 48.2, 43.1, 38.5, 31.7, 30.0, 26.4.C
36H
43N
2O
7S(MH
+)のHRMS(ESI)計算値 647.2785、実測値 647.2776。
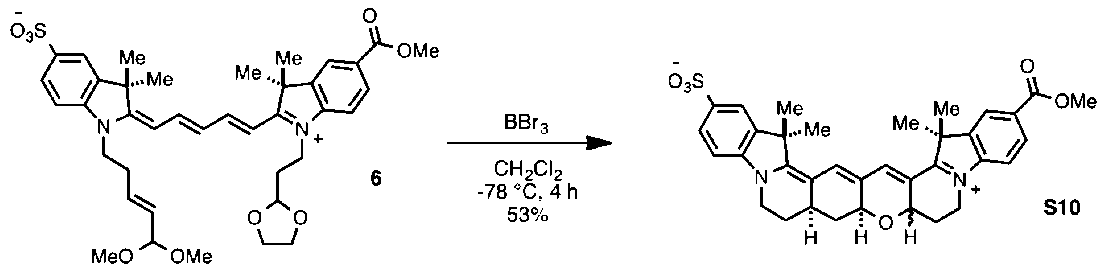
(10):5(120mg、0.19mmol、1.0当量)およびホベイダ(Hoyveyda)-グラブス触媒第2世代(58mg、0.093mmol、0.5当量)の無水CH
2Cl
2(9.3mL)中の溶液に、アクロレインジメチルアセタール(2、110μL、0.95mmol、5.0当量)をアルゴン下で添加した。青色の溶液を、閉じた還流冷却器を用いてアルゴンおよび静的真空(およそ30分ごとに再適用した)下で室温において5時間撹拌した。得られた青色の混合物を、1:1のジエチルエーテル/ヘキサン(45mL)中で沈殿させ、遠心分離し(7500rpm、5分)、デカンテーションした。この沈殿を繰り返した後、ペレットを高真空下で乾燥させて、6(130mg、0.18mmol、97%)を濃青色の固体として得、それをさらに精製することなく使用した。
1H NMR (500 MHz, CD
3OD) δ 8.36 (q, J = 13.7 Hz, 2H), 8.13 - 8.08 (m, 2H), 7.95 - 7.90 (m, 2H), 7.46 - 7.41 (m, 1H), 7.35 (dd, J = 8.3, 2.2 Hz, 1H), 6.72 (t, J = 12.4 Hz, 1H), 6.48 (d, J = 13.8 Hz, 1H), 6.35 (d, J = 13.4 Hz, 1H), 5.90 (ddd, J = 14.6, 7.6, 6.5 Hz, 1H), 5.49 - 5.37 (m, 1H), 4.99 (t, J = 3.8 Hz, 1H), 4.58 (dd, J = 5.3, 1.0 Hz, 1H), 4.32 (t, J = 6.9 Hz, 2H), 4.26 (t, J = 7.1 Hz, 2H), 4.01 - 3.79 (m, 7H), 3.37 (s, 3H), 2.66 (td, J = 8.0, 7.4, 5.3 Hz, 2H), 2.21 (td, J = 7.0, 4.0 Hz, 2H), 1.81 - 1.75 (m, 12H).
13C NMR (126 MHz, MeOD) δ 175.7, 172.7, 166.6, 155.7, 154.2, 146.3, 143.3, 142.7, 141.4, 141.2, 130.8, 130.6, 129.7, 126.7, 126.7, 125.9, 123.0, 120.0, 111.1, 110.1, 105.6, 103.7, 102.8, 101.6, 64.7, 51.3, 49.6, 48.6, 48.4, 48.2, 43.1, 38.6, 30.0, 26.3.C
39H
48N
2O
9S(MH
+)のHRMS(ESI)計算値 721.3153、実測値 721.3144。
(S10):6(0.13g、0.18mmol、1当量)のCH
2Cl
2(12mL、0.015M)中の濃青色の溶液を、-78℃に冷却し(ドライアイスおよびアセトン浴)、アルゴン下で脱気した。BBr
3(1.0M CH
2Cl
2溶液2.1mL、2.1mmol、12当量)をゆっくり添加すると、反応物の色が赤褐色に遷移した。温度を-78℃に維持しながら、反応物をアルゴン下で4時間にわたって撹拌し、その時点で、H
2O(10mL)を添加することによって反応をクエンチすると、すぐに青色に戻った。LC/MS分析によって、出発材料が完全に消費されたことが明らかになった。室温に加温した後、有機溶媒を減圧下で蒸発させた。残りの青色の水溶液を、逆相クロマトグラフィー(30gのC18金カラム、0~60%MeCN/H
2O)によって精製して、化合物S10のジアステレオマー混合物(57mg、0.094mmol、53%)を濃青色の固体として得た。C
35H
36N
2O
6S(MH
+)のHRMS(ESI)計算値 613.2367、実測値 613.2360。
(7):S10のジアステレオマー混合物(43mg、0.070mmol)を、MeOH(5.8mL)およびHCl水溶液(0.30M、17mL)に溶解させた。この反応混合物を60℃に7時間加熱し、その間に色が濃青色から緑青色に遷移した。室温に冷却した後、反応物を減圧下で乾燥させて、NMR分析によって確認される通り、S10の単一ジアステレオマーを得た。この平衡化したメチルエステル中間体の粗製固体を、MeOH(3.5mL)およびLiOH水溶液(2.0M、3.5mL)に再溶解させた。得られた青色の溶液を、室温で2.5時間撹拌し、その時点で、LC/MS分析によって7への完全な変換が明らかになった。飽和NaHCO
3水溶液(3.0mL)を添加して、反応をクエンチした。MeOHを真空中で除去した後、粗製の水性混合物を、逆相クロマトグラフィー(30gのC18金カラム、0~70%MeCNと0.05%ギ酸/H
2Oと0.05%ギ酸)によって精製して、7(34mg、0.057mmol、82%)を青色の固体として得た。
1H NMR (500 MHz, DMSO-d
6) δ 8.08 (s, 1H), 8.04 (s, 1H), 7.99 (s, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.89 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.23 (d, J = 8.3 Hz, 1H), 4.57 (dd, J = 11.4, 4.7 Hz, 1H), 4.51 (dd, J = 11.3, 4.6 Hz, 1H), 4.34 (d, J = 10.3 Hz, 1H), 4.24 (d, J = 9.5 Hz, 1H), 3.89 (t, J = 11.7 Hz, 1H), 3.80 (t, J = 12.9 Hz, 1H), 2.75 (t, J = 12.4 Hz, 1H), 2.43 (d, J = 12.2 Hz, 1H), 2.35 (dd, J = 7.0, 4.5 Hz, 1H), 2.28 (d, J = 12.9 Hz, 1H), 1.88 (dd, J = 12.2, 5.0 Hz, 1H), 1.77 - 1.62 (m, 13H), 1.34 (q, J = 11.8 Hz, 1H).
13C NMR (126 MHz, DMSO-d6) δ 169.4, 166.3, 162.4, 145.1, 143.9, 142.9, 140.4, 140.0, 139.3, 138.6, 129.2, 128.4, 124.9, 122.1, 118.8, 114.4, 109.7, 109.3, 107.9, 70.2, 68.4, 48.2, 46.3, 42.1, 39.4, 33.5, 29.2, 26.3, 25.9, 25.6, 25.4. C
35H
36N
2O
6S (MH
+)のHRMS(ESI)計算値 599.2210、実測値 599.2204.化合物7は、NMR分析によって帰属したsyn-syn環接合点立体化学を有する単一ジアステレオマーであった(図6)。環接合点(位置12、14、および15)に位置しているすべての3つのメチンプロトンは、syn,synジアステレオマーと合致する、高磁場軸方向のα-窒素プロトン(3.88(tq、J=13.3、4.5Hz、2H)-位置10および17)との1D-NOESY相互作用を示した。さらに、これらのプロトンはいずれも、低磁場エカトリアルα-窒素プロトン(4.40~4.24(m,2H)-位置10および17)に対してNOEを示さなかったが、それは任意の他のジアステレオマー構造のうちの少なくとも1つのメチンプロトンについても予測される。また、syn-synジアステレオマーの形成と合致して、α-酸素プロトン(位置14および15)は、ほぼ同じカップリング定数を示したが、このことは、これらのプロトンが類似の環系にあることを示している。
(S11):7(6.0mg、0.010mmol、1当量)のDMF(0.20mL、0.05M)中の溶液に、TSTU(4.5mg、0.015mmol、1.5当量)およびDIPEA(1.8μL、0.020mmol、2当量)を添加した。この青色の溶液を室温で1.5時間撹拌し、その時点で、LC/MS分析によってS11への完全な変換が明らかになった。次に、混合物をエーテル(14mL)中で沈殿させ、遠心分離し(6000rpm、5分)、上清をデカンテーションした。このエーテル洗浄を繰り返した後、濃青色の固体を真空中で乾燥させて、S11(6.9mg、0.0098mmol、98%)を得、それをさらに精製することなく使用した。
(8):S11(3.0mg、0.0043mmol、1当量)のDMF(0.15mL、0.03M)中の溶液に、NH
2-PEG
8-COOH(2.3mg、0.0053mmol、1.2当量)およびDIPEA(N,N-ジイソプロピルエチルアミン、3.8μL、0.022mmol、5当量)を添加した。この青色の溶液を室温で1.5時間撹拌し、その時点で、LC/MS分析によってS12の完全な消費が明らかになった。得られた溶液を、1:1のエーテルおよびヘキサン(14mL)中で沈殿させ、遠心分離し(4000rpm、5分)、上清を除去した。この磨砕を繰り返し、得られた青色の残留物を、逆相クロマトグラフィー(5.5gのC18Aq金カラム、0~70%MeCN/H
2Oと0.05%ギ酸)によって精製して、8(2.3mg、0.0023mmol、54%)を青色の固体として得た。C
53H
71N
3O
15S(MH
+)のHRMS(ESI)計算値 1022.4679、実測値 1022.4679。
(S12):8(3.4mg、0.0033mmol、1当量)のDMF(0.17mL、0.02M)中の溶液に、TSTU(1.5mg、0.0050mmol、1.5当量)およびDIPEA(1.7μL、0.010mmol、3当量)を添加した。この青色の反応混合物を、室温で2時間撹拌し、その時点で、LC/MS分析によって完全な変換が明らかになった。溶液を1:1のエーテル/ヘキサン(14mL)中で沈殿させ、遠心分離し(6500rpm、5分)、上清をデカンテーションした。この沈殿を繰り返した後、青色の残留物を減圧下で乾燥させて、S12(3.4mg、0.0030mmol、91%)を得、それをさらに精製することなく使用した。
(9):S12(0.98mg、8.8×10
-4mmol、1当量)のDMSO(0.12μL、0.008M)中の溶液に、アミノファロイジントシレート(0.80mg、8.8×10
-4mmol、1当量)およびDIPEA(0.73μL、0.0042mmol、5当量)を添加した。この溶液を室温で1時間撹拌し、その時点で、LC/MS分析によって、アミノファロイジントシレートの消費が明らかになった。青色の反応混合物を、1.5:1のエーテル/ヘキサン(1.4mL)中で沈殿させ、遠心分離し(6000rpm、30秒)、上清を除去した。この磨砕を合計3回実施した。減圧下で乾燥させた後、青色の残留物を、逆相分取HPLC(20~95%MeCN/H
2Oと0.1%ギ酸)によって精製し、凍結乾燥して、9(1.2mg、6.6×10
-4mmol、76%)を青色の固体として得た。C
88H
118N
12O
24S
2([M+2H
+]
+2)のHRMS(ESI)計算値 896.3984、実測値 896.3971。
(実施例2)
配座が束縛されたヘプタメチンシアニンフルオロフォアの例示的な合成
インドレニンおよびビスビニル性アミドを、10:1のEtOH/AcOH中で合わせ、70℃に加熱した。2時間後、混合物をCH
2Cl
2および飽和重炭酸ナトリウムで抽出した。有機層を硫酸ナトリウムで乾燥させ、濃縮し、次に順相クロマトグラフィーで精製して、生成物を提供した。
ケトンのCH
2Cl
2溶液に、10当量のアクロレインジメチルアセタールおよび0.4当量のHG-2を添加した。溶液を室温で18時間維持し、濃縮し、順相クロマトグラフィーによって精製して、生成物を提供した。
ケトンのTHF溶液を、1N HClで処理した。30分後、混合物をCH
2Cl
2および飽和重炭酸ナトリウムで抽出した。有機層を硫酸ナトリウムで乾燥させ、濃縮し、次に精製することなく次のステップで使用した。
ケトン、CH
2Cl
2、およびピリジンを-78℃に冷却し、Tf
2O(トリフルオロメタンスルホン酸無水物)を添加した。溶液を室温に加温し、CH
2Cl
2および飽和重炭酸ナトリウムで抽出した。順相クロマトグラフィーで精製して、生成物を提供した。
トリフレート、ボロン酸、およびPd(PPh3)4を、1:1のiPrOH:H2Oに溶解させ、90℃に加熱した。18時間後、混合物をCH2Cl2および飽和重炭酸ナトリウムで抽出した。有機層を硫酸ナトリウムで乾燥させ、濃縮し、次に順相クロマトグラフィーで精製して、生成物を提供した。
(実施例3)
配座が束縛されたシアニンフルオロフォアの特徴付け
配座が束縛された化合物4および7の分光学的特性を、制限されていない化合物10の分光学的特性と比較した。
aメタノール中、
bpH7.4のPBS中、
cH
2O中
化合物4および7は、配座の束縛の特徴的な特色を示した。量子収率は、化合物10による0.15(MeOH)から、それぞれ化合物4および7による0.69(MeOH)および0.55(PBS)に増大する(表1)。このことは、どちらの場合もおよそ25nmのλ
maxの偏移と共に生じる。蛍光寿命および量子収率は共に、光異性化を受ける従来のペンタメチンシアニン10とは異なり、大部分が溶媒粘度に非感受性である(表2)。
*積分球による参照Φ
F-他のΦ
F値は、比較方法によって算出した
さらに、化合物4の発光(四角)は、やはり化合物10(丸)とは異なり、温度に対して非感受性である(図7)。このことはやはり、高温であるほど効率的になる化合物10の光異性化に起因している。制限されていないシアニン10と比較して、化合物4および7の実質的により長い寿命は、蛍光寿命画像化顕微鏡法(FLIM)の著しい潜在可能性を示している。
(実施例4)
配座が束縛されたシアニンフルオロフォアを用いる単一分子局在性顕微鏡法
単一分子局在性顕微鏡法(SMLM)の中心的な特色は、蛍光性状態と非蛍光性状態との間のフルオロフォアの光活性化または変換である。この文脈では、(1)チオール-および(2)ホスフィン-ポリエン付加物の可逆的形成、ならびに(3)イミン様C2-N二重結合の逐次的な還元/酸化の3つの形式のシアニン反応性が適用される。
実施例3の化合物4、7、および10を評価した。最も際立つことには、NaBH4による還元後、UV光によって誘導された化合物4の再生は、制限されていないシアニンと比較して、劇的に増強された。2.0当量のNaBH4(1:1のDMSO:MeOH中2.5mM)による化合物4の還元、その後のUV光(365nm、5mW/cm2)を用いた4:1のPBS(50mM、pH7.4):DMSO中20μM溶液の光分解によって、5分後に化合物4(四角)で最大シアニン吸光度の38%が回収されたが、化合物10(丸)では、30分後の最大回収率はわずか6%であった(図8)。吸光度は、365nm照射(5mW/cm2)の時間関数として、λmax(化合物4については640nm、化合物10については660nm)で測定した。
ホスフィンおよびチオール付加物の形成を、化学的および単一分子画像化の文脈でも評価した。TCEP(トリス(2-カルボキシエチル)ホスフィン)を、化合物7およびAF647(Alexa Fluor 647、ThermoFisher Scientificを介して利用可能)(各10μM)に添加した(Vaughan et al.,JACS 2013,135(4):1197-1200)。Tris(0.20M、pH9.0)中のTCEP濃度の関数として、λmax(化合物7については670nm、AF647については650nm)における吸光度。化合物7(丸)は、ポリエン-ヘテロ原子付加物の形成に抵抗性であるように見えた(図9)。
(実施例5)
配座が束縛されたファロイジンコンジュゲートの結合および可視化
実施例3のファロイジンコンジュゲート9を初期広視野研究で適用して、細胞F-アクチンを可視化した。これらの研究には、広く使用されている商業的に利用可能なAlexa Fluor 647-ファロイジンコンジュゲート(AF647-ファロイジン)との比較が含まれていた。化合物9の還元およびUV-活性化(370nm)によって、AF647-ファロイジンと比較して回収が劇的に改善された。化合物9の光安定性は、AF647-ファロイジンの光安定性とほとんど区別することができなかった。
ファロイジンコンジュゲート9を、3D PALM様の超解画像化を用いて、二方向画像化スキーム(BP)(Ram et al.,J.,Biophys J 2008,95:6025)をTRABI(Franke et al.Nat Methods 2017,14:41)と組み合わせて利用して評価して、単一分子強度を正確に定量すると同時にTRABI-BP画像化を実施した。標識化および還元(26mM NaBH4)、その後の非脱気リン酸緩衝食塩水(PBS)中での画像化によって、U2OS(ヒト骨肉腫上皮)細胞におけるアクチン細胞骨格の高品質3D超解画像を提供した(図10)。フレーム当たり平均5181の光子(中央値)を、単一の活性化色素から検出すると同時に、連続フレームにおいて活性な放射体を追跡することによって、光退色または非蛍光性形態への変換の前に、6961の合成光子数(中央値)を得た。これは、実験的に測定された、横方向に5~7nmおよび軸方向に約20nmの位置決定精度に相当する。640nmまたは660nmのいずれかによる励起を用いることができ、後者は光子収率をいくらか改善する。UVレーザーは回収を促進することができるが、単に励起レーザー(640または660のいずれか)を使用して得られた光活性化は、SMLMに適した放射体密度を生じさせるのに十分であった。SMLM実験では、405nmの光を非常にごく短期間適用した場合、還元状態の回収は、ほぼ定量的に進行した。それに対して、AF647-ファロイジンを還元/回収の順に供した場合、再構築を得ることができなかった。還元的方法を使用して化合物9により得られた画像を、標準dSTORM緩衝液条件下でAF647と比較した。コンジュゲート9は、AF647-ファロイジンと比較して、わずかに改善された場合には類似の光子カウント(9:フレーム当たり3721、追跡5107、AF647:フレーム当たり3422、追跡3737)、ならびにそれぞれ5.2nmおよび5.9nmの位置決定精度を示した(図11~13)。図11は、図10の画像から算出した、横方向および軸方向の位置決定精度を示す。図12は、標準dSTORM光スイッチング緩衝液における、化合物93D(左)について示されたデータの単一フレーム(黒色)および追跡(灰色)中央値と、化合物9(中央)およびAF647(右)の2D画像モードの同等の測定値に関する、単一分子光子強度の比較を示す棒グラフである。図13は、化合物93D(6.9nm)、化合物92D(5.2nm)およびAF6472D(5.9nm)の実験的に決定された横方向の位置決定精度を示す棒グラフである。
(実施例6)
配座が束縛されたシアニンフルオロフォアを用いる腫瘍可視化
腫瘍を有する対象を、処置のために同定し、選択する。対象は、臨床所見に基づき、かつ/または腫瘍の存在を証明するための試験を実施することによって選択することができる。
式Iによる化合物、その薬学的に許容される塩、またはその医薬組成物を、臨床医によって有効であると決定された用量で投与することによって、対象を処置する。化合物を、任意の適切な手段、例えば静脈内または皮下注射によって投与する。一部の例では、化合物は、腫瘍に直接注射される。
可視化は、腫瘍に化合物を結合させるのに十分な時間が経過した後に実施することができる。例えば、照射は、化合物投与の数時間後から数日後、例えば化合物投与の1~7日後に実施することができる。対象の標的化部分に、シアニンフルオロフォアの蛍光を誘導するのに適した波長と選択された強度を有する有効量の光の適用を標的化し、それによってシアニンフルオロフォアを励起することによって、投与した化合物に照射する。有利には、照射の標的となる対象の部分は、腫瘍に近接している。化合物の蛍光は、蛍光画像化の分野の当業者に公知の任意の適切な方法によって検出される。蛍光ガイド手術を使用して、組織切除の位置および程度を決定する。
一部の場合では、対象は、腫瘍を有すると疑われ、腫瘍の存在は、対象に化合物を投与し、疑わしい腫瘍部位における化合物の蛍光をモニタリングすることによって確認される。疑わしい腫瘍部位における化合物および蛍光の蓄積により、腫瘍が存在すると診断される。
図14を参照すると、腫瘍110を有する対象100は、腫瘍細胞表面上の抗原または受容体を認識し、結合することができる抗体またはリガンドを含む式Iによる化合物で処置することができる。図14に示される例では、化合物120は、静脈内注射を介して投与される。抗体またはリガンド部分が腫瘍に結合するにつれて化合物が腫瘍部位に優先的に蓄積する時間が経過するのを待つ。その後、対象の標的部分に、外部光アプリケーター130を使用して、望ましい波長の有効量の遠赤またはNIR光エネルギーを選択的に照射する。光アプリケーター130は、腫瘍110の領域に限定された標的領域に光を適用し、それによって化合物の蛍光を生じさせる。腫瘍は、蛍光を検出することによって可視化される。
治療有効量の第2の薬剤を、式Iによる化合物またはその塩と併用投与することができる。化合物(またはその塩)および第2の薬剤は、別個に、または単一組成物中で一緒に投与することができる。第2の薬剤は、同じ経路または異なる経路によって投与することができる。同時に投与する場合、化合物(またはその塩)および第2の薬剤は、単一の医薬組成物に組み合わせることができ、または2つの医薬組成物として同時に投与することができる。第2の薬剤は、例えば、抗腫瘍剤または血管新生阻害剤であってもよい。
(実施例6)
ビス-スルホン化された配座が束縛されたシアニンフルオロフォアの合成
配座が束縛されたシアニン色素17の合成は、図15に記載されている。ラセミ体のメチル6-メチル-7-オキソオクタノエート11から出発して、フィッシャーインドール合成を、4-ヒドラジニルベンゼンスルホン酸(4-hydrazineylbenzenesulfonic acid)を用いて行って、12を収率76%で生成した。その後のN-アルキル化を、2-(2-ヨードエチル)-1,3-ジオキソランの存在下で、95℃において実施して、生成物3を収率51%で得た。シアニン骨格を、化合物13、1-(ブタ-3-エン-1-イル)-2,3,3-トリメチル-3H-インドール-1-イウム-5-スルホネートカリウム塩、およびN-((1E,3E)-3-(フェニルイミノ)プロパ-1-エン-1-イル)アニリンの3つの部分を反応させることによって形成して、所望の生成物14を収率24%で得た。交差メタセシスを、アクロレインジメチルアセタールを用いて、ホベイダ-グラブス触媒およびBu4NBrの下で実施して、15を生成した。非常に重要な分子内マイケル付加関連環化反応カスケードを、BBr3およびBu4NBrの存在下で進行させて、ジアステレオマー混合物を得、それをHCl/MeOHの平衡化条件下で60℃において2つのジアステレオマー16に変換した。収率は、3つのステップで41%であった。メチルエステルをLiOHで加水分解して、逆相C-18カラム精製後に、最終生成物17を収率43%で得た。
実験の詳細
ナトリウム3-(5-メトキシ-5-オキソペンチル)-2,3-ジメチル-3H-インドール5-スルホネート(12)。メチル6-メチル-7-オキソオクタノエート(1.86g、10mmol)を、撹拌したp-ヒドラジノベンゼンスルホン酸(2.24g、10mmol)の酢酸(6mL)中の溶液に添加した。溶液を4時間加熱還流し、次に室温に冷却した。溶媒を蒸発させた。Na2CO3(1.06g、10mmol)を、メタノール(15mL)に溶解させた残留物に添加した。得られた混合物を室温で15時間撹拌した。溶媒を蒸発させ、残留物を、逆相カラムクロマトグラフィーによって精製して、12(2.72g、7.6mmol、76%)を得た。1H NMR (400 MHz, CD3OD): δ 7.84-7.80 (m, 2H), 7.46 (d, 1H, J = 7.8 Hz), 3.57 (s, 3H), 2.28 (s, 3H), 2.16 (dt, 2H, J = 7.4, 2.3 Hz), 2.04-1.84 (m, 2H), 1.50-1.41 (m, 2H), 1.34 (s, 3H), 0.74-0.56(m, 2H). LC-MS (ESI) 340 (M+).
1-(2-(1,3-ジオキソラン-2-イル)エチル)-3-(5-メトキシ-5-オキソペンチル)-2,3-ジメチル-3H-インドール-1-イウム-5-スルホネート,ナトリウム塩(13)。密封容器中、化合物12(2.25g、6.23mmol)を、CH3CN(20mL)中、2-(2-ブロモエチル)-1,3-ジオキソラン(1.1mL、9.34mmol)、NaHCO3(1.57g、18.6mmol)、およびNaI(1.40g、9.34mmol)と合わせた。反応物を、95℃においてアルゴン雰囲気下で15時間撹拌すると、溶液の色が褐色に変化した。粗製生成物を減圧下で濃縮し、残留物を逆相カラムクロマトグラフィーによって精製して、13(1.45g、3.14mmol、51%)を得た。1H NMR (400 MHz, CD3OD): δ 7.61 (d, 1H, J = 7.8 Hz), 7.50 (s, 1H), 6.62 (d, 1H, J = 7.8 Hz), 4.97 (m, 1H), 4.03-3.93 (m, 4H), 3.71-3.65 (m, 2H), 3.58 (s, 3H), 2.19-2.10 (m, 3H), 2.06-2.20 (m, 2H), 1.96-1.90 (m, 2H), 1.56-1.46 (m, 2H), 1.43 (s, 3H), 1.05-0.82 (m, 2H). LC-MS (ESI) 440 (M+).
1-(2-(1,3-ジオキソラン-2-イル)エチル)-2-((1E,3E)-5-((E)-1-(ブタ-3-エン-1-イル)-3,3-ジメチル-5-スルホナトインドリン-2-イリデン)ペンタ-1,3-ジエン-1-イル)-3-(5-メトキシ-5-オキソペンチル)-3-メチル-3H-インドール-1-イウム-5-スルホネート(14)。無水酢酸(0.87mL、9.21mmol)を、13(2.10g、4.54mmol)、1-(ブタ-3-エン-1-イル)-2,3,3-トリメチル-3H-インドール-1-イウム-5-スルホネート,カリウム塩(1.51g、4.54mmol)、マロンアルデヒドビス(フェニルイミン)一塩酸塩(1.21g、5.45mmol)、およびEt3N(3.2mL、22.7mmol)のMeOH(23mL)中の溶液に添加した。室温で撹拌しながら、追加の無水酢酸(0.87mL、9.21)を、最初の1.5時間中30分ごとに添加し、合計3.48mL(36.8mmol)にした。反応中、溶液の色は、赤色から緑色、紫色、最後に濃青色に遷移した。合計15時間の反応時間が経過した後、LC/MS分析によって、3種類の可能なシアニン生成物の、およそ2:1:1比の非対称性の所望の生成物と望ましくない対称性シアニンの混合物が明らかになった。2:1のジエチルエーテル/ヘキサン溶液(180mL)を添加すると、沈殿物が形成され、それを遠心分離によって収集した。この固体を、2:1のジエチルエーテル/ヘキサンと共に3回磨砕し、真空中で乾燥させた後、濃青色の残留物を、逆相クロマトグラフィーを用いて精製して、14(946mg、1.09mmol、24%)を濃青色の固体として得た。1H NMR (400 MHz, CD3OD): δ 8.35-8.25 (m, 2H), 7.89-7.85 (m, 4H), 7.35 (dd, 2H, J = 20.1, 8.4 Hz ), 6.72 (m, 1H), 6.40 (t, 2H, J = 12.9 Hz ), 5.89 (m, 1H), 5.04-4.95 (m 3H), 4.26-4.23 (m, 4H), 3.96-3.93 (m, 2H), 3.85-3.81 (m, 2H), 3.53 (s, 3H), 2.62-2.57 (m, 2H), 2.47 (s, 1H), 2.23-2.12 (m, 5H), 1.74 (s, 3H), 1.73 (s, 3H), 1.70 (s, 3H), 1.50-1.42 (m, 2H), 0.93 (m, 1H), 0.63 (m, 1H). LC-MS (ESI) 769 (M+).
(7aS,8aR,9aS)-20-(5-メトキシ-5-オキソペンチル)-17,17,20-トリメチル-6,7,7a,8a,9,9a,10,11,17,20-デカヒドロベンゾ[2’,3’]インドリジノ[8’,7’:5,6]ピラノ[2,3-g]インドロ[2,1-a]イソキノリン-5-イウム-2,15-ジスルホネート(16)。アクロレインジメチルアセタール(1.16mL、0.98mmol)を、4(850mg、0.98mmol)、ホベイダ-グラブス触媒第2世代(313mg、0.50mmol)およびテトラブチルアンモニウムブロミド(946mg、2.94mmol)の無水CH2Cl2(50mL)中の溶液に添加した。フラスコを排気し、アルゴンで5時間、およそ30分ごとにフラッシュした。溶液をさらに10時間撹拌した。1:1のジエチルエーテル/ヘキサン(360mL)溶液を、青色の混合物に添加し、沈殿物を遠心分離によって収集した。2回目の沈殿および遠心分離の後、ペレットを高真空下で乾燥させて、15(694mg、収率75%)を濃青色の固体として得、それをさらに精製することなく使用した。15(694mg、0.74mmol)およびテトラブチルアンモニウムブロミド(714mg、2.22mmol)のCH2Cl2(49mL)中の濃青色の溶液を脱気し、アルゴン下で-78℃に冷却した。BBr3溶液(8.6mL、8.6mmol、CH2Cl2中1.0M)をゆっくり添加すると、反応物の色が赤褐色に遷移した。反応物を、アルゴン下で-78℃の温度において4時間撹拌し、その時点で、H2O(40mL)を添加することによって反応をクエンチすると、すぐに青色に戻った。室温に加温した後、有機溶媒を減圧下で蒸発させた。残りの青色の水溶液を、逆相クロマトグラフィーによって精製して、ジアステレオマー混合物の生成物(367mg)を得た。ジアステレオマー混合物(367mg、0.50mmol)を、MeOH(41mL)およびHCl水溶液(0.30M、120mL)に溶解させた。この反応混合物を、60℃に7時間加熱し、その間に色が濃青色から緑青色に遷移した。溶媒を蒸発させた後、粗製混合物を、逆相クロマトグラフィーによって精製して、6(294mg、0.40mmol、収率41%)を青色の固体として得た。1H NMR (400 MHz, d6-DMSO): δ 8.00 (s, 1H), 7.93 (s, 1H), 7.82 (d, 1H, J = 1.2 Hz), 7.72 (m, 1H), 7.64 (dd, 1H, J = 8.2, 1.6 Hz), 7.60 (dd, 1H, J = 8.2, 1.6 Hz), 7.26 (d, 1H, J = 8.2 Hz), 7.16 (dd, 1H, J = 8.2, 2.0 Hz), 4.56 (dd, 1H, J = 11.3, 5.1 Hz), 4.48 (m, 1H), 4.30-4.18 (m, 2H), 3.84-3.77 (m, 2H), 3.26 (s, -OMe), 3.45 (s, -OMe), 2.70 (m, 1H), 2.46-2.30 (m, 3H), 2.28-2.12 (m, 4H), 1.83 (m, 1H), 1.70 (s, 3H), 1.69 (s, 3H), 1.65 (s, 3H), 1.45-1.28 (m, 3H), 0.82 (m, 1H), 0.57 (m, 1H). LC-MS (ESI) 735 (M+).
5-((7aS,8aR,9aS)-17,17,20-トリメチル-2,15-ジスルホナト-6,7,7a,8a,9,9a,10,11,17,20-デカヒドロベンゾ[2’,3’]インドリジノ[8’,7’:5,6]ピラノ[2,3-g]インドロ[2,1-a]イソキノリン-5-イウム-20-イル)ペンタノエート(17)。化合物16(239mg、0.32mmol)を、MeOH(14mL)およびLiOH水溶液(2.0M、14mL)に溶解させた。得られた青色の溶液を、室温で2.5時間撹拌した。飽和NaHCO3水溶液(12.0mL)を添加して、反応をクエンチした。MeOHを真空中で除去した後、粗製の水性混合物を、逆相クロマトグラフィーによって精製して、17(101mg、0.14mmol、収率43%)を青色の固体として得た。1H NMR (400 MHz, d6-DMSO): δ 8.01 (d, 1H, J = 3.9 Hz), 7.92 (s, 1H), 7.82 (d, 1H, J = 1.2 Hz), 7.72 (m, 1H), 7.63 (dd, 1H, J = 8.2, 1.6 Hz), 7.60 (dd, 1H, J = 8.2, 1.6 Hz), 7.25 (d, 1H, J = 8.2 Hz), 7.16 (dd, 1H, J = 8.4, 3.7 Hz), 4.56 (m, 1H), 4.48 (td, 1H, ), 4.30-4.19 (m, 2H), 3.84-3.76 (m, 2H), 2.70 (m, 1H), 2.42-2.30 (m, 3H), 2.26-2.17 (m, 2H), 2.08-2.01 (m, 2H), 1.84 (m, 1H), 1.69 (s, 3H), 1.68 (s, 3H), 1.65 (s, 3H), 1.40-1.30 (m, 3H), 0.83 (m, 1H), 0.63 (m, 1H). LC-MS (ESI) 721 (M+).
VII.代表的な実施形態
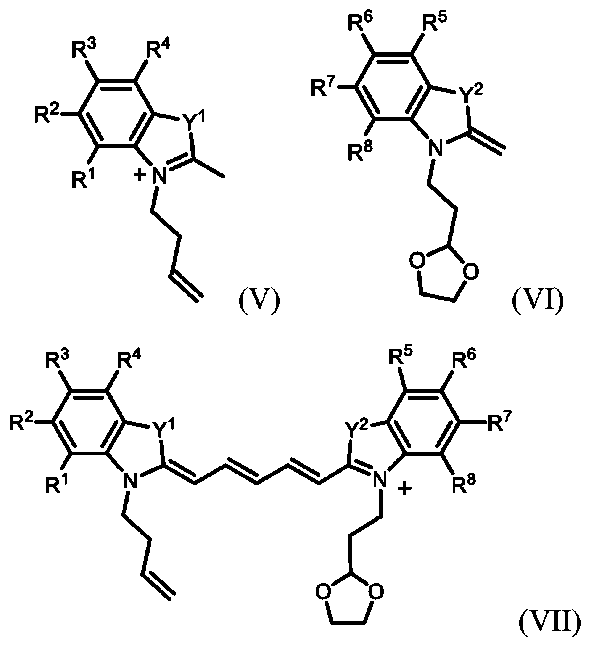
ある特定の代表的な実施形態を、以下の番号付きの項目に開示する。
1.式I:
[式中、Aは、
であり、各「
*」は、Aの結合点を示し、
によって表される結合は、原子価の要件を満たすために必要とされるように、単結合または二重結合であり、R
1~R
9およびR
11は、独立に、H、重水素、アルキル、ヘテロアルキル、-N(R
a)
2、スルホネート、アルキルスルホネート、アミノ、アミノアルキル、-C(O)OR
a、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基であり、ここで、R
aは、H、重水素、アルキル、またはヘテロアルキルであり、R
10は、H、重水素、O、アルキル、アリール、アミノ、スルホネート、トリフレート、-C(O)OR
b、-OR
b、-N(R
b)
2、ヘテロアルキル、ヘテロアリール、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基であり、ここで、各R
bは、独立に、H、重水素、アルキル、ヘテロアルキル、アリール、またはヘテロアリールであり、Y
1およびY
2は、独立に、C(R
c)
2、N(R
d)、S、O、またはSeであり、ここで、各R
cは、独立に、H、重水素、アルキル、-(OCH
2CH
2)
xOH(xは、≧2の整数である)、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基であり、各R
dは、独立に、H、重水素、アルキル、またはヘテロアルキルである]
による化学構造を有する化合物、またはその立体異性体もしくは薬学的に許容される塩。
2.式IA、IB、IC、またはID:
による化学構造を有する、項目1の化合物。
3.R3およびR6のうちの少なくとも1つが、スルホネート、-C(O)ORa、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基である、項目1または項目2の化合物。
4.Y1およびY2が、C(Rc)2であり、各Rcが、独立に、C1~C3アルキル、-(CH2)nC(O)Re、またはHであり、nが、≧1の整数であり、Reが、コンジュゲート可能な部分、標的化剤または薬物である、項目1~3のいずれか1つの化合物。
5.Y1およびY2が、C(CH3)2である、項目4の化合物。
6.R1、R2、R4、R5、R7、およびR8が、Hである、項目1~5のいずれか1つの化合物。
7.式IIまたは式III:
による化学構造を有する、項目1~6のいずれか1つの化合物。
8.各Rcが、-CH3である、項目7の化合物。
9.上記化合物が式IIによる化学構造を有し、R1~R10が、Hである、項目7または項目8の化合物。
10.R1、R2、R4、R5、およびR7~R11が、Hであり、R3およびR6が、独立に、-SO3または-CO2Raである、項目7または項目8の化合物。
11.上記化合物が式IIによる化学構造を有し、R9およびR10が、Hであり、R3およびR6のうちの少なくとも1つが、コンジュゲート可能な部分、標的化剤または薬物を含む基である、項目7または項目8の化合物。
12.上記化合物が式IIIによる化学構造を有し、R1~R9およびR11が、Hであり、R10が、H、O、トリフレート、アリール、-ORbまたは-N(Rb)2である、項目7または項目8の化合物。
13.上記化合物が式IIIによる化学構造を有し、R9およびR11が、Hであり、R3、R6、およびR10のうちの少なくとも1つが、コンジュゲート可能な部分、標的化剤または薬物を含む基である、項目7または項目8の化合物。
14.項目1~13のいずれか1つによる化合物および薬学的に許容される担体を含む、医薬組成物。
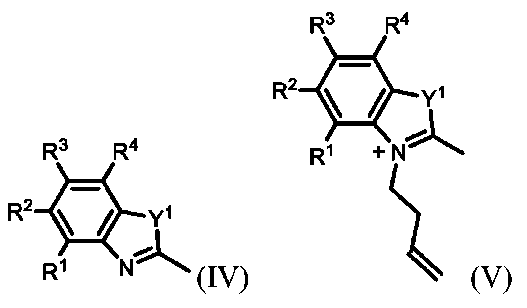
15.Aが
である、項目1による化合物を作製するための方法であって、
式IVによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式Vによる化合物を生成するステップと、
式Vによる化合物および式VIによる化合物を含む溶液を、N-((1E,3Z)-3-(フェニルアミノ)プロパ-1-エン-1-イル)アニリンと合わせて、式VIIによる化合物を形成するステップと、
式VIIによる化合物を含む溶液を、ルテニウム触媒の存在下で、3,3-ジメトキシ-1-プロペンと合わせて、式VIIIによる化合物を提供するステップと、
式VIIIによる化合物を、(i)CHCl
3およびH
2SO
4の混合物、または(ii)CH
2Cl
2中BBr
3と合わせて、式IXによる化合物を提供するステップと
を含む、方法。
16.ルテニウム触媒が、(1,3-ビス-(2,4,6-トリメチルフェニル)-2-イミダゾリジニリデン)ジクロロ(o-イソプロポキシフェニルメチレン)ルテニウムである、項目15の方法。
17.Aが
である、項目1による化合物を作製するための方法であって、
式IVによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式Vによる化合物を生成するステップと、
式Xによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式XIによる化合物を生成するステップと、
式Vによる化合物および式XIによる化合物を含む溶液(ここで、式Vおよび式XIによる化合物は、同じであっても異なっていてもよい)を、(1E,4E)-1,5-ビス(ジメチルアミノ)-ペンタ-1,4-ジエン-3-オンと合わせて、式XIIによる化合物を生成するステップと、
式XIIによる化合物を含む溶液を、ルテニウム触媒の存在下で、3,3-ジメトキシ-1-プロペンと合わせて、式XIIIによる化合物を提供するステップと、
式XIIIによる化合物を、テトラヒドロフラン中1NのHClの溶液と合わせて、式XIVによる化合物を提供するステップと
を含む、方法。
18.ルテニウム触媒が、(1,3-ビス-(2,4,6-トリメチルフェニル)-2-イミダゾリジニリデン)ジクロロ(o-イソプロポキシフェニル-メチレン)ルテニウムである、項目17の方法。
19.式XIVによる化合物を含む溶液を、トリフルオロメタンスルホン酸無水物(Tf
2O)と合わせて、式XVによる化合物を提供するステップ
をさらに含む、項目17の方法。
20.式XVによる化合物を含む溶液を、パラジウム触媒の存在下で、R
g-C
6H
4-B(OH)
2と合わせて、式XVIによる化合物を提供するステップ
(式中、R
gは、R
a、-COOR
a、または-OR
aであり、ここでR
aは、H、重水素、アルキルまたはヘテロアルキルである)
をさらに含む、項目19の方法。
21.式XVによる化合物を含む溶液を、式NH(R
20)(R
21)を有するアミンと合わせて、式XVIIによる化合物を提供するステップ
(式中、R
20およびR
21は、独立に、H、重水素、アルキル、ヘテロアルキル、アリールまたはヘテロアリールである)
をさらに含む、項目19の方法。
22.(i)R
20が、-(CR
h
2)
n-CH
2OHであり、ここで、各R
hが、独立に、H、重水素、ハロ、アルキル、またはアリールであり、nが、1、2、3、または4であり、(ii)R
21が、H、重水素、アルキル、ヘテロアルキル、アリール、またはヘテロアリールであり、上記方法が、式XVIIによる化合物を含む溶液を、求電子性基R
22を含む化合物と塩基条件下で合わせて、式XVIIIによる化合物を提供するステップ
をさらに含む、項目21の方法。
23.R1~R11のうちの少なくとも1つが標的化剤を含む項目1~13のいずれか1つによる化合物を使用するための方法であって、化合物を、標的化剤と結合することが可能である標的を含む試料と、標的化剤と標的を結合させるのに有効な条件下で合わせるステップと、標的に結合した化合物を可視化することによって、標的を画像化するステップとを含む、方法。
24.化合物を可視化することが、可視または近赤外範囲の波長と選択された強度を有する所定量の光の標的化された適用により、試料に照射することを含み、ここで、所定量の光は、化合物の蛍光を生じさせるのに十分であり、さらに、化合物によって放出された任意の蛍光を検出することを含む、項目23の方法。
25.化合物を試料と合わせるステップが、in vitro、ex vivo、またはin vivoで実施される、項目23または項目24の方法。
26.標的を画像化する前に、化合物を還元剤と合わせるステップをさらに含む、項目23~25のいずれか1つの方法。
27.試料が、組織試料、生物学的流体、または対象内の標的領域である、項目23~26のいずれか1つの方法。
28.試料が、対象内の標的領域であり、前記方法が、化合物、または化合物を含む医薬組成物を、対象に投与するステップと、その後、対象の標的化部分への所定量の光の標的化された適用によって、化合物に照射するステップと、対象の標的化部分における化合物からの任意の蛍光を検出するステップとをさらに含む、項目27の方法。
29.標的領域が、腫瘍部位であり、対象の標的化部分が、腫瘍部位を含み、対象の標的化部分における蛍光を検出した後に、対象から腫瘍の少なくとも一部を切除するステップをさらに含む、項目28の方法。
30.活性酸素種を検出するための方法であって、項目1~13のいずれか1つによる化合物を還元剤と合わせて、還元化合物を提供するステップと、試料を還元化合物と接触させるステップであって、試料中に活性酸素種(ROS)が存在する場合、それにより還元化合物が酸化されて、項目1~13のいずれか1つによる化合物が再生される、ステップと、試料に、可視または近赤外範囲の波長と選択された強度を有する所定量の光を照射するステップであって、所定量の光は、還元化合物がROSによって酸化されて、項目1~13のいずれか1つに記載の化合物が再生される場合に蛍光を生じさせるのに十分である、ステップと、項目1~13のいずれか1つに記載の化合物によって放出された任意の蛍光を検出するステップであって、ここで、蛍光は、試料中のROSの存在を示す、ステップを含む、方法。
開示される発明の原理を適用することができる多くの実施形態が存在する可能性を考慮して、例示される実施形態は、単に本発明の好ましい例であり、本発明の範囲を制限すると解釈されるべきでないことを認識されたい。むしろ、本発明の範囲は、以下の特許請求の範囲によって定義される。したがって、本発明者らは、本発明のすべてがこれらの特許請求の範囲および趣旨に含まれることを主張する。
本発明は、例えば、以下の項目を提供する。
(項目1)
式I:
[式中、Aは、
であり、各「
*
」は、Aの結合点を示し、
によって表される結合は、原子価の要件を満たすために必要とされるように、単結合または二重結合であり、
R
1
~R
9
およびR
11
は、独立に、H、スルホネート、-N(R
a
)
2
、重水素、アルキル、ヘテロアルキル、アルキルスルホネート、アミノアルキル、-C(O)OR
a
、トリチル、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基であり、ここで、各R
a
は、独立に、H、重水素、アルキル、またはヘテロアルキルであり、
R
10
は、H、重水素、O、アルキル、アリール、アミノ、スルホネート、トリフレート、-C(O)OR
b
、-OR
b
、-N(R
b
)
2
、ヘテロアルキル、ヘテロアリール、トリチル、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基であり、ここで、各R
b
は、独立に、H、重水素、アルキル、ヘテロアルキル、アリール、またはヘテロアリールであり、
Y
1
およびY
2
は、独立に、C(R
c
)
2
、N(R
d
)、S、O、またはSeであり、ここで、各R
c
は、独立に、アルキル、H、重水素、-(OCH
2
CH
2
)
x
OH[xは、≧2の整数である]、トリチル、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基であり、各R
d
は、独立に、H、重水素、アルキル、またはヘテロアルキルである]
による化学構造を有する化合物、またはその立体異性体もしくは薬学的に許容される塩。
(項目2)
R
3
およびR
6
のうちの少なくとも1つが、スルホネート、-C(O)OR
a
、またはコンジュゲート可能な部分、標的化剤もしくは薬物を含む基である、項目1に記載の化合物。
(項目3)
Y
1
およびY
2
が、C(R
c
)
2
であり、各R
c
が、独立に、C
1
~C
3
アルキル、-(CH
2
)
n
C(O)R
e
、またはHであり、nが、≧1の整数であり、R
e
が、コンジュゲート可能な部分、標的化剤または薬物である、項目1または項目2に記載の化合物。
(項目4)
式中、
Y
1
およびY
2
が、C(CH
3
)
2
であり、または
R
3
およびR
6
が、スルホネートであり、一方のR
c
が、-(CH
2
)
n
C(O)R
e
である、項目3に記載の化合物。
(項目5)
R
1
、R
2
、R
4
、R
5
、R
7
、およびR
8
が、Hである、項目1~4のいずれか一項に記載の化合物。
(項目6)
前記コンジュゲート可能な部分が、
(式中、yは、≧1の整数である)、またはホスホロアミダイト基である、項目1~5のいずれか一項に記載の化合物。
(項目7)
式IIまたは式III:
による化学構造を有する、項目1~6のいずれか一項に記載の化合物。
(項目8)
各R
c
が、-CH
3
であるか、
R
3
およびR
6
が、スルホネートであるか、
一方のR
c
が、コンジュゲート可能な部分、標的化剤または薬物を含む基であるか、または
R
3
およびR
6
が、スルホネートであり、一方のR
c
が、コンジュゲート可能な部分、標的化剤または薬物を含む基である、項目7に記載の化合物。
(項目9)
前記化合物が、式IIによる化学構造を有し、R
1
~R
10
が、Hであるか、
前記化合物が、式IIによる化学構造を有し、R
9
およびR
10
が、Hであり、R
3
およびR
6
のうちの少なくとも1つが、コンジュゲート可能な部分、標的化剤もしくは薬物を含む基であるか、
前記化合物が、式IIIによる化学構造を有し、R
1
~R
9
およびR
11
が、Hであり、R
10
が、H、O、トリフレート、アリール、-OR
b
もしくは-N(R
b
)
2
であるか、または
前記化合物が、式IIIによる化学構造を有し、R
9
およびR
11
が、Hであり、R
3
、R
6
、およびR
10
のうちの少なくとも1つが、コンジュゲート可能な部分、標的化剤もしくは薬物を含む基である、項目7または項目8に記載の化合物。
(項目10)
R
1
、R
2
、R
4
、R
5
、およびR
7
~R
11
が、Hであり、R
3
およびR
6
が、独立に、-SO
3
または-CO
2
R
a
である、項目7または8に記載の化合物。
(項目11)
項目1~10のいずれか一項に記載の化合物および薬学的に許容される担体を含む、医薬組成物。
(項目12)
Aが
である、項目1に記載の化合物を作製するための方法であって、
式IVによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式Vによる化合物を生成するステップと、
式Vによる化合物および式VIによる化合物を含む溶液を、N-((1E,3Z)-3-(フェニルアミノ)プロパ-1-エン-1-イル)アニリンまたはN-((1E,3E)-3-(フェニルイミノ)プロパ-1-エン-1-イル)アニリンと合わせて、式VIIによる化合物を形成するステップと、
式VIIによる化合物を含む溶液を、ルテニウム触媒の存在下で、3,3-ジメトキシ-1-プロペンと合わせて、式VIIIによる化合物を提供するステップと、
式VIIIによる化合物を、(i)酸性化CHCl
3
または(ii)CH
2
Cl
2
中BBr
3
と合わせて、式IXによる化合物を提供するステップと
を含む、方法。
(項目13)
Aが
である、項目1に記載の化合物を作製するための方法であって、
式IVによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式Vによる化合物を生成するステップと、
式Xによる化合物を含む溶液を、3-ブテン-1-イルトリフルオロメタンスルホネートと合わせて、式XIによる化合物を生成するステップと、
式Vによる化合物および式XIによる化合物を含む溶液[ここで、式Vおよび式XIによる化合物は、同じであっても異なっていてもよい]を、(1E,4E)-1,5-ビス(ジメチルアミノ)-ペンタ-1,4-ジエン-3-オンと合わせて、式XIIによる化合物を生成するステップと、
式XIIによる化合物を含む溶液を、ルテニウム触媒の存在下で、3,3-ジメトキシ-1-プロペンと合わせて、式XIIIによる化合物を提供するステップと、
式XIIIによる化合物を、酸性化テトラヒドロフランの溶液と合わせて、式XIVによる化合物を提供するステップと
を含む、方法。
(項目14)
式XIVによる化合物を含む溶液を、トリフルオロメタンスルホン酸無水物(Tf
2
O)と合わせて、式XVによる化合物を提供するステップ
をさらに含む、項目13に記載の方法。
(項目15)
式XVによる化合物を含む溶液を、パラジウム触媒の存在下で、R
g
-C
6
H
4
-B(OH)
2
と合わせて、式XVIによる化合物を提供するステップ
[式中、R
g
は、R
a
、-COOR
a
、または-OR
a
であり、ここでR
a
は、H、重水素、アルキルまたはヘテロアルキルである]、あるいは
式XVによる化合物を含む溶液を、式NH(R
20
)(R
21
)を有するアミンと合わせて、式XVIIによる化合物を提供するステップ
(式中、R
20
およびR
21
は、独立に、H、重水素、アルキル、ヘテロアルキル、アリールまたはヘテロアリールである)
をさらに含む、項目14に記載の方法。
(項目16)
R
1
~R
11
のうちの少なくとも1つが標的化剤を含む項目1~10のいずれか一項に記載の化合物を使用するための方法であって、
前記化合物を、前記標的化剤と結合することが可能である標的を含む試料と、前記標的化剤と前記標的を結合させるのに有効な条件下で合わせるステップと、
前記標的に結合した前記化合物を可視化することによって、前記標的を画像化するステップであって、好ましくは前記化合物を可視化することが、可視または近赤外範囲の波長と選択された強度を有する所定量の光の標的化された適用により、前記試料に照射することを含み、ここで、前記所定量の光は、前記化合物の蛍光を生じさせるのに十分であり、さらに、前記化合物によって放出された任意の蛍光を検出することを含む、ステップ
を含む、方法。
(項目17)
前記標的を画像化する前に、前記化合物を還元剤と合わせるステップをさらに含む、項目16に記載の方法。
(項目18)
前記試料が、対象内の標的領域であり、前記方法が、
前記化合物、または前記化合物を含む医薬組成物を、前記対象に投与するステップと、
その後、前記対象の標的化部分に、前記量の光の標的化された適用によって、前記化合物に照射するステップと、
前記対象の前記標的化部分における前記化合物からの任意の蛍光を検出するステップと
をさらに含む、項目16または項目17に記載の方法。
(項目19)
前記標的領域が、腫瘍部位であり、前記対象の前記標的化部分が、前記腫瘍部位を含み、前記方法が、前記対象の前記標的化部分における前記蛍光を検出した後に、前記対象から前記腫瘍の少なくとも一部を切除するステップをさらに含む、項目18に記載の方法。
(項目20)
活性酸素種を検出するための方法であって、
項目1~10のいずれか一項による化合物を還元剤と合わせて、還元化合物を提供するステップと、
試料を前記還元化合物と接触させるステップであって、前記試料中に活性酸素種(ROS)が存在する場合、それにより前記還元化合物が酸化されて、項目1~10のいずれか一項による化合物が再生される、ステップと、
前記試料に、可視または近赤外範囲の波長と選択された強度を有する所定量の光を照射するステップであって、前記所定量の光は、前記ROSによって前記還元化合物が酸化されて、項目1~10のいずれか一項に記載の化合物が再生される場合に蛍光を生じさせるのに十分である、ステップと、
項目1~10のいずれか一項に記載の化合物によって放出された任意の蛍光を検出するステップであって、ここで、蛍光は、前記試料中のROSの存在を示す、ステップと
を含む、方法。