JP7522097B2 - 抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ - Google Patents
抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ Download PDFInfo
- Publication number
- JP7522097B2 JP7522097B2 JP2021509568A JP2021509568A JP7522097B2 JP 7522097 B2 JP7522097 B2 JP 7522097B2 JP 2021509568 A JP2021509568 A JP 2021509568A JP 2021509568 A JP2021509568 A JP 2021509568A JP 7522097 B2 JP7522097 B2 JP 7522097B2
- Authority
- JP
- Japan
- Prior art keywords
- antibody
- amino acid
- acid sequence
- seq
- set forth
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images










































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/445—Non condensed piperidines, e.g. piperocaine
- A61K31/4523—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems
- A61K31/454—Non condensed piperidines, e.g. piperocaine containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. pimozide, domperidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/502—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with carbocyclic ring systems, e.g. cinnoline, phthalazine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/5025—Pyridazines; Hydrogenated pyridazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
- A61K31/5517—1,4-Benzodiazepines, e.g. diazepam or clozapine condensed with five-membered rings having nitrogen as a ring hetero atom, e.g. imidazobenzodiazepines, triazolam
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68035—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being a pyrrolobenzodiazepine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
- A61K47/6855—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell the tumour determinant being from breast cancer cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6875—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody being a hybrid immunoglobulin
- A61K47/6877—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody being a hybrid immunoglobulin the antibody being an immunoglobulin containing regions, domains or residues from different species
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/30—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants from tumour cells
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/32—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against translation products of oncogenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/46—Hybrid immunoglobulins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/40—Immunoglobulins specific features characterized by post-translational modification
- C07K2317/41—Glycosylation, sialylation, or fucosylation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/77—Internalization into the cell
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Cell Biology (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Peptides Or Proteins (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
ADCに用いられる薬物として有用なものの一つにピロロベンゾジアゼピン(PBD)が挙げられる。PBDはDNA小溝のPuGPu配列などに結合することによって細胞毒性を示す。天然由来のPBDであるanthramycinは1965年に初めて発見され、それ以降様々な天然由来、またその類縁体のPBDが発見された(非特許文献1~4)。
PBDの一般的な構造式は下式

PBDは二量体構造にすることにより、飛躍的に細胞毒性が向上することが知られており(非特許文献5、6)、二量体PBDをADC化したものも種々報告されている(特許文献1~15)。しかしながら、C2位においてスピロ環を有するPBD又はそのADC体は知られていない。
ポリ(ADP-リボース)ポリメラーゼ(PARP)阻害剤は、PARP(特にPARP-1及びPARP-2)を阻害することにより、一本鎖切断の修復を妨げる機能を有する薬剤である。乳がんや卵巣がん等の一部のがんでは、二本鎖切断の修復に異常があることが知られており、PARP阻害剤は、これらのがんに対し、合成致死による抗腫瘍効果が認められている(非特許文献7~11)。
PARP阻害剤としては、Olaparib(非特許文献12)、Rucaparib(非特許文献13)、Niraparib(非特許文献14)、及びTalazoparib(非特許文献15)等が知られている。
PARP阻害剤とPBDを用いたADCを併用することにより、合成致死に類似した効果を得られることも知られている。例えば、PARP阻害剤が有効性を示すBRCA2ノックアウトDLD1細胞のXenograftモデルにおいてOlaparibとADCの併用効果が確認されている。しかしながら、親株のDLD1細胞のXenograftモデルでは併用効果は認められなかった(非特許文献16)。また、上記スピロ環を有するPBDやその抗体-薬物コンジュゲートについてPARP阻害剤との併用効果については知られていない。
[1]抗体-ピロロベンゾジアゼピン誘導体コンジュゲート及び/又はPARP阻害剤を含むがん治療のための医薬組成物であって、該抗体-ピロロベンゾジアゼピン誘導体コンジュゲートと、該PARP阻害剤が、組み合わされて投与されることを特徴とし、
該コンジュゲートが、次式;




上記で示されるそれぞれの構造式において、m1は1又は2の整数であり、
Abは、抗体又は該抗体の機能性断片であり、
N297糖鎖は次式で示される構造を有するN297-(Fuc)MSG1、N297-(Fuc)MSG2もしくはそれらの混合物、又はN297-(Fuc)SGであり、



N297糖鎖中のL(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-であることを示し、
ここで、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-3鎖側又は/及び1-6鎖側の非還元末端のシアル酸の2位のカルボン酸とアミド結合を介して結合しており、左端のアステリスク*は、前記式中のトリアゾール環上の1位又は3位の窒素原子と結合していることを示す医薬組成物。
(a)配列番号9に記載のアミノ酸配列からなるCDRH1、配列番号10に記載のアミノ酸配列からなるCDRH2及び配列番号11に記載のアミノ酸配列からなるCDRH3、並びに、配列番号5に記載のアミノ酸配列からなるCDRL1、配列番号6に記載のアミノ酸配列からなるCDRL2及び配列番号7に記載のアミノ酸配列又は該アミノ酸配列において1又は2個のアミノ酸が置換されたアミノ酸配列からなるCDRL3、又は、
(b)配列番号15に記載のアミノ酸配列からなるCDRH1、配列番号16に記載のアミノ酸配列からなるCDRH2及び配列番号17に記載のアミノ酸配列からなるCDRH3、並びに、配列番号12に記載のアミノ酸配列からなるCDRL1、配列番号13に記載のアミノ酸配列からなるCDRL2及び配列番号14に記載のアミノ酸配列からなるCDRL3。
(a)配列番号9に記載のアミノ酸配列からなるCDRH1、配列番号10に記載のアミノ酸配列からなるCDRH2及び配列番号11に記載のアミノ酸配列からなるCDRH3、及び、配列番号5に記載のアミノ酸配列からなるCDRL1、配列番号6に記載のアミノ酸配列からなるCDRL2及び配列番号7に記載のアミノ酸配列又は配列番号8に記載のアミノ酸配列からなるCDRL3、又は、
(b)配列番号15に記載のアミノ酸配列からなるCDRH1、配列番号16に記載のアミノ酸配列からなるCDRH2及び配列番号17に記載のアミノ酸配列からなるCDRH3、及び、配列番号12に記載のアミノ酸配列からなるCDRL1、配列番号13に記載のアミノ酸配列からなるCDRL2及び配列番号14に記載のアミノ酸配列からなるCDRL3。
(a)配列番号21に記載のアミノ酸配列からなる重鎖可変領域、及び、配列番号19に記載のアミノ酸配列からなる軽鎖可変領域、又は、
(b)配列番号25に記載のアミノ酸配列からなる重鎖可変領域、及び、配列番号23に記載のアミノ酸配列からなる軽鎖可変領域。
(a)配列番号54に記載のアミノ酸配列、
(b)配列番号58に記載のアミノ酸配列、
(c)配列番号62に記載のアミノ酸配列、
(d)(a)~(c)の配列において各CDR配列以外のフレームワーク領域の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、
(e)(a)~(c)の配列における各CDR配列以外のフレームワーク領域の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列、
(f)配列番号38に記載のアミノ酸配列、
(g)配列番号42に記載のアミノ酸配列、
(h)配列番号46に記載のアミノ酸配列、
(i)配列番号50に記載のアミノ酸配列、
(j)(f)~(i)の配列において各CDR配列以外のフレームワーク領域の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、及び、
(k)(f)~(i)の配列における各CDR配列以外のフレームワーク領域の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列。
(a)配列番号54に記載のアミノ酸配列からなる重鎖可変領域および配列番号38に記載のアミノ酸配列からなる軽鎖可変領域、
(b)配列番号58に記載のアミノ酸配列からなる重鎖可変領域および配列番号42に記載のアミノ酸配列からなる軽鎖可変領域、
(c)配列番号54に記載のアミノ酸配列からなる重鎖可変領域および配列番号46に記載のアミノ酸配列からなる軽鎖可変領域、
(d)配列番号58に記載のアミノ酸配列からなる重鎖可変領域および配列番号50に記載のアミノ酸配列からなる軽鎖可変領域、及び、
(e)配列番号62に記載のアミノ酸配列からなる重鎖可変領域および配列番号46に記載のアミノ酸配列からなる軽鎖可変領域。
(a)配列番号52のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号36のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(b)配列番号56のアミノ酸番号20~471に記載のアミノ酸配列からなる、重鎖および配列番号40のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(c)配列番号52のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号44のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(d)配列番号56のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号48のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、及び、
(e)配列番号60のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号44のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖。
該コンジュゲートが、次式;




上記で示されるそれぞれの構造式において、m1は1又は2の整数であり、
Abは、抗体又は該抗体の機能性断片であり、
N297糖鎖は次式で示される構造を有するN297-(Fuc)MSG1、N297-(Fuc)MSG2もしくはそれらの混合物、又はN297-(Fuc)SGであり、



N297糖鎖中のL(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-であることを示し、
ここで、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-3鎖側又は/及び1-6鎖側の非還元末端のシアル酸の2位のカルボン酸とアミド結合を介して結合しており、左端のアステリスク*は、前記式中のトリアゾール環上の1位又は3位の窒素原子と結合していることを示す治療方法。
(a)配列番号9に記載のアミノ酸配列からなるCDRH1、配列番号10に記載のアミノ酸配列からなるCDRH2及び配列番号11に記載のアミノ酸配列からなるCDRH3、並びに、配列番号5に記載のアミノ酸配列からなるCDRL1、配列番号6に記載のアミノ酸配列からなるCDRL2及び配列番号7に記載のアミノ酸配列又は該アミノ酸配列において1又は2個のアミノ酸が置換されたアミノ酸配列からなるCDRL3、又は、
(b)配列番号15に記載のアミノ酸配列からなるCDRH1、配列番号16に記載のアミノ酸配列からなるCDRH2及び配列番号17に記載のアミノ酸配列からなるCDRH3、並びに、配列番号12に記載のアミノ酸配列からなるCDRL1、配列番号13に記載のアミノ酸配列からなるCDRL2及び配列番号14に記載のアミノ酸配列からなるCDRL3。
(a)配列番号9に記載のアミノ酸配列からなるCDRH1、配列番号10に記載のアミノ酸配列からなるCDRH2及び配列番号11に記載のアミノ酸配列からなるCDRH3、及び、配列番号5に記載のアミノ酸配列からなるCDRL1、配列番号6に記載のアミノ酸配列からなるCDRL2及び配列番号7に記載のアミノ酸配列又は配列番号8に記載のアミノ酸配列からなるCDRL3、又は、
(b)配列番号15に記載のアミノ酸配列からなるCDRH1、配列番号16に記載のアミノ酸配列からなるCDRH2及び配列番号17に記載のアミノ酸配列からなるCDRH3、及び、配列番号12に記載のアミノ酸配列からなるCDRL1、配列番号13に記載のアミノ酸配列からなるCDRL2及び配列番号14に記載のアミノ酸配列からなるCDRL3。
(a)配列番号21に記載のアミノ酸配列からなる重鎖可変領域、及び、配列番号19に記載のアミノ酸配列からなる軽鎖可変領域、又は、
(b)配列番号25に記載のアミノ酸配列からなる重鎖可変領域、及び、配列番号23に記載のアミノ酸配列からなる軽鎖可変領域。
(a)配列番号54に記載のアミノ酸配列、
(b)配列番号58に記載のアミノ酸配列、
(c)配列番号62に記載のアミノ酸配列、
(d)(a)~(c)の配列において各CDR配列以外のフレームワーク領域の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、
(e)(a)~(c)の配列における各CDR配列以外のフレームワーク領域の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列、
(f)配列番号38に記載のアミノ酸配列、
(g)配列番号42に記載のアミノ酸配列、
(h)配列番号46に記載のアミノ酸配列、
(i)配列番号50に記載のアミノ酸配列、
(j)(f)~(i)の配列において各CDR配列以外のフレームワーク領域の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、及び、
(k)(f)~(i)の配列における各CDR配列以外のフレームワーク領域の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列。
(a)配列番号54に記載のアミノ酸配列からなる重鎖可変領域および配列番号38に記載のアミノ酸配列からなる軽鎖可変領域、
(b)配列番号58に記載のアミノ酸配列からなる重鎖可変領域および配列番号42に記載のアミノ酸配列からなる軽鎖可変領域、
(c)配列番号54に記載のアミノ酸配列からなる重鎖可変領域および配列番号46に記載のアミノ酸配列からなる軽鎖可変領域、
(d)配列番号58に記載のアミノ酸配列からなる重鎖可変領域および配列番号50に記載のアミノ酸配列からなる軽鎖可変領域、及び、
(e)配列番号62に記載のアミノ酸配列からなる重鎖可変領域および配列番号46に記載のアミノ酸配列からなる軽鎖可変領域。
(a)配列番号52のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号36のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(b)配列番号56のアミノ酸番号20~471に記載のアミノ酸配列からなる、重鎖および配列番号40のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(c)配列番号52のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号44のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(d)配列番号56のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号48のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、及び、
(e)配列番号60のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号44のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖。
該ピロロベンゾジアゼピン誘導体がDNAのマイナーグルーブにおいてクロスリンクを形成しない医薬組成物。
がんがPARP阻害剤に非感受性である医薬組成物。
がんが、相同的組換え(HR)依存的DNA二本鎖破壊(DSB)修復経路に非依存的である医薬組成物。
[73][1]に記載の抗体-薬物コンジュゲートと、PARP阻害剤が、それぞれ別異の製剤に有効成分として含有され、同時に又は異なる時間に投与されることを特徴とする、[34]~[64]のいずれか一つに記載の治療方法。
本発明において使用される抗体-薬物コンジュゲートは、腫瘍細胞に発現している抗原を認識又は当該抗原に結合できる抗体にリンカー部分を介して抗腫瘍性化合物を結合させた抗腫瘍性薬物である。

m1は1又は2の整数(好ましくは、1)であり、Dは薬物、LはN297糖鎖とDを連結するリンカー、Abは抗体又は該抗体の機能性断片、N297糖鎖は前記抗体のAsn297の側鎖に結合する糖鎖を示す。N297糖鎖はリモデンリングされた糖鎖でも良い。
本発明の薬物Dは抗腫瘍性化合物であることが好ましい。本抗腫瘍性化合物は、本発明の抗体-薬物コンジュゲートのリンカーの一部又は全部が腫瘍細胞内で切断され抗腫瘍性化合物部分が遊離されて抗腫瘍効果が発現される。本発明の薬物Dとして、次式;



本発明のPBD誘導体の部分構造としては、上記I(a)が好ましい。好ましくは、以下の群から選択されるいずれか一つである。

本発明のリンカーLは、N297糖鎖とDを連結するリンカーである。
当該リンカーLは、次式で示される。
-Lb-La-Lp-NH-B-CH2-O(C=O)-*
アステリスク*は、薬物DのN10’位の窒素原子と結合していることを示し、Lbは、LaとN297糖鎖又はリモデリングされたN297糖鎖を結合するスペーサーを示す。
Lpは、2から7個(好ましくは、2から4個)のアミノ酸で構成されるペプチド残基である。すなわち、2から7個のアミノ酸がペプチド結合したオリゴペプチドの残基によって構成される。
Lpは、N末端においてLb-La-のLaのカルボニル基に結合し、C末端においてリンカーの-NH-B-CH2-O(C=O)-部分のアミノ基(-NH-)とアミド結合を形成する。前記エステラーゼ等の酵素によって、LpのC末端と-NH-間の結合が切断される。
これらのアミノ酸は重複してもよく、任意に選択されたアミノ酸を含むアミノ酸配列を有する。また、アミノ酸の種類によって、薬物遊離のパターンをコントロールすることができる。
-GGVA-、-GG-(D-)VA-、-VA-、-GGFG-、-GGPI-、-GGVCit-、-GGVK-、-GG(D-)PI-、-GGPL-、-EGGVA、-PI-、-GGF-、-DGGF-、(D-)D-GGF-、-EGGF-、-SGGF-、-KGGF-、-DGGFG-、-GGFGG-、-DDGGFG-、-KDGGFG-、-GGFGGGF-
を挙げることができる。
ここで、上記の『(D-)V』はD-バリン、『(D-)P』はD-プロリン、『(D-)D』はD-アスパラギン酸を意味する。
-GGVA-、-GG-(D-)VA-、-VA-、-GGFG-、-GGPI-、-GGVCit-、-GGVK-、-GG(D-)PI-、-GGPL-
-GGVA-、-GGVCit-、-VA-
-C(=O)-(CH2CH2)n2-C(=O)-、-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2)n3-C(=O)-、
-C(=O)-(CH2CH2)n2-C(=O)-NH-(CH2CH2O)n3-CH2-C(=O)-、
-C(=O)-(CH2CH2)n2-NH-C(=O)-(CH2CH2O)n3-CH2CH2-C(=O)-、-(CH2)n4-O-C(=O)-
ここで、式中、n2は1~3の整数(好ましくは、1又は2)、n3は1~5の整数(好ましくは、2~4の整数、より好ましくは、2又は4)、n4は0~2の整数(好ましくは、0又は1)を示す。
-C(=O)-CH2CH2-C(=O)-、-C(=O)-(CH2CH2)2-C(=O)-、
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2)2-C(=O)-
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)2-CH2-C(=O)-、
-C(=O)-CH2CH2-NH-C(=O)-(CH2CH2O)4-CH2CH2-C(=O)-、
-CH2-OC(=O)-、及び、-OC(=O)-
Laは、より好ましくは、-C(=O)-CH2CH2-C(=O)-、又は、-C(=O)-(CH2CH2)2-C(=O)-である。



上記で示されるLb(Lb-1、Lb-2又はLb-3)のそれぞれの構造式において、アジド基とDBCOのclick reactionで形成されるトリアゾール環部位は、幾何異性構造を有し、1つのLb中に、これら2種類の構造のいずれか一方、又は、それらの混合物として存在する。すなわち、本発明の抗体-薬物コンジュゲート1分子中には2又は4個(m1は1又は2)の『-L-D』が存在し、2又は4個それぞれの『-L-D』におけるL中のそれぞれのLb(Lb-1、Lb-2又はLb-3)は、これら2種類の構造のいずれか一方、又は、その両方が混在している。
Bは、1,4-フェニル基であり、
Lpは、以下の群から選択されるいずれか一つを示し、
-GGVA-、-GG-(D-)VA-、-VA-、-GGFG-、-GGPI-、-GGVCit-、-GGVK-、-GGPL-
Laは、以下の群から選択されるいずれか一つを示し、
-C(=O)-CH2CH2-C(=O)-、-C(=O)-(CH2CH2)2-C(=O)-、
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2)2-C(=O)-、
-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)2-CH2-C(=O)-、
-C(=O)-CH2CH2-NH-C(=O)-(CH2CH2O)4-CH2CH2-C(=O)-、-CH2-OC(=O)-、-OC(=O)-
Lbは、上記で示されるLbのいずれかの構造式を示す。
-Z1-C(=O)-CH2CH2-C(=O)-GGVA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GG-(D-)VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-(CH2CH2)2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GGPI-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GGFG-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GGVCit-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GGVK-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GGPL-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2)2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)2-CH2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-NH-C(=O)-(CH2CH2O)4-CH2CH2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z2-OC(=O)-GGVA-NH-B-CH2-OC(=O)-、-Z3-CH2-OC(=O)-GGVA-NH-B-CH2-OC(=O)-
ここで、Z1は、上記Lbの以下で示される構造式;



-Z1-C(=O)-CH2CH2-C(=O)-GGVA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-(CH2CH2)2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-GGVCit-NH-B-CH2-OC(=O)-、-Z1-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2)2-C(=O)-VA-NH-B-CH2-OC(=O)-、
-Z1-C(=O)-CH2CH2-C(=O)-NH-(CH2CH2O)2-CH2-C(=O)-VA-NH-B-CH2-OC(=O)-、及び
-Z1-C(=O)-CH2CH2-NH-C(=O)-(CH2CH2O)4-CH2CH2-C(=O)-VA-NH-B-CH2-OC(=O)-
ここで、Bは1,4-フェニル基であり、
Z1は上記Lbの以下で示される構造式:

本発明の抗体-薬物コンジュゲートの遊離薬物は、以下の群から選ばれる一つである。

本発明において、「がん」と「腫瘍」は同じ意味に用いている。
本発明において、「遺伝子」とは、蛋白質のアミノ酸をコードするヌクレオチド配列が含まれるヌクレオチドもしくはヌクレオチド配列、またはその相補鎖を意味し、例えば、蛋白質のアミノ酸をコードするヌクレオチド配列が含まれるヌクレオチド配列またはその相補鎖であるポリヌクレオチド、オリゴヌクレオチド、DNA、mRNA、cDNA、RNA等は「遺伝子」の意味に含まれる。「CLDN6遺伝子」としては、例えば、CLDN6蛋白質のアミノ酸配列をコードするヌクレオチド配列が含まれるDNA、mRNA、cDNA、cRNA等をあげることができる。
本発明において、「ヌクレオチド」、「ポリヌクレオチド」又は「ヌクレオチド配列」と「核酸」は同義であり、例えば、DNA、RNA、プローブ、オリゴヌクレオチド、ポリヌクレオチド、プライマー等も「ヌクレオチド」又は「ヌクレオチド配列」の意味に含まれる。
本発明においては、「ポリペプチド」、「ペプチド」、「蛋白質」は区別せずに用いている。
本発明において、「CLDN6」は、CLDN6蛋白質と同じ意味で用いている。
本発明において、「細胞傷害活性」とは、何らかの形で、細胞に病理的な変化を引き起こすことをいい、直接的な外傷にとどまらず、DNAの切断や塩基の二量体の形成、染色体の切断、細胞分裂装置の損傷、各種酵素活性の低下などあらゆる細胞の構造や機能上の損傷を引き起こすことをいう。
本発明の機能性断片は、IgG重鎖のFc領域においてよく保存されたN結合型糖鎖による修飾を受けるアスパラギン(Asn297)及びその周辺のアミノ酸を保持し、且つ抗原との結合能を有している機能性断片を含む。
本発明において、「1~数個」とは、1~10個、1~9個、1~8個、1~7個、1~6個、1~5個、1~4個、1~3個又は1~2個を意味する。
本発明の抗体-薬物コンジュゲートは抗腫瘍効果を発揮する化合物を結合させてあるので、抗体自体が抗腫瘍効果を有することは、好ましいが、必須ではない。抗腫瘍性化合物の細胞傷害性を腫瘍細胞において特異的・選択的に発揮させる目的からは、抗体又は抗体-薬物コンジュゲートが内在化して腫瘍細胞内に移行する性質を有することが重要であり、好ましい。抗腫瘍効果の発揮の点からは抗体又は抗体-薬物コンジュゲートが内在化して腫瘍細胞内に移行する性質を有することが、薬物によって腫瘍細胞を特異的・選択的に傷害を与える点で重要であり、好ましい。抗体の抗腫瘍活性は、腫瘍細胞への細胞傷害活性、抗細胞効果をいう。公知のin vitro又はIn vivoの評価系を用いて、抗腫瘍活性を確認することができる。抗体の内在化能は公知の評価系で測定することができる。
このような抗体として、腫瘍関連抗原に対する抗体が挙げられ、抗CLDN6抗体、抗CLDN9抗体、抗CLDN6/CLDN9抗体、抗HER2抗体、抗HER3抗体、抗DLL3(Delta like protein3)抗体、抗A33抗体、抗CanAg抗体、抗CD19抗体、抗CD20抗体、抗CD22抗体、抗CD25抗体、抗CD30抗体、抗CD33抗体、抗CD37抗体、抗CD56抗体、抗CD70抗体、抗CD98抗体、抗B7-H3(CD276)抗体、抗TROP2抗体、抗CEA 抗体、抗Cripto抗体、抗EphA2抗体、抗FGFR2抗体(WO201315206等)、抗G250抗体、抗MUC1抗体(WO2011012309等)、抗GPNMB抗体、抗Integrin抗体、抗PSMA抗体、抗Tenascin-C抗体、抗SLC44A4抗体、抗Mesothelin抗体、抗EGFR抗体、抗5T4 (oncofetal antigen 5T4; also TPBG and trophoblast glycoprotein)抗体、抗LRRC15 (Leucine-rich repeat-containing protein 15)抗体、抗DR5抗体、抗CDH3(cadherin 3)抗体、抗PDPN (podoplanin) 抗体、又は抗CD123抗体を例示できるがこれに限らない。
本発明の抗体として、好ましくは、抗CLDN6抗体、抗CLDN6/CLDN9抗体、抗HER2抗体、抗CD98抗体、抗TROP2抗体であり、さらに好ましくは抗CLDN6抗体、抗HER2抗体(例えば、Trastuzumab、Trastuzumab変異体、Trastuzumab変異体2)である。
1.CLDN6及びCLDN9
CLDN6は、Claudinファミリーに属する、220アミノ酸からなる4回膜貫通型の蛋白質であり、N末端及びC末端を細胞内に持つ。
ヒトCLDN6のアミノ酸配列及びDNA配列は公的データベース上に公開されており、例えばNP_067018(配列番号1)、NM_021195(配列番号2(ともにNCBI)等のアクセッション番号により参照可能である。
ヒトCLDN6蛋白質のアミノ酸配列(以下、「CLDN6アミノ酸配列」)について、細胞外領域は、配列表の配列番号1のアミノ酸番号29~81からなる細胞外ドメイン(EC1)、アミノ酸番号138~160からなる細胞外ドメイン(EC2)で構成されている。
CLDN9は、Claudinファミリーに属する、217アミノ酸からなる4回膜貫通型の蛋白質であり、N末端及びC末端を細胞内に持つ。CLDN9はCLDN6と高い相同性をもつ。
ヒトCLDN9のアミノ酸配列及びDNA配列は公的データベース上に公開されており、例えばNP_066192(配列番号3)、NM_020982(配列番号4)(ともにNCBI)等のアクセッション番号により参照可能である。
本発明の抗CLDN6抗体の一例として、配列表の配列番号1に示すCLDN6のN末端より29から81番目のアミノ酸配列、及び138から160番目のアミノ酸配列の2つの細胞外領域からなる高次構造を認識し、かつ内在化活性を有する抗CLDN6抗体を挙げることができる。
本発明の抗CLDN6抗体は腫瘍細胞を標的にできる抗体であり、すなわち腫瘍細胞を認識できる特性、腫瘍細胞に結合できる特性、そして腫瘍細胞内に取り込まれて内在化する特性等を備えている。したがって、本発明の抗CLDN6抗体と抗腫瘍活性を有する化合物を、リンカーを介して結合させて抗体-薬物コンジュゲートとすることができる。
本発明の抗CLDN6抗体は抗腫瘍活性を有していてもよい。
(a)CLDNファミリーを認識又は結合する。
本発明の抗体はCLDNファミリーを認識する。言い換えれば、本発明の抗体はCLDNファミリーに結合する。本発明の抗体は、好ましくはCLDN6に結合し、より好ましくは、CLDN6に特異的に結合する。更に、本発明の抗体はCLDN9を認識し又はCLDN9に結合してもよい。
本発明において「特異的な認識」、すなわち「特異的な結合」とは、非特異的な吸着ではない結合を意味する。結合が特異的であるか否かの判定基準としては、例えば、解離定数(Dissociation Constant:以下、「KDという」)をあげることができる。本発明の好適な抗体のCLDN6及び/又はCLDN9に対するKD値は1×10-5M以下、5×10-6M以下、2×10-6M以下または1×10-6M以下、より好適には5×10-7M以下、2×10-7M以下または1×10-7M以下である。
本発明における抗原と抗体の結合は、ELISA法、RIA法、Surface Plasmon Resonance(以下、「SPR」という)解析法等により測定または判定することができる。細胞表面上に発現している抗原と抗体との結合は、フローサートメトリー法等により測定することができる。
(b)CLDN6及び/又はCLDN9と結合することによってCLDN6及び/又はCLDN9発現細胞に内在化する活性を有する。
(2)CLDN6及び/又はCLDN9がヒトCLDN6及び/又はヒトCLDN9である上記(1)に記載の抗体。
B1抗体の重鎖可変領域の塩基配列は、配列表の配列番号20に、アミノ酸配列は配列番号21に記載されている。また、B1抗体の軽鎖可変領域の塩基配列は、配列表の配列番号18に、アミノ酸配列は配列番号19に記載されている。
B1抗体のCDRH1のアミノ酸配列は配列番号9に、CDRH2のアミノ酸配列は配列番号10に、CDRH3のアミノ酸配列は配列番号11に、CDRL1のアミノ酸配列は配列番号5に、CDRL2のアミノ酸配列は配列番号6に、CDRL3のアミノ酸配列は配列番号7に記載されている。
C7抗体の重鎖可変領域の塩基配列は、配列表の配列番号24に、アミノ酸配列は配列番号25に記載されている。また、C7抗体の軽鎖可変領域の塩基配列は、配列表の配列番号22に、アミノ酸配列は配列番号23に記載されている。
C7抗体のCDRH1のアミノ酸配列は配列番号15に、CDRH2のアミノ酸配列は配列番号16に、CDRH3のアミノ酸配列は配列番号17に、CDRL1のアミノ酸配列は配列番号12に、CDRL2のアミノ酸配列は配列番号13に、CDRL3のアミノ酸配列は配列番号14に記載されている。
キメラ抗体としては、抗体の可変領域と定常領域が互いに異種である抗体、例えばマウス又はラット由来抗体の可変領域をヒト由来の定常領域に接合したキメラ抗体を挙げることができる。
本発明のキメラ抗体として例示されるマウス抗ヒトCLDN6抗体B1抗体由来のキメラ抗体は、配列番号21に示されるアミノ酸配列からなる重鎖可変領域を含む重鎖及び配列番号19に示される軽鎖可変領域を含む軽鎖を含む抗体であり、任意のヒト由来の定常領域を有していてよい。
マウス抗ヒトCLDN6抗体B1抗体由来のキメラ抗体の具体例として、マウス抗ヒトCLDN6抗体B1抗体由来のキメラ抗体chB1抗体(以下、「chB1」とも記載する。)を挙げることができる。chB1抗体のアミノ酸配列は、配列表の配列番号32の20~471番目のアミノ酸残基からなるアミノ酸配列を有する重鎖及び配列表の配列番号28の21~234からなるアミノ酸配列を有する軽鎖を含む抗体を挙げることができる。
なお、配列表の配列番号32に示される重鎖配列中で、1~19番目のアミノ酸残基からなるアミノ酸配列はシグナル配列であり、20~141番目のアミノ酸残基からなるアミノ酸配列は重鎖可変領域であり、142~471番目の残基からなるアミノ酸配列は重鎖定常領域である。また、配列表の配列番号28に示される軽鎖配列中で、1~20番目のアミノ酸残基からなるアミノ酸配列はシグナル配列であり、21~127番目のアミノ酸残基からなるアミノ酸配列は軽鎖可変領域であり、128~234番目のアミノ酸残基からなるアミノ酸配列は軽鎖定常領域である。
chB1抗体の重鎖及び軽鎖の可変領域のアミノ酸配列は配列表の配列番号34、配列番号30に記載されている。
chB1抗体の重鎖アミノ酸配列は、配列表の配列番号33に示されるヌクレオチド配列によってコードされている。配列表の配列番号33に示されるヌクレオチド配列の1~57番目のヌクレオチドからなるヌクレオチド配列はchB1抗体重鎖のシグナル配列をコードしており、配列表の配列番号33に示されるヌクレオチド配列の58~423番目のヌクレオチドからなるヌクレオチド配列はchB1抗体の重鎖可変領域をコードしており、配列表の配列番号33に示されるヌクレオチド配列の424~1413番目のヌクレオチドからなるヌクレオチド配列はchB1抗体の重鎖定常領域をコードしている。
chB1抗体の重鎖可変領域の塩基配列は配列表の配列番号35に記載されている。
chB1抗体の軽鎖アミノ酸配列は、配列表の配列番号29に示されるヌクレオチド配列によってコードされている。配列表の配列番号29に示されるヌクレオチド配列の26~85番目のヌクレオチドからなるヌクレオチド配列はchB1抗体軽鎖のシグナル配列をコードしており、配列表の配列番号29に示されるヌクレオチド配列の86~406番目のヌクレオチドからなるヌクレオチド配列はchB1抗体の軽鎖可変領域をコードしており、配列表の配列番号29に示されるヌクレオチド配列の407~727番目のヌクレオチドからなるヌクレオチド配列はchB1抗体の軽鎖定常領域をコードしている。
chB1抗体の軽鎖可変領域の塩基配列は配列表の配列番号31に記載されている。
ヒト化抗体としては、相補性決定領域(CDR;complementarity determining region)のみをヒト由来の抗体に組み込んだ抗体(Nature(1986)321,p.522-525参照)、CDR移植法によって、CDRの配列に加え一部のフレームワークのアミノ酸残基もヒト抗体に移植した抗体(WO90/07861号)、更に、抗原に対する結合能を維持しつつ、一部のCDRのアミノ酸配列を改変した抗体を挙げることができる。
CDRのアミノ酸配列は、Kabatの定義、Chothiaの定義、Abmの定義、IMGT等公知の方法によって決めることができるが、本発明におけるCDRはいずれの方法によって定義されたものでもよい。
但し、B1抗体又はC1抗体由来のヒト化抗体としては、B1抗体又はC1抗体の6種全てのCDR配列を保持し、CLDN6結合活性を有する限り、特定のヒト化抗体に限定されず、更に1~数個(好ましくは、1~2個、より好ましくは1個)のCDRのアミノ酸配列を改変したヒト化抗体変異体もCLDN6蛋白質を認識する又は該抗体のCLDN6蛋白質結合活性を有する限り、特定のヒト化抗体に限定されない。
本発明の抗CLDN6ヒト化抗体又はその機能性断片としては、例えば、
配列表の配列番号9に示されるアミノ酸配列又は該アミノ酸配列の1~数個(好ましくは、1~2個)のアミノ酸が置換されたアミノ酸配列からなるCDRH1、
配列表の配列番号10に示されるアミノ酸配列又は該アミノ酸配列の1~数個(好ましくは、1~2個)のアミノ酸が置換されたアミノ酸配列からなるCDRH2、及び、
配列表の配列番号11に示されるアミノ酸配列又は該アミノ酸配列の1~数個(好ましくは、1~2個)のアミノ酸が置換されたアミノ酸配列からなるCDRH3を含む可変領域を有する重鎖、並びに、
配列表の配列番号5に示されるアミノ酸配列又は該アミノ酸配列の1~数個(好ましくは、1~2個)のアミノ酸が置換されたアミノ酸配列からなるCDRL1、
配列表の配列番号6に示されるアミノ酸配列又は該アミノ酸配列の1~数個(好ましくは、1~2個)のアミノ酸が置換されたアミノ酸配列からなるCDRL2、及び、
配列表の配列番号7に示されるアミノ酸配列又は該アミノ酸配列の1~数個(好ましくは、1~2個)のアミノ酸が置換されたアミノ酸配列からなるCDRL3を含む可変領域を有する軽鎖を含み、
本発明のCLDN6蛋白質を認識する又は該抗体のCLDN6蛋白質結合活性を保持している抗体又は該抗体の機能性断片等を挙げることができる。
上記抗CLDN6ヒト化抗体又はその機能性断片におけるCDRのアミノ酸置換の例としては、好ましくは、上記CDRL3の1~数個(好ましくは、1~2個)のアミノ酸置換が挙げられ、配列表の配列番号7のアミノ酸番号4番と5番のアミノ酸を置換した配列表の配列番号8に示されるCDRL3を例示できる。
配列表の配列番号54に示されるアミノ酸配列からなる重鎖可変領域、及び、配列表の配列番号38に示されるアミノ酸配列からなる軽鎖可変領域を含むことからなるヒト化抗体、
配列表の配列番号58に示されるアミノ酸配列からなる重鎖可変領域、及び、配列表の配列番号42に示されるアミノ酸配列からなる軽鎖可変領域を含むことからなるヒト化抗体、
配列表の配列番号54に示されるアミノ酸配列からなる重鎖可変領域、及び、配列表の配列番号46に示されるアミノ酸配列からなる軽鎖可変領域を含むことからなるヒト化抗体、
配列表の配列番号58に示されるアミノ酸配列からなる重鎖可変領域、及び、配列表の配列番号50に示されるアミノ酸配列からなる軽鎖可変領域を含むことからなるヒト化抗体、
配列表の配列番号62に示されるアミノ酸配列からなる重鎖可変領域、及び、配列表の配列番号46に示されるアミノ酸配列からなる軽鎖可変領域を含むことからなるヒト化抗体を好適に例示できる。
配列表の配列番号52のアミノ酸番号20~471に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号36のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H1L1)、
配列表の配列番号56のアミノ酸番号20~471に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号40のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H2L2)、
配列表の配列番号52のアミノ酸番号20~471に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号44のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H1L3)、
配列表の配列番号56のアミノ酸番号20~471に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号48のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H2L4)、又は、
配列表の配列番号60のアミノ酸番号20~471に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号44のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H3L3)を例示できる。
なお、配列表の配列番号52、56、又は60に示される重鎖アミノ酸配列中で、1~19番目のアミノ酸残基からなるアミノ酸配列はシグナル配列であり、20~141番目のアミノ酸残基からなるアミノ酸配列は重鎖可変領域であり、142~471番目のアミノ酸残基からなるアミノ酸配列は重鎖定常領域である。
また、配列表の配列番号36、40、44又は48に示される軽鎖アミノ酸配列中で、1~20番目のアミノ酸残基からなるアミノ酸配列はシグナル配列であり、21~127番目のアミノ酸残基からなるアミノ酸配列は軽鎖可変領域であり、128~234番目のアミノ酸残基からなるアミノ酸配列は軽鎖定常領域である。
欠失体の重鎖として、配列表の配列番号52、56、60のアミノ酸番号20~470番目に記載のアミノ酸配列を含む重鎖が挙げられる。
当該欠失体として、
配列表の配列番号52のアミノ酸番号20~470に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号36のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H1L1)、
配列表の配列番号56のアミノ酸番号20~470に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号40のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H2L2)、
配列表の配列番号52のアミノ酸番号20~470に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号44のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H1L3)、
配列表の配列番号56のアミノ酸番号20~470に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号48のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H2L4)、又は、
配列表の配列番号60のアミノ酸番号20~470に示されるアミノ酸配列からなる重鎖、及び、配列表の配列番号44のアミノ酸番号21~234に示されるアミノ酸配列からなる軽鎖を含むことからなるヒト化抗体(H3L3)を例示できる。
また、上記重鎖可変領域及び軽鎖可変領域の組合せを含むことからなる抗体、又は、上記重鎖及び軽鎖の組合せを含むことからなる抗体のCDRと同一のアミノ酸配列からなるCDRを有し、かつ該抗体のCDRのアミノ酸配列を除いたアミノ酸配列の同一性又は相同性が80%以上、好ましくは90%以上、より好ましくは95%以上、更に好ましくは97%以上、最も好ましくは99%以上である抗体もCLDN6への結合活性を有する限り、本発明の抗体に含まれる。
更に、重鎖又は軽鎖のアミノ酸配列に1~数個のアミノ酸残基が置換、欠失又は付加されたアミノ酸配列を組み合わせることによっても、上記の各抗体と同等の生物活性を有する抗体を選択することが可能である。また、本明細書中におけるアミノ酸の置換としては保存的アミノ酸置換が好ましい(WO2013154206)。
保存的アミノ酸置換とは、アミノ酸側鎖に関連のあるアミノ酸グループ内で生じる置換である。かかるアミノ酸置換は元のアミノ酸配列を有する物質の特性を低下させない範囲で行うのが好ましい。
二種類のアミノ酸配列間の相同性は、Blast algorithm version 2.2.2(Altschul, Stephen F., Thomas L.Madden, Alejandro A.Schaaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.Lipman(1997), 「Gapped BLAST and PSI-BLAST:a new generation of protein database search programs」, Nucleic Acids Res.25:3389-3402)のデフォルトパラメーターを使用することによって決定することができる。Blast algorithmは、インターネットでwww.ncbi.nlm.nih.gov/blastにアクセスすることによっても使用することができる。
本発明の抗体としては、さらに、CLDN6及び/又はCLDN9に結合する、ヒト抗体を挙げることができる。抗CLDN6及び/又はCLDN9ヒト抗体とは、ヒト染色体由来の抗体の遺伝子配列のみを有するヒト抗体を意味する。抗CLDN6ヒト抗体は、公知の方法によって得ることができる(Nature Genetics(1997)16,p.133-143、Nucl.Acids Res.(1998)26, p.3447-3448、Animal Cell Technology:Basic and Applied Aspects, vol.10, p.69-73、Kluwer Academic Publishers, 1999.、Proc. Natl. Acad. Sci. USA(2000) 97, p.722-727、Investigative Ophthalmology & Visual Science.(2002)43(7), p.2301-2308、Briefings in Functional Genomics and Proteomics(2002), 1(2), p.189-203、Ophthalmology(2002)109(3),p.427-431、WO92/01047、WO92/20791、WO93/06213、WO93/11236、WO93/19172、WO95/01438、WO95/15388、Annu.Rev.Immunol(1994)12, p.433-455、
Nature Biotechnology(2005)23(9), p.1105-1116)も知られている。
本発明の抗HER2抗体は、以下の特性を有する;
(1)以下の特性を有することを特徴とする抗HER2抗体;
(a)HER2に特異的に結合する。
(b)HER2と結合することによってHER2発現細胞に内在化する活性を有する。
(2)HER2の細胞外ドメインに結合する上記(1)に記載の抗体。
(3)前記抗体がモノクローナル抗体である上記(1)又は(2)に記載の抗体。
(4)抗体依存性細胞傷害(ADCC)活性及び/又は補体依存性細胞傷害(CDC)活性を有する上記(1)~(3)のいずれかに記載の抗体。
(5)マウスモノクローナル抗体、キメラモノクローナル抗体又はヒト化モノクローナル抗体である、上記(1)~(4)のいずれかに記載の抗体。
(6)重鎖定常領域がヒトIgG1の重鎖定常領域であり、ADCC及び/又はCDC活性の低減をもたらす変異を含む、上記(1)~(3)及び(5)のいずれかに記載の抗体。
(7)重鎖定常領域がヒトIgG1の重鎖定常領域であり、EU Indexにより示される234位及び235位のロイシンがアラニンに置換されている、上記(6)に記載の抗体。
(8)配列番号65に記載のアミノ酸配列からなる重鎖及び配列番号64に記載のアミノ酸配列からなる軽鎖を含んでなる抗体である、上記(1)~(5)のいずれかに記載の抗体。
(9)配列番号75のアミノ酸番号20~139に記載のアミノ酸配列からなる重鎖可変領域及び配列番号73のアミノ酸番号21~127に記載のアミノ酸配列からなる軽鎖可変領域を含む抗体である、上記(1)~(3)及び(5)~(7)のいずれかに記載の抗体。
(10)配列番号75のアミノ酸番号20~469に記載のアミノ酸配列からなる重鎖及び配列番号73のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖を含んでなる抗体である、上記(1)~(3)、(5)~(7)及び(9)のいずれかに記載の抗体。
(11)配列番号77のアミノ酸番号20~469に記載のアミノ酸配列からなる重鎖及び配列番号76のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖を含んでなる抗体である、上記(1)~(3)、(5)~(7)及び(9)のいずれかに記載の抗体。
(12)重鎖カルボキシル末端において1又は2つのアミノ酸が欠失している、上記(1)~(11)のいずれかに記載の抗体。
(13)配列番号65のアミノ酸番号1~449に記載のアミノ酸配列からなる重鎖及び配列番号64のアミノ酸番号1~214に記載のアミノ酸配列からなる軽鎖を含んでなる、上記(1)~(5)、(8)及び(12)のいずれかに記載の抗体。
(14)配列番号75のアミノ酸番号20~468に記載のアミノ酸配列からなる重鎖及び配列番号73のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖を含んでなる、上記(1)~(3)、(5)~(7)、(9)、(10)及び(12)のいずれかに記載の抗体。
(15)配列番号77のアミノ酸番号20~468に記載のアミノ酸配列からなる重鎖及び配列番号76のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖を含んでなる、上記(1)~(3)、(5)~(7)、及び(9)、(11)及び(12)のいずれかに記載の抗体。
(16)上記(1)~(15)のいずれかに記載の抗体をコードするポリヌクレオチドを含有する発現ベクターによって形質転換された宿主細胞を培養する工程及び当該工程で得られた培養物から目的の抗体を採取する工程を含む当該抗体の製造方法によって得られる抗体。
なお、本願において、trastuzumabの重鎖定常領域(配列番号65)のEU Indexにより示される234位及び235位のロイシンがアラニンに置換されている抗体をTrastuzumab変異体又はTrastuzumab変異体2と呼ぶ。
また、本発明の抗体に結合している糖鎖修飾を調節すること(グリコシル化、脱フコース化等)によって、抗体依存性細胞傷害活性を増強することが可能である。抗体の糖鎖修飾の調節技術としては、WO1999/54342、WO2000/61739、WO2002/31140、WO2007133855、WO2013120066等が知られているが、これらに限定されるものではない。本発明の抗体には当該糖鎖修飾を調節された抗体も含まれる。
かかる修飾には、抗体又はその機能性断片における任意の位置に、または所望の位置においても施されてもよく、1つ又は2つ以上の位置に同一又は2種以上の異なる修飾がなされていてもよい。
本発明において「抗体断片の修飾体」は「抗体の修飾体の断片」をもその意味に含むものである。
真核細胞を宿主として使用する場合、動物細胞、植物細胞、真核微生物を用いることができる。特に動物細胞としては、哺乳類細胞、例えば、サルの細胞であるCOS細胞(Cell(1981)23,p.175-182、ATCC CRL-1650)、マウス線維芽細胞NIH3T3(ATCC No.CRL-1658)やチャイニーズ・ハムスター卵巣細胞(CHO細胞、ATCC CCL-61)のジヒドロ葉酸還元酵素欠損株(Proc.Natl.Acad.Sci.U.S.A.(1980)77、 p.4126-4220)、FreeStyle 293F細胞(Invitrogen社)を挙げることができる。
原核細胞を使用する場合は、例えば、大腸菌、枯草菌を挙げることができる。
これらの細胞に目的とする抗体遺伝子を形質転換によって導入し、形質転換された細胞をin vitroで培養することによって抗体が得られる。当該培養においては抗体の配列によって収量が異なる場合があり、同等な結合活性を持つ抗体の中から収量を指標に医薬としての生産が容易なものを選別することが可能である。よって、本発明の抗体には、上記形質転換された宿主細胞を培養する工程、及び当該工程で得られた培養物から目的の抗体又は当該抗体の機能性断片を採取する工程を含むことを特徴とする当該抗体の製造方法によって得られる抗体も含まれる。
(a)B1もしくはC7抗体、chB1抗体、ヒト化抗体H1L1、H2L2、H1L3、H2L4、H3L3、 Trastuzumab及びその変異体のいずれか一つの抗体の重鎖アミノ酸配列をコードするポリヌクレオチドと軽鎖アミノ酸配列をコードするポリヌクレオチドの組み合わせ、
(b)B1もしくはC7抗体、chB1抗体、及び、ヒト化抗体H1L1、H2L2、H1L3、H2L4、H3L3、 Trastuzumab及びその変異体のいずれか一つの抗体のCDRH1~CDRH3を含む重鎖アミノ酸配列をコードするポリヌクレオチドとCDRL1~CDRL3を含む軽鎖アミノ酸配列をコードするポリヌクレオチドの組み合わせ
(c)B1もしくはC7抗体、chB1抗体、及び、ヒト化抗体H1L1、H2L2、H1L3、H2L4、H3L3、 Trastuzumab及びその変異体のいずれか一つの抗体の重鎖可変領域のアミノ酸配列を含む重鎖アミノ酸配列をコードするポリヌクレオチドと軽鎖可変領域のアミノ酸配列を含む軽鎖アミノ酸配列をコードするポリヌクレオチドの組み合わせ
(d)(a)~(c)のいずれか一つに記載のポリヌクレオチドに相補的なポリヌクレオチドからなるヌクレオチドとストリンジェントな条件下でハイブリダイズし、且つ、CDLN6又はHER2に結合する抗体のアミノ酸配列をコードするポリヌクレオチド、及び、
(e)(a)~(c)のいずれか一つに記載のポリヌクレオチドにおいて1~50個、1~45個、1~40個、1~35個、1~30個、1~25個、1~20個、1~15個、1~10個、1~8個、1~6個、1~5個、1~4個、1~3個、1若しくは2個、または1個のアミノ酸が置換、欠失、付加または挿入してなるポリペプチドのアミノ酸配列をコードし、且つ、CLDN6又はHER2に結合する抗体のアミノ酸配列をコードするポリヌクレオチド。
本発明は、本発明の抗体もしくはその機能性断片又はその修飾体をコードするヌクレオチド、該遺伝子が挿入された組換えベクター、該遺伝子又は該ベクターが導入された細胞を含む。
また、本発明は、前記細胞を培養する工程、及び、その培養物から抗体もしくはその機能性断片又はその修飾体を回収する工程を含む、抗体もしくはその機能性断片又はその修飾体の製造方法も含む。
近年、不均一な抗体の糖タンパク質を、酵素反応等によってリモデリングし、官能基を有する糖鎖を均一に導入する方法が報告されている(ACS Chemical Biology 2012, 7, 110、ACS Medicinal Chemistry Letters 2016, 7, 1005、Bioconjugate Chemistry 2015, 26, 2233、Angew. Chem. Int. Ed. 2016, 55, 2361-2367、US2016361436)。

N結合型糖鎖は、Nグリコシド結合、O結合型糖鎖はOグリコシド結合により、抗体のアミノ酸側鎖と結合している。


L(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-を示し、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-3鎖側の非還元末端のシアル酸の2位のカルボン酸とアミド結合を介して結合しており、左端のアステリスク*は、前記リンカーLにおけるLbの1,2,3-トリアゾール環上の1位又は3位の窒素原子と結合していることを示し、n5は2~10の整数であり、好ましくは、2~5の整数である。


L(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-を示し、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-6鎖側の非還元末端のシアル酸の2位のカルボン酸とアミド結合を介して結合しており、左端のアステリスク*は、前記リンカーLにおけるLbの1,2,3-トリアゾール環上の1位又は3位の窒素原子と結合していることを示し、n5は2~10の整数であり、好ましくは、2~5の整数である。


L(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-を示し、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-3鎖側及び1-6鎖側の両方の非還元末端のシアル酸の2位のカルボン酸とアミド結合していることを示し、左端のアステリスク*は、前記リンカーLにおけるLbの1,2,3-トリアゾール環上の1位又は3位の窒素原子と結合していることを示し、n5は2~10の整数であり、好ましくは、2~5の整数である。
例えば、実施例19:ADC1はN297糖鎖がN297-(Fuc)MSG1の場合である。
本発明の抗体-薬物コンジュゲートにおける抗体のN297糖鎖がN297-(Fuc)MSG1もしくはN297-(Fuc)MSG2又はN297-(Fuc)SGである場合、均一な品質のADCを取得することができる。
i)上述の宿主細胞(例えば、動物細胞(CHO細胞等))を培養し、得られた培養物から目的の抗体を採取する工程、
ii)工程i)で得られた抗体を加水分解酵素で処理し、N297糖鎖が(Fucα1,6)GlcNAcである抗体((Fucα1,6)GlcNAc-抗体)を製造する工程(図3A)、
好ましくは、更に当該反応液を、ハイドロキシアパタイトカラムによる精製を含む工程により(Fucα1,6)GlcNAc-抗体を精製する工程、及び、
iii)MSG(9)又はSG(10)のシアル酸の2位のカルボン酸のカルボニル基にアジド基を有するPEGリンカー-(N3-L(PEG))を導入し、且つ、還元末端をオキサゾリン化した糖鎖ドナー分子と、糖転移酵素存在下で(Fucα1,6)GlcNAc-抗体を反応させ、シアル酸にアジド基が導入された糖鎖リモデリング抗体を合成する工程。
また、かかる製造方法により得られた糖鎖リモデリング抗体もしくはその機能性断片、又はそれらの修飾体も本発明に含まれる。
このようなMSG(MSG1、MSG2)又はSG型N297糖鎖を有する糖鎖リモデリング抗体は、例えばWO2013/120066などに記載の方法に準じて、図3に示すような方法で製造することができる。公知の方法に準じて宿主として動物細胞を用いて、遺伝子組み換え蛋白質として抗体を産生させた場合(上記工程i)、N297糖鎖は、基本構造としてフコース付加したN結合型糖鎖構造を有するが、非還元末端の構造や構成糖に多様な修飾がされた様々な構造からなる糖鎖を有する抗体またはその断片の混合物として得られる(図3AのIV)。このように動物細胞で産生された抗体は、EndoSなどの加水分解酵素で処理することによって、還元末端のキトビオース構造のGlcNAcβ1-4GlcNAcの間のグリコシド結合が加水分解され、N297糖鎖として(Fucα1,6)GlcNAcのみを有する単一の糖鎖構造を有する抗体分子(「(Fucα1,6)GlcNAc-抗体」という、図2のA参照)が得られる(図3A)(上記工程ii))。




抗体Abは、抗CLDN6抗体、抗CLDN9抗体、抗CLDN6/CLDN9抗体、抗HER2抗体、抗HER3抗体、抗DLL3抗体、抗FAP抗体、抗CDH11抗体、抗A33抗体、抗CanAg抗体、抗CD19抗体、抗CD20抗体、抗CD22抗体、抗CD25抗体、抗CD30抗体、抗CD33抗体、抗CD37抗体、抗CD56抗体、抗CD70抗体、抗CD98抗体、抗B7-H3抗体、抗TROP2抗体、抗CEA抗体、抗Cripto抗体、抗EphA2抗体、抗FGFR2抗体、抗G250抗体、抗MUC1抗体、抗GPNMB抗体、抗Integrin抗体、抗PSMA抗体、抗Tenascin-C抗体、抗SLC44A4抗体、抗Mesothelin抗体、抗EGFR抗体、抗5T4抗体、抗LRRC15抗体、抗DR5抗体、抗CDH3抗体、抗PDPN抗体、又は抗CD123抗体であり(好ましくは、前記抗CLDN6抗体または抗HER2抗体)であり、
N297糖鎖は、N297-(Fuc)MSG1、N297-(Fuc)MSG2もしくはそれらの混合物又は N297-(Fuc)SG(好ましくは、N297-(Fuc)MSG1)のいずれか一つであることを示し、
L(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-であることを示し、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-3鎖側又は/及び1-6鎖側(好ましくは、1-3鎖側)の非還元末端のシアル酸の2位のカルボン酸とアミド結合を介して結合しており、左端のアステリスクは、前記構造式中のトリアゾール環上の1位又は3位の窒素原子と結合していることを示す。
便宜上、上記最も好ましい抗体-薬物コンジュゲートとして、コンジュゲート1分子中に「N297糖鎖がL中のLbのトリアゾール環上の1位の窒素原子と結合した『-(N297糖鎖)-L-D』(『(N297糖鎖)-(N1Lb)L-D』)」を2又は4個(m2=1又は2)有するか、「3位の窒素原子と結合した『-(N297糖鎖)-L-D』(『(N297糖鎖)-(N3Lb)L-D』)」を2又は4個(m2=1又は2)有する構造を記載しているが、コンジュゲート1分子中に『(N297糖鎖)-(N1Lb)L-D』(m2=1の場合、1個、m2=2の場合、1,2,3個)及び『(N297糖鎖)-(N3Lb)L-D』(m2=1の場合、1個、m2=2の場合、3,2,1個)の両方を有する抗体-薬物コンジュゲートも含む。すなわち、コンジュゲート1分子中に『(N297糖鎖)-(N1Lb)L-D』か『(N297糖鎖)- (N3Lb)L-D』のいずれか一方のみ、又は、その両方が混在している。
本発明の抗体-薬物コンジュゲートにおいて、抗体が、抗体のリモデリングされた糖鎖からLに結合している場合、抗体薬物コンジュゲートにおける抗体1分子あたりの薬物結合数m2は1または2の整数である。当該糖鎖がN297糖鎖であり、糖鎖がN297-(Fuc)MSG1、N297-(Fuc)MSG2又はN297-(Fuc)MSG1とN297-(Fuc)MSG2の混合物の場合、m2は1であり、DARは1~3の範囲(好ましくは、1.0~2.5の範囲、より好ましくは、1.2~2.2もしくは1.6~2.2の範囲)である。N297糖鎖が、N297-(Fuc)SGの場合、m2は2であり、DARは3~5の範囲(好ましくは、3.2~4.8の範囲であり、より好ましくは、3.5~4.2の範囲)である。
なお、当業者であれば本願の実施例の記載から抗体に必要な数の薬物を結合させる反応を設計することができ、ピロロベンゾジアゼピン誘導体の結合数をコントロールした抗体を取得することができる。
糖鎖リモデリング抗体、例えばWO2013/120066などに記載の方法に準じて、図3に示すような方法で製造することができる。
(共通操作A:抗体水溶液の濃縮)
Amicon Ultra(30,000乃至50,000 MWCO,Millipore Co.)の容器内に抗体又は抗体-薬物コンジュゲート溶液を入れ、遠心機(Allegra X-15R,Beckman Coulter,Inc.)を用いた遠心操作(2000G乃至4000Gで5乃至20分間遠心)にて、抗体および後述する抗体-薬物コンジュゲート溶液を濃縮した。
(共通操作B:抗体の濃度測定)
UV測定器(Nanodrop 1000, Thermo Fisher Scientific Inc.)を用いて、メーカー規定の方法に従い、抗体濃度の測定を行った。その際に、抗体ごとに異なる280nm吸光係数(1.3mLmg-1cm-1乃至1.8mLmg-1cm-1)を用いた。
(共通操作C:抗体のバッファー交換)
抗体水溶液は緩衝溶液(リン酸緩衝生理食塩水(pH6.0)、リン酸緩衝液(pH6.0)等)を加え共通操作Aを用いて濃縮した。この操作を数回行った後、共通操作Bを用いて抗体濃度の測定を行い、緩衝溶液(リン酸緩衝生理食塩水(pH6.0)、リン酸緩衝液(pH6.0)等)を用いて10mg/mLに抗体濃度を調整した。
本製造法は、上述の糖鎖リモデリング抗体と製造中間体(2)をSPAAC反応(strain-promoted alkyne azide cycloaddition: JACS. 2004, 126,15046-15047)により結合させ、抗体-薬物コンジュゲートを製造する方法である。

La’、Lp’、B’は、La、Lp、Bと同義であり、
Jは、以下で示されるいずれかの構造式を示し、
式中、アステリスクはLa’と結合していることを示す。

抗体1モルに対し、化合物(2)は2モルから過剰モル、好ましくは1モルから30モルであり、有機溶媒の比率は、抗体の緩衝液に対し1乃至200%v/vが好ましい。反応温度は0℃乃至37℃、好ましくは10℃から25℃であり、反応時間は1から150時間、好ましくは6時間から100時間である。反応時のpHは5乃至9が好ましい。
市販のSorbitol(5%)を含む酢酸緩衝液(10mM,pH5.5;本明細書でABSと称する)でNAP-25カラムを平衡化させた。このNAP-25カラムに、抗体-薬物コンジュゲート反応水溶液(約1.5~2.5mL)をのせ、メーカー規定の量の緩衝液で溶出させることで、抗体画分を分取した。この分取画分を再びNAP-25カラムにのせ、緩衝液で溶出させるゲルろ過精製操作を計2乃至3回繰り返すことで、未結合の薬物リンカーやジメチルスルホキシド、プロピレングリコールを除いた抗体-薬物コンジュゲートを得た。必要に応じて、共通操作AおよびCにより抗体-薬物コンジュゲート溶液の濃度を調製した。
共通操作E:抗体-薬物コンジュゲートにおける抗体濃度の測定
抗体-薬物コンジュゲートにおける結合薬物濃度は、下記に示すランベルト・ベールの法則を用いて、算出することができる。
以下にランベルト・ベールの法則を用いた式(I)を示す。

上記式(I)より抗体-薬物コンジュゲートのモル濃度C(mol・L-1)は以下の式(II)で求められる。


吸光度A280は、抗体-薬物コンジュゲート水溶液の280nmにおけるUV吸光度の実測値を用いた。モル質量MW(g・mol-1)は抗体のアミノ酸配列からより求められる抗体分子量の計算推定値を抗体-薬物コンジュゲートのモル質量の近似値として用いている。光路長l(cm)は1cmで測定した。
抗体薬物コンジュゲートのモル吸光係数ε280は、以下の式(IV)によって求めることができる。
εAb,280は抗体のアミノ酸配列から、既知の計算方法(Protein Science, 1995, vol.4, 2411-2423)によって推定することができる。実施例において、Trastuzumabのモル吸光係数は、εAb,280=215400(計算推定値)を用いた。CLDN6抗体のモル吸光係数は、εAb,280=221340(計算推定値)、TROP2抗体のモル吸光係数は、εAb,280=226400(計算推定値)、CD98抗体のモル吸光係数は、εAb,280=240400(計算推定値)、LPS抗体のモル吸光係数は、εAb,280=230300(計算推定値)を、Trastuzumab変異体のモル吸光係数は、εAb,280=215057(計算推定値)を用いた。
εDL,280は、都度UV測定で得た実測値より算出したものを使用した。すなわち、コンジュゲート前駆体(薬物)をあるモル濃度に溶解させた溶液の吸光度を測定し、ランベルト・ベールの法則、式(I)を適用することで得られる値を使用した。
抗体-薬物コンジュゲートにおける抗体一分子あたりの薬物平均結合数は、以下の方法を用いる高速液体クロマトグラフィー(HPLC)分析によって求めることができる。
[F-1.HPLC分析用サンプルの調製(抗体-薬物コンジュゲートの還元)]
抗体-薬物コンジュゲート溶液(約1mg/mL、60μL)をジチオトレイトール(DTT)水溶液(100mM、15μL)と混合する。混合物を37℃で30分インキュベートすることで、抗体-薬物コンジュゲートのL鎖及びH鎖間のジスルフィド結合を切断したサンプルを、HPLC分析に用いる。
HPLC分析を、下記の測定条件にて行う。
HPLCシステム:Agilent 1290 HPLCシステム(Agilent Technologies)
検出器:紫外吸光度計(測定波長:280nm、329nm)
カラム:BEH Phenyl(2.1×50mm、1.7μm、Waters Acquity)
カラム温度:75℃
移動相A:0.1%トリフルオロ酢酸(TFA),15%イソプロピルアルコール水溶液
移動相B:0.075%TFA、15%イソプロピルアルコールアセトニトリル溶液
グラジエントプログラム:14%-36%(0分-15分)、36%-80%(15-17分)、80%-14%(17分―17.1分)、14%-14%(17.1分―23分)
サンプル注入量:5μL
〔F-3-1〕薬物の結合していない抗体のH鎖(H0)に対して、薬物の結合したH鎖(薬物が一つ結合したH鎖:H1、薬物が二つ結合したH鎖:H2)は、結合した薬物の数に比例して疎水性が増して保持時間が大きくなることから、L0、H0、H1、H2、の順に溶出される。L0及びH0との保持時間比較により検出ピークをL0、H0、H1、H2のいずれかに割り当てることができる。また薬物の結合は、薬物の特長的な329nmの波長吸収でも確認できる。
〔F-3-2〕薬物リンカーにUV吸収があるため、薬物リンカーの結合数に応じて、L鎖、H鎖及び薬物リンカーのモル吸光係数を用いて下式に従ってピーク面積値の補正を行う。

本発明において「PARP阻害剤」とは、PARP(ポリアデノシン5’ニリン酸(ADP)リボースポリメラーゼ)を阻害することにより、一本鎖切断の修復を妨げる機能を有する薬剤である(Benafif S, et al., Onco. Targets Ther. (2015) 8, 519-528.)(Fong PC, et al., N. Engl. J. Med. (2009) 361, 123-134.)(Gelmon KA, et al., Lancet Oncol. (2011) 12, 852-861.)。PARPには複数のサブタイプが存在するが、本発明におけるPARP阻害剤は、好適には、PARP-1及びPARP-2を阻害する。本発明におけるPARP阻害剤は、PARPを阻害することにより、一本鎖切断の修復を妨げる機能を有する薬剤であれば限定はされないが、好適には、オラパリブ(Olaparib)(Menear KA, et al., J. Med. Chem. (2008) 51, 6581-6591.)、ルカパリブ(Rucaparib)(Gillmore AT, et al., Org. Process Res. Dev. (2012) 16, 1897-1904.)、ニラパリブ(Niraparib)(Jones P, et al., J. Med. Chem. (2009) 52, 7170-7185.)、タラゾパリブ(Talazoparib)(Shen Y, et al., Clin. Cancer Res. (2013) 19(18), 5003-15.)、ベリパリブ(Veliparib)、パミパリブ(Pamiparib)、及びフルゾパリブ(Fluzoparib)、並びにそれらの薬理上許容される塩を挙げることができ、より好適には、オラパリブ、ルカパリブ、ニラパリブ、及びタラゾパリブ、並びにそれらの薬理上許容される塩を挙げることができる。
以下、本発明に係る抗体-薬物コンジュゲートとPARP阻害剤が組み合わされて投与されることを特徴とする医薬組成物及び治療方法(予防も含む)について説明する。
がんはPARP阻害剤に非感受性であるか、相同的組換え(HR)依存的DNA二本鎖破壊(DSB)修復経路に非依存的であるか、PARP阻害剤に非感受性かつ相同的組換え(HR)依存的DNA二本鎖破壊(DSB)修復経路に非依存的である。
上述の方法により、本発明の医薬組成物及び治療方法の抗腫瘍効果について、既存のがん治療用医薬組成物及び治療方法に対する優位性を確認することができる。
本発明の医薬組成物及び治療方法は、外科手術と組み合わせた補助化学療法として使用することもできる。本発明の医薬組成物は外科手術の前に腫瘍の大きさを減じさせる目的で投与されてもよい(術前補助化学療法、又はネオアジュバント療法という)し、外科手術後に、腫瘍の再発を防ぐ目的で投与されてもよい(術後補助化学療法、又はアジュバント療法という)。
抗HER2抗体はUS5821337を参照して作製した。Trastuzumabの軽鎖及び重鎖のアミノ酸配列を配列番号64及び配列番号65に示した。
抗TROP2抗体はWO2003/074566、WO2015/098099(参考例1)を参照して作製した。hRS7の軽鎖及び重鎖のアミノ酸配列を配列番号68及び配列番号69に示した。
実施例1
[実施例1-1:中間体1]

5-ベンジル 6-メチル(6S)-5-アザスピロ[2.4]ヘプタン-5,6-ジカルボキシレート(1-1)(104mmol,WO2012087596)のテトラヒドロフラン(500mL)溶液に、水素化ホウ素リチウム(4.30g,178mmol)を0℃にて少量ずつ加えた。0℃にて30分撹拌した後、室温にて2時間撹拌した。0℃にて水(180mL)、2規定塩酸(186mL)を加え、減圧留去した。得られた残渣を酢酸エチルで4回抽出し、有機層を飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥した。減圧留去し、得られた残渣(1-2)(27.9g,90%)をそのまま次の反応に用いた。
上記工程1にて得られた化合物(1-2)(27.9g,107mmol)とイミダゾール(14.5g,214mmol)のジクロロメタン(300mL)溶液に、室温にてtert-ブチルジメチルシリルクロリド(24.2g,160mmol)を加え、室温にて18時間撹拌した。反応溶液を飽和クエン酸水溶液、飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した後、減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~50:50(v/v)]にて精製し、目的物(1-3)(32.5g,81%)を得た。
1H-NMR(CDCl3)δ:7.39-7.34(5H,m),5.23-5.11(2H,m),4.10-3.48(4H,m),3.16-3.14(1H,m),2.15-2.04(1H,m),1.81-1.77(1H,m),0.91-0.88(9H,m),0.65-0.55(4H,m),0.08-0.01(6H,m).
MS (APCI)m/z:376(M+H)+
上記工程2で得られた化合物(1-3)(32.5g,86.5mmol)のエタノール(400mL)溶液に、室温にて7.5%パラジウム炭素触媒(54%水分、5.00g)を加え、室温、水素雰囲気下にて6時間撹拌した。反応溶液をセライト濾過し、濾液を減圧留去し、目的物(1-4)(21.3g,定量的)を得た。
1H-NMR(CDCl3)δ:3.79-3.77(1H,m),3.71-3.69(1H,m),3.65-3.60(1H,m),3.01-2.98(2H,m),1.81-1.71(2H,m),0.90(9H,s),0.65-0.57(4H,m),0.08(3H,s),0.07(3H,s).
MS(APCI、ESI)m/z:242(M+H)+
5-メトキシ-2-ニトロ-4-{トリ(プロパン-2-イル)シリル]オキシ}安息香酸(52.2g,141mmol,US20150283262)と1-ヒドロキシベンゾトリアゾール一水和物(23.8g,155mmol)のジクロロメタン(500mL)溶液に、氷冷下にてN,N’-ジシクロヘキシルカルボジイミド(35.0g,170mmol)を加えた。反応混合物を室温にて撹拌した。カルボン酸消失後、-60℃にて上記工程3にて得られた化合物(1-4)(34.1g,141mmol)とトリエチルアミン(29.4mL,212mmol)のジクロロメタン(100mL)溶液をゆっくりと滴下した。反応溶液を室温にて一晩撹拌した後、反応混合物に飽和炭酸水素ナトリウム水溶液を加え、反応混合物をクロロホルムで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。減圧留去して得られた残渣に酢酸エチルとジエチルエ-テルを加え、固体成分を濾過により取り除き、濾液を減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~25:75(v/v)]にて精製し、目的物(1-5)(55.0g,66%)を得た。
1H-NMR(CDCl3)δ:7.72-7.66(1H,m),6.80-6.73(1H,m),4.53-4.49(1H,m),4.04-3.95(1H,m),3.91-3.88(3H,m),3.59-3.54(1H,m),3.36-3.25(0.5H,m),3.01-2.96(1.5H,m),2.24-2.20(0.3H,m),2.09-2.05(0.7H,m),2.00-1.97(0.7H,m),1.69-1.67(0.3H,m),1.32-1.24(3H,m),1.12-1.05(18H,m),0.93-0.91(6H,m),0.79-0.77(3H,m),0.71-0.62(2H,m),0.57-0.40(2H,m),0.12-0.10(4H,m),0.11-0.15(2H,m).
MS(APCI、ESI)m/z:593(M+H)+
上記工程4で得られた化合物(1-5)(55.0g,92.8mmol)のエタノール(300mL)溶液に、窒素雰囲気下にて、7.5%パラジウム炭素(10.0g)を加えた。窒素風船を直ちに水素風船に付け替え、反応混合物を水素雰囲気下、室温で激しく撹拌した。原料消失後、反応混合物を濾過し、濾液を減圧留去し、得られた目的物(1-6)(52.2g,100%)をそのまま次の反応に用いた。
1H-NMR(CDCl3)δ:6.71(1H,s),6.25(1H,s),4.55-4.28(2H,m),3.97(1H,m),3.75-3.62(3H,m),3.70(3H,s),3.09-3.07(1H,m),2.24-2.19(1H,m),1.81-1.68(1H,m),1.27-1.22(3H,m),1.09-1.05(18H,m),0.90(9H,s),0.65-0.46(4H,m),0.07-0.03(6H,m).
MS(APCI、ESI)m/z:563(M+H)+
上記工程5で得られた化合物(1-6)(18.6g,33.0mmol)およびトリエチルアミン(6.26mL,45.2mmol)のTHF(300mL)溶液に、エタノール-氷浴上にて、トリホスゲン(4.22g,14.2mmol)をゆっくりと添加した。添加後、氷冷した反応混合物に、N-[(プロプ-2-エン-1-イルオキシ)カルボニル]-L-バリル-N-[4-(ヒドロキシメチル)フェニル]-L-アラニンアミド(11.4g,30.2mmol,WO2011130598)とトリエチルアミン(6.26mL,45.2mmol)のテトラヒドロフラン(100mL)、N,N-ジメチルホルムアミド(30mL)混合溶液をゆっくりと滴下した。滴下後、氷浴をはずし、反応混合物を、窒素雰囲気下、40℃にて撹拌した。原料消失後、反応混合物に水を加え、反応混合物を酢酸エチルで抽出した。有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過後、減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~40:60(v/v)]にて精製し、目的物(1-7)(23.5g,74%)を得た。
1H-NMR(CDCl3)δ:8.99(1H,m),8.58(1H,s),7.80(1H,s),7.55-7.53(2H,m),7.34-7.32(2H,m),6.77-6.75(2H,m),5.94-5.87(1H,m),5.40-5.38(1H,m),5.33-5.29(1H,m),5.23-5.21(1H,m),5.13(1H,m),5.10(2H,m),4.69-4.64(1H,m),4.62-4.52(2H,m),4.06-4.03(1H,m),3.98(1H,m),3.76-3.65(6H,m),3.04(1H,m),2.28-2.26(1H,m),2.18-2.13(1H,m),1.46(3H,m),1.32-1.25(3H,m),1.11-1.09(18H,m),0.99-0.84(15H,m),0.65-0.40(4H,m),0.08-0.00(6H,m).
MS(APCI、ESI)m/z:966(M+H)+
上記工程6で得られた化合物(1-7)(23.5g,24.3mmol)のテトラヒドロフラン(50mL),メタノール(50mL),水(44mL)溶液に、室温下にて酢酸(200mL)を加えた。反応混合物を室温にて撹拌した。原料消失後、反応混合物を酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過後、減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~0:100(v/v)]にて精製し、目的物(1-8)(18.0g,87%)を得た。
1H-NMR(CDCl3)δ:8.64-8.62(1H,m),8.50(1H,m),7.69(1H,m),7.55-7.53(2H,m),7.34-7.32(2H,m),6.79-6.75(3H,m),5.91-5.89(1H,m),5.39(1H,m),5.32-5.29(1H,m),5.23-5.21(1H,m),4.68-4.54(4H,m),4.31(1H,m),4.06-4.04(1H,m),3.81-3.79(3H,m),3.76(3H,s),3.63-3.61(1H,m),3.13-3.11(1H,m),2.16-2.13(1H,m),1.87-1.81(2H,m),1.46-1.43(3H,m),1.30-1.24(3H,m),1.12-1.08(18H,m),0.98-0.91(6H,m),0.63-0.45(4H,m).
MS(APCI、ESI)m/z:852(M+H)+
ジメチルスルホキシド(3.75mL,52.8mmol)のジクロロメタン(300mL)溶液に、窒素雰囲気下、-78℃にて、塩化オキサリル(2.17mL,25.3mmol)をゆっくりと滴下した。滴下後、反応混合物を-78℃にて撹拌した。反応混合物に、上記工程7で得られた化合物(1-8)(18.0g,21.1mmol)のジクロロメタン(50.0mL)溶液をゆっくりと滴下した。反応溶液に-78℃にて、トリエチルアミン(14.6mL,105mmol)を加えた。添加後、冷媒浴をはずし、室温までゆっくりと昇温した。原料消失後、反応混合物に水を加え、反応混合物をクロロホルム(200mL)で抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥した。濾過後、減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~0:60(v/v)]にて精製し、目的物(1-9)(16.5g,92%)を得た。
1H-NMR(CDCl3)δ:8.51-8.36(1H,m),7.54-7.38(2H,m),7.22-7.07(3H,m),6.73-6.64(1H,m),5.94-5.87(2H,m),5.33-5.22(3H,m),5.09(1H,m),4.97(1H,m),4.64-4.58(4H,m),4.02-4.00(1H,m),3.86-3.83(3H,m),3.75-3.70(1H,m),3.61-3.54(2H,m),3.38-3.29(1H,m),2.40(1H,m),2.16-2.14(1H,m),1.74-1.71(1H,m),1.44(3H,m),1.18-1.16(3H,m),1.05-1.00(18H,m),0.97-0.92(6H,m),0.72-0.60(4H,m).
MS(APCI、ESI)m/z:850(M+H)+
上記工程8で得られた化合物(1-9)(12.0g,14.1mmol)および2,6-ルチジン(6.58mL,56.5mmol)のジクロロメタン(200mL)溶液に、窒素雰囲気下、0℃にて、トリフルオロメチルスルホン酸tert-ブチルジメチルシリル(9.73mL,42.3mmol)をゆっくりと滴下した。氷冷下、10分間撹拌した後に、氷浴をはずし、室温にて撹拌した。原料消失後、反応混合物に水を加え、反応混合物をクロロホルムで抽出した。水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過後、減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~25:75(v/v)]にて精製し、目的物(1-10)(8.12g,60%)を得た。
1H-NMR(CDCl3)δ:8.67-8.45(1H,m),7.50-7.44(2H,m),7.19(1H,s),7.13(2H,m),6.95(2H,m),6.62-6.57(2H,m),6.01(1H,m),5.95-5.86(1H,m),5.33-5.13(3H,m),4.82(1H,m),4.65-4.54(3H,m),4.03-4.01(1H,m),3.84-3.82(3H,m),3.73-3.66(1H,m),3.50-3.48(1H,m),3.27(1H,m),2.37-2.33(1H,m),2.19-2.13(1H,m),1.54-1.43(3H,m),1.22-1.13(3H,m),1.10-1.00(18H,m),0.97-0.91(6H,m),0.81(9H,s),0.76-0.59(4H,m),0.19-0.09(6H,m).
MS(APCI、ESI)m/z:964(M+H)+
上記工程9で得られた化合物(1-10)(8.12g,8.42mmol)のN,N-ジメチルホルムアミド(90mL)、水(2mL)溶液に、酢酸リチウム(0.611g,9.26mmol)を加え、室温下で撹拌した。原料消失後、反応混合物に水を加え、反応混合物を酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。濾過後、減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~0:100(v/v)]にて精製し、目的物(1-11)(5.48g,81%)を得た。
1H-NMR(400MHz,CDCl3,20.9℃)δ:8.76-8.60(1H,m),7.45-7.44(2H,m),7.21(1H,s),7.10-7.09(2H,m),6.81-6.74(1H,m),6.65(1H,s),6.23(1H,s),6.01-5.99(1H,m),5.95-5.84(1H,m),5.41-5.20(2H,m),5.16(1H,m),4.84(1H,m),4.67-4.54(4H,m),4.05-4.03(1H,m),3.87(3H,s),3.71(1H,m),3.55-3.51(1H,m),3.26(1H,m),2.35(1H,m),2.18-2.12(1H,m),1.55-1.42(4H,m),0.97-0.92(6H,m),0.81(9H,s),0.76-0.61(4H,m),0.20-0.06(6H,m)
MS(APCI、ESI)m/z:808(M+H)+


グリシルグリシン(0.328g,2.49mmol)、N,N-ジイソプロピルエチルアミン(0.433mL,2.49mmol)のN,N-ジメチルホルムアミド(20mL)溶液に、1-{[4-(11,12-ジデヒドロジベンゾ[b,f]アゾシン-5(6H)-イル)-4-オキソブタノイル]オキシ}ピロリジン-2,5-ジオン(2-1)(1.00g,2.49mmol,Click Chemistry Tools)、水(10mL)を室温にて加え、同温度にて一晩撹拌した。減圧留去し、得られた残渣を、シリカゲルカラムクロマトグラフィー[クロロホルム~クロロホルム:メタノ-ル:水=7:3:1(v/v/v)の分配有機層]にて精製し、目的物(0.930g,89%)を得た。
1H-NMR(DMSO-D6)δ:12.58(1H,s),8.14-8.12(1H,m),8.08-8.07(1H,m),7.69-7.68(1H,m),7.62-7.61(1H,m),7.53-7.45(3H,m),7.40-7.29(3H,m),5.05-5.01(1H,m),3.73-3.72(2H,m),3.66-3.60(3H,m),2.66-2.60(1H,m),2.33-2.24(1H,m),2.08-2.04(1H,m),1.81-1.77(1H,m).
MS(APCI、ESI)m/z:420[(M+H)+].
[実施例2-1:薬物リンカー1]

(2R,11aS)-8-(ベンジルオキシ)-2-{[tert-ブチル(ジメチル)シリル]オキシ}-7-メトキシ-10-{[2-(トリメチルシリル)エトキシ]メチル}-2,3-ジヒドロ-1H-ピロロ[2,1-c][1,4]ベンゾジアゼピン-5,11(10H,11aH)-ジオン(3-1)(25.5g,41.6mmol,WO2016149546)のテトラヒドロフラン(150mL)、エタノール(150mL)溶液に、窒素雰囲気下、5%パラジウム炭素(54%水分、10.0g)を加えた後、反応溶液を水素雰囲気下、室温にて三日間撹拌した。反応溶液にクロロホルムを加え、セライト濾過した後、濾液を減圧留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~50:50(v/v)]にて精製し、目的物(3-2)(19.4g,89%)を得た。
1H-NMR(CDCl3)δ:7.36(1H,s),7.25(1H,s),6.01(1H,s),5.45-5.43(1H,m),4.69-4.67(1H,m),4.60-4.55(1H,m),4.23-4.21(1H,m),3.96(3H,s),3.76-3.68(2H,m),3.63-3.61(1H,m),3.56-3.53(1H,m),2.88-2.83(1H,m),2.03-2.00(1H,m),1.00-0.98(2H,m),0.87(9H,s),0.10(6H,s),0.02(9H,s).
MS(APCI、ESI)m/z:523(M+H)+
上記工程1にて得られた化合物(3-2)(10.8g,20.7mmol)のN,N-ジメチルホルムアミド(30mL)溶液に、1,5-ジブロモペンタン(23.8g,103mmol)、炭酸カリウム(3.43g,24.8mmol)を室温で加えた。室温で3時間撹拌した後、反応溶液に水を加え、酢酸エチルで抽出した。得られた有機層を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥した後、減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=90:10(v/v)~50:50(v/v)]にて精製し、目的物(3-3)(14.5g,定量的)を得た。
1H-NMR(CDCl3)δ:7.34(1H,s),7.21(1H,s),5.52-5.49(1H,m),4.63-4.62(1H,m),4.58-4.55(1H,m),4.24-4.22(1H,m),4.07-4.04(2H,m),3.92(3H,s),3.82-3.64(3H,m),3.56-3.53(1H,m),3.45-3.43(2H,m),2.86-2.84(1H,m),2.04-2.00(1H,m),1.97-1.87(4H,m),1.66-1.62(2H,m),1.01-0.98(2H,m),0.87(9H,s),0.10(6H,s),0.04(9H,s)
MS(APCI、ESI)m/z:673[81Br,(M+H)+],671[79Br,(M+H)+].
上記工程2にて得られた化合物(3-3)(21.5mmol)のテトラヒドロフラン(40mL)溶液に、1mol/Lのテトラブチルアンモニウムフロリド テトラヒドロフラン溶液(28.0mL,28.0mmol)を0℃にて加えた。室温にて30分間撹拌した後、反応溶液に水を加え、酢酸エチルで抽出し、得られた有機層を飽和食塩水で洗浄した。硫酸ナトリウムで乾燥した後、減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー[クロロホルム:メタノール=97.5:2.5(v/v)~92.5:7.5(v/v)]にて精製し、目的物(3-4)(11.3g,94%)を得た。
1H-NMR(CDCl3)δ:7.34(1H,s),7.21(1H,s),5.53-5.50(1H,m),4.69-4.64(2H,m),4.32-4.30(1H,m),4.10-4.00(2H,m),3.91(3H,s),3.88-3.75(2H,m),3.73-3.64(2H,m),3.45-3.44(2H,m),2.99-2.96(1H,m),2.15-2.09(1H,m),1.99-1.85(5H,m),1.68-1.62(2H,m),1.01-0.95(2H,m),0.04(9H,s).
MS(APCI、ESI)m/z:559[81Br,(M+H)+],557[79Br,(M+H)+].
上記工程3にて得られた化合物(3-4)(11.3g,20.2mmol)、テトラブチルアンモニウムブロミド(0.325g,1.01mmol)、臭化カリウム(0.240g,2.02mmol,)を飽和炭酸水素ナトリウム水溶液(60mL)、ジクロロメタン(60mL)に溶解させ、nor-AZADO(0.0279g,0.202mmol)、次亜塩素酸ナトリウム五水和物(2.03g,27.2mmol)を0℃にて加え、0℃にて30分間撹拌した。原料が残存したため、次亜塩素酸ナトリウム五水和物(1.00g、13.4mmol)を0℃にて加え、0℃にて15分撹拌した。さらに次亜塩素酸ナトリウム五水和物(0.300g、4.03mmol)を0℃にて加え、0℃にて15分撹拌し、原料の消失をTLCにて確認した。反応溶液にチオ硫酸ナトリウム水溶液を加え、クロロホルムで抽出し、得られた有機層を硫酸ナトリウムで乾燥した。減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=75:25(v/v)~40:60(v/v)]にて精製し、目的物(3-5)(9.74g,87%)を得た。
1H-NMR(CDCl3)δ:7.33(1H,s),7.24(1H,s),5.56-5.53(1H,m),4.71-4.69(1H,m),4.66-4.63(1H,m),4.27-4.22(1H,m),4.12-4.02(2H,m),3.93-3.88(4H,m),3.82-3.75(1H,m),3.69-3.67(1H,m),3.61-3.56(1H,m),3.46-3.44(2H,m),2.82-2.77(1H,m),1.97-1.89(4H,m),1.68-1.64(2H,m),1.05-0.93(2H,m),0.04(9H,s).
MS(APCI、ESI)m/z:557[81Br,(M+H)+],555[79Br,(M+H)+].
上記工程4にて得られた化合物(3-5)(9.74g,17.5mmol)のジクロロメタン(160mL)溶液に、2,6-ルチジン(8.17mL,70.1mmol)を-40℃にて加え、-40℃にて10分間撹拌した。反応溶液に無水トリフルオロメタンスルホン酸(8.85mL,52.6mmol)を-40℃にて加え、-40℃にて30分間撹拌した。反応溶液に10%クエン酸水溶液を加え、クロロホルムで抽出し、得られた有機層を硫酸ナトリウムで乾燥した。減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=95:5→70:35]にて精製した後,NH2シリカゲルクロマトグラフィー[ヘキサン:酢酸エチル=95:5(v/v)~65:35(v/v)]にて精製し、目的物(3-6)(7.10g,59%)を得た。
1H-NMR(CDCl3)δ:7.32(1H,s),7.24(1H,s),7.15-7.14(1H,m),5.56-5.53(1H,m),4.70-4.68(1H,m),4.66-4.63(1H,m),4.11-4.01(2H,m),3.94-3.90(4H,m),3.84-3.75(1H,m),3.73-3.68(1H,m),3.46-3.44(2H,m),3.18-3.14(1H,m),1.96-1.88(4H,m),1.69-1.61(2H,m),1.02-0.92(2H,m),0.04(9H,s).
MS(APCI、ESI)m/z:689[81Br,(M+H)+],687[79Br,(M+H)+].
上記工程5にて得られた化合物(3-6)(2.00g,2.91mmol)、4-メトキシフェニルボロン酸(0.884g,5.82mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(0.336g,0.291mmol)、炭酸ナトリウム(1.23g,11.6mmol)の混合物にトルエン(20mL)、エタノール(10mL)、水(10mL)を室温にて加えた。反応溶液を室温にて30分撹拌した後、反応溶液を酢酸エチルで抽出し、水、飽和食塩水で洗浄した。有機層を硫酸ナトリウムで乾燥後、減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=90:10(v/v)~50:50(v/v)]にて精製し、目的物(3-7)(1.71g,91%)を得た。
1H-NMR(CDCl3)δ:7.38-7.37(3H,m),7.33(1H,s),7.25(1H,s),6.89-6.88(2H,m),5.56-5.54(1H,m),4.71-4.68(1H,m),4.65-4.62(1H,m),4.09-4.04(2H,m),3.96-3.91(4H,m),3.85-3.66(5H,m),3.46-3.45(2H,m),3.16-3.12(1H,m),1.99-1.94(4H,m),1.69-1.64(2H,m),1.00-0.98(2H,m),0.04(9H,s).
MS(APCI、ESI)m/z:647[81Br,(M+H)+],645[79Br,(M+H)+].
上記工程6にて得られた化合物(3-7)(0.789g,1.22mmol)をエタノール(10mL)、テトラヒドロフラン(10mL)に溶解し、2.0Mの水素化ホウ素リチウムテトラヒドロフラン溶液(6.11mL,12.2mmol)を0℃にて加え、0℃にて3時間撹拌した。反応溶液に水を加え、クロロホルムで抽出し、得られた有機層を硫酸ナトリウムで乾燥した。減圧留去し、得られた残渣をジクロロメタン(10mL)、エタノール(20mL)、水(10mL)に溶解し、シリカゲル(4g)を室温にて加え、室温で4日間撹拌した。シリカゲルをろ過により除き、水を加え、クロロホルムで抽出し、得られた有機層を硫酸ナトリウムで乾燥した。減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=60:40(v/v)~25:75(v/v)]にて精製し、目的物(3-8)(0.496g,81%)を得た。
1H-NMR(CDCl3)δ:7.90-7.89(1H,m),7.53(1H,s),7.40-7.40(1H,m),7.35-7.34(2H,m),6.92-6.90(2H,m),6.83-6.81(1H,m),4.43-4.40(1H,m),4.13-4.06(2H,m),3.96(3H,s),3.84(3H,s),3.61-3.57(1H,m),3.47-3.36(3H,m),2.00-1.92(4H,m),1.67-1.63(2H,m).
MS(APCI、ESI)m/z:501[81Br,(M+H)+],499[79Br,(M+H)+].
上記工程7にて得られた化合物(3-8)(0.496g,0.992mmol)のジクロロメタン(20mL)溶液にナトリウムトリアセトキシボロヒドリド(0.421g,1.99mmol)を0℃にて加えた。室温にて2時間撹拌した後、飽和炭酸水素ナトリウム水溶液を加え、クロロホルムで抽出した。有機層を硫酸ナトリウムで乾燥し、減圧留去した後、得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=60:40(v/v)~25:75(v/v)]にて精製し、目的物(3-9)(0.426g,86%)を得た。
1H-NMR(CDCl3)δ:7.53-7.53(2H,m),7.32-7.30(2H,m),6.89-6.87(2H,m),6.05(1H,s),4.33-4.27(2H,m),4.00-3.98(2H,m),3.86(3H,s),3.82(3H,s),3.57-3.55(2H,m),3.42-3.38(3H,m),2.76-2.72(1H,m),1.96-1.88(4H,m),1.65-1.62(2H,m).
MS(APCI、ESI)m/z:503[81Br,(M+H)+],501[79Br,(M+H)+].
上記工程8にて得られた化合物(3-9)(0.426g,0.849mmol)のジクロロメタン(30mL)溶液に、ピリジン(0.102mL1.27mmol)、クロロぎ酸アリル(0.374mL,3.54mmol)を0℃にて加え、0℃にて15分間撹拌した。反応溶液に10%クエン酸水溶液を加え、クロロホルムで抽出し、得られた有機層を飽和炭酸水素ナトリウム水溶液で洗浄後、硫酸ナトリウムで乾燥した。減圧留去し、得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=90:10(v/v)~50:50(v/v)]にて精製し、目的物(3-10)(0.465g,94%)を得た。
1H-NMR(CDCl3)δ:7.38(1H,s),7.31-7.29(2H,m),7.26-7.25(1H,m),6.89-6.87(2H,m),6.71(1H,s),5.80-5.78(1H,m),5.14-5.11(2H,m),4.65-4.62(1H,m),4.39-4.26(3H,m),4.03-4.01(2H,m),3.92(3H,s),3.82(3H,s),3.66-3.64(1H,m),3.46-3.44(2H,m),3.30-3.27(1H,m),2.72-2.68(1H,m),1.96-1.88(4H,m),1.68-1.60(2H,m).
MS(APCI、ESI)m/z:587[81Br,(M+H)+],585[79Br,(M+H)+].
実施例1-1工程10にて得られた化合物(1-11)(0.130g,0.161mmol)と上記工程9にて得られた化合物(3-10)(0.104g,0.177mmol)のN,N-ジメチルホルムアミド(3mL)溶液に炭酸カリウム(0.0266g,0.193mmol)を室温にて加え、室温にて一晩撹拌した。反応溶液を酢酸エチルで希釈し、水、飽和食塩水で洗浄した後、硫酸ナトリウムで乾燥した。減圧留去した後、得られた残渣をNH2-シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=70:30(v/v)~0:100(v/v)]にて精製し、目的物(3-11)(0.184g,87%)を得た。
1H-NMR(CDCl3)δ:8.76(1H,s),7.58-7.56(2H,m),7.39(1H,s),7.32-7.30(2H,m),7.26-7.24(2H,m),7.19-7.17(3H,m),6.90-6.88(2H,m),6.78(1H,s),6.68-6.66(1H,m),6.37(1H,s),5.99-5.93(3H,m),5.34-5.20(6H,m),4.66-4.01(11H,m),3.90(3H,s),3.89(3H,s),3.78-3.54(9H,m),3.31-3.28(2H,m),2.73-2.69(1H,m),2.38-2.35(1H,m),2.19-2.13(1H,m),1.82-1.80(2H,m),1.46-1.29(6H,m),0.98-0.90(6H,m),0.83(9H,s),0.69-0.63(4H,m),0.19-0.16(6H,m).
MS(APCI、ESI)m/z:1312(M+H)+
上記工程10にて得られた化合物(3-11)(0.1837g,0.140mmol)と酢酸(0.048mL,0.840mmol)のテトラヒドロフラン(5.00mL)溶液に1mol/Lのテトラブチルアンモニウムフロリド テトラヒドロフラン溶液(0.700mL,0.700mmol)を室温にて加え、室温にて3時間撹拌した。反応溶液を酢酸エチルで希釈し、有機層を飽和炭酸水素ナトリウム水溶液、飽和食塩水で洗浄した後、硫酸ナトリウムで乾燥した。減圧留去した後、得られた残渣をシリカゲルクロマトグラフィー[クロロホルム:メタノール=99.5:0.5(v/v)~95:5(v/v)]にて精製し、目的物(3-12)(0.178g、定量的)を得た。
1H-NMR(CDCl3)δ:8.86(1H,s),7.60-7.59(2H,m),7.39(1H,s),7.32-7.20(7H,m),6.90-6.88(2H,m),6.78(1H,s),6.68(1H,s),6.38(1H,s),5.90-5.87(3H,m),5.39-5.22(6H,m),4.72-4.02(11H,m),3.90(3H,s),3.88(3H,s),3.83(3H,s),3.70-3.63(6H,m),3.32-3.29(3H,m),2.73-2.69(1H,m),2.43-2.40(1H,m),2.12-2.06(1H,m),1.77-1.74(2H,m),1.39-1.25(6H,m),0.96-0.89(6H,m),0.73-0.66(4H,m).
MS(APCI、ESI)m/z:1198(M+H)+
上記工程11にて得られた化合物(3-12)(0.140mmol)のジクロロメタン(2mL)溶液に、室温にてピロリジン(0.0579mL, 0.700mmol)、テトラキス(トリフェニルホスフィン)パラジウム(0)(0.0162g, 0.0140mmol)を加え、室温にて15分撹拌した。減圧留去した後、得られた残渣をシリカゲルクロマトグラフィー[クロロホルム:メタノール=99.5:0.5(v/v)~92.5:7.5(v/v)]にて精製し、目的物(3-13)(0.143g, 99%)を得た。
1H-NMR(CDCl3)δ:9.12(1H,s),7.94-7.92(1H,m),7.57-7.53(4H,m),7.33-7.31(2H,m),7.20-7.18(3H,m),6.90-6.88(2H,m),6.36(1H,s),6.07(1H,s),5.91-5.88(1H,m),5.47-5.44(1H,m),5.21-5.13(1H,m),4.66-4.58(3H,m),4.32(1H,s),4.03-3.49(17H,m),3.38-3.29(4H,m),3.15-3.14(1H,m),2.77-2.73(1H,m),2.57(2H,s),2.43-2.40(1H,m),2.32-2.27(1H,m),1.81-1.39(8H,m),0.98-0.96(3H,m),0.85-0.83(3H,m),0.75-0.62(4H,m).
MS(APCI、ESI)m/z:1030(M+H)+
実施例1-2工程1にて得られた化合物(2-2)(0.0640g, 0.153mmol)、N‐エトキシカルボニル‐2-エトキシ‐1,2‐ジヒドロキノリン(0.0446g, 0.180mmol)の混合物にジクロロメタン(2mL)を室温にて加え、室温にて15分撹拌した。反応溶液に上記工程12にて得られた化合物(3-13)(0.143g, 0.139mmol)のジクロロメタン(2mL)溶液を加え、室温にて五時間撹拌した後、減圧留去した。得られた残渣をシリカゲルクロマトグラフィー[クロロホルム:メタノール=99.5:0.5(v/v)~92.5:7.5(v/v)]で精製し、目的物(3-14)(0.103g, 52%)を得た。
1H-NMR(DMSO-D6)δ:9.93(1H,s),8.21-8.16(2H,m),8.07-8.04(1H,m),7.83-7.64(2H,m),7.60-7.55(3H,m),7.51-7.28(10H,m),7.19-7.16(2H,m),7.10-7.04(1H,m),6.92-6.90(2H,m),6.76-6.70(1H,m),6.39(1H,s),5.77-5.75(1H,m),5.21-5.18(1H,m),5.03-4.99(1H,m),4.82-4.79(1H,m),4.37-4.35(1H,m),4.21-4.20(2H,m),4.02-3.24(26H,m),3.16-3.13(1H,m),2.79-2.59(2H,m),2.39-2.28(2H,m),2.05-1.97(2H,m),1.91-1.77(4H,m),1.57-1.54(3H,m),1.28-1.23(3H,m),0.85-0.80(6H,m),0.67-0.61(4H,m).
MS(APCI、ESI)m/z:1431(M+H)+

実施例2-1工程1にて得られた化合物(3-2)(5.06g,9.67mmol)と1,3-ジブロモプロパン(4.93mL,48.4mmol)を、実施例2-1工程2と同様に反応させ、目的物(4-1)(4.85g,78%)を得た。
MS(APCI、ESI)m/z:645[81Br,(M+H)+],643[79Br,(M+H)+].
上記工程1にて得られた化合物(4-1)(4.85g,7.54mmol)を、実施例2-1工程3と同様に反応させ、目的物(4-2)(4.05g,定量的)を得た。
MS(APCI、ESI)m/z:531[81Br,(M+H)+],529[79Br,(M+H)+].
上記工程2にて得られた化合物(4-2)(7.54mmol)を、実施例2-1工程4と同様に反応させ、目的物(4-3)(3.73g,93%)を得た。
1H-NMR(CDCl3)δ:7.34(1H,s),7.29(1H,s),5.56-5.53(1H,m),4.72-4.69(1H,m),4.67-4.61(1H,m),4.23-4.17(3H,m),3.97-3.88(4H,m),3.82-3.75(1H,m),3.74-3.56(4H,m),2.82-2.77(1H,m),2.43-2.38(2H,m),1.06-0.94(2H,m),0.08-0.00(9H,m).
上記工程3にて得られた化合物(4-3)(3.73g,7.08mmol)を、実施例2-1工程5と同様に反応させ、目的物(4-4)(3.27g,70%)を得た。
MS(APCI、ESI)m/z:661[81Br,(M+H)+],659[79Br,(M+H)+].
上記工程4にて得られた化合物(4-4)(3.27g,4.96mmol)を、実施例2-1工程6と同様に反応させ、目的物(4-5)(2.49g,81%)を得た。
MS(APCI、ESI)m/z:619[81Br,(M+H)+],617[79Br,(M+H)+].
上記工程5にて得られた化合物(4-5)(2.49g,4.04mmol)を、実施例2-1工程7と同様に反応させ、目的物(4-6)(1.59g,84%)を得た。
MS(APCI、ESI)m/z:473[81Br,(M+H)+],471[79Br,(M+H)+].
上記工程6にて得られた化合物(4-6)(1.59g,3.38mmol)を、実施例2-1工程8と同様に反応させ、目的物(4-7)(1.39g,87%)を得た。
MS(APCI、ESI)m/z:475[81Br,(M+H)+],473[79Br,(M+H)+].
上記工程7にて得られた化合物(4-7)(1.40g,2.95mmol)を、実施例2-1工程9と同様に反応させ、目的物(4-8)(0.885g,54%)を得た。
MS(APCI、ESI)m/z:559[81Br,(M+H)+],557[79Br,(M+H)+].
上記工程8にて得られた化合物(4-8)(0.0381g,0.0683mmol)と実施例1-1工程10で得られた化合物(1-11)(0.0552g、0.0683mmol)を、実施例2-1工程10と同様に反応させ、目的物(4-9)(0.0712g,81%)を得た。
MS(APCI、ESI)m/z:1284(M+H)+.
上記工程9にて得られた化合物(4-9)(0.0712g,0.0554mmol)を、実施例2-1工程11と同様に反応させ、目的物(4-10)(0.0671g,定量的)を得た。
MS(APCI、ESI)m/z:1170(M+H)+.
上記工程10にて得られた化合物(4-10)(0.0571mmol)を、実施例2-1工程12と同様に反応させ、目的物(4-11)(0.0574g,99%)を得た。
1H-NMR(CDCl3)δ:9.16(1H,s),7.93-7.91(1H,m),7.55-7.52(1H,m),7.50-7.47(3H,m),7.35-7.32(2H,m),7.21(1H,s),7.13-7.11(2H,m),6.90-6.87(2H,m),6.40(1H,s),6.08(1H,s),5.90-5.87(1H,m),5.37-5.34(1H,m),4.73-4.53(3H,m),4.23-4.08(5H,m),3.89(3H,s),3.82(3H,s),3.78-3.72(5H,m),3.57-3.51(3H,m),3.38-3.30(3H,m),2.76-2.71(1H,m),2.36-2.24(4H,m),1.78-1.42(6H,m),1.00-0.98(3H,m),0.87-0.84(3H,m),0.74-0.62(4H,m).
MS(APCI、ESI)m/z:1002(M+H)+.
上記工程11にて得られた化合物(4-11)(0.189g,0.189mmol)と実施例1-2工程1で得られた化合物(2-2)(0.087g、0.207mmol)を、実施例2-1工程13と同様に反応させ、目的物(4-12)(0.169g,64%)を得た。
MS(APCI、ESI)m/z:1402(M+H)+.

4,4’-[1,5-ペンタンジイルビス(オキシ)]ビス(5-メトキシ-2-ニトロ安息香酸)(5-1)(5.41g,10.9mmol,Journal of Medicinal Chemistry 2004,47,1161)のジクロロメタン(50mL)溶液に、0℃にて塩化オキサリル(5.63mL,65.7mmol)を加え、N,N-ジメチルホルムアミド(0.0844mL,1.09mmol)滴下した。反応溶液を室温まで昇温し、2時間撹拌した。減圧留去して得られた残渣をジクロロメタン(100mL)に溶解し、メチル(6S)-5-アザスピロ[2.4]ヘプタン-6-カルボキシレート塩酸塩(4.28g,24.1mmol,Tetrahedron Letters 2012. 53. 3847)とトリエチルアミン(6.07mL,43.8mmol)のジクロロメタン溶液(100mL)に窒素雰囲気下、-40℃で滴下した。反応溶液を0℃に昇温して2時間撹拌した。反応混合物に1規定塩酸(100mL)を加え、有機層を水、飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧留去し、目的物(5-2)(8.40g、定量的)を得た。
MS(APCI、ESI)m/z:769(M+H)+.
上記工程1にて得られた化合物(5-2)(8.40g,10.9mmol)のテトラヒドロフラン(100mL)溶液に、水素化ほう素リチウム(714mg,32.8mmol)を加え、0℃にて30分撹拌し、室温に昇温して1時間撹拌した。0℃にて1規定塩酸を加えた後、酢酸エチルで抽出し、飽和食塩水で洗浄した後、無水硫酸ナトリウムで乾燥させた。減圧下にて溶媒留去して、目的物(5-3)(7.70g,99%)を得た。
MS(APCI、ESI)m/z:713(M+H)+.
上記工程2にて得られた化合物(5-3)(7.70g、10.8mmol)をピリジン(20mL)及び無水酢酸(10mL,105.9mmol)に溶解して、室温にて撹拌した。減圧留去して、目的物(5-4)(8.38g,97%)を得た。
MS(APCI、ESI)m/z:797(M+H)+.
上記工程3にて得られた化合物(5-4)(8.28g,10.4mmol)のN,N-ジメチルホルムアミド(100mL)溶液に、5%パラジウム炭素(54%水分、1.00g)を加えた後、反応溶液を水素雰囲気下、室温にて6時間激しく撹拌した。セライトろ過した後、ろ液を減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー[クロロホルム:メタノール=100:0(v/v)~90:10(v/v)]にて精製し、目的物(5-5)(5.05g,66%)を得た。
MS(APCI、ESI)m/z:737(M+H)+.
上記工程4にて得られた化合物(5-5)(5.05g,6.85mmol)のジクロロメタン(100mL)溶液に、ピリジン(1.10mL,13.7mmol)を加え、窒素雰囲気下、-78℃にてクロロギ酸アリル(0.725mL,6.85mmol)を加え、2時間撹拌した。減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=70:30(v/v)~100:0(v/v)、クロロホルム:メタノール=100:0(v/v)~90:10(v/v)]にて精製し、目的物であるモノアリルオキシカルボニル体(5-6)(2.63g,47%)を得た。
MS(APCI、ESI)m/z:821(M+H)+.
上記工程5で得られたモノアリルオキシカルボニル体(5-6)(2.00g,2.44mmol)とN-[(プロプ-2-エン-1-イルオキシ)カルボニル]-L-バリル-N-[4-(ヒドロキシメチル)フェニル]-L-アラニンアミド(1.10g,2.92mmol,WO2011130598)を、実施例1-1工程6と同様に反応させ、目的物(5-7)(2.64g,89%)を得た。
MS(APCI、ESI)m/z:1224(M+H)+.
上記工程6にて得られた化合物(5-7)(2.64g,2.16mmol)のメタノール(10mL)溶液に、炭酸カリウム(1.49g,10.8mmol)を加え、室温で3時間撹拌した。反応混合物に飽和塩化アンモニウム水溶液(100mL)を加え、酢酸エチルで抽出した。有機層を無水硫酸ナトリウムで乾燥させた。減圧留去して、目的物(5-8)(2.21g,90%)を得た。
MS(APCI、ESI)m/z:1140(M+H)+.
上記工程7にて得られた化合物(5-8)(2.03g,1.78mmol)のジクロロメタン(50mL)溶液に、デスマーチンペルヨージナン(1.59g,3.74mmol)を加え、室温で一晩撹拌した。反応混合物に飽和炭酸水素ナトリウム水溶液(100mL)を加え、クロロホルムで抽出した。有機層を無水硫酸ナトリウムで乾燥させた。減圧留去して得られた残渣をシリカゲルカラムクロマトグラフィー[クロロホルム:メタノール=100:0(v/v)~90:10(v/v)]にて精製し、目的物(5-9)(2.05g,定量的)を得た。
MS(APCI、ESI)m/z:1136(M+H)+.
上記工程8にて得られた化合物(5-9)(2.05g,1.80mmol)を、実施例2-1工程12と同様に反応させ、目的物(5-10)(1.02g,60%)を得た。
MS(APCI、ESI)m/z:950(M+H)+.
上記工程9にて得られた化合物(5-10)(0.710g,0.747mmol)と実施例1-2工程1にて得られた化合物(2-2)(0.313g、0.747mmol)をジクロロメタン(1.5mL)とメタノール(0.1mL)の混合溶媒に溶解した。4-(4,6-ジメトキシ-1,3,5-トリアジン-2-イル)-4-メチルモルホリニウムクロリド(0.264g,0.897mmol)を加え、室温で1時間撹拌した。減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[クロロホルム:メタノール=100:0(v/v)~80:20(v/v)]にて精製し目的物(5-11)(0.671g,66%)を得た。
1H-NMR(DMSO-D6)δ:9.91(1H,s),8.32(1H,s),8.23-7.91(3H,m),7.81-7.19(14H,m),7.04(1H,m),6.80-6.62(3H,m),5.77-5.75(1H,m),5.20(1H,m),5.01(1H,m),4.79(1H,m),4.46-4.35(1H,m),4.04(4H,m),3.86-3.38(18H,m),3.22-3.15(2H,m),2.67-2.63(1H,m),2.46-2.23(3H,m),2.09-1.91(2H,m),1.80-1.78(5H,m),1.57(3H,m),1.27(3H,s),1.11-1.04(1H,m),0.87-0.79(6H,m),0.63-0.55(6H,m).
MS(APCI、ESI)m/z:1351(M+H)+.

4-(ベンジルオキシ)-5-メトキシ-2-ニトロ安息香酸(6-1)(6.07g,20.0mmol,Tetrahedron 1995,51,5617)、N,N-ジメチルホルムアミド(1.08mL,13.9mmol)のジクロロメタン(100mL)溶液に、氷冷下にて塩化オキサリル(3.43mL,40.0mmol)を5分間かけて滴下した。室温にて反応溶液を5時間撹拌した後、減圧留去し、得られた残渣をジクロロメタン(20mL)に溶解させ、減圧留去した。この操作を3回繰り返した後に、残渣をジクロロメタン(5mL)に懸濁させ、これに過剰のジエチルエ-テルとヘキサンを加え、ろ過し、減圧下に乾燥させることにより粗酸クロリドを得た。得られた酸クロリドをジクロロメタンに溶解させ、-40℃(ドライアイス-アセトニトリル浴)に冷却し、メチル(6S)-5-アザスピロ[2.4]ヘプタン-6-カルボキシレート塩酸塩(4.22g,22.0mmol,Tetrahedron Letters 2012.53.3847)、トリエチルアミン(3.36mL,24.2mmol)を徐々に加えた。反応混合物を一晩かけて室温にまで昇温した。反応混合物に1規定塩酸を加え、反応混合物をジクロロメタンで抽出した。有機層を水,飽和炭酸水素ナトリウム水溶液および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0~50:50]にて精製し、目的物(6-2)(6.55g,80%)を得た。
MS(APCI、ESI)m/z:441(M+H)+
上記工程1にて得られた化合物(6-2)(6.55g,16.0mmol)のエタノール(150mL),テトラヒドロフラン(150mL)溶液に窒素雰囲気下、ラネ-ニッケル(7.00g)を加えた。反応混合物にヒドラジン一水和物(7mL)を加え、50℃まで徐々に昇温した。50℃にて2時間撹拌した後にラネ-ニッケル(3.00g)、ヒドラジン一水和物(3mL)を加え、1時間撹拌した。反応混合物にTHF(100mL)を加えて、セライトろ過した。減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0~25:75]にて精製し、目的物(6-3)(4.42g,73%)を得た。
MS(APCI、ESI)m/z:379(M+H)+
上記工程2にて得られた化合物(6-3)(10.0g,26.4mmol)のテトラヒドロフラン(150mL)溶液に-40℃にて、2.6mol/Lのノルマルブチルリチウムノルマルヘキサン溶液(12.0mL,31.8mmol)をゆっくりと滴下した。反応溶液を-40℃にて15分間撹拌し後、2-(クロロメトキシ)エチルトリメチルシラン(5.57mL,31.7mmol)をゆっくりと滴下した。反応溶液を室温にて3時間撹拌した後、水を加え酢酸エチルで抽出した。有機層を水および飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥した。減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0~30:70]にて精製し、目的物(6-4)(11.8g,88%)を得た。
MS(APCI、ESI)m/z:509(M+H)+
上記工程3にて得られた化合物(6-4)(18.7g,36.8mmol)のテロラヒドロフラン(50mL)、エタノール(100mL)溶液に、窒素雰囲気下において5%のパラジウム炭素触媒(5.00g)を加えた。直ちに窒素風船を水素風船に付け替え、反応混合物を水素雰囲気下にて6時間撹拌した。反応混合物にクロロホルムを加えて希釈し、セライトろ過をした後、濾液を減圧留去して得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0~25:75]にて精製し、目的物(6-5)(15.1g,98%)を得た。
MS(APCI、ESI)m/z:419(M+H)+
上記工程4にて得られた化合物(6-5)(2.77g,6.62mmol)を、実施例2-1工程2と同様に反応させ、目的物(6-6)(3.31g,88%)を得た。
1H-NMR(CDCl3)δ:7.36(1H,s),7.25(1H,s),5.55(1H,m),4.65(1H,m),4.24-4.23(1H,m),4.11-4.03(2H,m),3.93(3H,s),3.85-3.78(1H,m),3.72-3.69(2H,m),3.46-3.39(3H,m),2.47-2.44(1H,m),2.25-2.22(1H,m),1.95-1.91(4H,m),1.67-1.59(1H,m),1.03-0.95(2H,m),0.90-0.85(1H,m),0.70-0.66(4H,m),0.05(9H,s).
上記工程5にて得られた化合物(6-6)(3.31g,5.83mmol)を、実施例2-1工程7と同様に反応させ、目的物(6-7)(1.11g,45%)を得た。
1H-NMR(CDCl3)δ:7.81(1H,m),7.53(1H,s),6.82(1H,s),4.13-4.06(2H,m),3.97(3H,s),3.88-3.83(1H,m),3.69(1H,m),3.52-3.39(3H,m),2.55-2.52(1H,m),2.06-1.89(5H,m),1.67-1.63(2H,m),0.76-0.72(4H,m).
上記工程6にて得られた化合物(6-7)(2.56g,6.08mmol)を、実施例2-1工程8と同様に反応させ、目的物(6-8)(1.15g,45%)を得た。
1H-NMR(CDCl3)δ:7.60(1H,s),6.07(1H,s),4.11-4.04(1H,m),3.99(2H,m),3.87-3.84(1H,m),3.85(3H,s),3.73(1H,m),3.58-3.53(2H,m),3.47-3.42(3H,m),2.03-1.78(6H,m),1.65-1.63(2H,m),0.77-0.56(4H,m).
上記工程7にて得られた化合物(6-8)(1.15g,2.72mmol)を、実施例2-1工程9と同様に反応させ、目的物(6-9)(1.14g,82%)を得た。
1H-NMR(CDCl3)δ:7.23(1H,s),6.69(1H,s),5.79(1H,s),5.13-5.10(2H,m),4.68-4.66(1H,m),4.48-4.45(2H,m),4.01(2H,m),3.92(3H,s),3.76(1H,m),3.54-3.37(3H,m),2.39(1H,m),1.95-1.90(4H,m),1.68-1.61(3H,m),1.44(1H,m),0.75-0.66(4H,m).
上記工程8にて得られた化合物(6-9)(0.374g,0.737mmol)と実施例1-1工程10にて得られた化合物(1-11)(0.452g、0.56mmol)を、実施例2-1工程10と同様に反応させ、目的物(6-10)(0.589g,65%)を得た。
MS(APCI、ESI)m/z:1234(M+H)+
上記工程9にて得られた化合物(6-10)(0.589g,0.477mmol)を、実施例2-1工程11と同様に反応させ、目的物(6-11)(0.382g,71%)を得た。
1H-NMR(CDCl3)δ:8.90(1H,s),7.55(2H,m),7.25-7.21(2H,m),6.74(2H,m),6.38(1H,s),5.90-5.87(5H,m),5.33-5.09(8H,m),4.66-4.60(8H,m),3.98-3.91(10H,m),3.77-3.30(12H,m),2.42-2.36(2H,m),1.77-1.39(6H,m),0.91-0.70(14H,m).
上記工程10にて得られた化合物(6-11)(0.382g,0.341mmol)を、実施例2-1工程12と同様に反応させ、目的物(6-12)(0.200g,62%)を得た。
MS(APCI、ESI)m/z:952(M+H)+
上記工程11にて得られた化合物(6-12)(0.0560g,0.0588mmol)と実施例1-2工程1にて得られた化合物(2-2)(0.022g、0.053mmol)を、実施例2-1工程13と同様に反応させ、目的物(6-13)(0.0500g,63%)を得た。
MS(APCI、ESI)m/z:1354(M+H)+
実施例3:[N3-PEG(3)]-MSG1-Ox

市販品であるmonosialo-Asn free (1S2G/1G2S-10NC-Asn、(株)糖鎖工学研究所製)(「(MSG-)Asn」と呼ぶ)(500mg)を以下の条件で逆相HPLC分離精製し、1st main peakとして溶出される(MSG1-)Asn(保持時間 15~19 min 付近)と2nd main peakとして溶出される(MSG2-)Asn(保持時間 21~26 min 付近)に分離した。0.1%ぎ酸水溶液を溶離液として使用し、装置にはELS-PDAトリガー分取システム(日本分光株式会社製)を使用し、カラムにはInertsil ODS-3(10um、 30Φx250mm、GLサイエンス社製)を使用し、流速を30mL/minとした。溶出中にUV検出(210nm)された最初のピ-クを分取し、凍結乾燥して、目的物(238mg)を得た。
上記工程1で得られた化合物(229mg)を200mMりん酸緩衝溶液(pH6.25)(1145μL)に溶解させ、EndoM(東京化成工業(株)製、1U/mL))水溶液(100μL)を加え、35℃で6日間インキュベートした。反応終了後、反応液をVIVASPIN 15R (Hydrosart膜、30K,6,000xG)を用いて限外ろ過し、得られた通過液を逆相HPLC分離精製した。0.1%トリフルオロ酢酸水溶液を溶離液として使用し、装置にはELS-PDA トリガー分取システム(日本分光株式会社製)、カラムにはInertsil ODS-3(GLサイエンス社製)を使用した。溶出中にUV検出(210nm)された目的物のピークを分取し、凍結乾燥し、目的物(117mg)を得た。
5mlサンプリングチューブ((株)イナ・オプティカ)に、11-アジド-3,6,9-トリオキサウンデカン-1-アミン(0.108mL,0.541mmol)と上記工程2で得られたMSG1(117mg,0.068mmol)の水溶液(1.2mL)を加え、1時間撹拌した後に、凍結乾燥した。凍結乾燥後の5mlサンプリングチューブに、O-(7-アザベンゾトリアゾ-ル-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスファート(103mg,0.27mmol)のN,N-ジメチルホルムアミド溶液(1.2mL)とジイソプロピルエチルアミン(0.046mL,0.27mmol)を加え、37℃で3時間撹拌した。反応終了後、あらかじめジエチルエ-テル(20ml)を加えた遠沈管(50ml)に反応液を移した。小型遠心機(日立工機、CF16RX)を利用して、固形物を沈殿させ、上澄みを取り除いた。さらにジエチルエ-テル(10ml)を加えて、遠心分離操作後、デカンテーションを行った。続いて、アセトニトリル(10mL)を加え、遠心分離後、デカンテーションする操作を二回繰り返した後、減圧下乾燥し、粗生成物を得た。得られた固形物を上記工程2と同様の条件にて逆相HPLC精製を行い、目的物(94.2mg)を得た。
5mLサンプリングチューブ((株)イナ・オプティカ製)に、上記工程3で合成した化合物(100mg)、2-クロロ-1,3-ジメチル-1H-ベンズイミダゾ-ル-3-イウム-クロライド(伏見製薬所製、56mg,0.257mmol)の水溶液(520μl)を加えた。氷冷後の反応液にりん酸三カリウム(165mg,0.78mmol)の水溶液(520μl)を加え、氷冷下で3時間撹拌した。得られた反応液を、アミコンウルトラ(ウルトラセル30K、Merck Millipore製)を用いて限外ろ過し、固形物を除去した。その通過液を、ゲルろ過クロマトグラフィーにて精製した。装置にはPurif-Rp2(昭光サイエンティフィック製)を使用し、カラムにはHiPrep 26/10 Desalting(GEヘルスケア製)を使用し、移動相に0.03% NH3水溶液を使用し、流速を10mL/min、分画容量を10mLとした。溶出中にUV検出(220nm)された目的物を含むフラクションを一つにまとめ、1N水酸化ナトリウム水溶液(104μl,0.104mmol)を加えて凍結乾燥し、目的物(84mg)を得た。

市販品である1S2G/1G2S-10NC-Asn-Fmoc((株)糖鎖工学研究所製)(「Fmoc-(MSG-)Asn」と呼ぶ)(1000mg)をエタノール/水(1/1)(10mL)に溶解させ、1規定の水酸化ナトリウム水溶液(1.75mL,4当量)を加え、室温で3時間撹拌した。反応終了後、反応液をアミコンウルトラ(30K、ミリポア社製)を用いて限外ろ過し、固形物を除去し、得られた通過液に1N塩酸(832μl,1.9当量)加えた。高速濃縮装置V-10(Biotage社製)を用いて、溶媒を除去した。アセトニトリルを加えて、高速濃縮装置V-10(Biotage社製)を用いて、溶媒を除去した後に、逆相HPLC分離精製 した。0.1%トリフルオロ酢酸水溶液と0.1%トリフルオロ酢酸アセトニトリル溶液を溶離液とし、装置にはPurif-Rp2(昭光サイエンティフィック製)を使用し、カラムにはInertsil ODS-3(GLサイエンス製)を使用した。溶出中にUV検出(220nm)された目的物を含むフラクションを一つにまとめ、凍結乾燥した。再び、純水に溶解させると、pH試験紙で酸性であることが確認されたので、18%アンモニア水(150μl)を加え、pH試験紙で塩基性になったことを確認し、再び凍結乾燥した。得られた目的物(840mg)をそのまま次の反応に用いた。
工程1にて得られた化合物(840mg)を200mMりん酸緩衝溶液(pH6.25)(6000μL)に溶解させ、EndoM(東京化成工業(株)製、1U/mL))水溶液(200μL)を加え、28℃で26時間インキュベートした。反応が完了していないため、EndoM(東京化成工業(株)製、1U/mL))水溶液(50μL)を加え、28℃で2時間インキュベートした後に、反応が完了するまで室温で放置した。反応終了後、反応液をアミコンウルトラ(30K、ミリポア社製)を用いて限外ろ過した。得られた通過液にトリフルオロ酢酸(80μl)加え、逆相HPLC分離精製した。0.1%トリフルオロ酢酸水溶液と0.1%トリフルオロ酢酸アセトニトリル溶液を溶離液とし、装置にはPurif-Rp2(昭光サイエンティフィック製)を使用し、カラムにはInertsil ODS-3 (GLサイエンス製)を使用した。溶出中にUV検出(220nm)された目的物を含むフラクションを一つにまとめ、凍結乾燥した。残留トリフルオロ酢酸を除く目的で、再度純水に溶解させ、目的化合物(618mg)を無色固体として得た。
ESI-MS: Calcd for C66H110N4O49 : [M+H]+ 1743.62, Found 1743.63
上記工程2にて得られた化合物(120mg)を用いて、実施例3工程3と同様の手法に従って、目的物(88.6mg)を得た。
ESI-MS: Calcd for C73H124N8O51 :[M+2H]2+ 965.37, Found 965.37
上記工程3にて得られた合成した化合物(100mg)を用いて、実施例3工程4と同様の手法に従って、目的物(88mg)を得た。

5mlサンプリングチューブ((株)イナ・オプティカ)に、11-アジド-3,6,9-トリオキサウンデカン-1-アミン(0.096mL,0.485mmol)、ジシアロオクタサッカリド(50 mg,0.24mmol)の水溶液(0.5mL)を加え、1時間撹拌した後に、凍結乾燥した。凍結乾燥後の5mlサンプリングチューブに、O-(7-アザベンゾトリアゾール-1-イル)-N,N,N’,N’-テトラメチルウロニウムヘキサフルオロホスファート(92mg,0.24mmol)のN,N-ジメチルホルムアミド溶液(0.6mL)とジイソプロピルエチルアミン(0.042mL,0.24mmol)を加え、37℃で4時間撹拌した。反応終了後、あらかじめジエチルエ-テル(20ml)を加えた遠沈管(50ml)に反応液を移した。小型遠心機(日立工機、CF16RX)を利用して、固形物を沈殿させ、上澄みを取り除いた。ジエチルエ-テル(20ml)を加えて、デカンテーションした。続いて、アセトニトリル(20mL)を加えて、デカンテーションした後に、減圧下乾燥し、粗生成物を得た。得られた固形物を適量の0.2%トリフルオロ酢酸水溶液に溶解させ、逆相HPLC分離精製した。0.1%トリフルオロ酢酸水溶液と0.1%トリフルオロ酢酸アセトニトリル溶液を溶離液とし、装置にはPurif-Rp2(昭光サイエンティフィック製)を使用し、カラムにはInertsil ODS-3(GLサイエンス製)を使用した。溶出中にUV検出(220nm)された目的物を含むフラクションを一つにまとめ、凍結乾燥して、目的物(42mg)を得た。
5mLサンプリングチューブ((株)イナ・オプティカ製)に、上記工程1で合成した化合物(40mg)、2-クロロ-1,3-ジメチル-1H-ベンズイミダゾール-3-イウム-クロライド(伏見製薬所製、17.9mg,0.083mmol)の水溶液(200μl)を加えた。氷冷後の反応液にりん酸三カリウム(52.6mg,0.25mmol)の水溶液(200μl)を加え、氷冷下で2時間撹拌した。得られた反応液を、アミコンウルトラ(ウルトラセル30K、 Merck Millipore製)を用いて限外ろ過し、固形物を除去した。その通過液を、ゲルろ過クロマトグラフィーにて精製した。装置にはPurif-Rp2(昭光サイエンティフィック製)を使用し、カラムにはHiPrep 26/10 Desalting(GEヘルスケア製)を使用し、移動相に0.03%-NH3水溶液を使用し、流速を10mL/min、分画容量を10mLとした。溶出中にUV検出(220nm)された目的物を含むフラクションを一つにまとめ、1N水酸化ナトリウム水溶液(33μl,0.033mmol)を加えて凍結乾燥し、目的物(34mg)を得た。
6-1.マウスの免疫とハイブリドーマの取得
1-1)マウス免疫に用いる細胞の準備
2×106個もしくは5×106個のNOR-P1細胞(ヒト膵がん細胞株、RIKEN RCB-2139)をRPMI-1640(Roswell Park Memorial Institute-1640)10%FBS(ウシ胎児血清)(+)培地(10mlもしくは20ml)で5日間培養後に回収し、PBS(リン酸緩衝生理食塩水)で2回洗浄し、PBS(300μl)中に再懸濁した。
BALB/cマウス(12週齢)に対し、1~5回目の免疫は約1週間間隔でNOR-P1細胞(2×106個)を腹腔内へ免疫した。5回目の免疫より約2週間後にNOR-P1細胞(5×106個)を腹腔内へ免疫した。6回目の免疫より約3週間後にNOR-P1細胞(2×106個)を腹腔内へ免疫した。8~10回目は約2週間間隔で2×106個のNOR-P1細胞を腹腔内へ免疫した。10回目の約3週間後(11回目)及びその3日後(12回目、最終免疫)では、5×106個のNOR-P1細胞を腹腔内へ免疫した。脾臓細胞は最終免疫から3日後に摘出した。
免疫済みマウスの脾臓を摘出し、すりつぶしてRPMI1640 10%FBS(+)培地に懸濁した。細胞懸濁液をセルストレイナー(70μm、BD Falcon社)に通した後、1500rpmで5分間、室温にて遠心して上清を廃棄した。Tris-NH4Cl溶液(20mM Tris-HCl pH7.2、77.6mM NH4Cl;20mL)を加え、室温で5分間処理した。PBS(20mL)を加え、1500rpmで5分間、室温にて遠心した。上清を廃棄した後に、RPMI1640 FBS(+)培地(10ml)を加えた。
P3U1細胞(マウスミエローマ細胞株)をRPMI1640 FBS(+)培地で5日間培養した後に回収し、RPMI1640 FBS(+)培地(20ml)に再懸濁した。
脾臓細胞とミエローマ細胞を5:1になるように混合し、1500rpmで5分間、室温にて遠心した。RPMI1640 FBS(-)培地(10ml)で2回洗浄した後に、遠心分離(1500rpm、5分間)した。得られた沈殿画分の細胞群をよくほぐした後、攪拌しながら、ポリエチレングリコール-1500(PEG-1500;1ml)を約1分かけて徐々に加えた。3分30秒間攪拌した後に、30秒間室温で静置した。その後、該細胞液に1分かけてRPMI培地 10%Low IgG FBS(+)(10ml)を加えた。該細胞懸濁液を遠心分離(1500rpm、5分間)し、得られた沈殿画分の細胞をゆるやかにほぐした後、HAT培地(10%Low IgG FBS、HAT Media Supplement、5%BriCloneを含むRPMI1640培地;200ml)中にゆるやかに懸濁した。該懸濁液を96ウェル培養用プレートに200μl/ウェルずつ分注し、37℃、5% CO2のインキュベーター中で、6日間培養した。
抗体の内在化及びイムノトキシン活性の測定を目的にリコンビナント複合蛋白質であるDT3Cを作製した。このDT3Cは、ジフテリア毒素(DT)の触媒領域と連鎖球菌プロテインGの抗体結合領域を遺伝子工学的に融合させた蛋白質である。DT3Cは抗体のFc部分に特異的に結合し、細胞内へ取り込まれると蛋白質合成阻害により細胞死を誘導する。この系を用いることで抗体の内在化とイムノトキシンによる殺細胞効果を同時に観察することができる(Yamaguchi, M. et al., Biochemical and Biophysical Research Communications 454 (2014) 600-603)。
96ウェルプレートに4μg/mlのDT3C(25μl)を加え、さらに工程1-5で取得されたハイブリドーマの培養上清(25μl)を加え、室温で30分インキュベーションした。2×105個/ml(RPMI培地 10%Low IgG FBS(+))のNOR-P1細胞(50μl)を播種し、37℃ CO2インキュベーターで3日間培養した。培養後の顕微鏡観察により、陰性対照抗体を用いた際の付着細胞数の約25%以下の付着細胞数を示したウェルを陽性と判定した。選抜したクローンは1~2回サブクローニングを行い、モノクローナルなハイブリドーマ細胞株8株を樹立した。
実施例6-1において調製されたハイブリドーマの産生する抗体のうち2つのクローン218B1及び218C7について抗原の同定を行った。
2-1)ビオチンラベルした細胞表面蛋白質の218B1抗体及び218C7抗体を用いた免疫沈降
2×106個のNTERA-2細胞(ヒト精巣がん細胞株、ATCC CRL-1973)の培養上清を除去し、PBSで2回洗浄した。EZ-Link Sulfo-NHS-Biotin(Thermo Fisher SCIENTIFIC社)を0.1mg/mlの濃度でPBSに懸濁した。PBSを除去した後に、Biotin/PBS溶液を加え、30分間振盪器上でインキュベーションした後に100mMグリシン/PBS溶液(25ml)で2回洗浄し、その後、PBS(10ml)で1回洗浄した。洗浄後の細胞を200μlの溶解バッファー(150mM NaCl、50mM Tris-HCl pH7.4、1%DDM、Protease inhibitor、Complete EDTA free(Roche社)1粒/50ml)中に再懸濁し、4℃で30分間処理した。遠心分離(13000rpm、20分間、4℃)して細胞溶解液を調製した。Protein G Sepharose(Protein G Sepharose 4 Fast Flow(GE Healthcare社))のバッファーを溶解バッファーに置換して得られたProtein G Sepharose/溶解バッファー(50%スラリー;30μl)を細胞溶解液に加え、4℃で30分間ローテートした後に4℃で1分間遠心し、上清を回収した。この上清に218B1抗体あるいは218C7抗体(約3μg)を加え、4℃で30分間ローテートした後にProtein G Sepharose/溶解バッファー(50%スラリー;60μl)を加え、4℃で1時間ローテートした。溶解バッファー(1ml)でProtein G Sepharoseを6回洗浄した後に1×SDSサンプルバッファー(BioRad社)に再懸濁した。100℃で5分間懸濁液を処理した後、溶液を回収し、SDS-PAGE(ポリアクリルアミドゲル電気泳動)のためのサンプルとした。
2-1)で調製したSDS-PAGEサンプルをSuperSep Ace 5ー20%(Wako社)を用いて、50mVで30分間スタッキングした後に、200mVで1時間泳動した後、ゲルよりメンブレンへ12mVで47分間ブロッティングした。メンブレンをPBS-T(PBS(-)-0.02% Tween 20)で洗浄した後、1時間ブロッキングを行った。メンブレンをPBS-Tで5分間3回洗浄した後にStreptavidin-horseradish peroxidase conjugate(GE Healthcare社;PBS-Tで2000倍に希釈して使用)と1時間反応させた。メンブレンをPBS-Tで5分間2回洗浄した後に、目的のバンドをenhanced chemiluminecscence(ECL)法を用いて検出した。218B1抗体及び218C7抗体を用いたいずれの場合においても、DTTの添加の有無によらず分子量18kDaのバンドが検出された。
2×107個のNTERA-2細胞を回収し、PBSで2回洗浄した。セルスクレーパーを用いて細胞を回収し、1500rpm、5分間遠心した。上清を除去した後に2mlの溶解バッファー中に再懸濁し、4℃で30分間処理した。遠心分離(13000rpm、20分間、4℃)して細胞溶解液を調製した。Protein G Sepharose/溶解バッファー(50%スラリー;180μl)を細胞溶解液に加え、4℃で30分間ローテートした後に4℃で1分間遠心し、上清を回収した。この上清に218B1抗体(約9μg)を加え、4℃で30分間ローテートした後にProtein G Sepharose/溶解バッファー(50%スラリー;180μl)を加え、4℃で1時間ローテートした。溶解バッファー(1ml)でProtein G Sepharoseを6回洗浄した後に1×SDSサンプルバッファーに再懸濁した。100℃で5分間懸濁液を処理した後、溶液を回収し、SDS-PAGEのためのサンプルとした。2-2)と同様の方法でSDS-PAGEを行い、泳動ゲルをCBB染色した。泳動ゲルより18kDaに相当する部分を切り出し、質量分析に供した。質量分析の結果、該ゲル片はClaudin-6を含むことが判明した。
質量分析から218B1抗体及び218C7抗体の抗原がClaudin-6であることが予測できたため、cDNAの遺伝子導入による強制発現解析を行った。FACS解析の結果、ヒトClaudin-6発現CHO-K1細胞では218B1抗体及び218C7抗体は強陽性反応を示したため、218B1抗体及び218C7抗体の抗原はClaudin-6であることが示された。
マウス抗CLDN6抗体B1産生ハイブリドーマ(218B1)及びマウス抗CLDN6抗体C7産生ハイブリドーマ(218C7)を、10%のFetal Bovine Serum, Ultra-Low IgG(Thermo Fisher Scientific)を含むHybridoma-SFM(Thermo Fisher Scientific)で培養した。培養上清を遠心により回収し0.45μmのフィルター(Corning社製)でろ過した。rProtein Aアフィニティークロマトグラフィー(4~6℃下)1段階工程で、培養上清から抗体を精製した。rProtein Aアフィニティークロマトグラフィー精製後のバッファー置換工程は4~6℃下で実施した。最初に培養上清をPBSで平衡化したMabSelectSuRe(GE Healthcare Bioscience社製)が充填されたカラムにアプライした。培養液がカラムに全て入ったのち、カラム容量2倍以上のPBSでカラムを洗浄した。次に2M アルギニン塩酸塩溶液(pH4.0)で溶出し、抗体の含まれる画分を集めた。その画分を透析(Thermo Scientific社、Slide-A-Lyzer Dialysis Cassette)によりPBS(-)への液置換を行った。最後にCentrifugal UF Filter Device VIVASPIN20(分画分子量UF10K,Sartorius社,4℃下)にて濃縮し、IgG濃度を1 mg/mL以上に調製した。Minisart-Plus filter(Sartorius社)でろ過し、精製サンプルとした。
7-1:フローサイトメトリーによるマウス抗CLDN6抗体の結合能評価
実施例6で作製したマウス抗CLDN6抗体のヒトCLDN6及びファミリー分子CLDN3、CLDN4、CLDN9結合性をフローサイトメトリー法により評価した。Origene社より購入したhuman CLDN3/pCMV6-Entry, human CLDN4/pCMV6-Entry, human CLDN6/pCMV-Entry, human CLDN9/pCMV6-Entry, またはpCMV6-Entryを、293T細胞 (Thermo Fisher Scientific HCL4517)にLipofectamine 2000 (Thermo Fisher Scientific)を用いて一過性に導入し、37℃、5%CO2の条件下で一晩培養した後、細胞懸濁液を調製した。遺伝子導入した293T細胞懸濁液を遠心し、上清を除去した後、マウス抗CLDN6抗体(クローン番号はB1、C7)、又はマウスIgG1コントロール抗体(R&D Systems)を終濃度30μg/mL, 10μg/mL, 3.3μg/mL, 1.1μg/mLで加えて懸濁し、4℃で1時間静置した。5%のウシ胎児血清(Hyclone)を含むDulbecco’s phosphate buffered saline (Sigma-Aldrich)(以下、5%FBS含有PBS)で2回洗浄した後、5%FBS含有PBSで500倍希釈したFLUORESCEIN-CONJUGATED GOAT IGG FRACTION TO MOUSE IGG (WHOLE MOLECULE) (MP BIOMEDICALS)を加えて懸濁し、4℃で1時間静置した。5%FBS含有PBSで2回洗浄した後、フローサイトメーター(FC500;Beckman Coulter)で検出を行った。データ解析はFlowJo(TreeStar)で行った。なお、各遺伝子導入の確認は、細胞を0.25%Tween20含有PBSで透過処理した後にマウス抗FLAG抗体(Sigma-Aldrich)を用いて行った。結果を図11に示す。図11のグラフにおいて、縦軸は抗体結合量を表すFITCの蛍光強度、横軸は抗体濃度を示す。作製したマウス抗CLDN6抗体はヒトCLDN6とヒトCLDN9に同程度に結合し、ヒトCLDN3、ヒトCLDN4には結合しなかった。マウスコントロールIgG1はいずれの細胞にも結合しなかった。
マウス抗CLDN6抗体B1およびC7の内在化活性は、蛋白質合成を阻害する毒素(サポリン)を結合させた抗マウスIgG試薬Mab-ZAP(ADVANCED TARGETING SYSTEMS)を用いて評価した。この評価では、マウス抗CLDN6抗体の内在化活性に依存してMab-ZAPが細胞内に取り込まれ、蛋白質合成を阻害するサポリンが細胞内に放出されることで、細胞増殖が抑制される。
ヒトCLDN6陽性細胞のヒト絨毛がん株であるJEG-3(ATCC HTB-36)、ヒトCLDN6陽性細胞のヒト卵巣がん株であるNIH:OVCAR-3 (ATCC HTB-161)、又はヒトCLDN6陰性細胞のヒト膵臓がん株であるBxPC-3(ATCC CRL-1687)を2x103 cells/wellで96穴細胞培養用マイクロプレートに播種して37℃、5% CO2の条件下で一晩培養した。翌日、マウス抗CLDN6抗体、又はマウスIgG1抗体(R&D Systems)を終濃度1nMとなるように、Mab-ZAP(終濃度:0.5nM)、又は毒素を結合していないAffiniPure Goat Anti-Mouse IgG, Fcγ Fragment Specific (Jackson ImmunoResearch) (終濃度:0.5 nM)と混合した混合溶液を添加して、37℃、5% CO2の条件下で5日間培養した。生存細胞数は、CellTiter-Glo Luminescent Cell Viability Assay (Promega)を用いたATP活性の定量で測定した。抗CLDN6抗体添加による細胞増殖抑制作用は、混合溶液非添加ウェルの値を100%とする相対生存率で求めた。図12に結果を示す。マウス抗CLDN6抗体(B1、C7)はヒトCLDN6陽性細胞株JEG-3とNIH:OVCAR-3に対して細胞増殖抑制効果を認めた。一方、ヒトCLDN6陰性細胞株BxPC-3に対しては細胞増殖抑制効果を認めなかった。また、マウスIgG1抗体はいずれの細胞株に対しても細胞増殖抑制効果を認めなかった。この結果、作製した抗CLDN6抗体(B1、C7)は内在化活性を有し、抗体―薬物コンジュゲート用の抗体として適すると考えられる。
8-1:B1抗体の可変領域をコードするcDNAのヌクレオチド配列の決定
8-1-1:B1抗体産生ハイブリドーマのtotal RNAの調製
B1抗体の可変領域をコードするcDNAを増幅するため、B1抗体産生ハイブリドーマよりTRIzol Reagent(Ambion社)を用いてtotal RNAを調製した。
8-1-2:5’-RACE PCRによるB1抗体の軽鎖可変領域をコードするcDNAの増幅と配列の決定
軽鎖可変領域をコードするcDNAの増幅は、実施例8-1-1で調製したtotal RNAの約1μgとSMARTer RACE 5’/3’ Kit(Clontech社)を用いて実施した。B1抗体の軽鎖遺伝子の可変領域をコードするcDNAをPCRで増幅するためのプライマーとして、UPM (Universal Primer A Mix:SMARTer RACE 5’/3’ Kitに付属)、及び公知のマウス軽鎖の定常領域の配列から設計したプライマーを用いた。
5’-RACE PCRで増幅した軽鎖の可変領域をコードするcDNAをプラスミドにクローニングし、次に軽鎖の可変領域をコードするcDNAのヌクレオチド配列のシークエンス解析を実施した。
決定されたB1抗体の軽鎖の可変領域をコードするcDNAのヌクレオチド配列を配列番号18に示し、アミノ酸配列を配列番号19に示す。
8-1-3:5’-RACE PCRによるB1抗体の重鎖可変領域をコードするcDNAの増幅と配列の決定
重鎖可変領域をコードするcDNAの増幅は、実施例8-1-1で調製したtotal RNAの約1μgとSMARTer RACE 5’/3’ Kit(Clontech社)を用いて実施した。LB1抗体の重鎖遺伝子の可変領域をコードするcDNAをPCRで増幅するためのプライマーとして、UPM (Universal Primer A Mix:SMARTer RACE 5’/3’ Kitに付属)、及び公知のマウス重鎖の定常領域の配列から設計したプライマーを用いた。
5’-RACE PCRで増幅した重鎖の可変領域をコードするcDNAをプラスミドにクローニングし、次に重鎖の可変領域をコードするcDNAのヌクレオチド配列のシークエンス解析を実施した。決定されたB1抗体の重鎖の可変領域をコードするcDNAのヌクレオチド配列を配列番号20に示し、アミノ酸配列を配列番号21に示す。
実施例8-1と同様の方法で実施した。決定されたC7抗体の軽鎖の可変領域をコードするcDNAのヌクレオチド配列を配列番号22に示し、アミノ酸配列を配列番号23に示す。重鎖の可変領域をコードするcDNAのヌクレオチド配列を配列番号24に示し、アミノ酸配列を配列番号25に示す。
9-1:キメラ化抗CLDN6抗体chB1の発現ベクターの構築
9-1-1:キメラ化及びヒト化軽鎖発現ベクターpCMA-LKの構築
プラスミドpcDNA3.3-TOPO/LacZ(Invitrogen社)を制限酵素XbaI及びPmeIで消化して得られる約5.4kbのフラグメントと、配列番号26に示すヒト軽鎖シグナル配列及びヒトκ鎖定常領域をコードするDNA配列を含むDNA断片をIn-Fusion HD PCRクローニングキット(Clontech社)を用いて結合して、pcDNA3.3/LKを作製した。pcDNA3.3/LKからネオマイシン発現ユニットを除去することによりpCMA-LKを構築した。
9-1-2:キメラ化及びヒト化IgG1LALAタイプ重鎖発現ベクターpCMA-G1LALAの構築
pCMA-LKをXbaI及びPmeIで消化して軽鎖シグナル配列及びヒトκ鎖定常領域を取り除いたDNA断片と、配列番号27で示されるヒト重鎖シグナル配列及びヒトIgG1LALA定常領域をコードするDNA配列を含むDNA断片をIn-Fusion HD PCRクローニングキット(Clontech社)を用いて結合して、pCMA-G1LALAを構築した。
9-1-3:キメラ化chB1重鎖発現ベクターの構築
配列番号33に示すchB1重鎖のヌクレオチド配列のヌクレオチド番号36乃至440に示されるDNA断片を合成した(GENEART社)。In-Fusion HD PCRクローニングキット(Clontech社)を用いて、pCMA-G1LALAを制限酵素BlpIで切断した箇所に合成したDNA断片を挿入することによりchB1重鎖発現ベクターを構築した。chB1重鎖のアミノ酸配列を配列番号32に示す。
9-1-4:キメラ化chB1軽鎖発現ベクターの構築
配列番号29に示すchB1軽鎖をコードするDNA配列を含むDNA断片を合成した(GENEART社)。In-Fusion HD PCRクローニングキット(Clontech社)を用いて、合成したDNA断片とpCMA-LKをXbaI及びPmeIで消化して軽鎖シグナル配列及びヒトκ鎖定常領域を取り除いたDNA断片を結合することにより、chB1軽鎖発現ベクターを構築した。chB1軽鎖のアミノ酸配列を配列番号28に示す。
9-2-1:キメラ化抗体chB1の生産
FreeStyle 293F細胞(Invitrogen社)はマニュアルに従い、継代、培養を行なった。対数増殖期の1.2×10 9 個のFreeStyle 293F細胞(Invitrogen社)を3L Fernbach Erlenmeyer Flask(CORNING社)に播種し、FreeStyle293 expression medium(Invitrogen社)で希釈して2.0×10 6 細胞/mLに調製した。40 mLのOpti-Pro SFM培地(Invitrogen社)に0.24mgの重鎖発現ベクターと0.36 mgの軽鎖発現ベクターと1.8 mgのPolyethyleneimine(Polyscience #24765)を加えて穏やかに攪拌し、さらに5分間放置した後にFreeStyle 293F細胞に添加した。37℃、8%CO2インキュベーターで4時間、90rpmで振とう培養後に600 mLのEX-CELL VPRO培地(SAFC Biosciences社)、18 mLのGlutaMAX I(GIBCO社)、及び30 mLのYeastolate Ultrafiltrate(GIBCO社)を添加し、37℃、8%CO2インキュベーターで7日間、90rpmで振とう培養して得られた培養上清をDisposable Capsule Filter (Advantec #CCS-045-E1H)でろ過した。取得したキメラ化抗CLDN6抗体を「chB1」と命名した。
9-2-2:キメラ化抗体chB1の精製
実施例9-2-1で得られた培養上清から抗体をrProtein Aアフィニティークロマトグラフィーの1段階工程で精製した。培養上清をPBSで平衡化したMabSelectSuReが充填されたカラム(GE Healthcare Bioscience社製)にアプライしたのちに、カラム容量の2倍以上のPBSでカラムを洗浄した。次に2Mアルギニン塩酸塩溶液(pH4.0)で溶出し、抗体の含まれる画分を集めた。その画分を透析(Thermo Scientific社、Slide-A-Lyzer Dialysis Cassette)によりPBS(-)へのバッファー置換を行った。Centrifugal UF Filter Device VIVASPIN20(分画分子量UF10K,Sartorius社)で抗体を濃縮し、IgG濃度を1mg/mL以上に調製した。最後にMinisart-Plus filter(Sartorius社)でろ過し、精製サンプルとした。
10-1:抗CLDN6抗体のヒト化体デザイン
10-1-1:キメラ化抗体chB1の可変領域の分子モデリング
chB1の可変領域の分子モデリングは、ホモロジーモデリングとして公知の方法(Methods in Enzymology,203,121-153,(1991))を利用した。chB1の重鎖と軽鎖の可変領域に対して高い配列同一性を有するProtein Data Bank(Nuc.Acid Res.35,D301-D303(2007))に登録されている構造(PDB ID:1XIW)を鋳型に、市販の蛋白質立体構造解析プログラムBioLuminate(Schrodinger社製)を用いて行った。
10-1-2:ヒト化アミノ酸配列の設計
chB1は、CDRグラフティング(Proc.Natl.Acad.Sci.USA 86,10029-10033(1989))によりヒト化した。Kabat et al.(Sequences of Proteins of Immunological Interest, 5th Ed.Public Health Service National Institutes of Health,Bethesda,MD.(1991))において既定されるヒトのgamma鎖サブグループ1およびkappa鎖サブグループ1のコンセンサス配列が、chB1のフレームワーク領域に対して高い同一性を有することから、それぞれ、重鎖と軽鎖のアクセプターとして選択された。アクセプター上に移入すべきドナー残基は、Queen et al.(Proc.Natl.Acad.Sci.USA 86,10029-10033(1989))によって与えられる基準などを参考に三次元モデルを分析することで選択された。なお、CDRL3が疎水的なアミノ酸に富んでいたため、CDRL3に変異を導入したヒト化軽鎖も設計した。
設計された3種の重鎖をhH1、hH2及びhH3と命名した。hH1の重鎖全長アミノ酸配列を、配列番号52に記載する。配列番号52のアミノ酸配列をコードするヌクレオチド配列を、配列番号53に記載する。hH2の重鎖全長アミノ酸配列を、配列番号56に記載する。配列番号56のアミノ酸配列をコードするヌクレオチド配列を、配列番号57に記載する。hH3の重鎖全長アミノ酸配列を、配列番号60に記載する。配列番号60のアミノ酸配列をコードするヌクレオチド配列を、配列番号61に記載する。
設計された4種の軽鎖をhL1、hL2、hL3及びhL4と命名した。hL1の軽鎖全長アミノ酸配列を、配列番号36に記載する。配列番号36のアミノ酸配列をコードするヌクレオチド配列を、配列番号37に記載する。hL2の軽鎖全長アミノ酸配列を、配列番号40に記載する。配列番号40のアミノ酸配列をコードするヌクレオチド配列を、配列番号41に記載する。hL3の軽鎖全長アミノ酸配列を、配列番号44に記載する。配列番号44のアミノ酸配列をコードするヌクレオチド配列は、配列番号45に記載する。hL4の軽鎖全長アミノ酸配列を、配列番号48に記載する。配列番号48のアミノ酸配列をコードするヌクレオチド配列を、配列番号49に記載する。
hH1及びhL1からなる抗体を「H1L1抗体」又は「H1L1」と称する。hH2及びhL2からなる抗体を「H2L2抗体」又は「H2L2」と称する。hH1及びhL3からなる抗体を「H1L3抗体」又は「H1L3」と称する。hH2及びhL4からなる抗体を「H2L4抗体」又は「H2L4」と称する。hH3及びhL3からなる抗体を「H3L3抗体」又は「H3L3」と称する。
10-5-1:ヒト化重鎖発現ベクターの構築
10-5-1-1:hH1発現ベクターの構築
配列番号53に示すhH1のヌクレオチド配列のヌクレオチド番号36乃至440に示されるDNA断片を合成した(GENEART社)。実施例9-1-3と同様の方法でhH1発現ベクターを構築した。
10-5-1-2:hH2発現ベクターの構築
配列番号57に示すhH2のヌクレオチド配列のヌクレオチド番号36乃至440に示されるDNA断片を合成した(GENEART社)。実施例9-1-3と同様の方法でhH2発現ベクターを構築した。
10-5-1-3:hH3発現ベクターの構築
配列番号61に示すhH2のヌクレオチド配列のヌクレオチド番号36乃至440に示されるDNA断片を合成した(GENEART社)。実施例9-1-3と同様の方法でhH3発現ベクターを構築した。
10-5-2:ヒト化軽鎖発現ベクターの構築
10-5-2-1:hL1発現ベクターの構築
配列番号37に示すhL1のヌクレオチド配列のヌクレオチド番号37乃至402に示されるDNA断片を合成した(GENEART社)。In-Fusion HD PCRクローニングキット(Clontech社)を用いて、pCMA-LKを制限酵素BsiWIで切断した箇所に合成したDNA断片を挿入することによりhL1発現ベクターを構築した。
10-5-2-2:hL2発現ベクターの構築
配列番号41に示すhL2のヌクレオチド配列のヌクレオチド番号37乃至402に示されるDNA断片を合成した(GENEART社)。実施例10-5-2-1と同様の方法でhL2発現ベクターを構築した。
10-5-2-3:hL3発現ベクターの構築
配列番号45に示すhL3のヌクレオチド配列のヌクレオチド番号37乃至402に示されるDNA断片を合成した(GENEART社)。実施例10-5-2-1と同様の方法でhL3発現ベクターを構築した。
10-5-2-4:hL4発現ベクターの構築
配列番号49に示すhL4のヌクレオチド配列のヌクレオチド番号37乃至402に示されるDNA断片を合成した(GENEART社)。実施例10-5-2-1と同様の方法でhL4発現ベクターを構築した。
10-5-3:ヒト化抗体の調製
10-5-3-1:ヒト化抗体H1L1、H2L2、H1L3、H2L4、及びH3L3の生産
実施例9-2-1と同様の方法で生産した。実施例10-4に示した重鎖と軽鎖の組み合わせに対応する重鎖発現ベクターと軽鎖発現ベクターの組み合わせにより、H1L1、H2L2、H1L3、H2L4、及びH3L3を生産した。
10-5-3-2:ヒト化抗体H1L1、H2L2、H1L3、H2L4、及びH3L3の2step精製
実施例10-5-3-1で得られた培養上清をrProtein Aアフィニティークロマトグラフィーとセラミックハイドロキシアパタイトの2段階工程で精製した。培養上清をPBSで平衡化したMabSelectSuReが充填されたカラム(GE Healthcare Bioscience社製)にアプライした後に、カラム容量の2倍以上のPBSでカラムを洗浄した。次に2Mアルギニン塩酸塩溶液(pH4.0)で抗体を溶出した。抗体の含まれる画分を透析(Thermo Scientific社、Slide-A-Lyzer Dialysis Cassette)によりPBSへのバッファー置換を行い、5mMリン酸ナトリウム/50mM MES/pH7.0のバッファーで5倍希釈した後に、5mM NaPi/50mM MES/30mM NaCl/pH7.0のバッファーで平衡化したセラミックハイドロキシアパタイトカラム(日本バイオラッド、Bio-Scale CHT Type-1 Hydroxyapatite Column)にアプライした。塩化ナトリウムによる直線的濃度勾配溶出を実施し、抗体の含まれる画分を集めた。その画分を透析(Thermo Scientific社、Slide-A-Lyzer Dialysis Cassette)によりHBSor(25mM ヒスチジン/5% ソルビトール、pH6.0)へのバッファー置換を行った。Centrifugal UF Filter Device VIVASPIN20(分画分子量UF10K,Sartorius社)にて抗体を濃縮し、IgG濃度を50mg/mLに調製した。最後にMinisart-Plus filter(Sartorius社)でろ過し、精製サンプルとした。
実施例10で作製したヒト化抗CLDN6抗体のヒトCLDN6及びファミリー分子CLDN3、CLDN4、CLDN9結合性をフローサイトメトリー法により評価した。実施例7-1と同様の手法にて一過性に遺伝子導入した293T細胞を使用した。ヒトCLDN6又はヒトCLDN9遺伝子を導入した細胞には、ヒト化抗CLDN6抗体H1L1、H2L2、H1L3、H2L4、及びH3L3、又はヒトIgG1コントロール抗体(CALBIOCHEM)を終濃度100nM、20nM、4nM、0.8nMで加えて懸濁し、4℃で30分静置した。ヒトCLDN3、ヒトCLDN4遺伝子、又は空のベクターを導入した細胞には、ヒト化抗CLDN6抗体H1L1、H2L2、H1L3、H2L4、及びH3L3を終濃度100 nMで加えて懸濁し、4℃で30分静置した。5%のウシ胎児血清(Hyclone)を含むDulbecco’s phosphate buffered saline (Sigma-Aldrich)(以下、5%FBS含有PBS)で洗浄した後、5%FBS含有PBSで150倍希釈したFITC AffiniPureF(ab’)2 Fragment Goat Anti-Human IgG(H+L) (Jackson ImmunoResearch)を加えて懸濁し、4℃で30分静置した。5%FBS含有PBSで洗浄した後、フローサイトメーター(FC500;Beckman Coulter)で検出を行った。データ解析はFlowJo(TreeStar)で行い、抗体の結合量を示すFITCの平均蛍光強度(MFI)を算出した。結果を図13に示す。図13のグラフにおいて、横軸は抗体濃度、縦軸はMFIを示す。作製したヒト化抗CLDN6抗体はヒトCLDN6とヒトCLDN9に同程度に結合し、ヒトCLDN3、ヒトCLDN4には結合しなかった。ヒトコントロールIgG1はいずれの細胞にも結合しなかった。
実施例12:糖鎖変換1(T-SG)
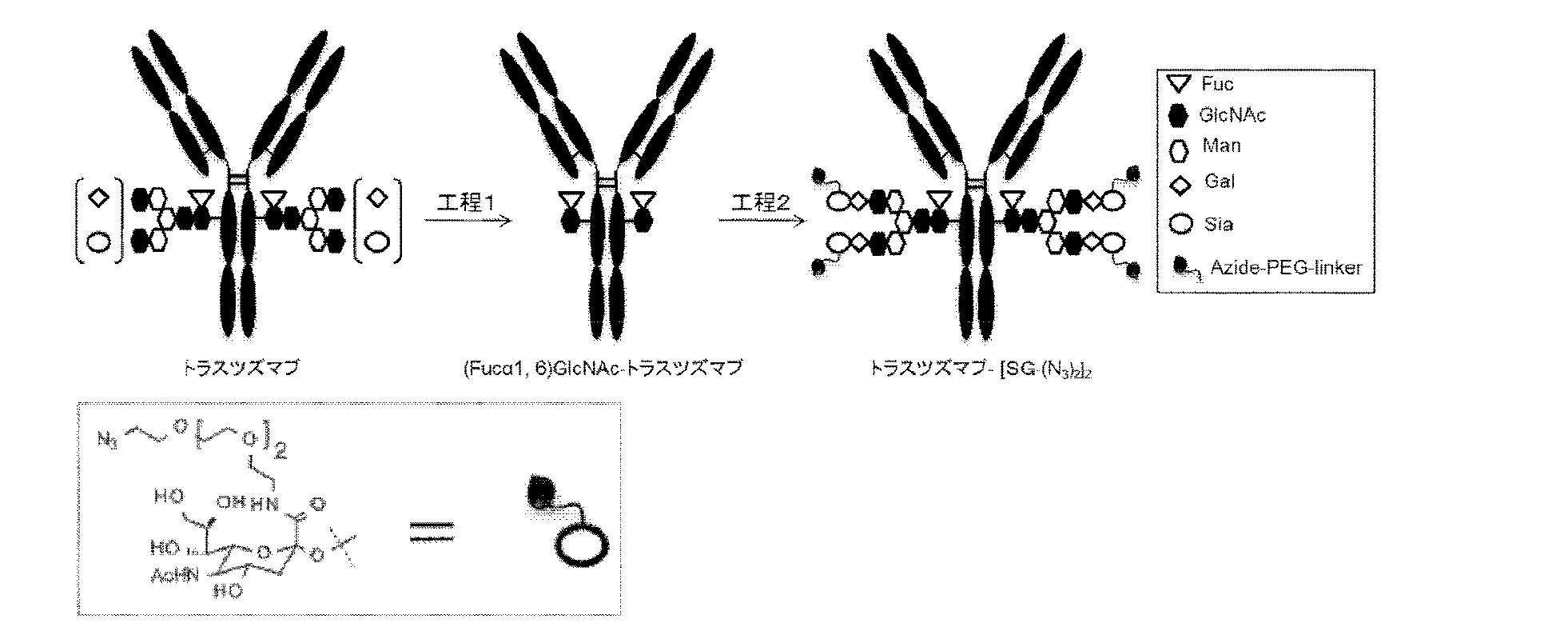
参考例1で調製したTrastuzumab溶液22mg/mLの(25mMヒスチジン溶液(pH6.0)、5%ソルビトール溶液)(45.5mL)を共通操作Cを用いて、50mMりん酸緩衝液(pH6.0)への緩衝液交換を二回に分けて行った。得られた28.1mg/mLのTrastuzumab溶液(50mMりん酸緩衝液(pH6.0)(18mL)と28.0mg/mLの同溶液(18mL)に、2.0mg/mL野生型EndoS溶液(PBS)をそれぞれ1.26mL、1.27mL加え、37℃で4時間インキュベートした。反応の進行具合をExperion電気泳動ステーション(BIO-RAD製)を用いて確認した。反応終了後、以下の方法に従い、アフィニティークロマトグラフィーによる精製とハイドロキシアパタイトカラムによる精製を行った。
精製装置:AKTA pure150(GE ヘルスケア製)
カラム:HiTrap rProtein A FF (5mL)(GE ヘルスケア製)
流速:5mL/min(チャージ時は1.25mL/min)
上記で得た反応液を複数回に分けて精製した。カラムは2連結し、カラムへの結合時は、反応液をカラム上部へ添加し、結合バッファー(20mMりん酸緩衝液(pH6.0))を1.25mL/minで2CV流し、更に5mL/minで5CV流した。中間洗浄時は、洗浄溶液(20mMりん酸緩衝液(pH7.0)、0.5M 塩化ナトリウム溶液)を15CV流した。溶出時は、溶出バッファー(ImmunoPure IgG Eution buffer、PIERCE製)を6CV流した。溶出液を1M トリス緩衝液(pH9.0)で直ちに中和した。溶出中にUV検出(280nm)されたフラクションについて、微量分光光度計Xpose(Trinean製)、及びExperion電気泳動ステーション(BIO-RAD製)を用いて確認した。目的物を含むフラクションを共通操作Cを用いて、5mMりん酸緩衝液50mM2-モルホリノエタンスルホン酸(MES)溶液(pH6.8)への緩衝液交換を行った。
精製装置:AKTA avant25(GE ヘルスケア製)
カラム:Bio-Scale Mini CHT Type Iカートリッジ(5mL)(BIO-RAD製)
流速:5mL/min(チャージ時は1.25mL/min)
カラムを2連結し、上記(1)で得られた溶液を複数回に分けて精製した。溶液をカラムの上部へ添加し、A液(5mMりん酸緩衝液50mM-モルホリノエタンスルホン酸(MES)溶液(pH6.8))を1.25mL/minで2CV流し、更に5mL/minで3CV流した。その後、A液とB液(5mMりん酸緩衝液50mM-モルホリノエタンスルホン酸(MES)溶液(pH6.8)、2M塩化ナトリウム溶液)を用いて、溶出した。溶出条件は、A液:B液=100:0~0:100(15CV)である。さらに、洗浄溶液(500mMりん酸緩衝液(pH6.5))を5CV流した。
目的物を含むフラクションを共通操作Cを用いて緩衝液交換を行い、25.5mg/mLの(Fucα1,6)GlcNAc-Trastuzumab溶液(50mMりん酸緩衝液(pH6.0))(35mL)を得た。
上記工程1にて得られた23.9mg/mL (Fucα1,6)GlcNAc-Trastuzumab溶液 (50mMりん酸緩衝液(pH6.0))(3.37mL)に、実施例5工程2で合成した化合物(12.9mg)の50mMりん酸緩衝液(pH6.0)溶液(0.258mL)、4.90mg/mLのEndoS D233Q/Q303L溶液(PBS)(0.328mL)を加えて、30℃で4.5時間インキュベートした。以上の操作を2ロット行った。反応の進行具合をExperion電気泳動ステーション(BIO-RAD製)を用いて確認した。反応終了後、上記工程1と同様にアフィニティークロマトグラフィーによる精製とハイドロキシアパタイトクロマトグラフィーによる精製を行った後、目的物を含むフラクションを共通操作Cを用いてりん酸緩衝生理食塩水(pH6.0)へ緩衝液交換を行い、10.0mg/mLのTrastuzumab[SG-(N3)2]2溶液(りん酸緩衝生理食塩水(pH6.0))(15.5mL)を得た。

以下の操作を5ロット行った。実施例12工程1にて得られた化合物(20mg/mL,15.0mL)を用いて、実施例4工程4にて得られた化合物(25.5mg)を糖鎖供与体として使い、30℃で3時間インキュベートし、実施例12工程2と同様の操作を行った。5ロット合わせて、14.4mg/mL Trastuzumab[MSG-N3]2溶液(りん酸緩衝生理食塩水(pH6.0))(93.5mL)を得た。

以下の操作を2ロット行った。実施例12工程1で得られた化合物(25.5mL,7.8mL)を用いて、実施例3工程4にて得られた化合物(25.5mg)を糖鎖供与体として使い、30℃で3時間インキュベートし、実施例12工程2と同様の操作を行った。2ロット合わせて、10.6mg/mL Trastuzumab[MSG1-N3]2溶液(りん酸緩衝生理食塩水(pH6.0))(31mL)を得た。

実施例10で調製した抗CLDN6抗体(H1L1)溶液ca.37.7mg/mL(25mMヒスチジン溶液(pH6.0)、5%ソルビトール溶液)(2.5mL)を用いて、実施例12工程1と同様の操作を行い、19.2mg/mLの(Fucα1,6)GlcNAc-抗CLDN6抗体(H1L1)溶液(50mMりん酸緩衝液(pH6.0))(4.8mL)を得た。
上記工程1で得られた19.2mg/mL(Fucα1,6)GlcNAc-抗CLDN6(H1L1)抗体溶液(50mMりん酸緩衝液(pH6.0))(4.8mL)を用いて、実施例3工程4にて得られた化合物(25.5mg)を糖鎖供与体として使い、実施例12工程2と同様の操作を行うことによって、10.2mg/mL抗CLDN6抗体(H1L1)-[MSG1-N3]2溶液(リン酸緩衝生理食塩水(pH6.0))(7.2mL)を得た。
工程1:(Fucα1,6)GlcNAc-抗CLDN6抗体(H2L2)
実施例10で調製した抗CLDN6抗体(H2L2)溶液ca.20mg/mL(25mMヒスチジン溶液(pH6.0)、5%ソルビトール溶液)(6mL)を用いて、実施例12工程1と同様の操作を行い、21.84mg/mLの(Fucα1,6)GlcNAc-抗CLDN6抗体(H2L2)溶液(50mMりん酸緩衝液(pH6.0))(5.7mL)を得た。
上記工程1で得られた21.8mg/mL(Fucα1,6)GlcNAc-抗CLDN6(H2L2)抗体溶液(50mMりん酸緩衝液(pH6.0))(5.7mL)を用いて、実施例3工程4にて得られた化合物(25.5mg)を糖鎖供与体として使い、実施例12工程2と同様の操作を行うことによって、10.2mg/mL抗CLDN6抗体(H2L2)-[MSG1-N3]2溶液(リン酸緩衝生理食塩水(pH6.0))(11.1mL)を得た。
工程1:(Fucα1,6)GlcNAc-抗CLDN6抗体(H1L3)
実施例10で調製した抗CLDN6抗体(H1L3)溶液ca.39.4mg/mL(25mMヒスチジン溶液(pH6.0)、5%ソルビトール溶液)(3mL)を用いて、実施例12工程1と同様の操作を行い、39.2mg/mLの(Fucα1,6)GlcNAc-抗CLDN6抗体(H1L3)溶液(50mMりん酸緩衝液(pH6.0))(4.5mL)を得た。
上記工程1で得られた39.2mg/mL(Fucα1,6)GlcNAc-抗CLDN6(H1L3)抗体溶液(50mMりん酸緩衝液(pH6.0))(4.5mL)を用いて、実施例3工程4にて得られた化合物(25.5mg)を糖鎖供与体として使い、実施例12工程2と同様の操作を行うことによって、9.83mg/mL抗CLDN6抗体(H1L3)-[MSG1-N3]2溶液(リン酸緩衝生理食塩水(pH6.0))(7.2mL)を得た。
工程1:(Fucα1,6)GlcNAc-抗Trop2抗体
参考例2で調製した抗Trop2抗体溶液ca.20mg/mL(25mMヒスチジン溶液(pH6.0)、5%ソルビトール溶液)(6mL)を用いて、実施例12工程1と同様の操作を行い、21.69mg/mLの(Fucα1,6)GlcNAc-抗Trop2抗体溶液(50mMりん酸緩衝液(pH6.0))(3.3mL)を得た。
上記工程1で得られた21.69mg/mL(Fucα1,6)GlcNAc-抗Trop2抗体溶液(50mMりん酸緩衝液(pH6.0))(3.35mL)を用いて、実施例3工程4にて得られた化合物(25.5mg)を糖鎖供与体として使い、実施例12工程2と同様の操作を行うことによって、10.3mg/mLの抗Trop2抗体-[MSG1-N3]2溶液(リン酸緩衝生理食塩水(pH6.0))(6.4mL)を得た。
実施例19~23にADC1~ADC6の調製方法を示す。実施例19~23の反応式中のR基はいずれも次式に示すものである。


実施例15工程2にて得られた抗体のリン酸緩衝生理食塩水(pH6.0)溶液(10.2mg/mL,2.50mL)に、室温にて1,2-プロパンジオール(2.29mL)、実施例2-1工程13にて得られた化合物(3-14)を10mM含むジメチルスルホキシド溶液(0.206mL;抗体1分子に対して12当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて48時間反応させた。
精製操作:上記溶液を共通操作Dを用いて精製を行い、目的の化合物を有する溶液を14.5mL得た。
特性評価:共通操作E、Fを用いて下記の特性値を得た。
抗体濃度:1.54mg/mL,抗体収量:22.3mg(89%),抗体一分子あたりの薬物平均結合数(n):1.9

実施例14工程1にて得られた抗体のリン酸緩衝生理食塩水(pH6.0)溶液(10.2mg/mL,1.00mL)に、室温にて1,2-プロパンジオール(0.917mL)、実施例2-1工程13にて得られた化合物(3-14)を10mM含むジメチルスルホキシド溶液(0.0825mL;抗体1分子に対して12当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて2日間反応させた。
精製操作:上記溶液を共通操作Dを用いて精製を行い、目的の化合物を有する溶液を6.00mL得た。
特性評価:共通操作E、Fを用いて下記の特性値を得た。
抗体濃度:1.41mg/mL,抗体収量:8.45mg(85%),抗体一分子あたりの薬物平均結合数(n):1.9

実施例18工程2にて得られた抗体のリン酸緩衝生理食塩水(pH6.0)溶液(10mg/mL,1.00mL)に、室温にて1,2-プロパンジオール(0.917mL)、実施例2-1工程13にて得られた化合物(3-14)を10mM含むジメチルスルホキシド溶液(0.0825mL;抗体1分子に対して12当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて2日間反応させた。
精製操作:上記溶液を共通操作Dを用いて精製を行い、目的の化合物を有する溶液を6.00mL得た。
特性評価:共通操作E、Fを用いて下記の特性値を得た。
抗体濃度:1.47mg/mL,抗体収量:8.8mg(88%),抗体一分子あたりの薬物平均結合数(n):1.9
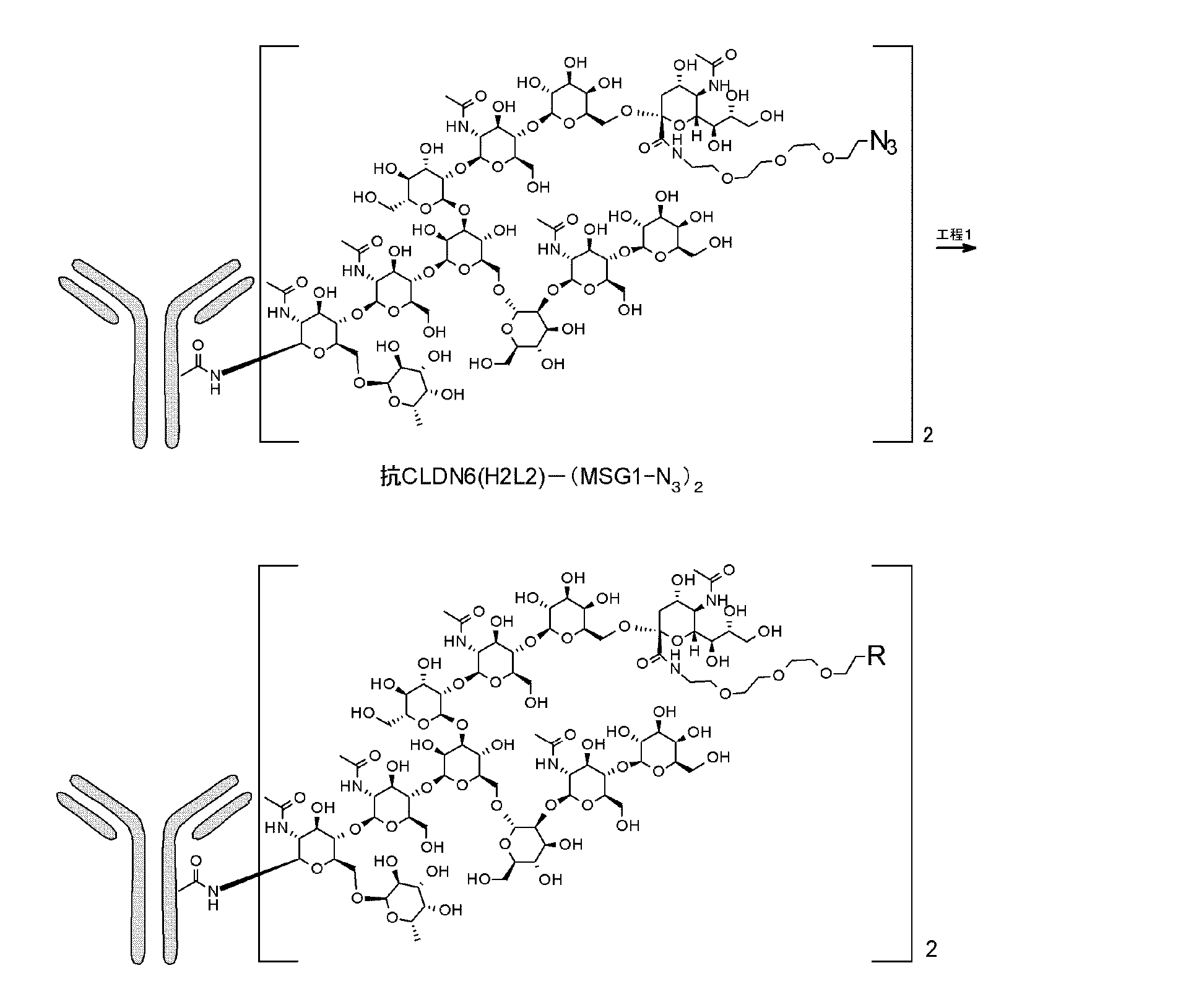
実施例16工程2にて得られた抗体のリン酸緩衝生理食塩水(pH6.0)溶液(9.96mg/mL,2.50mL)に、室温にて1,2-プロパンジオール(2.29mL)、実施例2-1工程13にて得られた化合物(3-14)を10mM含むジメチルスルホキシド溶液(0.206mL;抗体1分子に対して12当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて48時間反応させた。
精製操作:上記溶液を共通操作Dを用いて精製を行い、目的の化合物を有する溶液を14.5mL得た。
特性評価:共通操作E、Fを用いて下記の特性値を得た。
抗体濃度:1.52mg/mL,抗体収量:22.0mg(88%),抗体一分子あたりの薬物平均結合数(n):1.9

実施例17工程2にて得られた抗体のリン酸緩衝生理食塩水(pH6.0)溶液(9.83mg/mL,2.50mL)に、室温にて1,2-プロパンジオール(2.29mL)、実施例2-1工程13にて得られた化合物(3-14)を10mM含むジメチルスルホキシド溶液(0.206mL;抗体1分子に対して12当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて48時間反応させた。
精製操作:上記溶液を共通操作Dを用いて精製を行い、目的の化合物を有する溶液を14.5mL得た。
特性評価:共通操作E、Fを用いて下記の特性値を得た。
抗体濃度:1.45mg/mL,抗体収量:21.0mg(84%),抗体一分子あたりの薬物平均結合数(n):1.9
実施例24:ピロロベンゾジアゼピン誘導体A
薬物1を下記のスキームに従って合成した。

実施例1-1工程5にて得られた化合物(1-6)を(4.59g,8.15mmol)を、実施例2-1工程9と同様に反応させ、目的物(7-1)(4.86g,92%)を得た。
MS(APCI、ESI)m/z:647(M+H)+
上記工程1にて得られた化合物(7-1)(4.86g,7.51mmol)を、実施例1-1工程7と同様に反応させ、目的物(7-2)(3.42g,86%)を得た。
MS(APCI、ESI)m/z:533(M+H)+
上記工程2にて得られた化合物(7-2)(6.68g,12.5mmol)を、実施例1-1工程8と同様に反応させ、目的物(7-3)(6.44g,97%)を得た。
MS(APCI、ESI)m/z:531(M+H)+
上記工程3にて得られた化合物(7-3)(3.24g,6.10mmol)を、実施例1-1工程9と同様に反応させ、目的物(7-4)(3.86g,98%)を得た。
MS(APCI、ESI)m/z:645(M+H)+
上記工程4にて得られた化合物(7-4)(4.49g,6.96mmol)を、実施例1-1工程10と同様に反応させ、目的物(7-5)(3.24g,95%)を得た。
MS(APCI、ESI)m/z:489(M+H)+
上記工程5にて得られた化合物(7-5)(0.080g,0.164mmol)を、実施例2-1工程10と同様に反応させ、目的物(7-6)(0.160g,98%)を得た。
MS(APCI、ESI)m/z:993(M+H)+
上記工程6にて得られた化合物(7-6)(160mg,0.161mmol)を、実施例2-1工程11と同様に反応させ、目的物(7-7)(141mg,定量的)を得た。
MS(APCI、ESI)m/z:879(M+H)+
上記工程7にて得られた化合物(7-7)(141mg,0.161mmol)を、実施例2-1工程12と同様に反応させ、目的物(7-8)(109.8mg,99%)を得た。
1H-NMR(DMSO-D6)δ:7.92-7.91(1H,m),7.45(1H,s),7.39-7.37(2H,m),7.33(1H,s),7.29(1H,s),6.92-6.89(2H,m),6.85(1H,s),6.56-6.54(1H,m),6.31(1H,s),4.19-4.12(2H,m),4.05-3.99(1H,m),3.95-3.93(2H,m),3.82-3.79(4H,m),3.76(3H,s),3.66(3H,s),3.52-3.46(3H,m),3.30-3.21(2H,m),2.78-2.74(1H,m),2.45-2.42(1H,m),2.06-2.05(1H,m),1.89-1.82(4H,m),1.60-1.58(2H,m),0.80-0.63(4H,m).
MS(APCI、ESI)m/z:693(M+H)+

実施例2-4工程1にて得られた化合物(6-2)(6.49g,14.7mmol)のテトラヒドロフラン(147mL)溶液に、水素化ホウ素リチウム(0.642g,29.5mmol)を0℃で加え、室温で2時間撹拌した。反応溶液に1規定塩酸を加え、酢酸エチルで抽出した。得られた有機層を、飽和食塩水で洗浄し、硫酸マグネシウムで乾燥した後、減圧留去した。得られた残渣(6.94g,定量的)は精製せず次工程で使用した。
MS(APCI、ESI)m/z:413(M+H)+
上記工程1にて得られた化合物(8-1)(4.50g,11.0mmol)を、実施例1工程8と同様に反応させ、目的物(8-2)(1.94g,43%)を得た。
MS(APCI、ESI)m/z:411(M+H)+
上記工程2にて得られた化合物(8-2)(1.94g,4.73mmol)のテトラヒドロフラン(25mL)、酢酸エチル(25mL)、メタノール(25mL)の混合溶液に、窒素雰囲気下、5%パラジウム炭素(54%水分、1.0g)を加えた後、反応溶液を水素雰囲気下、室温にて22時間撹拌した。反応溶液をセライト濾過した後、濾液を減圧留去した。得られた残渣を、シリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=80:20(v/v)~0:100(v/v)]にて精製し、目的物(8-3)(1.20g,93%)を得た。
MS(APCI、ESI)m/z:275(M+H)+
実施例24工程5にて得られた化合物(7-5)(0.300g,0.614mmol)を、実施例2-1工程2と同様に反応させ、目的物(8-4)(0.388g,99%)を得た。
MS(APCI、ESI)m/z:639[81Br,(M+H)+],637[79Br,(M+H)+].
上記工程4にて得られた化合物(8-4)(0.203g, 0.318mmol)と上記工程3にて得られた化合物(0.131g,0.478mmol)を、実施例2-1工程10と同様に反応させ、目的物(8-5)(0.0880g,33%)を得た。
MS(APCI、ESI)m/z:831(M+H)+
上記工程5にて得られた化合物(8-5)(0.0880g,0.106mmol)を、実施例2-1工程11と同様に反応させ、目的物(8-6)(0.0500g、66%)を得た。
MS(APCI、ESI)m/z:717(M+H)+
上記工程6にて得られた化合物(8-6)(0.0500g、0.0698mmol)を実施例2-1工程12と同様に反応させ、目的物(8-7)(0.0330g,77%)を得た。
1H-NMR(CDCl3)δ:7.80(1H,m),7.58(1H,s),7.52(1H,s),6.81(1H,s),6.05(1H,s),4.17-3.97(5H,m),3.94(3H,s),3.87(1H,m),3.84(3H,s),3.72-3.68(3H,m),3.51-3.45(5H,m),2.54-2.51(1H,m),2.03-1.90(6H,m),1.75-1.68(2H,m),0.66(8H,m).
MS(APCI、ESI)m/z:615(M+H)+

実施例2-1工程1で得られた化合物(3-2)(5.00g,9.66mmol)を実施例2-1工程3と同様に反応させ目的物(9-1)(3.95g,100%)を得た。
MS(APCI,ESI)m/z:409(M+H)+
上記工程1で得られた化合物(9-1)(3.95g,9.67mmol)のジクロロメタン(97mL)溶液に、イミダゾール(1.65g,24.2mmol)、トリイソプロピルシリルクロリド(2.46mL,11.6mmol)とジメチルホルムアミド(5mL)を加え、室温で21時間攪拌した。反応溶液に水を加え、クロロホルムで抽出し、得られた有機層を水で洗浄し、減圧留去した。得られた残渣をシリカゲルカラムクロマトグラフィー[ヘキサン:酢酸エチル=100:0(v/v)~20:80(v/v)]にて精製し、目的物(9-2)(4.78g,87%)を得た。
MS(APCI,ESI)m/z:565(M+H)+
上記工程2にて得られた化合物(9-2)(4.78g,8.43mmol)を、実施例2-1工程4と同様に反応させ、目的物(9-3)(2.36g,50%)を得た。
MS(APCI,ESI)m/z:563(M+H)+
上記工程3にて得られた化合物(9-3)(1.53g,2,72mmol)を実施例2-1工程5と同様に反応させ、目的化合物(9-4)(1.27g,69%)を得た。
1H-NMR(CDCl3)δ:7.31(2H,s),7.15(1H,m),5.52(1H,m),4.65(1H,m),4.57(1H,m),3.95-3.89(1H,m),3.87(3H,s),3.75-3.58(2H,m),3.18-3.14(1H,m),1.33-1.25(3H,m),1.10(18H,m),1.00-0.96(2H,m),0.03(9H,s).
上記工程4で得られた化合物(9-4)(0.519g,0.747mmol)を、実施例2-1工程6と同様に反応させ、目的化合物(9-5)(0.511g,定量的)を得た。
MS(APCI,ESI)m/z:653[(M+H)+]
上記工程5で得られた化合物(9-5)(0.178g,0.272mmol)を、実施例2-1工程7と同様に反応させ、目的化合物(9-6)(0.094g,68%)を得た。
MS(APCI,ESI)m/z:507[(M+H)+]
上記工程6で得られた化合物(9-6)(0.063g,0.124mmol)を用いて、実施例2-1工程8と同様に反応させ、目的化合物(9-7)(0.046g,72%)を得た。
MS(APCI,ESI)m/z:509[(M+H)+]
上記工程7で得られた化合物(10-7)(0.046g,0.090mmol)を用いて、実施例2-1工程9と同様に反応させ、目的化合物(9-8)(0.03g,56%)を得た。
MS(APCI,ESI)m/z:593[(M+H)+]
上記工程8で得られた化合物(10-8)(0.030g,0.050mmol)を、実施例2-1工程10と同様に反応させ、目的化合物(9-9)(0.015g,0.034mmol)を得た。
1H-NMR(CDCl3)δ:7.39-7.25(4H,m),6.92-6.78(3H,m),6.03-5.92(1H,m),5.86-5.68(1H,m),5.20-5.07(2H,m),4.66-4.57(1H,m),4.52-4.40(1H,m),4.40-4.27(1H,m),4.27-4.16(1H,m),3.95(3H,s),3.82(3H,s),3.66-3.59(1H,m),3.32-3.21(1H,m),2.74-2.64(1H,m).
MS(APCI,ESI)m/z:437[(M+H)+]
実施例7工程5で得られた化合物(7-5)(0.131g,0.268mmol)を実施例2-2工程1と同様に反応させ、目的物(9-10)(0.086g,52%)を得た。
MS(APCI,ESI)m/z:611[81Br,(M+H)+],609[79Br,(M+H)+]
上記工程10で得られる化合物(10-10)(0.015g,0.034mmol)と、上記工程9で得られる化合物(10-9)(0.030g,0.048mmol)を用いて、実施例2-1工程10と同様に反応させ、目的化合物(9-11)(0.032g,96%)を得た。
MS(APCI,ESI)m/z:965[(M+H)+]
上記工程11で得られた化合物(9-11)(0.031g,0.032mmol)を実施例2-1工程11と同様に反応させ、目的物(9-12)(0.026g,95%)を得た。
MS(APCI,ESI)m/z:851[(M+H)+]
上記工程12で得られた化合物(10-12)(0.026g,0.030mmol)を実施例2-1工程12と同様に反応させ、目的物(9-13)(0.018g,88%)を得た。
1H-NMR(CDCl3)δ:7.80(1H,m),7.54-7.51(3H,m),7.33-7.29(2H,m),6.91-6.85(3H,m),6.14(1H,s),4.35-4.17(6H,m),3.95(3H,s),3.85(3H,s),3.82(3H,s),3.76-3.25(5H,m),2.79-2.69(1H,m),2.52(1H,m),2.45-2.35(1H,m),2.03-1.96(1H,m),1.28-1.23(2H,m),0.78-0.69(4H,m).
MS(APCI,ESI)m/z:665[(M+H)+]
ATCC(American Type Culture Collection)より入手したヒト肺がん細胞株Calu-6を評価に使用した。10%のウシ胎児血清(GE Healthcare)とMEM Non-Essential Amino Acids Solution(Thermo Fisher Scientific)およびSodium Pyruvate(Thermo Fisher Scientific)を含むMEM (Thermo Fisher Scientific;以下、EMEM培地)で1.25×104 Cells/mLになるように調製し、96穴細胞培養用マイクロプレートに80μLずつ添加した。細胞添加後、37℃、5%CO2下で一晩培養した。
翌日、EMEM培地で100pM、50pM、25pM、12.5pM、6.25pM、3.13pM、1.56pM、0.78pMに希釈したピロロベンゾジアゼピン誘導体A、B、又はCをマイクロプレートに10μLずつ添加した。ピロロベンゾジアゼピン誘導体の非添加ウェルにはEMEM培地を10μLずつ添加した。さらに、EMEM培地で2μMに調製したオラパリブをマイクロプレートに10μLずつ添加した。オラパリブ非添加ウェルにはEMEM培地を10μLずつ添加した。その後、マイクロプレートを37℃、5%CO2下で6日間培養した。培養後、マイクロプレートをインキュベーターから取り出して室温で30分間静置した。培養液と等量のCellTiter-Glo Luminescent Cell Viability Assay (Promega)を添加してプレートミキサーで攪拌した。室温で10分間静置後にプレートリーダー(PerkinElmer)で発光量を計測した。
生細胞率(%)=a÷b×100
a:誘導体A、B、又はC添加ウェルの発光量の平均値
b:培地添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体A、B、又はCの濃度a
b:誘導体A、B、又はCの濃度b
c:濃度aの誘導体A、B、又はCを添加した時の生細胞率
d:濃度bの誘導体A、B、又はCを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
生細胞率(%)=a÷b×100
a:誘導体A、B、又はCとオラパリブ2μM同時添加ウェルの発光量の平均値
b:オラパリブ2μM添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体A、B、又はCの濃度a
b:誘導体A、B、又はCの濃度b
c:濃度aの誘導体A、B、又はCとオラパリブ2μMを添加した時の生細胞率
d:濃度bの誘導体A、B、又はCとオラパリブ2μMを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
ATCC(American Type Culture Collection)より入手したヒト咽頭がん細胞株FaDuを評価に使用した。10%のウシ胎児血清(GE Healthcare)とMEM Non-Essential Amino Acids Solution(Thermo Fisher Scientific)およびSodium Pyruvate(Thermo Fisher Scientific)を含むMEM (Thermo Fisher Scientific;以下、EMEM培地)で6.25×103 Cells/mLになるように調製し、96穴細胞培養用マイクロプレートに80μLずつ添加した。細胞添加後、37℃、5%CO2下で一晩培養した。
翌日、EMEM培地で100pM、50pM、25pM、12.5pM、6.25pM、3.13pM、1.56pM、0.78pMに希釈したピロロベンゾジアゼピン誘導体A、B、又はCをマイクロプレートに10μLずつ添加した。ピロロベンゾジアゼピン誘導体の非添加ウェルにはEMEM培地を10μLずつ添加した。さらに、EMEM培地で1μMに調製したオラパリブをマイクロプレートに10μLずつ添加した。オラパリブ非添加ウェルにはEMEM培地を10μLずつ添加した。その後、マイクロプレートを37℃、5%CO2下で6日間培養した。培養後、マイクロプレートをインキュベーターから取り出して室温で30分間静置した。培養液と等量のCellTiter-Glo Luminescent Cell Viability Assay (Promega)を添加してプレートミキサーで攪拌した。室温で10分間静置後にプレートリーダー(PerkinElmer)で発光量を計測した。
生細胞率(%)=a÷b×100
a:誘導体A、B、又はC添加ウェルの発光量の平均値
b:培地添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体A、B、又はCの濃度a
b:誘導体A、B、又はCの濃度b
c:濃度aの誘導体A、B、又はCを添加した時の生細胞率
d:濃度bの誘導体A、B、又はCを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
生細胞率(%)=a÷b×100
a:誘導体A、B、又はCとオラパリブ1μM同時添加ウェルの発光量の平均値
b:オラパリブ1μM添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体A、B、又はCの濃度a
b:誘導体A、B、又はCの濃度b
c:濃度aの誘導体A、B、又はCとオラパリブ1μMを添加した時の生細胞率
d:濃度bの誘導体A、B、又はCとオラパリブ1μMを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
ATCC(American Type Culture Collection)より入手したヒト咽頭がん細胞株FaDuを評価に使用した。10%のウシ胎児血清(GE Healthcare)とMEM Non-Essential Amino Acids Solution(Thermo Fisher Scientific)およびSodium Pyruvate(Thermo Fisher Scientific)を含むMEM (Thermo Fisher Scientific;以下、EMEM培地)で6.25×103 Cells/mLになるように調製し、96穴細胞培養用マイクロプレートに80μLずつ添加した。細胞添加後、37℃、5%CO2下で一晩培養した。
翌日、EMEM培地で100pM、50pM、25pM、12.5pM、6.25pM、3.13pM、1.56pM、0.78pMに希釈したピロロベンゾジアゼピン誘導体A、B、又はCをマイクロプレートに10μLずつ添加した。ピロロベンゾジアゼピン誘導体の非添加ウェルにはEMEM培地を10μLずつ添加した。さらに、EMEM培地で2nMに調製したタラゾパリブをマイクロプレートに10μLずつ添加した。タラゾパリブ非添加ウェルにはEMEM培地を10μLずつ添加した。その後、マイクロプレートを37℃、5%CO2下で6日間培養した。培養後、マイクロプレートをインキュベーターから取り出して室温で30分間静置した。培養液と等量のCellTiter-Glo Luminescent Cell Viability Assay (Promega)を添加してプレートミキサーで攪拌した。室温で10分間静置後にプレートリーダー(PerkinElmer)で発光量を計測した。
生細胞率(%)=a÷b×100
a:誘導体A、B、又はC添加ウェルの発光量の平均値
b:培地添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体A、B、又はCの濃度a
b:誘導体A、B、又はCの濃度b
c:濃度aの誘導体A、B、又はCを添加した時の生細胞率
d:濃度bの誘導体A、B、又はCを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
生細胞率(%)=a÷b×100
a:誘導体A、B、又はCとタラゾパリブ2nM同時添加ウェルの発光量の平均値
b:タラゾパリブ2nM添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体A、B、又はCの濃度a
b:誘導体A、B、又はCの濃度b
c:濃度aの誘導体A、B、又はCとタラゾパリブ2nMを添加した時の生細胞率
d:濃度bの誘導体A、B、又はCとタラゾパリブ2nMを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
ATCC(American Type Culture Collection)から購入したヒト卵巣がん株SK-OV-3細胞を評価に使用した。10%のウシ胎児血清(GE Healthcare)を含むMcCoy‘s 5A(Modified)Medium(Thermo Fisher Scientific;以下、McCoy’s 5A培地)で1.25×104 C ells/mLになるように調製し、96穴細胞培養用マイクロプレートに80μLずつ添加した。細胞添加後、37℃、5%CO2下で一晩培養した。
翌日、McCoy’s 5A培地で100pM、50pM、25pM、12.5pM、6.25pM、3.13pM、1.56pM、0.78pMに希釈したピロロベンゾジアゼピン誘導体Aをマイクロプレートに10μLずつ添加した。ピロロベンゾジアゼピン誘導体A非添加ウェルにはMcCoy’s 5A培地を10μLずつ添加した。さらに、McCoy’s 5A培地で5nMに調製したタラゾパリブをマイクロプレートに10μLずつ添加した。タラゾパリブ非添加ウェルにはMcCoy’s 5A培地を10μLずつ添加した。その後、マイクロプレートを37℃、5%CO2下で6日間培養した。培養後、マイクロプレートをインキュベーターから取り出して室温で30分間静置した。培養液と等量のCellTiter-Glo Luminescent Cell Viability Assay (Promega)を添加してプレートミキサーで攪拌した。室温で10分間静置後にプレートリーダー(PerkinElmer)で発光量を計測した。
生細胞率(%)=a÷b×100
a:誘導体A添加ウェルの発光量の平均値
b:培地添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体Aの濃度a
b:誘導体Aの濃度b
c:濃度aの誘導体Aを添加した時の生細胞率
d:濃度bの誘導体Aを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
生細胞率(%)=a÷b×100
a:誘導体Aとタラゾパリブ5nM同時添加ウェルの発光量の平均値
b:タラゾパリブ5nM添加ウェルの発光量の平均値
IC50値は次式で算出した。
IC50(nM) =antilog((50-d)×(LOG10(b)-LOG10(a))÷(d-c)+LOG10(b))
a:誘導体Aの濃度a
b:誘導体Aの濃度b
c:濃度aの誘導体Aとタラゾパリブ5nMを添加した時の生細胞率
d:濃度bの誘導体Aとタラゾパリブ5nMを添加した時の生細胞率
a、bは生細胞率50%を挟む2点でa>b。
マウス:5-6週齢の雌BALB/c ヌードマウス(日本チャールス・リバー社)を実験に供した。
測定・計算式:全ての研究において、腫瘍の長径及び短径を電子式デジタルキャリパー(CD-15CX,Mitutoyo Corp.)で1週間に2回測定し、腫瘍体積(mm3)を計算した。計算式は以下に示す通り。
腫瘍体積(mm3)=1/2×長径(mm)×[短径(mm)]2
抗CLDN6抗体-薬物コンジュゲートADC1、抗HER2抗体-薬物コンジュゲートADC2および抗TROP2抗体-薬物コンジュゲートADC3はABS buffer (10mM酢酸緩衝液 (pH5.5), 5%ソルビトール)で希釈し、10mL/kgの液量を尾静脈内投与した。オラパリブはDimethyl sulfoxide(DMSO)で溶解し、10% 2-hydroxy-propyl-β-cyclodextrin(SIGMA-ALDRICH)/Dulbecco’s Phosphate-Buffered Salineで希釈した後に10mL/kgの液量を腹腔内投与した。タラゾパリブはDMSOで溶解し、10%N,N-ジメチルアセトアミド/5%Kolliphor HS15(SIGMA-ALDRICH)/Dulbecco’s Phosphate-Buffered Salineで希釈し、10mL/kgの液量を経口投与した。ニラパリブはDMSOで溶解し、0.5% methylcelluloseで希釈し、10mL/kgの液量を経口投与した。以上の方法は、実施例34~37で共通である。
ATCC(American Type Culture Collection)から購入したヒト膵臓がん細胞株CFPAC-1を生理食塩水に懸濁し、5.0×106cellsを雌ヌードマウスの右体側部に皮下移植し、移植10日後に無作為に群分けを実施した(Day0)。ADC2はDay0に0.2mg/kgの用量で投与した。タラゾパリブはDay0から5日間、0.8mg/kgの用量で投与した。それぞれの単剤投与群と併用投与群および、コントロール群として薬物処置なし(No treatment)の群を設定した。
ADC2とタラゾパリブの併用効果を図1に示す。図中、横軸は細胞移植後の日数、縦軸は腫瘍体積を示す。タラゾパリブ単剤投与の試験最終日(Day35)における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は17%であった。ADC2の単剤投与によるTGIは94%であった。一方、ADC2とタラゾパリブとの併用投与では、タラゾパリブ単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められた(P<0.005。Dunnett’s testにより算出。以下同様。)。また、ADC2単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められ(P<0.05)、腫瘍増殖抑制率は(TGI,99%)であった。また、いずれの単剤および併用投与群において、体重減少等の特に目立った所見は認められなかった。
DSMZ(Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH)から購入したヒト乳がん株JIMT-1細胞を生理食塩水に懸濁して5×106cellsを雌ヌードマウスの右体側部に皮下移植し、移植10日後に無作為に群分けを実施した(Day0)。抗HER2抗体-薬物コンジュゲートADC2はDay0に0.2mg/kgの用量で投与した。PARP阻害剤はオラパリブを50mg/kgの用量またはタラゾパリブを0.8mg/kgの用量でDay0から5日間投与した。それぞれの単剤投与群、ADC2とPARP阻害剤の併用投与群および、コントロール群としてABS buffer投与群を設定した。
ADC2とオラパリブの併用効果を図2に示す。図中、横軸は細胞移植後の日数、縦軸は腫瘍体積を示す。オラパリブ単剤投与では腫瘍増殖抑制効果を認めなかった。ADC2の単剤投与による試験最終日(Day43)における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は75%であった。一方、ADC2とオラパリブとの併用投与では、オラパリブ単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められた(P<0.005)。また、ADC2単剤投与よりも腫瘍増殖抑制率は高く(TGI,82%)、強い併用効果が認められた。また、いずれの単剤および併用投与群において、体重減少等の特に目立った所見は認められなかった。
ADC2とタラゾパリブの併用効果を図3に示す。図中、横軸は細胞移植後の日数、縦軸は腫瘍体積を示す。タラゾパリブ単剤投与では腫瘍増殖抑制効果を認めなかった。ADC2の単剤投与による試験最終日(Day43)における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は75%であった。一方、ADC2とタラゾパリブとの併用投与では、タラゾパリブ単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められた(P<0.005)。また、ADC2単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められ(P<0.05)、腫瘍増殖抑制率は(TGI,91%)であった。また、いずれの単剤および併用投与群において、体重減少等の特に目立った所見は認められなかった。
ATCC(American Type Culture Collection)より入手したヒト咽頭がん細胞株FaDuを生理食塩水に懸濁し、3.0×106cellsを雌ヌードマウスの右体側部に皮下移植し、移植10日後に無作為に群分けを実施した(Day0)。抗TROP2抗体-薬物コンジュゲートADC3はDay0に0.2mg/kgの用量で投与した。PARP阻害剤はオラパリブを50mg/kgの用量またはタラゾパリブを0.8mg/kgの用量でDay0から5日間投与した。それぞれの単剤投与群、ADC3とPARP阻害剤の併用投与群および、コントロール群としてABS buffer投与群を設定した。
ADC3とオラパリブの併用効果を図4に示す。図中、横軸は細胞移植後の日数、縦軸は腫瘍体積を示す。オラパリブ単剤投与の試験最終日(Day25)における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は7%であった。ADC3の単剤投与による試験最終日における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は67%であった。一方、ADC3とオラパリブとの併用投与では、オラパリブ単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められた(P<0.005)。また、ADC3単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められ(P<0.05)、腫瘍増殖抑制率は(TGI,76%)であった。また、いずれの単剤および併用投与群において、体重減少等の特に目立った所見は認められなかった。
ADC3とタラゾパリブの併用効果を図5に示す。図中、横軸は細胞移植後の日数、縦軸は腫瘍体積を示す。タラゾパリブ単剤投与の試験最終日(Day25)における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は33%であった。ADC3の単剤投与による試験最終日における腫瘍増殖抑制率(Tumor Growth Inhibition,TGI)は67%であった。一方、ADC3とタラゾパリブとの併用投与では、タラゾパリブ単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められた(P<0.005)。また、ADC3単剤投与よりも有意に優れた腫瘍増殖抑制効果が認められ(P<0.005)、腫瘍増殖抑制率は(TGI,83%)であった。また、いずれの単剤および併用投与群において、体重減少等の特に目立った所見は認められなかった。
ATCC(American Type Culture Collection)から購入したヒト卵巣がん細胞株OV-90をマトリゲル(CORNING)に懸濁し、2.5×106cellsを雌ヌードマウスの右体側部に皮下移植し、移植18日後に無作為に群分けを実施した(Day0)。抗CLDN6抗体-薬物コンジュゲートADC1はDay0に0.3mg/kgの用量で投与した。ニラパリブはDay0から5日間、75mg/kgの用量で投与した。それぞれの単剤投与群と併用投与群および、コントロール群として薬物処置なし(No treatment)の群を設定した。
ADC1とニラパリブの併用結果を図42に示す。図中、横軸は細胞移植後の日数、縦軸は腫瘍体積を示す。ニラパリブ単剤投与によるDay21時点の腫瘍増殖抑制率(Tumor Growth Inhibition、TGI)は1%で、Day21時点で体重減少等の目立った所見は認められなかった。ADC1単剤投与群およびADC1とニラパリブの併用投与群におけるDay21時点のTGIはいずれも96%であり、Day21時点で体重減少等の目立った所見は認められなかった。一方、Day53時点ではADC1とニラパリブの併用投与では、ADC1単剤投与と比較して腫瘍増殖抑制効果が認められた。Day53時点でADC1単剤投与、およびADC1とニラパリブの併用投与による体重減少等の目立った所見は認められなかった。
なお、本実施例で使用したADC1は、抗CLDN6(H1L1)抗体を用いて、実施例15及び実施例19と同様の方法を従い作製した。
ATCC(American Type Culture Collection)から購入したヒト卵巣がん細胞株OV-90をマトリゲル(CORNING)に懸濁し、2.5×106cellsを雌ヌードマウスの右体側部に皮下移植し、移植13~18日後に無作為に群分けを実施する(Day0)。抗CLDN6抗体-薬物コンジュゲートADC1はDay0に0.2mg/kgの用量で投与する。PARP阻害薬はオラパリブを50mg/kgの用量またはタラゾパリブを0.8mg/kgの用量でDay0から5日間投与する。それぞれの単剤投与群、ADC1とPARP阻害薬の併用投与群および、コントロール群として薬物処置なし(No treatment)の群を設定する。
工程1:(Fucα1,6)GlcNAc-Trastuzumab変異体の調製
Trastuzumab 変異体(軽鎖:配列番号73, 重鎖:配列番号75)溶液ca.22.3mg/mL(50mMりん酸緩衝液(pH6.0))(2.69mL)に、7.7mg/mL野生型EndoS溶液(PBS)を0.156mL加え、37℃で4時間インキュベートした。反応の進行具合をExperion電気泳動ステーション(BIO-RAD製)を用いて確認した。反応終了後、以下の方法に従い、アフィニティークロマトグラフィーによる精製とハイドロキシアパタイトカラムによる精製を行った。
(1)アフィニティークロマトグラフィーによる精製
精製装置:AKTA avant(GEヘルスケア製)
カラム:HiTrap rProtein A FF(5mL)(GEヘルスケア製)
流速:5mL/min(チャージ時は1.25mL/min)
上記で得た反応液を複数回に分けて精製した。カラムへの結合時は、反応液をカラム上部へ添加し、結合バッファー(20mMりん酸緩衝液(pH6.0))を1.25mL/minで4CV(Column Volume)流し、更に5mL/minで5CV流した。中間洗浄時は、洗浄溶液(20mMりん酸緩衝液(pH7.0)、0.5M塩化ナトリウム溶液)を15CV流した。溶出時は、溶出バッファー(ImmunoPure IgG Eution buffer、PIERCE製)を6CV流した。溶出液を1Mトリス緩衝液(pH9.0)で直ちに中和した。目的物を含むフラクションを共通操作Cを用いて、5mMりん酸緩衝液50mM-モルホリノエタンスルホン酸(MES)溶液(pH6.8)への緩衝液交換を行った。
(2)ハイドロキシアパタイトクロマトグラフィーによる精製
精製装置:AKTA avant(GE ヘルスケア製)
カラム:Bio-Scale Mini CHT Type Iカートリッジ(5mL)(BIO-RAD製)
流速:5mL/min(チャージ時は1.25mL/min)
上記(1)で得られた溶液をカラムの上部へ添加し、A液(5mMりん酸緩衝液50mM-モルホリノエタンスルホン酸(MES)溶液(pH6.8))を1.25mL/minで4CV流し、更に5mL/minで3CV流した。その後、A液とB液(5mMりん酸緩衝液50mM-モルホリノエタンスルホン酸(MES)溶液(pH6.8)、2M塩化ナトリウム溶液)を用いて、溶出した。溶出条件は、A液:B液=100:0~0:100(15CV)である。さらに、洗浄溶液(500mMりん酸緩衝液(pH6.5))を5CV流した。
目的物を含むフラクションを共通操作Cを用いて緩衝液交換を行い、6.08mg/mLの(Fucα1,6)GlcNAc-Trastuzumab変異体溶液(50mMリン酸緩衝液(pH6.0))(6.10mL)を得た。
上記工程1にて得られた6.08mg/mL(Fucα1,6)GlcNAc-Trastuzumab変異体溶液(50mMりん酸緩衝液(pH6.0))(6.10mL)を用いて、実施例3工程4で合成した化合物(9.78mg)の50mMりん酸緩衝液(pH6.0)溶液(0.200mL)、5.80mg/mLのEndoS D233Q/Q303L溶液(PBS)(0.128mL)を加えて、30℃で3時間インキュベートした。反応の進行具合をExperion電気泳動ステーション(BIO-RAD製)を用いて確認した。反応終了後、上記工程1と同様にアフィニティークロマトグラフィーによる精製とハイドロキシアパタイトクロマトグラフィーによる精製を行った後、目的物を含むフラクションを共通操作Cを用いてりん酸緩衝生理食塩水(pH6.0)へ緩衝液交換を行い、10.2mg/mLのTrastuzumab 変異体-[MSG1-N3]2溶液(りん酸緩衝生理食塩水(pH6.0))(3.65mL)を得た。
Trastuzumab変異体2(軽鎖:配列番号76, 重鎖:77)を用いて、実施例38の工程1及び2と同様の操作を行うことによってTrastuzumab 変異体2- [MSG1-N3]2を得た。
実施例38工程2にて得られたTrastuzumab 変異体- [MSG1-N3]2のリン酸緩衝生理食塩水(pH6.0)溶液(10.0mg/mL,0.40mL)に、室温にて1,2-プロパンジオール(0.767mL)、実施例2-1工程13にて得られた化合物(3-14)の10mMジメチルスルホキシド溶液(0.033mL;抗体1分子に対して12当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて48時間反応させた。
精製操作:上記溶液を後述の共通操作Dを用いて精製を行い、目的の化合物を有する溶液を7.00mL得た。
特性評価:共通操作E,Fを用いて下記の特性値を得た。
抗体濃度:0.48mg/mL,抗体収量:3.39mg(85%),抗体一分子あたりの平均薬物結合数(n):1.7
工程1-1:抗体と薬物リンカーのコンジュゲーション(ADC7)
実施例38にて得られたTrastuzumab 変異体2- [MSG1-N3]2のリン酸緩衝生理食塩水(pH6.0)溶液(10.0mg/mL,0.50mL)に、室温にて1,2-プロパンジオール(0.486mL)、実施例2-1工程13にて得られた化合物(3-14)の10mMジメチルスルホキシド溶液(0.014mL;抗体1分子に対して4当量)を加え、チューブローテーター(MTR-103、アズワン株式会社)を用いて、室温にて40時間反応させた。
精製操作:上記溶液を後述の共通操作Dを用いて精製を行い、目的の化合物を有する溶液を2.50mL得た。
特性評価:共通操作E,Fを用いて下記の特性値を得た。
抗体濃度:1.12mg/mL,抗体収量:2.80mg(56%),抗体一分子あたりの平均薬物結合数(n):1.8
配列番号2:ヒトCLDN6のアミノ酸配列をコードするcDNAのヌクレオチド配列
配列番号3:ヒトCLDN9のアミノ酸配列
配列番号4:ヒトCLDN9のアミノ酸配列をコードするcDNAのヌクレオチド配列
配列番号5:B1抗体軽鎖のCDRL1のアミノ酸配列
配列番号6:B1抗体軽鎖のCDRL2のアミノ酸配列
配列番号7:B1抗体軽鎖のCDRL3のアミノ酸配列
配列番号8:ヒト化B1抗体軽鎖L4のCDRL3のアミノ酸配列
配列番号9:B1抗体重鎖のCDRH1のアミノ酸配列
配列番号10:B1抗体重鎖のCDRH2のアミノ酸配列
配列番号11:B1抗体重鎖のCDRH3のアミノ酸配列
配列番号12:C7抗体軽鎖のCDRL1のアミノ酸配列
配列番号13:C7抗体軽鎖のCDRL2のアミノ酸配列
配列番号14:C7抗体軽鎖のCDRL3のアミノ酸配列
配列番号15:C7抗体重鎖のCDRH1のアミノ酸配列
配列番号16:C7抗体重鎖のCDRH2のアミノ酸配列
配列番号17:C7抗体重鎖のCDRH3のアミノ酸配列
配列番号18:B1抗体軽鎖の可変領域をコードするcDNAのヌクレオチド配列
配列番号19:B1抗体軽鎖の可変領域のアミノ酸配列
配列番号20:B1抗体重鎖の可変領域をコードするcDNAのヌクレオチド配列
配列番号21:B1抗体重鎖の可変領域のアミノ酸配列
配列番号22:C7抗体軽鎖の可変領域をコードするcDNAのヌクレオチド配列
配列番号23:C7抗体軽鎖の可変領域のアミノ酸配列
配列番号24:C7抗体重鎖の可変領域をコードするcDNAのヌクレオチド配列
配列番号25:C7抗体重鎖の可変領域のアミノ酸配列
配列番号26:ヒト軽鎖シグナル配列及びヒトκ鎖定常領域をコードするDNA配列を含むDNA断片
配列番号27:ヒト重鎖シグナル配列及びヒトIgG1LALA定常領域をコードするDNA配列を含むDNA断片
配列番号28:chB1軽鎖のアミノ酸配列
配列番号29:chB1軽鎖のアミノ酸配列をコードするDNA配列を含むDNA断片
配列番号30:chB1軽鎖の可変領域のアミノ酸配列
配列番号31:chB1軽鎖可変領域をコードするヌクレオチド配列
配列番号32:chB1重鎖のアミノ酸配列
配列番号33:chB1重鎖をコードするヌクレオチド配列
配列番号34:chB1重鎖の可変領域のアミノ酸配列
配列番号35:chB1重鎖の可変領域をコードするヌクレオチド配列
配列番号36:ヒト化抗体軽鎖hL1のアミノ酸配列
配列番号37:ヒト化抗体軽鎖hL1をコードするヌクレオチド配列
配列番号38:ヒト化抗体軽鎖hL1の可変領域のアミノ酸配列
配列番号39:ヒト化抗体軽鎖hL1の可変領域をコードするヌクレオチド配列
配列番号40:ヒト化抗体軽鎖hL2のアミノ酸配列
配列番号41:ヒト化抗体軽鎖hL2をコードするヌクレオチド配列
配列番号42:ヒト化抗体軽鎖hL2の可変領域のアミノ酸配列
配列番号43:ヒト化抗体軽鎖hL2の可変領域をコードするヌクレオチド配列
配列番号44:ヒト化抗体軽鎖hL3のアミノ酸配列
配列番号45:ヒト化抗体軽鎖hL3をコードするヌクレオチド配列
配列番号46:ヒト化抗体軽鎖hL3の可変領域のアミノ酸配列
配列番号47:ヒト化抗体軽鎖hL3の可変領域をコードするヌクレオチド配列
配列番号48:ヒト化抗体軽鎖hL4のアミノ酸配列
配列番号49:ヒト化抗体軽鎖hL4をコードするヌクレオチド配列
配列番号50:ヒト化抗体軽鎖hL4の可変領域のアミノ酸配列
配列番号51:ヒト化抗体軽鎖hL4の可変領域をコードするヌクレオチド配列
配列番号52:ヒト化抗体重鎖hH1のアミノ酸配列
配列番号53:ヒト化抗体重鎖hH1をコードするヌクレオチド配列
配列番号54:ヒト化抗体重鎖hH1の可変領域のアミノ酸配列
配列番号55:ヒト化抗体重鎖hH1の可変領域をコードするヌクレオチド配列
配列番号56:ヒト化抗体重鎖hH2のアミノ酸配列
配列番号57:ヒト化抗体重鎖hH2をコードするヌクレオチド配列
配列番号58:ヒト化抗体重鎖hH2の可変領域のアミノ酸配列
配列番号59:ヒト化抗体重鎖hH2の可変領域をコードするヌクレオチド配列
配列番号60:ヒト化抗体重鎖hH3のアミノ酸配列
配列番号61:ヒト化抗体重鎖hH3をコードするヌクレオチド配列
配列番号62:ヒト化抗体重鎖hH3の可変領域のアミノ酸配列
配列番号63:ヒト化抗体重鎖hH3の可変領域をコードするヌクレオチド配列
配列番号64:Trastuzumab軽鎖のアミノ酸配列
配列番号65:Trastuzumab重鎖のアミノ酸配列
配列番号66:抗LPS抗体(h#1G5-H1L1)軽鎖のアミノ酸配列
配列番号67:抗LPS抗体(h#1G5-H1L1)重鎖のアミノ酸配列
配列番号68:抗TROP2抗体(hRS7)軽鎖のアミノ酸配列
配列番号69:抗TROP2抗体(hRS7)重鎖のアミノ酸配列
配列番号70:抗CD98抗体(hM23-H1L1)軽鎖のアミノ酸配列
配列番号71:抗CD98抗体(hM23-H1L1)重鎖のアミノ酸配列
配列番号72:Trastuzumab変異体軽鎖をコードするヌクレオチド配列
配列番号73:Trastuzumab変異体軽鎖のアミノ酸配列
配列番号74:Trastuzumab変異体重鎖をコードするヌクレオチド配列
配列番号75:Trastuzumab変異体重鎖のアミノ酸配列
配列番号76:Trastuzumab変異体2軽鎖のアミノ酸配列
配列番号77:Trastuzumab変異体2重鎖のアミノ酸配列
Claims (40)
- 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート及び/又はPARP阻害剤を含むがん治療のための医薬組成物であって、該抗体-ピロロベンゾジアゼピン誘導体コンジュゲートと、該PARP阻害剤が、組み合わされて投与されることを特徴とし、
該コンジュゲートが、次式;




上記で示されるそれぞれの構造式において、m1は1又は2の整数であり、
Abは、抗体又は該抗体の機能性断片であり、
N297糖鎖は次式で示される構造を有するN297-(Fuc)MSG1、N297-(Fuc)MSG2もしくはそれらの混合物、又はN297-(Fuc)SGであり、



N297糖鎖中のL(PEG)は、*-(CH2CH2-O)3-CH2CH2-NH-であることを示し、
ここで、右端のアミノ基がN297糖鎖のβ-Manの分岐鎖の1-3鎖側又は/及び1-6鎖側の非還元末端のシアル酸の2位のカルボン酸とアミド結合を介して結合しており、左端のアステリスク*は、前記式中のトリアゾール環上の1位又は3位の窒素原子と結合していることを示す医薬組成物。 - 抗体が、腫瘍細胞に発現している抗原に結合し、腫瘍細胞内に取り込まれて内在化することを特徴とする、請求項1に記載の医薬組成物。
- 抗体が、抗腫瘍効果を有することを特徴とする、請求項1又は2に記載の医薬組成物。
- 抗体が、抗CLDN6抗体、抗CLDN9抗体、抗CLDN6/CLDN9抗体、抗HER2抗体、抗HER3抗体、抗DLL3抗体、抗FAP抗体、抗CDH11抗体、抗A33抗体、抗CanAg抗体、抗CD19抗体、抗CD20抗体、抗CD22抗体、抗CD25抗体、抗CD30抗体、抗CD33抗体、抗CD37抗体、抗CD56抗体、抗CD70抗体、抗CD98抗体、抗B7-H3抗体、抗TROP2抗体、抗CEA抗体、抗Cripto抗体、抗EphA2抗体、抗FGFR2抗体、抗G250抗体、抗MUC1抗体、抗GPNMB抗体、抗Integrin抗体、抗PSMA抗体、抗Tenascin-C抗体、抗SLC44A4抗体、抗Mesothelin抗体、抗EGFR抗体、抗5T4抗体、抗LRRC15抗体、抗DR5抗体、抗CDH3抗体、抗PDPN 抗体、又は抗CD123抗体である、請求項1~3のいずれか一項に記載の医薬組成物。
- 抗体が、CLDN6及び/又はCLDN9に特異的に結合することを特徴とする、請求項1~4のいずれか一項に記載の医薬組成物。
- 抗体が、以下の(a)又は(b)に記載のCDRH1、CDRH2及びCDRH3を含む重鎖、並びに、CDRL1、CDRL2及びCDRL3を含む軽鎖を含んでなる、請求項5に記載の医薬組成物;
(a)配列番号9に記載のアミノ酸配列からなるCDRH1、配列番号10に記載のアミノ酸配列からなるCDRH2及び配列番号11に記載のアミノ酸配列からなるCDRH3、並びに、配列番号5に記載のアミノ酸配列からなるCDRL1、配列番号6に記載のアミノ酸配列からなるCDRL2及び配列番号7に記載のアミノ酸配列又は該アミノ酸配列において1又は2個のアミノ酸が置換されたアミノ酸配列からなるCDRL3、又は、
(b)配列番号15に記載のアミノ酸配列からなるCDRH1、配列番号16に記載のアミノ酸配列からなるCDRH2及び配列番号17に記載のアミノ酸配列からなるCDRH3、並びに、配列番号12に記載のアミノ酸配列からなるCDRL1、配列番号13に記載のアミノ酸配列からなるCDRL2及び配列番号14に記載のアミノ酸配列からなるCDRL3。 - 抗体が、以下の(a)又は(b)に記載のCDRH1、CDRH2及びCDRH3を含む重鎖、並びに、CDRL1、CDRL2及びCDRL3を含む軽鎖を含んでなる、請求項6に記載の医薬組成物;
(a)配列番号9に記載のアミノ酸配列からなるCDRH1、配列番号10に記載のアミノ酸配列からなるCDRH2及び配列番号11に記載のアミノ酸配列からなるCDRH3、及び、配列番号5に記載のアミノ酸配列からなるCDRL1、配列番号6に記載のアミノ酸配列からなるCDRL2及び配列番号7に記載のアミノ酸配列又は配列番号8に記載のアミノ酸配列からなるCDRL3、又は、
(b)配列番号15に記載のアミノ酸配列からなるCDRH1、配列番号16に記載のアミノ酸配列からなるCDRH2及び配列番号17に記載のアミノ酸配列からなるCDRH3、及び、配列番号12に記載のアミノ酸配列からなるCDRL1、配列番号13に記載のアミノ酸配列からなるCDRL2及び配列番号14に記載のアミノ酸配列からなるCDRL3。 - 抗体が、以下の(a)又は(b)に記載の重鎖可変領域、及び、軽鎖可変領域を含む、請求項5~7のいずれか一項に記載の医薬組成物;
(a)配列番号21に記載のアミノ酸配列からなる重鎖可変領域、及び、配列番号19に記載のアミノ酸配列からなる軽鎖可変領域、又は、
(b)配列番号25に記載のアミノ酸配列からなる重鎖可変領域、及び、配列番号23に記載のアミノ酸配列からなる軽鎖可変領域。 - 抗体が、以下の(a)~(e)からなる群から選択されるアミノ酸配列からなる重鎖可変領域、及び(f)~(k)からなる群から選択されるアミノ酸配列からなる軽鎖可変領域を含む、請求項5~8のいずれか一項に記載の医薬組成物;
(a)配列番号54に記載のアミノ酸配列、
(b)配列番号58に記載のアミノ酸配列、
(c)配列番号62に記載のアミノ酸配列、
(d)(a)~(c)の配列において各CDR配列以外のフレームワーク領域の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、
(e)(a)~(c)の配列における各CDR配列以外のフレームワーク領域の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列、
(f)配列番号38に記載のアミノ酸配列、
(g)配列番号42に記載のアミノ酸配列、
(h)配列番号46に記載のアミノ酸配列、
(i)配列番号50に記載のアミノ酸配列、
(j)(f)~(i)の配列において各CDR配列以外のフレームワーク領域の配列に対して少なくとも95%以上の相同性を有するアミノ酸配列、及び、
(k)(f)~(i)の配列における各CDR配列以外のフレームワーク領域の配列において1又は数個のアミノ酸が欠失、置換又は付加されたアミノ酸配列。 - 抗体が、以下の(a)~(e)からなる群から選択される重鎖可変領域及び軽鎖可変領域を含む、請求項9に記載の医薬組成物;
(a)配列番号54に記載のアミノ酸配列からなる重鎖可変領域および配列番号38に記載のアミノ酸配列からなる軽鎖可変領域、
(b)配列番号58に記載のアミノ酸配列からなる重鎖可変領域および配列番号42に記載のアミノ酸配列からなる軽鎖可変領域、
(c)配列番号54に記載のアミノ酸配列からなる重鎖可変領域および配列番号46に記載のアミノ酸配列からなる軽鎖可変領域、
(d)配列番号58に記載のアミノ酸配列からなる重鎖可変領域および配列番号50に記載のアミノ酸配列からなる軽鎖可変領域、及び、
(e)配列番号62に記載のアミノ酸配列からなる重鎖可変領域および配列番号46に記載のアミノ酸配列からなる軽鎖可変領域。 - 抗体が、キメラ抗体である、請求項5~10のいずれか一項に記載の医薬組成物。
- 抗体が、ヒト化抗体である、請求項5~10のいずれか一項に記載の医薬組成物。
- 抗体が、ヒトIgG1、ヒトIgG2又はヒトIgG4の重鎖定常領域を含む、請求項5~12のいずれか一項に記載の医薬組成物。
- 抗体が、以下の(a)~(e)からなる群から選択される重鎖及び軽鎖を含む、請求項12又は13に記載の医薬組成物;
(a)配列番号52のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号36のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(b)配列番号56のアミノ酸番号20~471に記載のアミノ酸配列からなる、重鎖および配列番号40のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(c)配列番号52のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号44のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、
(d)配列番号56のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号48のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖、及び、
(e)配列番号60のアミノ酸番号20~471に記載のアミノ酸配列からなる重鎖および配列番号44のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖。 - 抗体が、請求項6~10及び14のいずれか一項に記載の抗体と、CLDN6及び/又はCLDN9への結合において競合するか、又は、請求項6~10及び14のいずれか一項に記載の抗体が認識するCLDN6及び/又はCLDN9上の部位に結合する、請求項5に記載の医薬組成物。
- 抗体が、HER2に特異的に結合する請求項1~4のいずれか一項に記載の医薬組成物。
- 抗体依存性細胞傷害(ADCC)活性及び/又は補体依存性細胞傷害(CDC)活性を有する請求項16に記載の医薬組成物。
- 抗体の重鎖定常領域がヒトIgG1の重鎖定常領域であり、ADCC及び/又はCDC活性の低減をもたらす変異を含む、請求項16に記載の医薬組成物。
- 抗体の重鎖定常領域がヒトIgG1の重鎖定常領域であり、該重鎖定常領域においてEU Indexにより示される234位及び235位のロイシンがアラニンに置換されている、請求項18に記載の医薬組成物。
- 配列番号65に記載のアミノ酸配列からなる重鎖及び配列番号64に記載のアミノ酸配列からなる軽鎖を含んでなる抗体である、請求項16又は17に記載の医薬組成物。
- 配列番号75のアミノ酸番号20~139に記載のアミノ酸配列からなる重鎖可変領域及び配列番号73のアミノ酸番号21~127に記載のアミノ酸配列からなる軽鎖可変領域を含む抗体である、請求項16、18及び19のいずれか一項に記載の医薬組成物。
- 配列番号75のアミノ酸番号20~469に記載のアミノ酸配列からなる重鎖及び配列番号73のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖を含む抗体である、請求項16、18、19及び21のいずれか一項に記載の医薬組成物。
- 配列番号77のアミノ酸番号20~469に記載のアミノ酸配列からなる重鎖及び配列番号76のアミノ酸番号21~234に記載のアミノ酸配列からなる軽鎖を含む抗体である、請求項16、18、19及び21のいずれか一項に記載の医薬組成物。
- 抗体が、N-結合型糖鎖付加、O-結合型糖鎖付加、N末のプロセッシング、C末のプロセッシング、脱アミド化、アスパラギン酸の異性化、メチオニンの酸化、N末におけるメチオニン残基の付加、プロリン残基のアミド化、及び重鎖のカルボキシル末端における1つ又は2つのアミノ酸残基の欠失からなる群より選択される1又は2以上の修飾を含む、請求項5~23のいずれか一項に記載の医薬組成物。
- 抗体の重鎖のカルボキシル末端において1つ又は数個のアミノ酸残基が欠失している、請求項24に記載の医薬組成物。
- 抗体の2本の重鎖の双方のカルボキシル末端において1つのアミノ酸残基が欠失している、請求項24又は25に記載の医薬組成物。
- 抗体の重鎖のカルボキシル末端のプロリン残基が更にアミド化されている、請求項24~26のいずれか一項に記載の医薬組成物。
- N297糖鎖が、N297-(Fuc)MSG1である、請求項1~27のいずれか一項に記載の医薬組成物。
- m1が、1の整数である、請求項1~28のいずれか一項に記載の医薬組成物。
- 抗体―ピロロベンゾジアゼピン誘導体コンジュゲートにおける抗体1分子あたりの平均薬物結合数が、1~3又は3~5である、請求項1~29のいずれか一項に記載の医薬組成物。
- PARP阻害剤が、オラパリブ、ルカパリブ、ニラパリブ、若しくはタラゾパリブ、又はそれらの薬理上許容される塩である、請求項1~30のいずれか一項に記載の医薬組成物。
- 抗体-薬物コンジュゲートと、PARP阻害剤が、それぞれ別異の製剤に有効成分として含有され、同時に又は異なる時間に投与されることを特徴とする、請求項1~31のいずれか一項に記載の医薬組成物。
- 肺がん(非小細胞肺がん、小細胞肺がん等)、腎がん、尿路上皮がん、大腸がん、前立腺がん、多形神経膠芽腫、卵巣がん(表層上皮性腫瘍、間質性腫瘍、胚細胞腫瘍等)、膵がん、乳がん、メラノーマ、肝がん、膀胱がん、胃がん、食道がん、子宮体がん、精巣がん(セミノーマ、非セミノーマ)、子宮頸がん、胎盤絨毛がん、脳腫瘍、頭頚部がんならびにそれらの転移性形態からなる群より選択される少なくとも一つのがんの治療のための、請求項1~32のいずれか一項に記載の医薬組成物。
- PARP阻害剤と併用するための、請求項1に記載の抗体-ピロロベンゾジアゼピン誘導体コンジュゲートを含む医薬組成物。
- PARP阻害剤を含み、請求項1に記載の抗体-ピロロベンゾジアゼピン誘導体コンジュゲートと併用することにより、該コンジュゲートの作用を上昇させる医薬組成物。
- 請求項1に記載の抗体-ピロロベンゾジアゼピン誘導体コンジュゲートと併用するための、PARP阻害剤を含む医薬組成物。
- 請求項1に記載の抗体-ピロロベンゾジアゼピン誘導体コンジュゲートを含み、PARP阻害剤と併用することより、PARP阻害剤の作用を上昇させる医薬組成物。
- 請求項1に記載の医薬組成物であって、
該ピロロベンゾジアゼピン誘導体がDNAのマイナーグルーブにおいてクロスリンクを形成しない医薬組成物。 - 請求項1に記載の医薬組成物であって、
がんが、PARP阻害剤に非感受性である医薬組成物。 - 請求項1に記載の医薬組成物であって、
がんが、相同的組換え(HR)依存的DNA二本鎖破壊(DSB)修復経路に非依存的である医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019061761 | 2019-03-27 | ||
| JP2019061761 | 2019-03-27 | ||
| PCT/JP2020/013555 WO2020196712A1 (ja) | 2019-03-27 | 2020-03-26 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JPWO2020196712A1 JPWO2020196712A1 (ja) | 2020-10-01 |
| JPWO2020196712A5 JPWO2020196712A5 (ja) | 2023-10-17 |
| JP7522097B2 true JP7522097B2 (ja) | 2024-07-24 |
Family
ID=72611051
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021509568A Active JP7522097B2 (ja) | 2019-03-27 | 2020-03-26 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20220168438A1 (ja) |
| EP (1) | EP3949988A4 (ja) |
| JP (1) | JP7522097B2 (ja) |
| KR (1) | KR20210143839A (ja) |
| CN (1) | CN113631191B (ja) |
| BR (1) | BR112021017350A2 (ja) |
| CA (1) | CA3139180A1 (ja) |
| TW (1) | TWI841715B (ja) |
| WO (1) | WO2020196712A1 (ja) |
Families Citing this family (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ762505A (en) | 2017-09-29 | 2026-03-27 | Daiichi Sankyo Co Ltd | Antibody-pyrrolobenzodiazepine derivative conjugate |
| SG11202104993SA (en) * | 2018-11-14 | 2021-06-29 | Daiichi Sankyo Co Ltd | Anti-cdh6 antibody-pyrrolobenzodiazepine derivative conjugate |
| WO2020196475A1 (ja) * | 2019-03-25 | 2020-10-01 | 第一三共株式会社 | 抗her2抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| CN113631229B (zh) * | 2019-03-25 | 2025-09-23 | 第一三共株式会社 | 抗体-吡咯并苯并二氮杂卓衍生物偶联物 |
| JP7847132B2 (ja) * | 2020-10-14 | 2026-04-16 | 江蘇恒瑞医薬股▲ふん▼有限公司 | 抗her3抗体と抗her3抗体薬物複合体及びその医薬用途 |
| EP4265275A4 (en) * | 2020-12-18 | 2024-03-20 | Shanghai Fudan-Zhangjiang Bio-Pharmaceutical Co., Ltd. | ANTIBODY-DRUG CONJUGATE TARGETING TROP2, PREPARATION METHOD AND USE THEREOF |
| CA3204628A1 (en) * | 2021-01-13 | 2022-07-21 | John T. POIRIER | Antibody-pyrrolobenzodiazepine derivative conjugate |
| US11739144B2 (en) | 2021-03-09 | 2023-08-29 | Xencor, Inc. | Heterodimeric antibodies that bind CD3 and CLDN6 |
| WO2023053282A1 (ja) * | 2021-09-29 | 2023-04-06 | 中外製薬株式会社 | がんの治療に用いるための細胞傷害誘導治療剤 |
| AU2022353890A1 (en) * | 2021-09-30 | 2024-04-04 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Pyrrolo benzodiazepine derivative, and conjugate, preparation method and use thereof |
| CA3240378A1 (en) * | 2021-12-23 | 2023-06-29 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Anti-dll3 antibody and pharmaceutical use thereof, and antibody-drug conjugate containing anti-dll3 antibody |
| EP4534103A1 (en) * | 2022-05-31 | 2025-04-09 | CSPC Megalith Biopharmaceutical Co., Ltd. | Pharmaceutical composition of recombinant anti-human cldn18.2 monoclonal antibody-mmae conjugate |
| EP4548932A1 (en) | 2022-06-30 | 2025-05-07 | Toray Industries, Inc. | Pharmaceutical composition for treating and/or preventing cancer |
| TW202508595A (zh) | 2023-05-04 | 2025-03-01 | 美商銳新醫藥公司 | 用於ras相關疾病或病症之組合療法 |
| CN120957754A (zh) * | 2023-05-12 | 2025-11-14 | 四川科伦博泰生物医药股份有限公司 | 稠环化合物及其制备方法和用途 |
| IL324834A (en) | 2023-05-25 | 2026-01-01 | Univ California | Dosage of claudin-6 conjugates for cancer treatment |
| US20250049810A1 (en) | 2023-08-07 | 2025-02-13 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| AU2024360465A1 (en) | 2023-10-12 | 2026-04-09 | Revolution Medicines, Inc. | Macrocyclic ras inhibitors |
| WO2025171296A1 (en) | 2024-02-09 | 2025-08-14 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2025217307A1 (en) | 2024-04-09 | 2025-10-16 | Revolution Medicines, Inc. | Methods for predicting response to a ras(on) inhibitor and combination therapies |
| TW202547461A (zh) | 2024-05-17 | 2025-12-16 | 美商銳新醫藥公司 | Ras抑制劑 |
| WO2025255438A1 (en) | 2024-06-07 | 2025-12-11 | Revolution Medicines, Inc. | Methods of treating a ras protein-related disease or disorder |
| WO2025265060A1 (en) | 2024-06-21 | 2025-12-26 | Revolution Medicines, Inc. | Therapeutic compositions and methods for managing treatment-related effects |
| WO2026006747A1 (en) | 2024-06-28 | 2026-01-02 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026015790A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015796A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015801A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Methods of treating a ras related disease or disorder |
| WO2026015825A1 (en) | 2024-07-12 | 2026-01-15 | Revolution Medicines, Inc. | Use of ras inhibitor for treating pancreatic cancer |
| WO2026050446A1 (en) | 2024-08-29 | 2026-03-05 | Revolution Medicines, Inc. | Ras inhibitors |
| WO2026072904A2 (en) | 2024-09-26 | 2026-04-02 | Revolution Medicines, Inc. | Compositions and methods for treating lung cancer |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090149449A1 (en) | 2003-10-22 | 2009-06-11 | The United States Of America, As Represented By The Secretary, Dept. Of Health And Human Service | Pyrrolobenzodiazepine derivatives, compositions comprising the same and methods related thereto |
| JP2015534996A (ja) | 2012-10-23 | 2015-12-07 | シンアフィックス ビー.ブイ. | 修飾抗体、抗体コンジュゲート及びそれらを調製する方法 |
| JP2016512540A (ja) | 2013-03-13 | 2016-04-28 | シアトル ジェネティックス, インコーポレイテッド | シクロデキストリンおよび抗体−薬物結合体製剤 |
| WO2017137556A1 (en) | 2016-02-10 | 2017-08-17 | Adc Therapeutics Sa | Pyrrolobenzodiazepine anti-her2 antibody conjugates |
| JP7133079B2 (ja) | 2017-09-29 | 2022-09-07 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
Family Cites Families (52)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3101690B2 (ja) | 1987-03-18 | 2000-10-23 | エス・ビィ・2・インコーポレイテッド | 変性抗体の、または変性抗体に関する改良 |
| IL162181A (en) | 1988-12-28 | 2006-04-10 | Pdl Biopharma Inc | A method of producing humanized immunoglubulin, and polynucleotides encoding the same |
| DK0585287T3 (da) | 1990-07-10 | 2000-04-17 | Cambridge Antibody Tech | Fremgangsmåde til fremstilling af specifikke bindingsparelementer |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| EP1400536A1 (en) | 1991-06-14 | 2004-03-24 | Genentech Inc. | Method for making humanized antibodies |
| EP0605522B1 (en) | 1991-09-23 | 1999-06-23 | Medical Research Council | Methods for the production of humanized antibodies |
| PT1024191E (pt) | 1991-12-02 | 2008-12-22 | Medical Res Council | Produção de auto-anticorpos a partir de reportórios de segmentos de anticorpo e exibidos em fagos |
| CA2131151A1 (en) | 1992-03-24 | 1994-09-30 | Kevin S. Johnson | Methods for producing members of specific binding pairs |
| US5885573A (en) | 1993-06-01 | 1999-03-23 | Arch Development Corporation | Methods and materials for modulation of the immunosuppressive activity and toxicity of monoclonal antibodies |
| AU691811B2 (en) | 1993-06-16 | 1998-05-28 | Celltech Therapeutics Limited | Antibodies |
| GB9313509D0 (en) | 1993-06-30 | 1993-08-11 | Medical Res Council | Chemisynthetic libraries |
| WO1995015388A1 (en) | 1993-12-03 | 1995-06-08 | Medical Research Council | Recombinant binding proteins and peptides |
| DK1071700T3 (da) | 1998-04-20 | 2010-06-07 | Glycart Biotechnology Ag | Glykosylerings-modifikation af antistoffer til forbedring af antistofafhængig cellulær cytotoksicitet |
| CA2704600C (en) | 1999-04-09 | 2016-10-25 | Kyowa Kirin Co., Ltd. | A method for producing antibodies with increased adcc activity |
| CA2953239A1 (en) | 2000-10-06 | 2002-04-18 | Kyowa Hakko Kirin Co., Ltd. | Antibody composition-producing cell |
| CA2478047C (en) | 2002-03-01 | 2014-01-21 | Immunomedics, Inc. | Rs7 antibodies |
| US7449464B2 (en) * | 2003-03-12 | 2008-11-11 | Kudos Pharmaceuticals Limited | Phthalazinone derivatives |
| EP1976515A2 (en) * | 2006-01-17 | 2008-10-08 | Abbott Laboratories | Combination therapy with parp inhibitors |
| US7763534B2 (en) | 2007-10-26 | 2010-07-27 | Tela Innovations, Inc. | Methods, structures and designs for self-aligning local interconnects used in integrated circuits |
| WO2007133855A2 (en) | 2006-03-27 | 2007-11-22 | University Of Maryland Biotechnology Institute | Glycoprotein synthesis and remodeling by enzymatic transglycosylation |
| EP2281844A1 (en) | 2009-07-31 | 2011-02-09 | Glycotope GmbH | MUC 1 antibodies |
| CA2795349C (en) | 2010-04-15 | 2016-11-29 | Seattle Genetics, Inc. | Targeted pyrrolobenzodiazepine conjugates |
| SI2528625T1 (sl) | 2010-04-15 | 2013-11-29 | Spirogen Sarl | Pirolobenzodiazepini in njihovi konjugati |
| US20130273005A1 (en) | 2010-12-20 | 2013-10-17 | Gilead Sciences, Inc. | Methods for treating hcv |
| JP2014200118A (ja) | 2011-07-28 | 2014-10-23 | 三洋電機株式会社 | 電池駆動機器及び電池パック |
| IN2014DN06806A (ja) | 2012-02-10 | 2015-05-22 | Univ Maryland | |
| AU2013247645B2 (en) | 2012-04-09 | 2019-02-14 | Daiichi Sankyo Company, Limited | Anti-FGFR2 antibody |
| US20130309223A1 (en) | 2012-05-18 | 2013-11-21 | Seattle Genetics, Inc. | CD33 Antibodies And Use Of Same To Treat Cancer |
| HRP20182129T1 (hr) | 2012-10-12 | 2019-02-08 | Adc Therapeutics Sa | Konjugati protutijelo - pirolobenzodiazepin |
| ES2731681T3 (es) | 2013-02-22 | 2019-11-18 | Abbvie Stemcentrx Llc | Conjugados de anticuerpo anti-DLL3 y PBD y usos de los mismos |
| KR20160047567A (ko) | 2013-08-28 | 2016-05-02 | 스템센트알엑스 인코포레이티드 | 조작된 항-dll3 접합체 및 사용 방법 |
| GB201317981D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| WO2015052534A1 (en) | 2013-10-11 | 2015-04-16 | Spirogen Sàrl | Pyrrolobenzodiazepine-antibody conjugates |
| GB201317982D0 (en) | 2013-10-11 | 2013-11-27 | Spirogen Sarl | Pyrrolobenzodiazepines and conjugates thereof |
| KR102337414B1 (ko) | 2013-12-16 | 2021-12-10 | 제넨테크, 인크. | 펩타이드 모방체 화합물 및 이의 항체-약물 컨쥬게이트 |
| AU2014371934B2 (en) | 2013-12-25 | 2020-01-23 | Daiichi Sankyo Company, Limited | Anti-TROP2 antibody-drug conjugate |
| AU2015311987B2 (en) * | 2014-09-03 | 2020-06-04 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| WO2016036804A1 (en) | 2014-09-03 | 2016-03-10 | Immunogen, Inc. | Cytotoxic benzodiazepine derivatives |
| MX2017003472A (es) * | 2014-09-17 | 2017-10-31 | Genentech Inc | Inmunoconjugados que comprenden anticuerpos anti-her2. |
| US11559581B2 (en) | 2015-01-09 | 2023-01-24 | Oxford University Innovation Limited | Antibody conjugates and methods of making the antibody conjugates |
| MX2017009145A (es) | 2015-01-14 | 2017-11-22 | Bristol Myers Squibb Co | Dimeros de benzodiazepina, conjugados de estos, y sus metodos de preparacion y uso. |
| US9504694B2 (en) | 2015-03-19 | 2016-11-29 | Cellerant Therapeutics, Inc. | Isoquinolidinobenzodiazepines |
| SMT202000614T1 (it) | 2015-06-29 | 2021-01-05 | Immunogen Inc | Coniugati di anticorpi ingegnerizzati con cisteina |
| SG10202005460RA (en) | 2015-06-30 | 2020-07-29 | Seattle Genetics Inc | Anti-ntb-a antibodies and related compositions and methods |
| CN108026518B (zh) | 2015-07-16 | 2022-09-06 | 第一三共株式会社 | 新型endos突变型酶 |
| GB201513607D0 (en) | 2015-07-31 | 2015-09-16 | Feingold Jay M | Pyrrolobenzodiazepine-antibody conjugates |
| CR20180243A (es) * | 2015-10-02 | 2018-07-31 | Genentech Inc | Conjugados de anticuerpo-fármaco de pirrolobenzodiazepina y métodos de uso |
| WO2018003983A1 (ja) * | 2016-07-01 | 2018-01-04 | 第一三共株式会社 | hANP-Fc含有分子コンジュゲート |
| WO2018102212A1 (en) * | 2016-12-01 | 2018-06-07 | Immunomedics, Inc. | Therapy for metastatic urothelial cancer with the antibody-drug conjugate, sacituzumab govitecan (immu-132) |
| SG11202104993SA (en) * | 2018-11-14 | 2021-06-29 | Daiichi Sankyo Co Ltd | Anti-cdh6 antibody-pyrrolobenzodiazepine derivative conjugate |
| WO2020196475A1 (ja) * | 2019-03-25 | 2020-10-01 | 第一三共株式会社 | 抗her2抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| CN113631229B (zh) * | 2019-03-25 | 2025-09-23 | 第一三共株式会社 | 抗体-吡咯并苯并二氮杂卓衍生物偶联物 |
-
2020
- 2020-03-26 CA CA3139180A patent/CA3139180A1/en active Pending
- 2020-03-26 WO PCT/JP2020/013555 patent/WO2020196712A1/ja not_active Ceased
- 2020-03-26 JP JP2021509568A patent/JP7522097B2/ja active Active
- 2020-03-26 KR KR1020217034025A patent/KR20210143839A/ko active Pending
- 2020-03-26 BR BR112021017350A patent/BR112021017350A2/pt unknown
- 2020-03-26 US US17/442,608 patent/US20220168438A1/en active Pending
- 2020-03-26 CN CN202080024555.3A patent/CN113631191B/zh active Active
- 2020-03-26 EP EP20778479.4A patent/EP3949988A4/en active Pending
- 2020-03-26 TW TW109110172A patent/TWI841715B/zh active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090149449A1 (en) | 2003-10-22 | 2009-06-11 | The United States Of America, As Represented By The Secretary, Dept. Of Health And Human Service | Pyrrolobenzodiazepine derivatives, compositions comprising the same and methods related thereto |
| JP2015534996A (ja) | 2012-10-23 | 2015-12-07 | シンアフィックス ビー.ブイ. | 修飾抗体、抗体コンジュゲート及びそれらを調製する方法 |
| JP2016512540A (ja) | 2013-03-13 | 2016-04-28 | シアトル ジェネティックス, インコーポレイテッド | シクロデキストリンおよび抗体−薬物結合体製剤 |
| WO2017137556A1 (en) | 2016-02-10 | 2017-08-17 | Adc Therapeutics Sa | Pyrrolobenzodiazepine anti-her2 antibody conjugates |
| JP7133079B2 (ja) | 2017-09-29 | 2022-09-07 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
| JP7273716B2 (ja) | 2017-09-29 | 2023-05-15 | 第一三共株式会社 | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート |
Non-Patent Citations (1)
| Title |
|---|
| ZHONG Haihong et al.,Improved Therapeutic Window in BRCA-mutant Tumors with Antibody-linked Pyrrolobenzodiazepine Dimers,Molecular Cancer Therapeutics,2018年10月23日,Vol.18, No.1,pp.89-99 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20210143839A (ko) | 2021-11-29 |
| US20220168438A1 (en) | 2022-06-02 |
| EP3949988A1 (en) | 2022-02-09 |
| CN113631191A (zh) | 2021-11-09 |
| WO2020196712A1 (ja) | 2020-10-01 |
| CN113631191B (zh) | 2024-09-27 |
| EP3949988A4 (en) | 2022-11-16 |
| TWI841715B (zh) | 2024-05-11 |
| BR112021017350A2 (pt) | 2021-11-16 |
| JPWO2020196712A1 (ja) | 2020-10-01 |
| CA3139180A1 (en) | 2020-10-01 |
| TW202102226A (zh) | 2021-01-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7522097B2 (ja) | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲートとparp阻害剤の組み合わせ | |
| JP7627655B2 (ja) | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート | |
| JP7615189B2 (ja) | 抗体-ピロロベンゾジアゼピン誘導体コンジュゲート | |
| JP7529654B2 (ja) | 抗her2抗体-ピロロベンゾジアゼピン誘導体コンジュゲート | |
| CN113166764B (zh) | 抗cdh6抗体-吡咯并苯并二氮杂环庚三烯衍生物缀合物 | |
| US20190127471A1 (en) | Novel b7-h3 binding molecules, antibody drug conjugates thereof and methods of use thereof | |
| HK40067376A (en) | Combination of antibody-pyrrolobenzodiazepine derivative conjugate and parp inhibitor | |
| HK40068280A (en) | Antibody-pyrrolobenzodiazepine derivative conjugate | |
| HK40054688A (en) | (anti-cdh6 antibody)-(pyrrolobenzodiazepine derivative) conjugate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210524 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230110 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20231006 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240123 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240321 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20240618 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240711 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7522097 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |