JP7601462B2 - Method for preparing levoterbutaline using chiral auxiliary - Google Patents
Method for preparing levoterbutaline using chiral auxiliary Download PDFInfo
- Publication number
- JP7601462B2 JP7601462B2 JP2023558989A JP2023558989A JP7601462B2 JP 7601462 B2 JP7601462 B2 JP 7601462B2 JP 2023558989 A JP2023558989 A JP 2023558989A JP 2023558989 A JP2023558989 A JP 2023558989A JP 7601462 B2 JP7601462 B2 JP 7601462B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- terbutaline
- chiral auxiliary
- preparing
- hours
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/64—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms
- C07C217/66—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain
- C07C217/70—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain linked by carbon chains having two carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C313/00—Sulfinic acids; Sulfenic acids; Halides, esters or anhydrides thereof; Amides of sulfinic or sulfenic acids, i.e. compounds having singly-bound oxygen atoms of sulfinic or sulfenic groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C313/02—Sulfinic acids; Derivatives thereof
- C07C313/06—Sulfinamides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/09—Geometrical isomers
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Chemical Kinetics & Catalysis (AREA)
Description
本発明は医薬技術分野に属し、具体的にはL-テルブタリンのキラル調製方法に関する。 The present invention belongs to the pharmaceutical technology field, and specifically relates to a method for the chiral preparation of L-terbutaline.
テルブタリンは速効性で効果の短い副腎受容体アゴニストであり、β2受容体を選択的に活性化することができ、気管支の平滑筋を弛緩させ、内因性けいれん物質の放出を抑制して内因性伝達物質および粘膜繊毛を抑制することで活発化によって引き起こされる水腫を解消する(非特許文献1:Kyeong H.K., Hyun J.K., Seon-Pyo H., Sang D.S. , Arch. Pharm. Res. 2000, 23:441-445)。臨床的には主に気管支喘息 、喘息型気管支炎、肺気腫および慢性閉塞性肺部疾患時の気管支痙攣の治療などに用いられる。テルブタリンは節位第二級アルコールのキラル中心を有するが、現在臨床で使用されているのはすべてラセミ体である。研究によると、テルブタリンの左旋体は薬効を発揮する有効成分(非特許文献2:J. Med. Chem., 1972, 15, 1182-1183)であり、右旋体は無効であるだけでなく、さらに毒性の副作用を有するため、L-テルブタリンの調製方法を研究し、このような左旋体を再び上市することは、重要な臨床的応用の価値を有し、例えばCN110156614ではレボ(-)テルブタリンと市販のラセミ体を比較すると、抗喘息の薬効が二倍向上し、喘息関連疾患に新たな好ましい治療方法を提供することが開示されている。 Terbutaline is a fast-acting, short-acting adrenal receptor agonist that can selectively activate β2 receptors, relax bronchial smooth muscles, inhibit the release of endogenous spasmodic substances, and suppress endogenous transmitters and mucociliary activation, thereby relieving edema caused by activation (Non-Patent Document 1: Kyeon H.K., Hyun J.K., Seon-Pyo H., Sang D.S., Arch. Pharm. Res. 2000, 23:441-445). Clinically, it is mainly used to treat bronchial spasms in bronchial asthma, asthmatic bronchitis, emphysema, and chronic obstructive pulmonary disease. Terbutaline has a chiral center of a secondary alcohol, but all of the compounds currently used in clinical practice are racemic. According to research, the levo form of terbutaline is the active ingredient that exerts its medicinal effects (Non-Patent Document 2: J. Med. Chem., 1972, 15, 1182-1183), and the dextro form is not only ineffective but also has toxic side effects. Therefore, researching methods for preparing L-terbutaline and re-marketing this levo form has important clinical value. For example, CN110156614 discloses that when comparing levo(-)terbutaline with the commercially available racemate, the anti-asthma efficacy is doubled, providing a new and preferable treatment method for asthma-related diseases.
開示されたL-テルブタリンの調製方法は主に以下のようなものがある:1、分解法。酒石酸または酒石酸誘導体を使用してラセミ化したテルブタリンを分解するのは最も簡単で直観的なL-テルブタリンを調製する方法である(特許文献1-2:CN1273966A, CN201810147612. 1)。しかしながらこのような方法は往々にして需要複数回結晶化しなければ満足する鏡像体純度が得られず、プロセス全体の操作が複雑で、収率が低く、かつ少なくとも半分の生成物が廃棄され、現在提唱されている環境にやさしい化学という原則に合致しない。 The disclosed methods for preparing L-terbutaline are mainly as follows: 1. Decomposition method. Using tartaric acid or tartaric acid derivatives to decompose racemized terbutaline is the simplest and most intuitive method for preparing L-terbutaline (Patent Documents 1-2: CN1273966A, CN201810147612.1). However, such methods often require multiple crystallizations to obtain satisfactory enantiomeric purity, the overall process is complicated, the yield is low, and at least half of the product is discarded, which does not comply with the currently advocated principle of environmentally friendly chemistry.
2.酵素法。酵素還元方法はプロキラルケトンによってキラル第二級アルコールを調製する一般的な方法であるが、最適な酵素触媒を選別する必要があり、且つしばしば還元効率が低いなどの欠陥が存在する(非特許文献3:Journal of Molecular Catalysis B: Enzymatic, 84, (2012), 83 - 88)。 2. Enzymatic method. The enzymatic reduction method is a common method for preparing chiral secondary alcohols from prochiral ketones, but it requires the selection of an optimal enzyme catalyst and often has drawbacks such as low reduction efficiency (Non-Patent Document 3: Journal of Molecular Catalysis B: Enzymatic, 84, (2012), 83-88).
3、ボランをキラル触媒(Corey-Bakshi-Shibata触媒または類似物)の作用でプロキラルケトンを還元することには、ボランの毒性および後処理が困難であるという欠陥が存在し、工業化された生産を実現することが困難である。 3. The reduction of prochiral ketones using borane in the presence of a chiral catalyst (Corey-Bakshi-Shibata catalyst or similar) has drawbacks, such as the toxicity of borane and the difficulty of post-treatment, making it difficult to realize industrialized production.
4、キラルルテニウム触媒の転移水素化法によってクロロアセトフェノンの非対象還元を実現し、合成L-テルブタリンの重要な中間体(非特許文献4:Chem. Pharm. Bull. 65, (2017), 389-395)を得る。このような方法は高価な貴金属を使用し、且つ生成物のeeは91%しかなく、要件を満たすことができない。 4. The asymmetric reduction of chloroacetophenone is realized by the chiral ruthenium catalyzed transfer hydrogenation method to obtain a key intermediate for the synthesis of L-terbutaline (Non-Patent Document 4: Chem. Pharm. Bull. 65, (2017), 389-395). This method uses expensive precious metals, and the ee of the product is only 91%, which cannot meet the requirements.
従来技術には様々な欠陥が存在するため、新たに操作が簡単で、高効率で、低コストな方法でL-テルブタリンを調製する方法を研究開発することは重要な意義を有する。 Because conventional technology has various deficiencies, it is of great significance to research and develop a new method for preparing L-terbutaline that is simple to operate, highly efficient, and low-cost.
本発明の目的はL-テルブタリンを調製する方法を提供することである;本方法は簡単で信頼性が高く、調製されたコストが安価で、キラル生成物のeeは99.9%に達する。 The object of the present invention is to provide a method for preparing L-terbutaline; the method is simple and reliable, has low preparation cost, and the ee of the chiral product reaches 99.9%.
本発明の技術的解決手段は以下のとおりである:不斉補助剤を使用してL-テルブタリンを調製する方法であって、パラジウム触媒と塩酸の存在において、化合物7をアルコール溶剤で水素化分解し、L-テルブタリンを得る;ここで、化合物7の化学構造式は以下のとおりである。 The technical solution of the present invention is as follows: A method for preparing L-terbutaline using an asymmetric auxiliary, which comprises hydrogenolyzing compound 7 with an alcohol solvent in the presence of a palladium catalyst and hydrochloric acid to obtain L-terbutaline; where the chemical structure of compound 7 is as follows:


前記技術的解決手段において、パラジウム触媒はパラジウム炭素触媒である;アルコール溶剤は低分子アルコールであり、好ましくはメタノールである。具体的には、化合物7をメタノールに溶解させ、パラジウム炭素触媒を加え、塩酸を滴下し、常圧水素で1~3時間水素化分解し、濾過してパラジウム炭素を除去し、濾液を減圧蒸留し、残った個体を結晶化し、生成物の塩酸L-テルブタリン8を得る。
In the above technical solution, the palladium catalyst is a palladium carbon catalyst; the alcohol solvent is a low molecular weight alcohol, preferably methanol. Specifically, compound 7 is dissolved in methanol, palladium carbon catalyst is added, hydrochloric acid is added dropwise, hydrogenolysis is carried out with hydrogen at normal pressure for 1 to 3 hours, palladium carbon is removed by filtration, the filtrate is distilled under reduced pressure, and the remaining solid is crystallized to obtain the product L-
本発明では、S-(-)-tert-ブチルスルフィンアミドを原料とし、順次2-ブロモ-2-メチルプロパン、3,5-ジベンジルオキシブロモアセトフェノンと反応させ、化合物5を得る;化合物5は第四級アンモニウム塩触媒下で、還元反応させて化合物6を得る;化合物6はtert-ブチルスルフィニルを除去して保護し、中間体7を得る。具体的なステップおよび化合物の構造式は以下のとおりである:(1)tert-ブチルスルフィンアミド置換アセトフェノン中間体(化合物5)を調製する。tert-ブチルスルフィンアミド1
を有機溶剤に溶解させ、アルカリを加え、その後2-ブロモ-2-メチルプロパンを滴下し、0~60℃で2~5時間反応させ、その後3,5-ジベンジルオキシブロモアセトフェノン4を加え、引き続き30~80℃で2~8時間反応させる;反応終了後、濾過して固体物質を除去し、その後減圧蒸留して溶剤を除去し、化合物5を得て、直接次の反応に使用する;前記有機溶剤はアセトニトリル、THF、DMF、アセトン、トルエンまたは酢酸エチルであり、好ましくはアセトニトリル、THFおよびDMFであり、より好ましくはアセトニトリルである;前記アルカリは炭酸カリウム、炭酸ナトリウム、水酸化ナトリウム、水酸化カリウム、リン酸カリウム、フッ化カリウム、水素化ナトリウムまたはカリウムtert-ブトキシドであり、好ましくは炭酸カリウムおよび炭酸ナトリウムであり、より好ましくは炭酸カリウムである;反応は以下に示すとおりである。
In the present invention, S-(-)-tert-butylsulfinamide is used as the raw material, which is reacted with 2-bromo-2-methylpropane and 3,5-dibenzyloxybromoacetophenone in sequence to obtain
is dissolved in an organic solvent, an alkali is added, then 2-bromo-2-methylpropane is dropped, and the mixture is reacted at 0-60°C for 2-5 hours, and then 3,5-

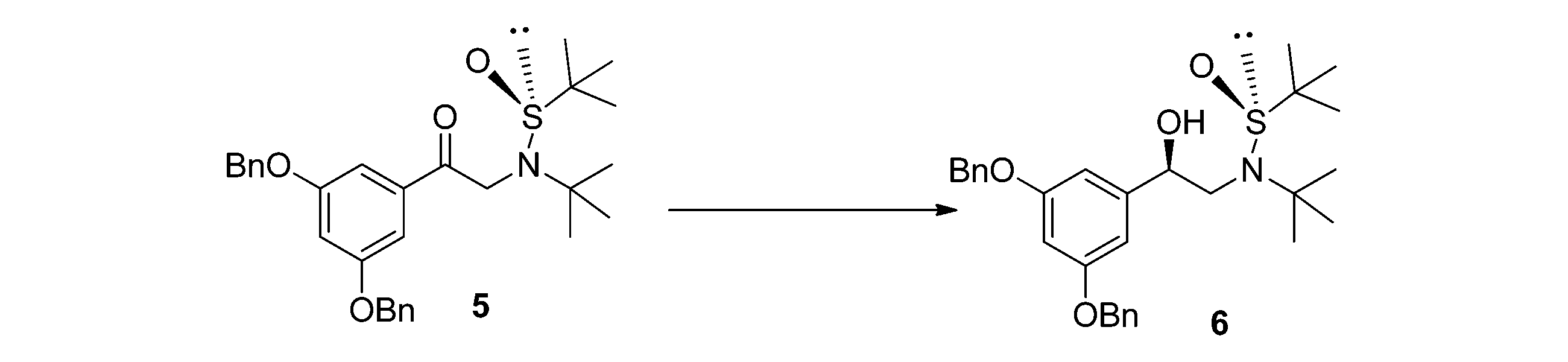
(3)tert-ブチルスルフィニル保護を取り除いて中間体(化合物7)を得る。化合物6をメタノールに溶解させ、濃塩酸を加え、0~60℃で2~5時間反応させ、減圧蒸留して溶剤を除去し、残留物に炭酸水素ナトリウム飽和溶液を加え、有機溶剤で抽出し、乾燥させ、減圧蒸留し、化合物7を得て、直接次の反応に使用する。反応は以下に示すとおりである。
(3) The tert-butylsulfinyl protection is removed to obtain an intermediate (compound 7).

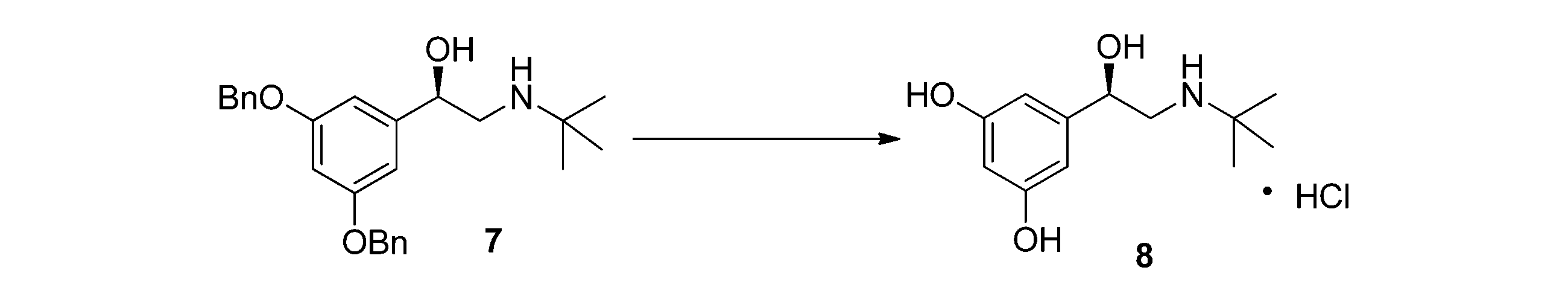
(4)ベンジル保護を除去して目標生成物のL-テルブタリンを得る。化合物7をメタノールに溶解させ、パラジウム炭素触媒を加え、塩酸を滴下し、常圧水素で1~3時間水素化分解し、濾過してパラジウム炭素を除去し、濾液を減圧蒸留し、残った個体を結晶化し、R-塩酸テルブタリン8を得る。反応は以下に示すとおりである。
(4) The benzyl protection is removed to obtain the target product L-terbutaline. Compound 7 is dissolved in methanol, a palladium carbon catalyst is added, hydrochloric acid is added dropwise, and hydrogenolysis is carried out at normal pressure hydrogen for 1 to 3 hours. The palladium carbon is removed by filtration, and the filtrate is distilled under reduced pressure. The remaining solid is crystallized to obtain R-
前記技術的解決手段を運用するため、本発明は従来技術に比べて以下の利点を有する:本発明は初めて商品化された安価なキラル源のtert-ブチルスルフィンアミドを補欠分子団として使用することでケトンの非対象還元を制御し、少量の非エナンチオマーを容易に結晶化によって除去し、生成物のeeは99.9%に達する;プロセス全体の操作が非常に簡単で、使用する試薬がいずれも安価で入手しやすく、毒がなく、工業生産に非常に適する。 To implement the above technical solution, the present invention has the following advantages over the prior art: the present invention uses the first commercialized and inexpensive chiral source tert-butylsulfinamide as the prosthetic group to control the asymmetric reduction of ketones, and the small amount of non-enantiomers can be easily removed by crystallization, with the ee of the product reaching 99.9%; the operation of the entire process is very simple, and all the reagents used are inexpensive, readily available, non-toxic, and very suitable for industrial production.
本発明の具体的な調製方法と試験方法は一般的な方法であり、例えば一般的な液体クロマトグラム(HPLC+キラルカラム)によって純度とee値を試験する。以下、実施例を組み合わせて本発明をより詳細に説明する。 The specific preparation and testing methods of the present invention are general methods, for example, the purity and ee value are tested by general liquid chromatography (HPLC + chiral column). The present invention will be described in more detail below in combination with examples.
本発明の調製方法は以下のように示される。 The preparation method of the present invention is as follows:
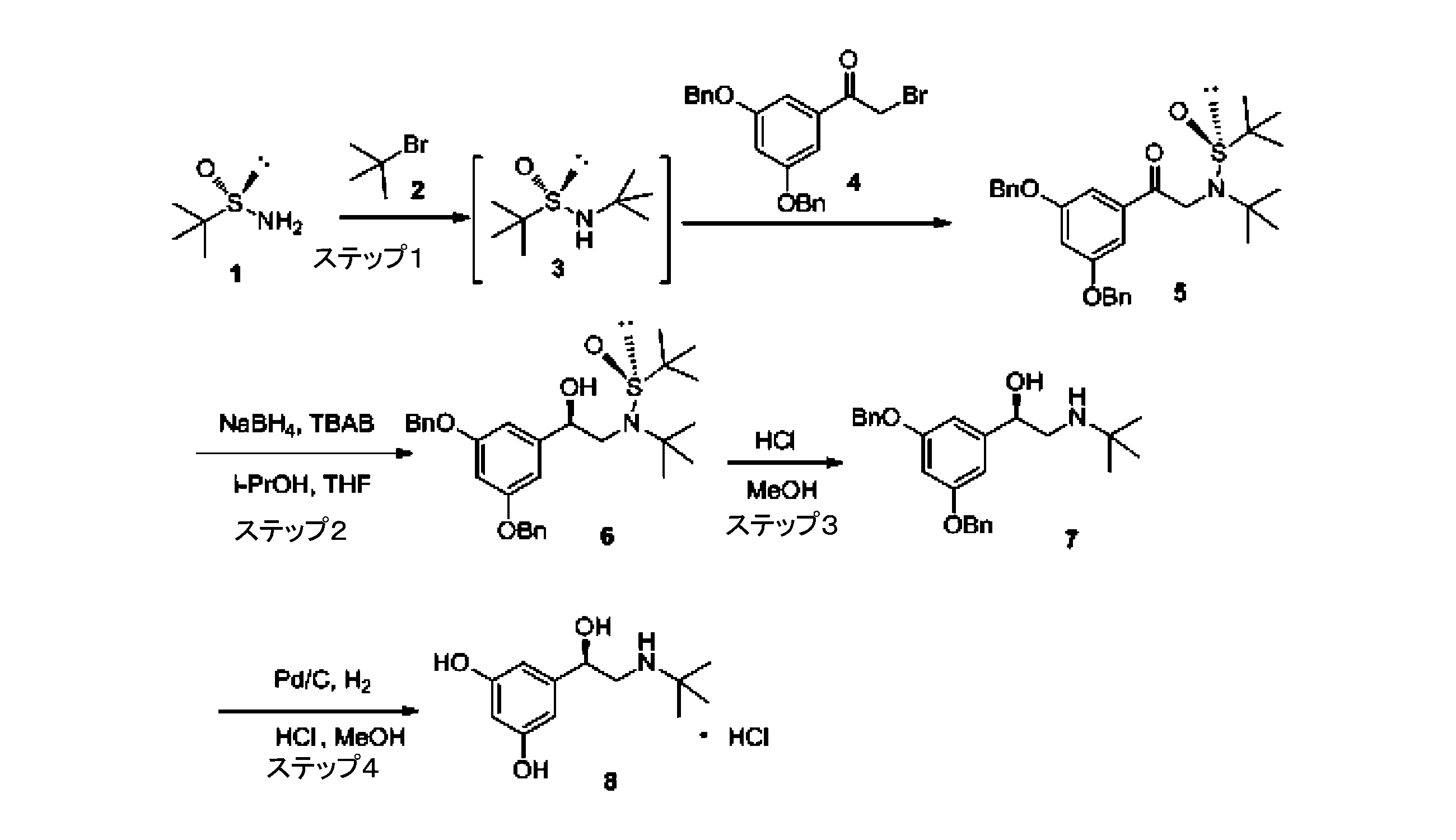
実施例1。 Example 1.
S-(-)-tert-ブチルスルフィンアミド1 (14.8g、110mmol)をアセトニトリル(300mL)に溶解させ、炭酸カリウム(18.0g、130mmol)を加え、その後2-ブロモ-2-メチルプロパン(17.8g、130mmol)を滴下し、さらに50℃に加熱して4時間反応させ、その後3,5-ジベンジルオキシブロモアセトフェノン4 (41.1g、100mmol)を加え、その後60℃で6時間させる。反応終了後、濾過して固体物質を除去し、その後減圧蒸留して溶剤を除去し、化合物5を得て、直接次の反応に使用する。
S-(-)-tert-butylsulfinamide 1 (14.8 g, 110 mmol) was dissolved in acetonitrile (300 mL), potassium carbonate (18.0 g, 130 mmol) was added, then 2-bromo-2-methylpropane (17.8 g, 130 mmol) was added dropwise, and the mixture was heated to 50°C for 4 hours, after which 3,5-dibenzyloxybromoacetophenone 4 (41.1 g, 100 mmol) was added, and the mixture was heated to 60°C for 6 hours. After the reaction was completed, the solid material was removed by filtration, and then the solvent was removed by vacuum distillation to obtain
前記で得られた化合物5をTHF (200mL)およびイソプロパノール(200mL)の混合溶剤に溶解させ、テトラブチルアンモニウムブロミド(3.22g、10mmol)を加え、反応系を0℃に冷却し、その後1.5時間以内に三回に分けて水素化ホウ素ナトリウム(3.02g、80mmol)を均一に加え、転化が完了したら引き続き0℃で1時間撹拌し、塩化アンモニウム水溶液(50mL)を加えてクエンチし、酢酸エチルを用いて抽出し、抽出液を会わせ、硫酸ナトリウムを乾燥させ、減圧蒸留し、残りの固体をエタノール(90mL)で結晶化させ、化合物6の白色固体37.1gを得て、2段階の収率は71%である。
The
化合物6 (26.1g、50mmol)をメタノール(150mL)に溶解させ、濃塩酸(12.5mL, 37.5wt%) を加え、50℃で反応3時間させ、反応終了後、減圧蒸留して溶剤を除去し、残留物に炭酸水素ナトリウム飽和溶液(80mL)を加え、酢酸エチルで三回抽出し、有機層を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留して溶剤を除去し、化合物7を得て、直接次の反応に使用する。 Compound 6 (26.1 g, 50 mmol) was dissolved in methanol (150 mL), concentrated hydrochloric acid (12.5 mL, 37.5 wt%) was added, and the mixture was allowed to react at 50°C for 3 hours. After the reaction was completed, the solvent was removed by distillation under reduced pressure, and the residue was added with saturated sodium bicarbonate solution (80 mL), extracted three times with ethyl acetate, the organic layers were combined, the sodium sulfate was dried, and the solvent was removed by distillation under reduced pressure to obtain compound 7, which was used directly in the next reaction.
上記で得られた化合物7をメタノール(200mL)に溶解させ、10wt%のパラジウム炭素触媒(2.0g)を加え、2 mol/Lの塩化水素メタノール溶液(25mL)を滴下し、その後常圧水素で2時間水素化分解し、その後濾過してパラジウム炭素を除去し、濾液に一般的な減圧蒸留を行って溶剤を除去し、残りの固体をテトラヒドロフラン(100mL)で結晶化させ、R-テルブタリンの塩酸塩8を計11.7g得て、二段階の収率は85%であり、純度は99.7%であり、 eeは99.9%であり、旋光度[a ] D20 = -39.2 (c =1.0 in MeOH)である。
The compound 7 obtained above was dissolved in methanol (200 mL), 10 wt% palladium carbon catalyst (2.0 g) was added, 2 mol/L hydrogen chloride methanol solution (25 mL) was added dropwise, and then hydrogenolysis was performed under normal pressure hydrogen for 2 hours, followed by filtration to remove palladium carbon, and the filtrate was subjected to general reduced pressure distillation to remove the solvent, and the remaining solid was crystallized from tetrahydrofuran (100 mL), obtaining a total of 11.7 g of R-
実施例2。 Example 2.
実施例1に基づき、炭酸カリウム(18.0g、130mmol)を炭酸ナトリウム(130mmol)に変更し、残りは変更せず、R-テルブタリンの塩酸塩8を得て、純度は99.6%、 eeは99.7%である。
Based on Example 1, potassium carbonate (18.0 g, 130 mmol) was replaced with sodium carbonate (130 mmol), and the rest was left unchanged to obtain R-
実施例1に基づき、テトラブチルアンモニウムブロミド(3.22g、10mmol)をテトラブチル塩化アンモニウム(10mmol)に変更し、残りは変更せず、R-テルブタリンの塩酸塩8を得て、純度は99.5%、eeは99.1%である。
Based on Example 1, tetrabutylammonium bromide (3.22 g, 10 mmol) was replaced with tetrabutylammonium chloride (10 mmol), and the rest was left unchanged to obtain R-
実施例1に基づき、イソプロパノール(200mL)をエタノール(200mL)に変更し、残りは変更せず、R-テルブタリンの塩酸塩8を得て、純度は99.1%、eeは99.2%である。
Based on Example 1, but changing isopropanol (200 mL) to ethanol (200 mL) and leaving the rest unchanged, R-
比較例1。 Comparative Example 1.
実施例1に基づき、テトラブチルアンモニウムブロミドを省き、残りは変更せず、化合物6を得る二段階の収率は55%であり、さらにR-テルブタリンの塩酸塩8を得て、 eeは95.2%である。
Based on Example 1, omitting tetrabutylammonium bromide and leaving the rest unchanged, the two-step yield of
実施例1に基づき、THF (200mL)およびイソプロパノール(200mL)の混合溶剤をイソプロパノール(400mL)に変更し、残りは変更せず、化合物6を得る二段階の収率は60%であり、さらにR-テルブタリンの塩酸塩8を調製し、eeは98.8%である。
Based on Example 1, the mixed solvent of THF (200 mL) and isopropanol (200 mL) was changed to isopropanol (400 mL), and the rest was left unchanged. The two-step yield of
実施例1に基づき、水素化ホウ素ナトリウム(3.02g、80mmol)を水素化ホウ素カリウム(80mmol)に変更し、残りは変更せず、化合物6を得る二段階の収率は59%であり、さらにR-テルブタリンの塩酸塩8を調製し、eeは98.3%である。
Based on Example 1, sodium borohydride (3.02 g, 80 mmol) was changed to potassium borohydride (80 mmol) and the rest was left unchanged, the two-step yield of
実施例3。 Example 3.
S-(-)-tert-ブチルスルフィンアミド1 (14.8g、110mmol)をアセトニトリル(300mL)に溶解させ、炭酸カリウム(18.0g、130mmol)を加え、その後2-ブロモ-2-メチルプロパン(17.8g、130mmol)を滴下し、さらに40℃に加熱して3時間反応させ、その後3,5-ジベンジルオキシブロモアセトフェノン4 (41.1g、100mmol)を加え、その後60℃で6時間反応させる。反応終了後、濾過して固体物質を除去し、その後減圧蒸留して溶剤を除去し、化合物5を得て、直接次の反応に使用する。
S-(-)-tert-butylsulfinamide 1 (14.8 g, 110 mmol) is dissolved in acetonitrile (300 mL), potassium carbonate (18.0 g, 130 mmol) is added, then 2-bromo-2-methylpropane (17.8 g, 130 mmol) is added dropwise, and the mixture is heated to 40°C and reacted for 3 hours, after which 3,5-dibenzyloxybromoacetophenone 4 (41.1 g, 100 mmol) is added, and the mixture is reacted at 60°C for 6 hours. After the reaction is complete, the solid material is removed by filtration, and then the solvent is removed by vacuum distillation to obtain
前記で得られた化合物5をTHF (200mL)およびイソプロパノール(200mL)の混合溶剤に溶解させ、テトラブチルアンモニウムブロミド(3.22g、10mmol)を加え、反応系を0℃に冷却し、その後1.5時間以内に三回に分けて水素化ホウ素ナトリウム(3.02g、80mmol)を均一に加え、添加が完了したら引き続き0℃で1時間撹拌し、塩化アンモニウム水溶液(50mL)を加えてクエンチし、酢酸エチルで抽出し、抽出液を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留し、残りの固体をエタノール(90mL)で結晶化させ、白色の固体化合物6を得る。
The
化合物6 (26.1g、50mmol)をメタノール(150mL)に溶解させ、濃塩酸(12.5mL, 37.5wt%)を加え、50℃で3時間反応させ、反応終了後、減圧蒸留して溶剤を除去し、残留物に炭酸水素ナトリウム飽和溶液(80mL)を加え、酢酸エチルで三回抽出し、有機層を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留して溶剤を除去し、化合物7を得て、直接次の反応に使用する。 Compound 6 (26.1 g, 50 mmol) was dissolved in methanol (150 mL), concentrated hydrochloric acid (12.5 mL, 37.5 wt%) was added, and the mixture was reacted at 50°C for 3 hours. After the reaction was completed, the solvent was removed by distillation under reduced pressure, and the residue was added with saturated sodium bicarbonate solution (80 mL), extracted three times with ethyl acetate, the organic layers were combined, the sodium sulfate was dried, and the solvent was removed by distillation under reduced pressure to obtain compound 7, which was directly used in the next reaction.
上記で得られた化合物7をメタノール(200mL)に溶解させ、10wt%のパラジウム炭素触媒(2.0g)を加え、2 mol/Lの塩化水素メタノール溶液(25mL)を滴下し、その後常圧水素で2時間水素化分解し、その後濾過してパラジウム炭素を除去し、濾液に一般的な減圧蒸留を行って溶剤を除去し、残りの固体をテトラヒドロフラン(100mL)で結晶化させ、R-テルブタリンの塩酸塩8を得る。
The compound 7 obtained above is dissolved in methanol (200 mL), 10 wt% palladium carbon catalyst (2.0 g) is added, 2 mol/L hydrogen chloride methanol solution (25 mL) is added dropwise, and then hydrogenolysis is performed under normal pressure hydrogen for 2 hours, followed by filtration to remove the palladium carbon, and the filtrate is subjected to general reduced pressure distillation to remove the solvent, and the remaining solid is crystallized from tetrahydrofuran (100 mL) to obtain R-
実施例4。 Example 4.
S-(-)-tert-ブチルスルフィンアミド1 (14.8g、110mmol)をアセトニトリル(300mL)に溶解させ、炭酸カリウム(18.0g、130mmol)を加え、その後2-ブロモ-2-メチルプロパン(17.8g、130mmol)を滴下し、さらに50℃に加熱して4時間反応させ、その後3,5-ジベンジルオキシブロモアセトフェノン4 (41.1g、100mmol)を加え、その後60℃で6時間させる。反応終了後、濾過して固体物質を除去し、その後減圧蒸留して溶剤を除去し、化合物5を得て、直接次の反応に使用する。
S-(-)-tert-butylsulfinamide 1 (14.8 g, 110 mmol) was dissolved in acetonitrile (300 mL), potassium carbonate (18.0 g, 130 mmol) was added, then 2-bromo-2-methylpropane (17.8 g, 130 mmol) was added dropwise, and the mixture was heated to 50°C for 4 hours, after which 3,5-dibenzyloxybromoacetophenone 4 (41.1 g, 100 mmol) was added, and the mixture was heated to 60°C for 6 hours. After the reaction was completed, the solid material was removed by filtration, and then the solvent was removed by vacuum distillation to obtain
前記得られた化合物5をTHF (200mL)およびイソプロパノール(200mL)の混合溶剤に溶解させ、テトラブチルアンモニウムブロミド(3.22g、10mmol)を加え、反応系を5℃に冷却し、その後1時間内に二回に分けて水素化ホウ素ナトリウム(3.02g、80mmol)を均一に加え、添加が完了したら0℃で1時間撹拌し、塩化アンモニウム水溶液(50mL)を加えてクエンチし、酢酸エチルで抽出し、抽出液を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留し、残りの固体をエタノール(90mL)で結晶化させ、白色の固体化合物6を得る。
The obtained
化合物6 (26.1g、50mmol)をメタノール(150mL)に溶解させ、濃塩酸(12.5mL,37.5wt%)を加え、50℃で3時間反応させ、反応終了後、減圧蒸留して溶剤を除去し、残留物に炭酸水素ナトリウム飽和溶液(80mL)を加え、酢酸エチルで三回抽出し、有機層を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留して溶剤を除去し、化合物7を得て、直接次の反応に使用する。 Compound 6 (26.1 g, 50 mmol) is dissolved in methanol (150 mL), concentrated hydrochloric acid (12.5 mL, 37.5 wt%) is added, and the mixture is reacted at 50°C for 3 hours. After the reaction is completed, the solvent is removed by distillation under reduced pressure, a saturated solution of sodium bicarbonate (80 mL) is added to the residue, and the mixture is extracted three times with ethyl acetate. The organic layers are combined, the sodium sulfate is dried, and the solvent is removed by distillation under reduced pressure to obtain compound 7, which is used directly in the next reaction.
上記で得られた化合物7をメタノール(200mL)に溶解させ、10wt%のパラジウム炭素触媒(2.0 g)を加え、2 mol/Lの塩化水素メタノール溶液(25mL)滴下し、その後常圧水素で水素化分解2.5時間,その後濾過してパラジウム炭素を除去し、濾液に一般的な減圧蒸留を行って溶剤を除去し、残りの固体をテトラヒドロフラン(100mL)結晶化させ、R-テルブタリンの塩酸塩8を得る。
The compound 7 obtained above is dissolved in methanol (200 mL), 10 wt% palladium carbon catalyst (2.0 g) is added, 2 mol/L hydrogen chloride methanol solution (25 mL) is added dropwise, and then hydrogenolysis is performed with hydrogen at normal pressure for 2.5 hours, followed by filtration to remove the palladium carbon, and the filtrate is subjected to general reduced pressure distillation to remove the solvent. The remaining solid is crystallized in tetrahydrofuran (100 mL) to obtain R-
実施例5。 Example 5.
S-(-)-tert-ブチルスルフィンアミド1 (14.8g、110mmol)をアセトニトリル(300mL)に溶解させ、炭酸カリウム(18.0g、130mmol)を加え、その後2-ブロモ-2-メチルプロパン(17.8g、130mmol)を滴下し、さらに50℃に加熱して4時間反応させ、その後3,5-ジベンジルオキシブロモアセトフェノン4 (41.1g、100mmol)を加え、その後60℃で6時間させる。反応終了後、濾過して固体物質を除去し、その後減圧蒸留して溶剤を除去し、化合物5を得て、直接次の反応に使用する。
S-(-)-tert-butylsulfinamide 1 (14.8 g, 110 mmol) was dissolved in acetonitrile (300 mL), potassium carbonate (18.0 g, 130 mmol) was added, then 2-bromo-2-methylpropane (17.8 g, 130 mmol) was added dropwise, and the mixture was heated to 50°C for 4 hours, after which 3,5-dibenzyloxybromoacetophenone 4 (41.1 g, 100 mmol) was added, and the mixture was heated to 60°C for 6 hours. After the reaction was completed, the solid material was removed by filtration, and then the solvent was removed by vacuum distillation to obtain
前記得られた化合物5をTHF (200mL)およびイソプロパノール(200mL)の混合溶剤に溶解させ、テトラブチルアンモニウムブロミド(3.22g、10mmol)を加え、反応系を0℃に冷却し、その後1.5時間以内に三回に分けて水素化ホウ素ナトリウム(3.02g、80mmol)を均一に加え、転化が完了したら引き続き0℃で1時間撹拌し、塩化アンモニウム水溶液(50mL)を加えてクエンチし、酢酸エチルで抽出し、抽出液を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留し、残りの固体をエタノール(90mL)で結晶化させ、白色の固体化合物6を得る。
The obtained
化合物6を(26.1g、50mmol)メタノール(150mL)に溶解させ、濃塩酸(12.5mL, 37. 5wt%)を加え、 50℃で4時間反応させ、反応終了後、減圧蒸留して溶剤を除去し、残留物に炭酸水素ナトリウム飽和溶液(80mL)を加え、酢酸エチルで三回抽出し、有機層を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留して溶剤を除去し、化合物7を得て、直接次の反応に使用する。 Compound 6 (26.1 g, 50 mmol) was dissolved in methanol (150 mL), concentrated hydrochloric acid (12.5 mL, 37.5 wt%) was added, and the mixture was reacted at 50°C for 4 hours. After the reaction was completed, the solvent was removed by distillation under reduced pressure, and the residue was added with saturated sodium bicarbonate solution (80 mL), extracted three times with ethyl acetate, the organic layers were combined, dried over sodium sulfate, and the solvent was removed by distillation under reduced pressure to obtain compound 7, which was directly used in the next reaction.
上記で得られた化合物7をメタノール(200mL)に溶解させ、10wt%のパラジウム炭素触媒(2.0 g)を加え、2 mol/Lの塩化水素メタノール溶液(25mL),その後常圧水素で2時間水素化分解し、その後濾過してパラジウム炭素を除去し、濾液に一般的な減圧蒸留を行って溶剤を除去し、残りの固体をテトラヒドロフラン(100mL)結晶化させ、R-テルブタリンの塩酸塩8を得る。
The compound 7 obtained above is dissolved in methanol (200 mL), 10 wt% palladium carbon catalyst (2.0 g) is added, and the mixture is hydrogenolyzed with 2 mol/L hydrogen chloride methanol solution (25 mL) and then at atmospheric pressure for 2 hours. The mixture is then filtered to remove the palladium carbon, and the filtrate is subjected to a general vacuum distillation to remove the solvent. The remaining solid is crystallized in tetrahydrofuran (100 mL) to obtain R-
実施例6。 Example 6.
S-(-)-tert-ブチルスルフィンアミド1 (14.8g、110mmol)をアセトニトリル(300mL)に溶解させ、炭酸カリウム(18.0g、130mmol)を加え、その後2-ブロモ-2-メチルプロパン(17.8g、130mmol)を滴下し、さらに50℃に加熱して4時間反応させ、その後3,5-ジベンジルオキシブロモアセトフェノン4(41.1g、100mmol)を加え、その後60℃で6時間反応させる。反応終了後、濾過して固体物質を除去し、その後減圧蒸留して溶剤を除去し、化合物5を得て、直接次の反応に使用する。
S-(-)-tert-butylsulfinamide 1 (14.8 g, 110 mmol) is dissolved in acetonitrile (300 mL), potassium carbonate (18.0 g, 130 mmol) is added, then 2-bromo-2-methylpropane (17.8 g, 130 mmol) is added dropwise, and the mixture is heated to 50°C and reacted for 4 hours, after which 3,5-dibenzyloxybromoacetophenone 4 (41.1 g, 100 mmol) is added, and the mixture is reacted at 60°C for 6 hours. After the reaction is complete, the solid material is removed by filtration, and then the solvent is removed by vacuum distillation to obtain
前記得られた化合物5をTHF (200mL)およびイソプロパノール(200mL)の混合溶剤中に溶解させ、テトラブチルアンモニウムブロミド(3.22g、10mmol)を加え、反応系を0℃に冷却し、その後1.5時間以内に三回に分けて水素化ホウ素ナトリウム(3.02g、80mmol)を均一に加え、転化が完了したら引き続き0℃で1時間撹拌し、塩化アンモニウム水溶液(50mL)を加えてクエンチし、酢酸エチルで抽出し、抽出液を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留し、残りの固体をエタノール(90mL)で結晶化させ、白色の固体化合物6を得る。
The obtained
化合物6 (26.1g、50mmol)をメタノール(150mL)に溶解させ、濃塩酸(12.5mL,37.5wt%)を加え、50℃で3時間反応させ、反応終了後、減圧蒸留して溶剤を除去し、残留物に炭酸水素ナトリウム飽和溶液(80mL)を加え、酢酸エチルで三回抽出し、有機層を合わせ、硫酸ナトリウムを乾燥させ、減圧蒸留して溶剤を除去し、化合物7を得て、直接次の反応に使用する。 Compound 6 (26.1 g, 50 mmol) is dissolved in methanol (150 mL), concentrated hydrochloric acid (12.5 mL, 37.5 wt%) is added, and the mixture is reacted at 50°C for 3 hours. After the reaction is completed, the solvent is removed by distillation under reduced pressure, a saturated solution of sodium bicarbonate (80 mL) is added to the residue, and the mixture is extracted three times with ethyl acetate. The organic layers are combined, the sodium sulfate is dried, and the solvent is removed by distillation under reduced pressure to obtain compound 7, which is used directly in the next reaction.
上記で得られた化合物7をメタノール(200mL)に溶解させ、10wt%のパラジウム炭素触媒(2.0 g)を加え、2 mol/Lの塩化水素メタノール溶液(25mL)滴下し、その後常圧水素で水素化分解2.5時間,その後濾過してパラジウム炭素を除去し、濾液に一般的な減圧蒸留を行って溶剤を除去し、残りの固体をテトラヒドロフラン(100mL)結晶化させ、R-テルブタリンの塩酸塩8を得る。
The compound 7 obtained above is dissolved in methanol (200 mL), 10 wt% palladium carbon catalyst (2.0 g) is added, 2 mol/L hydrogen chloride methanol solution (25 mL) is added dropwise, and then hydrogenolysis is performed with hydrogen at normal pressure for 2.5 hours, followed by filtration to remove the palladium carbon, and the filtrate is subjected to general reduced pressure distillation to remove the solvent. The remaining solid is crystallized in tetrahydrofuran (100 mL) to obtain R-
本発明が提供する方法は安価で入手しやすいキラルtert-ブチルスルフィンアミドを不斉補助剤として、ケトン非対象還元に必要なキラル第二級アルコールを制御し、不斉補助剤は簡単な酸性条件下で除去できる。 The method provided by the present invention uses inexpensive and readily available chiral tert-butylsulfinamide as an asymmetric auxiliary to control the chiral secondary alcohol required for asymmetric reduction of ketones, and the asymmetric auxiliary can be removed under simple acidic conditions.
Claims (10)

パラジウム触媒と塩酸の存在下で、化合物7を、アルコール溶剤で水素化分解し、
下記のL-テルブタリン:

ことを特徴とする不斉補助剤を使用してL-テルブタリンを調製する方法。 Compound 7:
Compound 7 is hydrogenolyzed in an alcohol solvent in the presence of a palladium catalyst and hydrochloric acid ;
L-Terbutaline as follows:

化合物5は第四級アンモニウム塩の存在下で水素化し、還元反応させて下記化合物6:

化合物6のtert-ブチルスルフィニル保護を取り除いて化合物7を得る
請求項1に記載の不斉補助剤を使用してL-テルブタリンを調整する方法。 Using S- (−) -tert-butylsulfinamide as a chiral auxiliary as a raw material , the compound was reacted with 2-bromo-2-methylpropane and 3,5-dibenzyloxybromoacetophenone in this order to obtain the following compound 5:
Compound 5 is hydrogenated in the presence of a quaternary ammonium salt and reduced to give compound 6:
A method for preparing L-terbutaline using the chiral auxiliary of claim 1, which comprises removing the tert -butylsulfinyl protection of compound 6 to give compound 7.
請求項2に記載の不斉補助剤を使用してL-テルブタリンを生成する方法。 The method for producing L-terbutaline using the chiral auxiliary according to claim 2, comprising dissolving tert-butylsulfinamide in an organic solvent, adding an alkali, then dropping 2-bromo-2-methylpropane, reacting at 0 to 60°C for 2 to 5 hours, then adding 3,5-dibenzyloxybromoacetophenone, and continuing to react at 30 to 80°C for 2 to 8 hours to obtain compound 5.
請求項2に記載の不斉補助剤を使用してL-テルブタリンを調製する方法。 A method for preparing L-terbutaline using the chiral auxiliary according to claim 2, comprising dissolving compound 5 in a solvent, adding a quaternary ammonium salt, and then adding sodium borohydride at 0 to 10°C to carry out a reduction reaction to obtain compound 6.
請求項2に記載の不斉補助剤を使用してL-テルブタリンを調製する方法。 A method for preparing L-terbutaline using the chiral auxiliary according to claim 2, comprising dissolving compound 6 in an alcohol solvent, adding concentrated hydrochloric acid, and reacting at 0 to 60° C. for 2 to 5 hours to obtain compound 7.
アルコール溶剤がメタノールである
請求項1に記載の不斉補助剤を使用してL-テルブタリンを調製する方法。 the palladium catalyst is an inorganic palladium catalyst;
2. The method for preparing L-terbutaline using the chiral auxiliary according to claim 1, wherein the alcohol solvent is methanol .
アルコール溶剤がメタノールである
請求項6に記載の不斉補助剤を使用してL-テルブタリンを調製する方法。 the palladium catalyst is a palladium on carbon catalyst;
The method for preparing L-terbutaline using the chiral auxiliary according to claim 6, wherein the alcohol solvent is methanol.
請求項1に記載の不斉補助剤を使用してL-テルブタリンを調製する方法。 The method for preparing L-terbutaline using the chiral auxiliary according to claim 1, comprising dissolving compound 7 in an alcohol solvent, adding a palladium catalyst, adding hydrochloric acid dropwise, and hydrogenolysis under normal pressure of hydrogen for 1 to 3 hours to obtain the product L-terbutaline.

ことを特徴とする化合物。 The structural formula:
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/CN2021/090369 WO2022226812A1 (en) | 2021-04-27 | 2021-04-27 | Method for preparing l-terbutaline by using chiral auxiliary group |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2024514774A JP2024514774A (en) | 2024-04-03 |
| JP7601462B2 true JP7601462B2 (en) | 2024-12-17 |
Family
ID=83847702
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2023558989A Active JP7601462B2 (en) | 2021-04-27 | 2021-04-27 | Method for preparing levoterbutaline using chiral auxiliary |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20240190807A1 (en) |
| EP (1) | EP4332085A4 (en) |
| JP (1) | JP7601462B2 (en) |
| WO (1) | WO2022226812A1 (en) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1273966A (en) | 1999-10-19 | 2000-11-22 | 中国科学院成都有机化学研究所 | Process for preparing adrenin beta-excitomotors by combinaion and disconnection method |
| CN105330553A (en) | 2015-12-02 | 2016-02-17 | 湖南理工学院 | Method for separating terbutaline enantiomers in extracted mode through hydrophobicity phase transferring chirality |
| CN106631831A (en) | 2015-10-29 | 2017-05-10 | 北京盈科瑞药物研究院有限公司 | A method of preparing R-terbutaline |
| CN110156614A (en) | 2018-02-13 | 2019-08-23 | 东莞市凯法生物医药有限公司 | Preparation method of levo (-) terbutaline and application of levo (-) terbutaline in resisting asthma |
| CN110950765A (en) | 2019-12-10 | 2020-04-03 | 浙江医药高等专科学校 | A kind of preparation method of terbutaline sulfate |
| CN112250586A (en) | 2020-10-21 | 2021-01-22 | 福安药业集团宁波天衡制药有限公司 | Preparation method of terbutaline sulfate and B crystal form thereof |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101391965A (en) * | 2008-11-02 | 2009-03-25 | 李勤耕 | Method for preparing terbutaline sulphate crystal B fulfilling medicinal requirements |
| CN108129367B (en) * | 2018-02-05 | 2020-10-02 | 南开大学 | A kind of construction method of chiral quaternary carbon at α position of chiral sulfinyl imide and its product and application |
| CN110734382B (en) * | 2019-10-09 | 2024-02-20 | 苏州弘森药业股份有限公司 | Method for synthesizing terbutaline |
-
2021
- 2021-04-27 WO PCT/CN2021/090369 patent/WO2022226812A1/en not_active Ceased
- 2021-04-27 EP EP21938298.3A patent/EP4332085A4/en active Pending
- 2021-04-27 JP JP2023558989A patent/JP7601462B2/en active Active
- 2021-04-27 US US18/282,913 patent/US20240190807A1/en active Pending
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1273966A (en) | 1999-10-19 | 2000-11-22 | 中国科学院成都有机化学研究所 | Process for preparing adrenin beta-excitomotors by combinaion and disconnection method |
| CN106631831A (en) | 2015-10-29 | 2017-05-10 | 北京盈科瑞药物研究院有限公司 | A method of preparing R-terbutaline |
| CN105330553A (en) | 2015-12-02 | 2016-02-17 | 湖南理工学院 | Method for separating terbutaline enantiomers in extracted mode through hydrophobicity phase transferring chirality |
| CN110156614A (en) | 2018-02-13 | 2019-08-23 | 东莞市凯法生物医药有限公司 | Preparation method of levo (-) terbutaline and application of levo (-) terbutaline in resisting asthma |
| CN110950765A (en) | 2019-12-10 | 2020-04-03 | 浙江医药高等专科学校 | A kind of preparation method of terbutaline sulfate |
| CN112250586A (en) | 2020-10-21 | 2021-01-22 | 福安药业集团宁波天衡制药有限公司 | Preparation method of terbutaline sulfate and B crystal form thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4332085A1 (en) | 2024-03-06 |
| JP2024514774A (en) | 2024-04-03 |
| US20240190807A1 (en) | 2024-06-13 |
| WO2022226812A1 (en) | 2022-11-03 |
| EP4332085A4 (en) | 2025-07-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5343925B2 (en) | Method for producing (2R) -2-propyloctanoic acid | |
| CA2790519A1 (en) | Improved resolution methods for isolating desired enantiomers of tapentadol intermediates and use thereof for the preparation of tapentadol | |
| KR100812046B1 (en) | PROCESS FOR PREPARATION OF 1-2S,3S-2-BENZHYDRYL-N-5-tert-BUTYL-2-METHOXYBENZYLQUINUCLIDIN-3-AMINE | |
| KR100704590B1 (en) | Novel Process for Making (+)-cis-sertraline | |
| CN106631831B (en) | A kind of preparation method of left-handed Terbutaline | |
| JP2009062360A6 (en) | Cinacalcet manufacturing method | |
| JP2009062360A (en) | Method for producing cinacalcet | |
| JP7601462B2 (en) | Method for preparing levoterbutaline using chiral auxiliary | |
| JP2011515453A (en) | Method for producing donepezil hydrochloride | |
| CN114805167A (en) | A kind of preparation method of briracetam | |
| CN113264839B (en) | Method for preparing levo-terbutaline by using chiral prosthetic group | |
| EP2012777A2 (en) | Process for the synthesis of (+) and (-)-1-(3,4-dichlorophenyl)-3-azabicyclo[3.1.0]hexane | |
| CN112300150B (en) | Preparation method of milpitant and intermediate thereof | |
| EP2545028A1 (en) | A novel stereospecific synthesis of (-) (2s,3s)-1-dimethylamino-3-(3-methoxyphenyl)-2-methyl pentan-3-ol | |
| WO2003051852A1 (en) | Intermediate and process for producing optically active compound from the intermediate | |
| JP3902384B2 (en) | Method for purifying optically active α-methyl-bis-3,5- (trifluoromethyl) benzylamines | |
| CN111377850B (en) | Chiral N-substituted-3,3-difluoro-4-hydroxypiperidine derivative and preparation method thereof | |
| JP3982991B2 (en) | Process for producing optically active 1- (trifluoromethylmono-substituted phenyl) ethylamine | |
| WO2008001719A1 (en) | Method for producing optically active 1-(fluoro-, trifluoromethyl- or trifluoromethoxy-substituted phenyl)alkylamine n-monoalkyl derivative | |
| WO2007026373A2 (en) | Process for preparing rivastigmine | |
| WO2020165672A1 (en) | Process for preparation of highly pure fingolimod hydrochloride | |
| JP4260941B2 (en) | Azetidine-3-ol | |
| CN110922354B (en) | Chemical resolution preparation method of 1-R-3-haloperidol-4-carboxylic acid and product thereof | |
| JP4350530B2 (en) | Process for producing optically active 3,3,3-trifluoro-2-hydroxypropionic acid derivative | |
| JP5510040B2 (en) | Optical resolution to obtain optically active (R) -1- (4-fluorophenyl) ethylamine |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20230925 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20240813 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20240815 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20241023 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20241112 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20241128 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7601462 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |