JP7647101B2 - 繊維強化複合材料の成形方法、およびそれに用いられるエポキシ樹脂組成物 - Google Patents
繊維強化複合材料の成形方法、およびそれに用いられるエポキシ樹脂組成物 Download PDFInfo
- Publication number
- JP7647101B2 JP7647101B2 JP2020564680A JP2020564680A JP7647101B2 JP 7647101 B2 JP7647101 B2 JP 7647101B2 JP 2020564680 A JP2020564680 A JP 2020564680A JP 2020564680 A JP2020564680 A JP 2020564680A JP 7647101 B2 JP7647101 B2 JP 7647101B2
- Authority
- JP
- Japan
- Prior art keywords
- epoxy resin
- fiber
- resin composition
- reinforced composite
- composite material
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/161—Catalysts containing two or more components to be covered by at least two of the groups C08G18/166, C08G18/18 or C08G18/22
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/166—Catalysts not provided for in the groups C08G18/18 - C08G18/26
- C08G18/168—Organic compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/18—Catalysts containing secondary or tertiary amines or salts thereof
- C08G18/1875—Catalysts containing secondary or tertiary amines or salts thereof containing ammonium salts or mixtures of secondary of tertiary amines and acids
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/18—Catalysts containing secondary or tertiary amines or salts thereof
- C08G18/20—Heterocyclic amines; Salts thereof
- C08G18/2045—Heterocyclic amines; Salts thereof containing condensed heterocyclic rings
- C08G18/2063—Heterocyclic amines; Salts thereof containing condensed heterocyclic rings having two nitrogen atoms in the condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/08—Processes
- C08G18/16—Catalysts
- C08G18/18—Catalysts containing secondary or tertiary amines or salts thereof
- C08G18/20—Heterocyclic amines; Salts thereof
- C08G18/2045—Heterocyclic amines; Salts thereof containing condensed heterocyclic rings
- C08G18/2072—Heterocyclic amines; Salts thereof containing condensed heterocyclic rings having at least three nitrogen atoms in the condensed ring system
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/28—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the compounds used containing active hydrogen
- C08G18/40—High-molecular-weight compounds
- C08G18/58—Epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G18/00—Polymeric products of isocyanates or isothiocyanates
- C08G18/06—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen
- C08G18/70—Polymeric products of isocyanates or isothiocyanates with compounds having active hydrogen characterised by the isocyanates or isothiocyanates used
- C08G18/72—Polyisocyanates or polyisothiocyanates
- C08G18/74—Polyisocyanates or polyisothiocyanates cyclic
- C08G18/76—Polyisocyanates or polyisothiocyanates cyclic aromatic
- C08G18/7657—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings
- C08G18/7664—Polyisocyanates or polyisothiocyanates cyclic aromatic containing two or more aromatic rings containing alkylene polyphenyl groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J5/00—Manufacture of articles or shaped materials containing macromolecular substances
- C08J5/04—Reinforcing macromolecular compounds with loose or coherent fibrous material
- C08J5/0405—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres
- C08J5/042—Reinforcing macromolecular compounds with loose or coherent fibrous material with inorganic fibres with carbon fibres
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2363/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2463/00—Characterised by the use of epoxy resins; Derivatives of epoxy resins
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Manufacturing & Machinery (AREA)
- Materials Engineering (AREA)
- Epoxy Resins (AREA)
- Reinforced Plastic Materials (AREA)
Description
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
(ここで、前記の吸光度比は、FT-IR(ATR法)において、オキサゾリドン環のカルボキシル基のC=O二重結合に起因する吸収の吸光度Daと、イソシアヌレート環のカルボキシル基のC=O二重結合に起因する吸収の吸光度Dbから吸光度比Da/(Da+Db)を算出することにより特定される。)
繊維強化複合材料の成形方法についての第2の態様として、少なくとも、強化繊維[A]およびエポキシ樹脂組成物[B]の硬化物からなる繊維強化複合材料の成形方法であって、エポキシ樹脂組成物[B]が次の構成要素[a]、[b]、[c]を含み、かつエポキシ樹脂組成物[B]をゴム状態弾性率(Gr)とガラス転移温度(Tg)の関係が式1を満たすように硬化して繊維強化複合材料を得る、繊維強化複合材料の成形方法である。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
Tg≧10×Gr+120 (式1)
繊維強化複合材料用エポキシ樹脂組成物についての第1の態様として、次の構成要素[a]、[b]、[c]を含み、30℃から10℃/分で昇温しながら硬化した際に、硬化度X%における吸光度比Da/(Da+Db)が0.4~1の範囲となるある特定の硬化度Xが85~95%の範囲に存在する、繊維強化複合材料用エポキシ樹脂組成物である。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
(ここで、前記の硬化度は、DSCにより得られるエポキシ樹脂組成物の総発熱量QTと、エポキシ樹脂組成物の硬化物の残存発熱量QRから硬化度(%)=(QT-QR)/QT×100で算出することにより特定される。)
繊維強化複合材料用エポキシ樹脂組成物についての第2の態様として、次の構成要素[a]、[b]、[c]を含み、30℃から10℃/分で昇温しながら硬化した際に、硬化度Xにおけるゴム状態弾性率(Gr)とガラス転移温度(Tg)の関係が式1を満たすある特定の硬化度Xが85~95%の範囲に存在する、繊維強化複合材料用エポキシ樹脂組成物である。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
Tg≧10×Gr+120 (式1)
さらに、これらのエポキシ樹脂組成物の硬化物、およびそれを用いた繊維強化複合材料である。
Tg≧10×Gr+120 (式1)
Tg≧10×Gr+140 (式1a)
Tg≧10×Gr+160 (式1b)
Tg≦10×Gr+230 (式1’)
本発明の繊維強化複合材料の成形方法についての第2の態様では、エポキシ樹脂組成物[B]をゴム状態弾性率(Gr)が式2を満たすように硬化して繊維強化複合材料を得ることが好ましい。
0.5≦Gr≦15 (式2)
0.5≦Gr≦10 (式2a)
0.5≦Gr≦5 (式2b)
ここでガラス転移温度は、エポキシ樹脂硬化物を、示差走査熱量測定装置を用いて、10℃/分の昇温速度で30℃から350℃まで昇温測定し、JIS K7121:1987に基づいて求めた中間点温度である。
Vf(%)=(Af×N)/(ρf×h)/10
Af:強化繊維[A]からなる基材1枚・1m2当たりの質量(g/m2)
N:強化繊維[A]からなる基材の積層枚数(枚)
ρf:強化繊維[A]の密度(g/cm3)
h:繊維強化複合材料(試験片)の厚み(mm)。
Tg≧10×Gr+120 (式1)
Tg≧10×Gr+140 (式1a)
Tg≧10×Gr+160 (式1b)
Tg≦10×Gr+230 (式1’)
また、本発明の繊維強化複合材料用エポキシ樹脂組成物についての第2の態様において、30℃から10℃/分で昇温しながら硬化した際に、硬化度Xにおけるゴム状態弾性率が0.5~15MPaの範囲となるある特定の硬化度Xが85~95%の範囲に存在することが好ましく、硬化度Xにおけるゴム状態弾性率が0.5~10MPaの範囲となるある特定の硬化度Xが85~95%の範囲に存在することがより好ましい。すなわち、本発明の繊維強化複合材料用エポキシ樹脂組成物についての第2の態様において、30℃から10℃/分で昇温しながら硬化した際に、硬化度85~95%の範囲のいずれか(例えば、硬化度90%)におけるゴム状態弾性率が0.5~15MPaの範囲となることが好ましい。
COH=(Σ(wn/wnOH))/W×1000 ・・・(式3)
COH:エポキシ樹脂組成物中の水酸基量(mol/kg)
wn:各成分の質量部
wnOH:各成分の水酸基当量(g/eq)
W:全成分の質量部の和。
実施例のエポキシ樹脂組成物を得るために、以下の原料を用いた。
・“jER(登録商標)”828(ビスフェノールA型エポキシ樹脂液状、三菱ケミカル(株)製)
・“ルプラネート(登録商標)”M20S(ポリメリックMDI、BASF INOAC ポリウレタン(株)製)
・“DBU(登録商標)”(1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン、サンアプロ(株)製))
・3,3’-DAS(3,3’-ジアミノジフェニルスルホン、三井化学ファイン(株)製)。
実施例1~3、参考例1は、エポキシ樹脂として、“jER(登録商標)”828 100質量部、“DBU(登録商標)” 4質量部を投入し、混練し、透明な粘調液を得た。その後、“ルプラネート(登録商標)”M20S 72質量部添加し、さらに混練し、エポキシ樹脂組成物を得た。
上記(2)で作製したエポキシ樹脂硬化物を真空中で脱泡した後、予備加熱したプレートに注型し、表1に記載した硬化条件で、動的粘弾性試験装置(ATD:アルファテクノロジーズLLC製)を用いてエポキシ樹脂硬化板を作製した。
上記(2)で調製したエポキシ樹脂組成物を5mg採取し、示差走査熱量測定装置(DSC2910:TAインスツルメンツ社製)を用いて、10℃/分の昇温速度で30℃から350℃まで昇温測定し、発熱カーブを取得し、その発熱ピークを積分することにより、熱硬化性樹脂の総発熱量QTを算出した。分解反応などによる発熱または吸熱のピークが見られる場合は、それらピーク以下の温度範囲で測定を行った。
上記(3)で作製したエポキシ樹脂硬化板から10mg採取し、示差走査熱量測定装置(DSC2910:TAインスツルメンツ社製)を用いて、10℃/分の昇温速度で30℃から350℃まで昇温測定し、JIS K7121:1987に基づいて求めた中間点温度をガラス転移温度Tgとし、耐熱性を評価した。
上記(3)で作製したエポキシ樹脂硬化板を#240、#800、#2000のサンドペーパーで表面を研磨させ、厚さ2mmのエポキシ樹脂硬化板を得た。次に、得られたエポキシ樹脂硬化板から、幅10mm、長さ60mmの試験片を切り出し、スパン間32mmの3点曲げを測定し、JIS K7171:1994に従い、樹脂靭性の指標となる曲げ撓み量を求めた。
上記(3)で作製したエポキシ樹脂硬化板、または硬化度15~25%の範囲内のある特定の硬化度(本実施例では、硬化度20%)のエポキシ樹脂硬化板を採取し、FT-IR装置(7000FT-IR:Varian製)を用いて、FT-IR(ATR法)を実施した。測定条件は、分解能を4cm-1、積算回数を32回とした。
上記(3)で作製したエポキシ樹脂硬化板を10mg採取し、熱重量分析機(TGA7:パーキンエルマー社製)を用いて、窒素(純度:99.99%以上)気流下、プログラム温度50℃で1分保持、プログラム温度50℃から800℃まで昇温速度20℃/分で昇温の条件にて質量減少率の測定を行った。
350mm×700mm×2mmの板状キャビティーを持つ金型に、強化繊維として炭素繊維織物CO6343(炭素繊維:T300-3K、組織:平織、目付:198g/m2、東レ(株)製)をキャビティー内に9枚積層し、プレス装置で型締めを行った。次に、100℃(成形温度)に保持した金型内を、真空ポンプにより、大気圧-0.1MPaに減圧し、あらかじめ、それぞれに50℃に加温しておいたエポキシ樹脂組成物を、樹脂注入機を用いて混合し、0.2MPaの圧力で注入した。その後、表1に記載の硬化条件で硬化し、脱型して、繊維強化複合材料を得た。
上記(9)で作製した繊維強化複合材料から10mg採取し、示差走査熱量測定装置(DSC2910:TAインスツルメンツ社製)を用いて、10℃/分の昇温速度で30℃から350℃まで昇温測定し、JIS K7121:1987に基づいて求めた中間点温度をガラス転移温度Tgとし、耐熱性を評価した。
前記のようにして、表1に記載の硬化条件でエポキシ樹脂組成物の硬化物および繊維強化複合材料を作製した。かかるエポキシ樹脂組成物の硬化物は、耐熱性とΔWrは問題ないレベルであり、靭性は優れていた。
実施例1から硬化条件を変更した。かかるエポキシ樹脂組成物の硬化物は、耐熱性は問題ないレベルであり、靭性とΔWrは優れていた。
実施例1から硬化条件を変更した。かかるエポキシ樹脂組成物の硬化物は、耐熱性、靭性とΔWrは優れていた。
実施例1から硬化条件を変更した。かかるエポキシ樹脂組成物の硬化物はDa/(Da+Db)が劣っており、耐熱性は優れているものの、靭性およびΔWrに劣っていた。
構成要素[b]以外の硬化剤として、アミン化合物を配合した。かかるエポキシ樹脂組成物の硬化物はオキサゾリドン環が形成されず、耐熱性とΔWrに劣っていた。

実施例のエポキシ樹脂組成物を得るために、以下の原料を用いた。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
・“jER(登録商標)”828(ビスフェノールA型エポキシ樹脂、三菱ケミカル(株)製)
・“エポトート(登録商標)”YD-8125(ビスフェノールA型エポキシ樹脂、日鉄ケミカル&マテリアル(株)製)
・“EPICLON(登録商標)”830(ビスフェノールF型エポキシ樹脂、DIC(株)製)
・YD-8125変性品
100質量部の“エポトート(登録商標)”YD-8125に無水酢酸を10質量部添加し、110℃で1時間加熱撹拌し、YD-8125に少量含まれる水酸基をアセチル化した。その後、110℃で真空加熱することにより、余剰の酢酸と生成した酢酸を除去し、YD-8125変性品を得た。
・“デナコール”EX-313(グリセリン型エポキシ樹脂、1,3-ビス(オキシラニルメトキシ)プロパン-2-オール、ナガセケムテックス(株)製)
・“アラルダイド(登録商標)”MY0510(トリグリシジル-p-アミノフェノール、ハンツマン・アドバンスト・マテリアルズ社製)
・“アラルダイド(登録商標)”MY721(テトラグリシジルジアミノジフェニルメタン、ハンツマン・アドバンスト・マテリアルズ社製)。
・“ルプラネート(登録商標)”M20S(ポリメリックMDI、BASF INOAC ポリウレタン(株)製)
・“ルプラネート(登録商標)”MI(モノメリックMDI、BASF INOAC ポリウレタン(株)製)。
・“DBU(登録商標)”(1,8-ジアザビシクロ[5.4.0]ウンデカ-7-エン、サンアプロ(株)製、pKb=25)
・“DBU”/フタル酸(東京化成工業(株)製、pKa=3)塩
・“DBU”/ジクロロ酢酸(東京化成工業(株)製、pKa=1.5)塩
・“DBU”/p-トルエンスルホン酸(東京化成工業(株)製、pKa=-3)塩
・“DBN(登録商標)”(1,5-ジアザビシクロ[4.3.0]-5-ノネン、サンアプロ(株)製、pKb=24)/フタル酸塩
・TBD(1,5,7-トリアザビシクロ[4.4.0]デカ-5-エン、東京化成工業(株)製、pKb=26)/ジクロロ酢酸塩
・TBAB(テトラブチルアンモニウムブロミド、東京化成工業(株)製)
・“ホクコー TBP-BB(登録商標)”(テトラブチルホスホニウムブロミド、北興化学工業(株)製)。
・BGE(4-tert-ブチルフェニルグリシジルエーテル、東京化成工業(株)製)。
・2-フェニルエチルイソシアナート(東京化成工業(株)製)。
・ポリプロピレングリコール(富士フイルム和光純薬(株)製)。
・“ロンザキュア(登録商標)”M-DEA(ハンツマン・アドバンスト・マテリアルズ社製)。
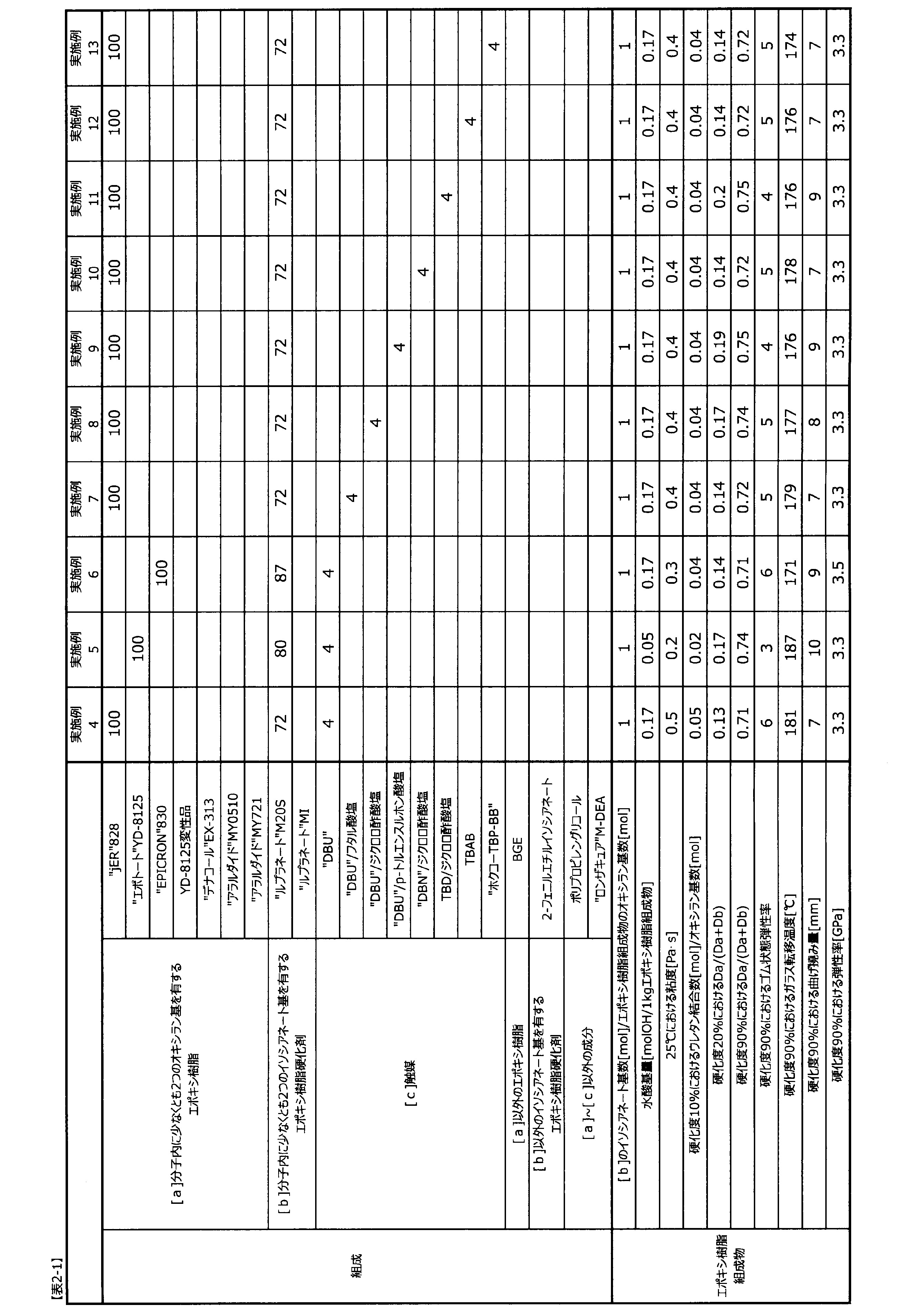
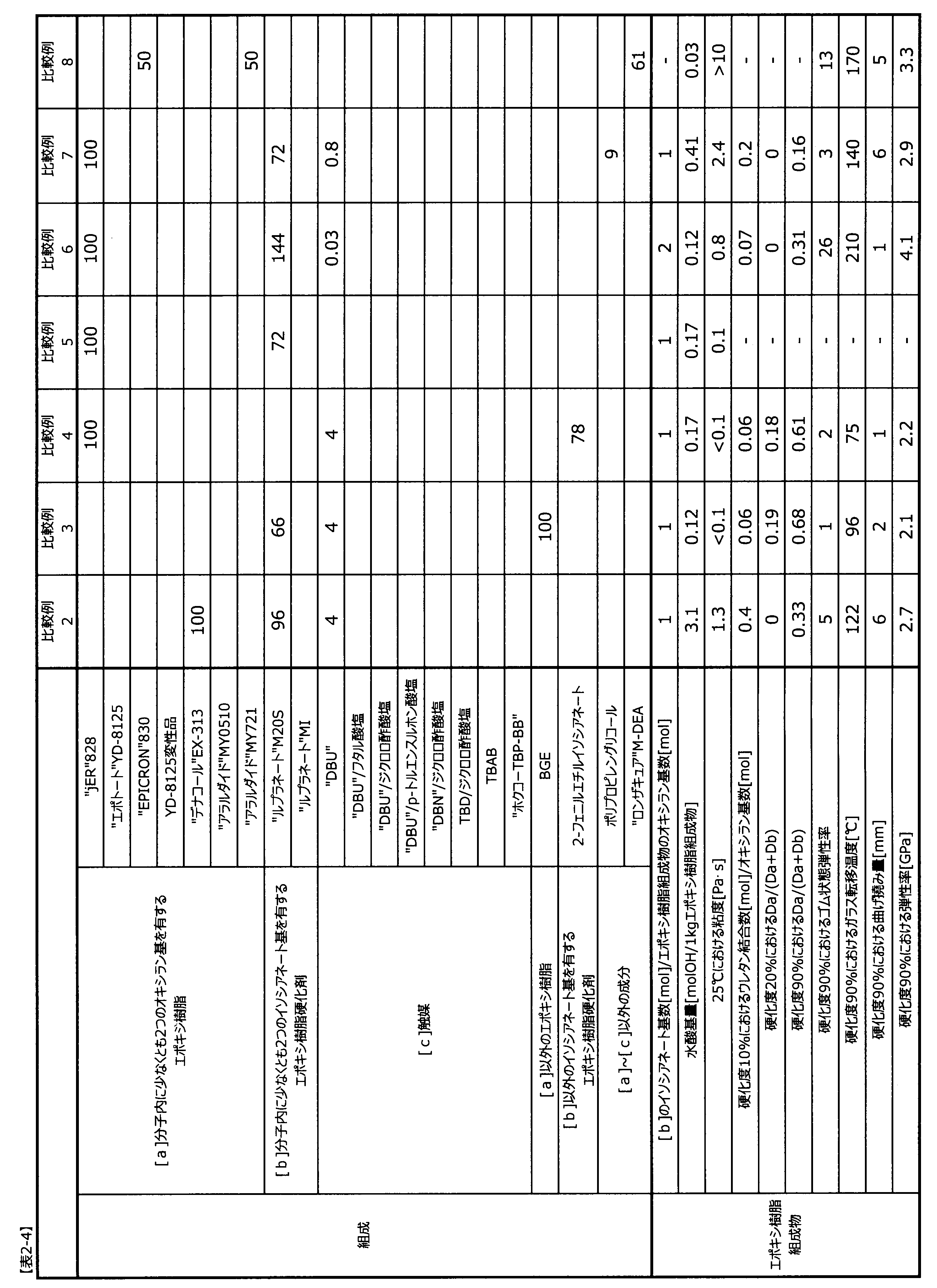
表2-1~表2-4に記載した配合比(質量比)でエポキシ樹脂と触媒を配合し、位相差顕微鏡にて溶解を確認した後に、エポキシ樹脂硬化剤を配合してエポキシ樹脂組成物を調製した。
エポキシ樹脂組成物中の水酸基量は、構成要素の成分ごとに水酸基当量を用いて、式3で算出した。
COH=(Σ(wn/wnOH))/W×1000 ・・・(式3)
COH:エポキシ樹脂組成物中の水酸基量(mol/kg)
wn:各成分の質量部
wnOH:各成分の水酸基当量(g/eq)
W:全成分の質量部の和。
動的粘弾性測定装置(ARES:TAインスツルメント社製)を用い、直径40mmのパラレルプレートを用い、昇温速度1.5℃/minで単純昇温し、周波数1Hz、Gap 1mmの測定条件で得られた、複素粘性率η*の25℃における値を採用した。
上記(2)で調製したエポキシ樹脂組成物を真空中で脱泡した後、予備加熱したプレートに注型し、動的粘弾性試験装置(ATD:アルファテクノロジーズLLC製)を用いて、30℃から(6)項に記載の測定で得た硬化度が90%となる温度まで10℃/分で昇温することでエポキシ樹脂硬化板を作製した。
上記(2)で調製したエポキシ樹脂組成物を5mg採取し、示差走査熱量測定装置(DSC2910:TAインスツルメンツ社製)を用いて、10℃/分の昇温速度で30℃から350℃まで昇温測定し、発熱カーブを取得し、その発熱ピークを積分することにより、熱硬化性樹脂の総発熱量QTを算出した。分解反応などによる発熱または吸熱のピークが見られる場合は、それらピーク以下の温度範囲で測定を行った。
上記(5)で作製した特定の硬化度Z(本実施例では、硬化度10%)のエポキシ樹脂硬化物を採取し、重水素化クロロホルム溶媒中、500MHz 1H-NMRを用い、積算回数128回により測定した。2.7ppmにエポキシ樹脂のオキシラン基における炭素に隣接するプロトンと、5.4ppmにウレタン結合の窒素に隣接するプロトンの面積値より存在比率を算出した。
上記(5)で作製した特定の硬化度X(本実施例では、硬化度90%)および特定の硬化度Y(本実施例では、硬化度20%)のエポキシ樹脂硬化物を採取し、FT-IR装置(7000FT-IR:Varian製)を用いて、FT-IR(ATR法)を実施した。測定条件は、分解能を4cm-1、積算回数を32回とした。
上記(5)で作製した硬化度X(本実施例では、硬化度90%)のエポキシ樹脂硬化物から10mg採取し、示差走査熱量測定装置(DSC2910:TAインスツルメンツ社製)を用いて、10℃/分の昇温速度で30℃から350℃まで昇温測定し、JIS K7121:1987に基づいて求めた中間点温度をガラス転移温度Tgとし、耐熱性を評価した。
上記(5)で作製した硬化度X(本実施例では、硬化度90%)のエポキシ樹脂硬化物からから、幅10mm、長さ40mmの試験片を切り出し、動的粘弾性測定装置(ARES:TAインスツルメント社製)を用い、固体ねじり治具に試験片をセットし、昇温速度5℃/分、周波数1Hz、歪み量0.1%にて30~300℃の温度範囲について測定を行った。架橋密度の指標となるゴム状態弾性率は、動的粘弾性測定で得られるガラス転移温度を50℃上回った温度における貯蔵弾性率を採用した。なお、動的粘弾性測定で得られるガラス転移温度は、温度-貯蔵弾性率曲線において、ガラス領域に引いた接線と、ガラス転移領域に引いた接線との交点における温度とした。
上記(5)で作製した硬化度X(本実施例では、硬化度90%)のエポキシ樹脂硬化物を#240、#800、#2000のサンドペーパーで表面を研磨させ、厚さ2mmのエポキシ樹脂硬化板を得た後に、得られたエポキシ樹脂硬化板から、幅10mm、長さ60mmの試験片を切り出し、スパン間32mmの3点曲げを測定し、JIS K7171:1994に従い、曲げ弾性率と樹脂靭性の指標となる曲げ撓み量を求めた。
前記のようにして、表2-1に記載した含有割合でエポキシ樹脂組成物を調製した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性、靭性、弾性率は優れていた。
実施例4から構成要素[a]を水酸基量の少ないエポキシ樹脂に変更した。かかるエポキシ樹脂組成物は25℃における粘度は特に優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性、弾性率は優れており、靭性は特に優れていた。
実施例4から構成要素[a]をビスフェノールF型エポキシ樹脂に変更した。かかるエポキシ樹脂組成物は25℃における粘度は特に優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性は問題ないレベルであり、靭性と弾性率は優れていた。
実施例4から構成要素[c]をブレンステッド塩基とブレンステッド酸の塩に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性は問題ないレベルであり、靭性と弾性率は優れていた。
実施例4から構成要素[c]をハロゲン化オニウム塩に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性は問題ないレベルであり、靭性と弾性率は優れていた。
実施例4から構成要素[c]の量を1質量部に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は問題ないレベルであり、靭性は優れていた。
実施例4から構成要素[c]の量を10質量部に変更した。かかるエポキシ樹脂組成物は25℃における粘度は問題ないレベルであった。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は優れており、靭性は問題ないレベルであった。
実施例4から構成要素[b]のイソシアネート基数とエポキシ樹脂組成物のオキシラン基数の比を0.8に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は問題ないレベルであり、靭性は優れていた。
実施例4から構成要素[b]のイソシアネート基数とエポキシ樹脂組成物のオキシラン基数の比を0.5に変更した。かかるエポキシ樹脂組成物は25℃における粘度は問題ないレベルであった。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は劣るものの、靭性は問題ないレベルであった。
実施例4から構成要素[b]のイソシアネート基数とエポキシ樹脂組成物のオキシラン基数の比を1.1に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は優れており、靭性は問題ないレベルであった。
実施例4から構成要素[b]のイソシアネート基数とエポキシ樹脂組成物のオキシラン基数の比を1.4に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は優れており、靭性は許容されるレベルであった。
実施例4から構成要素[b]のイソシアネート基数とエポキシ樹脂組成物のオキシラン基数の比を1.7に変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は優れており、靭性は許容されるレベルであった。
実施例4から構成要素[b]の種類を変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は問題ないレベルであり、靭性は優れていた。
実施例4から構成要素[a]の3割を水酸基量の少ないエポキシ樹脂に変更した。かかるエポキシ樹脂組成物は25℃における粘度はやや優位となり、エポキシ樹脂組成物の硬化度90%における耐熱性と靭性もそれぞれ向上した。
実施例22に対し、構成要素[a]の水酸基量の少ないエポキシ樹脂を7割に増量した。かかるエポキシ樹脂組成物は25℃における粘度はさらに優位となり、エポキシ樹脂組成物の硬化度90%における耐熱性と靭性もそれぞれさらに向上した。
実施例4から構成要素[a]を4官能アミン型エポキシ樹脂に変更した。かかるエポキシ樹脂組成物は25℃における粘度は特に優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は特に優れており、靭性は許容されるレベルであった。
実施例4から構成要素[a]をビスフェノールF型エポキシと3官能アミン型エポキシ樹脂の組み合わせに変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は特に優れており、靭性は優れていた。
実施例4から構成要素[a]をビスフェノールF型エポキシと4官能アミン型エポキシ樹脂の組み合わせに変更した。かかるエポキシ樹脂組成物は25℃における粘度は優れていた。エポキシ樹脂組成物の硬化度90%における耐熱性と弾性率は特に優れており、靭性は優れていた。
実施例5から構成要素[a]をさらに水酸基量の少ないエポキシ樹脂に変更した。かかるエポキシ樹脂組成物は25℃における粘度はさらに優位となり、エポキシ樹脂組成物の硬化度90%における耐熱性と靭性もそれぞれさらに向上した。
構成要素[a]としてグリセリン型エポキシ樹脂を配合し、25℃における粘度、硬化度90%における耐熱性、弾性率に劣っていた。
構成要素[a]として単官能エポキシ樹脂を配合し、25℃における粘度、硬化度90%における耐熱性、靭性、弾性率に劣っていた。
構成要素[b]として単官能イソシアネートを配合し、25℃における粘度、硬化度90%における耐熱性、靭性、弾性率に劣っていた。
構成要素[c]を配合しないことで、指定の条件では硬化度90%の硬化物が得られなかった。
特許文献1(国際公開第2014/184082号)の実施例I12に類似したものである。イソシアヌレート環が多く形成され、靭性に劣っていた。
特許文献2(国際公開第2016/102358号)の実施例1に類似したものである。ポリオールを配合した樹脂組成物とすることで水酸基量が大幅に増え、ウレタン結合が多く形成され、25℃における粘度、硬化度90%における耐熱性、弾性率に劣っていた。
構成要素[c]を含まず、かつ構成要素[b]の代わりにアミン硬化剤を配合した結果、25℃における粘度、硬化度90%における耐熱性、弾性率に劣っていた。


Claims (28)
- 少なくとも、強化繊維[A]およびエポキシ樹脂組成物[B]の硬化物からなる繊維強化複合材料の成形方法であって、エポキシ樹脂組成物[B]が次の構成要素[a]、[b]、[c]を含み水酸基量が0.09mol/kg以下であり、かつエポキシ樹脂組成物[B]を吸光度比Da/(Da+Db)が0.4~1の範囲となるように硬化して繊維強化複合材料を得る、繊維強化複合材料の成形方法。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
(ここで、前記の吸光度比は、FT-IR(ATR法)において、オキサゾリドン環のカルボキシル基のC=O二重結合に起因する吸収の吸光度Daと、イソシアヌレート環のカルボキシル基のC=O二重結合に起因する吸収の吸光度Dbから吸光度比Da/(Da+Db)を算出することにより特定される。) - エポキシ樹脂組成物[B]を硬化度15~25%の範囲内のある特定の硬化度における吸光度比Da/(Da+Db)が0.01~1の範囲となるように硬化する、請求項1に記載の繊維強化複合材料の成形方法。
(ここで、前記の硬化度は、昇温速度10℃/分でのDSCにより得られるエポキシ樹脂組成物の総発熱量QTと、その硬化物の残存発熱量QRから硬化度(%)=(QT-QR)/QT×100を算出することにより特定される。) - エポキシ樹脂組成物[B]を吸光度比が0.7~1の範囲となるように硬化して繊維強化複合材料を得る、請求項1または2に記載の繊維強化複合材料の成形方法。
- 少なくとも、強化繊維[A]およびエポキシ樹脂組成物[B]の硬化物からなる繊維強化複合材料の成形方法であって、エポキシ樹脂組成物[B]が次の構成要素[a]、[b]、[c]を含み水酸基量が0.09mol/kg以下であり、かつエポキシ樹脂組成物[B]をゴム状態弾性率(Gr)とガラス転移温度(Tg)の関係が式1を満たすように硬化して繊維強化複合材料を得る、繊維強化複合材料の成形方法。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
Tg≧10×Gr+120 (式1) - さらにエポキシ樹脂組成物[B]を、式2を満たすように硬化して繊維強化複合材料を得る、請求項4記載の繊維強化複合材料の成形方法。
0.5≦Gr≦15 (式2) - 繊維強化複合材料を構成するエポキシ樹脂組成物[B]の硬化物は、質量減少率△Wrが10%以下の範囲となるものである、請求項1~5のいずれかに記載の繊維強化複合材料の成形方法。
(ここで、前記の質量減少率は、常圧の非酸化性雰囲気下で50℃から800℃の温度まで昇温速度10℃/分で熱重量分析を行った際に、70℃到達時点の質量W1と、320℃到達時の試料質量W2から質量減少率△Wr(%)=(W1-W2)/W1×100を算出することにより特定される。) - エポキシ樹脂組成物[B]を、100~200℃に加熱した成形型内に配置した強化繊維[A]からなる基材に注入し、含浸させ、該成形型内で硬化する、請求項1~6のいずれかに記載の繊維強化複合材料の成形方法。
- 30~80℃に加温したエポキシ樹脂組成物[B]を、120~180℃に加熱した成形型内に配置した強化繊維[A]からなる基材に注入し、含浸させ、該成形型内で硬化する、請求項1~7のいずれかに記載の繊維強化複合材料の成形方法。
- エポキシ樹脂組成物[B]を、成形型内に配置した強化繊維[A]からなる基材に注入するに際して、該樹脂を該成形型に設けられた複数の箇所から注入する、請求項7または8に記載の繊維強化複合材料の成形方法。
- 次の構成要素[a]、[b]、[c]を含み水酸基量が0.09mol/kg以下であり、30℃から10℃/分で昇温しながら硬化した際に、硬化度Xにおける吸光度比Da/(Da+Db)が0.4~1の範囲となるある特定の硬化度Xが85~95%の範囲に存在する、繊維強化複合材料用エポキシ樹脂組成物。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
(ここで、前記の吸光度比は、FT-IR(ATR法)において、オキサゾリドン環のカルボキシル基のC=O二重結合に起因する吸収の吸光度Daと、イソシアヌレート環のカルボキシル基のC=O二重結合に起因する吸収の吸光度Dbから吸光度比Da/(Da+Db)を算出することにより特定される。また、前記の硬化度は、昇温速度10℃/分でのDSCにより得られるエポキシ樹脂組成物の総発熱量QTと、その硬化物の残存発熱量QRから硬化度(%)=(QT-QR)/QT×100を算出することにより特定される。) - 30℃から10℃/分で昇温しながら硬化した際に、硬化度Yにおける吸光度比Da/(Da+Db)が0.01~1の範囲となるある特定の硬化度Yが15~25%の範囲に存在する、請求項10に記載の繊維強化複合材料用エポキシ樹脂組成物。
- 次の構成要素[a]、[b]、[c]を含み水酸基量が0.09mol/kg以下であり、30℃から10℃/分で昇温しながら硬化した際に、硬化度Xにおけるゴム状態弾性率(Gr)とガラス転移温度(Tg)の関係が式1を満たすある特定の硬化度Xが85~95%の範囲に存在する、繊維強化複合材料用エポキシ樹脂組成物。
[a]分子内に少なくとも2つのオキシラン基を有するエポキシ樹脂
[b]分子内に少なくとも2つのイソシアネート基を有するエポキシ樹脂硬化剤
[c]触媒
Tg≧10×Gr+120 (式1) - 30℃から10℃/分で昇温しながら硬化した際に、硬化度Xにおけるゴム状態弾性率が0.5~15MPaの範囲となるある特定の硬化度Xが85~95%の範囲に存在する、請求項12に記載の繊維強化複合材料用エポキシ樹脂組成物。
- 構成要素[a]として1種類以上のアミン型エポキシ樹脂を含む、請求項10~13のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 構成要素[a]として1種類以上のビスフェノール型エポキシ樹脂を含む、請求項10~14のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- エポキシ樹脂組成物に含まれる全エポキシ樹脂のオキシラン基のmol数に対する、構成要素[b]のイソシアネート基のmol数の比率が0.5~1.8である、請求項10~15のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 構成要素[c]として、アセトニトリル中での塩基解離定数pKbが20以上のブレンステッド塩基とブレンステッド酸からなる塩を含む、請求項10~16のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- ブレンステッド酸の水中での酸解離定数pKaが5以下である、請求項17に記載の繊維強化複合材料用エポキシ樹脂組成物。
- ブレンステッド塩基がアミン化合物およびイミダゾール化合物からなる群から選択される少なくとも1種類である、請求項17または18に記載の繊維強化複合材料用エポキシ樹脂組成物。
- ブレンステッド酸がカルボン酸、スルホン酸およびハロゲン化水素からなる群から選択される少なくとも1種類である、請求項17~19のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 構成要素[c]として、アニオンがハロゲン化物イオンであるオニウム塩を含む、請求項10~16のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- オニウム塩が四級アンモニウム塩および四級ホスホニウム塩からなる群から選択される少なくとも1種類である、請求項21に記載の繊維強化複合材料用エポキシ樹脂組成物。
- 構成要素[c]が構成要素[a]の総量100質量部に対して1質量部以上10質量部以下である、請求項10~22のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 構成要素[c]は、構成要素[a]に溶解し得る、請求項10~23のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 30℃から10℃/分で昇温しながら硬化した際に、硬化度Zにおけるウレタン結合とオキシラン基の存在比率が0.10以下となるある特定の硬化度Zが5~15%の範囲に存在する、請求項10~24のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 25℃における粘度が0.1~1.0Pa・sである、請求項10~25のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物。
- 請求項10~26のいずれかに記載の繊維強化複合材料用エポキシ樹脂組成物の硬化物。
- 請求項27に記載の硬化物と、強化繊維を含んでなる繊維強化複合材料。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2019207633 | 2019-11-18 | ||
| JP2019207633 | 2019-11-18 | ||
| JP2020096754 | 2020-06-03 | ||
| JP2020096754 | 2020-06-03 | ||
| PCT/JP2020/042584 WO2021100649A1 (ja) | 2019-11-18 | 2020-11-16 | 繊維強化複合材料の成形方法、およびそれに用いられるエポキシ樹脂組成物 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JPWO2021100649A1 JPWO2021100649A1 (ja) | 2021-05-27 |
| JPWO2021100649A5 JPWO2021100649A5 (ja) | 2023-11-20 |
| JP7647101B2 true JP7647101B2 (ja) | 2025-03-18 |
Family
ID=75980754
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2020564680A Active JP7647101B2 (ja) | 2019-11-18 | 2020-11-16 | 繊維強化複合材料の成形方法、およびそれに用いられるエポキシ樹脂組成物 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20220396696A1 (ja) |
| EP (1) | EP4063436A4 (ja) |
| JP (1) | JP7647101B2 (ja) |
| CN (1) | CN114729133B (ja) |
| AU (1) | AU2020387250A1 (ja) |
| WO (1) | WO2021100649A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20230406994A1 (en) * | 2020-11-16 | 2023-12-21 | Toray Industries, Inc. | Thermosetting epoxy resin composition, molded article of same, fiber-reinforced composite material, molding material for fiber-reinforced composite materials, and method for producing fiber-reinforced composite material |
| JPWO2022124191A1 (ja) * | 2020-12-08 | 2022-06-16 | ||
| CN115436220B (zh) * | 2022-09-19 | 2025-06-24 | 东方电气集团东方电机有限公司 | 一种适用于发电机定子绕组绝缘交联固化程度的评价方法 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018501391A (ja) | 2014-12-22 | 2018-01-18 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェンHenkel AG & Co. KGaA | 繊維強化材料用オキサゾリジノンおよびイソシアヌレート架橋マトリックス |
| JP2019521216A (ja) | 2016-06-20 | 2019-07-25 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェンHenkel AG & Co. KGaA | エポキシ樹脂及びポリイソシアネートをベースとする高衝撃強度及び耐高温性を有する硬化組成物 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3323122A1 (de) * | 1983-06-27 | 1985-05-23 | Siemens AG, 1000 Berlin und 8000 München | Verfahren zur herstellung von reaktionsharzformstoffen |
| DE3323084A1 (de) * | 1983-06-27 | 1985-01-10 | Siemens AG, 1000 Berlin und 8000 München | Verfahren zur herstellung von formstoffen |
| GB8912952D0 (en) * | 1989-06-06 | 1989-07-26 | Dow Rheinmuenster | Epoxy-terminated polyoxazolidones,process for the preparation thereof and electrical laminates made from the epoxy-terminated polyoxazolidones |
| EP2803684A1 (de) | 2013-05-13 | 2014-11-19 | Basf Se | Isocyanat-Epoxid-Hybridharze |
| DE102014226842A1 (de) | 2014-12-22 | 2016-06-23 | Henkel Ag & Co. Kgaa | Katalysator-Zusammensetzung zur Härtung von Epoxidgruppen-haltigen Harzen |
| CN111065664A (zh) | 2017-09-01 | 2020-04-24 | 陶氏环球技术有限责任公司 | 热固化组合物 |
-
2020
- 2020-11-16 US US17/775,628 patent/US20220396696A1/en active Pending
- 2020-11-16 EP EP20890479.7A patent/EP4063436A4/en active Pending
- 2020-11-16 JP JP2020564680A patent/JP7647101B2/ja active Active
- 2020-11-16 CN CN202080078489.8A patent/CN114729133B/zh active Active
- 2020-11-16 WO PCT/JP2020/042584 patent/WO2021100649A1/ja not_active Ceased
- 2020-11-16 AU AU2020387250A patent/AU2020387250A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018501391A (ja) | 2014-12-22 | 2018-01-18 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェンHenkel AG & Co. KGaA | 繊維強化材料用オキサゾリジノンおよびイソシアヌレート架橋マトリックス |
| JP2019521216A (ja) | 2016-06-20 | 2019-07-25 | ヘンケル・アクチェンゲゼルシャフト・ウント・コムパニー・コマンディットゲゼルシャフト・アウフ・アクチェンHenkel AG & Co. KGaA | エポキシ樹脂及びポリイソシアネートをベースとする高衝撃強度及び耐高温性を有する硬化組成物 |
Also Published As
| Publication number | Publication date |
|---|---|
| CN114729133B (zh) | 2024-09-20 |
| EP4063436A4 (en) | 2023-12-06 |
| US20220396696A1 (en) | 2022-12-15 |
| JPWO2021100649A1 (ja) | 2021-05-27 |
| EP4063436A1 (en) | 2022-09-28 |
| AU2020387250A1 (en) | 2022-06-02 |
| CN114729133A (zh) | 2022-07-08 |
| WO2021100649A1 (ja) | 2021-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2834308B1 (en) | Benzoxazine resin composition, prepreg, and fiber-reinforced composite material | |
| JP5733301B2 (ja) | 繊維強化複合材料rtm成形用エポキシ樹脂組成物、繊維強化複合材料およびその製造方法 | |
| JP7647101B2 (ja) | 繊維強化複合材料の成形方法、およびそれに用いられるエポキシ樹脂組成物 | |
| EP3345949B1 (en) | Epoxy resin composition, prepreg, and carbon fiber-reinforced composite material | |
| US11319435B2 (en) | Heat-curable resin composition, prepreg, and fiber-reinforced composite material | |
| US11661484B2 (en) | Epoxy resin composition for fiber-reinforced composite materials, and fiber-reinforced composite material | |
| JP7694381B2 (ja) | 成形材料および繊維強化複合材料 | |
| WO2020123640A1 (en) | Polyisocyanurate based polymers and fiber reinforced composites | |
| CN112955495A (zh) | 热固性成型材料、纤维增强复合材料、纤维增强塑料用热固性环氧树脂组合物、热固性成型材料的制造方法、纤维增强塑料 | |
| US20230406994A1 (en) | Thermosetting epoxy resin composition, molded article of same, fiber-reinforced composite material, molding material for fiber-reinforced composite materials, and method for producing fiber-reinforced composite material | |
| JP2018053065A (ja) | 繊維強化複合材料用エポキシ樹脂組成物、および繊維強化複合材料 | |
| JP2018135496A (ja) | 繊維強化複合材料用2液型エポキシ樹脂組成物および繊維強化複合材料 | |
| JP2016138205A (ja) | プリプレグ及び繊維強化複合材料 | |
| JP2016525605A (ja) | イソシアネートエポキシフォーム系 | |
| WO2020217894A1 (ja) | エポキシ樹脂組成物、中間基材および繊維強化複合材料 | |
| EP3763765A1 (en) | Resin composition for fiber-reinforced composite materials, and fiber-reinforced composite material using same | |
| CN112218904A (zh) | 环氧树脂组合物、预浸体及纤维强化复合材料 | |
| JP2020158716A (ja) | 硬化性樹脂組成物、及びそれを用いたトゥプリプレグ | |
| WO2023219007A1 (ja) | 熱硬化性樹脂組成物、成形品、繊維強化複合材料用成形材料および繊維強化複合材料 | |
| JP7160219B1 (ja) | 熱硬化性エポキシ樹脂組成物とその成形品、繊維強化複合材料、繊維強化複合材料用成形材料、および繊維強化複合材料の製造方法 | |
| JP7424990B2 (ja) | 繊維強化複合材料用樹脂組成物及びそれを用いた繊維強化複合材料 | |
| JPH05170862A (ja) | 多液型熱硬化性樹脂組成物及び硬化樹脂成形物の製造方法 | |
| JP2025054937A (ja) | 熱硬化性樹脂組成物、繊維強化複合材料用成形材料および繊維強化複合材料 | |
| JP2024064038A (ja) | 硬化性エポキシ樹脂組成物、及びそれを用いた繊維強化複合材料 | |
| WO2024024677A1 (ja) | プリプレグ、繊維強化複合材料、および繊維強化複合材料の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20231110 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20231110 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250204 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250217 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7647101 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |