JP7655728B2 - 化学変性ミクロフィブリルセルロース繊維の製造方法 - Google Patents
化学変性ミクロフィブリルセルロース繊維の製造方法 Download PDFInfo
- Publication number
- JP7655728B2 JP7655728B2 JP2021006910A JP2021006910A JP7655728B2 JP 7655728 B2 JP7655728 B2 JP 7655728B2 JP 2021006910 A JP2021006910 A JP 2021006910A JP 2021006910 A JP2021006910 A JP 2021006910A JP 7655728 B2 JP7655728 B2 JP 7655728B2
- Authority
- JP
- Japan
- Prior art keywords
- chemically modified
- pulp
- raw material
- beating
- cellulose fibers
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 229920003043 Cellulose fiber Polymers 0.000 title claims description 45
- 238000004519 manufacturing process Methods 0.000 title claims description 38
- 210000001724 microfibril Anatomy 0.000 title claims description 33
- 238000010009 beating Methods 0.000 claims description 71
- 239000000835 fiber Substances 0.000 claims description 57
- 239000002994 raw material Substances 0.000 claims description 52
- 238000000034 method Methods 0.000 claims description 49
- 239000007787 solid Substances 0.000 claims description 27
- 238000011282 treatment Methods 0.000 claims description 25
- 238000007385 chemical modification Methods 0.000 claims description 20
- 239000000203 mixture Substances 0.000 claims description 14
- 150000001875 compounds Chemical class 0.000 claims description 9
- 238000007254 oxidation reaction Methods 0.000 claims description 8
- 125000002057 carboxymethyl group Chemical group [H]OC(=O)C([H])([H])[*] 0.000 claims description 6
- 230000003647 oxidation Effects 0.000 claims description 5
- 150000003842 bromide salts Chemical class 0.000 claims description 3
- 150000004694 iodide salts Chemical class 0.000 claims description 3
- 230000004048 modification Effects 0.000 claims description 3
- 238000012986 modification Methods 0.000 claims description 3
- 239000007800 oxidant agent Substances 0.000 claims description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 42
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 21
- 229920002678 cellulose Polymers 0.000 description 19
- 239000001913 cellulose Substances 0.000 description 19
- 238000012545 processing Methods 0.000 description 19
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 16
- 239000006185 dispersion Substances 0.000 description 13
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 12
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 12
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 12
- 238000006266 etherification reaction Methods 0.000 description 10
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 9
- 239000001768 carboxy methyl cellulose Substances 0.000 description 9
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 9
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- 239000002121 nanofiber Substances 0.000 description 8
- 239000000123 paper Substances 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 229920001131 Pulp (paper) Polymers 0.000 description 6
- 239000002002 slurry Substances 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 239000006228 supernatant Substances 0.000 description 6
- 238000009826 distribution Methods 0.000 description 5
- 239000002655 kraft paper Substances 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 229920002201 Oxidized cellulose Polymers 0.000 description 4
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 4
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- 239000003513 alkali Substances 0.000 description 4
- 210000000988 bone and bone Anatomy 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 239000011121 hardwood Substances 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- 238000000691 measurement method Methods 0.000 description 4
- 229910052751 metal Inorganic materials 0.000 description 4
- 239000002184 metal Substances 0.000 description 4
- 229940107304 oxidized cellulose Drugs 0.000 description 4
- 238000011084 recovery Methods 0.000 description 4
- 239000011122 softwood Substances 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical group OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 3
- 239000005708 Sodium hypochlorite Substances 0.000 description 3
- 230000000996 additive effect Effects 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 125000000129 anionic group Chemical group 0.000 description 3
- 206010061592 cardiac fibrillation Diseases 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- 230000002600 fibrillogenic effect Effects 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- -1 recycled pulp Substances 0.000 description 3
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 3
- 239000008399 tap water Substances 0.000 description 3
- 235000020679 tap water Nutrition 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical group OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 238000010306 acid treatment Methods 0.000 description 2
- 239000000853 adhesive Substances 0.000 description 2
- 230000001070 adhesive effect Effects 0.000 description 2
- 125000002947 alkylene group Chemical group 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000007942 carboxylates Chemical group 0.000 description 2
- 238000005119 centrifugation Methods 0.000 description 2
- 239000000084 colloidal system Substances 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 238000010924 continuous production Methods 0.000 description 2
- 238000007865 diluting Methods 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 239000002612 dispersion medium Substances 0.000 description 2
- 230000005611 electricity Effects 0.000 description 2
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 239000003973 paint Substances 0.000 description 2
- KJFMBFZCATUALV-UHFFFAOYSA-N phenolphthalein Chemical compound C1=CC(O)=CC=C1C1(C=2C=CC(O)=CC=2)C2=CC=CC=C2C(=O)O1 KJFMBFZCATUALV-UHFFFAOYSA-N 0.000 description 2
- 150000003839 salts Chemical class 0.000 description 2
- JHJLBTNAGRQEKS-UHFFFAOYSA-M sodium bromide Chemical compound [Na+].[Br-] JHJLBTNAGRQEKS-UHFFFAOYSA-M 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 235000011149 sulphuric acid Nutrition 0.000 description 2
- UZFMOKQJFYMBGY-UHFFFAOYSA-N 4-hydroxy-TEMPO Chemical compound CC1(C)CC(O)CC(C)(C)N1[O] UZFMOKQJFYMBGY-UHFFFAOYSA-N 0.000 description 1
- 235000017166 Bambusa arundinacea Nutrition 0.000 description 1
- 235000017491 Bambusa tulda Nutrition 0.000 description 1
- 240000006248 Broussonetia kazinoki Species 0.000 description 1
- 244000025254 Cannabis sativa Species 0.000 description 1
- 235000012766 Cannabis sativa ssp. sativa var. sativa Nutrition 0.000 description 1
- 235000012765 Cannabis sativa ssp. sativa var. spontanea Nutrition 0.000 description 1
- 240000000491 Corchorus aestuans Species 0.000 description 1
- 235000011777 Corchorus aestuans Nutrition 0.000 description 1
- 235000010862 Corchorus capsularis Nutrition 0.000 description 1
- 241001265525 Edgeworthia chrysantha Species 0.000 description 1
- 240000000797 Hibiscus cannabinus Species 0.000 description 1
- 241000243251 Hydra Species 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 244000082204 Phyllostachys viridis Species 0.000 description 1
- 235000015334 Phyllostachys viridis Nutrition 0.000 description 1
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000003905 agrochemical Substances 0.000 description 1
- 125000003172 aldehyde group Chemical group 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 239000011425 bamboo Substances 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical group OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 235000013361 beverage Nutrition 0.000 description 1
- 235000009120 camo Nutrition 0.000 description 1
- 235000005607 chanvre indien Nutrition 0.000 description 1
- 239000012459 cleaning agent Substances 0.000 description 1
- 238000013329 compounding Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000002537 cosmetic Substances 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 238000007278 cyanoethylation reaction Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000007599 discharging Methods 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 239000012776 electronic material Substances 0.000 description 1
- 230000006203 ethylation Effects 0.000 description 1
- 238000006200 ethylation reaction Methods 0.000 description 1
- 239000003063 flame retardant Substances 0.000 description 1
- 238000007667 floating Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013373 food additive Nutrition 0.000 description 1
- 239000002778 food additive Substances 0.000 description 1
- 239000003205 fragrance Substances 0.000 description 1
- 239000003349 gelling agent Substances 0.000 description 1
- 125000002791 glucosyl group Chemical group C1([C@H](O)[C@@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 1
- 239000011487 hemp Substances 0.000 description 1
- 235000008216 herbs Nutrition 0.000 description 1
- 239000011346 highly viscous material Substances 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 1
- 238000004898 kneading Methods 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- KJYQVRBDBPBZTD-UHFFFAOYSA-N methanol;nitric acid Chemical compound OC.O[N+]([O-])=O KJYQVRBDBPBZTD-UHFFFAOYSA-N 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 239000010893 paper waste Substances 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- ABLZXFCXXLZCGV-UHFFFAOYSA-N phosphonic acid group Chemical group P(O)(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 1
- OJMIONKXNSYLSR-UHFFFAOYSA-N phosphorous acid Chemical group OP(O)O OJMIONKXNSYLSR-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920000137 polyphosphoric acid Polymers 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 239000013055 pulp slurry Substances 0.000 description 1
- 239000011541 reaction mixture Substances 0.000 description 1
- 238000007670 refining Methods 0.000 description 1
- 239000013049 sediment Substances 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 125000001424 substituent group Chemical group 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- YSGSDAIMSCVPHG-UHFFFAOYSA-N valyl-methionine Chemical compound CSCCC(C(O)=O)NC(=O)C(N)C(C)C YSGSDAIMSCVPHG-UHFFFAOYSA-N 0.000 description 1
Images

Landscapes
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Paper (AREA)
Description
(1) BET比表面積が50m2/g以上、平均繊維幅が500nm以上である化学変性ミクロフィブリルセルロース繊維の製造方法であって、原料パルプを化学変性して化学変性パルプを得る化学変性工程と、前記化学変性工程で得た化学変性パルプを固形分濃度15重量%以下の条件で、2個のディスクとしてDAおよびDBと、その間に存在するディスクとしてDMとを備え、前記DAおよび前記DB又は前記DMの何れか一方が固定され、他方が回転するダブルディスクリファイナーを用いて叩解処理する叩解処理工程と、を含む化学変性ミクロフィブリルセルロース繊維の製造方法。
(2) 前記叩解処理工程におけるパス数が、30回以下である(1)に記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
(3) 前記化学変性が、N-オキシル化合物と、臭化物、ヨウ化物およびこれらの混合物からなる群から選択される化合物と、酸化剤を用いて実施する酸化である、(1)又は(2)記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
(4) 前記化学変性が、カルボキシメチル変性である、(1)又は(2)記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
(5) 前記ダブルディスクリファイナーは、原料の流れ方式がモノフロー式であり、前記DAおよび前記DBの、原料流入側から数えて第1のディスクの刃幅X1が、第2のディスクの刃幅X2よりも大きいことを特徴とする、(1)~(4)の何れかに記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
(6) 前記ダブルディスクリファイナーは、原料の流れ方式がモノフロー式であり、前記DAおよび前記DBの、原料流入側から数えて第1のディスクの刃幅X1が、第2のディスクの刃幅X2よりも小さいことを特徴とする、(1)~(4)の何れかに記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
(7) 前記ダブルディスクリファイナーは、原料の流れ方式がモノフロー式であり、前記DAおよび前記DBの、原料流入側から数えて第1のディスクの刃幅X1が、第2のディスクの刃幅X2と同一であることを特徴とする、(1)~(4)の何れかに記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
ミクロフィブリルセルロース繊維(以下「MFC」ともいう)とは、パルプ等のセルロース系原料を解繊して得られる500nm以上の平均繊維幅を有する繊維であり、化学変性ミクロフィブリルセルロース繊維(以下「化学変性MFC」ともいう)とは、化学変性セルロース系原料を解繊して得られるMFCである。本発明において平均繊維幅とは長さ加重平均繊維幅であり、当該繊維幅はABB株式会社製ファイバーテスターやバルメット社製フラクショネータで測定できる。当該繊維径の下限は好ましくは500nm以上であり、上限は特に限定されないが60μm以下程度である。MFCは、セルロース系原料をビーターやディスパーザーなどで比較的弱く解繊または叩解処理して得られる。したがってMFCは、高圧ホモジナイザーなどでセルロース系原料を強く解繊処理して得られるセルロースナノファイバーと比較して繊維幅が大きく、また繊維自体の微細化を抑制しながら効率的に繊維表面を毛羽立たせた(外部フィブリル化した)形状を有する。
本発明の製造方法により得られる化学変性ミクロフィブリルセルロース繊維は、BET比表面積が50m2/g以上であり、好ましくは70m2/g以上である。BET比表面積が高いと、例えば製紙用添加剤として用いた場合にパルプに結合しやすくなり、歩留まりが向上する、紙への強度付与の効果が高まるなどの利点がある。BET比表面積は、窒素ガス吸着法(JISZ8830)を参考に以下の方法により測定できる:
(1)化学変性ミクロフィブリルセルロース繊維の約2%スラリー(分散媒:水)を、固形分が約0.1gとなるように取り分け遠心分離の容器に入れ、100mLのエタノールを加える。
(2)攪拌子を入れ、500rpmで30分以上攪拌する。
(3)撹拌子を取り出し、遠心分離機で、7000G、30分、30℃の条件で化学変性ミクロフィブリルセルロース繊維を沈降させる。
(4)化学変性ミクロフィブリルセルロース繊維をできるだけ除去しないようにしながら、上澄みを除去する。
(5)100mLエタノールを加え、撹拌子を加え、(2)の条件で攪拌、(3)の条件で遠心分離、(4)の条件で上澄み除去をし、これを3回繰り返す。
(6)(5)の溶媒をエタノールからt-ブタノールに変え、t-ブタノールの融点以上の室温下で、(5)と同様にして撹拌、遠心分離、上澄み除去を3回繰り返す。
(7)最後の溶媒除去後、t-ブタノールを30mL加え、軽く混ぜた後ナスフラスコに移し、氷浴を用いて凍結させる。
(8)冷凍庫で30分以上冷却する。
(9)凍結乾燥機に取り付け、3日間凍結乾燥する。
(10)BET測定装置(Micromeritics(マイクロメリティックス)社製)を用いてBET測定を行う(前処理条件:窒素気流下105℃2時間、相対圧0.01~0.30、サンプル量30mg程度)。
本発明の製造方法により得られる化学変性ミクロフィブリルセルロース繊維は、1重量%、60rpm、25℃の条件におけるB型粘度が、解繊の進み度合の観点から、好ましくは10~6000mPa・s、より好ましくは20~5000mPa・s、さらに好ましくは50~4000mPa・sである。
化学変性工程では、原料パルプを化学変性して化学変性パルプを得る。
原料パルプとしては、針葉樹未漂白クラフトパルプ(NUKP)、針葉樹漂白クラフトパルプ(NBKP)、広葉樹未漂白クラフトパルプ(LUKP)、広葉樹漂白クラフトパルプ(LBKP)、針葉樹未漂白サルファイトパルプ(NUSP)、針葉樹漂白サルファイトパルプ(NBSP)、広葉樹未漂白サルファイトパルプ(LUSP)、広葉樹漂白サルファイトパルプ(LBSP)、サーモメカニカルパルプ(TMP)、加圧砕木パルプ(PGW)、リファイナーグラウンドウッドパルプ(RGP)、アルカリ過酸化水素メカニカルパルプ(APMP)、アルカリ過酸化水素サーモメカニカルパルプ(APTMP)、リンター、ジュート、麻、コウゾ、ミツマタ、ケナフ等の草本由来のパルプ、竹由来のパルプ、再生パルプ、古紙パルプ等が挙げられるが、これらに限定されない。
化学変性とはパルプに官能基を導入することであり、化学変性はアニオン変性であることが好ましい、すなわち化学変性パルプはアニオン性基を有することが好ましい。アニオン性基としてはカルボキシル基、カルボキシル基含有基、リン酸基、リン酸基含有基、硫酸エステル基等の酸基が挙げられる。カルボキシル基含有基としては、-COOH基、-R-COOH(Rは炭素数が1~3のアルキレン基)、-O-R-COOH(Rは炭素数が1~3のアルキレン基)が挙げられる。リン酸基含有基としては、ポリリン酸基、亜リン酸基、ホスホン酸基、ポリホスホン酸基等が挙げられる。これらの酸基は反応条件によっては、塩の形態(例えばカルボキシレート基(-COOM、Mは金属原子))で導入されることもある。本発明において化学変性は酸化またはエーテル化が好ましい。
カルボキシル基量〔mmol/g酸化セルロース〕=a〔mL〕×0.05/酸化セルロース重量〔g〕
A=[(100×F’-(0.1NのH2SO4)(mL)×F)×0.1]/(水素型カルボキシメチル化セルロースの絶乾重量(g))
DS=0.162×A/(1-0.058×A)
A:水素型カルボキシメチル化セルロースの1gの中和に要する1NのNaOH量(mL)
F:0.1NのH2SO4のファクター
F’:0.1NのNaOHのファクター
本工程では、固形分濃度を15重量%以下に調整した化学変性パルプに対して、ダブルディスクリファイナーを用いて叩解処理を行う。化学変性パルプに対して叩解処理を行うと、繊維長、繊維幅が小さくなる微細化、および繊維の毛羽立ちが多くなるフィブリル化が進行する。本発明の叩解処理工程においては、ダブルディスクリファイナーを用いた叩解処理を循環処理としてもよいし、複数台のダブルディスクリファイナーを用いて叩解処理を連続して行う連続処理としてもよい。本発明において、ダブルディスクリファイナーを用いた叩解処理を循環処理または連続処理とした場合における叩解処理のパス数は、所望の化学変性ミクロフィブリルセルロース繊維が得られる限り特に制限はないが、生産性の観点、および繊維の過剰な短小化や処理時に発生する熱による劣化を抑える観点から、30回以下が好ましく、20回以下がより好ましく、10回以下がさらに好ましく、5回以下がさらに好ましい。
n=1/(1-a)
ここで、a=循環割合である。(循環率50%の場合、a=0.5であり、循環率75%の場合、a=0.75である)
ディスクリファイナーとは、叩解刃のついた円盤(ディスクプレート(単に「ディスク」ということがある。))が至近距離で向い合い、一方のみまたは相互に逆方向に所定の回転数で回転して、その間を通過するスラリーに対して加圧叩解の効果と遠心力による連続送り出し効果とを与える装置をいう。ディスクリファイナーのうち、ディスクプレートによって形成される叩解間隙の数が一つのものを、シングルディスクリファイナー(「SDR」と略記することがある。)といい、ディスクプレートによって形成される叩解間隙の数が二つのものを、ダブルディスクリファイナー(「DDR」と略記することがある。)という。
ダブルディスクリファイナーは、2個のディスクDAおよびDBと、その間にディスクDMを備え、DAおよびDBまたはDMの何れか一方が固定され、他方が回転する構成、もしくは、DAおよびDBとDMとが逆方向に回転する構成をとる。ダブルディスクリファイナーとして、2個のディスクDA、DBが固定ディスクであり、DMがその間で自由に回転するフローティングディスクである構成をとるものとしては、例えば、相川鉄工株式会社製のダブルディスクリファイナー、三菱重工業/ベロイト(ジョーンズ)製のダブルディスクリファイナー、石川島産業機械/ブラック・クローソン製のツインハイドラディスク、日立造船(日立造船富岡機械)/エッシャーウイス製のツインディスクリファイナー等が挙げられる。
X1=X2:得られるMFCの繊維幅、繊維長がそろいやすい。
X1>X2:原料が、広い刃上で叩解されフィブリル化された後に狭い刃で叩解されてカッティングが進むため、原料が未解繊のまま通過しにくくなる。
X1<X2:原料が幅の狭い刃で叩解されてカッティングが進んだ後に、広い刃上で叩解されてフィブリル化が進むため、フィブリル化が進んだ微細な繊維分が得られやすい。
Y1=Y2:得られるMFCの繊維幅、繊維長がそろいやすい。
Y1>Y2:原料が、広い叩解間隙を通過した後に狭い叩解間隙を通過するため、余剰な負荷がかかりにくく、粘度が高いものを処理するのに適する。
Y1<Y2:最初に狭い叩解間隙を通過する際に解繊が進み、粘度が上がりやすいものの、次に広い叩解間隙を通過することになり、原料が滞ることなく通過できる。
<化学変性パルプの調製1>
針葉樹由来の漂白済み未叩解クラフトパルプ(白色度85%:日本製紙株式会社製)40kg(絶乾)をTEMPO(Sigma Aldrich社製)312g(絶乾1gのセルロースに対し0.05mmol)と臭化ナトリウム4112g(絶乾1gのセルロースに対し1.0mmol)を溶解した水溶液4000Lに加え、パルプが均一に分散するまで撹拌した。反応系に次亜塩素酸ナトリウム水溶液を次亜塩素酸ナトリウムが5.5mmol/gになるように添加し、室温にて酸化反応を開始した。反応中は系内のpHが低下するが、3M水酸化ナトリウム水溶液を逐次添加し、pH10に調整した。次亜塩素酸ナトリウムを消費し、系内のpHが変化しなくなった時点で反応を終了した。反応混合物に塩酸を添加してpH2に調整した後、脱水と水での希釈を繰り返してパルプを十分に水洗し、最終的にパルプ固形分濃度が20重量%となるまで脱水して化学変性パルプ(TEMPO酸化パルプ)を得た。パルプ収率は90%であり、カルボキシル基量は1.41mmol/gであった。
得られたTEMPO酸化パルプの水分散液3000kgをモノフロー式のダブルディスクリファイナー(相川鉄工株式会社製 AWN20、プレート:刃幅(X1):0.8mm、溝幅(Y1):1.3mm、刃幅(X2):0.6mm、溝幅(Y2):1.0mm)を用い、クリアランス:0.4mm以下、循環率75.5%の条件で45分間運転を行い、叩解処理して、TEMPO酸化パルプをMFCとした。なお、叩解処理のパス数は8.1回であった。次いで、後述する方法によって、当該MFCを評価した。結果を表1に示す。
実施例1で得られた固形分濃度20重量%のTEMPO酸化パルプを水道水に分散させ、水酸化ナトリウムを加えて攪拌することにより、pH7.6、固形分濃度1.1重量%のTEMPO酸化パルプの水分散液を得た。
このTEMPO酸化パルプの水分散液3000kgを、プレートを刃幅(X1):0.6mm、溝幅(Y1):1.0mm、刃幅(X2):0.6mm、溝幅(Y2):1.0mmのものに変更したこと以外は、実施例1と同様に叩解処理することによりMFCを得た。次いで、後述する方法によって、当該MFCを評価した。結果を表1に示す。
実施例1で得られた固形分濃度20重量%のTEMPO酸化パルプを水道水に分散させ、水酸化ナトリウムを加えて攪拌することにより、pH7.6、固形分濃度1.1重量%のTEMPO酸化パルプの水分散液を得た。
このTEMPO酸化パルプの水分散液3000kgを、プレートを刃幅(X1):0.6mm、溝幅(Y1):1.0mm、刃幅(X2):0.8mm、溝幅(Y2):1.3mmのものに変更したこと以外は、実施例1と同様に叩解処理することによりMFCを得た。次いで、後述する方法によって、当該MFCを評価した。結果を表1に示す。
実施例1で得られた固形分濃度20重量%のTEMPO酸化パルプをイオン交換水に分散させ、水酸化ナトリウムを加えて攪拌することにより、pH8.8、固形分濃度2重量%のTEMPO酸化パルプの水分散液を得た。
このTEMPO酸化パルプの水分散液74kgを、シングルディスクリファイナー(相川鉄工株式会社製 14インチラボリファイナー(RF-14型)、プレート:刃幅:0.6mm、溝幅:1.0mm)を用い、クリアランス0.23~0.25mmの条件で10分間循環運転を行い、叩解処理して、TEMPO酸化MFCとした。次いで、後述する方法によって、当該MFCを評価した。結果を表1に示す。
実施例1で得られた固形分濃度20重量%のTEMPO酸化パルプをイオン交換水に分散させ、水酸化ナトリウムを加えて攪拌することにより、pH8.6、固形分濃度4重量%のTEMPO酸化パルプの水分散液を得た。
このTEMPO酸化パルプの水分散液58kgを、シングルディスクリファイナー(相川鉄工株式会社製 14インチラボリファイナー(RF-14型)、プレート:刃幅:0.8mm、溝幅:1.5mm)を用い、クリアランス0.23~0.25mmの条件で10分間循環運転を行い、叩解処理して、TEMPO酸化MFCとした。次いで、後述する方法によって、当該MFCを評価した。結果を表1に示す。

<平均繊維長、平均繊維幅(測定方法A)>
化学変性ミクロフィブリルセルロース繊維のスラリーを固形分濃度が0.25%となるように水で希釈し、流速5.7L/min、水温25±1℃、全流出量22Lの条件で約250gずつ(うち50gが測定に供される)2回フラクショネータにかけ、フラクショネータに付属のCCDカメラで装置内部にて、流量で分級された化学変性ミクロフィブリルセルロース繊維の画像およそ2000枚を取得した。
解析ソフトIMG(Metso社)の繊維解析パラメーターを表2~4のように設定し、取得したおよそ2000枚の画像を解析し、平均繊維長・平均繊維幅、繊維長分布等のデータを得た。2回測定・解析を行った平均値を測定データとして採用した。

・平均繊維長(Length):長さ加重平均繊維長
・平均繊維幅(Width):長さ加重平均繊維幅
・繊維長分布(Fraction percentage of length weighted distribution):長さ加重繊維長分布(各フラクションの設定は表2に記載した通り。)
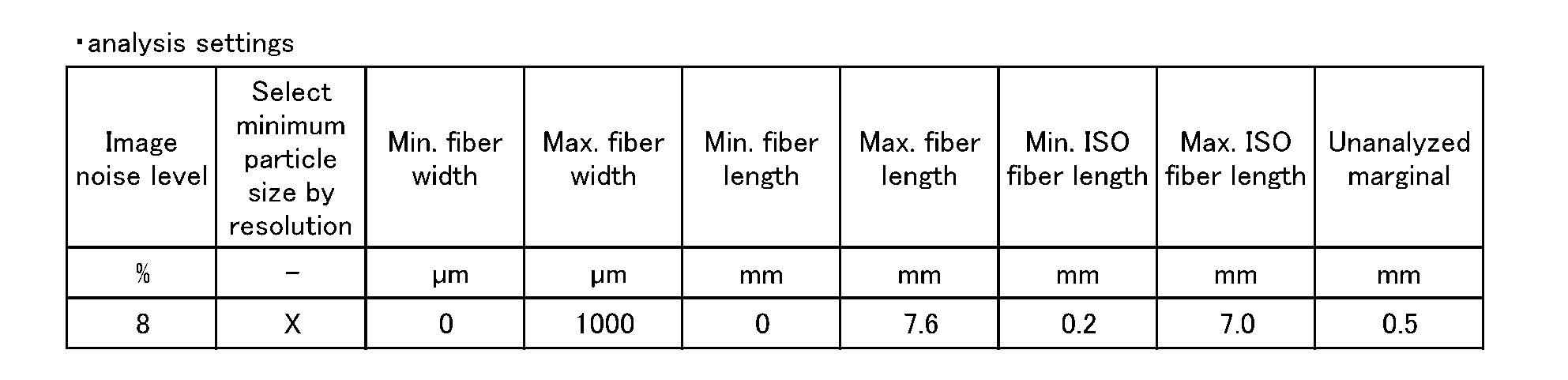

解析ソフトIMG(Metso社)の繊維解析パラメーターを表5~7の通り設定した以外は、測定条件Aと同様にして実施した。



処理後の分散液にイオン交換水を加えて1重量%スラリーを調製し、25℃で3時間放置した後、PRIMIX社製ホモディスパー(3000rpm)で5分間攪拌し、攪拌直後にB型粘度計(東機産業社製)を用いて、No.1~4のうち適切なローターを使用して回転数60rpmで1分後の粘度を測定した。
BET比表面積は、窒素ガス吸着法(JIS Z 8830)を参考に以下の方法により測定した:
(1)処理後の分散液に、必要に応じてイオン交換水を加えて約2%スラリー(分散媒:水)を調製し、これを固形分が約0.1gとなるように取り分け遠心分離の容器に入れ、100mLのエタノールを加えた。
(2)攪拌子を入れ、500rpmで30分以上攪拌した。
(3)撹拌子を取り出し、遠心分離機で、7000G、30分、30℃の条件でフィブリル化された化学変性セルロース繊維を沈降させた。
(4)フィブリル化された化学変性セルロース繊維をできるだけ除去しないようにしながら、上澄みを除去した。
(5)100mLエタノールを加え、撹拌子を加え、(2)の条件で攪拌、(3)の条件で遠心分離、(4)の条件で上澄み除去をし、これを3回繰り返した。
(6)(5)の溶媒をエタノールからt-ブタノールに変え、t-ブタノールの融点以上の室温下で、(5)と同様にして撹拌、遠心分離、上澄み除去を3回繰り返した。
(7)最後の溶媒除去後、t-ブタノールを30mL加え、軽く混ぜた後ナスフラスコに移し、氷浴を用いて凍結させた。
(8)冷凍庫で30分以上冷却した。
(9)凍結乾燥機に取り付け、3日間凍結乾燥した。
(10)BET測定装置(Micromeritics(マイクロメリティックス)社製)を用いてBET測定を行った(前処理条件:窒素気流下105℃2時間、相対圧0.01~0.30、サンプル量30mg程度)。
生産性は、下記式の通り求めた。
生産性(BDkg/h)= 処理量(有姿kg)×処理時濃度(%)×0.01÷処理時間(h)
電力原単位は、下記式の通り求めた。
電力原単位(kwh/BDT)=処理時負荷(kw)/生産性(BDkg/h)×1000
Claims (6)
- BET比表面積が50m2/g以上、平均繊維幅が500nm以上である化学変性ミクロフィブリルセルロース繊維の製造方法であって、
原料パルプを化学変性して化学変性パルプを得る化学変性工程と、
前記化学変性工程で得た化学変性パルプを固形分濃度0.3~10重量%の条件で、2個のディスクとしてDAおよびDBと、その間に存在するディスクとしてDMとを備え、前記DAおよび前記DB又は前記DMの何れか一方が固定され、他方が回転するダブルディスクリファイナーを用いて叩解処理する叩解処理工程と、を含み、
前記叩解処理工程におけるパス数が10回未満である
化学変性ミクロフィブリルセルロース繊維の製造方法。 - 前記化学変性が、N-オキシル化合物と、臭化物、ヨウ化物およびこれらの混合物からなる群から選択される化合物と、酸化剤を用いて実施する酸化である、請求項1記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
- 前記化学変性が、カルボキシメチル変性である、請求項1記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
- 前記ダブルディスクリファイナーは、原料の流れ方式がモノフロー式であり、前記DAおよび前記DBの、原料流入側から数えて第1のディスクの刃幅X1が、第2のディスクの刃幅X2よりも大きいことを特徴とする、請求項1~3の何れか一項に記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
- 前記ダブルディスクリファイナーは、原料の流れ方式がモノフロー式であり、前記DAおよび前記DBの、原料流入側から数えて第1のディスクの刃幅X1が、第2のディスクの刃幅X2よりも小さいことを特徴とする、請求項1~3の何れか一項に記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
- 前記ダブルディスクリファイナーは、原料の流れ方式がモノフロー式であり、前記DAおよび前記DBの、原料流入側から数えて第1のディスクの刃幅X1が、第2のディスクの刃幅X2と同一であることを特徴とする、請求項1~3の何れか一項に記載の化学変性ミクロフィブリルセルロース繊維の製造方法。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021006910A JP7655728B2 (ja) | 2021-01-20 | 2021-01-20 | 化学変性ミクロフィブリルセルロース繊維の製造方法 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021006910A JP7655728B2 (ja) | 2021-01-20 | 2021-01-20 | 化学変性ミクロフィブリルセルロース繊維の製造方法 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2022111469A JP2022111469A (ja) | 2022-08-01 |
| JP7655728B2 true JP7655728B2 (ja) | 2025-04-02 |
Family
ID=82655793
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021006910A Active JP7655728B2 (ja) | 2021-01-20 | 2021-01-20 | 化学変性ミクロフィブリルセルロース繊維の製造方法 |
Country Status (1)
| Country | Link |
|---|---|
| JP (1) | JP7655728B2 (ja) |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004009902A1 (ja) | 2002-07-18 | 2004-01-29 | Japan Absorbent Technology Institute | 超微細セルロース繊維の製造方法および製造装置 |
| WO2019189588A1 (ja) | 2018-03-30 | 2019-10-03 | 日本製紙株式会社 | 酸化ミクロフィブリルセルロース繊維およびその組成物 |
| WO2019221272A1 (ja) | 2018-05-18 | 2019-11-21 | 日本製紙株式会社 | カルボキシメチル化パルプの粉砕物及び該粉砕物を含む添加剤 |
| WO2020059860A1 (ja) | 2018-09-20 | 2020-03-26 | 日本製紙株式会社 | 微細セルロース繊維の製造方法およびそれを含有する紙 |
| WO2020195671A1 (ja) | 2019-03-28 | 2020-10-01 | 日本製紙株式会社 | フィブリル化された化学変性セルロース繊維 |
| JP2020532661A (ja) | 2017-09-01 | 2020-11-12 | ストラ エンソ オーワイジェイ | 板紙の製造方法、板紙、および段ボール |
-
2021
- 2021-01-20 JP JP2021006910A patent/JP7655728B2/ja active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004009902A1 (ja) | 2002-07-18 | 2004-01-29 | Japan Absorbent Technology Institute | 超微細セルロース繊維の製造方法および製造装置 |
| JP2020532661A (ja) | 2017-09-01 | 2020-11-12 | ストラ エンソ オーワイジェイ | 板紙の製造方法、板紙、および段ボール |
| WO2019189588A1 (ja) | 2018-03-30 | 2019-10-03 | 日本製紙株式会社 | 酸化ミクロフィブリルセルロース繊維およびその組成物 |
| WO2019221272A1 (ja) | 2018-05-18 | 2019-11-21 | 日本製紙株式会社 | カルボキシメチル化パルプの粉砕物及び該粉砕物を含む添加剤 |
| WO2020059860A1 (ja) | 2018-09-20 | 2020-03-26 | 日本製紙株式会社 | 微細セルロース繊維の製造方法およびそれを含有する紙 |
| WO2020195671A1 (ja) | 2019-03-28 | 2020-10-01 | 日本製紙株式会社 | フィブリル化された化学変性セルロース繊維 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022111469A (ja) | 2022-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7395491B2 (ja) | 微細セルロース繊維の製造方法およびそれを含有する紙 | |
| JP5895834B2 (ja) | 微細セルロース繊維の製造方法 | |
| CN106170593B (zh) | 制备纳米原纤维纤维素和纳米原纤维纤维素产品的方法 | |
| JP7194503B2 (ja) | セルロースナノファイバーの製造方法 | |
| CN103930615A (zh) | 一种用于生产纳米纤丝纤维素的方法 | |
| JP7554770B2 (ja) | 変性セルロースマイクロフィブリルの製造方法 | |
| JP6839511B2 (ja) | セルロースナノファイバーの製造装置及びセルロースナノファイバーの製造方法 | |
| JP5988843B2 (ja) | 複合材料 | |
| JP2014125689A (ja) | 微細セルロース繊維の製造方法 | |
| JP7402154B2 (ja) | セルロースナノファイバー分散液の製造方法 | |
| CN113728138B (zh) | 化学改性微原纤纤维素纤维的制造方法 | |
| JP7655728B2 (ja) | 化学変性ミクロフィブリルセルロース繊維の製造方法 | |
| WO2017057710A1 (ja) | セルロースナノファイバー分散液及びその製造方法 | |
| JP2023013443A (ja) | 化学変性ミクロフィブリルセルロース繊維の製造方法 | |
| JP6619576B2 (ja) | セルロースナノファイバーの製造方法 | |
| JP6670059B2 (ja) | セルロースナノファイバーの製造方法 | |
| JP7769475B2 (ja) | フィブリル化された化学変性セルロース繊維含有クリア塗工層を備える紙 | |
| JP7252975B2 (ja) | 微細繊維状セルロース分散体の製造方法 | |
| JP2024138894A (ja) | ミクロフィブリルセルロースの製造方法及びダブルディスクリファイナー | |
| JP7574553B2 (ja) | 化学変性ミクロフィブリルセルロース繊維の製造方法 | |
| WO2021201114A1 (ja) | 繊維含有クリア塗工層を備える紙 | |
| JP2024081949A (ja) | ミクロフィブリルセルロース | |
| JP2023148586A (ja) | 微細セルロース繊維 | |
| JP7690729B2 (ja) | 化学変性ミクロフィブリルセルロース繊維及びその製造方法 | |
| JP6797320B2 (ja) | セルロースナノファイバーの製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20231221 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20240909 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20241001 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20241121 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20241217 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20250206 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250311 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250321 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7655728 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |