JP7538155B2 - Novel polymerizable liquid crystals with a quinoxaline-hydrazone core - Google Patents
Novel polymerizable liquid crystals with a quinoxaline-hydrazone core Download PDFInfo
- Publication number
- JP7538155B2 JP7538155B2 JP2021577566A JP2021577566A JP7538155B2 JP 7538155 B2 JP7538155 B2 JP 7538155B2 JP 2021577566 A JP2021577566 A JP 2021577566A JP 2021577566 A JP2021577566 A JP 2021577566A JP 7538155 B2 JP7538155 B2 JP 7538155B2
- Authority
- JP
- Japan
- Prior art keywords
- group
- carbon atoms
- substituted
- unsubstituted
- lcp
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/06—Non-steroidal liquid crystal compounds
- C09K19/34—Non-steroidal liquid crystal compounds containing at least one heterocyclic ring
- C09K19/3441—Non-steroidal liquid crystal compounds containing at least one heterocyclic ring having nitrogen as hetero atom
- C09K19/345—Non-steroidal liquid crystal compounds containing at least one heterocyclic ring having nitrogen as hetero atom the heterocyclic ring being a six-membered aromatic ring containing two nitrogen atoms
- C09K19/3452—Pyrazine
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/38—Polymers
- C09K19/3833—Polymers with mesogenic groups in the side chain
- C09K19/3842—Polyvinyl derivatives
- C09K19/3852—Poly(meth)acrylate derivatives
- C09K19/3861—Poly(meth)acrylate derivatives containing condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D241/00—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings
- C07D241/36—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings condensed with carbocyclic rings or ring systems
- C07D241/38—Heterocyclic compounds containing 1,4-diazine or hydrogenated 1,4-diazine rings condensed with carbocyclic rings or ring systems with only hydrogen or carbon atoms directly attached to the ring nitrogen atoms
- C07D241/40—Benzopyrazines
- C07D241/44—Benzopyrazines with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F122/00—Homopolymers of compounds having one or more unsaturated aliphatic radicals each having only one carbon-to-carbon double bond, and at least one being terminated by a carboxyl radical and containing at least one other carboxyl radical in the molecule; Salts, anhydrides, esters, amides, imides or nitriles thereof
- C08F122/10—Esters
- C08F122/12—Esters of phenols or saturated alcohols
- C08F122/22—Esters containing nitrogen
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D4/00—Coating compositions, e.g. paints, varnishes or lacquers, based on organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond ; Coating compositions, based on monomers of macromolecular compounds of groups C09D183/00 - C09D183/16
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/06—Non-steroidal liquid crystal compounds
- C09K19/08—Non-steroidal liquid crystal compounds containing at least two non-condensed rings
- C09K19/10—Non-steroidal liquid crystal compounds containing at least two non-condensed rings containing at least two benzene rings
- C09K19/24—Non-steroidal liquid crystal compounds containing at least two non-condensed rings containing at least two benzene rings linked by a chain containing nitrogen-to-nitrogen bonds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/06—Non-steroidal liquid crystal compounds
- C09K19/34—Non-steroidal liquid crystal compounds containing at least one heterocyclic ring
- C09K19/3441—Non-steroidal liquid crystal compounds containing at least one heterocyclic ring having nitrogen as hetero atom
- C09K19/345—Non-steroidal liquid crystal compounds containing at least one heterocyclic ring having nitrogen as hetero atom the heterocyclic ring being a six-membered aromatic ring containing two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K19/38—Polymers
- C09K19/3804—Polymers with mesogenic groups in the main chain
-
- G—PHYSICS
- G02—OPTICS
- G02F—OPTICAL DEVICES OR ARRANGEMENTS FOR THE CONTROL OF LIGHT BY MODIFICATION OF THE OPTICAL PROPERTIES OF THE MEDIA OF THE ELEMENTS INVOLVED THEREIN; NON-LINEAR OPTICS; FREQUENCY-CHANGING OF LIGHT; OPTICAL LOGIC ELEMENTS; OPTICAL ANALOGUE/DIGITAL CONVERTERS
- G02F1/00—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics
- G02F1/01—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour
- G02F1/13—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells
- G02F1/137—Devices or arrangements for the control of the intensity, colour, phase, polarisation or direction of light arriving from an independent light source, e.g. switching, gating or modulating; Non-linear optics for the control of the intensity, phase, polarisation or colour based on liquid crystals, e.g. single liquid crystal display cells characterised by the electro-optical or magneto-optical effect, e.g. field-induced phase transition, orientation effect, guest-host interaction or dynamic scattering
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K2019/0444—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit characterized by a linking chain between rings or ring systems, a bridging chain between extensive mesogenic moieties or an end chain group
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09K—MATERIALS FOR MISCELLANEOUS APPLICATIONS, NOT PROVIDED FOR ELSEWHERE
- C09K19/00—Liquid crystal materials
- C09K19/04—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit
- C09K2019/0444—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit characterized by a linking chain between rings or ring systems, a bridging chain between extensive mesogenic moieties or an end chain group
- C09K2019/0448—Liquid crystal materials characterised by the chemical structure of the liquid crystal components, e.g. by a specific unit characterized by a linking chain between rings or ring systems, a bridging chain between extensive mesogenic moieties or an end chain group the end chain group being a polymerizable end group, e.g. -Sp-P or acrylate
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Crystallography & Structural Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Physics & Mathematics (AREA)
- Nonlinear Science (AREA)
- Polymers & Plastics (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Health & Medical Sciences (AREA)
- General Physics & Mathematics (AREA)
- Optics & Photonics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Wood Science & Technology (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Polarising Elements (AREA)
- Liquid Crystal (AREA)
Description
本発明は、キノキサリン-ヒドラゾンコアを有する新規な重合性異方性液晶(LCP)化合物、そのようなLCP化合物を含む組成物、及びそのようなLCP化合物又は組成物を含む光学フィルムに関する。本発明のLCPを含む光学フィルムは、広い波長帯域にわたる偏光の逆リタデーションパターンを示した。最後に、本発明は、例えば、フラットディスプレイ、テレビ、スマートフォン、タブレットなどの、そのようなLCP化合物を含む、又はそのようなLCP化合物を含む光学フィルムを含む、光学的異方性製品に関する。 The present invention relates to novel polymerizable anisotropic liquid crystal (LCP) compounds having a quinoxaline-hydrazone core, compositions comprising such LCP compounds, and optical films comprising such LCP compounds or compositions. Optical films comprising the LCPs of the present invention exhibited reverse retardation patterns of polarized light over a wide wavelength range. Finally, the present invention relates to optically anisotropic products comprising such LCP compounds or comprising optical films comprising such LCP compounds, such as, for example, flat displays, televisions, smartphones, tablets, etc.
硬化性LCP化合物から調製された光学フィルム(LCPフィルム)は、当業者には周知であり、光学デバイスの調製に使用される。光学フィルムの例は、リタデーションフィルム又は偏光子である。リタデーションフィルムは、それを通過する光の偏光状態を変化させる一種の光学素子である。光がリタデーションフィルムを通過すると、複屈折及びフィルムの厚さのため、光の偏光方向が変化する。4分の1波長リタデーション板は、直線偏光を円偏光に変換し、半波長リタデーション板は、直線偏光の振動面を90°変換する。このようなリタデーションフィルムは、λ/4又はλ/2リタデーションが発生するように、特定の単色光の変換を達成できる。しかしながら、既知のリタデーションフィルムには、通過する偏光が着色偏光に変換されるという欠点がある。また、偏光した白色光については、各波長に対応する偏光状態分布が生じる。したがって、波長帯域全体にわたって正確なλ/4又はλ/2リタデーションを実現することは不可能である。このような欠点を改善するために、短波長よりも長波長でより大きな波長分散を有するリターダフィルムを開発する必要がある。リターダとしても知られるリタデーションフィルムの調製における別の問題は、少量の材料で高性能フィルムを調製することである。 Optical films (LCP films) prepared from curable LCP compounds are well known to those skilled in the art and are used in the preparation of optical devices. An example of an optical film is a retardation film or polarizer. A retardation film is a type of optical element that changes the polarization state of light passing through it. When light passes through a retardation film, the polarization direction of the light changes due to birefringence and the thickness of the film. A quarter-wave retardation plate converts linearly polarized light into circularly polarized light, and a half-wave retardation plate converts the vibration plane of linearly polarized light by 90°. Such retardation films can achieve the conversion of a specific monochromatic light so that λ/4 or λ/2 retardation occurs. However, known retardation films have the disadvantage that the polarized light passing through them is converted into colored polarized light. Also, for polarized white light, a polarization state distribution corresponding to each wavelength occurs. Therefore, it is impossible to achieve exact λ/4 or λ/2 retardation over the entire wavelength band. To improve such disadvantages, it is necessary to develop a retarder film with a larger wavelength dispersion at longer wavelengths than at shorter wavelengths. Another problem in preparing retardation films, also known as retarders, is preparing high-performance films with small amounts of material.
したがって、上記のような光学フィルムの調製に使用できる新規なLCP化合物が必要であり、これにより前述の欠点が大幅に軽減される。本発明はその必要性に取り組む。 Therefore, there is a need for new LCP compounds that can be used to prepare such optical films, which significantly reduce the aforementioned disadvantages. The present invention addresses that need.
幾つかの異方性LCP化合物が当技術分野において既知であるが、より広い波長にわたって偏光の均一な変換が改善された新規なLCP化合物を開発する要求が依然として存在する。そのような異方性LCP化合物の幾つかの例は、WO2012/147904、WO2016/104317、WO2017/043437及びJP2016/128403に開示されている。 Although several anisotropic LCP compounds are known in the art, there remains a need to develop new LCP compounds with improved uniform conversion of polarized light over a wider range of wavelengths. Some examples of such anisotropic LCP compounds are disclosed in WO2012/147904, WO2016/104317, WO2017/043437 and JP2016/128403.
LCPフィルムは、一般に、当業者には周知の方法で製造される。これは、架橋可能なLCP又はLCP混合物の有機溶液を、配向層を備えた基板上、又は前もってラビング法によって処理された基板上にコーティングすることを伴う。あるいは液晶用の他のアライニング法を使用してもよい。続いて、有機溶媒を除去することにより、配向性が高く、無溶媒のLCP層を与え、これが次に架橋されて、液晶特性の秩序構造を固定する。このようなフィルムの望ましい光学性能は、異方性LCP材料が同時に満たさなければならない幾つかの物理パラメーターに決定的に依存する。このような特性とは、低融点又は融点未満に冷却されたとき(過冷却)の低い結晶化傾向、有機溶媒への良好な溶解性、他のLCPとの良好な混和性、配向層での良好なアライニング特性、及び本質的に傾斜ドメイン及び回位のない基板面からの調整可能な傾斜の形成能である。傾斜ドメインとは、LCP分子の長軸が、同じサイズで反対方向の基板面から傾斜角を形成する、LCPフィルム内の領域である。回位とは、反対の傾斜角のLCP分子が近接する隣接傾斜ドメインの境界線である。これらの傾斜ドメイン及び回位は、フィルムの均一な外観の乱れと不均質な光学性能との両方をもたらす。 LCP films are generally produced by methods well known to those skilled in the art. It involves coating an organic solution of a crosslinkable LCP or LCP mixture onto a substrate with an alignment layer or onto a substrate previously treated by a rubbing method. Alternatively, other aligning methods for liquid crystals may be used. The organic solvent is subsequently removed to give a highly oriented, solvent-free LCP layer, which is then crosslinked to fix the ordered structure of liquid crystal properties. The desired optical performance of such films crucially depends on several physical parameters that the anisotropic LCP material must simultaneously satisfy: a low melting point or a low tendency to crystallize when cooled below the melting point (supercooling), good solubility in organic solvents, good miscibility with other LCPs, good aligning properties in the alignment layer, and the ability to form essentially tilted domains and tunable tilts from the substrate plane without disclinations. Tilted domains are regions in the LCP film where the long axes of the LCP molecules form tilt angles from the substrate plane of the same size and opposite directions. Disclinations are the boundaries between adjacent tilt domains where LCP molecules with opposite tilt angles are in close proximity. These tilt domains and disclinations cause both disruptions to the uniform appearance of the film and inhomogeneous optical performance.
発明の要約
本発明の第1の目的は、式(I)により記述される異方性LCP化合物、並びに少なくとも1種の前記化合物と少なくとも1種の添加剤及び/又は溶媒とを含む組成物を提供することである。
SUMMARY OF THEINVENTION It is a first object of the present invention to provide anisotropic LCP compounds described by formula (I), as well as compositions comprising at least one said compound and at least one additive and/or solvent.
本発明の更なる目的は、少なくとも1種の前記異方性LCP化合物を含む光学フィルム及びその調製方法、偏光の均一な変換を達成するリタデーションフィルムとしての前記光学フィルムの使用、並びに前記光学フィルムを含むデバイス及びその製造法を提供することである。 A further object of the present invention is to provide an optical film comprising at least one of said anisotropic LCP compounds and a method for preparing the same, the use of said optical film as a retardation film to achieve uniform transformation of polarized light, and a device comprising said optical film and a method for manufacturing the same.
発明の詳細な説明
本発明の第1の態様は、式(I)の化合物を提供する。

式(I)の化合物において、R1、R2及びR3は、互いに独立して、水素、C1~C12直鎖又は分岐のアルキル鎖、C3~C12アルケニル、C1~C12アルコキシ、C3~C12アルケニルオキシ、-(CH2)m-C(CH3)3、NO2、CN、COR、-COOR、-OCOR、-CONR’R、-NR’COR、OCOOR、-OCONR’R、-NR’COOR、-F、-Cl、-CF3及び-OCF3からなる群より選択され;
ここで、
mは、0~12の整数であり;
Rは、水素、C1~18アルキル基、3位以上に二重結合を持つC3~18アルケニル基、-(CH2)p-C-(CF3)3、CN及び非置換又は置換のフェニル環からなる群より選択され、ここで、フェニル環の置換基は、C1~C6直鎖又は分岐のアルキル鎖、C1~C6アルコキシ、-C-(CH3)3、ハロゲン、-CF3、NO2、CN、COR'''、-COOR'''、-OCOR'''、-CONR''R'''、-NR''COR'''、OCOOR'''、-OCONR''R'''、-NR''COOR'''、-F、-Cl、-CF3及び-OCF3からなる群より選択され;
ここで、
R''は、水素、低級アルキル基及び低級アルケニル基からなる群より選択され;
R'''は、水素、C1~18アルキル基及び3位以上に二重結合を持つC3~18アルケニル基からなる群より選択され;
pは、0~12の整数であり;
R’は、水素、低級アルキル、低級アルケニル及び低級アルコキシからなる群より選択され;そして
ここで、
nは、0、1、2、又は3である。
In the compounds of formula (I), R 1 , R 2 and R 3 are each independently selected from the group consisting of hydrogen, C 1 -C 12 straight or branched alkyl chain, C 3 -C 12 alkenyl, C 1 -C 12 alkoxy, C 3 -C 12 alkenyloxy, -(CH 2 ) m -C(CH 3 ) 3 , NO 2 , CN, COR, -COOR, -OCOR, -CONR'R, -NR'COR, OCOOR, -OCONR'R, -NR'COOR, -F, -Cl, -CF 3 and -OCF 3 ;
Where:
m is an integer from 0 to 12;
R is selected from the group consisting of hydrogen, a C 1-18 alkyl group, a C 3-18 alkenyl group having a double bond at position 3 or higher, -(CH 2 ) p -C-(CF 3 ) 3 , CN, and an unsubstituted or substituted phenyl ring, where the substituents on the phenyl ring are selected from the group consisting of a C 1 -C 6 straight or branched alkyl chain, a C 1 -C 6 alkoxy, -C-(CH 3 ) 3 , halogen, -CF 3 , NO 2 , CN, COR''', -COOR'''', -OCOR'''', -CONR''R'''', -NR''COR'''', OCOOR'''', -OCONR''R'''', -NR''COOR'''', -F, -Cl, -CF 3 , and -OCF 3 ;
Where:
R″ is selected from the group consisting of hydrogen, lower alkyl, and lower alkenyl;
R''' is selected from the group consisting of hydrogen, a C1-18 alkyl group, and a C3-18 alkenyl group having a double bond at the 3rd or higher position;
p is an integer from 0 to 12;
R' is selected from the group consisting of hydrogen, lower alkyl, lower alkenyl, and lower alkoxy; and
n is 0, 1, 2, or 3.
好ましくは、R1、R2及びR3は、互いに独立して、水素、低級アルキル、低級アルケニル、低級アルコキシ、低級アルケニルオキシ、-F及び-CF3からなる群より選択される。 Preferably, R 1 , R 2 and R 3 are each independently selected from the group consisting of hydrogen, lower alkyl, lower alkenyl, lower alkoxy, lower alkenyloxy, -F and -CF 3 .
最も好ましくは、R1、R2及びR3は、互いに独立して、水素、メチル、メトキシ、-F、-C-(CH3)3及び-CF3からなる群より選択される。 Most preferably, R 1 , R 2 and R 3 are independently selected from the group consisting of hydrogen, methyl, methoxy, -F, -C-(CH 3 ) 3 and -CF 3 .
Yは、H、又は1~12個の炭素原子を有する置換若しくは非置換のアルキル基からなる群より選択される。 Y is selected from the group consisting of H or a substituted or unsubstituted alkyl group having 1 to 12 carbon atoms.
Zは、水素、1~20個の炭素原子を有する置換又は非置換のアルキル基、2~20個の炭素原子を有する置換又は非置換のアルケニル基、3~12個の炭素原子を有する置換又は非置換のシクロアルキル基、2~20個の炭素原子を有する置換又は非置換のアルキニル基からなる群より選択されるが、ここで、1個以上の炭素原子は、-O-、-COO-、-OCO-、-OOC-、-O(CO)O-、-N-、-NRa-、-CON-、-CO-Rb、-NH-Rcにより置き換えられていてもよく、ここで、Raは、C1~C12アルキル基であり、Rb及びRcは、互いに独立して、1~20個の炭素原子を有する置換若しくは非置換のアルキル基、又は少なくとも1個の芳香環を包含する2~30個の炭素原子を有する有機基、又は2~20個の炭素原子を有する置換若しくは非置換のアルケニル基、又は3~12個の炭素原子を有する置換若しくは非置換のシクロアルキル基である。 Z is selected from the group consisting of hydrogen, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, a substituted or unsubstituted alkenyl group having 2 to 20 carbon atoms, a substituted or unsubstituted cycloalkyl group having 3 to 12 carbon atoms, a substituted or unsubstituted alkynyl group having 2 to 20 carbon atoms, where one or more carbon atoms may be replaced by -O-, -COO-, -OCO-, -OOC-, -O(CO)O-, -N-, -NR a -, -CON-, -CO-R b , -NH-R c , where R a is a C 1 to C 12 alkyl group, R b and R c are each independently a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, or an organic group having 2 to 30 carbon atoms containing at least one aromatic ring, or a substituted or unsubstituted alkenyl group having 2 to 20 carbon atoms, or a substituted or unsubstituted cycloalkyl group having 3 to 12 carbon atoms.
好ましくは、Zは、水素及び1~12個の炭素原子を有する置換又は非置換のアルキル基からなる群より選択される。 Preferably, Z is selected from the group consisting of hydrogen and substituted or unsubstituted alkyl groups having 1 to 12 carbon atoms.
環C及びDは、互いに独立して、フェニル、ビフェニル、ナフチル、シクロアルキル、好ましくはシクロヘキシル、ビシクロアルキル、好ましくはビシクロヘキシル、下記式:

で示される基からなる群より選択される(ただし、環C又はDの少なくとも一方は、芳香環を含有する)。
Rings C and D are each independently phenyl, biphenyl, naphthyl, cycloalkyl, preferably cyclohexyl, bicycloalkyl, preferably bicyclohexyl, the following formula:
(with the proviso that at least one of rings C and D contains an aromatic ring).
好ましくは、環C及びDは、互いに独立してフェニル又はシクロヘキシルであり、最も好ましくは、環C及びDの両方は、フェニル環である。 Preferably, rings C and D are each independently phenyl or cyclohexyl, and most preferably, both rings C and D are phenyl rings.
環Eは、フェニル、ビフェニル及びナフチルからなる群より選択される。 Ring E is selected from the group consisting of phenyl, biphenyl and naphthyl.
好ましくは、環Eは、フェニル環である。 Preferably, ring E is a phenyl ring.
置換基X1及びX2は、互いに独立して、水素、C1~C12の置換又は非置換の直鎖又は分岐のアルキル鎖、C3~C12の置換又は非置換の直鎖又は分岐のアルケニル鎖及びC1~C12アルコキシからなる群より選択されるが、ここで、1個以上の炭素原子は、-O-、-COO-、-OCO-、-OOC-、-O(CO)O-、-N-、-NRa-、-CON-により置き換えられていてもよく、ここで、Raは、C1~C12アルキル基であるか;あるいは置換基X1及びX2は、互いに独立して、式(II):
![]()
で示される基により表される。
The substituents X 1 and X 2 are, independently of one another, selected from the group consisting of hydrogen, a C 1 -C 12 substituted or unsubstituted linear or branched alkyl chain, a C 3 -C 12 substituted or unsubstituted linear or branched alkenyl chain and a C 1 -C 12 alkoxy, in which one or more carbon atoms may be replaced by -O-, -COO-, -OCO-, -OOC-, -O(CO)O-, -N-, -NR a -, -CON-, in which R a is a C 1 -C 12 alkyl group; or the substituents X 1 and X 2 are, independently of one another, selected from the group consisting of hydrogen, a C 1 -C 12 substituted or unsubstituted linear or branched alkyl chain, a C 3 -C 12 substituted or unsubstituted linear or branched alkenyl chain and a C 1 -C 12 alkoxy, in which one or more carbon atoms may be replaced by -O-, -COO- , -OCO-, -OOC-, -O(CO)O-, -N-, -NR a -, -CON-, in which R a is a C 1 -C 12 alkyl group;
![]()
The compound is represented by the group represented by the formula:
式(II)の基において、nは、0~24の整数であるが、ここで、1個以上の炭素原子は、-O-、-COO-、-OCO-、-OOC-、-O(CO)O-、-N-、-NRa-、-CON-により置き換えられていてもよく、ここで、Raは、C1~C12アルキル基である。 In the group of formula (II), n is an integer from 0 to 24, wherein one or more carbon atoms may be replaced by -O-, -COO-, -OCO-, -OOC-, -O(CO)O-, -N-, -NR a -, -CON-, where R a is a C 1 to C 12 alkyl group.
式(II)の基の置換基PGは、CH2=C(Ph)-、CH2=CW-COO-、CH2=CH-COO-Ph-、CH2=CW-CO-NH-、CH2=CH-O-、CH2=CH-OOC-、Ph-CH=CH-、CH2=CH-Ph-、CH2=CH-Ph-O-、Rb-Ph-CH=CH-COO-、Rb-OOC-CH=CH-Ph-O-及び2-W-エポキシエチルからなる群より選択される重合性基を表すが;ここで、Wは、H、Cl、Ph又は低級アルキルを表し、そしてRbは、低級アルキルを表すが、ただし、Rbは、フェニレン基(-Ph-)に結合しているとき、水素又は低級アルコキシをも表してよい。好ましくは、PGは、アクリラート又はメタクリラート基を表す。 The substituent PG in the radical of formula (II) represents a polymerizable group selected from the group consisting of CH 2 ═C(Ph)-, CH 2 ═CW-COO-, CH 2 ═CH-COO-Ph-, CH 2 ═CW-CO-NH-, CH 2 ═CH-O-, CH 2 ═CH-OOC-, Ph-CH═CH-, CH 2 ═CH-Ph-, CH 2 ═CH-Ph-O-, R b -Ph-CH═CH-COO-, R b -OOC-CH═CH-Ph-O- and 2-W-epoxyethyl; where W represents H, Cl, Ph or lower alkyl and R b represents lower alkyl, with the proviso that R b , when attached to the phenylene group (-Ph-), may also represent hydrogen or lower alkoxy. Preferably, PG represents an acrylate or methacrylate group.
環C及びDが、互いに独立してシクロヘキシルであるか、又はシクロヘキシルを含有するとき、好ましくは、置換基X1及びX2は、水素、C1~C12の置換又は非置換の直鎖又は分岐のアルキル鎖、C3~C12の置換又は非置換の直鎖又は分岐のアルケニル鎖及びC1~C12アルコキシからなる群より選択される。 When rings C and D are, independently of one another, cyclohexyl or contain cyclohexyl, preferably the substituents X1 and X2 are selected from the group consisting of hydrogen, C1 - C12 substituted or unsubstituted linear or branched alkyl chains, C3 - C12 substituted or unsubstituted linear or branched alkenyl chains and C1 - C12 alkoxy.
好ましくは、環C又はDが、互いに独立して芳香環であるか、又は芳香環を含有する場合、より好ましくは、C又はDが、互いに独立してフェニル環であるか、又はフェニル環を含有する場合、式(II)の基は、下記式:

[式中、nは、上記と同じ意味を有する]で示される基か、又はそれらの対応するメタクリラートから選択される。
Preferably, when rings C or D are, independently of one another, aromatic rings or contain an aromatic ring, more preferably when C or D are, independently of one another, phenyl rings or contain a phenyl ring, the group of formula (II) is
wherein n has the same meaning as above, or the corresponding methacrylates thereof.
「低級アルキル」という用語は、C1~6のアキラルの分岐又は直鎖のアルキル基を包含するものと理解されるべきである。本発明の化合物中に存在し得る低級アルキル基の例は、メチル、エチル、プロピル、ブチル、ペンチル、ヘキシルなどを包含する。 The term "lower alkyl" should be understood to include C 1-6 achiral branched or straight chain alkyl groups. Examples of lower alkyl groups that may be present in the compounds of the invention include methyl, ethyl, propyl, butyl, pentyl, hexyl, and the like.
「低級アルケニル」という用語は、二重結合が2位以上にある、C3~6のアキラルの分岐又は直鎖のアルケニル基を包含するものと理解されるべきである。本発明の化合物中に存在し得る低級アルケニル基の例は、2-プロペニル、3-ブテニル、3-イソペンテニル、4-ペンテニル、5-ヘキセニル、4-イソヘキセニルなどを包含する。 The term "lower alkenyl" should be understood to include C 3-6 achiral branched or straight chain alkenyl groups in which the double bond is at position 2 or higher. Examples of lower alkenyl groups which may be present in the compounds of the invention include 2-propenyl, 3-butenyl, 3-isopentenyl, 4-pentenyl, 5-hexenyl, 4-isohexenyl, and the like.
「低級アルコキシ」という用語は、C1~6のアキラルの分岐又は直鎖のアルコキシ基を包含するものと理解されるべきである。本発明の化合物中に存在し得る低級アルコキシ基の例は、メトキシ、エトキシ、プロポキシ、ブトキシ、ペントキシ、ヘキソキシなどを包含する。 The term "lower alkoxy" should be understood to include C 1-6 achiral branched or straight chain alkoxy groups. Examples of lower alkoxy groups that may be present in the compounds of the invention include methoxy, ethoxy, propoxy, butoxy, pentoxy, hexoxy, and the like.
「低級アルケニルオキシ」という用語は、二重結合が2位以上にある、C3~6のアキラルの分岐又は直鎖のアルケニルオキシ基を包含するものと理解されるべきである。本発明の化合物中に存在し得る低級アルケニルオキシ基の例は、2-プロペニルオキシ、3-ブテニルオキシ、4-ペンテニルオキシ、5-ヘキセニルオキシなどを包含する。 The term "lower alkenyloxy" should be understood to include C 3-6 achiral branched or straight chain alkenyloxy groups in which the double bond is at position 2 or higher. Examples of lower alkenyloxy groups which may be present in the compounds of the invention include 2-propenyloxy, 3-butenyloxy, 4-pentenyloxy, 5-hexenyloxy, and the like.
本発明の重合性異方性LCP化合物は、当業者には周知の手順を用いて容易に調製され
得るが、僅かな限定されない手順が実施例に提供されている。
The polymerizable anisotropic LCP compounds of the present invention can be readily prepared using procedures well known to those skilled in the art, a few non-limiting procedures being provided in the examples.
出発物質は、市販されているか、又は容易に調製され得るもので、当業者には周知のものである。 The starting materials are commercially available or can be readily prepared and are well known to those skilled in the art.
本出願に関連して使用される重合性異方性LCP化合物材料とは、液晶モノマー及び/又は液晶オリゴマー及び/又は液晶ポリマー及び/又は架橋液晶を含む、液晶材料を意味するものとする。液晶材料が液晶モノマーを含む場合、そのようなモノマーは、典型的には、例えば、配向層との接触に起因して又はラビングによって、LCP材料に異方性が生じた後に重合され得る。重合は、熱処理によって、及び/又は化学線(好ましくはUV光を含む)への曝露によって開始され得る。異方性LCP材料は、単一種の液晶化合物のみを含んでもよいが、また追加の重合性及び/又は非重合性の化合物を含んでもよく、ここで、全ての化合物が液晶化合物である必要はない。光学フィルムの場合、LCPモノマーは、光配向層の上に又はラビング処理表面の上に適用される。光配向層の又はラビング処理表面のアラインメント情報がLCPモノマーに転移された後、LCP材料を固化するために、モノマーが重合及び/又は架橋される。本発明の重合又は架橋ポリマーは、式(I)の異方性LCP化合物のみを単独で含有してもよく、この場合、ポリマーはホモポリマーであるか、あるいは重合又は架橋ポリマーは更に異なるモノマーを含有してもよく、この場合、ポリマーはコポリマーであることが理解される。更に異なるモノマーは、LCP特性を有していても有していなくともよい。 Polymerizable anisotropic LCP compound material as used in the context of this application shall mean a liquid crystal material comprising liquid crystal monomers and/or liquid crystal oligomers and/or liquid crystal polymers and/or crosslinked liquid crystals. When the liquid crystal material comprises liquid crystal monomers, such monomers can typically be polymerized after the LCP material is rendered anisotropic, for example, due to contact with an alignment layer or by rubbing. Polymerization can be initiated by heat treatment and/or exposure to actinic radiation (preferably including UV light). Anisotropic LCP materials may comprise only a single type of liquid crystal compound, but may also comprise additional polymerizable and/or non-polymerizable compounds, where not all compounds need to be liquid crystal compounds. In the case of optical films, the LCP monomers are applied on top of a photoalignment layer or on top of a rubbed surface. After the alignment information of the photoalignment layer or of the rubbed surface is transferred to the LCP monomers, the monomers are polymerized and/or crosslinked to solidify the LCP material. It is understood that the polymerized or crosslinked polymer of the present invention may contain only the anisotropic LCP compound of formula (I) alone, in which case the polymer is a homopolymer, or the polymerized or crosslinked polymer may contain further different monomers, in which case the polymer is a copolymer. The further different monomers may or may not have LCP properties.
本発明の異方性LCP化合物は、従来技術のLCP化合物について以前に記載された欠点を克服する。更に、本発明の異方性LCP化合物は、優れた溶解性及び低温加工性を有する。 The anisotropic LCP compounds of the present invention overcome the disadvantages previously described for prior art LCP compounds. Furthermore, the anisotropic LCP compounds of the present invention have excellent solubility and low temperature processability.
本発明の更なる目的は、式(I)によって記述される異方性LCP化合物及び少なくとも1種の添加剤を含む組成物に関する。添加剤は、以下から選択することができる:酸化防止剤、光開始剤などの開始剤、促進剤、染料、阻害剤、活性剤、充填剤、連鎖移動阻害剤、顔料、帯電防止剤、難燃剤、増粘剤、チキソトロピー剤、界面活性剤、粘度調整剤、伸展油、可塑剤、粘着付与剤、触媒、増感剤、安定剤、平滑剤、分散剤、ポリマー結合剤及び/又は重合によってポリマー結合剤に変換できるモノマー化合物、又はエマルションコーティング及び印刷インクの場合には、分散助剤、疎水化剤、接着剤、流動性向上剤、消泡剤、脱気剤、希釈剤、助剤、着色剤、染料及び顔料、硬化防止剤、キラル添加剤、等方性又は異方性の蛍光及び/又は非蛍光染料、特に二色性染料。このような液晶組成物の調製に使用され得る溶媒は、アセトン、シクロペンタノン(CP)、シクロヘキサノン(CH)、メチルイソブチルケトン(MIBK)、メチルエチルケトン(MEK)、N,N-ジメチルホルムアミド(DMF)、N-メチルピロリドン(NMP)、N-エチルピロリドン、N-ビニルピロリドン、N,N-ジメチルアセトアミド、(AN)、テトラヒドロフラン(THF)、1,3-ジオキソラン(DXG)、エチレングリコール、ジプロピレングリコール、ブチルカルビトール、エチルカルビトールアセタート、ジプロピレングリコールモノメチルエーテル、酢酸エチル(EA)、1-メトキシ-2-プロパノールアセタート(MPA)、γ-ブチロラクトン(BL)、プロピレングリコールモノアセタート、プロピレングリコールジアセタート、ジプロピレングリコールモノメチルエーテル、ジメチルスルホキシド(DMSO)を包含するが、これらに限定されない。最も好ましい溶媒は、シクロペンタノン(CP)、シクロヘキサノン(CH)、メチルイソブチルケトン(MIBK)、メチルエチルケトン(MEK)、酢酸エチル(EA)、1-メトキシ-2-プロパノールアセタート(MPA)、1,3-ジオキソラン(DXG)、ジメチルスルホキシド(DMSO)である。 A further object of the present invention relates to a composition comprising an anisotropic LCP compound described by formula (I) and at least one additive. The additive can be selected from: antioxidants, initiators such as photoinitiators, accelerators, dyes, inhibitors, activators, fillers, chain transfer inhibitors, pigments, antistatic agents, flame retardants, thickeners, thixotropic agents, surfactants, viscosity modifiers, extender oils, plasticizers, tackifiers, catalysts, sensitizers, stabilizers, leveling agents, dispersants, polymeric binders and/or monomeric compounds that can be converted into polymeric binders by polymerization, or in the case of emulsion coatings and printing inks, dispersing aids, hydrophobizing agents, adhesives, flow improvers, defoamers, degassing agents, diluents, auxiliaries, colorants, dyes and pigments, hardening inhibitors, chiral additives, isotropic or anisotropic fluorescent and/or non-fluorescent dyes, in particular dichroic dyes. Solvents that can be used in the preparation of such liquid crystal compositions include, but are not limited to, acetone, cyclopentanone (CP), cyclohexanone (CH), methyl isobutyl ketone (MIBK), methyl ethyl ketone (MEK), N,N-dimethylformamide (DMF), N-methylpyrrolidone (NMP), N-ethylpyrrolidone, N-vinylpyrrolidone, N,N-dimethylacetamide, (AN), tetrahydrofuran (THF), 1,3-dioxolane (DXG), ethylene glycol, dipropylene glycol, butyl carbitol, ethyl carbitol acetate, dipropylene glycol monomethyl ether, ethyl acetate (EA), 1-methoxy-2-propanol acetate (MPA), γ-butyrolactone (BL), propylene glycol monoacetate, propylene glycol diacetate, dipropylene glycol monomethyl ether, and dimethyl sulfoxide (DMSO). The most preferred solvents are cyclopentanone (CP), cyclohexanone (CH), methyl isobutyl ketone (MIBK), methyl ethyl ketone (MEK), ethyl acetate (EA), 1-methoxy-2-propanol acetate (MPA), 1,3-dioxolane (DXG), and dimethyl sulfoxide (DMSO).
本発明の更なる好ましい目的は、式(I)の異方性LCP化合物、LCP混合物、並びにシクロヘキサノン、トルエン及びシクロペンタノンから選択される少なくとも1種の溶媒;並びに/又は添加剤を含む組成物に関する。 A further preferred object of the present invention relates to a composition comprising an anisotropic LCP compound of formula (I), an LCP mixture, and at least one solvent selected from cyclohexanone, toluene and cyclopentanone; and/or an additive.
本発明の更なる好ましい実施態様は、式(I)の異方性化合物を含む組成物、LCP混合物であり、これは、好ましくは、結晶化なしに3か月を超える貯蔵寿命安定性を有する。 A further preferred embodiment of the present invention is a composition, an LCP mixture, comprising an anisotropic compound of formula (I), which preferably has a shelf life stability of more than 3 months without crystallization.
本発明は、本発明の異方性LCP化合物又は組成物のうちの少なくとも一方を含む光学フィルムに関する。光学フィルムの一例は、本発明の光学フィルムを偏光子と組合せることによって製造される反射防止フィルムである。 The present invention relates to an optical film comprising at least one of the anisotropic LCP compounds or compositions of the present invention. One example of an optical film is an anti-reflective film produced by combining the optical film of the present invention with a polarizer.
異方性LCP化合物又は異方性LCP化合物を含む組成物は、支持体上に適用することができる。支持体は、剛性であっても柔軟であってもよく、任意の形態又は外形を有することができる。原則として、それは任意の材料で構成されていてよい。好ましくは、支持体は、プラスチック、ガラス若しくは金属を含むか、又はシリコンウェーハである。支持体が柔軟である場合、支持体はプラスチック又は金属箔であることが好ましい。好ましくは、支持体の表面は平坦である。幾つかの用途では、支持体は、マイクロレンズ若しくはマイクロプリズムのような微細構造などのトポグラフィー表面構造、又は矩形構造などの外形の急激な変化を示す構造を含んでいてもよい。好ましくは、支持体は透明である。支持体はまた、本発明の異方性LCP化合物でコーティングする前に処理を受けていてもよい。 The anisotropic LCP compound or a composition comprising an anisotropic LCP compound can be applied onto a support. The support can be rigid or flexible and can have any form or geometry. In principle, it can be made of any material. Preferably, the support comprises plastic, glass or metal, or is a silicon wafer. If the support is flexible, it is preferred that the support is a plastic or metal foil. Preferably, the surface of the support is flat. In some applications, the support may include topographical surface structures, such as microstructures like microlenses or microprisms, or structures that exhibit abrupt changes in geometry, such as rectangular structures. Preferably, the support is transparent. The support may also be subjected to a treatment before coating with the anisotropic LCP compound of the present invention.
支持体は、異方性LCP化合物又は異方性LCP化合物を含む組成物の堆積中に移動してもよい。例えば、LCP混合物の層は、好ましくはプラスチック又は金属である移動する柔軟な箔上に材料組成物を堆積させることによって、連続ロールツーロールプロセスで製造され得る。次に、得られたフィルムを支持箔と一緒にロールに巻いてもよいし、又はフィルムを支持体から解放してもよく、次に支持体なしで独立フィルムとして巻く。 The support may move during deposition of the anisotropic LCP compound or a composition comprising an anisotropic LCP compound. For example, a layer of an LCP mixture may be produced in a continuous roll-to-roll process by depositing the material composition onto a moving flexible foil, preferably plastic or metal. The resulting film may then be wound into a roll together with the support foil, or the film may be released from the support and then wound as a free-standing film without the support.
支持体は、光配向層、有機層、絶縁層又は金属層などの追加の層を有してよい。層は異なる機能を有することができ、例えば、有機層は、コーティングされる材料の支持体との適合性を高めるプライマー層としてコーティングすることができる。金属層は、例えば、ディスプレイなどの電気光学デバイスで使用されるときに電極として使用され得るか、又は反射体としての機能を有することができる。支持体はまた、例えば、薄膜トランジスタ、電極又はカラーフィルターを含み得る、LCD用の基板などの特定の機能を有する光学素子又はデバイスであってもよい。別の例では、支持体は、OLED層構造を含むデバイスである。支持体はまた、偏光フィルム又はシート偏光子などの偏光子、市販のVikuity(商標)DBEFフィルムなどの反射偏光子であることができる。 The support may have additional layers such as photoalignment layers, organic layers, insulating layers or metal layers. The layers may have different functions, for example an organic layer may be coated as a primer layer to enhance the compatibility of the coated material with the support. The metal layer may be used as an electrode, for example when used in an electro-optical device such as a display, or may have the function of a reflector. The support may also be an optical element or device with a specific function, such as a substrate for an LCD, which may include thin film transistors, electrodes or color filters. In another example, the support is a device that includes an OLED layer structure. The support may also be a polarizer, such as a polarizing film or sheet polarizer, a reflective polarizer such as the commercially available Vikuity™ DBEF film.
本発明に関連して、「光配向層」は、アライニング光への曝露時に異方性特性が誘起され得る、光配向可能な物質である材料から作製される。更に、「光配向層」という用語は、アライニング光への曝露によって整列した層のことをいう。本発明の場合、誘起される異方性は、それが、例えば、式(I)の異方性LCP化合物を含む隣接する層にアラインメント能を提供するようなものでなければならない。「アラインメント方向」という用語は、隣接する層に誘起される好ましい方向のことをいうものであり、例えば、アラインメント方向は、LCP化合物が整列するであろう方向である。 In the context of the present invention, a "photoalignment layer" is made from a material that is a photoalignable substance in which anisotropic properties can be induced upon exposure to aligning light. Furthermore, the term "photoalignment layer" refers to a layer that is aligned by exposure to aligning light. In the present case, the induced anisotropy must be such that it provides alignment capabilities to an adjacent layer that contains, for example, an anisotropic LCP compound of formula (I). The term "alignment direction" refers to the preferred direction that is induced in the adjacent layer, for example, the alignment direction is the direction in which the LCP compound will align.
光配向可能な物質は、光配向可能な部分構造を組み込んでおり、これが、アライニング光への曝露時に好ましい方向を発現させ、そして異方性特性を作り出すことができる。そのような光配向可能な部分構造は、好ましくは異方性吸収特性を有する。典型的には、そのような部分構造は、230~500nmの波長範囲内で吸収を示す。好ましくは、光配向可能な部分構造は、300~450nmの波長範囲で光の吸収を示し、より好ましいのは、310~380nmの波長範囲で吸収を示す部分構造である。 The photoalignable material incorporates photoalignable substructures that can develop a preferred direction and produce anisotropic properties upon exposure to aligning light. Such photoalignable substructures preferably have anisotropic absorption properties. Typically, such substructures exhibit absorption in the wavelength range of 230-500 nm. Preferably, the photoalignable substructures exhibit absorption of light in the wavelength range of 300-450 nm, and more preferably, in the wavelength range of 310-380 nm.
好ましくは、光配向可能な部分構造は、炭素-炭素、炭素-窒素、又は窒素-窒素の二重結合を有する。 Preferably, the photoalignable substructure has a carbon-carbon, carbon-nitrogen, or nitrogen-nitrogen double bond.
例えば、光配向可能な部分構造は、置換又は非置換のアゾ染料、アントラキノン、クマリン、メリシアニン、2-フェニルアゾチアゾール、2-フェニルアゾベンゾチアゾール、スチルベン、シアノスチルベン、フルオロスチルベン、シンナモニトリル、カルコン、シンナマート、シアノシンナマート、スチルバゾリウム、1,4-ビス(2-フェニルエチレニル)ベンゼン、4,4’-ビス(アリールアゾ)スチルベン、ペリレン、4,8-ジアミノ-1,5-ナフトキノン染料、アリールオキシカルボン酸誘導体、アリールエステル、N-アリールアミド、ポリイミド、2個の芳香環と結合したケトン部分構造又はケトン誘導体を有するジアリールケトン(例えば、置換ベンゾフェノン、ベンゾフェノンイミン、フェニルヒドラゾン、及びセミカルバゾンなど)である。 For example, the photo-orientable moieties are substituted or unsubstituted azo dyes, anthraquinones, coumarins, melissyanins, 2-phenylazothiazoles, 2-phenylazobenzothiazoles, stilbenes, cyanostilbenes, fluorostilbenes, cinnamonitriles, chalcones, cinnamates, cyanocinnamates, stilbazoliums, 1,4-bis(2-phenylethylenyl)benzenes, 4,4'-bis(arylazo)stilbenes, perylenes, 4,8-diamino-1,5-naphthoquinone dyes, aryloxycarboxylic acid derivatives, aryl esters, N-arylamides, polyimides, diaryl ketones having ketone moieties or ketone derivatives bonded to two aromatic rings (e.g., substituted benzophenones, benzophenone imines, phenylhydrazones, and semicarbazones).
上に列挙された異方性吸収材料の調製法は、例えば、Hoffmanらの米国特許第4,565,424号、Jonesらの米国特許第4,401,369号、Cole, Jr.らの米国特許第4,122,027号、Etzbachらの米国特許第4,667,020号、及びShannonらの米国特許第5,389,285号に示されるとおり周知である。 Methods for preparing the anisotropic absorbing materials listed above are well known, as shown, for example, in U.S. Pat. No. 4,565,424 to Hoffman et al., U.S. Pat. No. 4,401,369 to Jones et al., U.S. Pat. No. 4,122,027 to Cole, Jr. et al., U.S. Pat. No. 4,667,020 to Etzbach et al., and U.S. Pat. No. 5,389,285 to Shannon et al.
好ましくは、光配向可能な部分構造は、アリールアゾ、ポリ(アリールアゾ)、スチルベン、シアノスチルベン、シンナマート又はカルコンを含む。 Preferably, the photoalignable moiety includes an arylazo, a poly(arylazo), a stilbene, a cyanostilbene, a cinnamate, or a chalcone.
光配向可能な物質は、具体的にはモノマー、オリゴマー又はポリマーであってよい。光配向可能な部分構造は、例えば、ポリマー又はオリゴマーの主鎖内又は側鎖内で共有結合することができるか、あるいはそれらは、モノマー又は重合性ではない他の化合物の一部であってもよい。光配向可能な物質は、更に、異なる種類の光配向可能な部分構造を含むコポリマーであってもよく、又は光配向可能な部分構造を伴う及び伴わない側鎖を含むコポリマーであってもよい。 The photoalignable material may in particular be a monomer, oligomer or polymer. The photoalignable moieties may be covalently bonded, for example, in the main chain or in the side chains of a polymer or oligomer, or they may be part of a monomer or other compound that is not polymerizable. The photoalignable material may furthermore be a copolymer comprising different types of photoalignable moieties, or a copolymer comprising side chains with and without photoalignable moieties.
ポリマーは、例えば、ポリアクリラート、ポリメタクリラート、ポリイミド、ポリウレタン、ポリアミド酸、ポリマレインイミド、ポリ-2-クロロアクリラート、ポリ-2-フェニルアクリラート;非置換若しくはC1~C6アルキル置換のポリアクリルアミド、ポリメタクリルアミド、ポリ-2-クロロアクリルアミド、ポリ-2-フェニルアクリルアミド、ポリエーテル、ポリビニルエーテル、ポリエステル、ポリビニルエステル、ポリスチレン誘導体、ポリシロキサン、ポリアクリル酸若しくはポリメタクリル酸の直鎖若しくは分岐のアルキルエステル;ポリフェノキシアルキルアクリラート、ポリフェノキシアルキルメタクリラート、1~20個の炭素原子のアルキル残基を有するポリフェニルアルキルメタクリラート;ポリアクリロニトリル、ポリメタクリロニトリル、シクロオレフィンポリマー、ポリスチレン、ポリ-4-メチルスチレン又はそれらの混合物を意味する。 Polymers mean, for example, polyacrylates, polymethacrylates, polyimides, polyurethanes, polyamic acids, polymaleimides, poly-2-chloroacrylates, poly-2-phenylacrylates; unsubstituted or C 1 -C 6 alkyl substituted polyacrylamides, polymethacrylamides, poly-2-chloroacrylamides, poly-2-phenylacrylamides, polyethers, polyvinyl ethers, polyesters, polyvinyl esters, polystyrene derivatives, polysiloxanes, linear or branched alkyl esters of polyacrylic or polymethacrylic acid; polyphenoxyalkylacrylates, polyphenoxyalkylmethacrylates, polyphenylalkylmethacrylates with alkyl residues of 1 to 20 carbon atoms; polyacrylonitriles, polymethacrylonitriles, cycloolefin polymers, polystyrene, poly-4-methylstyrene or mixtures thereof.
光配向可能な物質はまた、光増感剤、例えば、ケトクマリン及びベンゾフェノンを含んでもよい。 The photoalignable material may also include photosensitizers, such as ketocoumarins and benzophenones.
更に、好ましい光配向可能なモノマー又はオリゴマー又はポリマーは、米国特許第5,539,074号、米国特許第6,201,087号、米国特許第6,107,427号、米国特許第6,632,909号及び米国特許第7,959,990号に記載されている。 Further preferred photoalignable monomers, oligomers or polymers are described in U.S. Pat. Nos. 5,539,074, 6,201,087, 6,107,427, 6,632,909 and 7,959,990.
LCPのアラインメントは、液晶を整列させるための他の既知の手段によって達成することができる。例えば、支持体は、アライニング表面を有してもよく、このことは、表面が液晶を整列させる能力を有することを意味するものである。支持体は、それ以上の処理なしに既にアラインメントを提供していてもよい。例えば、プラスチック基板が支持体として使用される場合、それは、製造方法、例えば、基板の押し出し又は延伸に起因して、表面上でアラインメントを提供し得る。また、支持体にブラシをかけたり、又は方向性のある微細構造をインプリントして、アラインメント能を作り出すことも可能である。 Alignment of LCPs can be achieved by other known means for aligning liquid crystals. For example, the support may have an aligning surface, meaning that the surface has the ability to align liquid crystals. The support may already provide alignment without further processing. For example, if a plastic substrate is used as the support, it may provide alignment on the surface due to the manufacturing method, e.g., extrusion or stretching of the substrate. It is also possible to brush the support or imprint a directional microstructure to create alignment capabilities.
LCP化合物の重合及びアライニング光への曝露の工程は、任意の順序であってよい。重合は、アライニング光への曝露の前又は後に開始させることができる。あるいは重合と曝露とは同時に行われてもよい。 The steps of polymerizing the LCP compound and exposing it to the aligning light may be in any order. Polymerization may be initiated before or after exposure to the aligning light. Alternatively, polymerization and exposure may occur simultaneously.
本発明の更なる実施態様は、本発明の式(I)の異方性化合物、LCP混合物又はLCPネットワークを含む光学フィルムを製造する方法であって、好ましくは<200mJ、より好ましくは<150mJ、そしてより好ましくは<100mJのエネルギーによるアライニング光への曝露による方法に関する。 A further embodiment of the present invention relates to a method for producing an optical film comprising an anisotropic compound of formula (I) of the present invention, an LCP blend or an LCP network, preferably by exposure to aligning light with an energy of <200 mJ, more preferably <150 mJ and more preferably <100 mJ.
LCP混合物は、押し出し、注型、成形、2D若しくは3D印刷又はコーティングなどの任意の適切な方法によって支持体に適用することができる。適切なコーティング方法は、例えば、スピンコーティング、ブレードコーティング、ナイフコーティング、キスロールコーティング、ダイコーティング、ディッピング、ブラッシング、バーによる注型、ローラーコーティング、フローコーティング、ワイヤーコーティング、スプレーコーティング、ディップコーティング、カーテンコーティング、エアナイフコーティング、リバースロールコーティング、グラビアコーティング、メータリングロッド(マイヤーバー)コーティング、スロットダイ(押し出し)コーティング、ローラーコーティング、フレキソコーティングである。適切な印刷方法は、シルクスクリーン印刷、フレキソ印刷などの凸版印刷、ジェット印刷、直接グラビア印刷若しくはオフセットグラビア印刷などの凹版印刷、オフセット印刷などの平版印刷、又はスクリーン印刷などの孔版印刷を包含する。 The LCP mixture can be applied to the substrate by any suitable method, such as extrusion, casting, molding, 2D or 3D printing or coating. Suitable coating methods are, for example, spin coating, blade coating, knife coating, kiss roll coating, die coating, dipping, brushing, casting with a bar, roller coating, flow coating, wire coating, spray coating, dip coating, curtain coating, air knife coating, reverse roll coating, gravure coating, metering rod (Meyer bar) coating, slot die (extrusion) coating, roller coating, flexo coating. Suitable printing methods include letterpress printing such as silk screen printing, flexography, jet printing, intaglio printing such as direct gravure printing or offset gravure printing, lithography printing such as offset printing, or stencil printing such as screen printing.
本発明の更なる実施態様は、本発明の式(I)の異方性化合物、又はLCP混合物、又はLCPネットワークを含む光学フィルムである。好ましくは、光学フィルムは、整列した式(I)の異方性化合物、又はLCP混合物、又はLCPネットワークを含む。より好ましくは、アラインメント品質が均一であり、傾斜ドメインがないことである。 A further embodiment of the present invention is an optical film comprising an anisotropic compound of formula (I) of the present invention, or an LCP mixture, or an LCP network. Preferably, the optical film comprises an aligned anisotropic compound of formula (I), or an LCP mixture, or an LCP network. More preferably, the alignment quality is uniform and free of tilted domains.
更に好ましいのは、広い波長帯域にわたって偏光の逆リタデーションパターンを示す本発明のLCPを含む光学フィルムである。本発明の光学フィルムは、好ましくは、逆波長分散を伴う複屈折を有する:Re450/Re550は0.88未満であり、好ましくは、Re450/Re550は<0.85であるが;一方、Re650/Re550は1.01を上回り、好ましくはRe650/Re550は>1.03である(Re450は波長450nmでのフィルムのリタデーションを、Re550は波長550nmでのフィルムのリタデーションを、そしてRe650は波長650nmでのフィルムのリタデーションを表す)。 Further preferred are optical films comprising the LCP of the present invention that exhibit a reverse retardation pattern of polarized light over a wide wavelength band.The optical films of the present invention preferably have birefringence with reverse wavelength dispersion: Re450 / Re550 is less than 0.88, preferably Re450/ Re550 is <0.85; while Re650 / Re550 is greater than 1.01, preferably Re650 / Re550 is >1.03 ( Re450 represents the retardation of the film at a wavelength of 450 nm, Re550 represents the retardation of the film at a wavelength of 550 nm, and Re650 represents the retardation of the film at a wavelength of 650 nm).
次に、以下の限定されない実施例を参照して本発明を説明する。これらの実施例は、説明のみを目的として提供される。本発明の範囲内にあるこれらの実施例の変形は、当業者には明らかとなろう。 The present invention will now be described with reference to the following non-limiting examples. These examples are provided for illustrative purposes only. Variations of these examples that fall within the scope of the invention will be apparent to those of ordinary skill in the art.
以下の実施例は、本発明を更に説明し、理解を容易にするために提供されており、決して本発明を限定することを意図するものではない。 The following examples are provided to further illustrate and facilitate understanding of the present invention and are not intended to limit the invention in any way.
実施例1
化合物1の合成
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-ホルミル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル]の合成:
塩化オキサリル(2.5mL、29mmol)を、無水トルエン 48mL、4-メトキシフェノール 4mg及び無水DMF 1mL中の4-(6-アクリロイルオキシ-ヘキサ-1-イルオキシ)安息香酸(6.98g、24mmol)の溶液に45℃で滴下した。2時間後、反応混合物を0℃に冷却し、0~5℃で無水DMA 40mL及びN,N-ジメチルシクロヘキシルアミン(7.8g、52mmol)中の2,5-ジヒドロキシベンズアルデヒド(1.5g、11mmol)の溶液に滴下した。反応混合物を周囲温度で一晩撹拌した。橙色の溶液を、水15mLを加えることによりクエンチし、ジクロロメタンで抽出し、水、食塩水で連続して洗浄し、硫酸ナトリウムで乾燥させて真空下で濃縮した。ジクロロメタン/メタノールからの再結晶後、標題化合物を白色の固体(4.19g、56%)として得た。
1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.08 (m, 4H), 7.82 (d,1H), 7.71 (dd, 1H), 7.54 (d, 1H), 7.10 (m, 4H), 6.26 (d, 1H), 6.15 (dd, 1H), 5.90 (d, 1H), 4.09 (m, 8H), 1.72 (m, 4H), 1.60 (m, 4H), 1.39 (m, 8H).
Example 1
Synthesis of Compound 1 Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-formyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl]:
Oxalyl chloride (2.5 mL, 29 mmol) was added dropwise to a solution of 4-(6-acryloyloxy-hex-1-yloxy)benzoic acid (6.98 g, 24 mmol) in 48 mL anhydrous toluene, 4 mg 4-methoxyphenol and 1 mL anhydrous DMF at 45° C. After 2 h, the reaction mixture was cooled to 0° C. and added dropwise to a solution of 2,5-dihydroxybenzaldehyde (1.5 g, 11 mmol) in 40 mL anhydrous DMA and N,N-dimethylcyclohexylamine (7.8 g, 52 mmol) at 0-5° C. The reaction mixture was stirred overnight at ambient temperature. The orange solution was quenched by the addition of 15 mL water, extracted with dichloromethane, washed successively with water, brine, dried over sodium sulfate and concentrated in vacuo. After recrystallization from dichloromethane/methanol, the title compound was obtained as a white solid (4.19 g, 56%).
1 H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.08 (m, 4H), 7.82 (d,1H), 7.71 (dd, 1H), 7.54 (d, 1H), 7.10 (m, 4H), 6.26 (d, 1H), 6.15 (dd, 1H) ), 5.90 (d, 1H), 4.09 (m, 8H), 1.72 (m, 4H), 1.60 (m, 4H), 1.39 (m, 8H).
2-ヒドラジニルキノキサリンの合成:
マイクロ波反応容器に、無水エタノール(15mL)中の2-クロロキノキサリン(1.70g、15mmol)、ヒドラジン一水和物(1.82mL、37.5mmol)を仕込んだ。混合物をマイクロ波反応器中で90℃まで20分間加熱した。反応混合物を冷却し、沈殿物を濾過し、ヘキサンで洗浄して、標題化合物を橙色の固体(2.07g、86%)として与えた。1H-NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1H), 8.34 (s, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.52-7.58 (m, 2H), 7.30-7.34 (m, 1H), 4.43 (s, 2H).
Synthesis of 2-hydrazinylquinoxaline:
A microwave reactor was charged with 2-chloroquinoxaline (1.70 g, 15 mmol), hydrazine monohydrate (1.82 mL, 37.5 mmol) in absolute ethanol (15 mL). The mixture was heated to 90 °C for 20 min in a microwave reactor. The reaction mixture was cooled and the precipitate was filtered and washed with hexane to give the title compound as an orange solid (2.07 g, 86%). 1 H-NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1H), 8.34 (s, 1H), 7.76 (d, J = 8.2 Hz, 1H), 7.52-7.58 (m, 2H), 7.30-7.34 (m, 1H), 4.43 (s, 2H).
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-3-[(E)-(キノキサリン-2-イルヒドラゾノ)メチル]フェニル](化合物1)の合成:
(±)10-カンファースルホン酸(23.2mg、0.1mmol)を無水テトラヒドロフラン10mL中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-ホルミル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](0.69g、1.0mmol)及び2-ヒドラジニルキノキサリン(0.19g、1.2mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で3時間撹拌した。反応終了後、反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/ヘキサンからの再結晶後、標題化合物を黄色の固体(0.67g、81%)として得た。
1H NMR (400 MHz, CDCl3) δ 9.11 (s, 1H), 8.63 (s, 1H), 8.17-8.20 (m, 4H), 7.97 (d, J = 8.2 Hz, 1H), 7.92-7.94 (m, 2H), 7.59-7.68 (m, 2H), 7.46-7.52 (m, 1H), 7.29 (s, 2H), 6.98-7.02 (m, 4H), 6.38-6.43 (m, 2H), 6.10-6.17 (m, 2H), 5.81-5.84 (m, 2H), 4.19 (t, J = 6.6 Hz, 4H), 4.06-4.11 (m, 4H), 1.83-1.90 (m, 4H), 1.71-1.78 (m, 4H), 1.45-1.53 (m, 8H) ; MALDI-TOF (CHCA) 851.33 (M++Na).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-3-[(E)-(quinoxalin-2-ylhydrazono)methyl]phenyl] (Compound 1)
(±)10-Camphorsulfonic acid (23.2 mg, 0.1 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-formyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (0.69 g, 1.0 mmol) and 2-hydrazinylquinoxaline (0.19 g, 1.2 mmol) in 10 mL of anhydrous tetrahydrofuran under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 3 h. After completion of the reaction, the reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. After recrystallization from dichloromethane/hexane, the title compound was obtained as a yellow solid (0.67 g, 81%).
1 H NMR (400 MHz, CDCl 3 ) δ 9.11 (s, 1H), 8.63 (s, 1H), 8.17-8.20 (m, 4H), 7.97 (d, J = 8.2 Hz, 1H), 7.92-7.94 (m, 2H), 7.59-7.68 (m, 2H), 7 .46-7.52 (m, 1H), 7.29 (s, 2H), 6.98-7.02 (m, 4H), 6.38-6.43 (m, 2H), 6.10-6.17 (m, 2H), 5.81-5.84 (m, 2H), 4.19 (t, J = 6.6 Hz, 4H), 4.06-4.11 (m, 4H), 1.83-1.90 (m, 4H), 1.71-1.78 (m, 4H), 1.45-1.53 (m, 8H); MALDI-TOF (CHCA) 851.33 (M + +Na).
実施例2
化合物2の合成:
N-(キノキサリン-2-イルアミノ)カルバミン酸tert-ブチルの合成:
無水THF(9mL)中の二炭酸ジ-tert-ブチル(3.9g、18mmol)の溶液を、無水THF(30mL)中の2-ヒドラジニルキノキサリン(2.4g、15mmol)の溶液に加えた。反応混合物を50℃で2時間撹拌した。反応混合物を室温まで冷却して、重炭酸ナトリウムの飽和水溶液でクエンチして、酢酸エチルとヘキサンの1:2混合物で2回抽出した。有機層を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去して、標題化合物を褐色の固体(4.11g、定量的)として与えた。
1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 9.02 (s, 1H), 8.35 (s, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.60-7.63 (m, 2H), 7.41-7.45 (m, 1H), 1.46 (s, 9H).
Example 2
Synthesis of compound 2:
Synthesis of tert-butyl N-(quinoxalin-2-ylamino)carbamate:
A solution of di-tert-butyl dicarbonate (3.9 g, 18 mmol) in anhydrous THF (9 mL) was added to a solution of 2-hydrazinylquinoxaline (2.4 g, 15 mmol) in anhydrous THF (30 mL). The reaction mixture was stirred at 50 °C for 2 h. The reaction mixture was cooled to room temperature, quenched with a saturated aqueous solution of sodium bicarbonate, and extracted twice with a 1:2 mixture of ethyl acetate and hexane. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure to give the title compound as a brown solid (4.11 g, quantitative).
1 H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 9.02 (s, 1H), 8.35 (s, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.60-7.63 (m, 2H), 7.41-7.45 (m, 1H), (s, 9H).
N-[メチル(キノキサリン-2-イル)アミノ]カルバミン酸tert-ブチルの合成:
ヨードメタン(0.4mL、6.3mmol)を無水DMF(10mL)中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-ホルミル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](1.5g、5.8mmol)の溶液に0℃で加えた。反応混合物を周囲温度で3時間撹拌した。反応混合物を水でクエンチして、酢酸エチルとヘキサンの1:2混合物で2回抽出した。有機層を水及び食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。酢酸エチル/ヘキサンの1:2混合物を用いて、残渣をシリカゲルのフラッシュカラムクロマトグラフィーにより精製して、標題化合物を赤色の油状物(0.46g、21%)として与えた。1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.54 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.68-7.71 (m, 1H), 7.63-7.67 (m, 1H), 7.45-7.49 (m, 1H), 3.29 (s, 3H), 1.44 (s, 9H).
Synthesis of tert-butyl N-[methyl(quinoxalin-2-yl)amino]carbamate:
Iodomethane (0.4 mL, 6.3 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-formyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (1.5 g, 5.8 mmol) in anhydrous DMF (10 mL) at 0° C. The reaction mixture was stirred at ambient temperature for 3 h. The reaction mixture was quenched with water and extracted twice with a 1:2 mixture of ethyl acetate and hexanes. The organic layer was washed with water and brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel using a 1:2 mixture of ethyl acetate/hexanes to give the title compound as a red oil (0.46 g, 21%). 1 H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.54 (s, 1H), 7.87 (d, J = 7.8 Hz, 1H), 7.68-7.71 (m, 1H), 7.63-7.67 (m, 1H), 7.45-7.49 (m, 1H), 3.29 (s, 3H), 1.44 (s, 9H).
1-メチル-1-キノキサリン-2-イル-ヒドラジンの合成:
トリフルオロ酢酸(TFA)(3mL)をジクロロメタン(3mL)中のN-[メチル(キノキサリン-2-イル)アミノ]カルバミン酸tert-ブチル(0.43g、1.6mmol)の溶液に加えた。反応混合物を周囲温度で3時間撹拌した。減圧下で溶媒を除去して、残渣をジクロロメタンに溶解し、重炭酸ナトリウムの飽和水溶液及び食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去して、標題化合物を黄色の固体(0.25g、93%)として与えた。1H NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 7.90 (dd, J = 8.2, 1.4 Hz, 1H), 7.69 (dd, J = 8.2, 0.9 Hz, 1H), 7.55-7.59 (m, 1H), 7.37-7.42 (m, 1H), 4.14 (s, 2H), 3.47 (s, 3H).
Synthesis of 1-methyl-1-quinoxalin-2-yl-hydrazine:
Trifluoroacetic acid (TFA) (3 mL) was added to a solution of tert-butyl N-[methyl(quinoxalin-2-yl)amino]carbamate (0.43 g, 1.6 mmol) in dichloromethane (3 mL). The reaction mixture was stirred at ambient temperature for 3 h. The solvent was removed under reduced pressure and the residue was dissolved in dichloromethane, washed with a saturated aqueous solution of sodium bicarbonate and brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure to give the title compound as a yellow solid (0.25 g, 93%). 1 H NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 7.90 (dd, J = 8.2, 1.4 Hz, 1H), 7.69 (dd, J = 8.2, 0.9 Hz, 1H), 7.55-7.59 (m, 1H), 7.37-7.42 (m, 1H) , 4.14 (s, 2H), 3.47 (s, 3H).
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-[(E)-[メチル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](化合物2)の合成:
(±)10-カンファースルホン酸(32mg、0.14mmol)を無水テトラヒドロフラン 14mL中のビス[4-(6-アクリロイルオキシヘキシルオキシ)安息香酸]2-ホルミル-1,4-フェニレン(0.95g、1.4mmol)及びN-メチル-N-(キノキサリン-2-イル)ヒドラジン(0.25g、1.5mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で3時間撹拌した。反応終了後、反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/ヘキサンからの再結晶後、標題化合物を黄色の固体(0.92g、79%)として得た。
1H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.12-8.17 (m, 4H), 8.08 (d, J = 2.7 Hz, 1H), 7.93 (s, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.74-7.77 (m, 1H), 7.66-7.70 (m, 1H), 7.50-7.55 (m, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.37 (dd, J = 8.7, 2.7 Hz, 1H), 7.12-7.15 (m, 4H), 6.29-6.34 (m, 2H), 6.14-6.20 (m, 2H), 5.92-5.95 (m, 2H), 4.08-4.13 (m, 8H), 3.63 (s, 3H), 1.76 (s, 4H), 1.60-1.68 (m, 4H), 1.40-1.46 (m, 8H).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-[(E)-[methyl(quinoxalin-2-yl)hydrazono]methyl]-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (Compound 2)
(±)10-Camphorsulfonic acid (32 mg, 0.14 mmol) was added to a solution of bis[4-(6-acryloyloxyhexyloxy)benzoic acid]2-formyl-1,4-phenylene (0.95 g, 1.4 mmol) and N-methyl-N-(quinoxalin-2-yl)hydrazine (0.25 g, 1.5 mmol) in 14 mL of anhydrous tetrahydrofuran under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 3 h. After completion of the reaction, the reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. After recrystallization from dichloromethane/hexane, the title compound was obtained as a yellow solid (0.92 g, 79%).
1 H NMR (400 MHz, DMSO-d6) δ 9.44 (s, 1H), 8.12-8.17 (m, 4H), 8.08 (d, J = 2.7 Hz, 1H), 7.93 (s, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.74-7.77 (m, 1H), 7.66-7.70 (m, 1H), 7.50-7.55 (m, 1H), 7.43 (d, J = 8.7 Hz, 1H), 7.37 (dd, J = 8.7, 2.7 Hz, 1H), 7.12-7.15 (m, 4H), 6.29-6.34 (m, 2H) , 6.14-6.20 (m, 2H), 5.92-5.95 (m, 2H), 4.08-4.13 (m, 8H), 3.63 (s, 3H), 1.76 (s, 4H), 1.60-1.68 (m, 4H), 1.40-1.46 (m, 8H).
実施例2A
化合物2Aの合成:
N-[ヘキシル(キノキサリン-2-イル)アミノ]カルバミン酸tert-ブチルの合成:
丸底フラスコにN-(キノキサリン-2-イルアミノ)カルバミン酸tert-ブチル(1.36g、5mmol)、1-ヨードヘキサン(0.88mL、6mmol)、炭酸カリウム(1.04g、7.5mmol)、及び無水DMF(10mL)を仕込んだ。混合物を50℃で窒素雰囲気下18時間撹拌した。反応混合物を周囲温度まで冷却し、酢酸エチル/ヘキサン(1/2)の混合物で希釈し、水で3回及び食塩水で1回洗浄した。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。酢酸エチル/ヘキサンを用いて、残渣をシリカゲルのフラッシュカラムクロマトグラフィーにより精製して、標題化合物を褐色の油状物(443mg、26%)として与えた。
1H-NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.46 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.61-7.67 (m, 2H), 7.43-7.47 (m, 1H), 1.57-1.64 (m, 2H), 1.45 (s, 9H), 1.24-1.33 (m, 8H), 0.83-0.87 (m, 3H)
Example 2A
Synthesis of compound 2A:
Synthesis of tert-butyl N-[hexyl(quinoxalin-2-yl)amino]carbamate:
A round-bottom flask was charged with tert-butyl N-(quinoxalin-2-ylamino)carbamate (1.36 g, 5 mmol), 1-iodohexane (0.88 mL, 6 mmol), potassium carbonate (1.04 g, 7.5 mmol), and anhydrous DMF (10 mL). The mixture was stirred at 50° C. under nitrogen atmosphere for 18 h. The reaction mixture was cooled to ambient temperature, diluted with a mixture of ethyl acetate/hexane (1/2), washed three times with water and once with brine. The organic layer was dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with ethyl acetate/hexane to give the title compound as a brown oil (443 mg, 26%).
1 H-NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.46 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.61-7.67 (m, 2H), 7.43-7.47 (m, 1H), 1.57-1.64 (m, 2H) , 1.45 (s, 9H), 1.24-1.33 (m, 8H), 0.83-0.87 (m, 3H)
1-ヘキシル-1-キノキサリン-2-イル-ヒドラジンの合成:
トリフルオロ酢酸(TFA)(2.5mL)をジクロロメタン(2.5mL)中のN-[ヘキシル(キノキサリン-2-イル)アミノ]カルバミン酸tert-ブチル(0.44g、1.3mmol)の溶液に加えた。反応混合物を周囲温度で19時間撹拌した。減圧下で溶媒を除去して、残渣を酢酸エチルに溶解し、重炭酸ナトリウムの飽和水溶液及び食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去して、標題化合物をベージュ色の油状物(0.31g、定量的)として与えた。
1H-NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.49-7.55 (m, 2H), 7.29-7.33 (m, 1H), 4.86 (s, 2H), 3.74-3.77 (m, 2H), 1.64-1.71 (m, 2H), 1.23-1.35 (m, 6H), 0.83-0.87 (m, 3H)
Synthesis of 1-hexyl-1-quinoxalin-2-yl-hydrazine:
Trifluoroacetic acid (TFA) (2.5 mL) was added to a solution of tert-butyl N-[hexyl(quinoxalin-2-yl)amino]carbamate (0.44 g, 1.3 mmol) in dichloromethane (2.5 mL). The reaction mixture was stirred at ambient temperature for 19 h. The solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate, washed with a saturated aqueous solution of sodium bicarbonate and brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure to give the title compound as a beige oil (0.31 g, quantitative).
1 H-NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 7.76 (d, J = 7.8 Hz, 1H), 7.49-7.55 (m, 2H), 7.29-7.33 (m, 1H), 4.86 (s, 2H), 3.74-3.77 (m, 2H) , 1.64-1.71 (m, 2H), 1.23-1.35 (m, 6H), 0.83-0.87 (m, 3H)
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-[(E)-[ヘキシル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](化合物2A)の合成:
(±)10-カンファースルホン酸(26mg、0.11mmol)を無水テトラヒドロフラン(10mL)中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-[(E)-[メチル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](0.74g、1.1mmol)及び1-ヘキシル-1-キノキサリン-2-イル-ヒドラジン(0.31g、1.3mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で18時間撹拌した。反応終了後、反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/ヘキサンからの再結晶後、標題化合物を淡黄色の固体(0.70g、72%)として得た。
1H-NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 8.18 (d, J = 9.1 Hz, 2H), 8.13 (d, J = 9.1 Hz, 2H), 8.10 (d, J = 2.7 Hz, 1H), 7.88-7.91 (m, 2H), 7.73 (dd, J = 8.5, 1.1 Hz, 1H), 7.65-7.69 (m, 1H), 7.50-7.54 (m, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.38 (dd, J = 8.7, 2.7 Hz, 1H), 7.15 (d, J = 2.7 Hz, 2H), 7.12 (d, J = 3.2 Hz, 2H), 6.32 (dd, J = 17.4, 1.8 Hz, 2H), 6.17 (ddd, J = 17.4, 10.3, 1.1 Hz, 2H), 5.93 (dd, J = 10.5, 1.8 Hz, 2H), 4.24-4.28 (m, 2H), 4.07-4.13 (m, 8H), 1.72-1.80 (m, 4H), 1.59-1.68 (m, 4H), 1.36-1.50 (m, 10H), 0.95-1.11 (m, 6H), 0.72 (t, J = 7.1 Hz, 3H).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-[(E)-[hexyl(quinoxalin-2-yl)hydrazono]methyl]-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (Compound 2A)
(±) 10-Camphorsulfonic acid (26 mg, 0.11 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-[(E)-[methyl(quinoxalin-2-yl)hydrazono]methyl]-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (0.74 g, 1.1 mmol) and 1-hexyl-1-quinoxalin-2-yl-hydrazine (0.31 g, 1.3 mmol) in anhydrous tetrahydrofuran (10 mL) under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 18 hours. After completion of the reaction, the reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. After recrystallization from dichloromethane/hexanes, the title compound was obtained as a pale yellow solid (0.70 g, 72%).
1 H-NMR (400 MHz, DMSO-d6) δ 9.47 (s, 1H), 8.18 (d, J = 9.1 Hz, 2H), 8.13 (d, J = 9.1 Hz, 2H), 8.10 (d, J = 2.7 Hz, 1H), 7.88-7.91 (m, 2H), 3 (dd, J = 8.5, 1.1 Hz, 1H), 7.65-7.69 (m, 1H), 7.50-7.54 (m, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.38 (dd, J = 8.7, 2.7 Hz, 1H), 7.15 (d, J = 2.7 Hz , 2H), 7.12 (d, J = 3.2 Hz, 2H), 6.32 (dd, J = 17.4, 1.8 Hz, 2H), 6.17 (ddd, J = 17.4, 10.3, 1.1 Hz, 2H), 5.93 (dd, J = 10.5, 1.8 Hz, 2H), 4.24-4.28 (m, 2 H), 4.07-4.13 (m, 8H), 1.72-1.80 (m, 4H), 1.59-1.68 (m, 4H), 1.36-1.50 (m, 10H), 0.95-1.11 (m, 6H), 0.72 (t, J = 7.1 Hz, 3H).
実施例2B
化合物2Bの合成:
4-プロピルシクロヘキサンカルボン酸(3-ホルミル-4-ヒドロキシ-フェニル)の合成:
無水ジクロロメタン(20mL)中の2,5-ジヒドロキシベンズアルデヒド(1.38g、10mmol)、trans-4-プロピルシクロヘキサンカルボン酸(1.79g、10.5mmol)及び4-ジメチルアミノピリジン(DMAP)(122mg、1mmol)の溶液に、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド(EDC)(1.94mL、11mmol)をゆっくり加えた。反応混合物を周囲温度で22時間撹拌した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。酢酸エチル/ヘキサンを用いて、残渣をシリカゲルのフラッシュカラムクロマトグラフィーにより精製して、標題化合物を黄色の固体(1.31g、45%)として与えた。
1H-NMR (400 MHz, CDCl3) δ 10.90 (s, 1H), 9.85 (s, 1H), 7.31 (d, J = 2.7 Hz, 1H), 7.23 (dd, J = 9.1, 2.7 Hz, 1H), 6.99 (d, J = 9.1 Hz, 1H), 2.44-2.52 (m, 1H), 2.11-2.15 (m, 2H), 1.86-1.90 (m, 2H), 1.50-1.57 (m, 2H), 1.29-1.39 (m, 3H), 1.17-1.23 (m, 2H), 0.97-1.04 (m, 3H), 0.87-0.92 (m, 3H)
Example 2B
Synthesis of compound 2B:
Synthesis of 4-propylcyclohexanecarboxylic acid (3-formyl-4-hydroxy-phenyl)
To a solution of 2,5-dihydroxybenzaldehyde (1.38 g, 10 mmol), trans-4-propylcyclohexanecarboxylic acid (1.79 g, 10.5 mmol) and 4-dimethylaminopyridine (DMAP) (122 mg, 1 mmol) in anhydrous dichloromethane (20 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) (1.94 mL, 11 mmol) slowly. The reaction mixture was stirred at ambient temperature for 22 h. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with ethyl acetate/hexane to give the title compound as a yellow solid (1.31 g, 45%).
1 H-NMR (400 MHz, CDCl 3 ) δ 10.90 (s, 1H), 9.85 (s, 1H), 7.31 (d, J = 2.7 Hz, 1H), 7.23 (dd, J = 9.1, 2.7 Hz, 1H), 6.99 (d, J = 9.1 Hz, 1H), -2.52 (m, 1H), 2.11-2.15 (m, 2H), 1.86-1.90 (m, 2H), 1.50-1.57 (m, 2H), 1.29-1.39 (m, 3H), 1.17-1.23 (m, 2H), 0.97-1.04 (m, 3H), 0. 87-0.92 (m, 3H)
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[2-ホルミル-4-(4-プロピルシクロヘキサンカルボニル)オキシ-フェニル]の合成:
無水ジクロロメタン(9mL)中の4-プロピルシクロヘキサンカルボン酸(3-ホルミル-4-ヒドロキシ-フェニル)(1.3g、4.5mmol)、4-(6-アクリロイルオキシ-ヘキサ-1-イルオキシ)安息香酸(1.38g、4.70mmol)及び4-ジメチルアミノピリジン(DMAP)(55mg、0.45mmol)の溶液に、1-(3-ジメチルアミノプロピル)-3-エチルカルボジイミド(EDC)(0.87mL、4.93mmol)を加えた。反応混合物を周囲温度で5時間撹拌した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、酢酸エチルで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。酢酸エチル/ヘキサンを用いて、残渣をシリカゲルのフラッシュカラムクロマトグラフィーにより精製して、標題化合物をベージュ色の少量の不純物を含有する油状物(2.73g)として与え、これを更に精製することなく次の反応に使用した。
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [2-formyl-4-(4-propylcyclohexanecarbonyl)oxy-phenyl]:
To a solution of 4-propylcyclohexanecarboxylic acid (3-formyl-4-hydroxy-phenyl) (1.3 g, 4.5 mmol), 4-(6-acryloyloxy-hex-1-yloxy)benzoic acid (1.38 g, 4.70 mmol) and 4-dimethylaminopyridine (DMAP) (55 mg, 0.45 mmol) in anhydrous dichloromethane (9 mL) was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) (0.87 mL, 4.93 mmol). The reaction mixture was stirred at ambient temperature for 5 h. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with ethyl acetate. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with ethyl acetate/hexane to give the title compound as a beige oil (2.73 g) containing minor impurities, which was used in the next reaction without further purification.
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[2-[(E)-[メチル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-(4-プロピルシクロヘキサンカルボニル)オキシ-フェニル](化合物2B)の合成:
(±)10-カンファースルホン酸(105mg、0.45mmol)を無水テトラヒドロフラン(45mL)中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-ホルミル-4-(4-プロピルシクロヘキサンカルボニル)オキシ-フェニル](2.73g、4.5mmol)、1-メチル-1-キノキサリン-2-イル-ヒドラジン(0.82g、4.7mmol)、及び4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン 1-オキシル フリーラジカル(TEMPO-OH)(9mg、0.05mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で24時間撹拌した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/ヘキサンからの再結晶後、標題化合物を灰色の固体(1.61g、50%)として得た。
1H-NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.14 (d, J = 9.1 Hz, 2H), 7.90-7.92 (m, 2H), 7.85-7.86 (m, 1H), 7.75-7.77 (m, 1H), 7.67-7.71 (m, 1H), 7.52-7.56 (m, 1H), 7.39 (d, J = 8.7 Hz, 1H), 7.20 (dd, J = 8.7, 2.7 Hz, 1H), 7.12 (d, J = 8.7 Hz, 2H), 6.32 (dd, J = 17.2, 1.6 Hz, 1H), 6.17 (dd, J = 17.4, 10.5 Hz, 1H), 5.93 (dd, J = 10.3, 1.6 Hz, 1H), 4.07-4.13 (m, 4H), 3.62 (s, 3H), 2.11-2.15 (m, 2H), 1.72-1.84 (m, 4H), 1.61-1.68 (m, 2H), 1.37-1.54 (m, 7H), 1.27-1.35 (m, 3H), 1.16-1.21 (m, 2H), 0.95-1.04 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H)
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [2-[(E)-[methyl(quinoxalin-2-yl)hydrazono]methyl]-4-(4-propylcyclohexanecarbonyl)oxy-phenyl] (compound 2B)
(±) 10-Camphorsulfonic acid (105 mg, 0.45 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-formyl-4-(4-propylcyclohexanecarbonyl)oxy-phenyl] (2.73 g, 4.5 mmol), 1-methyl-1-quinoxalin-2-yl-hydrazine (0.82 g, 4.7 mmol), and 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl free radical (TEMPO-OH) (9 mg, 0.05 mmol) in anhydrous tetrahydrofuran (45 mL) under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 24 h. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. After recrystallization from dichloromethane/hexanes, the title compound was obtained as a grey solid (1.61 g, 50%).
1 H-NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.14 (d, J = 9.1 Hz, 2H), 7.90-7.92 (m, 2H), 7.85-7.86 (m, 1H), 7.75-7.77 (m, 1H), 7.67-7.71 (m , 1H), 7.52-7.56 (m, 1H), 7.39 (d, J = 8.7 Hz, 1H), 7.20 (dd, J = 8.7, 2.7 Hz, 1H), 7.12 (d, J = 8.7 Hz, 2H), 6.32 (dd, J = 17.2, 1.6 Hz, 1H), 6.17 (dd, J = 17.4, 10.5 Hz, 1H), 5.93 (dd, J = 10.3, 1.6 Hz, 1H), 4.07-4.13 (m, 4H), 3.62 (s, 3H), 2.11-2.15 (m, 2H), 1.72-1.84 (m, 4H), 1.61-1.6 8 (m, 2H), 1.37-1.54 (m, 7H), 1.27-1.35 (m, 3H), 1.16-1.21 (m, 2H), 0.95-1.04 (m, 2H), 0.87 (t, J = 7.1 Hz, 3H)
実施例2C
化合物2Cの合成:
2-[アミノ(キノキサリン-2-イル)アミノ]エタノールの合成:
マイクロ波反応容器に、無水EtOH(2.5mL)中の2-クロロキノキサリン(0.82g、5mmol)及び2-ヒドラジノエタノール(0.85mL、12.5mmol)を仕込んだ。混合物をマイクロ波反応器中で90℃まで10分間加熱した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。残渣をヘキサンで洗浄して、標題化合物を黄色の固体(0.59g、58%)として与えた。
1H-NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.51-7.54 (m, 2H), 7.30-7.34 (m, 1H), 4.91 (s, 2H), 4.79 (t, J = 5.3 Hz, 1H), 3.86 (t, J = 5.9 Hz, 2H), 3.73 (q, J = 5.6 Hz, 2H)
Example 2C
Synthesis of compound 2C:
Synthesis of 2-[amino(quinoxalin-2-yl)amino]ethanol:
A microwave reaction vessel was charged with 2-chloroquinoxaline (0.82 g, 5 mmol) and 2-hydrazinoethanol (0.85 mL, 12.5 mmol) in absolute EtOH (2.5 mL). The mixture was heated to 90 °C for 10 min in a microwave reactor. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent removed under reduced pressure. The residue was washed with hexane to give the title compound as a yellow solid (0.59 g, 58%).
1 H-NMR (400 MHz, DMSO-d6) δ 9.05 (s, 1H), 7.77 (d, J = 8.2 Hz, 1H), 7.51-7.54 (m, 2H), 7.30-7.34 (m, 1H), 4.91 (s, 2H), 4.79 (t, J = 5.3 Hz, 1H), 3.86 (t, J = 5.9 Hz, 2H), 3.73 (q, J = 5.6 Hz, 2H)
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-[(E)-[2-ヒドロキシエチル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](化合物2C)の合成:
(±)10-カンファースルホン酸(46mg、0.2mmol)を無水テトラヒドロフラン(20mL)中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-ホルミル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](1.37g、2mmol)、2-[アミノ(キノキサリン-2-イル)アミノ]エタノール(0.43g、2.1mmol)、及び4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン 1-オキシル フリーラジカル(TEMPO-OH)(3mg、0.02mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で23時間撹拌した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/メタノールからの再結晶後、標題化合物を黄色の固体(1.46g、83%)として得た。
1H-NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.26 (s, 1H), 8.12-8.16 (m, 4H), 8.05 (d, J = 2.7 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.74 (dd, J = 8.2, 0.9 Hz, 1H), 7.65-7.69 (m, 1H), 7.50-7.54 (m, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.37 (dd, J = 8.7, 2.7 Hz, 1H), 7.10-7.15 (m, 4H), 6.32 (dd, J = 17.2, 1.6 Hz, 2H), 6.17 (dd, J = 17.4, 10.1 Hz, 2H), 5.93 (dd, J = 10.1, 1.8 Hz, 2H), 4.86 (t, J = 5.3 Hz, 1H), 4.41 (t, J = 5.9 Hz, 2H), 4.07-4.14 (m, 8H), 3.63 (q, J = 5.6 Hz, 2H), 1.72-1.80 (m, 4H), 1.59-1.68 (m, 4H), 1.37-1.49 (m, 8H).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-[(E)-[2-hydroxyethyl(quinoxalin-2-yl)hydrazono]methyl]-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (Compound 2C)
(±)10-Camphorsulfonic acid (46 mg, 0.2 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-formyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (1.37 g, 2 mmol), 2-[amino(quinoxalin-2-yl)amino]ethanol (0.43 g, 2.1 mmol), and 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl free radical (TEMPO-OH) (3 mg, 0.02 mmol) in anhydrous tetrahydrofuran (20 mL) under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 23 h. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. After recrystallization from dichloromethane/methanol the title compound was obtained as a yellow solid (1.46 g, 83%).
1 H-NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.26 (s, 1H), 8.12-8.16 (m, 4H), 8.05 (d, J = 2.7 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.74 (dd, J = 8. 2, 0.9 Hz, 1H), 7.65-7.69 (m, 1H), 7.50-7.54 (m, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.37 (dd, J = 8.7, 2.7 Hz, 1H), 7.10-7.15 (m, 4H), 6.32 (dd , J = 17.2, 1.6 Hz, 2H), 6.17 (dd, J = 17.4, 10.1 Hz, 2H), 5.93 (dd, J = 10.1, 1.8 Hz, 2H), 4.86 (t, J = 5.3 Hz, 1H), 4.41 (t, J = 5.9 Hz, 2H), 4.07-4.14 (m, 8H), 3.63 (q, J = 5.6 Hz, 2H), 1.72-1.80 (m, 4H), 1.59-1.68 (m, 4H), 1.37-1.49 (m, 8H).
実施例2D
化合物2Dの合成:
1-シクロヘキシル-1-キノキサリン-2-イル-ヒドラジンの合成:
マイクロ波反応容器に、トリエチルアミン(10mL)中の2-クロロキノキサリン(1.65g、10mmol)及びシクロヘキシルヒドラジン塩酸塩(1.81g、12mmol)を仕込んだ。混合物をマイクロ波反応器中で90℃まで20分間加熱した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、酢酸エチルで抽出した。有機相を水及び食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。残渣をヘキサンで洗浄して、標題化合物を黄色の固体(0.70g、29%)として与えた。
1H-NMR (400 MHz, CDCl3) δ 8.98 (s, 1H), 7.86-7.89 (m, 1H), 7.65-7.68 (m, 1H), 7.53-7.57 (m, 1H), 7.35-7.39 (m, 1H), 4.46-4.53 (m, 1H), 3.85 (s, 2H), 1.88-1.92 (m, 2H), 1.64-1.81 (m, 5H), 1.45-1.56 (m, 2H), 1.14-1.26 (m, 1H)
Example 2D
Synthesis of compound 2D:
Synthesis of 1-cyclohexyl-1-quinoxalin-2-yl-hydrazine:
A microwave reactor was charged with 2-chloroquinoxaline (1.65 g, 10 mmol) and cyclohexylhydrazine hydrochloride (1.81 g, 12 mmol) in triethylamine (10 mL). The mixture was heated to 90° C. for 20 min in a microwave reactor. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with ethyl acetate. The organic phase was washed with water and brine, dried over anhydrous sodium sulfate and the solvent removed under reduced pressure. The residue was washed with hexane to give the title compound as a yellow solid (0.70 g, 29%).
1 H-NMR (400 MHz, CDCl 3 ) δ 8.98 (s, 1H), 7.86-7.89 (m, 1H), 7.65-7.68 (m, 1H), 7.53-7.57 (m, 1H), 7.35-7.39 (m, 1H), 4.46-4.53 (m, 1H), 3.85 (s, 2H), 1.88-1.92 (m, 2H), 1.64-1.81 (m, 5H), 1.45-1.56 (m, 2H), 1.14-1.26 (m, 1H)
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-[(E)-[シクロヘキシル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](化合物2D)の合成:
(±)10-カンファースルホン酸(33mg、0.14mmol)を無水テトラヒドロフラン(14mL)中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[3-ホルミル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](0.97g、1.42mmol)、1-シクロヘキシル-1-キノキサリン-2-イル-ヒドラジン(0.36g、1.5mmol)、及び4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン 1-オキシル フリーラジカル(TEMPO-OH)(2mg、0.01mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で23時間撹拌した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/メタノールからの再結晶後、標題化合物を黄色の固体(1.18g、91%)として得た。
1H-NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.26 (s, 1H), 8.17 (d, J = 6.9 Hz, 2H), 8.13 (d, J = 9.1 Hz, 2H), 8.01 (d, J = 2.7 Hz, 1H), 7.87 (d, J = 9.1 Hz, 1H), 7.69 (dd, J = 8.2, 1.4 Hz, 1H), 7.65 (td, J = 7.5, 1.4 Hz, 1H), 7.51-7.55 (m, 1H), 7.45 (d, J = 8.7 Hz, 1H), 7.38 (dd, J = 8.9, 3.0 Hz, 1H), 7.13-7.15 (m, 2H), 7.11-7.12 (m, 2H), 6.32 (dd, J = 17.4, 1.8 Hz, 2H), 6.13-6.20 (m, 2H), 5.93 (dd, J = 10.3, 1.6 Hz, 2H), 4.86-4.94 (m, 1H), 4.07-4.13 (m, 8H), 2.15-2.25 (m, 2H), 1.72-1.79 (m, 4H), 1.61-1.68 (m, 8H), 1.37-1.50 (m, 9H), 1.21 (q, J = 13.0 Hz, 2H), 0.68-0.77 (m, 1H).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-[(E)-[cyclohexyl(quinoxalin-2-yl)hydrazono]methyl]-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (Compound 2D)
(±) 10-Camphorsulfonic acid (33 mg, 0.14 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [3-formyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (0.97 g, 1.42 mmol), 1-cyclohexyl-1-quinoxalin-2-yl-hydrazine (0.36 g, 1.5 mmol), and 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl free radical (TEMPO-OH) (2 mg, 0.01 mmol) in anhydrous tetrahydrofuran (14 mL) under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 23 h. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure to give the title compound as a yellow solid (1.18 g, 91%) after recrystallization from dichloromethane/methanol.
1 H-NMR (400 MHz, DMSO-d6) δ 9.34 (s, 1H), 8.26 (s, 1H), 8.17 (d, J = 6.9 Hz, 2H), 8.13 (d, J = 9.1 Hz, 2H), 8.01 (d, J = 2.7 Hz, 1H), 7.87 (d, J = 9.1 Hz, 1H), 7.69 (dd, J = 8.2, 1.4 Hz, 1H), 7.65 (td, J = 7.5, 1.4 Hz, 1H), 7.51-7.55 (m, 1H), 7.45 (d, J = 8.7 Hz, 1H), 7.38 (dd, J = 8.9, 3.0Hz, 1H), 7.13-7.15 (m, 2H), 7.11-7.12 (m, 2H), 6.32 (dd, J = 17.4, 1.8 Hz, 2H), 6.13-6.20 (m, 2H), 5.93 (dd, J = 10.3, 1.6 Hz, 2H), 4.86-4.94 (m, 1H), 4.07-4.13 (m, 8H), 2.15-2.25 (m, 2H), 1.72-1.79 (m, 4H), 1.61-1.68 (m, 8H), 1.37-1.50 (m, 9H), 1.21 (q, J = 13.0 Hz, 2H), 0.77 (m, 1H).
実施例2E
化合物2Eの合成:
2,5-ジヒドロキシ-4-メチル-ベンズアルデヒドの合成:
無水アセトニトリル(15mL)中のパラホルムアルデヒド(2.1g、70mmol)、塩化マグネシウム(1.43g、15mmol)、トリエチルアミン(5.6mL、40mmol)の懸濁液に、メチルヒドロキノン(1.24g、10mmol)を加えた。混合物を窒素雰囲気下で撹拌しながら17時間還流した。反応混合物を周囲温度まで冷却し、1N HCl水溶液でクエンチし、酢酸エチルで抽出して、食塩水で洗浄した。有機層を無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。酢酸エチル/ヘキサンを用いて、残渣をシリカゲルのフラッシュカラムクロマトグラフィーにより精製して、標題化合物を黄色の固体(0.31g、20%)として与えた。
1H-NMR (400 MHz, MeOD) δ 9.85 (s, 1H), 6.95 (s, 1H), 6.70 (s, 1H), 2.21 (s, 3H).
Example 2E
Synthesis of compound 2E:
Synthesis of 2,5-dihydroxy-4-methyl-benzaldehyde:
To a suspension of paraformaldehyde (2.1 g, 70 mmol), magnesium chloride (1.43 g, 15 mmol), triethylamine (5.6 mL, 40 mmol) in anhydrous acetonitrile (15 mL) was added methylhydroquinone (1.24 g, 10 mmol). The mixture was refluxed with stirring under nitrogen atmosphere for 17 h. The reaction mixture was cooled to ambient temperature, quenched with 1N aqueous HCl, extracted with ethyl acetate, and washed with brine. The organic layer was dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with ethyl acetate/hexanes to give the title compound as a yellow solid (0.31 g, 20%).
1H -NMR (400 MHz, MeOD) δ 9.85 (s, 1H), 6.95 (s, 1H), 6.70 (s, 1H), 2.21 (s, 3H).
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[2-ホルミル-5-メチル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル]の合成:
無水トルエン(15mL)及び無水ジメチルホルムアミド(DMF)(0.3mL)中の4-(6-アクリロイルオキシ-ヘキサ-1-イルオキシ)安息香酸(2.19g、7.5mmol)及び4-メトキシフェノール(47mg、0.4mmol)の溶液に、塩化オキサリル(0.77mL、9mmol)を45℃にて15分間で滴下した。19時間撹拌後、反応混合物を周囲温度まで冷却し、無水ジメチルアセトアミド(11mL)中のBuilding block K(0.51g、3.4mmol)及びN,N-ジメチルシクロヘキシルアミン(2.5mL、17mmol)の溶液に0~5℃にて20分間で滴下した。反応混合物を周囲温度で3時間撹拌した。反応混合物を1M HCl水溶液及び氷水浴の添加によりクエンチし、ジクロロメタンで抽出して、水、食塩水で連続して洗浄し、硫酸ナトリウムで乾燥させ、真空下で濃縮した。ジクロロメタン/メタノールからの再結晶後、標題化合物を桃色の固体(1.38g、59%)として得た。
1H-NMR (400 MHz, CDCl3) δ 10.15 (s, 1H), 8.15-8.18 (m, 4H), 7.71 (s, 1H), 6.98-7.01 (m, 5H), 6.41 (dd, J = 17.4, 1.4 Hz, 2H), 6.13 (dd, J = 17.4, 10.5 Hz, 2H), 5.83 (dd, J = 10.3, 1.6 Hz, 2H), 4.15-4.21 (m, 8H), 4.07 (t, J = 5.9 Hz, 8H), 2.32 (s, 3H), 1.81-1.89 (m, 4H), 1.69-1.77 (m, 4H), 1.48-1.54 (m, 8H).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [2-formyl-5-methyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl]
To a solution of 4-(6-acryloyloxy-hex-1-yloxy)benzoic acid (2.19 g, 7.5 mmol) and 4-methoxyphenol (47 mg, 0.4 mmol) in anhydrous toluene (15 mL) and anhydrous dimethylformamide (DMF) (0.3 mL) was added oxalyl chloride (0.77 mL, 9 mmol) dropwise over 15 min at 45° C. After stirring for 19 h, the reaction mixture was cooled to ambient temperature and added dropwise over 20 min to a solution of building block K (0.51 g, 3.4 mmol) and N,N-dimethylcyclohexylamine (2.5 mL, 17 mmol) in anhydrous dimethylacetamide (11 mL) at 0-5° C. The reaction mixture was stirred at ambient temperature for 3 h. The reaction mixture was quenched by the addition of 1 M aqueous HCl and an ice-water bath, extracted with dichloromethane, washed successively with water, brine, dried over sodium sulfate and concentrated in vacuo. After recrystallization from dichloromethane/methanol, the title compound was obtained as a pink solid (1.38 g, 59%).
1 H-NMR (400 MHz, CDCl 3 ) δ 10.15 (s, 1H), 8.15-8.18 (m, 4H), 7.71 (s, 1H), 6.98-7.01 (m, 5H), 6.41 (dd, J = 17.4, 1.4 Hz, 2H), 6.13 (dd, J = 1 7.4, 10.5 Hz, 2H), 5.83 (dd, J = 10.3, 1.6 Hz, 2H), 4.15-4.21 (m, 8H), 4.07 (t, J = 5.9 Hz, 8H), 2.32 (s, 3H), 1.81-1.89 (m, 4H), 1.69-1. 77 (m, 4H), 1.48-1.54 (m, 8H).
4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[5-メチル-2-[(E)-[メチル(キノキサリン-2-イル)ヒドラゾノ]メチル]-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](化合物2E)の合成:
(±)10-カンファースルホン酸(44mg、0.19mmol)を無水テトラヒドロフラン(20mL)中の4-(6-プロパ-2-エノイルオキシヘキソキシ)安息香酸[2-ホルミル-5-メチル-4-[4-(6-プロパ-2-エノイルオキシヘキソキシ)ベンゾイル]オキシ-フェニル](1.3g、1.9mmol)、1-メチル-1-キノキサリン-2-イル-ヒドラジン(0.34g、2.0mmol)、及び4-ヒドロキシ-2,2,6,6-テトラメチルピペリジン 1-オキシル フリーラジカル(TEMPO-OH)(4mg、0.02mmol)の溶液に窒素雰囲気下で加えた。得られた溶液を周囲温度で22時間撹拌した。反応混合物を重炭酸ナトリウムの飽和水溶液でクエンチして、ジクロロメタンで抽出した。有機相を食塩水で洗浄し、無水硫酸ナトリウムで乾燥させ、減圧下で溶媒を除去した。ジクロロメタン/ヘキサンからの再結晶後、標題化合物を黄色の固体(1.20g、75%)として得た。
1H-NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 8.14-8.16 (m, 4H), 8.01 (s, 1H), 7.87-7.89 (m, 2H), 7.73-7.75 (m, 1H), 7.65-7.69 (m, 1H), 7.49-7.53 (m, 1H), 7.34 (s, 1H), 7.11-7.15 (m, 4H), 6.32 (dd, J = 17.2, 1.6 Hz, 2H), 6.17 (dd, J = 17.4, 10.5 Hz, 2H), 5.93 (dd, J = 10.3, 1.6 Hz, 2H), 4.07-4.14 (m, 8H), 3.61 (s, 3H), 2.20 (s, 3H), 1.72-1.80 (m, 4H), 1.59-1.68 (m, 4H), 1.36-1.51 (m, 8H).
Synthesis of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [5-methyl-2-[(E)-[methyl(quinoxalin-2-yl)hydrazono]methyl]-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (Compound 2E)
(±) 10-Camphorsulfonic acid (44 mg, 0.19 mmol) was added to a solution of 4-(6-prop-2-enoyloxyhexoxy)benzoic acid [2-formyl-5-methyl-4-[4-(6-prop-2-enoyloxyhexoxy)benzoyl]oxy-phenyl] (1.3 g, 1.9 mmol), 1-methyl-1-quinoxalin-2-yl-hydrazine (0.34 g, 2.0 mmol), and 4-hydroxy-2,2,6,6-tetramethylpiperidine 1-oxyl free radical (TEMPO-OH) (4 mg, 0.02 mmol) in anhydrous tetrahydrofuran (20 mL) under nitrogen atmosphere. The resulting solution was stirred at ambient temperature for 22 h. The reaction mixture was quenched with a saturated aqueous solution of sodium bicarbonate and extracted with dichloromethane. The organic phase was washed with brine, dried over anhydrous sodium sulfate and the solvent was removed under reduced pressure to give the title compound as a yellow solid (1.20 g, 75%) after recrystallization from dichloromethane/hexanes.
1 H-NMR (400 MHz, DMSO-d6) δ 9.41 (s, 1H), 8.14-8.16 (m, 4H), 8.01 (s, 1H), 7.87-7.89 (m, 2H), 7.73-7.75 (m, 1H), 7.65-7.69 (m, 1H), 7. 49-7.53 (m, 1H), 7.34 (s, 1H), 7.11-7.15 (m, 4H), 6.32 (dd, J = 17.2, 1.6 Hz, 2H), 6.17 (dd, J = 17.4, 10.5 Hz, 2H), 5.93 (dd, J = 10.3, 1.6 Hz, 2H), 4.07-4.14 (m, 8H), 3.61 (s, 3H), 2.20 (s, 3H), 1.72-1.80 (m, 4H), 1.59-1.68 (m, 4H), 1.36-1.51 (m, 8H).
実施例3
相転移温度及び中間相テクスチャーは、実施例1~2Eの化合物を使用して以下に記載されるとおり示差走査熱量測定法及び偏光顕微鏡法によって求められ、公開特許WO2012/141245に記載された化合物R1と比較された。
Example 3
The phase transition temperatures and mesophase textures were determined by differential scanning calorimetry and polarized light microscopy as described below using the compounds of Examples 1-2E and compared with compound R1 described in published patent WO2012/141245.
偏光顕微鏡法:
各化合物3mgをホットステージオーブン内の2枚のスライドガラスの間に置き、25℃から200℃まで加熱し、次に再び25℃まで冷却した。特徴的な複屈折テクスチャーは、偏光顕微鏡を使用して分析された。ラビング処理ポリイミド層でコーティングされたガラス基板も、化合物の中間相挙動を確認するために使用された。
Polarized light microscopy:
Three milligrams of each compound was placed between two glass slides in a hot stage oven, heated from 25°C to 200°C, and then cooled again to 25°C. The characteristic birefringent texture was analyzed using a polarized light microscope. Glass substrates coated with a rubbed polyimide layer were also used to confirm the mesophase behavior of the compounds.
示差走査熱量測定法:
密封されたアルミニウムパンに入れられた各化合物約5mgを、10℃/分のスキャン速度で2回の連続した加熱-冷却サークルに付した。転移温度は、得られたDSCサーモグラムの吸熱/発熱ピークから求められた。
Differential Scanning Calorimetry:
Approximately 5 mg of each compound in a sealed aluminum pan was subjected to two successive heating-cooling cycles at a scan rate of 10° C./min. Transition temperatures were determined from the endothermic/exothermic peaks of the resulting DSC thermograms.
全ての相転移の結果は表1に要約される。化合物の状態は、固体結晶相には「Cr」、ネマチックには「N」、そして等方性液体状態には「Iso」として示される。 All phase transition results are summarized in Table 1. The state of the compound is indicated as "Cr" for solid crystalline phase, "N" for nematic, and "Iso" for isotropic liquid state.

実施例4
光学フィルムの調製
ラビング処理基板上でのLCPコーティングフィルムの調製及び評価の一般的な手順
液晶化合物0.26gをシクロペンタノン830μLに溶解した。BYK361の1重量% シクロペンタノン溶液13μLを加え、得られた溶液を0.45μm PTFEフィルターで濾過した。Irgacure907 13mgを加えて、重合性組成物(1~2)を得た。
Example 4
Preparation of Optical Films General Procedure for Preparation and Evaluation of LCP Coating Films on Rubbed Substrates 0.26 g of liquid crystal compound was dissolved in 830 μL of cyclopentanone. 13 μL of 1 wt% cyclopentanone solution of BYK361 was added, and the resulting solution was filtered through a 0.45 μm PTFE filter. 13 mg of Irgacure907 was added to obtain polymerizable compositions (1-2).
各重合性組成物は、ラビング処理ポリイミド層(EHC製)でコーティングされたガラス基板に適用された。表2に示される温度で4分間フィルムを乾燥/アニーリングした後、1000mJ/cm2の線量でUVスポットキュア(ウシオ製のSP-7)を使用して、フィルムを光硬化させた。表2に記載された温度でUV曝露を行った。 Each polymerizable composition was applied to a glass substrate coated with a rubbed polyimide layer (EHC). After drying/annealing the film for 4 minutes at the temperature indicated in Table 2, the film was photocured using a UV spot cure (Ushio SP-7) at a dose of 1000 mJ/ cm2 . UV exposure was performed at the temperature listed in Table 2.

実施例5
可視領域(400~700nm)のリタデーションは、偏光顕微鏡、セナルモン(Senarmont)補償板及び光学フィルターを使用した偏光回転法によって測定された。面内リタデーションR0は、特定波長λでの消光位置θから次の式を使用して求められる。

The retardation in the visible region (400-700 nm) was measured by the polarization rotation method using a polarizing microscope, a Senarmont compensator and an optical filter. The in-plane retardation R0 can be calculated from the extinction position θ at a specific wavelength λ using the following equation:
フィルム厚さdは、BrukerのDektak(登録商標)スタイラスプロファイラーを使用して測定される。フィルムの複屈折Δnは、次の式から導かれる:R0=Δn.d The film thickness d is measured using a Bruker Dektak® stylus profiler. The birefringence Δn of the film is derived from the following equation: R 0 =Δn.d
結果は、液晶フィルムが、可視光領域の波長の増加に関連して増加するリタデーションで製造されたことを示している。結果を図1に示すが、ここで、実施例1、2、2B及び2Dの複屈折の波長分散がプロットされている。 The results show that liquid crystal films were produced with increasing retardation relative to increasing wavelengths in the visible light range. The results are shown in Figure 1, where the wavelength dispersion of birefringence for Examples 1, 2, 2B and 2D is plotted.
実施例6
化合物R1、1、2、2A、2B、2D及び2Eについての光配向層上のLCPコーティングフィルム調製の一般的な手順:
光配向材料を使用した配向層の調製:
ガラス基板に光配向組成物(公開特許WO2012/085048の40ページの適用例に記載されているシクロペンタノン中の2%固形分の光配向ポリマー)をスピンコートした。フィルムを80℃で30秒間乾燥させ、得られたフィルムの厚さは約100nmであった。次にフィルムをアライニング光に曝露したが、この光は、500mJ/cm2の平行及び直線偏光UV(LPUV)光(280~320nm)であった。偏光面は、基板上の基準エッジに対して0°であった。
Example 6
General procedure for preparation of LCP coating film on photoalignment layer for compounds R1, 1, 2, 2A, 2B, 2D and 2E:
Preparation of alignment layer using photoalignment material:
A glass substrate was spin-coated with a photoalignment composition (a photoalignment polymer at 2% solids in cyclopentanone as described in the application example on page 40 of published patent WO2012/085048). The film was dried at 80°C for 30 seconds, and the resulting film had a thickness of about 100 nm. The film was then exposed to aligning light, which was 500 mJ/ cm2 of parallel and linearly polarized UV (LPUV) light (280-320 nm). The polarization plane was at 0° with respect to a reference edge on the substrate.
LCP配合物の調製:
LCPの15.0重量%溶液は、シクロヘキサノン中の14.325重量%のLCP、0.075重量%の阻害剤2,6-ジ-tert-ブチル-4-メチルフェノール(未成熟重合を防ぐため)、0.45重量%の光開始剤Irgacure 369、0.15重量%のTinuvin 123を混合することによって調製され、固体が室温で完全に溶解するまで十分に撹拌される。上記配向層を有するガラス板上にコーティング液を塗布し、スピンコーティングにより液晶フィルムを形成した。表3に示される時間と温度でフィルムを乾燥させた後、試料を室温まで冷却する。次に、表3に示されるおおよその時間と温度で、UV光(水銀灯を用いて)を照射することにより、それを光重合させた。
Preparation of LCP formulations:
A 15.0 wt% solution of LCP was prepared by mixing 14.325 wt% LCP, 0.075 wt% inhibitor 2,6-di-tert-butyl-4-methylphenol (to prevent premature polymerization), 0.45 wt% photoinitiator Irgacure 369, and 0.15 wt% Tinuvin 123 in cyclohexanone and thoroughly stirred until the solids were completely dissolved at room temperature. The coating solution was applied onto a glass plate having the above alignment layer, and a liquid crystal film was formed by spin coating. After drying the film at the time and temperature shown in Table 3, the sample was cooled to room temperature. It was then photopolymerized by irradiating it with UV light (using a mercury lamp) at the approximate time and temperature shown in Table 3.

実施例7
実施例6で製造されたフィルムのリタデーションは、エリプソメトリーによって測定された。図2は、実施例6で製造されたフィルムのリタデーション分散を示す。
Example 7
The retardation of the film prepared in Example 6 was measured by ellipsometry. Figure 2 shows the retardation dispersion of the film prepared in Example 6.
上記の結果は、例示された化合物で製造された液晶フィルムが、可視光領域の波長の増加に関連して増加するリタデーションを示すことを示した。 The above results show that liquid crystal films prepared with the exemplified compounds exhibit increasing retardation with increasing wavelength in the visible light range.
表4は、各試料のRe450/Re550及びRe650/Re550の値を示す。Re450は波長450nmでのフィルムのリタデーションを、Re550は波長550nmでのフィルムのリタデーションを、そしてRe650は波長650nmでのフィルムのリタデーションを表す。 Table 4 shows the values of Re 450 /Re 550 and Re 650 /Re 550 for each sample, where Re 450 represents the retardation of the film at a wavelength of 450 nm, Re 550 represents the retardation of the film at a wavelength of 550 nm, and Re 650 represents the retardation of the film at a wavelength of 650 nm.

表4は、液晶組成物フィルムのリタデーション特性の結果を示す。色を改善するには、Re650/Re550の値が1.00より大きく、かつ、1.2未満であることが好ましい。実施例1、実施例2、実施例2A、実施例2B、実施例2D、実施例2Eからの化合物で製造された液晶重合フィルムのRe650/Re550は、比較例R1の液晶組成物のRe650/Re550よりも有意に大きく、全てのRe650/Re550値が1.01を上回り最大1.09であることが見出された。更には、0.90未満のRe450/Re550値が好ましい。 Table 4 shows the results of retardation properties of liquid crystal composition film. To improve color, it is preferable that Re 650 /Re 550 value is greater than 1.00 and less than 1.2. It is found that Re 650 /Re 550 of liquid crystal polymer film made with compound from Example 1, Example 2, Example 2A, Example 2B, Example 2D, Example 2E is significantly greater than Re 650 /Re 550 of liquid crystal composition of Comparative Example R1, and all Re 650 /Re 550 values are greater than 1.01 and up to 1.09. Furthermore, Re 450 / Re 550 value less than 0.90 is preferable.
実施例8
化合物2Bの30.0重量%溶液は、シクロヘキサノン中の22.635重量%の化合物2B、0.015重量%の阻害剤2,6-ジ-tert-ブチル-4-メチルフェノール、0.9重量%の光開始剤Irgacure(登録商標)Oxe 03(BASF Corporation製)、0.45重量%のTego(登録商標)flow 425、6重量%のO1-(2-プロパ-2-エノイルオキシエチル)ペンタン二酸O5-[4-[3-メチル-4-[4-[5-オキソ-5-(2-プロパ-2-エノイルオキシエトキシ)ペンタノイル]オキシベンゾイル]オキシ-フェノキシ]カルボニルフェニル](LCP1、米国特許US2012114907に記載)を混合することによって調製され、固体が室温で完全に溶解するまで十分に撹拌された。フィルムを実施例20に記載されるとおり調製して、温度制御ホットプレートで97℃にて2分間乾燥させた。得られたフィルムは、良好なアラインメント品質を示した。
Example 8
A 30.0 wt% solution of compound 2B was prepared by mixing 22.635 wt% compound 2B, 0.015 wt% inhibitor 2,6-di-tert-butyl-4-methylphenol, 0.9 wt% photoinitiator Irgacure® Oxe 03 (BASF Corporation), 0.45 wt% Tego® flow 425, 6 wt% O1-(2-prop-2-enoyloxyethyl)pentanedioic acid O5-[4-[3-methyl-4-[4-[5-oxo-5-(2-prop-2-enoyloxyethoxy)pentanoyl]oxybenzoyl]oxy-phenoxy]carbonylphenyl] (LCP1, described in US Patent US2012114907) in cyclohexanone and thoroughly stirred until the solids were completely dissolved at room temperature. The film was prepared as described in Example 20 and dried on a temperature controlled hot plate at 97° C. for 2 minutes. The resulting film showed good alignment quality.
実施例9
化合物2Bの30.0重量%溶液は、シクロヘキサノン中の25.635重量%の化合物2B、0.015重量%の阻害剤2,6-ジ-tert-ブチル-4-メチルフェノール、0.90重量%のIrgacure(登録商標)Oxe03、0.45重量%のTego(登録商標)flow 425、3重量%の6-(4-プロパ-2-エノイルオキシブトキシカルボニルオキシ)ナフタレン-2-カルボン酸[4-[6-(4-プロパ-2-エノイルオキシブトキシカルボニルオキシ)ナフタレン-2-カルボニル]オキシフェニル](LCP2、US 7,670,505 B2に記載)を混合することによって調製され、固体が室温で完全に溶解するまで十分に撹拌された。フィルムを実施例20に記載されるとおり調製して、温度制御ホットプレートで105℃にて2分間乾燥させた。得られたフィルムは、良好なアラインメント品質を示した。
Example 9
A 30.0 wt% solution of compound 2B was prepared by mixing 25.635 wt% compound 2B, 0.015 wt% inhibitor 2,6-di-tert-butyl-4-methylphenol, 0.90 wt% Irgacure® Oxe03, 0.45 wt% Tego® flow 425, 3 wt% 6-(4-prop-2-enoyloxybutoxycarbonyloxy)naphthalene-2-carboxylic acid [4-[6-(4-prop-2-enoyloxybutoxycarbonyloxy)naphthalene-2-carbonyl]oxyphenyl] (LCP2, described in US 7,670,505 B2) in cyclohexanone and thoroughly stirred until the solids were completely dissolved at room temperature. A film was prepared as described in Example 20 and dried on a temperature controlled hot plate at 105°C for 2 minutes. The resulting film showed good alignment quality.
実施例10
実施例8及び9に記載された試料のリタデーションは、エリプソメーターで測定された。表5は、液晶組成物に依存する様々なフィルムのリタデーション特性の結果を示す。
Example 10
The retardation of the samples described in Examples 8 and 9 was measured by an ellipsometer. Table 5 shows the results of the retardation properties of various films depending on the liquid crystal composition.
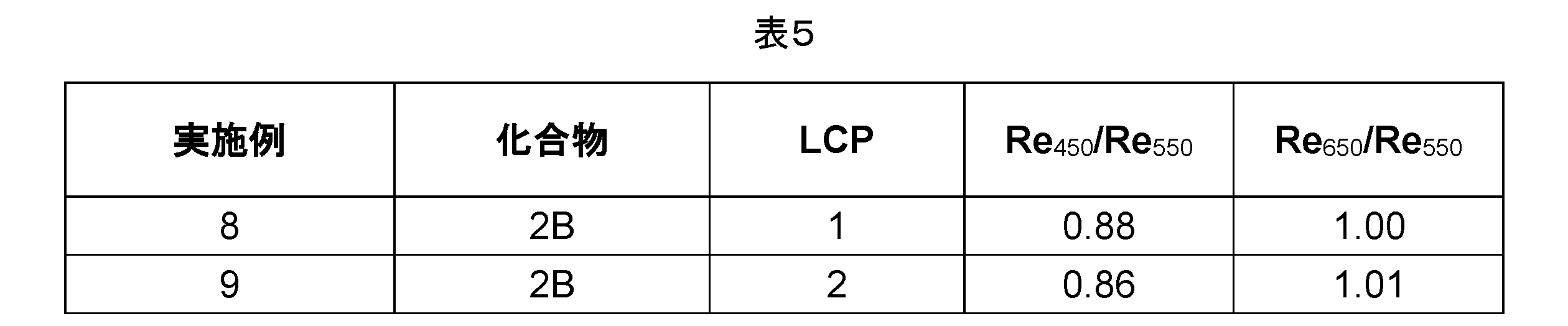
液晶組成物を使用する場合、実施例8及び9に記載されたフィルムは、液晶組成物の様々な成分の割合を調節することにより、リタデーションを微調整し、そして要求に応じてRe450/Re550及びRe650/Re550値を得ることが可能であることが見出された。 When using a liquid crystal composition, it was found that the films described in Examples 8 and 9 allow fine tuning of the retardation and obtaining Re450 / Re550 and Re650 / Re550 values as required by adjusting the ratio of various components of the liquid crystal composition.
Claims (13)

[式中、
R1、R2及びR3は、互いに独立して、水素、C1~C12直鎖又は分岐のアルキル鎖、C3~C12アルケニル、C1~C12アルコキシ、C3~C12アルケニルオキシ、-(CH2)m-C(CH3)3、NO2、CN、COR、-COOR、-OCOR、-CONR’R、-NR’COR、OCOOR、-OCONR’R、-NR’COOR、-F、-Cl、-CF3及び-OCF3からなる群より選択され;
ここで、
mは、0~12の整数であり;
Rは、水素、C1~18アルキル基、3位以上に二重結合を持つC3~18アルケニル基、-(CH2)p-C-(CF3)3、CN及び非置換又は置換のフェニル環からなる群より選択され、ここで、フェニル環の置換基は、C1~C6直鎖又は分岐のアルキル鎖、C1~C6アルコキシ、-C-(CH3)3、ハロゲン、-CF3、NO2、CN、COR'''、-COOR'''、-OCOR'''、-CONR''R'''、-NR''COR'''、OCOOR'''、-OCONR''R'''、-NR''COOR'''、-F、-Cl、-CF3及び-OCF3からなる群より選択され;
ここで、
R''は、水素、低級アルキル基及び低級アルケニル基からなる群より選択され、低級アルキル基は、メチル、エチル、プロピル、ブチル、ペンチル又はヘキシルであり、低級アルケニル基は、2-プロペニル、3-ブテニル、3-イソペンテニル、4-ペンテニル、5-ヘキセニル又は4-イソヘキセニルであり;
R'''は、水素、C1~18アルキル基及び3位以上に二重結合を持つC3~18アルケニル基からなる群より選択され;
pは、0~12の整数であり;
R’は、水素、低級アルキル、低級アルケニル及び低級アルコキシからなる群より選択され、低級アルキルは、メチル、エチル、プロピル、ブチル、ペンチル又はヘキシルであり、低級アルケニルは、2-プロペニル、3-ブテニル、3-イソペンテニル、4-ペンテニル、5-ヘキセニル又は4-イソヘキセニルであり、低級アルコキシは、メトキシ、エトキシ、プロポキシ、ブトキシ、ペントキシ又はヘキソキシであり;
nは、0、1、2又は3であり;
Yは、H、又は1~12個の炭素原子を有する置換若しくは非置換のアルキル基からなる群より選択され;
Zは、水素、1~20個の炭素原子を有する置換又は非置換のアルキル基、2~20個の炭素原子を有する置換又は非置換のアルケニル基、3~12個の炭素原子を有する置換又は非置換のシクロアルキル基、2~20個の炭素原子を有する置換又は非置換のアルキニル基からなる群より選択されるが、ここで、1個以上の炭素原子は、-O-、-COO-、-OCO-、-OOC-、-O(CO)O-、-N-、-NRa-、-CON-、-CO-Rb、-NH-Rcにより置き換えられていてもよく、ここで、Raは、C1~C12アルキル基であり、Rb及びRcは、互いに独立して、1~20個の炭素原子を有する置換若しくは非置換のアルキル基、又は少なくとも1個の芳香環を包含する2~30個の炭素原子を有する有機基、又は2~20個の炭素原子を有する置換若しくは非置換のアルケニル基、又は3~12個の炭素原子を有する置換若しくは非置換のシクロアルキル基であり;
環C及びDは、互いに独立して、フェニル環又はシクロアルキル環であり、ただし、環C又はDの少なくとも一方は、フェニル環であり;
環Eは、フェニル環であり;
置換基X 1 は、C 1~C12の置換又は非置換の直鎖又は分岐のアルキル鎖及びC1~C12アルコキシからなる群より選択されるが、ここで、1個以上の炭素原子は、-O-、-COO-、-OOC-、-O(CO)O-により置き換えられていてもよいか;あるいは
置換基X 1 は、式(II):
で示される基により表され;
置換基X 2 は、式(II):

で示される基により表され;
ここで、式(II)の基において、nは、0~24の整数であり、そして式(II)の-(CH 2 ) n -部分において、1個以上の炭素原子は、-O-、-COO-、-OOC-、-O(CO)O-により置き換えられていてもよく;
式(II)の基のPGは、CH2=CW-COO-からなる群より選択される重合性基を表すが;ここで、Wは、H、メチル、エチル、プロピル、ブチル、ペンチル又はヘキシルを表す]で示される異方性化合物。 Formula (I):
[Wherein,
R 1 , R 2 and R 3 are each independently selected from the group consisting of hydrogen, C 1 -C 12 straight or branched alkyl chain, C 3 -C 12 alkenyl, C 1 -C 12 alkoxy, C 3 -C 12 alkenyloxy, -(CH 2 ) m -C(CH 3 ) 3 , NO 2 , CN, COR, -COOR, -OCOR, -CONR'R, -NR'COR, OCOOR, -OCONR'R, -NR'COOR, -F, -Cl, -CF 3 and -OCF 3 ;
Where:
m is an integer from 0 to 12;
R is selected from the group consisting of hydrogen, a C 1-18 alkyl group, a C 3-18 alkenyl group having a double bond at position 3 or higher, -(CH 2 ) p -C-(CF 3 ) 3 , CN, and an unsubstituted or substituted phenyl ring, where the substituents on the phenyl ring are selected from the group consisting of a C 1 -C 6 straight or branched alkyl chain, a C 1 -C 6 alkoxy, -C-(CH 3 ) 3 , halogen, -CF 3 , NO 2 , CN, COR''', -COOR'''', -OCOR'''', -CONR''R'''', -NR''COR'''', OCOOR'''', -OCONR''R'''', -NR''COOR'''', -F, -Cl, -CF 3 , and -OCF 3 ;
Where:
R″ is selected from the group consisting of hydrogen, a lower alkyl group, and a lower alkenyl group , the lower alkyl group being methyl, ethyl, propyl, butyl, pentyl, or hexyl, and the lower alkenyl group being 2-propenyl, 3-butenyl, 3-isopentenyl, 4-pentenyl, 5-hexenyl, or 4-isohexenyl ;
R''' is selected from the group consisting of hydrogen, a C1-18 alkyl group, and a C3-18 alkenyl group having a double bond at the 3rd or higher position;
p is an integer from 0 to 12;
R' is selected from the group consisting of hydrogen, lower alkyl, lower alkenyl and lower alkoxy , where lower alkyl is methyl, ethyl, propyl, butyl, pentyl or hexyl, lower alkenyl is 2-propenyl, 3-butenyl, 3-isopentenyl, 4-pentenyl, 5-hexenyl or 4-isohexenyl and lower alkoxy is methoxy, ethoxy, propoxy, butoxy, pentoxy or hexoxy ;
n is 0, 1, 2 or 3;
Y is selected from the group consisting of H or a substituted or unsubstituted alkyl group having 1 to 12 carbon atoms;
Z is selected from the group consisting of hydrogen, a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, a substituted or unsubstituted alkenyl group having 2 to 20 carbon atoms, a substituted or unsubstituted cycloalkyl group having 3 to 12 carbon atoms, a substituted or unsubstituted alkynyl group having 2 to 20 carbon atoms, where one or more carbon atoms may be replaced by -O-, -COO-, -OCO-, -OOC-, -O(CO)O-, -N-, -NR a -, -CON-, -CO-R b , -NH-R c , where R a is a C 1 to C 12 alkyl group, R b and R c are each independently a substituted or unsubstituted alkyl group having 1 to 20 carbon atoms, or an organic group having 2 to 30 carbon atoms containing at least one aromatic ring, or a substituted or unsubstituted alkenyl group having 2 to 20 carbon atoms, or a substituted or unsubstituted cycloalkyl group having 3 to 12 carbon atoms;
Rings C and D are, independently of each other, a phenyl ring or a cycloalkyl ring , provided that at least one of rings C or D is a phenyl ring ;
Ring E is a phenyl ring ;
The substituent X 1 is selected from the group consisting of C 1 -C 12 substituted or unsubstituted linear or branched alkyl chains and C 1 -C 12 alkoxy, where one or more carbon atoms may be replaced by -O-, -COO-, -OOC-, -O(CO)O-; or the substituent X 1 is selected from the group consisting of C 1 -C 12 substituted or unsubstituted linear or branched alkyl chains and C 1 -C 12 alkoxy, where one or more carbon atoms may be replaced by -O-, -COO- , -OOC-, -O(CO)O-;
Represented by the group represented by:
The substituent X2 is represented by the formula (II):
Represented by the group represented by:
wherein in the group of formula (II), n is an integer from 0 to 24, and in the -(CH2)n- moiety of formula (II) , one or more carbon atoms may be replaced by -O-, -COO- , -OOC-, -O(CO)O- ;
An anisotropic compound of formula (II) wherein the group PG represents a polymerizable group selected from the group consisting of C H 2 ═CW—COO—; where W represents H, methyl, ethyl, propyl, butyl, pentyl or hexyl .

[式中、nは、0~24の整数である]で示される基並びにそれらの対応するメタクリラートからなる群より選択される、請求項1~3のいずれか一項に記載の化合物。 When both rings C and D are phenyl rings , the groups of formula (II) representing X1 and X2 are each independently of one another represented by the following formula:
4. The compound according to claim 1 , wherein n is an integer from 0 to 24, and the corresponding methacrylates thereof.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP19183356 | 2019-06-28 | ||
| EP19183356.5 | 2019-06-28 | ||
| EP19194041.0 | 2019-08-28 | ||
| EP19194041 | 2019-08-28 | ||
| PCT/EP2020/068081 WO2020260621A1 (en) | 2019-06-28 | 2020-06-26 | New polymerizable liquid crystal having a quinoxaline-hydrazone core |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022539176A JP2022539176A (en) | 2022-09-07 |
| JP7538155B2 true JP7538155B2 (en) | 2024-08-21 |
Family
ID=71138765
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021577566A Active JP7538155B2 (en) | 2019-06-28 | 2020-06-26 | Novel polymerizable liquid crystals with a quinoxaline-hydrazone core |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US12006463B2 (en) |
| EP (1) | EP3990568B1 (en) |
| JP (1) | JP7538155B2 (en) |
| KR (1) | KR102857085B1 (en) |
| CN (1) | CN114026074B (en) |
| WO (1) | WO2020260621A1 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US12006463B2 (en) * | 2019-06-28 | 2024-06-11 | Rolic Technologies AG | Polymerizable liquid crystal having a quinoxaline-hydrazone core |
| JP7564192B2 (en) * | 2019-08-28 | 2024-10-08 | ロリク・テクノロジーズ・アーゲー | Polymerizable liquid crystal composition |
| TW202448972A (en) | 2023-05-12 | 2024-12-16 | 瑞士商羅立克科技股份公司 | Polymerizable liquid crystal composition |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012141245A1 (en) | 2011-04-15 | 2012-10-18 | 日本ゼオン株式会社 | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic body |
| WO2012147904A1 (en) | 2011-04-27 | 2012-11-01 | 日本ゼオン株式会社 | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic material |
| WO2013180217A1 (en) | 2012-05-30 | 2013-12-05 | 日本ゼオン株式会社 | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic material |
| JP2017125009A (en) | 2015-12-21 | 2017-07-20 | Jnc株式会社 | Polymerizable liquid crystal compound, polymerizable liquid crystal composition, liquid crystal polymer thereof and method for producing liquid crystal polymer |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4122027A (en) | 1976-11-08 | 1978-10-24 | General Electric Company | Dichroic liquid crystal composition with 4,4-bis (substituted naphthylazo)azobenzene dichroic dyes |
| US4565424A (en) | 1980-12-12 | 1986-01-21 | Minnesota Mining And Manufacturing Company | Asymmetric dichroic dye molecules having poly(arylazo) linking groups, a bis-substituted aryl thiazyl end group, and another end group |
| US4401369A (en) | 1981-10-19 | 1983-08-30 | Electronic Display Systems, Inc. | Class of dichroic dyes for use with liquid crystals |
| DE3406209A1 (en) | 1984-02-21 | 1985-08-29 | Basf Ag, 6700 Ludwigshafen | AZO DYES AND LIQUID CRYSTAL MATERIALS CONTAINING THESE DYES |
| US5389285A (en) | 1989-12-11 | 1995-02-14 | Hercules Incorporated | Liquid crystal coupled dichroic dyes |
| DE59408097D1 (en) | 1993-02-17 | 1999-05-20 | Rolic Ag | Orientation layer for liquid crystals |
| US6201087B1 (en) | 1994-09-29 | 2001-03-13 | Rolic Ag | Coumarin and quinolinone derivatives for the production of orienting layers for liquid crystals |
| US6107427A (en) | 1995-09-15 | 2000-08-22 | Rolic Ag | Cross-linkable, photoactive polymer materials |
| DE59814236D1 (en) | 1997-02-24 | 2008-07-17 | Rolic Ag | Photocrosslinkable polymers |
| DE102005022642A1 (en) | 2005-05-11 | 2006-11-16 | Basf Ag | 2,6-naphthyl radicals containing compounds |
| EP1764405A1 (en) | 2005-09-20 | 2007-03-21 | Rolic AG | Functionalized photoreactive compounds |
| TWI490316B (en) | 2009-07-09 | 2015-07-01 | Rolic Ag | Ester group containing liquid crystals for optical or electro optical devices |
| CN105504273B (en) | 2010-12-23 | 2019-08-23 | 罗利克有限公司 | Photoactive polymer materials |
| JP6597244B2 (en) | 2014-12-02 | 2019-10-30 | Jnc株式会社 | Liquid crystal compound, liquid crystal composition and polymer thereof |
| WO2016104317A1 (en) | 2014-12-25 | 2016-06-30 | Dic株式会社 | Polymerizable compound and optically anisotropic object |
| JP6432690B2 (en) | 2015-09-11 | 2018-12-05 | 株式会社村田製作所 | Infusion device |
| US12006463B2 (en) * | 2019-06-28 | 2024-06-11 | Rolic Technologies AG | Polymerizable liquid crystal having a quinoxaline-hydrazone core |
| JP7564192B2 (en) * | 2019-08-28 | 2024-10-08 | ロリク・テクノロジーズ・アーゲー | Polymerizable liquid crystal composition |
-
2020
- 2020-06-26 US US17/611,644 patent/US12006463B2/en active Active
- 2020-06-26 EP EP20734551.3A patent/EP3990568B1/en active Active
- 2020-06-26 WO PCT/EP2020/068081 patent/WO2020260621A1/en not_active Ceased
- 2020-06-26 JP JP2021577566A patent/JP7538155B2/en active Active
- 2020-06-26 KR KR1020227003620A patent/KR102857085B1/en active Active
- 2020-06-26 CN CN202080045703.XA patent/CN114026074B/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012141245A1 (en) | 2011-04-15 | 2012-10-18 | 日本ゼオン株式会社 | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic body |
| WO2012147904A1 (en) | 2011-04-27 | 2012-11-01 | 日本ゼオン株式会社 | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic material |
| WO2013180217A1 (en) | 2012-05-30 | 2013-12-05 | 日本ゼオン株式会社 | Polymerizable compound, polymerizable composition, polymer, and optically anisotropic material |
| JP2017125009A (en) | 2015-12-21 | 2017-07-20 | Jnc株式会社 | Polymerizable liquid crystal compound, polymerizable liquid crystal composition, liquid crystal polymer thereof and method for producing liquid crystal polymer |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2020260621A1 (en) | 2020-12-30 |
| EP3990568B1 (en) | 2023-11-15 |
| US20220251450A1 (en) | 2022-08-11 |
| KR102857085B1 (en) | 2025-09-10 |
| CN114026074B (en) | 2024-02-09 |
| KR20220029727A (en) | 2022-03-08 |
| CN114026074A (en) | 2022-02-08 |
| JP2022539176A (en) | 2022-09-07 |
| EP3990568A1 (en) | 2022-05-04 |
| US12006463B2 (en) | 2024-06-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7389147B2 (en) | Novel polymerizable liquid crystal | |
| JP7564192B2 (en) | Polymerizable liquid crystal composition | |
| JP7578614B2 (en) | Novel polymerizable liquid crystals with a carbazole core. | |
| JP7538155B2 (en) | Novel polymerizable liquid crystals with a quinoxaline-hydrazone core | |
| CN113631686B (en) | Liquid crystal compounds | |
| TW202330873A (en) | Liquid crystal compounds | |
| TW202321422A (en) | Liquid crystal compounds | |
| WO2024068311A1 (en) | Liquid crystal compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230313 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20240307 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240312 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20240509 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20240716 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20240808 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7538155 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |