JP7702973B2 - Glp-1rアゴニストの塩及び結晶形態、並びにそれらの使用 - Google Patents
Glp-1rアゴニストの塩及び結晶形態、並びにそれらの使用 Download PDFInfo
- Publication number
- JP7702973B2 JP7702973B2 JP2022572686A JP2022572686A JP7702973B2 JP 7702973 B2 JP7702973 B2 JP 7702973B2 JP 2022572686 A JP2022572686 A JP 2022572686A JP 2022572686 A JP2022572686 A JP 2022572686A JP 7702973 B2 JP7702973 B2 JP 7702973B2
- Authority
- JP
- Japan
- Prior art keywords
- salt
- compound
- tris salt
- tris
- single crystalline
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images












Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C55/00—Saturated compounds having more than one carboxyl group bound to acyclic carbon atoms
- C07C55/22—Tricarboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
本出願は、2020年5月27日に出願された国際特許出願第PCT/CN2020/092530号に対する優先権の利益を主張する。前述の出願の全容は、参照により本明細書に組み込まれる。

一実施形態では、1:1の化合物Iトリス塩は、2θにおいて17.5°、20.1°、20.7°、21.1°、及び22.6°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Aである。別の実施形態では、形態Aは、2θにおいて17.5°、20.1°、20.7°、21.1°、及び22.6°±0.2から選択される少なくとも3つのピーク(又は4つのピーク)を含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Aは、2θにおいて4.1°、14.8°、17.5°、18.8°、20.1°、20.7°、21.1°、及び22.6°±0.2にピークを含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Aは、2θにおいて4.1°、8.1°、12.8°、14.8°、16.3°、17.5°、18.8°、19.3°、20.1°、20.7°、21.1°、22.6°、25.1°、及び25.8°±0.2にピークを含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Aは、図1と実質的に同様のX線粉末回折パターンを特徴とする。
一実施形態では、1:1の化合物IIクエン酸塩は、2θにおいて5.4°、9.4°、12.4°、14.3°、及び17.8°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Aである。別の実施形態では、形態Aは、2θにおいて5.4°、9.4°、12.4°、14.3°、及び17.8°±0.2から選択される少なくとも3つのピーク(又は4つのピーク)を含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Aは、2θにおいて5.4°、9.4°、10.8°、12.4°、14.3°、16.2°、17.8°、19.6°、及び24.9°±0.2にピークを含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Aは、2θにおいて5.4°、9.4°、10.8°、12.4°、14.3°、16.2°、17.8°、18.8°、19.6°、23.6°、及び24.9°±0.2にピークを含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Aは、図4と実質的に同様のX線粉末回折パターンを特徴とする。
一実施形態では、1:1の化合物IIトリス塩は、2θにおいて4.1°、14.7°、18.8°、20.1°、及び23.1°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Bである。別の実施形態では、形態Bは、2θにおいて4.1°、14.7°、18.8°、20.1°、及び23.1°±0.2から選択される少なくとも3つのピーク(又は4つのピーク)を含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Bは、2θにおいて4.1°、8.2°、14.7°、16.4°、18.8°、20.1°、20.7°、21.3°、及び23.1°±0.2にピークを含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Bは、2θにおいて4.1°、8.2°、14.7°、16.4°、18.8°、19.1°、20.1°、20.7°、21.3°、23.1°、24.1°、及び25.4°±0.2にピークを含むX線粉末回折パターンを特徴とする。更に別の実施形態では、形態Bは、図6と実質的に同様のX線粉末回折パターンを特徴とする。
別の実施形態では、医薬組成物が本明細書に開示される。そのような医薬組成物は、本明細書に記載の化合物I(又は化合物II)の塩と、薬学的に許容される担体と、を含む。他の薬理学的に活性な物質も存在し得る。
「対象」は、哺乳動物、好ましくはヒトであるが、獣医学的治療を必要とする動物、例えば、コンパニオンアニマル(例えば、イヌ、ネコ等)、家畜(例えば、ウシ、ヒツジ、ブタ、ウマ等)及び実験動物(例えば、ラット、マウス、モルモット等)であってもよい。
典型的には、本開示の化合物は、本明細書に記載されるような症状を治療するのに有効な量で投与される。本開示の化合物は、化合物それ自体として、又は代替的に、薬学的に許容される塩として投与することができる。投与及び投薬目的のために、化合物それ自体又はその薬学的に許容される塩は、単に本開示の化合物と称される。
本開示の化合物は、単独で、又は他の治療剤と組み合わせて使用することができる。本開示は、本明細書中で定義される使用、方法又は組成物のいずれかを提供し、本明細書中の上記の式のいずれか1つの任意の実施形態の化合物、又はその薬学的に許容される塩、あるいは当該化合物又は塩の薬学的に許容される溶媒和物は、本明細書中で考察される1つ以上の他の治療剤と組み合わせて使用される。
本開示の別の態様は、上記の式のいずれか1つの化合物を含むキット、又は本開示の上記の式のいずれか1つの化合物を含む医薬組成物を提供する。キットは、上記の式のいずれか1つの化合物に加えて、本開示又はその医薬組成物の診断薬又は治療薬を含み得る。キットはまた、診断又は治療方法で使用するための説明書を含み得る。いくつかの実施形態では、キットは、上記の式のいずれか1つの化合物、又はその医薬組成物及び診断薬を含む。他の実施形態では、キットは、上記の式のいずれか1つの化合物、又はその医薬組成物を含む。
上記の式のいずれか1つの化合物は、合成有機化学の当業者の共通の一般知識を使用して、以下に記載される一般的かつ特定の方法によって調製することができる。そのような共通の一般知識は、Comprehensive Organic Chemistry,Ed.Barton and Ollis,Elsevier、Comprehensive Organic Transformations:A Guide to Functional Group Preparations,Larock,John Wiley and Sons、及びCompendium of Organic Synthetic Methods,Vol.I-XII(published by Wiley-Interscience)などの標準的な参考文献に見出すことができる。本明細書で使用される出発物質は、市販されているか、又は当技術分野で既知の通常の方法によって調製することができる。
溶媒の略語を以下の表に列記する。

X線粉末回折(XRPD)
XRPD分析については、PANalytical Empyrean/X’ Pert3 X線粉末回折計を使用した。使用するXRPDパラメータを以下の表に列記する。


熱重量分析及び示差走査熱量測定(TGA&DSC)
TGAデータを、TA InstrumentsのTA Q5000/Discovery 5500 TGAを使用して収集した。DSCを、TA InstrumentsのTA Q2000/Discovery 2500 DSCを使用して行った。使用される詳細なパラメーを以下の表に列記する。

1H溶液NMRを、DMSO-d6を使用してBruker 400M NMR分光計で収集した。
DVSを、SMS(表面測定システム)DVS Intrinsicを介して測定した。25℃での相対湿度を、LiCl、Mg(NO3)2、及びKClの潮解点に対して較正した。DVS試験のパラメータを以下の表に列記する。

Agilent 1260 HPLCを使用し、詳細なクロマトグラフィ条件を以下の表に列記する。


ステップ1
1,4-ジオキサン(50mL)中の3-ブロモフェノール(1.0g、5.8mmol)の溶液に、tert-ブチル4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(1.9g、6.4mmol)、並びにCs2CO3(3.8g、11.6mmol)及びPd(dppf)Cl2(416mg、0.58mmol)を添加した。混合物を窒素下、90℃で8時間撹拌した。混合物をセライトで濾過して溶液を得て、水(150mL)で希釈し、酢酸エチル(150mL×3)で抽出し、合わせた有機物をブライン(150mL×3)で洗浄し、乾燥させ、真空中で濃縮して粗生成物を得た。粗生成物を分取TLC(PE:EA=2:1)によって精製すると、tert-ブチル4-(3-ヒドロキシフェニル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(151g、収率94%)が白色の固体として得られた。LCMS:[M+H]+=221、保持時間(10mM NH4HCO3)=1.81分。
1,4-ジオキサン(10mL)中のtert-ブチル4-(3-ヒドロキシフェニル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(275mg、1.0mmol)の溶液に、1-(ブロモメチル)-4-クロロ-2-フルオロベンゼン(223mg、1.0mmol)及びPd2(dba)3(91.5mg、0.1mmol)及びBINAP(62.2mg、0.1mmol)を添加した。混合物を窒素下、100℃で8時間撹拌した。反応物を室温まで冷却し、反応物を水(150mL)で希釈し、酢酸エチル(150mL×3)で抽出し、合わせた有機物をブライン(150mL×3)で洗浄し、乾燥させ、真空中で濃縮して粗生成物を得た。次いで、粗生成物を分取TLC(PE:EA=3:1)によって精製すると、tert-ブチル4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(241mg、収率57.8%)が黄色油状物として得られた。LCMS:[M+H]+=363、保持時間(10mM NH4HCO3)=2.04分。
DCM(10mL)中のtert-ブチル4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(322mg、0.77mmol)の溶液に、HCl/1,4-ジオキサン(1.2mL)を添加した。混合物を室温で2時間撹拌した。反応物を水(50mL)で希釈し、酢酸エチル(50mL×3)で抽出し、合わせた有機物をブライン(50mL×3)で洗浄し、乾燥させ、真空中で濃縮して粗生成物を得て、これを分取TLC(PE:EA=5:1)によって精製すると、4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-1,2,3,6-テトラヒドロピリジン(198mg)が黄色油状物として得られた。LCMS:[M+H]+=318、保持時間(10mM NH4HCO3)=1.61分。
1,4-ジオキサン(10mL)中の4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-1,2,3,6-テトラヒドロピリジン(90mg、0.28mmol)の溶液に、tert-ブチル(S)-2-(クロロメチル)-1-(オキセタン-2-イルメチル)-1H-ベンゾ[d]イミダゾール-6-カルボキシラート(85mg、0.28mmol)及びDIPEA(0.3mL、1.4mol)を添加した。混合物を90℃で3時間撹拌した。反応物を水(100mL)で希釈し、酢酸エチル(100mL×3)で抽出し、合わせた有機層をブライン(100mL×3)で洗浄し、乾燥させ、真空中で濃縮して粗生成物を得て、これを分取TLCによって精製すると、tert-ブチル(S)-2-((4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-3,6-ジヒドロピリジン-1(2H)-イル)メチル)-1-(オキセタン-2-イルメチル)-1H-ベンゾ[d]イミダゾール-6-カルボキシラート(80mg、収率46%)が黄色の固体として得られた。
DCM(12mL)中のtert-ブチル(S)-2-((4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-3,6-ジヒドロピリジン-1(2H)-イル)メチル)-1-(オキセタン-2-イルメチル)-1H-ベンゾ[d]イミダゾール-6-カルボキシラート(80mg、0.13mmol)の溶液に、TFA(2mL、26.93mmol)を添加した。混合物を室温で3時間撹拌した。反応物を真空中で濃縮して粗生成物を得て、これを分取HPLC(NH4HCO3)によって精製すると、(S)-2-((4-(3-((4-クロロ-2-フルオロベンジル)オキシ)フェニル)-3,6-ジヒドロピリジン-1(2H)-イル)メチル)-1-(オキセタン-2-イルメチル)-1H-ベンゾ[d]イミダゾール-6-カルボン酸(18.5mg、収率25%)が白色固体として得られた。LCMS:[M+H]+=562.0、保持時間(10mM NH4HCO3)=1.40分。

ステップ1
THF(500mL)中のt-BuOK(31.3g、279.7mmol)の撹拌懸濁液に、3-フルオロ-4-(ヒドロキシメチル)ベンゾニトリル(28.1g、186.5mmol)を10~15℃で少しずつ添加した。混合物を15℃で45分間撹拌し、2,6-ジクロロピリジン(23.0g、155.4mmol)を反応混合物に15℃で数回に分けて添加し、混合物を15℃で18時間撹拌した。混合物を水溶液NH4Cl(1000mL)に注いだ。EtOAc(1000mL)を添加し、混合物を15分間撹拌した。混合物をセライトのパッドを通して濾過した。有機層を分離し、水層をEtOAc(2×600mL)で抽出した。合わせた有機層をブライン(500mL)で洗浄し、Na2SO4で乾燥させ、濾過し、減圧下で濃縮した。粗生成物をシリカゲルカラムクロマトグラフィ(PE/EtOAc=10/1)によって精製すると、4-((6-クロロピリジン-2-イルオキシ)メチル)-3-フルオロベンゾニトリル(26.0g、収率64%)が淡黄色固体として得られた。
ジオキサン(20mL)及びH2O(4g、222.22mmol)中の4-[(6-クロロ-2-ピリジル)オキシメチル]-3-フルオロ-ベンゾニトリル(1g、3.81mmol)、tert-ブチル4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロ-2H-ピリジン-1-カルボキシラート(1.29g、4.19mmol)、Pd(dppf)Cl2(278.3mg、0.38mmol)及びNaHCO3(479.69mg、5.71mmol)の混合物を、LCMSによって示されるように反応が完了するまで、N2下、90℃で2時間撹拌した。反応混合物を、EtOAcを用いたセライトパッドで濾過し、合わせた有機物を真空中で濃縮し、シリカゲルクロマトグラフィ(ヘキサン/EtOAc=0~11%)によって精製すると、所望の生成物tert-ブチル4-[6-[(4-シアノ-2-フルオロ-フェニル)メトキシ]-2-ピリジル]-3,6-ジヒドロ-2H-ピリジン-1-カルボキシラート(1.5g、3.63mmol、収率95.4%)が淡黄色液体として得られた。LCMS:[M+H]+=410.1、保持時間(10mM NH4HCO3)=2.22分。
DCM(20mL)中のtert-ブチル4-[6-[(4-シアノ-2-フルオロ-フェニル)メトキシ]-2-ピリジル]-3,6-ジヒドロ-2H-ピリジン-1-カルボキシラート(1.5g、3.66mmol)の溶液に、TFA(7.40g、64.90mmol、5mL)をゆっくり添加した。反応物を28℃で2時間撹拌した。LCMSによって判定した反応の完了後、反応混合物を真空中で濃縮すると、3-フルオロ-4-[[6-(1,2,3,6-テトラヒドロピリジン-4-イル)-2-ピリジル]オキシメチル]ベンゾニトリル(1.8g、3.94mmol)TFA塩が淡黄色がかった液体として得られた。粗生成物を更に精製することなく次のステップで直接使用した。LCMS:[M+H]+=310.1、保持時間(0.01%TFA)=1.42分。
ジオキサン(10mL)中の3-フルオロ-4-[[6-(1,2,3,6-テトラヒドロピリジン-4-イル)-2-ピリジル]オキシメチル]ベンゾニトリル(340mg、0.80mmol)と、tert-ブチル2-(クロロメチル)-3-[[(2S)-オキセタン-2-イル]メチル]ベンズイミダゾール-5-カルボキシラート(225mg、670mmol、本合成は、参照により本明細書に組み込まれる国際出願第2018/109607号に開示されている)と、DIPEA(216.42mg、1.67mmol)との混合物を、LCMSによって示されるように反応が完了するまで、90℃で1時間撹拌し、反応混合物を真空中で濃縮し、シリカゲルクロマトグラフィ(ヘキサン/EtOAc=20:1)によって精製すると、所望の生成物tert-ブチル2-[[4-[6-[(4-シアノ-2-フルオロ-フェニル)メトキシ]-2-ピリジル]-3,6-ジヒドロ-2H-ピリジン-1-イル]メチル]-3-[[(2S)-オキセタン-2-イル]メチル]ベンズイミダゾール-5-カルボキシラート(366mg、0.31mmol)が淡褐色の固体として得られた。LCMS:[M+H]+=610.0、保持時間(10mM NH4H CO3)=1.87分。
DCM(6mL)中のtert-ブチル2-[[4-[6-[(4-シアノ-2-フルオロ-フェニル)メトキシ]-2-ピリジル]-3,6-ジヒドロ-2H-ピリジン-1-イル]メチル]-3-[[(2S)-オキセタン-2-イル]メチル]ベンズイミダゾール-5-カルボキシラート(180mg、0.30mmol)の溶液に、DCM(2mL)中のTFA(2.96g、25.96mmol、2mL)を28℃でゆっくり添加し、1時間撹拌した。LCMSによって判定された反応の完了後、反応混合物を真空中で濃縮し、粗生成物を分取HPLC(10mM NH4HCO3)によって精製すると、(S)-2-((6-((4-シアノ-2-フルオロベンジル)オキシ)-3’,6’-ジヒドロ-[2,4’-ビピリジン]-1’(2’H)-イル)メチル)-1-(オキセタン-2-イルメチル)-1H-ベンゾ[d]イミダゾール-6-カルボン酸(54mg、収率33%)が白色の固体として得られた。LCMS:[M+H]+=554.2、保持時間(10mM NH4HCO3)=1.42分。
トリス塩(形態A)
3.1.1調製方法
500.9mgの化合物Iの遊離形態の溶液を、20mLのアセトン/H2O(9:1、v:v)に溶解した。その間に、等モルのトリス(107.9mg)を2.5mLのH2Oに溶解した。得られたトリス溶液を化合物I溶液の遊離形態に滴下し、室温で撹拌(RT、約1000rpm)した。得られたスラリーをRTで17時間継続的に撹拌し、次いで5℃まで冷却し、更に4時間撹拌した。混合物中の固体を遠心分離によって単離し、RTで20時間真空乾燥させると、445.9mgの所望の生成物が得られた(収率約73.2%)。

1:1の化合物Iトリス塩(形態A)の調製を、スラリー/溶液結晶化を介して数回試みた。調製の結果を表A及び表Bに要約した。詳細な特徴評価の結果を表Cに要約した。



SCXRDによって特徴付けられた1:1の化合物Iトリス塩(形態A)の単結晶サンプルを、ゆっくりとした蒸発実験から得た。実験の詳細を、以下に詳しく説明する。
I)生体関連媒体及びpH緩衝液の調製
模擬胃液(SGF)
100mgの塩化ナトリウム及び50mgのTriton X-100を50mLのメスフラスコに量り入れる。適切な量の精製水を添加し、全ての固体が完全に溶解するまで超音波処理をする。約68μLの12MHCl及び十分な精製水を目標量に近づけて添加し、pH1.8に調整する。精製水で定容し、よく混合し、pHメーターでpHを確認する。
170mgのリン酸ナトリウム一塩基、21mgの水酸化ナトリウム、及び310mgの塩化ナトリウム、110mgのSIF粉末を50mLのメスフラスコに量り入れる。適切な量の精製水を添加し、全ての固体が完全に溶解するまで超音波処理をする。十分な精製水を目標量に近づけて添加し、pH6.5に調整する。精製水で定容し、よく混合し、pHメーターでpHを確認する。
0.41mLの氷酢酸、202mgの水酸化ナトリウム、及び594mgの塩化ナトリウム、560mgのSIF粉末を50mLのメスフラスコに量り入れる。適切な量の精製水を添加して、固体を溶解する。次いで、十分な精製水を目標量に近づけて添加し、pH5.0に調整する。精製水で定容し、よく混合し、pHメーターでpHを確認する。
302.5mgのトリスを50mLのメスフラスコに量り入れる。十分な精製水を目標量に近づけて添加し、pH8.0に調整する。精製水で定容し、よく混合し、pHメーターでpHを確認する。
動的可溶度を、水及び3つの生体関連媒体(SGF、FaSSIF、及びFeSSIF)において1/4/24時間評価した。3~4mgの固体を3mLの各培地に懸濁すると(遊離形態を使用して計算された固体負荷:1mg/mL)、懸濁液が得られ、続いて37℃で1/4/24時間、ローリング(25rpm)した。各時点で、約1.0mLの懸濁液を、遠心分離(12000rpm、2分)及び0.45μmのPTFE膜を通す濾過のためにサンプリングすると、HPLC及びpH試験用の上清が得られ、残留固体をXRPDによって分析した。詳細な結果を表2に要約した。

1:1の化合物Iトリス塩(形態A)及び非晶質遊離形態の物理的及び化学的安定性を評価するために、サンプルを60℃で1日間、及び25℃/60%RH又は40℃/75%RHでそれぞれ1週間の条件下で保存した。XRPD及びHPLCを使用して固体を特徴付け、結果を表3に要約した。1週間後も形態変化は観察されなかった。1:1の化合物Iトリス塩(形態A)は、全ての条件で非晶質遊離形態よりも良好な化学的安定性を示した。

1:1の化合物Iトリス塩(形態A)の化学的安定性を固体状態で更に評価し、サンプルを周囲条件又は40℃/75%RHの下でそれぞれ1日間及び3日間保存した。HPLC純度及び重量純度を、HPLCを使用して3回試験し、結果を表4に要約した。1:1の化合物Iトリス塩(形態A)は、全ての条件でより良好な化学的安定性を示した。

SGF(pH1.8)及びpH8.0の緩衝液中の溶液の安定性を、RTで1:1の化合物Iトリス塩(形態A)について評価した。透明な溶液を0.01mg/mLの濃度で調製し、次いでRTで6時間、1日間、及び5日間置いた。1日後、pH1.8の溶液については著しい分解は観察されなかったが、pH8.0の溶液の純度はわずかに減少した。HPLC純度及びpH値を表5及び表6に要約した。


湿度の関数としての固体形態の安定性を調査するために、1:1の化合物Iトリス塩(形態A)のDVS等温線プロットを、25℃で0%RHと95%RHとの間で収集した。25℃/80%RHで2.77%の吸水率が観察された。1:1の化合物Iトリス塩(形態A)が水和物であり、低湿度下で脱水し得ることを考慮して、DVSを20及び30℃でも試験し、好適な保存条件を更に確認し、脱水を避けた。詳細な結果を表7に要約した。DVS試験後も形態変化は観察されなかった。VH-XRPD及びDVSの結果を組み合わせると、1:1の化合物Iトリス塩(形態A)の保存には、20~30℃及び相対湿度≧40%RHの条件が好適である。

1:1の化合物Iトリス塩(形態A)における多形スクリーニング実験を、抗溶媒添加、逆抗溶媒添加、ゆっくりとした蒸発、ゆっくりとした冷却、RTでのスラリー、50℃でのスラリー、5~50℃でのスラリー循環、蒸気-固体拡散、蒸気-溶液拡散、ポリマー誘起結晶化、及び粉砕を含む異なる溶液結晶化又は固体転移法を使用して、様々な条件下で行った。
4.1.1調製方法
15mgの化合物IIの遊離形態及び5.21mgのアセトン中のクエン酸を混合した。得られたスラリーをRTで3日間撹拌し、続いてRTで約12時間真空乾燥させた。

1:1の化合物IIクエン酸塩(形態A)の調製を、スラリー/溶液結晶化を介して数回試みた。調製結果を表9及び表10に要約した。詳細な特徴評価結果を表11に要約した。



5.1.1調製方法
バッチA:
1)N2雰囲気下で、反応器にアセトン(10L、10.0V)を充填した。化合物IIの遊離形態1.0Kg(1.0当量)をかき混ぜながら反応器に添加した。温度を23℃に調整し、得られた混合物を0.5時間撹拌した後、混合物を0.2μmの微孔性フィルターで濾過した。濾液を収集した。
2)N2雰囲気下で、濾液を反応器に移した。10LのEA(10.0V)を反応器に添加した。温度を21℃に調整した。トリス溶液(2Lの軟水(2.0V)に溶解した218.8gのトリス(1.0当量))を1.5時間以内に反応器に添加した。混合物を1時間撹拌した。
3)次いで、混合物を遠心分離によって単離した。濾塊をEA(2L、2.0V)で洗浄し、フィルター濾塊を収集した。
4)フィルター濾塊を真空下、70℃で16時間乾燥させると、1.03Kgの1:1の化合物IIトリス塩(形態B)がオフホワイトの固体として得られた。
1)N2雰囲気下で、反応器にアセトン(21.5L、10.0V)を充填した。化合物IIの遊離形態2.15Kg(1.0当量)をかき混ぜながら反応器に添加した。温度を25℃に調整し、得られた混合物を0.5時間撹拌した後、混合物を0.2μmの微孔性フィルターで濾過した。濾液を収集した。
2)N2雰囲気下で、濾液を反応器に移した。21.5LのEA(10.0V)を反応器に添加した。温度を21℃に調整した。トリス溶液(4.3Lの軟水(2.0V)に溶解した470.4gのトリス(1.0当量))を3時間以内に反応器に添加した。混合物を1.5時間撹拌した。
3)次いで、混合物を遠心分離によって単離した。濾塊をEA(4.3L、2.0V)で洗浄し、フィルター濾塊を収集した。
4)フィルター濾塊を真空下、70℃で16時間乾燥させると、2.45Kgの1:1の化合物IIトリス塩(形態B)が白色の固体として得られた。
1)バッチA及びバッチBから得られた3.45Kg(1.0eq)の1:1の化合物IIトリス塩(形態B)を3.45Lの軟水(1V)と混合した。混合物を、かき混ぜながらN2雰囲気下で44.9LのEA(13V)を充填した反応器に添加した。温度を25℃に調整し、得られた混合物を1時間撹拌した。
2)次いで、混合物を遠心分離によって単離した。濾塊をEA(6.9L、2.0V)で洗浄し、フィルター濾塊を収集した。
3)フィルター濾塊を真空下、70℃で16時間乾燥させると、3.45Kgの1:1の化合物IIトリス塩(形態B)がオフホワイトの固体として得られた。

THF/MTBE(1:4、v:v)中の化合物IIとトリスの遊離形態との混合物(1:0.95のモル比)を7日間撹拌し、続いてRTで約3時間真空乾燥させると、1:1の化合物IIトリス塩(形態B)が得られた。
6.1調製方法
15mgの1:1の化合物IIトリス塩(形態B)を0.1mLのNMPに溶解して、透明な溶液を形成した。CH3CNを溶液に一滴ずつ添加した。沈殿物が観察された。遠心分離を介して固体を単離し、105℃まで加熱し、次いでRTまで冷却すると、1:1の化合物IIトリス塩(形態G)が得られた。

1:1の化合物IIトリス塩(形態B)及び1:1の化合物IIクエン酸塩(形態A)を、動的可溶度、固体安定性、及び溶液安定性によって評価した。遊離形態化合物IIもまた比較のために評価した。更に、両方の塩の吸湿性をDVSによって評価した。
動的可溶度を、水及び3つの生体関連媒体(SGF、FaSSIF、及びFeSSIF)中の1:1の化合物IIクエン酸塩(形態A)、1:1の化合物IIトリス塩(形態B)、及び化合物IIの遊離形態について、1/4/24時間評価した。3~4mgの固体を3mLの各培地に懸濁すると(遊離形態を使用して計算された固体負荷:1mg/mL)、懸濁液が得られ、続いて37℃で1/4/24時間、ローリング(25rpm)した。各時点で、約1.0mLの懸濁液を、遠心分離(12000rpm、2分)及び0.45μmのPTFE膜を通す濾過のためにサンプリングすると、HPLC及びpH試験用の上清が得られ、残留固体をXRPDによって分析した。詳細な結果を表7-1に要約した。可溶度評価の結果に基づいて、1:1の化合物IIクエン酸塩(形態A)及び1:1の化合物IIトリス塩(形態B)は、SGF、FaSSIF、及びFeSSIFにおいて同様の可溶度プロファイルを示し、これは、1時間以内の遊離形態の可溶度プロファイルよりも高かった。1:1の化合物IIトリス塩(形態B)は、H2Oでより良好な可溶度を示した。サンプルは全て懸濁液であったため、純度の結果は、単に参考値であった。

1:1の化合物IIクエン酸塩(形態A)、1:1の化合物IIトリス塩(形態B)、及び遊離形態の物理的及び化学的安定性を評価するために、サンプルを60℃で1日間、及び25℃/60%RH又は40℃/75%RHでそれぞれ1週間の条件下で保存した。XRPD及びHPLCを使用して固体を特徴付け、結果を表7-2-1に要約した。XRPDの結果及びHPLCクロマトグラムによって証明されるように、1週間後に形態変化は観察されなかった。1:1の化合物IIクエン酸塩(形態A)及び1:1の化合物IIトリス塩(形態B)は、化合物IIの遊離形態よりも良好な化学的安定性を示した。

1:1の化合物IIクエン酸塩(形態A)及び1:1の化合物IIトリス塩(形態B)の化学的安定性を固体状態で更に評価し、サンプルを周囲条件又は40℃/75%RHの下でそれぞれ1日間及び3日間保存した。HPLC純度及び重量純度を、HPLCを使用して3回試験し、結果を表7-2-2に要約した。1:1の化合物IIクエン酸塩(形態A)及び1:1の化合物IIトリス塩(形態B)は、評価条件において同様の化学的安定性を示した。

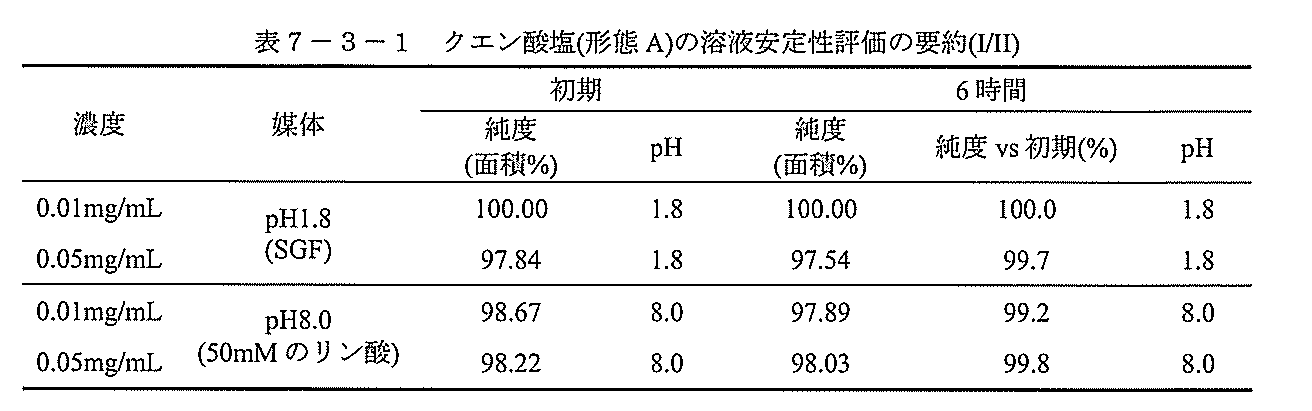
溶液安定性を、SGF(pH1.8)及びpH8.0の緩衝液中の1:1の化合物IIクエン酸塩(形態A)及び1:1の化合物IIトリス塩(形態B)について、RTで評価した。透明な溶液を2つの濃度(0.01mg/mL、0.05mg/mL)で調製し、次いでRTで6時間、1/5日、又は6時間、1/3/7日間置いた。両方の塩とも、pH8.0の緩衝液中でより良好な安定性を示したが、pH1.8の溶液の純度は、1日後にわずかに減少した。HPLC純度及びpH値を試験し、結果を表7-3-1~表7-3-4に要約した。



湿度の関数としての固体形態の安定性を調査するために、1:1の化合物IIクエン酸塩(形態A)及び1:1の化合物IIトリス塩(形態B)のDVS等温線プロットを、25℃、0%RH~95%RHで収集した。結果を表7-4に要約した。1:1の化合物IIクエン酸塩(形態A)については、0.71%の吸水率が25℃/80%RHで観察された。1:1の化合物IIトリス塩(形態B)については、吸水率は、サイクル2収着曲線において0%~10%RHで2.47%増加した。吸水率は、10%~80%RHで0.97%増加し、プラットフォームが観察された。両方の塩について、DVS試験後も形態変化は観察されなかった。

Claims (40)
- 化合物Iのトリス塩であって、化合物Iが、以下の構造式によって表され、
- 前記トリス塩が、結晶性である、請求項1に記載のトリス塩。
- 前記トリス塩が、単結晶性形態にある、請求項1に記載のトリス塩。
- 前記トリス塩が、一水和物である、請求項2又は3に記載のトリス塩。
- 前記トリス塩が、非溶媒和である、請求項2又は3に記載のトリス塩。
- 前記トリス塩が、2θにおいて17.5°、20.1°、20.7°、21.1°、及び22.6°±0.2から選択される少なくとも3つのピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物Iのトリス塩形態Aである、請求項4に記載のトリス塩。
- 前記トリス塩が、2θにおいて17.5°、20.1°、20.7°、21.1°、及び22.6°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物Iのトリス塩形態Aである、請求項4に記載のトリス塩。
- 前記トリス塩が、2θにおいて4.1°、14.8°、17.5°、18.8°、20.1°、20.7°、21.1°、及び22.6°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物Iのトリス塩形態Aである、請求項4に記載のトリス塩。
- 前記トリス塩が、2θにおいて4.1°、8.1°、12.8°、14.8°、16.3°、17.5°、18.8°、19.3°、20.1°、20.7°、21.1°、22.6°、25.1°、及び25.8°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物Iのトリス塩形態Aである、請求項4に記載のトリス塩。
- 前記トリス塩が、173±3℃の示差走査熱量計(DSC)ピーク相転移温度を特徴とする単結晶性形態の化合物Iのトリス塩形態Aである、請求項6~9のいずれか一項に記載のトリス塩。
- 前記トリス塩の少なくとも90重量%が、単結晶性形態の化合物Iのトリス塩形態Aである、請求項6~10のいずれか一項に記載のトリス塩。
- 化合物IIのトリス塩であって、化合物IIが、以下の構造式によって表され、
- 前記トリス塩が、結晶性である、請求項12に記載のトリス塩。
- 前記トリス塩が、単結晶性形態にある、請求項13に記載のトリス塩。
- 前記トリス塩が、一水和物である、請求項13又は14に記載のトリス塩。
- 前記トリス塩が、非溶媒和である、請求項13又は14に記載のトリス塩。
- 前記トリス塩が、2θにおいて4.1°、14.7°、18.8°、20.1°、及び23.1°±0.2から選択される少なくとも3つのピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Bにある、請求項14又は15に記載のトリス塩。
- 前記トリス塩が、2θにおいて4.1°、14.7°、18.8°、20.1°、及び23.1°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Bにある、請求項14又は15に記載のトリス塩。
- 前記トリス塩が、2θにおいて4.1°、4.1°、8.2°、14.7°、16.4°、18.8°、20.1°、20.7°、21.3°、及び23.1°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Bにある、請求項14又は15に記載のトリス塩。
- 前記トリス塩が、2θにおいて4.1°、8.2°、14.7°、16.4°、18.8°、19.1°、20.1°、20.7°、21.3°、23.1°、24.1°、及び25.4°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Bにある、請求項14又は15に記載のトリス塩。
- 前記トリス塩が、168±4℃の示差走査熱量計(DSC)ピーク相転移温度を特徴とする単結晶性形態、形態Bにある、請求項17~20のいずれか一項に記載のトリス塩。
- 前記トリス塩の少なくとも90重量%が、単結晶性形態Bにある、請求項17~21のいずれか一項に記載のトリス塩。
- 前記トリス塩が、2θにおいて6.2°、7.6°、13.1°、13.4°、及び18.5°±0.2から選択される少なくとも3つのピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Gにある、請求項14に記載のトリス塩。
- 前記トリス塩が、2θにおいて6.2°、7.6°、13.1°、13.4°、及び18.5°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Gにある、請求項14に記載のトリス塩。
- 前記トリス塩が、2θにおいて6.2°、7.6°、13.1°、13.4°、18.5°、21.5°、23.7°、及び24.1°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Gにある、請求項14に記載のトリス塩。
- 前記トリス塩が、2θにおいて6.2°、7.6°、13.1°、13.4°、18.0°、18.5°、20.8°、21.5°、23.7°、及び24.1°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態、形態Gにある、請求項14に記載のトリス塩。
- 前記トリス塩が、129.5±4℃の示差走査熱量計(DSC)ピーク相転移温度を特徴とする単結晶性形態、形態Gにある、請求項23~26のいずれか一項に記載のトリス塩。
- 前記トリス塩の少なくとも90重量%が、単結晶性形態Gにある、請求項23~27のいずれか一項に記載のトリス塩。
- 化合物IIのクエン酸塩であって、化合物IIが、以下の構造式によって表され、
- 前記クエン酸塩が、結晶性である、請求項29に記載のクエン酸塩。
- 前記クエン酸塩が、単結晶性形態にある、請求項30に記載のクエン酸塩。
- 前記クエン酸塩が、2θにおいて5.4°、9.4°、12.4°、14.3°、及び17.8°±0.2から選択される少なくとも3つのピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物IIのクエン酸塩形態Aである、請求項31に記載のクエン酸塩。
- 前記クエン酸塩が、2θにおいて5.4°、9.4°、12.4°、14.3°、及び17.8°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物IIのクエン酸塩形態Aである、請求項31に記載のクエン酸塩。
- 前記クエン酸塩が、2θにおいて5.4°、9.4°、10.8°、12.4°、14.3°、16.2°、17.8°、19.6°、及び24.9°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物IIのクエン酸塩形態Aである、請求項31に記載のクエン酸塩。
- 前記クエン酸塩が、2θにおいて5.4°、9.4°、10.8°、12.4°、14.3°、16.2°、17.8°、18.8°、19.6°、23.6°、及び24.9°±0.2にピークを含むX線粉末回折パターンを特徴とする単結晶性形態の化合物IIのクエン酸塩形態Aである、請求項31に記載のクエン酸塩。
- 前記クエン酸塩が、170±3℃の示差走査熱量計(DSC)ピーク相転移温度を特徴とする単結晶性形態の化合物IIのクエン酸塩形態Aである、請求項32~35のいずれか一項に記載のクエン酸塩。
- 前記クエン酸塩の少なくとも90重量%が、単結晶性形態の化合物IIのクエン酸塩形態Aである、請求項32~36のいずれか一項に記載のクエン酸塩。
- 請求項1~37のいずれか一項に記載の化合物と、薬学的に許容される担体と、を含む、医薬組成物。
- 心血管代謝及び関連疾患を治療するための医薬組成物であって、前記医薬組成物が、治療有効量の請求項1~37のいずれか一項に記載の塩を含み、前記疾患が、1型糖尿病、2型糖尿病、前糖尿病、特発性1型糖尿病、成人の潜在的自己免疫性糖尿病、早期発症型2型糖尿病、若年発症型非定型糖尿病、若年発症型成人型糖尿病、栄養失調関連糖尿病、妊娠糖尿病、高血糖、インスリン抵抗性、肝臓インスリン抵抗性、グルコース耐性障害、糖尿病性神経障害、糖尿病性腎症、腎臓病、糖尿病性網膜症、脂肪細胞機能障害、睡眠時無呼吸、肥満症、摂食障害、脂質異常症、高インスリン血症、非アルコール性脂肪性肝疾患、非アルコール性脂肪性肝炎、線維症、肝硬変、肝細胞がん、心血管疾患、アテローム性動脈硬化症、冠状動脈疾患、末梢血管疾患、高血圧、内皮障害、血管コンプライアンス障害、うっ血性心不全、心筋梗塞、脳卒中、出血性脳卒中、虚血性脳卒中、外傷性脳傷害、肺高血圧症、血管形成術後の再狭窄、間欠性跛行、食後脂肪血症、代謝性アシドーシス、ケトーシス、関節炎、骨粗鬆症、パーキンソン病、左心室肥大、末梢動脈疾患、黄斑変性症、白内障、糸球体硬化症、慢性腎不全、メタボリックシンドローム、シンドロームX、月経前症候群、狭心症、血栓症、アテローム性動脈硬化症、一過性虚血性発作、血管再狭窄、グルコース代謝障害、高尿酸血症、痛風、勃起障害、皮膚及び結合組織障害、乾癬、足潰瘍、潰瘍性大腸炎、高アポBリポタンパク血症、アルツハイマー病、統合失調症、認知障害、炎症性腸疾患、短腸症候群、クローン病、大腸炎、過敏性腸症候群、多嚢胞性卵巣症候群の予防又は治療、並びに中毒の治療である、前記医薬組成物。
- 請求項39に記載の医薬組成物であって、前記疾患が、アルツハイマー病、1型糖尿病、2型糖尿病、高血糖、非アルコール性脂肪性肝炎、肥満、、非アルコール性脂肪性肝疾患、またはパーキンソン病である、前記医薬組成物。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2020092530 | 2020-05-27 | ||
| CNPCT/CN2020/092530 | 2020-05-27 | ||
| PCT/US2021/034191 WO2021242817A1 (en) | 2020-05-27 | 2021-05-26 | Salt and crystal forms of glp-1r agonists and uses thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2023527997A JP2023527997A (ja) | 2023-07-03 |
| JP7702973B2 true JP7702973B2 (ja) | 2025-07-04 |
Family
ID=76502864
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022572686A Active JP7702973B2 (ja) | 2020-05-27 | 2021-05-26 | Glp-1rアゴニストの塩及び結晶形態、並びにそれらの使用 |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US20230203023A1 (ja) |
| EP (1) | EP4157832B1 (ja) |
| JP (1) | JP7702973B2 (ja) |
| CN (1) | CN115867546B (ja) |
| AU (1) | AU2021281237A1 (ja) |
| CA (1) | CA3179890A1 (ja) |
| DK (1) | DK4157832T3 (ja) |
| ES (1) | ES2985435T3 (ja) |
| FI (1) | FI4157832T3 (ja) |
| HU (1) | HUE068535T2 (ja) |
| PL (1) | PL4157832T3 (ja) |
| PT (1) | PT4157832T (ja) |
| WO (1) | WO2021242817A1 (ja) |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CR20210341A (es) * | 2018-11-22 | 2021-11-25 | Qilu Regor Therapeutics Inc | Agonistas de glp-ir y usos de los mismos |
| CA3157525A1 (en) | 2019-10-25 | 2021-04-29 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| WO2021154796A1 (en) | 2020-01-29 | 2021-08-05 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| EP4247804A1 (en) | 2020-11-20 | 2023-09-27 | Gilead Sciences, Inc. | Polyheterocyclic glp-1 r modulating compounds |
| WO2022192430A1 (en) | 2021-03-11 | 2022-09-15 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| EP4304711A1 (en) | 2021-03-11 | 2024-01-17 | Gilead Sciences, Inc. | Glp-1r modulating compounds |
| WO2022202864A1 (ja) | 2021-03-24 | 2022-09-29 | 塩野義製薬株式会社 | 縮合環を有するglp-1受容体作動薬を含有する医薬組成物 |
| PH12023552860A1 (en) | 2021-04-21 | 2024-05-20 | Gilead Sciences Inc | Carboxy-benzimidazole glp-1r modulating compounds |
| IL310377A (en) | 2021-08-30 | 2024-03-01 | Mindrank Ai Ltd | Novel aryl ether substituted heterocyclic compound as glp1r agonist |
| WO2023038039A1 (ja) | 2021-09-08 | 2023-03-16 | 塩野義製薬株式会社 | 抗肥満作用の関与する疾患の予防及び治療用医薬 |
| KR20240068737A (ko) | 2021-09-27 | 2024-05-17 | 테른스 파마슈티칼스, 인크. | Glp-1r 효능제로서의 벤즈이미다졸 카복실산 |
| AU2022375634A1 (en) | 2021-10-25 | 2024-06-06 | Terns Pharmaceuticals, Inc. | Compounds as glp-1r agonists |
| EP4725484A2 (en) * | 2021-12-23 | 2026-04-15 | Jiangsu Hengrui Pharmaceuticals Co., Ltd. | Crystalline form of glp-1 receptor agonist and preparation method therefor |
| KR20240150488A (ko) | 2022-02-23 | 2024-10-15 | 테른스 파마슈티칼스, 인크. | Glp-1r 작용제로서의 화합물 |
| CN117362282B (zh) * | 2022-07-07 | 2026-04-21 | 杭州德睿智药科技有限公司 | Glp-1r激动剂的盐及其制备方法和应用 |
| AU2023310481A1 (en) * | 2022-07-18 | 2025-03-06 | Mindrank Therapeutics (Suzhou) New Drug Research And Development Co., Ltd | Polymorphic form of glp-1r agonist, preparation method therefor and use thereof |
| US20260097037A1 (en) | 2022-09-22 | 2026-04-09 | Shionogi & Co., Ltd. | Fused ring compound having glp-1 receptor agonist effect |
| US20240398794A1 (en) | 2023-04-07 | 2024-12-05 | Terns Pharmaceuticals, Inc. | COMBINATIONS OF GLP-1R AND THRß AGONISTS AND METHODS OF USE THEREOF |
| TW202521533A (zh) | 2023-09-14 | 2025-06-01 | 香港商歌禮製藥(中國)有限公司 | Glp-1r 激動劑及其治療方法 |
| US12291530B1 (en) | 2023-11-24 | 2025-05-06 | Ascletis Pharma (China) Co., Limited | GLP-1R agonist and therapeutic method thereof |
| WO2025158275A1 (en) | 2024-01-24 | 2025-07-31 | Pfizer Inc. | Combination therapy using glucose-dependent insulinotropic polypeptide receptor antagonist compounds and glp-1 receptor agonist compounds |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018109607A1 (en) | 2016-12-16 | 2018-06-21 | Pfizer Inc. | Glp-1 receptor agonists and uses thereof |
| JP2022508203A (ja) | 2018-11-22 | 2022-01-19 | キル・レガー・セラピューティクス・インコーポレーテッド | Glp-1rアゴニスト及びその使用 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3773919A (en) | 1969-10-23 | 1973-11-20 | Du Pont | Polylactide-drug mixtures |
| US4485045A (en) | 1981-07-06 | 1984-11-27 | Research Corporation | Synthetic phosphatidyl cholines useful in forming liposomes |
| US4544545A (en) | 1983-06-20 | 1985-10-01 | Trustees University Of Massachusetts | Liposomes containing modified cholesterol for organ targeting |
| US5013556A (en) | 1989-10-20 | 1991-05-07 | Liposome Technology, Inc. | Liposomes with enhanced circulation time |
| TW202144340A (zh) * | 2020-04-03 | 2021-12-01 | 大陸商江蘇恆瑞醫藥股份有限公司 | 稠合咪唑類衍生物、其製備方法及其在醫藥上的應用 |
| CN111978313B (zh) * | 2020-08-28 | 2022-01-18 | 深圳大学 | 具备聚集诱导发光性质的多模态光诊疗剂及其制备与应用 |
-
2021
- 2021-05-26 FI FIEP21733318.6T patent/FI4157832T3/fi active
- 2021-05-26 PL PL21733318.6T patent/PL4157832T3/pl unknown
- 2021-05-26 CA CA3179890A patent/CA3179890A1/en active Pending
- 2021-05-26 ES ES21733318T patent/ES2985435T3/es active Active
- 2021-05-26 EP EP21733318.6A patent/EP4157832B1/en active Active
- 2021-05-26 PT PT217333186T patent/PT4157832T/pt unknown
- 2021-05-26 JP JP2022572686A patent/JP7702973B2/ja active Active
- 2021-05-26 WO PCT/US2021/034191 patent/WO2021242817A1/en not_active Ceased
- 2021-05-26 CN CN202180041199.0A patent/CN115867546B/zh active Active
- 2021-05-26 AU AU2021281237A patent/AU2021281237A1/en active Pending
- 2021-05-26 HU HUE21733318A patent/HUE068535T2/hu unknown
- 2021-05-26 DK DK21733318.6T patent/DK4157832T3/da active
- 2021-05-26 US US17/927,502 patent/US20230203023A1/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018109607A1 (en) | 2016-12-16 | 2018-06-21 | Pfizer Inc. | Glp-1 receptor agonists and uses thereof |
| JP2022508203A (ja) | 2018-11-22 | 2022-01-19 | キル・レガー・セラピューティクス・インコーポレーテッド | Glp-1rアゴニスト及びその使用 |
Also Published As
| Publication number | Publication date |
|---|---|
| PT4157832T (pt) | 2024-07-30 |
| US20230203023A1 (en) | 2023-06-29 |
| ES2985435T3 (es) | 2024-11-05 |
| CN115867546A (zh) | 2023-03-28 |
| DK4157832T3 (da) | 2024-09-23 |
| CA3179890A1 (en) | 2021-12-02 |
| EP4157832B1 (en) | 2024-06-19 |
| WO2021242817A1 (en) | 2021-12-02 |
| AU2021281237A1 (en) | 2023-01-19 |
| TW202210473A (zh) | 2022-03-16 |
| HUE068535T2 (hu) | 2025-01-28 |
| PL4157832T3 (pl) | 2024-12-16 |
| JP2023527997A (ja) | 2023-07-03 |
| FI4157832T3 (fi) | 2024-08-29 |
| CN115867546B (zh) | 2024-10-18 |
| EP4157832A1 (en) | 2023-04-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7702973B2 (ja) | Glp-1rアゴニストの塩及び結晶形態、並びにそれらの使用 | |
| JP7769695B2 (ja) | Glp-1rアゴニストの結晶形態およびその使用 | |
| TWI809334B (zh) | 2-((4-((S)-2-(5-氯吡啶-2-基)-2-甲基苯并〔d〕〔1,3〕二氧呃-4-基)哌啶-1-基)甲基)-1-(((S)-氧呾-2-基)甲基)-1H-苯并〔d〕咪唑-6-羧酸1,3-二羥基-2-(羥基甲基)丙烷-2-胺鹽之固體形式 | |
| TWI839416B (zh) | Glp-1r促效劑及其用途 | |
| JP6637641B1 (ja) | Glp−1受容体アゴニストおよびその使用 | |
| CN112533674B (zh) | Glp-1受体激动剂及其用途 | |
| TW202214622A (zh) | Glp-1r促效劑及其用途 | |
| HK40087545B (en) | Salt and crystal forms of glp-1r agonists and uses thereof | |
| HK40087545A (en) | Salt and crystal forms of glp-1r agonists and uses thereof | |
| TWI913272B (zh) | Glp-1r促效劑之鹽和晶型及其用途 | |
| HK40089377A (en) | Crystal forms of glp-1r agonists and uses thereof | |
| HK40089377B (en) | Crystal forms of glp-1r agonists and uses thereof | |
| HK40082417A (en) | Solid forms of 2-((4-((s)-2-(5-chloropyridin-2-yl)-2-methylbenzo[d] [1,3]dioxol-4-yl)piperidin-1-yl)methyl)-1-(((s)-oxetan-2-yl)methyl)-1h-benzo[d] imidazole-6-carboxylic acid, 1,3-dihydroxy-2-(hydroxymethyl)propan-2-amine salt |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20240523 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20250127 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20250425 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250526 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250624 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7702973 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |