JP7635741B2 - シリコンウェーハ用の研磨スラリーの分析方法 - Google Patents
シリコンウェーハ用の研磨スラリーの分析方法 Download PDFInfo
- Publication number
- JP7635741B2 JP7635741B2 JP2022038640A JP2022038640A JP7635741B2 JP 7635741 B2 JP7635741 B2 JP 7635741B2 JP 2022038640 A JP2022038640 A JP 2022038640A JP 2022038640 A JP2022038640 A JP 2022038640A JP 7635741 B2 JP7635741 B2 JP 7635741B2
- Authority
- JP
- Japan
- Prior art keywords
- slurry
- solution
- film
- drying
- analyzing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 239000002002 slurry Substances 0.000 title claims description 154
- 235000012431 wafers Nutrition 0.000 title claims description 72
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 title claims description 49
- 229910052710 silicon Inorganic materials 0.000 title claims description 49
- 239000010703 silicon Substances 0.000 title claims description 49
- 238000005498 polishing Methods 0.000 title claims description 30
- 238000004458 analytical method Methods 0.000 title description 40
- 238000000034 method Methods 0.000 claims description 50
- 238000001035 drying Methods 0.000 claims description 48
- 239000010408 film Substances 0.000 claims description 48
- 239000005416 organic matter Substances 0.000 claims description 29
- 239000010409 thin film Substances 0.000 claims description 27
- 239000012535 impurity Substances 0.000 claims description 24
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 16
- 239000008119 colloidal silica Substances 0.000 claims description 14
- 229910052751 metal Inorganic materials 0.000 claims description 14
- 239000002184 metal Substances 0.000 claims description 14
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 claims description 12
- 238000010438 heat treatment Methods 0.000 claims description 12
- 238000004876 x-ray fluorescence Methods 0.000 claims description 7
- 230000001678 irradiating effect Effects 0.000 claims description 5
- 239000000243 solution Substances 0.000 description 36
- 101100208102 Brassica napus TRXF gene Proteins 0.000 description 30
- 238000011109 contamination Methods 0.000 description 27
- 238000000354 decomposition reaction Methods 0.000 description 12
- 238000012360 testing method Methods 0.000 description 12
- 230000000052 comparative effect Effects 0.000 description 11
- 230000000694 effects Effects 0.000 description 11
- 238000001704 evaporation Methods 0.000 description 10
- 238000007796 conventional method Methods 0.000 description 9
- 238000012864 cross contamination Methods 0.000 description 9
- 230000008020 evaporation Effects 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- 238000001228 spectrum Methods 0.000 description 9
- 238000004140 cleaning Methods 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 239000007788 liquid Substances 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 229910017604 nitric acid Inorganic materials 0.000 description 4
- 229910052684 Cerium Inorganic materials 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 239000002738 chelating agent Substances 0.000 description 3
- 229910052804 chromium Inorganic materials 0.000 description 3
- 229910052802 copper Inorganic materials 0.000 description 3
- 229910052742 iron Inorganic materials 0.000 description 3
- 229910052759 nickel Inorganic materials 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 229910052719 titanium Inorganic materials 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 150000004697 chelate complex Chemical class 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 238000000605 extraction Methods 0.000 description 2
- 229920011301 perfluoro alkoxyl alkane Polymers 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- 238000005979 thermal decomposition reaction Methods 0.000 description 2
- 229910021642 ultra pure water Inorganic materials 0.000 description 2
- 239000012498 ultrapure water Substances 0.000 description 2
- 229910003638 H2SiF6 Inorganic materials 0.000 description 1
- 229910004014 SiF4 Inorganic materials 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000004380 ashing Methods 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 238000009614 chemical analysis method Methods 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229910052681 coesite Inorganic materials 0.000 description 1
- 229910052906 cristobalite Inorganic materials 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000007654 immersion Methods 0.000 description 1
- 238000009616 inductively coupled plasma Methods 0.000 description 1
- 238000001095 inductively coupled plasma mass spectrometry Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- QSHDDOUJBYECFT-UHFFFAOYSA-N mercury Chemical compound [Hg] QSHDDOUJBYECFT-UHFFFAOYSA-N 0.000 description 1
- 229910052753 mercury Inorganic materials 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 238000010943 off-gassing Methods 0.000 description 1
- 229920013653 perfluoroalkoxyethylene Polymers 0.000 description 1
- 238000007517 polishing process Methods 0.000 description 1
- -1 polytetrafluoroethylene Polymers 0.000 description 1
- 229920001343 polytetrafluoroethylene Polymers 0.000 description 1
- 239000004810 polytetrafluoroethylene Substances 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000009738 saturating Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 238000010206 sensitivity analysis Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- ABTOQLMXBSRXSM-UHFFFAOYSA-N silicon tetrafluoride Chemical compound F[Si](F)(F)F ABTOQLMXBSRXSM-UHFFFAOYSA-N 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 229910052682 stishovite Inorganic materials 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- ZEFWRWWINDLIIV-UHFFFAOYSA-N tetrafluorosilane;dihydrofluoride Chemical compound F.F.F[Si](F)(F)F ZEFWRWWINDLIIV-UHFFFAOYSA-N 0.000 description 1
- 229910052905 tridymite Inorganic materials 0.000 description 1
Landscapes
- Analysing Materials By The Use Of Radiation (AREA)
- Mechanical Treatment Of Semiconductor (AREA)
Description
そして本発明は、TRXF分析における上記問題点に鑑みてなされたものであり、剥離することなく安定してスラリー薄膜を形成し、分析することができる方法を提供することを目的とする。
測定対象の研磨スラリーにフッ酸を添加して、前記研磨スラリー中のコロイダルシリカを溶解してスラリー溶液とする工程Aと、
該工程A後のスラリー溶液で、シリコンウェーハ上に溶液膜を形成する工程Bと、
該工程Bで形成した溶液膜にUV光を照射して溶液膜中の有機物を分解する工程Cと、
該工程Cで前記有機物を分解した溶液膜を前記シリコンウェーハ上で乾燥させてスラリー薄膜を形成する工程Dと、
該工程D後のスラリー薄膜から前記金属不純物の分析を全反射蛍光X線分析装置を用いて行う工程Eと、
を備えることを特徴とするシリコンウェーハ用の研磨スラリーの分析方法を提供する。
また、ICP-MS等での化学分析法で必要となる複雑な工程も不要とすることができ、スラリー中のコロイダルシリカや有機物の分解や除去が完全でなくとも、また、キレート錯体による抽出を行わずとも分析することが可能であり、実に簡便である。ICP-MS等を用いなくとも、高感度で分析することができる。
波長が344nm~179nmのUV光を前記溶液膜に10~15分間照射することができる。
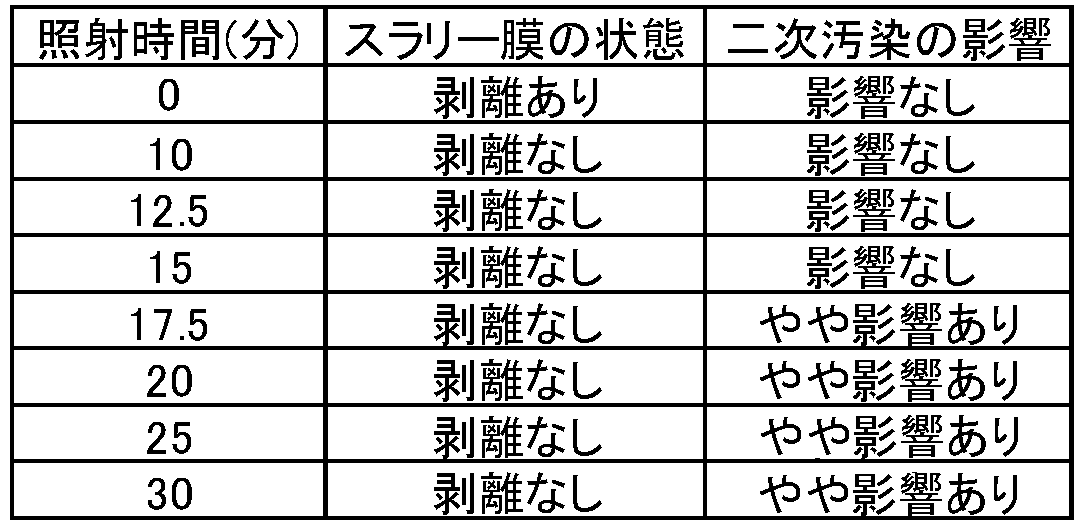
また、上記のような照射時間であれば、スラリー薄膜の剥離をより確実に防ぐことができるし、UV光照射でのクロスコンタミネーション等による二次汚染の影響をより効果的に抑えることができる。
70℃~100℃で加熱して乾燥させるか、または、150Torr(19998.3Pa)以下で減圧乾燥させることができる。
前記工程A後のスラリー溶液を、前記シリコンウェーハ上に滴下して形成することができる。
図15の方法について蒸発乾燥温度を50℃から100℃、乾燥時間を8.5~1.5分の間で振ったときのスラリー薄膜の状態を表1に示す。後述する本発明のフローのように有機物分解処理を行わず、そのまま蒸発乾燥した場合は、すべての条件において、ウェーハ表面からスラリー薄膜が剥離し、TRXF分析ができないことから、有機物分解工程が必須であることがわかった。

図1に示す本発明のスラリーの分析方法では、工程A:シリコン成分溶解工程、工程B:溶液膜形成工程(スラリー滴下工程)、工程C:有機物分解工程、工程D:スラリー薄膜形成工程(蒸発乾燥工程)、工程E:TRXF分析工程、からなっている。そして本発明では、図1のスラリー滴下工程と蒸発乾燥工程との間に、スラリー溶液からなる溶液膜にUV光(例えば、波長185nmの光)を照射して溶液膜中の有機物を分解する工程(上記の有機物分解工程)を含むことに特徴がある。
工程Aでは、測定対象の研磨スラリーにフッ酸を添加して、研磨スラリー中のコロイダルシリカを溶解してスラリー溶液とする。また、工程Bでは、工程A後のスラリー溶液で、シリコンウェーハ上に溶液膜を形成する。
これらの工程は、例えば、前述した図15におけるシリコン成分溶解工程、スラリー滴下工程と同様の手順で行うことができる。
なお、工程Aに関して、HFとHNO3の混酸を用いても良いが、前述したようにさらに熱分解処理を要するため、この加熱時のクロスコンタミネーションの影響も考慮すると、HF単独での添加がより好ましい。
また、工程Bに関して、ここではスラリー溶液を滴下することでシリコンウェーハ上に溶液膜を形成する例について説明するが、溶液膜の形成方法自体は特に限定されない。例えば塗布や浸漬などの方法を用いても良く、その場合にクロスコンタミネーションを起こさないように適宜工夫することができる。滴下による方法であれば、クロスコンタミネーションの発生を効果的に防ぐことができるので好ましい。
ここで光の波長とエネルギーの関係について記す。UV光の波長と光エネルギーとの関係は次式で表される。
E=hc/λ
E=エネルギー (J)
h=プランク定数 6.626×10-34 (Js)
c=光速度 2.9979×108 (m/s)
λ=波長 185×10-9 (m)
この式から、例えば波長185nmのエネルギーは1.074×10-18Jとなる。
これを光子1molに換算すると、アボガドロ数Na=6.022×1023/molより、647KJ/molとなる。
また、市販のUV光照射装置には波長179nmのエキシマレーザーを用いたものもあるが、波長185nmのUV光照射装置は低圧水銀ランプを用いるため一般に入手が容易であるため好ましい。
以上より、波長344nm~179nmのUV光を照射するのが特に好ましい。
またさらにはUV光照射中に二次汚染を抑制できると好ましい。
また、UV光照射と二次汚染の関係についての試験2も行った。上記の試験1とは別にウェーハ製造工程の最終洗浄まで行ったシリコンウェーハに対してスラリーを滴下せずに、前記試験1と同様の環境に10分~30分間暴露した後、ウェーハ表面の不純物をTRXF分析してUV光照射中の二次汚染の影響を調査した。
その結果を表2、表3、および図2に示す。なお、UV光照射時間が0分(照射なし)の場合についても併せて示す。
このときの二次汚染量が1.7×1011atoms/cm3以下であれば、デバイスに影響を及ぼすことはないので特に問題はない。なお、本発明の分析方法において、二次汚染量については上記数値範囲に特に限定されないものの、二次汚染量が小さくなるように、適宜、UV光の照射条件等の設定を行うことができる。

他方、UV光照射中の二次汚染の影響調査では、図2からも分かるように暴露時間が長くなるほど不純物の濃度が高くなっている。
工程CにおけるUV光照射時間は特に限定されるものではないが、例えば、上記のように10分間以上とすることができ、30分間もすれば十分である。また、スラリー薄膜の剥離の抑制効果の他に、UV光照射によるクロスコンタミネーション等による二次汚染の影響もさらに考慮すると、UV光照射時間は特に10分間から15分間の範囲が好ましい。
この工程の好ましい条件を検討するため、蒸発乾燥とスラリー薄膜の剥離の関係に関する試験3を行った。ウェーハ製造工程の最終洗浄まで行ったシリコンウェーハにスラリーを滴下し、UV光照射時間を10分間行った後、ホットプレートによるスラリーの蒸発乾燥の条件を温度50℃~100℃、時間8.5分~1.5分の間で振って乾燥させ、その後のスラリー薄膜の状態を確認した。
また、蒸発乾燥と二次汚染の関係に関する試験4を行った。上記の試験3とは別にウェーハ製造工程の最終洗浄まで行ったシリコンウェーハに対してスラリーを滴下せずにシリコンウェーハをホットプレートの上方に配置させ、ホットプレートを前記試験3と同様の条件で加熱した際のホットプレートからのアウトガスをシリコンウェーハに付着させ、その後、ホットプレート側のウェーハ表面の不純物をTRXF分析して、蒸発乾燥中の二次汚染の影響を調査した。
その結果を表4、表5、および、図3に示す。なお、表4に記載されているように、ここでは温度が高くなるに従い、短時間で乾燥させることができるので、それぞれの温度において、最短の乾燥時間に設定した。


そして、図3から分かるように温度が90℃以上、つまり、90℃(2分)~100℃(1.5分)では時間が短くても不純物濃度が高くなっているのに対して、温度が90℃より低い条件、つまり、80℃(2.5分)~50℃(8.5分)では、温度に関係なく時間が長くなるに従い不純物濃度が高くなっている。
工程Dにおける蒸発乾燥温度は特に限定されないが、例えば上記のように50℃から100℃の範囲とすることができ、二次汚染の影響も考慮すると60℃から100℃の範囲、さらには70℃から100℃の範囲が好ましい。
全反射蛍光X線分析装置自体は市販のものを用いることができ、例えば、テクノス製TREX630Tなどが挙げられる。このような装置を用いた分析手順自体は従来法と同様に行うことができる。
前述したように、従来法では溶液膜の乾燥後のスラリー薄膜がシリコンウェーハから剥離してしまっていたが、乾燥前にUV光照射を溶液膜に対して行う本発明ではスラリー薄膜が剥離するのを防ぐことができ、そのため、より確実にTRXF分析を行うことができる。しかもICP-MS等を用いた分析方法よりも簡便な工程で行うことができる。また、前述したように各工程での条件調整により二次汚染の抑制を図ることも十分可能であり、従来法と同レベルの高精度で分析することも可能である。
図4に示すスラリーの分析方法は、工程Dのスラリー薄膜形成工程として、図1の蒸発乾燥(加熱)工程の代わりに減圧による溶液膜の乾燥工程を行っている(蒸発乾燥(減圧)工程)。例えば、チャンバー内に溶液膜を有するウェーハを入れ、チャンバー内を減圧することによって行うことができる。
減圧時での乾燥工程の好ましい条件を検討するため、減圧乾燥とスラリー薄膜の剥離の関係に関する試験5を行った。ウェーハ製造工程の最終洗浄まで行ったシリコンウェーハにスラリーを滴下し、減圧乾燥の圧力値を15Torr~760Torr(1999.83Pa~101325Pa)、時間を2.1分~120分の間で振って減圧乾燥を行い、乾燥後のスラリー薄膜の状態を確認した。
また、減圧乾燥と二次汚染の関係に関する試験6を行った。試験5とは別にウェーハ製造工程の最終洗浄まで行ったシリコンウェーハに対してスラリーを滴下せずにシリコンウェーハを前記試験5と同じ減圧環境中に暴露し、ウェーハ表面の不純物をTRXF分析して減圧乾燥中の二次汚染の影響を調査した。
その結果を表6、表7、および、図5に示す。減圧乾燥条件は圧力が高くなるに従い、乾燥するまでの時間が長くなるので、それぞれの圧力値において、最短の乾燥時間に設定した。


また、図5に示すように、大気圧中に放置する場合は暴露時間が長くクロスコンタミネーション等による二次汚染の影響が見られたが、150Torr(19998.3Pa)以下の減圧環境では乾燥時間は10分以下と短く、クロスコンタミネーション等による二次汚染の影響はないと考えられる。
工程Dにおける減圧乾燥での圧力は特に限定されないが、例えば760Torr(101325Pa)未満とすることができ、二次汚染の影響も考慮すると上記のように150Torr(19998.3Pa)以下が特に好ましい。また、例えば上記のように15Torr(1999.83Pa)以上とすることができる。
(実施例1)
シリコンウェーハ研磨工程で用いられるニッタ・デュポン製の研磨スラリー3種類(スラリーA、B、C)について、図4に示すような本発明の分析方法により分析を行う。具体的な工程を以下に示す。
研磨スラリーを滴下・乾燥させるウェーハには、ウェーハ製造工程の最終洗浄まで行った直径200mmの抵抗率9~12Ωcmのp型シリコンPWウェーハを用いた。研磨スラリーを0.2mLと希釈用の超純水を9mL、および、スラリー中のコロイダルシリカを溶解するため、多摩化学製の超純度フッ化水素酸(Tapapure-AA100 HF 38%)の0.8mLを清浄なPFA(パーフルオロアルコキシエチレン共重合体)製のボトルに入れ攪拌(トータル50倍希釈)し、静置した。
その後、このスラリー溶液をマイクロピペット(エッペンドルフ製)で0.2mLを取り出し、洗浄済みウェーハの中心付近に滴下する。滴下したスラリー溶液は直径約40mmの大きさに広がる(溶液膜の形成)。
その後、ダイヤフラムポンプを有する減圧チャンバーに当該ウェーハを移し、150Torr(19998.3Pa)まで減圧したまま10分間放置し、スラリーを減圧により蒸発乾燥させる。
乾燥させると直径40mmのスラリー薄膜が生成されるため、その個所を全反射蛍光X線分析装置(テクノス製TREX630T)で測定した。


実施例1とは別の形態の分析方法(図1参照)により分析を行う。
使用するスラリー、ウェーハや、スラリー中のシリコン成分分解から有機物分解に至る工程は実施例1と同様であり記載は省略する。
有機物分解後、クラス100の環境に設置されたホットプレートにウェーハを移し、90℃で2分間放置し、スラリーを蒸発乾燥させる。
乾燥させると直径40mmのスラリー薄膜が生成されるため、その個所を全反射蛍光X線分析装置(テクノス製TREX630T)で測定した。

特許文献1に記載の研磨スラリーの分析方法により分析を行う。
まず、実施例1と同じ3種類の研磨スラリー1mLをフッ素樹脂(PFA製)ビーカーに入れ、多摩化学製高純度フッ化水素酸(Tapapure-AA100 HF 38%)1.5mLと多摩化学製超高純度硝酸(Tapapure-AA100 HNO3 68%)0.5mLを添加して研磨スラリー中のコロイダルシリカを溶解する。
つぎに、最大出力1000W、2450MHzのマイクロウェーブ湿式灰化装置(パーキンエルマー社製)を用い4段階分解レシピでスラリー中の有機物を分解する。
つぎに、清浄な直径200mmのシリコンPWウェーハ上に、有機物分解後のスラリーを1mL滴下し、70℃で30分間蒸発乾固した。つぎに、蒸発乾固したウェーハを350℃のホットプレートで10分間加熱し、乾燥残渣中のシリコン成分を揮発させた。
つぎに、残った乾燥残渣を上記HFと多摩化学製の超高純度過酸化水素水(Tapapure-AA100 H2O2 30%)を2wt%/2wt%の水溶液に調製し、乾燥残渣を1mLの2wt%/2wt%の水溶液で回収し、回収液とした。
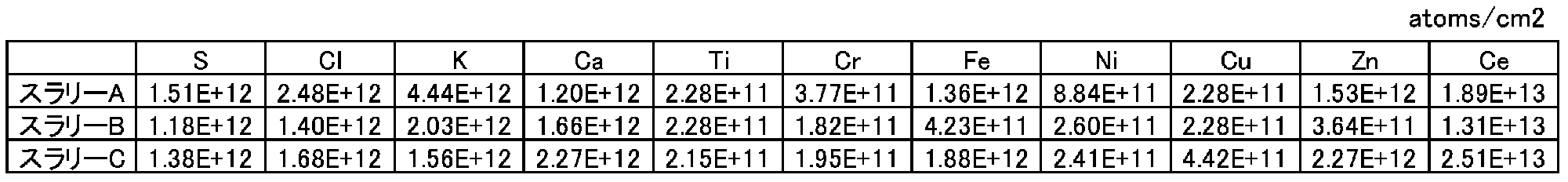
最後に回収液を超純水で200倍に希釈した後、誘導結合プラズマ質量分析(ICP-MS)装置(サーモフィッシャーサイエンティフィック社製ELEMENT2)で、K、Ca、Ti、Cr、Fe、Ni、Cu、Zn、Ceを分析した。
3種類のスラリーの分析結果を表12に示す。

実施例1、2の分析方法とは異なり、UV光照射を行わない場合の分析方法を行う。
まず、実施例1と同じウェーハ、および3種類の研磨スラリーを準備する。スラリーは原液では濃度が高いため、実施例1と同様にスラリーを希釈して、そこから0.2mLを取り出し、洗浄済みウェーハの中心付近に滴下する。
つぎにクラス100の環境に設置されたホットプレートにウェーハを移し、90℃で2分間放置し、スラリーを蒸発乾燥させた。
この結果、乾燥させたスラリー薄膜は剥離し反り返ってしまい、TRXF分析は不可であった(表13)。
また、ホットプレートによる蒸発乾燥の代わりに150Torrで10分間の減圧乾燥を行った場合も、乾燥させたスラリー膜は剥離し反り返ってしまい、TRXF分析は不可であった(表14)。


また、比較例2において、UV光照射による有機物分解処理を行わないと、乾燥後のスラリー薄膜が剥離し反り返ることによりTRXF分析できない問題がある。
Claims (4)
- シリコンウェーハ用の研磨スラリー中の金属不純物を分析する方法であって、
測定対象の研磨スラリーにフッ酸を添加して、前記研磨スラリー中のコロイダルシリカを溶解してスラリー溶液とする工程Aと、
該工程A後のスラリー溶液で、シリコンウェーハ上に溶液膜を形成する工程Bと、
該工程Bで形成した溶液膜にUV光を照射して溶液膜中の有機物を分解する工程Cと、
該工程Cで前記有機物を分解した溶液膜を前記シリコンウェーハ上で乾燥させてスラリー薄膜を形成する工程Dと、
該工程D後のスラリー薄膜から前記金属不純物の分析を全反射蛍光X線分析装置を用いて行う工程Eと、
を備えることを特徴とするシリコンウェーハ用の研磨スラリーの分析方法。 - 前記工程Cにおいて前記UV光を照射するとき、
波長が344nm~179nmのUV光を前記溶液膜に10~15分間照射することを特徴とする請求項1に記載のシリコンウェーハ用の研磨スラリーの分析方法。 - 前記工程Dにおいて前記溶液膜を乾燥させるとき、
70℃~100℃で加熱して乾燥させるか、または、150Torr(19998.3Pa)以下で減圧乾燥させることを特徴とする請求項1または請求項2に記載のシリコンウェーハ用の研磨スラリーの分析方法。 - 前記工程Bにおいて前記溶液膜を形成するとき、
前記工程A後のスラリー溶液を、前記シリコンウェーハ上に滴下して形成することを特徴とする請求項1から請求項3のいずれか一項に記載のシリコンウェーハ用の研磨スラリーの分析方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022038640A JP7635741B2 (ja) | 2022-03-11 | 2022-03-11 | シリコンウェーハ用の研磨スラリーの分析方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2022038640A JP7635741B2 (ja) | 2022-03-11 | 2022-03-11 | シリコンウェーハ用の研磨スラリーの分析方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2023133001A JP2023133001A (ja) | 2023-09-22 |
| JP7635741B2 true JP7635741B2 (ja) | 2025-02-26 |
Family
ID=88064863
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022038640A Active JP7635741B2 (ja) | 2022-03-11 | 2022-03-11 | シリコンウェーハ用の研磨スラリーの分析方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP7635741B2 (ja) |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5662769A (en) | 1995-02-21 | 1997-09-02 | Advanced Micro Devices, Inc. | Chemical solutions for removing metal-compound contaminants from wafers after CMP and the method of wafer cleaning |
| JP2001237203A (ja) | 2000-02-24 | 2001-08-31 | Mitsubishi Materials Silicon Corp | シリコンウェーハの研磨液及びこれを用いた研磨方法 |
| JP2004109072A (ja) | 2002-09-20 | 2004-04-08 | Sumitomo Mitsubishi Silicon Corp | 液中の金属不純物分析方法 |
| JP2005244123A (ja) | 2004-02-27 | 2005-09-08 | Fujimi Inc | 研磨用組成物 |
| JP2008020339A (ja) | 2006-07-13 | 2008-01-31 | Sumco Corp | シリコンウェーハの研磨スラリーの分析方法 |
| JP2009141077A (ja) | 2007-12-05 | 2009-06-25 | Toppan Printing Co Ltd | 有機エレクトロルミネッセンス素子、その製造方法および表示装置 |
| JP2011038843A (ja) | 2009-08-07 | 2011-02-24 | Siltronic Ag | シリコン研磨用スラリー中および研磨後スラリー中の金属分析方法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH0964133A (ja) * | 1995-08-29 | 1997-03-07 | Mitsubishi Materials Shilicon Corp | 半導体基板内部のCu濃度の検出方法 |
-
2022
- 2022-03-11 JP JP2022038640A patent/JP7635741B2/ja active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5662769A (en) | 1995-02-21 | 1997-09-02 | Advanced Micro Devices, Inc. | Chemical solutions for removing metal-compound contaminants from wafers after CMP and the method of wafer cleaning |
| JP2001237203A (ja) | 2000-02-24 | 2001-08-31 | Mitsubishi Materials Silicon Corp | シリコンウェーハの研磨液及びこれを用いた研磨方法 |
| JP2004109072A (ja) | 2002-09-20 | 2004-04-08 | Sumitomo Mitsubishi Silicon Corp | 液中の金属不純物分析方法 |
| JP2005244123A (ja) | 2004-02-27 | 2005-09-08 | Fujimi Inc | 研磨用組成物 |
| JP2008020339A (ja) | 2006-07-13 | 2008-01-31 | Sumco Corp | シリコンウェーハの研磨スラリーの分析方法 |
| JP2009141077A (ja) | 2007-12-05 | 2009-06-25 | Toppan Printing Co Ltd | 有機エレクトロルミネッセンス素子、その製造方法および表示装置 |
| JP2011038843A (ja) | 2009-08-07 | 2011-02-24 | Siltronic Ag | シリコン研磨用スラリー中および研磨後スラリー中の金属分析方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2023133001A (ja) | 2023-09-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6118031B2 (ja) | 不純物分析装置及び方法 | |
| JP3494102B2 (ja) | シリコンウエーハ中の金属不純物濃度評価方法 | |
| JPH10199855A (ja) | 基板の乾燥方法及び装置 | |
| KR101668494B1 (ko) | 반도체 장치의 제조 방법 및 반도체 기판의 세정 방법 | |
| JP5904512B1 (ja) | シリコン基板の分析方法 | |
| JP7635741B2 (ja) | シリコンウェーハ用の研磨スラリーの分析方法 | |
| Hannon et al. | Oxidative Removal of Photoresist by Oxygen/Freon® 116 Discharge Products | |
| US20030011774A1 (en) | Methods and systems for monitoring process fluids | |
| CN110223927A (zh) | 硅晶片的金属污染分析方法 | |
| JP2002368052A (ja) | 珪素脱離方法及びシリコンウェーハの不純物分析方法 | |
| US5695569A (en) | Removal of metal contamination | |
| JP4857973B2 (ja) | シリコンウェーハの研磨スラリーの分析方法 | |
| JPH11344440A (ja) | 有機物中の不純物分析方法及び分析装置 | |
| CN117732770A (zh) | 改善硅抛光片边缘金属污染的加工方法 | |
| US5695570A (en) | Method for the photo-stimulated removal of trace metals from a semiconductor surface | |
| US6146909A (en) | Detecting trace levels of copper | |
| Lu et al. | Evaluation of cleaning efficiency with a radioactive tracer and development of a microwave digestion method for semiconductor processes | |
| KR101064842B1 (ko) | 분석 방법 및 분석 장치 | |
| JP2006032859A (ja) | シリコンウェハの不純物の除去方法及び分析方法 | |
| CN111989565A (zh) | 硅基板的分析方法 | |
| EP0571950A2 (en) | Removal of metal contamination | |
| Le Tulzo et al. | Developing TSV wet cleaning chemistry for quantum computing application | |
| Rizquez et al. | Comparison study between optical emission spectroscopy and x-ray photoelectron spectroscopy techniques during process etch plasma | |
| Pfarr et al. | The use of plasma chemistry in failure analysis | |
| CN119694916A (zh) | 残余离子的测试方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20240327 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20241212 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250114 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250127 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7635741 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |