KR20130143084A - 마크로락탐의 제조 방법 및 중간체 - Google Patents
마크로락탐의 제조 방법 및 중간체 Download PDFInfo
- Publication number
- KR20130143084A KR20130143084A KR1020137015211A KR20137015211A KR20130143084A KR 20130143084 A KR20130143084 A KR 20130143084A KR 1020137015211 A KR1020137015211 A KR 1020137015211A KR 20137015211 A KR20137015211 A KR 20137015211A KR 20130143084 A KR20130143084 A KR 20130143084A
- Authority
- KR
- South Korea
- Prior art keywords
- compound
- salt
- formula
- alkyl
- mmol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 0 CC[C@](C(C)(C)COC(N[C@@](*)C(N(C[C@@](C1)OC(N2CC3=C(C)C=CC[C@]3C2)=O)[C@]1C(*)=O)O)=O)C=C* Chemical compound CC[C@](C(C)(C)COC(N[C@@](*)C(N(C[C@@](C1)OC(N2CC3=C(C)C=CC[C@]3C2)=O)[C@]1C(*)=O)O)=O)C=C* 0.000 description 8
- AMXFGLZWKBYNGE-UHFFFAOYSA-N Brc1cccc2c1CNC2 Chemical compound Brc1cccc2c1CNC2 AMXFGLZWKBYNGE-UHFFFAOYSA-N 0.000 description 1
- GTUAOVPQKDAPQF-LOSJGSFVSA-N CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(O)=O)=O)NC(OCC(C)(C)CC(C(Cc1cccc(C2)c1CN2C(O)=O)=C)=C)=O Chemical compound CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(O)=O)=O)NC(OCC(C)(C)CC(C(Cc1cccc(C2)c1CN2C(O)=O)=C)=C)=O GTUAOVPQKDAPQF-LOSJGSFVSA-N 0.000 description 1
- JVZOIBPRFRUCPQ-PZUNEJSGSA-N CC(C)(C)[C@@H](C(N(C[C@@H](C1)OC(N(C2)Cc3c2cccc3CC=C)=O)[C@@H]1C(O)=O)=O)NC(OCC(C)(C)CC=C)=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](C1)OC(N(C2)Cc3c2cccc3CC=C)=O)[C@@H]1C(O)=O)=O)NC(OCC(C)(C)CC=C)=O JVZOIBPRFRUCPQ-PZUNEJSGSA-N 0.000 description 1
- FNCIBXCDJLBQKC-AKIFATBCSA-N CC(C)(C)[C@@H](C(N(C[C@@H](C1)OC(N(C2)Cc3c2cccc3CCCCC(C)(C)CO2)=O)[C@@H]1C(O)=O)=O)NC2=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](C1)OC(N(C2)Cc3c2cccc3CCCCC(C)(C)CO2)=O)[C@@H]1C(O)=O)=O)NC2=O FNCIBXCDJLBQKC-AKIFATBCSA-N 0.000 description 1
- JIZOCWLGHJFRID-ZKABDWDDSA-N CC(C)(C)[C@@H](C(N(C[C@@H](CC1)OC(N(CC2=C)C/C2=C(\CCCCC(C)(C)CO2)/C=C\C)=O)[C@@H]1C(O)=O)=O)NC2=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](CC1)OC(N(CC2=C)C/C2=C(\CCCCC(C)(C)CO2)/C=C\C)=O)[C@@H]1C(O)=O)=O)NC2=O JIZOCWLGHJFRID-ZKABDWDDSA-N 0.000 description 1
- JSSMVRYDELYMIR-SSDOTTSWSA-N CCC[C@H](C(O)=O)NC(OCC=C)=O Chemical compound CCC[C@H](C(O)=O)NC(OCC=C)=O JSSMVRYDELYMIR-SSDOTTSWSA-N 0.000 description 1
- SCNYGLGKLIHYDR-UHFFFAOYSA-N N#CC1=CCCC(Br)=C1 Chemical compound N#CC1=CCCC(Br)=C1 SCNYGLGKLIHYDR-UHFFFAOYSA-N 0.000 description 1
- JCKKLQQRXDXCHV-UHFFFAOYSA-N OC(c1c2c(Br)ccc1)NC2=O Chemical compound OC(c1c2c(Br)ccc1)NC2=O JCKKLQQRXDXCHV-UHFFFAOYSA-N 0.000 description 1
- OJOYGIJPPABLBZ-UHFFFAOYSA-N [Cl-]CN(C1)Cc2c1cccc2Br Chemical compound [Cl-]CN(C1)Cc2c1cccc2Br OJOYGIJPPABLBZ-UHFFFAOYSA-N 0.000 description 1
- PAFZNILMFXTMIY-UHFFFAOYSA-O [NH3+]C1CCCCC1 Chemical compound [NH3+]C1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/16—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/10—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C271/22—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atoms of the carbamate groups bound to hydrogen atoms or to acyclic carbon atoms to carbon atoms of hydrocarbon radicals substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/08—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms
- C07C271/24—Esters of carbamic acids having oxygen atoms of carbamate groups bound to acyclic carbon atoms with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C271/00—Derivatives of carbamic acids, i.e. compounds containing any of the groups, the nitrogen atom not being part of nitro or nitroso groups
- C07C271/06—Esters of carbamic acids
- C07C271/40—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of six-membered aromatic rings
- C07C271/56—Esters of carbamic acids having oxygen atoms of carbamate groups bound to carbon atoms of six-membered aromatic rings with the nitrogen atom of at least one of the carbamate groups bound to a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/78—Halides of sulfonic acids
- C07C309/79—Halides of sulfonic acids having halosulfonyl groups bound to acyclic carbon atoms
- C07C309/80—Halides of sulfonic acids having halosulfonyl groups bound to acyclic carbon atoms of a saturated carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C309/00—Sulfonic acids; Halides, esters, or anhydrides thereof
- C07C309/78—Halides of sulfonic acids
- C07C309/85—Halides of sulfonic acids having halosulfonyl groups bound to carbon atoms of rings other than six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/30—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/45—Sulfonamides, the carbon skeleton of the acid part being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups at least one of the singly-bound nitrogen atoms being part of any of the groups, X being a hetero atom, Y being any atom, e.g. N-acylaminosulfonamides
- C07C311/47—Y being a hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/50—Compounds containing any of the groups, X being a hetero atom, Y being any atom
- C07C311/51—Y being a hydrogen or a carbon atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/16—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/44—Iso-indoles; Hydrogenated iso-indoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D498/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06008—Dipeptides with the first amino acid being neutral
- C07K5/06017—Dipeptides with the first amino acid being neutral and aliphatic
- C07K5/06034—Dipeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Veterinary Medicine (AREA)
- Virology (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pyrrole Compounds (AREA)
- Indole Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
본 발명은 마크로락탐 화합물, 마크로락탐의 제조에 유용한 중간체, 중간체의 제조 방법, 및 마크로락탐의 제조 및 변형 방법에 관한 것이다. 본원에 기재된 화합물 및 방법의 한 용도는 HCV NS3 프로테아제 활성의 억제가 가능한 마크로락탐 화합물의 제조에 있다. 본원에 기재된 절차를 사용하여 합성될 수 있는 HCV 억제 화합물의 예는 화합물 A 및 그의 유도체이다.
Description
본 발명은 마크로락탐을 제조하고, 마크로락탐을 변형시키는데 사용될 수 있는 방법 및 화합물에 관한 것이다. 본원에 기재된 방법 및 화합물의 한 용도는 HCV NS3 프로테아제 활성의 억제가 가능한 마크로락탐 화합물의 제조에 있다.
C형 간염 바이러스(HCV) 감염은 상당수의 감염된 개체들에 있어서, 만성 간 질환, 예컨대 간경화 및 간세포 암종을 초래하는 주요 건강 문제점이다. HCV 감염에 대한 현재의 치료법은 재조합 인터페론-α 단독에 의한 또는 뉴클레오시드 유사체 리바비린과 병용한 면역요법을 포함한다.
몇몇 바이러스적으로-암호화된 효소는 메탈로프로테아제(NS2-3), 세린 프로테아제(NS3), 헬리카제(NS3), 및 RNA-의존성 RNA 폴리머라제(NS5B)를 포함한, 치료학적 중재를 위해 추정되는 표적들이다. NS3 프로테아제는 NS3 단백질의 N-말단 도메인에 위치한다. NS4A는 NS3 활성에 대한 보조인자(cofactor)를 제공한다.
HCV 감염에 대한 잠재적인 치료법이 문헌 ([Balsano, mini Rev . Med . Chem . 8(4):307-318, 2008], [Roenn et al., Current Topics in Medicinal Chemistry 8: 533-562, 2008], [Sheldon et al., Expert Opin . Investig . Drugs 16(8):1171-1181, 2007] 및 [De Francesco et al., Antiviral Research 58:1-16, 2003])을 포함한 상이한 참조 문헌에 논의되어 왔다.
HCV 프로테아제 활성의 억제가 가능한 마크로락탐 화합물을 기재하는 간행물의 예는 다음을 포함한다: [Holloway et al., 미국 특허 번호 7,470,664], [Harper et al., WO2010011566]; [Liverton et al., WO2009134624]; [McCauley et al., WO2009108507]; Liverton et al., WO2009010804]; [Liverton et al., WO2008057209]; [Liverton et al., WO2008051477]; [Liverton et al., WO2008051514]; [Liverton et al., WO2008057208]; [Crescenzi et al., WO2007148135]; [Di Francesco et al., WO2007131966]; [Holloway et al., WO2007015855]; [Holloway et al., WO2007015787]; Holloway et al., WO2007016441]; [Holloway et al., WO2006119061]; [Liverton et al., J. Am . Chem. Soc., 130:4607-4609, 2008]; [McCauley et al., Abstracts of Papers , 235 th ACS National Meeting, New Orleans, LA, United States, April 6-10, 2008]; [Liverton et al., Antimicrobial Agents and Chemotherapy 54:305-311, 2009 (온라인에 게재됨)]; 및 [McCauley et al., Journal of Medicinal Chemistry, 53(6):2443-2463, 2010].
발명의 개요
본 발명은 마크로락탐 화합물, 마크로락탐의 제조에 유용한 중간체, 중간체의 제조 방법, 및 마크로락탐의 제조 및 변형 방법에 관한 것이다. 본원에 기재된 화합물 및 방법의 한 용도는 HCV NS3 프로테아제 활성의 억제가 가능한 마크로락탐 화합물의 제조에 있다. 본원에 기재된 절차를 사용하여 합성될 수 있는 HCV 억제 화합물의 예는 화합물 A 및 그의 유도체이다. 화합물 A는 하기 구조를 갖는다:
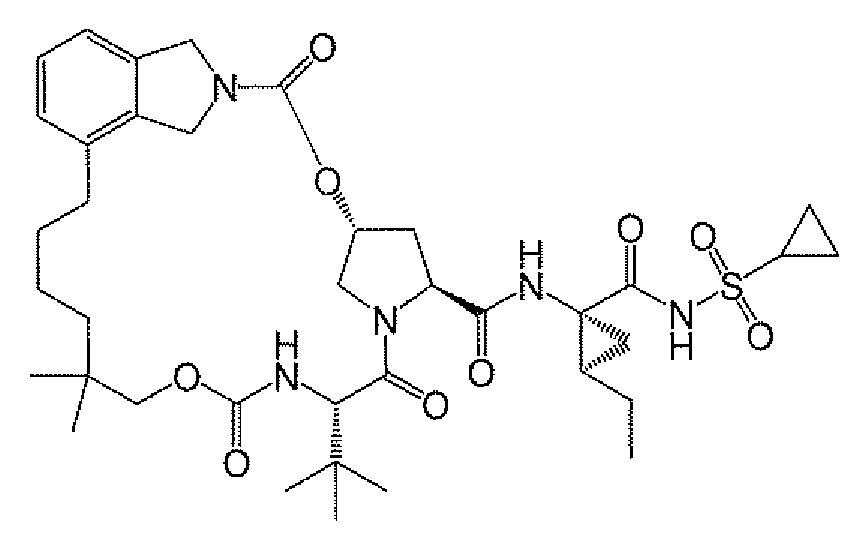
따라서, 본 발명의 제1 측면은 하기 화학식 I의 화합물, 하기 화학식 II의 화합물 또는 그의 염, 하기 화학식 III의 화합물 또는 그의 염, 하기 화합물 6A 또는 그의 염, 하기 화합물 3 또는 그의 염, 하기 화합물 A-2, 하기 화합물 A-4, 하기 화합물 A-10 또는 그의 염, 및 하기 화합물 B-6 또는 그의 염으로 이루어진 군으로부터 선택된 화합물에 관한 것이고,
<화학식 I>
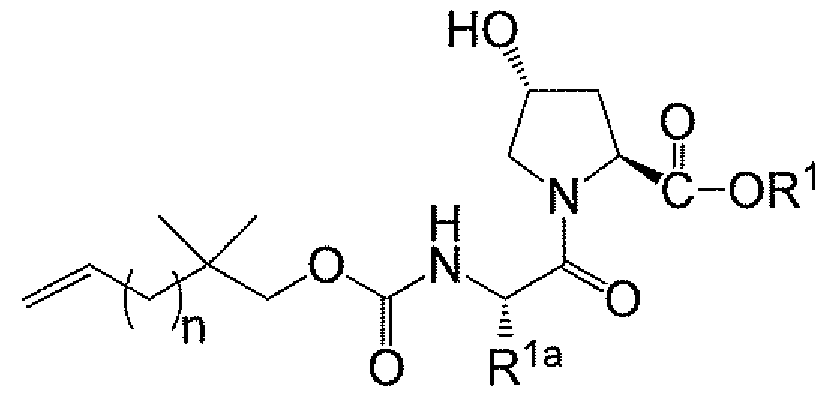
<화학식 II>

<화학식 III>
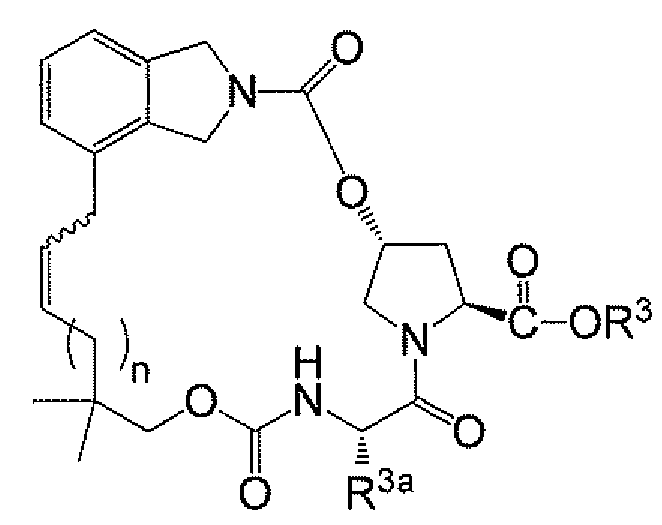
<화합물 6A>

<화합물 3>

<화합물 A-2>

<화합물 A-4>

<화합물 A-10>

<화합물 B-6>

여기서 상이한 기가 본원에 기재되어 있다. (예를 들어, 하기 섹션 I. 중간체 참조) 화학식 II 또는 III 화합물의 염은 상응하는 카르복실산 (즉, R2 또는 R3이 수소임)으로부터 용이하게 제조될 수 있다.
본 발명의 또 다른 측면은 화학식 I의 화합물을 화합물 3 또는 그의 염과 커플링시키는 단계를 포함하는, 화학식 II의 화합물 또는 그의 염의 제조 방법에 관한 것이다.
또 다른 측면은 화학식 II의 화합물 또는 그의 염을 폐환 및 수소화시켜 하기 화학식 IV의 화합물 또는 그의 염을 형성하는 단계를 포함하는, 화학식 IV의 화합물 또는 그의 염의 제조 방법에 관한 것이다:
<화학식 IV>
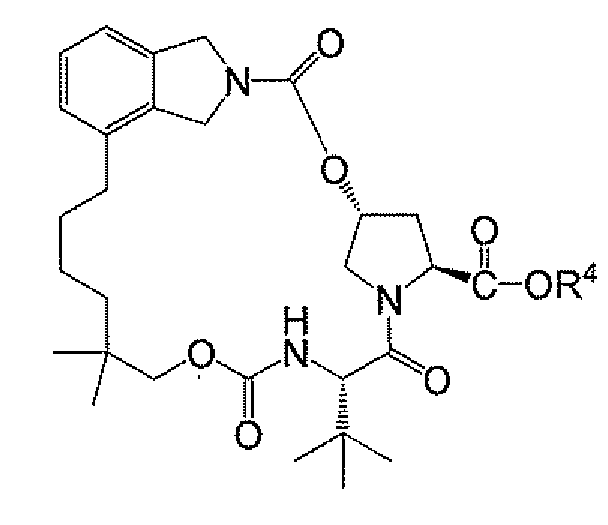
화학식 IV 염은 상응하는 카르복실산 (즉, R4)으로부터 용이하게 제조될 수 있다.
또 다른 측면은
a) 화학식 II의 화합물 또는 그의 염을 폐환 및 수소화시켜 화학식 IV의 화합물 또는 그의 염을 형성하는 단계를 포함하는, 화학식 IV의 화합물 또는 그의 염을 제조하는 단계;
b) 화학식 IV의 화합물 또는 그의 염을 가수분해시켜 하기 화합물 11 또는 그의 염을 형성하는 단계;
c) 화합물 11 또는 그의 염을 하기 화합물 A-11 또는 그의 염에 커플링시켜 화합물 A 또는 그의 염을 형성하는 단계; 및
d) 임의로, 화합물 A 또는 그의 염을 제약상 허용되는 염으로 전환시키는 단계
를 포함하는, 화합물 A 또는 그의 제약상 허용되는 염의 제조 방법에 관한 것이다:
<화합물 11>

<화합물 A-11>

본 발명의 또 다른 측면은 다음 단계를 포함하는 화합물 3 또는 그의 염의 제조 방법에 관한 것이다:

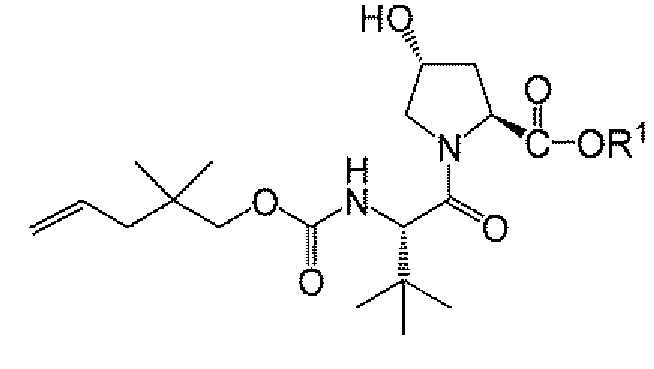
또 다른 측면은 하기 단계를 포함하는, 화합물 A-8 또는 그의 염의 제조 방법에 관한 것이다:

그의 염에 관한 언급은 화합물 A-7 및 A-8이 염으로서 제공될 수 있음을 나타낸다.
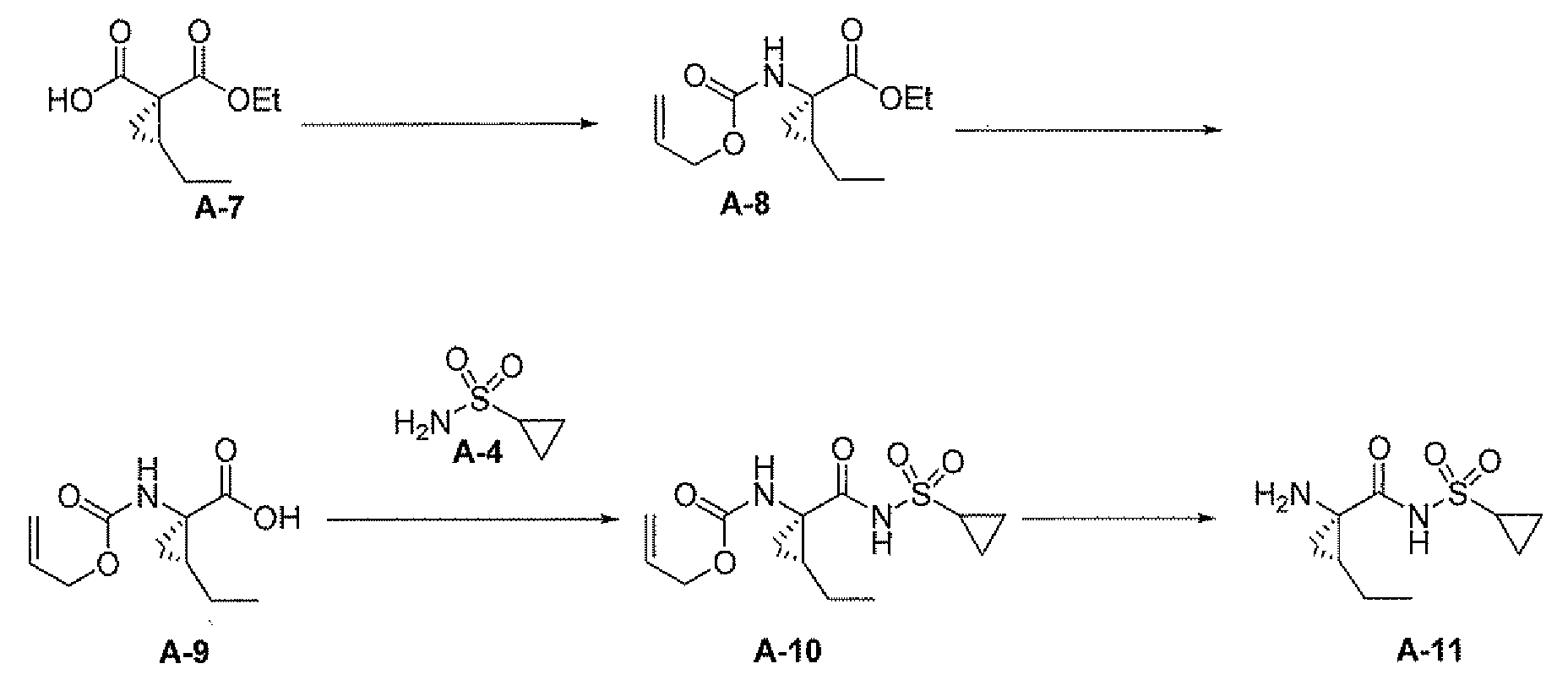
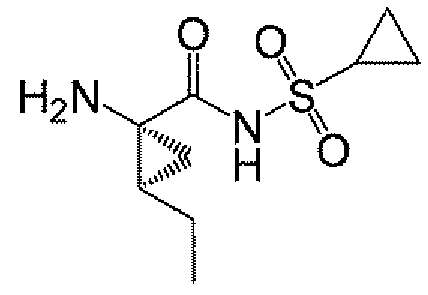
또 다른 측면은 다음을 포함하는, 화합물 A-11 또는 그의 염의 제조 방법에 관한 것이다:

그의 염에 관한 언급은 화합물 A-10 및 A-11이 염으로서 제공될 수 있음을 나타낸다.
또 다른 측면은 또는 그의 염을
또는 그의 염을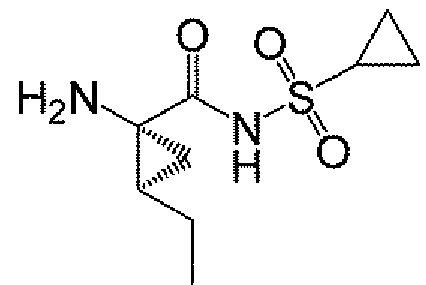 또는 그의 염에 커플링시켜 화합물 A 또는 그의 염을 형성하는 단계를 포함하고, 여기서 반응은 커플링 시약 및 피리딘 또는 피리딘 유도체의 사용을 포함하는 것인, 화합물 A 또는 그의 염의 제조 방법에 관한 것이다.
또는 그의 염에 커플링시켜 화합물 A 또는 그의 염을 형성하는 단계를 포함하고, 여기서 반응은 커플링 시약 및 피리딘 또는 피리딘 유도체의 사용을 포함하는 것인, 화합물 A 또는 그의 염의 제조 방법에 관한 것이다.
본 발명의 다른 실시양태, 측면 및 특징은 본원에 추가로 기재되거나 다음의 설명, 실시예 및 첨부된 특허청구범위로부터 명백할 것이다.
본원에 기재된 방법 및 중간체를 사용하여 마크로락탐, 예컨대 화합물 A 및 화합물 A에 존재하는 하나 이상의 관능기에 의해 화합물 A와 차이가 나는 화합물을 합성할 수 있다. 변형될 수 있는 관능기에는 상이한 헤테로사이클기, t-부틸기 대신에 상이한 알킬, 및 시클로프로필술포닐 관능기의 변경 (예를 들어, 에틸렌을 대체하는 에틸기 및/또는 시클로프로필기를 대체하는 메틸시클로프로필기로)이 포함된다. 화합물 A의 일부 유도체를 포함하는 구조의 예는 다음과 같다:
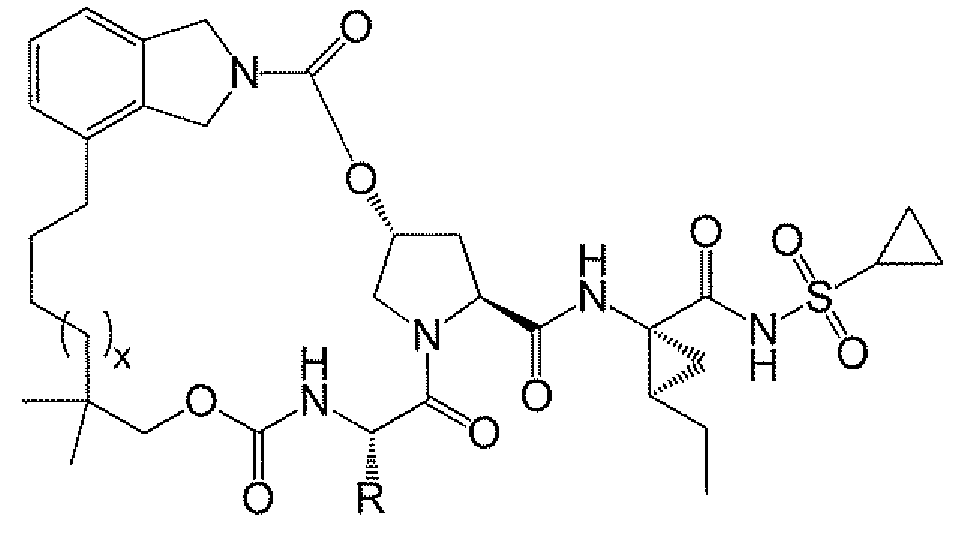
상기 식에서, x는 0 내지 5이고, R은 C1 -6 알킬 또는 C3-C8 시클로알킬이다. 바람직하게는, x는 0 내지 2, 더 바람직하게는 1이다. 바람직하게는 R은 t-부틸 또는 시클로헥실이다.
궁극적으로 화합물 A가 수득되는, 상이한 중간체 및 합성 프로토콜이 본원에서 설명된다. 그러나, 본원에 제공된 지침을 기반으로 적절한 중간체를 사용하고 상이한 관능기를 첨가하거나 변형시켜 다른 마크로락탐이 제조될 수 있는 것으로 이해된다. 상이한 관능기를 갖는 상이한 마크로락탐의 예는 문헌 ([Holloway et al., 미국 특허 번호 7,470,664], [Harper et al., WO2010011566]; [Liverton et al., WO2009134624]; [McCauley et al., WO2009108507]; Liverton et al., WO2009010804]; [Liverton et al., WO2008057209]; [Liverton et al., WO2008051477]; [Liverton et al., WO2008051514]; [Liverton et al., WO2008057208]; [Crescenzi et al., WO2007148135]; [Di Francesco et al., WO2007131966]; [Holloway et al., WO2007015855]; [Holloway et al ., WO2007015787]; Holloway et al., WO2007016441]; [Holloway et al., WO2006119061]; [Liverton et al., J. Am . Chem . Soc., 130:4607-4609, 2008]; [McCauley et al., Abstracts of Papers , 235 th ACS National Meeting, New Orleans, LA, United States, April 6-10, 2008]; [Liverton et al., Antimicrobial Agents and Chemotherapy 54:305-311, 2009 (온라인에 게재됨)]; 및 [McCauley et al., Journal of Medicinal Chemistry, 53(6):2443-2463, 2010])에서 제공되어 있다.
문헌 ([McCauley et al., Abstracts of Papers , 235 th ACS National Meeting, New Orleans, LA, United States, April 6-10, 2008]; [Liverton et al., Antimicrobial Agents and Chemotherapy 54:305-311, 2009 (온라인에 게재됨)]; [McCauley et al., Journal of Medicinal Chemistry, 53(6):2443-2463, 2010]; [Holloway et al., 미국 특허 번호 7,470,664]; [Holloway et al., WO2007015855]; 및 [Holloway et al., WO2007015787])은 화합물 A 및 화합물 A의 대안적 제조 방법을 기재한다.
HCV 활성의 억제가 가능한 마크로락탐 화합물은 생체내 HCV 활성 억제, 시험관내 HCV 활성 억제, 및 HCV NS3 효소 활성 억제를 포함한 상이한 용도를 갖는다. HCV 활성의 생체내 억제는 치료에의 응용에 사용될 수 있다. 시험관내 HCV 활성 억제는 HCV 내성 변이체를 수득하는데 사용되는 것, HCV 레플리콘 또는 효소 활성을 억제하는 관능기의 능력을 추가의 특징으로 하는 것, 및 HCV 복제 또는 프로테아제 활성을 연구하는 것을 포함한 상이한 응용을 갖는다.
I. 중간체
화합물 A와 같은 마크로락탐 화합물을 제조하는데 사용할 수 있는 상이한 화합물이 본 출원에서 본 섹션 등을 비롯하여 본원에 기재되어 있다. 상이한 중간체에 관한 제1 측면에서, 화합물은 하기 화학식 I의 화합물, 하기 화학식 II의 화합물 또는 그의 염, 하기 화학식 III의 화합물 또는 그의 염, 하기 화합물 6A 또는 그의 염, 하기 화합물 3 또는 그의 염, 하기 화합물 A-2, 하기 화합물 A-4, 하기 화합물 A-10 또는 그의 염, 및 하기 화합물 B-6 또는 그의 염으로 이루어진 군으로부터 선택된다:
<화학식 I>
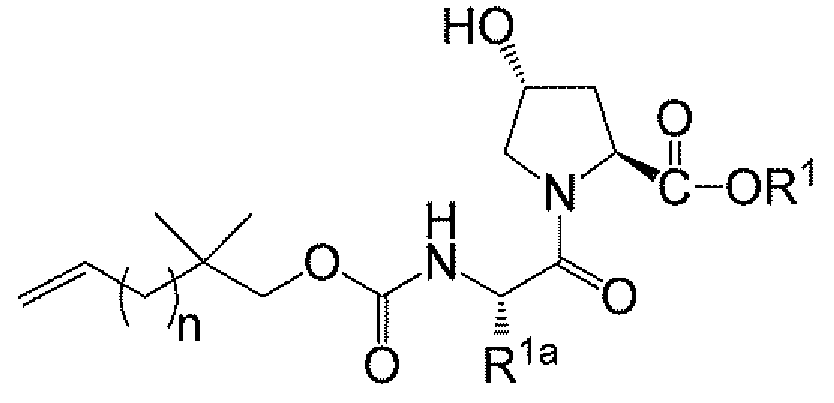
<화학식 II>

<화학식 III>

<화합물 6A>

<화합물 3>

<화합물 A-2>

<화합물 A-4>

<화합물 A-10>

<화합물 B-6>
상기 식에서,
R1은 C1 -6 알킬, C3-C8 시클로알킬, 또는 아릴이고;
R2 및 R3은 각각 H, C1 -6 알킬, C3-C8 시클로알킬, 또는 아릴이고;
R1a, R2a, 및 R3a는 각각 C1 -6 알킬 또는 C3-C8 시클로알킬이고;
n은 0 내지 5이고;
아릴은 페닐, 치환된 페닐, 나프틸, 또는 치환된 나프틸이되, 단, 치환된 페닐 및 치환된 나프틸은 각각
(1) C1 -6 알킬,
(2) OH, O-C1 -6 알킬, O-C1 -6 할로알킬, CN, NO2, N(RA)RB, C(O)N(RA)RB, C(O)RA, CO2RA, SRA, SORA, SO2RA, SO2N(RA)RB, N(RA)C(O)RB, N(RA)CO2RB, N(RA)SO2RB, N(RA)SO2N(RA)RB, OC(O)N(RA)RB, N(RA)C(O)N(RA)RB, 또는 N(RA)C(O)C(O)N(RA)RB로 치환된 C1 -6 알킬,
(3) O-C1 -6 알킬,
(4) C1 -6 할로알킬,
(5) O-C1 -6 할로알킬,
(6) OH,
(7) 할로겐,
(8) CN,
(9) NO2,
(10) N(RA)RB,
(11) C(O)N(RA)RB,
(12) C(O)RA,
(13) C(O)-C1 -6 할로알킬,
(14) C(O)ORA,
(15) OC(O)N(RA)RB,
(16) SRA,
(17) SORA,
(18) SO2RA,
(19) SO2N(RA)RB,
(20) N(RA)SO2RB,
(21) N(RA)SO2N(RA)RB,
(22) N(RA)C(O)RB,
(23) N(RA)C(O)N(RA)RB,
(24) N(RA)C(O)C(O)N(RA)RB, 또는
(25) N(RA)CO2RB
로 이루어진 군으로부터 독립적으로 선택된 1 내지 5개의 치환기 (여기서 RA 및 RB는 각각 독립적으로 H 또는 C1 -6 알킬임)를 갖는다.
제2 측면에서, 화합물은 하기 화학식 I의 화합물이고, 여기서 바람직한 하위부류는 하기 화학식 Ia의 화합물이다:
<화학식 I>
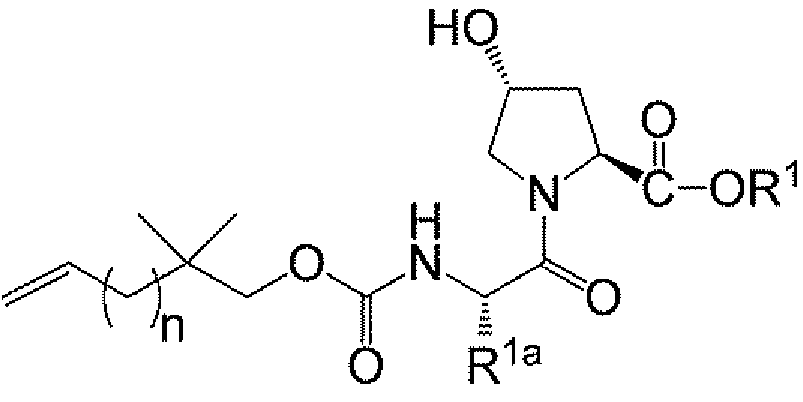
<화학식 Ia>

상기 식에서, R1, R1a, 및 n은 제1 측면에서 정의된 바와 같다.
제1 실시양태에서, R1은 C1 -6 알킬, C3-C8 시클로알킬, 페닐 또는 나프틸이다.
제2 실시양태에서, R1은 C1 -6 알킬 또는 C3-C8 시클로알킬이다.
제3 실시양태에서, R1은 C1 -6 알킬이다.
제4 실시양태에서, R1a는 t-부틸 또는 시클로헥실이고, R1은 제1 측면 또는 실시양태 1 내지 3에 제공된 바와 같다.
제5 실시양태에서, R1a는 t-부틸이고, R1은 제1 측면 또는 실시양태 1 내지 3 중 임의의 것에 제공된 바와 같다.
제6 실시양태에서, n은 0 내지 2이고, R1 및 R1a는 제1 측면 또는 실시양태 1 내지 4 중 임의의 것에 제공된 바와 같다.
제7 실시양태에서, n은 1이고, R1 및 R1a는 제1 측면 또는 실시양태 1 내지 5 중 임의의 것에 제공된 바와 같다.
제8 실시양태에서, 화학식 I 화합물은 하기 화합물 7이다:
<화합물 7>

제3 측면에서, 화합물은 하기 화학식 II의 화합물 또는 그의 염이고, 여기서 바람직한 하위부류는 하기 화학식 IIa의 화합물 또는 그의 염이다:
<화학식 II>

<화학식 IIa>

상기 식에서, R2, R2a, 및 n은 제1 측면에서 정의된 바와 같다. R2가 H인 경우 염은 용이하게 제조될 수 있다.
제1 실시양태에서, R2는 H, C1 -6 알킬, C3-C8 시클로알킬, 페닐 또는 나프틸이다.
제2 실시양태에서, R2는 H, C1 -6 알킬 또는 C3-C8 시클로알킬이다.
제3 실시양태에서, R2는 C1 -6 알킬이다.
제4 실시양태에서, 화합물은 화학식 II 또는 IIa의 염이다. 추가 실시양태에서, 염은 칼륨, 나트륨, 리튬, 1급 아민 (NH3 +-RC), 2급 아민 (NH2 +-(RC)2), 또는 3급 아민 (NH+-(RC)3)이고; 여기서 각각의 RC는 독립적으로 C1 -6 알킬, C3 -C8 시클로알킬, 또는 아릴이되; 단, 2개의 RC는 함께 NH+ 및 -(CH2)n- (여기서 n은 2 내지 7, 바람직하게는 5 또는 6임)을 함유하는 3원 내지 8원 헤테로시클릭기를 형성할 수 있다.
제5 실시양태에서, R2a는 t-부틸 또는 시클로헥실이고, R2는 제1 측면 또는 실시양태 1 내지 4 중 임의의 것에 제공된 바와 같다.
제6 실시양태에서, R2a는 t-부틸이고, R2는 제1 측면 또는 실시양태 1 내지 4 중 임의의 것에 제공된 바와 같다.
제7 실시양태에서, n은 0 내지 2이고, R2a 및 R2는 제1 측면 또는 실시양태 1 내지 6 중 임의의 것에 제공된 바와 같다.
제8 실시양태에서, n은 1이고, R2 및 R2a는 제1 측면 또는 실시양태 1 내지 6 중 임의의 것에 제공된 바와 같다.
제9 실시양태에서, 화합물은 하기 화합물 8이다:
<화합물 8>

제10 실시양태에서, 화합물은 하기 화합물 8B이다:
<화합물 8B>
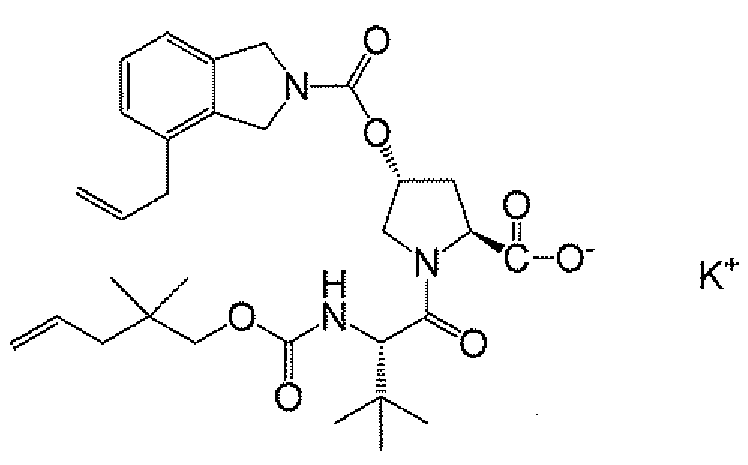
제11 실시양태에서, 화합물은 하기 화합물 8C이다:
<화합물 8C>

제4 측면에서, 화합물은 하기 화학식 III의 화합물 또는 그의 염이고, 여기서 바람직한 하위부류는 하기 화학식 IIIa의 화합물 또는 그의 염이다:
<화학식 III>

<화학식 IIIa>

상기 식에서, R3a, R3, 및 n은 제1 측면에서 정의된 바와 같다. R3이 H인 경우 염은 용이하게 제조될 수 있다. 화학식 III 및 IIIa의 화합물은 시스 및 트랜스 배열을 둘 다 포함한다. 본원에 기재된 방법은 시스와 트랜스의 혼합물을 제공한다.
제1 실시양태에서, R3은 H, C1 -6 알킬, C3-C8 시클로알킬, 페닐 또는 나프틸이다.
제2 실시양태에서, R3은 H, C1 -6 알킬 또는 C3-C8 시클로알킬이다.
제3 실시양태에서, R3은 C1 -6 알킬이다.
제4 실시양태에서, 화합물은 화학식 III 또는 IIIa의 염이다. 추가 실시양태에서, 염은 칼륨, 나트륨, 리튬, 1급 아민 (NH3 +-RC), 2급 아민 (NH2 +-(RC)2), 또는 3급 아민 (NH+-(RC)3)이고; 여기서 각각의 RC는 독립적으로 C1 -6 알킬, C3 -C8 시클로알킬, 또는 아릴이되; 단, 2개의 RC는 함께 NH+ 및 -(CH2)n- (여기서 n은 2 내지 7, 바람직하게는 5 또는 6임)을 함유하는 3원 내지 8원 헤테로시클릭기를 형성할 수 있다.
제5 실시양태에서, R3a는 t-부틸 또는 시클로헥실이고, R3은 제1 측면 또는 실시양태 1 내지 4 중 임의의 것에 제공된 바와 같다.
제6 실시양태에서, R3a는 t-부틸이고, R3은 제1 측면 또는 실시양태 1 내지 4 중 임의의 것에 제공된 바와 같다.
제7 실시양태에서, n은 0 내지 2이고, R3a 및 R3은 제1 측면 또는 실시양태 1 내지 6 중 임의의 것에 제공된 바와 같다.
제8 실시양태에서, n은 1이고, R3 및 R3a는 제1 측면 또는 실시양태 1 내지 6 중 임의의 것에 제공된 바와 같다.
제9 실시양태에서, 화학식 III 화합물은 하기 화합물 9이다:
<화합물 9>

제5 측면에서, 화합물은 하기 화합물 6A 또는 그의 염이다:
<화합물 6A>

추가 실시양태에서, 염은 칼륨, 나트륨, 리튬, 1급 아민 (NH3 +-RC), 2급 아민 (NH2 +-(RC)2), 또는 3급 아민 (NH+-(RC)3)이고; 여기서 각각의 RC는 독립적으로 C1 -6 알킬, C3 -C8 시클로알킬, 또는 아릴이되; 단, 2개의 RC는 함께 NH+ 및 -(CH2)n- (여기서 n은 2 내지 7, 바람직하게는 5 또는 6임)을 함유하는 3원 내지 8원 헤테로시클릭기를 형성할 수 있다. 추가 실시양태에서, 화합물은 화합물 6A의 시클로헥실아민 또는 디시클로헥실아민 염이다.
제6 측면에서, 화합물은 하기 화합물 3 또는 그의 염이다:
<화합물 3>

한 실시양태에서, 염은 HCl, HBr, HI, H3PO4, H2SO4, TsOH (파라-톨루엔술폰산), MsOH (메탄술폰산), 벤젠술폰산, AcOH, Cl3CCO2H, Cl2CHCO2H. ClCH2CO2H, 또는 CF3CO2H이다.
제7 실시양태에서, 화합물은 하기 화합물 A-2이다:
<화합물 A-2>

제8 실시양태에서, 화합물은 하기 화합물 A-4이다:
<화합물 A-4>
제9 실시양태에서, 화합물은 하기 화합물 A-10 또는 그의 염이다:
<화합물 A-10>
제10 실시양태에서, 화합물은 하기 화합물 B-6 또는 그의 염이다:
<화합물 B-6>

본원에서 사용된 용어는 그의 통상적인 의미를 갖고 그러한 용어의 의미는 각 경우에 독립적이다. 그럼에도 불구하고 달리 명시되어 있는 경우를 제외하고, 하기 정의가 명세서 및 특허청구범위 전반에 걸쳐 적용된다. 화학명, 통칭, 및 화학 구조는 동일 구조를 기술하는데 교체사용될 수 있다. 화합물은 화학 구조 및 화학명 둘 다를 사용하여 언급되며 구조와 명칭 사이에 다의성(ambiguity)이 존재할 경우, 구조가 우위를 차지한다. 이들 정의는 달리 지시되지 않는 한, 용어가 그 자체로든 또는 다른 용어와 함께 사용되든 상관없이 적용된다. 그런 이유로, "알킬"의 정의는 "알킬"뿐만 아니라 "히드록시알킬", "할로알킬", "-O-알킬" 등의 "알킬" 부분에 적용된다.
용어 "알킬"은 특정된 범위로 탄소 원자의 수를 갖는 1가 직쇄 또는 분지쇄, 포화 지방족 탄화수소 라디칼을 지칭한다. 따라서, 예를 들어, "C2 -6 알킬" (또는 "C2-C6 알킬")은 헥실 알킬 및 펜틸 알킬 이성질체뿐만 아니라 n-, 이소-, sec- 및 tert- 또는 t-부틸, n- 및 이소프로필, 및 에틸 중 임의의 것을 지칭한다. 또 다른 예로서, "C1 -4 알킬"은 n-, 이소-, sec- 및 t-부틸, n- 및 이소프로필, 에틸, 및 메틸을 지칭한다.
용어 "아릴"은 페닐, 치환된 페닐, 나프틸, 또는 치환된 나프틸을 지칭하되, 단, 치환된 페닐 및 치환된 나프틸은 각각은 1 내지 5개의 독립적으로 선택된 치환기를 갖는다. 아릴 치환기는 상기 제1 측면에 설명되어 있다.
용어 "시클로알킬"은 특정된 범위로 탄소 원자의 수를 갖는 알칸의 임의의 모노시클릭 환을 지칭한다. 따라서, 예를 들어, "C3 -8 시클로알킬" (또는 "C3-C8 시클로알킬")은 시클로프로필, 시클로부틸, 시클로펜틸, 시클로헥실, 시클로헵틸, 및 시클로옥틸을 지칭한다.
용어 "할로겐" (또는 "할로")은 플루오린, 염소, 브로민 및 아이오딘을 지칭한다 (대안적으로 플루오로, 클로로, 브로모, 및 아이오도로 지칭됨).
용어 "할로알킬"은 1개 이상의 수소 원자가 할로겐 (즉, F, Cl, Br 및/또는 I)으로 대체되어 있는 상기 정의된 바와 같은 알킬기를 지칭한다. 따라서, 예를 들어, "C1 -6 할로알킬" (또는 "C1-C6 할로알킬")은 1개 이상의 할로겐 치환기를 갖는 상기 정의된 바와 같은 C1 내지 C6 선형 또는 분지형 알킬기를 지칭한다. 용어 "플루오로알킬"은 할로겐 치환기가 플루오로로 제한되는 것을 제외하고는 유사한 의미를 갖는다. 적합한 플루오로알킬에는 시리즈 (CH2)0-4CF3 (즉, 트리플루오로메틸, 2,2,2-트리플루오로에틸, 3,3,3-트리플루오로-n-프로필 등)가 포함된다.
본원에 기재된 화합물에서의 원자는 그의 본질적 동위원소 존재율을 나타낼 수 있거나, 1개 이상의 원자는 동일 원자수를 갖지만, 주로 자연 상태에서 발견되는 원자 질량 또는 질량수와 상이한 원자 질량 또는 질량수를 갖는 특정 동위 원소에서 인위적으로 농축(enrich)될 수 있다. 본 발명은 본원에 기재된 화합물의 모든 적합한 동위 원소 변형을 포함하는 것으로 여겨진다. 예를 들어, 수소 (H)의 상이한 동위 원소 형태에는 경수소 (1H) 및 중수소 (2H)가 포함된다. 경수소는 자연에서 발견되는 지배적인 수소 동위 원소이다. 중수소의 농축은 치료상 이점, 예컨대 생체내 반감기 증가 또는 투여 요건 감소를 제공해 줄 수 있거나, 생체 시료의 특성화의 표준으로서 유용한 화합물을 제공할 수 있다. 동위 원소-농축된 화합물은 과도한 실험 없이 당업자에게 주지된 통상적인 기술에 의해 또는 본원에서 반응식 및 실시예에 기재된 것과 유사한 공정에 의해 적절한 동위 원소-농축된 시약 및/또는 중간체를 사용하여 제조될 수 있다.
II
.
헤테로사이클
합성
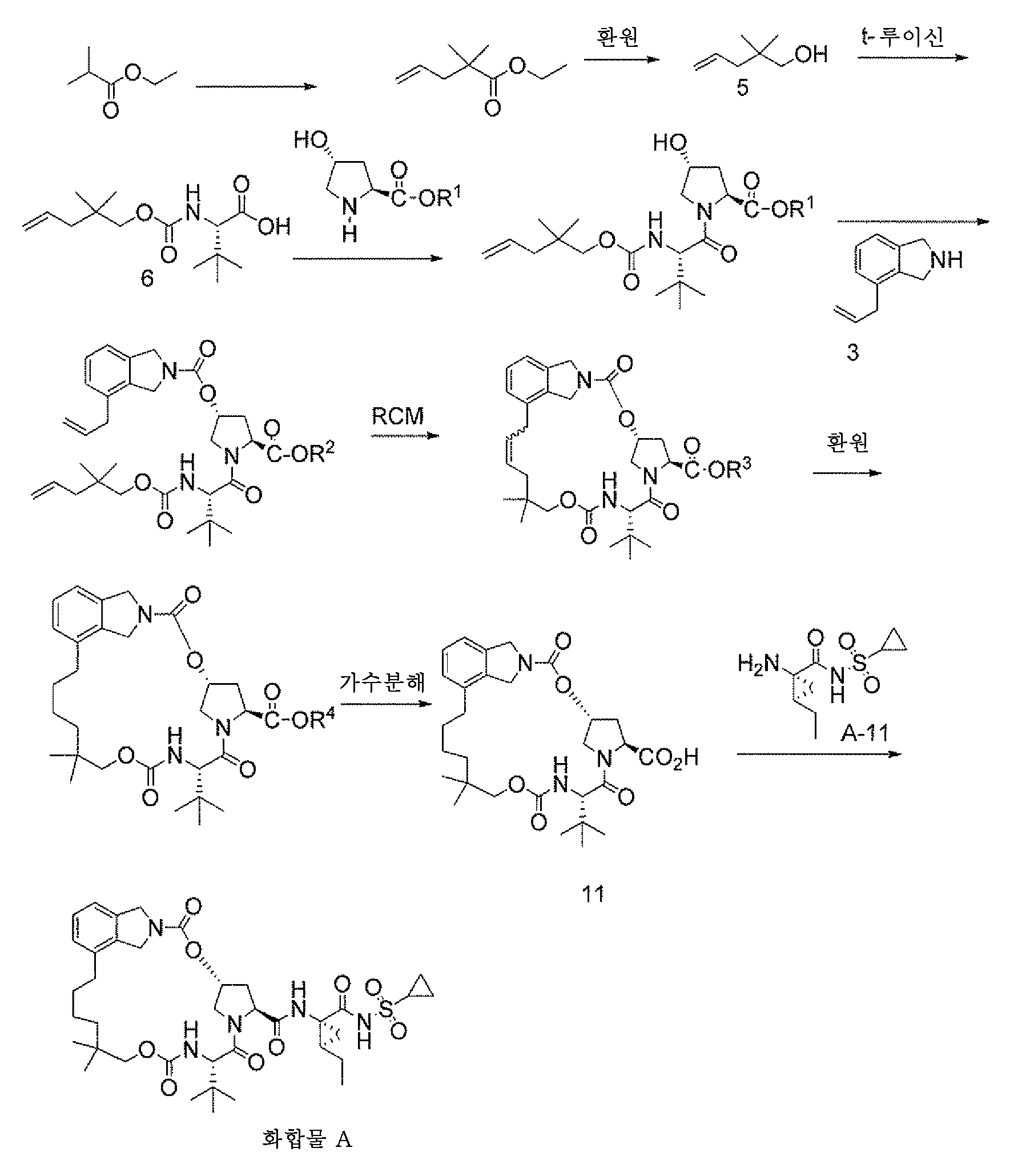
반응식 A는 알릴-이소인돌린 (화합물 5), 및 화합물 5를 제조하는데 사용될 수 있는 상이한 화합물의 제조를 설명한다:
<반응식 A>

헤테로시클릭 형성에 관한 제1 측면에서, 화합물 3 또는 그의 염은 다음 단계를 포함하는 방법에 의해 제조된다:

적합한 반응 조건은 Pd-촉매 조건하에 화합물 2와 알릴 마그네슘 클로라이드의 교차-커플링을 포함한다. 또한, 반응은 문헌 [Michael J. Zacuto et al., "Preparation of 4- Allylisoindoline via a Kumada Coupling with Allylmagnesium Chloride," 15(1) Organic Process Research 158 (2011, 온라인에 게재됨 (2010년 12월 6일)]에 설명되어 있다. (청구된 발명에 대한 선행 기술로 인정되지 않음.)
제1 실시양태에서, 화합물 2 또는 그의 염은 다음을 포함하는 방법에 의해 제조된다:

제1 반응은 염기 및 알킬포르메이트를 사용하여 수행한다. 상이한 염기의 예는 리튬 디이소프로필 아미드(LDA); 및 리튬, 나트륨, 또는 칼륨 헥사메틸디실라잔을 포함한다. 적합한 용매는 에테르 용매, 예컨대 디에틸에테르, 테트라히드로푸란(THF), 메틸-THF, 메틸-t-부틸 에테르(MTBE), 디글림, 및 디메톡시에탄을 포함한다. 일반적 온도 범위는 -20℃ 내지 -78℃이다.
화합물 2를 제공하는 화합물 1의 후속 환원에 적합한 반응 조건은 BF3 또는 에테레이트의 존재하에 수소화붕소나트륨을 사용하는 것을 포함한다. 적합한 용매는 비양성자성 유기물이다. 비양성자성 용매의 예는 예컨대 톨루엔, 크실렌, 클로로벤젠, 및 디클로로벤젠을 포함한다. 일반적 온도 범위는 약 100℃ 내지 약 130℃이다.
또 다른 실시양태에서, 화합물 2 또는 그의 염은  이다.
이다.
III
.
측쇄
합성
반응식 B, C, 및 D는 상이한 화합물의 제조를 설명한다. 이들 반응식에 제공되는 단계 각각은 상이한 실시양태를 나타낸다. 추가 실시양태는 상류(upstream) 및/또는 하류(downstream) 단계의 임의의 단계에 의해 제공된다.
<반응식 B>

반응식 B에 관한 측면에서, 화합물 A-3은 다음 단계를 포함하는 방법에 의해 제조된다:

적합한 반응 조건은 60℃ 내지 100℃에서 비양성자성 용매, 예컨대 N,N-디메틸포름아미드(DMF), 디메틸아세트아미드(DMAc), N-메틸 피롤리돈(NMP), 또는 디메틸술폭시드(DMSO) 중 무기 염기, 예컨대 K2CO3, Cs2CO3, CsF, 및 K3PO4의 존재하에 A-2를 가열하는 것을 포함한다.
<반응식 C>

반응식 C의 상이한 측면 및 실시양태는 상이한 단계 각각의 단독, 또는 상류 또는 하류 단계와의 임의의 조합에 관한 것이다. 예를 들어, 한 실시양태는 다음에 관한 것이다:

적합한 조건은 알릴 또는 벤질 알콜, 및 촉매량의 Ti(OtBu)4의 사용을 포함한다. 적합한 용매는 비양성자성 용매, 예컨대 톨루엔, 벤젠, 및 크실렌, 및 클로로벤젠을 포함한다. 일반적 온도는 65℃ 내지 100℃이다.
또 다른 실시양태는 다음과 같다:

적합한 반응 조건은 알콜성 용매, 예컨대 메탄올, 에탄올, 프로판올 및 부탄올의 사용을 포함한다. 일반적 온도 범위는 20℃ 내지 50℃이다.
추가적 단계를 포함하는 실시양태는 다음과 같다:
추가적 단계에 관한 적합한 조건은 이하의 실시예에 제공되어 있다.
추가적 측면은 화합물 A-8, A-9, 및 A-10 또는 그의 염; 및 실질적으로 입체화학적으로 순수한 A-6, A-7, A-8, A-9, 또는 A-10 또는 그의 염에 관한 것이다. 실질적으로 입체화학적으로 순수한은 명시된 입체이성질체가 다른 이성질체보다 상당한 정도로 존재한다는 것을 의미한다. 상이한 실시양태에서, 명시된 입체이성질체는 존재할 수 있는 다른 입체이성질체에 비해 적어도 80%, 적어도 85%, 적어도 90%, 적어도 95% 또는 적어도 99% 초과를 이룬다.
대안적 반응식은 반응식 D에 의해 제공된다:
<반응식 D>

반응식 D의 상이한 측면 및 실시양태는 상이한 단계 각각의 단독, 또는 상류 또는 하류 단계와의 임의의 조합에 관한 것이다. 추가적 측면은 벤조아민 염으로서의 화합물 B5, 및 화합물 B6, B7, 및 B8 및 그의 염을 포함한다.
IV
.
마크로락탐
제조
마크로락탐 형성 방법, 마크로락탐 형성을 위한 중간체 제조 방법, 및 측쇄 첨가 방법이 반응식 E에 설명되어 있다. 반응식 E는 바람직한 기를 사용하는 마크로락탐 제조를 설명한다. 예를 들어, 화학식 I, II 또는 III 화합물을 사용하는 대안적인 마크로락탐은, 본원에 제공된 지침을 기반으로 하여 제조될 수 있다.
<반응식 E>
마크로락탐 형성에 관한 제1 측면은
a) 하기 화학식 IIa의 화합물 또는 그의 염을 폐환 및 수소화시켜 하기 화학식 IV의 화합물 또는 그의 염을 형성하는 단계를 포함하는 방법에 관한 것이다:
<화학식 IIa>

<화학식 IV>

상기 식에서, R2는 상기 섹션 I. 중간체의 제1 측면에 정의된 바와 같고, R4는 H, C1 -6 알킬, C3-C8 시클로알킬, 또는 아릴이다.
제2 측면은
a) 화학식 IIa의 화합물 또는 그의 염을 폐환 및 수소화시켜 화학식 IV의 화합물 또는 그의 염을 형성하는 단계를 포함하고, 추가로
b) 화학식 IV의 화합물 또는 그의 염을 가수분해시켜 하기 화합물 1 또는 그의 염을 형성하는 단계;
c) 화합물 1 또는 그의 염을 하기 화합물 A-11 또는 그의 염에 커플링시켜 화합물 A 또는 그의 염을 형성하는 단계; 및
d) 임의로, 화합물 A 또는 그의 염을 제약상 허용되는 염으로 전환시키는 단계
를 포함하는, 화합물 A의 제조 방법에 관한 것이다:
<화합물 1>

<화합물 A-11>
폐환에 적합한 조건은 비양성자성 용매, 예컨대 IPAc, 톨루엔, 크실렌, 메시틸렌, 및 벤젠을 포함한다. 일반적 온도 범위는 80℃ 내지 120℃이다.
가수분해를 위한 적합한 조건은 알콜성 용매 중 0℃ 내지 50℃ (바람직하게는 실온)의 온도 범위에서 가성 염기를 사용하는 것을 포함한다. 적합한 염기의 예는 수산화리튬, 수산화칼륨, 및 수산화나트륨을 포함한다. 적합한 알콜성 용매는 메탄올, 에탄올, 프로판올, 및 부탄올을 포함한다.
화합물 A-11을 커플링시키기에 적합한 조건은 커플링 시약, 비양성자성 유기 용매 및 피리딘 또는 피리딘 유도체를 사용하는 것을 포함한다. 일반적 온도는 0℃ 내지 50℃ (바람직하게는 실온)이다. 커플링 시약의 예는 디시클로헥실카르보디이미드(DCC), N,N'-디이소프로필카르보디이미드(DIC), 1-에틸-3-(3-디메틸아미노프로필) 카르보디이미드 염화수소 (EDC-HCl) 및 1-에틸-3-(3-디메틸아미노프로필) 카르보디이미드 (EDC)를 포함한다. 비양성자성 유기 용매의 예는 아세토니트릴, THF, IPAc 및 톨루엔을 포함한다. 한 실시양태에서, EDC가 사용된다.
커플링을 위해 HOBt 대신에 피리딘 또는 피리딘 유도체를 사용하는 것은 더 높은 수율 및 프롤린 α-중심에서의 더 적은 에피머화를 비롯한 몇몇 이점을 제공해 준다. 게다가, HOBt는 건조 상태에서 충격 민감성이다.
바람직한 피리딘 유도체는 3 및 4 위치에서 전자 공여 또는 중성 R기를 갖는다. 피리딘 및 유도체를 포함하는 일반 구조의 예는 다음을 포함한다:

상기 식에서, R5는 수소, 아릴, 할로겐, C1 -6 알킬, O-C1 -6 알킬 또는 C3-C8 시클로알킬이다.
바람직한 시약은 피리딘 및 4-페닐피리딘, 4-알킬피리딘, 메틸피리딘, 3- 또는 4-모노 또는 디알킬피리딘이고, 여기서 알킬기는 C1 -6 알킬일 수 있다.
제3 측면은 화합물 1을 피리딘 또는 피리딘 유도체를 사용하여 화합물 A-11과 커플링시키는 단계를 포함하는, 화합물 A의 제조 방법에 관한 것이다. 바람직하게는, 검출가능한 HOBt가 전혀 존재하지 않는다.
추가적 실시양태는 다음을 포함한다:
제1 실시양태에서, 화학식 IIa 화합물의 R2는 H, C1 -6 알킬, C3-C8 시클로알킬, 페닐 또는 나프틸이다.
제2 실시양태에서, 화학식 IIa 화합물의 R2는 H, C1 -6 알킬 또는 C3-C8 시클로알킬이다.
제3 실시양태에서, 화학식 IIa 화합물의 R2는 C1 -6 알킬이다.
제4 실시양태에서, 화합물은 화학식 IIa의 염이다. 추가 실시양태에서, 염은 칼륨, 나트륨, 리튬, 1급 아민 (NH3 +-RC), 2급 아민 (NH2 +-(RC)2), 또는 3급 아민 (NH+-(RC)3)이고; 여기서 각각의 RC는 독립적으로 C1 -6 알킬, C3 -C8 시클로알킬, 또는 아릴이되; 단, 2개의 RC는 함께 N 및 -(CH2)n- (여기서 n은 2 내지 7, 바람직하게는 5 또는 6임)을 함유하는 3원 내지 8원 헤테로시클릭기를 형성할 수 있다.
제5 실시양태에서, 화학식 IIa 화합물 또는 그의 염은 화합물 8이다.
제6 실시양태에서, 화학식 IIa 화합물 또는 그의 염은 화합물 8B이다.
제7 실시양태에서, 화학식 II 화합물 또는 그의 염은 화합물 8C이다.
제8 실시양태에서, 화학식 IV 화합물의 R4는 H, C1 -6 알킬, C3 -C8 시클로알킬, 페닐 또는 나프틸이고, R2는 제1 측면 또는 실시양태 1 내지 8 중 임의의 것에 제공된 바와 같다.
제9 실시양태에서, 화학식 IV 화합물의 R4는 C1 -6 알킬이고, 화학식 IIa 화합물 또는 그의 염은 제1 측면 또는 제2 측면, 또는 실시양태 1 내지 8 중 임의의 것에 제공된 바와 같다.
제10 실시양태에서, 화학식 IV의 화합물 또는 그의 염은 하기 화합물 11 또는 그의 염이고, 화학식 IIa 화합물 또는 그의 염은 제1 측면 또는 제2 측면, 또는 실시양태 1 내지 8 중 임의의 것에 제공된 바와 같다:
<화합물 11>

R2 및 R4는 바람직하게는 특정 폐환 및 수소화 반응을 위해 동일하다. 그러나 R2 및 R4는, 예를 들어, R2가 폐환 후 및 환원 이전에 변형될 경우, 상이할 수 있다.
제11 실시양태에서, 방법은 하기 화학식 Ia의 화합물을 또는 그의 염과 커플링시키는 단계를 포함하는, 화학식 IIa의 화합물 또는 그의 염을 제조하는 단계를 추가로 포함한다:
또는 그의 염과 커플링시키는 단계를 포함하는, 화학식 IIa의 화합물 또는 그의 염을 제조하는 단계를 추가로 포함한다:
<화학식 Ia>
상기 식에서, R1은 상기 섹션 I. 중간체에서 정의된 바와 같다.
제12 실시양태에서, 방법은 하기 화합물 6A 또는 그의 염과  또는 그의 염 (여기서 R1은 상기 섹션 I. 중간체에서 정의된 바와 같음)을 커플링시켜 화학식 I의 화합물을 제조하는 단계를 추가로 포함한다:
또는 그의 염 (여기서 R1은 상기 섹션 I. 중간체에서 정의된 바와 같음)을 커플링시켜 화학식 I의 화합물을 제조하는 단계를 추가로 포함한다:
<화합물 6A>

제13 실시양태에서, 제11 또는 제12 실시양태에 관한 R1은 H, C1 -6 알킬, 또는 C3 -C8 시클로알킬이다. 추가 실시양태에서, R1은 메틸이다.
제14 실시양태에서, 화합물 6A 또는 그의 염은

제15 실시양태에서,
 은 하기 단계를 포함하는 공정에 의해 제조된다:
은 하기 단계를 포함하는 공정에 의해 제조된다:
제16 실시양태에서, 폐환은, 촉매 및 화학식 IIa의 화합물을 용매에 대략 동시에 서서히 동시 첨가함으로써 수행하고, 여기서
용매는 기질 1 Kg 당 약 5 내지 25 리터, 바람직하게는 기질 1 Kg 당 약 10 L로 제공되고;
촉매는 촉매 1 Kg 당 약 250 ml 내지 3 L, 바람직하게는 촉매 1 Kg 당 약 1 L의 농도로 제공되고;
화합물은 기질 1 Kg 당 약 500 ml 내지 6 L, 바람직하게는 기질 1 Kg 당 약 2 L의 농도로 제공되고;
화합물-용액, 촉매-용액 및 용매를 0.5 내지 2.5시간의 기간에 걸쳐, 바람직하게는 약 1.25시간에 걸쳐 함께 배합한다.
상이한 용매, 촉매, 및 온도 범위를 사용하여 반응을 수행할 수 있다. 일반적 온도 범위는 50℃ 내지 150℃이다. 상이한 유형의 유기 및 무기 용매를 이용할 수 있다. 용매의 예는 톨루엔, 벤젠, 아세토니트릴, 디클로로에탄, 디클로로메탄, 이소프로필아세테이트, 에틸아세테이트, 및 알콜 (예를 들어, 이소프로판올, 메탄올, 및 에탄올)을 포함한다. 적합한 촉매의 예는 N-헤테로시클릭 카르벤 루테늄-알킬리덴, 포스폰 루테늄-알킬리덴 몰리브덴-알킬리덴, 루테늄-카르벤, 및 몰리브덴-카르벤을 포함한다. 조건의 바람직한 설정은 80℃ 내지 110℃의 온도 범위에서 톨루엔, 및 촉매 그럽스-호베이다(Grubbs-Hoveyda) II를 사용하는 것이다.
V. 염
적절한 관능기를 갖는 본원에 기재된 화합물을 염으로서 제공할 수 있다. 환자를 치료하기 위해 제약상 허용되는 염을 화합물과 함께 사용할 수 있다. 그러나, 비-제약상 염은 중간체 화합물의 제조에 유용할 수 있다.
제약상 허용되는 염은 환자, 바람직하게는 인간에게 투여하기에 적합하다. 적합한 염은, 예를 들어, 화합물의 용액을 제약상 허용되는 산, 예컨대 염산, 황산, 아세트산, 트리플루오로아세트산, 또는 벤조산의 용액을 혼합함으로써 형성될 수 있는 산 부가염을 포함한다. 산성 모이어티를 갖는 화합물은 적합한 제약상 허용되는 염과 혼합하여, 예를 들어, 알칼리 금속염 (예를 들어, 나트륨 또는 칼륨 염), 알칼리 토금속염 (예를 들어, 칼슘 또는 마그네슘 염), 및 적합한 유기 리간드와 함께 형성된 염, 예컨대 4급 암모늄 염을 제공할 수 있다. 또한, 산 (-COOH) 또는 알콜기가 존재하는 경우에, 화합물의 용해도 또는 가수분해 특성을 개질시키기 위하여 제약상 허용되는 에스테르가 사용될 수 있다.
VI
. 투여 및 조성물
치료에의 응응을 갖는 화합물, 예컨대 화합물 A를 HCV로 감염된 환자에게 투여할 수 있다. 용어 "투여" 및 그의 변형 (예를 들어, 화합물을 "투여함")은 치료를 필요로 하는 개체에게 화합물 또는 화합물의 전구약물을 제공함을 의미한다. 화합물이 1종 이상의 다른 활성제 (예를 들어, HCV 감염을 치료하는데 유용한 항바이러스제)와 병용하여 제공되는 경우에, "투여" 및 그의 변형은 각각 화합물 또는 염 및 다른 작용제의 동시 및 순차적 제공을 포함하는 것으로 이해된다.
본원에 사용된 바와 같이, 용어 "전구약물"은 그것이 투여되는 개체의 신체에서 효소, 화학 물질 또는 대사과정의 작용에 의해 활성 약물 형태 또는 화합물로 전환되는 비활성 약물 형태 또는 화합물을 포함하는 것으로 의도된다.
본원에 사용된 바와 같이, 용어 "조성물"은 특정된 성분을 포함하는 생성물뿐만 아니라, 특정된 성분을 배합함으로써 직접 또는 간접적으로 생성되는 임의의 생성물을 포함하는 것으로 의도된다.
"제약상 허용되는"이란 대상체에게 투여하기에 적합함을 의미한다.
본원에 사용된 바와 같은 용어 "대상체" (대안으로 본원에서 "환자"로 지칭됨)는 치료, 관찰 또는 실험의 대상이 되는 동물, 바람직하게는 포유류, 가장 바람직하게는 인간을 지칭한다.
용어 "유효량"은 치료학적 또는 예방학적 효과를 발휘하기에 충분한 양을 나타낸다. HCV로 감염된 환자의 경우, 유효량은 다음 효과 중 하나 이상을 달성하기에 충분하다: 복제하는 HCV의 능력을 감소시키고, HCV 농도(load)를 감소시키고, 바이러스 제거(clearance)를 증가시킨다. HCV에 감염되지 않은 환자의 경우, 유효량은 다음 중 하나 이상을 달성하기에 충분하다: HCV 감염에 대한 감수성(susceptibility) 감소, 및 만성 질환에 관한 지속적인 감염을 확립하는 감염 바이러스의 능력의 감소.
HCV NS3 프로테아제를 억제하고 HCV 감염을 치료하고/하거나 HCV 감염의 증상의 가능성 또는 중증도를 저하시키기 위하여, 화합물은, 임의로 염의 형태로, 활성제를 활성제의 작용 부위와 접촉하게 하는 수단에 의해 투여될 수 있다. 이들은 개별적인 치료제로서 또는 치료제의 병용물로서 제약과 함께 사용하기에 유용한 통상적인 수단에 의해 투여할 수 있다. 이들은 단독으로 투여될 수 있지만, 전형적으로는 선택된 투여 경로 및 표준 제약 실무에 근거하여 선택되는 제약 담체와 함께 투여된다.
화합물은, 예를 들어, 다음의 경로들 중 하나 이상에 의해 투여될 수 있다: 유효량의 화합물 및 통상적인 무독성의 제약상 허용되는 담체, 아쥬반트 및 비히클을 함유하는 제약 조성물의 단위 투여 형태로, 경구, 비경구 (피하 주사, 정맥내, 근육내, 흉골내 주사 또는 주입 기술을 포함함), 흡입에 의한 투여 (예컨대 분무 형태로), 또는 직장내. 경구 투여에 적합한 액체 제제 (예를 들어, 현탁액, 시럽, 엘릭시르 등)는 당 분야에 공지된 기술에 따라 제조할 수 있으며, 통상적인 매질, 예컨대 물, 글리콜, 오일, 알콜 등 중의 임의의 것을 사용할 수 있다. 경구 투여에 적합한 고체 제제 (예를 들어, 분말, 환제, 캡슐 및 정제)는 당 분야에 공지된 기술에 따라 제조할 수 있으며, 고체 부형제, 예컨대 전분, 당, 카올린, 윤활제, 결합제, 붕해제 등을 사용할 수 있다. 비경구용 조성물은 당 분야에 공지된 기술에 따라 제조할 수 있으며, 전형적으로는 담체로서 멸균수 및 임의로 다른 성분, 예컨대 용해도 보조제를 사용할 수 있다. 주사용 액제는 당 분야에 공지된 방법에 따라 제조할 수 있으며, 이때 담체는 생리 식염수, 글루코스 용액 또는 생리 식염수와 글루코스의 혼합물을 함유하는 용액을 포함한다. 제약 조성물의 제조시 사용하기에 적합한 방법에 대한 추가 지침이 문헌 [Remington's Pharmaceutical Sciences, 20th edition (ed. A.R. Gennaro, Mack Publishing Co., 2000)]에 제공되어 있다.
치료 화합물은 단일 용량으로 또는 분할 용량으로 하루에 포유동물 (예: 인간) 체중 1 ㎏당 0.001 내지 1000 ㎎의 투여량 범위로 경구 투여할 수 있다. 한 투여량 범위는 단일 용량으로 또는 분할 용량으로 경구로 하루에 체중 1 ㎏당 0.01 내지 500 ㎎이다. 또 다른 투여량 범위는 단일 또는 분할 용량으로 경구로 하루에 체중 1 ㎏당 0.1 내지 100 ㎎이다. 경구 투여시, 조성물은 치료할 환자에 대한 투여량의 증상적 조정을 위해 활성 성분 1.0 내지 500 ㎎, 특히 활성 성분 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 및 750 ㎎을 함유하는 정제 또는 캡슐의 형태로 제공될 수 있다. 임의의 특정 환자의 경우 구체적 용량 수준 및 투여의 빈도를 달리할 수 있고, 사용된 구체적 화합물의 활성, 대사적 안정성 및 화합물의 작용 기간, 연령, 체중, 전체적인 건강, 성별, 식이, 투여 형태와 시간, 배설률, 약물 병용, 특정 병태의 중증도, 및 치료를 수행하는 숙주를 포함한 다양한 요인에 따라 좌우될 것이다.
VII
.
HCV
억제 활성
HCV NS3 활성, HCV 레플리콘 활성, 및 HCV 복제 활성을 억제하는 화합물의 능력은 당 분야에 주지된 기술을 사용하여 평가될 수 있다. (예를 들어, 문헌 [Carroll et al., J. Biol . Chem . 278: 11979-11984, 2003] 참조.) 한 그러한 검정법은 하기 기재 및 문헌 ([Mao et al., Anal . Biochem . 373:1-8, 2008] 및 [Mao et al., WO2006/102087])에 기재된 바와 같은 HCV NS3 프로테아제 시간-분해 형광 (TRF) 검정법이다.
VIII
.
실시예
하기 제공된 실시예는 본 발명 및 그의 실시를 설명하고자 하는 것이다. 특허청구범위에 달리 제공되지 않는 한, 실시예는 발명의 범위 또는 취지에 대한 제한으로서 해석되지 않는다.
본원에서 사용된 약어는 다음을 포함한다:
MTBE = 메틸-tert-부틸 에테르
CPME= 시클로펜틸 메틸 에테르
DMAC = 디메틸아세트아미드
DCM = 디클로로메탄
DMF = 디메틸포름아미드
THF = 테트라히드로푸란
DPPM = 디페닐포스피노메탄
DPPE = 디페닐포스피노에탄
DPPP = 디페닐포스피노프로판
LDA = 리튬 디이소프로필아미드
PhMe = 톨루엔
IPA = 이소프로필 알콜
IPAc = 이소프로필 아세테이트
RB = 환저
TEA = 트리에틸아민
CDI = 1,1'-카르보닐디이미다졸
EDC-HCl = 1-에틸-3-(3-디메틸아미노프로필)카르보디이미드 히드로클로라이드
DI = 탈이온화됨
GH-II = 그럽스-호베이다 제2 세대 촉매 - (1,3-비스-(2,4,6-트리메틸페닐)-2-이미다졸리디닐리덴)디클로로(o-이소프로폭시페닐메틸렌)루테늄)
DIPEA = 후니그(hunig's) 염기 = 디이소프로필에틸아민
실시예
1:
측쇄
합성
화합물 A11을 본 실시예에 기재된 방법을 사용하여 제조하였다. 본 실시예에 기재된 화합물 및 방법은 본 발명의 상이한 측면 및 실시양태를 제공한다.
1. 활성화
메틸-tert-부틸 에테르(MTBE) (25.00 ml) 및 물 (15.00 ml) 중 플루오린화칼륨 (3.28 g, 56.5 mmol)의 혼합물에 3-클로로프로판술포닐 클로라이드 (5.0 g, 28.2 mmol)를 첨가하였다. 혼합물을 주위 온도에서 12시간 동안 교반하였다. MTBE 층을 분리하고 물 (25.00 ml)로 세척하고 농축하여 액체: 3-클로로프로판술포닐 플루오라이드 (A-2, 4.54 g, 28.2 mmol, 98% 수율)를 수득하였다.
2.
환화
및
아미드화

디메틸 아세트아미드 (DMAc, 16.00 ml) 중 클로로프로판술포닐 플루오라이드 (3.2 g, 19.93 mmol) 및 K2CO3 (5.5 g, 40 mmol)의 혼합물을 65℃ 내지 75℃에서 12시간 동안 가열하여 반응을 완료시켰다. 혼합물을 실온으로 냉각하고, 여과하고 무기물 케이크를 DMAc (8 mL)로 세척하였다. 여액 및 세척액을 합한 후, 수성 암모니아 (11.31 g, 199 mmol)를 첨가하였다. 혼합물을 밀폐 용기에서 또 다른 12시간 동안 65℃ 내지 75℃에서 가열하여 시클로프로필 술포닐 아미드 A-4 (85% 분석 수율(assayed yield))를 수득하였다.
3.
디올
보호

절차 A
:
디클로로메탄(DCM) (0.3 mL) 중 (S)-1,2-부탄디올 (100 mg, 1.1 mmol)의 빙냉 용액에 DCM (0.2 mL) 중 티오닐 클로라이드 (0.1 mL, 1.35 mmol)를 충전한 다음, 빙조를 제거하고 반응물을 2시간 동안 주위 온도에서 숙성시켜 1H NMR 관측에 의해 완료되었다. 반응물을 냉각조로 물에 의해 켄칭하여 온도를 < 25℃로 유지하였다. 유기층을 물로 2회 세척하고 그 다음 단계에서 직접 사용하였다.
절차 B
:
빙냉 순수 (S)-1,2-부탄디올 (10.0 g, 110 mmol)에 냉각조로 서서히 티오닐 클로라이드 (8.42 mL, 115 mmol)를 충전하고, 제1 절반 첨가는 발열성으로서, 냉각조 및 첨가속도로 T < 40℃를 유지하고, 첨가의 제2 절반은 흡열성으로서, 냉각조를 제거하고 온조에 두어, T를 10℃ 내지 20℃로 유지하고, 첨가 동안 많은 HCl 가스가 형성되었고, 잘 환기하고 2N NaOH 용액으로 소거하였다. 실온에서 30분 동안 숙성시켰다. 반응이 완료되었고 NMR 또는 GC (2:3 dr 비)에 의해 관측하였다. 반응물을 EtOAc (80 mL)로 희석하고, 빙조와 함께 물 (80 mL)로 켄칭하고, T를 ~20℃ 내지 25℃로 유지하였다. 수성층을 분리(cut)하고, 유기층을 물 (100 mL)로 1회 세척 (최후 수성층 pH ~1 내지 2)하였다.
4. 산화
절차 A
:

MeCN (1.5 mL) 및 DCM (1.5 mL) 중 화합물 A-5 (0.5 g, 3.6 mmol)의 빙냉 용액에 물 (3 mL), 삼염화루테늄(III) (0.075 mg, 0.0036 mmol), 이어서 과아이오딘산나트륨 (0.85 g, 3.96 mmol)을 첨가하였다. 빙조를 제거하고 반응 혼합물을 슬러리화하여 주위 온도에서 2.5시간 후 반응이 완료되었다. NMR 또는 GC에 의해 반응을 관측하였다. 반응 슬러리를 솔카-플록(SOLKA-FLOC)을 통해 여과하여 침전물을 제거하고, 10 vol MTBE로 세정하였다. 유기층을 염수 (2 x 3 mL)로 세척하여 0.53 g 생성물 A-6 (97.4%, NMR에 의한 분석 수율)을 수득하였다.
절차 B
:

80 mL EtOAc 중 화합물 A-5 (110 mmol)의 상기 유기 용액에 물 (80 mL)을 충전하고, RuCl3ㆍH2O (11 mg, 0.055 mmol)를 충전하였다. 혼합물을 혼합물이 모두 용해될 때까지 ~10분 동안 교반하고 NaBrO3 (6.63 g, 44 mmol)를 ~40분 내에 조금씩 첨가 (온도 증가 지연 ~10분)하고, T <40℃를 유지하였다. 첨가 후, ~1 내지 2시간 동안 30℃에서 숙성시켜 NMR 또는 GC에 의해 관측된 바와 같이 완료시켰다. 유기층을 분리하고 수성층을 제거하고 EtOAc (30 mL)로 1회 역추출하였다. 유기층을 합하고 5 wt% 수성 NaHSO3 (60 mL) 및 염수 (60 mL)로 세척하였다. 유기층을 농축하고 그 다음 단계에서 MTBE 용액로서 사용하였다. 전형적인 NMR 또는 GC 전체 분석 수율: 85 내지 89%.
5. 알킬화

30 ml 아세토니트릴 (N2 하/기계 교반/수조) 중 LiOtBu (5.3 g, 66.2 mmol)의 슬러리에 10 mL 아세토니트릴 중 화합물 A-6 및 디에틸 말로네이트 (5.05 g, 31.5 mmol)의 용액을 첨가 깔대기를 통해 30분에 걸쳐 서서히 충전하였다 (반응 온도를 30℃ 미만으로 제어함). 반응물을 주위 온도에서 1시간 동안 및 40℃에서 2시간 동안 교반하였다. GC에 의해 관측된 바와 같이 반응물은 ~95% 전환을 제공하였다.
반응 혼합물을 40 mL 물로 켄칭하고, 40 mL MTBE로 추출하고 수성층을 20 mL MTBE로 1회 역추출하였다. 합해진 유기층을 농축하여 화합물 A-7을 투명 오일 (NMR 분석 ~89% 수율)로서 수득하였다.
비키랄(achiral) GC 조건: 레스텍(Restek) RTX-1 (15 m x 320 x 1 um) 등온성 130℃ 검출기 및 250℃로 설정된 주입 히터(inlet heater), 100:1 분할비, 9 psi로설정된 정압 모드 (유속 ~54 cm/초) 총 시행시간은 5분이었다.

6. 가수분해

150 mL MeOH 중 화합물 A-7 (52 g, 81% wt, 198 mmol)의 빙냉 용액에 물 (150 mL) 중 NaOH (10.75 g, 262 mmol)의 용액을 첨가 깔대기를 통해 20분에 걸쳐 서서히 충전하였다 (반응 온도를 20℃ 미만으로 제어함). 반응 슬러리를 서서히 가온하고 실온에서 밤새 교반하였다. 반응물을 빙조를 사용하여 13℃로 냉각하였다. 물 (250 mL), 이어서 에틸 아세테이트 (250 mL)를 충전하였다. 수성층을 에틸 아세테이트 (100 mL)로 추출하고, 수성층을 농염산 (25 mL)을 사용하여 pH 2.1로 산성화하였다. 생성된 수성층을 에틸 아세테이트 (2 x 200 mL)로 추출하였다. 합해진 유기층을 염수 (2 x 100 mL)로 세척하고, 진공하에 농축하여 화합물 A-8을 투명 오일 (35.6 g, 94 wt%, 90% 수율)로서 수득하였다.
HPLC 방법: 아센티스(Ascentis) R 익스프레스(Express) C18, 10 cm x 4.6 mm, 2.7 μ; 표준 구배: 6분 내에 10 내지 95%의 B (A=0.1% 인산, B= 아세토니트릴), 2분 유지, 2분 후(post); 유속: 1.8 ml/분; 210 nm, 40℃에서 UV 검출.

7.
쿠르티우스
배열

산 클로라이드 A-8a의 제조: 화합물 A-7 (2.0 g, 10 mmol)을 20 ml의 톨루엔에 용해시키고, 빙조를 사용하여 5℃로 냉각하고, DMF (0.079 g, 0.083 mmol)를 첨가한 다음, (COCl)2 (1.91 g, 15 mL)를 서서히 첨가하고 반응 온도를 <20℃로 유지하였다. 첨가 후, 반응물을 ~30분 내지 60분 동안 또는 GC 분석이 완전 전환을 나타낼 때까지 주위 온도에서 숙성시켰다. 반응물을 빙조로 냉각하고 물 (20 mL)로 켄칭하였다. 유기층을 10 wt% NaHCO3로 2회 (2 x 10 mL) 세척하여 pH를 ~8.0으로 만들었다. 톨루엔 함유 산 클로라이드 용액을 그 다음 단계에 직접 취하였다.
아실 아지드 A-8b의 제조: 플라스크 중 물 (12 mL)에 나트륨 아지드 및 황산수소테트라부틸암모늄 (0.18 g, 0.535 mmol)을 첨가하였다. 톨루엔 중 산 클로라이드 (A-8a)의 용액을 당해 나트륨 아지드 용액에 격렬히 교반 (>400 RPM)하면서 30 내지 60분에 걸쳐 첨가하였다. GC 분석이 완전 전환을 나타낼 때까지 혼합물을 주위 온도에서 ~1 내지 2시간 동안 교반하였다. 유기층을 분리하고, 1 M NaHCO3 (60 mL), 물 (50 mL) 및 염수로 순차적으로 세척하여 KF ~700 ppm으로 수분 함량을 갖는 용액을 수득하였다. 용액을 추가로 MgSO4 상에서 건조시키고, 여과하여 톨루엔 중 아실 아지드 용액 (KF ~100 ppm)을 수득하고 이를 다음 단계에 직접 취하였다.
화합물 A-8의 제조: 첨가 깔대기 및 응축기가 연결된 3-구 플라스크 (500 mL)를 진공화하고/N2로 3회 플러싱하였다. 톨루엔 (10 mL, 50 ppm 하에 KF)을 충전하고 95℃ (내부 온도)로 가열하였다. 가열된 톨루엔에 아실 아지드 용액을 60분에 걸쳐 충전하고 온도를 90℃ 내지 100℃에서 유지하였다. 첨가 후, 반응 용액을 당해 온도에서 ~1시간 동안 숙성시켰다. 반응물을 ~20℃로 냉각하고, 알릴 알콜 (0.94 g, 16.11 mmol), 이어서 Ti(Ot/Bu)4 (0.18 g, 0.54 mmol)를 첨가하고, GC 분석이 완전 전환을 나타낼 때까지 반응물을 주위 온도에서 교반하였다. 반응물을 1 N HCl (44 mL)로 켄칭하였다. 유기층을 물 및 염수로 세척하고, 농축하여 화합물 A-8을 담황색 액체로서 수득하였다.
HPLC 조건: 애질런트 이클립스(Aglient Eclipse) 플러스 C18, 4.6 x 50 mm, 1.8 μ; RT, 선형 구배: 5분 내에 10 내지 90%의 B (MeCN), 2분까지 유지; A: 0.1% H3PO4의 수용액; 유속: 1.0 ml/분; 210 nm에서 UV 검출.

8. 가수분해

톨루엔 중 화합물 A-8 (7.79 g, 32.3 mmol, 14 mL, ~2 용적)의 용액에 물 (10 mL) 중 NaOH (3.95 g, 96.9 mmol)의 용액을 주위 온도에서 충전하였다 (내부 온도가 28℃로 상승됨). 생성된 용액을 40℃에서 4시간 동안 가열 (90% 전환)한 다음, 주위 온도에서 밤새 교반하였다 (95% 전환).
반응물을 빙조를 사용하여 6℃로 냉각하고; 물 (70 mL) 및 톨루엔 (34 mL)을 충전하였다 (온도가 15℃로 상승됨). 수성층을 IPAc (30 mL)로 추출하였다. 잔류 수성층을 빙조를 사용하여 냉각하고 5N HCl (35 mL)을 사용하여 pH 2.1로 산성화하고 에틸 아세테이트로 2회 (1 x 50 ml, 1 x 30 mL) 추출하였다. 합해진 유기층을 물 30 mL 및 염수 30 mL (pH 1.9)로 세척하고, MgSO4 상에서 건조시키고, 여과하고 농축하여 화합물 A-9를 투명 액체로서 수득하였다. (5.93 g, NMR에 의한 화합물 A-8로부터의 86% 분석 수율).
HPLC 방법: 아센티스 R 익스프레스 C18, 10 cm x 4.6 mm, 2.7 μ; 표준 구배: 6분 내에 10 내지 95%의 B (A=0.1% 인산, B= 아세토니트릴), 2분 유지, 2분 후; 유속: 1.8 ml/분; 210 nm, 40℃에서 UV 검출.
9.
CDI
커플링

무수 에틸 아세테이트 (33 mL) 중 화합물 A-9 (3.24 g, 97W%, 14.74 mmol)의 용액에 N2 하 CDI (2.92 g, 17.69 mmol, 1.1 eq)를 충전하였다. 3분 후, 반응물을 1시간 동안 40℃로 가열하였다. 또 다른 0.1 eq CDI를 충전하고 혼합물을 또 다른 1시간 동안 가열하여 완료시켰다. 반응물을 빙조에서 12℃로 냉각하였다. DBU (2.98 g, 19.16 mmol), 이어서 시클로프로필 술폰아미드 A-4 (1.88 g, 15.48 mmol)를 충전하였다. 반응 혼합물을 1시간 동안 40℃에서 가열하고, 빙조에서 3℃로 냉각하고, 3 N HCl (20 mL)로 켄칭하여 pH를 2.5로 만들었다. 화합물 A-10이 부분적으로 침전되었다. 침전물을 여과에 의해 수집하고, 물로 세정하였다. 여과 후 유기층을 물 (15 mL) 및 염수 (15 mL)로 세척하였다. 유기층 및 침전물을 합하고, 진공하에 농축하여 화합물 A-10을 백색 분말 (~4.2g, 90 내지 92% 수율)로서 수득하였다.
HPLC 조건: 애질런트 이클립스 플러스 C18, 4.6 x 50 mm, 1.8 μ; RT, 선형 구배: 5분 내에 10 내지 90%의 B (MeCN), 2분까지 유지; A: 수용액의 0.1% H3PO4; 유속: 1.0 ml/분; 210 nm에서 UV 검출.

10.
탈보호
및 염 형성

촉매 활성화: Pd(OAc)2 (6.4 mg, 0.028 mmol)에 2-Me-THF 또는 THF (0.1 mL), DPPM (10.9 mg, 0.0284 mmol), 이어서 N-메틸시클로헥실아민 (11 μl)을 충전하였다. 생성된 슬러리를 주위 온도에서 30분 동안 숙성시켰다. 그 다음 10 μl의 당해 용액을 다음 용액에 사용하였다.
반응 절차: 8 ml 바이알에 화합물 A-10 (100 mg, 0.284 mmol) 및 1-프로판올 (2.5 mL)을 충전하였다. 당해 슬러리를 40℃에서 30분 동안 숙성시키고, 냉각하였다. NaBH4 (21.52 mg, 0.568 mmol)를 첨가한 후, 활성 촉매 (0.01 mL)의 용액을 첨가하였다. 반응 혼합물을 주위 온도에서 5분 동안 교반하고, 40℃로 가온하고 당해 온도에서 16.5시간 동안 숙성시켰다. (NMR에 의한 ~90% 분석 수율). 반응물을 냉각하고, TsOH (57 mg, 0.3 mmol, 1 eq)를 충전하고 주위 온도에서 교반하였다. 헵탄을 첨가하여 생성물을 결정화시켰다.
HPLC 조건: 애질런트 이클립스 XDB, 4.6 x 50 mm; RT, 선형 구배: 5분 내에 5 내지 95%의 B(MeOH), 8분까지 유지; A: pH 3.5 (1 L로 희석된 10 ml의 원액(stock solution) + 200 mM 과염소산나트륨 일수화물 (원액: 12.6 g 암모늄 포름산 포르메이트+ 7.9 ml 포름산); 유속: 1.0 ml/분; 210 nm에서 UV 검출.

11.
쿠르티우스
배열
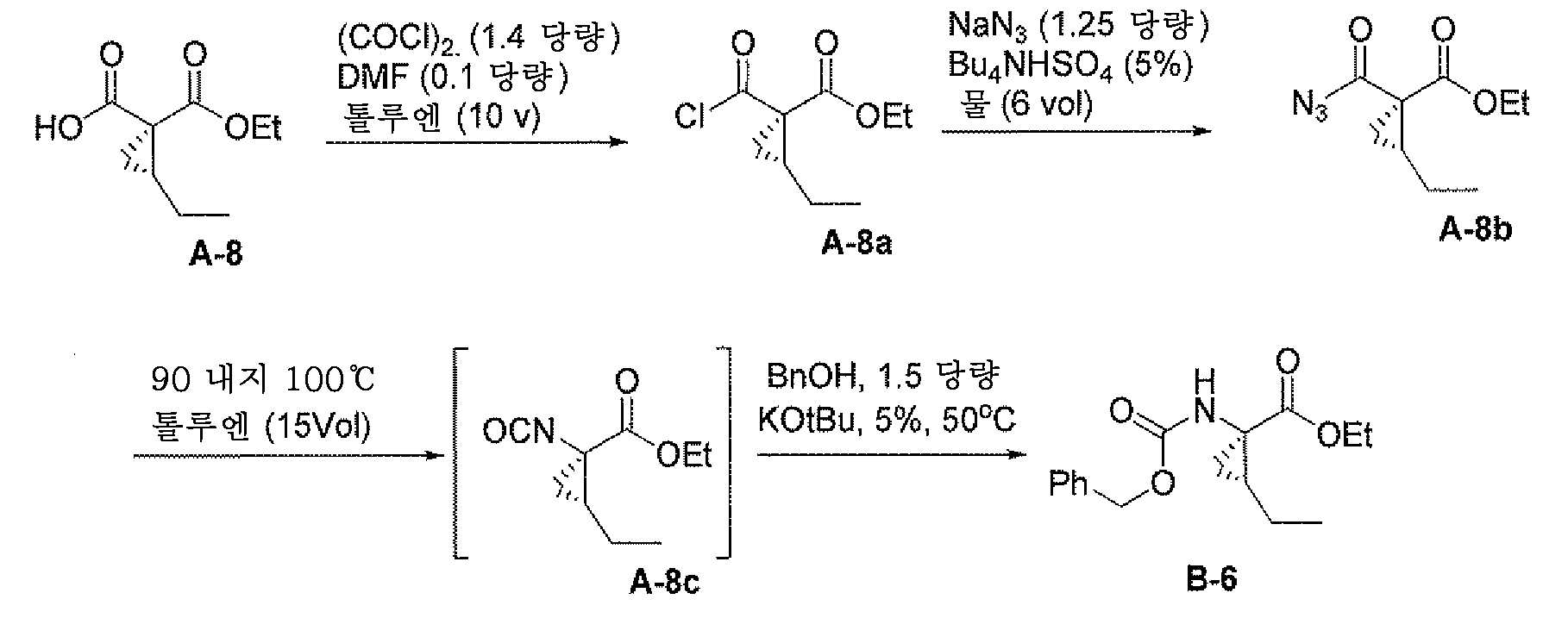
산 클로라이드 A-8a의 제조: 화합물 A-8 (2.0 g, 10 mmol)을 20 ml의 톨루엔에 용해시키고, 빙조를 사용하여 5℃로 냉각하고, DMF (0.079 g, 0.083 mmol), 그 다음 (COCl)2 (1.91 g, 15 mL)를 서서히 첨가하고 반응 온도를 <20℃로 유지하였다. 첨가 후, 반응물을 ~30분 내지 60분 동안 또는 GC 분석이 완전 전환을 나타낼 때까지 주위 온도에서 숙성시켰다. 반응물을 빙조로 냉각하고 물 (20 mL)로 켄칭하였다. 유기층을 10 wt% NaHCO3로 2회 (2 x 10 mL) 세척하여 pH를 ~8.0으로 만들었다. 톨루엔 함유 산 클로라이드 용액을 그 다음 단계에 직접 취하였다.
아실 아지드 A-8b의 제조: 플라스크 중 물 (12 mL)에 나트륨 아지드 및 황산수소테트라부틸암모늄 (0.18 g, 0.535 mmol)을 첨가하였다. 톨루엔 중 산 클로라이드 (A-8a)의 용액을 당해 나트륨 아지드 용액에 격렬히 교반 (>400 RPM)하면서 30 내지 60분에 걸쳐 첨가하였다. GC 분석이 완전 전환을 나타낼 때까지 혼합물을 주위 온도에서 ~1 내지 2시간 동안 교반하였다. 유기층을 분리하고, 1 M NaHCO3 (60 mL), 물 (50 mL) 및 염수로 순차적으로 세척하여 KF ~700 ppm으로 수분 함량을 갖는 용액을 수득하였다. 용액을 추가로 MgSO4 상에서 건조시키고, 여과하여 톨루엔 중 아실 아지드 용액 (KF <100 ppm)을 수득하고 이를 다음 단계에 직접 취하였다.
화합물 B-6의 제조: 첨가 깔대기 및 응축기가 연결된 3-구 플라스크 (500 mL)를 진공화하고/N2로 3회 플러싱하였다. 톨루엔 (10 mL, 50 ppm 하에 KF)을 충전하고 95℃ (내부 온도)로 가열하였다. 가열된 톨루엔에 아실 아지드 용액을 60분에 걸쳐 충전하고 온도를 90℃ 내지 100℃에서 유지하였다. 첨가 후, 반응 용액을 당해 온도에서 ~1시간 동안 숙성시켰다. 반응물을 ~20℃로 냉각하였다.
BnOH (15 mmol), KOtBu (0.5 mmol) 및 6 ml 톨루엔이 충전된 또 다른 플라스크에, 상기 이소시아네이트 톨루엔 용액을 1시간 내에 첨가 깔대기를 통해 30℃에서 첨가하고 약간 발열성으로서, T <50℃를 유지하고, HPLC 또는 GC에 의해 완료될 때까지 50℃에서 2 내지 4시간 숙성시켰다. 반응물을 물 (44 mL)로 켄칭하고, 물로 1회 세척하였다. 화합물 B-6의 분석 수율은 ~85%이었다. 화합물 B-6 톨루엔 용액의 용매를 MeOH로 교체하고 그 다음 단계에서 사용하였다.
HPLC 조건: 애질런트 이클립스 플러스 C18, 4.6 x 50 mm, 1.8 μ; RT, 선형 구배: 5분 내에 10 내지 90%의 B (MeCN), 2분까지 유지; A: 0.1% H3PO4의 수용액; 유속: 1.0 ml/분; 210 nm에서 UV 검출.

12. 가수분해

MeOH (5v) 중 화합물 B-6의 용액에 물 (5v) 중 NaOH (10 N, 3.5 당량)을 주위 온도에서 첨가하고 온도가 ~28℃로 상승되었다. 생성된 용액을 40℃에서 8 내지 12시간 동안 가열하여 HPLC에 의한 완전 전환을 수득하였다.
반응물을 빙조를 사용하여 6℃로 냉각하고; 물 (20 mL), MTBE (10 mL) 및 헵탄 (20 mL)을 충전하고, 유기층을 분리해 내어 재배열 단계로부터 거의 모든 BnOH를 제거하였다. 수성층을 12N HCl을 사용하여 산성화하여 pH를 2.1로 만들고 IPAc로 2회 추출하였다. 합해진 유기층을 물 30 mL 및 염수 30 mL (pH 1.9)로 세척하였다. 유기상을 공비혼합하고 IPAc로 플러싱하여 KF <200 ppm으로 만들었다. IPAc 4 v를 유지하고 헵탄 8 내지 10v를 40℃에서 첨가하고, 실온으로 냉각하고 2℃에서 2시간 동안 숙성시켰다. 고체를 여과에 의해 수집하고 헵탄으로 세척하여 B-7 고체를 90 내지 94% 수율로 수득하였다.
HPLC 방법: 아센티스 R 익스프레스 C18, 10 cm x 4.6 mm, 2.7 μ; 표준 구배: 6분 내에 10 내지 95%의 B (A=0.1% 인산, B= 아세토니트릴), 2분 유지, 2분 후; 유속: 1.8 ml/분; 210 nm, 40℃에서 UV 검출.

13.
CDI
커플링
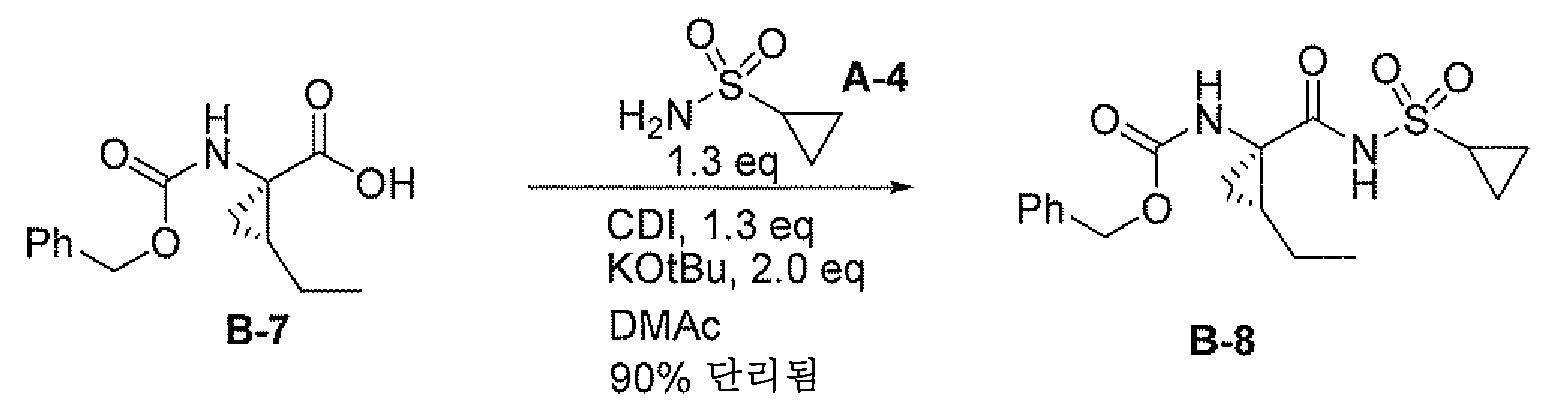
건조 DMAc (10 mL) 중 화합물 B-7 (1.0 g, 3.8 mmol)의 용액에 N2 하 CDI (0.83 g, 5.1 mmol, 1.3 eq)를 충전하였다. HPLC (CH3CN 중 nBuNH2로 켄칭됨)가 완료를 나타낼 때까지 반응물을 30분 내지 1시간 동안 40℃로 가열하였다. 반응물을 빙조 중에서 20℃로 냉각하였다. KOtBu (0.85 g, 7.6 mmol), 이어서 시클로프로필 술폰아미드 A-4 (0.59 g, 4.9 mmol)를 충전하였다. HPLC가 완료를 나타낼 때까지 반응 혼합물을 40℃에서 가열하고 실온으로 냉각하고 2 N HCl (10 mL)로 켄칭하여 pH를 ~2로 만들었다. 30분 내에 20 ml를 첨가하고 실온에서 2시간 동안 숙성시켰다. 고체를 여과에 의해 수집하고 DMAc/물 (1:2, 10 mL), 물 (10 mL) 및 헵탄으로 세척하고 N2 퍼지와 함께 진공하에 건조시키고 B-8 (1.26 g 고체, ~90% 단리 수율)을 수득하였다.
HPLC 방법: 아센티스 익스프레스 C18, 10 cm x 4.6 mm, 2.7 μ; 표준 구배: 6분 내에 10 내지 95%의 B (A=0.1% 인산, B= 아세토니트릴), 2분 유지, 2분 후; 유속: 1.8 ml/분; 210 nm, 40℃에서 UV 검출.

14.
탈보호
및 염 형성

플라스크에 화합물 B-8 (2.12 g, 5.79 mmol), Pd/탄소 (0.106 g, 5 wt%) 및 암모늄 포르메이트 (1.82 g, 28.9 mmol) 및 MeOH (21 mL)를 충전하였다. HPLC가 완전 완료를 나타낼 때까지 혼합물을 50℃에서 1 내지 2시간 동안 가열하였다. 혼합물을 실온으로 냉각하고 셀라이트(CELITE)를 통해 여과하고 MeOH 10 mL로 세척하고, 여액을 n-PrOH로 용매 교체하고 n-PrOH를 ~20 mL로 유지하였다. n-PrOH 중 혼합물을 60℃로 가열하고 p-TSA (1.1 g, 5.79 mmol)를 첨가하였다. 혼합물을 60℃에서 1시간 동안 교반하고 실온으로 냉각하였다. 헵탄 (10 mL)을 30분에 걸쳐 첨가하고, 슬러리를 2.5시간 동안 교반하고 여과하였다. 케이크를 n-PrOH/헵탄 (2:1 10 mL)으로 세척하고 건조시켜 ~90% 수율 염 생성물을 수득하였다.
HPLC 방법: 아센티스 R 익스프레스 C18, 10 cm x 4.6 mm, 2.7 μ; 표준 구배: 6분 내에 10 내지 95%의 B (A=0.1% 인산, B= 아세토니트릴), 2분 유지, 2분 후; 유속: 1.8 ml/분; 210 nm, 40℃에서 UV 검출.

실시예
2:
헤테로사이클
합성
화합물 3을 당해 실시예에 기재된 방법을 사용하여 제조하였다. 실시예에 기재된 화합물 및 방법은 본 발명의 상이한 측면 및 실시양태를 제공한다.
1. 배치 반응: 3-
브로모벤조니트릴
및
포르메이트
켄치
(
quench
) (1)의
오르토
-리튬치환반응(
lithiation
)
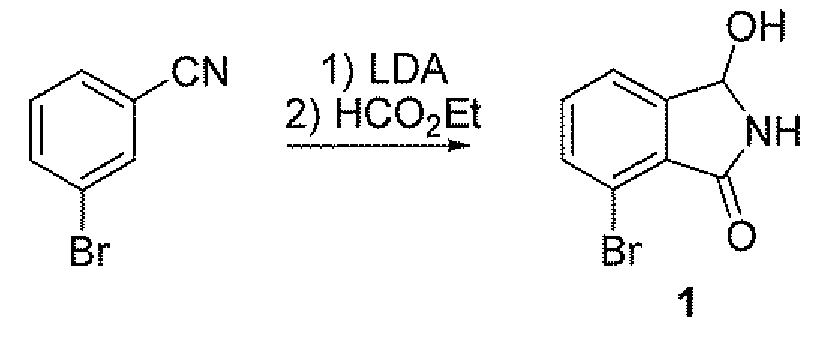
기계 교반기, 열전대, 질소 주입구 및 냉각조가 장착된 500 mL 3-구 환저 플라스크에 디이소프로필아민 (6.12 g, 8.61 mL, 60.4 mmol, 1.1 eq) 및 THF (50 mL)를 충전하였다. 혼합물을 -20℃로 냉각하고, n-부틸리튬 (24.17 mL, 60.4 mmol, 1.1 eq)을 첨가하고, 온도를 0℃ 미만으로 유지하였다. 용액을 5분 동안 숙성시킨 다음, CO2/아세톤 중 -70℃로 냉각하였다.
분리하기 위하여, 육안상 깨끗한 환저 플라스크에 3-브로모벤조니트릴 (10 g, 54.9 mmol, 1.0 eq) 및 THF (20 mL)를 충전하였다. 니트릴의 용액을 캐뉼라를 통해 리튬-아미드 용액에 옮기고, 내부 온도를 -65℃ 미만으로 유지하였다. 생성 용액을 -70℃에서 5분 숙성시켰다.
에틸 포르메이트 (6.04 mL, 74.2 mmol, 1.35 eq)를 반응 혼합물에 서서히 첨가하고, 내부 용액을 -65℃ 미만으로 유지하였다. 생성 용액을 -70℃에서 5분 숙성시켰다.
생성 용액은 역 켄칭된 (첨가된) 빙냉수 (50 mL)이고, 내부 온도를 5℃ 미만으로 유지하였다. EtOAc (50 mL)에 이어서 농 HCl (9 mL)을 첨가하여 pH가 ~4인 2상 혼합물을 수득하였다. 혼합물을 분액 깔대기에 옮기고, 수성층을 제거한 다음, EtOAc (50 mL)로 2회 역-추출하였다. 합해진 유기층을 MgSO4 상에서 건조시키고, 여과하고 진공 중에 농축하여 1 (11.63 g, 93% 단리 수율)을 수득하였다.
HPLC 방법: 칼럼: 이클립스 C18 플러스, 4.6 x 100 mm; 1.5 mL/분; 210 nm, 40℃, MeCN/물에 용해시킨 샘플. 이동상 A: 물 중 0.1% H3PO4; 상 B: MeCN. 5분에 걸쳐 20% B 내지 95% B의 시행 구배, 2분 유지.

1. 흐름 반응: 3-
브로모벤조니트릴
및
포르메이트
켄치
(1)의
오르토
-리튬치환반응

1.00 M 디이소프로필아민의 원액 A를 다음과 같이 제조하였다: 100 mL 메스 플라스크에 디이소프로필아민 (10.12 g, 14.25 mL, 100 mmol, 1.0 eq)을 충전하고THF로 희석하여 총 용적을 100 mL로 만들었다. 1.00 M 3-브로모벤조니트릴의 원액 B를 다음과 같이 제조하였다: 100 mL 메스 플라스크에 3-브로모벤조니트릴 (18.2 g, 100 mmol, 1.0 eq)을 충전하고 THF로 희석하여 총 용적을 100 mL로 만들었다. 4.00 M 에틸 포르메이트의 원액 C를 다음과 같이 제조하였다: 50 mL 메스 플라스크에 에틸 포르메이트 (14.8 g, 16.1 mL, 200 mmol, 1.0 eq)를 충전하고 THF로 희석하여 총 용적을 100 mL로 만들었다. 시판 n-부틸리튬 (40 mL, 100 mmol, 2.5 M)을 수령된 바와 같이 사용하고, 1회용 플라스틱 시린지에 충전하고 시린지 펌프를 통해 펌핑하였다. 모든 다른 원액을 HPLC (크나우어(Knauer)) 펌프를 통해 펌핑하고, 펌프와 반응기 사이에 100 psi 배압(back-pressure) 조절기를 포함하여 일관된 유속을 보장하였다.
HPLC 방법: 칼럼: 이클립스 C18 플러스, 4.6 x 100 mm; (1.5 mL/분; 210 nm, 40℃, MeCN/물에 용해시킨 샘플. 이동상 A: 물 중 0.1% H3PO4; 상 B: MeCN. 5분에 걸쳐 20% B 내지 95% B의 시행 구배, 2분 유지.

2. 이소-
인다논의
이소-인돌 (2)로의 환원

기계 교반기, 열전대, 질소 주입구 및 환류-응축기가 장착된 50 mL 환저 플라스크에 수소화붕소나트륨 (0.5 g, 13.16 mmol, 6 eq) 및 THF (10 mL)를 충전하였다. 삼플루오린화붕소 에테레이트 (1.67 mL, 13.16 mmol, 6 eq)를 첨가하고 혼합물을 5분 동안 실온에서 숙성시켰다.
조 이소-인다논 (0.5 g, 2.193 mmol, 1 eq)을 첨가하여 슬러리를 수득하고 이를 후속적으로 2시간 동안 60℃로 가열하였다. 실온으로 냉각 후, 혼합물을 EtOAc (5 mL)로 희석한 다음 50 wt% 수성 NaOH (~1.5 mL)를 첨가하여 염기성화하여 pH를 ~12로 만들었다. 2상 혼합물을 분액 깔대기에 옮기고, 수성층을 제거하였다. 유기층을 수집하고, MgSO4 상에서 건조시키고, 여과하고 진공 중에 농축하고 265 mg의 2 (60% 단리 수율)를 수득하였다.
HPLC 방법: 칼럼: 이클립스 C18 플러스, 4.6 x 100 mm; 1.0 mL/분; 210 nm, 40℃, MeCN/물에 용해시킨 샘플. 이동상 A: 물 중 0.1% H3PO4; 상 B: MeCN. 20분에 걸쳐 5% B 내지 95% B의 시행 구배, 5분 유지.

3.
브로모
-이소-
인돌의
쿠마다
커플링

오버헤드 교반하면서 3-구 플라스크를 vac/N2 백필(backfill)로 3회 퍼지하였다. N2 정압(positive pressure)하에, 플라스크에 브로모이소인돌린 HCl 염 (25 g, 106.6 mmol), Pd(OAc)2 (0.199 g, 0.533 mmol), 및 리간드 (디-tert-부틸네오펜틸포스포늄 테트라플루오로보레이트, 0.325 g, 1.066 mmol)를 충전하였다. 그 다음 PhMe (톨루엔) (450 mL, N2 살포를 통해 산소가 제거됨)를 첨가한 다음, 외부 욕을 사용하여, 생성된 슬러리를 Ti = 5℃로 냉각하였다. 알릴마그네슘 클로라이드 (THF 중 1.7 N, 207 mL, 351.8 mmol)를 캐뉼라를 통해 첨가 깔대기에 충전한 다음, Ti < 20℃이 되도록 하는 속도로 첨가하였다. 그 다음 생성된 용액을 Ti = 45 내지 50℃로 가열하였다.
16시간 후, LC는 출발 물질의 >99% 전환을 나타내었다. 반응물을 실온으로 냉각한 다음, 250 mL의 15% 수성 시트르산으로 역(inverse)-켄칭하였다. 상을 분리하고, 생성물 함유 수성상을 유지하고, 한편 암색(dark) 유기상을 폐기하였다. 수성상을 함유한 추출기에 125 mL의 PhMe를 충전하였다. 수성상의 pH는 115 mL의 NH4OH를 첨가하여 조정하였다. 상을 분리하고, 생성물 함유 유기상을 유지하고, 한편 수성상을 폐기하였다. 유기상을 20 mL의 15% 수성 NaCl로 세척하였다. PhMe 용액을 공비혼합물로 농축하여 10 용적 용액 (KF < 2000 ppm H2O)을 수득하였다.
생성물의 당해 PhMe 용액을 오버헤드 교반하면서 250 mL 플라스크에 옮겼다. 첨가 깔대기에 IPA (5.3 M) 중 17.5 ml의 5.33 M HCl을 충전하고, 이를 20분에 걸쳐 서서히 첨가하였다. 생성된 슬러리를 Ti = 40℃에서 30분 동안 숙성시킨 다음, 30분에 걸쳐 Ti = 20 내지 22℃로 서서히 냉각하였다. 1시간 동안 숙성시킨 후, 슬러리를 외부욕을 사용하여 30분에 걸쳐 Ti = 0℃로 서서히 냉각하였다. 30분 후, 슬러리를 여과하였다. 케이크를 16 mL의 냉 (Ti = -10vC) 14:2 PhMe:IPA로 세척하였다. 그 다음 케이크를 15 mL의 주위 온도 (Ti = 22℃) MTBE로 세척하였다. 건조 후, 7.6 g의 알릴 이소인돌린을 회백색(off white) 고체로서 단리하였다. 단리 생성물은 98.7 wt%로 분석되었다.
실시예
3:
tert
-루이신 단위
화합물 6을 당해 실시예에 기재된 방법을 사용하여 제조하였다. 실시예에 기재된 화합물 및 방법은 본 발명의 상이한 측면 및 실시양태를 제공한다.
에틸
이소부티레이트의
알릴 브로마이드 (4)를 사용한 알킬화

2 L 3-구 RB 플라스크에 THF (406 mL), 디이소프로필아민 (157 mL, 111 g, 1.1 mol)을 충전하고 약 -30℃로 냉각하였다. n-헥실리튬 (2.3 M/헥산, 457 mL, 1.05 mol)을 -20℃ 내지 -10℃에서 15분에 걸쳐 첨가하고, 첨가 완료 후 추가의 10분 동안 숙성시켰다. 온도를 -5℃ 내지 -10℃로 유지하면서 에틸 이소부티레이트 (135 mL, 116 g, 1.0 mol)를 15분에 걸쳐 첨가하였다. 첨가 말미에, DMPU (60.3 mL, 64.1 g, 0.5 mol)를 몇분에 걸쳐 첨가하고, 생성된 용액을 -10℃ 내지 -20℃에서 15분 동안 숙성시켰다. 그 다음 온도를 약 -10℃로 유지하면서 알릴 브로마이드 (91 mL, 127 g, 1.05 mol)를 15 내지 30분에 걸쳐 적가하였다. 생성된 용액 (용액으로부터 LiBr)을 실온으로 가온하고 n-헵탄 (696 mL, 6 용적)과 2.5 N 수성 HCl (580 mL)의 2상 혼합물로 역(reverse) 켄칭하였다. 층을 분리하고 (pH ~1 내지 2), 유기층을 물 (2 x 348 mL, 2 x 3 용적)로 세척하였다. 그 다음 유기층을 증류하여 +50℃ 내지 +50℃로 이루어진 내부 온도 및 +250 내지 +400 mm Hg의 압력에서 대부분의 용매 (THF, 헥산, 헵탄)를 제거하였다. 농도가 약 1 몰 (156 g/L)일 때 증류를 중지하였다. 조 황색 농축물을 그 다음 단계에서 그대로 사용하였다.
GC 방법: 칼럼: 모세관; 고정상: HP-1 메틸 실록산 (30 m x 250 μm x 0.25 μm); 검출기: FID; 캐리어 가스: He= 3.0 mL/분, 일정 유량 [P~25 psig]; 오븐 온도= 50℃ 3분 유지, 그 다음 20℃/분으로 280℃까지; 주입기 온도= 250℃; 검출기 온도= 300℃; 검출기 가스 유량: 40 mL/분으로 H2; 400 mL/분으로 공기; 메이크업(make-up) 가스: 25 mL/분으로 He.

비트리드
(
VITRIDE
)® 환원 - 2,2-디메틸-
펜트
-4-엔-1-올 (5)의 제조.

2 L RB 플라스크에 조 알릴 에스테르의 약 1 M 헵탄 용액 (156.2 g 분석, 약 740 mL의 헵탄 중 1.0 mol)을 충전하고, 약 +10℃로 냉각하였다. 내부 온도를 +30 내지 +35℃로 유지하면서 비트리드® (311 g, 301 mL, 1.2 mol)을 30분에 걸쳐 첨가하였다. 배치를 +30℃에서 1시간 동안 숙성시키고, +10℃로 냉각하고 5분에 걸쳐 IPA (77 mL)를 서서히 첨가함으로써 가수분해시켰다. 온도를 +30℃ 미만으로 유지하면서 반응 혼합물을 냉각하는 동안 실온에서 6N HCl (1350 mL)로 역으로 켄칭하였다.
2상 혼합물을 주위 온도에서 1시간 동안 숙성시키고, 층을 분리하였다. 유기층을 물 (2 x 500 mL)로 세척하고, 감압 (25℃에서 35 mmHg)하에 농축하였다. 조 농축물 생성물 (105 g 분석, 92% AY)을 그 다음 단계에서 그대로 사용하였다.
GC 방법: 칼럼: 모세관; 고정상: HP-1 메틸 실록산 (30 m x 250 μm x 0.25 μm); 검출기: FID; 캐리어 가스: He= 3.0 mL/분, 일정 유량 [P~25 psig]; 오븐 온도= 50℃ 3분 유지, 그 다음 20℃/분으로 280℃까지; 주입기 온도= 250℃; 검출기 온도= 300℃; 검출기 가스 유량: 40 mL/분으로 H2; 400 mL/분으로 공기; 메이크업 가스: 25 mL/분으로 He.

카르바메이트
/루이신 형성 -
CHA
(
시클로헥실아민
) 염 제조 (6)
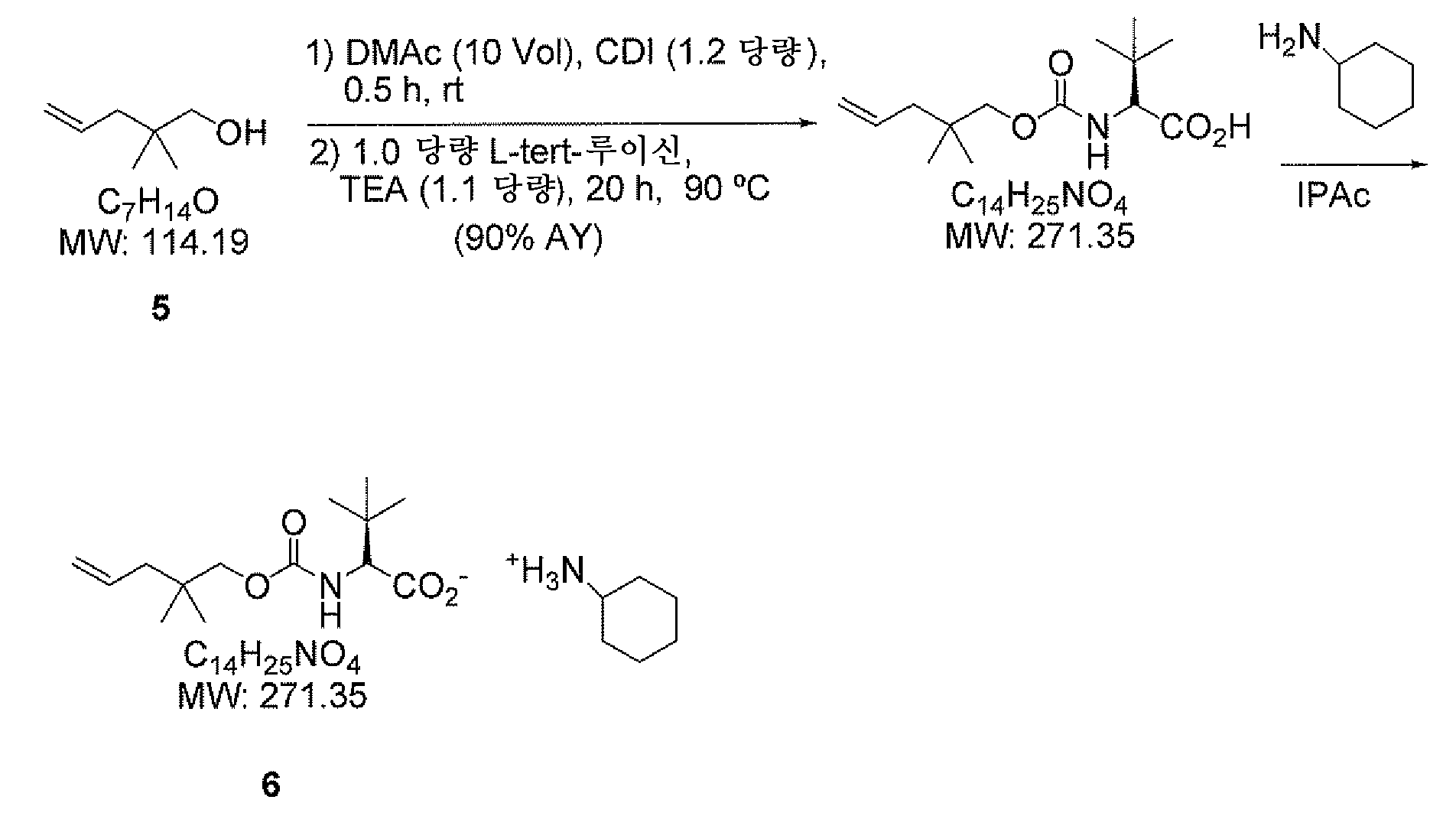
절차 A
50 mL RB 플라스크에 DMF (18 mL) 및 조 알콜 (5.179 g, 약 45 내지 50 wt%, 약 2.4 g 분석, 18.7 mmol)을 충전하고, 약 +10℃로 냉각하였다. CDI (3.0 g, 18.7 mmol)를 15분에 걸쳐 조금씩 첨가하였다. 생성된 균질 혼합물을 주위 온도에서 30분 동안 교반하였다.
L-tert-루이신 (2.45 g, 18.7 mmol)을 한번에 첨가한 후 트리에틸아민 (2.85 mL, 20.5 mmol)을 첨가하였다. 생성된 슬러리를 12시간 동안 90℃로 가열하고, 실온으로 냉각하였다. 용액을 n-헵탄 (15 mL), 및 물 (18 mL)에 분배하였다. 층을 분리하고, 유기층을 폐기하였다.
DMF 수성 염기성 층을 MTBE (22 mL)로 분배하고 (12 N) 농 HCl 용액 (약 5.5 mL)을 사용하여 중화시켜 pH를 ~1 내지 2로 만들었다. 층을 분리하고, 유기층을 물 (2 x 15 mL)로 세척하였다. 유기 용액을 농축하고, 아세토니트릴로 교체하여 KF<500 ppm으로 건조시켰다. 생성된 조 카르바메이트를 100 mL 플라스크에 넣고, 아세토니트릴 (65 mL)에 용해시키고, 45℃로 가열하였다. 디시클로헥실아민 (3.72 mL, 18.7 mmol)을 1시간에 걸쳐 첨가하여 염을 결정화시켰다. 슬러리를 45℃에서 2시간 동안 교반하고, 주위 온도로 냉각하고, 여과하고, 아세토니트릴 (10 mL)로 세정하였다. 생성된 백색 염을 오븐에서 45℃에서 24시간 동안 건조시켜 6.1 g의 생성물 (74% 전체 수율)을 수득하였다.
절차 B
50 mL RB 플라스크에 DMF (18 mL) 및 조 알콜 (5.179 g, 약 45 내지 50 wt%, 약 2.4 g 분석, 18.7 mmol)을 충전하고, 약 +10℃로 냉각하였다. CDI (3.0 g, 18.7 mmol)를 15분에 걸쳐 조금씩 첨가하였다. 생성된 균질 혼합물을 주위 온도에서 30분 동안 교반하였다.
L-tert-루이신 (2.45 g, 18.7 mmol)을 한번에 첨가한 후 트리에틸아민 (2.85 mL, 20.5 mmol)을 첨가하였다. 생성된 슬러리를 12시간 동안 90℃로 가열하고, 실온으로 냉각하였다. 용액을 n-헵탄 (15 mL), 및 물 (18 mL)에 분배하였다. 층을 분리하고, 유기층을 폐기하였다.
DMF 수성 염기성 층을 MTBE (22 mL)로 분배하고 (12 N) 농 HCl 용액 (약 5.5 mL)을 사용하여 중화시켜 pH를 ~1 내지 2로 만들었다. 층을 분리하고, 유기층을 물 (2 x 15 mL)로 세척하였다. 유기 용액을 농축하고, 아세토니트릴로 교체하여 KF<500 ppm으로 건조시켰다. 생성된 조 카르바메이트를 100 mL 플라스크에 넣고, IPAc (65 mL)에 용해시키고, 45℃로 가열하였다. 시클로헥실아민 (3.72 mL, 18.7 mmol)을 1시간에 걸쳐 첨가하여 염을 결정화시켰다. 슬러리를 45℃에서 2시간 동안 교반하고, 주위 온도로 냉각하고, 여과하고, IPAC (10 mL)로 세정하였다. 생성된 백색 염을 오븐에서 45℃에서 24시간 동안 건조시켜 6.1 g의 생성물 (74% 전체 수율)을 수득하였다.
실시예
4:
디엔
-에스테르
디엔-에스테르를 당해 실시예에 기재된 방법을 사용하여 제조하였다. 실시예에 기재된 화합물 및 방법은 본 발명의 상이한 측면 및 실시양태를 제공한다.
디엔
-에스테르 형성
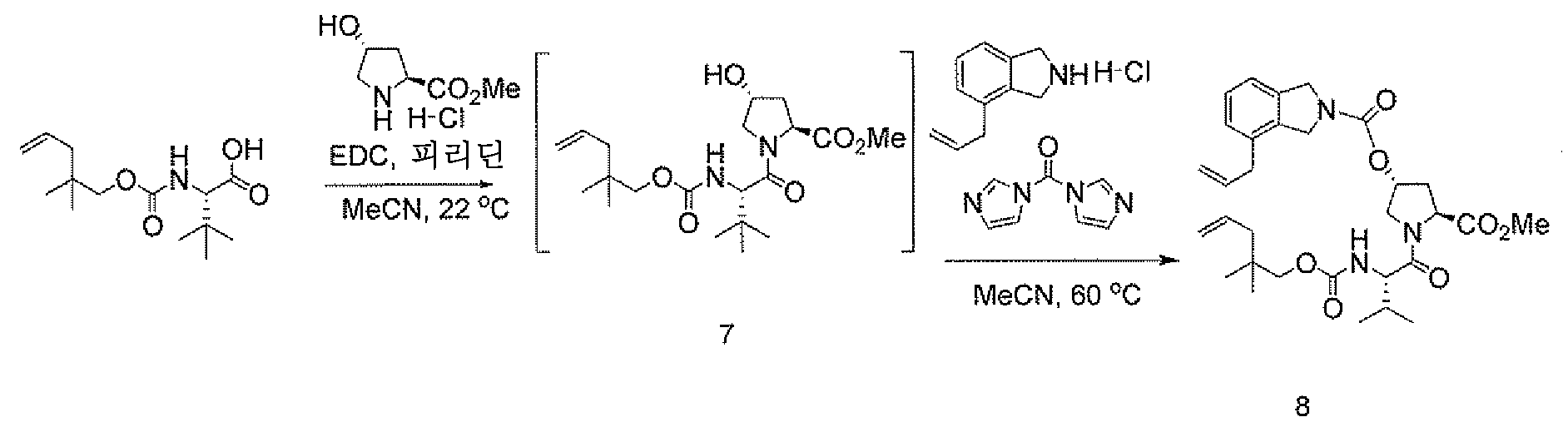
오버헤드 교반하면서 100 mL 플라스크에 "엔-산" (5.0 g, 18.43 mmol), 이어서 MeCN (KF = 135 ppm)을 충전하였다. 트랜스-4-히드록시-L-프롤린 메틸 에스테르 히드로클로라이드 (3.87 g, 95 W%, 20.26 mmol), 이어서 피리딘 (1.6 g, 20.27 mmol)을 충전하였다. 45분 숙성 후, EDC-HCl (4.42 g, 23 mmol)을 단일 분량으로 고체로서 충전하였다. 3.5시간 후, LC는 목적 생성물로의 >98% 전환을 나타내었다.
CDI (3.45 g, 21.2 mmol)를 첨가하였다. 그 다음 반응물을 Ti = 55℃로 가열하였다. 1시간 후, LC는 상당한 개선이지만 여전히 불완전한 알콜 활성화를 나타내었다 (이미다졸 카르바메이트:sm의 LCAP 비 = 80:20). 4시간 후, <97% 전환이 관찰되었다. 이 시점에서, 1.4 당량의 4-알릴이소인돌린 HCl 염 (5.05 g, 21.2 mmol)을 첨가하고 반응물을 Ti = 55 내지 60℃에서 밤새 교반하였다.
16시간 후, 균질 반응 혼합물을 60 mL의 물 및 40 mL의 MTBE로 역 켄칭하였다. 수성상을 폐기하고, 유기상을 50 mL의 15% 시트르산 (39 mmol의 시트르산)으로 세척하였다. 유기상을 15 mL의 4% 수성 Na2CO3, 그 다음 10 mL의 H2O로 세척하였다. 유기상을 건조시키고 9.09 g의 목적 생성물 (15.58 mmol, 84.5% AY)로 분석되었다.
"
디엔
-K 염" 형성
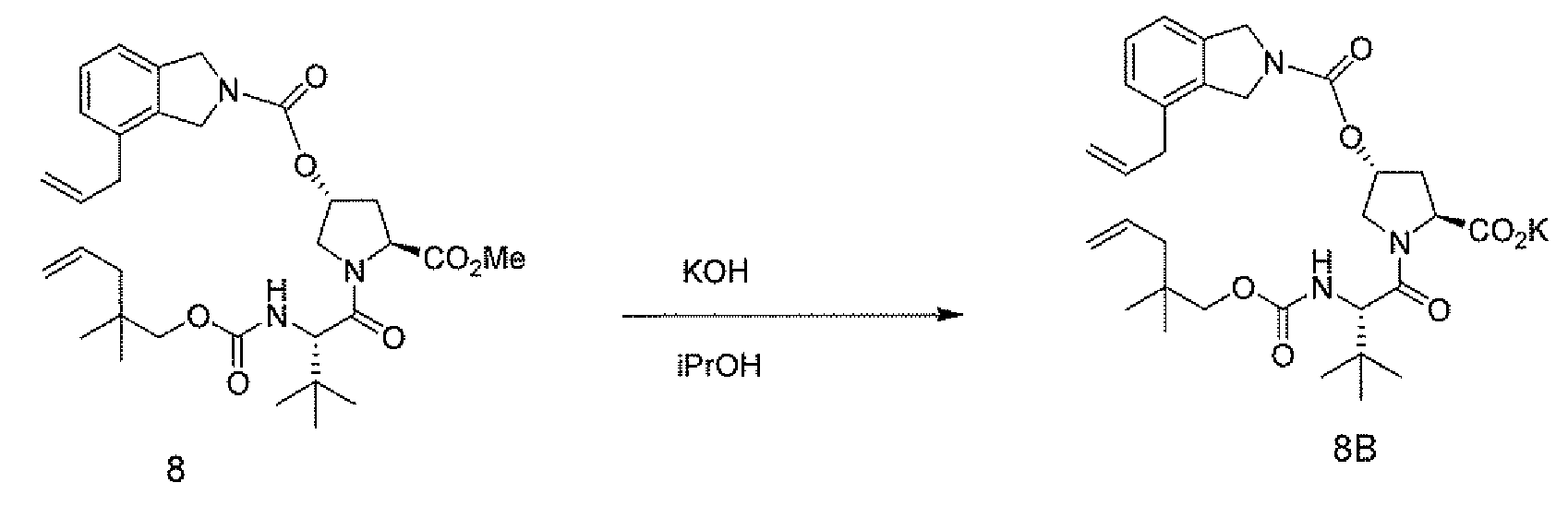
100 mL IPA (KF < 1000 ppm) 중 "디엔 에스테르" (9.09 g, 15.57 mmol)에 고체 KOH (85 W%, 1.44 g, 21.8 mmol)를 첨가하였다. 1.5시간 후, 용액을 50 mg의 시드로 처리하고, 생성된 슬러리를 16시간 동안 숙성시켰다.
그 다음 슬러리를 여과하고, 30 mL의 iPrOH로 세척하였다. 케이크를 흡입 건조시키고, 그로부터 8.81 g의 디엔산 칼륨 염을 단리하였다.
"
디엔
-산"형성

250 mL 플라스크에 75 mL의 PhMe 중 "디엔-K 염" (9.5 g, 15.63 mmol)을 충전하였다. 35 mL의 15% 시트르산 (27.3 mmol의 시트르산)을 첨가하였다. 1시간 후, 상을 분리하였다. 유기상을 10 mL의 H2O로 세척하였다. 유기상을 일정 용적 조건하에 PhMe와의 공비혼합물을 통해 건조시킨 다음, 여과하고 농축하였다.
실시예
5:
마크로락탐
형성 (화합물 A)
화합물 A (본원에서 화합물 12로도 칭해짐)를 당해 실시예에 기재된 방법을 사용하여 제조하였다. 실시예에 기재된 화합물 및 방법은 본 발명의 상이한 측면 및 실시양태를 제공한다.
디엔
-산을 사용한
RCM

환류 응축기를 갖춘 500 mL 3구 RB 플라스크에 1,6-디클로로퀴논 (0.105 g, 0.595 mmol) 및 톨루엔 (17 0 mL, 10 Vol)을 실온에서 충전하였다. 반응 용액을 온화한 질소 가스 버블링으로 107℃로 가열하였다. 한편, 톨루엔 원액 (45 wt%) 중 디엔-산 (16.94 g, 29.7 mmol)을 17 mL의 탈기된 톨루엔으로 희석하고 그럽스-호베이다-II 촉매 (0.037 g, 0.059 mmol)를 17 mL의 탈기된 톨루엔에 용해시켰다. 10 v%의 디엔-산 원액을 반응 용기에 첨가하였다. 반응 온도가 약 107℃에 이르렀을 때, 잔류 디엔-산 원액 및 그럽스-호베이다 제2 세대 촉매를 각각 58분 및 60분 동안 반응 용액에 동시에 첨가하였다. 촉매 첨가가 완료된 후, 반응 혼합물을 1시간 더 교반하여 디엔-산 기질의 완전 소모를 달성하였다. 반응 혼합물을 실온으로 냉각하였다. 톨루엔 용액을 수소화를 위한 고압-실험실(High-Pressure-Lab)에 옮겼다.
HPLC 조건: 아센티스 익스프레스 C18 (150 mm x 4.6 mm; 2.7 um), 1.0 mL/분, 220 nm, 40℃에서 검출, 표준 구배: 0분: 40%의 B, 15분: 95% B, 20분: 95% B, 20.1분: 40% B (A= 0.1% H3PO4 함유 물, B= 아세토니트릴).
19-원 RCM-에스테르-생성물: 7.986분 (시스) 및 8.137분 (트랜스).
RCM-산 목적 생성물: 8.917분 (시스) 및 9.236분 (트랜스).
디엔-산 출발 물질: 11.252분.
시클릭 이량체: 12.436분 (넓은).
RCM
-산 생성물의 수소화

톨루엔 (17.0 g, 31.4 mmol) 중 RCM-산 생성물을 고압 반응기에 옮기고 잔류물을 25.5 mL IPA로 세척하고 반응기에 옮겼다. 20 wt%의 5% Pd/C-쉘 촉매를 반응 용액에 첨가하였다. 반응 용기를 질소 가스로 3회 퍼지, 이어서 수소 가스로 100 psi에서 3회 퍼지하였다. 반응 혼합물을 100 psi 수소하에 24시간 동안 교반하였다. 반응이 완료된 후, 촉매를 여과하고 IPA (410 mL, 5 vol)로 세척하였다. 결정화용으로 용매를 IPA (300 mL, 3 vol)로 교체하였다.
결정화 절차
0.56 mL IPAc를 IPA 중 조 Mac-산 원액에 첨가하였다. 그 다음 암갈색 용액을 40℃로 가열하고 40℃에서 15분에 걸쳐 숙성시켰다. 1.78 mL DI수를 고온(hot) 용액에 40℃에서 10분에 걸쳐 서서히 첨가하고, 생성된 용액을 추가로 15분에 걸쳐 교반하였다. 용액을 22℃로 냉각하였다. 그 시점에서, 1 wt%의 시드를 균질 용액에 첨가하였다. 그 다음 용액을 3시간에 걸쳐 0℃로 냉각하였다.
슬러리를 0℃에서 14시간 동안 숙성시켰다. 슬러리를 냉장실 (약 3℃)에서 여과하고 0.75 mL의 미리-냉각된 IPA-물로 2회 세척하였다. 고체를 진공하에 (~30 mmHg) 45℃에서 24시간에 걸쳐 건조시켰다. 650 mg의 목적 생성물 (조 Mac-산으로부터의 40% 단리 수율)을 99% 초과의 HPLC 순도를 갖는 백색 고체로서수득하였다.
HPLC 조건: 아센티스 익스프레스 C18 (150 mm x 4.6 mm; 2.7 um), 1.0 mL/분, 220 nm, 40℃에서 검출, 표준 구배: 0분: 40%의 B, 15분: 95% B, 20분: 95% B, 20.1분: 40% B (A= 0.1% H3PO4 함유 물, B= 아세토니트릴).
19-원 Mac-에스테르: 8.382분.
Mac-산 목적 생성물: 9.614분.
시클릭 이량체: 13.185분.
가수분해

오버헤드 교반기 및 열전대가 장착된 100-L 추출 용기에 THF (96 mL) 중 에스테르 (17.8 g, 31.91 mmol)의 용액을 충전하고 5℃로 냉각하였다. 온도를 15℃ 미만으로 유지하면서 수산화리튬 (1 N, 96 mL, 96 mmol)의 수용액을 첨가 깔대기를 통해 30분에 걸쳐 적가하였다. 동일한 첨가 깔대기를 사용하여, 메탄올을 15℃에서 10분에 걸쳐 첨가한 후, 백색, 불균질 혼합물을 실온으로 가온하였다. 가온하면, 용액은 균질하게 되었다. 약 30분 후, 용액은 담황색에서 암갈색으로 변하였다. 이 시점에서 샘플링된 반응이 HPLC 분석에 의해 완료된 것으로 판단되었다 (>99.9A% 전환).
배치를 5℃로 냉각하고 1 N HCl (112 mL)로 처리하여 과량의 LiOH를 켄칭하였다. 첨가 후, 용액을 20℃로 가온하고 IPAc (180 mL, 10 vol)로 희석하였다. 15분 동안 교반 후, 층을 분리하고 유기층을 수집하였다 (170 g, 98% 분석 수율).
IPAc 용액 (~340 mL)을 20℃에서 10분 동안 다르코(Darco) KB-G (40 wt%, 7 g)로 처리하고, 용액을 솔카-플록을 통해 여과한 후 5 um 인-라인(in-line) 필터 (170 g, >99% 회수)를 통해 여과하였다. 온도를 25℃ 미만으로 유지하면서 IPAc 용액을 감압하에 100 mL로 농축하였다. 추가의 100 mL의 IPAc를 첨가하고 배치를 100 mL로 농축하였다. 용액을 DMF (80 mL)로 희석하고 최종 배치 용적이 80 mL일 때까지 농축을 계속하였다. 배치를 DMF (20 mL) 및 IPAc (80 mL)로 희석하였다.
HPLC 방법: 칼럼: Ace 3 C8 (3 mm x 150 mm, 3 μm) (0.75 mL/분; 215 nm, 35℃, MeCN/물에 용해시킨 샘플. 이동상 A: 물 중 0.1% H3PO4; 상 B: MeCN. 12분에 걸쳐 20% B 내지 90% B의 시행 구배, 3분 유지.

커플링 (
HOBt
)

오버헤드 교반기, 질소 주입구, 열전대가 장착된 1-L 플라스크에 168 mL IPAc 중 마크로시클릭 산 용액 (16.8 g, 30.8 mmol)을 충전하였다. 용액을 교반하고 토실레이트 P1 단편(piece) (14 g, 34.6 mmol)을 고체로서 첨가하였다. 용해시킨 후 (<10분), HOBt (4.7 g, 31 mmol)를 고체로서 첨가하였다. 배치를 15℃로 냉각하고 온도를 20℃ 미만으로 유지하면서 DIPEA (8.0 g, 61.8 mmol)를 첨가 깔대기를 통해 첨가하였다. 고체 EDC HCl (8.3 g, 43 mmol)을 첨가하였다. 온도의 어떠한 변화도 관찰되지 않았다. 3시간 후, 반응이 HPLC에 의해 완료된 것으로 판단되었다 (>99.8 A% 전환, 91% 분석 수율, 210 g).
배치를 1-L 추출 용기에 옮기고, 10℃로 냉각하고, IPAc (16.8 L) 및 물 (33.6 L)로 희석하였다. 혼합물을 10분 동안 교반하였다. 층을 분리하고, 수성층을 폐기하였다 (pH = 6 내지 7). 수성 HCl (1 N, 168 mL)을 IPAc 층에 첨가하고 용액을 10분 동안 교반하였다. 층을 분리하고, 수성층을 폐기하였다 (pH = 1 내지 2). 그 다음 IPAc 용액을 물/염수 (150 mL/170 mL)로 처리하였다. 교반 10분 후, 층을 상 분리하고, 수성층을 폐기하였다 (pH = 2 내지 3). IPAc 용액을 농축하고 1H NMR 분광학에 의해 판단된 바와 같이, 에탄올 중 2.5 mol% IPAc가 존재할 때까지 에탄올 (500 mL)로 플러싱하였다. 수율 = 202 g, 87% 분석 수율.
HPLC 방법: 칼럼: Ace 3 C8 (3 mm x 150 mm, 3 μm) (0.75 mL/분; 215 nm, 35℃, MeCN/물에 용해시킨 샘플. 이동상 A: 물 중 0.1% H3PO4; 상 B: MeCN. 12분에 걸쳐 20% B 내지 90% B의 시행 구배, 3분 유지.

대안적 커플링 (
EDC
-피리딘)

자기 교반기, 질소 주입구 및 열전대가 장착된 250 mL 3-구 환저 플라스크에 IPAc (16 mL) 및 MeCN (36 mL) 중 마크로시클릭 산 (62.55 g, 26.9 mmol)을 충전하여 완전한 이송을 보장하였다. 아민-토실레이트 (11.44 g, 28.3 mmol) 및 피리딘 (3.27 mL, 40.5 mL)을 혼합물에 첨가하여, 회백색 슬러리를 수득하였다. 생성 슬러리를 5분 동안 질소 표면아래(sub-surface) 퍼지함으로써 탈기시켰다. EDC-HCl (6.72 g, 35.0 mmol)을 실온에서 플라스크에 첨가하였다. ~15분 후, 슬러리가 투명한, 호박색(amber) 용액이 되는 것으로 관찰되었다. 용액을 실온에서 연속 표면아래 N2-퍼지로 숙성시켜 산화적 분해를 방지하였다. EDC-HCl 첨가 후 1 내지 2℃의 아주 적은 정도의 발열이 관찰되었다. 첨가 완료 5분 후 조 반응 혼합물의 분취액은 80% 전환을 나타내었다. 첨가 완료 75분 후 조 반응 혼합물의 분취액은 >99% 전환을 나타내었다. IPAc (45 mL) 및 DI-수 (45 mL)를 반응 혼합물에 첨가하여 2상 혼합물을 수득하였다. 혼합물을 분액 깔대기에 옮기고, 격렬하게 혼합한 후, 수성 (기저)층을 제거하였다. 유기상을 1 N HCl로 세척한 다음, 솔라-플록 상에서 여과하고 진공 중에 농축하고, IPAc (2x 100 mL)로 플러싱하여 임의의 잔류 물로부터 공비혼합물을 제거하였다. 물질을 농축하여 40.0 g 담황색 오일을 수득하고, 이는 HPLC 분석에 의해 49 wt% 아미드 A (96% 분석 수율)인 것으로 결정되었다.
화합물 A의 재결정화

18 ml IPAc (1 vol) 및 24 ml n-헵탄 (1.25 vol)을 충전하여 57:43 v/v 믹스를 창출함으로써 시드층을 제조하였다. 그 다음, 1.9 g 무수 화합물 A (PSD - MV > 16 um, MV가 <16 um인 경우, 대안적 시드 원숙(seed ripening) 절차는 하기에 제공됨)를 헵탄/IPAc 믹스에 충전하고, 15 내지 25℃에서 교반하고 30분 동안 회전시켜 화합물 A 헵탄 용매화물을 형성하였다. 시드층은 IKA 밀(mill) (정밀(fine)/초정밀(superfine) 회전자-고정자(rotor-stator), 40 내지 60회 회전)을 사용하여 습윤 밀링할 수 있었다. 그 다음, 시드층을 50℃로 가온하였다.
시드 원숙: 1.9 g 무수 건조 케이크를 21 ml의 45/55 (v/v)으로 n-헵탄/IPAc에 충전하여 슬러리를 형성하고 헵탄 용매화물로의 전환을 위해 30분 이상 동안 교반시켰다. 슬러리를 55 내지 65℃로 만들고 여기서 11.4 ml의 n-헵탄을 3시간에 걸쳐 슬러리에 충전하였다. 일단 완료되면, 4.7 ml의 건조 IPAc를 충전하여, 슬러리를 47 g/L 농도 및 57/43 v/v 헵탄/IPAc로 만들었다. 그 다음, 층을 12시간 이상에 걸쳐 45℃로, 그 다음 적어도 3시간 이상에 걸쳐 주위 온도로 냉각하였다. 그 다음, 시드층을 필요에 따라 밀링할 수 있다.
대안으로, 시드층은, 이전 시행으로부터의 최종 결정화 슬러리로부터 제조할 수 있다. 27 ml의 결정화 후의 슬러리 (~70 g/L. 1.9 g 분석, 65/35 (v/v) n-헵탄/IPAc)를 남겨 두었다. 5.6 ml n-헵탄 및 7.7 ml 건조 IPAc를 첨가하고 교반하였다. 목표 슬러리 조성물은 47 g/L 및 57/43 v/v n-헵탄/IPAc이었다. 그 다음, 층을 상기한 바와 같이 밀링하고 50℃로 가열하였다.
12시간에 걸쳐, IPAc (커플링 생성물, 87% 분석 용적 IPAc, 18.67 g 분석) 중 92 ml의 화합물 A 조 스트림을 50℃ 시드층에 첨가하였다. 동시에, 103 ml (5.8 vol)의 n-헵탄을 시드층에 첨가하고 57:43 v/v 헵탄/IPAc를 유지하였다. 12시간 첨가 말미에, 추가의 52 ml의 n-헵탄을 3시간에 걸쳐 첨가하고, 헵탄:IPAc 비를 65/35 v/v로 만들었다. 일단 헵탄 첨가가 완료되면, 배치를 3시간에 걸쳐 20℃로 냉각하고 여과하였다.
케이크 세척은 1회 세척의 37 ml (2 vol) 65/35 v/v n-헵탄/IPAc 믹스로 이루어졌다. 그 후 37 ml 각각 (2 vol 각각)의 순수 n-헵탄의 세척을 2회 더 행하였다. 그 다음 습윤 케이크 (화합물 A 헵탄 용매화물)의 벌크액(bulk liquor)을 드라이로 말리고 진공하에 70℃에서 건조시켜 화합물 A 유리 산 무수물(anhydrate)을 생성하였다.
다른 실시양태는 특허청구범위 이내에 있다. 몇몇 실시양태를 나타내고 기재하였지만, 본 발명의 취지 및 범위를 벗어남이 없이 다양한 변형이 이루어질 수 있다.
Claims (23)
- 하기 화학식 I의 화합물, 하기 화학식 II의 화합물 또는 그의 염, 하기 화학식 III의 화합물 또는 그의 염, 하기 화합물 6A 또는 그의 염, 하기 화합물 3 또는 그의 염, 하기 화합물 A-2, 하기 화합물 A-4, 하기 화합물 A-10 또는 그의 염, 및 하기 화합물 B-6 또는 그의 염으로 이루어진 군으로부터 선택된 화합물:
<화학식 I>

<화학식 II>
<화학식 III>

<화합물 6A>

<화합물 3>

<화합물 A-2>

<화합물 A-4>

<화합물 A-10>
<화합물 B-6>

상기 식에서,
R1은 C1 -6 알킬, C3-C8 시클로알킬, 또는 아릴이고;
R2 및 R3은 각각 H, C1 -6 알킬, C3-C8 시클로알킬, 또는 아릴이고;
R1a, R2a, 및 R3a는 각각 C1 -6 알킬 또는 C3-C8 시클로알킬이고;
n은 0 내지 5이고;
아릴은 페닐, 치환된 페닐, 나프틸, 또는 치환된 나프틸이되, 단, 치환된 페닐 및 치환된 나프틸은 각각
(1) C1 -6 알킬,
(2) OH, O-C1 -6 알킬, O-C1 -6 할로알킬, CN, NO2, N(RA)RB, C(O)N(RA)RB, C(O)RA, CO2RA, SRA, SORA, SO2RA, SO2N(RA)RB, N(RA)C(O)RB, N(RA)CO2RB, N(RA)SO2RB, N(RA)SO2N(RA)RB, OC(O)N(RA)RB, N(RA)C(O)N(RA)RB, 또는 N(RA)C(O)C(O)N(RA)RB로 치환된 C1 -6 알킬,
(3) O-C1 -6 알킬,
(4) C1 -6 할로알킬,
(5) O-C1 -6 할로알킬,
(6) OH,
(7) 할로겐,
(8) CN,
(9) NO2,
(10) N(RA)RB,
(11) C(O)N(RA)RB,
(12) C(O)RA,
(13) C(O)-C1 -6 할로알킬,
(14) C(O)ORA,
(15) OC(O)N(RA)RB,
(16) SRA,
(17) SORA,
(18) SO2RA,
(19) SO2N(RA)RB,
(20) N(RA)SO2RB,
(21) N(RA)SO2N(RA)RB,
(22) N(RA)C(O)RB,
(23) N(RA)C(O)N(RA)RB,
(24) N(RA)C(O)C(O)N(RA)RB, 또는
(25) N(RA)CO2RB
로 이루어진 군으로부터 독립적으로 선택된 1 내지 5개의 치환기 (여기서 RA 및 RB는 각각 독립적으로 H 또는 C1 -6 알킬임)를 갖는다. - 제1항에 있어서, 구조:
(화학식 IIa) 또는 그의 염을 갖고, R2가 제1항에 정의된 바와 같은 것인 화합물.
- 제2항에 있어서, 구조:
(화학식 II) 또는 그의 염을 갖고, R2가 H 또는 C1 -6 알킬인 화합물.
- 제3항에 있어서,
의 염인 화합물.
- 제4항에 있어서,
인 화합물.
- 하기 화학식 Ia의 화합물과또는 그의 염을 커플링시켜 하기 화학식 IIa의 화합물 또는 그의 염을 형성하는 단계를 포함하는, 제2항의 화학식 IIa 화합물의 제조 방법:

<화학식 Ia>

<화학식 IIa>
상기 식에서, R1 및 R2는 제1항에 정의된 바와 같다. - 제6항에 있어서, R1이 C1 -6 알킬이고, R2가 H 또는 C1 -6 알킬인 방법.
- a) 하기 화학식 IIa의 화합물 또는 그의 염을 폐환 및 수소화시켜 하기 화학식 IV의 화합물 또는 그의 염을 형성하는 단계를 포함하는, 제1항의 화학식 IV 화합물 또는 그의 염의 제조 방법:
<화학식 IIa>

<화학식 IV>

상기 식에서, R2는 제1항에 정의된 바와 같고, R4는 H, C1 -6 알킬, C3-C8 시클로알킬 또는 아릴이되, 단, 상기 아릴은 제1항에 정의된 바와 같다. - a) 하기 화학식 IIa의 화합물 또는 그의 염을 폐환 및 수소화시켜 하기 화학식 IV의 화합물 또는 그의 염을 형성하는 단계;
b) 화학식 IV의 화합물 또는 그의 염을 가수분해시켜 하기 화합물 11 또는 그의 염을 형성하는 단계;
c) 화합물 11 또는 그의 염을 하기 화합물 A-11 또는 그의 염에 커플링시켜 하기 화합물 A 또는 그의 염을 형성하는 단계; 및
d) 임의로, 화합물 A 또는 그의 염을 제약상 허용되는 염으로 전환시키는 단계
를 포함하는, 화합물 A 또는 그의 제약상 허용되는 염의 제조 방법:
<화학식 IIa>
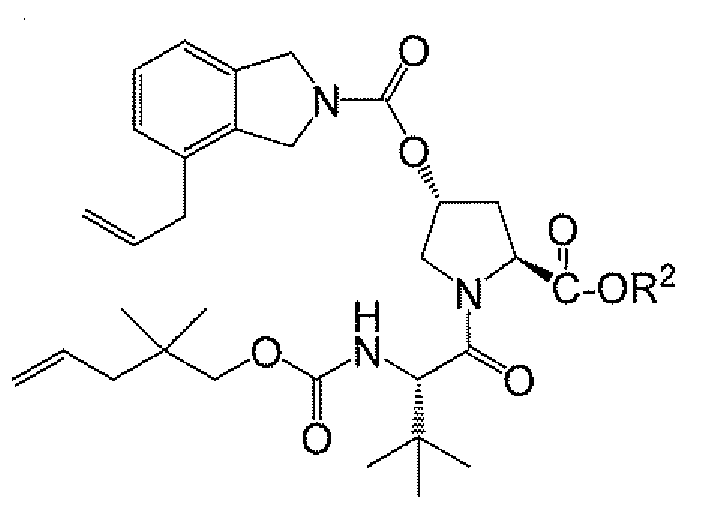
<화학식 IV>

<화합물 11>
<화합물 A-11>

<화합물 A>

상기 식에서, R2는 제1항에 정의된 바와 같고, R4는 H, C1 -6 알킬, C3-C8 시클로알킬 또는 아릴이되, 단, 상기 아릴은 제1항에 정의된 바와 같다. - 제9항에 있어서, 상기 단계 C 커플링을 EDC 및 피리딘 또는 피리딘 유도체를 사용하여 수행하는 것인 방법.
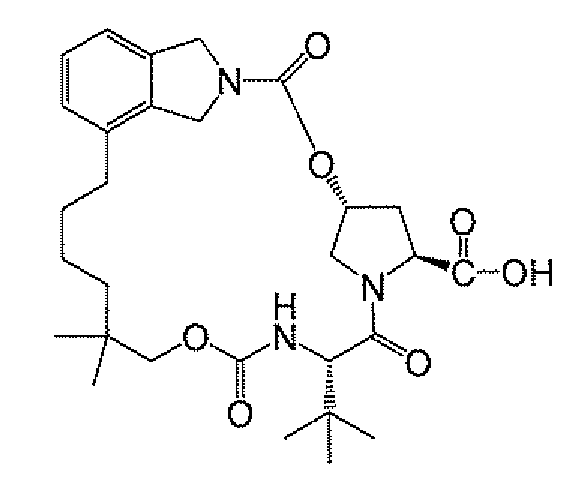
- 제8항 내지 제10항 중 어느 한 항에 있어서, 화학식 IIa의 화합물이
의 염인 방법.
- 제8항 내지 제11항 중 어느 한 항에 있어서, 화학식 IIa의 화합물이
인 방법.
- 제8항 내지 제12항 중 어느 한 항에 있어서, R4가 H 또는 C1 -6 알킬인 방법.
- 제8항 내지 제13항 중 어느 한 항에 있어서, 하기 화학식 Ia의 화합물을또는 그의 염과 커플링시키는 단계를 포함하는, 화학식 IIa의 화합물 또는 그의 염을 제조하는 단계를 추가로 포함하는 방법:

<화학식 Ia>
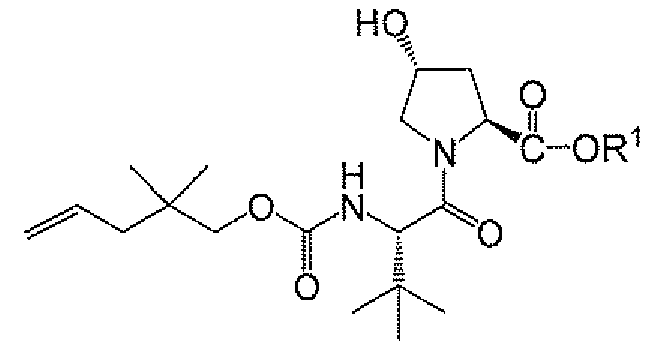
상기 식에서, R1은 제1항에 정의된 바와 같다. - 제14항에 있어서,또는 그의 염과
 또는 그의 염 (여기서 R1은 제1항에 정의된 바와 같음)을 커플링시켜 화학식 Ia의 화합물을 제조하는 단계를 추가로 포함하는 방법.
또는 그의 염 (여기서 R1은 제1항에 정의된 바와 같음)을 커플링시켜 화학식 Ia의 화합물을 제조하는 단계를 추가로 포함하는 방법.
- 제15항에 있어서,또는 그의 염이

 인 방법.
인 방법.
- 제13항 내지 제16항 중 어느 한 항에 있어서, R1이 H 또는 C1 -6 알킬인 방법.
- 제17항에 있어서,
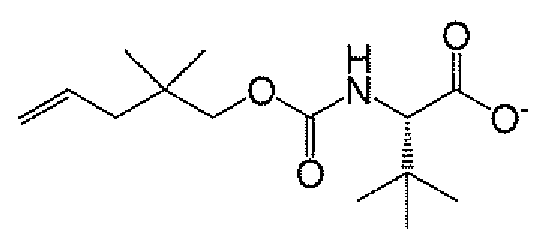 을 하기 단계를 포함하는 공정에 의해 제조하는 것인 방법:
을 하기 단계를 포함하는 공정에 의해 제조하는 것인 방법:

- 제8항 내지 제18항 중 어느 한 항에 있어서, 상기 폐환을 촉매 및 화학식 IIa의 화합물을 용매에 대략 동시에 서서히 첨가함으로써 수행하고, 여기서 상기 용매는 기질 1 Kg 당 약 5 내지 25 리터로 제공되고; 상기 촉매는 촉매 1 Kg 당 약 250 ml 내지 3 L의 농도로 제공되고; 상기 화합물은 기질 1 Kg 당 약 500 ml 내지 6 L의 농도로 제공되고; 상기 화합물-용액, 상기 촉매-용액 및 상기 용매를 0.5 내지 2.5시간의 기간에 걸쳐 함께 배합하는 것인 방법.
- 제9항 내지 제18항 중 어느 한 항에 있어서, 화합물 A-11 또는 그의 염을 하기 단계를 포함하는 공정에 의해 제조하는 것인 방법:
; 또는 그의 염.
- 하기 단계를 포함하는, 화합물 A-8 또는 그의 염의 제조 방법:

- 하기를 포함하는, 화합물 A-11 또는 그의 염의 제조 방법:
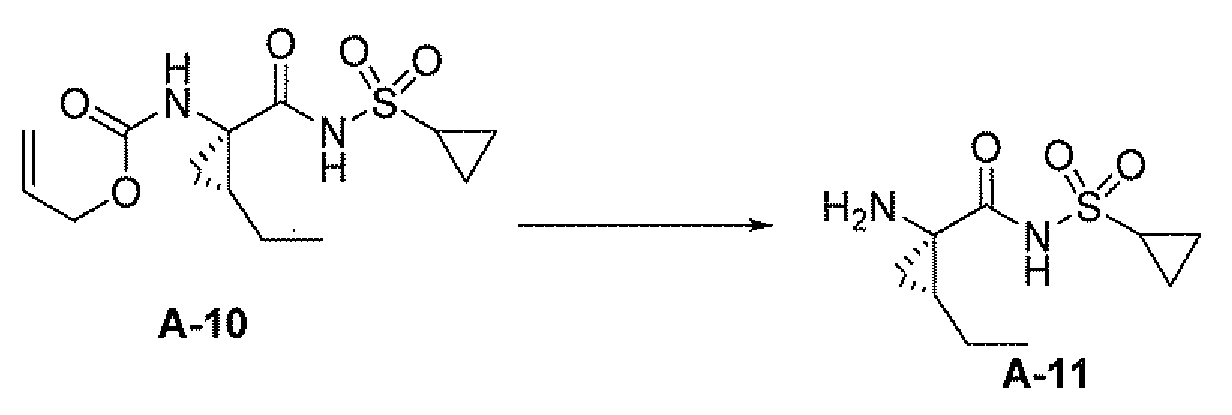
- 또는 그의 염을
 또는 그의 염에 커플링시켜 화합물 A 또는 그의 염을 형성하는 단계를 포함하고, 여기서 상기 반응은 커플링 시약 및 피리딘 또는 피리딘 유도체의 사용을 포함하는 것인, 화합물 A 또는 그의 염의 제조 방법.
또는 그의 염에 커플링시켜 화합물 A 또는 그의 염을 형성하는 단계를 포함하고, 여기서 상기 반응은 커플링 시약 및 피리딘 또는 피리딘 유도체의 사용을 포함하는 것인, 화합물 A 또는 그의 염의 제조 방법.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US42290710P | 2010-12-14 | 2010-12-14 | |
| US61/422,907 | 2010-12-14 | ||
| PCT/US2011/064521 WO2012082672A2 (en) | 2010-12-14 | 2011-12-13 | Process and intermediates for preparing macrolactams |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20130143084A true KR20130143084A (ko) | 2013-12-30 |
Family
ID=46245309
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020137015211A Withdrawn KR20130143084A (ko) | 2010-12-14 | 2011-12-13 | 마크로락탐의 제조 방법 및 중간체 |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US9120818B2 (ko) |
| EP (1) | EP2651884A2 (ko) |
| JP (1) | JP6034802B2 (ko) |
| KR (1) | KR20130143084A (ko) |
| CN (1) | CN103717067A (ko) |
| AU (1) | AU2011344075A1 (ko) |
| BR (1) | BR112013010372A2 (ko) |
| CA (1) | CA2817365A1 (ko) |
| WO (1) | WO2012082672A2 (ko) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| UA119315C2 (uk) | 2012-07-03 | 2019-06-10 | Гіліад Фармассет Елелсі | Інгібітори вірусу гепатиту с |
| EA025560B1 (ru) | 2012-10-19 | 2017-01-30 | Бристол-Майерс Сквибб Компани | Ингибиторы вируса гепатита с |
| EP2914613B1 (en) | 2012-11-02 | 2017-11-22 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| JP6342922B2 (ja) | 2013-03-07 | 2018-06-13 | ブリストル−マイヤーズ スクイブ カンパニーBristol−Myers Squibb Company | C型肝炎ウイルス阻害剤 |
| WO2014145095A1 (en) | 2013-03-15 | 2014-09-18 | Gilead Sciences, Inc. | Macrocyclic and bicyclic inhibitors of hepatitis c virus |
| CN103450070B (zh) * | 2013-08-13 | 2015-06-24 | 张家港威胜生物医药有限公司 | 一种二氢异吲哚的合成工艺 |
| WO2015095430A1 (en) * | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Methods and intermediates for the preparation of macrolactams |
| WO2015095437A1 (en) * | 2013-12-20 | 2015-06-25 | Merck Sharp & Dohme Corp. | Methods and intermediates for the preparation of macrolactams |
| ES2990061T3 (es) | 2016-05-10 | 2024-11-28 | C4 Therapeutics Inc | Degronímeros espirocíclicos para la degradación de proteínas diana |
| CN109641874A (zh) | 2016-05-10 | 2019-04-16 | C4医药公司 | 用于靶蛋白降解的c3-碳连接的戊二酰亚胺降解决定子体 |
| EP3454856B1 (en) | 2016-05-10 | 2024-09-11 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| CN109790143A (zh) | 2016-05-10 | 2019-05-21 | C4医药公司 | 用于靶蛋白降解的胺连接的c3-戊二酰亚胺降解决定子体 |
| CN110769822A (zh) | 2017-06-20 | 2020-02-07 | C4医药公司 | 用于蛋白降解的n/o-连接的降解决定子和降解决定子体 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3085041A (en) * | 1959-01-23 | 1963-04-09 | Du Pont | Aliphatic sulfonyl fluoride fungicide |
| DE10127294A1 (de) * | 2001-06-05 | 2002-12-12 | Martin Ziegler | Strömungskraftmaschine zur Nutzung von Potentialdifferenzen ruhender Fluide im Gleichgewicht |
| GB0118238D0 (en) * | 2001-07-26 | 2001-09-19 | Smithkline Beecham Plc | Medicaments |
| MY140680A (en) | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| US7834145B2 (en) | 2005-03-22 | 2010-11-16 | Merck Sharp & Dohme Corp. | HCV protease substrates |
| AU2006242475B2 (en) | 2005-05-02 | 2011-07-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| TWI387603B (zh) * | 2005-07-20 | 2013-03-01 | Merck Sharp & Dohme | Hcv ns3蛋白酶抑制劑 |
| UA90909C2 (en) * | 2005-07-20 | 2010-06-10 | Мерк Шарп Энд Домэ Корп. | Hcv ns3 protease inhibitors |
| BRPI0614205A2 (pt) | 2005-08-01 | 2016-11-22 | Merck & Co Inc | composto, composição farmacêutica, e, uso de composto |
| GB0609492D0 (en) | 2006-05-15 | 2006-06-21 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| GB0612423D0 (en) | 2006-06-23 | 2006-08-02 | Angeletti P Ist Richerche Bio | Therapeutic agents |
| TW200815428A (en) * | 2006-08-15 | 2008-04-01 | Wyeth Corp | Oxazolidone derivatives as PR modulators |
| CN101583372A (zh) | 2006-10-24 | 2009-11-18 | 默克公司 | Hcv ns3蛋白酶抑制剂 |
| JP2010507656A (ja) | 2006-10-24 | 2010-03-11 | メルク エンド カムパニー インコーポレーテッド | Hcvns3プロテアーゼ阻害剤 |
| AU2007318164B2 (en) | 2006-10-27 | 2013-02-07 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| WO2008057209A1 (en) * | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2008116054A1 (en) * | 2007-03-20 | 2008-09-25 | Trustees Of Tufts College | Inhibitors of fibroblast activation protein, and methods of use thereof |
| EP2160392A2 (en) * | 2007-05-03 | 2010-03-10 | Intermune, Inc. | Novel macrocyclic inhibitors of hepatitis c virus replication |
| AU2008277377B2 (en) | 2007-07-19 | 2013-08-01 | Msd Italia S.R.L. | Macrocyclic compounds as antiviral agents |
| AU2009217551B2 (en) | 2008-02-25 | 2014-07-31 | Msd Italia S.R.L. | Therapeutic compounds |
| AU2009241445A1 (en) | 2008-04-28 | 2009-11-05 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| PT2540350E (pt) | 2008-07-22 | 2014-08-27 | Merck Sharp & Dohme | Combinações de um composto de quinoxalina macrocílico o qual é um inibidor da protease ns3 do hcv com outros agentes do hcv |
-
2011
- 2011-12-13 EP EP11848832.9A patent/EP2651884A2/en not_active Withdrawn
- 2011-12-13 WO PCT/US2011/064521 patent/WO2012082672A2/en not_active Ceased
- 2011-12-13 BR BR112013010372A patent/BR112013010372A2/pt not_active IP Right Cessation
- 2011-12-13 CA CA2817365A patent/CA2817365A1/en not_active Abandoned
- 2011-12-13 KR KR1020137015211A patent/KR20130143084A/ko not_active Withdrawn
- 2011-12-13 AU AU2011344075A patent/AU2011344075A1/en not_active Abandoned
- 2011-12-13 CN CN201180060220.8A patent/CN103717067A/zh active Pending
- 2011-12-13 JP JP2013544661A patent/JP6034802B2/ja not_active Expired - Fee Related
- 2011-12-13 US US13/993,541 patent/US9120818B2/en not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| CA2817365A1 (en) | 2012-06-21 |
| JP2014508116A (ja) | 2014-04-03 |
| AU2011344075A1 (en) | 2013-05-09 |
| CN103717067A (zh) | 2014-04-09 |
| JP6034802B2 (ja) | 2016-11-30 |
| EP2651884A2 (en) | 2013-10-23 |
| BR112013010372A2 (pt) | 2016-07-05 |
| US20130274463A1 (en) | 2013-10-17 |
| WO2012082672A3 (en) | 2013-12-27 |
| US9120818B2 (en) | 2015-09-01 |
| WO2012082672A2 (en) | 2012-06-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20130143084A (ko) | 마크로락탐의 제조 방법 및 중간체 | |
| US9238604B2 (en) | Process and intermediates for preparing macrolactams | |
| WO2022222994A1 (zh) | 一种核苷类化合物及其在治疗猫传染性腹膜炎中的应用 | |
| CN102140100A (zh) | 高效抑制丙型肝炎病毒的多环化合物及其制备方法和用途 | |
| EP1709047A2 (en) | Azabenzofuran substituted thioureas as inhibitors of viral replication | |
| CN112592331B (zh) | 一种奥司他韦protac化合物及其制备方法与在抗流感病毒药物中的应用 | |
| CN103387601B (zh) | 抗登革热病毒(denv)杂环肽类化合物及其制备方法和用途 | |
| WO2015095430A1 (en) | Methods and intermediates for the preparation of macrolactams | |
| CN118908937B (zh) | 一种具有sting蛋白降解作用的化合物及其制备方法和用途 | |
| CN108610332B (zh) | 诱导MDM2自我降解E3泛素连接酶二聚体酯类小分子PROTACs | |
| WO2017173960A1 (zh) | 一类抑制丙肝病毒的大环状杂环化合物及其制备和用途 | |
| CN109134511B (zh) | C19位氟代的Largazole类似物、其制备方法和用途 | |
| CN107074876B (zh) | 一类抑制丙肝病毒的大环状杂环化合物及其制备和用途 | |
| Michel et al. | EX-CHIRAL POOL SYNTHESIS OF AMINOOXAZEPINONES AS CONFORMATIONALLY RESTRICTED 8-AMINO ACID ANALOGS | |
| WO2015095437A1 (en) | Methods and intermediates for the preparation of macrolactams |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20130613 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| PC1203 | Withdrawal of no request for examination | ||
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |