JP7536016B2 - 無制御細胞成長の阻害用の化合物 - Google Patents
無制御細胞成長の阻害用の化合物 Download PDFInfo
- Publication number
- JP7536016B2 JP7536016B2 JP2021535210A JP2021535210A JP7536016B2 JP 7536016 B2 JP7536016 B2 JP 7536016B2 JP 2021535210 A JP2021535210 A JP 2021535210A JP 2021535210 A JP2021535210 A JP 2021535210A JP 7536016 B2 JP7536016 B2 JP 7536016B2
- Authority
- JP
- Japan
- Prior art keywords
- cancer
- compound
- formula
- reaction mixture
- benzo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images




































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D407/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00
- C07D407/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings
- C07D407/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having oxygen atoms as the only ring hetero atoms, not provided for by group C07D405/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/08—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms
- C07D295/084—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms with the ring nitrogen atoms and the oxygen or sulfur atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
- C07D295/088—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms with the ring nitrogen atoms and the oxygen or sulfur atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings to an acyclic saturated chain
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/50—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to atoms of the carbocyclic ring
- C07D317/54—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Epidemiology (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Description
EとZはC、O、N、S、Nの塩(例えばN.HCl)から選択され、
QはO、S、-CH2O-、-NY’であり、Y’は-H、アルキル、SOOCH3から選択され、
R5は-H、又は-Clであり、
R6とR7は、各々独立して、-H、アルコキシ、アルキル、置換又は非置換芳香族基、-NH2、-NO2、-NHCOCH3、-CN、-O-、ハロゲン、-OCF3から選択されるか、又はR6とR7が一緒になって複素環を形成し、
R8は-H、又は-Clであり、
R10は下記式から選択され、
R11とR12は、各々独立して、-Hから選択されるか、又はR11とR12はラクトン等の置換又は非置換の5員環又は6員環、-C(O)OC2H5等の-C(O)O-アルキルとすることができ、
Rは、-NR13R14、NR13CO(CH2)nNR13R14、-NR13R14.HCl又は酸塩、-O-R13R14、-CO-R13、-NR13CO-NR13R14、-NR13R14SOO-NR13R14、任意で少なくとも1つのヘテロ原子を有する置換又は未置換のシクロアルカン、スルホンアミド、-(CH2)nNH2、-(CH2)nOH、-NR13CO-R15、(但し、R15は任意で少なくとも1つのヘテロ原子を有する、置換又は未置換の五員環又は六員環)、-CH-R16R17(R16、R17は、各々独立して、シクロアルカン又はアリールから選択され)、-O-(CH2)nR18(但し、R18は-OH、-NH2、置換又は未置換のアリール、置換又は未置換のヘテロ環基、置換又は未置換のシクロアルカン)から選択され、
但しR13又はR14は、各々独立して、-H、置換又は未置換のアルキル、アルケン、アルコキシ、置換又は未置換のアリール、ヘテロアリール基、置換又は未置換のヘテロ環基、アルキルアミン及び置換アリールアミン、アミド、スルホンアミド、-OH、-(CH2)n-O-、及び下記式:
但し、R19は-OH、-NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R20はアルコキシ、-OMe、-OH、NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R21はアルコキシ、-OMe、-OH、-H、Br、NH2、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R22は-H、-CH2OH、-OH、アルキル、アルコキシから選択され、そして
nは1~10である。)
QはO、S、-CH2O-、又は-NY’であり、Y’は-H、アルキル、SOOCH3から選択され、
R5は-H又は-Clであり、
R6とR7は、各々独立して、-H、アルコキシ、アルキル、置換又は非置換芳香族基、-NH2、-NO2、-NHCOCH3、-CN、-O-、ハロゲン、-OCF3から選択されるか、又はR6とR7が一緒になって複素環を形成し、
R8は-H又は-Clであり、
R10は下記式から選択され、
R11とR12は、各々独立して、-Hから選択されるか、或いは、R11とR12は、ラクトン等の置換又は非置換の5員環又は6員環、-C(O)OC2H5等の-C(O)O-アルキルでもよく、
Rは、-NR13R14、NR13CO(CH2)nNR13R14、-NR13R14.HCl又は酸塩、-O-R13R14、-CO-R13、-NR13CO-NR13R14、-NR13R14SOO-NR13R14、任意で少なくとも1つのヘテロ原子を有する置換又は未置換のシクロアルカン、スルホンアミド、-(CH2)nNH2、-(CH2)nOH、-NR13CO-R15、(但し、R15は任意で少なくとも1つのヘテロ原子を有する、置換又は未置換の五員環又は六員環)、-CH-R16R17(R16、R17は、各々独立して、シクロアルカン又はアリールから選択され)、-O-(CH2)nR18(但し、R18は-OH、-NH2、置換又は未置換のアリール、置換又は未置換のヘテロ環基、置換又は未置換のシクロアルカン)から選択され、
但しR13又はR14は、各々独立して、-H、置換又は未置換のアルキル、アルケン、アルコキシ、置換又は未置換のアリール、ヘテロアリール基、置換又は未置換のヘテロ環基、アルキルアミン及び置換アリールアミン、アミド、スルホンアミド、-OH、-(CH2)n-O-、及び下記式:
但し、R19は-OH、-NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R20はアルコキシ、-OMe、-OH、NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R21はアルコキシ、-OMe、-OH、-H、Br、NH2、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R22は-H、-CH2OH、-OH、アルキル、アルコキシから選択され、そして
nは1~10である。)
EとZはC、O、N、S、Nの塩(例えばN.HCl)から選択され、
QはO、S、-CH2O-、又は-NY’であり、Y’は-H、アルキル、SOOCH3から選択され、
R5は-H又は-Clであり、
R6とR7は、各々独立して、-H、アルコキシ、アルキル、置換又は非置換芳香族基、-NH2、-NO2、-NHCOCH3、-CN、-O-、ハロゲン、-OCF3から選択されるか、又はR6とR7が一緒になって複素環を形成し、
R8は-H又は-Clであり、
R10は下記式から選択され、
R11とR12は、各々独立して、-Hから選択されるか、又はR11とR12はラクトン等の置換又は非置換の5員環又は6員環、-C(O)OC2H5等の-C(O)O-アルキルとすることができ、
Rは、-NR13R14、NR13CO(CH2)nNR13R14、-NR13R14.HCl又は酸塩、-O-R13R14、-CO-R13、-NR13CO-NR13R14、-NR13R14SOO-NR13R14、任意で少なくとも1つのヘテロ原子を有する置換又は未置換のシクロアルカン、スルホンアミド、-(CH2)nNH2、-(CH2)nOH、-NR13CO-R15、(但し、R15は任意で少なくとも1つのヘテロ原子を有する、置換又は未置換の五員環又は六員環)、-CH-R16R17(R16、R17は、各々独立して、シクロアルカン又はアリールから選択され)、-O-(CH2)nR18(但し、R18は-OH、-NH2、置換又は未置換のアリール、置換又は未置換のヘテロ環基、置換又は未置換のシクロアルカン)から選択され、但しR13又はR14は、各々独立して、-H、置換又は未置換のアルキル、アルケン、アルコキシ、置換又は未置換のアリール、ヘテロアリール基、置換又は未置換のヘテロ環基、アルキルアミン及び置換アリールアミン、アミド、スルホンアミド、-OH、-(CH2)n-O-、及び下記式:
R19は-OH、-NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R20はアルコキシ、-OMe、-OH、NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R21はアルコキシ、-OMe、-OH、-H、Br、NH2、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R22は-H、-CH2OH、-OH、アルキル、アルコキシから選択され、そして
nは1~10である。)
EとZはC、O、N、S、Nの塩(例えばN.HCl)から選択され、
QはO、S、-CH2O-、又は-NY’であり、Y’は-H、アルキル、SOOCH3から選択され、
R5は-H又は-Clであり、
R6とR7は、各々独立して、-H、アルコキシ、アルキル、置換又は非置換芳香族基、-NH2、-NO2、-NHCOCH3、-CN、-O-、ハロゲン、-OCF3から選択されるか、又はR6とR7が一緒になって複素環を形成し、
R8は-H又は-Clであり、
R10は下記式から選択され、
R11とR12は、各々独立して、-H、ラクトン等の置換又は非置換の5員環又は6員環、-C(O)OC2H5等の-C(O)O-アルキルから選択され、
Rは、-NR13R14、NR13CO(CH2)nNR13R14、-NR13R14.HCl又は酸塩、-O-R13R14、-CO-R13、-NR13CO-NR13R14、-NR13R14SOO-NR13R14、任意で少なくとも1つのヘテロ原子を有する置換又は未置換のシクロアルカン、スルホンアミド、-(CH2)nNH2、-(CH2)nOH、-NR13CO-R15、(但し、R15は任意で少なくとも1つのヘテロ原子を有する、置換又は未置換の五員環又は六員環)、-CH-R16R17(R16、R17は、各々独立して、シクロアルカン又はアリールから選択され)、-O-(CH2)nR18(但し、R18は-OH、-NH2、置換又は未置換のアリール、置換又は未置換のヘテロ環基、置換又は未置換のシクロアルカン)から選択され、
但しR13又はR14は、各々独立して、-H、置換又は未置換のアルキル、アルケン、アルコキシ、置換又は未置換のアリール、ヘテロアリール基、置換又は未置換のヘテロ環基、アルキルアミン及び置換アリールアミン、アミド、スルホンアミド、-OH、-(CH2)n-O-、及び下記式:
R19は-OH、-NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R20はアルコキシ、-OMe、-OH、NH2、-NHCOCH3、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R21はアルコキシ、-OMe、-OH、-H、Br、NH2、X=F、Cl、Br、アルキル、アセチル、C3-C8アシル基から選択され、
R22は-H、-CH2OH、-OH、アルキル、アルコキシから選択され、そして
nは1~10である。)
イマチニブ、ニロチニブ、ゲフィチニブ、スニチニブ、カルフィルゾミブ、サリノスポラミドA、レチノイン酸、シスプラチン、カルボプラチン、オキサリプラチン、メクロレタミン、シクロホスファミド、クロラムブシル、イホスファミド、アザチオプリン、メルカプトプリン、ドキシフルリジン、フルオロウラシル、ゲムシタビン、メトトレキサート、チオグアニン、ビンクリスチン、ビンブラスチン、ビノレルビン、ビンデシン、ポドフィロトキシン、エトポシド、テニポシド、タフルポシド、パクリタキセル、ドセタキセル、イリノテカン、トポテカン、アムサクリン、アクチノマイシン、ドキソルビシン、ダウノルビシン、バルルビシン、イダルビシン、エピルビシン、プリカマイシン、マイトマイシン、ミトキサントロン、メルファラン、ブスルファン、カペシタビン、ペメトレキセド、エポチロン、13-シス-レチノイン酸、2-CdA、2-クロロデオキシアデノシン、5-アザシチジン、5-フルオロウラシル、5-FU、6-メルカプトプリン、6-MP、6-TG、6-チオグアニン、
スキーム1は、下記スキーム中でそれぞれ5a、5b1、5b2、5c及び6cとして表される式X、VIII、XIV、IV及びVの化合物の合成を示す。
180mlのACN中のナフトール(1)(10g、69.4mmol)の溶液(200ml)にNBS(12.36g、69.4mmol)をRTで1時間かけて添加した。反応混合物をRTで更に30分間撹拌し、TLCを用いてモニターした。反応の完了後、反応混合物を氷冷水(100ml)に注いだ。反応混合物を酢酸エチルで抽出した(100ml×3回)。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、PETエーテル中の2%酢酸エチル及び100~200メッシュサイズのシリカゲルを用いたカラムクロマトグラフィーで精製し、白色固体として4-ブロモナフタレン-1-オール(2)を得た。
純粋化合物=8.83gm.%収率=57%.
化合物2(2.0g、8.96mmol)をトルエン(40ml)に溶解した。水(7.0ml)中のNaOH(0.717gm、17.93mmol)の溶液及び3、4-(メチレンジオキシ)フェニルホウ酸(2.23g、13.45mmol)を反応混合物にRT、N2条件下で加えた。15分後にPd(PPh3)4(0.517g、0.44mmol)をRT、N2条件下で加えた。反応混合物を110℃で8時間還流させ、TLCを用いてモニターした。反応の完了後、反応混合物をRTまで冷却し、反応混合物を水(100ml)に注ぎ、酢酸エチルで抽出した(50ml×3回)。有機層を水(50ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物をカラムクロマトグラフィー(ヘキサン:酢酸エチル=90:10)で精製し、白色固体として4-(ベンゾ[d][1,3]ジオキソ-5-イル)ナフタレン-1-オール(3)を得た。
純粋化合物=1.5gm.%収率=70%.
ジブロモブタン(16.35gm、75.75mol)を化合物3(2.0g、7.57mol)、NaOH(0.606g、15.15mol)、TBAB(0.244g、0.75mol)及び水(40ml)の溶液に滴下した。反応混合物を60℃で5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水(100ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが90:10)で精製し、白色固体として5-(1-(4-ブロモブトキシ)ナフタレン-4-イル)ベンゾ[d][1,3]ジオキソール(4b)を得た。
純粋化合物=1.49gm.%収率=50%.
ジブロモヘキサン(27.72g、113.6mol)を化合物3(5.0g、18.9mol)、NaOH(1.512g、37.8mol)、TBAB(0.60g、1.89mol)及び水(250ml)の溶液に滴下した。反応混合物を50℃で5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水(100ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが90:10)で精製し、白色固体として5-(1-(6-ブロモヘキシロキシ)ナフタレン-4-イル)ベンゾ[d][1,3]ジオキソール(4c)を得た。
純粋化合物=6.035gm.%収率=75%.
4-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-1-オール(3)(0.5g、1.89mmol)を室温で乾燥DMF(10ml)に溶解した。炭酸カリウム(0.654g、4.73mmol)及びヨウ化カリウム(0.314g、1.89mmol)を反応混合物に加えた。反応混合物をRTで15分間撹拌した。4-(2-クロロエチル)モルホリン(0.528g、2.84mmol)を反応混合物に添加した。反応混合物を100℃で8時間加熱した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(2-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)エチル)モルホリン5a)を得た。
純粋化合物=0.120gm.%収率=17%.
5-(1-(4-ブロモブトキシ)ナフタレン-4-イル)ベンゾ[d][1,3]ジオキソール(4b)(0.5g、1.25mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(1.732g、12.5mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。モルホリン(1.09g、12.5mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ブチル)モルホリン(5b1)を得た。
純粋化合物=0.190gm.%収率=37%.
5-(1-(4-ブロモブトキシ)ナフタレン-4-イル)ベンゾ[d][1,3]ジオキソール(4b)(0.45g、1.13mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(1.565g、11.3mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。シクロヘキシルアミン(1.12g、11.3mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチル(100ml×3回)で抽出した。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体としてN-(4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ブチル)シクロヘキサンアミン(5b2)を得た。
純粋化合物=0.135gm.%収率=32%.
5-(1-(6-ブロモヘキシロキシ)ナフタレン-4-イル)ベンゾ[d][1,3]ジオキソール(4c)(0.4gm、0.936mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(2.58g、18.7mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。モルホリン(1.0g、9.36mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(6-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ヘキシル)モルホリン(5c)を得た。
純粋化合物=0.172gm.%収率=43%.
4-(6-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ヘキシル)モルホリン(5c)(100mg)をジクロロメタン(10ml)に溶解した。メタノール(2ml)中のHClを反応混合物に0℃で添加した。反応混合物はRTで2時間撹拌した。反応をTLCでモニターした。反応完了後、反応混合物をロータリーエバポレーターで蒸発させ、高真空下で乾燥させた。化合物を酢酸エチルで結晶化させた。
純粋化合物=85mg,%収率=75%.
スキーム2は、下記スキーム中でそれぞれ15a、15b、16a及び16bとして表される式VI、III、VII及びIXの化合物の合成を示す。
滴下漏斗、マグネチックスターラー及びストッパーを備えた三口丸底フラスコ(500mL)にベラトルムアルデヒド又は4,5-ジメトキシベンズアルデヒド(7、15g、0.090mol)と酢酸(210mL)を投入した。この溶液に酢酸(60mL)中の臭素(9.67mL)を0.5時間に亘って撹拌し続けながら滴下し、撹拌を室温で更に3時間続けた。TLC(3:7、EtOAc:ヘキサン)で確認されたように、この間に全ての出発材料が消費された。水(250mL)を反応混合物に添加し、0℃まで冷却した。沈殿した固体を濾別し、冷水で洗浄し、真空下で乾燥させて白色固体の2-ブロモ-4,5-ジメトキシベンズアルデヒド(8)を得た。
三口丸底フラスコ(250mL)にディーンスターク装置と還流凝縮器を取り付け、8(19.0g、0.07mol)、トルエン(200mL)、エチレングリコール(1.8mL、0.21mol)及び触媒量のp-トルエンスルホン酸を投入した。この反応フラスコを油浴に浸漬し、(水が全て除去されるまで)還流下で9時間(90~95℃で)加熱した。TLC(2:8、EtOAc:ヘキサン)で反応の終了を判断した後、反応混合物を室温まで冷却し、重炭酸ナトリウム溶液で中和し、酢酸エチルで抽出した(3×100mL)。全ての有機層を混合し、無水硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮した。溶離液として酢酸エチル(5~10%)のヘキサン溶液を使用したシリカゲルでのカラムクロマトグラフィーによって粗製塊を精製し、2-(2-ブロモ-4,5-ジメトキシフェニル)-1,3-ジオキソラン(9)を白色固体として得た。
火炎乾燥した三口丸底フラスコ(100mL)に窒素雰囲気下で9(1.0g、0.0034モル)と無水THF(25mL)を添加した。フラスコをドライアイス-アセトン浴で-78℃に冷却し、n-BuLi(5.3mL、0.005mol)を-78℃で撹拌しながら滴下し、15分間撹拌した。別の火炎乾燥したフラスコにピペロナール(0.517g、0.0034mol)と乾燥THF(6mL)を投入した。ピペロナール溶液を反応混合物に30分間カニューレ挿入し、添加後、反応混合物をゆっくりと室温まで温め、更に2.5時間撹拌した。TLC(5:5、EtOAc:ヘキサン)で全てのブロモ化合物が消費されたことを確認した後、飽和塩化アンモニウム溶液を添加して反応混合物の反応を停止させ、酢酸エチルで抽出した(3×20mL)。全ての有機層を混合し、無水硫酸ナトリウムで乾燥させ、濾過し、濃縮した。ヘプタンを用いた滴定によって粗生成物を精製し、(2-(1,3-ジオキソラン-2-イル)-4,5-ジメトキシフェニル)(ベンゾ[d][1,3]ジオキソール-5-イル)-メタノール(11)は次の段階に進むのに十分に純粋であった。
密閉管に11(0.30g、0.833mmol)、アセチレンジカルボン酸ジエチル(0.141g、0.833mol)、ジクロロメタン(0.4mL)及び氷酢酸(0.242mL)を投入し、混合物を140℃で1時間加熱した。TLC(5:5、EtOAc:ヘキサン)で反応の終了を判断した後、反応混合物を室温まで冷却し、ジクロロメタン(10mL)で希釈し、5%重炭酸ナトリウム溶液で洗浄し(3×10mL)、有機層を無水硫酸ナトリウムで乾燥させ、濾過し、濃縮した。EtOAc:ヘキサン(15:85)を使用したシリカゲルでのフラッシュカラムクロマトグラフィーによって粗反応塊を精製し、1-(3’,4’-メチレンジオキシフェニル)-4-ヒドロキシ-6,7-ジメトキシ-ナフタレン-2,3-ジカルボン酸ジエチル(12)を白色固体として得た。
二口丸底フラスコ(25mL)にLAH(0.032g、0.852mmol)と無水THF(4mL)を投入し、混合物を撹拌しながら0℃に冷却した。この懸濁液に12(0.200g、0.426mmol)のTHF(4mL)溶液を0℃で滴下し、同じ温度で撹拌を2時間続けた。TLC(1:9、MeOH:DCM)で反応の終了を判断した後、飽和硫酸ナトリウム溶液で反応混合物の反応を停止させ、t-ブタノールで抽出した(4×20mL)。有機層を無水硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮した。シリカゲルでのフラッシュカラムクロマトグラフィーによって粗残渣を精製し、黄色固体の9-(3’,4’-メチレンジオキシフェニル)-4-ヒドロキシ-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(13)を得た。
LC-MS(ESI)m/z:381[M+H]+
ジブロモブタン(11.36g、52.63mol)をDMSO(50ml)中の化合物13(2.0g、5.26mol)、NaOH(1.26gm、31.57mol)の溶液に滴下した。反応混合物は40℃で5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水(100ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが90:10)で精製し、白色固体として4-(4-ブロモブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(14a)を得た。
純粋化合物=1.2gm.%収率=45%.
ジブロモヘキサン(38.52g、157.8mol)をDMSO(100ml)中の化合物13(6.0g、15.78mol)、NaOH(3.8gm、95.4mol)の溶液に滴下した。反応混合物は40℃で5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(150ml×3回)。有機層を水(150ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが90:10)で精製し、白色固体として4-(6-ブロモヘキシロキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(14b)を得た。
純粋化合物=5.0gm.%収率=58%.
4-(4-ブロモブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(14a)(0.5g、0.970mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(1.341g、9.70mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。モルホリン(0.844g、9.70mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(4-モルホリノブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(15a)を得た。
純粋化合物=0.335gm.%収率=66%.
4-(6-ブロモヘキシロキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(14b)(0.3g、0.553mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(1.43g、5.53mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。ピペラジン-1-カルボン酸エチル(0.874g、5.53mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(6-(9-(ベンゾ[d][1,3]ジオキソール-5-イル)-1,3-ジヒドロ-6,7-ジメトキシ-1-オキソナフト[2,3-c]フラン-4-イルオキシ)ヘキシル)ピペラジン-1ーカルボン酸エチル(15b)を得た。
純粋化合物=0.120gm.%収率=47%.
4-(4-モルホリノブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-ジメトキシナフト[2,3-c]フラン-1(3H)-オン(15a)(100mg)をジクロロメタン(10ml)に溶解した。メタノール(2ml)中のHClを0℃で反応混合物に添加した。反応混合物はRTで2時間撹拌した。反応をTLCでモニターした。反応完了後、反応混合物をロータリーエバポレーターで蒸発させ、高真空下で乾燥させた。化合物を酢酸エチルで結晶化させた。
純粋化合物=98mg,%収率=91%.
4-(6-(9-(ベンゾ[d][1,3]ジオキソール-5-イル)-1,3-ジヒドロ-6,7-ジメトキシ-1-オキソナフト[2,3-c]フラン-4-イルオキシ)ヘキシル)ピペラジン-1ーカルボン酸エチル(15b)(73mg)をジクロロメタン(10ml)に溶解した。メタノール(2ml)中のHClを0℃で反応混合物に添加した。反応混合物はRTで2時間撹拌した。反応をTLCでモニターした。反応完了後、反応混合物をロータリーエバポレーターで蒸発させ、高真空下で乾燥させた。化合物を酢酸エチルで結晶化させた。
純粋化合物=56mg,%収率=80%.
スキーム3は、下記スキーム中で18aとして表される式XIIの化合物の合成を示す。
ナフトール(1)(10g、69.4mmol)の溶液に、180mlのACN(200ml)、NBS(12.36g、69.4mmol)を1時間かけて室温で添加した。反応混合物をRTでさらに30分間撹拌し、TLCでモニターした。反応完了後、反応混合物を氷冷水(100ml)に注いだ。反応混合物を酢酸エチルで抽出した(100ml×3回)。有機層を無水硫酸ナトリウム上で乾燥させ、減圧下で濃縮した。粗化合物をpetエーテル中の2%酢酸エチル及び100~200メッシュサイズのシリカゲルを用いたクロマトグラフィーで精製し、白色固体として4-ブロモナフタレノ-1-オール(2)を得た。
純粋化合物=8.83gm.%収率=57%.
化合物2(2.0g、8.96mmol)をトルエン(40ml)に溶解した。NaOH(0.717g、17.93mmol)の水溶液(7.0ml)及び3-メトキシフェニルボロン酸(2.04g、13.45mmol)を反応混合物にRT、N2条件下で添加した。15分後、Pd(PPh3)4(0.517g、0.44mmol)をRT、N2条件下で添加した。反応混合物を110℃で8時間還流させ、TLCでモニターした。完了後、反応混合物をRTまで冷却し、反応混合物を水(100ml)に注ぎ、酢酸エチルで抽出した(50ml×3回)。有機層を水(50ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが90:10)で精製し、白色固体として4-(3-メトキシフェニル)ナフタレン-1-オール(17)を得た。
純粋化合物=1.3gm.%収率=55%.
4-(3-メトキシフェニル)ナフタレン-1-オール(17)(0.5g、2.0mmol)を室温で乾燥DMF(20ml)に溶解した。炭酸カリウム(0.691g、5.0mmol)及びヨウ化カリウム(0.352g、2.0mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。4-(2-クロロエチル)モルホリン(0.55g、3.0mmol)を反応混合物に添加した。反応混合物を100℃で8時間加熱した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(2-(1-(3-メトキシフェニル)ナフタレン-4-イルオキシ)エチル)モルホリン(18a)を得た。
純粋化合物=0.70gm.%収率=96%.
スキーム4は、下記スキーム中で30a及び30bとして表される式XI及びXIIIの化合物の合成を示す。
滴下漏斗、マグネチックスターラー及びストッパーを備えた三口丸底フラスコ(500ml)にベンゾ[d][1,3]ジオキソール-5-カルバアルデヒド(22、17g、0.12mol)及び酢酸(130ml)を投入した。この溶液を常に撹拌しながら、酢酸(60ml)中の臭素(12.3ml)を0.5時間かけて滴下し、さらに撹拌を室温で3時間続した。この間に全ての出発材料が消費され、それをTLC(3:7、EtOAc:ヘキサン)で確認した。水(250mL)を反応混合物に添加し、0℃まで冷却した。沈殿した固体をろ過で回収し、冷水で洗浄し、真空下で乾燥させて白色固体の6-ブロモベンゾ[d][1,3]ジオキソール-5-カルバアルデヒド(23)を得た。
純粋化合物=18gm.%収率=70%.
三口丸底フラスコ(250mL)にディーンスターク装置と還流凝縮器を取り付け、23(15.0g、0.065mol)、トルエン(150ml)、エチレングリコール(10.9ml、0.196mol)及び触媒量のp-トルエンスルホン酸を投入した。この反応フラスコを油浴に浸漬し、還流下で9時間加熱(90~95℃)した(水が全て除去されるまで行った)。TLC(2:8、EtOAc:ヘキサン)で反応の終了を判断した後、反応混合物を室温まで冷却し、重炭酸ナトリウム溶液で中和し、酢酸エチルで抽出した(3×100mL)。全ての有機層を混合し、無水硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮した。溶離液として酢酸エチル(5~10%)のヘキサン溶液を使用したシリカゲルでのカラムクロマトグラフィーによって粗製塊を精製し、白色固体として5-ブロモ-6-(1,3-ジオキソールアニ-2-イル)ベンゾ[d][1,3]ジオキソール(24)を得た。
純粋化合物=17gm.%収率=87%.
火炎乾燥した三口丸底フラスコ(100mL)に窒素雰囲気下で24(15g、0.0549mole)及び無水THF(150ml)を添加した。フラスコをドライアイス-アセトン浴で-78℃に冷却し、n-BuLi(52ml、0.082mol)を-78℃で撹拌しながら滴下し、15分間撹拌した。別の火炎乾燥したフラスコにピペロナール(8.24g、0.054mol)及び乾燥THF(50ml)を投入した。ピペロナール溶液を反応混合物に30分間カニューレ挿入し、添加後、反応混合物をゆっくりと室温まで温め、更に2.5時間撹拌した。TLC(5:5、EtOAc:ヘキサン)で全てのブロモ化合物が消費されたことを確認した後、飽和塩化アンモニウム溶液を添加して反応混合物を反応停止させ、酢酸エチルで抽出した(3×20mL)。全ての有機層を混合し、無水硫酸ナトリウムで乾燥させ、濾過し、濃縮した。ヘプタンを用いた滴定によって粗生成物を精製し、(5-(1,3-ジオキソールアニ-2-イル)ベンゾ[d][1,3]ジオキソール-6-イル)(ベンゾ[d][1,3]ジオキソール-5-イル)メタノール(26)は次の段階に進むのに十分に純粋であった。
純粋化合物=18gm.%収率=90%.
密閉管に26(18g、0.052mol)、アセチリンジカルボン酸ジエチル(8.8g、0.052mol)、ジクロロメタン(500ml)及び氷酢酸(21ml)を投入し、混合物を140℃で1時間加熱した。TLC(5:5、EtOAc:ヘキサン)で反応の終了を判断した後、反応混合物を室温まで冷却し、ジクロロメタン(500ml)で希釈し、5%重炭酸ナトリウム溶液で洗浄し(3×500ml)、有機層を無水硫酸ナトリウムで乾燥させ、濾過し、濃縮した。EtOAc:ヘキサン(15:85)を使用したシリカゲルでのフラッシュカラムクロマトグラフィーによって粗反応塊を精製し、白色固体として5-(ベンゾ[d][1,3]ジオキソール-5-イル)-8-ヒドロキシナフト[2,3-d][1,3]ジオキソール-6,7-ジカルボン酸ジエチル(27)を得た。
純粋化合物=11.97gm.%収率=50.7%.
二口丸底フラスコ(25mL)にLAH(2.52g、0.066mol)及び無水THF(100ml)を投入し、混合物を撹拌しながら0℃に冷却した。この懸濁液に27(12g、0.026mol)のTHF(100ml)溶液を0℃で滴下し、同じ温度で撹拌を2時間続けた。TLC(1:9、MeOH:DCM)で反応の終了を判断した後、反応混合物を飽和硫酸ナトリウム溶液で反応停止させ、酢酸エチルで抽出した(4×200ml)。有機層を無水硫酸ナトリウムで乾燥させ、濾過し、減圧下で濃縮した。シリカゲルでのフラッシュカラムクロマトグラフィーによって粗残渣を精製し、黄色固体の9-(3’,4’-メチレンジオキシフェニル)-4-ヒドロキシ-6,7-メチレンジオキシナフト[2,3-c]フラン-l(3H)-オン(28)を得た。
純粋化合物=2.1gm.%収率=17%.
ジブロモブタン(11.86g、0.0549mol)を水(40ml)中の化合物28(2.0g、0.00549mol)、NaOH(0.439g、0.0109mol)、TBAB(0.177g、0.00054mol)の溶液に滴下した。反応混合物はRTで5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水(100ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが90:10)で精製し、白色固体として4-(4-ブロモブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-メチレンジオキシナフト[2,3-c]フラン-1(3H)-オン(29b)を得た。
純粋化合物=1.09gm.%収率=26%.
化合物28(0.5g、1.37mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(0.227g、1.64mmol)及びヨウ化カリウム(0.228g、1.37mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。4-(2-クロロエチル)モルホリン(0.383g、2.06mmol)を反応混合物に添加した。反応混合物を100℃で8時間加熱した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(2-モルフォリノエトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-メチレンジオキシナフト[2,3-c]フラン-1(3H)-オン(30a)を得た。
純粋化合物=0.08gm.%収率=13%.
4-(4-ブロモブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-メチレンジオキシナフト[2,3-c]フラン-1(3H)-オン(29b)(0.3g、0.601mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(0.830g、6.01mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。モルホリン(0.523g、6.01mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として4-(4-モルホリノブトキシ)-9-(ベンゾ[d][1,3]ジオキソール-5-イル)-6,7-メチレンジオキシナフト[2,3-c]フラン-1(3H)-オン(30b)を得た。
純粋化合物=0.3gm.%収率=95%.
スキーム5は、下記スキーム中でそれぞれ21a、21b、21c、21c1及び21c2として表される式XXI、XIX、XVIII、XX及びXXIIの化合物の合成を示す。
3(1g、3.78mmol)と、50mlのEtOH:EtOAc(1:1)混合物中の10%Pd/Cとを50℃で60~80psiの震盪水素付加装置に投入した。反応をTLCでモニターした。完了後、Pd/Cをろ過で除き、濾液を蒸発させた。得られた固体をカラムクロマトグラフィー(ヘキサン:酢酸エチル、95:5)で精製し、液体として4-(ベンゾ[d][1,3]ジオキソール-5-イル)-5,6,7,8-テトラヒドロナフタレン-1-オール(19)を得た。
純粋化合物=0.5gm.%収率=51%.
ジブロモブタン(8.05g、37.3mmol)を化合物19(1.0g、3.73mmol)、NaOH(0.23g、7.46mmol)、TBAB(0.12g、0.37mmol)及び水(50ml)の溶液に滴下した。反応混合物を35℃で4時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×1回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが95:5)で精製し、液体として5-(5-(4-ブロモブトキシ)-1,2,3,4-テトラヒドロナフタレン-8-イル)ベンゾ[d][1,3]ジオキソール(20b)を得た。
純粋化合物=1.0gm.%収率=67%.
ジブロモヘキサン(5.45g、22.3mmol)を化合物19(1.0g、3.72mmol)、NaOH(0.8g、22.3mmol)及びDMSO(50ml)の溶液に滴下した。反応混合物を40℃で2時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが95:5)で精製し、液体として5-(5-(6-ブロモヘキシロキシ)-1,2,3,4-テトラヒドロナフタレン-8-イル)ベンゾ[d][1,3]ジオキソール(20c)を得た。
純粋化合物=2.1gm.%収率=87%.
4-(ベンゾ[d][1,3]ジオキソール-5-イル)-5,6,7,8-テトラヒドロナフタレン-1-オール(19)(0.5g、1.86mmol)を乾燥DMF(10ml)に室温で溶解した。炭酸カリウム(0.654g、4.73mmol)及びヨウ化カリウム(0.309g、1.86mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。4-(2-クロロエチル)モルホリン(0.416g、2.23mmol)を反応混合物に添加した。反応混合物を100℃で8時間加熱した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウム上で乾燥させ、減圧下で濃縮させた。得られた粗化合物をカラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、液体として4-(2-(5-(ベンゾ[d][1,3]ジオキソール-5-イル)-1,2,3,4-テトラヒドロナフタレン-8-イルオキシ)エチル)モルホリン(21a)を得た。
純粋化合物=0.1gm.%収率=15%.
5-(5-(4-ブロモブトキシ)-1,2,3,4-テトラヒドロナフタレン-8-イル)ベンゾ[d][1,3]ジオキソール(20b)(1.0g、2.48mmol)を室温で乾燥DMF(50ml)に溶解した。炭酸カリウム(3.42g、24.8mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。N-メチルピペラジン(2.48g、24.8mmol)を反応混合物に添加した。反応混合物をRTで12時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが20:80)で精製した。
純粋化合物=0.4gm.%収率=38%.
5-(5-(6-ブロモヘキシロキシ)-1,2,3,4-テトラヒドロナフタレン-8-イル)ベンゾ[d][1,3]ジオキソール(20c)(0.5g、1.15mmol)を乾燥DMF(30ml)に室温で溶解した。炭酸カリウム(1.6g、11.5mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。シクロヘキシルアミン(0.511g、6.27mmol)を反応混合物に添加した。反応混合物をRTで12時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが50:50)で精製した。
純粋化合物=0.1gm.%収率=17%.
5-(5-(6-ブロモヘキシロキシ)-1,2,3,4-テトラヒドロナフタレン-8-イル)ベンゾ[d][1,3]ジオキソール(20c)(0.5g、1.15mmol)を乾燥DMF(30ml)に室温で溶解した。炭酸カリウム(0.16g、1.15mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。モルホリン(0.6g、6.95mmol)を反応混合物に添加した。反応混合物をRTで12時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが30:70)で精製した。
純粋化合物=0.242gm.%収率=47%.
5-(5-(6-ブロモヘキシロキシ)-1,2,3,4-テトラヒドロナフタレン-8-イル)ベンゾ[d][1,3]ジオキソール(20c)(0.5g、1.15mmol)を乾燥DMF(30ml)に室温で溶解した。炭酸カリウム(1.6g、11.5mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。アリルアミン(0.66g、11.5mmol)を反応混合物に添加した。反応混合物をRTで12時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが10:90)で精製した。
純粋化合物=0.2gm.%収率=43%.
スキーム6は、下記スキーム中で33aとして表される式XVの化合物の合成を示す。
4-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-1-オール(3)(4.1gm、15.5mmol)を乾燥DMF(150ml)に室温で溶解した。炭酸カリウム(4.29gm、31.1mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。4-ブロモブタン酸エチル(6.06gm、31.06mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(150ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが95:5)で精製し、白色固体として4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ブタン酸エチル(31a)を得た。
純粋化合物=5.49gm.%収率=93%.
4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ブタン酸エチル(31a)(5.0gm、13.2mmol)をRTでTHF(100ml)及び水(100ml)に溶解した。水酸化ナトリウム(1.06gm、26.45mmol)を反応混合物にRTで添加した。反応混合物をRTで12時間撹拌した。反応をTLCでモニターした。完了後、反応混合物を氷冷水に注ぎ、希釈HClで酸性にした。反応混合物を酢酸エチルで抽出した(150ml×3回)。有機層を水で洗浄した(100ml×1回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが50:50)で精製し、灰白色の固体として4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ブタン酸(32a)を得た。
純粋化合物=4.4gm.%収率=95%.
4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)ブタン酸(32a)(0.3gm、0.85mmol)をDIPEA(0.332gm、2.57mmol)及びDMF(20ml)に溶解した。モルホリン(0.089gm、1.02mmol)及びHATU(0.448gm、1.28mmol)を反応混合物にRTで添加した。反応混合物をRTで12時間撹拌した。反応をTLCでモニターした。完了後、反応混合物を水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが30:70)で精製し、灰白色の固体として4-(1-(ベンゾ[d][1,3]ジオキソール-5-イル)ナフタレン-4-イルオキシ)-1-モルホリノブタン-1-オン(33a)を得た。
純粋化合物=0.330gm.%収率=92%.
スキーム7は、下記スキーム中でそれぞれ40b及び40cとして表される式XVII及びXVIの化合物の合成を示す。
滴下漏斗、マグネチックスターラー及びガード管を備えた三口丸底フラスコ(500mL)に8-ヒドロキシキノリン(34、5g、0.0344mol)、炭酸カリウム(9.5gm、0.0688mol)及びDMF(100mL)を投入した。この溶液に臭化ベンジル(6.13mL、0.0516mol)を0.5時間に亘って攪拌し続けながら滴下し、撹拌を室温で更に12時間続けた。この間に全ての出発材料が消費されたことをTLC(3:9、EtOAc:ヘキサン)で確認した。反応混合物を冷水(250mL)に添加した。反応混合物を酢酸エチルで抽出した(3×100mL)。全ての有機層を混合し、水で洗浄した(3×100mL)。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。溶離液として酢酸エチル(5~10%)のヘキサン溶液を使用したシリカゲルのカラムクロマトグラフィーによって粗製塊を精製して、8-(ベンジルオキシ)キノリン(35)を白色固体として得た(収量=5.2gm)。
マグネチックスターラーとガード管を備えた一口丸底フラスコ(250mL)にDCM(100mL)中の8-ベンジルオキシキノリン(35、4g、0.016mol)を投入した。この溶液にN-ブロモコハク酸イミド(3.02gm、0.016mol)を10℃で0.5時間に亘って撹拌し続けながら少しずつ添加し、撹拌を室温で更に1時間続けた。この間に全ての出発材料が消費されたことをTLC(2:8、EtOAc:ヘキサン)で確認した。反応混合物を冷水(150mL)に添加した。反応混合物をジクロロメタンで抽出した(3×100mL)。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。溶離液としてヘキサン中の酢酸エチル(5~10%)を使用したシリカゲルのカラムクロマトグラフィーによって粗製塊を精製して、8-(ベンジルオキシ)-5-ブロモキノリン(36)を白色固体として得た(収量=3.9gm)。
マグネチックスターラー、凝縮器及びガード管を備えた一口丸底フラスコ(250mL)にDME(20mL)中の8-(ベンジルオキシ)-5-ブロモキノリン(36、1g、0.00318mol)と3,4(メチレンジオキシ)フェニルボロン酸(0.79gm、0.00477mol)を投入した。この溶液に水(3.2mL)に溶解した炭酸ナトリウム(0.673gm、0.00636mol)を添加した。反応混合物を室温で10分間撹拌した後、テトラキスパラジウム(0)(0.183gm、0.000159mol)を添加した。反応混合物を12時間還流させた。この間に全ての出発材料が消費されたことをTLC(3:7、EtOAc:ヘキサン)で確認した。反応混合物を水(100mL)に添加した。反応混合物を酢酸エチルで抽出した(3×100mL)。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。溶離液として酢酸エチル(10~20%)のヘキサン溶液を使用したシリカゲルでのカラムクロマトグラフィーによって粗製塊を精製して、37を白色固体として得た(収量=0.94gm)。
マグネチックスターラー、凝縮器、とガード管を備えた一口丸底フラスコ(250mL)に5-(ベンゾ[d][1,3]ジオキソール-5-イル)-8-(ベンジルオキシ)キノリン(37、3g、0.00845モル)及びエチレングリコール(55mL)を投入した。この溶液に濃塩酸(55ml)をRTで添加した。反応混合物を12時間還流させた。この間に全ての出発材料が消費されたことをTLC(3:7、EtOAc:ヘキサン)で確認した。反応混合物を氷冷水(200mL)に添加した。反応混合物を重炭酸ナトリウムで中和し、酢酸エチルで抽出した(3×100mL)。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。溶離液としてヘキサン中の酢酸エチル(5~20%)を使用したシリカゲルでのカラムクロマトグラフィーによって粗製塊を精製し、5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノリ-8-ノール(38)を灰白色固体として得た(収量=1.21gm)。
ジブロモブタン(12.22gm、56.6mmol)を化合物38(1.5g、5.66mmol)、NaOH(1.358g、33.9mmol)及びDMSO(60ml)の溶液に滴下した。反応混合物を45℃で5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが50:50)で精製し、灰白色の固体として8-(4-ブロモブトキシ)-5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノリン(39b)を得た。
純粋化合物=1.34gm.%収率=59%.
ジブロモヘキサン(1.0g、3.76mmol)を化合物38(5.5g、22.6mmol)、NaOH(0.308g、7.58mmol)、TBAB(0.121g、3.76mmol)及び水(100ml)の溶液に滴下した。反応混合物は40℃で5時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水(100ml)で洗浄した。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(ヘキサン:酢酸エチルが50:50)で精製し、粘性の油として8-(6-ブロモヘキシロキシ)-5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノリン(39c)を得た。
純粋化合物=0.475gm.%収率=30%.
8-(4-ブロモブトキシ)-5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノロン(39b)(0.4g、1.0mmol)を乾燥DMF(20ml)に室温で溶解した。炭酸カリウム(1.38g、10mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。シクロヘキシルアミン(0.991g、10mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。合わせた有機層を無水硫酸ナトリウムで乾燥させ、減圧下で濃縮した。得られた粗化合物を、カラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、灰白色固体としてN-(4-(5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノリン-8-イルオキシ)ブチル)シクロヘキサンアミン(40b)を得た。
純粋化合物=0.1gm.%収率=29%.
8-(6-ブロモヘキシロキシ)-5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノリン(39c)(0.45gm、1.05mmol)を乾燥DMF(30ml)に室温で溶解した。炭酸カリウム(1.5g、10.5mmol)を反応混合物に添加した。反応混合物をRTで15分間撹拌した。モルホリン(0.91g、10.5mmol)を反応混合物に添加した。反応混合物をRTで16時間撹拌した。反応をTLCでモニターした。反応の完了後、反応混合物を氷冷水に注ぎ、酢酸エチルで抽出した(100ml×3回)。有機層を水で洗浄した(100ml×3回)。混合した有機層を無水硫酸ナトリウム上で乾燥させ、減圧下で濃縮させた。得られた粗化合物をカラムクロマトグラフィー(メタノール:酢酸エチルが5:95)で精製し、白色固体として8-(6-モルフォリノヘキシルオキシ)-5-(ベンゾ[d][1,3]ジオキソール-5-イル)キノリン(40c)を得た。
純粋化合物=0.157gm.%収率=36%.
化合物の効率及び非毒性を決定するために、以下の試験を実施した。
1.インビトロ抗増殖アッセイ(MTTアッセイ)
MTTアッセイは細胞の代謝抑制活性を測定する簡易で高感度のアッセイである。時間に対するこの活性の増加を細胞増殖のパラメータとする。薬物による治療によってこの増加が弱まる場合、その作用は増殖阻害、細胞殺傷又はその両方の結果である。乳癌細胞系、前立腺癌細胞系及び口腔癌細胞系を使用して、本発明の化合物と標準的な細胞毒性薬(例えば、シスプラチン)を様々な濃度(1、0.1、0.01、0.001mM)で試験した。全ての細胞系を5%CO2環境の37℃のインキュベーターで培養した。化合物を濃度0.1MのDMSOに溶解した(原液)。細胞を適切なプレーティング効率で96ウェルプレートに播種した。
ソフトアガーコロニー形成アッセイは、ソフトアガーにおける足場非依存性の増殖アッセイであり、細胞の悪性形質転換を検出するための最も厳密なアッセイの1種である。このアッセイでは、悪性細胞をソフトアガー培地で1~2週間、適切な制御下で培養する。このインキュベーション期間の後、形成されたコロニーを細胞染色によって形態学的に解析するか、形成されたコロニーの数を定量化することができる。このアッセイの結果はヌードマウスに腫瘍形成細胞を注入した後に得られる結果に匹敵し、インビトロでの細胞の腫瘍形成性(癌幹細胞(CSC)の重要な特徴の1種)を試験するための「至適基準」と見なされる。
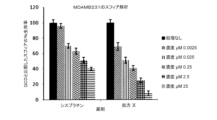
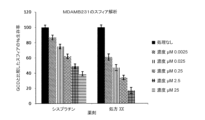
In vitroのスフィア形成アッセイ: スフィアッセイでは、特別に設計された無血清培地で癌幹細胞がスフィアを形成する能力を測定する。このアッセイを用いて、標準化学療法薬シスプラチンと比較した試験化合物の殺傷効率を測定した。
Claims (17)
- 式IVで表される化合物。

- 式Vで表される化合物。

- 式VIIIで表される化合物。

- 式Xで表される化合物。

- 式XIで表される化合物。

- 式XIIで表される化合物。

- 式XIIIで表される化合物。

- 式XIVで表される化合物。

- 式XVで表される化合物。

- 式XVIで表される化合物。

- 式XVIIで表される化合物。

- 式XVIII、XIX、XX、XXI及びXXIIからなる群より選ばれる式で表される化合物。



- 請求項1~12のいずれか一項に記載の化合物と、少なくとも1種の薬学的に許容される賦形剤と、所望により少なくとも1種の活性成分とを含む医薬組成物。
- 無制御の細胞増殖の治療又は阻害、あるいは癌幹細胞を含む癌細胞を効果的に標的化するための、請求項1~12のいずれか一項に記載の化合物。
- 前記無制御の細胞増殖が癌細胞の増殖である、請求項14に記載の化合物。
- 前記癌は、乳癌、前立腺癌、脳癌、血液癌、骨髄癌、肝臓癌、膵臓癌、皮膚癌、腎臓癌、結腸癌、卵巣癌、肺癌、精巣癌、陰茎癌、甲状腺癌、副甲状腺癌、下垂体癌、胸腺癌、網膜癌、ブドウ膜癌、結膜癌、脾臓癌、頭部癌、頸部癌、気管癌、胆嚢癌、直腸癌、唾液腺癌、副腎癌、咽頭癌、食道癌、リンパ節癌、汗腺癌、皮脂腺癌、筋肉癌、心臓癌又は胃癌である、請求項14又は15に記載の化合物。
- 前記癌は、乳癌、又は前立腺癌である、請求項16に記載の化合物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN201821047582 | 2018-12-17 | ||
| IN201821047582 | 2018-12-17 | ||
| PCT/IN2019/050926 WO2020129082A1 (en) | 2018-12-17 | 2019-12-16 | Compounds for the inhibition of unregulated cell growth |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022514329A JP2022514329A (ja) | 2022-02-10 |
| JP7536016B2 true JP7536016B2 (ja) | 2024-08-19 |
Family
ID=69770987
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021535210A Active JP7536016B2 (ja) | 2018-12-17 | 2019-12-16 | 無制御細胞成長の阻害用の化合物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US12459930B2 (ja) |
| EP (1) | EP3897592B1 (ja) |
| JP (1) | JP7536016B2 (ja) |
| CN (1) | CN113365615A (ja) |
| ES (1) | ES3050624T3 (ja) |
| WO (1) | WO2020129082A1 (ja) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111171011B (zh) * | 2020-01-23 | 2021-12-10 | 南通大学 | 一种山荷叶素杂环衍生物及其制备方法和应用 |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001514176A (ja) | 1997-08-27 | 2001-09-11 | ファイザー・プロダクツ・インク | Nos阻害剤であり縮合環置換基を含む2−アミノピリジン類 |
| JP2007505127A (ja) | 2003-09-11 | 2007-03-08 | ケミア,インコーポレイテッド | サイトカイン阻害剤 |
| WO2012081038A2 (en) | 2010-12-17 | 2012-06-21 | Godavari Biorefineries Limited | Anticancer compounds and targeting cancer with the same |
| WO2015153653A1 (en) | 2014-03-31 | 2015-10-08 | Ohio State Innovation Foundation | Arylnaphthalene lactone derivatives and methods of making and using thereof |
| WO2017126635A1 (ja) | 2016-01-22 | 2017-07-27 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| WO2017147624A1 (en) | 2016-02-26 | 2017-08-31 | Ohio State Innovation Foundation | Antitumor arylnaphthalene ligand glycosides |
| WO2018193476A2 (en) | 2017-04-20 | 2018-10-25 | Godavari Biorefineries Limited | Anticancer compounds |
| WO2019182947A1 (en) | 2018-03-19 | 2019-09-26 | Purdue Research Foundation | Arylnaphthalene compounds as vacuolar-atpase inhibitors and the use thereof |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2737721B1 (fr) * | 1995-08-08 | 1997-09-05 | Roussel Uclaf | Nouveaux composes biphenyles, leur procede de preparation et les intermediaires de ce procede, leur application a titre de medicament et les compositions pharmaceutiques les contenant |
| BRPI1008117A2 (pt) * | 2009-02-05 | 2015-08-25 | Godavari Biorefineries Ltd | Processo para a sintese de cleistantina |
| EP2726458B1 (en) | 2011-06-30 | 2017-01-04 | Godavari Biorefineries Ltd. | Synthesis of cleistanthin a and derivatives thereof |
| AU2013365649A1 (en) * | 2012-12-18 | 2015-07-30 | Godavari Biorefineries Limited | Agents for eliminating tumour-initiating cells |
| CN103467463B (zh) | 2013-09-18 | 2016-06-22 | 南开大学 | 一类木脂素类衍生物及其制备方法和用途 |
-
2019
- 2019-12-16 JP JP2021535210A patent/JP7536016B2/ja active Active
- 2019-12-16 US US17/415,676 patent/US12459930B2/en active Active
- 2019-12-16 ES ES19858686T patent/ES3050624T3/es active Active
- 2019-12-16 WO PCT/IN2019/050926 patent/WO2020129082A1/en not_active Ceased
- 2019-12-16 CN CN201980090916.1A patent/CN113365615A/zh active Pending
- 2019-12-16 EP EP19858686.9A patent/EP3897592B1/en active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001514176A (ja) | 1997-08-27 | 2001-09-11 | ファイザー・プロダクツ・インク | Nos阻害剤であり縮合環置換基を含む2−アミノピリジン類 |
| JP2007505127A (ja) | 2003-09-11 | 2007-03-08 | ケミア,インコーポレイテッド | サイトカイン阻害剤 |
| WO2012081038A2 (en) | 2010-12-17 | 2012-06-21 | Godavari Biorefineries Limited | Anticancer compounds and targeting cancer with the same |
| WO2015153653A1 (en) | 2014-03-31 | 2015-10-08 | Ohio State Innovation Foundation | Arylnaphthalene lactone derivatives and methods of making and using thereof |
| WO2017126635A1 (ja) | 2016-01-22 | 2017-07-27 | 武田薬品工業株式会社 | 複素環化合物およびその用途 |
| WO2017147624A1 (en) | 2016-02-26 | 2017-08-31 | Ohio State Innovation Foundation | Antitumor arylnaphthalene ligand glycosides |
| WO2018193476A2 (en) | 2017-04-20 | 2018-10-25 | Godavari Biorefineries Limited | Anticancer compounds |
| WO2019182947A1 (en) | 2018-03-19 | 2019-09-26 | Purdue Research Foundation | Arylnaphthalene compounds as vacuolar-atpase inhibitors and the use thereof |
Non-Patent Citations (10)
| Title |
|---|
| ACS Chemical Biology,2018年11月21日,14(1),p.20-26 |
| Australian Journal of Chemistry,1963年,16(5),p.845-53 |
| Bioorganic & Medicinal Chemistry Letters,2001年,11(13),p.1713-1716 |
| Chemical Biology & Drug Design,2016年,88(4),p.562-567 |
| ChemMedChem,2018年10月18日,13(24),p.2664-2676 |
| Chinese Journal of Organic Chemistry,2013年,33(1),p.169-173 |
| Dalton Transactions,2015年,44(1),p.401-410 |
| Dyes and Pigments,2015年,113,p.174-180 |
| Journal of Organic Chemistry,2003年,68(18),p.7101-7103 |
| Journal of Photochemistry,1982年,20(4),p.341-54 |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020129082A1 (en) | 2020-06-25 |
| JP2022514329A (ja) | 2022-02-10 |
| ES3050624T3 (en) | 2025-12-22 |
| US12459930B2 (en) | 2025-11-04 |
| EP3897592A1 (en) | 2021-10-27 |
| CN113365615A (zh) | 2021-09-07 |
| US20220073501A1 (en) | 2022-03-10 |
| EP3897592B1 (en) | 2025-09-24 |
| EP3897592C0 (en) | 2025-09-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9174969B2 (en) | Indoline scaffold SHP-2 inhibitors and cancer treatment method | |
| DK2718290T3 (en) | Compositions and Methods for Modulating a Kinase | |
| CA2854836C (en) | Tricyclic amino containing compounds for treatment or prevention of symptoms associated with endocrine dysfunction | |
| US11680078B2 (en) | Anticancer compounds | |
| CA2901155C (en) | Camkii inhibitors and uses thereof | |
| JP2016503005A (ja) | 腫瘍始原細胞を除去するための薬剤 | |
| AU2015311730A1 (en) | Camkii inhibitors and uses thereof | |
| JP7536016B2 (ja) | 無制御細胞成長の阻害用の化合物 | |
| JP2023550014A (ja) | がんの治療に使用するための5-ヒドロキシ-1,4-ナフタレンジオン | |
| JP2017506617A (ja) | 未制御細胞成長の阻害のための化合物 | |
| EA042939B1 (ru) | Ингибиторы арил-гидрокарбонового рецептора (ahr) и их применение | |
| HK1191324B (en) | Compositions and methods for modulating a kinase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20221208 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20231129 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20231219 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240314 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240416 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240711 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20240723 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240806 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7536016 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |