JP7587681B2 - 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 - Google Patents
含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 Download PDFInfo
- Publication number
- JP7587681B2 JP7587681B2 JP2023509346A JP2023509346A JP7587681B2 JP 7587681 B2 JP7587681 B2 JP 7587681B2 JP 2023509346 A JP2023509346 A JP 2023509346A JP 2023509346 A JP2023509346 A JP 2023509346A JP 7587681 B2 JP7587681 B2 JP 7587681B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- fluorine
- solvent
- mass
- general formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images


Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/78—Halides of sulfonic acids
- C07C309/79—Halides of sulfonic acids having halosulfonyl groups bound to acyclic carbon atoms
- C07C309/80—Halides of sulfonic acids having halosulfonyl groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/78—Halides of sulfonic acids
- C07C309/79—Halides of sulfonic acids having halosulfonyl groups bound to acyclic carbon atoms
- C07C309/84—Halides of sulfonic acids having halosulfonyl groups bound to acyclic carbon atoms of a carbon skeleton substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D285/00—Heterocyclic compounds containing rings having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by groups C07D275/00 - C07D283/00
- C07D285/01—Five-membered rings
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/052—Li-accumulators
- H01M10/0525—Rocking-chair batteries, i.e. batteries with lithium insertion or intercalation in both electrodes; Lithium-ion batteries
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0566—Liquid materials
- H01M10/0567—Liquid materials characterised by the additives
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0566—Liquid materials
- H01M10/0568—Liquid materials characterised by the solutes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M10/00—Secondary cells; Manufacture thereof
- H01M10/05—Accumulators with non-aqueous electrolyte
- H01M10/056—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes
- H01M10/0564—Accumulators with non-aqueous electrolyte characterised by the materials used as electrolytes, e.g. mixed inorganic/organic electrolytes the electrolyte being constituted of organic materials only
- H01M10/0566—Liquid materials
- H01M10/0569—Liquid materials characterised by the solvents
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
-
- H—ELECTRICITY
- H01—ELECTRIC ELEMENTS
- H01M—PROCESSES OR MEANS, e.g. BATTERIES, FOR THE DIRECT CONVERSION OF CHEMICAL ENERGY INTO ELECTRICAL ENERGY
- H01M2300/00—Electrolytes
- H01M2300/0017—Non-aqueous electrolytes
- H01M2300/0025—Organic electrolyte
- H01M2300/0028—Organic electrolyte characterised by the solvent
- H01M2300/0037—Mixture of solvents
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E60/00—Enabling technologies; Technologies with a potential or indirect contribution to GHG emissions mitigation
- Y02E60/10—Energy storage using batteries
Landscapes
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Electrochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Inorganic Chemistry (AREA)
- Condensed Matter Physics & Semiconductors (AREA)
- Physics & Mathematics (AREA)
- Materials Engineering (AREA)
- Secondary Cells (AREA)
- Sealing Battery Cases Or Jackets (AREA)
Description
ところで、化合物(1A)を製造する方法としては、FSO2CF2COOHの電解カップリング反応により合成したFO2SCF2CF2SO2Fとアンモニアを反応させ、含フッ素環状スルホニルイミドアンモニウム塩を得た(環化反応の)後、アルカリ金属の水酸化物や炭酸塩と反応させて(カチオン交換反応)を経る方法が知られている(以下の特許文献3参照)。
特許文献3に従い一般式(1A):

H(CR2)m-SO2NHM2 (2)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてM2は、Li、Na、K、H又はNH4である。}で表される含フッ素スルホンアミド化合物(以下、化合物(2)ともいう。)が、最終生成物に副生成物(不純物)として混入し、結果として化合物(1A)の所望の耐熱特性が得られない。
<1>
非水系電溶媒及びリチウム塩を含有する非水系電解液であって、
前記非水系溶媒はカーボネート溶媒を含み、
前記リチウム塩が、下記一般式(1):

で表される含フッ素環状スルホニルイミド塩を含有し、かつ
下記一般式(2):
H(CR2)m-SO2NHM2 (2)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、M2は、Li、Na、K、H又はNH4であり、そしてmは、1又は2である}
で表される含フッ素スルホンアミド化合物を前記非水系電解液の全量に対して、1000質量ppm以下含有する、非水系電解液。
<2>
前記一般式(1)で表される含フッ素環状スルホニルイミド塩が、下記式(1-1)~(1-5):

<3>
前記一般式(2)で表される含フッ素スルホンアミド化合物を、前記非水系電解液の全量に対して、0.1質量ppm以上含有する、項目1又は2に記載の非水系電解液。
<4>
前記非水系溶媒が、アセトニトリルを、前記非水系溶媒の全量に対して、3体積%以上97体積%以下含有する、項目1~3のいずれか一項に記載の非水系電解液。
<5>
前記アセトニトリルの含有量が、前記非水系溶媒の全量に対して、20体積%以上95体積%以下である、項目4に記載の非水系電解液。
<6>
前記一般式(2)で表される含フッ素スルホンアミド化合物を、前記非水系電解液の全量に対して、1質量ppm以上200質量ppm以下含有する、項目1~5のいずれか一項に記載の非水系電解液。
<7>
前記一般式(1)で表される含フッ素環状スルホニルイミド塩の含有量が、前記非水系溶媒1Lに対して、0.8モル以上1.5モル以下である、項目1~6のいずれか一項に記載の非水系電解液。
<8>
前記一般式(2)で表される含フッ素スルホンアミド化合物を、前記非水系電解液の全量に対して、80質量ppm以上120質量ppm以下含有する、項目1~7のいずれか一項に記載の非水系電解液。
<9>
前記リチウム塩が、前記一般式(1)で表される含フッ素環状スルホニルイミド塩と、LiPF6とを含有し、
前記非水系溶媒が、ビニレンカーボネート及び/またはフルオロエチレンカーボネートを含有し、
前記一般式(1)で表される含フッ素環状スルホニルイミド塩の含有量は、前記LiPF6の含有量に対してモル比で2.5以上であり、かつ
前記LiPF6の含有量は、ビニレンカーボネート及びフルオロエチレンカーボネートの含有量に対してモル比で0.01以上4以下である、項目1~8のいずれか一項に記載の非水系電解液。
<10>
前記一般式(1)で表される含フッ素環状スルホニルイミド塩の含有量は、前記LiPF6の含有量に対してモル比で10より大きい、項目1~9のいずれか一項に記載の非水系電解液。
<11>
項目1~10のいずれか一項の非水系電解液を備えた、非水系二次電池。
<12>
前記非水系二次電池は電池外装を備え、前記電池外装が、正極側の、前記非水系電解液との接液層の少なくとも一部にアルミニウムを含む、項目11に記載の非水系二次電池。
<13>
下記一般式(3):
MO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてMは、H、Li、Na、K又はNH4である}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、前記一般式(1)で表される含フッ素環状スルホニルイミド塩を製造し、
前記一般式(1)で表される含フッ素環状スルホニルイミド塩と非水系溶媒とを混合する混合工程を含み、
該電解カップリング反応工程後に下記一般式(5):
H-(CR2)m-SO2F (5)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である}で表されるフルオロスルホニル基含有化合物を、該反応液中0.1質量%以下まで低下させる精製工程、を含むことを特徴とする項目1~10のいずれか一項に記載の非水系電解液の製造方法。
<14>
下記一般式(3):
MO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてMは、H、Li、Na、K又はNH4である}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、前記一般式(1)で表される含フッ素環状スルホニルイミド塩を製造し、
前記カチオン交換工程後に得られる前記一般式(1)で表される含フッ素環状スルホニルイミド塩を、単座配位性の鎖状エーテル溶媒に、溶解させた後に晶析して、精製された含フッ素環状スルホニルイミド塩を得る晶析工程と、
前記晶析工程で得られた前記一般式(1)で表される含フッ素環状スルホニルイミド塩と非水系溶媒とを混合する混合工程とを含む、項目1~10のいずれか一項に記載の非水系電解液の製造方法。
<15>
下記一般式(1A):

窒素気流下、100℃より10℃/分の昇温速度で加熱したときに、質量減少率が2質量%となる温度が385℃以上であることを特徴とする含フッ素環状スルホニルイミド塩組成物。
<16>
下記一般式(2):
H(CR2)m-SO2NHM2 (2)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、M2は、Li、Na、K、H又はNH4であり、そしてmは、1又は2である}
で表される含フッ素スルホンアミド化合物を5000質量ppm以下含有する、項目15に記載の含フッ素環状スルホニルイミド塩組成物。
<17>
前記一般式(1A)中、nが2である、項目16に記載の含フッ素環状スルホニルイミド塩組成物。
<18>
前記一般式(1A)と(2)中、Rがフッ素原子である、項目16又は17に記載の含フッ素環状スルホニルイミド塩組成物。
<19>
前記一般式(1A)中、M1がLiであり、かつ、前記一般式(2)中、M2がLiである、項目16~18のいずれか一項に記載の含フッ素環状スルホニルイミド塩組成物。
<20>
前記含フッ素環状スルホニルイミド塩が、下記式:

<21>
下記一般式(3):
MO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてMは、H、Li、Na、K又はNH4である}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、下記一般式(1A):

H-(CR2)m-SO2F (5)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である}で表されるフルオロスルホニル基含有化合物を、該反応液中0.1質量%以下まで低下させる精製工程を特徴とする前記製造方法。
<22>
下記一般式(3):
MO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてMは、H、Li、Na、K又はNH4である}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、下記一般式(1A):

<23>
前記晶析に用いる単座配位性の鎖状エーテル溶媒が、エチルエーテル、プロピルメチルエーテル、プロピルエチルエーテル、プロピルエーテル、イソプロピルメチルエーテル、イソプロピルエチルエーテル、イソプロピルエーテル、ブチルメチルエーテル、ブチルエチルエーテル、イソブチルメチルエーテル、イソブチルエチルエーテル、sec-ブチルメチルエーテル、sec-ブチルエチルエーテル、tert-ブチルメチルエーテル、tert-ブチルエチルエーテル、ペンチルメチルエーテル、イソペンチルメチルエーテル、sec-ペンチルメチルエーテル、tert-ペンチルメチルエーテル、ネオペンチルメチルエーテル、及びシクロペンチルメチルエーテルからなる群から選ばれる少なくとも1種の溶媒である、項目22に記載の方法。
<24>
前記一般式(1A)中のnが2である、項目22又は23に記載の方法。
<25>
前記一般式(1A)中のRがFである、項目22~24のいずれか一項に記載の方法。
<26>
前記一般式(1A)中のMがLiである、項目22~25のいずれか一項に記載の方法。
<27>
前記電解カップリング反応工程において、上記一般式(3)で表されるフルオロスルホニル基含有カルボン酸化合物を、水の質量(α)とニトリル基含有溶媒の質量(β)の比率(β/α)が0.80以下である水とニトリル基含有溶媒との混合溶媒の存在下、電解カップリング反応させる、項目21~26のいずれか一項に記載の方法。
<28>
前記水の質量(α)とニトリル基含有溶媒の質量(β)の比率(β/α)が0.75以下である、項目27に記載の方法。
<29>
前記電解カップリング反応工程において、上記一般式(3)で表されるフルオロスルホニル基含有カルボン酸化合物を、水とニトリル基含有溶媒との混合溶媒の存在下、反応容器中で、電解カップリング反応させ、
反応中、前記反応容器底部から反応液の液面までの高さ(距離)TLと前記反応容器底部から電極上部(最高位)までの高さ(距離)TEとが、TL>TEの関係を満たし、かつ、反応後には、前記反応容器底部から、有機相と水相の間の界面までの高さ(距離)TSと、前記反応容器底部から電極下部(最低位)までの高さ(距離)TE’とが、TE’>TSの関係を満たす、項目21~28のいずれか一項に記載の方法。
<30>
前記TE’と前記TSの関係が、TE’×0.6>TSの関係である、項目29に記載の方法。
<31>
前記電解カップリング反応における反応液中のアルカリ金属の含有量が2000質量ppm以下である、項目27~30のいずれか一項に記載の方法。
<32>
前記電解カップリング反応工程において、上記一般式(5)で表される化合物の生成モル比が、下記一般式(4):
FO2S-(CR2)m-SO2F (4)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である}で表されるビス(フルオロスルホニル)化合物(4)の生成量1モルに対し、0.01以下とする、項目27~31のいずれか一項に記載の方法。
<33>
前記電解カップリング反応工程において、上記一般式(3)で表されるフルオロスルホニル基含有カルボン酸化合物を、水とニトリル基含有溶媒との混合溶媒存在下、電解カップリング反応させ、該フルオロスルホニル基含有カルボン酸化合物の質量(γ)と、該混合溶媒の質量(δ)の比率(δ/γ)が1.5以下であることを特徴とする、項目27~32のいずれか一項に記載の方法。
<34>
前記電解カップリング反応において、陽極と陰極との極板間隔が0.001mm~9.9mmである、項目27~33のいずれか一項に記載の方法。
<35>
前記電解カップリング反応において、前記化合物(4)の収率を、70%以上とする、項目33又は34に記載の方法。
<36>
前記ニトリル基含有溶媒が、アセトニトリル、プロピオニトリル、ブチロニトリル、及びベンゾニトリルからなる群より選ばれる少なくとも1種である、項目27~35のいずれか一項に記載の方法。
<37>
前記ニトリル基含有溶媒がアセトニトリルである、項目36に記載の方法。
<38>
前記電解カップリング反応が、-35℃以上10℃以下の範囲の反応温度で行われる、項目27~37のいずれか一項に記載の方法。

以下、化合物(1A)について詳細に説明する。
一般式(1A)中、nは、入手又は製造が容易であり、経済性に優れる観点から1~4の整数であり、同様の観点から、1~3の整数であることが好ましく、1又は2であることがより好ましく、2であることが最も好ましい。
Rはそれぞれ同一であっても異なっていてもよく、入手又は製造が容易であり、経済性に優れる観点から、フッ素原子又はトリフルオロメチル基であり、フッ素原子が最も好ましい。
M1は、入手又は製造が容易であり、経済性に優れる観点から、Li、Na、K又はNH4(すなわち、アルカリ金属イオン、アンモニウムイオン)であり、Li又はNaが好ましく、Liが最も好ましい。
化合物(1A)の具体例としては、以下の構造式:


これらの中でも、以下の構造式:

上記2%質量減少温度の測定に使用可能な装置としては、示差熱熱重量同時測定装置(例えば、株式会社島津製作所製の「DTG-60A」)等が挙げられる。
H(CR2)m-SO2NHM2 (2)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、M2は、Li、Na、K、H又はNH4である。}で表される含フッ素スルホンアミド化合物の含有量が10000質量ppm(1質量%)以下であることが好ましい。化合物(2)の含有量が上記範囲内にあると、化合物(1A)を含む組成物の耐熱特性がより高まるとともに、金属腐食性が低減され、正極Al集電体の腐食による充放電時の不可逆容量の増加及びサイクル性能の低下を抑制できる。同様の観点から、化合物(2)の含有量は、含フッ素環状スルホニルイミド塩(1A)の全量に対して、5000質量ppm以下であることがより好ましく、1000質量ppm以下であることが最も好ましい。化合物(2)の含有量は、その下限値は特に限定されないが、含フッ素環状スルホニルイミド塩(1A)の全量に対して、0質量ppm以上であってもよく、0.001質量ppm以上であってもよく、10質量ppm以上であってもよく、100質量ppm以上であってもよい。
化合物(2)の含有量(質量%)=(Im×Ci×Cmw)/(Ii×Ia)×100
(式中、
Im=内部標準物質モル数=ベンゾトリフルオリドの測定サンプル内の質量/ベンゾトリフルオリドの分子量
Ii=内部標準物質の1Fあたりの積分値=ベンゾトリフルオリドの積分値/ベンゾトリフルオリドのフッ素原子数
Ci=化合物(2)の1Fあたりの積分値=化合物(2)の積分値/化合物(2)のフッ素原子数
Cmw=化合物(2)の分子量
Ia=測定サンプル内の含フッ素環状スルホニルイミド塩を含む組成物の総質量)
例えば、非水系電解液中の化合物(2)の含有量が10質量ppm以下であり、上記の内部標準法での検出が困難な場合は、100mLの非水系電解液を溶媒留去して得られた固体サンプルを0.5mLの溶媒に再溶解したサンプルに関して、上記の内部標準法を用いて化合物(2)の含有量を仮に算出し、その値を200倍することで、元の非水系電解液中の化合物(2)の含有量を算出できる。または、非水系電解液の原料に用いられた化合物(1A)中の化合物(2)の含有量を上記の内部標準法を用いて算出し、化合物(1A)を非水系電解液中の化合物(1A)の濃度に希釈した際の希釈倍率で除することで算出できる。
化合物(2)の構造式の同定は、質量分析法、核磁気共鳴法、赤外分光法、元素分析、イオンクロマトグラフィ―法、ICP発光分光分析法を含む一般的な分析手法のうち一つまたは組合せによって可能である。
また、化合物(2)の含有量が所望の範囲の含フッ素環状スルホニルイミド塩組成物および非水系電解液は、化合物(2)を含フッ素環状スルホニルイミド塩組成物および非水系電解液に後添加することでも得られる。
化合物(2)の具体例としては、下記の構造式で表される化合物が挙げられる。これらの中でも、HCF2SO2NHLi(化合物(2-1))、HF2CSO2NHNH4(化合物(2-2))が好ましく、HCF2SO2NHLi(化合物(2-1))が最も好ましい。

前記したように、化合物(1A)を製造する方法として、従来公知の方法、例えば、特許文献3に記載の方法を採用することができる。以下に、Rがフッ素原子、nが2である場合を例にとり、化合物(1A)の反応スキームを示す。かかる方法は、以下の反応スキームの上段に示すように、下記一般式(3):
HO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である。}で表されるフルオロスルホニル基含有カルボン酸化合物(以下、化合物(3)ともいう。)から出発して、電解カップリング反応工程により化合物(4)を得て、次いで、環化工程により化合物(1-N)を得て、最後にカチオン交換工程を経て、化合物(1A)を製造する方法である。化合物(1A)においてM1=NH4である場合は、カチオン交換工程を省略できる。

本願発明者らは、鋭意検討し実験を重ねた結果、最終生成物である化合物(1A)組成物の耐熱特性を高め、金属腐食性を低減するためには、前記したように、化合物(1A)組成物中の化合物(2)の含有量を所定値以下にすればよいことを見出した。
さらに、そうするためには、上記反応スキームの下段に示すように、電解カップリング反応工程において、副生成物として生じるフルオロスルホニル基含有化合物(化合物(5))を、該工程で得られる反応液中0.1質量%以下まで低下させればよいことを見出し、本発明を完成する至ったもののである。
HO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である。}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、下記一般式(1A):

H-(CR2)m-SO2F (5)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である。}で表されるフルオロスルホニル基含有化合物を、該反応液中0.1質量%以下まで低下させる精製工程を特徴とする前記製造方法である。なお、化合物(5)は、該電解カップリング反応工程において得られる反応液を精製して得られる、下記一般式(4):
FO2S-(CR2)m-SO2F (4)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である。}で表されるビス(フルオロスルホニル)化合物の副生成物である。
前記一般式(3)で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、前記一般式(1A)で表される含フッ素環状スルホニルイミド塩を製造する方法において、カチオン交換工程後に得られる含フッ素環状スルホニルイミド塩を、単座配位性の鎖状エーテル溶媒に、溶解させた後に晶析して、精製された含フッ素環状スルホニルイミド塩を得る晶析工程を含む、ものである。
<電解カップリング反応工程>
化合物(3)を電解カップリング反応させることにより、ビス(フルオロスルホニル)化合物(化合物(4))を製造することができる。化合物(3)は市販のものを使用しても、公知の方法、例えば、テトラフルオロエチレンにSO3を付加して、サルトンを得、NEt3の存在下でサルトン開環物を得、これを加水分解して、サルトン加水分解物として化合物(3)を得る方法を使用してもよい。
化合物(3)の具体例としては、下記の構造式で表される化合物が挙げられる。これらの中でも、FO2SCF2CO2H(2,2-ジフルオロ-2-(フルオロスルホニル)酢酸、化合物(3-1))が好ましい。

上記溶媒としては、一般的に使用されるものであれば特に限定されないが、具体例としては、ベンゼン、トルエン、クロロベンゼン、及び1,2-ジクロロベンゼン等の芳香族系溶媒、1,2-ジメトキシエタン、1,2-ジエトキシエタン、ジエチレングリコールジメチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,4-ジオキサン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、及びアニソール等のエーテル基含有溶媒、アセトニトリル、プロピオニトリル、ブチロニトリル、及びベンゾニトリル等のニトリル基含有溶媒、水、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、及び2-ブタノール等の水酸基含有溶媒、ジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート、プロピレンカーボネート等の炭酸エステル基含有溶媒等が挙げられる。中でも、化合物(4)の収率が高まる傾向にあることから、アセトニトリル、プロピオニトリル、ブチロニトリル、及びベンゾニトリル等のニトリル溶媒、水、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、及び2-ブタノール等の水酸基含有溶媒、及びジメチルカーボネート、ジエチルカーボネート、エチルメチルカーボネート、プロピレンカーボネート等の炭酸エステル基含有溶媒が好ましい。
これらの溶媒は単独で用いてもよいし、複数種を組み合わせてもよく、化合物(4)の収率がより高まる傾向にあることから、水とニトリル基含有溶媒との混合溶媒、又は水と炭酸エステル基含有溶媒との混合溶媒が好ましく、水とニトリル基含有溶媒との混合溶媒がより好ましく、水とアセトニトリルとの混合溶媒が最も好ましい。溶媒中のアセトニトリルの量は、0.5~90質量%が好ましい。
同様の観点から、TE’とTSの関係は、TE’×0.8>TSの関係を満たすことがより好ましく、TE’×0.6>TSをさらに満たすことが最も好ましい。
これらの方法は、単独で用いてもよいし、複数種の方法を組み合わせて用いてもよい。

化合物(5)の具体例としては、下記の構造式で表される化合物が挙げられる。これらの中でも、HCF2SO2F(1,1-ジフルオロメタンスルホニルフルオリド、化合物(5-1))が好ましい。

本実施形態において使用される電解槽は有機電解反応において通常用いられるものであり、バッチ式であっても、化合物(3)等を連続的に供給しながら行う連続式であってもよい。
電解カップリング反応工程の圧力は、反応を行う温度によるが、通常用いられる範囲であれば特に限定されないが、10kPa~5000kPaが好ましい。
反応雰囲気は、通常用いられる雰囲気であれば特に限定されないが、通常は大気雰囲気、窒素雰囲気、及びアルゴン雰囲気等が用いられる。これらの中でも、より安全に化合物(4)を製造できる傾向にあることから、窒素雰囲気、及びアルゴン雰囲気が好ましい。また、より経済性に優れる製造方法となる傾向にあることから、窒素雰囲気がさらに好ましい。
反応雰囲気は、単独で用いてもよいし、複数種の反応雰囲気を組み合わせて用いてもよい。
化合物(4)と、アンモニアを反応させることにより、下記一般式(1-N):


上記溶媒としては、反応時に不活性であり、一般的に使用されるものであれば特に限定されないが、具体例としては、ベンゼン、トルエン、クロロベンゼン、及び1,2-ジクロロベンゼン等の芳香族系溶媒、1,2-ジメトキシエタン、1,2-ジエトキシエタン、ジエチレングリコールジメチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,4-ジオキサン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、及びアニソール等のエーテル基含有溶媒、アセトニトリル、プロピオニトリル、及びベンゾニトリル等のニトリル基含有溶媒、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、及び2-ブタノール等の水酸基含有溶媒等が挙げられる。中でも、化合物(1-N)の収率が高まる傾向にあることから、1,2-ジメトキシエタン、1,2-ジエトキシエタン、ジエチレングリコールジメチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,4-ジオキサン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、及びアニソール等のエーテル基含有溶媒がより好ましい。同様の観点から、1,2-ジメトキシエタン、テトラヒドロフラン、1,4-ジオキサン、及び4-メチルテトラヒドロピランがより好ましく、1,2-ジメトキシエタン、テトラヒドロフラン、及び4-メチルテトラヒドロピランがさらに好ましい。
これらの有機溶媒は単独で用いてもよいし、複数種の溶媒を組み合わせて用いてもよい。
環化工程の圧力は、反応を行う温度によるが、通常用いられる範囲であれば特に限定されないが、10kPa~5000kPaが好ましい。
反応雰囲気は、通常用いられる雰囲気であれば特に限定されないが、通常は大気雰囲気、窒素雰囲気、及びアルゴン雰囲気等が用いられる。これらの中でも、より安全に化合物(1-N)を製造できる傾向にあることから、窒素雰囲気、及びアルゴン雰囲気が好ましい。また、より経済性に優れる製造方法となる傾向にあることから、窒素雰囲気がさらに好ましい。
反応雰囲気は、単独で用いてもよいし、複数種の反応雰囲気を組み合わせて用いてもよい。
これらの方法は、単独で用いてもよいし、複数種の方法を組み合わせて用いてもよい。
化合物(1-N)と、下記一般式(6):
M1 pX (6)
(式中、M1は、Li、Na又はKであり、Xは、OH又はCO3であり、そしてpは、XがOHの場合、1であり、XがCO3の場合、2である。}で表されるアルカリ金属塩(以下、化合物(6)ともいう。)とを、反応させることにより、化合物(1A)を製造することができる。


上記溶媒としては、反応時に不活性であり、一般的に使用されるものであれば特に限定されないが、具体例としては、ベンゼン、トルエン、クロロベンゼン、及び1,2-ジクロロベンゼン等の芳香族系溶媒、1,2-ジメトキシエタン、1,2-ジエトキシエタン、ジエチレングリコールジメチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,4-ジオキサン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、及びアニソール等のエーテル基含有溶媒、アセトニトリル、プロピオニトリル、及びベンゾニトリル等のニトリル基含有溶媒、メタノール、エタノール、1-プロパノール、2-プロパノール、1-ブタノール、及び2-ブタノール等の水酸基含有溶媒等が挙げられる。中でも、化合物(1A)の収率が高まる傾向にあることから、1,2-ジメトキシエタン、1,2-ジエトキシエタン、ジエチレングリコールジメチルエーテル、トリエチレングリコールジメチルエーテル、テトラエチレングリコールジメチルエーテル、テトラヒドロフラン、2-メチルテトラヒドロフラン、1,4-ジオキサン、4-メチルテトラヒドロピラン、シクロペンチルメチルエーテル、及びアニソール等のエーテル基含有溶媒がより好ましい。同様の観点から、1,2-ジメトキシエタン、テトラヒドロフラン、1,4-ジオキサン、及び4-メチルテトラヒドロピランがより好ましく、1,2-ジメトキシエタン、テトラヒドロフラン、及び4-メチルテトラヒドロピランがさらに好ましい。
これらの有機溶媒は単独で用いてもよいし、複数種の溶媒を組み合わせて用いてもよい。
カチオン交換工程の圧力は、反応を行う温度によるが、通常用いられる範囲であれば特に限定されないが、10kPa~5000kPaが好ましい。
反応雰囲気は、通常用いられる雰囲気であれば特に限定されないが、通常は大気雰囲気、窒素雰囲気、及びアルゴン雰囲気等が用いられる。これらの中でも、より安全に化合物(1A)を製造できる傾向にあることから、窒素雰囲気、及びアルゴン雰囲気が好ましい。また、より経済性に優れる製造方法となる傾向にあることから、窒素雰囲気がさらに好ましい。
反応雰囲気は、単独で用いてもよいし、複数種の反応雰囲気を組み合わせて用いてもよい。
含フッ素環状スルホニルイミド塩は、一般に、水及び配位性の各種有機溶媒への溶解性が極めて高く、このことが帯電防止剤、有機電解質等のイオン電導材料や難燃剤といった幅広い用途での利用に有利に働いている。他方、この高い溶解性は、低い結晶性及び乾燥時の溶媒残存量の増大と直接的に結びついている。含フッ素環状スルホニルイミド塩のような粉体物質を高純度化するための最も一般的で簡便な製造方法の一つとして晶析が挙げられるが、この低い結晶性と溶媒残存量の増大が問題となり、含フッ素環状スルホニルイミド塩の晶析による高純度化に係る既存技術は限られている。
結晶化を促すために貧溶媒を殊更に添加する場合には、用いる貧溶媒は特に限定されないが、含フッ素環状スルホニルイミド塩を溶解せず、単座配位性の鎖状エーテル溶媒と混和し、かつ低沸点であることが望ましく、ペンタン、ヘキサン、シクロヘキサン、ヘプタン、クロロホルム、ジクロロメタン、1,2-ジクロロエタン等が挙げられる。
次に、捕集後の含フッ素環状スルホニルイミド塩中の余剰の溶媒の除去のために乾燥工程を行なうことが好ましい。乾燥工程では一般に行なわれるように加熱減圧乾燥を行なうことができる。加熱温度は好ましくは40℃、より好ましくは60℃であるが、含フッ素環状スルホニルイミド塩の分解が進行しない限りにおいては100℃以上の温度で行なってもよく、含フッ素環状スルホニルイミド塩の熱安定性を考慮すると上限は200℃とするのが好ましい。減圧時の圧力は好ましくは100hPa以下、より好ましくは50hPa以下、さらに好ましくは10hPa以下であり、最も好ましくは1hPa以下である。
乾燥を行なう際は、上記工程で捕集されたそのままの形態の結晶を乾燥しても充分に残存溶媒を除去できるが、より速やかに乾燥を進行させる目的で、結晶の破砕、撹拌操作を加えてもよく、この場合は更なる熱履歴、時間、及びコストの低減が期待できる。本実施形態の製造方法によれば、得られる結晶は残存溶媒量が少なく、結晶同士の固着や容器への固着を抑制できることから、結晶の破砕や粉砕工程には特殊な製造装置は必要なく、一般に広く用いられる撹拌翼やミルを用いることができる。

で表される含フッ素環状スルホニルイミド塩を用いることによって、初回充放電効率とサイクル性能が向上する。その詳細な機構は不明だが、含フッ素環状スルホニルイミド塩が解離して生成した環状アニオンが負極で還元分解され、負極に堆積してSEIとして働くためだと推測される。また、含フッ素環状スルホニルイミド塩は金属腐食性が低く、正極集電体のAlイオンの溶出が抑制されたためだと推測される。

前記非水系溶媒はカーボネート溶媒を含み、
前記リチウム塩が、上記一般式(1)
で表される含フッ素環状スルホニルイミド塩を含有し、
下記一般式(2):
H(CR2)m-SO2NHM2 (2)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、M2は、Li、Na、K、H又はNH4であり、そしてmは、1又は2である}
で表される含フッ素スルホンアミド化合物を前記非水系電解液の全量に対して、1000質量ppm以下含有する。
本実施形態の非水系溶媒はカーボネート溶媒を含む。カーボネート溶媒としては、環状カーボネート、フルオロエチレンカーボネート、鎖状カーボネート、前記カーボネート性溶媒のH原子の一部または全部をハロゲン原子で置換した化合物等が挙げられる。
を挙げることができる。
Rcc-O-C(O)O-Rdd
{式中、Rcc及びRddは、CH3、CH2CH3、CH2CH2CH3、CH(CH3)2、及びCH2Rf eeから成る群より選択される少なくとも一つであり、Rf eeは、少なくとも1つのフッ素原子で水素原子が置換された炭素数1~3のアルキル基であり、そしてRcc及び/又はRddは、少なくとも1つのフッ素原子を含有する}
で表すことができる。
本実施形態におけるアセトニトリル以外のカーボネート溶媒は、1種を単独で使用することができ、又は2種以上を組み合わせて使用してよい。
を挙げることができる。
Rff-C(O)O-Rgg
{式中、Rffは、CH3、CH2CH3,CH2CH2CH3、CH(CH3)2、CF3CF2H、CFH2、CF2Rf hh、CFHRf hh、及びCH2Rf iiから成る群より選択される少なくとも一つであり、Rggは、CH3、CH2CH3、CH2CH2CH3、CH(CH3)2、及びCH2Rf iiから成る群より選択される少なくとも一つであり、Rf hhは、少なくとも1つのフッ素原子で水素原子が置換されてよい炭素数1~3のアルキル基であり、Rf iiは、少なくとも1つのフッ素原子で水素原子が置換された炭素数1~3のアルキル基であり、そしてRff及び/又はRggは、少なくとも1つのフッ素原子を含有し、RffがCF2Hである場合、RggはCH3ではない}
で表すことができる。
(含フッ素環状スルホニルイミド塩(1))
前記リチウム塩が、上記一般式(1)で表される含フッ素環状スルホニルイミド塩である。
前記式(1)で表される含フッ素環状スルホニルイミド塩は、上記項目<含フッ素環状スルホニルイミド塩(化合物(1A))を含む組成物>に記載の内容と同じでよい。
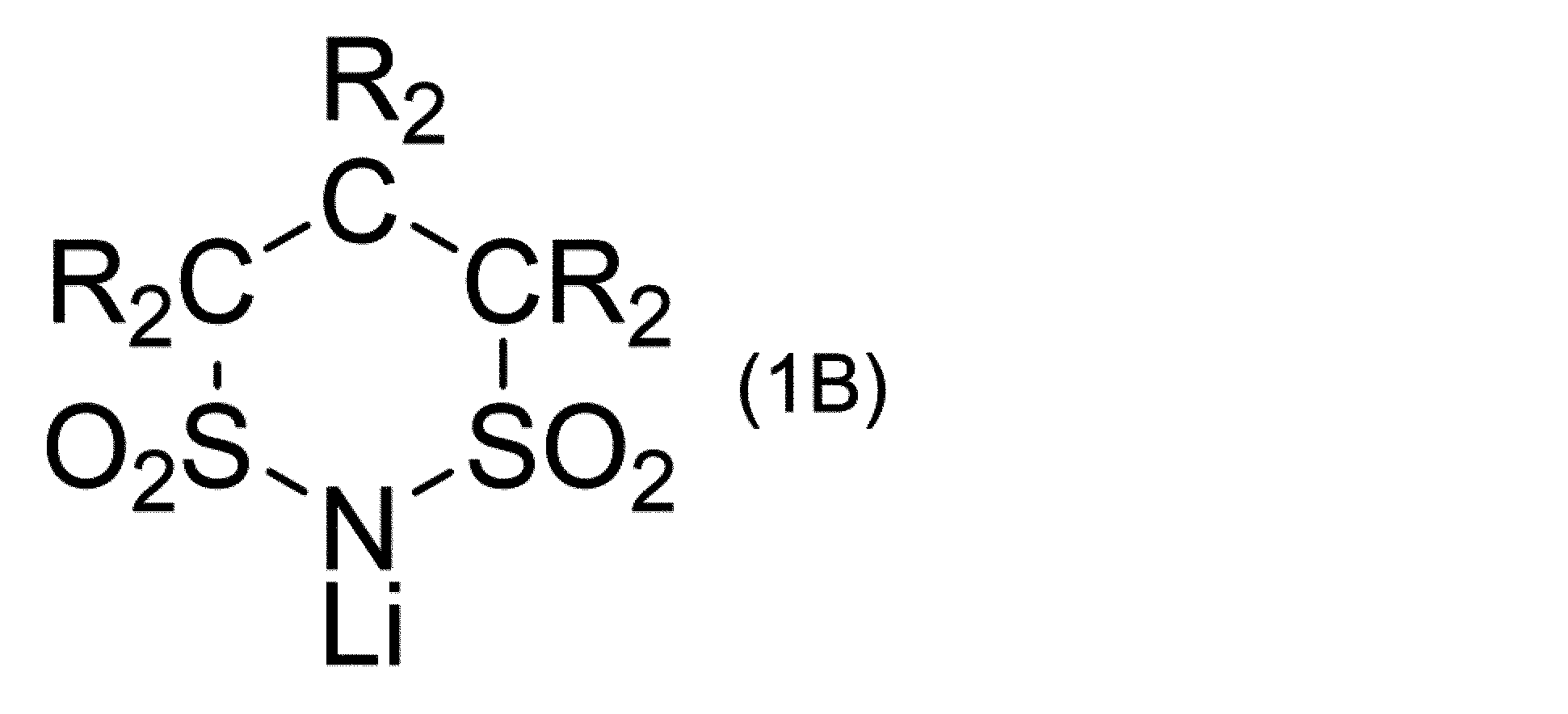
上記式(1)で表される含フッ素環状スルホニルイミド塩(化合物(1))の製造方法は、上記項目<含フッ素環状スルホニルイミド塩(化合物(1A))の製造方法>に記載の内容と同じでよい。
本実施形態の非水系電解液は、リチウム塩として、LiPF6をさらに含有してもよい。本実施形態の非水系電解液におけるLiPF6の含有量については、非水系溶媒1L当たりの量として、0.5モル未満であることが好ましく、0.1モル未満であることがより好ましく、0.01モル未満であることが更に好ましい。LiPF6の含有量が上述の範囲内にある場合、LiPF6の熱分解反応による酸成分の生成を抑えることができ、負極SEIにおける無機成分の過剰な堆積による負極の抵抗増加を最小限に留めることが出来る。
イミド塩としては、具体的には、LiN(SO2F)2、及びLiN(SO2CF3)2のうち少なくとも1種を含むことが好ましい。
本実施形態の非水系電解液で用いるリチウム塩は、LiPF6以外のフッ素含有無機リチウム塩を含んでもよく、例えば、LiBF4、LiAsF6、Li2SiF6、LiSbF6、Li2B12FbH12-b〔式中、bは0~3の整数である〕等のフッ素含有無機リチウム塩を含んでもよい。ここで「無機リチウム塩」とは、炭素原子をアニオンに含まず、アセトニトリルに可溶なリチウム塩をいう。また、「フッ素含有無機リチウム塩」とは、炭素原子をアニオンに含まず、フッ素原子をアニオンに含み、アセトニトリルに可溶なリチウム塩をいう。フッ素含有無機リチウム塩は、正極集電体であるアルミニウム箔の表面に不働態被膜を形成し、正極集電体の腐食を抑制する点で優れている。これらのフッ素含有無機リチウム塩は、1種を単独で又は2種以上を組み合わせて用いられる。フッ素含有無機リチウム塩として、LiFとルイス酸との複塩である化合物が好ましく、中でも、リン原子を有するフッ素含有無機リチウム塩を用いると、遊離のフッ素原子を放出し易くなることからより好ましい。フッ素含有無機リチウム塩として、ホウ素原子を有するフッ素含有無機リチウム塩を用いた場合には、電池劣化を招くおそれのある過剰な遊離酸成分を捕捉し易くなることから好ましく、このような観点からはLiBF4が特に好ましい。
本実施形態におけるリチウム塩は、有機リチウム塩を含んでよい。「有機リチウム塩」とは、炭素原子をアニオンに含み、アセトニトリルに可溶な、イミド塩以外のリチウム塩をいう。
本実施形態におけるリチウム塩は、上記以外に、その他のリチウム塩を含んでよい。
LiClO4、LiAlO4、LiAlCl4、LiB10Cl10、クロロボランLi等のフッ素原子をアニオンに含まない無機リチウム塩;
LiCF3SO3、LiCF3CO2、Li2C2F4(SO3)2、LiC(CF3SO2)3、LiCnF(2n+1)SO3{式中、n≧2}、低級脂肪族カルボン酸Li、四フェニルホウ酸Li、LiB(C3O4H2)2等の有機リチウム塩;
LiPF5(CF3)等のLiPFn(CpF2p+1)6-n〔式中、nは1~5の整数、pは1~8の整数である〕で表される有機リチウム塩;
LiBF3(CF3)等のLiBFq(CsF2s+1)4-q〔式中、qは1~3の整数、sは1~8の整数である〕で表される有機リチウム塩;
多価アニオンと結合されたリチウム塩;
下記式(XXa):
LiC(SO2Rjj)(SO2Rkk)(SO2Rll) (XXa)
{式中、Rjj、Rkk、及びRllは、互いに同一でも異なっていてもよく、炭素数1~8のパーフルオロアルキル基を示す。}、
下記式(XXb):
LiN(SO2ORmm)(SO2ORnn) (XXb)
{式中、Rmm、及びRnnは、互いに同一でも異なっていてもよく、炭素数1~8のパーフルオロアルキル基を示す。}、及び
下記式(XXc):
LiN(SO2Roo)(SO2ORpp) (XXc)
{式中、Roo、及びRppは、互いに同一でも異なっていてもよく、炭素数1~8のパーフルオロアルキル基を示す。}
のそれぞれで表される有機リチウム塩等が挙げられ、これらのうちの1種又は2種以上を、フッ素含有無機リチウム塩と共に使用することができる。
本実施形態に係る非水系電解液は、電極を保護するための添加剤(電極保護用添加剤)を含んでよい。電極保護用添加剤は、リチウム塩を溶解させるための溶媒としての役割を担う物質(すなわち上記の非水系溶媒)と実質的に重複してよい。電極保護用添加剤は、非水系電解液及び非水系二次電池の性能向上に寄与する物質であることが好ましいが、電気化学的な反応には直接関与しない物質をも包含する。
4-フルオロ-1,3-ジオキソラン-2-オン、4,4-ジフルオロ-1,3-ジオキソラン-2-オン、シス-4,5-ジフルオロ-1,3-ジオキソラン-2-オン、トランス-4,5-ジフルオロ-1,3-ジオキソラン-2-オン、4,4,5-トリフルオロ-1,3-ジオキソラン-2-オン、4,4,5,5-テトラフルオロ-1,3-ジオキソラン-2-オン、及び4,4,5-トリフルオロ-5-メチル-1,3-ジオキソラン-2-オンに代表されるフルオロエチレンカーボネート;
ビニレンカーボネート、4,5-ジメチルビニレンカーボネート、及びビニルエチレンカーボネートに代表される不飽和結合含有環状カーボネート;
γ-ブチロラクトン、γ-バレロラクトン、γ-カプロラクトン、δ-バレロラクトン、δ-カプロラクトン、及びε-カプロラクトンに代表されるラクトン;
1,4-ジオキサンに代表される環状エーテル;
エチレンサルファイト、プロピレンサルファイト、ブチレンサルファイト、ペンテンサルファイト、スルホラン、3-スルホレン、3-メチルスルホラン、1,3-プロパンスルトン、1,4-ブタンスルトン、1-プロペン1,3-スルトン、及びテトラメチレンスルホキシドに代表される環状硫黄化合物;
が挙げられ、これらは1種を単独で又は2種以上を組み合わせて用いられる。
本実施形態に係る非水系二次電池は、初回充電のときに非水系電解液の一部が分解し、負極表面にSEIを形成することにより安定化する。このSEIをより効果的に強化するため、非水系電解液、電池部材、又は非水系二次電池に、酸無水物を添加することができる。非水系溶媒としてアセトニトリルを含む場合には、温度上昇に伴いSEIの強度が低下する傾向にあるが、酸無水物の添加によってSEIの強化が促進される。よって、このような酸無水物を用いることにより、効果的に熱履歴による経時的な内部抵抗の増加を抑制することができる。
本実施形態においては、非水系二次電池の充放電サイクル特性の改善、高温貯蔵性、安全性の向上(例えば過充電防止等)等の目的で、非水系電解液に、任意的添加剤(酸無水物、及び電極保護用添加剤以外の添加剤)を適宜含有させることもできる。
化合物(2)は、上記一般式(2)で表される含フッ素スルホンアミド化合物である。
反応式a:Al+3(H(CR2)m-SO2NHM)→[Al(H(CR2)m-SO2NHM)3]3++3e-
反応式b:[Al(H(CR2)m-SO2NHM)3]3++3e-→Al+3(H(CR2)m-SO2NHM)
化合物(2)の含有量を低減することで、充電時の正極でのAl腐食反応(反応式a)及び負極でのAlの還元析出反応(反応式b)を抑制でき、充放電時の不可逆容量が低下し、サイクル性能が向上する。これらの現象は、本発明者らの検討により新たに判明し、特許文献1~4には一切記載されていない。
本実施形態の非水系電解液は、非水系二次電池に用いることができる。
本実施形態に係る非水系二次電池は、特に制限を与えるものではないが、正極、負極、セパレータ、及び非水系電解液が、適当な電池外装中に収納されて構成される。
以下、本実施形態に係る非水系二次電池を構成する各要素について、順に説明する。
正極150は、正極合剤から作製した正極活物質層と、正極集電体とから構成される。正極合剤は、正極活物質を含有し、必要に応じて、導電助剤及びバインダーを含有する。
LipNiqCorMnsMtOu・・・・・(at)
{式中、Mは、Al、Sn、In、Fe、V、Cu、Mg、Ti、Zn、Mo、Zr、Sr、及びBaから成る群から選ばれる少なくとも1種の金属であり、且つ、0<p<1.3、0<q<1.2、0<r<1.2、0≦s<0.5、0≦t<0.3、0.7≦q+r+s+t≦1.2、1.8<u<2.2の範囲であり、そしてpは、電池の充放電状態により決まる値である。}
で表されるリチウム(Li)含有金属酸化物から選ばれる少なくとも1種のLi含有金属酸化物が好適である。
LiwMIIPO4 (Xba)
{式中、MIIは、Feを含む少なくとも1種の遷移金属元素を含む1種以上の遷移金属元素を示し、そしてwの値は、電池の充放電状態により決まり、0.05~1.10の数を示す}
で表されるはオリビン構造を有するリチウムリン金属酸化物を用いることがより好ましい。これらのリチウム含有金属酸化物は、構造を安定化させる等の目的から、Al、Mg、又はその他の遷移金属元素により遷移金属元素の一部を置換したもの、これらの金属元素を結晶粒界に含ませたもの、酸素原子の一部をフッ素原子等で置換したもの、正極活物質表面の少なくとも一部に他の正極活物質を被覆したもの等であってもよい。
LivMID2 (Xa)
{式中、Dは、カルコゲン元素を示し、MIは、少なくとも1種の遷移金属元素を含む1種以上の遷移金属元素を示し、そしてvの値は、電池の充放電状態により決まり、0.05~1.10の数を示す}、
で表される化合物を使用してもよい。
集電体の片面に電極活物質層を形成する場合の目付量は、下記式(12)により算出することができる。
目付量[mg/cm2]=(電極質量[mg]-電極集電体質量[mg])÷電極面積[cm2] ・・・・・(12)
負極160は、負極合剤から作製した負極活物質層と、負極集電体とから構成される。負極160は、非水系二次電池の負極として作用することができる。
本実施形態における非水系二次電池100は、正極150及び負極160の短絡防止、シャットダウン等の安全性付与の観点から、正極150と負極160との間にセパレータ170を備えることが好ましい。セパレータ170としては、イオン透過性が大きく、機械的強度に優れる絶縁性の薄膜が好ましい。セパレータ170としては、例えば、織布、不織布、合成樹脂製微多孔膜等が挙げられ、これらの中でも、合成樹脂製微多孔膜が好ましい。
本実施形態における非水系二次電池100の電池外装110の構成は、例えば、電池缶(図示せず)、及びラミネートフィルム外装体のいずれかの電池外装を用いることができる。電池缶としては、例えば、スチール、ステンレス(SUS)、アルミニウム、又はクラッド材等から成る角型、角筒型、円筒型、楕円型、扁平型、コイン型、又はボタン型等の金属缶を用いることができる。ラミネートフィルム外装体としては、例えば、熱溶融樹脂/金属フィルム/樹脂の3層構成から成るラミネートフィルムを用いることができる。
本実施形態における非水系二次電池100は、上述の非水系電解液、集電体の片面又は両面に正極活物質層を有する正極150、集電体の片面又は両面に負極活物質層を有する負極160、及び電池外装110、並びに必要に応じてセパレータ170を用いて、公知の方法により作製される。
<分析方法>
実施例及び比較例において使用した分析方法、原材料、反応条件等は、以下のとおりのものであった。
実施例、及び比較例で得られた生成物について、1H-NMR(400MHz)、及び19F-NMR(337MHz)を用いて、下記測定条件にて分子構造解析を行った。
[測定条件]
測定装置:JNM-ECZ400S型核磁気共鳴装置(日本電子株式会社製)
観測核:1H、19F
溶媒:重クロロホルム、重ジメチルスルホキシド
基準物質:テトラメチルシラン(1H、0.00ppm)、トリクロロフルオロメタン(19F、0.00ppm)
基準物質濃度:5質量%
測定試料濃度:20質量%
パルス幅:6.5μ秒
待ち時間:2秒
積算回数:8回(1H)、1024回(19F)
<化合物(2)の含有量>
19F-NMRの測定結果から、化合物(1A)及び化合物(2)の代表的なピークの積分値より化合物(2)の含有量を測定した。
本実施例においては、下記構造式:

実施例、及び比較例で得られた生成物について、下記測定条件にて分子構造解析を行った。
[測定条件]
測定装置:SPS3520UV-DD(株式会社日立ハイテクサイエンス製)
測定原子:Li
サンプル調製条件:生成物0.1gを超純水9.9gと混合した濃度1質量%の一時希釈液を得た。次いで、一時希釈液1.5gを1質量%硝酸水溶液28.5gと混合し、測定試料とした。
高周波パワー:1.2kW
プラズマガス(アルゴン)流量:16L/分
補助ガス(アルゴン)流量:0.5L/分
キャリヤーガス(アルゴン)圧力:0.24MPa
キャリヤーガス(アルゴン)流量:0.3L/分
測光高さ:12mm
チャンバーガス(アルゴン)流量:0.6L/分
実施例、及び比較例で得られた生成物について、下記測定条件にて質量減少率の測定を行った。
アルミパン(「クリンプセル(オートサンプラ用)品番346-66963-91」、株式会社島津製作所製、尚、測定時にはカバーを使用しなかった。)に5mgの試料を秤量し、示差熱熱重量同時測定装置(「DTG-60A」、株式会社島津製作所製)を使用し、窒素気流下(流量50mL/分)、25℃~100℃まで昇温速度10℃/分で昇温し、100℃で30分間保持した後、測定温度範囲100℃~500℃、昇温速度10℃/分で加熱し、質量減少の様子を観察した。
上述の100℃で30分間保持した後の100℃からの昇温開始時の質量を基準(100質量%)として、測定温度範囲100℃~500℃、昇温速度10℃/分で加熱する過程で1質量%、2質量%の質量減少率が確認される温度(℃)を観測した。
実施例、及び比較例で使用した原材料を以下に示す。
(フルオロスルホニル基含有カルボン酸(化合物(3)))
・2,2-ジフルオロ-2-(フルオロスルホニル)酢酸(富士フイルム和光純薬株式会社製)(化合物(3-1))
・水酸化リチウム一水和物(富士フイルム和光純薬株式会社製)
(溶媒)
・テトラヒドロフラン(富士フイルム和光純薬株式会社製、乾燥したモレキュラーシーブ3A 1/16(富士フイルム和光純薬株式会社製)を加え、脱水し、モレキュラーシーブ3A 1/16を除去することにより水分量を調整した)
反応温度は、外部加熱冷却装置を用いず、室温である場合は、室温である。また、ウォーターバスやオイルバス等の外部加熱冷却装置を利用する場合には、外部加熱冷却装置に用いられている媒体の温度が反応温度である。
(電解カップリング反応工程)
200mLの三口フラスコに窒素雰囲気下、攪拌子、アセトニトリル(59g)、水(75g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、25.7g、144.3mmоl)を加えた。陽極、及び陰極として、白金板電極(25mm×50mm)を6mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5Aの電流を4時間通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。下層を分取すると7.9gの液体が得られた。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))が90.3質量%(収率37.1%)、HCF2SO2F(化合物(5-1))が1.2質量%含まれていることが確認された。
上記操作により得られた粗製のFO2SCF2CF2SO2F(化合物(4-1))を常圧単蒸留で精製することにより、95.6質量%のFO2SCF2CF2SO2F(化合物(4-1))、及び0.042質量%のHCF2SO2F(化合物(5-1))を含む液体6.9gが得られた。
FO2SCF2CF2SO2F(化合物(4-1))
19F-NMR:δ(ppm)46.1(2F)、-108.7(4F)
HCF2SO2F(化合物(5-1))
1H-NMR:δ(ppm)7.0ppm(1H)
19F-NMR:36.0(1F)、-122.0(2F)
3Lオートクレーブを-78℃に冷却し、アンモニアガス(250g、14680mmоl)、及びテトラヒドロフラン(222.3g)を添加した後、前記電解カップリング反応工程で得られたFO2SCF2CF2SO2F(化合物(4-1)、208g、純度95.6質量%、747.2mmоl)のテトラヒドロフラン(222.3g)溶液を3時間かけて滴下した。滴下終了後、室温で12時間攪拌した。攪拌終了後、反応液を加圧濾過し、不溶固体を除去し、濾液を減圧濃縮、乾燥し、肌色固体193.0gを得た。得られた固体をサンプリングし、19F-NMRで分析すると、下記構造式:

化合物(1-N-2)
19F-NMR:δ(ppm)-115.4(4F)
HF2CSO2NHNH4(化合物(2-2))
1H-NMR:δ(ppm)6.0ppm(1H)
19F-NMR:36.0(1F)、-122.6(2F)
1Lの三口フラスコに、窒素雰囲気下、攪拌子、テトラヒドロフラン(400g)、実施例I-1で得られた含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2)、192.7g、純度99.0質量%、733.2mmоl)、及び水酸化リチウム一水和物(34.2g、815.1mmоl)を添加し、70℃で4時間攪拌した。得られた反応液を減圧濃縮した後、水(500mL)、活性炭(65.8g)を添加し、105℃で3時間攪拌した。得られた反応液を加圧濾過し不溶固体を除去した後、減圧濃縮し肌色固体を得た。得られた肌色固体にテトラヒドロフラン(889g)を添加し、50℃で30分攪拌した後加圧濾過により不溶固体を除去し、減圧濃縮することで177.9gの白色固体を得た。得られた固体をサンプリングし、ICP発光分光分析法により分析を行ったところ、アンモニウムカチオンがリチウムカチオンに交換されていることを確認した。さらに、19F-NMRで分析すると、下記構造式:

化合物(1-2)
19F-NMR:δ(ppm)-115.4(4F)
HCF2SO2NHLi(化合物(2-1))
1H-NMR:δ(ppm)6.0ppm(1H)
19F-NMR:36.0(1F)、-122.6(2F)
(電解カップリング反応工程)
電解反応後に蒸留精製を行わなかったこと以外は、実施例I-1と同様にして、FO2SCF2CF2SO2F(化合物(4-1))の製造を行い、90.3質量%のFO2SCF2CF2SO2F(化合物(4-1))、及び1.2質量%のHCF2SO2F(化合物(5-1))を含む液体7.9g(収率37.2%)を得た。
前記電解カップリング反応工程で得られたFO2SCF2CF2SO2F(化合物(4-1))を用い、実施例I-1と同様に、含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2))の製造を行い、96.2質量%の含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2))、及び1.43質量%のHCF2SO2NHNH4(化合物(2-2))を含む肌色固体188.0g(収率98.3%)を得た。
前記環化工程で得られた含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2))を用い、実施例I-1と同様にして含フッ素環状スルホンイミドリチウム塩(化合物(1-2))の製造を行い、98.3質量%の含フッ素環状スルホンイミドリチウム塩(化合物(1-2))、及び1.22質量%のHCF2SO2NHLi(化合物(2-1))を含む白色固体170.3g(収率97.2%)を得た。

不活性雰囲気下、各種非水系溶媒を、それぞれが所定の濃度になるよう混合した。更に、各種リチウム塩をそれぞれ所定の濃度になるよう添加することにより、非水系電解液(S01)~(S20)を調製した。また、(S01)を母電解液とし、式(2-1)で表される化合物が所定の質量部になるよう添加することにより、非水系電解液(S21)を調製した。これらの非水系電解液組成を表2に示す。
(非水系溶媒)
AN:アセトニトリル
PC:プロピレンカーボネート
EMC:エチルメチルカーボネート
ES:エチレンサルファイト
VC:ビニレンカーボネート
FEC:4-フルオロ-1,3-ジオキソラン-2-オン
(リチウム塩)
(1-2):上記式(1-2)で表される化合物
(1-6):下記式(1-6)で表される化合物

LiTFSI:リチウムビス(トリフルオロメタンスルホニル)イミド(LiN(SO2CF3)2)
(含フッ素スルホンアミド化合物)
(2-1):上記式(2-1):HCF2SO2NHLiで表される含フッ素スルホンアミド化合物
<分析方法>
化合物(1-2)の作製において使用した分析方法、原材料、反応条件等は、以下のとおりのものであった。
化合物(1-2)の作製過程で得られた生成物について、1H-NMR(400MHz)、及び19F-NMR(337MHz)を用いて、下記測定条件にて分子構造解析を行った。
[測定条件]
測定装置:JNM-ECZ400S型核磁気共鳴装置(日本電子株式会社製)
観測核:1H、19F
溶媒:重クロロホルム、重ジメチルスルホキシド
基準物質:テトラメチルシラン(1H、0.00ppm)、トリクロロフルオロメタン(19F、0.00ppm)
基準物質濃度:5質量%
測定試料濃度:20質量%
パルス幅:6.5μ秒
待ち時間:2秒
積算回数:8回(1H)、1024回(19F)
19F-NMRの測定結果から、化合物(1-2)及び化合物(2-1)の代表的なピークの積分値より化合物(2-1)の含有量を測定した。

作製した化合物(1-2)について、下記測定条件にて分子構造解析を行った。
[測定条件]
測定装置:SPS3520UV-DD(株式会社日立ハイテクサイエンス製)
測定原子:Li
サンプル調製条件:生成物0.1gを超純水9.9gと混合した濃度1質量%の一時希釈液を得た。次いで、一時希釈液1.5gを1質量%硝酸水溶液28.5gと混合し、測定試料とした。
高周波パワー:1.2kW
プラズマガス(アルゴン)流量:16L/分
補助ガス(アルゴン)流量:0.5L/分
キャリヤーガス(アルゴン)圧力:0.24MPa
キャリヤーガス(アルゴン)流量:0.3L/分
測光高さ:12mm
チャンバーガス(アルゴン)流量:0.6L/分
化合物(1-2)の作製で使用した原材料を以下に示す。
・2,2-ジフルオロ-2-(フルオロスルホニル)酢酸(化合物(3-1)、富士フイルム和光純薬株式会社製)
・テトラヒドロフラン(富士フイルム和光純薬株式会社製、乾燥したモレキュラーシーブ3A 1/16(富士フイルム和光純薬株式会社製)を加え、脱水し、モレキュラーシーブ3A 1/16を除去することにより水分量を調整した)
反応温度は、外部加熱冷却装置を用いない場合は、室温である。また、ウォーターバスやオイルバス等の外部加熱冷却装置を利用する場合には、外部加熱冷却装置に用いられている媒体の温度が、反応温度である。
(電解カップリング反応工程)
200mLの三口フラスコに窒素雰囲気下、攪拌子、アセトニトリル(59g)、水(75g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、富士フイルム和光純薬株式会社製、25.7g、144.3mmоl)を加えた。陽極、及び陰極として、白金板電極(25mm×50mm)を6mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5Aの電流を4時間通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。下層を分取すると7.9gの液体が得られた。得られた液体を水洗し、モレキュラーシーブで乾燥することにより、FO2SCF2CF2SO2F(化合物(4-1))を含む液体6.9gが得られた。
FO2SCF2CF2SO2F(化合物(4-1))
19F-NMR(重クロロホルム):δ(ppm)46.1(2F)、-108.7(4F)
3Lオートクレーブを-78℃に冷却し、アンモニアガス(250g、14.68mоl)、及びテトラヒドロフラン(250mL)を添加した後、オートクレーブ内の温度を-55℃以下に保ちつつ、FO2SCF2CF2SO2F(化合物(4-1)、208g、0.71mоl)のテトラヒドロフラン(250.0mL)溶液を滴下した。滴下終了後、室温で終夜攪拌した。その後、攪拌を維持しつつ、内圧を常圧に戻し、アルゴンを0.5L/分の速度で1.5時間オートクレーブに吹きこみ、アンモニアを排出した。反応液を濾過することで白色固体を除去し、テトラヒドロフランで数回洗浄した。得られた濾液を3Lのフラスコに移し、減圧濃縮し、さらに40℃で12時間真空乾燥させることで固体188.0gを得た。得られた固体をサンプリングし、19F-NMRで分析すると、下記構造式:

19F-NMR(重ジメチルスルホキシド):δ(ppm)-115.4(4F)
1Lの三口フラスコに、窒素雰囲気下、攪拌子、テトラヒドロフラン(450mL)、含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2)、187.0g、0.69mоl)、及び水酸化リチウム一水和物(32.2g、0.77mоl)を添加し、70℃で4時間攪拌した。得られた反応液を減圧濃縮した後、イオン交換水(500mL)、および活性炭(65.8g)を添加し、105℃で3時間攪拌した。得られた反応液を加圧濾過して不溶固体を除去した後、減圧濃縮して固体(205.8g)を得た。得られた肌色固体にテトラヒドロフラン(1L)を添加し、50℃で30分攪拌した後、加圧濾過により不溶固体を除去し、減圧濃縮した後、70℃で16時間、90℃で10時間真空乾燥することで170.3gの微褐色固体を得た。得られた固体をサンプリングし、ICP発光分光分析法により分析を行ったところ、アンモニウムカチオンがリチウムカチオンに交換されていることを確認した。さらに、19F-NMRで分析すると、下記構造式:

化合物(1-2)
19F-NMR(重ジメチルスルホキシド):δ(ppm)-115.4(4F)
HCF2SO2NHLi(化合物(2-1))
1H-NMR(重ジメチルスルホキシド):δ(ppm)6.0(1H)
19F-NMR(重ジメチルスルホキシド):δ(ppm)-122.6(2F)
撹拌子を備えた100mLナスフラスコに、合成した化合物(1-2)10gとtert-ブチルメチルエーテル27gとを秤量し、密栓を取り付けて30分撹拌した。得られた混合液を減圧濾過後、-20℃の冷凍庫で30分間冷却したところ、無色の結晶が生じた。デカンテーションによる液部の除去後、-35℃のtert-ブチルメチルエーテルを添加して薬さじで混ぜることにより結晶を洗浄し、液部をデカンテーションによって除去した。得られた結晶を、無撹拌条件かつ60℃、および40hPaの条件で3時間減圧乾燥した。この結晶を乳鉢と乳棒を用いて粉砕し、バートレル(三井・ケマーズフロロプロダクツ株式会社製、主成分:1,1,1,2,2,3,4,5,5,5-デカフルオロペンタン)20mLを添加し、無撹拌条件かつ100℃、および15hPaの条件で2時間減圧乾燥する操作を4回繰り返す追乾燥工程を行なうことで、4.9g(収率49.0%)の結晶が得られた。得られた結晶をサンプリングし、19F-NMRで分析すると、化合物(1-2)が99.9質量%、HCF2SO2NHLi(化合物(2-1))が462質量ppm含まれていることが確認された(ロットA)。
化合物(1-2)
19F-NMR(重ジメチルスルホキシド):δ(ppm)-115.4(4F)
HCF2SO2NHLi(化合物(2-1))
1H-NMR(重ジメチルスルホキシド):δ(ppm)6.0(1H)
19F-NMR(重ジメチルスルホキシド):δ(ppm)-122.6(2F)
(電解カップリング反応工程)
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(4.5g)、水(22.6g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、30.0g、168.2mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を3時間通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。下層を分取すると17.2gの液体が得られた。得られた液体をモレキュラーシーブで乾燥後、蒸留精製し、FO2SCF2CF2SO2F(化合物(4-1))を含む液体12.2gが得られた。
FO2SCF2CF2SO2F(化合物(4-1))
19F-NMR(重クロロホルム):δ(ppm)46.1(2F)、-108.7(4F)
3Lオートクレーブを-78℃に冷却し、アンモニアガス(240g、14.09mоl)、及びテトラヒドロフラン(200mL)を添加した後、オートクレーブ内の温度を-55℃以下に保ちつつ、FO2SCF2CF2SO2F(化合物(4-1)、173g、0.59mоl)のテトラヒドロフラン(200.0mL)溶液を滴下した。滴下終了後、室温で終夜攪拌した。その後、攪拌を維持しつつ、内圧を常圧に戻し、アルゴンを0.5L/分の速度で1.5時間オートクレーブに吹きこみ、アンモニアを排出した。反応液を濾過することで白色固体を除去し、テトラヒドロフランで数回洗浄した。得られた濾液を3Lのフラスコに移し、減圧濃縮し、さらに40℃で12時間真空乾燥させることで固体160.2gを得た。得られた固体をサンプリングし、19F-NMRで分析すると、下記構造式:

19F-NMR(重ジメチルスルホキシド):δ(ppm)-115.4(4F)
1Lの三口フラスコに、窒素雰囲気下、攪拌子、テトラヒドロフラン(564mL)、含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2)、242.0g、0.93mоl)、及び水酸化リチウム一水和物(42.9g、1.02mоl)を添加し、75℃で4時間攪拌した。得られた反応液を減圧濃縮した後、イオン交換水(628mL)、および活性炭(82.7g)を添加し、105℃で3時間攪拌した。得られた反応液を加圧濾過して不溶固体を除去した後、減圧濃縮して固体(245g)を得た。得られた肌色固体にテトラヒドロフラン(1.27L)を添加し、50℃で30分攪拌した後、加圧濾過により不溶固体を除去し、減圧濃縮した後、70℃で16時間、90℃で6時間真空乾燥することで212gの白色固体を得た。得られた固体をサンプリングし、ICP発光分光分析法により分析を行ったところ、アンモニウムカチオンがリチウムカチオンに交換されていることを確認した。さらに、19F-NMRで分析すると、下記構造式:

化合物(1-2)
19F-NMR(重ジメチルスルホキシド):δ(ppm)-115.4(4F)
HCF2SO2NHLi(化合物(2-1))
1H-NMR(重ジメチルスルホキシド):δ(ppm)6.0(1H)
19F-NMR(重ジメチルスルホキシド):δ(ppm)-122.6(2F)

(2-1)正極(P1)の作製
正極活物質として、リチウム、ニッケル、マンガン及びコバルトの複合酸化物(LiNi0.33Mn0.33Co0.33O2)と、導電助剤として、カーボンブラック粉末と、バインダーとして、ポリフッ化ビニリデン(PVDF)とを、94:3:3の質量比で混合し、正極合剤を得た。
負極活物質として、グラファイト粉末と、導電助剤として、カーボンブラック粉末と、バインダーとして、ポリフッ化ビニリデン(PVDF)とを、90:3:7の質量比で混合し、負極合剤を得た。
CR2032タイプの電池ケース(SUS304/Alクラッド)にポリプロピレン製ガスケットをセットし、その中央に、上述のようにして得られた正極(P1)を直径15.958mmの円盤状に打ち抜いたものを、正極活物質層を上向きにしてセットした。その上から、ガラス繊維濾紙(GA-100;アドバンテック社製)を直径16.156mmの円盤状に打ち抜いたものをセットして、非水系電解液(S01~S21)を150μL注入した後、上述のようにして得られた負極(N1)を直径16.156mmの円盤状に打ち抜いたものを、負極活物質層を下向きにしてセットした。さらに、電池ケース内にスペーサーとスプリングをセットした後に電池キャップをはめ込み、カシメ機でかしめた。溢れた非水系電解液はウエスで拭き取った。正極とガラス繊維濾紙と負極の積層体、及び非水系電解液を含むアセンブリを25℃で12時間保持し、積層体に非水系電解液を十分馴染ませてコイン型非水系二次電池を得た。
上述のようにして得られたコイン型非水系二次電池について、まず、下記(3-1)の手順に従って初回充電処理及び初回充放電容量測定を行った。次に下記(3-2)の手順に従って、それぞれの非水系二次電池を評価した。なお、充放電はアスカ電子(株)製の充放電装置ACD-M01A(商品名)及びヤマト科学(株)製のプログラム恒温槽IN804(商品名)を用いて行った。
ここで、1Cとは、満充電状態の電池を定電流で放電して1時間で放電終了となることが期待される電流値を意味する。下記(3-1)~(3-2)の評価では、1Cは、具体的には、4.2Vの満充電状態から定電流で3.0Vまで放電して1時間で放電終了となることが期待される電流値を意味する。
コイン型非水系二次電池の周囲温度を25℃に設定し、0.025Cに相当する0.075mAの定電流で充電して3.1Vに到達した後、4.2Vの定電圧で電流が0.025mAに減衰するまで充電を行った。続いて3時間休止後、0.05Cに相当する0.15mAの定電流で電池を充電して4.2Vに到達した後、4.2Vの定電圧で電流が0.025Cに減衰するまで充電を行った。このときの充電容量(3.1Vまでの充電容量と4.2Vまでの充電容量の和)を初回充電容量(X)とした。その後、0.15Cに相当する0.45mAの定電流で3.0Vまで電池を放電した。このときの放電容量を初回放電容量(Y)とした。また、以下の式に基づき、初回充放電効率を算出した。
初回充放電効率=(初回放電容量(Y)/初回充電容量(X))×100[%]
次に、0.2Cに相当する0.6mAの定電流で4.2Vに到達した後、4.2Vの定電圧で電流が0.025Cに減衰するまで充電を行った。その後、0.2Cに相当する0.6mAの電流値で3Vまで放電した。その後、上記と同様の充放電を1サイクル行った。
上記(3-1)に記載の方法で初回充放電処理を行ったコイン型非水系二次電池について、周囲温度を25℃に設定し、1Cに相当する3mAの定電流で充電して4.2Vに到達した後、4.2Vの定電圧で電流が0.025Cに減衰するまで充電を行った。その後、1Cに相当する3mAの定電流で電池電圧3.0Vまで放電した。充電と放電とを各々1回ずつ行うこの工程を1サイクルとし、100サイクルの充放電を行った。このときの放電容量を100サイクル目放電容量(以後、(T)と表記する場合がある)とした。以下の式に基づき、サイクル容量維持率を算出した。
サイクル容量維持率=(25℃サイクル試験での100サイクル目放電容量(T)/初回充放電処理における初回放電容量(Y))×100[%]
なお、25℃サイクル試験前に電圧が2V以下に低下していた場合、電圧異常としてサイクル試験を行わなかった。
25℃サイクル試験終了後、25℃環境下で電池電圧が2.5Vとなるまで0.1Cに相当する定電流で放電を行った。その後、アルゴン雰囲気下でコイン型非水系二次電池を解体して負極を取り出した。アセトニトリルで負極を洗浄した後、乾燥した。さらに、以下に示される前処理を行なった後に、負極のICP発光分光分析を行った。
装置:SII(エスアイアイ)ナノテクノロジー社製SPS3520UV-DD
高周波パワー:1.2(Kw)
プラズマガス(Ar)流量:16(L/min)
補助ガス(Ar)流量 :0.5(L/min)
キャリヤーガス(Ar)圧力:0.24(MPa)
キャリヤーガス(Ar)流量:0.3(L/min)
チャンバーガス(Ar)流量:0.6(L/min)
パージガス(N2)流量:5(L/min)
測光高さ:12(mm)
元素:Al
分光器:R
波長:167.079(nm)
パージガス:ON
検量線は、Al標準液100mg/Lを、酸水溶液(試料の分解液と同様の酸濃度になるように、超純水に68%硝酸と98%硫酸を加えて調製)で希釈して調製した、0、0.01、0.05、0.1、0.5、1、2mg/Lの6点の検量線用標準液を用いて作成した。
25℃サイクル試験終了後、25℃環境下で電池電圧が2.5Vとなるまで0.1Cに相当する定電流で放電を行った。その後、アルゴン雰囲気下でコイン型非水系二次電池を解体して正極を取り出した。正極Al集電体上の正極活物質を、N-メチル-2-ピロリドン(NMP)に浸した綿棒で除去した。アセトニトリルで正極Al集電体を洗浄した後、乾燥した。その後、アルゴン雰囲気下で正極Al集電体を適切な大きさに切り出し、走査電子顕微鏡(SEM)観察用試料台に固定し、表面SEM観察試料とした。試料は専用のトランスファーベッセルを用いて、雰囲気遮断した状態でSEM装置に導入した。
従って、25℃サイクル容量維持率は70%以上であることが望ましい。
また、25℃サイクル試験前に電圧が2V以下に低下した場合、電池内で微短絡が生じていると考えられ、長期使用を目的とする用途に使用するのは困難であると考えられる。
従って、負極Al溶出量は1000質量ppm以下であることが望ましく、500質量ppm以下であることがさらに望ましい。

各サイクルにおいて、放電開始前の電圧と放電開始から10秒後の電圧の差(単位:V)を、放電電流値である3.0(単位:mA)で割ることで求められる値(単位:kΩ)を各サイクルにおける放電IRドロップとし、内部抵抗の指標とした。
100サイクル後IRドロップ増加率=(25℃サイクル試験での100サイクル目放電IRドロップ/1サイクル目放電IRドロップ)×100[%]

(7-1)正極(P2)の作製
正極活物質として、リチウム、ニッケル、マンガン及びコバルトの複合酸化物(LiNi0.8Mn0.1Co0.1O2)と、導電助剤として、カーボンブラック粉末と、バインダーとして、ポリフッ化ビニリデン(PVDF)とを、94:3:3の質量比で混合し、正極合剤を得た。
(A)負極活物質として、黒鉛粉末と、(B)導電助剤としてカーボンブラック粉末と、(C)バインダーとして、ポリフッ化ビニリデン(PVDF)とを、90:3:7の固形分質量比で混合し、負極合剤を得た。
上述のようにして得られた正極(P2)を直径18mmの円盤状に打ち抜いたものと、上述のようにして得られた負極(N2)を直径18mmの円盤状に打ち抜いたものと、セパレータ内蔵絶縁スリーブ(EL-Cell社製、ECC1-00-0210-W/X)の両側に重ね合わせ、非水系電解液(S19~S20)を120μL注入した後、正極側をアルミニウム製プランジャー、負極側をSUS製プランジャーで押さえることで積層体を得た。その積層体を電池ケース(EL-Cell社製、PAT-Cell)に挿入して組み立てた後、電池ケースを密閉して25℃で12時間保持し、積層体に非水系電解液を十分馴染ませて小型非水系二次電池を得た。
上述のようにして得られた小型非水系二次電池について、まず、下記(8-1)の手順に従って初回充放電処理、及び初回充放電容量測定を行った。次に下記(8-2)の手順に従って、それぞれの小型非水系二次電池を評価した。なお、充放電はアスカ電子(株)製の充放電装置ACD-M01A(商品名)、及びヤマト科学(株)製のプログラム恒温槽IN804(商品名)を用いて行った。
小型非水系二次電池の周囲温度を25℃に設定し、0.025Cに相当する0.185mAの定電流で充電して3.1Vに到達した後、3.1Vの定電圧で1.5時間充電を行った。続いて3時間休止後、0.05Cに相当する0.37mAの定電流で電池を充電して4.2Vに到達した後、4.2Vの定電圧で1.5時間充電を行った。その後、0.15Cに相当する1.11mAの定電流で3.0Vまで電池を放電した。
上記(8-1)に記載の方法で初回充放電処理を行った小型非水系二次電池について、周囲温度を50℃に設定し、0.5Cに相当する3.7mAの定電流で充電して4.2Vに到達した後、4.2Vの定電圧で1.5時間充電を行った。その後、0.5Cに相当する3.7mAの電流値で3.0Vまで電池を放電した。充電と放電とを各々1回ずつ行うこの工程を1サイクルとし、100サイクルの充放電を行った。
100サイクル後IRドロップ増加率=(50℃サイクル試験での100サイクル目放電IRドロップ/1サイクル目放電IRドロップ)×100[%]

実施例及び比較例において使用し分析方法、原材料、反応条件等は、以下の通りのものであった。
(核磁気共鳴分析(NMR):1H-NMR、及び19F-NMRによる分子構造解析)
実施例、及び比較例で得られた生成物について、1H-NMR(400MHz)、及び19F-NMR(337MHz)を用いて、下記測定条件にて分子構造解析を行った。
[測定条件]
測定装置:JNM-ECZ400S型核磁気共鳴装置(日本電子株式会社製)
観測核:1H、19F
溶媒:重クロロホルム、重ジメチルスルホキシド
基準物質:テトラメチルシラン(1H、0.00ppm)、トリクロロフルオロメタン(19F、0.00ppm)
基準物質濃度:5質量%
測定試料濃度:20質量%
パルス幅:6.5μ秒
待ち時間:2秒
積算回数:8回(1H)、1024回(19F)
実施例及び比較例で使用した原材料を以下に示す。
(フルオロスルホニル基含有カルボン酸化合物(3))
・2,2-ジフルオロ-2-(フルオロスルホニル)酢酸(富士フイルム和光純薬株式会社製)(化合物(3-1))
・アンモニア(住友精化株式会社製)
(アルカリ金属塩(6))
・水酸化リチウム一水和物(富士フイルム和光純薬株式会社製)
・アセトニトリル(富士フイルム和光純薬株式会社製、乾燥したモレキュラーシーブ3A 1/16(富士フイルム和光純薬株式会社製)を加え、脱水し、モレキュラーシーブ3A 1/16を除去することにより水分量を調整した)
・テトラヒドロフラン(富士フイルム和光純薬株式会社製、乾燥したモレキュラーシーブ3A 1/16(富士フイルム和光純薬株式会社製)を加え、脱水し、モレキュラーシーブ3A 1/16を除去することにより水分量を調整した)
・オクタフルオロトルエン(東京化成工業株式会社製)
反応温度は、外部加熱冷却装置を用いず、室温である場合は、室温である。また、ウォーターバスやオイルバス等の外部加熱冷却装置を利用する場合には、外部加熱冷却装置に用いられている媒体の温度が反応温度である。
[実施例III-1]
100mLの三口フラスコに窒素雰囲気下、攪拌子、アセトニトリル(12.9g)、水(17.1g)を添加した後(β/α=0.75)、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.0g、28.1mmоl)を加えた。陽極、及び陰極として、白金板電極(25mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5Aの電流を0.5時間通電した。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.9であった。
FO2SCF2CF2SO2F(化合物(4-1))
19F-NMR:δ(ppm)46.1(2F)、-108.7(4F)
HCF2SO2F(化合物(5-1))
1H-NMR:δ(ppm)7.7ppm(1H)
19F-NMR:37.0(1F)、-120.3(2F)
アセトニトリル(10.0g)、水(20.0g)を使用したこと以外は、実施例III-1と同様にして通電を行った(β/α=0.50)。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.8であった。
アセトニトリル(5.0g)、水(25.0g)を使用したこと以外は、実施例III-1と同様にして通電を行った(β/α=0.20)。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.4であった。
(電解カップリング反応)
アセトニトリル(2.5g)、水(27.5g)を使用したこと以外は、実施例III-1と同様にして通電を行った(β/α=0.09)。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.3であった。
1Lオートクレーブを-78℃に冷却し、アンモニアガス(60.0g、3523.2mmоl)、及びテトラヒドロフラン(110.0g)を添加した後、電解カップリング反応で得られたFO2SCF2CF2SO2F(化合物(3-1)、50g、187.5mmоl)のテトラヒドロフラン(50.0g)溶液を3時間かけて滴下した。滴下終了後、室温で12時間攪拌した。攪拌終了後、反応液を加圧濾過し、不溶固体を除去し、濾液を減圧濃縮、乾燥し、肌色固体49.1gを得た。得られた固体をサンプリングし、19F-NMRで分析すると、下記構造式:

19F-NMR:δ(ppm)-115.4(4F)
1Lの三口フラスコに、窒素雰囲気下、攪拌子、テトラヒドロフラン(100g)、環化工程で得られた含フッ素環状スルホニルイミドアンモニウム塩(化合物(1-N-2)、45.0g、170.4mmоl)、及び水酸化リチウム一水和物(8.1g、193.0mmоl)を添加し、70℃で4時間攪拌した。得られた反応液を減圧濃縮した後、水(120mL)、活性炭(16.5g)を添加し、105℃で3時間攪拌した。得られた反応液を加圧濾過し不溶固体を除去した後、減圧濃縮し肌色固体を得た。得られた肌色固体にテトラヒドロフラン(220g)を添加し、50℃で30分攪拌した後加圧濾過により不溶固体を除去し、減圧濃縮することで40.9gの白色固体を得た。得られた固体をサンプリングし、ICP発光分光分析法により分析を行ったところ、アンモニウムカチオンがリチウムカチオンに交換されていることを確認した。さらに、19F-NMRで分析すると、下記構造式:

19F-NMR:δ(ppm)-115.4(4F)
アセトニトリル(13.0g)、水(17.0g)を使用したこと以外は、実施例III-1と同様にして通電を行った(β/α=0.76)。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:1.6であった。
アセトニトリル(15.0g)、水(15.0g)を使用したこと以外は、実施例III-1と同様にして通電を行った(β/α=1.00)。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:4.4であった。
アセトニトリル(20.0g)、水(10.0g)を使用したこと以外は、実施例III-1と同様にして通電を行った(β/α=2.00)。通電終了後、得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:8.4であった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(5g)、水(25g)を添加した後(β/α=0.2)、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.2g、29.4mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を0.5時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、HCF2SO2F(化合物(5-1))が収率0.4%、HO2CCF2SO3Hが収率0.2%生成し、FO2SCF2CO2Hが23%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率49%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、49%であった。FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.8であった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(10g)、水(20g)を添加した後(β/α=0.5)、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.1g、28.7mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を0.5時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率0.5%、FO2SCF2CF2SO3Hが収率0.8%、HCF2SO2F(化合物(5-1))が収率1%、HO2CCF2SO3Hが収率0.2%生成し、FO2SCF2CO2H(化合物(3-1))が16%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率53%、HCF2SO2Fが痕跡量生成していることが明らかになった。電流効率は、53%であった。FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:1.9であった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(4.17g)、水(20.85g)を添加した後(β/α=0.2)、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、10.1g、56.9mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を1時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、HCF2SO2F(化合物(5-1))が収率0.3%、HO2CCF2SO3Hが収率0.6%生成し、FO2SCF2CO2H(化合物(3-1))が17%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率57%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、57%であった。FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.5であった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(3.3g)、水(16.7g)を添加した後(β/α=0.2)、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、15.2g、85.2mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を1.5時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、HCF2SO2F(化合物(5-1))が収率0.1%、HO2CCF2SO3Hが収率0.6%生成し、FO2SCF2CO2H(化合物(3-1))が13%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率62%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、62%であった。FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.2であった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(4.5g)、水(22.5g)を添加した後(β/α=0.2)、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、30.0g、168.5mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を3時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、HCF2SO2F(化合物(5-1))が収率0.1%、HO2CCF2SO3Hが収率0.7%生成し、FO2SCF2CO2H(化合物(3-1))が9%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率72%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、72%であった。FO2SCF2CF2SO2F(化合物(4-1))とHCF2SO2F(化合物(5-1))の生成比は100:0.1であった。
[実施例III-13]
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(15g)、水(15g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.0g、28.3mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を0.25時間(0.5F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率8%、FO2SCF2CF2SO3Hが収率1%、HCF2SO2F(化合物(5-1))が収率3%、HO2CCF2SO3Hが収率0.3%生成し、FO2SCF2CO2H(化合物(3-1))が41%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率22%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、60%であった。反応容器と電極の関係について、高さTLは75mmであり、最高位TEが65mm、高さTSが8mm、最低位TE’が15mmであったため、TL>TEを満たし、TE’>TSを満たしていた。反応中、電圧が急激に上昇することはなく、ハンチングは発生しなかった。
FO2SCF2CF2SO3H
19F-NMR:δ(ppm)44.3(1F)、-106.2(2F)、-113.7(2F)
HCF2SO2F(化合物(5-1))
19F-NMR:δ(ppm)37.0(1F)、-120.3(2F)
1H-NMR:δ(ppm)6.5(1H)
HO2CCF2SO3H
19F-NMR:δ(ppm)-111.5(2F)
FO2SCF2CO2H(化合物(3-1))
19F-NMR:δ(ppm)37.1(1F)、-102.0(2F)
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(15g)、水(15g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.1g、28.4mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を0.5時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率8%、FO2SCF2CF2SO3Hが収率2%、HCF2SO2F(化合物(5-1))が収率2%生成し、FO2SCF2CO2Hが11%残存していることが明らかになった。下相を分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率41%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、49%であった。反応容器と電極の関係について、高さTLは75mmであり、最高位TEが65mm、高さTSが8mm、最低位TE’が15mmであったため、TL>TEを満たし、TE’>TSを満たしていた。反応中、電圧が急激に上昇することはなく、ハンチングは発生しなかった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(15g)、水(15g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.1g、28.5mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を0.75時間(1.5F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率7%、FO2SCF2CF2SO3Hが収率3%、HCF2SO2F(化合物(5-1))が収率1%、HO2CCF2SO3Hが痕跡量生成し、FO2SCF2CO2H(化合物(3-1))が5%残存していることが明らかになった。下相を分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率38%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、30%であった。反応容器と電極の関係について、高さTLは75mmであり、最高位TEが65mm、高さTSが5mm、最低位TE’が15mmであったため、TL>TEを満たし、TE’>TSを満たしていた。反応中、電圧が急激に上昇することはなく、ハンチングは発生しなかった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(15g)、水(15g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、5.0g、28.3mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を1時間(2F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率7%、FO2SCF2CF2SO3Hが収率3%、HCF2SO2F(化合物(5-1))が収率1%、HO2CCF2SO3Hが痕跡量生成し、FO2SCF2CO2H(化合物(3-1))が2%残存していることが明らかになった。下相を19F-NMRで分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率44%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、26%であった。反応容器と電極の関係について、高さTLは75mmであり、最高位TEが65mm、高さTSが8mm、最低位TE’が15mmであったため、TL>TEを満たし、TE’>TSを満たしていた。反応中、電圧が急激に上昇することはなく、ハンチングは発生しなかった。
[貯蔵安定性評価]
30mLのスクリュー管にFO2SCF2CO2H(化合物(3-1)、10.0g)、アセトニトリル(1.5g)、水(7.5g)、及び19F―NMR用内部標準物質としてオクタフルオロトルエン(0.3g)を添加し、振とうした後19F-NMRを測定し、オクタフルオロトルエンの-57.4ppmの三重線のピークの積分値を3、としたときの化合物(3-1)の-102.0ppmの二重線のピークの積分値を算出した結果、88.72であった。本数値を経過時間0時間の化合物(3-1)の基準含有量、とした。続いて、上記溶液を3本の10mLスクリュー管に分割し、それぞれのスクリュー管を-20℃、-3℃、23℃で静置し、所定時間経過後に19F-NMRを測定し、オクタフルオロトルエンの-57.4ppmの三重線のピークの積分値を3、としたときの化合物(3-1)の-102.0ppmの二重線のピークの積分値を算出し下式より化合物(3-1)の残存率を算出した。
化合物(3-1)の残存率=(-102.0ppmの二重線のピークの積分値)/88.72×100

[実施例IV-1]
100mLの三口フラスコに窒素雰囲気下、攪拌子、アセトニトリル(4.0g)、水(18.5g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(3-1)、15.0g、84.2mmоl)を加えた(δ/γ=1.5)。陽極、及び陰極として、白金板電極(25mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5Aの電流を1.5時間通電した。通電終了後、攪拌を止めると反応液が2相に分離した。このうち下相を分液し、9.0gの液体を得た。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1)、純度90.5%、収率72.7%)であることが分かった。
FO2SCF2CF2SO2F(化合物(4-1))
19F-NMR:δ(ppm)46.1(2F)、-108.7(4F)
アセトニトリル(3.3g)、水(16.7g)、FO2SCF2CO2H(化合物(3-1)、15.0g、84.2mmоl)を使用したこと以外は、実施例IV-1と同様にして通電を行った(δ/γ=1.3)。通電終了後、攪拌を止めると反応液が2相に分離した。このうち下相を分液し、9.4gの液体を得た。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1)、純度92.4%、収率77.5%)であることが分かった。
(電解カップリング反応)
アセトニトリル(4.5g)、水(22.5g)、FO2SCF2CO2H(化合物(3-1)、30.0g、168.5mmоl)を使用し、通電時間を3時間としたこと以外は、実施例IV-1と同様にして通電を行った(δ/γ=0.9)。通電終了後、攪拌を止めると反応液が2相に分離した。このうち下相を分液し、20.6gの液体を得た。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1)、純度92.4%、収率84.9%)であることが分かった。
1Lオートクレーブを-78℃に冷却し、アンモニアガス(60.0g、3523.2mmоl)、及びテトラヒドロフラン(110.0g)を添加した後、電解カップリング反応で得られたFO2SCF2CF2SO2F(化合物(4-1)、50g、純度92.4%、173.6mmоl)のテトラヒドロフラン(50.0g)溶液を3時間かけて滴下した。滴下終了後、室温で12時間攪拌した。攪拌終了後、反応液を加圧濾過し、不溶固体を除去し、濾液を減圧濃縮、乾燥し、肌色固体45.4gを得た。得られた固体をサンプリングし、19F-NMRで分析すると、下記構造式:

化合物(1-N-2)
19F-NMR:δ(ppm)-115.4(4F)

化合物(1-2)
19F-NMR:δ(ppm)-115.4(4F)
アセトニトリル(4.1g)、水(20.9g)、FO2SCF2CO2H(化合物(3-1)、10.0g、56.2mmоl)を使用し(δ/γ=2.5)、通電時間を1時間としたこと以外は、実施例IV-1と同様にして通電を行った。通電終了後、攪拌を止めると反応液が2相に分離した。このうち下相を分液し、4.8gの液体を得た。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1)、純度91.0%、収率58.5%)であることが分かった。
アセトニトリル(5.0g)、水(25.0g)、FO2SCF2CO2H(化合物(3-1)、5.0g、28.1mmоl)を使用し(δ/γ=6.0)、通電時間を0.5時間としたこと以外は、実施例IV-1と同様にして通電を行った。通電終了後、攪拌を止めると反応液が2相に分離した。このうち下相を分液し、2.2gの液体を得た。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1)、純度89.1%、収率51.5%)であることが分かった。
アセトニトリル(4.0g)、水(20.0g)、FO2SCF2CO2H(化合物(3-1)、15.0g、84.2mmоl)を使用したこと以外は、実施例IV-1と同様にして通電を行った(δ/γ=1.6)。通電終了後、攪拌を止めると反応液が2相に分離した。このうち下相を分液し、8.3gの液体を得た。得られた液体をサンプリングし、19F-NMRで測定すると、FO2SCF2CF2SO2F(化合物(4-1)、純度91.1%、収率67.5%)であることが分かった。
外形が、直径30mm、高さ170mmであり、容量が50mLのシュレンク管に窒素雰囲気下、攪拌子、アセトニトリル(4.5g)、水(22.5g)を添加した後、0℃に冷却し、FO2SCF2CO2H(化合物(4-1)、30.0g、168.5mmоl)を加えた。陽極、及び陰極として、白金板電極(13mm×50mm)を3mmの間隔で設置し、溶液中に浸漬させた。0℃冷却下で攪拌を維持しつつ、電極に1.5A(231mA/cm2)の電流を3時間(1F/mоl)通電した。通電終了後、攪拌を止めると、反応液が2相に分離した。上層を分析すると、HCF2SO2F(化合物(5-1)が収率0.1%、HO2CCF2SO3Hが収率0.7%生成し、FO2SCF2CO2H(化合物(3-1))が9%残存していることが明らかになった。下相を分析すると、FO2SCF2CF2SO2F(化合物(4-1))が収率72%、HCF2SO2F(化合物(5-1))が痕跡量生成していることが明らかになった。電流効率は、72%であった。
(環化工程)
3Lオートクレーブを-78℃に冷却し、アンモニアガス(250g、14.68mоl)、及びテトラヒドロフラン(250mL)を添加した後、オートクレーブ内の温度を-55℃以下に保ちつつ、実施例IV-3の電解カップリング反応で得られたFO2SCF2CF2SO2F(化合物(4-1)、208g、0.71mоl)のテトラヒドロフラン(250.0mL)溶液を滴下した。滴下終了後、室温で終夜攪拌した。その後、攪拌を維持しつつ、内圧を常圧に戻し、アルゴンを0.5L/分の速度で1.5時間オートクレーブに吹きこみ、アンモニアを排出した。反応液を濾過することで白色固体を除去し、テトラヒドロフランで数回洗浄した。得られた濾液を3Lのフラスコに移し、減圧濃縮し、さらに40℃で12時間真空乾燥させることで固体188.0gを得た。得られた固体をサンプリングし、19F-NMRで分析すると、下記構造式:


<分析方法>
実施例及び比較例において使用した分析方法は、以下のとおりのものであった。
実施例及び比較例で得られた生成物について、1H-NMR(400MHz)、19F-NMR(376MHz)を用いて、下記測定条件にて分子構造解析を行った。
[測定条件]
測定装置:JNM-ECZ400S型核磁気共鳴装置(日本電子株式会社製)
観測核:1H又は19F
溶媒:重クロロホルム、重ジメチルスルホキシド
基準物質:テトラメチルシラン(1H,0.00ppm),トリクロロフルオロメタン(19F,0.00ppm)
基準物質濃度:5質量%
測定試料濃度:20質量%
パルス幅:6.5μ秒
待ち時間:2秒
積算回数:8回(1H)又は1024回(19F)。
尚、以下の実施例において、19F-NMR測定によって得られたスペクトルにおいて、含フッ素環状スルホニルイミド塩のピークの積分値の和をT1と、-115ppm~-125ppmの領域に単一のまたは複数の二重線ピークとして現れることに特徴的な不純物のピークの積分値の和をT2、T1とT2との比をT1/T2と表記し、これを含フッ素環状スルホニルイミドの純度を示す指標として用いる。また、19F-NMRにおける含フッ素環状スルホニルイミド塩、及び不純物のピークは、純度や残存溶媒量、測定条件等によって化学シフト値が±5ppm程度変動する場合がある。本発明に係る含フッ素環状スルホニルイミド塩は、このような化学シフト値の変動に伴う帰属の誤りに制限を受けるものではない。
特許文献2(国際公開第2006/106960号)の実施例[例2]及び[例4]から[例6]に記載された方法に従い、市販の2,2-ジフルオロ-2-フルオロスルホニル酢酸(富士フイルム和光純薬株式会社製)を原料として、電解カップリング反応工程、アンモニアによる環化工程、水酸化リチウムによるカチオン交換工程を経て、下記構造式:

実施例V-1と同様に化合物(1-2)を合成したが、その後に晶析による精製を行なわなかった。19F-NMRにおいて、化合物(1-2)のピーク(δ(ppm)-115.4(4F))と、-122ppm~-125ppmの領域に複数の二重線として見られた不純物のピークの積分値の和に関して、T1/T2は215と低純度であり、破砕乾燥後に得られた粉体は褐色に着色していた。
実施例V-1と同様に化合物(1-2)を合成した。次いで、20mLナスフラスコに合成した化合物(1-2)1gとアセトン(沸点56℃)2gを秤量し、密栓を取り付けて30分撹拌した。混合液を-50℃で一晩冷却したが、結晶は生じず、高純度化はできなかった。回収のため60℃、10hPaの条件で3時間減圧乾燥したところ、内容物は壁面の一部が固化したのみで、底部には高粘性の液体が残存しており、薬さじで固体として回収することは不可能であった。低沸点のアセトンを用いても多量の溶媒が残存したのは、アセトンとリチウムとの高い親和性により、アセトンの留去が阻害されたためと考えられる。
実施例V-1と同様に化合物(1-2)を合成した。次いで、50mLナスフラスコに合成した化合物(1-2)1gと炭酸ジメチル2gを秤量し、密栓を取り付けて30分撹拌した。混合物を濾過後、得られた液にシクロヘキサン20gを添加したところ、内容液は2相に分離し、結晶は生じなかった。相分離した内容液を-20℃で一晩冷却したが、化合物(1-2)の結晶は生じず、高純度化はできなかった。炭酸ジメチルとリチウムとの高い親和性により結晶化が抑制されたためと考えられる。
実施例V-1と同様に化合物(1-2)を合成した。次いで、200mLナスフラスコに合成した化合物(1-2)10gと炭酸ジメチル20gを秤量し、密栓を取り付けて30分撹拌した。混合物を濾過後、液を-50℃で一晩冷却したが、結晶は生じなかった。回収のため100℃、5hPaの条件で10時間減圧乾燥したところ、混合物全体が白濁したゲル状物となり、その後更に乾燥を続けることで高粘性の固体状となった。ここにtert-ブチルメチルエーテル27gを加えて溶解させた後に、-50℃で一晩冷却したところ、無色の結晶が生じた。液部をデカンテーションにより除去した後、-35℃のtert-ブチルメチルエーテルを添加して薬さじで混ぜることにより結晶を洗浄し、液部をデカンテーションによって除去した。得られた結晶を無撹拌条件で60℃、40hPaの条件で3時間減圧乾燥した。得られた結晶を回収すべく薬さじで砕いて取り出すことを試みたが、ナスフラスコ底部への固着が顕著であり、薬さじでの粉砕で得られた結晶は2.7g(収率27%)に留まった。固着による損失は2.0gであった。リチウムとの親和性の高い炭酸ジメチルが残存したことで乾燥時に結晶が部分溶解し、ナスフラスコ壁面に固着したものと考えられる。結晶同士の固着による試料の不均一性が予想されたことから、回収できた結晶を更に100℃、20hPaの条件で3時間乾燥させ、乳棒で粉砕することで最終的に結晶2.4gを取得した。19F-NMRによるT1/T2は1550であった。ベンゾトリフルオリドを内部標準とした1H-NMRの測定から、tert-ブチルメチルエーテルの残存量は0.29質量%、炭酸ジメチルの残存量は0.13質量%であり、これらの結果から化合物(1-2)の純度は99.5質量%であった。
実施例V-1と同様に化合物(1-2)を合成した。次いで、20mLナスフラスコに合成した化合物(1-2)1.0gとテトラヒドロフラン(沸点65℃)1.3gを秤量し、密栓を取り付けて30分撹拌した。混合液を-50℃で一晩冷却したが結晶は生成せず、高純度化はできなかった。回収のため60℃、10hPaの条件で3時間減圧乾燥したところ、内容物は壁面の一部が固化したのみで、底部には高粘性の液体が残存しており、薬さじで固体として回収することは不可能であった。低沸点のエーテル溶媒であるテトラヒドロフランを用いても多量の溶媒が残存したのは、テトラヒドロフランの環状構造に伴うリチウムへの強い配位力により、テトラヒドロフランの留去が阻害されたためと考えられる。
実施例V-1と同様に化合物(1-2)の合成、晶析および乾燥を行い、4.9gの結晶を得た。次いで、1,1,1,2,2,3,4,5,5,5-デカフルオロペンタン30mLを添加して薬さじで混ぜることにより結晶を洗浄し、100℃、15hPa条件で7時間乾燥した。この洗浄および乾燥操作を計5回繰り返し、最終的に結晶4.6gを取得した。19F-NMRによるT1/T2は3355であった。ベンゾトリフルオリドを内部標準とした1H-NMRの測定から、tert-ブチルメチルエーテルの残存量は0.072質量%であり、これらの結果から化合物(1-2)の純度は99.9質量%であった。得られた乾燥後の結晶は薬さじで砕くことで容易に全量をナスフラスコから取り出すことが可能であった。
2 一対の電極
3 水相(上層)
4 有機相(下層)
TL 反応容器底部から反応液の液面までの高さ(距離)
TE 反応容器底部から電極上部(最高位)までの高さ(距離)
TE’ 反応容器底部から電極下部(最低位)までの高さ(距離)
TS 反応容器底部から、有機相と水相の間の界面までの高さ(距離)
110 電池外装
120 電池外装の空間
130 正極リード体
140 負極リード体
150 正極
160 負極
170 セパレータ
Claims (13)
- 非水系溶媒及びリチウム塩を含有する非水系電解液であって、
前記非水系溶媒はカーボネート溶媒を含み、
前記リチウム塩が、下記一般式(1):
で表される含フッ素環状スルホニルイミド塩を含有し、かつ
下記一般式(2):
H(CR2)m-SO2NHM2 (2)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、M2は、Li、Na、K、H又はNH4であり、そしてmは、1又は2である}
で表される含フッ素スルホンアミド化合物を前記非水系電解液の全量に対して、1質量ppm以上200質量ppm以下含有する、非水系電解液。 - 前記一般式(1)で表される含フッ素環状スルホニルイミド塩が、下記式(1-1)~(1-5):
- 前記一般式(2)で表される含フッ素スルホンアミド化合物を、前記非水系電解液の全量に対して、0.1質量ppm以上含有する、請求項1又は2に記載の非水系電解液。
- 前記非水系溶媒が、アセトニトリルを、前記非水系溶媒の全量に対して、3体積%以上97体積%以下含有する、請求項1~3のいずれか一項に記載の非水系電解液。
- 前記アセトニトリルの含有量が、前記非水系溶媒の全量に対して、20体積%以上95体積%以下である、請求項4に記載の非水系電解液。
- 前記一般式(1)で表される含フッ素環状スルホニルイミド塩の含有量が、前記非水系溶媒1Lに対して、0.8モル以上1.5モル以下である、請求項1~5のいずれか一項に記載の非水系電解液。
- 前記一般式(2)で表される含フッ素スルホンアミド化合物を、前記非水系電解液の全量に対して、80質量ppm以上120質量ppm以下含有する、請求項1~6のいずれか一項に記載の非水系電解液。
- 前記リチウム塩が、前記一般式(1)で表される含フッ素環状スルホニルイミド塩と、LiPF6とを含有し、
前記非水系溶媒が、ビニレンカーボネート及び/またはフルオロエチレンカーボネートを含有し、
前記一般式(1)で表される含フッ素環状スルホニルイミド塩の含有量は、前記LiPF6の含有量に対してモル比で2.5以上であり、かつ
前記LiPF6の含有量は、ビニレンカーボネート及びフルオロエチレンカーボネートの含有量に対してモル比で0.01以上4以下である、請求項1~7のいずれか一項に記載の非水系電解液。 - 前記一般式(1)で表される含フッ素環状スルホニルイミド塩の含有量は、前記LiPF6の含有量に対してモル比で10より大きい、請求項8に記載の非水系電解液。
- 請求項1~9のいずれか一項の非水系電解液を備えた、非水系二次電池。
- 前記非水系二次電池は電池外装を備え、前記電池外装が、正極側の、前記非水系電解液との接液層の少なくとも一部にアルミニウムを含む、請求項10に記載の非水系二次電池。
- 下記一般式(3):
MO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてMは、H、Li、Na、K又はNH4である}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、前記一般式(1)で表される含フッ素環状スルホニルイミド塩を製造し、
前記一般式(1)で表される含フッ素環状スルホニルイミド塩と非水系溶媒とを混合する混合工程を含み、
該電解カップリング反応工程後に下記一般式(5):
H-(CR2)m-SO2F (5)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2である}で表されるフルオロスルホニル基含有化合物を、該電解カップリング反応工程で得られる反応液中0.1質量%以下まで低下させる精製工程、を含むことを特徴とする請求項1~9のいずれか一項に記載の非水系電解液の製造方法。 - 下記一般式(3):
MO2C-(CR2)m-SO2F (3)
{式中、Rは、それぞれ、同一であっても異なっていてもよく、フッ素原子又はトリフルオロメチル基であり、mは、1又は2であり、そしてMは、H、Li、Na、K又はNH4である}で表されるフルオロスルホニル基含有カルボン酸化合物から出発して、電解カップリング反応工程、環化工程、及びカチオン交換工程を経て、前記一般式(1)で表される含フッ素環状スルホニルイミド塩を製造し、
前記カチオン交換工程後に得られる前記一般式(1)で表される含フッ素環状スルホニルイミド塩を、単座配位性の鎖状エーテル溶媒に、溶解させた後に晶析して、精製された含フッ素環状スルホニルイミド塩を得る晶析工程と、
前記晶析工程で得られた前記一般式(1)で表される含フッ素環状スルホニルイミド塩と非水系溶媒とを混合する混合工程とを含む、請求項1~9のいずれか一項に記載の非水系電解液の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2024176033A JP2025001013A (ja) | 2021-03-26 | 2024-10-07 | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 |
Applications Claiming Priority (17)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021053666 | 2021-03-26 | ||
| JP2021053666 | 2021-03-26 | ||
| JP2021108912 | 2021-06-30 | ||
| JP2021108939 | 2021-06-30 | ||
| JP2021108940 | 2021-06-30 | ||
| JP2021108912 | 2021-06-30 | ||
| JP2021108939 | 2021-06-30 | ||
| JP2021108940 | 2021-06-30 | ||
| JP2021140158 | 2021-08-30 | ||
| JP2021140158 | 2021-08-30 | ||
| JP2021161468 | 2021-09-30 | ||
| JP2021161369 | 2021-09-30 | ||
| JP2021161468 | 2021-09-30 | ||
| JP2021161369 | 2021-09-30 | ||
| JP2021201100 | 2021-12-10 | ||
| JP2021201100 | 2021-12-10 | ||
| PCT/JP2022/014660 WO2022203074A1 (ja) | 2021-03-26 | 2022-03-25 | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2024176033A Division JP2025001013A (ja) | 2021-03-26 | 2024-10-07 | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JPWO2022203074A1 JPWO2022203074A1 (ja) | 2022-09-29 |
| JP7587681B2 true JP7587681B2 (ja) | 2024-11-20 |
Family
ID=83397576
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023509346A Active JP7587681B2 (ja) | 2021-03-26 | 2022-03-25 | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 |
| JP2024176033A Pending JP2025001013A (ja) | 2021-03-26 | 2024-10-07 | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2024176033A Pending JP2025001013A (ja) | 2021-03-26 | 2024-10-07 | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20240194948A1 (ja) |
| EP (1) | EP4317129A4 (ja) |
| JP (2) | JP7587681B2 (ja) |
| KR (1) | KR20230146646A (ja) |
| WO (1) | WO2022203074A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20230167411A (ko) * | 2021-04-08 | 2023-12-08 | 시온 파워 코퍼레이션 | 리튬 배터리용 전해액 |
| WO2024184682A1 (en) * | 2023-03-08 | 2024-09-12 | Ses Holdings Pte. Ltd. | Electrolytes containing sulfonamide-type cyclic salts, and energy-storage cells and batteries made therewith |
| CN119976754B (zh) * | 2023-11-13 | 2025-12-05 | 浙江省化工研究院有限公司 | 一种双氟磺酰亚胺钠的制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006106960A1 (ja) | 2005-04-01 | 2006-10-12 | Asahi Glass Company, Limited | ジスルホニルフロリド化合物の製造方法 |
| WO2009025246A1 (ja) | 2007-08-17 | 2009-02-26 | Asahi Glass Company, Limited | 精製された含フッ素ビススルホニルイミドのアンモニウム塩の製造方法 |
| WO2010110388A1 (ja) | 2009-03-27 | 2010-09-30 | 旭硝子株式会社 | 蓄電デバイス用電解液および蓄電デバイス |
| WO2020262670A1 (ja) | 2019-06-28 | 2020-12-30 | 旭化成株式会社 | 非水系電解液、及び非水系二次電池 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008021517A (ja) * | 2006-07-12 | 2008-01-31 | Sony Corp | 非水電解質二次電池 |
| JP6085456B2 (ja) * | 2012-12-04 | 2017-02-22 | 三星エスディアイ株式会社Samsung SDI Co., Ltd. | リチウムイオン二次電池用電解液及びリチウムイオン二次電池 |
-
2022
- 2022-03-25 US US18/283,573 patent/US20240194948A1/en active Pending
- 2022-03-25 EP EP22775855.4A patent/EP4317129A4/en active Pending
- 2022-03-25 JP JP2023509346A patent/JP7587681B2/ja active Active
- 2022-03-25 KR KR1020237032198A patent/KR20230146646A/ko active Pending
- 2022-03-25 WO PCT/JP2022/014660 patent/WO2022203074A1/ja not_active Ceased
-
2024
- 2024-10-07 JP JP2024176033A patent/JP2025001013A/ja active Pending
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006106960A1 (ja) | 2005-04-01 | 2006-10-12 | Asahi Glass Company, Limited | ジスルホニルフロリド化合物の製造方法 |
| WO2009025246A1 (ja) | 2007-08-17 | 2009-02-26 | Asahi Glass Company, Limited | 精製された含フッ素ビススルホニルイミドのアンモニウム塩の製造方法 |
| WO2010110388A1 (ja) | 2009-03-27 | 2010-09-30 | 旭硝子株式会社 | 蓄電デバイス用電解液および蓄電デバイス |
| WO2020262670A1 (ja) | 2019-06-28 | 2020-12-30 | 旭化成株式会社 | 非水系電解液、及び非水系二次電池 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4317129A4 (en) | 2025-05-07 |
| EP4317129A1 (en) | 2024-02-07 |
| US20240194948A1 (en) | 2024-06-13 |
| JPWO2022203074A1 (ja) | 2022-09-29 |
| WO2022203074A1 (ja) | 2022-09-29 |
| JP2025001013A (ja) | 2025-01-07 |
| KR20230146646A (ko) | 2023-10-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6258584B2 (ja) | 非水系電解液及び非水系二次電池 | |
| JP6879811B2 (ja) | リチウム塩錯化合物、電池用添加剤、電池用非水電解液、及びリチウム二次電池 | |
| CN112602224A (zh) | 非水系二次电池 | |
| JP7587681B2 (ja) | 含フッ素環状スルホニルイミド塩、及びその製法、非水系電解液、非水系二次電池 | |
| JP5977573B2 (ja) | 非水系二次電池 | |
| JP7016460B1 (ja) | 非水系電解液、セルパック、及びセルパックの製造方法 | |
| JP6087075B2 (ja) | 複合酸化物及びその製造方法、並びに非水系二次電池 | |
| JP7587020B2 (ja) | 非水系電解液及び非水系二次電池 | |
| WO2020121850A1 (ja) | 電池用非水電解液及びリチウム二次電池 | |
| JP7689636B2 (ja) | 非水系電解液及び非水系二次電池 | |
| JP6231775B2 (ja) | 正極活物質及びその製造方法、正極、並びに非水系二次電池 | |
| JP7233323B2 (ja) | 非水系電解液、及び非水系二次電池 | |
| JP6564336B2 (ja) | 非水系電解液及び非水系二次電池 | |
| JP7628451B2 (ja) | 非水系電解液及び非水系二次電池 | |
| JP2017045722A (ja) | 電池用非水電解液及びリチウム二次電池 | |
| JP7098276B2 (ja) | 電池用非水電解液及びリチウム二次電池 | |
| JP7339921B2 (ja) | 非水系電解液及び非水系二次電池 | |
| CN113646931B (zh) | 电池用非水电解液及锂二次电池 | |
| CN111066191B (zh) | 非水电解液用添加剂、非水电解液及蓄电装置 | |
| JP6879799B2 (ja) | 電池用非水電解液及びリチウム二次電池 | |
| JP7160461B2 (ja) | リチウム二次電池の製造方法 | |
| JP7514113B2 (ja) | 非水系二次電池 | |
| JP2024060500A (ja) | 非水系電解液及び非水系二次電池 | |
| CN117121258A (zh) | 含氟环状磺酰亚胺盐和其制备方法、非水系电解液、非水系二次电池 | |
| JP7825439B2 (ja) | 非水系電解液及び非水系二次電池 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230607 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240806 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20241007 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20241029 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20241108 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7587681 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |