JP7706475B2 - 三環式化合物を含む組み合わせ及びhbv治療薬物の製造におけるその使用 - Google Patents
三環式化合物を含む組み合わせ及びhbv治療薬物の製造におけるその使用 Download PDFInfo
- Publication number
- JP7706475B2 JP7706475B2 JP2022570093A JP2022570093A JP7706475B2 JP 7706475 B2 JP7706475 B2 JP 7706475B2 JP 2022570093 A JP2022570093 A JP 2022570093A JP 2022570093 A JP2022570093 A JP 2022570093A JP 7706475 B2 JP7706475 B2 JP 7706475B2
- Authority
- JP
- Japan
- Prior art keywords
- compound
- hbv
- added
- hepatitis
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Images






Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
- A61K31/522—Purines, e.g. adenine having oxo groups directly attached to the heterocyclic ring, e.g. hypoxanthine, guanine, acyclovir
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5365—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/554—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one sulfur as ring hetero atoms, e.g. clothiapine, diltiazem
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/683—Diesters of a phosphorus acid with two hydroxy compounds, e.g. phosphatidylinositols
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Description
本発明は医薬組み合わせに関し、具体的には三環式構造を有する化合物又はその薬学的に許容される塩、及び任意選択でB型肝炎表面抗原阻害剤及び/又は逆転写酵素阻害剤の一つ又は二つを含む組み合わせ、及びB型肝炎治療薬物の製造における当該組み合わせの使用に関する。具体的には式(I)の化合物又はその薬学的に許容される塩、及び任意選択でB型肝炎表面抗原阻害剤及び/又は逆転写酵素阻害剤の一つ又は二つを含む組み合わせ、及びB型肝炎治療薬物の製造における当該組み合わせの使用を開示する。
a、B型肝炎表面抗原阻害剤、
b、逆転写酵素阻害剤、
c、B型肝炎表面抗原阻害剤及び逆転写酵素阻害剤。

L1は単結合又はC1-6アルキルであり、
R1はH、Cl、F、Br、I、或いは任意選択で1、2又は3個のRaで置換されたC1-3アルキルであり、
環Aは、4~8員ヘテロシクロアルキル又はC3-8シクロアルキルであり、
R2はH及びC1-3アルキルであり、
R3はH及びC1-3アルキルであり、
Raはそれぞれ独立してH、F、Cl、Br、I、NH2、OH、CN、C1-6アルキル又はC1-6ヘテロアルキル、或いは任意選択で1、2又は3個のR’で置換されたC1-6アルキル及びC1-6ヘテロアルキルであり、
R’はそれぞれ独立してCl、F、Br、I、NH2、CH3、CN及び-N(CH3)2から選択され、
前記4~8員ヘテロシクロアルキル及びC1-6ヘテロアルキルはそれぞれ、1、2、3又は4個の独立して-O-、-NH-、-S-及びNから選択されるヘテロ原子又はヘテロ原子団を含む。
ここで、Raはそれぞれ独立してH、F、Cl、Br、I、NH2、OH又はCNである。
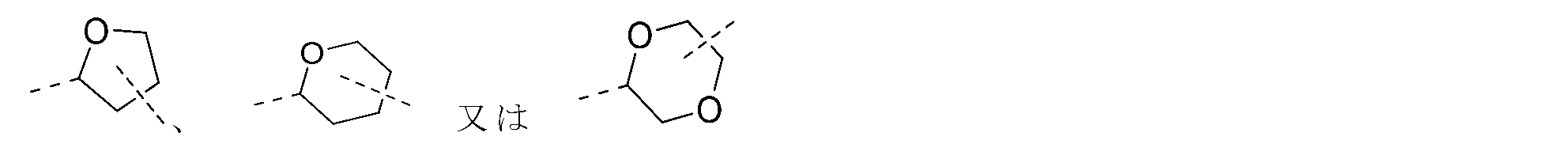



R1はH、OH、CN、NH2から選択され、或いは任意選択で1、2又は3個のRで置換されたC1-6アルキル、C1-6ヘテロアルキル、C2-5アルケニル、C2-5ヘテロアルケニル、C3-6シクロアルキル又は3~6員ヘテロシクロアルキルから選択される。
R2はH、OH、CN、NH2、ハロゲンから選択され、或いは任意選択で1、2又は3個のRで置換されたC1-3アルキル、C1-3ヘテロアルキル、C3-6シクロアルキル又は3~6員ヘテロシクロアルキルから選択され、
R3は任意選択で1、2又は3個のRで置換されたC1-6アルキル、C3-6シクロアルキルから選択され、
mは、0、1、2、3、4又は5から選択され、
mが0の場合、R1はOH、CN、NH2から選択されなく、
RはH、ハロゲン、OH、CN、NH2から選択され、或いは任意選択で1、2又は3個のR’で置換されたC1-3アルキル、C1-3ヘテロアルキルから選択され、
R’はF、Cl、Br、I、OH、CN、NH2、CH3、CH3CH2、CH3O、CF3、CHF2、CH2Fから選択され、
「ヘテロ」は、ヘテロ原子又はヘテロ原子団を表し、前記C1-6ヘテロアルキル、C2-5ヘテロアルケニル、3~6員ヘテロシクロアルキル、C1-3ヘテロアルキルの「ヘテロ」は、それぞれ独立して-C(=O)N(R)-、-N(R)-、-C(=NR)-、-(R)C=N-、-S(=O)2N(R)-、-S(=O)N(R)-、N、-O-、-S-、=O、=S、-C(=O)O-、-C(=O)-、-C(=S)-、-S(=O)-、-S(=O)2-、-N(R)C(=O)N(R)-から選択され、
上記の場合のいずれにおいても、ヘテロ原子又はヘテロ原子団の数は、それぞれ独立して1、2又は3から選択される。







好ましくは、前記B型肝炎表面抗原阻害剤は、下記の式(IIb)で表される構造を有する。


R1はH、OH、CN、NH2から選択され、或いは任意選択で1、2又は3個のRで置換されたC1-5アルキル、C1-5ヘテロアルキル、C2-5アルキニル、C3-6シクロアルキル及び3~6員ヘテロシクロアルキルから選択され、
R2はH、ハロゲンから選択され、或いは任意選択で1、2又は3個のRで置換されたC1-3アルキル及びC1-3ヘテロアルキルから選択され、
mは、0、1、2、3、4及び5から選択され、
Aは任意選択で1、2又は3個のRで置換されたフェニル又は5~6員ヘテロアリールから選択され、
RはH、ハロゲン、OH、CN、NH2、=O、CH3、CH3CH2、CH3O、CF3、CHF2、CH2Fから選択され、
前記C1-5ヘテロアルキル、3~6員ヘテロシクロアルキル、C1-3ヘテロアルキル、5~6員ヘテロアリールの「ヘテロ」は、それぞれ独立してN、-O-、=O、-S-、-NH-、-(C=O)-、-(S=O)-、-(S=O)2-から選択され、
上記の場合のいずれにおいても、ヘテロ原子又はヘテロ原子団の数は、それぞれ独立して1、2又は3から選択される。









以下の内容は、本発明の組み合わせの投与方法を限定するものではない。
本発明の式(I)の化合物は、B型肝炎コアタンパク質阻害剤であり、ウイルスcccDNAライブラリーを妨害し、HBVウイルスの複製を阻害することができ;本発明の式(II)及び式(III)の化合物は、B型肝炎表面抗原阻害剤であり、HBsAgを効果的に低減することができ、式(I)の化合物と前記B型肝炎表面抗原阻害剤を併用するか、又は式(I)の化合物とヌクレオシド系又はヌクレオチド系逆転写酵素阻害剤を併用するか、又はこれら3種類のメカニズムの薬物を併用することにより、HBVウイルスの複製プロセスをマルチチャンネルで阻害し、治療効果の向上、毒性副作用の低減、治療サイクル及び投与量の短縮、薬剤耐性の低減等の目的を達成することができ、HBV負荷量の低下、HBsAgの減少、さらには除去の効果を達成することができる。
別途に説明しない限り、本明細書で用いられる以下の用語及び連語は以下の意味を含む。一つの特定の用語又は連語は、特別に定義されない場合、不確定又は不明瞭ではなく、普通の定義として理解されるべきである。本明細書で商品名が出た場合、相応の商品又はその活性成分を指す。
(1)哺乳動物における疾患又は疾患状態の出現を予防すること、特にそのような哺乳動物が当該疾患状態にかかりやすいが、その疾患状態に罹患したと診断されていない場合、
(2)疾患又は疾患状態を阻害すること、即ち、その進行を阻止すること、
(3)疾患又は疾患状態を緩和すること、即ち、疾患又は疾患状態を退行させること。
楔形点線結合
棒状実線結合
棒状点線結合
波線
楔形実線結合
楔形点線結合
波線
棒状実線結合
棒状点線結合



直線実線結合
波線







乾いた10Lの三口フラスコに無水ジクロロメタン(5L)を加え、攪拌を開始し、化合物1-SMA(500.00g)とニトロメタンを三口フラスコに順次添加し、反応系をドライアイスエタノール浴に入れ、-10℃に冷却させた。温度を-10℃~0℃に制御し、三塩化アルミニウム(1.15kg)を反応フラスコにゆっくりと添加し、温度を-0℃以下に制御し、α,α-ジクロロジメチルメチルエーテル(495.00g)を反応釜にゆっくりと加え、室温までゆっくりと上昇させ、18時間攪拌した。TLC(PE:EA3:1)モニタリングにより、原料点が消失し、極性の大きい新しい点が生成された。反応液を抜き出し、10%硫酸水素カリウム溶液(3L)にゆっくりと滴下し、20分間攪拌し、過熱を防ぐためにクラッシュアイスを加えた。混合溶液を25L分液ロートに移し、静置して分離させ、ジクロロメタン層を分離して得、水相をジクロロメタン(2L×2)で抽出した。有機相を10%硫酸水素カリウム溶液(5L×2)で洗浄し、有機相を分離し、無水硫酸ナトリウム(1kg)で乾燥させた。有機相を減圧濃縮して、暗緑色固体化合物1-Aを得た。
化合物1-A(2kg、11.96mol)のTHF(20L)溶液にp-トルエンスルホニルヒドラジド(2.23kg、11.96mol)を加えた。20℃で約1時間攪拌した。TLCで原料の消失を確認した後、反応系を60℃に加熱し、その後、シアノ水素化ホウ素ナトリウム(902g、14.36mol)をバッチで添加し、添加完了後、反応を70℃に加熱して3時間攪拌した。加熱を停止し、室温まで冷却させた後、5Lの水を加えて反応をクエンチし、THCの大部分を減圧下で除去し、残留物をEA(1.5L×3)で抽出した。有機相を合わせ、飽和塩化ナトリウムで洗浄し、無水硫酸ナトリウムで乾燥させた。ろ過し、溶媒を減圧下で除去し、粗生成物をカラムクロマトグラフィーで分離して淡黄色固体化合物1-Bを得た。
メタノール(32L)を50Lジャケット釜に加え、攪拌を開始し、化合物1-SMB(4000.00g)とジイソプロピルエチルアミン(5.25L)を順次に加え、内部温度を5~10℃に下げ、ベンジルメルカプタン(2490.00g)をゆっくりと滴下し、内部温度を5~15℃に維持させた。滴下終了後、冷却システムを閉じ、自然に昇温させ、2.5時間攪拌を続けた。攪拌を停止し、回転速度を100rpmに調節し、反応液を放出し、卓上フィルターで濾過し、ケーキを水(5L)で3回洗浄し、次いでEtOH(3L)を加えて1回洗浄し、ケーキが粘稠でなくなるまで吸引濾過して、淡黄色固体化合物1-Cを得た。
ジクロロメタン(7.5L)を50Lのジャケット釜に加え、攪拌を開始し、化合物1-C(1500g)を加え、内部温度を0~10℃に下げ、HCl溶液(6M、4.12L)を加えた。0~10℃の条件で、次亜塩素酸ナトリウム溶液(市販の8%溶液、23.0kg)を開口で滴下し、滴下終了後、冷却システムを閉じ、開口で約17時間攪拌を続けた。次いで亜硫酸水素ナトリウム溶液(1000g、5L水溶液)をそれに滴下し、ヨウ化カリウムデンプン試験紙で水相に酸化剤が残っていないことを検出した。攪拌を停止し、静置して分離し、ジクロロメタン層を収集し、水層をジクロロメタン(2.5L)で抽出し、ジクロロメタン層を合わせた。有機相を無水硫酸ナトリウムで乾燥させ、濾過し、溶媒を減圧下で除去して白色固体化合物1-Dを得た。
テトラヒドロフラン(10L)を乾いた50Lジャケット釜に加え、攪拌を開始し、化合物1-B(2000g)を加え、内部温度を0~10℃に下げた。約1.5時間以内で、温度を0~15℃に維持して、カリウムtert-ブトキシド(1MのTHF溶液、15.67L)を加え、終了後、温度を約20℃まで上げ、1時間攪拌を続けた。温度を0~10℃に下げ、化合物1-D(4380g)のテトラヒドロフラン(10L)溶液をゆっくりと加えた。滴下終了後、ゆっくりと15℃まで昇温させ、約16時間攪拌を続けた。酢酸エチル(10L)を加えて抽出し、有機相を飽和塩化ナトリウム溶液(10L)で2回洗浄し、水相を合わせ、EA(5L)抽出し、有機相を合わせた。有機相を減圧下で溶媒を除去して、淡黄色固体化合物1-Eを得た。
化合物1-E(1000.0g)を乾いた10L三口フラスコに加え、攪拌を開始し、氷酢酸(5L)を加え、反応内温を25~30℃に制御した。鉄粉末(1eq、140.9g)をゆっくりと加え、30分攪拌した後、第2バッチの鉄粉末(0.5eq、70.44g)をゆっくりと加え、30分攪拌を続けた後、第3バッチの鉄粉末(0.5eq、70.44g)をゆっくりと加え、再び30分攪拌した後、第4バッチの鉄粉末(0.5eq、70.44g)を加え、原料が消失するまで攪拌を続けて反応させ、極性の大きな新たな点を生成した。攪拌を停止し、反応液を25L分液フラスコに移し、10L酢酸エチルを加え、飽和硫酸水素ナトリウム水溶液5L×2で洗浄し、分離し、水相を酢酸エチル5Lで逆抽出した。有機相を合わせ、10%NaOH水溶液でpH>8まで洗浄し、有機相を分離して収集した。有機相を減圧濃縮して、白色固体化合物1-SM1を得た。
トルエン(12L)を乾いた50Lジャケット釜に加え、攪拌を開始し、2-ブロモエタノール(9930g)を加え、次いで三フッ化ホウ素エチルエーテル(268g)を加え、反応を30~35℃に昇温させた。化合物1-SMC(3500g)をゆっくりと滴下し、約1.5時間で滴下を完了させ、この時、反応の内部温度は約55~65℃に上げ、ヒーター温度を60℃に調節し、内部温度を55~65℃に1時間維持させた。反応系の内部温度を約10℃に下げ、反応系内に約20℃の水酸化ナトリウム水溶液(3783g、水17.5L)をゆっくりと加え、内部温度を10~20℃に維持させた。NaOH溶液を加えた後、温度制御器を停止させ、反応を約16時間攪拌し続けた。撹拌を停止させ、静置し、分離し、水層を2-メチルテトラヒドロフラン(10L)で抽出し、有機相を合わせ、水(10L)で洗浄し、静置し、分離し、有機相を収集した。有機相を減圧濃縮して、無色の油状化合物1-Fを得た。
水酸化ナトリウム(3240g、水15L)水溶液を50Lジャケット釜に加え、化合物1-F(4430g)を加え、加熱を開始し、反応を90℃に昇温させた後、1時間攪拌を続けた。冷却を開始し、約15℃まで下げ、p-トルエンスルホニルクロリドのテトラヒドロフラン溶液(6180g、テトラヒドロフラン15L)を加え、温度制御器を閉じ、反応を約15℃で約16時間攪拌を続けた。攪拌を停止し、静置し、分離し、水相を2-メチルテトラヒドロフラン(10L)で抽出し、2-メチルテトラヒドロフラン相(白色不溶物があり、水で洗浄した後消失した)を水(5L)で洗浄し、有機相を合わせた。有機相にDMAP(500g)、トリエチルアミン(2.5L)を加え、30分間攪拌し、飽和塩化ナトリウム溶液(10L)を加えて洗浄し、静置して分離し、水相を廃棄した。有機相を硫酸水素カリウム溶液(3800g、水15L)、飽和塩化ナトリウム溶液(5L×2)で洗浄し、静置して分離し、有機相を収集した。有機相を減圧濃縮して溶媒を除去し、粗生成物化合物1-Gを得た。
アセトン(30L)を清浄な50Lジャケット釜に加え、攪拌を開始し、化合物1-G(4500g)を加え、次いでヨウ化ナトリウム(6190g)を加え、加熱を開始し、反応を75℃に昇温させた後、16時間攪拌を続けた。室温まで下げた後で濾過し、濾液を50℃で減圧濃縮した。濃縮後の粗生成物に酢酸エチル(15L)、水(10L)を加え、攪拌し、静置して分離し、有機相を0.5Mチオ硫酸ナトリウム(10L)で洗浄した。水相とチオ硫酸ナトリウム溶液を合わせた後、EtOAc(5L)で抽出した。有機相を合わせ、飽和塩化ナトリウム溶液(10L)で洗浄し、静置し、分離し、有機相を収取した。有機相を減圧濃縮して溶媒を除去し、粗生成物化合物1-Hを得た。
DMSO(20L)を清浄な50Lジャケット釜に加え、攪拌を開始し、化合物1-H(4700g)を加え、温度を35℃に上げ、次いでシアン化ナトリウム(1010g)を加え、反応の内部温度を20分以内に約60℃まで上げ、その後徐々に35℃まで下げ、約16時間攪拌を続けた。反応系内に炭酸水素ナトリウム溶液(炭酸水素ナトリウム2000g、水10L)を加え、約5分間攪拌を続け、EtOAc:MeOH(20L、2L)を加え、2分間攪拌を続け、約1時間静置した。分離し、下層の溶液を約30L分離し、EtOAc:MeOH(1回目は15L:1.5L、2回目は5L:0.5L)で2回抽出した。抽出後の上層有機相及び残りの反応液上層を合わせ、飽和塩化ナトリウム溶液(各10L)で3回洗浄し、静置し、分離し、水相を廃棄し、有機相を収取した。有機相を減圧して溶媒を除去し、粗生成物をカラムクロマトグラフィーで分離して無色の油状物化合物1-Iを得た。
アルゴンガスの保護下で、ラネーニッケル(10.00g、116.73mmol)とEtOH(150mL)を乾いた水素化フラスコに加え、さらに1-I(20g、157.31mmol)、NH3.H2O(13.65g、97.36mmol、15.00mL、25%純度)を反応系に加え、その後置換し、反応を50psi、50℃で3.5時間攪拌した。反応液を珪藻土で濾過し、濾液を減圧濃縮して黄色の油状物化合物1-Jを得た。
1-J(800.00g)を5L三口フラスコに加え、攪拌を開始し、酢酸エチル(800mL)を約0.5時間以内に加え、HCl/EtOAc(1.6L)4Mを反応系のpH<5までゆっくりと滴下し、内部温度を5~15℃に維持させた。冷却システムを閉じ、室温まで昇温させ、1時間攪拌を続けた。撹拌を停止し、卓上フィルターで濾過して、ケーキを得た。ケーキを減圧濃縮し(40~45℃)て、粗生成物を得た。上記生成物にアセトニトリル(2mL/g)を加え、1時間スラリー化させた。卓上フィルターで濾過し、ケーキを収取し、有機溶液を減圧下で除去して、白色固体化合物1-SM2を得た。
トルエン(20L)を乾いた50Lジャケット釜に加え、攪拌を開始し、化合物1-SM1(2500g)を加え、内部温度を30~35℃に上げた。窒素ガスでパージングして釜内を不活性ガス雰囲気を維持させ、トリメチルアルミニウム(3.0L、Al(CH3)3添加によりケトルの内部温度度が緩やかな上昇)を滴下し、終了後、窒素ガスを閉じ、温度を80~85℃まで上げ、約16時間攪拌を続けた。冷却を開始し、反応温度を20~30℃に下げ、反応液の半分の約12Lを移し、EToAc(10L)を加え、均一に混合した。攪拌しながら、10%KHSO4溶液(10L)に混合液を加え、2分間攪拌し、静置し、分離し、有機層をさらに10%KHSO4溶液(10L)で洗浄し、水相を合わせ、DCM(各7.5L)を用いて2回抽出した。残りの半分の反応液約12Lを移し、処理方法は上記と同様であり、有機相を合わせ、有機相を減圧濃縮して粗生成物を得、2倍体積のn-ヘプタンを加えて1時間スラリー化させた。濾過し、真空乾燥させた(>12時間、温度40℃、P≦-0.1MPa)。化合物1-1-A及び化合物1-1-Bの混合物を得た。
テトラヒドロフラン(3840mL)を10L三口フラスコに加え、攪拌を開始し、化合物1-1-Aと化合物1-1-Bの混合物(480.00g)を加え、LiOH.H2O(118.84g)のH2O(960mL)溶液をゆっくりと滴下した。滴下終了後、60℃まで昇温させ、1時間攪拌し、反応液に濃HClを加え、反応系pHを2に調節し、攪拌を停止した。静置し、分離した。水相をTHF(600mL)で2回抽出し、有機相を合わせた。有機相を減圧濃縮(40~45℃)し、固体を純水(2mL/g)で0.5時間スラリー化させ、ろ過し、ケーキを真空乾燥させて(>12時間、温度40℃、P≦-0.1MPa)、化合物1-2を得た。
DMF(2.25L)を5L三口フラスコに加え、開始攪拌し、化合物1-2(400.00g)及びHATU(744.83g)を順次に加えて30分間攪拌し、次いで化合物1-SM2(229.86g)を加えてた。1時間以内に、DIPEA(568.68mL)を室温でゆっくりと滴下し、滴下終了後、室温で攪拌し、16時間攪拌した。反応液を分液ロートに移し、酢酸エチル(2L)と純水(1L)を加えて2分間攪拌した。静置し、水相を分離した。さらに純水(1L)を加えて洗浄し、攪拌し、静置し、分離した。合わせた水相をEtOAc(500mL)で3回抽出し、有機相を合わせた。有機相を炭酸ナトリウム溶液(1.5L)で2回、硫酸水素カリウム溶液(1L)で2回、純水(1L)で2回順次に洗浄した。有機相を減圧濃縮して(40~45℃)、粗生成物を得た。粗生成物に酢酸エチル(2mL/g)を加え、1時間スラリー化させた。濾過してケーキを収集し、化合物1を得た。
1.1 1日目に、HepG2.2.15細胞を96ウェル細胞培養プレートに40,000細胞/ウェルの密度で播種し、その後、細胞を5%CO2、37℃で一晩培養した。

化合物1とTDFの体外併用投与によるHBVの阻害活性をHepG 2.2.15細胞を用いて評価した。HBVの阻害活性と細胞毒性に対する医薬の併用投与結果のまとめは表2に示される通りであり、併用投与の効果図は図1に示される通りである。

薬物併用の総合指数の説明:指数の絶対値<25の場合、即ち、プラス効果であり、指数の絶対値が25~50の範囲の場合、即ち、軽度であるが明確な相乗効果又は拮抗効果であり、指数の絶対値が50~100の範囲の場合、即ち、中程度の相乗作用又は拮抗作用であり、体内作用に重要な意義を持つ可能性がある。指数の絶対値>100の場合、即ち、高度の相乗効果又は拮抗効果であり、体内作用に重要な意義を持つ可能性が高い。

化合物1とTDFの体外併用投与はHBVの阻害活性にプラス効果を示し、化合物はテスト濃度内で細胞毒性を示さなかった。テスト結果は慢性HBV感染患者の治療における化合物1とTDFの臨床併用を支持した。
5週齢の特定病原体を含まないオスのC57BL/6マウスであり、SHANGHAI SLAC LABORATORY ANIMAL CO. LTDから購入した。
溶媒:
10%ソリュートール水溶液
適量の化合物1を上記溶媒に加え、ボルテックスした後、粒子の均一な懸濁液が得られ、製造した濃度は1.0、3.0及び10.0mg/mLであった。使用まで4℃に保存した。化合物1を製造時には、1.0の塩係数、100%の純度に従って計算された。
テノホビルジソプロキシルフマル酸塩(TDF)は、Shanghai Panhong Chemical Technology Co., Ltd.から購入した。適量のTDFを秤量して、生理食塩水に溶解させ、1mg/mLの母液に調製し、TDFが完全に溶解するまでボルテックスし、1mL仕様に分注し、-20℃で保存した。毎回投与する前に1mLの母液を採取し、生理食塩水で10倍で0.1mg/mLの作業液に希釈し、当日の投与に用いた。
rAAV8-1.3HBV(タイプD、ayw)はBeijing FivePlus Molecular Medicine Institute Co. Ltd.,5+MMIから購入し、バッチ番号がx2018032301であり、1×1012viral genome(v.g.)/mLであった。実験前に無菌PBSで5×1011v.g./mLに希釈させた。各マウスに200μLを注射し、即ち、各マウスに1×1011v.g.を注射した。
AAV/HBVマウスモデルの構築
AAV/HBV注射:rAAV8-1.3HBVは、注射前に予め無菌PBSを用いて1×1011v.g./200μLの濃度の溶液に調製した。
ウイルス注射後14日目と21日目に、血清の採取のためにすべての感染マウスの顎下静脈から~120μLの血液を採取した。全血を37℃インキュベーターに入れて30分間培養した後、13,200×g、4℃で3分間遠心分離して血清~30μLを収集した。血清は-80℃に保存され、HBV DNA、HBeAg及びHBsAgの検出に使用された。
ウイルス注射後28日目に、ウイルス注射後14日目及び21日目の血清試料中のHBV DNA、HBsAg及びHBeAgのレベル、及びマウス体重に基づいて群を分けた。
初回投与日を実験0日目とした。
体内実験の投与とサンプリングのスキームは表4に示される通りである。

血液試料を37℃で~30分間培養した後、4℃、13,200gの条件で3分間遠心分離し、分離した上清をドライアイスで急速凍結した。
血液試料をK2EDTAで抗凝固処理した後、4℃、7,000gの条件で10分間遠心分離して上清を分離した。分離した血漿に1:20の比で沈殿剤を加え、[メタノール:アセトニトリル(v:v、50:50)溶液]、ボルテックスして混合し、ドライアイスで急速凍結した。
体内実験の過程で、マウスの状態を定期的に観察し、感染、投与、採血、及び実験が終了した日にマウスの体重を記録した。
1.HBsAg ELISA(Antu Bio、CL 0310)キット説明書で、マウス血清中のHBsAg含有量を検出した。

データは、各群の試料の平均±標準誤差として表され、特に明記しない限り、第1~6群:n=8である。Student’s t testを用いて統計分析を行った。
1)AAV/HBVマウス実験における血清HBV DNAに対する被験化合物の影響
各群のマウスの血清におけるHBV DNAの含有量は図2にまとめられた通りである。
溶媒群(Group1)のマウスの血清HBV DNA含有量は投与後安定を維持し、化合物1の単剤群は:溶媒群(Group1)と比較して、化合物1の低(10mpk、Group2)、中(30mpk)、高(100mpk)の3つの用量群のマウス血清中のHBV DNA含有量は投与3日後に用量依存的な低下を示し始め、低、中用量群は投与7日後にHBV DNA含有量が安定性を維持し、高用量群は投与7~28日間血清中のHBV DNA含有量は持続的に低下し、溶媒群(p<0.01)よりも有意に低かった。
各群のマウスの肝臓におけるHBV DNAの含有量は図3にまとめられた通りである。
溶媒群(Group1)と比較して、投与28日後、化合物1の低(10mpk)、中(30mpk)、高(100mpk)の3つの用量群のマウス肝臓におけるHBV DNA含有量は低下した。化合物1とTDFとの併用投与群(Group6、10+1mpk)は、投与28日後、肝臓におけるHBV DNA含有量は化合物1の10mpk単剤群より有意に低かった(p<0.001)。
各群のマウス血清におけるHBV RNAの含有量は図4にまとめられる通りである。
溶媒群(Group1)と比較して、投与28日後、化合物1の高(100mpk)用量投与群のマウス血清におけるHBV RNA含有量は低下した。
第2群のマウス血清中の薬物濃度は図5及び表6にまとめられる通りである。
27日目の、最初の投与後0時間(投与前)、1、4、8、9、12、24時間後、マウス血清における薬物濃度の平均値は、それぞれ78、122200、1630、605、10300、1280及び66nMであった。薬物吸収は投与後1時間でピークに達し、血漿半減期は2.23時間であり、AUC0-infは47600nM.hであった。

実験中、マウスの健康状態及び体重を定期的にモニタリングした。
0日目の体重を基準として比較し、マウスの体重変化結果は図6にまとめられる通りである。
投与期間中、全部のマウスの体重は安定しており、有意な減少はなく、マウスの状態は良好であった。
マウスの体重変化を-28日目から28日目に記録した(最初の投与日を0日目とした)。0日目の体重を基準として比較し、IACUCの規定により20%の体重減少を人道的エンドポイントとし、マウス体重が20%以上低下した場合、実験から除去する必要があった。
溶媒対照群と比較して、化合物1の治療後のマウスの血清及び肝臓のHBV DNAは有意に減少し、血清の中で用量依存的な傾向を示した。化合物1とTDFとの併用後、効果が化合物1単独治療よりも有意に優れており、良好な併用投与効果を示した。実験期間中、マウスの体重が有意に減少せず、被験化合物に対するマウスの耐性が良好であることを示した。
上記実験におけるTDFをETV(エンテカビル)に置き換えて、化合物1とETVとを併用した体内抗ウイルス効果を得、マウス血清及び肝臓HBV DNAは有意に低下し、化合物1単独治療よりも優れていた。

細胞株:
HepG2.2.15細胞はWuXi AppTecによって構築・提供され、細胞培地はDMEM/F12培地に2%ウシ胎児血清、2mMグルタミン、1×非必須アミノ酸、100U/mLペニシリン及び100g/mLストレプトマイシンを添加したものであった。
本研究で使用された主な試薬には、FastStart Universal Probe MasterMaster(Roche、カタログ番号 04914058001)、DNA抽出キットKit(Qiagen、カタログ番号:51162)、CellTiter-Glo(Promega-G7573)が含まれた。
本研究で使用された主な機器は、7900リアルタイムPCR装置(Applied Biosystems)、多機能マイクロプレートリーダー(BioTek、Synergy2)、QuantStudioTM6Flex System(Applied Biosystems)であった。
化合物1、式(IIb)の化合物、式(IIIb)の化合物。



a=化合物1、b=式(IIb)の化合物又は式(IIIb)の化合物であり、0は化合物がないことを表し、1~7は7個の濃度を表す。
HepG2.2.15細胞を用いて、HBVに対する化合物1と式(IIb)の化合物、化合物1と式(IIIb)の化合物の体外併用投与の阻害活性を評価した。HBVに対する化合物単剤の阻害活性及び細胞毒性は表10にまとめられる通りであり、HBVに対する併用投与の阻害活性及び細胞毒性の結果は表11~表13にまとめられる通りである。


薬物併用指数の説明:指数の絶対値<25の場合、即ち、プラス効果であり、指数の絶対値が25~50の範囲の場合、即ち、軽度であるが明確な相乗作用又は拮抗効果を示し、指数の絶対値が50~100の範囲の場合、即ち、中程度の相乗作用又は拮抗効果を示し、体内効果に重要な意義を持つ可能性がある。指数の絶対値>100の場合、即ち、高度の相乗又は拮抗効果を示し、体内効果に重要な意義を持つ可能性が高い。


HepG2.2.15体外感染HBVモデルにおいて、被験化合物は用量依存的にHBV DNAを阻害し、化合物1と式(IIb)の化合物、化合物1と式(IIIb)の化合物はプラスの併用結果を示し、併用試験濃度内では細胞毒性を示さなかった。試験結果は、慢性HBV感染の治療における化合物1と前記式(IIb)化合物、式(IIIb)化合物などのB型肝炎表面抗原阻害剤の、臨床的に併用を支持した。
上記実施形態によれば、化合物1を化合物2


Claims (6)
- 式(I)の化合物

a、B型肝炎表面抗原阻害剤、
b、逆転写酵素阻害剤、
c、B型肝炎表面抗原阻害剤及び逆転写酵素阻害剤、
のいずれか1つと、からなる医薬組成物。
(ここで、式(I)の化合物は、以下の化合物

前記B型肝炎表面抗原阻害剤は、以下の化合物

前記逆転写酵素阻害剤は、ラミブジン、アデホビルジピボキシル、エンテカビル、テノホビルジソプロキシルフマル酸塩又はテノホビルアラフェナミドフマル酸塩から選択される。) - 前記逆転写酵素阻害剤は、エンテカビル及びテノホビルジソプロキシルフマル酸塩から選択される請求項1に記載の組成物。
- 請求項1に記載の組成物を含む、B型肝炎ウイルス感染の治療のための医薬。
- 請求項1に記載の組成物と少なくとも一つの薬学的に許容される担体及び/又は賦形剤とを含むこと特徴とする、医薬組成物。
- 請求項1に記載の組成物、又は、請求項4に記載の組成物を含むこと特徴とする、キット。
- 請求項4に記載の組成物、又は、請求項5に記載のキットを含む、B型肝炎の治療のための医薬。
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202010412760.9 | 2020-05-15 | ||
| CN202010412760 | 2020-05-15 | ||
| CN202011633373 | 2020-12-31 | ||
| CN202011633373.4 | 2020-12-31 | ||
| PCT/CN2021/093769 WO2021228213A1 (zh) | 2020-05-15 | 2021-05-14 | 包含三并环类化合物的组合及其在制备治疗hbv药物中的应用 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| JP2023526346A JP2023526346A (ja) | 2023-06-21 |
| JP7706475B2 true JP7706475B2 (ja) | 2025-07-11 |
Family
ID=78525325
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2022570093A Active JP7706475B2 (ja) | 2020-05-15 | 2021-05-14 | 三環式化合物を含む組み合わせ及びhbv治療薬物の製造におけるその使用 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US12569500B2 (ja) |
| EP (1) | EP4151221A4 (ja) |
| JP (1) | JP7706475B2 (ja) |
| CN (1) | CN115515596A (ja) |
| WO (1) | WO2021228213A1 (ja) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA3109746C (en) * | 2018-08-23 | 2023-07-11 | Fujian Cosunter Pharmaceutical Co., Ltd. | Crystal form of tri-cycle compound and application thereof |
| CN116514698A (zh) * | 2023-04-10 | 2023-08-01 | 佛山奕安赛医药科技有限公司 | 一种沃诺拉赞中间体的制备方法及其应用 |
| WO2026052081A1 (zh) * | 2024-09-06 | 2026-03-12 | 福建广生中霖生物科技有限公司 | 用于治疗乙型肝炎的药物及组合 |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019501202A (ja) | 2016-01-08 | 2019-01-17 | アルブータス・バイオファーマー・コーポレイション | B型肝炎を治療するための治療用組成物及び方法 |
| JP2019511542A (ja) | 2016-04-15 | 2019-04-25 | ヤンセン・サイエンシズ・アイルランド・アンリミテッド・カンパニー | カプシド集合阻害剤を含む組み合わせ及び方法 |
| JP2019526562A (ja) | 2016-08-24 | 2019-09-19 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | Hbvキャプシドアセンブリ阻害剤とヌクレオシ(チ)ド類似体の併用療法 |
| CA3109746A1 (en) | 2018-08-23 | 2020-02-27 | Fujian Cosunter Pharmaceutical Co., Ltd. | Crystal form of tri-cycle compound and application thereof |
| JP2020508342A (ja) | 2017-02-23 | 2020-03-19 | フーチェン・コサンター・ファーマスーティカル・カンパニー・リミテッドFujian Cosunter Pharmaceutical Co., Ltd. | 三環式化合物及びその応用 |
| JP2020509075A (ja) | 2017-03-09 | 2020-03-26 | フーチェン・コサンター・ファーマスーティカル・カンパニー・リミテッドFujian Cosunter Pharmaceutical Co., Ltd. | B型肝炎ウイルス表面抗原阻害剤 |
| EP3632914A1 (en) | 2017-05-22 | 2020-04-08 | Fujian Cosunter Pharmaceutical Co., Ltd. | Hepatitis b virus surface antigen inhibitor |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102060786A (zh) * | 2011-01-20 | 2011-05-18 | 天津大学 | 4-(取代-1,3-二炔基)-4-(三氟甲基)-3,4-二氢取代喹唑啉-2-酮类化合物及其制备方法和应用 |
| MX353412B (es) * | 2013-04-03 | 2018-01-10 | Janssen Sciences Ireland Uc | Derivados de n-fenil-carboxamida y su uso como medicamentos para el tratamiento de la hepatitis b. |
| WO2015073774A1 (en) * | 2013-11-14 | 2015-05-21 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis b infections |
| KR20160148715A (ko) * | 2014-05-13 | 2016-12-26 | 에프. 호프만-라 로슈 아게 | B형 간염 바이러스 감염의 치료 또는 예방을 위한 신규한 다이하이드로퀴놀리진온 |
| WO2018022282A1 (en) * | 2016-07-29 | 2018-02-01 | Newave Pharmaceutical Inc. | Novel therapeutic agents for the treatment of hbv infection |
-
2021
- 2021-05-14 JP JP2022570093A patent/JP7706475B2/ja active Active
- 2021-05-14 WO PCT/CN2021/093769 patent/WO2021228213A1/zh not_active Ceased
- 2021-05-14 CN CN202180034217.2A patent/CN115515596A/zh active Pending
- 2021-05-14 US US17/925,075 patent/US12569500B2/en active Active
- 2021-05-14 EP EP21804833.8A patent/EP4151221A4/en active Pending
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2019501202A (ja) | 2016-01-08 | 2019-01-17 | アルブータス・バイオファーマー・コーポレイション | B型肝炎を治療するための治療用組成物及び方法 |
| JP2019511542A (ja) | 2016-04-15 | 2019-04-25 | ヤンセン・サイエンシズ・アイルランド・アンリミテッド・カンパニー | カプシド集合阻害剤を含む組み合わせ及び方法 |
| JP2019526562A (ja) | 2016-08-24 | 2019-09-19 | エフ.ホフマン−ラ ロシュ アーゲーF. Hoffmann−La Roche Aktiengesellschaft | Hbvキャプシドアセンブリ阻害剤とヌクレオシ(チ)ド類似体の併用療法 |
| JP2020508342A (ja) | 2017-02-23 | 2020-03-19 | フーチェン・コサンター・ファーマスーティカル・カンパニー・リミテッドFujian Cosunter Pharmaceutical Co., Ltd. | 三環式化合物及びその応用 |
| JP2020509075A (ja) | 2017-03-09 | 2020-03-26 | フーチェン・コサンター・ファーマスーティカル・カンパニー・リミテッドFujian Cosunter Pharmaceutical Co., Ltd. | B型肝炎ウイルス表面抗原阻害剤 |
| EP3632914A1 (en) | 2017-05-22 | 2020-04-08 | Fujian Cosunter Pharmaceutical Co., Ltd. | Hepatitis b virus surface antigen inhibitor |
| CA3109746A1 (en) | 2018-08-23 | 2020-02-27 | Fujian Cosunter Pharmaceutical Co., Ltd. | Crystal form of tri-cycle compound and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2021228213A1 (zh) | 2021-11-18 |
| EP4151221A1 (en) | 2023-03-22 |
| US12569500B2 (en) | 2026-03-10 |
| EP4151221A4 (en) | 2023-11-15 |
| JP2023526346A (ja) | 2023-06-21 |
| CN115515596A (zh) | 2022-12-23 |
| US20230190768A1 (en) | 2023-06-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7773987B2 (ja) | スピロ環含有キナゾリン化合物 | |
| CN108290893B (zh) | 二氢蝶啶酮类衍生物、其制备方法及其用途 | |
| JP7706475B2 (ja) | 三環式化合物を含む組み合わせ及びhbv治療薬物の製造におけるその使用 | |
| JP2023508482A (ja) | スピロ環含有キナゾリン化合物 | |
| JP6283033B2 (ja) | ウイルス感染症およびさらなる疾患の処置のためのアシルアミノピリミジン誘導体 | |
| JP7624924B2 (ja) | ニトロキソリンプロドラッグ及びその使用 | |
| WO2023232135A1 (zh) | Pde4b抑制剂及其用途 | |
| EP2867219B1 (en) | Asymmetrical reversible neuromuscular blocking agents of ultra-short, short, or intermediate duration | |
| CN108727378A (zh) | 新型异喹啉类化合物及其医药用途 | |
| WO2011134283A1 (zh) | 苦参酸/碱衍生物及其制备方法和用途 | |
| JP2025528804A (ja) | イソインドリン置換グルタルイミド骨格に基づく化合物、及びその応用 | |
| JP7561285B2 (ja) | 複素環置換の縮合γ-カルボリン誘導体、その製造方法、中間体及び使用 | |
| JP2012516333A (ja) | Hivインテグラーゼ阻害剤としての架橋化合物 | |
| EP2139889B1 (en) | Amino-naphthyridine derivatives | |
| CN121449548B (zh) | 一种哌啶衍生物及其药物组合物、用途以及制备方法 | |
| CN111574533A (zh) | 柠檬苦素a环开环胺化衍生物或其药学上可接受的盐、制备方法及用途 | |
| JP7603320B2 (ja) | 環内チアミジノアミド-アリールアミド系化合物及びb型肝炎を治療するためのその使用 | |
| WO2025001680A1 (zh) | 新型GalNAc的靶向递送片段及其制备和应用 | |
| JPH0611758B2 (ja) | ベンジル酸−4−ピペリジルエステル誘導体 | |
| EP0826686A2 (en) | Tricyclic compounds, their production and use | |
| US8741893B2 (en) | 6,7-dihydro-[1,3,4]thiadiazolo-[3,2-a][1,3]diazepin derivatives and pharmaceutical compositions containing the same as hypnotic or anesthetic agent and method for their preparation | |
| JP2024105489A (ja) | Μgaτ2阻害活性を有する縮合環誘導体の製造方法 | |
| WO1993007122A1 (en) | 4-diphenylacetoxy-n-substituted piperidines with antimuscarinic activity | |
| EP4384161A1 (en) | Inhibitors of 12/15-lipoxygenase | |
| CN121824494A (zh) | 哒嗪酮类化合物及其药物组合物和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20221227 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20240312 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20250228 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20250304 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20250603 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20250610 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A821 Effective date: 20250604 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20250701 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 7706475 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |