KR20170087907A - 시르투인 조정제로서의 치환된 가교된 우레아 유사체 - Google Patents
시르투인 조정제로서의 치환된 가교된 우레아 유사체 Download PDFInfo
- Publication number
- KR20170087907A KR20170087907A KR1020177016279A KR20177016279A KR20170087907A KR 20170087907 A KR20170087907 A KR 20170087907A KR 1020177016279 A KR1020177016279 A KR 1020177016279A KR 20177016279 A KR20177016279 A KR 20177016279A KR 20170087907 A KR20170087907 A KR 20170087907A
- Authority
- KR
- South Korea
- Prior art keywords
- branched
- straight
- chain
- sirtuin
- heterocyclyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
- 102000011990 Sirtuin Human genes 0.000 title abstract description 175
- 108050002485 Sirtuin Proteins 0.000 title abstract description 175
- 150000003672 ureas Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 417
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 152
- 238000000034 method Methods 0.000 claims abstract description 131
- -1 urea analog compounds Chemical class 0.000 claims abstract description 89
- 201000010099 disease Diseases 0.000 claims abstract description 83
- 150000003839 salts Chemical class 0.000 claims abstract description 74
- 208000035475 disorder Diseases 0.000 claims abstract description 68
- 238000011282 treatment Methods 0.000 claims abstract description 58
- 239000003814 drug Substances 0.000 claims abstract description 49
- 230000001965 increasing effect Effects 0.000 claims abstract description 44
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 43
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 36
- 230000035882 stress Effects 0.000 claims abstract description 31
- 206010061218 Inflammation Diseases 0.000 claims abstract description 25
- 230000032683 aging Effects 0.000 claims abstract description 25
- 230000004054 inflammatory process Effects 0.000 claims abstract description 25
- 230000004770 neurodegeneration Effects 0.000 claims abstract description 25
- 208000015122 neurodegenerative disease Diseases 0.000 claims abstract description 25
- 208000024172 Cardiovascular disease Diseases 0.000 claims abstract description 20
- 238000002360 preparation method Methods 0.000 claims abstract description 20
- 230000000694 effects Effects 0.000 claims description 153
- 229910052739 hydrogen Inorganic materials 0.000 claims description 87
- 239000001257 hydrogen Substances 0.000 claims description 86
- 125000003118 aryl group Chemical group 0.000 claims description 64
- 125000000623 heterocyclic group Chemical group 0.000 claims description 58
- 150000002431 hydrogen Chemical class 0.000 claims description 54
- 229910052736 halogen Inorganic materials 0.000 claims description 45
- 150000002367 halogens Chemical class 0.000 claims description 45
- 206010022489 Insulin Resistance Diseases 0.000 claims description 37
- 102000000344 Sirtuin 1 Human genes 0.000 claims description 37
- 108010041191 Sirtuin 1 Proteins 0.000 claims description 37
- 125000001072 heteroaryl group Chemical group 0.000 claims description 37
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 36
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 29
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 26
- 125000001424 substituent group Chemical group 0.000 claims description 25
- 230000002829 reductive effect Effects 0.000 claims description 18
- 238000004519 manufacturing process Methods 0.000 claims description 16
- 208000001145 Metabolic Syndrome Diseases 0.000 claims description 15
- 201000004681 Psoriasis Diseases 0.000 claims description 15
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 claims description 15
- 230000003818 metabolic dysfunction Effects 0.000 claims description 14
- 125000004452 carbocyclyl group Chemical group 0.000 claims description 12
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 11
- 239000013543 active substance Substances 0.000 claims description 10
- 238000002560 therapeutic procedure Methods 0.000 claims description 9
- 206010003246 arthritis Diseases 0.000 claims description 8
- 208000002874 Acne Vulgaris Diseases 0.000 claims description 7
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 7
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 7
- 208000001132 Osteoporosis Diseases 0.000 claims description 7
- 206010000496 acne Diseases 0.000 claims description 7
- 201000008937 atopic dermatitis Diseases 0.000 claims description 7
- 230000014509 gene expression Effects 0.000 claims description 7
- 208000027866 inflammatory disease Diseases 0.000 claims description 7
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 7
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 6
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 6
- 241000220317 Rosa Species 0.000 claims description 6
- 201000011529 cardiovascular cancer Diseases 0.000 claims description 6
- 210000000936 intestine Anatomy 0.000 claims description 6
- 206010040047 Sepsis Diseases 0.000 claims description 5
- 239000003937 drug carrier Substances 0.000 claims description 5
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 3
- 230000002438 mitochondrial effect Effects 0.000 abstract description 28
- 206010028980 Neoplasm Diseases 0.000 abstract description 25
- 208000008589 Obesity Diseases 0.000 abstract description 21
- 235000020824 obesity Nutrition 0.000 abstract description 21
- 201000011510 cancer Diseases 0.000 abstract description 19
- 229940124597 therapeutic agent Drugs 0.000 abstract description 19
- 230000008901 benefit Effects 0.000 abstract description 15
- 230000023555 blood coagulation Effects 0.000 abstract description 13
- 206010053567 Coagulopathies Diseases 0.000 abstract description 11
- 230000006806 disease prevention Effects 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 110
- 210000004027 cell Anatomy 0.000 description 109
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 75
- 239000000203 mixture Substances 0.000 description 75
- 239000000243 solution Substances 0.000 description 64
- 206010057190 Respiratory tract infections Diseases 0.000 description 60
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 57
- 235000019439 ethyl acetate Nutrition 0.000 description 50
- 239000011541 reaction mixture Substances 0.000 description 50
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 48
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 47
- 230000000875 corresponding effect Effects 0.000 description 44
- 238000006243 chemical reaction Methods 0.000 description 40
- 239000003795 chemical substances by application Substances 0.000 description 40
- 239000007787 solid Substances 0.000 description 40
- 125000004429 atom Chemical group 0.000 description 37
- 239000000758 substrate Substances 0.000 description 35
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 34
- 108090000623 proteins and genes Proteins 0.000 description 34
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 32
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 30
- 102000000478 Sirtuin 3 Human genes 0.000 description 30
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 30
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 29
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 29
- 108091005770 SIRT3 Proteins 0.000 description 27
- 238000011010 flushing procedure Methods 0.000 description 27
- 230000015572 biosynthetic process Effects 0.000 description 26
- 238000012360 testing method Methods 0.000 description 26
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 25
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 25
- 238000006467 substitution reaction Methods 0.000 description 25
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 24
- 239000012043 crude product Substances 0.000 description 24
- 239000000047 product Substances 0.000 description 24
- 102000004169 proteins and genes Human genes 0.000 description 23
- 101000654471 Mus musculus NAD-dependent protein deacetylase sirtuin-1 Proteins 0.000 description 22
- 239000003153 chemical reaction reagent Substances 0.000 description 22
- 125000002619 bicyclic group Chemical group 0.000 description 21
- 230000006378 damage Effects 0.000 description 21
- 229940079593 drug Drugs 0.000 description 20
- 238000009472 formulation Methods 0.000 description 20
- 239000011734 sodium Substances 0.000 description 20
- 238000003786 synthesis reaction Methods 0.000 description 20
- 239000002904 solvent Substances 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 230000006907 apoptotic process Effects 0.000 description 18
- 210000003169 central nervous system Anatomy 0.000 description 17
- 239000013058 crude material Substances 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 16
- 238000003381 deacetylation reaction Methods 0.000 description 16
- 238000002347 injection Methods 0.000 description 16
- 239000007924 injection Substances 0.000 description 16
- 241000894007 species Species 0.000 description 16
- 239000000126 substance Substances 0.000 description 16
- 210000001519 tissue Anatomy 0.000 description 16
- 208000006011 Stroke Diseases 0.000 description 15
- 230000006196 deacetylation Effects 0.000 description 15
- 201000006417 multiple sclerosis Diseases 0.000 description 15
- 229910052757 nitrogen Inorganic materials 0.000 description 15
- 230000002265 prevention Effects 0.000 description 15
- 108090000765 processed proteins & peptides Proteins 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 14
- 125000003275 alpha amino acid group Chemical group 0.000 description 14
- 239000002246 antineoplastic agent Substances 0.000 description 14
- 239000012267 brine Substances 0.000 description 14
- 239000010410 layer Substances 0.000 description 14
- 102000039446 nucleic acids Human genes 0.000 description 14
- 108020004707 nucleic acids Proteins 0.000 description 14
- 150000007523 nucleic acids Chemical class 0.000 description 14
- 239000012071 phase Substances 0.000 description 14
- 208000011580 syndromic disease Diseases 0.000 description 14
- 102000003964 Histone deacetylase Human genes 0.000 description 13
- 108090000353 Histone deacetylase Proteins 0.000 description 13
- 241001465754 Metazoa Species 0.000 description 13
- 238000003556 assay Methods 0.000 description 13
- 125000000392 cycloalkenyl group Chemical group 0.000 description 13
- 125000000753 cycloalkyl group Chemical group 0.000 description 13
- 229940127089 cytotoxic agent Drugs 0.000 description 13
- 210000001508 eye Anatomy 0.000 description 13
- 239000000706 filtrate Substances 0.000 description 13
- 210000003205 muscle Anatomy 0.000 description 13
- 238000004809 thin layer chromatography Methods 0.000 description 13
- 241000196324 Embryophyta Species 0.000 description 12
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 12
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 12
- 208000007536 Thrombosis Diseases 0.000 description 12
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 12
- 150000003973 alkyl amines Chemical class 0.000 description 12
- 125000004432 carbon atom Chemical group C* 0.000 description 12
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 12
- 238000002648 combination therapy Methods 0.000 description 12
- 208000037765 diseases and disorders Diseases 0.000 description 12
- 229950006238 nadide Drugs 0.000 description 12
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 12
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 12
- 208000024891 symptom Diseases 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 11
- 125000000217 alkyl group Chemical group 0.000 description 11
- 229940024606 amino acid Drugs 0.000 description 11
- 150000001413 amino acids Chemical class 0.000 description 11
- 210000004369 blood Anatomy 0.000 description 11
- 239000008280 blood Substances 0.000 description 11
- 230000001419 dependent effect Effects 0.000 description 11
- YZTFIKANQHTFDZ-UHFFFAOYSA-N diazocine Chemical compound C1=CC=CN=NC=C1 YZTFIKANQHTFDZ-UHFFFAOYSA-N 0.000 description 11
- 239000007788 liquid Substances 0.000 description 11
- 208000033808 peripheral neuropathy Diseases 0.000 description 11
- 239000012453 solvate Substances 0.000 description 11
- LAMQVIQMVKWXOC-UHFFFAOYSA-N 4-methyl-n-[2-[3-(morpholin-4-ylmethyl)imidazo[2,1-b][1,3]thiazol-6-yl]phenyl]-2-pyridin-3-yl-1,3-thiazole-5-carboxamide Chemical compound CC=1N=C(C=2C=NC=CC=2)SC=1C(=O)NC1=CC=CC=C1C(N=C1SC=2)=CN1C=2CN1CCOCC1 LAMQVIQMVKWXOC-UHFFFAOYSA-N 0.000 description 10
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 10
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 10
- LQZMLBORDGWNPD-UHFFFAOYSA-N N-iodosuccinimide Chemical compound IN1C(=O)CCC1=O LQZMLBORDGWNPD-UHFFFAOYSA-N 0.000 description 10
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 10
- 125000003342 alkenyl group Chemical group 0.000 description 10
- 125000000304 alkynyl group Chemical group 0.000 description 10
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 10
- 238000010168 coupling process Methods 0.000 description 10
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 10
- 230000007423 decrease Effects 0.000 description 10
- 239000002552 dosage form Substances 0.000 description 10
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 10
- 239000000543 intermediate Substances 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- 229960001866 silicon dioxide Drugs 0.000 description 10
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 10
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 9
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 9
- 241000282412 Homo Species 0.000 description 9
- 101000616727 Homo sapiens NAD-dependent protein deacylase sirtuin-5, mitochondrial Proteins 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 9
- 238000005859 coupling reaction Methods 0.000 description 9
- 235000019441 ethanol Nutrition 0.000 description 9
- 239000008103 glucose Substances 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 230000009467 reduction Effects 0.000 description 9
- 229920006395 saturated elastomer Polymers 0.000 description 9
- 210000003491 skin Anatomy 0.000 description 9
- 210000000278 spinal cord Anatomy 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 238000005406 washing Methods 0.000 description 9
- SBTYJUZFGGEWNU-UHFFFAOYSA-N 1,4-diazocine Chemical compound C1=CN=CC=NC=C1 SBTYJUZFGGEWNU-UHFFFAOYSA-N 0.000 description 8
- 101000616738 Homo sapiens NAD-dependent protein deacetylase sirtuin-6 Proteins 0.000 description 8
- 101000863629 Homo sapiens NAD-dependent protein lipoamidase sirtuin-4, mitochondrial Proteins 0.000 description 8
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 8
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 8
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 8
- 208000022873 Ocular disease Diseases 0.000 description 8
- 102000000477 Sirtuin 2 Human genes 0.000 description 8
- 230000021736 acetylation Effects 0.000 description 8
- 238000006640 acetylation reaction Methods 0.000 description 8
- 229910002091 carbon monoxide Inorganic materials 0.000 description 8
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 8
- 239000006184 cosolvent Substances 0.000 description 8
- 239000012634 fragment Substances 0.000 description 8
- 230000000302 ischemic effect Effects 0.000 description 8
- 125000006574 non-aromatic ring group Chemical group 0.000 description 8
- 229910052763 palladium Inorganic materials 0.000 description 8
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 8
- 230000008569 process Effects 0.000 description 8
- 102000004196 processed proteins & peptides Human genes 0.000 description 8
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 8
- 230000001225 therapeutic effect Effects 0.000 description 8
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 7
- 241000206602 Eukaryota Species 0.000 description 7
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 7
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 7
- 206010038848 Retinal detachment Diseases 0.000 description 7
- 208000017442 Retinal disease Diseases 0.000 description 7
- 229910052786 argon Inorganic materials 0.000 description 7
- 210000004556 brain Anatomy 0.000 description 7
- 235000013877 carbamide Nutrition 0.000 description 7
- 229940000425 combination drug Drugs 0.000 description 7
- 230000008878 coupling Effects 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- 208000002780 macular degeneration Diseases 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 201000001119 neuropathy Diseases 0.000 description 7
- 239000002773 nucleotide Substances 0.000 description 7
- 125000003729 nucleotide group Chemical group 0.000 description 7
- 210000001428 peripheral nervous system Anatomy 0.000 description 7
- 230000009885 systemic effect Effects 0.000 description 7
- 230000000699 topical effect Effects 0.000 description 7
- 208000029257 vision disease Diseases 0.000 description 7
- 230000004584 weight gain Effects 0.000 description 7
- 235000019786 weight gain Nutrition 0.000 description 7
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 6
- 208000024827 Alzheimer disease Diseases 0.000 description 6
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- 241000283690 Bos taurus Species 0.000 description 6
- 229920000858 Cyclodextrin Polymers 0.000 description 6
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 6
- 206010015150 Erythema Diseases 0.000 description 6
- 241000282326 Felis catus Species 0.000 description 6
- 241000238631 Hexapoda Species 0.000 description 6
- 101000654472 Homo sapiens NAD-dependent protein deacetylase sirtuin-1 Proteins 0.000 description 6
- 101000863566 Homo sapiens NAD-dependent protein deacetylase sirtuin-3, mitochondrial Proteins 0.000 description 6
- 101000709248 Homo sapiens NAD-dependent protein deacetylase sirtuin-7 Proteins 0.000 description 6
- 206010020772 Hypertension Diseases 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N N-phenyl amine Natural products NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 6
- 102100021839 NAD-dependent protein deacylase sirtuin-5, mitochondrial Human genes 0.000 description 6
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 6
- 206010038923 Retinopathy Diseases 0.000 description 6
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 150000001408 amides Chemical class 0.000 description 6
- 239000000010 aprotic solvent Substances 0.000 description 6
- 239000004202 carbamide Substances 0.000 description 6
- 238000004113 cell culture Methods 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 239000007789 gas Substances 0.000 description 6
- 230000023597 hemostasis Effects 0.000 description 6
- 102000056482 human SIRT1 Human genes 0.000 description 6
- 150000004677 hydrates Chemical class 0.000 description 6
- 239000000411 inducer Substances 0.000 description 6
- 208000015181 infectious disease Diseases 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 208000014674 injury Diseases 0.000 description 6
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 6
- 239000002502 liposome Substances 0.000 description 6
- FPYJFEHAWHCUMM-UHFFFAOYSA-N maleic anhydride Chemical compound O=C1OC(=O)C=C1 FPYJFEHAWHCUMM-UHFFFAOYSA-N 0.000 description 6
- 230000004065 mitochondrial dysfunction Effects 0.000 description 6
- 230000007823 neuropathy Effects 0.000 description 6
- 235000005152 nicotinamide Nutrition 0.000 description 6
- 239000011570 nicotinamide Substances 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 229920001184 polypeptide Polymers 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 230000004264 retinal detachment Effects 0.000 description 6
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 6
- 231100001274 therapeutic index Toxicity 0.000 description 6
- 231100000331 toxic Toxicity 0.000 description 6
- 230000002588 toxic effect Effects 0.000 description 6
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 6
- 239000003981 vehicle Substances 0.000 description 6
- BFNOPXRXIQJDHO-YDKGJHSESA-N 2''-O-acetyl-ADP-D-ribose Chemical compound O[C@H]1[C@@H](OC(=O)C)C(O)O[C@@H]1COP(O)(=O)OP(O)(=O)OC[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C3=NC=NC(N)=C3N=C2)O1 BFNOPXRXIQJDHO-YDKGJHSESA-N 0.000 description 5
- 241000282472 Canis lupus familiaris Species 0.000 description 5
- 206010050389 Cerebral ataxia Diseases 0.000 description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- 101000825628 Homo sapiens NAD-dependent protein deacetylase sirtuin-2 Proteins 0.000 description 5
- 241000725303 Human immunodeficiency virus Species 0.000 description 5
- 208000023105 Huntington disease Diseases 0.000 description 5
- 208000031226 Hyperlipidaemia Diseases 0.000 description 5
- 206010021143 Hypoxia Diseases 0.000 description 5
- 102000004877 Insulin Human genes 0.000 description 5
- 108090001061 Insulin Proteins 0.000 description 5
- 102100021840 NAD-dependent protein deacetylase sirtuin-6 Human genes 0.000 description 5
- 206010033307 Overweight Diseases 0.000 description 5
- 208000018737 Parkinson disease Diseases 0.000 description 5
- 241001494479 Pecora Species 0.000 description 5
- QNVSXXGDAPORNA-UHFFFAOYSA-N Resveratrol Natural products OC1=CC=CC(C=CC=2C=C(O)C(O)=CC=2)=C1 QNVSXXGDAPORNA-UHFFFAOYSA-N 0.000 description 5
- 108010041216 Sirtuin 2 Proteins 0.000 description 5
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 5
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 5
- LUKBXSAWLPMMSZ-OWOJBTEDSA-N Trans-resveratrol Chemical compound C1=CC(O)=CC=C1\C=C\C1=CC(O)=CC(O)=C1 LUKBXSAWLPMMSZ-OWOJBTEDSA-N 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 239000012190 activator Substances 0.000 description 5
- 206010064930 age-related macular degeneration Diseases 0.000 description 5
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 description 5
- 235000020827 calorie restriction Nutrition 0.000 description 5
- 230000030833 cell death Effects 0.000 description 5
- 125000001309 chloro group Chemical group Cl* 0.000 description 5
- 239000006071 cream Substances 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 239000003480 eluent Substances 0.000 description 5
- 239000000499 gel Substances 0.000 description 5
- 230000030279 gene silencing Effects 0.000 description 5
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 5
- 102000055034 human SIRT4 Human genes 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000001802 infusion Methods 0.000 description 5
- 229940125396 insulin Drugs 0.000 description 5
- 208000030159 metabolic disease Diseases 0.000 description 5
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 5
- 125000002950 monocyclic group Chemical group 0.000 description 5
- 229960003966 nicotinamide Drugs 0.000 description 5
- 210000000056 organ Anatomy 0.000 description 5
- 239000012044 organic layer Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 239000001301 oxygen Substances 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 230000000069 prophylactic effect Effects 0.000 description 5
- 230000005855 radiation Effects 0.000 description 5
- 230000004044 response Effects 0.000 description 5
- 235000021283 resveratrol Nutrition 0.000 description 5
- 229940016667 resveratrol Drugs 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- 101150089009 sir2 gene Proteins 0.000 description 5
- 238000011287 therapeutic dose Methods 0.000 description 5
- 239000003053 toxin Substances 0.000 description 5
- 231100000765 toxin Toxicity 0.000 description 5
- 108700012359 toxins Proteins 0.000 description 5
- 238000002054 transplantation Methods 0.000 description 5
- 230000004393 visual impairment Effects 0.000 description 5
- LPNUQEZRCLLNOW-ZETCQYMHSA-N (9S)-5-chloro-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound Clc1ccc2N3CCC[C@@H](C3)Nc2n1 LPNUQEZRCLLNOW-ZETCQYMHSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 4
- ALYNCZNDIQEVRV-UHFFFAOYSA-N 4-aminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1 ALYNCZNDIQEVRV-UHFFFAOYSA-N 0.000 description 4
- QRXMUCSWCMTJGU-UHFFFAOYSA-N 5-bromo-4-chloro-3-indolyl phosphate Chemical compound C1=C(Br)C(Cl)=C2C(OP(O)(=O)O)=CNC2=C1 QRXMUCSWCMTJGU-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- 208000030507 AIDS Diseases 0.000 description 4
- 206010003694 Atrophy Diseases 0.000 description 4
- 208000023275 Autoimmune disease Diseases 0.000 description 4
- 206010007559 Cardiac failure congestive Diseases 0.000 description 4
- 208000002177 Cataract Diseases 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 4
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- 239000007821 HATU Substances 0.000 description 4
- 206010019280 Heart failures Diseases 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 108010042046 Mitochondrial processing peptidase Proteins 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- 208000021642 Muscular disease Diseases 0.000 description 4
- 201000009623 Myopathy Diseases 0.000 description 4
- 102100034376 NAD-dependent protein deacetylase sirtuin-7 Human genes 0.000 description 4
- MWUXSHHQAYIFBG-UHFFFAOYSA-N Nitric oxide Chemical compound O=[N] MWUXSHHQAYIFBG-UHFFFAOYSA-N 0.000 description 4
- 208000002193 Pain Diseases 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- 206010057430 Retinal injury Diseases 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 206010047571 Visual impairment Diseases 0.000 description 4
- 208000027418 Wounds and injury Diseases 0.000 description 4
- 230000003213 activating effect Effects 0.000 description 4
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical compound C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 4
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 4
- 230000037444 atrophy Effects 0.000 description 4
- 230000001363 autoimmune Effects 0.000 description 4
- 230000009286 beneficial effect Effects 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- 210000004958 brain cell Anatomy 0.000 description 4
- 244000309466 calf Species 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- 238000002512 chemotherapy Methods 0.000 description 4
- 235000012000 cholesterol Nutrition 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 230000007850 degeneration Effects 0.000 description 4
- 238000002651 drug therapy Methods 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 239000003925 fat Substances 0.000 description 4
- 235000019197 fats Nutrition 0.000 description 4
- 125000000524 functional group Chemical group 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 125000005842 heteroatom Chemical group 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 4
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 150000007529 inorganic bases Chemical class 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- 239000006210 lotion Substances 0.000 description 4
- 210000004962 mammalian cell Anatomy 0.000 description 4
- RUZLIIJDZBWWSA-INIZCTEOSA-N methyl 2-[[(1s)-1-(7-methyl-2-morpholin-4-yl-4-oxopyrido[1,2-a]pyrimidin-9-yl)ethyl]amino]benzoate Chemical group COC(=O)C1=CC=CC=C1N[C@@H](C)C1=CC(C)=CN2C(=O)C=C(N3CCOCC3)N=C12 RUZLIIJDZBWWSA-INIZCTEOSA-N 0.000 description 4
- 210000003470 mitochondria Anatomy 0.000 description 4
- 208000010125 myocardial infarction Diseases 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- UMRZSTCPUPJPOJ-KNVOCYPGSA-N norbornane Chemical compound C1C[C@H]2CC[C@@H]1C2 UMRZSTCPUPJPOJ-KNVOCYPGSA-N 0.000 description 4
- 239000002674 ointment Substances 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 230000000750 progressive effect Effects 0.000 description 4
- 238000007670 refining Methods 0.000 description 4
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- RTKIYFITIVXBLE-QEQCGCAPSA-N trichostatin A Chemical compound ONC(=O)/C=C/C(/C)=C/[C@@H](C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-QEQCGCAPSA-N 0.000 description 4
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 4
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 4
- UFYBTLOLWSABAU-UHFFFAOYSA-N (2-methylpyridin-4-yl)boronic acid Chemical compound CC1=CC(B(O)O)=CC=N1 UFYBTLOLWSABAU-UHFFFAOYSA-N 0.000 description 3
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- CMJNIYUZFQWYCK-UHFFFAOYSA-N 2,6-dichloro-4-methyl-3-nitropyridine Chemical compound CC1=CC(Cl)=NC(Cl)=C1[N+]([O-])=O CMJNIYUZFQWYCK-UHFFFAOYSA-N 0.000 description 3
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- HCWXUWGQMOEJJJ-UHFFFAOYSA-N 3-(trifluoromethyl)pyrrolidine Chemical compound FC(F)(F)C1CCNC1 HCWXUWGQMOEJJJ-UHFFFAOYSA-N 0.000 description 3
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 3
- 201000001320 Atherosclerosis Diseases 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 201000004569 Blindness Diseases 0.000 description 3
- 206010006187 Breast cancer Diseases 0.000 description 3
- 208000026310 Breast neoplasm Diseases 0.000 description 3
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 3
- 241000283707 Capra Species 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 3
- 201000004624 Dermatitis Diseases 0.000 description 3
- 208000002249 Diabetes Complications Diseases 0.000 description 3
- 241000283086 Equidae Species 0.000 description 3
- 208000010412 Glaucoma Diseases 0.000 description 3
- 102000006947 Histones Human genes 0.000 description 3
- 108010033040 Histones Proteins 0.000 description 3
- 206010060378 Hyperinsulinaemia Diseases 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 229910010082 LiAlH Inorganic materials 0.000 description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 3
- 208000001344 Macular Edema Diseases 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 206010028289 Muscle atrophy Diseases 0.000 description 3
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 3
- 102100030709 NAD-dependent protein lipoamidase sirtuin-4, mitochondrial Human genes 0.000 description 3
- 208000028389 Nerve injury Diseases 0.000 description 3
- 206010029240 Neuritis Diseases 0.000 description 3
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- 206010036105 Polyneuropathy Diseases 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 206010060862 Prostate cancer Diseases 0.000 description 3
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 3
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 230000001154 acute effect Effects 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 239000003146 anticoagulant agent Substances 0.000 description 3
- 235000019789 appetite Nutrition 0.000 description 3
- 230000036528 appetite Effects 0.000 description 3
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 3
- 210000002565 arteriole Anatomy 0.000 description 3
- 230000007845 axonopathy Effects 0.000 description 3
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 3
- 230000004071 biological effect Effects 0.000 description 3
- 210000000601 blood cell Anatomy 0.000 description 3
- 208000015294 blood coagulation disease Diseases 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 229910052796 boron Inorganic materials 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 208000015114 central nervous system disease Diseases 0.000 description 3
- 229910052801 chlorine Inorganic materials 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 230000035602 clotting Effects 0.000 description 3
- 101150018117 cobB gene Proteins 0.000 description 3
- 230000001276 controlling effect Effects 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- 208000029078 coronary artery disease Diseases 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- 230000008034 disappearance Effects 0.000 description 3
- 238000012377 drug delivery Methods 0.000 description 3
- 230000004064 dysfunction Effects 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 231100000321 erythema Toxicity 0.000 description 3
- 229910052731 fluorine Inorganic materials 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 230000002140 halogenating effect Effects 0.000 description 3
- 230000026030 halogenation Effects 0.000 description 3
- 238000005658 halogenation reaction Methods 0.000 description 3
- 102000055198 human SIRT5 Human genes 0.000 description 3
- 102000049192 human SIRT6 Human genes 0.000 description 3
- 238000009396 hybridization Methods 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 230000003451 hyperinsulinaemic effect Effects 0.000 description 3
- 201000008980 hyperinsulinism Diseases 0.000 description 3
- 230000007954 hypoxia Effects 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000002513 implantation Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- 150000003951 lactams Chemical class 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000012280 lithium aluminium hydride Substances 0.000 description 3
- 206010025135 lupus erythematosus Diseases 0.000 description 3
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 3
- 239000004530 micro-emulsion Substances 0.000 description 3
- 230000020763 muscle atrophy Effects 0.000 description 3
- 201000000585 muscular atrophy Diseases 0.000 description 3
- 210000005036 nerve Anatomy 0.000 description 3
- 230000008764 nerve damage Effects 0.000 description 3
- 230000001537 neural effect Effects 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 229960003512 nicotinic acid Drugs 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 210000001328 optic nerve Anatomy 0.000 description 3
- 201000008482 osteoarthritis Diseases 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 235000019271 petrolatum Nutrition 0.000 description 3
- 230000036470 plasma concentration Effects 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 230000002062 proliferating effect Effects 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- HFHDHCJBZVLPGP-UHFFFAOYSA-N schardinger α-dextrin Chemical compound O1C(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC(C(O)C2O)C(CO)OC2OC(C(C2O)O)C(CO)OC2OC2C(O)C(O)C1OC2CO HFHDHCJBZVLPGP-UHFFFAOYSA-N 0.000 description 3
- 238000007423 screening assay Methods 0.000 description 3
- 229940126570 serotonin reuptake inhibitor Drugs 0.000 description 3
- 239000003772 serotonin uptake inhibitor Substances 0.000 description 3
- 238000010898 silica gel chromatography Methods 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 229910052938 sodium sulfate Inorganic materials 0.000 description 3
- 235000011152 sodium sulphate Nutrition 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 238000007920 subcutaneous administration Methods 0.000 description 3
- 238000001356 surgical procedure Methods 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 239000012049 topical pharmaceutical composition Substances 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 230000032258 transport Effects 0.000 description 3
- 230000008733 trauma Effects 0.000 description 3
- 230000000472 traumatic effect Effects 0.000 description 3
- 229930195735 unsaturated hydrocarbon Natural products 0.000 description 3
- 235000015112 vegetable and seed oil Nutrition 0.000 description 3
- 239000008158 vegetable oil Substances 0.000 description 3
- 230000009385 viral infection Effects 0.000 description 3
- 239000003643 water by type Substances 0.000 description 3
- 210000005253 yeast cell Anatomy 0.000 description 3
- ZWMGBALOCKIARD-ZDUSSCGKSA-N (9S)-5-[3-(trifluoromethyl)phenyl]-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound FC(F)(F)c1cccc(c1)-c1ccc2N3CCC[C@@H](C3)Nc2n1 ZWMGBALOCKIARD-ZDUSSCGKSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- FPIRBHDGWMWJEP-UHFFFAOYSA-N 1-hydroxy-7-azabenzotriazole Chemical compound C1=CN=C2N(O)N=NC2=C1 FPIRBHDGWMWJEP-UHFFFAOYSA-N 0.000 description 2
- GBBSAMQTQCPOBF-UHFFFAOYSA-N 2,4,6-trimethyl-1,3,5,2,4,6-trioxatriborinane Chemical compound CB1OB(C)OB(C)O1 GBBSAMQTQCPOBF-UHFFFAOYSA-N 0.000 description 2
- SHCWQWRTKPNTEM-UHFFFAOYSA-N 2,6-dichloro-3-nitropyridine Chemical compound [O-][N+](=O)C1=CC=C(Cl)N=C1Cl SHCWQWRTKPNTEM-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 2
- JOHFJTBDUSVGQB-UHFFFAOYSA-N 3-(trifluoromethyl)piperidine Chemical compound FC(F)(F)C1CCCNC1 JOHFJTBDUSVGQB-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- RMFVVPBBEDMZQI-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1,3-benzothiazole Chemical compound C1CCCC2=C1N=CS2 RMFVVPBBEDMZQI-UHFFFAOYSA-N 0.000 description 2
- QSVDFJNXDKTKTJ-UHFFFAOYSA-N 4,5,6,7-tetrahydro-1h-indene Chemical compound C1CCCC2=C1CC=C2 QSVDFJNXDKTKTJ-UHFFFAOYSA-N 0.000 description 2
- 206010001605 Alcohol poisoning Diseases 0.000 description 2
- 208000000044 Amnesia Diseases 0.000 description 2
- 206010002383 Angina Pectoris Diseases 0.000 description 2
- 206010002660 Anoxia Diseases 0.000 description 2
- 241000976983 Anoxia Species 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 229940127291 Calcium channel antagonist Drugs 0.000 description 2
- 208000031229 Cardiomyopathies Diseases 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 208000020406 Creutzfeldt Jacob disease Diseases 0.000 description 2
- 208000003407 Creutzfeldt-Jakob Syndrome Diseases 0.000 description 2
- 208000010859 Creutzfeldt-Jakob disease Diseases 0.000 description 2
- 206010048843 Cytomegalovirus chorioretinitis Diseases 0.000 description 2
- 206010067276 Cytotoxic oedema Diseases 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 230000033616 DNA repair Effects 0.000 description 2
- 206010012442 Dermatitis contact Diseases 0.000 description 2
- 208000032131 Diabetic Neuropathies Diseases 0.000 description 2
- 206010012689 Diabetic retinopathy Diseases 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- 208000032928 Dyslipidaemia Diseases 0.000 description 2
- 206010013935 Dysmenorrhoea Diseases 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 206010014733 Endometrial cancer Diseases 0.000 description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 description 2
- 206010014824 Endotoxic shock Diseases 0.000 description 2
- 241000282324 Felis Species 0.000 description 2
- 208000024412 Friedreich ataxia Diseases 0.000 description 2
- 241000233866 Fungi Species 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 201000005569 Gout Diseases 0.000 description 2
- 102000015779 HDL Lipoproteins Human genes 0.000 description 2
- 108010010234 HDL Lipoproteins Proteins 0.000 description 2
- 229910004373 HOAc Inorganic materials 0.000 description 2
- 208000032843 Hemorrhage Diseases 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 241000257303 Hymenoptera Species 0.000 description 2
- 208000035150 Hypercholesterolemia Diseases 0.000 description 2
- XQFRJNBWHJMXHO-RRKCRQDMSA-N IDUR Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(I)=C1 XQFRJNBWHJMXHO-RRKCRQDMSA-N 0.000 description 2
- 208000017170 Lipid metabolism disease Diseases 0.000 description 2
- 206010025415 Macular oedema Diseases 0.000 description 2
- 208000002720 Malnutrition Diseases 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- 102100026934 Mitochondrial intermediate peptidase Human genes 0.000 description 2
- 108010047660 Mitochondrial intermediate peptidase Proteins 0.000 description 2
- 241000699666 Mus <mouse, genus> Species 0.000 description 2
- 206010049565 Muscle fatigue Diseases 0.000 description 2
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 2
- 208000012902 Nervous system disease Diseases 0.000 description 2
- 208000025966 Neurological disease Diseases 0.000 description 2
- 208000003435 Optic Neuritis Diseases 0.000 description 2
- 206010034719 Personality change Diseases 0.000 description 2
- 239000004264 Petrolatum Substances 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- REFJWTPEDVJJIY-UHFFFAOYSA-N Quercetin Chemical compound C=1C(O)=CC(O)=C(C(C=2O)=O)C=1OC=2C1=CC=C(O)C(O)=C1 REFJWTPEDVJJIY-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 206010038910 Retinitis Diseases 0.000 description 2
- 206010040070 Septic Shock Diseases 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 241000282887 Suidae Species 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 238000006069 Suzuki reaction reaction Methods 0.000 description 2
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 2
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 206010052779 Transplant rejections Diseases 0.000 description 2
- RTKIYFITIVXBLE-UHFFFAOYSA-N Trichostatin A Natural products ONC(=O)C=CC(C)=CC(C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-UHFFFAOYSA-N 0.000 description 2
- MFUPLHQOVIUESQ-JEDNCBNOSA-N [(2s)-1,5-dimethoxy-1,5-dioxopentan-2-yl]azanium;chloride Chemical compound Cl.COC(=O)CC[C@H](N)C(=O)OC MFUPLHQOVIUESQ-JEDNCBNOSA-N 0.000 description 2
- SORGEQQSQGNZFI-UHFFFAOYSA-N [azido(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(N=[N+]=[N-])OC1=CC=CC=C1 SORGEQQSQGNZFI-UHFFFAOYSA-N 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 150000001298 alcohols Chemical class 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 229960004050 aminobenzoic acid Drugs 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000007953 anoxia Effects 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000002785 anti-thrombosis Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229940127219 anticoagulant drug Drugs 0.000 description 2
- 239000000935 antidepressant agent Substances 0.000 description 2
- 229940005513 antidepressants Drugs 0.000 description 2
- 239000003429 antifungal agent Substances 0.000 description 2
- 229940121375 antifungal agent Drugs 0.000 description 2
- 239000000164 antipsychotic agent Substances 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 210000001367 artery Anatomy 0.000 description 2
- 230000003143 atherosclerotic effect Effects 0.000 description 2
- 210000003403 autonomic nervous system Anatomy 0.000 description 2
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 description 2
- 229940049706 benzodiazepine Drugs 0.000 description 2
- RPZUBXWEQBPUJR-UHFFFAOYSA-N bicyclo[4.2.0]octane Chemical compound C1CCCC2CCC21 RPZUBXWEQBPUJR-UHFFFAOYSA-N 0.000 description 2
- 230000008827 biological function Effects 0.000 description 2
- 230000008512 biological response Effects 0.000 description 2
- 230000017531 blood circulation Effects 0.000 description 2
- 210000004204 blood vessel Anatomy 0.000 description 2
- 210000001124 body fluid Anatomy 0.000 description 2
- 239000010839 body fluid Substances 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 206010006451 bronchitis Diseases 0.000 description 2
- 239000006172 buffering agent Substances 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 230000001201 calcium accumulation Effects 0.000 description 2
- 239000000480 calcium channel blocker Substances 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000000747 cardiac effect Effects 0.000 description 2
- 239000002327 cardiovascular agent Substances 0.000 description 2
- 229940125692 cardiovascular agent Drugs 0.000 description 2
- 210000001715 carotid artery Anatomy 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000032677 cell aging Effects 0.000 description 2
- 238000002659 cell therapy Methods 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 201000005667 central retinal vein occlusion Diseases 0.000 description 2
- 201000001883 cholelithiasis Diseases 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 238000013270 controlled release Methods 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 210000004087 cornea Anatomy 0.000 description 2
- 210000004351 coronary vessel Anatomy 0.000 description 2
- 239000002537 cosmetic Substances 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 229940097362 cyclodextrins Drugs 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 208000001763 cytomegalovirus retinitis Diseases 0.000 description 2
- 230000000850 deacetylating effect Effects 0.000 description 2
- 230000007812 deficiency Effects 0.000 description 2
- 230000003111 delayed effect Effects 0.000 description 2
- 230000003831 deregulation Effects 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 235000007882 dietary composition Nutrition 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- KZLCDAUOZNWNDS-ZETCQYMHSA-N dimethyl (2S)-2-[(6-chloro-3-nitropyridin-2-yl)amino]pentanedioate Chemical compound COC(=O)CC[C@H](Nc1nc(Cl)ccc1[N+]([O-])=O)C(=O)OC KZLCDAUOZNWNDS-ZETCQYMHSA-N 0.000 description 2
- GUVUOGQBMYCBQP-UHFFFAOYSA-N dmpu Chemical compound CN1CCCN(C)C1=O GUVUOGQBMYCBQP-UHFFFAOYSA-N 0.000 description 2
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 description 2
- XMOCLSLCDHWDHP-IUODEOHRSA-N epi-Gallocatechin Chemical compound C1([C@H]2OC3=CC(O)=CC(O)=C3C[C@H]2O)=CC(O)=C(O)C(O)=C1 XMOCLSLCDHWDHP-IUODEOHRSA-N 0.000 description 2
- 206010015037 epilepsy Diseases 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- MMXKVMNBHPAILY-UHFFFAOYSA-N ethyl laurate Chemical compound CCCCCCCCCCCC(=O)OCC MMXKVMNBHPAILY-UHFFFAOYSA-N 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 230000008020 evaporation Effects 0.000 description 2
- 230000003492 excitotoxic effect Effects 0.000 description 2
- 231100000063 excitotoxicity Toxicity 0.000 description 2
- 208000030533 eye disease Diseases 0.000 description 2
- 239000000945 filler Substances 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 208000001130 gallstones Diseases 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 238000012226 gene silencing method Methods 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 230000005484 gravity Effects 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical compound O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 230000035876 healing Effects 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 208000002672 hepatitis B Diseases 0.000 description 2
- 235000010299 hexamethylene tetramine Nutrition 0.000 description 2
- VKYKSIONXSXAKP-UHFFFAOYSA-N hexamethylenetetramine Chemical compound C1N(C2)CN3CN1CN2C3 VKYKSIONXSXAKP-UHFFFAOYSA-N 0.000 description 2
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 2
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 2
- 229940088597 hormone Drugs 0.000 description 2
- 239000005556 hormone Substances 0.000 description 2
- 102000050401 human SIRT2 Human genes 0.000 description 2
- 102000045076 human SIRT3 Human genes 0.000 description 2
- 102000044649 human SIRT7 Human genes 0.000 description 2
- 244000052637 human pathogen Species 0.000 description 2
- 201000001421 hyperglycemia Diseases 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 150000002475 indoles Chemical class 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 210000000554 iris Anatomy 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 210000003734 kidney Anatomy 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 150000002596 lactones Chemical class 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 229920006008 lipopolysaccharide Polymers 0.000 description 2
- 238000004811 liquid chromatography Methods 0.000 description 2
- 244000144972 livestock Species 0.000 description 2
- 230000033001 locomotion Effects 0.000 description 2
- 201000010230 macular retinal edema Diseases 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 235000000824 malnutrition Nutrition 0.000 description 2
- 230000001071 malnutrition Effects 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 238000004949 mass spectrometry Methods 0.000 description 2
- 235000013372 meat Nutrition 0.000 description 2
- LULAYUGMBFYYEX-UHFFFAOYSA-N metachloroperbenzoic acid Natural products OC(=O)C1=CC=CC(Cl)=C1 LULAYUGMBFYYEX-UHFFFAOYSA-N 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229960004011 methenamine Drugs 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 230000008437 mitochondrial biogenesis Effects 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 201000005518 mononeuropathy Diseases 0.000 description 2
- 230000004118 muscle contraction Effects 0.000 description 2
- 230000004220 muscle function Effects 0.000 description 2
- 201000006938 muscular dystrophy Diseases 0.000 description 2
- 125000001624 naphthyl group Chemical group 0.000 description 2
- 230000017074 necrotic cell death Effects 0.000 description 2
- 230000006764 neuronal dysfunction Effects 0.000 description 2
- 230000002981 neuropathic effect Effects 0.000 description 2
- 239000011664 nicotinic acid Substances 0.000 description 2
- 235000001968 nicotinic acid Nutrition 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 208000015380 nutritional deficiency disease Diseases 0.000 description 2
- 208000001797 obstructive sleep apnea Diseases 0.000 description 2
- GLDOVTGHNKAZLK-UHFFFAOYSA-N octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCCCO GLDOVTGHNKAZLK-UHFFFAOYSA-N 0.000 description 2
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 239000003883 ointment base Substances 0.000 description 2
- 230000008816 organ damage Effects 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 150000007530 organic bases Chemical class 0.000 description 2
- 125000002524 organometallic group Chemical group 0.000 description 2
- 238000012856 packing Methods 0.000 description 2
- MUJIDPITZJWBSW-UHFFFAOYSA-N palladium(2+) Chemical compound [Pd+2] MUJIDPITZJWBSW-UHFFFAOYSA-N 0.000 description 2
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 2
- 230000000737 periodic effect Effects 0.000 description 2
- 229940066842 petrolatum Drugs 0.000 description 2
- RLOWWWKZYUNIDI-UHFFFAOYSA-N phosphinic chloride Chemical compound ClP=O RLOWWWKZYUNIDI-UHFFFAOYSA-N 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 201000010065 polycystic ovary syndrome Diseases 0.000 description 2
- 108010040003 polyglutamine Proteins 0.000 description 2
- 229920000155 polyglutamine Polymers 0.000 description 2
- 230000007824 polyneuropathy Effects 0.000 description 2
- 108091033319 polynucleotide Proteins 0.000 description 2
- 102000040430 polynucleotide Human genes 0.000 description 2
- 239000002157 polynucleotide Substances 0.000 description 2
- 238000002600 positron emission tomography Methods 0.000 description 2
- 229910000027 potassium carbonate Inorganic materials 0.000 description 2
- 229910000160 potassium phosphate Inorganic materials 0.000 description 2
- 235000011009 potassium phosphates Nutrition 0.000 description 2
- 229920001592 potato starch Polymers 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 230000002035 prolonged effect Effects 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- 230000000241 respiratory effect Effects 0.000 description 2
- 230000002207 retinal effect Effects 0.000 description 2
- 208000004644 retinal vein occlusion Diseases 0.000 description 2
- SOEDEYVDCDYMMH-UHFFFAOYSA-N robinetin Chemical compound C=1C(O)=CC=C(C(C=2O)=O)C=1OC=2C1=CC(O)=C(O)C(O)=C1 SOEDEYVDCDYMMH-UHFFFAOYSA-N 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 230000009758 senescence Effects 0.000 description 2
- 230000035945 sensitivity Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000000377 silicon dioxide Substances 0.000 description 2
- 210000002027 skeletal muscle Anatomy 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 235000010356 sorbitol Nutrition 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 230000037078 sports performance Effects 0.000 description 2
- 230000000087 stabilizing effect Effects 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical compound C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- 230000000451 tissue damage Effects 0.000 description 2
- 231100000827 tissue damage Toxicity 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 208000016261 weight loss Diseases 0.000 description 2
- 230000004580 weight loss Effects 0.000 description 2
- 229930014124 (-)-epigallocatechin gallate Natural products 0.000 description 1
- 235000004911 (-)-epigallocatechin gallate Nutrition 0.000 description 1
- VINAMCOZNJHNIH-BYPYZUCNSA-N (2s)-2-(trifluoromethyl)pyrrolidine Chemical compound FC(F)(F)[C@@H]1CCCN1 VINAMCOZNJHNIH-BYPYZUCNSA-N 0.000 description 1
- WSYRBHQWMXTCHQ-SFIKJRKMSA-N (2s)-n-[(2r)-1-[[(2s)-1-[[(2r)-1-[[(2r)-1-[[(2s)-1-amino-4-methylsulfanyl-1-oxobutan-2-yl]amino]-3-(1h-indol-3-yl)-1-oxopropan-2-yl]amino]-3-(1h-indol-3-yl)-1-oxopropan-2-yl]amino]-1-oxo-3-phenylpropan-2-yl]amino]-3-(1h-indol-3-yl)-1-oxopropan-2-yl]-2-[[( Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)[C@@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C1=CC=CC=C1 WSYRBHQWMXTCHQ-SFIKJRKMSA-N 0.000 description 1
- AQZSYDUADJTDCR-ZETCQYMHSA-N (3S)-6-chloro-3-propyl-1,2,3,4-tetrahydropyrido[2,3-b]pyrazine Chemical compound ClC=1C=CC2=C(N[C@H](CN2)CCC)N=1 AQZSYDUADJTDCR-ZETCQYMHSA-N 0.000 description 1
- ZQJIIBUKDBKENC-ZDUSSCGKSA-N (9S)-5-(2-methylpyridin-4-yl)-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound Cc1cc(ccn1)-c1ccc2N3CCC[C@@H](C3)Nc2n1 ZQJIIBUKDBKENC-ZDUSSCGKSA-N 0.000 description 1
- WRPHKGYYKLIMHC-WSYDAFBYSA-N (9S)-5-[(2R)-2-(trifluoromethyl)pentyl]-1,6,8-triazatricyclo[7.3.1.02,7]trideca-3,5-diene Chemical compound FC([C@@H](CC=1C=CC2C(N[C@H]3CCCN2C3)N=1)CCC)(F)F WRPHKGYYKLIMHC-WSYDAFBYSA-N 0.000 description 1
- ZLLDNSCUHVRIBO-CMPLNLGQSA-N (9S)-5-[(2R)-2-(trifluoromethyl)pyrrolidin-1-yl]-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound FC(F)(F)[C@H]1CCCN1c1ccc2N3CCC[C@@H](C3)Nc2n1 ZLLDNSCUHVRIBO-CMPLNLGQSA-N 0.000 description 1
- ZLLDNSCUHVRIBO-JQWIXIFHSA-N (9S)-5-[(2S)-2-(trifluoromethyl)pyrrolidin-1-yl]-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound FC(F)(F)[C@@H]1CCCN1c1ccc2N3CCC[C@@H](C3)Nc2n1 ZLLDNSCUHVRIBO-JQWIXIFHSA-N 0.000 description 1
- NMJZXBBKPVRPCD-DTIOYNMSSA-N (9S)-5-[3-(trifluoromethyl)pyrrolidin-1-yl]-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound FC(F)(F)C1CCN(C1)c1ccc2N3CCC[C@@H](C3)Nc2n1 NMJZXBBKPVRPCD-DTIOYNMSSA-N 0.000 description 1
- UGLBFIWMBPZYOY-QMMMGPOBSA-N (9S)-5-chloro-3-methyl-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound Cc1cc(Cl)nc2N[C@H]3CCCN(C3)c12 UGLBFIWMBPZYOY-QMMMGPOBSA-N 0.000 description 1
- PQLZWPYKRISZBV-LURJTMIESA-N (9S)-5-chloro-4-iodo-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2,4,6-triene Chemical compound ClC=1C(=CC2=C(N[C@H]3CCCN2C3)N=1)I PQLZWPYKRISZBV-LURJTMIESA-N 0.000 description 1
- NDJNNUJKBIYGED-QMMMGPOBSA-N (9S)-5-chloro-4-methyl-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound Cc1cc2N3CCC[C@@H](C3)Nc2nc1Cl NDJNNUJKBIYGED-QMMMGPOBSA-N 0.000 description 1
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 1
- DSSYKIVIOFKYAU-XCBNKYQSSA-N (R)-camphor Chemical class C1C[C@@]2(C)C(=O)C[C@@H]1C2(C)C DSSYKIVIOFKYAU-XCBNKYQSSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- UWINQFQLMRKGBB-UHFFFAOYSA-N 1,3-bis[2,6-di(propan-2-yl)phenyl]imidazolidine Chemical compound CC(C)C1=CC=CC(C(C)C)=C1N1CN(C=2C(=CC=CC=2C(C)C)C(C)C)CC1 UWINQFQLMRKGBB-UHFFFAOYSA-N 0.000 description 1
- VYXHVRARDIDEHS-UHFFFAOYSA-N 1,5-cyclooctadiene Chemical compound C1CC=CCCC=C1 VYXHVRARDIDEHS-UHFFFAOYSA-N 0.000 description 1
- 239000004912 1,5-cyclooctadiene Substances 0.000 description 1
- LMVVXYPKRHWZLV-UHFFFAOYSA-N 1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5,8,10-pentaene Chemical compound N1=CC=CC2=C1N=C1C=CCN2C1 LMVVXYPKRHWZLV-UHFFFAOYSA-N 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- JVCBVWTTXCNJBJ-UHFFFAOYSA-N 1-azabicyclo[2.2.1]heptane Chemical compound C1CC2CCN1C2 JVCBVWTTXCNJBJ-UHFFFAOYSA-N 0.000 description 1
- HCYLFYASAYLVBO-UHFFFAOYSA-N 1-oxa-7-azaspiro[3.4]octane Chemical compound O1CCC11CNCC1 HCYLFYASAYLVBO-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- GTHRJKYVJZJPCF-UHFFFAOYSA-N 2,6-dichloro-4-methylpyridine Chemical compound CC1=CC(Cl)=NC(Cl)=C1 GTHRJKYVJZJPCF-UHFFFAOYSA-N 0.000 description 1
- COAWNPJQKJEHPG-UHFFFAOYSA-N 2-(3,4-dihydroxyphenyl)-3,5,7-trihydroxy-1lambda^{4}-chromen-1-ylium chloride Chemical compound [Cl-].[O+]=1C2=CC(O)=CC(O)=C2C=C(O)C=1C1=CC=C(O)C(O)=C1 COAWNPJQKJEHPG-UHFFFAOYSA-N 0.000 description 1
- PAIGGJZZAKARDX-VIFPVBQESA-N 2-[[(4S)-2,2-dimethyl-1,3-dioxolan-4-yl]methoxy]pyridine Chemical compound CC1(OC[C@@H](O1)COC1=CC=CC=N1)C PAIGGJZZAKARDX-VIFPVBQESA-N 0.000 description 1
- PAIGGJZZAKARDX-SECBINFHSA-N 2-[[(4r)-2,2-dimethyl-1,3-dioxolan-4-yl]methoxy]pyridine Chemical compound O1C(C)(C)OC[C@H]1COC1=CC=CC=N1 PAIGGJZZAKARDX-SECBINFHSA-N 0.000 description 1
- TYYHDKOVFSVWON-UHFFFAOYSA-N 2-butyl-2-methoxy-1,3-diphenylpropane-1,3-dione Chemical compound C=1C=CC=CC=1C(=O)C(OC)(CCCC)C(=O)C1=CC=CC=C1 TYYHDKOVFSVWON-UHFFFAOYSA-N 0.000 description 1
- FNRMMDCDHWCQTH-UHFFFAOYSA-N 2-chloropyridine;3-chloropyridine;4-chloropyridine Chemical compound ClC1=CC=NC=C1.ClC1=CC=CN=C1.ClC1=CC=CC=N1 FNRMMDCDHWCQTH-UHFFFAOYSA-N 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- HVCOBJNICQPDBP-UHFFFAOYSA-N 3-[3-[3,5-dihydroxy-6-methyl-4-(3,4,5-trihydroxy-6-methyloxan-2-yl)oxyoxan-2-yl]oxydecanoyloxy]decanoic acid;hydrate Chemical compound O.OC1C(OC(CC(=O)OC(CCCCCCC)CC(O)=O)CCCCCCC)OC(C)C(O)C1OC1C(O)C(O)C(O)C(C)O1 HVCOBJNICQPDBP-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- MTJGVAJYTOXFJH-UHFFFAOYSA-N 3-aminonaphthalene-1,5-disulfonic acid Chemical compound C1=CC=C(S(O)(=O)=O)C2=CC(N)=CC(S(O)(=O)=O)=C21 MTJGVAJYTOXFJH-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-N 4-bromobenzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-N 0.000 description 1
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- YVVUSIMLVPJXMY-UHFFFAOYSA-N 4-phenylcyclohexan-1-ol Chemical compound C1CC(O)CCC1C1=CC=CC=C1 YVVUSIMLVPJXMY-UHFFFAOYSA-N 0.000 description 1
- OGELTFZRACUWNA-UHFFFAOYSA-N 5,6,7,8-tetramethyl-3,4-dihydro-2h-chromene-2-carboxylic acid Chemical compound O1C(C(O)=O)CCC2=C(C)C(C)=C(C)C(C)=C21 OGELTFZRACUWNA-UHFFFAOYSA-N 0.000 description 1
- LPNUQEZRCLLNOW-UHFFFAOYSA-N 5-chloro-1,6,8-triazatricyclo[7.3.1.02,7]trideca-2(7),3,5-triene Chemical compound Clc1ccc2N3CCCC(C3)Nc2n1 LPNUQEZRCLLNOW-UHFFFAOYSA-N 0.000 description 1
- UTVQGMITLJXYNP-UHFFFAOYSA-N 6-chloro-8-methyl-1,2,3,4-tetrahydropyrido[2,3-b]pyrazine Chemical compound ClC=1C=C(C2=C(NCCN2)N=1)C UTVQGMITLJXYNP-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 102000009062 ADP Ribose Transferases Human genes 0.000 description 1
- 108010049290 ADP Ribose Transferases Proteins 0.000 description 1
- 208000010444 Acidosis Diseases 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 208000030090 Acute Disease Diseases 0.000 description 1
- 208000004142 Acute Retinal Necrosis Syndrome Diseases 0.000 description 1
- 208000001395 Acute radiation syndrome Diseases 0.000 description 1
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 description 1
- 208000026872 Addison Disease Diseases 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 201000010053 Alcoholic Cardiomyopathy Diseases 0.000 description 1
- 235000019489 Almond oil Nutrition 0.000 description 1
- 201000004384 Alopecia Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 208000031091 Amnestic disease Diseases 0.000 description 1
- 108010064733 Angiotensins Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 208000032467 Aplastic anaemia Diseases 0.000 description 1
- 108091023037 Aptamer Proteins 0.000 description 1
- 241000180579 Arca Species 0.000 description 1
- 206010003210 Arteriosclerosis Diseases 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010061666 Autonomic neuropathy Diseases 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 208000023328 Basedow disease Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102100022548 Beta-hexosaminidase subunit alpha Human genes 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 206010065687 Bone loss Diseases 0.000 description 1
- 241000283725 Bos Species 0.000 description 1
- 208000003174 Brain Neoplasms Diseases 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- 206010006458 Bronchitis chronic Diseases 0.000 description 1
- 239000007848 Bronsted acid Substances 0.000 description 1
- 239000003341 Bronsted base Substances 0.000 description 1
- 206010068597 Bulbospinal muscular atrophy congenital Diseases 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- DCERHCFNWRGHLK-UHFFFAOYSA-N C[Si](C)C Chemical compound C[Si](C)C DCERHCFNWRGHLK-UHFFFAOYSA-N 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- 101001059929 Caenorhabditis elegans Forkhead box protein O Proteins 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 208000002061 Cardiac Conduction System Disease Diseases 0.000 description 1
- 206010007637 Cardiomyopathy alcoholic Diseases 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 101000832687 Cavia porcellus 3-alpha-hydroxysteroid sulfotransferase Proteins 0.000 description 1
- 101000643834 Cavia porcellus 3-beta-hydroxysteroid sulfotransferase Proteins 0.000 description 1
- 101710150820 Cellular tumor antigen p53 Proteins 0.000 description 1
- 208000003569 Central serous chorioretinopathy Diseases 0.000 description 1
- 206010008025 Cerebellar ataxia Diseases 0.000 description 1
- 208000010693 Charcot-Marie-Tooth Disease Diseases 0.000 description 1
- 241000606161 Chlamydia Species 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 208000017667 Chronic Disease Diseases 0.000 description 1
- 206010008909 Chronic Hepatitis Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010009900 Colitis ulcerative Diseases 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010741 Conjunctivitis Diseases 0.000 description 1
- 206010010904 Convulsion Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- 241000195493 Cryptophyta Species 0.000 description 1
- IVOMOUWHDPKRLL-KQYNXXCUSA-N Cyclic adenosine monophosphate Chemical compound C([C@H]1O2)OP(O)(=O)O[C@H]1[C@@H](O)[C@@H]2N1C(N=CN=C2N)=C2N=C1 IVOMOUWHDPKRLL-KQYNXXCUSA-N 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- 206010058202 Cystoid macular oedema Diseases 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 230000005778 DNA damage Effects 0.000 description 1
- 231100000277 DNA damage Toxicity 0.000 description 1
- GCPYCNBGGPHOBD-UHFFFAOYSA-N Delphinidin Natural products OC1=Cc2c(O)cc(O)cc2OC1=C3C=C(O)C(=O)C(=C3)O GCPYCNBGGPHOBD-UHFFFAOYSA-N 0.000 description 1
- 206010012289 Dementia Diseases 0.000 description 1
- 206010012434 Dermatitis allergic Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 206010012655 Diabetic complications Diseases 0.000 description 1
- 206010012688 Diabetic retinal oedema Diseases 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 208000012661 Dyskinesia Diseases 0.000 description 1
- 208000000059 Dyspnea Diseases 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- 208000030814 Eating disease Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 208000032274 Encephalopathy Diseases 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283087 Equus Species 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- GKPYMCCCQRIAJW-UHFFFAOYSA-N FC(F)(F)C1=C(C(=C(C=C1)B(O)O)C(F)(F)F)C(F)(F)F Chemical compound FC(F)(F)C1=C(C(=C(C=C1)B(O)O)C(F)(F)F)C(F)(F)F GKPYMCCCQRIAJW-UHFFFAOYSA-N 0.000 description 1
- 208000019454 Feeding and Eating disease Diseases 0.000 description 1
- 241000282323 Felidae Species 0.000 description 1
- 102100028075 Fibroblast growth factor 6 Human genes 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 206010017533 Fungal infection Diseases 0.000 description 1
- WMBWREPUVVBILR-UHFFFAOYSA-N GCG Natural products C=1C(O)=C(O)C(O)=CC=1C1OC2=CC(O)=CC(O)=C2CC1OC(=O)C1=CC(O)=C(O)C(O)=C1 WMBWREPUVVBILR-UHFFFAOYSA-N 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 208000031448 Genomic Instability Diseases 0.000 description 1
- 241000250507 Gigaspora candida Species 0.000 description 1
- 208000002705 Glucose Intolerance Diseases 0.000 description 1
- 206010018429 Glucose tolerance impaired Diseases 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-M Glycolate Chemical compound OCC([O-])=O AEMRFAOFKBGASW-UHFFFAOYSA-M 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 206010018612 Gonorrhoea Diseases 0.000 description 1
- ZHPLPRUARZZBET-UHFFFAOYSA-N Gossypetin Natural products O1C2=C(O)C(O)=CC(O)=C2C(=O)C(O)C1C1=CC=C(O)C(O)=C1 ZHPLPRUARZZBET-UHFFFAOYSA-N 0.000 description 1
- 208000015023 Graves' disease Diseases 0.000 description 1
- 208000035895 Guillain-Barré syndrome Diseases 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101150021206 HST3 gene Proteins 0.000 description 1
- 101150077931 HST4 gene Proteins 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 208000030836 Hashimoto thyroiditis Diseases 0.000 description 1
- 208000016988 Hemorrhagic Stroke Diseases 0.000 description 1
- 208000024659 Hemostatic disease Diseases 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 208000005176 Hepatitis C Diseases 0.000 description 1
- 206010019728 Hepatitis alcoholic Diseases 0.000 description 1
- 208000003809 Herpes Zoster Ophthalmicus Diseases 0.000 description 1
- 241000228402 Histoplasma Species 0.000 description 1
- 201000002563 Histoplasmosis Diseases 0.000 description 1
- 241000714260 Human T-lymphotropic virus 1 Species 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- 206010058222 Hypertensive cardiomyopathy Diseases 0.000 description 1
- 206010020880 Hypertrophy Diseases 0.000 description 1
- 206010021283 IIIrd nerve paralysis Diseases 0.000 description 1
- 206010061598 Immunodeficiency Diseases 0.000 description 1
- 208000029462 Immunodeficiency disease Diseases 0.000 description 1
- 206010061216 Infarction Diseases 0.000 description 1
- 241000712431 Influenza A virus Species 0.000 description 1
- 206010048858 Ischaemic cardiomyopathy Diseases 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- 208000012659 Joint disease Diseases 0.000 description 1
- 208000027747 Kennedy disease Diseases 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 206010024229 Leprosy Diseases 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- GSDSWSVVBLHKDQ-JTQLQIEISA-N Levofloxacin Chemical compound C([C@@H](N1C2=C(C(C(C(O)=O)=C1)=O)C=C1F)C)OC2=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-JTQLQIEISA-N 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 208000009564 MELAS Syndrome Diseases 0.000 description 1
- 208000002569 Machado-Joseph Disease Diseases 0.000 description 1
- 208000001940 Massive Hepatic Necrosis Diseases 0.000 description 1
- 241001599018 Melanogaster Species 0.000 description 1
- 208000026139 Memory disease Diseases 0.000 description 1
- 208000037093 Menstruation Disturbances Diseases 0.000 description 1
- 206010070909 Metabolic cardiomyopathy Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000019695 Migraine disease Diseases 0.000 description 1
- 206010049567 Miller Fisher syndrome Diseases 0.000 description 1
- 201000002169 Mitochondrial myopathy Diseases 0.000 description 1
- 206010027910 Mononeuritis Diseases 0.000 description 1
- 208000026072 Motor neurone disease Diseases 0.000 description 1
- 101100149522 Mus musculus Sirt1 gene Proteins 0.000 description 1
- 101100477601 Mus musculus Sirt3 gene Proteins 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 206010028372 Muscular weakness Diseases 0.000 description 1
- 208000031888 Mycoses Diseases 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000009525 Myocarditis Diseases 0.000 description 1
- IKMDFBPHZNJCSN-UHFFFAOYSA-N Myricetin Chemical compound C=1C(O)=CC(O)=C(C(C=2O)=O)C=1OC=2C1=CC(O)=C(O)C(O)=C1 IKMDFBPHZNJCSN-UHFFFAOYSA-N 0.000 description 1
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 1
- DTERQYGMUDWYAZ-ZETCQYMHSA-N N(6)-acetyl-L-lysine Chemical compound CC(=O)NCCCC[C@H]([NH3+])C([O-])=O DTERQYGMUDWYAZ-ZETCQYMHSA-N 0.000 description 1
- GXCLVBGFBYZDAG-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]-N-methylprop-2-en-1-amine Chemical compound CN(CCC1=CNC2=C1C=CC=C2)CC=C GXCLVBGFBYZDAG-UHFFFAOYSA-N 0.000 description 1
- JLEBZPBDRKPWTD-TURQNECASA-O N-ribosylnicotinamide Chemical compound NC(=O)C1=CC=C[N+]([C@H]2[C@@H]([C@H](O)[C@@H](CO)O2)O)=C1 JLEBZPBDRKPWTD-TURQNECASA-O 0.000 description 1
- 102100030710 NAD-dependent protein deacetylase sirtuin-3, mitochondrial Human genes 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 206010064997 Necrotising retinitis Diseases 0.000 description 1
- 206010029148 Nephrolithiasis Diseases 0.000 description 1
- 208000001738 Nervous System Trauma Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 102000007999 Nuclear Proteins Human genes 0.000 description 1
- 108010089610 Nuclear Proteins Proteins 0.000 description 1
- REYJJPSVUYRZGE-UHFFFAOYSA-N Octadecylamine Chemical class CCCCCCCCCCCCCCCCCCN REYJJPSVUYRZGE-UHFFFAOYSA-N 0.000 description 1
- YBGZDTIWKVFICR-JLHYYAGUSA-N Octyl 4-methoxycinnamic acid Chemical compound CCCCC(CC)COC(=O)\C=C\C1=CC=C(OC)C=C1 YBGZDTIWKVFICR-JLHYYAGUSA-N 0.000 description 1
- 208000014723 Oculomotor Nerve disease Diseases 0.000 description 1
- 206010061323 Optic neuropathy Diseases 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 241000283898 Ovis Species 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 241001631646 Papillomaviridae Species 0.000 description 1
- 101150003085 Pdcl gene Proteins 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241000721454 Pemphigus Species 0.000 description 1
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 1
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 1
- 206010034620 Peripheral sensory neuropathy Diseases 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 1
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 208000002787 Pregnancy Complications Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 208000024777 Prion disease Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 206010037075 Protozoal infections Diseases 0.000 description 1
- 201000001263 Psoriatic Arthritis Diseases 0.000 description 1
- 208000036824 Psoriatic arthropathy Diseases 0.000 description 1
- 208000010378 Pulmonary Embolism Diseases 0.000 description 1
- 102000000033 Purinergic Receptors Human genes 0.000 description 1
- 108010080192 Purinergic Receptors Proteins 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- ZVOLCUVKHLEPEV-UHFFFAOYSA-N Quercetagetin Natural products C1=C(O)C(O)=CC=C1C1=C(O)C(=O)C2=C(O)C(O)=C(O)C=C2O1 ZVOLCUVKHLEPEV-UHFFFAOYSA-N 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- 206010068142 Radiation sickness syndrome Diseases 0.000 description 1
- 208000003782 Raynaud disease Diseases 0.000 description 1
- 208000012322 Raynaud phenomenon Diseases 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 208000001647 Renal Insufficiency Diseases 0.000 description 1
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 description 1
- 208000002367 Retinal Perforations Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 206010038933 Retinopathy of prematurity Diseases 0.000 description 1
- 206010039085 Rhinitis allergic Diseases 0.000 description 1
- HWTZYBCRDDUBJY-UHFFFAOYSA-N Rhynchosin Natural products C1=C(O)C(O)=CC=C1C1=C(O)C(=O)C2=CC(O)=C(O)C=C2O1 HWTZYBCRDDUBJY-UHFFFAOYSA-N 0.000 description 1
- 235000019485 Safflower oil Nutrition 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- 208000021811 Sandhoff disease Diseases 0.000 description 1
- 206010039710 Scleroderma Diseases 0.000 description 1
- 206010039793 Seborrhoeic dermatitis Diseases 0.000 description 1
- 208000000453 Skin Neoplasms Diseases 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 201000002661 Spondylitis Diseases 0.000 description 1
- 206010066218 Stress Urinary Incontinence Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical class OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- 201000009594 Systemic Scleroderma Diseases 0.000 description 1
- 206010042953 Systemic sclerosis Diseases 0.000 description 1
- 208000022292 Tay-Sachs disease Diseases 0.000 description 1
- 208000024313 Testicular Neoplasms Diseases 0.000 description 1
- 206010057644 Testis cancer Diseases 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 241000223996 Toxoplasma Species 0.000 description 1
- 201000005485 Toxoplasmosis Diseases 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- GLEVLJDDWXEYCO-UHFFFAOYSA-N Trolox Chemical compound O1C(C)(C(O)=O)CCC2=C1C(C)=C(C)C(O)=C2C GLEVLJDDWXEYCO-UHFFFAOYSA-N 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 201000006704 Ulcerative Colitis Diseases 0.000 description 1
- 206010046851 Uveitis Diseases 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 206010047139 Vasoconstriction Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 208000034698 Vitreous haemorrhage Diseases 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- 208000006269 X-Linked Bulbo-Spinal Atrophy Diseases 0.000 description 1
- 102100036976 X-ray repair cross-complementing protein 6 Human genes 0.000 description 1
- 101710124907 X-ray repair cross-complementing protein 6 Proteins 0.000 description 1
- ZZXDRXVIRVJQBT-UHFFFAOYSA-M Xylenesulfonate Chemical compound CC1=CC=CC(S([O-])(=O)=O)=C1C ZZXDRXVIRVJQBT-UHFFFAOYSA-M 0.000 description 1
- 210000001015 abdomen Anatomy 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- IPBVNPXQWQGGJP-UHFFFAOYSA-N acetic acid phenyl ester Natural products CC(=O)OC1=CC=CC=C1 IPBVNPXQWQGGJP-UHFFFAOYSA-N 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 230000007950 acidosis Effects 0.000 description 1
- 208000026545 acidosis disease Diseases 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 208000009621 actinic keratosis Diseases 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 239000012042 active reagent Substances 0.000 description 1
- 201000001509 acute urate nephropathy Diseases 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 210000001789 adipocyte Anatomy 0.000 description 1
- 201000000028 adult respiratory distress syndrome Diseases 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 208000002353 alcoholic hepatitis Diseases 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 150000001345 alkine derivatives Chemical class 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 208000002029 allergic contact dermatitis Diseases 0.000 description 1
- 201000010105 allergic rhinitis Diseases 0.000 description 1
- 239000008168 almond oil Substances 0.000 description 1
- 231100000360 alopecia Toxicity 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 125000006242 amine protecting group Chemical group 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000006986 amnesia Effects 0.000 description 1
- 239000002280 amphoteric surfactant Substances 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 239000002870 angiogenesis inducing agent Substances 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 239000003945 anionic surfactant Substances 0.000 description 1
- RWZYAGGXGHYGMB-UHFFFAOYSA-N anthranilic acid Chemical class NC1=CC=CC=C1C(O)=O RWZYAGGXGHYGMB-UHFFFAOYSA-N 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000001430 anti-depressive effect Effects 0.000 description 1
- 239000000883 anti-obesity agent Substances 0.000 description 1
- 230000000692 anti-sense effect Effects 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 239000003529 anticholesteremic agent Substances 0.000 description 1
- 229940127226 anticholesterol agent Drugs 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 229940125710 antiobesity agent Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229940005529 antipsychotics Drugs 0.000 description 1
- 229940111121 antirheumatic drug quinolines Drugs 0.000 description 1
- 229960004676 antithrombotic agent Drugs 0.000 description 1
- 210000000709 aorta Anatomy 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 238000009360 aquaculture Methods 0.000 description 1
- 244000144974 aquaculture Species 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 208000011775 arteriosclerosis disease Diseases 0.000 description 1
- 230000002917 arthritic effect Effects 0.000 description 1
- 150000001500 aryl chlorides Chemical class 0.000 description 1
- 150000008378 aryl ethers Chemical class 0.000 description 1
- 150000001502 aryl halides Chemical class 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 230000000386 athletic effect Effects 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 208000010668 atopic eczema Diseases 0.000 description 1
- 201000003639 autosomal recessive cerebellar ataxia Diseases 0.000 description 1
- 229960005193 avobenzone Drugs 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 201000007917 background diabetic retinopathy Diseases 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 235000013405 beer Nutrition 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- 125000003310 benzodiazepinyl group Chemical class N1N=C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000012965 benzophenone Substances 0.000 description 1
- 150000008366 benzophenones Chemical class 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- 102000007478 beta-N-Acetylhexosaminidases Human genes 0.000 description 1
- 108010085377 beta-N-Acetylhexosaminidases Proteins 0.000 description 1
- 235000013361 beverage Nutrition 0.000 description 1
- JBFDZEJAJZJORO-UHFFFAOYSA-N bicyclo[4.1.0]hept-3-ene Chemical compound C1C=CCC2CC21 JBFDZEJAJZJORO-UHFFFAOYSA-N 0.000 description 1
- DCRRIOWFXXDTHV-UHFFFAOYSA-N bicyclo[4.2.0]oct-3-ene Chemical compound C1C=CCC2CCC21 DCRRIOWFXXDTHV-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 1
- 229960001467 bortezomib Drugs 0.000 description 1
- 210000003461 brachial plexus Anatomy 0.000 description 1
- 201000006431 brachial plexus neuropathy Diseases 0.000 description 1
- 230000003925 brain function Effects 0.000 description 1
- 235000008429 bread Nutrition 0.000 description 1
- 150000001649 bromium compounds Chemical class 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- CDQSJQSWAWPGKG-UHFFFAOYSA-N butane-1,1-diol Chemical compound CCCC(O)O CDQSJQSWAWPGKG-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000006227 byproduct Substances 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- FUFJGUQYACFECW-UHFFFAOYSA-L calcium hydrogenphosphate Chemical compound [Ca+2].OP([O-])([O-])=O FUFJGUQYACFECW-UHFFFAOYSA-L 0.000 description 1
- 239000007894 caplet Substances 0.000 description 1
- 229940077731 carbohydrate nutrients Drugs 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 230000009084 cardiovascular function Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 239000003093 cationic surfactant Substances 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000005779 cell damage Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 208000037887 cell injury Diseases 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000004663 cell proliferation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000019522 cellular metabolic process Effects 0.000 description 1
- 230000010094 cellular senescence Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 229920002301 cellulose acetate Polymers 0.000 description 1
- 208000025434 cerebellar degeneration Diseases 0.000 description 1
- 210000003591 cerebellar nuclei Anatomy 0.000 description 1
- 210000001638 cerebellum Anatomy 0.000 description 1
- 201000004559 cerebral degeneration Diseases 0.000 description 1
- 230000002490 cerebral effect Effects 0.000 description 1
- 206010008129 cerebral palsy Diseases 0.000 description 1
- 210000001175 cerebrospinal fluid Anatomy 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 150000001793 charged compounds Chemical class 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 230000002113 chemopreventative effect Effects 0.000 description 1
- 230000000973 chemotherapeutic effect Effects 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 210000000038 chest Anatomy 0.000 description 1
- 235000013330 chicken meat Nutrition 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 210000003161 choroid Anatomy 0.000 description 1
- 208000007451 chronic bronchitis Diseases 0.000 description 1
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N cinnamic acid Chemical class OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000010367 cloning Methods 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 230000015271 coagulation Effects 0.000 description 1
- 238000005345 coagulation Methods 0.000 description 1
- 239000011436 cob Substances 0.000 description 1
- 230000006999 cognitive decline Effects 0.000 description 1
- 208000010877 cognitive disease Diseases 0.000 description 1
- 230000003920 cognitive function Effects 0.000 description 1
- 230000001332 colony forming effect Effects 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 108091036078 conserved sequence Proteins 0.000 description 1
- 208000010247 contact dermatitis Diseases 0.000 description 1
- 230000008602 contraction Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 210000000695 crystalline len Anatomy 0.000 description 1
- 239000002178 crystalline material Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 150000003950 cyclic amides Chemical class 0.000 description 1
- 150000001925 cycloalkenes Chemical group 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 201000010206 cystoid macular edema Diseases 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007547 defect Effects 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- FFNDMZIBVDSQFI-UHFFFAOYSA-N delphinidin chloride Chemical compound [Cl-].[O+]=1C2=CC(O)=CC(O)=C2C=C(O)C=1C1=CC(O)=C(O)C(O)=C1 FFNDMZIBVDSQFI-UHFFFAOYSA-N 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 201000001981 dermatomyositis Diseases 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000000368 destabilizing effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 150000001975 deuterium Chemical group 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 125000004431 deuterium atom Chemical group 0.000 description 1
- 201000011190 diabetic macular edema Diseases 0.000 description 1
- NZZIMKJIVMHWJC-UHFFFAOYSA-N dibenzoylmethane Chemical compound C=1C=CC=CC=1C(=O)CC(=O)C1=CC=CC=C1 NZZIMKJIVMHWJC-UHFFFAOYSA-N 0.000 description 1
- 235000019700 dicalcium phosphate Nutrition 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 230000000378 dietary effect Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 230000010339 dilation Effects 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 235000014632 disordered eating Nutrition 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 229960003638 dopamine Drugs 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 235000013399 edible fruits Nutrition 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 229940037395 electrolytes Drugs 0.000 description 1
- 230000004970 emotional disturbance Effects 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 150000002085 enols Chemical class 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- VFSWRBJYBQXUTE-UHFFFAOYSA-N epi-Gallocatechin 3-O-gallate Natural products Oc1ccc2C(=O)C(OC(=O)c3cc(O)c(O)c(O)c3)C(Oc2c1)c4cc(O)c(O)c(O)c4 VFSWRBJYBQXUTE-UHFFFAOYSA-N 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 125000005469 ethylenyl group Chemical group 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 230000004438 eyesight Effects 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 150000002191 fatty alcohols Chemical class 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- GIVLTTJNORAZON-HDBOBKCLSA-N ganglioside GM2 (18:0) Chemical compound O[C@@H]1[C@@H](O)[C@H](OC[C@H](NC(=O)CCCCCCCCCCCCCCCCC)[C@H](O)\C=C\CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@]2(O[C@H]([C@H](NC(C)=O)[C@@H](O)C2)[C@H](O)[C@H](O)CO)C(O)=O)[C@@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](CO)O1 GIVLTTJNORAZON-HDBOBKCLSA-N 0.000 description 1
- 238000001030 gas--liquid chromatography Methods 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 208000007565 gingivitis Diseases 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 230000001434 glomerular Effects 0.000 description 1
- JFCQEDHGNNZCLN-UHFFFAOYSA-L glutarate(2-) Chemical compound [O-]C(=O)CCCC([O-])=O JFCQEDHGNNZCLN-UHFFFAOYSA-L 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 208000001786 gonorrhea Diseases 0.000 description 1
- YRRAGUMVDQQZIY-UHFFFAOYSA-N gossypetin Chemical compound C1=C(O)C(O)=CC=C1C1=C(O)C(=O)C2=C(O)C=C(O)C(O)=C2O1 YRRAGUMVDQQZIY-UHFFFAOYSA-N 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 230000008821 health effect Effects 0.000 description 1
- 210000002216 heart Anatomy 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 230000004217 heart function Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 230000002439 hemostatic effect Effects 0.000 description 1
- 230000002440 hepatic effect Effects 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- WZHKDGJSXCTSCK-UHFFFAOYSA-N hept-3-ene Chemical compound CCCC=CCC WZHKDGJSXCTSCK-UHFFFAOYSA-N 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- KKLGDUSGQMHBPB-UHFFFAOYSA-N hex-2-ynedioic acid Chemical compound OC(=O)CCC#CC(O)=O KKLGDUSGQMHBPB-UHFFFAOYSA-N 0.000 description 1
- 239000004312 hexamethylene tetramine Substances 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003707 hexyloxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 235000012907 honey Nutrition 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000004678 hydrides Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 1
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 1
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 1
- 230000003345 hyperglycaemic effect Effects 0.000 description 1
- 208000020346 hyperlipoproteinemia Diseases 0.000 description 1
- 208000010522 hyperproinsulinemia Diseases 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 208000022368 idiopathic cardiomyopathy Diseases 0.000 description 1
- 210000003090 iliac artery Anatomy 0.000 description 1
- 210000001822 immobilized cell Anatomy 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 230000000984 immunochemical effect Effects 0.000 description 1
- 230000007813 immunodeficiency Effects 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 229940124589 immunosuppressive drug Drugs 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000007574 infarction Effects 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000010954 inorganic particle Substances 0.000 description 1
- 239000002917 insecticide Substances 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 230000003914 insulin secretion Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 208000020658 intracerebral hemorrhage Diseases 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007914 intraventricular administration Methods 0.000 description 1
- 150000004694 iodide salts Chemical class 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 208000002551 irritable bowel syndrome Diseases 0.000 description 1
- 208000001875 irritant dermatitis Diseases 0.000 description 1
- 208000023589 ischemic disease Diseases 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- MWDZOUNAPSSOEL-UHFFFAOYSA-N kaempferol Natural products OC1=C(C(=O)c2cc(O)cc(O)c2O1)c3ccc(O)cc3 MWDZOUNAPSSOEL-UHFFFAOYSA-N 0.000 description 1
- 206010023332 keratitis Diseases 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 201000006370 kidney failure Diseases 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 229960003376 levofloxacin Drugs 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 230000004132 lipogenesis Effects 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 239000003509 long acting drug Substances 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 208000018769 loss of vision Diseases 0.000 description 1
- 231100000864 loss of vision Toxicity 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 208000029233 macular holes Diseases 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- LDDHMLJTFXJGPI-UHFFFAOYSA-N memantine hydrochloride Chemical compound Cl.C1C(C2)CC3(C)CC1(C)CC2(N)C3 LDDHMLJTFXJGPI-UHFFFAOYSA-N 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000004379 membrane Anatomy 0.000 description 1
- 230000006984 memory degeneration Effects 0.000 description 1
- 208000023060 memory loss Diseases 0.000 description 1
- 230000009245 menopause Effects 0.000 description 1
- 231100000544 menstrual irregularity Toxicity 0.000 description 1
- 230000036630 mental development Effects 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 239000002207 metabolite Chemical class 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- ICFZETOOUNWSOC-ZETCQYMHSA-N methyl 3-[(3S)-6-chloro-2-oxo-3,4-dihydro-1H-pyrido[2,3-b]pyrazin-3-yl]propanoate Chemical compound COC(=O)CC[C@@H]1Nc2nc(Cl)ccc2NC1=O ICFZETOOUNWSOC-ZETCQYMHSA-N 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 206010027599 migraine Diseases 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 230000006676 mitochondrial damage Effects 0.000 description 1
- 230000008811 mitochondrial respiratory chain Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 125000001620 monocyclic carbocycle group Chemical group 0.000 description 1
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 208000013734 mononeuritis simplex Diseases 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 210000002161 motor neuron Anatomy 0.000 description 1
- 208000005264 motor neuron disease Diseases 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 230000003387 muscular Effects 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000036473 myasthenia Effects 0.000 description 1
- 206010028417 myasthenia gravis Diseases 0.000 description 1
- 230000002107 myocardial effect Effects 0.000 description 1
- PCOBUQBNVYZTBU-UHFFFAOYSA-N myricetin Natural products OC1=C(O)C(O)=CC(C=2OC3=CC(O)=C(O)C(O)=C3C(=O)C=2)=C1 PCOBUQBNVYZTBU-UHFFFAOYSA-N 0.000 description 1
- 229940105132 myristate Drugs 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- GOQYKNQRPGWPLP-UHFFFAOYSA-N n-heptadecyl alcohol Natural products CCCCCCCCCCCCCCCCCO GOQYKNQRPGWPLP-UHFFFAOYSA-N 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N n-hexadecyl alcohol Natural products CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000030524 negative regulation of hemostasis Effects 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 208000028412 nervous system injury Diseases 0.000 description 1
- 230000000626 neurodegenerative effect Effects 0.000 description 1
- 210000004498 neuroglial cell Anatomy 0.000 description 1
- 230000002232 neuromuscular Effects 0.000 description 1
- 208000018360 neuromuscular disease Diseases 0.000 description 1
- 230000016273 neuron death Effects 0.000 description 1
- 230000009223 neuronal apoptosis Effects 0.000 description 1
- 231100000189 neurotoxic Toxicity 0.000 description 1
- 230000002887 neurotoxic effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 235000020956 nicotinamide riboside Nutrition 0.000 description 1
- 239000011618 nicotinamide riboside Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 238000007899 nucleic acid hybridization Methods 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 235000018343 nutrient deficiency Nutrition 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 229960001679 octinoxate Drugs 0.000 description 1
- 229960003921 octisalate Drugs 0.000 description 1
- WCJLCOAEJIHPCW-UHFFFAOYSA-N octyl 2-hydroxybenzoate Chemical compound CCCCCCCCOC(=O)C1=CC=CC=C1O WCJLCOAEJIHPCW-UHFFFAOYSA-N 0.000 description 1
- 230000009965 odorless effect Effects 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 206010030875 ophthalmoplegia Diseases 0.000 description 1
- 208000020911 optic nerve disease Diseases 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 230000008723 osmotic stress Effects 0.000 description 1
- 230000016087 ovulation Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 230000004792 oxidative damage Effects 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- DXGLGDHPHMLXJC-UHFFFAOYSA-N oxybenzone Chemical compound OC1=CC(OC)=CC=C1C(=O)C1=CC=CC=C1 DXGLGDHPHMLXJC-UHFFFAOYSA-N 0.000 description 1
- 229960001173 oxybenzone Drugs 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 208000027232 peripheral nervous system disease Diseases 0.000 description 1
- 239000003209 petroleum derivative Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- AHWALFGBDFAJAI-UHFFFAOYSA-N phenyl carbonochloridate Chemical compound ClC(=O)OC1=CC=CC=C1 AHWALFGBDFAJAI-UHFFFAOYSA-N 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- 229940049953 phenylacetate Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 235000021317 phosphate Nutrition 0.000 description 1
- 239000008055 phosphate buffer solution Substances 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004585 polycyclic heterocycle group Chemical group 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 102000054765 polymorphisms of proteins Human genes 0.000 description 1
- 208000019629 polyneuritis Diseases 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001296 polysiloxane Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000029279 positive regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000002980 postoperative effect Effects 0.000 description 1
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 1
- 244000144977 poultry Species 0.000 description 1
- 235000013594 poultry meat Nutrition 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 208000012113 pregnancy disease Diseases 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Chemical class 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 201000007914 proliferative diabetic retinopathy Diseases 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000001814 protein method Methods 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 208000005069 pulmonary fibrosis Diseases 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000004944 pyrazin-3-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- LJXQPZWIHJMPQQ-UHFFFAOYSA-N pyrimidin-2-amine Chemical compound NC1=NC=CC=N1 LJXQPZWIHJMPQQ-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- 229960001285 quercetin Drugs 0.000 description 1
- 235000005875 quercetin Nutrition 0.000 description 1
- 150000003248 quinolines Chemical class 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 230000000306 recurrent effect Effects 0.000 description 1
- 230000011514 reflex Effects 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000025915 regulation of apoptotic process Effects 0.000 description 1
- 230000034986 regulation of gene silencing Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 210000002254 renal artery Anatomy 0.000 description 1
- 230000003252 repetitive effect Effects 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 230000009933 reproductive health Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 208000019793 rhegmatogenous retinal detachment Diseases 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 235000005713 safflower oil Nutrition 0.000 description 1
- 239000003813 safflower oil Substances 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 201000000306 sarcoidosis Diseases 0.000 description 1
- 230000036186 satiety Effects 0.000 description 1
- 235000019627 satiety Nutrition 0.000 description 1
- 230000002000 scavenging effect Effects 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 208000008864 scrapie Diseases 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 208000008742 seborrheic dermatitis Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000001235 sensitizing effect Effects 0.000 description 1
- 201000005572 sensory peripheral neuropathy Diseases 0.000 description 1
- 239000003215 serotonin 5-HT2 receptor antagonist Substances 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000035936 sexual power Effects 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 101150006137 sir gene Proteins 0.000 description 1
- 101150084733 sir-2.1 gene Proteins 0.000 description 1
- 201000000849 skin cancer Diseases 0.000 description 1
- 210000004927 skin cell Anatomy 0.000 description 1
- 210000002807 slow-twitch muscle fiber Anatomy 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- 229940080313 sodium starch Drugs 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000012265 solid product Substances 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 210000005070 sphincter Anatomy 0.000 description 1
- OGNAOIGAPPSUMG-UHFFFAOYSA-N spiro[2.2]pentane Chemical compound C1CC11CC1 OGNAOIGAPPSUMG-UHFFFAOYSA-N 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 238000009168 stem cell therapy Methods 0.000 description 1
- 238000011476 stem cell transplantation Methods 0.000 description 1
- 238000009580 stem-cell therapy Methods 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 208000003265 stomatitis Diseases 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-L sulfite Chemical class [O-]S([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-L 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 230000008833 sun damage Effects 0.000 description 1
- 230000000475 sunscreen effect Effects 0.000 description 1
- 239000000516 sunscreening agent Substances 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- FIAFUQMPZJWCLV-UHFFFAOYSA-N suramin Chemical compound OS(=O)(=O)C1=CC(S(O)(=O)=O)=C2C(NC(=O)C3=CC=C(C(=C3)NC(=O)C=3C=C(NC(=O)NC=4C=C(C=CC=4)C(=O)NC=4C(=CC=C(C=4)C(=O)NC=4C5=C(C=C(C=C5C(=CC=4)S(O)(=O)=O)S(O)(=O)=O)S(O)(=O)=O)C)C=CC=3)C)=CC=C(S(O)(=O)=O)C2=C1 FIAFUQMPZJWCLV-UHFFFAOYSA-N 0.000 description 1
- 229960005314 suramin Drugs 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 230000009747 swallowing Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 208000006379 syphilis Diseases 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 229960001603 tamoxifen Drugs 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- 108091035539 telomere Proteins 0.000 description 1
- 102000055501 telomere Human genes 0.000 description 1
- 210000003411 telomere Anatomy 0.000 description 1
- VCKUSRYTPJJLNI-UHFFFAOYSA-N terazosin Chemical compound N=1C(N)=C2C=C(OC)C(OC)=CC2=NC=1N(CC1)CCN1C(=O)C1CCCO1 VCKUSRYTPJJLNI-UHFFFAOYSA-N 0.000 description 1
- 229960001693 terazosin Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 201000003120 testicular cancer Diseases 0.000 description 1
- TUNFSRHWOTWDNC-UHFFFAOYSA-N tetradecanoic acid Chemical compound CCCCCCCCCCCCCC(O)=O TUNFSRHWOTWDNC-UHFFFAOYSA-N 0.000 description 1
- 150000004685 tetrahydrates Chemical class 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000004305 thiazinyl group Chemical group S1NC(=CC=C1)* 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001730 thiiranyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000000115 thoracic cavity Anatomy 0.000 description 1
- 210000003448 thoracic nerve Anatomy 0.000 description 1
- 201000005665 thrombophilia Diseases 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 239000003860 topical agent Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000000844 transformation Methods 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 150000008648 triflates Chemical class 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- XZZNDPSIHUTMOC-UHFFFAOYSA-N triphenyl phosphate Chemical compound C=1C=CC=CC=1OP(OC=1C=CC=CC=1)(=O)OC1=CC=CC=C1 XZZNDPSIHUTMOC-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 201000008827 tuberculosis Diseases 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 208000019808 uric acid nephrolithiasis Diseases 0.000 description 1
- 208000019553 vascular disease Diseases 0.000 description 1
- 230000025033 vasoconstriction Effects 0.000 description 1
- 229940124549 vasodilator Drugs 0.000 description 1
- 239000003071 vasodilator agent Substances 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 230000004304 visual acuity Effects 0.000 description 1
- 210000004127 vitreous body Anatomy 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 238000004260 weight control Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 230000037303 wrinkles Effects 0.000 description 1
- 229940071104 xylenesulfonate Drugs 0.000 description 1
- 244000000190 yeast pathogen Species 0.000 description 1
- 235000013618 yogurt Nutrition 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/18—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4995—Pyrazines or piperazines forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
- A61K31/5513—1,4-Benzodiazepines, e.g. diazepam or clozapine
- A61K31/5517—1,4-Benzodiazepines, e.g. diazepam or clozapine condensed with five-membered rings having nitrogen as a ring hetero atom, e.g. imidazobenzodiazepines, triazolam
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/18—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D519/00—Heterocyclic compounds containing more than one system of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring system not provided for in groups C07D453/00 or C07D455/00
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
본 발명은 화학식 I의 신규 치환된 가교된 우레아 유사체 화합물 또는 그의 제약상 허용되는 염, 상응하는 제약 조성물, 제조 방법, 및 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하기에 유용한 시르투인 조정제로서의, 단독으로의 또는 다른 치료제와 조합된 이러한 화합물의 용도에 관한 것이다.
<화학식 I>
<화학식 I>
Description
일반적으로, 본 발명은 화학식 I 내지 VI의 치환된 가교된 우레아 유사체 화합물, 그의 상응하는 유사체 또는 유도체 또는 그의 제약상 허용되는 염, 상응하는 제약 조성물, 제조 방법, 및 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하기에 유용한 시르투인 조정제로서의, 단독으로의 또는 다른 치료제와 조합된 이러한 화합물의 방법 또는 용도에 관한 것이다.
유전자의 침묵 정보 조절인자 (SIR) 패밀리는 아르카에박테리아로부터 진핵생물까지 이르는 유기체의 게놈에 존재하는 고도로 보존된 유전자 군을 나타낸다. 코딩된 SIR 단백질은 유전자 침묵화의 조절로부터 DNA 복구까지의 다양한 과정에 수반된다. 이러한 패밀리에서 잘 특징화된 유전자는 에스. 세레비지아에(S. cerevisiae) SIR2이며, 이는 효모 교배 유형, 텔로미어 위치 효과 및 세포 노화를 명시하는 정보를 함유하는 HM 유전자좌를 침묵화시키는데 수반된다. 효모 Sir2 단백질은 히스톤 데아세틸라제의 패밀리에 속한다. SIR 유전자 패밀리의 구성원에 의해 코딩되는 단백질은 250개 아미노산 코어 도메인에서 고도의 서열 보존을 나타낸다. 살모넬라 티피뮤리움(Salmonella typhimurium)에서의 Sir2 상동체, CobB는 NAD (니코틴아미드 아데닌 디뉴클레오티드)-의존성 ADP-리보실 트랜스퍼라제로서 기능한다.
Sir2 단백질은 NAD를 공동기질로서 사용하는 클래스 III 데아세틸라제이다. 다수가 유전자 침묵화에 수반되는 다른 데아세틸라제와는 달리, Sir2는 클래스 I 및 II 히스톤 데아세틸라제 억제제 예컨대 트리코스타틴 A (TSA)에 대해 비감수성이다.
Sir2에 의한 아세틸-리신의 탈아세틸화는 NAD-가수분해에 밀접하게 커플링되어 있으며, 니코틴아미드 및 신규 아세틸-ADP 리보스 화합물을 생산한다. Sir2의 NAD-의존성 데아세틸라제 활성은 그의 기능에 필수적이며, 그의 생물학적 역할을 효모에서의 세포 대사와 연결시킬 수 있다. 포유동물 Sir2 상동체는 NAD-의존성 히스톤 데아세틸라제 활성을 갖는다.
생화학적 연구는 Sir2가 히스톤 H3 및 H4의 아미노-말단 꼬리를 용이하게 탈아세틸화시켜 2'/3'-O-아세틸-ADP-리보스 (OAADPR) 및 니코틴아미드를 형성할 수 있다는 것을 제시한 바 있다. SIR2의 추가 카피를 갖는 균주는 증가된 rDNA 침묵화 및 30% 더 긴 수명을 나타낸다. 또한, 씨. 엘레간스(C. elegans) SIR2 상동체, sir-2.1 및 디. 멜라노가스터(D. melanogaster) dSir2 유전자의 추가 카피는 이들 유기체에서 수명을 연장시키는 것으로 제시된 바 있다. 이는 노화에 대한 SIR2-의존성 조절 경로가 진화에서의 초기에 발생하였으며 잘 보존되어 왔다는 것을 암시한다. 오늘날, Sir2 유전자는 유기체의 건강 및 스트레스 저항성을 증진시켜 그의 역경 극복 기회를 증가시키기 위해 진화되어 온 것으로 여겨진다.
인간에는, Sir2의 보존된 촉매 도메인을 공유하는 7종의 Sir2-유사 유전자 (SIRT1-SIRT7)가 존재한다. SIRT1은 Sir2와 가장 높은 정도의 서열 유사성을 갖는 핵 단백질이다. SIRT1은 종양 억제자 p53, 세포 신호전달 인자 NF-κB 및 FOXO 전사 인자를 포함한 탈아세틸화에 의한 다중 세포 표적을 조절한다.
SIRT3은 원핵생물 및 진핵생물에서 보존되는 SIRT1의 상동체이다. SIRT3 단백질은 N-말단에 위치하는 독특한 도메인에 의해 미토콘드리아 크리스타에 대해 표적화된다. SIRT3은 NAD+-의존성 단백질 데아세틸라제 활성을 가지며, 특히 대사적 활성 조직에서 편재적으로 발현된다. 미토콘드리아로의 전달 시에, SIRT3은 미토콘드리아 매트릭스 프로세싱 펩티다제 (MPP)에 의해 더 작은 활성 형태로 절단되는 것으로 여겨진다.
칼로리 제한은 70년이 넘게 포유동물의 건강을 개선시키고 수명을 연장시키는 것으로 공지되어 왔다. 효모 수명은, 후생동물의 수명과 마찬가지로, 또한 칼로리 제한과 유사한 개입, 예컨대 낮은 글루코스에 의해 연장된다. SIR2 유전자가 결여된 효모 및 파리가 둘 다 칼로리 제한 시에 더 오래 살지는 않는다는 발견은 SIR2 유전자가 제한된 칼로리 식이의 유익한 건강 효과를 매개한다는 증거를 제공한다. 더욱이, 효모 글루코스-반응성 cAMP (아데노신 3',5'-모노포스페이트)-의존성 (PKA) 경로의 활성을 감소시키는 돌연변이는 야생형 세포에서는 수명을 연장시키지만, 돌연변이 sir2 균주에서는 그렇지 않으며, 이는 SIR2가 칼로리 제한 경로의 주요 하류 성분일 가능성이 있다는 것을 증명한다.
치료 잠재력 이외에도, SIRT1 활성 및 소분자 시르투인 조정제에 의한 활성화의 구조적 및 생물물리학적 연구는 시르투인의 생물학적 기능의 이해, 추가로 시르투인 활성화의 작용 메카니즘의 이해를 발전시키고, 신규 시르투인 조정제를 확인하는 검정의 개발을 보조하기에 유용할 것이다.
본 발명은 관련 기술분야에서 직면하는 이들 및 다른 문제의 극복에 관한 것이다.
일반적으로, 본 발명은 화학식 I 내지 VI의 치환된 가교된 우레아 유사체 화합물, 그의 상응하는 유사체 또는 유도체 또는 그의 제약상 허용되는 염, 상응하는 제약 조성물, 제조 방법, 및 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하기에 유용한 시르투인 조정제로서의, 단독으로의 또는 다른 치료제와 조합된 이러한 화합물의 용도에 관한 것이다.
특히, 본 발명은 화학식 I 내지 VI의 신규 화합물, 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 및 화학식 I 내지 VI의 화합물을 포함하는 상응하는 제약 조성물 각각에 관한 것이다.
본 발명은 또한 화학식 I 내지 VI의 화합물 및 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 각각의 제조 방법에 관한 것이다.
본 발명은 또한 본원에 정의된 바와 같은 시르투인 조정제 화합물을, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않으며, 추가로 건선, 아토피성 피부염, 여드름, 장미증, 염증성 장 질환, 골다공증, 패혈증, 관절염, COPD, 전신 홍반성 루푸스 및 안부 염증으로부터 선택되거나 또는 그를 포함할 수 있으나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하는데 사용하는 방법 또는 그의 용도에 관한 것이다.
일반적으로, 본 발명은 화학식 I 내지 VI의 치환된 가교된 우레아 유사체 화합물, 그의 상응하는 유사체 또는 유도체 또는 그의 제약상 허용되는 염, 상응하는 제약 조성물, 제조 방법, 및 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하기에 유용한 시르투인 조정제로서의, 단독으로의 또는 다른 치료제와 조합된 이러한 화합물의 용도에 관한 것이다.
화합물
특히, 본 발명은 화학식 I 내지 VI의 신규 화합물, 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 및 화학식 I 내지 VI의 화합물을 포함하는 상응하는 제약 조성물 각각에 관한 것이다.
국제 출원일: 2014년 5월 13일의 국제 특허 출원 번호 WO09/061879는 화학식 (I)의 신규 시르투인-조정 치환된 가교된 우레아 및 관련 유사체 화합물 또는 그의 제약상 허용되는 염, 상응하는 제약 조성물, 다른 치료제와의 조합물, 제조 방법, 및 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함한 매우 다양한 질환 및 장애를 치료 및/또는 예방하는 방법 또는 그를 위한 용도를 개시하고 있다.
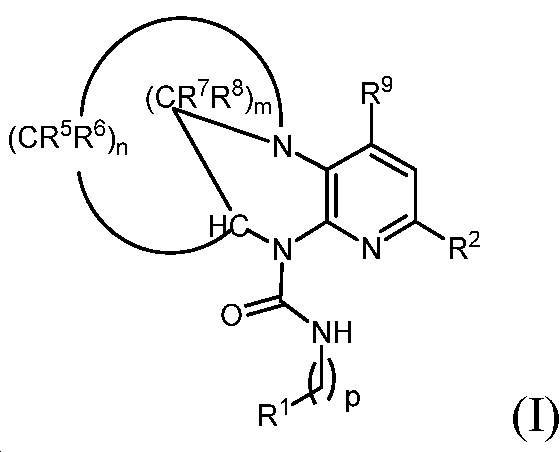
한 측면에서, 본 발명은 하기에 상세하게 기재된 바와 같은 구조 화학식 I 내지 VI의 신규 시르투인-조정 화합물, 각각 상응하는 유사체 (즉, R2 위치에서의 수소 치환을 갖는 것)를 제공한다.
한 측면에서, 본 발명은 화학식 I의 화합물 또는 그의 제약상 염에 관한 것이다.
<화학식 I>
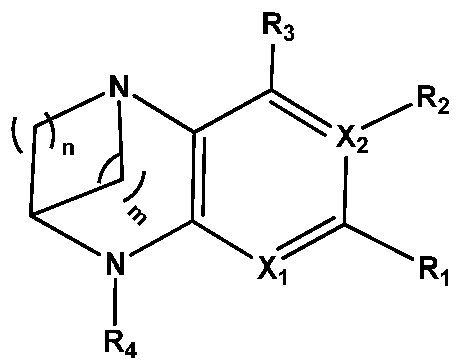
여기서
X1 또는 X2는 독립적으로 -N 또는 -C로부터 선택되고;
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
R4는 수소 또는 -C(O)NRbRc이고;
여기서
X2가 -N인 경우에, R2는 존재하지 않거나; 또는
X2가 -C인 경우에, R2는 상기 정의된 바와 같고;
상기 정의된 바와 같은 각각의 R1, R2, R3 또는 R4는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, 또는 Rf는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
Rg는 -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬이고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, 또는 Rf는 수소, 할로겐, -OH, -C≡N, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, 카르보시클릴, -(CH2)x-카르보시클릴, -헤테로시클릴, -O-헤테로시클릴 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)-OH로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
m은 1 내지 3의 정수이고;
n은 1 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다.
또 다른 측면에서, 본 발명은 상기 및 본 출원 전반에 걸쳐 정의된 바와 같은 본 발명의 화합물 (즉, 구조 화학식 I 내지 VI의 화합물, 각각 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것))에 관한 것이며, 여기서 단
n = 1인 경우에, m ≠ 1이고;
n = 3인 경우에, m ≠ 3이다.
또 다른 측면에서, 본 발명은 R2는 C(O)-NRbRc이고; 여기서 Rb 및 Rc는 상기 및 본 출원 전반에 걸쳐 정의된 바와 같은 것인 본 발명의 화합물에 관한 것이다.
또 다른 측면에서, 본 발명은 화학식 I 내지 VI의 화합물에 관한 것이며, 여기서
m은 1이고;
n은 2 또는 3이고;
R4는 수소이다.
또 다른 측면에서, 본 발명은 화학식 I 내지 VI의 화합물에 관한 것이며, 여기서
m은 1이고;
n은 2 또는 3이고;
R4는 -C(O)NRbRc이고, 여기서 각각의 Rb 및 Rc는 상기 정의된 바와 같다.
또 다른 측면에서, 본 발명은 화학식 I 내지 VI의 화합물에 관한 것이며, 여기서
m은 1이고;
n은 3이고;
R4는 수소 또는 -C(O)NRbRc이고, 여기서 Rb 및 Rc는 상기 정의된 바와 같다.
한 측면에서, 본 발명은 화학식 II의 화합물 또는 그의 제약상 염에 관한 것이다.
<화학식 II>

여기서
X1 또는 X2는 독립적으로 -N 또는 -C로부터 선택되고;
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴 또는 헤테로아릴이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
R4는 수소 또는 -C(O)NRbRc이고;
여기서
X2가 -N인 경우에, R2는 존재하지 않거나; 또는
X2가 -C인 경우에, R2는 상기 정의된 바와 같고;
상기 정의된 바와 같은 각각의 R1, R2, R3 또는 R4는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, 또는 Rf는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
Rg는 -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬이고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, 또는 Rf는 수소, 할로겐, -OH, -C≡N, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, 카르보시클릴, -(CH2)x-카르보시클릴, -헤테로시클릴, -O-헤테로시클릴 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)-OH로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
m은 1 내지 3의 정수이고;
n은 1 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다.
한 측면에서, 본 발명은 화학식 III의 화합물 또는 그의 제약상 염에 관한 것이다.
<화학식 III>
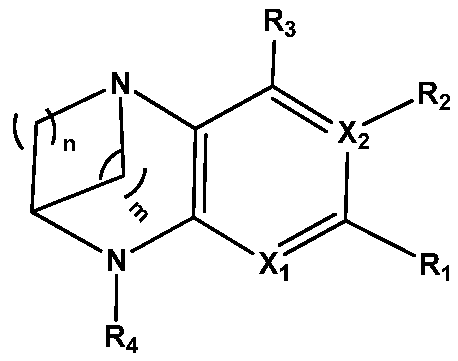
여기서
X1 또는 X2는 독립적으로 -N 또는 -C로부터 선택되고;
R1은 -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
R4는 수소 또는 -C(O)NRbRc이고;
여기서
X2가 -N인 경우에, R2는 존재하지 않거나; 또는
X2가 -C인 경우에, R2는 상기 정의된 바와 같고;
상기 정의된 바와 같은 각각의 R1, R2, R3 또는 R4는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, 또는 Rf는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
Rg는 -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬이고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, 또는 Rf는 수소, 할로겐, -OH, -C≡N, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, 카르보시클릴, -(CH2)x-카르보시클릴, -헤테로시클릴, -O-헤테로시클릴 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)-OH로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
m은 1 내지 3의 정수이고;
n은 1 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다.
또 다른 측면에서, 본 발명은 상기 및 본 출원 전반에 걸쳐 정의된 바와 같은 본 발명의 화합물 (즉, 구조 화학식 I 내지 VI의 화합물, 각각 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것)에 관한 것이며, 여기서 단
n = 1인 경우에, m ≠ 1이고;
n = 3인 경우에, m ≠ 3이다.
또 다른 측면에서, 본 발명은 R2는 C(O)-NRbRc이고; 여기서 Rb 및 Rc는 상기 및 본 출원 전반에 걸쳐 정의된 바와 같은 것인 본원에 기재된 바와 같은 본 발명의 화합물에 관한 것이다.
또 다른 측면에서, 본 발명은 화학식 I 내지 VI의 화합물 또는 본원에 기재된 바와 같은 임의의 화합물에 관한 것이며, 여기서
m은 1이고;
n은 2 또는 3이고;
R4는 수소이다.
또 다른 측면에서, 본 발명은 화학식 I 내지 VI의 화합물에 관한 것이며, 여기서
m은 1이고;
n은 2 또는 3이고;
R4는 -C(O)NRbRc이고, 여기서 각각의 Rb 및 Rc는 상기 정의된 바와 같다.
또 다른 측면에서, 본 발명은 화학식 IV의 화합물 또는 그의 제약상 염에 관한 것이다.
<화학식 IV>

여기서
X1 또는 X2는 독립적으로 -N 또는 -C로부터 선택되고;
여기서
X2가 -N인 경우에, R2는 존재하지 않거나; 또는
X2가 -C인 경우에, R2는 상기 정의된 바와 같고;
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
각각의 R5 및 R6은 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6시클로알킬, -(CH2)xC1-C6시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 R1, R2, R3, R5 및 R6은 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, Rf 또는 Rg는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, Rf 또는 Rg는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, NRhRi, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형-C1-C6 할로알콕시, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, -헤테로시클릴, -O-헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)ORj로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
여기서
각각의 Rh, Ri 및 Rj는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬 또는 -직쇄형 또는 분지형-C1-C6 할로알킬로부터 선택되고;
m은 1 내지 3의 정수이고;
n은 2 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다.
또 다른 측면에서, 본 발명은 n은 2 또는 3이고, m은 1인 본 발명의 화합물에 관한 것이다.
또 다른 측면에서, 본 발명은 화학식 V의 화합물 또는 그의 제약상 염에 관한 것이다.
<화학식 V>

여기서
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
R4는 수소 또는 -C(O)-NRbRc이고;
여기서
상기 정의된 바와 같은 각각의 R1, R2, R3 또는 R4는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, 또는 Rf는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, 또는 Rf는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRgRh, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형-C1-C6 할로알콕시, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, -헤테로시클릴, -O-헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)ORi로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
여기서
상기 정의된 바와 같은 각각의 Rg, Rh 및 Ri는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬 또는 -직쇄형 또는 분지형-C1-C6 할로알킬로부터 선택되고;
m은 1 내지 3의 정수이고;
n은 2 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다.
또 다른 측면에서, 본 발명은 화학식 VI의 화합물 또는 그의 제약상 염에 관한 것이다.
<화학식 VI>

여기서
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
각각의 R5 및 R6은 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6시클로알킬, -(CH2)xC1-C6시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 R1, R2, R3, R5 및 R6은 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, Rf 또는 Rg는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, Rf 또는 Rg는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, NRhRi, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형-C1-C6 할로알콕시, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, -헤테로시클릴, -O-헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)ORj로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
여기서
각각의 Rh, Ri 및 Rj는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬 또는 -직쇄형 또는 분지형-C1-C6 할로알킬로부터 선택되고;
m은 1 내지 3의 정수이고;
n은 2 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다.
또 다른 측면에서, 본 발명은 화학식 I 내지 IV의 화합물 각각에 관한 것이며, 여기서 R1은 하기로부터 선택된다.



또 다른 측면에서, 본 발명은 화학식 I 내지 IV의 화합물(들) 각각에 관한 것이며, 여기서 R1은 하기이다.

또 다른 측면에서, 본 발명은 화학식 I 내지 IV의 화합물(들) 각각에 관한 것이며, 여기서 R4는 하기로부터 선택된다.
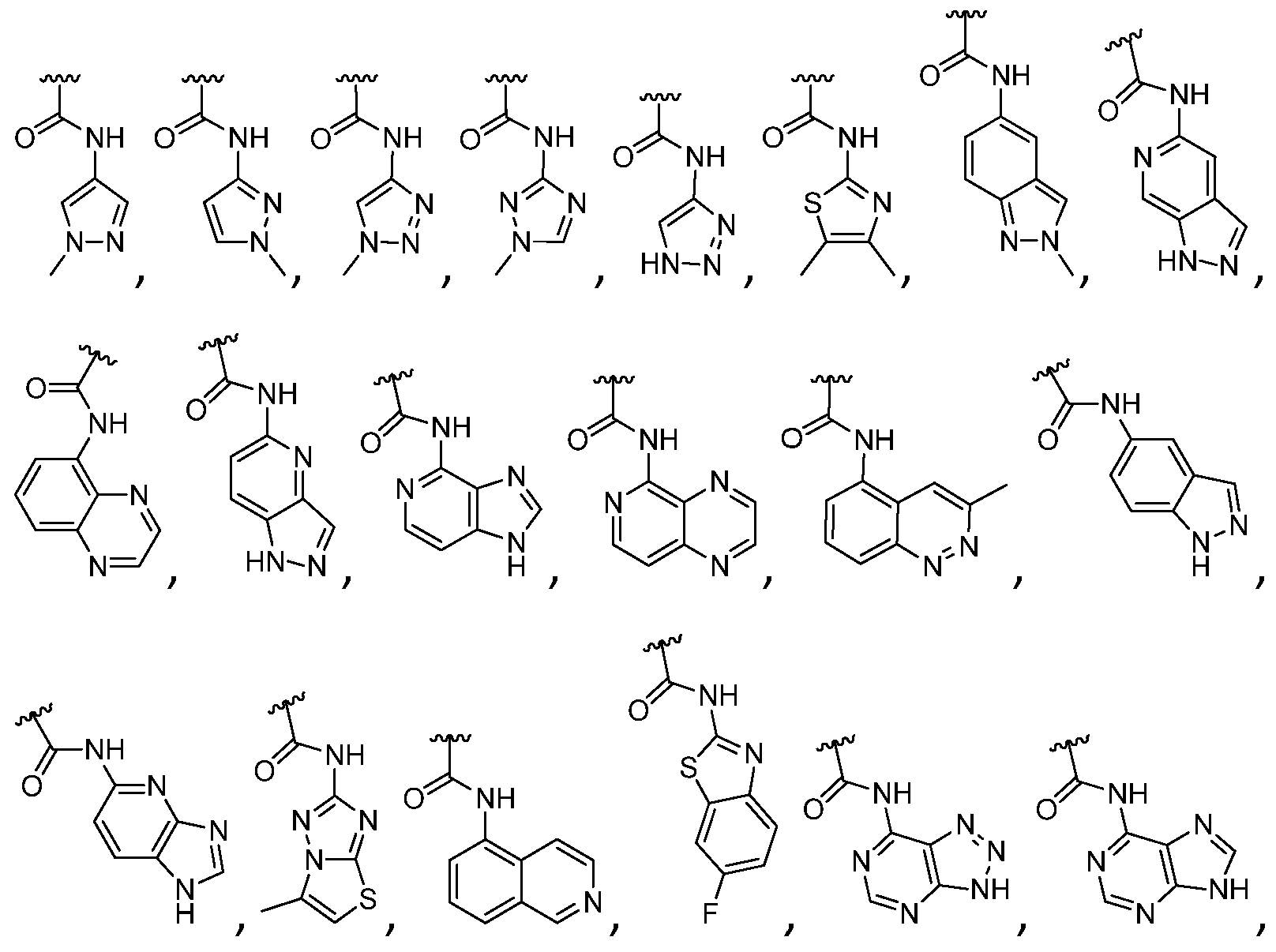

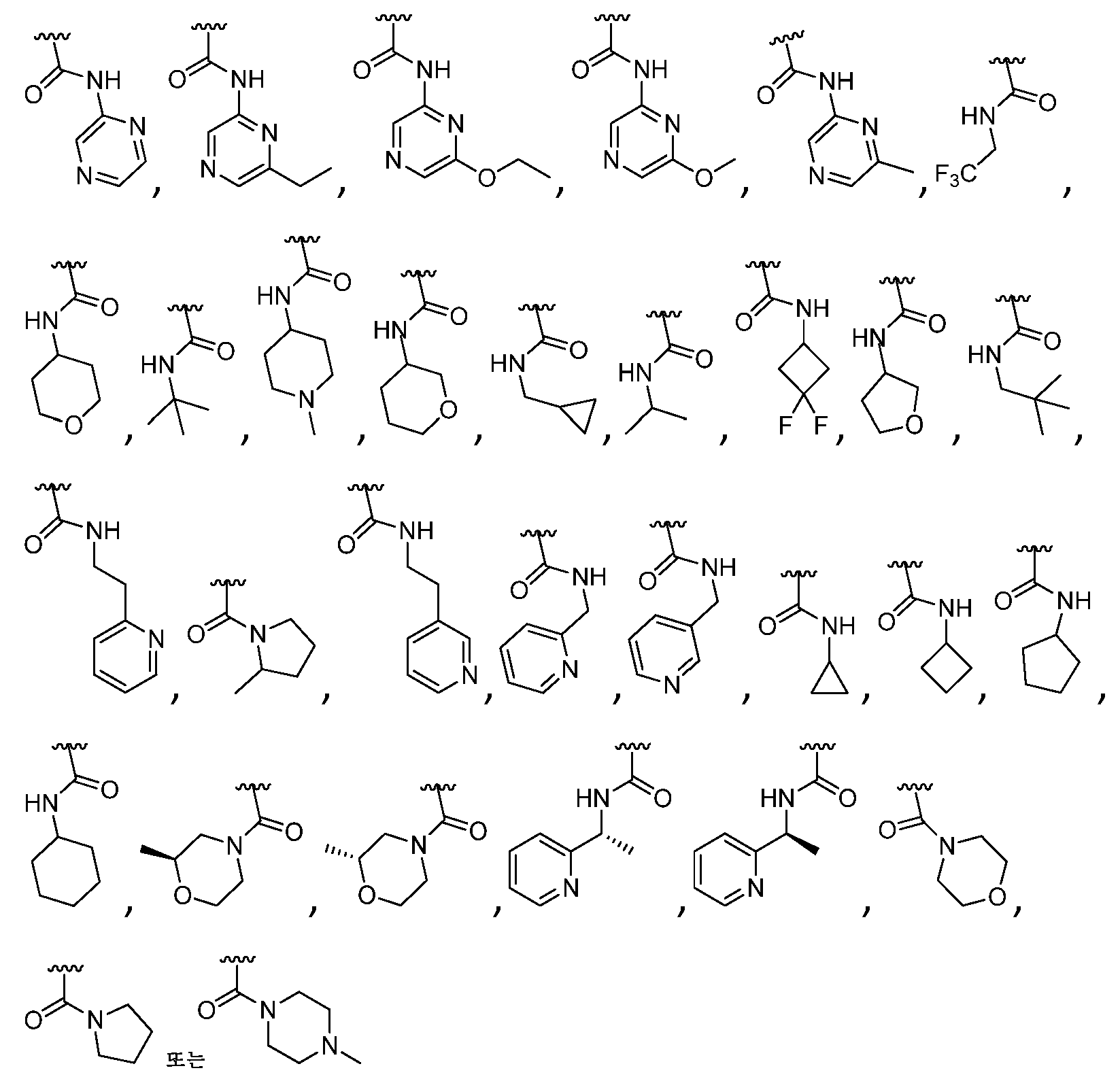
또 다른 측면에서, 본 발명은 화학식 I 내지 IV의 화합물(들) 각각에 관한 것이며, 여기서 R4는 하기이다.


또 다른 측면에서, 본 발명은 본 명세서의 표 1 (영문 명세서 페이지 303에서 시작함)에 정의된 바와 같은 것인 화합물에 관한 것이다.
또 다른 측면에서, 본 발명은 하기인 화합물에 관한 것이다.
차트 3
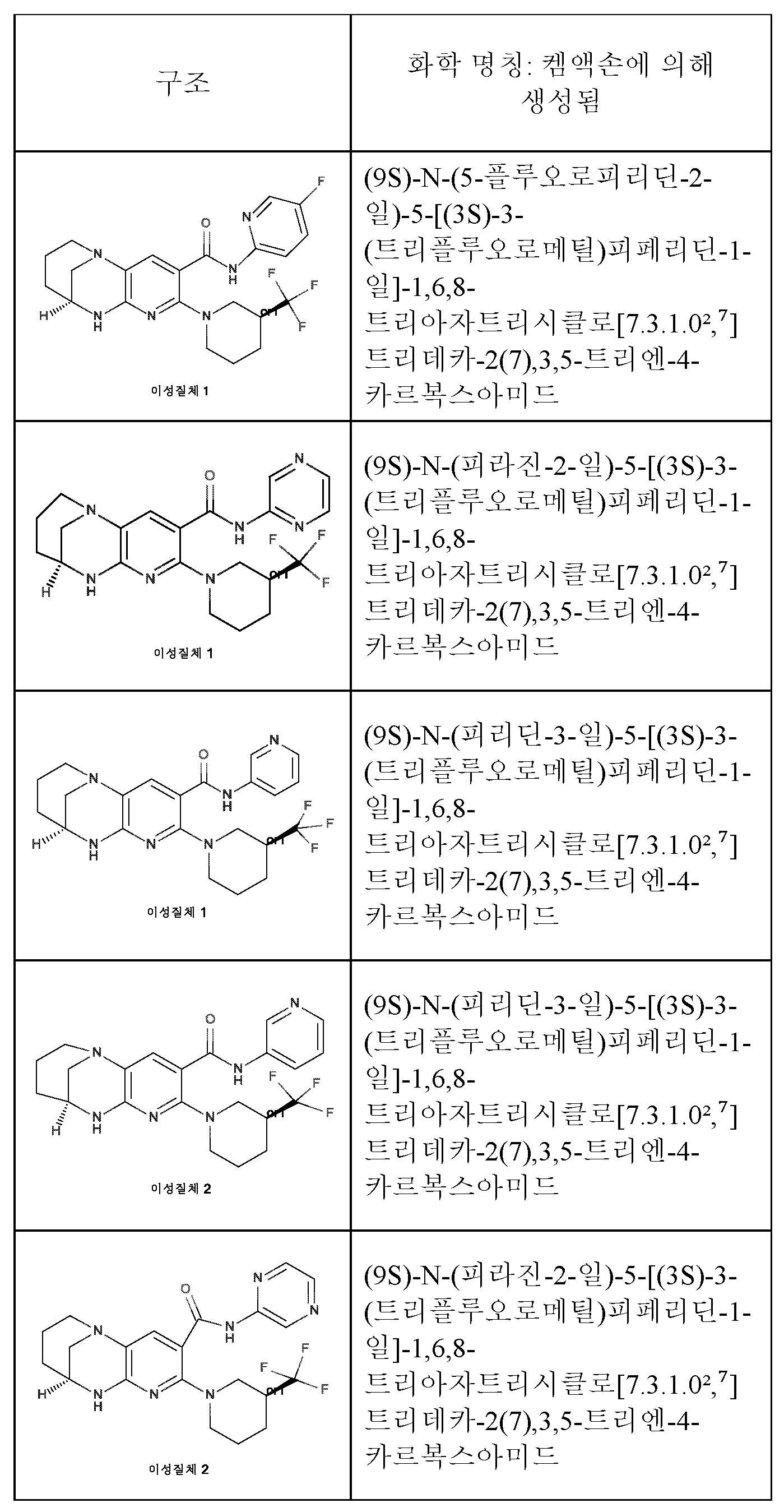
또 다른 측면에서, 본 발명은 본 발명의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것)인 화합물에 관한 것이다.
차트 4 - 3개 탄소 단위 및 피리딘 - 메타 치환 없음

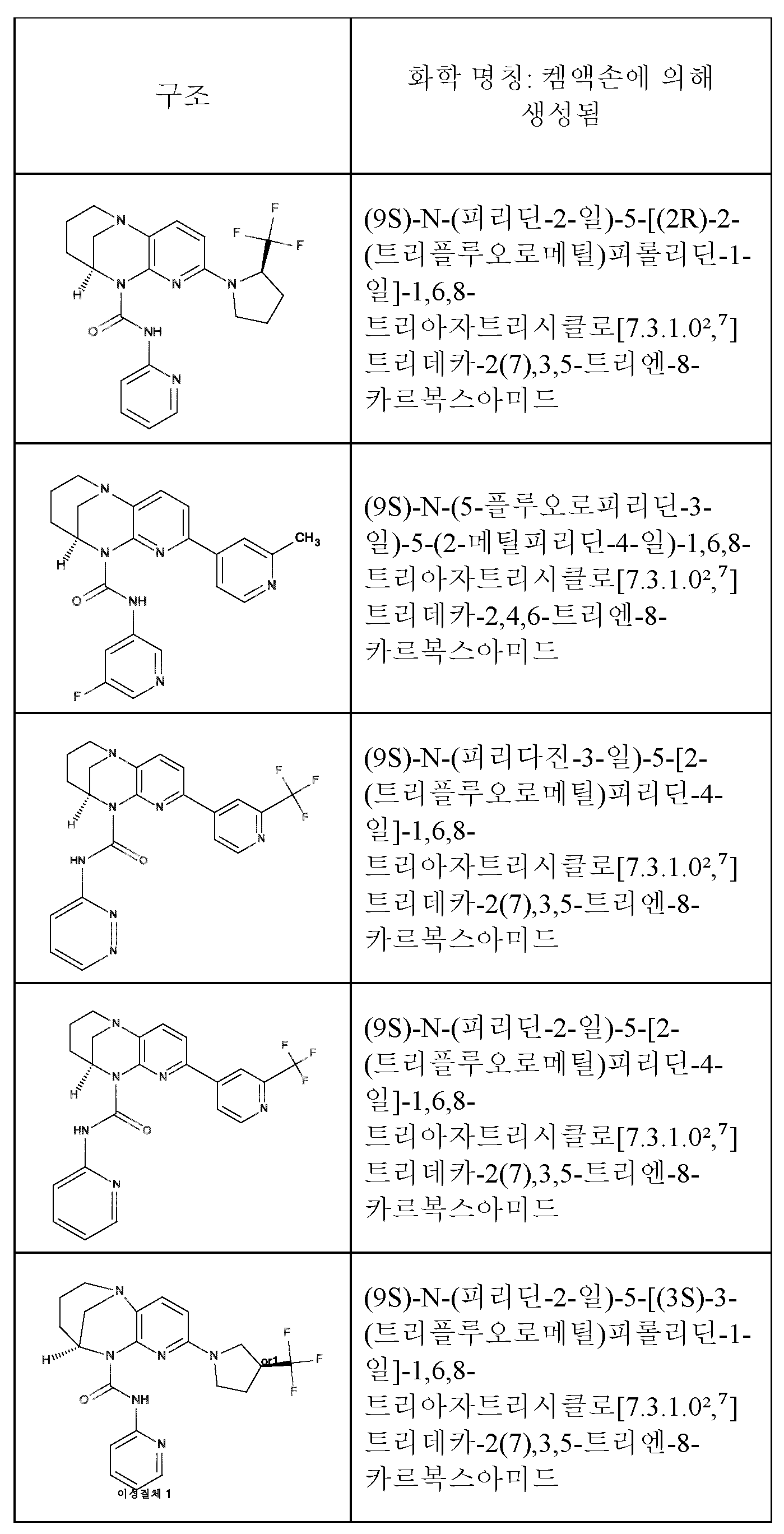





용어 및 정의
섹션 1
본 발명의 특정 화합물은 특정한 기하 또는 입체이성질체 형태로 존재할 수 있다. 본 발명은 시스- 및 트랜스-이성질체, (R)- 및 (S)-거울상이성질체, 부분입체이성질체, (D)-이성질체, (L)-이성질체, 그의 라세미 혼합물, 및 그의 다른 혼합물을 포함한 모든 이러한 화합물을 본 발명의 범주 내에 해당하는 것으로서 고려한다. 추가의 비대칭 탄소 원자는 치환기 예컨대 알킬 기 내에 존재할 수 있다. 모든 이러한 이성질체 뿐만 아니라 그의 혼합물은 본 발명에 포함되도록 의도된다.
본원에 기재된 화합물 및 그의 염은 또한 상응하는 수화물 (예를 들어, 반수화물, 1수화물, 2수화물, 3수화물, 4수화물) 및 용매화물로서 존재할 수 있다. 용매화물 및 수화물의 제조에 적합한 용매는 일반적으로 통상의 기술자에 의해 선택될 수 있다.
화합물 및 그의 염은 무정형 또는 결정질 (공-결정질 및 다형체 포함) 형태로 존재할 수 있다.
본 발명의 시르투인-조정 화합물은 시르투인 단백질의 수준 및/또는 활성, 특히 시르투인 단백질의 데아세틸라제 활성을 유리하게 조정한다.
상기 특성에 대해 개별적으로 또는 추가로, 본 발명의 특정 시르투인-조정 화합물은 시르투인 단백질 (예를 들어, 예컨대 SIRT1 및/또는 SIRT3 단백질)의 탈아세틸화 활성을 조정하기에 효과적인 화합물의 농도에서, 하기 활성 중 1종 이상을 실질적으로 갖지 않는다: PI3-키나제의 억제, 알도리덕타제의 억제, 티로신 키나제의 억제, EGFR 티로신 키나제의 전사활성화, 관상동맥 확장 또는 연축완화 활성.
"알킬" 기 또는 "알칸"은 완전 포화인 직쇄형 또는 분지형 비-방향족 탄화수소이다. 전형적으로, 직쇄형 또는 분지형 알킬 기는 달리 정의되지 않는 한, 1 내지 약 20개의 탄소 원자, 바람직하게는 1 내지 약 10개를 갖는다. 직쇄형 및 분지형 알킬 기의 예는 메틸, 에틸, n-프로필, 이소-프로필, n-부틸, sec-부틸, tert-부틸, 펜틸, 헥실, 펜틸 및 옥틸을 포함한다. C1-C4 직쇄형 또는 분지형 알킬 기는 또한 "저급 알킬" 기로 지칭된다.
임의의 상기 실시양태에서, C1-C4 알콕시-치환된 기는, 예를 들어 1개 이상의 알콕시 치환기 예컨대 1, 2 또는 3개의 메톡시 기, 또는 메톡시 기 및 에톡시 기를 포함할 수 있다. 예시적인 C1-C4 알콕시 치환기는 메톡시, 에톡시, 이소프로폭시, 및 tert-부톡시를 포함한다.
임의의 상기 실시양태에서, 히드록시-치환된 기는 1개 이상의 히드록시 치환기, 예컨대 2 또는 3개의 히드록시 기를 포함할 수 있다.
"할로겐"은 F, Cl, Br 또는 I를 지칭한다.
"할로겐-치환" 또는 "할로" 치환은 1개 이상의 수소의 F, Cl, Br 또는 I로의 대체를 나타낸다.
한 측면에서, 용어 할로알킬은 1개 이상의 수소 원자가 할로겐 원자에 의해 대체된 임의의 알킬 라디칼로서 정의된다. 임의의 상기 실시양태에서, "할로-치환된" 기는 1개의 할로 치환기로부터 퍼할로 치환까지 포함한다. 예시적인 할로-치환된 C1-C4 알킬은 CFH2, CClH2 , CBrH2 , CF2H, CCl2H, CBr2H, CF3, CCl3 , CBr3 , CH2CH2F, CH2CH2Cl, CH2CH2Br, CH2CHF2, CHFCH3, CHClCH3 , CHBrCH3 , CF2CHF2, CF2CHCl2 , CF2CHBr2, CH(CF3)2, 및 C(CF3)3을 포함한다. 퍼할로-치환된 C1-C4 알킬은, 예를 들어 CF3, CCl3, CBr3, CF2CF3, CCl2CF3 및 CBr2CF3을 포함한다.
용어 "알케닐" ("알켄") 및 "알키닐" ("알킨")은 길이 및 가능한 치환에 있어서 상기 기재된 알킬 기와 유사하지만, 각각 적어도 1개의 이중 또는 삼중 결합을 함유하는 불포화 지방족 기를 지칭한다.
임의의 상기 실시양태에서, "카르보사이클" 기는 모노시클릭 카르보사이클 실시양태 및/또는 폴리시클릭 카르보사이클 실시양태, 예컨대 융합된, 가교된 또는 비시클릭 카르보사이클 실시양태를 지칭할 수 있다. 본 발명의 "카르보사이클" 기는 추가로 방향족 카르보사이클 실시양태 및/또는 비-방향족 카르보사이클 실시양태, 또는 폴리시클릭 실시양태의 경우에는, 1개 이상의 방향족 고리 및/또는 1개 이상의 비-방향족 고리 둘 다를 갖는 카르보사이클을 지칭할 수 있다. 폴리시클릭 카르보사이클 실시양태는 비시클릭 고리, 융합된 고리 또는 가교된 비사이클일 수 있다. 비제한적 예시적인 카르보사이클은 페닐, 시클로헥산, 시클로펜탄 또는 시클로헥센, 아만타딘, 시클로펜탄, 시클로헥산, 비시클로[2.2.1]헵탄, 1,5-시클로옥타디엔, 1,2,3,4-테트라히드로나프탈렌, 비시클로[4.2.0]옥트-3-엔, 나프탈렌, 아다만탄, 데칼린, 나프탈렌, 1,2,3,4-테트라히드로나프탈렌, 노르보르난, 데칼린, 스피로펜탄, 메만틴, 비페리덴, 리만타딘, 캄포르, 콜레스테롤, 4-페닐시클로헥산올, 비시클로[4.2.0]옥탄, 메만틴 및 4,5,6,7-테트라히드로-1H-인덴 및 비시클로[4.1.0]헵트-3-엔을 포함한다.
임의의 상기 실시양태에서, "헤테로사이클" 기는 모노시클릭 헤테로사이클 실시양태 및/또는 폴리시클릭 헤테로시클릭 실시양태, 예컨대 융합된, 가교된 또는 비시클릭 헤테로사이클 실시양태를 지칭할 수 있다. 본 발명의 "헤테로사이클" 기는 추가로 방향족 헤테로사이클 실시양태 및/또는 비-방향족 헤테로사이클 실시양태, 또는 폴리시클릭 실시양태의 경우에는, 1개 이상의 방향족 고리 및/또는 1개 이상의 비-방향족 고리 둘 다를 갖는 헤테로사이클을 지칭할 수 있다. 폴리시클릭 헤테로사이클 실시양태는 비시클릭 고리, 융합된 고리 또는 가교된 비사이클일 수 있다. 비제한적 예시적인 헤테로사이클은 피리딜, 피롤리딘, 피페리딘, 피페라진, 피롤리딘, 모르폴린, 피리미딘, 벤조푸란, 인돌, 퀴놀린, 락톤, 락탐, 벤조디아제핀, 인돌, 퀴놀린, 퓨린, 아데닌, 구아닌, 4,5,6,7-테트라히드로벤조[d]티아졸, 헥사민 및 메텐아민을 포함한다.
"알케닐"은 명시된 수의 구성원 탄소 원자를 갖고 쇄 내에 1개 이상의 탄소-탄소 이중 결합을 갖는 불포화 탄화수소 쇄를 지칭한다. 예를 들어, C2-C6 알케닐은 2 내지 6개의 구성원 탄소 원자를 갖는 알케닐 기를 지칭한다. 특정 실시양태에서, 알케닐 기는 쇄 내에 1개의 탄소-탄소 이중 결합을 갖는다. 다른 실시양태에서, 알케닐 기는 쇄 내에 1개 초과의 탄소-탄소 이중 결합을 갖는다. 알케닐 기는 본원에 정의된 바와 같은 1개 이상의 치환기로 임의로 치환될 수 있다. 알케닐 기는 선형 또는 분지형일 수 있다. 대표적인 분지형 알케닐 기는 1, 2, 또는 3개의 분지를 갖는다. 알케닐은 에틸레닐, 프로페닐, 부테닐, 펜테닐, 및 헥세닐을 포함한다.
"알콕시"는 산소 가교를 통해 부착된 알킬 모이어티 (즉, -O-C1-C6 알킬 기, 여기서 C1-C6은 본원에 정의되어 있음)를 지칭한다. 이러한 기의 예는 메톡시, 에톡시, 프로폭시, 부톡시, 펜톡시 및 헥속시를 포함한다.
"알키닐"은 명시된 수의 구성원 탄소 원자를 갖고 쇄 내에 1개 이상의 탄소-탄소 삼중 결합을 갖는 불포화 탄화수소 쇄를 지칭한다. 예를 들어, C2-C6 알키닐은 2 내지 6개의 구성원 원자를 갖는 알키닐 기를 지칭한다. 특정 실시양태에서, 알키닐 기는 쇄 내에 1개의 탄소-탄소 삼중 결합을 갖는다. 다른 실시양태에서, 알키닐 기는 쇄 내에 1개 초과의 탄소-탄소 삼중 결합을 갖는다. 명확성을 위해, 쇄 내에 1개 이상의 탄소-탄소 삼중 결합을 갖고 쇄 내에 1개 이상의 탄소-탄소 이중 결합을 갖는 불포화 탄화수소 쇄는 알키닐 기로 지칭된다. 알키닐 기는 본원에 정의된 바와 같은 1개 이상의 치환기로 임의로 치환될 수 있다. 대표적인 분지형 알키닐 기는 1, 2, 또는 3개의 분지를 갖는다. 알키닐은 에티닐, 프로피닐, 부티닐, 펜티닐, 및 헥시닐을 포함한다.
용어 "방향족 카르보사이클"은 적어도 1개의 방향족 고리를 함유하는 방향족 탄화수소 고리계를 지칭한다. 고리는 다른 방향족 카르보시클릭 고리 또는 비-방향족 카르보시클릭 고리에 융합 또는 달리 부착될 수 있다. 방향족 카르보사이클 기의 예는 카르보시클릭 방향족 기 예컨대 페닐, 나프틸, 및 안트라실을 포함한다.
"아자비시클로"는 고리 골격 내에 질소 원자를 함유하는 비시클릭 분자를 지칭한다. 비사이클의 2개의 고리는 2개의 서로 결합된 원자에서 융합되며, 예를 들어 인돌일 수 있거나, 원자의 순서를 가로질러 융합되며, 예를 들어 아자비시클로[2.2.1]헵탄)일 수 있거나, 또는 단일 원자에서 연결되며, 예를 들어 스피로사이클일 수 있다.
"비사이클" 또는 "비시클릭"은 1, 2 또는 3개 또는 그 초과의 원자가 2개의 고리 사이에 공유된 것인 2-고리계를 지칭한다. 비사이클은 2개의 인접한 원자가 각각의 2개의 고리에 의해 공유된 것인 융합된 비사이클, 예를 들어 데칼린, 인돌을 포함한다. 비사이클은 2개의 고리가 단일 원자를 공유하는 것인 스피로 비사이클, 예를 들어 스피로[2.2]펜탄, 1-옥사-6-아자스피로[3.4]옥탄을 또한 포함한다. 비사이클은 적어도 3개의 원자가 2개의 고리 사이에 공유된 것인 가교된 비사이클, 예를 들어 노르보르난을 추가로 포함한다.
"가교된 비사이클" 화합물은 적어도 3개의 원자가 계의 둘 다의 고리에 의해 공유된 것인 비시클릭 고리계이며, 즉 이들은 2개의 브리지헤드 원자를 연결시키는 1개 이상의 원자의 적어도 1개의 브리지를 포함한다. 가교된 아자비시클로는 고리 중 적어도 1개 내에 질소 원자를 함유하는 가교된 비시클릭 분자를 지칭한다.
용어 "Boc"는 tert-부틸옥시카르보닐 기 (통상의 아민 보호기)를 지칭한다.
본원에 사용된 용어 "카르보사이클" 및 "카르보시클릭"은 고리의 각각의 원자가 탄소인 포화 또는 불포화 고리를 지칭한다. 용어 카르보사이클은 방향족 카르보사이클 및 비-방향족 카르보사이클 둘 다를 포함한다. 비-방향족 카르보사이클은 모든 탄소 원자가 포화인 시클로알칸 고리, 및 적어도 1개의 이중 결합을 함유하는 시클로알켄 고리 둘 다를 포함한다. "카르보사이클"은 5-7원 모노시클릭 및 8-12원 비시클릭 고리를 포함한다. 비시클릭 카르보사이클의 각각의 고리는 비-방향족 및 방향족 고리로부터 선택될 수 있다. 카르보사이클은 1, 2 또는 3개 또는 그 초과의 원자가 2개의 고리 사이에 공유된 것인 비시클릭 분자를 포함한다. 용어 "융합된 카르보사이클"은 각각의 고리가 다른 고리와 2개의 인접한 원자를 공유하는 것인 비시클릭 카르보사이클을 지칭한다. 융합된 카르보사이클의 각각의 고리는 비-방향족 및 방향족 고리로부터 선택될 수 있다. 예시적 실시양태에서, 방향족 고리, 예를 들어 페닐은 비-방향족 또는 방향족 고리, 예를 들어 시클로헥산, 시클로펜탄 또는 시클로헥센에 융합될 수 있다. 원자가가 허용하는 한, 비-방향족 및 방향족 비시클릭 고리의 임의의 조합이 카르보시클릭의 정의에 포함된다. 예시적인 "카르보사이클"은 시클로펜탄, 시클로헥산, 비시클로[2.2.1]헵탄, 1,5-시클로옥타디엔, 1,2,3,4-테트라히드로나프탈렌, 비시클로[4.2.0]옥트-3-엔, 나프탈렌 및 아다만탄을 포함한다. 예시적인 융합된 카르보사이클은 데칼린, 나프탈렌, 1,2,3,4-테트라히드로나프탈렌, 비시클로[4.2.0]옥탄, 4,5,6,7-테트라히드로-1H-인덴 및 비시클로[4.1.0]헵트-3-엔을 포함한다. "카르보사이클"은 수소 원자를 보유할 수 있는 임의의 1개 이상의 위치에서 치환될 수 있다.
"시클로알킬" 기는 완전 포화 (비-방향족)인 명시된 수의 구성원 탄소 원자를 갖는 시클릭 탄화수소 고리이다. 전형적으로, 시클로알킬 기는 달리 정의되지 않는 한, 3 내지 약 10개의 탄소 원자, 보다 전형적으로 3 내지 8개의 탄소 원자를 갖는다. 시클로알킬 기는 모노시클릭 고리계이다. 예를 들어, C3-C6 시클로알킬은 3 내지 6개의 구성원 원자를 갖는 시클로알킬 기를 지칭한다. 시클로알킬 기는 본원에 정의된 바와 같은 1개 이상의 치환기로 임의로 치환될 수 있다. 시클로알킬은 시클로프로필, 시클로부틸, 시클로펜틸, 및 시클로헥실을 포함한다.
"시클로알케닐" 기는 고리 내에 1개 이상의 이중 결합을 함유하는 시클릭 탄화수소 고리이다. 예를 들어, C3-C6 시클로알케닐은 3 내지 6개의 구성원 탄소 원자를 갖는 시클로알케닐 기를 지칭한다. 특정 실시양태에서, 시클로알케닐 기는 고리 내에 1개의 탄소-탄소 이중 결합을 갖는다. 다른 실시양태에서, 시클로알케닐 기는 고리 내에 1개 초과의 탄소-탄소 이중 결합을 갖는다. 시클로알케닐 고리는 방향족이 아니다. 시클로알케닐 기는 모노시클릭 고리계이다. 시클로알케닐 기는 본원에 정의된 바와 같은 1개 이상의 치환기로 임의로 치환될 수 있다. 시클로알케닐은 시클로프로페닐, 시클로부테닐, 시클로펜테닐, 시클로헥세닐, 및 시클로헥사디에닐을 포함한다.
"아릴"은 방향족 탄화수소 고리계를 지칭한다. 아릴 기는 모노시클릭 고리계 또는 비시클릭 고리계이다. 모노시클릭 아릴 고리는 페닐을 지칭한다. 비시클릭 아릴 고리는 나프틸, 및 페닐이 5, 6 또는 7개의 구성원 탄소 원자를 갖는 시클로알킬 또는 시클로알케닐 고리에 융합된 것인 고리를 지칭한다. 아릴 기는 본원에 정의된 바와 같이 1개 이상의 치환기로 임의로 치환될 수 있다.
용어 "헤테로아릴" 또는 "방향족 헤테로사이클"은 고리 구조가 적어도 1개의 헤테로원자, 바람직하게는 1 내지 4개의 헤테로원자, 보다 바람직하게는 1 또는 2개의 헤테로원자를 포함하는 것인 치환 또는 비치환된 방향족 단일 고리 구조, 바람직하게는 5- 내지 7-원 고리, 보다 바람직하게는 5- 내지 6-원 고리를 포함한다. 용어 "헤테로아릴"은 고리 중 적어도 1개는 헤테로방향족이고, 예를 들어 다른 시클릭 고리는 시클로알킬, 시클로알케닐, 시클로알키닐, 방향족 카르보사이클, 헤테로아릴 및/또는 헤테로시클릴일 수 있는 것인 1 또는 2개의 고리를 갖는 고리계를 또한 포함한다. 헤테로아릴 기는, 예를 들어 피롤, 푸란, 티오펜, 이미다졸, 옥사졸, 티아졸, 피라졸, 피리딘, 피라진, 피리다진, 및 피리미딘을 포함한다.
본원에 사용된 용어 "헤테로사이클" 및 "헤테로시클릭"은 N, O, B 및 S 원자, 바람직하게는 N, O, 또는 S로부터 선택된 1개 이상의 헤테로원자를 포함하는 비-방향족 또는 방향족 고리를 지칭한다. 용어 "헤테로사이클"은 "방향족 헤테로사이클" 및 "비-방향족 헤테로사이클" 둘 다를 포함한다. 헤테로사이클은 4-7원 모노시클릭 및 8-12원 비시클릭 고리를 포함한다. 헤테로사이클은 1, 2 또는 3개 또는 그 초과의 원자가 2개의 고리 사이에 공유된 것인 비시클릭 분자를 포함한다. 비시클릭 헤테로사이클의 각각의 고리는 비-방향족 및 방향족 고리로부터 선택될 수 있다. 용어 "융합된 헤테로사이클"은 각각의 고리가 다른 고리와 2개의 인접한 원자를 공유하는 것인 비시클릭 헤테로사이클을 지칭한다. 융합된 헤테로사이클의 각각의 고리는 비-방향족 및 방향족 고리로부터 선택될 수 있다. 예시적 실시양태에서, 방향족 고리, 예를 들어 피리딜은 비-방향족 또는 방향족 고리, 예를 들어 시클로헥산, 시클로펜탄, 피롤리딘, 2,3-디히드로푸란 또는 시클로헥센에 융합될 수 있다. "헤테로사이클" 기는, 예를 들어 피페리딘, 피페라진, 피롤리딘, 모르폴린, 피리미딘, 벤조푸란, 인돌, 퀴놀린, 락톤, 및 락탐을 포함한다. 예시적인 "융합된 헤테로사이클"은 벤조디아제핀, 인돌, 퀴놀린, 퓨린, 및 4,5,6,7-테트라히드로벤조[d]티아졸을 포함한다. "헤테로사이클"은 수소 원자를 보유할 수 있는 임의의 1개 이상의 위치에서 치환될 수 있다.
"모노시클릭 고리"는 5-7원 방향족 카르보사이클 또는 헤테로아릴, 3-7원 시클로알킬 또는 시클로알케닐, 및 5-7원 비-방향족 헤테로시클릴을 포함한다. 예시적인 모노시클릭 기는 치환 또는 비치환된 헤테로사이클 또는 카르보사이클 예컨대 티아졸릴, 옥사졸릴, 옥사지닐, 티아지닐, 디티아닐, 디옥사닐, 이속사졸릴, 이소티아졸릴, 트리아졸릴, 푸라닐, 테트라히드로푸라닐, 디히드로푸라닐, 피라닐, 테트라졸릴, 피라졸릴, 피라지닐, 피리다지닐, 이미다졸릴, 피리디닐, 피롤릴, 디히드로피롤릴, 피롤리디닐, 피페리디닐, 피페라지닐, 피리미디닐, 모르폴리닐, 테트라히드로티오페닐, 티오페닐, 시클로헥실, 시클로펜틸, 시클로프로필, 시클로부틸, 시클로헵타닐, 아제티디닐, 옥세타닐, 티이라닐, 옥시라닐, 아지리디닐, 및 티오모르폴리닐을 포함한다.
"구성원 원자"는 쇄 또는 고리를 형성하는 원자 또는 원자들을 지칭한다. 1개 초과의 구성원 원자가 쇄 및 고리 내에 존재하는 경우에, 각각의 구성원 원자는 쇄 또는 고리 내의 인접한 구성원 원자에 공유 결합된다. 쇄 또는 고리 상의 치환기 군을 구성하는 원자는 쇄 또는 고리 내의 구성원 원자가 아니다.
"임의로 치환된"은 알킬, 알케닐, 알키닐, 아릴, 시클로알킬, 시클로알케닐, 헤테로시클로알킬, 또는 헤테로아릴과 같은 기가 비치환될 수 있거나, 또는 상기 기가 본원에 정의된 바와 같은 1개 이상의 치환기로 치환될 수 있는 것을 나타낸다.
본원에 사용된 "치환된"은 구조 내의 수소 원자를 수소 이외의 원자 또는 분자로 치환하는 것을 의미한다. 기에 대한 언급에서 "치환된"은 기 내의 구성원 원자에 부착된 1개 이상의 수소 원자가 정의된 치환기의 군으로부터 선택된 치환기로 대체된 것을 나타낸다. 치환가능한 원자 예컨대 "치환가능한 질소"는 수소 원자를 적어도 1종의 공명 형태로 보유하는 원자이다. 수소 원자는 또 다른 원자 또는 CH3 또는 OH 기와 같은 기로 치환될 수 있다. 예를 들어, 피페리딘 분자 내의 질소가 수소 원자에 결합된 경우에, 상기 질소는 치환가능하다. 예를 들어 피페리딘의 질소가 수소 이외의 원자에 결합된 경우에, 상기 질소는 치환가능하지 않다. 수소 원자를 임의의 공명 형태로 보유할 수 없는 원자는 치환가능하지 않다. 용어 "치환된"은 이러한 치환이 치환된 원자 및 치환기의 허용되는 원자가에 따르며, 치환이 안정한 화합물 (즉, 가수분해, 재배열, 고리화 또는 제거에 의한 것과 같은 변환이 자발적으로 일어나지 않으며, 반응 혼합물로부터의 단리를 견디기에 충분히 강건한 것)을 생성한다는 내포적 단서를 포함하는 것으로 이해되어야 한다. 기가 1개 이상의 치환기를 함유할 수 있는 것으로 언급되는 경우에, 기 내의 1개 이상 (적절한 경우)의 구성원 원자는 치환될 수 있다. 추가로, 기 내의 단일 구성원 원자는 치환이 원자의 허용된 원자가에 따르는 한, 1개 초과의 치환기로 치환될 수 있다. 적합한 치환기는 각각의 치환된 또는 임의로 치환된 기에 대해 본원에 정의되어 있다.
본 발명에 의해 고려되는 치환기 및 가변기의 조합은 단지 안정한 화합물의 형성을 일으키는 것들이다. 본원에 사용된 용어 "안정한"은 제조를 가능하게 하기에 충분한 안정성을 보유하며, 본원에 상세화된 목적에 유용하도록 충분한 기간 동안 화합물의 완전성을 유지하는 화합물을 지칭한다.
중수소화 화합물
본원에 개시된 화합물은 부분 및 완전 중수소화 변이체를 또한 포함한다. 특정 실시양태에서, 중수소화 변이체는 동역학적 연구에 사용될 수 있다. 관련 기술분야의 통상의 기술자는 이러한 중수소 원자가 존재하는 부위를 선택할 수 있다.
본 발명은 화학식 I의 화합물 또는 그의 제약상 허용되는 염의 다양한 중수소화 형태를 또한 포함한다. 탄소 원자에 부착된 각각의 이용가능한 수소 원자는 독립적으로 중수소 원자로 대체될 수 있다. 관련 기술분야의 통상의 기술자는 본 발명의 화학식 I 내지 II의 화합물의 중수소화 형태를 합성하는 방법을 알 것이다. 예를 들어, 중수소화 물질, 예컨대 알킬 기는 통상적인 기술에 의해 제조될 수 있다 (예를 들어, 위스콘신주 밀워키 소재의 알드리치 케미칼 캄파니(Aldrich Chemical Co.)로부터 입수가능한 메틸-d3-아민, Cat. No.489,689-2 참조).
동위원소
본 발명은 1개 이상의 원자가 자연에서 가장 흔히 발견되는 원자 질량 또는 질량수와 상이한 원자 질량 또는 질량수를 갖는 원자에 의해 대체된 사실을 제외하고는, 화학식 I 및 II에 열거된 것들과 동일한 동위원소-표지된 화합물을 또한 포함한다. 본 발명의 화합물에 혼입될 수 있는 동위원소의 예는 수소, 탄소, 질소, 산소, 플루오린, 아이오딘 및 염소의 동위원소, 3H, 11C, 14C, 18F, 123I 또는 125I를 포함한다.
상기 언급된 동위원소 및/또는 다른 원자의 다른 동위원소를 함유하는 본 발명의 화합물 및 상기 화합물의 제약상 허용되는 염은 본 발명의 범주 내이다. 동위원소 표지된 본 발명의 화합물, 예를 들어 방사성 동위원소 예컨대 3H 또는 14C가 혼입된 것들은 약물 및/또는 기질 조직 분포 검정에 유용하다. 삼중수소, 즉 3H, 및 탄소-14, 즉 14C 동위원소는 그의 제조 용이성 및 검출감도에 있어서 특히 바람직하다. 11C 및 18F 동위원소는 PET (양전자 방출 단층촬영)에 특히 유용하다.
순도
본 발명의 화합물은 제약 조성물에 사용하도록 의도되기 때문에, 이들이 각각 바람직하게는 실질적으로 순수한 형태, 예를 들어 적어도 60% 순도, 보다 적합하게는 적어도 75% 순도, 바람직하게는 적어도 85%, 특히 적어도 98% 순도 (%는 중량 대 중량 기준임)로 제공되는 것으로 용이하게 이해될 것이다. 화합물의 불순한 제제는 제약 조성물에 사용되는 더 순수한 형태를 제조하는데 사용될 수 있다.
염
특정 실시양태에서, 화학식 I에 따른 화합물 또는 그의 제약상 허용되는 염은 산성 관능기를 함유할 수 있다. 특정의 다른 실시양태에서, 화학식 I에 따른 화합물은 염기성 관능기를 함유할 수 있다. 따라서, 통상의 기술자는 화학식 I에 따른 화합물의 염이 제조될 수 있다는 것을 인지할 것이다. 사실상, 본 발명의 특정 실시양태에서, 화학식 I에 따른 화합물의 염은 각각의 유리 염기 또는 유리 산에 비해 바람직할 수 있으며, 이는 예를 들어 이러한 염이 분자에 더 큰 안정성 또는 용해도를 부여하여 투여 형태로의 제제화를 용이하게 할 수 있기 때문이다.
의약에서의 그의 잠재적인 용도 때문에, 화학식 I의 화합물의 염은 적합하게는 제약상 허용되는 염이다. 적합한 제약상 허용되는 염은 문헌 [Berge, Bighley and Monkhouse J.Pharm.Sci (1977) 66, pp 1-19]에 의해 기재된 것들을 포함한다.
본원에 기재된 화합물의 염, 특히 제약상 허용되는 염이 본 발명에 또한 포함된다. 충분히 산성, 충분히 염기성, 또는 이들 둘 다인 관능기를 보유하는 본 발명의 화합물은 다수의 무기 염기, 및 무기 및 유기 산 중 임의의 것과 반응하여 염을 형성할 수 있다. 대안적으로, 본래 하전되어 있는 화합물, 예컨대 4급 질소를 갖는 것들은 적절한 반대이온 (예를 들어, 할라이드 예컨대 브로마이드, 클로라이드 또는 플루오라이드, 특히 브로마이드)과 함께 염을 형성할 수 있다.
산 부가염을 형성하는데 흔히 사용되는 산은 무기 산 예컨대 염산, 브로민화수소산, 아이오딘화수소산, 황산, 인산 등, 및 유기 산 예컨대 p-톨루엔술폰산, 메탄술폰산, 옥살산, p-브로모페닐-술폰산, 탄산, 숙신산, 시트르산, 벤조산, 아세트산 등이다. 이러한 염의 예는 술페이트, 피로술페이트, 비술페이트, 술파이트, 비술파이트, 포스페이트, 모노히드로겐포스페이트, 디히드로겐포스페이트, 메타포스페이트, 피로포스페이트, 클로라이드, 브로마이드, 아이오다이드, 아세테이트, 프로피오네이트, 데카노에이트, 카프릴레이트, 아크릴레이트, 포르메이트, 이소부티레이트, 카프로에이트, 헵타노에이트, 프로피올레이트, 옥살레이트, 말로네이트, 숙시네이트, 수베레이트, 세바케이트, 푸마레이트, 말레에이트, 부틴-1,4-디오에이트, 헥신-1,6-디오에이트, 벤조에이트, 클로로벤조에이트, 메틸벤조에이트, 디니트로벤조에이트, 히드록시벤조에이트, 메톡시벤조에이트, 프탈레이트, 술포네이트, 크실렌술포네이트, 페닐아세테이트, 페닐프로피오네이트, 페닐부티레이트, 시트레이트, 락테이트, 감마-히드록시부티레이트, 글리콜레이트, 타르트레이트, 메탄술포네이트, 프로판술포네이트, 나프탈렌-1-술포네이트, 나프탈렌-2-술포네이트, 만델레이트 등을 포함한다.
염기 부가염은 무기 염기, 예컨대 암모늄 또는 알칼리 금속 또는 알칼리 토금속 히드록시드, 카르보네이트, 비카르보네이트 등으로부터 유도된 것들을 포함한다. 따라서, 본 발명의 염을 제조하기에 유용한 이러한 염기는 수산화나트륨, 수산화칼륨, 수산화암모늄, 탄산칼륨 등을 포함한다.
"거울상이성질체 과잉률" 또는 "ee"는 백분율로서 표현된 1종의 거울상이성질체의 다른 것에 대한 과잉률이다. 그 결과, 라세미 혼합물에서 둘 다의 거울상이성질체는 동등량으로 존재하기 때문에, 거울상이성질체 과잉률은 0 (0% ee)이다. 그러나, 1종의 거울상이성질체가 생성물의 95%를 구성할 정도로 풍부한 경우에, 거울상이성질체 과잉률은 90% ee일 것이다 (풍부한 거울상이성질체의 양 95% 마이너스 다른 거울상 이성질체의 양 5%)
"거울상이성질체적으로 풍부한"은 거울상이성질체 과잉률이 0 초과인 생성물을 지칭한다. 예를 들어, "거울상이성질체적으로 풍부한"은 거울상이성질체 과잉률이 50% ee 초과, 75% ee 초과, 또는 90% ee 초과인 생성물을 지칭한다.
"거울상이성질체적으로 순수한"은 거울상이성질체 과잉률이 99% ee 이상인 생성물을 지칭한다.
"제약상 허용되는"은 합리적 이익/위험 비에 상응하는, 타당한 의학적 판단의 범주 내에서, 과도한 독성, 자극, 또는 다른 문제 또는 합병증 없이 인간 또는 동물의 조직과 접촉시켜 사용하기에 적합한 이들 화합물, 물질, 조성물, 및 투여 형태를 지칭한다.
화학식 I에 따른 화합물 또는 그의 제약상 허용되는 염은 1개 이상의 비대칭 중심 (키랄 중심으로도 지칭됨)을 함유할 수 있으며, 따라서 개별 거울상이성질체, 부분입체이성질체, 또는 다른 입체이성질체 형태로서, 또는 그의 혼합물로서 존재할 수 있다.
키랄 중심, 예컨대 키랄 탄소 원자는 또한 치환기 예컨대 알킬 기에 존재할 수 있다. 화학식 I 또는 본원에 예시된 임의의 화학 구조에 존재하는 키랄 중심의 입체화학이 명시되지 않은 경우에, 구조는 모든 개별 입체이성질체 및 그의 모든 혼합물을 포괄하도록 의도된다.
따라서, 1개 이상의 키랄 중심을 함유하는 화학식 I에 따른 화합물 또는 그의 제약상 허용되는 염은 라세미 혼합물, 부분입체이성질체 혼합물, 거울상이성질체적으로 풍부한 혼합물, 부분입체이성질체적으로 풍부한 혼합물로서, 또는 거울상이성질체적으로 및 부분입체이성질체적으로 순수한 개별 입체이성질체로서 사용될 수 있다.
1개 이상의 비대칭 중심을 함유하는 화학식 I에 따른 화합물 또는 그의 제약상 허용되는 염의 개별 입체이성질체는 관련 기술분야의 통상의 기술자에게 공지된 방법에 의해 분해될 수 있다. 예를 들어, 이러한 분해는 (1) 부분입체이성질체 염, 착물 또는 다른 유도체의 형성에 의해; (2) 입체이성질체-특이적 시약과의 선택적 반응에 의해, 예를 들어 효소적 산화 또는 환원에 의해; 또는 (3) 키랄 환경 하의, 예를 들어 키랄 지지체 예컨대 결합된 키랄 리간드를 갖는 실리카 상의 또는 키랄 용매의 존재 하의 기체-액체 또는 액체 크로마토그래피에 의해 수행될 수 있다. 통상의 기술자는 원하는 입체이성질체가 부분입체이성질체 염, 착물 또는 유도체로 전환되는 경우에, 추가의 단계가 원하는 형태를 유리시키기 위해 필요하다는 것을 인지할 것이다. 대안적으로, 특정한 입체이성질체는 광학 활성 시약, 기질, 촉매 또는 용매를 사용하는 비대칭 합성에 의해, 또는 1종의 거울상이성질체를 비대칭 변환에 의해 다른 것으로 전환시킴으로써 합성될 수 있다.
개시된 화합물 또는 그의 염이 명명되거나 또는 구조에 의해 도시되는 경우에, 용매화물 (특히, 수화물)을 포함한 화합물 또는 염은 결정질 형태, 비-결정질 형태 또는 그의 혼합물로 존재할 수 있는 것으로 이해되어야 한다. 화합물 또는 염 또는 그의 용매화물 (특히, 수화물)은 또한 다형성 (즉, 상이한 결정질 형태로 발생하는 것에 대한 능력)을 나타낼 수 있다. 이들 상이한 결정질 형태는 전형적으로 "다형체"로서 공지되어 있다.
이에 비추어, 본 발명의 염 형태 (즉, 이는 그의 상이한 다형체, 무수 형태, 용매화물 또는 수화물을 포함할 수 있음)는 특징적 다형성을 나타낼 수 있다. 관련 기술분야에서 통상적으로 이해되는 바와 같이, 다형성은 1종 초과의 별개의 결정질 또는 "다형성" 종으로서 결정화하는 것에 대한 화합물의 능력으로서 정의된다. 다형체는 적어도 2종의 상이한 배열을 갖는 화합물의 고체 결정질 상 또는 고체 상태에서의 그 화합물 분자의 다형체 형태로서 정의된다.
본 발명의 것들을 포함한 임의의 주어진 화합물의 다형체 형태는 동일한 화학식 또는 조성에 의해 정의되며, 2종의 상이한 화학적 화합물의 결정질 구조처럼 화학 구조에 있어서 별개이다. 이러한 화합물은 패킹, 각각의 결정질 격자의 기하학적 배열 등에 있어서 상이할 수 있다.
명명되거나 또는 구조에 의해 도시되는 경우에, 개시된 화합물 또는 그의 용매화물 (특히, 수화물)은 그의 모든 다형체를 또한 포함하는 것으로 이해되어야 한다. 다형체는 동일한 화학적 조성을 갖지만, 패킹, 기하학적 배열, 및 결정질 고체 상태의 다른 서술적 특성에 있어서 상이하다.
상기에 비추어, 화학적 및/또는 물리적 특성 또는 특징은 각각의 별개의 다형체 형태에 따라 달라지며, 이는 용해도, 융점, 밀도, 경도, 결정 형상, 광학적 및 전기적 특성, 증기압, 안정성 등에서의 변경을 포함할 수 있다.
본 발명의 결정질 염 형태의 용매화물 및/또는 수화물은 또한 용매 분자가 결정화 과정 동안 화합물 분자의 결정질 격자 구조에 혼입되는 경우에 형성될 수 있다. 예를 들어, 본 발명의 용매화물 형태에는 하기 본원에 기재된 바와 같은 비수성 용매 예컨대 메탄올 등이 혼입될 수 있다. 수화물 형태는 용매로서의 물이 결정질 격자에 혼입된 용매화물 형태이다.
고체 상태 다형성에 대한 무수는 격자 내에 반복 결정질 용매를 함유하지 않는 결정질 구조를 지칭한다. 그러나, 결정질 물질은 다공성일 수 있으며, 물의 가역적 표면 흡착을 나타낼 수 있다.
용어 및 정의
섹션 2
1. 정의
본원에 사용된 하기 용어 및 어구는 하기 제시된 의미를 가질 것이다. 달리 정의되지 않는 한, 본원에 사용된 모든 기술 과학 용어는 관련 기술분야의 통상의 기술자에 의해 흔히 이해되는 바와 동일한 의미를 갖는다.
용어 "작용제"는 본원에서 화학적 화합물, 화학적 화합물의 혼합물, 생물학적 거대분자 (예컨대 핵산, 항체, 단백질 또는 그의 부분, 예를 들어 펩티드), 또는 생물학적 물질 예컨대 박테리아, 식물, 진균 또는 동물 (특히 포유동물) 세포 또는 조직으로부터 제조된 추출물을 나타내는데 사용된다.
화합물에 대해 언급되는 경우에 용어 "생체이용가능한"은 관련 기술분야에 인식되어 있으며, 투여되는 화합물의 양의 전부 또는 일부가, 투여되는 대상체 또는 환자에 의해 흡수되거나, 그에 혼입되거나, 또는 달리 생리학상 이용가능하도록 하는 화합물의 형태를 지칭한다.
"시르투인의 생물학적 활성 부분"은 생물학적 활성, 예컨대 탈아세틸화 능력 ("촉매 활성")을 갖는 시르투인 단백질의 부분을 지칭한다. 시르투인의 촉매 활성 부분은 시르투인의 코어 도메인을 포함할 수 있다. NAD+ 결합 도메인 및 기질 결합 도메인을 포괄하는 진뱅크 수탁 번호 NP_036370을 갖는 SIRT1의 촉매 활성 부분은, 예를 들어 비제한적으로 진뱅크 수탁 번호 NM_012238의 폴리뉴클레오티드에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 아미노산 240-664 또는 240-505를 포함할 수 있다. 따라서, 이러한 영역은 때때로 코어 도메인으로 지칭된다. 때때로 코어 도메인으로도 지칭되는 SIRT1의 다른 촉매 활성 부분은 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 834 내지 1394에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 대략 아미노산 261 내지 447; 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 777 내지 1532에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 대략 아미노산 242 내지 493; 또는 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 813 내지 1538에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 대략 아미노산 254 내지 495를 포함한다. SIRT1의 또 다른 "생물학적 활성" 부분은 화합물 결합 부위에 중요한 코어 도메인에 대한 도메인 N-말단을 포함하는 진뱅크 등록 번호 NP_036370의 아미노산 62-293 또는 183-225이다.
용어 "반려 동물"은 고양이 및 개를 지칭한다. 본원에 사용된 용어 "개(들)"는 다수의 상이한 품종이 존재하는 카니스 파밀리아리스(Canis familiaris) 종의 임의의 구성원을 나타낸다. 용어 "고양이(들)"는 집고양이 및 펠리다에(Felidae) 과, 펠리스(Felis) 속의 다른 구성원을 포함한 고양이과 동물을 지칭한다.
"당뇨병"은 고혈당 또는 케톤산증, 뿐만 아니라 장기간 고혈당 상태 또는 글루코스 내성의 감소로부터 발생하는 만성 전신 대사 이상을 지칭한다. "당뇨병"은 질환의 제I형 및 제II형 (비 인슐린 의존성 당뇨병 또는 NIDDM) 둘 다의 형태를 포괄한다. 당뇨병에 대한 위험 인자는 하기 인자를 포함한다: 남성의 경우에 40 인치 초과 또는 여성의 경우에 35 인치 초과의 허리둘레, 130/85 mmHg 이상의 혈압, 150 mg/dl 초과의 트리글리세리드, 100 mg/dl 초과의 공복 혈액 글루코스, 또는 남성에서는 40 mg/dl 미만 또는 여성에서는 50 mg/dl 미만의 고밀도 지단백질.
용어 "ED50"은 관련 기술분야에 인식된 유효 용량의 척도를 지칭한다. 특정 실시양태에서, ED50은 그의 최대 반응 또는 효과의 50%를 생성하는 약물의 용량, 또는 대안적으로 시험 대상체 또는 제제, 예컨대 단리된 조직 또는 세포의 50%에서 미리 결정된 반응을 생성하는 용량을 의미한다. 용어 "LD50"은 관련 기술분야에 인식된 치사 용량의 척도를 지칭한다. 특정 실시양태에서, LD50은 시험 대상체의 50%에서 치사성인 약물의 용량을 의미한다. 용어 "치료 지수"는 LD50/ED50으로서 정의되는, 약물의 치료 지수를 지칭하는 관련 기술분야에 인식된 용어이다.
용어 "고인슐린혈증"은 혈액 중 인슐린의 수준이 정상보다 더 높은 개체에서의 상태를 지칭한다.
용어 "인슐린 저항성"은 정상적인 양의 인슐린이 인슐린 저항성을 갖지 않는 대상체에서의 생물학적 반응에 비해 정상 미만의 생물학적 반응을 생성하는 상태를 지칭한다.
본원에 논의된 바와 같은 "인슐린 저항성 장애"는 인슐린 저항성에 의해 유발되거나 또는 그에 기인하는 임의의 질환 또는 상태를 지칭한다. 예는 하기를 포함한다: 당뇨병, 비만, 대사 증후군, 인슐린-저항성 증후군, 증후군 X, 인슐린 저항성, 높은 혈압, 고혈압, 높은 혈액 콜레스테롤, 이상지혈증, 고지혈증, 아테롬성동맥경화성 질환 예컨대 졸중, 관상 동맥 질환 또는 심근경색, 고혈당증, 고인슐린혈증 및/또는 고프로인슐린혈증, 글루코스 내성 장애, 지연형 인슐린 방출, 당뇨병성 합병증, 예컨대 관상동맥 심장 질환, 협심증, 울혈성 심부전, 졸중, 치매에서의 인지 기능, 망막병증, 말초 신경병증, 신병증, 사구체신염, 사구체경화증, 신증후군, 고혈압성 신경화증, 일부 유형의 암 (예컨대 자궁내막암, 유방암, 전립선암, 및 결장암), 임신 합병증, 불량한 여성 생식 건강 (예컨대 월경 불순, 불임, 불규칙 배란, 다낭성 난소 증후군 (PCOS)), 지방이영양증, 콜레스테롤-관련 장애, 예컨대 담석, 담낭염 및 담석증, 통풍, 폐쇄성 수면 무호흡 및 호흡기 문제, 골관절염, 및 골 손실, 예를 들어 특히 골다공증.
용어 "가축 동물"은 가축화된 네발동물을 지칭하며, 이는 고기 및 다양한 부산물을 위해 길러지는 것들, 예를 들어 소 및 보스(Bos) 속의 다른 구성원을 포함한 소과 동물, 집돼지 및 수스(Sus) 속의 다른 구성원을 포함한 돼지과 동물, 양 및 오비스(Ovis) 속의 다른 구성원을 포함한 양과 동물, 집염소 및 카프라(Capra) 속의 다른 구성원; 특수 과제 예컨대 짐 운반용 짐승으로서의 용도를 위해 길러지는 가축화된 네발동물, 예를 들어 집말 및 에퀴다에(Equidae) 과, 에쿠스(Equus) 속의 다른 구성원을 포함한 말과 동물을 포함한다.
용어 "포유동물"은 관련 기술분야에 공지되어 있으며, 예시적인 포유동물은 인간, 영장류, 가축 동물 (소, 돼지 등 포함), 반려 동물 (예를 들어, 개, 고양이 등) 및 설치류 (예를 들어, 마우스 및 래트)를 포함한다.
"비만" 개체 또는 비만을 앓고 있는 개체는 일반적으로 적어도 25 이상의 체질량 지수 (BMI)를 갖는 개체이다. 비만은 인슐린 저항성과 연관되거나 또는 연관되지 않을 수 있다.
용어 "비경구 투여" 및 "비경구로 투여된"은 관련 기술분야에 인식되어 있으며, 통상적으로 주사에 의한 경장 및 국소 투여 이외의 투여 방식을 지칭하고, 비제한적으로 정맥내, 근육내, 동맥내, 척수강내, 피막내, 안와내, 심장내, 피내, 복강내, 경기관, 피하, 표피하, 관절내, 피막하, 지주막하, 척수내 및 흉골내 주사 및 주입을 포함한다.
"환자", "대상체", "개체" 또는 "숙주"는 인간 또는 비-인간 동물을 지칭한다.
용어 "제약상 허용되는 담체"는 관련 기술분야에 인식되어 있으며, 임의의 대상 조성물 또는 그의 성분의 운반 또는 수송에 수반되는 제약상 허용되는 물질, 조성물 또는 비히클, 예컨대 액체 또는 고체 충전제, 희석제, 부형제, 용매 또는 캡슐화 물질을 지칭한다. 각각의 담체는 대상 조성물 및 그의 성분과 상용성이고 환자에게 유해하지 않다는 관점에서 "허용"되어야 한다. 제약상 허용되는 담체로서 기능할 수 있는 물질의 일부 예는 하기를 포함한다: (1) 당, 예컨대 락토스, 글루코스 및 수크로스; (2) 전분, 예컨대 옥수수 전분 및 감자 전분; (3) 셀룰로스, 및 그의 유도체, 예컨대 소듐 카르복시메틸 셀룰로스, 에틸 셀룰로스 및 셀룰로스 아세테이트; (4) 분말화 트라가칸트; (5) 맥아; (6) 젤라틴; (7) 활석; (8) 부형제, 예컨대 코코아 버터 및 좌제 왁스; (9) 오일, 예컨대 땅콩 오일, 목화씨 오일, 홍화 오일, 참깨 오일, 올리브 오일, 옥수수 오일 및 대두 오일; (10) 글리콜, 예컨대 프로필렌 글리콜; (11) 폴리올, 예컨대 글리세린, 소르비톨, 만니톨 및 폴리에틸렌 글리콜; (12) 에스테르, 예컨대 에틸 올레에이트 및 에틸 라우레이트; (13) 한천; (14) 완충제, 예컨대 수산화마그네슘 및 수산화알루미늄; (15) 알긴산; (16) 발열원-무함유 물; (17) 등장성 염수; (18) 링거액; (19) 에틸 알콜; (20) 포스페이트 완충제 용액; 및 (21) 제약 제제에 사용되는 다른 비-독성 상용성 물질.
용어 "예방하는"은 관련 기술분야에 인식되어 있으며, 상태, 예컨대 국부 재발 (예를 들어, 통증), 질환 예컨대 암, 증후군 복합증 예컨대 심부전 또는 임의의 다른 의학적 상태와 관련하여 사용되는 경우에, 관련 기술분야에서 널리 이해되어 있으며, 조성물을 투여받지 않은 대상체에 비해 대상체에서의 의학적 상태의 증상의 빈도를 감소시키거나 또는 그의 발병을 지연시키는 조성물의 투여를 포함한다. 따라서, 암의 예방은, 예를 들어 통계적으로 및/또는 임상적으로 유의한 양만큼, 예를 들어 비치료 대조군 집단에 비해 예방적 치료를 받은 환자의 집단에서의 검출가능한 암성 성장의 수를 감소시키고/거나, 비치료 대조군 집단에 대비하여 치료 집단에서의 검출가능한 암성 성장의 출현을 지연시키는 것을 포함한다. 감염의 예방은, 예를 들어 비치료 대조군 집단에 대비하여 치료 집단에서의 감염의 진단 수를 감소시키고/거나, 비치료 대조군 집단에 대비하여 치료 집단에서의 감염의 증상의 발병을 지연시키는 것을 포함한다. 통증의 예방은, 예를 들어 비치료 대조군 집단에 대비하여 치료 집단에서 대상체가 경험하는 통각의 크기를 감소시키거나, 또는 대안적으로 통각을 지연시키는 것을 포함한다.
용어 "예방적" 또는 "치유적" 치료는 관련 기술분야에 인식되어 있으며, 숙주에 대한 약물의 투여를 지칭한다. 원치 않는 상태 (예를 들어, 숙주 동물의 질환 또는 다른 원치 않는 상태)의 임상적 징후 전에 투여되는 경우에, 치료는 예방적이며, 즉 이는 원치 않는 상태가 발생하는 것에 대해 숙주를 보호하는 반면에, 원치 않는 상태의 징후 후에 투여되는 경우에, 치료는 치유적이다 (즉, 존재하는 원치 않는 상태 또는 그로부터의 부작용을 감소, 호전 또는 유지하도록 의도됨).
조성물과 관련된 용어 "발열원-무함유"는 조성물이 투여된 대상체에서 부작용 (예를 들어, 자극, 열, 염증, 설사, 호흡 곤란, 내독소성 쇼크 등)으로 이어질 양으로 발열원을 함유하지 않는 조성물을 지칭한다. 예를 들어, 상기 용어는 내독소, 예컨대 예를 들어 리포폴리사카라이드 (LPS)를 함유하지 않거나 또는 실질적으로 함유하지 않는 조성물을 포괄하도록 의도된다.
세포의 "복제 수명"은 개별 "모세포"에 의해 생산되는 딸세포의 수를 지칭한다. 다른 한편으로, "생활 연령" 또는 "생활 수명"은 영양소 결핍 시에 비-분할 세포 집단이 생존한 채로 남아있는 시간의 길이를 지칭한다. 세포 또는 유기체에 적용 시에 "세포의 수명을 증가시키는" 또는 "세포의 수명을 연장시키는"은 1개의 세포에 의해 생산되는 딸세포의 수를 증가시키는 것; 스트레스에 대처하고, 예를 들어 DNA, 단백질에 대한 손상을 방지하는 것에 대한 세포 또는 유기체의 능력을 증가시키는 것; 및/또는 특정한 조건, 예를 들어 스트레스 (예를 들어, 열 쇼크, 삼투 스트레스, 고에너지 방사선, 화학-유발 스트레스, DNA 손상, 불충분한 염 수준, 불충분한 질소 수준, 또는 불충분한 영양소 수준) 하에 더 장기간 동안 생존하고 살아있는 상태로 존재하는 것에 대한 세포 또는 유기체의 능력을 증가시키는 것을 지칭한다. 수명은 본원에 기재된 방법을 사용하여 적어도 약 10%, 20%, 30%, 40%, 50%, 60%, 또는 20% 내지 70%, 30% 내지 60%, 40% 내지 60% 또는 그 초과만큼 증가될 수 있다.
"시르투인-조정 화합물"은 시르투인 단백질의 수준을 증가시키고/거나 시르투인 단백질의 적어도 1종의 활성을 증가시키는 화합물을 지칭한다. 예시적 실시양태에서, 시르투인-조정 화합물은 시르투인 단백질의 적어도 1종의 생물학적 활성을 적어도 약 10%, 25%, 50%, 75%, 100%, 또는 그 초과만큼 증가시킬 수 있다. 시르투인 단백질의 예시적인 생물학적 활성은, 예를 들어 히스톤 및 p53의 탈아세틸화; 수명을 연장시키는 것; 게놈 안정성을 증가시키는 것; 전사를 침묵화시키는 것; 및 모세포와 딸세포 사이의 산화된 단백질의 분리를 제어하는 것을 포함한다.
단백질은, 예를 들어 아세틸화 펩티드 기질의 탈아세틸화를 포함한다.
"시르투인 단백질"은 시르투인 데아세틸라제 단백질 패밀리의 구성원, 또는 바람직하게는 sir2 패밀리를 지칭하며, 이는 효모 Sir2 (진뱅크 수탁 번호 P53685), 씨. 엘레간스 Sir-2.1 (진뱅크 수탁 번호 NP_501912), 및 인간 SIRT1 (진뱅크 수탁 번호 NM_012238 및 NP_036370 (또는 AF083106)) 및 SIRT2 (진뱅크 수탁 번호 NM_012237, NM_030593, NP_036369, NP_085096, 및 AF083107) 단백질을 포함한다. 다른 패밀리 구성원은 "HST 유전자" (Sir2의 상동체)로 칭하는 4종의 추가의 효모 Sir2-유사 유전자인 HST1, HST2, HST3 및 HST4, 및 5종의 다른 인간 상동체인 hSIRT3, hSIRT4, hSIRT5, hSIRT6 및 hSIRT7을 포함한다 (Brachmann et al. (1995) Genes Dev. 9:2888 및 Frye et al. (1999) BBRC 260:273).
"SIRT1 단백질"은 시르투인 데아세틸라제의 sir2 패밀리의 구성원을 지칭한다. 특정 실시양태에서, SIRT1 단백질은 효모 Sir2 (진뱅크 수탁 번호 P53685), 씨. 엘레간스 Sir-2.1 (진뱅크 수탁 번호 NP_501912), 인간 SIRT1 (진뱅크 수탁 번호 NM_012238 또는 NP_036370 (또는 AF083106)), 마우스 SIRT1 (진뱅크 수탁 번호 NM_019812 또는 NP_062786), 및 그의 등가물 및 단편을 포함한다. 또 다른 실시양태에서, SIRT1 단백질은 진뱅크 수탁 번호 NP_036370, NP_501912, NP_085096, NP_036369, 또는 P53685에 제시된 아미노산 서열로 이루어지거나 또는 그로 본질적으로 이루어진 서열을 포함하는 폴리펩티드를 포함한다. SIRT1 단백질은 진뱅크 수탁 번호 NP_036370, NP_501912, NP_085096, NP_036369, 또는 P53685에 제시된 아미노산 서열의 전부 또는 일부; 진뱅크 수탁 번호 NP_036370, NP_501912, NP_085096, NP_036369, 또는 P53685에 제시된, 1 내지 약 2, 3, 5, 7, 10, 15, 20, 30, 50, 75개 또는 그 초과의 보존적 아미노산 치환을 갖는 아미노산 서열; 진뱅크 수탁 번호 NP_036370, NP_501912, NP_085096, NP_036369, 또는 P53685와 적어도 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 또는 99% 동일한 아미노산 서열을 포함하는 폴리펩티드, 및 그의 기능적 단편을 포함한다. 본 발명의 폴리펩티드는 진뱅크 수탁 번호 NP_036370, NP_501912, NP_085096, NP_036369, 또는 P53685의 상동체 (예를 들어, 오르토로그 및 파라로그), 변이체, 또는 단편을 또한 포함한다.
본원에 사용된 "SIRT2 단백질", "SIRT3 단백질", "SIRT4 단백질", "SIRT5 단백질", "SIRT6 단백질", 및 "SIRT7 단백질"은 특히 대략 275개 아미노산 보존된 촉매 도메인에서, SIRT1 단백질과 상동성인 다른 포유동물, 예를 들어 인간 시르투인 데아세틸라제 단백질을 지칭한다. 예를 들어, "SIRT3 단백질"은 SIRT1 단백질과 상동성인 시르투인 데아세틸라제 단백질 패밀리의 구성원을 지칭한다. 특정 실시양태에서, SIRT3 단백질은 인간 SIRT3 (진뱅크 수탁 번호 AAH01042, NP_036371, 또는 NP_001017524) 및 마우스 SIRT3 (진뱅크 수탁 번호 NP_071878) 단백질, 및 그의 등가물 및 단편을 포함한다. 특정 실시양태에서, SIRT4 단백질은 인간 SIRT4 (진뱅크 수탁 번호 NM_012240 또는 NP_036372)를 포함한다. 특정 실시양태에서, SIRT5 단백질은 인간 SIRT5 (진뱅크 수탁 번호 NM_012241 또는 NP_036373)를 포함한다. 특정 실시양태에서, SIRT6 단백질은 인간 SIRT6 (진뱅크 수탁 번호 NM_016539 또는 NP_057623)을 포함한다. 또 다른 실시양태에서, SIRT3 단백질은 진뱅크 수탁 번호 AAH01042, NP_036371, NP_001017524, 또는 NP_071878에 제시된 아미노산 서열로 이루어지거나 또는 그로 본질적으로 이루어진 서열을 포함하는 폴리펩티드를 포함한다. SIRT3 단백질은 진뱅크 수탁 번호 AAH01042, NP_036371, NP_001017524, 또는 NP_071878에 제시된 아미노산 서열의 전부 또는 일부; 진뱅크 수탁 번호 AAH01042, NP_036371, NP_001017524, 또는 NP_071878에 제시된, 1 내지 약 2, 3, 5, 7, 10, 15, 20, 30, 50, 75개 또는 그 초과의 보존적 아미노산 치환을 갖는 아미노산 서열; 진뱅크 수탁 번호 AAH01042, NP_036371, NP_001017524, 또는 NP_071878과 적어도 60%, 70%, 80%, 90%, 95%, 96%, 97%, 98%, 또는 99% 동일한 아미노산 서열을 포함하는 폴리펩티드, 및 그의 기능적 단편을 포함한다. 본 발명의 폴리펩티드는 진뱅크 수탁 번호 AAH01042, NP_036371, NP_001017524, 또는 NP_071878의 상동체 (예를 들어, 오르토로그 및 파라로그), 변이체, 또는 단편을 또한 포함한다. 특정 실시양태에서, SIRT3 단백질은 미토콘드리아 매트릭스 프로세싱 펩티다제 (MPP) 및/또는 미토콘드리아 중간 펩티다제 (MIP)로의 절단에 의해 생산되는 SIRT3 단백질의 단편을 포함한다.
본원에 사용된 용어 "입체이성질체"는 관련 기술분야에 인식되어 있으며, 동일한 분자 구조를 갖고 단지 그의 원자 공간군의 3차원 배열에 있어서 상이한 2종 이상의 이성질체 중 임의의 것을 지칭한다. 본원에서 화합물 또는 화합물 군을 기재하는데 사용되는 경우에, 입체이성질체는 화합물의 임의의 부분 또는 화합물 전체를 포함한다. 예를 들어, 부분입체이성질체 및 거울상이성질체는 입체이성질체이다.
용어 "전신 투여" 및 "전신 투여된"은 관련 기술분야에 인식되어 있으며, 대상 조성물, 치료 또는 다른 물질을 경장으로 또는 비경구로 투여하는 것을 지칭한다.
본원에 사용된 용어 "호변이성질체"는 관련 기술분야에 인식되어 있으며, 구조가 특히 산소에 결합된 수소의 위치에 대해 2종 이상의 구조적 배열로 존재할 수 있는 것인 구조 이성질현상의 형태를 지칭하는 호변이성질현상의 결과로서 존재할 수 있는, 가능한 대안적 구조 중 임의의 1종을 지칭한다. 본원에서 화합물 또는 화합물 군을 기재하는데 사용되는 경우에, "호변이성질체"는 용이하게 상호전환가능하고 평형으로 존재하는 것으로 추가로 이해된다. 예를 들어, 케토 및 엔올 호변이성질체는 임의의 주어진 조건 또는 조건의 세트 동안 평형 위치에 의해 결정되는 비율로 존재한다.
용어 "치료제"는 관련 기술분야에 인식되어 있으며, 대상체에서 국부 또는 전신 작용하는 임의의 생물학적, 생리학적, 또는 약물학적 활성 물질을 지칭한다. 상기 용어는 또한 동물 또는 인간에서 질환의 진단, 치유, 완화, 치료 또는 예방, 또는 바람직한 신체 또는 정신 발달 및/또는 상태의 증진에 사용하기 위해 의도된 임의의 물질을 의미한다.
용어 "치료 효과"는 관련 기술분야에 인식되어 있으며, 약리학적 활성 물질에 의해 유발된 동물, 특히 포유동물, 보다 특히 인간에서의 유익한 국부 또는 전신 효과를 지칭한다. 어구 "치료 유효량"은 일부 원하는 국부 또는 전신 효과를 임의의 치료에 적용가능한 합리적 이익/위험 비로 생성하는 물질의 양을 의미한다. 이러한 물질의 치료 유효량은 치료되는 대상체 및 질환 상태, 대상체의 체중 및 연령, 질환 상태의 중증도, 투여 방식 등에 따라 달라질 것이며, 이는 관련 기술분야의 통상의 기술자에 의해 용이하게 결정될 수 있다. 예를 들어, 본원에 기재된 특정 조성물은 원하는 효과를 이러한 치료에 적용가능한 합리적 이익/위험 비로 생성하기에 충분한 양으로 투여될 수 있다.
상태 또는 질환을 "치료하는"은 상태 또는 질환의 적어도 1종의 증상을 치유 뿐만 아니라 호전시키는 것을 지칭한다.
용어 "시각 장애"는 치료 (예를 들어, 수술) 시에 종종 단지 부분 가역적 또는 비가역적인 저하된 시각을 지칭한다. 특히 중증 시각 장애는 "실명" 또는 "시각 상실"로 칭하며, 이는 시각의 완전 상실, 교정 렌즈에 의해 개선될 수 없는 20/200보다 더 나쁜 시력, 또는 20도 직경 (10도 반경) 미만의 시야를 지칭한다.
약어 및 기호
본 발명을 기재함에 있어서, 화학 원소는 원소 주기율표에 따라 확인된다. 본원에 이용된 약어 및 기호는 화학 및 생물 기술분야의 통상의 기술자에 의한 이러한 약어 및 기호의 통상의 용법에 따른다.
구체적으로, 하기 약어가 실시예에서 및 명세서 전반에 걸쳐 사용될 수 있다:
g (그램); mg (밀리그램);
kg (킬로그램); μg (마이크로그램);
L (리터); mL (밀리리터);
μL (마이크로리터); psi (제곱 인치당 파운드);
M (몰); mM (밀리몰);
μM (마이크로몰); nM (나노몰);
pM (피코몰); nm (나노미터);
mm (밀리미터); wt (중량);
N (노르말); CFU (콜로니 형성 단위);
I. V. (정맥내); Hz (헤르츠);
MHz (메가헤르츠); mol (몰);
mmol (밀리몰); RT (실온);
min (분); h (시간);
b.p. (비점); TLC (박층 크로마토그래피);
Tr (체류 시간); RP (역상);
MeOH (메탄올); i-PrOH (이소프로판올);
TEA (트리에틸아민); TFA (트리플루오로아세트산);
TFAA (트리플루오로아세트산 무수물); THF (테트라히드로푸란);
DMSO (디메틸술폭시드); EtOAc (에틸 아세테이트);
DME (1,2-디메톡시에탄); DCM (디클로로메탄);
DCE (디클로로에탄); DMF (N,N-디메틸포름아미드);
DMPU (N,N'-디메틸프로필렌우레아); CDI (1,1-카르보닐디이미다졸);
IBCF (이소부틸 클로로포르메이트); AcOH (아세트산);
HOAt (1-히드록시-7-아자벤조트리아졸);
THP (테트라히드로피란); NMM (N-메틸모르폴린);
Pd/C (탄소 상 팔라듐); MTBE (tert-부틸 메틸 에테르);
HOBT (1-히드록시벤조트리아졸); mCPBA (메타-클로로퍼벤조산);
EDC (1-[3-디메틸아미노) 프로필]-3-에틸카르보디이미드 히드로클로라이드);
Boc (tert-부틸옥시카르보닐); FMOC (9-플루오레닐메톡시카르보닐);
DCC (디시클로헥실카르보디이미드); CBZ (벤질옥시카르보닐);
Ac (아세틸); atm (기압);
TMSE (2-(트리메틸실릴)에틸); TMS (트리메틸실릴);
TIPS (트리이소프로필실릴); TBS (t-부틸디메틸실릴);
DMAP (4-디메틸아미노피리딘); BSA (소 혈청 알부민)
NAD (니코틴아미드 아데닌 디뉴클레오티드);
HPLC (고압 액체 크로마토그래피);
LC/MS (액체 크로마토그래피/질량 분광측정법);
BOP (비스(2-옥소-3-옥사졸리디닐)포스핀산 클로라이드);
TBAF (테트라-n-부틸암모늄 플루오라이드);
HBTU (O-벤조트리아졸-1-일-N,N,N',N'-테트라메틸우로늄헥사플루오로 포스페이트);
HEPES (4-(2-히드록시에틸)-1-피페라진 에탄 술폰산);
DPPA (디페닐포스포릴 아지드); LAH (수소화알루미늄리튬);
fHNO3 (발연 HNO3); NaOMe (소듐 메톡시드);
EDTA (에틸렌디아민테트라아세트산);
TMEDA (N,N,N',N'-테트라메틸-1,2-에탄디아민);
NBS (N-브로모숙신이미드); DIPEA (디이소프로필에틸아민);
dppf (1,1'-비스(디페닐포스피노)페로센); 및
NIS (N-아이오도숙신이미드).
에테르에 대한 모든 언급은 디에틸 에테르에 대한 것이고, 염수는 NaCl의 포화 수용액을 지칭한다.
합성 반응식 및 일반적 제조 방법
본 발명은 또한 화학식 I 내지 IV의 화합물, 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 및/또는 그의 중간체 화합물 각각의 제조 방법에 관한 것이다.
화학식 I 내지 IV의 화합물, 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 및/또는 그의 중간체 화합물 또는 그의 제약상 허용되는 염은 하기 반응식에 예시된 합성 절차를 사용하거나 또는 숙련된 유기 화학자의 지식에서 도출함으로써 수득될 수 있다.
이들 반응식 I 내지 VI에 제공된 합성은 본원에 개략화된 반응과의 상용성을 달성하기 위해 필요한 경우에 적합하게 보호되는 적절한 전구체를 사용하여 다양한 상이한 관능기를 갖는 본 발명의 화합물을 제조하는데 적용가능하다. 후속 탈보호는 필요한 경우에 일반적으로 개시된 성질의 화합물을 제공한다. 반응식은 화합물을 사용하여 제시되며, 이들은 본 발명의 화합물을 제조하는데 사용될 수 있는 방법의 예시이다.
중간체 (본 발명의 화합물의 제조에 사용되는 화합물)는 또한 염으로서 존재할 수 있다. 따라서, 중간체에 대한 언급에서, 어구 "화학식 (번호)의 화합물(들)"은 그 구조 화학식을 갖는 화합물 또는 그의 제약상 허용되는 염을 의미한다.
본 발명은 또한 화학식 I 내지 IV의 화합물, 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 및/또는 그의 중간체 화합물 각각 또는 그의 제약상 허용되는 염의 제조 방법에 관한 것이다.
화학식 I 내지 II에 따른 화합물 각각, 본 발명은 또한 화학식 I 내지 IV의 화합물, 상응하는 유사체 (즉, R2 위치에서 수소 치환을 갖는 것) 및/또는 그의 중간체 화합물 각각 또는 그의 제약상 허용되는 염의 제조 방법에 관한 것이며, 통상적인 유기 합성을 사용하여 제조된다.
본 발명의 화합물은 하기 반응식에 예시된 합성 절차를 사용하거나, 또는 숙련된 유기 화학자의 지식에서 도출함으로써 수득될 수 있다.
적합한 합성 경로는 하기 반응식에 하기 도시되어 있다.
화합물 제조
또 다른 실시양태에 따르면, 본 발명은 상기 정의된 화합물의 제조 방법을 제공한다. 화합물은 통상적인 기술을 사용하여 합성될 수 있다. 유리하게, 이들 화합물은 용이하게 입수가능한 출발 물질로부터 편리하게 합성된다.
본원에 기재된 화합물을 합성하기에 유용한 합성 화학 변형 및 방법론은 관련 기술분야에 공지되어 있으며, 예를 들어 문헌 [R. Larock, Comprehensive Organic Transformations (1989); T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 2d. Ed. (1991); L. Fieser and M. Fieser, Fieser and Fieser's Reagents for Organic Synthesis (1994); 및 L. Paquette, ed., Encyclopedia of Reagents for Organic Synthesis (1995)]에 기재된 것들을 포함한다.
일반적 절차
반응식 I

시약: (a) THF, NaHCO3, 60℃; (b) Fe, i-PrOH, HOAc, 80℃; (c) AlCl3, LiAlH4, THF, RT; (d) POCl3, DEA, DCM, 0℃.
상업적 클로로피리딘 I-1을 비양성자성 용매 (예를 들어, THF, DMF, 디옥산) 중에서 염기 (HCl을 스캐빈징하기 위함)의 존재 하에 친핵성 아민 I-2와 반응시켜 위치선택적 부가 생성물 I-3을 제공하였다. 종 I-3의 니트로 관능기를 브뢴스테드 산 (HCl, HOAc) 및 양성자성 용매의 존재 하에 Fe(0)를 사용하여 환원시켰다 (문헌 [Bechamp reduction, Org React. 2, 428, 1944] 참조). 다른 금속 예컨대 Sn이 이러한 환원을 실시하는데 사용될 수 있다. 계내 형성된 생성된 중간체 아민 종을 승온 하에 에스테르 관능기와 반응시켜 시클릭 아미드 I-4를 형성하였다. 강한 히드라이드 환원제, 예컨대 LiAlH4를 화합물 I-4와 반응시켜 에스테르의 상응하는 알콜로의 환원과 동시에 락탐의 시클릭 아민으로의 환원을 유발하였다. 이러한 유형의 환원은 관련 기술분야의 통상의 기술자에게 널리 공지되어 있으며, 문헌 [H.C. Brown and S.Krishnamurthy, Tetrahedron, 1979, 35, 567]을 참조한다. 용이한 이탈기를 형성할 수 있는 알콜 I-5와 활성화 기 (예컨대 POCl3)의 반응은 비시클릭 아민 화합물 I-6을 제공하였다.
반응식 II

시약: (a) TEA, CH3OH, 120℃, 300 psi CO, PdCl2(dppf); (b) NaH, THF, 3-(피리딘-2-일)-2H-피리도[1,2a][1,3,5]트리아진-2,4(3H)-디온, 65℃, 이어서 H2O; (c) DIPEA, DMF, HATU, 알킬 아민, RT.
아릴 클로라이드 I-6을 알콜의 존재 하에 압력 및 승온 하에 CO와 반응시켜 에스테르 II-1을 생성하였다. 카르보닐화 반응은 문헌에 기재되어 있으며 (문헌 [Principles and Applications of Organotransition Metal Chemistry. Sausalito, CA: University Science Books; 1987] 참조), 관련 기술분야의 통상의 기술자에게 널리 공지되어 있다. 아민 II-1을 비양성자성 용매 (DCM, CHCl3, THF 등) 중에서 아실화 시약, 예컨대 트리포스겐 또는 카르보닐 디이미다졸과 반응시키고, 이어서 3급 알킬 아민 염기의 존재 하에 아닐린 화합물 또는 알킬 아민으로 계내 처리하였다. 계내 발생된 NaOH를 통해 일어나는 에스테르 관능기의 병행 가수분해는 우레아 종 II-2를 형성하였다. 카르복실산 II-2를 극성 비양성자성 용매 중에서 커플링 시약 (HATU)의 존재 하에 알킬 아민과 반응시켜 상응하는 아미드 II-3을 제공하였다. 다양한 아미드 커플링 시약 예컨대 EDC, PyBrop 등은 상업적으로 입수가능하다. 아미드 커플링 반응은 일반적으로 유기 염기 예컨대 Et3N 또는 (i-Pr)2NEt를 이용하여 용매 예컨대 DCM 또는 DMF 중에서 실행된다.
반응식 III
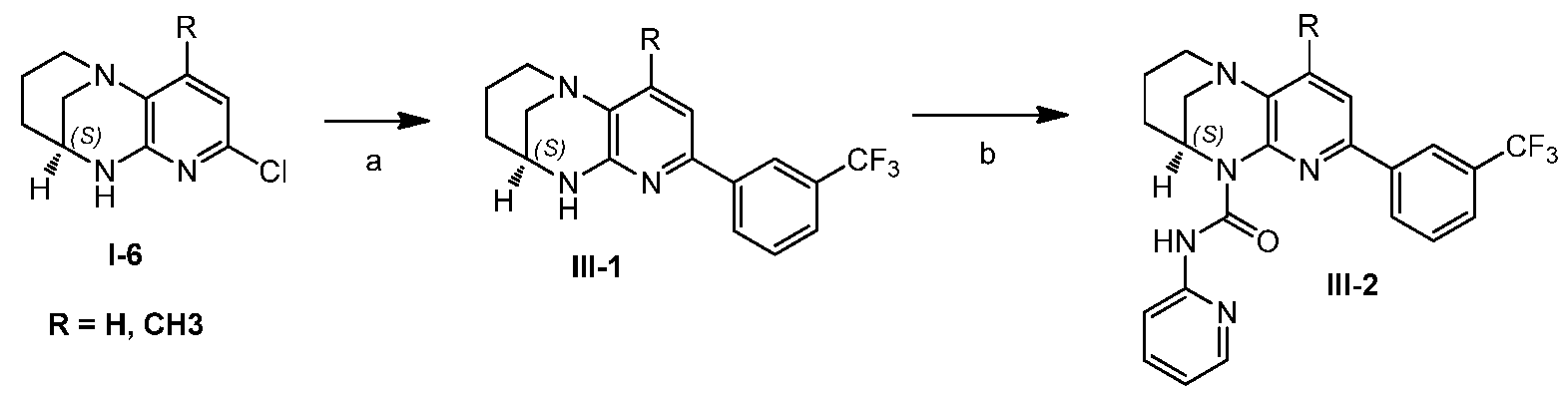
시약: (a) Pd2(dba)3, X-Phos, Cs2CO3, 디옥산/H2O, 90℃; (b) TEA, 트리포스겐, THF; 이어서 2-아미노피리딘, 65℃.
화합물 I-6의 클로로 관능기를 스즈키 커플링 화학을 사용하여 보론산과 커플링시켜 III-1을 제공하였다. 스즈키-유사 커플링은 전형적으로 에테르성 용매 예컨대 DME, 디옥산 또는 THF를 함유하는 수성 혼합물 중에서 무기 염기, 예를 들어 K2CO3, Na2CO3 또는 K3PO4와 함께 팔라듐(0) 촉매 예컨대 Pd(PPh3)4를 사용하여 실행된다. 팔라듐-매개 커플링 방법은 표준 참고 문헌, 예컨대 문헌 [Schlosser "Organometallics in Synthesis" (published by Wiley and sons)]에 기재되어 있다. 화합물 III-1은 비양성자성 용매 (DCM, CHCl3, THF 등) 중에서 아실화 시약, 예컨대 트리포스겐 또는 카르보닐 디이미다졸과 반응시켜 반응성 아실 중간체 종을 제공하고, 이를 3급 알킬 아민 염기의 존재 하에 아닐린 화합물 또는 알킬 아민으로 계내 처리하여 우레아 종 III-2를 형성하였다.
반응식 IV

시약: (a) 3-트리플루오로메틸피롤리딘, (1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일일리덴)클로로) (3-페닐알릴)팔라듐, KOt-Bu, DME, 80℃; (b) TEA, 트리포스겐, THF; 이어서 2-아미노피리딘, 65℃.
우레아 I-6의 클로로 관능기를 부흐발트-하르트비히 아미노화 조건을 사용하여 알킬 아민에 의해 선택적으로 대체하여 IV-1을 제공하였다. 부흐발트-하르트비히 반응은 전형적으로 에테르성 용매 예컨대 DME, 디옥산 또는 THF를 함유하는 벌키 브뢴스테드 염기, 예를 들어 KOt-Bu 또는 KHMDS와 함께 팔라듐(0) 촉매 예컨대 Pd(PPh3)4를 사용하여 실행된다. 팔라듐-매개 아민 커플링 방법은 문헌 [Hartwig, J.F. (1998), Transition Metal Catalyzed Synthesis of Arylamines and Aryl Ethers from Aryl Halides and Triflates: Scope and Mechanism, Angew. Chem. Int. Ed. 37: 2046-2067]에 기재되어 있다. 화합물 IV-1을 비양성자성 용매 (DCM, CHCl3, THF 등) 중에서 아실화 시약, 예컨대 트리포스겐 또는 카르보닐 디이미다졸과 반응시켜 반응성 아실 중간체 종을 제공하고, 이를 3급 알킬 아민 염기의 존재 하에 아닐린 화합물로 계내 처리하여 우레아 종 IV-2를 형성하였다.
반응식 V

시약: (a) NBS 또는 NCS, CHCl3, 60℃; (b) NaH, THF, 3-(피리딘-2-일)-2H-피리도[1,2a][1,3,5]트리아진-2,4(3H)-디온, 65℃, 이어서 H2O; (c) DIPEA, DMF, HATU, 알킬 아민, RT.
아민 II-1을 적절한 용매 중에서 친전자성 할로겐화 시약, 예컨대 NCS 또는 NBS와 반응시켜 상응하는 할로겐화 종 V-1을 위치선택적 방식으로 제공하였다. 방향족 고리의 할로겐화를 실시하는 많은 방법이 존재하며, 관련 기술분야의 통상의 기술자에게 널리 공지되어 있다. 에스테르 V-1을 비양성자성 용매 (DCM, CHCl3, THF 등) 중에서 아실화 시약, 예컨대 트리포스겐 또는 카르보닐 디이미다졸과 반응시키고, 이어서 3급 알킬 아민 염기의 존재 하에 아닐린 화합물 또는 알킬 아민에 의해 처리하였다. 계내 발성된 NaOH를 통해 일어나는 에스테르 관능기의 병행 가수분해는 우레아 종 V-2를 형성하였다. 카르복실산 V-2를 극성 비양성자성 용매 중에서 커플링 시약 (HATU)의 존재 하에 알킬 아민과 반응시켜 상응하는 아미드 V-3을 제공하였다. 다양한 아미드 커플링 시약 예컨대 EDC, PyBrop 등은 상업적으로 입수가능하다. 아미드 커플링 반응은 일반적으로 유기 염기 예컨대 Et3N 또는 (i-Pr)2NEt를 이용하여 용매 예컨대 DCM 또는 DMF 중에서 실행된다.
반응식 VI

시약: (a) NBS 또는 NCS, CHCl3, 60℃; (b) TEA, 트리포스겐, THF; 이어서 2-아미노피리딘, 65℃.
화합물 III-1을 적절한 용매 중에서 친전자성 할로겐화 시약, 예컨대 NCS 또는 NBS와 반응시켜 상응하는 할로겐화 종 VI-1을 위치선택적 방식으로 제공하였다. 방향족 고리의 할로겐화를 실시하는 많은 방법이 존재하며, 관련 기술분야의 통상의 기술자에게 널리 공지되어 있다. 화합물 VI-1을 비양성자성 용매 (DCM, CHCl3, THF 등) 중에서 아실화 시약, 예컨대 트리포스겐 또는 카르보닐 디이미다졸과 반응시켜 반응성 아실 중간체 종을 제공하고, 이를 3급 알킬 아민 염기의 존재 하에 아닐린 화합물 또는 알킬 아민으로 계내 처리하여 우레아 종 VI-2를 형성하였다.
반응식 VII

시약: (a) NIS, CHCl3, 60℃; (b) 트리메틸보록신, Pd(PPh3)4, K2CO3, 디옥산/H2O, 90℃;
화합물 I-6을 적절한 용매 중에서 친전자성 할로겐화 시약 (NIS)과 반응시켜 상응하는 할로겐화 종 VII-1을 위치선택적 방식으로 제공하였다. 방향족 고리의 할로겐화를 실시하는 많은 방법이 존재하며, 관련 기술분야의 통상의 기술자에게 널리 공지되어 있다. 화합물 VII-1의 아이오도 관능기를 스즈키 커플링 화학을 사용하여 알킬보론산과 커플링시켜 VII-2를 제공하였다. 스즈키-유사 커플링은 전형적으로 에테르성 용매 예컨대 DME, 디옥산 또는 THF를 함유하는 수성 혼합물 중에서 무기 염기, 예를 들어 K2CO3, Na2CO3 또는 K3PO4와 함께 팔라듐(0) 촉매 예컨대 Pd(PPh3)4를 사용하여 실행된다. 팔라듐-매개 커플링 방법은 표준 참고 문헌, 예컨대 문헌 [Schlosser "Organometallics in Synthesis" (published by Wiley and sons)]에 기재되어 있다. 메틸화 종 VII-2를 화합물 I-6에 대한 대용물로서 반응식 II 및 III에 사용하여 유사한 메틸화 생성물을 생성하였다.
화합물 특징 및 특성
예시적 실시양태에서, 치료 화합물은 세포의 세포질 막을 가로지를 수 있다. 예를 들어, 화합물은 적어도 약 20%, 50%, 75%, 80%, 90% 또는 95%의 세포-투과성을 가질 수 있다.
본원에 기재된 화합물은 또한 하기 특징 중 1종 이상을 가질 수 있다: 화합물은 세포 또는 대상체에 대해 본질적으로 비-독성일 수 있고; 화합물은 2000 amu 이하, 1000 amu 이하의 유기 분자 또는 소분자일 수 있고; 화합물은 적어도 약 30일, 60일, 120일, 6개월 또는 1년의 정상 대기 조건 하의 반감기를 가질 수 있고; 화합물은 적어도 약 30일, 60일, 120일, 6개월 또는 1년의 용액 중에서의 반감기를 가질 수 있고; 화합물은 용액 중에서 레스베라트롤보다 적어도 약 50%, 2배, 5배, 10배, 30배, 50배 또는 100배의 배수만큼 더 안정할 수 있고; 화합물은 DNA 복구 인자 Ku70의 탈아세틸화를 촉진할 수 있고; 화합물은 RelA/p65의 탈아세틸화를 촉진할 수 있고; 화합물은 일반적인 전환율을 증가시키고, TNF-유발 아폽토시스에 대한 세포의 감수성을 증진시킬 수 있다.
특정 실시양태에서, 시르투인-조정 화합물은 시르투인의 데아세틸라제 활성을 조정하기에 효과적인 농도 (예를 들어, 생체내)에서, 히스톤 데아세틸라제 (HDAC) 클래스 I 및 /또는 HDAC 클래스 II를 억제하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다. 예를 들어, 바람직한 실시양태에서, 상기 시르투인-조정 화합물은 시르투인-조정 화합물이며, HDAC I 및/또는 HDAC II의 억제에 대한 EC50보다 적어도 5배 더 적은, 보다 더 바람직하게는 적어도 10배, 100배 또는 심지어 1000배 더 적은, 시르투인 데아세틸라제 활성의 활성화에 대한 EC50을 갖도록 선택된다. HDAC I 및/또는 HDAC II 활성을 검정하는 방법은 관련 기술분야에 널리 공지되어 있으며, 이러한 검정을 수행하기 위한 키트는 상업적으로 구입할 수 있다. 예를 들어, 바이오비전, 인크.(BioVision, Inc.) (캘리포니아주 마운틴 뷰; biovision.com의 월드 와이드 웹) 및 토마스 사이언티픽(Thomas Scientific) (뉴저지주 스웨데스보로; tomassci.com의 월드 와이드 웹)을 참조한다.
특정 실시양태에서, 시르투인-조정 화합물은 시르투인 상동체를 조정하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다. 특정 실시양태에서, 인간 시르투인 단백질의 활성화제는 인간 시르투인의 데아세틸라제 활성을 활성화시키기에 효과적인 농도 (예를 들어, 생체내)에서, 하등 진핵생물, 특히 효모 또는 인간 병원체로부터 시르투인 단백질을 활성화시키는 것에 대해 어떠한 실질적인 능력도 갖지 않을 수 있다. 예를 들어, 시르투인-조정 화합물은 효모 시르투인, 예컨대 Sir2 (예컨대 칸디다(Candida), 에스. 세레비지아에 등)의 활성화에 대한 EC50보다 적어도 5배 더 적은, 보다 더 바람직하게는 적어도 10배, 100배 또는 심지어 1000배 더 적은, 인간 시르투인, 예컨대 SIRT1 및/또는 SIRT3 데아세틸라제 활성의 활성화에 대한 EC50을 갖도록 선택될 수 있다. 또 다른 실시양태에서, 하등 진핵생물, 특히 효모 또는 인간 병원체로부터의 시르투인 단백질의 억제제는 하등 진핵생물로부터의 시르투인 단백질의 데아세틸라제 활성을 억제하기에 효과적인 농도 (예를 들어, 생체내)에서, 인간으로부터의 시르투인 단백질을 억제하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다. 예를 들어, 시르투인-억제 화합물은 효모 시르투인, 예컨대 Sir2 (예컨대 칸디다, 에스. 세레비지아에 등)의 억제에 대한 IC50보다 적어도 5배 더 적은, 보다 더 바람직하게는 적어도 10배, 100배 또는 심지어 1000배 더 적은, 인간 시르투인, 예컨대 SIRT1 및/또는 SIRT3 데아세틸라제 활성의 억제에 대한 IC50을 갖도록 선택될 수 있다.
특정 실시양태에서, 시르투인-조정 화합물은 1종 이상의 시르투인 단백질 상동체, 예컨대 예를 들어 인간 SIRT1, SIRT2, SIRT3, SIRT4, SIRT5, SIRT6 또는 SIRT7 중 1종 이상을 조정하는 능력을 가질 수 있다. 일부 실시양태에서, 시르투인-조정 화합물은 SIRT1 및 SIRT3 단백질 둘 다를 조정하는 능력을 갖는다.
다른 실시양태에서, SIRT1 조정제는 인간 SIRT1의 데아세틸라제 활성을 조정하기에 효과적인 농도 (예를 들어, 생체내)에서, 다른 시르투인 단백질 상동체, 예컨대 예를 들어 인간 SIRT2, SIRT3, SIRT4, SIRT5, SIRT6 또는 SIRT7 중 1종 이상을 조정하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다. 예를 들어, 시르투인-조정 화합물은 인간 SIRT2, SIRT3, SIRT4, SIRT5, SIRT6 또는 SIRT7 중 1종 이상의 조정에 대한 ED50보다 적어도 5배 더 적은, 보다 더 바람직하게는 적어도 10배, 100배 또는 심지어 1000배 더 적은, 인간 SIRT1 데아세틸라제 활성의 조정에 대한 ED50을 갖도록 선택될 수 있다. 일부 실시양태에서, SIRT1 조정제는 SIRT3 단백질을 조정하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다.
다른 실시양태에서, SIRT3 조정제는 인간 SIRT3의 데아세틸라제 활성을 조정하기에 효과적인 농도 (예를 들어, 생체내)에서, 다른 시르투인 단백질 상동체, 예컨대 예를 들어 인간 SIRT1, SIRT2, SIRT4, SIRT5, SIRT6 또는 SIRT7 중 1종 이상을 조정하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다. 예를 들어, 시르투인-조정 화합물은 인간 SIRT1, SIRT2, SIRT4, SIRT5, SIRT6 또는 SIRT7 중 1종 이상의 조정에 대한 ED50보다 적어도 5배 더 적은, 보다 더 바람직하게는 적어도 10배, 100배 또는 심지어 1000배 더 적은, 인간 SIRT3 데아세틸라제 활성의 조정에 대한 ED50을 갖도록 선택될 수 있다. 일부 실시양태에서, SIRT3 조정제는 SIRT1 단백질을 조정하는 것에 대해 어떠한 실질적인 능력도 갖지 않는다.
특정 실시양태에서, 시르투인-조정 화합물은 약 10-9M, 10-10M, 10-11M, 10-12M 또는 그 미만의 시르투인 단백질에 대한 결합 친화도를 가질 수 있다. 시르투인-조정 화합물은 그의 기질 또는 NAD+ (또는 다른 보조인자)에 대한 시르투인 단백질의 겉보기 Km을 적어도 약 2, 3, 4, 5, 10, 20, 30, 50 또는 100의 배수만큼 감소 (활성화제) 또는 증가 (억제제)시킬 수 있다. 특정 실시양태에서, Km 값은 본원에 기재된 질량 분광측정법 검정을 사용하여 결정된다. 바람직한 활성화 화합물은 그의 기질 또는 보조인자에 대한 시르투인의 Km을 유사한 농도의 레스베라트롤에 의해 유발된 것보다 더 큰 정도로 감소시키거나, 또는 그의 기질 또는 보조인자에 대한 시르투인의 Km을 더 낮은 농도의 레스베라트롤에 의해 유발된 것과 유사하게 감소시킨다. 시르투인-조정 화합물은 시르투인 단백질의 Vmax를 적어도 약 2, 3, 4, 5, 10, 20, 30, 50 또는 100의 배수만큼 증가시킬 수 있다. 시르투인-조정 화합물은 약 1 nM 미만, 약 10 nM 미만, 약 100 nM 미만, 약 1 μM 미만, 약 10 μM 미만, 약 100 μM 미만, 또는 약 1-10 nM, 약 10-100 nM, 약 0.1-1 μM, 약 1-10 μM 또는 약 10-100 μM의 SIRT1 및/또는 SIRT3 단백질의 데아세틸라제 활성의 조정에 대한 ED50을 가질 수 있다. 시르투인-조정 화합물은 세포 검정 또는 세포 기반 검정으로 측정 시에, SIRT1 및/또는 SIRT3 단백질의 데아세틸라제 활성을 적어도 약 5, 10, 20, 30, 50 또는 100의 배수만큼 조정할 수 있다. 시르투인-조정 화합물은 동일한 농도의 레스베라트롤에 비해 적어도 약 10%, 30%, 50%, 80%, 2배, 5배, 10배, 50배 또는 100배 더 큰 시르투인 단백질의 데아세틸라제 활성의 유도를 유발할 수 있다. 시르투인-조정 화합물은 SIRT1 및/또는 SIRT3의 조정에 대한 것보다 적어도 약 10배, 20배, 30배, 50배 더 큰, SIRT5의 조정에 대한 ED50을 가질 수 있다.
예시적인 용도
특정 측면에서, 본 발명은 시르투인 단백질의 수준 및/또는 활성을 조정하는 방법 또는 그를 위한 용도 및 그의 사용 방법을 제공한다.
특정 실시양태에서, 본 발명은 시르투인-조정 화합물이 시르투인 단백질을 활성화시키며, 예를 들어 시르투인 단백질의 수준 및/또는 활성을 증가시키는 것인 시르투인-조정 화합물을 사용하는 방법 또는 그를 위한 용도를 제공한다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은, 예를 들어 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 등을 포함한 매우 다양한 질환 및 장애를 치료 및/또는 예방하는 것을 포함한 다양한 치료 용도에 유용할 수 있다. 방법 또는 용도는 그를 필요로 하는 대상체에게 제약 유효량의 시르투인-조정 화합물, 예를 들어 시르투인-조정 화합물을 투여하는 것을 포함한다.
이론에 얽매이는 것을 원하지는 않지만, 본 발명의 활성화제는 시르투인 단백질 내의 동일한 위치 (예를 들어, 활성 부위, 또는 활성 부위의 Km 또는 Vmax에 영향을 미치는 부위)에서 시르투인과 상호작용할 수 있는 것으로 여겨진다. 이러한 이유로 인해 특정 클래스의 시르투인 활성화제 및 억제제가 실질적인 구조적 유사성을 가질 수 있는 것으로 여겨진다.
특정 실시양태에서, 본원에 기재된 시르투인-조정 화합물은 단독으로 또는 다른 화합물과 조합되어 취해질 수 있다. 특정 실시양태에서, 2종 이상의 시르투인-조정 화합물의 혼합물이 그를 필요로 하는 대상체에게 투여될 수 있다. 또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 하기 화합물 중 1종 이상과 함께 투여될 수 있다: 레스베라트롤, 부테인, 피세틴, 피세아타놀, 또는 퀘르세틴. 예시적 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 니코틴산 또는 니코틴아미드 리보시드와 조합되어 투여될 수 있다. 또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 감소시키는 시르투인-조정 화합물은 하기 화합물 중 1종 이상과 함께 투여될 수 있다: 니코틴아미드 (NAM), 수라민; NF023 (G-단백질 길항제); NF279 (퓨린성 수용체 길항제); 트롤록스 (6-히드록시-2,5,7,8,테트라메틸크로만-2-카르복실산); (-)-에피갈로카테킨 (부위 3,5,7,3',4', 5' 상의 히드록시); (-)-에피갈로카테킨 갈레이트 (부위 5,7,3',4',5' 상의 히드록시 및 부위 3 상의 갈레이트 에스테르); 시아니딘 클로라이드 (3,5,7,3',4'-펜타히드록시플라빌리움 클로라이드); 델피니딘 클로라이드 (3,5,7,3',4',5'-헥사히드록시플라빌리움 클로라이드); 미리세틴 (칸나비스세틴; 3,5,7,3',4',5'-헥사히드록시플라본); 3,7,3',4',5'-펜타히드록시플라본; 고시페틴 (3,5,7,8,3',4'-헥사히드록시플라본), 시르티놀; 및 스플리토미신. 또 다른 실시양태에서, 1종 이상의 시르투인-조정 화합물은, 예를 들어 암, 당뇨병, 신경변성 질환, 심혈관 질환, 혈액 응고, 염증, 홍조, 비만, 노화, 스트레스 등을 포함한 다양한 질환의 치료 또는 예방을 위한 1종 이상의 치료제와 함께 투여될 수 있다. 다양한 실시양태에서, 시르투인-조정 화합물을 포함하는 조합 요법은 (1) 1종 이상의 시르투인-조정 화합물을 1종 이상의 치료제 (예를 들어, 본원에 기재된 1종 이상의 치료제)와 조합하여 포함하는 제약 조성물; 및 (2) 1종 이상의 시르투인-조정 화합물과 1종 이상의 치료제의 공-투여로서, 여기서 시르투인-조정 화합물 및 치료제는 동일한 조성물로 제제화된 것은 아닌 것 (그러나 동일한 키트 또는 패키지, 예컨대 블리스터 팩 또는 다른 다중-챔버 패키지; 사용자에 의해 분리될 수 있는 연결된 개별 밀봉 용기 (예를 들어, 호일 파우치); 또는 화합물(들) 및 다른 치료제(들)가 개별 용기에 존재하는 키트 내에 존재할 수 있음)을 지칭할 수 있다. 개별 제제를 사용하는 경우에, 시르투인-조정 화합물은 또 다른 치료제의 투여와 동시에, 간헐적으로, 시차를 두고, 그 전에, 그 후에, 또는 그의 조합으로 투여될 수 있다.
특정 실시양태에서, 본원에 기재된 화합물을 사용하여 질환 또는 장애를 감소, 예방 또는 치료하는 방법 또는 그를 위한 용도는 시르투인, 예컨대 인간 SIRT1, SIRT2 및/또는 SIRT3, 또는 그의 상동체의 단백질 수준을 증가시키는 것을 또한 포함할 수 있다. 단백질 수준을 증가시키는 것은 시르투인을 코딩하는 핵산의 1개 이상의 카피를 세포에 도입함으로써 달성될 수 있다. 예를 들어, 시르투인의 수준은 시르투인을 코딩하는 핵산을 포유동물 세포에 도입함으로써 포유동물 세포에서 증가될 수 있으며, 예를 들어 진뱅크 수탁 번호 NP_036370에 제시된 아미노산 서열을 코딩하는 핵산을 도입함으로써 SIRT1의 수준을 증가시킬 수 있고/거나 진뱅크 수탁 번호 AAH01042에 제시된 아미노산 서열을 코딩하는 핵산을 도입함으로써 SIRT3의 수준을 증가시킬 수 있다.
시르투인의 단백질 수준을 증가시키기 위해 세포에 도입되는 핵산은 시르투인, 예를 들어 SIRT1 및/또는 SIRT3 단백질의 서열과 적어도 약 80%, 85%, 90%, 95%, 98%, 또는 99% 동일한 단백질을 코딩할 수 있다. 예를 들어, 단백질을 코딩하는 핵산은 SIRT1 (예를 들어, 진뱅크 수탁 번호 NM_012238) 및/또는 SIRT3 (예를 들어, 진뱅크 수탁 번호 BC001042) 단백질을 코딩하는 핵산과 적어도 약 80%, 85%, 90%, 95%, 98%, 또는 99% 동일할 수 있다. 핵산은 또한, 바람직하게는 엄격한 혼성화 조건 하에 야생형 시르투인, 예를 들어 SIRT1 및/또는 SIRT3 단백질을 코딩하는 핵산에 혼성화되는 핵산일 수 있다. 엄격한 혼성화 조건은 65℃에서 0.2 x SSC 중에서의 혼성화 및 세척을 포함할 수 있다. 야생형 시르투인 단백질, 예컨대 야생형 시르투인의 단편인 단백질과 상이한 단백질을 코딩하는 핵산을 사용하는 경우에, 상기 단백질은 바람직하게는 생물학적으로 활성이며, 예를 들어 탈아세틸화될 수 있다. 단지, 생물학적으로 활성인 시르투인의 부분을 세포에서 발현시키는 것이 필요하다. 예를 들어, 진뱅크 수탁 번호 NP_036370을 갖는 야생형 SIRT1과 상이한 단백질은 바람직하게는 그의 코어 구조를 함유한다. 상기 코어 구조는 때때로 NAD 결합 뿐만 아니라 기질 결합 도메인을 포괄하는 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 237 내지 932에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 아미노산 62-293을 지칭한다. SIRT1의 코어 도메인은 또한 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 834 내지 1394에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 대략 아미노산 261 내지 447; 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 777 내지 1532에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 대략 아미노산 242 내지 493; 또는 진뱅크 수탁 번호 NM_012238의 뉴클레오티드 813 내지 1538에 의해 코딩되는 진뱅크 수탁 번호 NP_036370의 대략 아미노산 254 내지 495를 지칭할 수 있다. 단백질이 생물학적 기능, 예컨대 탈아세틸화 능력을 유지하는지의 여부는 관련 기술분야에 공지된 방법에 따라 결정될 수 있다.
특정 실시양태에서, 시르투인-조정 화합물을 사용하여 질환 또는 장애를 감소, 예방 또는 치료하는 방법 또는 그를 위한 용도는 시르투인, 예컨대 인간 SIRT1, SIRT2 및/또는 SIRT3, 또는 그의 상동체의 단백질 수준을 감소시키는 것을 또한 포함할 수 있다. 시르투인 단백질 수준을 감소시키는 것은 관련 기술분야에 공지된 방법에 따라 달성될 수 있다. 예를 들어, 시르투인에 대해 표적화된 siRNA, 안티센스 핵산 또는 리보자임을 세포에서 발현시킬 수 있다. 우성 음성 시르투인 돌연변이체, 예를 들어 탈아세틸화될 수 없는 돌연변이체가 또한 사용될 수 있다. 예를 들어, 문헌 [Luo et al. (2001) Cell 107:137]에 기재된 SIRT1의 돌연변이체 H363Y가 사용될 수 있다. 대안적으로, 전사를 억제하는 작용제가 사용될 수 있다.
시르투인 단백질 수준을 조정하는 방법 또는 그를 위한 용도는 시르투인을 코딩하는 유전자의 전사를 조정하는 방법 또는 그를 위한 용도, 상응하는 mRNA를 안정화/탈안정화시키는 방법, 및 관련 기술분야에 공지된 다른 방법을 또한 포함한다.
노화/스트레스
한 측면에서, 본 발명은 세포를 시르투인 단백질의 수준 및/또는 활성을 증가시키는 본 발명의 시르투인-조정 화합물과 접촉시킴으로써, 세포의 수명을 연장시키거나, 세포의 증식 능력을 연장시키거나, 세포의 노화를 저속화시키거나, 세포의 생존을 촉진하거나, 세포에서 세포성 노쇠를 지연시키거나, 칼로리 제한의 효과를 모방하거나, 스트레스에 대한 세포의 저항성을 증가시키거나, 또는 세포의 아폽토시스를 방지하는 방법을 제공한다. 예시적 실시양태에서, 방법 또는 용도는 세포를 시르투인-조정 화합물과 접촉시키는 것을 포함한다.
본원에 기재된 방법 또는 용도는 세포, 특히 1차 세포 (즉, 유기체, 예를 들어 인간으로부터 수득된 세포)가 세포 배양 시에 생존하도록 유지될 수 있는 시간의 양을 증가시키는데 사용될 수 있다. 배아 줄기 (ES) 세포 및 만능 세포, 및 그로부터 분화된 세포를 또한 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 처리하여 배양 시에 세포 또는 그의 자손을 더 장기간 동안 유지할 수 있다. 이러한 세포는 또한, 예를 들어 생체외 변형 후, 대상체로의 이식에 사용될 수 있다.
한 측면에서, 장기간 동안 보존되도록 의도된 세포는 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 처리될 수 있다. 세포는 현탁액 (예를 들어, 혈액 세포, 혈청, 생물학적 성장 배지 등), 또는 조직 또는 기관에 존재할 수 있다. 예를 들어, 수혈의 목적을 위해 개체로부터 수집된 혈액을 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 처리하여 혈액 세포를 더 장기간 동안 보존할 수 있다. 추가로, 법의학적 목적에 사용될 혈액을 또한 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 사용하여 보존할 수 있다. 수명을 연장시키거나 또는 아폽토시스에 대해 보호하도록 처리될 수 있는 다른 세포는 소비를 위한 세포, 예를 들어 비-인간 포유동물로부터의 세포 (예컨대 고기) 또는 식물 세포 (예컨대 채소)를 포함한다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한, 예를 들어 발생 및/또는 성장 과정을 변경, 지연 또는 촉진하기 위해 포유동물, 식물, 곤충 또는 미생물에서의 발생기 및 성장기 동안 적용될 수 있다.
또 다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은, 예를 들어 고형 조직 이식편, 기관 이식물, 세포 현탁액, 줄기 세포, 골수 세포 등을 포함한 이식 또는 세포 요법에 유용한 세포를 처리하는데 사용될 수 있다. 세포 또는 조직은 자가이식편, 동종이식편, 동계이식편 또는 이종이식편일 수 있다. 세포 또는 조직은 대상체로의 투여/이식 전에, 투여/이식과 공동으로, 및/또는 투여/이식 후에 시르투인-조정 화합물로 처리될 수 있다. 세포 또는 조직은 공여자 개체로부터의 세포의 제거 전에, 공여자 개체로부터의 세포 또는 조직의 제거 후에 생체외에서, 또는 수용자로의 이식 후에 처리될 수 있다. 예를 들어, 공여자 또는 수용자 개체는 시르투인-조정 화합물로 전신으로 처리될 수 있거나, 또는 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 사용하여 국부로 처리된 세포/조직의 하위세트를 가질 수 있다. 특정 실시양태에서, 세포 또는 조직 (또는 공여자/수용자 개체)은 이식편 생존을 연장시키기에 유용한 또 다른 치료제, 예컨대 예를 들어 면역억제제, 시토카인, 혈관형성 인자 등으로 추가로 처리될 수 있다.
또 다른 실시양태에서, 세포를 생체내 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 처리하여, 예를 들어 그의 수명을 증가시키거나 또는 아폽토시스를 방지할 수 있다. 예를 들어, 피부 또는 상피 세포를 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 처리함으로써 피부를 노화 (예를 들어, 주름 발생, 탄력 손실 등)로부터 보호할 수 있다. 예시적 실시양태에서, 피부를 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 포함하는 제약 또는 화장품 조성물과 접촉시킨다. 본원에 기재된 방법 또는 용도에 따라 치료될 수 있는 예시적인 피부 고통 또는 피부 상태는 염증, 일광 손상 또는 자연 노화와 연관되거나 또는 그에 의해 유발된 장애 또는 질환을 포함한다. 예를 들어, 상기 조성물은 접촉성 피부염 (자극성 접촉성 피부염 및 알레르기성 접촉성 피부염 포함), 아토피성 피부염 (알레르기성 습진으로도 공지됨), 광선 각화증, 각질화 장애 (습진 포함), 수포성 표피박리 질환 (천포창 포함), 박탈성 피부염, 지루성 피부염, 홍반 (다형성 홍반 및 결절성 홍반 포함), 일광 또는 다른 광원에 의해 유발된 손상, 원판상 홍반성 루푸스, 피부근염, 건선, 피부암 및 자연 노화 영향의 예방 또는 치료에 유용하다. 또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은, 예를 들어 1도, 2도 또는 3도 화상 및/또는 열, 화학 또는 전기 화상을 포함한 상처 및/또는 화상을 치료하여 치유를 촉진하는데 사용될 수 있다. 제제는 피부 또는 점막 조직에 국소로 투여될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 1종 이상의 시르투인-조정 화합물을 포함하는 국소 제제는 또한 예방적, 예를 들어 화학예방적 조성물로서 사용될 수 있다. 화학예방적 방법에 사용되는 경우에, 감수성 피부는 특정한 개체에서의 임의의 가시적 상태 전에 처리된다.
시르투인-조정 화합물은 대상체에게 국부로 또는 전신으로 전달될 수 있다. 특정 실시양태에서, 시르투인-조정 화합물은 주사, 국소 제제 등에 의해 대상체의 조직 또는 기관에 국부로 전달된다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 대상체에서 세포성 노쇠에 의해 유발 또는 악화되는 질환 또는 상태를 치료 또는 예방하기 위해; 예를 들어 노쇠의 발병 후, 대상체의 노쇠 속도를 감소시키는 방법 또는 그를 위한 용도; 대상체의 수명을 연장시키는 방법; 수명과 관련된 질환 또는 상태를 치료 또는 예방하는 방법 또는 그를 위한 용도; 세포의 증식성 능력과 관련된 질환 또는 상태를 치료 또는 예방하는 방법 또는 그를 위한 용도; 및 세포 손상 또는 사멸로부터 유발된 질환 또는 상태를 치료 또는 예방하는 방법 또는 그를 위한 용도에 사용될 수 있다. 특정 실시양태에서, 방법은 대상체의 수명을 단축시키는 질환의 발생률을 감소시킴으로써 작용하지는 않는다. 특정 실시양태에서, 방법은 질환, 예컨대 암에 의해 유발된 치사율을 감소시킴으로써 작용하지는 않는다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 일반적으로 대상체의 세포의 수명을 증가시키고, 대상체의 세포를 스트레스 및/또는 아폽토시스에 대해 보호하기 위해 대상체에게 투여될 수 있다. 대상체를 본원에 기재된 화합물로 치료하는 것은 대상체를 호르메시스, 즉 유기체에 유익하며 그의 수명을 연장시킬 수 있는 가벼운 스트레스에 적용하는 것과 유사한 것으로 여겨진다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 노화 및 노화-관련 예후 또는 질환, 예컨대 졸중, 심장 질환, 심부전, 관절염, 고혈압 및 알츠하이머병을 예방하기 위해 대상체에게 투여될 수 있다. 치료될 수 있는 다른 상태는, 예를 들어 눈의 노화와 연관된 안구 장애, 예컨대 백내장, 녹내장 및 황반 변성을 포함한다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 세포 사멸과 연관된 질환, 예를 들어 만성 질환을 치료하여 세포를 세포 사멸로부터 보호하기 위해 대상체에게 투여될 수 있다. 예시적인 질환은 신경 세포 사멸, 뉴런 기능장애, 또는 근육 세포 사멸 또는 기능장애와 연관된 것들, 예컨대 파킨슨병, 알츠하이머병, 다발성 경화증, 근위축성 측삭 경화증, 및 근육 이영양증; AIDS; 전격성 간염; 뇌의 변성에 연관된 질환, 예컨대 크로이츠펠트-야콥병, 색소성 망막염 및 소뇌 변성; 골수이형성증 예컨대 재생불량성 빈혈; 허혈성 질환 예컨대 심근경색 및 졸중; 간 질환 예컨대 알콜성 간염, B형 간염 및 C형 간염; 관절-질환 예컨대 골관절염; 아테롬성동맥경화증; 탈모증; UV 광으로 인한 피부에 대한 손상; 편평 태선; 피부의 위축; 백내장; 및 이식편 거부를 포함한다. 세포 사멸은 또한 수술, 약물 요법, 화학물질 노출 또는 방사선 노출에 의해 유발될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 급성 질환, 예컨대 기관 또는 조직 손상을 앓고 있는 대상체, 예를 들어 졸중 또는 심근경색을 앓고 있는 대상체 또는 척수 손상을 앓고 있는 대상체에게 투여될 수 있다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 알콜성 간을 복구하는데 사용될 수 있다.
심혈관 질환
또 다른 실시양태에서, 본 발명은 심혈관 질환의 치료 및/또는 예방을 필요로 하는 대상체에게 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 투여함으로써, 심혈관 질환을 치료 및/또는 예방하는 방법을 제공한다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 사용하여 치료 또는 예방될 수 있는 심혈관 질환은 심근병증 또는 심근염; 예컨대 특발성 심근병증, 대사 심근병증, 알콜성 심근병증, 약물-유발 심근병증, 허혈성 심근병증, 및 고혈압 심근병증을 포함한다. 본원에 기재된 화합물 및 방법 또는 용도를 사용하여 또한 치료가능 또는 예방가능한 것은 주요 혈관, 예컨대 대동맥, 관상 동맥, 경동맥, 뇌혈관 동맥, 신동맥, 장골 동맥, 대퇴 동맥 및 슬와 동맥의 아테롬성 장애 (대혈관 질환)이다. 치료 또는 예방될 수 있는 다른 혈관 질환은 혈소판 응집, 망막 세동맥, 사구체 세동맥, 신경벽 혈관, 심장 세동맥, 및 눈, 신장, 심장, 및 중추 및 말초 신경계의 연합 모세혈관상과 관련된 것들을 포함한다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 개체의 혈장 중 HDL 수준을 증가시키는데 사용될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 치료될 수 있는 또 다른 장애는, 예를 들어 관상동맥 개입 후, 재협착, 및 고밀도 및 저밀도 콜레스테롤의 비정상적 수준과 관련된 장애를 포함한다.
특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또 다른 심혈관제와의 조합 요법의 일부로서 투여될 수 있다. 특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 항부정맥제와의 조합 요법의 일부로서 투여될 수 있다. 또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또 다른 심혈관제와의 조합 요법의 일부로서 투여될 수 있다.
세포 사멸/암
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 소정 용량의 방사선 또는 독소를 최근에 수용하였거나 또는 수용할 가능성이 있는 대상체에게 투여될 수 있다. 특정 실시양태에서, 상기 용량의 방사선 또는 독소는 직업-관련 또는 의료 절차의 일부로서 수용되며, 예를 들어 예방적 조치로서 투여된다. 또 다른 실시양태에서, 방사선 또는 독소 노출은 의도치 않게 수용된다. 이러한 경우에, 화합물은 바람직하게는 아폽토시스 및 급성 방사선 증후군의 후속 발생을 억제하기 위해 노출 후 가능한 한 빨리 투여된다.
시르투인-조정 화합물은 또한 암을 치료 및/또는 예방하는데 사용될 수 있다. 특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 암을 치료 및/또는 예방하는데 사용될 수 있다. 칼로리 제한은 암을 포함한 연령-관련 장애의 발생의 감소와 연관된 바 있다. 따라서, 시르투인 단백질의 수준 및/또는 활성에서의 증가는 연령-관련 장애, 예컨대 예를 들어 암의 발생을 치료 및/또는 예방하기에 유용할 수 있다. 시르투인-조정 화합물을 사용하여 치료될 수 있는 예시적인 암은 뇌 및 신장의 암; 유방암, 전립선암, 고환암 및 난소암을 포함한 호르몬-의존성 암; 림프종, 및 백혈병이다. 고형 종양과 연관된 암에서, 조정 화합물은 종양에 직접 투여될 수 있다. 혈액 세포의 암, 예를 들어 백혈병은 조정 화합물을 혈류 또는 골수에 투여함으로써 치료될 수 있다. 양성 세포 성장, 예를 들어 사마귀가 또한 치료될 수 있다. 치료될 수 있는 다른 질환은 자가면역 세포가 제거되어야 하는 자가면역 질환, 예를 들어 전신 홍반성 루푸스, 경피증 및 관절염을 포함한다. 바이러스 감염 예컨대 포진, HIV, 아데노바이러스 및 HTLV-1 연관 악성 및 양성 장애는 또한 시르투인-조정 화합물의 투여에 의해 치료될 수 있다. 대안적으로, 세포를 대상체로부터 수득하고, 생체외 처리하여 특정의 바람직하지 않은 세포, 예를 들어 암 세포를 제거하고, 동일하거나 상이한 대상체에게 다시 투여할 수 있다.
화학요법제는 항암 활성을 갖는 것으로서 본원에 기재된 조정 화합물, 예를 들어 아폽토시스를 유발하는 화합물, 수명을 감소시키는 화합물, 또는 세포를 스트레스에 대해 감수성으로 만드는 화합물과 공-투여될 수 있다. 화학요법제는 그 자체로 세포 사멸을 유발하거나, 수명을 감소시키거나, 또는 스트레스에 대한 감수성을 증가시키는 것으로 본원에 기재된 시르투인-조정 화합물과 함께 및/또는 다른 화학요법제와 조합되어 사용될 수 있다. 통상적인 화학요법제 이외에도, 본원에 기재된 시르투인-조정 화합물은 또한 원치 않는 세포 증식에 기여하는 세포 성분의 발현을 억제하기 위해 안티센스 RNA, RNAi 또는 다른 폴리뉴클레오티드와 함께 사용될 수 있다.
시르투인-조정 화합물 및 통상적인 화학요법제를 포함하는 조합 요법은 관련 기술분야에 공지된 조합 요법에 비해 유리할 수 있으며, 이는 조합이 통상적인 화학요법제가 더 낮은 투여량에서 더 큰 효과를 발휘하는 것을 가능하게 하기 때문이다. 바람직한 실시양태에서, 시르투인-조정 화합물과 조합되어 사용되는 경우에, 화학요법제 또는 통상적인 화학요법제의 조합에 대한 유효 용량 (ED50)은 화학요법제 단독에 대한 ED50보다 적어도 2배 더 적으며, 보다 더 바람직하게는 5배, 10배 또는 심지어 25배 더 적다. 반대로, 본원에 기재된 시르투인-조정 화합물과 조합되어 사용되는 경우에, 이러한 화학요법제 또는 이러한 화학요법제의 조합의 치료 지수 (TI)는 통상적인 화학치료 요법 단독에 대한 TI보다 적어도 2배 더 크며, 보다 더 바람직하게는 5배, 10배 또는 심지어 25배 더 클 수 있다.
뉴런 질환/장애
특정 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 신경변성 질환, 및 중추 신경계 (CNS), 척수 또는 말초 신경계 (PNS)에 대한 외상성 또는 기계적 손상을 앓고 있는 환자를 치료하는데 사용될 수 있다. 신경변성 질환은 전형적으로 뇌 세포의 위축 및/또는 사멸로 인한 것일 수 있는 인간 뇌의 질량 및 부피의 감소를 수반하며, 이는 노화에 기인하는 건강한 사람에서의 것들보다 훨씬 더 심하다. 신경변성 질환은 특정한 뇌 영역의 진행성 퇴행 (예를 들어, 신경 세포 기능장애 및 사멸)으로 인해 장기간의 정상적인 뇌 기능 후에 점진적으로 발생할 수 있다. 대안적으로, 외상 또는 독소와 연관된 것들과 같은 신경변성 질환은 급속한 발병을 가질 수 있다. 뇌 변성의 실제 발병은 임상적 발현보다 수년만큼 선행할 수 있다. 신경변성 질환의 예는 알츠하이머병 (AD), 파킨슨병 (PD), 헌팅톤병 (HD), 근위축성 측삭 경화증 (ALS; 루게릭병), 미만성 루이 소체 질환, 무도병-유극적혈구증가증, 원발성 측삭 경화증, 안구 질환 (안구 신경염), 화학요법-유발 신경병증 (예컨대 빈크리스틴, 파클리탁셀, 보르테조밉으로부터), 당뇨병-유발 신경병증 및 프리드라이히 운동실조를 포함하나, 이에 제한되지는 않는다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 이들 장애 및 하기 기재된 바와 같은 다른 것을 치료하는데 사용될 수 있다.
AD는 기억 상실, 비통상적 행동, 인격 변화, 및 지적 능력의 저하를 일으키는 CNS 장애이다. 이들 손실은 특정한 유형의 뇌 세포의 사멸, 및 이들 사이의 연결 및 그의 지지 네트워크 (예를 들어, 신경교 세포)의 파괴와 관련되어 있다. 가장 초기 증상은 최근 기억의 상실, 판단 오류, 및 인격 변화를 포함한다. PD는 비제어된 신체 운동, 경직, 진전 및 이상운동증을 일으키는 CNS 장애이며, 도파민을 생산하는 뇌 영역에서의 뇌 세포의 사멸과 연관되어 있다. ALS (운동 뉴런 질환)는 뇌를 골격근과 연결시키는 CNS의 성분인 운동 뉴런을 공격하는 CNS 장애이다.
HD는 비제어된 운동, 지적 능력의 상실, 및 정서 장애를 유발하는 또 다른 신경변성 질환이다. 테이-작스병 및 샌드호프병은 GM2 강글리오시드 및 β-헥소사미니다제에 대한 관련 당지질 기질이 신경계에 축적되고 급성 신경변성을 촉발하는 당지질 축적 질환이다.
아폽토시스는 면역계에서 AIDS 발병기전에서 역할을 하는 것으로 널리 공지되어 있다. 그러나, HIV-1은 또한 신경계 질환을 유발하며, 이는 본 발명의 시르투인-조정 화합물로 치료될 수 있다.
뉴런 손실은 프리온 질환, 예컨대 인간에서의 크로이츠펠트-야콥병, 소에서의 BSE (광우병), 양 및 염소에서의 스크래피 질환, 및 고양이에서의 고양이 해면상 뇌병증 (FSE)의 두드러진 특색이다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 이들 이전 질환으로 인한 뉴런 손실을 치료 또는 예방하기에 유용할 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 축삭병증을 수반하는 임의의 질환 또는 장애를 치료 또는 예방하는데 사용될 수 있다. 원위 축삭병증은 말초 신경계 (PNS) 뉴런의 일부 대사 또는 독성 장애로부터 유발된 유형의 말초 신경병증이다. 이는 대사 또는 독성 장애에 대한 신경의 가장 흔한 반응이며, 이와 같이 대사 질환 예컨대 당뇨병, 신부전, 결핍 증후군, 예컨대 영양실조 및 알콜중독, 또는 독소 또는 약물의 영향에 의해 유발될 수 있다. 원위 축삭병증을 갖는 것들은 통상적으로 대칭적 글로브-스타킹 감각-운동 장애를 제시한다. 심부건 반사 및 자율 신경계 (ANS) 기능은 또한 이환 부위에서 상실 또는 감소된다.
당뇨병성 신경병증은 당뇨병과 연관된 신경병증성 장애이다. 당뇨병성 신경병증과 연관될 수 있는 비교적 흔한 상태는 제3 신경 마비; 단일신경병증; 다발성 단일신경염; 당뇨병성 근위축; 통증성 다발신경병증; 자율 신경병증; 및 흉복부 신경병증을 포함한다.
말초 신경병증은 신경의 질환에 의해 또는 전신 질병의 부작용으로부터 유발될 수 있는 말초 신경계의 신경 손상에 대한 의학 용어이다. 당뇨병이 가능성이 가장 큰 원인이지만, 말초 신경병증의 주요 원인은 발작, 영양 결핍, 및 HIV를 포함한다.
예시적 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 재발성 MS 및 단일증상 MS를 포함한 다발성 경화증 (MS), 및 다른 탈수초성 상태, 예컨대 예를 들어 만성 염증성 탈수초성 다발신경병증 (CIDP), 또는 그와 연관된 증상을 치료 또는 예방하는데 사용될 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 질환으로 인한 외상, 상해 (외과적 개입 포함), 또는 환경적 외상 (예를 들어, 신경독소, 알콜중독 등)을 포함한 신경에 대한 외상을 치료하는데 사용될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 다양한 PNS 장애의 증상을 예방, 치료 및 완화하기에 유용할 수 있다. 용어 "말초 신경병증"은 뇌 및 척수 외부의 신경-말초 신경-이 손상된 광범위한 장애를 포괄한다. 말초 신경병증은 또한 말초 신경염으로도 지칭될 수 있거나, 또는 많은 신경이 수반되는 경우에는, 용어 다발신경병증 또는 다발신경염이 사용될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물로 치료가능한 PNS 질환은 하기를 포함한다: 당뇨병, 나병, 샤르코-마리-투스병, 길랑-바레 증후군 및 상완 신경총 신경병증 (상완 신경총의 경부 및 제1 흉부 신경근, 신경 줄기, 신경삭 및 말초 신경 성분의 질환).
또 다른 실시양태에서, 시르투인-조정 화합물은 폴리글루타민 질환을 치료 또는 예방하는데 사용될 수 있다. 예시적인 폴리글루타민 질환은 척수연수성 근육 위축 (케네디병), 헌팅톤병 (HD), 치상핵적핵-담창구시상하핵 위축 (하우 리버 증후군), 척수소뇌성 운동실조 제1형, 척수소뇌성 운동실조 제2형, 척수소뇌성 운동실조 제3형 (마차도-요셉병), 척수소뇌성 운동실조 제6형, 척수소뇌성 운동실조 제7형, 및 척수소뇌성 운동실조 제17형을 포함한다.
특정 실시양태에서, 본 발명은 세포로의 혈류의 감소에 반응하는 손상을 예방하기 위해 중추 신경계 세포를 치료하는 방법을 제공한다. 전형적으로, 예방될 수 있는 손상의 중증도는 주로 세포로의 혈류의 감소 정도 및 감소 지속기간에 따라 달라질 것이다. 특정 실시양태에서, 아폽토시스성 또는 괴사성 세포 사멸이 예방될 수 있다. 추가 실시양태에서, 허혈-매개 손상, 예컨대 세포독성 부종 또는 중추 신경계 조직 무산소혈증이 예방될 수 있다. 각 실시양태에서, 중추 신경계 세포는 척수 세포 또는 뇌 세포일 수 있다.
또 다른 측면은 중추 신경계 허혈성 상태를 치료하기 위해 대상체에게 시르투인-조정 화합물을 투여하는 것을 포괄한다. 다수의 중추 신경계 허혈성 상태는 본원에 기재된 시르투인-조정 화합물에 의해 치료될 수 있다. 특정 실시양태에서, 허혈성 상태는 임의의 유형의 허혈성 중추 신경계 손상, 예컨대 아폽토시스성 또는 괴사성 세포 사멸, 세포독성 부종 또는 중추 신경계 조직 무산소증을 일으키는 졸중이다. 졸중은 뇌의 임의의 영역에 충격을 주거나, 또는 졸중의 발생을 일으키는 것으로 통상적으로 공지된 임의의 병인에 의해 유발될 수 있다. 이러한 실시양태의 한 대안에서, 졸중은 뇌간 졸중이다. 이러한 실시양태의 또 다른 대안에서, 졸중은 소뇌 졸중이다. 또 다른 실시양태에서, 졸중은 색전성 졸중이다. 또 다른 대안에서, 졸중은 출혈성 졸중일 수 있다. 추가 실시양태에서, 졸중은 혈전성 졸중이다.
또 다른 측면에서, 시르투인-조정 화합물은 중추 신경계 허혈성 상태 후 허혈성 코어의 경색 크기를 감소시키기 위해 투여될 수 있다. 더욱이, 시르투인-조정 화합물은 또한 유익하게는 중추 신경계 허혈성 상태 후 허혈성 반음영 또는 전이 구역의 크기를 감소시키기 위해 투여될 수 있다.
특정 실시양태에서, 조합 약물 요법은 신경변성 장애 또는 이들 상태와 연관된 속발성 상태의 치료 또는 예방을 위한 약물 또는 화합물을 포함할 수 있다. 따라서, 조합 약물 요법은 1종 이상의 시르투인 활성화제 및 1종 이상의 항신경변성제를 포함할 수 있다.
혈액 응고 장애
다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 혈액 응고 장애 (또는 지혈 장애)를 치료 또는 예방하는데 사용될 수 있다. 교환가능하게 본원에 사용된 용어 "지혈", "혈액 응고" 및 "혈병 형성"은 혈관수축 및 응고의 생리적 특성을 포함한 출혈의 제어를 지칭한다. 혈액 응고는 손상, 염증, 질환, 선천성 결함, 기능장애 또는 다른 파괴 후 포유동물 순환의 완전성을 유지하는 것을 돕는다. 추가로, 혈병의 형성은 상해의 경우에 출혈을 제한할 뿐만 아니라 (지혈), 중요 동맥 또는 정맥의 폐쇄에 의해 아테롬성동맥경화성 질환의 맥락에서 심각한 기관 손상 및 사망으로 이어질 수 있다. 따라서, 혈전증은 잘못된 시간 및 장소에서의 혈병 형성이다.
따라서, 본 발명은 혈액 응고 장애, 예컨대 심근경색, 졸중, 말초 동맥 질환에 의한 사지의 손실 또는 폐 색전증을 예방 또는 치료하기 위해 혈병의 형성을 억제하는 것을 목표로 하는 항응고 및 항혈전 치료를 제공한다.
상호교환가능하게 본원에 사용된 "지혈을 조정하는 또는 그의 조정" 및 "지혈을 조절하는 또는 그의 조절"은 지혈의 유도 (예를 들어, 자극 또는 증가), 뿐만 아니라 지혈의 억제 (예를 들어, 감소 또는 저하)를 포함한다.
한 측면에서, 본 발명은 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 투여함으로써, 대상체에서 지혈을 감소 또는 억제하는 방법을 제공한다. 본원에 개시된 조성물 및 방법 또는 용도는 혈전성 장애의 치료 또는 예방에 유용하다. 본원에 사용된 용어 "혈전성 장애"는 과도하거나 또는 원치 않는 응고 또는 지혈 활성을 특징으로 하는 임의의 장애 또는 상태, 또는 응고항진 상태를 포함한다. 혈전성 장애는 혈소판 부착 및 혈전 형성을 수반하는 질환 또는 장애를 포함하며, 혈전의 형성에 대한 증가된 성향, 예를 들어 증가된 수의 혈전, 이른 연령에서의 혈전증, 혈전증에 대한 가족성 경향, 및 비통상적 부위에서의 혈전증으로서 나타날 수 있다.
또 다른 실시양태에서, 조합 약물 요법은 혈액 응고 장애 또는 이들 상태와 연관된 속발성 상태의 치료 또는 예방을 위한 약물 또는 화합물을 포함할 수 있다. 따라서, 조합 약물 요법은 시르투인 단백질의 수준 및/또는 활성을 증가시키는 1종 이상의 시르투인-조정 화합물 및 1종 이상의 항응고제 또는 항혈전제를 포함할 수 있다.
체중 제어
또 다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 대상체에서 체중 증가 또는 비만을 치료 또는 예방하는데 사용될 수 있다. 예를 들어, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은, 예를 들어 유전성 비만, 식이성 비만, 호르몬 관련 비만, 의약의 투여와 관련된 비만을 치료 또는 예방하거나, 대상체의 체중을 감소시키거나, 또는 대상체에서의 체중 증가를 감소 또는 예방하는데 사용될 수 있다. 이러한 치료를 필요로 하는 대상체는 비만이거나, 비만이 될 가능성이 있거나, 과체중이거나, 또는 과체중이 될 가능성이 있는 대상체일 수 있다. 비만 또는 과체중이 될 가능성이 있는 대상체는, 예를 들어 가족력, 유전, 식이, 활동 수준, 의약 섭취, 또는 그의 다양한 조합을 기준으로 하여 확인될 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 대상체에서 체중 감소를 촉진함으로써 치료 또는 예방될 수 있는 다양한 다른 질환 및 상태를 앓고 있는 대상체에게 투여될 수 있다. 이러한 질환은, 예를 들어 높은 혈압, 고혈압, 높은 혈액 콜레스테롤, 이상지혈증, 제2형 당뇨병, 인슐린 저항성, 글루코스 불내성, 고인슐린혈증, 관상동맥 심장 질환, 협심증, 울혈성 심부전, 졸중, 담석, 담낭염 및 담석증, 통풍, 골관절염, 폐쇄성 수면 무호흡 및 호흡기 문제, 일부 유형의 암 (예컨대 자궁내막암, 유방암, 전립선암 및 결장암), 임신 합병증, 불량한 여성 생식 건강 (예컨대 월경 불순, 불임, 불규칙 배란), 방광 제어 문제 (예컨대 복압성 요실금); 요산 신석증; 심리 장애 (예컨대 우울증, 섭식 장애, 왜곡된 신체상 및 낮은 자존감)를 포함한다. 최종적으로, AIDS를 갖는 환자에서는 AIDS에 대한 조합 요법에 반응하여 지방이영양증 또는 인슐린 저항성이 발생할 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 시험관내이든지 또는 생체내이든지 간에 지방생성 또는 지방 세포 분화를 억제하는데 사용될 수 있다. 이러한 방법 또는 용도는 비만을 치료 또는 예방하는데 사용될 수 있다.
다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 식욕을 감소시키고/거나 포만감을 증가시켜 체중 감소 또는 체중 증가의 회피를 유발하는데 사용될 수 있다. 이러한 치료를 필요로 하는 대상체는 과체중 또는 비만인 대상체, 또는 과체중 또는 비만이 될 가능성이 있는 대상체일 수 있다. 방법은 대상체에게 용량을, 예를 들어 환제 형태로 매일, 또는 격일, 또는 1주 1회 투여하는 것을 포함할 수 있다. 용량은 "식욕 감소 용량"일 수 있다.
예시적 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 체중 증가 또는 비만을 치료 또는 예방하기 위한 조합 요법으로서 투여될 수 있다. 예를 들어, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 1종 이상의 시르투인-조정 화합물은 1종 이상의 항비만제와 조합되어 투여될 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 약물-유발 체중 증가를 감소시키기 위해 투여될 수 있다. 예를 들어, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 식욕을 자극하거나, 또는 체중 증가, 특히 수분 저류 이외의 요인으로 인한 체중 증가를 유발할 수 있는 의약과의 조합 요법으로서 투여될 수 있다.
대사 장애/당뇨병
또 다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 대사 장애, 예컨대 인슐린-저항성, 당뇨병전기 상태, 제II형 당뇨병, 및/또는 그의 합병증을 치료 또는 예방하는데 사용될 수 있다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물의 투여는 대상체에서 인슐린 감수성을 증가시키고/거나 인슐린 수준을 감소시킬 수 있다. 이러한 치료를 필요로 하는 대상체는 인슐린 저항성 또는 제II형 당뇨병의 다른 전구 증상을 갖거나, 제II형 당뇨병을 갖거나, 또는 이들 상태 중 임의의 것이 발생할 가능성이 있는 대상체일 수 있다. 예를 들어, 대상체는 인슐린 저항성을 가지며, 예를 들어 높은 순환 수준의 인슐린 및/또는 연관 상태, 예컨대 고지혈증, 이상지방형성증, 고콜레스테롤혈증, 글루코스 내성 장애, 높은 혈액 글루코스 당 수준, 증후군 X의 다른 징후, 고혈압, 아테롬성동맥경화증 및 지방이영양증을 갖는 대상체일 수 있다.
예시적 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 대사 장애를 치료 또는 예방하기 위한 조합 요법으로서 투여될 수 있다. 예를 들어, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 1종 이상의 시르투인-조정 화합물은 1종 이상의 항당뇨병제와 조합되어 투여될 수 있다.
염증성 질환
다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 염증과 연관된 질환 또는 장애를 치료 또는 예방하는데 사용될 수 있다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 염증의 발병 전에, 염증의 개시 시에, 또는 그 후에 투여될 수 있다. 예방적으로 사용되는 경우에, 화합물은 바람직하게는 임의의 염증 반응 또는 증상에 선행하도록 제공된다. 화합물의 투여는 염증 반응 또는 증상을 예방 또는 감쇠시킬 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 천식, 기관지염, 폐 섬유증, 알레르기성 비염, 산소 독성, 기종, 만성 기관지염, 급성 호흡 곤란 증후군, 및 임의의 만성 폐쇄성 폐 질환 (COPD)을 포함한 알레르기 및 호흡기 상태를 치료 또는 예방하는데 사용될 수 있다. 화합물은 B형 간염 및 C형 간염을 포함한 만성 간염 감염을 치료하는데 사용될 수 있다.
추가로, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 자가면역 질환, 및/또는 자가면역 질환과 연관된 염증, 예컨대 류마티스 관절염, 건선성 관절염 및 강직성 척추염을 포함한 관절염, 뿐만 아니라 기관-조직 자가면역 질환 (예를 들어, 레이노 증후군), 궤양성 결장염, 크론병, 구강 점막염, 경피증, 중증 근무력증, 이식 거부, 내독소 쇼크, 패혈증, 건선, 습진, 피부염, 다발성 경화증, 자가면역 갑상선염, 포도막염, 전신 홍반성 루푸스, 애디슨병, 자가면역 다선성 질환 (자가면역 다선성 증후군으로도 공지됨), 및 그레이브스병을 치료하는데 사용될 수 있다.
특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 1종 이상의 시르투인-조정 화합물은 단독으로 또는 염증을 치료 또는 예방하기에 유용한 다른 화합물과 조합되어 취해질 수 있다.
홍조
또 다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 장애의 증상인 홍조 및/또는 안면 홍조의 발생률 또는 중증도를 감소시키는데 사용될 수 있다. 예를 들어, 본 발명의 방법은 암 환자에서 홍조 및/또는 안면 홍조의 발생률 또는 중증도를 감소시키기 위해, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물을 단독으로 또는 다른 작용제와 조합하여 사용하는 것을 포함한다. 다른 실시양태에서, 방법은 폐경기 및 폐경후 여성에서 홍조 및/또는 안면 홍조의 발생률 또는 중증도를 감소시키기 위한, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물의 용도를 제공한다.
또 다른 측면에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또 다른 약물 요법의 부작용인 홍조 및/또는 안면 홍조, 예를 들어 약물-유발 홍조의 발생률 또는 중증도를 감소시키기 위한 요법으로서 사용될 수 있다. 특정 실시양태에서, 약물-유발 홍조를 치료 및/또는 예방하는 방법은 그를 필요로 하는 환자에게 적어도 1종의 홍조 유발 화합물, 및 시르투인 단백질의 수준 및/또는 활성을 증가시키는 적어도 1종의 시르투인-조정 화합물을 포함하는 제제를 투여하는 것을 포함한다. 다른 실시양태에서, 약물 유발 홍조를 치료하는 방법은 홍조를 유발하는 1종 이상의 화합물 및 1종 이상의 시르투인-조정 화합물을 개별적으로 투여하는 것을 포함하며, 예를 들어 여기서 시르투인-조정 화합물 및 홍조 유발제는 동일한 조성물로 제제화된 것은 아니다. 개별 제제를 사용하는 경우에, 시르투인-조정 화합물은 (1) 홍조 유발제의 투여와 동시에, (2) 홍조 유발제와 간헐적으로, (3) 홍조 유발제의 투여에 대해 시차를 두고, (4) 홍조 유발제의 투여 전에, (5) 홍조 유발제의 투여 후에, 및 (6) 그의 다양한 조합으로 투여될 수 있다. 예시적인 홍조 유발제는, 예를 들어 니아신, 랄록시펜, 항우울제, 항정신병제, 화학요법제, 칼슘 채널 차단제, 및 항생제를 포함한다.
특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 혈관확장제 또는 항고지혈증제 (항콜레스테롤혈증제 및 친지방제 포함)의 홍조 부작용을 감소시키는데 사용될 수 있다. 예시적 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 니아신의 투여와 연관된 홍조를 감소시키는데 사용될 수 있다.
또 다른 실시양태에서, 본 발명은 감소된 홍조 부작용을 갖는 고지혈증을 치료 및/또는 예방하는 방법을 제공한다. 또 다른 대표적 실시양태에서, 방법은 랄록시펜의 홍조 부작용을 감소시키기 위한, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물의 용도를 수반한다. 또 다른 대표적 실시양태에서, 방법은 항우울제 또는 항정신병제의 홍조 부작용을 감소시키기 위한, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물의 용도를 수반한다. 예를 들어, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 세로토닌 재흡수 억제제 또는 5HT2 수용체 길항제와 함께 사용 (개별적으로 또는 함께 투여)될 수 있다.
특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 홍조를 감소시키기 위해 세로토닌 재흡수 억제제 (SRI)로의 치료의 일부로서 사용될 수 있다. 또 다른 대표적 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 화학요법제, 예컨대 시클로포스파미드 및 타목시펜의 홍조 부작용을 감소시키는데 사용될 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 칼슘 채널 차단제, 예컨대 암로디핀의 홍조 부작용을 감소시키는데 사용될 수 있다.
또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 항생제의 홍조 부작용을 감소시키는데 사용될 수 있다. 예를 들어, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 레보플록사신과 조합되어 사용될 수 있다.
안구 장애
본 발명의 한 측면은 환자에게 치료 투여량의 본원에 개시된 화합물 또는 그의 제약상 허용되는 염, 전구약물 또는 대사 유도체로부터 선택된 시르투인 조정제를 투여함으로써, 시각 장애를 억제, 감소 또는 달리 치료하는 방법이다.
본 발명의 특정 측면에서, 시각 장애는 시신경 또는 중추 신경계에 대한 손상에 의해 유발된다. 특정한 실시양태에서, 시신경 손상은 높은 안내압, 예컨대 녹내장에 의해 생성된 것에 의해 유발된다. 다른 특정한 실시양태에서, 시신경 손상은 시신경염에서와 같이 종종 감염 또는 면역 (예를 들어, 자가면역) 반응과 연관된 신경의 팽윤에 의해 유발된다.
본 발명의 특정 측면에서, 시각 장애는 망막 손상에 의해 유발된다. 특정한 실시양태에서, 망막 손상은 눈으로의 혈류에서의 장애 (예를 들어, 동맥경화증, 혈관염)에 의해 유발된다. 특정한 실시양태에서, 망막 손상은 황반의 파괴 (예를 들어, 삼출성 또는 비-삼출성 황반 변성)에 의해 유발된다.
예시적인 망막 질환은 삼출성 연령 관련 황반 변성, 비삼출성 연령 관련 황반 변성, 망막 전자 인공삽입물 및 RPE 이식 연령 관련 황반 변성, 급성 다초점성 판상 색소 상피병증, 급성 망막 괴사, 베스트병, 분지 망막 동맥 폐쇄, 분지 망막 정맥 폐쇄, 암 연관 및 관련 자가면역 망막병증, 중심 망막 동맥 폐쇄, 중심 망막 정맥 폐쇄, 중심 장액성 맥락망막병증, 일스병, 황반전막, 격자 변성, 대혈관류, 당뇨병성 황반 부종, 어바인-가스 황반 부종, 황반 원공, 망막하 신생혈관막, 미만성 단안 아급성 시신경망막염, 비인공수정체성 낭포양 황반 부종, 추정 안구 히스토플라스마 증후군, 삼출성 망막 박리, 수술후 망막 박리, 증식성 망막 박리, 열공성 망막 박리, 견인성 망막 박리, 색소성 망막염, CMV 망막염, 망막모세포종, 미숙아 망막병증, 산탄 망막병증, 배경 당뇨병성 망막병증, 증식성 당뇨병성 망막병증, 혈색소병증성 망막병증, 푸르처 망막병증, 발살바 망막병증, 소아 망막층간분리, 노인성 망막층간분리, 터슨 증후군 및 백반 증후군을 포함한다.
다른 예시적인 질환은 안구 박테리아 감염 (예를 들어, 결막염, 각막염, 결핵, 매독, 임질), 바이러스 감염 (예를 들어, 안구 단순 포진 바이러스, 수두 대상포진 바이러스, 시토메갈로바이러스 망막염, 인간 면역결핍 바이러스 (HIV)) 뿐만 아니라 HIV 또는 다른 HIV-연관 및 다른 면역결핍-연관 안구 질환에 대해 속발성인 진행성 외부 망막 괴사를 포함한다. 추가로, 안구 질환은 진균 감염 (예를 들어, 칸디다 맥락막염, 히스토플라스마증), 원충 감염 (예를 들어, 톡소플라스마증) 및 다른 것 예컨대 안구 톡소카라증 및 사르코이드증을 포함한다.
본 발명의 한 측면은 시각 장애의 치료를 필요로 하는 대상체에게 치료 투여량의 본원에 개시된 시르투인 조정제를 투여함으로써, 화학요법 약물 (예를 들어, 신경독성 약물, 또는 안내압을 상승시키는 약물, 예컨대 스테로이드)로의 치료를 겪는 대상체에서 시각 장애를 억제, 감소 또는 치료하는 방법이다.
본 발명의 또 다른 측면은 시각 장애의 치료를 필요로 하는 대상체에게 치료 투여량의 본원에 개시된 시르투인 조정제를 투여함으로써, 안구 수술 또는 복와위에서 수행되는 다른 수술, 예컨대 척수 수술을 포함한 수술을 겪는 대상체에서 시각 장애를 억제, 감소 또는 치료하는 방법이다. 안구 수술은 백내장, 홍채절개 및 수정체 대체를 포함한다.
본 발명의 또 다른 측면은 백내장, 안구 건조, 연령-관련 황반 변성 (AMD), 망막 손상 등을 포함한 연령 관련 안구 질환의 치료를 필요로 하는 대상체에게 치료 투여량의 본원에 개시된 시르투인 조정제를 투여하는 것에 의한 상기 질환의 억제 및 예방적 치료를 포함한 치료이다.
본 발명의 또 다른 측면은 스트레스, 화학적 손상 또는 방사선에 의해 유발된 눈에 대한 손상의 치료를 필요로 하는 대상체에게 치료 투여량의 본원에 개시된 시르투인 조정제를 투여함으로써, 상기 손상의 예방 또는 치료이다. 눈에 대한 방사선 또는 전자기 손상은 CRT, 또는 태양광 또는 UV에 대한 노출에 의해 유발된 것을 포함할 수 있다.
특정 실시양태에서, 조합 약물 요법은 안구 장애 또는 이들 상태와 연관된 속발성 상태의 치료 또는 예방을 위한 약물 또는 화합물을 포함할 수 있다. 따라서, 조합 약물 요법은 1종 이상의 시르투인 활성화제, 및 안구 장애의 치료를 위한 1종 이상의 치료제를 포함할 수 있다.
특정 실시양태에서, 시르투인 조정제는 안내압을 감소시키기 위한 요법과 함께 투여될 수 있다. 또 다른 실시양태에서, 시르투인 조정제는 녹내장을 치료 및/또는 예방하기 위한 요법과 함께 투여될 수 있다. 또 다른 실시양태에서, 시르투인 조정제는 시신경염을 치료 및/또는 예방하기 위한 요법과 함께 투여될 수 있다. 특정 실시양태에서, 시르투인 조정제는 CMV 망막병증을 치료 및/또는 예방하기 위한 요법과 함께 투여될 수 있다. 또 다른 실시양태에서, 시르투인 조정제는 다발성 경화증을 치료 및/또는 예방하기 위한 요법과 함께 투여될 수 있다.
미토콘드리아-연관 질환 및 장애
특정 실시양태에서, 본 발명은 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 치료하는 방법 또는 그를 위한 용도를 제공한다. 방법 또는 용도는 치료 유효량의 시르투인-조정 화합물을 그를 필요로 하는 대상체에게 투여하는 것을 수반한다. 증가된 미토콘드리아 활성은 미토콘드리아의 전체 수 (예를 들어, 미토콘드리아 질량)를 유지하면서 미토콘드리아 활성을 증가시키는 것, 미토콘드리아의 수를 증가시켜 미토콘드리아 활성을 증가시키는 것 (예를 들어, 미토콘드리아 생물발생을 자극함으로써), 또는 그의 조합을 지칭한다. 특정 실시양태에서, 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 및 장애는 미토콘드리아 기능장애와 연관된 질환 또는 장애를 포함한다.
특정 실시양태에서, 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 치료하는 방법 또는 그를 위한 용도는 미토콘드리아 기능장애를 앓고 있는 대상체를 확인하는 것을 포함할 수 있다. 미토콘드리아 기능장애를 진단하는 방법 또는 그를 위한 용도는 분자 유전학적, 병리학적 및/또는 생화학적 분석을 수반할 수 있다. 미토콘드리아 기능장애와 연관된 질환 및 장애는 미토콘드리아 호흡 사슬 활성의 결핍이 포유동물에서의 이러한 질환 또는 장애의 병리생리상태의 발생에 기여하는 질환 및 장애를 포함한다. 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애는 일반적으로, 예를 들어 자유 라디칼 매개 산화성 상해가 조직 변성으로 이어지는 질환, 세포가 부적절하게 아폽토시스를 겪는 질환, 및 세포가 아폽토시스를 겪는데 실패하는 질환을 포함한다.
특정 실시양태에서, 본 발명은 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애의 치료를 필요로 하는 대상체에게 1종 이상의 시르투인-조정 화합물을 또 다른 치료제, 예컨대 예를 들어 미토콘드리아 기능장애를 치료하기에 유용한 작용제 또는 미토콘드리아 기능장애를 수반하는 질환 또는 장애와 연관된 증상을 감소시키기에 유용한 작용제와 조합하여 투여하는 것을 포함하는, 상기 질환 또는 장애를 치료하는 방법 또는 그를 위한 용도를 제공한다.
예시적 실시양태에서, 본 발명은 대상체에게 치료 유효량의 시르투인-조정 화합물을 투여함으로써, 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 치료하는 방법 또는 그를 위한 용도를 제공한다. 예시적인 질환 또는 장애는, 예를 들어 신경근육 장애 (예를 들어, 프리드라이히 운동실조, 근육 이영양증, 다발성 경화증 등), 뉴런 불안정성 장애 (예를 들어, 발작 장애, 편두통 등), 발달 지체, 신경변성 장애 (예를 들어, 알츠하이머병, 파킨슨병, 근위축성 측삭 경화증 등), 허혈, 신세관성 산증, 연령-관련 신경변성 및 인지 저하, 화학요법 피로, 연령-관련 또는 화학요법-유발 폐경기, 또는 월경 주기 또는 배란의 불순, 미토콘드리아 근병증, 미토콘드리아 손상 (예를 들어, 칼슘 축적, 흥분독성, 산화질소 노출, 저산소증 등), 및 미토콘드리아 탈조절을 포함한다.
근육 이영양증은 종종 골격근의 위축 및 심근 기능장애, 예컨대 뒤시엔느 근육 이영양증을 일으키는, 신경근육 구조 및 기능의 열화를 수반하는 질환의 패밀리를 지칭한다. 특정 실시양태에서, 시르투인-조정 화합물은 근육 이영양증을 갖는 환자에서 근육 기능 능력의 저하 속도를 감소시키고, 근육 기능 상태를 개선시키는데 사용될 수 있다.
특정 실시양태에서, 시르투인-조정 화합물은 미토콘드리아 근병증 치료에 유용할 수 있다. 미토콘드리아 근병증은 경증의 천천히 진행되는 외안근의 약화로부터 중증의 치명적 영아 근병증 및 다계통 뇌근병증까지 이른다. 일부 증후군이 정의된 바 있으며, 일부는 그들 사이에서 중복되어 있다. 근육에 이환되는 확립된 증후군은 진행성 외안근마비, 컨스-세이어 증후군 (안근마비, 색소성 망막병증, 심장 전도 결손, 소뇌 운동실조, 및 감각신경성 난청 동반), MELAS 증후군 (미토콘드리아 뇌근병증, 락트산 산증, 및 졸중-유사 에피소드), MERFF 증후군 (근대간성 간질 및 불균일 적색 근섬유), 지대 분포 약화, 및 영아 근병증 (양성 또는 중증 및 치명적)을 포함한다.
특정 실시양태에서, 시르투인-조정 화합물은 미토콘드리아에 대한 독성 손상, 예컨대 칼슘 축적, 흥분독성, 산화질소 노출, 약물 유발 독성 손상 또는 저산소증으로 인한 독성 손상을 앓고 있는 환자를 치료하기에 유용할 수 있다.
특정 실시양태에서, 시르투인-조정 화합물은 미토콘드리아 탈조절과 연관된 질환 또는 장애를 치료하기에 유용할 수 있다.
근육 수행능
다른 실시양태에서, 본 발명은 치료 유효량의 시르투인-조정 화합물을 투여함으로써, 근육 수행능을 증진시키는 방법 또는 그를 위한 용도를 제공한다. 예를 들어, 시르투인-조정 화합물은 신체 지구력 (예를 들어, 신체적 과제, 예컨대 운동, 육체 노동, 스포츠 활동 등을 수행하는 능력)을 개선시키고/거나, 신체적 피로를 억제 또는 지연시키고/거나, 혈액 산소 수준을 증진시키고/거나, 건강한 개체에서 에너지를 증진시키고/거나, 작업 용량 및 지구력을 증진시키고/거나, 근육 피로를 감소시키고/거나, 스트레스를 감소시키고/거나, 심장 및 심혈관 기능을 증진시키고/거나, 성적 능력을 개선시키고/거나, 근육 ATP 수준을 증가시키고/거나, 혈액 중 락트산을 감소시키기에 유용할 수 있다. 특정 실시양태에서, 방법 또는 용도는 미토콘드리아 활성을 증가시키고/거나, 미토콘드리아 생물발생을 증가시키고/거나, 미토콘드리아 질량을 증가시키는 양의 시르투인-조정 화합물을 투여하는 것을 수반한다.
스포츠 수행능은 스포츠 활동에 참여하는 경우에 운동선수의 근육이 수행하는 능력을 지칭한다. 증진된 스포츠 수행능, 강도, 속도 및 지구력은 근육 수축 강도의 증가, 근육 수축 폭의 증가, 자극과 수축 사이의 근육 반응 시간의 단축에 의해 측정된다. 운동선수는 임의의 수준의 스포츠에 참여하며 그의 수행능에 있어서 개선된 수준의 강도, 속도 및 지구력을 달성하는 것을 추구하는 개체, 예컨대 예를 들어 보디 빌더, 사이클선수, 장거리 달리기 선수, 단거리 달리기 선수 등을 지칭한다. 증진된 스포츠 수행능은 근육 피로를 극복하는 능력, 더 장기간 동안 활동을 유지하는 능력, 및 더 효과적인 활동을 하는 것에 의해 나타난다.
운동선수 근육 수행능의 장에서, 장기간 동안 더 높은 수준의 저항성으로 경쟁 또는 훈련을 허용하는 상태를 생성하는 것이 바람직하다.
본 발명의 방법 또는 용도는 급성 근육감소증, 예를 들어 화상, 침상 안정, 사지 고정화, 또는 주요 흉부, 복부 및/또는 정형외과 수술과 연관된 근육 위축 및/또는 악액질을 포함한 근육 관련 병리학적 상태의 치료에 또한 효과적일 것으로 고려된다.
특정 실시양태에서, 본 발명은 시르투인 조정제를 포함하는 신규 식이성 조성물, 그의 제조 방법, 및 스포츠 수행능의 개선을 위한 상기 조성물의 사용 방법을 제공한다. 따라서, 지구력을 필요로 하는 스포츠 및 반복적인 근육 운동을 필요로 하는 노동을 포함한 광범위하게 정의된 활동에 관련된 이들 사람들을 위해, 신체 지구력을 개선시키고/거나 신체 피로를 억제하는 작용을 갖는 치료 조성물, 식품 및 음료가 제공된다. 이러한 식이성 조성물은 전해질, 카페인, 비타민, 탄수화물 등을 추가로 포함할 수 있다.
다른 용도
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 바이러스 감염 (예컨대 인플루엔자, 포진 또는 유두종 바이러스에 의한 감염)을 치료 또는 예방하는데 사용되거나 또는 항진균제로서 사용될 수 있다. 특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 바이러스성 질환의 치료를 위한 또 다른 치료제와의 조합 약물 요법의 일부로서 투여될 수 있다. 또 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또 다른 항진균제와의 조합 약물 요법의 일부로서 투여될 수 있다.
본원에 기재된 바와 같이 치료될 수 있는 대상체는 진핵생물, 예컨대 포유동물, 예를 들어 인간, 양, 소, 말, 돼지, 개, 고양이, 비-인간 영장류, 마우스 및 래트를 포함한다. 치료될 수 있는 세포는, 예를 들어 상기 기재된 대상체로부터의 진핵 세포, 또는 식물 세포, 효모 세포, 및 원핵 세포, 예를 들어 박테리아 세포를 포함한다. 예를 들어, 조정 화합물은 사육 조건을 더 장기간 견디는 능력을 개선시키기 위해 사육 동물에게 투여될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 식물에서 수명, 스트레스 저항성 및 아폽토시스에 대한 저항성을 증가시키는데 사용될 수 있다. 특정 실시양태에서, 화합물은, 예를 들어 주기적 방식으로 식물에, 또는 진균에 적용된다. 또 다른 실시양태에서, 식물은 화합물을 생산하도록 유전자 변형된다. 또 다른 실시양태에서, 식물 및 과일은 선박운송 동안의 손상에 대한 저항성을 증가시키기 위해 수확 및 선박운송 전에 화합물로 처리된다. 식물 종자는 또한, 예를 들어 이들을 보존하기 위해 본원에 기재된 화합물과 접촉될 수 있다.
다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 효모 세포에서 수명을 조정하는데 사용될 수 있다. 효모 세포의 수명을 연장시키는 것이 바람직할 수 있는 상황은, 효모가 사용되는 임의의 과정, 예들 들어 맥주, 요구르트, 및 제빵 물품, 예를 들어 빵의 제조를 포함한다. 연장된 수명을 갖는 효모의 사용은 더 적은 효모를 사용하는 것 또는 효모가 더 장기간 동안 활성을 갖도록 하는 것을 유발할 수 있다. 효모 또는 단백질을 재조합 생산하는데 사용되는 다른 포유동물 세포는 또한 본원에 기재된 바와 같이 처리될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 곤충에서 수명, 스트레스 저항성 및 아폽토시스에 대한 저항성을 증가시키는데 사용될 수 있다. 이러한 실시양태에서, 화합물은 유용한 곤충, 예를 들어 벌 및 식물의 수분에 수반되는 다른 곤충에 적용될 것이다. 구체적 실시양태에서, 화합물은 꿀의 생산에 수반되는 벌에 적용될 것이다. 일반적으로, 본원에 기재된 방법 또는 용도는 상업적 중요성을 가질 수 있는 모든 유기체, 예를 들어 진핵생물에 적용될 수 있다. 예를 들어, 이들은 어류 (수산양식) 및 조류 (예를 들어, 닭 및 가금류)에 적용될 수 있다.
시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물의 더 높은 용량이 또한 발생 동안 침묵화된 유전자의 조절 및 아폽토시스의 조절을 방해함으로써 살충제로서 사용될 수 있다. 이러한 실시양태에서, 화합물은, 화합물이 곤충 유충에 대해서는 생체-이용가능하고 식물에 대해서는 그렇지 않은 것을 보장하는 관련 기술분야에 공지된 방법을 사용하여 식물에 적용될 수 있다.
적어도 생식과 장수명 사이의 연관성의 관점에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 유기체, 예컨대 곤충, 동물 및 미생물의 생식에 영향을 미치기 위해 적용될 수 있다.
추가의 실시양태
한 측면에서, 본 발명은 세포를 화학식 I의 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각과 접촉시키는 단계를 포함하는, 세포에서 시르투인-1 활성을 증가시키는 방법에 관한 것이다.
한 측면에서, 본 발명은 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증의 치료 또는 인슐린 감수성의 증가를 필요로 하는 대상체에게 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각을 투여하는 것을 포함하는, 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증을 치료하거나 또는 인슐린 감수성을 증가시키는 방법에 관한 것이다.
한 측면에서, 본 발명은 대사 기능장애의 치료를 필요로 하는 대상체에게 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각을 투여하는 것을 포함하는, 대사 기능장애를 치료하는 방법에 관한 것이다.
한 측면에서, 본 발명은 감소된 SIRT1 발현 또는 활성으로부터 유발된 질환 또는 장애의 치료를 필요로 하는 대상체에게 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각을 투여하는 것을 포함하는, 상기 질환 또는 장애를 치료하는 방법에 관한 것이다.
한 측면에서, 본 발명은 감소된 SIRT1 발현 또는 활성으로부터 유발된 질환 또는 장애가 노화 또는 스트레스, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환으로부터 선택되나, 이에 제한되지는 않는 것인 방법에 관한 것이다.
한 측면에서, 본 발명은 노화 또는 스트레스와 관련된 질환, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환이 건선, 아토피성 피부염, 여드름, 장미증, 염증성 장 질환, 골다공증, 패혈증, 관절염, COPD, 전신 홍반성 루푸스 및 안부 염증으로부터 선택된 것인 방법에 관한 것이다.
한 측면에서, 본 발명은 노화 또는 스트레스와 관련된 질환, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환이 건선, 아토피성 피부염, 여드름, 장미증, 염증성 장 질환, 골다공증, 패혈증, 관절염, COPD, 전신 홍반성 루푸스 및 안부 염증으로부터 선택된 것인 방법에 관한 것이다.
한 측면에서, 본 발명은 건선의 치료를 필요로 하는 대상체에게 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각을 투여하는 것을 포함하는, 건선을 치료하는 방법에 관한 것이다.
한 측면에서, 본 발명은 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증을 앓고 있거나 또는 그에 걸리기 쉬운 대상체의 치료에 사용하기 위해 또는 대상체에서 인슐린 감수성을 증가시키기 위해, 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각을 투여하는 것에 관한 것이다.
한 측면에서, 본 발명은 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증의 치료에 사용하기 위한 또는 대상체에서 인슐린 감수성을 증가시키기 위한 의약의 제조에서의, 화합물 또는 그의 제약상 허용되는 염 또는 상응하는 제약 조성물 각각을 투여하는 것의 용도에 관한 것이다.
검정
본원에서 고려되는 또 다른 방법 또는 용도는 시르투인을 조정하는 화합물 또는 작용제를 확인하기 위한 스크리닝 방법을 포함한다. 작용제는 핵산, 예컨대 압타머일 수 있다. 검정은 세포 기반 또는 무세포 포맷으로 수행될 수 있다. 예를 들어, 검정은 시르투인을 조정하는 것으로 공지된 작용제에 의해 시르투인이 조정될 수 있는 조건 하에 시르투인을 시험 작용제와 함께 인큐베이션 (또는 접촉)하고, 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 시르투인의 조정 수준을 모니터링 또는 결정하는 것을 포함할 수 있다. 시르투인의 조정 수준은 기질을 탈아세틸화시키는 것에 대한 그의 능력을 결정함으로써 결정될 수 있다. 예시적인 기질은 바이오몰 (펜실베니아주 플리머스 미팅)에서 입수될 수 있는 아세틸화 펩티드이다. 바람직한 기질은 p53의 펩티드, 예컨대 아세틸화 K382를 포함하는 것들을 포함한다. 특히 바람직한 기질은 플루오르 드 Lys-SIRT1 (바이오몰), 즉 아세틸화 펩티드 Arg-His-Lys-Lys이다. 다른 기질은 인간 히스톤 H3 및 H4로부터의 펩티드 또는 아세틸화 아미노산이다. 기질은 형광원성일 수 있다. 시르투인은 SIRT1, Sir2, SIRT3, 또는 그의 부분일 수 있다. 예를 들어, 재조합 SIRT1은 바이오몰로부터 입수될 수 있다. 반응은 약 30분 동안 수행되고, 예를 들어 니코틴아미드를 사용하여 중지될 수 있다. HDAC 형광 활성 검정/약물 발견 키트 (AK-500, 바이오몰 리서치 래보러토리즈)가 아세틸화의 수준을 결정하는데 사용될 수 있다. 유사한 검정은 문헌 [Bitterman et al. (2002) J. Biol. Chem. 277:45099]에 기재되어 있다. 검정에서의 시르투인의 조정 수준은 양성 또는 음성 대조군으로서 기능할 수 있는 본원에 기재된 1종 이상의 (개별적으로 또는 동시에) 화합물의 존재 하의 시르투인의 조정 수준과 비교될 수 있다. 검정에 사용하기 위한 시르투인은 전장 시르투인 단백질 또는 그의 부분일 수 있다. 활성화 화합물이 SIRT1의 N-말단과 상호작용하는 것으로 보인다는 것은 본원에 제시된 바 있기 때문에, 검정에 사용하기 위한 단백질은 시르투인의 N-말단 부분, 예를 들어 SIRT1의 대략 아미노산 1-176 또는 1-255; Sir2의 대략 아미노산 1-174 또는 1-252를 포함한다.
특정 실시양태에서, 스크리닝 검정은 (i) 시험 작용제의 부재 하에 시르투인이 기질을 탈아세틸화시키기에 적절한 조건 하에 시르투인을 시험 작용제 및 아세틸화된 기질과 접촉시키고; (ii) 기질의 아세틸화 수준을 결정하는 것을 포함하며, 여기서 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 더 낮은 수준의 기질의 아세틸화는 시험 작용제가 시르투인에 의한 탈아세틸화를 자극한다는 것을 나타내는 반면에, 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 더 높은 수준의 기질의 아세틸화는 시험 작용제가 시르투인에 의한 탈아세틸화를 억제한다는 것을 나타낸다.
또 다른 실시양태에서, 스크리닝 검정은 시르투인-매개 NAD-의존성 탈아세틸화의 2'/3'-O-아세틸-ADP-리보스 산물의 형성을 검출할 수 있다. 이러한 O-아세틸-ADP-리보스 산물은 시르투인 탈아세틸화 반응의 탈아세틸화 펩티드 산물과 등몰량으로 형성된다. 따라서, 스크리닝 검정은 (i) 시험 작용제의 부재 하에 시르투인이 기질을 탈아세틸화시키기에 적절한 조건 하에 시르투인을 시험 작용제 및 아세틸화된 기질과 접촉시키고; (ii) O-아세틸-ADP-리보스 형성의 양을 결정하는 것을 포함하며, 여기서 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 O-아세틸-ADP-리보스 형성에서의 증가는 시험 작용제가 시르투인에 의한 탈아세틸화를 자극한다는 것을 나타내는 반면에, 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 O-아세틸-ADP-리보스 형성에서의 감소는 시험 작용제가 시르투인에 의한 탈아세틸화를 억제한다는 것을 나타낸다.
생체내 시르투인을 조정, 예를 들어 자극하는 작용제를 확인하는 방법 또는 그를 위한 용도는 (i) 시험 작용제의 부재 하에 시르투인이 기질을 탈아세틸화시키기에 적절한 조건 하에 클래스 I 및 클래스 II HDAC 억제제의 존재 하에 세포를 시험 작용제 및 세포에 진입할 수 있는 기질과 접촉시키고; (ii) 기질의 아세틸화 수준을 결정하는 것을 포함할 수 있으며, 여기서 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 기질의 아세틸화의 더 낮은 수준은 시험 작용제가 시르투인에 의한 탈아세틸화를 자극한다는 것을 나타내는 반면에, 시험 작용제의 부재에 비해 시험 작용제의 존재 하의 기질의 아세틸화의 더 높은 수준은 시험 작용제가 시르투인에 의한 탈아세틸화를 억제한다는 것을 나타낸다. 바람직한 기질은 아세틸화 펩티드이며, 이는 또한 바람직하게는 본원에 추가로 기재된 바와 같이 형광원성이다. 방법은 세포를 용해시켜 기질의 아세틸화 수준을 결정하는 것을 추가로 포함할 수 있다. 기질은 약 1μM 내지 약 10mM 범위, 바람직하게는 약 10μM 내지 1mM, 보다 더 바람직하게는 약 100μM 내지 1mM, 예컨대 약 200μM의 농도로 세포에 첨가될 수 있다. 바람직한 기질은 아세틸화 리신, 예를 들어 ε-아세틸 리신 (플루오르 드 Lys, FdL) 또는 플루오르 드 Lys-SIRT1이다. 클래스 I 및 클래스 II HDAC의 바람직한 억제제는 트리코스타틴 A (TSA)이며, 이는 약 0.01 내지 100μM 범위, 바람직하게는 약 0.1 내지 10μM, 예컨대 1μM의 농도로 사용될 수 있다. 세포와 시험 화합물 및 기질과의 인큐베이션은 약 10분 내지 5시간, 바람직하게는 약 1-3시간 동안 수행될 수 있다. TSA는 모든 클래스 I 및 클래스 II HDAC를 억제하고, 그 특정 기질, 예를 들어 플루오르 드 Lys는 SIRT2에 대해 불량한 기질이고 SIRT3-7에 대해서는 보다 더 불량한 기질이기 때문에, 이러한 검정은 생체내 SIRT1의 조정제를 확인하는데 사용될 수 있다.
방법
본 발명은 또한, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않으며, 추가로 건선, 아토피성 피부염, 여드름, 장미증, 염증성 장 질환, 골다공증, 패혈증, 관절염, COPD, 전신 홍반성 루푸스 및 안부 염증으로부터 선택되거나 또는 그를 포함할 수 있으나, 이에 제한되지는 않는 매우 다양한 질환 및 장애의 치료 및/또는 예방에 본원에 정의된 바와 같은 시르투인 조정제 화합물을 사용하는 방법 또는 그를 위한 용도에 관한 것이다.
또 다른 측면에서, 본 발명은 시르투인-조정 화합물, 또는 시르투인-조정 화합물을 포함하는 조성물을 사용하는 방법 또는 그를 위한 용도를 제공한다. 특정 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은, 예를 들어 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 화학요법-유발 신경병증, 허혈성 사건과 연관된 신경병증, 안구 질환 및/또는 장애, 심혈관 질환, 혈액 응고 장애, 염증 및/또는 홍조 등을 포함한 매우 다양한 질환 및 장애를 치료 및/또는 예방하는 것을 포함한 다양한 치료 용도에 사용될 수 있다. 시르투인 단백질의 수준 및/또는 활성을 증가시키는 시르투인-조정 화합물은 또한 대상체에서 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 치료하거나, 근육 수행능을 증진시키거나, 근육 ATP 수준을 증가시키거나, 또는 저산소증 또는 허혈과 연관된 근육 조직 손상을 치료 또는 예방하는데 사용될 수 있다. 다른 실시양태에서, 시르투인 단백질의 수준 및/또는 활성을 감소시키는 시르투인-조정 화합물은, 예를 들어 스트레스에 대한 세포 감수성의 증가, 아폽토시스의 증가, 암의 치료, 식욕의 자극 및/또는 체중 증가의 자극 등을 포함한 다양한 치료 용도에 사용될 수 있다. 하기에 추가로 기재된 바와 같이, 방법 또는 용도는 그를 필요로 하는 대상체에게 제약 유효량의 시르투인-조정 화합물을 투여하는 것을 포함한다.
특정 측면에서, 시르투인-조정 화합물은 단독으로 투여되거나 또는 다른 시르투인-조정 화합물을 포함한 다른 화합물, 또는 다른 치료제와 조합되어 투여될 수 있다.
또 다른 측면에서, 본 발명은 세포를 본 발명의 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염과 접촉시키는 단계를 포함하는, 세포에서 시르투인-1 활성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 세포를 본원에 정의된 바와 같은 본 발명의 제약 조성물과 접촉시키는 단계를 포함하는, 세포에서 시르투인-1 활성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증의 치료 또는 인슐린 감수성의 증가를 필요로 하는 대상체에게 본 발명의 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것)를 투여하는 것을 포함하는, 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증을 치료하거나 또는 인슐린 감수성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증의 치료 또는 인슐린 감수성의 증가를 필요로 하는 대상체에게 본 발명의 제약 조성물을 투여하는 것을 포함하는, 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증을 앓고 있거나 또는 그에 걸리기 쉬운 대상체를 치료하거나 또는 대상체에서 인슐린 감수성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증의 치료 또는 인슐린 감수성의 증가를 필요로 하는 대상체에게 본 발명의 제약 조성물을 투여하는 것을 포함하는, 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증을 치료하거나 또는 인슐린 감수성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 세포를 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염과 접촉시키는 단계를 포함하는, 세포에서 시르투인-1 활성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 세포를 본 발명의 제약 조성물과 접촉시키는 단계를 포함하는, 세포에서 시르투인-1 활성을 증가시키는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 대사 기능장애의 치료를 필요로 하는 대상체에게 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염을 투여하는 것을 포함하는, 대사 기능장애를 치료하는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 대사 기능장애의 치료를 필요로 하는 대상체에게 본 발명의 제약 조성물을 투여하는 것을 포함하는, 대사 기능장애를 치료하는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 감소된 SIRT1 발현 또는 활성으로부터 유발된 질환 또는 장애의 치료를 필요로 하는 대상체에게 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염을 투여하는 것을 포함하는, 상기 질환 또는 장애를 치료하는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 감소된 SIRT1 발현 또는 활성으로부터 유발된 질환 또는 장애가 노화 또는 스트레스, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환으로부터 선택되나, 이에 제한되지는 않는 것인 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 노화 또는 스트레스와 관련된 질환, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환이 건선, 아토피성 피부염, 여드름, 장미증, 염증성 장 질환, 골다공증, 패혈증, 관절염, COPD, 전신 홍반성 루푸스 및 안부 염증으로부터 선택된 것인 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 건선의 치료를 필요로 하는 대상체에게 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염을 투여하는 것을 포함하는, 건선을 치료하는 방법에 관한 것이다.
또 다른 측면에서, 본 발명은 건선의 치료를 필요로 하는 대상체에게 본 발명의 제약 조성물을 투여하는 것을 포함하는, 건선을 치료하는 방법에 관한 것이다.
제약 조성물 및 제제
일반적으로, 본 발명은 화학식 I 내지 IV의 치환된 가교된 우레아 유사체 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염, 상응하는 제약 조성물, 제조 방법, 및 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하기에 유용한 시르투인 조정제로서의, 단독으로의 또는 다른 치료제와 조합된 이러한 화합물의 용도에 관한 것이다.
특히, 본 발명은 화학식 I 내지 IV의 신규 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염 및 화학식 I 내지 IV의 화합물을 포함하는 제약 조성물 각각에 관한 것이다.
또 다른 측면에서, 본 발명은 제약상 허용되는 담체 또는 희석제, 및 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염을 포함하는 제약 조성물에 관한 것이다.
또 다른 측면에서, 본 발명은 추가의 활성제를 추가로 포함하는, 본 발명의 제약 조성물에 관한 것이다.
또 다른 측면에서, 본 발명은 화학식 I 내지 IV의 화합물, 그의 상응하는 유사체 또는 유도체 (즉, R2 위치에서 수소 치환을 갖는 것) 또는 그의 제약상 허용되는 염, 및 적어도 1종의 제약상 허용되는 담체를 포함하는 제약 조성물에 관한 것이다.
본원에 기재된 화합물은 1종 이상의 생리학상 또는 제약상 허용되는 담체 또는 부형제를 사용하여 통상적인 방식으로 제제화될 수 있다. 예를 들어, 화합물 및 그의 제약상 허용되는 염 및 용매화물은, 예를 들어 주사 (예를 들어, SubQ, IM, IP), 흡입 또는 취입 (구강 또는 코를 통함)에 의한 투여, 또는 경구, 협측, 설하, 경피, 비강, 비경구 또는 직장 투여를 위해 제제화될 수 있다. 특정 실시양태에서, 화합물은 표적 세포가 존재하는 부위, 즉 특정한 조직, 기관 또는 체액 (예를 들어, 혈액, 뇌척수액 등)에 국부로 투여될 수 있다.
화합물은 전신 및 국소 또는 국부 투여를 포함한 다양한 투여 방식을 위해 제제화될 수 있다. 기술 및 제제는 일반적으로 문헌 [Remington's Pharmaceutical Sciences, Meade Publishing Co., Easton, PA]에서 찾아볼 수 있다. 비경구 투여를 위해, 근육내, 정맥내, 복강내 및 피하를 포함한 주사가 바람직하다. 주사를 위해, 화합물은 바람직하게는 생리학상 상용성인 완충제 예컨대 행크 용액 또는 링거액 중 액체 용액으로 제제화될 수 있다. 추가로, 화합물은 고체 형태로 제제화되고, 사용 직전에 재용해 또는 현탁될 수 있다. 동결건조 형태가 또한 포함된다.
경구 투여를 위해, 제약 조성물은, 예를 들어 제약상 허용되는 부형제 예컨대 결합제 (예를 들어, 예비젤라틴화 옥수수 전분, 폴리비닐피롤리돈 또는 히드록시프로필 메틸셀룰로스); 충전제 (예를 들어, 락토스, 미세결정질 셀룰로스 또는 인산수소칼슘); 윤활제 (예를 들어, 스테아르산마그네슘, 활석 또는 실리카); 붕해제 (예를 들어, 감자 전분 또는 소듐 스타치 글리콜레이트); 또는 습윤제 (예를 들어, 소듐 라우릴 술페이트)를 사용하여 통상적인 수단에 의해 제조된 정제, 로젠지 또는 캡슐의 형태를 취할 수 있다. 정제는 관련 기술분야에 널리 공지된 방법에 의해 코팅될 수 있다. 경구 투여를 위한 액체 제제는, 예를 들어 용액, 시럽 또는 현탁액의 형태를 취할 수 있거나, 또는 이들은 사용 전에 물 또는 다른 적합한 비히클로 구성하기 위한 건조 생성물로서 제시될 수 있다. 이러한 액체 제제는 제약상 허용되는 첨가제 예컨대 현탁화제 (예를 들어, 소르비톨 시럽, 셀룰로스 유도체 또는 수소화 식용 지방); 유화제 (예를 들어, 레시틴 또는 아카시아); 비-수성 비히클 (예를 들어, 아몬드 오일, 유성 에스테르, 에틸 알콜 또는 분별 식물성 오일); 및 보존제 (예를 들어, 메틸 또는 프로필-p-히드록시벤조에이트 또는 소르브산)를 사용하여 통상적인 수단에 의해 제조될 수 있다. 제제는 적절한 경우에 완충 염, 향미제, 착색제 및 감미제를 또한 함유할 수 있다. 경구 투여를 위한 제제는 활성 화합물의 제어 방출을 제공하도록 적합하게 제제화될 수 있다.
흡입 (예를 들어, 폐 전달)에 의한 투여를 위해, 화합물은 적합한 추진제, 예를 들어 디클로로디플루오로메탄, 트리클로로플루오로메탄, 디클로로테트라플루오로에탄, 이산화탄소 또는 다른 적합한 기체의 사용과 함께, 가압 팩 또는 네뷸라이저로부터의 에어로졸 스프레이 제공물의 형태로 편리하게 전달될 수 있다. 가압 에어로졸의 경우에, 투여 단위는 계량된 양을 전달하기 위해 밸브를 제공함으로써 결정될 수 있다. 흡입기 또는 취입기에 사용하기 위한, 예를 들어 젤라틴의 캡슐 및 카트리지는 화합물의 분말 믹스 및 적합한 분말 베이스 예컨대 락토스 또는 전분을 함유하도록 제제화될 수 있다.
화합물은 주사, 예를 들어 볼루스 주사 또는 연속 주입에 의한 비경구 투여를 위해 제제화될 수 있다. 주사를 위한 제제는, 예를 들어 첨가된 보존제와 함께, 앰플 또는 다중-용량 용기 내에 단위 투여 형태로 제시될 수 있다. 조성물은 유성 또는 수성 비히클 중 현탁액, 용액 또는 에멀젼과 같은 형태를 취할 수 있으며, 제제화제 예컨대 현탁화제, 안정화제 및/또는 분산제를 함유할 수 있다. 대안적으로, 활성 성분은 사용 전에 적합한 비히클, 예를 들어 멸균 발열원-무함유 물로 구성하기 위한 분말 형태일 수 있다.
화합물은 또한, 예를 들어 통상적인 좌제 베이스 예컨대 코코아 버터 또는 다른 글리세리드를 함유하는 직장 조성물 예컨대 좌제 또는 정체 관장제로 제제화될 수 있다.
상기 기재된 제제 이외에도, 화합물은 또한 데포 제제로서 제제화될 수 있다. 이러한 장기 작용 제제는 이식에 의해 (예를 들어, 피하로 또는 근육내로), 또는 근육내 주사에 의해 투여될 수 있다. 따라서, 예를 들어 화합물은 적합한 중합체 또는 소수성 물질 (예를 들어, 허용되는 오일 중 에멀젼으로서) 또는 이온 교환 수지를 사용하여, 또는 난용성 유도체, 예를 들어 난용성 염으로서 제제화될 수 있다. 제어 방출 제제는 패치를 또한 포함한다.
특정 실시양태에서, 본원에 기재된 화합물은 중추 신경계 (CNS)로의 전달을 위해 제제화될 수 있다 (문헌 [Begley, Pharmacology & Therapeutics 104: 29-45 (2004)]에서 검토됨). CNS로의 약물 전달을 위한 통상적인 접근법은 하기를 포함한다: 신경외과적 전략 (예를 들어, 뇌내 주사 또는 뇌실내 주입); BBB의 내인성 수송 경로 중 1개를 이용하기 위한 시도에서의 작용제의 분자 조작 (예를 들어, 내피 세포 표면 분자에 대한 친화도를 갖는 수송 펩티드를, 그 자체로는 BBB를 횡단할 수 없는 작용제와 조합하여 포함하는 키메라 융합 단백질의 생산); 작용제의 지질 용해도를 증가시키도록 설계된 약리학적 전략 (예를 들어, 지질 또는 콜레스테롤 담체에 대한 수용성 작용제의 접합); 및 고삼투압 파괴에 의한 BBB의 완전성의 일시적 파괴 (경동맥으로의 만니톨 용액의 주입, 또는 생물학적 활성제 예컨대 안지오텐신 펩티드의 사용으로부터 유발됨).
리포솜은 용이하게 주사가능한 추가의 약물 전달 시스템이다. 따라서, 본 발명의 방법에서, 활성 화합물은 또한 리포솜 전달 시스템의 형태로 투여될 수 있다. 리포솜은 관련 기술분야의 통상의 기술자에 의해 널리 공지되어 있다. 리포솜은 다양한 인지질, 예컨대 콜레스테롤, 포스파티딜콜린의 스테아릴아민으로부터 형성될 수 있다. 본 발명의 방법에 사용가능한 리포솜은 소형 단층 소포, 대형 단층 소포 및 다층 소포를 포함하나, 이에 제한되지는 않는 모든 유형의 리포솜을 포괄한다.
본원에 기재된 화합물의 제제, 특히 용액을 제조하기 위한 또 다른 방식은 시클로덱스트린의 사용을 통한 것이다. 시클로덱스트린은 α-, β-, 또는 γ-시클로덱스트린을 의미한다. 시클로덱스트린은 피타 등의 미국 특허 번호 4,727,064에 상세하게 기재되어 있다. 시클로덱스트린은 글루코스의 시클릭 올리고머이며; 이들 화합물은 분자가 시클로덱스트린 분자의 친지성기-추구 캐비티 내로 피팅될 수 있는 임의의 약물과의 포접 복합체를 형성한다.
급속 붕해 또는 용해 투여 형태는 제약 활성제의 급속 흡수, 특히 협측 및 설하 흡수에 유용하다. 신속 용융 투여 형태는 전형적인 고체 투여 형태, 예컨대 캐플릿 및 정제를 삼키기에 어려움이 있는 환자, 예컨대 노령 및 소아과 환자에게 유익하다. 추가로, 신속 용융 투여 형태는 예를 들어 저작성 투여 형태와 연관된 결점을 회피하며, 여기서 활성제가 환자의 구강에 남아있는 시간의 길이는 미각 차폐의 양, 및 환자가 활성제의 인후 껄끄러움을 거부할 수 있는 정도를 결정함에 있어서 중요한 역할을 한다.
제약 조성물 (화장품 제제 포함)은 중량 기준으로 약 0.00001 내지 100% 예컨대 0.001 내지 10% 또는 0.1% 내지 5%의 본원에 기재된 1종 이상의 화합물을 포함할 수 있다. 다른 실시양태에서, 제약 조성물은 하기를 포함한다: (i) 0.05 내지 1000 mg의 본 발명의 화합물 또는 그의 제약상 허용되는 염, 및 (ii) 0.1 내지 2 그램의 1종 이상의 제약상 허용되는 부형제.
일부 실시양태에서, 본원에 기재된 화합물은 국소 약물 투여에 일반적으로 적합한 국소 담체를 함유하고 관련 기술분야에 공지된 임의의 이러한 물질을 포함하는 국소 제제에 혼입된다. 국소 담체는 조성물을 원하는 형태로, 예를 들어 연고, 로션, 크림, 마이크로에멀젼, 겔, 오일, 용액 등으로서 제공하도록 선택될 수 있으며, 자연 발생 또는 합성 기원의 물질로 구성될 수 있다. 선택된 담체는 활성제 또는 국소 제제의 다른 성분에 부정적인 영향을 미치지 않는 것이 바람직하다. 본원에 사용하기에 적합한 국소 담체의 예는 물, 알콜 및 다른 비독성 유기 용매, 글리세린, 미네랄 오일, 실리콘, 석유 젤리, 라놀린, 지방산, 식물성 오일, 파라벤, 왁스 등을 포함한다.
제제는 무색, 무취 연고, 로션, 크림, 마이크로에멀젼 및 겔일 수 있다.
화합물은, 전형적으로 페트롤라툼 또는 다른 석유 유도체를 기재로 하는 일반적으로 반고체 제제인 연고에 혼입될 수 있다. 관련 기술분야의 통상의 기술자에 의해 인지될 바와 같이, 사용되는 특정한 연고 베이스는 최적의 약물 전달을 제공할 것이고, 바람직하게는 다른 바람직한 특징, 예를 들어 연화성 등을 또한 제공할 것들이다. 다른 담체 또는 비히클과 같이, 연고 베이스는 불활성이고 안정하고 비자극성 및 비감작성이어야 한다.
화합물은, 일반적으로 마찰 없이 피부 표면에 적용되는 제제이며 전형적으로 활성제를 포함한 고체 입자가 물 또는 알콜 베이스 중에 존재하는 액체 또는 반액체 제제인 로션에 혼입될 수 있다. 로션은 통상적으로 고체의 현탁액이며, 수중유 형태의 액체 유성 에멀젼을 포함할 수 있다.
화합물은, 일반적으로 수중유 또는 유중수의 점성 액체 또는 반고체 에멀젼인 크림에 혼입될 수 있다. 크림 베이스는 물-세척가능하며, 오일 상, 유화제 및 수성 상을 함유한다. 오일 상은 일반적으로 페트롤라툼 및 지방 알콜 예컨대 세틸 또는 스테아릴 알콜로 구성되고; 수성 상은 반드시는 아니지만 통상적으로 부피에 있어서 오일 상을 초과하고, 일반적으로 함습제를 함유한다. 크림 제제 중 유화제는 상기 문헌 [Remington's]에 설명된 바와 같이, 일반적으로 비이온성, 음이온성, 양이온성 또는 양쪽성 계면활성제이다.
화합물은, 일반적으로 계면활성제 분자의 경계면 필름에 의해 안정화된 2종의 불혼화성 액체, 예컨대 오일 및 물의 열역학적으로 안정하고 등방성으로 투명한 분산액인 마이크로에멀젼에 혼입될 수 있다 (Encyclopedia of Pharmaceutical Technology (New York: Marcel Dekker, 1992), volume 9).
화합물은, 일반적으로 소형 무기 입자로 구성된 현탁액 (2-상 시스템) 또는 담체 액체 전반에 걸쳐 실질적으로 균일하게 분포된 대형 유기 분자 (단일 상 겔)로 이루어진 반고체 시스템인 겔 제제에 혼입될 수 있다. 겔은 통상적으로 수성의 담체 액체를 사용하지만, 알콜 및 오일이 또한 담체 액체로서 사용될 수 있다.
다른 활성제, 예를 들어 다른 항염증제, 진통제, 항미생물제, 항진균제, 항생제, 비타민, 항산화제, 및 안트라닐레이트, 벤조페논 (특히 벤조페논-3), 캄포르 유도체, 신나메이트 (예를 들어, 옥틸 메톡시신나메이트), 디벤조일 메탄 (예를 들어, 부틸 메톡시디벤조일 메탄), p-아미노벤조산 (PABA) 및 그의 유도체, 및 살리실레이트 (예를 들어, 옥틸 살리실레이트)를 포함하나, 이에 제한되지는 않는 선스크린 제제에서 흔히 발견되는 선블록제가 또한 제제에 포함될 수 있다.
특정 국소 제제에서, 활성제는 제제의 대략 0.25 wt.% 내지 75 wt.% 범위, 바람직하게는 제제의 대략 0.25 wt.% 내지 30 wt.% 범위, 보다 바람직하게는 제제의 대략 0.5 wt.% 내지 15 wt.% 범위, 가장 바람직하게는 제제의 대략 1.0 wt.% 내지 10 wt.% 범위의 양으로 존재한다.
눈의 상태는, 예를 들어 화합물의 전신, 국소, 안내 주사에 의해, 또는 화합물을 방출하는 지속 방출 디바이스의 삽입에 의해 치료 또는 예방될 수 있다. 화합물이 예를 들어 전방, 후방, 유리체, 방수, 유리체액, 각막, 홍채/섬모체, 수정체, 맥락막/망막 및 공막과 같은 눈의 각막 및 내부 영역을 관통하는 것을 가능하게 하기에 충분한 시간 동안 화합물이 눈 표면과 접촉하여 유지되도록, 화합물은 제약상 허용되는 안과용 비히클 중에 전달될 수 있다. 제약상 허용되는 안과용 비히클은, 예를 들어 연고, 식물성 오일 또는 캡슐화 물질일 수 있다. 대안적으로, 본 발명의 화합물은 유리체액 및 방수에 직접 주사될 수 있다. 추가의 대안에서, 화합물은 눈의 치료를 위해 전신으로, 예컨대 정맥내 주입 또는 주사에 의해 투여될 수 있다.
본원에 기재된 화합물은 무산소 환경 하에 저장될 수 있다. 예를 들어, 조성물은 경구 투여를 위한 기밀 캡슐, 예컨대 화이자, 인크.(Pfizer, Inc.)로부터의 캡슈겔 내에 제조될 수 있다.
예를 들어 본원에 기재된 바와 같은 화합물로 생체외 처리된 세포는 대상체에게 이식편을 투여하는 방법 또는 그를 위한 용도에 따라 투여될 수 있으며, 이에는 예를 들어 면역억제 약물, 예를 들어 시클로스포린 A의 투여가 동반될 수 있다. 의약 제제에서의 일반적인 원리에 대해서는, 문헌 [Cell Therapy: Stem Cell Transplantation, Gene Therapy, and Cellular Immunotherapy, by G. Morstyn & W. Sheridan eds, Cambridge University Press, 1996; 및 Hematopoietic Stem Cell Therapy, E. D. Ball, J. Lister & P. Law, Churchill Livingstone, 2000]을 참조한다.
화합물의 독성 및 치료 효능은 세포 배양 또는 실험 동물에서의 표준 제약 절차에 의해 결정될 수 있다. LD50은 집단의 50%에 대해 치명적인 용량이다. ED50은 집단의 50%에서 치료상 유효한 용량이다. 독성과 치료 효과 사이의 용량 비 (LD50/ED50)는 치료 지수이다. 큰 치료 지수를 나타내는 화합물이 바람직하다. 독성 부작용을 나타내는 화합물이 사용될 수 있지만, 비감염된 세포에 대한 잠재적 손상을 최소화시키고, 그에 의해 부작용을 감소시키기 위해, 이러한 화합물을 이환 조직의 부위에 표적화하는 전달 시스템을 설계하는 것에 주의를 기울여야 한다.
세포 배양 검정 및 동물 연구로부터 수득되는 데이터는 인간에 사용하기 위한 투여량 범위를 제제화하는데 사용될 수 있다. 이러한 화합물의 투여량은 독성이 거의 또는 전혀 없으면서 ED50을 포함하는 순환 농도 범위 내일 수 있다. 투여량은 사용되는 투여 형태 및 이용되는 투여 경로에 따라 이러한 범위 내에서 달라질 수 있다. 임의의 화합물에 대해, 치료 유효 용량은 초기에 세포 배양 검정으로부터 추정될 수 있다. 용량은 세포 배양으로 결정 시에 IC50 (즉, 증상의 반수-최대 억제를 달성하는 시험 화합물의 농도)을 포함하는 순환 혈장 농도 범위를 달성하도록 동물 모델에서 제제화될 수 있다. 이러한 정보는 인간에서의 유용한 용량을 더 정확하게 결정하는데 사용될 수 있다. 혈장 중 수준은, 예를 들어 고성능 액체 크로마토그래피에 의해 측정될 수 있다.
키트
키트, 예를 들어 치료 목적을 위한 키트, 또는 세포의 수명을 조정하거나 또는 아폽토시스를 조정하기 위한 키트가 또한 본원에 제공된다. 키트는 본원에 기재된 바와 같은 1종 이상의 화합물을, 예를 들어 사전측정된 용량으로 포함할 수 있다. 키트는 세포를 화합물과 접촉시키기 위한 디바이스 및 사용 지침서를 임의로 포함할 수 있다. 디바이스는 화합물을 대상체 (예를 들어, 대상체의 혈관)에 도입하거나 또는 대상체의 피부에 적용하기 위한 시린지, 스텐트 및 다른 디바이스를 포함한다.
또 다른 실시양태에서, 본 발명은 개별 투여 형태이지만 서로 연관된 본 발명의 화합물 및 또 다른 치료제 (조합 요법 및 조합 조성물에 사용되는 동일한 것들)를 포함하는 물질의 조성물을 제공한다. 본원에 사용된 용어 "서로 연관된"은 개별 투여 형태를 동일한 요법의 일부로서 판매 및 투여되도록 의도한 것이 용이하게 명백하도록, 개별 투여 형태가 함께 포장되거나 또는 달리 서로 부착되어 있는 것을 의미한다. 화합물 및 다른 작용제는 바람직하게는 블리스터 팩 또는 다른 다중-챔버 패키지 내에, 또는 사용자에 의해 (예를 들어, 2개의 용기 사이의 절개선 위를 찢음으로써) 분리될 수 있는 연결된 개별 밀봉 용기 (예컨대 호일 파우치 등)로서 함께 포장된다.
또 다른 실시양태에서, 본 발명은 개별 용기 내에 a) 본 발명의 화합물; 및 b) 또 다른 치료제 예컨대 본 명세서의 다른 곳에 기재된 것들을 포함하는 키트를 제공한다.
본 발명의 방법의 실시는 달리 나타내지 않는 한, 관련 기술분야의 기술 내인 세포 생물학, 세포 배양, 분자 생물학, 트랜스제닉 생물학, 미생물학, 재조합 DNA 및 면역학의 통상적인 기술을 사용할 것이다. 이러한 기술은 문헌에 완전히 설명되어 있다. 예를 들어, 문헌 [Molecular Cloning A Laboratory Manual, 2nd Ed., ed. by Sambrook, Fritsch and Maniatis (Cold Spring Harbor Laboratory Press: 1989); DNA Cloning, Volumes I and II (D. N. Glover ed., 1985); Oligonucleotide Synthesis (M. J. Gait ed., 1984); Mullis et al. U.S. Patent No: 4,683,195; Nucleic Acid Hybridization (B. D Hames & S. J. Higgins eds. 1984); Transcription And Translation (B. D. Hames & S. J. Higgins eds. 1984); Culture Of Animal Cells (R. I. Freshney, Alan R. Liss, Inc., 1987); Immobilized Cells And Enzymes (IRL Press, 1986); B. Perbal, A Practical Guide To Molecular Cloning (1984); the treatise, Methods In Enzymology (Academic Press, Inc., N.Y.); Gene Transfer Vectors For Mammalian Cells (J. H. Miller and M. P. Calos eds., 1987, Cold Spring Harbor Laboratory); Methods In Enzymology, Vols. 154 and 155 (Wu et al. eds.), Immunochemical Methods In Cell And Molecular Biology (Mayer and Walker, eds., Academic Press, London, 1987); Handbook Of Experimental Immunology, Volumes I-IV (D. M. Weir and C. C. Blackwell, eds., 1986); Manipulating the Mouse Embryo, (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y., 1986)]을 참조한다. 하기 제시된 실시예는 본 발명의 예시이며, 어떠한 방식으로든 본 발명의 범주를 제한하도록 의도된 것은 아니다.
하기 제시된 실시예는 본 발명의 예시이며, 어떠한 방식으로든 본 발명의 범주를 제한하도록 의도된 것은 아니다.
실시예
하기 제시된 실시예는 본 발명의 예시이며, 어떠한 방식으로든 본 발명의 범주를 제한하도록 의도된 것은 아니며, 오히려 통상의 기술자에게 본 발명의 화합물, 조성물 및 방법 또는 용도를 제조 및 사용하는 것에 대한 지침을 제공하도록 의도된 것이다. 본 발명의 특정한 실시양태가 기재되어 있지만, 통상의 기술자는 본 발명의 취지 및 범주로부터 벗어나지 않으면서 다양한 변화 및 변형이 이루어질 수 있다는 것을 인지할 것이다.
본원에 사용된 바와 같이, 이들 과정, 반응식 및 실시예에 사용된 기호 및 규정은 현대 과학 문헌, 예를 들어 문헌 [Journal of the American Chemical Society 또는 Journal of Biological Chemistry]에 사용된 것들과 일치한다. 달리 나타내지 않는 한, 표준 단일-문자 또는 3-문자 약어가 일반적으로 L-배위인 것으로 추정되는 아미노산 잔기를 지정하는데 사용된다. 달리 나타내지 않는 한, 모든 출발 물질은 상업적 공급업체로부터 입수하고, 추가 정제 없이 사용하였다.
에테르에 대한 모든 언급은 디에틸 에테르에 대한 것이고; 염수는 NaCl의 포화 수용액을 지칭한다. 달리 나타내지 않는 한, 모든 온도는 ℃ (섭씨 온도)로 표현된다. 모든 반응은 달리 나타내지 않는 한, 불활성 분위기 하에 실온에서 수행되고, 모든 용매는 달리 나타내지 않는 한, 가장 높은 입수가능한 순도이다.
사용된 기기
LCMS와 PDA:
워터스 얼라이언스2695-2996/쿼트로마이크로
애질런트-1200/SQD
정제용 LC와 UV 검출기 (정제용 HPLC):
워터스-2545/2998 PDA 및 2487 UV
시마즈 -LC-20AP/20AV-UV
길슨-333,334/115-UV
키랄 HPLC:
워터스 얼라이언스-2695/2998 &2996
SFC 정제 시스템:
타르 - SFC-80
워터스 SFC - 200
NMR (400 MHz):
배리안-400 MHz
1H NMR 도표화는 2014 ACD 랩스 소프트웨어로 작성하였다.
1H NMR (이하 또한 "NMR") 스펙트럼을 배리안-400 MHz 분광계 상에서 기록하였다. 화학적 이동은 백만분율 (ppm, δ 단위)로 표현된다. 커플링 상수는 헤르츠 (Hz) 단위이다. 분할 패턴은 겉보기 다중도를 기재하고, s (단일선), d (이중선), t (삼중선), q (사중선), quint (오중선), m (다중선), br (넓음)로서 나타내어진다.
사용된 LCMS 방법
획득 방법 조건: RND-ABC-6-분
칼럼: 엑스브리지 BEH C18 (50mmx4.6mm, 2.5μm)
이동상: A: 물 중 5mM 중탄산암모늄 (PH-10, 암모니아 함유): ACN
시간 (분) /%ACN: 0/5, 0.5/5, 1/15, 3.3/98, 5.2/98, 5.5/5, 6.0/5
칼럼 온도: 35℃, 유량: 1.3 ml/분
MS 파라미터:
질량 범위: 100-1000
스캔 시간: 0.5초
스캔간 지연: 0.1초
실행 시간: 6.0분
획득 방법 조건: RND-FA-4.5-분
칼럼: 액퀴티 BEH C18 (50mmx2.1mm, 1.7um)
이동상: A: 물 중 0.1% FA; B: ACN 중 0.1% FA
시간 (분) /%B: 0/3, 0.4/3, 3.2/98, 3.8/98, 4.2/3, 4.5/3
칼럼 온도: 35℃, 유량: 0.6mL/분
MS 파라미터:
질량 범위: 100-1000
스캔 시간: 0.5초
스캔간 지연: 0.1초
실행 시간: 4.5분
획득 방법 조건: RND-FA-4.5-분
칼럼: 액퀴티 BEH C18 (50mmx2.1mm, 1.7um)
이동상: A: 물 중 0.1% FA; B: ACN 중 0.1% FA
시간 (분) /%B: 0/3, 0.4/3, 3.2/98, 3.8/98, 4.2/3, 4.5/3
칼럼 온도: 35℃, 유량: 0.6mL/분
MS 파라미터:
질량 범위: 100-1000
프래그멘터: 100
스텝 크기: 0.1
실행 시간: 4.5분
획득 방법 조건: RND-ABC-6.5-분
칼럼: 엑스브리지 BEH C18 (50mmx4.6mm, 2.5μm)
물 중 이동상: A: 5mM 중탄산암모늄 (PH-10, 암모니아 함유): ACN
시간 (분) /%ACN: 0/5, 0.5/5, 1/15, 3.3/98, 6.0/98, 6.1/5, 6.5/5
칼럼 온도: 35℃, 유량: 1.3 ml/분
MS 파라미터:
질량 범위: 100-1000
프래그멘터: 100
스텝 크기: 0.1
실행 시간: 6.5분
획득 방법 조건: RND-ABC-10-분
칼럼: 엑스브리지 BEH C18 (50mmx4.6mm, 2.5μm)
물 중 이동상: A: 5mM 중탄산암모늄 (PH-10, 암모니아 포함): ACN
시간 (분) /%ACN: 0/5, 0.5/5, 1.5/15, 7/98, 9.0/98, 9.5/5, 10/5
칼럼 온도: 35℃, 유량: 1.3 ml/분
MS 파라미터:
질량 범위: 100-1000
프래그멘터: 100
스텝 크기: 0.1
실행 시간: 10.0분
중간체
(S)-디메틸 2-((6-클로로-3-니트로피리딘-2-일)아미노)펜탄디오에이트의 합성

2,6-디클로로-3-니트로피리딘, 87.7 g (414 40.0 g (207 mmol), L-글루탐산 디메틸 에스테르 히드로클로라이드 mmol), 및 NaHCO3 69.6 g (829 mmol)의 혼합물에 테트라히드로푸란 600 mL를 첨가하였다. 혼합물을 40℃에서 24시간 동안 교반하면서, 2,6-디클로로-3-니트로피리딘의 소멸에 대해 HPLC에 의해 모니터링하였다. 반응이 완결된 후, 고체를 여과하고, 에틸 아세테이트 (3 x 100 mL)로 세척하였다. 합한 여과물 및 세척물을 진공 하에 농축시킨 다음, 잔류물을 실리카 겔 크로마토그래피에 의해, 10/1 (v/v) 헥산/에틸 아세테이트로 용리시키면서 정제하여 생성물을 황색 고체로서 수득하였다 (60 g, 87%), LCMS (m/z) 332.1 [M+H]+.
(S)-메틸 3-(6-클로로-2-옥소-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로파노에이트의 합성

(S)-디메틸 2-((6-클로로-3-니트로피리딘-2-일)아미노)펜탄디오에이트 20 g (60.2 mmol) 및 철 분말 16.8 g (301 mmol)의 혼합물에 2-프로판올 375 mL에 이어서 물 125 mL를 첨가하였다. 교반 혼합물에 아세트산 5.5 g (90.3 mmol)을 첨가한 다음, 반응물을 환류 하에 1시간 동안 교반하였다. 반응물을 출발 물질의 소멸을 HPLC에 의해 모니터링하였다. 반응이 완결된 후, 고체를 여과하고, 2-프로판올 (3 x 50 mL)로 세척하였다. 합한 여과물 및 세척물을 농축 건조시킨 다음, 잔류물을 진공 하에 건조시켜 생성물 15 g (81%)을 암황색 고체로서 수득하였다. 이를 추가 정제 없이 후속 단계에 사용하였다, LCMS (m/z) 270.1 [M+H]+.
(S)-3-(6-클로로-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로판-1-올의 합성

N2 하에 테트라히드로푸란 (THF) 260 mL 중 AlCl3 17.78 g (133.3 mmol)에 THF 중 2M LiAlH4 200 mL의 용액을 기체 발생을 제어하는 속도로 적가하였다. 이와 같이 하여 THF 중 알란 (AlH3)의 용액을 수득하였다. 분리형 플라스크에서, THF 460 mL 중 (S)-메틸 3-(6-클로로-2-옥소-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로파노에이트 26.0 g (96.4 mmol)의 용액을 N2 하에 제조한 다음, 드라이 아이스/아세톤 조로 냉각시켰다. 이에 교반 하에 알란 용액을 2시간에 걸쳐 적가하였다. 첨가가 완결되었을 때, 냉각 조를 제거하고, 반응물을 주위 온도로 가온되도록 하였다. 1.5시간 후, LCMS 분석은 반응이 완결되었음을 나타내었다. 다음에, 물 65 mL 중 17.6 g NaOH의 용액을 H2의 발생이 제어되도록 천천히 첨가하였다. 현탁액을 18시간 동안 교반되도록 한 다음, 고체를 여과하였다. 침전물을 에틸 아세테이트로 세척한 다음, 여과물 및 세척물을 진공 하에 농축시켰다. 생성물을 실리카 겔 크로마토그래피에 의해 CH2Cl2에 이어서 CH2Cl2 중 0에서 10% 메탄올의 구배로 용리시키면서 정제하여 황색빛 오렌지색 고체 15.21 g (69%)을 수득하였다, LCMS (m/z) 228.1 [M+H]+.
(9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

(S)-3-(6-클로로-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로판-1-올 12 g (52.7 mmol)에 48% (w/w) 수성 HBr 160 mL를 첨가한 다음, 반응물을 90℃에서 18시간 동안 교반하였다. 반응물을 출발 알콜의 소멸에 대해 HPLC에 의해 모니터링하였다. 반응이 완결된 후, 이를 주위 온도로 냉각시킨 다음, 1.2 M 수성 NaHCO3을 pH = 8까지 첨가하였다. 혼합물을 에틸 아세테이트 (3 x 100 mL)로 추출한 다음, 합한 유기 상을 염수 (1 x 100 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 여과물을 농축 건조시켰다. 잔류물을 실리카 겔 크로마토그래피에 의해 2/1 (v/v) 헥산/에틸 아세테이트로 용리시키면서 정제하여 생성물 6.0 g (55%)을 담황색 고체로서 수득하였다, LCMS (m/z) 210.1 [M+H]+.
2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

실온에서 1,4-디옥산 (60 ml) 및 물(6 ml) 중 2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (3 g, 14.31 mmol), 4-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)-2-(트리플루오로메틸)피리딘 (3.91 g, 14.31 mmol) 및 탄산세슘 (4.66 g, 14.31 mmol)의 용액에 첨가하고, 반응물을 아르곤으로 20분 동안 탈기하였다. 다음에, 반응 혼합물에 고체 아세트산팔라듐(II) (3.21 g, 14.31 mmol) 및 x-phos (6.82 g, 14.31 mmol)를 1회 충전량으로 첨가하였다. 반응 혼합물을 105℃에서 3-4시간 동안 교반하였다. 반응물을 셀라이트 층을 통해 여과하고, 농축시켰다. 조 물질을 에틸 아세테이트에 녹이고, 용해시키고, 중탄산나트륨 용액 및 물로 세척하였다. 유기 상을 황산나트륨 상에서 건조시키고, 농축시켜 수득하였다. 잔류물을 n-펜탄(3x 50 mL)으로 연화처리하였다. 생성된 고체를 부흐너 깔때기를 통해 여과하고, n-펜탄으로 헹궈 목적 생성물 (4 g, 86%)로서 수집하였다, LCMS (m/z) 321.3 (M+H)+.
(9S)-2-(5-(트리플루오로메틸)피리딘-3-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

질소 하에 25℃에서 교반하는 테트라히드로푸란 (THF) (60 ml), 물(4ml) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (3 g, 14.31 mmol), (5-(트리플루오로메틸)피리딘-3-일)보론산 (5.46 g, 28.6 mmol) 및 Cs2CO3 (13.99 g, 42.9 mmol)의 용액에 20분 동안 아르곤 기체로 퍼징하였다. 이어서, 아세트산팔라듐(II) (0.080 g, 0.358 mmol) 및 X-Phos (227mg)를 첨가하였다. 반응 혼합물을 110℃에서 16시간 동안 교반하였다. 다음에, 반응 혼합물을 농축시키고, 잔류물을 DCM (100 mL)에 녹였다. 용액을 물 및 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 조 생성물을 실리카 겔 칼럼에 첨가하고, Hex/EtOAc (1:1)로 용리시켰다. 수집된 분획을 증발시켜 회백색 고체로서 수득하였다 (3.6g, 11.1mmol 76%), LCMS (m/z) 321.2 [M+H]+.
(9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

25℃에서 질소 하에 교반하고, 20분 동안 아르곤 기체로 퍼징된 1-부탄올 (300 ml), 물(100ml) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (10 g, 47.7 mmol), (2-메틸피리딘-4-일)보론산 (8.49 g, 62.0 mmol) 및 인산칼륨 (30.4 g, 143 mmol)의 용액에 X-Phos (2.274 g, 4.77 mmol), Pd2(dba)3 (2.184 g, 2.385 mmol)을 첨가하였다. 반응 혼합물을 120℃에서 16시간 동안 교반하였다. 농축시키기 전에 잔류물을 DCM (700 mL)에 녹였다. 용액을 물 및 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 갈색 반고체 조 생성물을 수득하였다. 조 생성물을 헥산으로 세척함으로써 정제하여 회백색 고체 생성물을 수득하였다(9 g, 32.4 mmol, 67.9% 수율), LCMS (m/z): 267.3 (M+H)+.
(9S)-2-((R)-2-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

실온에서 질소 분위기 하에 1,2-디메톡시에탄 (DME) (10 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5 g, 2.385 mmol), (R)-2-(트리플루오로메틸)피롤리딘 (0.663 g, 4.77 mmol) 및 KOtBu (0.535 g, 4.77 mmol)의 현탁액에 고체 [1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일리덴]클로로][3-페닐알릴]팔라듐(II) (1.549 g, 2.385 mmol)을 첨가하고, 80℃에서 16시간 동안 교반하였다. 반응물을 실온으로 냉각시키고, 셀라이트를 통해 여과하고, 용매를 감압 하에 증발시켜 조 잔류물을 수득하였다. 조 잔류물을 EtOAc (100 mL)로 희석하고, 물 (50mL x2)에 이어서 염수 용액으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 증발시켜 조 생성물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카 겔: 100-200 메쉬, 1:1 Hex/EtOAc로 용리시킴)에 의해 정제하여 (9S)-2-((2R)-2-(트리플루오로메틸)시클로펜틸)-6,7,8,9,10,10a-헥사히드로-4aH-5,9-메타노피리도[2,3-b][1,4]디아조신을 담녹색 고체로서 수득하였다 (500mg, 1.596 mmol, 64.6% 수율), (TLC: Rf 0.3, 용리액: 헥산 중 80% EtOAc), LCMS (m/z) 313.2 [M+H]+.
(9S)-2-((S)-2-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성
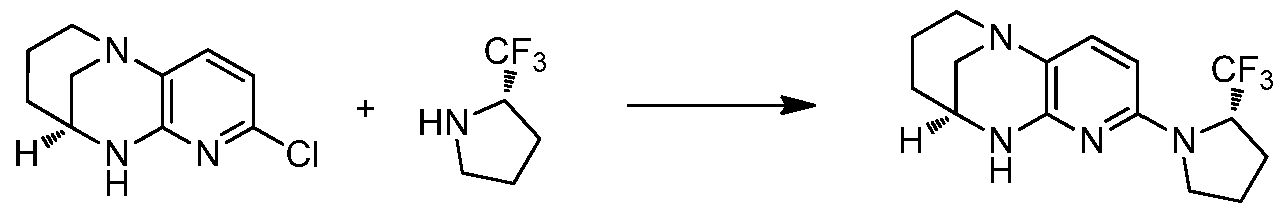
1,2-디메톡시에탄 (DME) (10 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5 g, 2.385 mmol), (S)-2-(트리플루오로메틸)피롤리딘 (0.663 g, 4.77 mmol) 및 KOtBu (0.535 g, 4.77 mmol)의 현탁액을 교반하고, 아르곤으로 실온에서 15분 동안 탈기하였다. 다음에, [1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일리덴]클로로][3-페닐알릴]팔라듐(II) (1.549 g, 2.385 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 완결된 후, 반응물을 셀라이트를 통해 여과하고, 농축 건조시켰다. 생성된 잔류물을 EtOAc로 희석하고, 물에 이어서 염수 용액으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 증발시켰다. 조 생성물을 실리카 겔 칼럼에 첨가하고, Hex/EtOAc (1:1)로 용리시켰다. 수집된 분획을 증발시켜 (9S)-2-((S)-2-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 회백색 고체로서 수득하였다 (0.6 g, 1.868 mmol, 78% 수율), LCMS (m/z) 313.3 [M+H]+.
(9S)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성
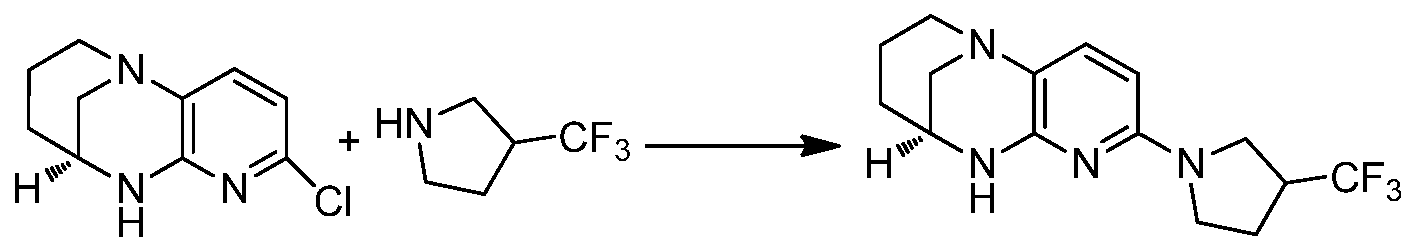
KOtBu (0.321 g, 2.86 mmol)을 1,2-디메톡시에탄 (DME) (10 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.3 g, 1.431 mmol), 및 3-(트리플루오로메틸)피롤리딘 (0.398 g, 2.86 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 교반하고, 아르곤으로 실온에서 15분 동안 탈기한 다음, (1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일리덴)클로로) (3-페닐알릴)팔라듐(2) (0.037 g, 0.057 mmol)을 반응 혼합물에 첨가하였다. 반응물을 80℃에서 16시간 동안 교반하였다. 반응물을 실온으로 냉각시키고, 셀라이트를 통해 여과하고, 감압 하에 증발시켰다. 반응 혼합물을 물 (20 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 80% EtOAc: Rf-0.4; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 헥산 중 50% EtOAc로 용리시켜 (85: 15) 혼합물을 수득하고, 추가로 SFC에 의해 정제하여 순수한 (9S)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 회백색 고체로서 수득하였다 (0.151 g, 0.473 mmol, 33.1% 수율).
분석용 SFC 조건: 피크-II 칼럼/치수:
키랄팩 AD-H (250 X 4.6)mm,5u
% CO2: 70.0%
% 공용매: 30.0% (MeOH 중 0.5% DEA)
총 유량: 3.0g/분
배압: 100 bar
온도: 26.8℃
UV: 212nm
정제용 SFC 조건
칼럼/치수: 키랄팩 AD-H (250 X 30)mm
% CO2: 75%
% 공용매: 25.0% (MeOH)
총 유량: 100.0g/분
배압: 100bar
UV: 212nm
스택 시간: 1.8분
로드/주입: 5.5mg
LCMS (m/z) 313.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 6.87 (d, J = 8.1 Hz, 1H), 6.49 (d, J = 4.9 Hz,1H), 5.58 (d, J = 8.1 Hz, 1H), 3.60 (dd, J = 10.8, 8.4 Hz, 1H), 3.47 (s, 1H), 3.39 (dd, J = 10.6, 5.8 Hz, 2H), 3.30 (s, 2H), 3.14 - 2.98 (m, 2H), 2.90 (d, J = 4.5 Hz, 1H), 2.50 (qd, J = 3.2, 2.2, 1.6 Hz,1H), 2.31 - 2.13 (m, 1H), 2.05 (d, J = 7.1 Hz, 1H), 1.72 (dt, J = 7.0, 3.0 Hz, 2H), 1.44 (s, 1H), 1.16 (d, J = 14.4 Hz, 1H).
(9S)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

(1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일리덴)클로로) (3-페닐알릴) 팔라듐 (0.062 g, 0.095 mmol)을 실온에서 1,2-디메톡시에탄 (20 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5 g, 2.385 mmol), 3-(트리플루오로메틸)피롤리딘 (0.663 g, 4.77 mmol) 및 KOtBu (0.535 g, 4.77 mmol)의 탈기된 현탁액에 첨가하였다. 반응 혼합물을 추가로 10분 동안 탈기하고, 16시간 동안 80℃에서 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 셀라이트의 패드를 통해 여과하였다. 여과물을 증발시켜 갈색 잔류물을 수득하였다. 잔류물을 물 (15 mL)과 EtOAc (2X 25 mL) 사이에 분배하였다. 유기 층을 물에 이어서 염수 용액으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다 (TLC 용리액: 80% EtOAc/헥산, Rf 값: 0.4, UV 활성). 조 물질을 칼럼 크로마토그래피 (100-200 메쉬)에 의해 실리카 겔을 사용하여 정제하고, 생성물을 석유 에테르 중 50% 에틸 아세테이트로 용리시켜 거울상이성질체의 (30:70) 혼합물을 수득하였으며, 이를 추가로 SFC 정제하여 (9S)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 갈색 고체로서 수득하였다 (0.125 g, 0.393 mmol, 16.46% 수율).
분석용 SFC 조건: 피크 I (주요)
키랄팩 AD-H (250 X 4.6)mm,5u
% CO2: 70.0%
% 공용매: 30.0% (MeOH 중 0.5% DEA)
총 유량: 3.0g/분
배압: 100 bar
온도: 26.8℃
UV: 212nm
정제용 SFC 조건
칼럼/치수: 키랄팩 AD-H (250 X 30)mm
% CO2: 75%
% 공용매: 25.0% (MeOH)
총 유량: 100.0g/분
배압: 100bar
UV: 212nm
스택 시간: 1.8분
로드/주입: 5.5mg
LCMS (m/z) 313.31[M+H]+.
1H NMR (400 MHz, CDCl3): δ 7.03 (dd, J = 8.2, 0.6 Hz, 1H), 5.67 (d, J = 8.2 Hz, 1H), 4.91 (s, 1H), 3.71 (dd, J = 10.8, 8.5 Hz, 1H), 3.62 (s, 1H), 3.52 (dd, J = 10.8, 7.2 Hz, 2H), 3.43 (dt, J = 9.6, 7.5 Hz, 1H), 3.24 - 3.08 (m, 3H), 2.99 (d, J = 8.2 Hz, 1H), 2.83 (d, J = 12.0 Hz, 1H), 2.27 - 2.08 (m, 2H), 1.91 - 1.75 (m, 2H), 1.65 (d, J = 5.0 Hz, 2H), 1.26 (t, J = 6.6 Hz, 1H).
(9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성.

1,2-디메톡시에탄 (DME) (30 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.750g, 3.58 mmol), 3-(트리플루오로메틸)피페리딘 (1.096 g, 7.15 mmol) 및 KOtBu (1.204 g, 10.73 mmol)의 현탁액을 교반하고, 아르곤으로 실온에서 15분 동안 탈기한 다음, (1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일리덴)클로로) (3-페닐알릴)팔라듐(2) (0.093 g, 0.143 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 반응물을 셀라이트를 통해 여과하고, 감압 하에 완전히 증발시켰다. 반응 혼합물을 EtOAc (50 ml)로 희석하고, 물에 이어서 염수 용액으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 증발시켰다. 조 생성물을 실리카 겔 칼럼에 첨가하고, Hex/EtOAc (1:1)로 용리시켰다. 수집된 분획을 증발시키고, 생성된 잔류물을 키랄 SFC 분리에 의해 정제하여 순수한 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 회백색 고체로서 수득하였다 (0.450 g, 1.371 mmol, 38.3% 수율), (TLC: Rf 값: 0.4, 80% EtOAc/헥산).
분석용 SFC 조건: 피크-I
칼럼/치수: 키랄팩 AD-H (250 X 4.6)mm,5u
% CO2: 60.0%
% 공용매: 40.0% (MeOH 중 0.5% DEA)
총 유량: 4.0g/분
배압: 100 bar
온도: 26.8℃
UV: 215nm
정제용 SFC 조건
칼럼/치수: 키랄팩 AD-H (250 X 30)mm
% CO2: 65.0%
% 공용매: 35.0% (100% MeOH)
총 유량: 100.0g/분
배압: 100bar
UV: 215nm
스택 시간: 2.2분
로드/주입: 50.0mg
LCMS (m/z) 327.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 6.89 (dd, J = 8.2, 0.7 Hz, 1H), 6.56 (d, J = 4.6 Hz, 1H), 5.89 (d, J = 8.3 Hz, 1H), 4.58 - 4.33 (m, 1H), 3.97 (d, J = 12.8 Hz, 1H), 3.48 (s, 1H), 3.17 - 2.98 (m, 2H), 2.92 (d, J = 4.4 Hz, 1H), 2.76 - 2.57 (m, 2H), 2.50 (p, J = 1.9 Hz, 2H), 1.96 (d, J = 10.6 Hz, 1H), 1.74 (dd, J = 9.4, 3.3 Hz, 3H), 1.61 - 1.32 (m, 3H), 1.25 - 1.08 (m, 1H).
(9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성
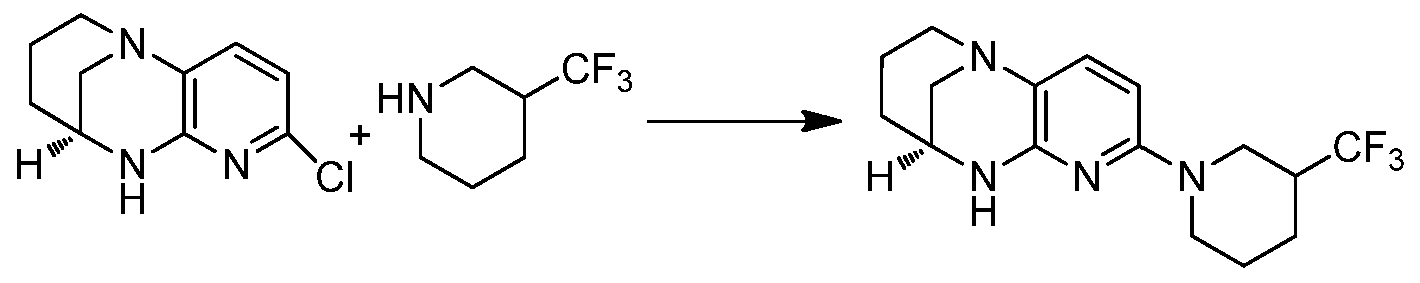
KOtBu (0.803 g, 7.15 mmol)를 1,2-디메톡시에탄 (DME) (20 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.750 g, 3.58 mmol), 및 3-(트리플루오로메틸)피페리딘 (1.096 g, 7.15 mmol)의 교반 용액에 첨가하였다. 반응물을 교반하고, 아르곤으로 실온에서 15분 동안 탈기한 다음, (1,3-비스(2,6-디-이소프로필페닐)-4,5-디히드로이미다졸-2-일리덴)클로로) (3-페닐알릴)팔라듐(2) (0.093 g, 0.143 mmol)을 반응 혼합물에 첨가하였다. 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 반응물을 실온으로 냉각시키고, 셀라이트를 통해 여과하고, 완전히 증발시키고, 물 (20 mL)와 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 80% EtOAc: Rf-0.4; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 헥산 중 50% EtOAc로 용리시켜 (69: 31) 혼합물을 수득하고, 추가로 SFC에 의해 정제하여 순수한 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 연황색 고체로서 수득하였다 (450 mg, 1.310 mmol, 36.6% 수율).
분석용 SFC 조건: 피크-II
칼럼/치수: 키랄팩 AD-H (250 X 4.6)mm,5u
% CO2: 60.0%
% 공용매: 40.0% (MeOH 중 0.5% DEA)
총 유량: 4.0g/분
배압: 100 bar
온도: 26.8℃
UV: 215nm
정제용 SFC 조건
칼럼/치수: 키랄팩 AD-H (250 X 30)mm
% CO2: 65.0%
% 공용매: 35.0% (100% MeOH)
총 유량: 100.0g/분
배압: 100bar
UV: 215nm
스택 시간: 2.2분
로드/주입: 50.0mg
LCMS (m/z) 327.2 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ ppm 6.89 (d, J=8.11 Hz, 1 H), 6.55 (br d, J=3.95 Hz, 1 H), 5.89 (d, J=8.33 Hz, 1 H), 4.49 (dt, J=12.44, 1.78 Hz, 1 H), 3.96 (d, J=13.15 Hz, 1 H), 3.48 (br s, 1 H), 3.15 - 2.96 (m, 2 H), 2.94 - 2.80 (m, 1 H), 2.74 - 2.56 (m, 2 H), 2.55 - 2.41 (m, 2 H), 2.12 - 1.84 (m, 1 H), 1.82 - 1.64 (m, 3 H), 1.58 - 1.32 (m, 3 H), 1.32 - 1.08 (m, 1 H).
(9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성
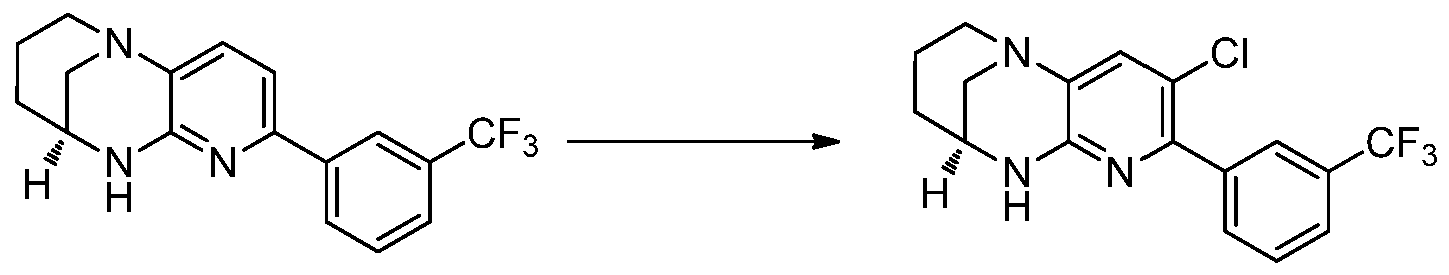
0℃에서 질소 하에 교반하는 클로로포름 (20 mL) 중 (9S)-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (2 g, 6.26 mmol)의 용액에 NCS (1.004 g, 7.52 mmol)를 첨가하였다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 반응 혼합물을 물로 켄칭하고, DCM 2x 25 ml로 추출하고, 유기 층을 Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피에 의해 100-200 실리카 겔을 사용하여 정제하고, 석유 에테르 중 70% 에틸 아세테이트로 용리시켜 순수한 화합물 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 연황색 반고체로서 수득하였다 (800 mg, 1.859 mmol, 29.7% 수율), LCMS (m/z): 354.22 [M+H]+.
(9S)-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

교반하고 10분 동안 아르곤으로 탈기된 1,4-디옥산 (160 mL) 및 물 (40 mL) 중 고체 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (5 g, 23.85 mmol), (3-(트리플루오로메틸)페닐)보론산 (6.79 g, 35.8 mmol) 및 삼염기성 인산칼륨 (15.19 g, 71.5 mmol)에 고체 x-phos (0.689 g, 4.77 mmol) 및 Pd2(dba)3 (2.184 g, 2.385 mmol)을 첨가한 다음, 다시 반응 혼합물을 5분 동안 탈기하고, 16시간 동안 110℃에서 교반하였다. 유기 상을 물 50 mL, 포화 염수 100 mL로 세척하고, Na2SO4 상에서 건조시키고, 진공 하에 증발시켜 조 생성물을 갈색 고체로서 수득하였다. 조 생성물을 디에틸 에테르 및 펜탄으로 세척하고, 여과하고, 디에틸 에테르로 세척하여 순수한 화합물 (9S)-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 수득하였다 (4.8 g, 13.81 mmol, 57.9% 수율), LCMS (m/z): 320.11 [M+H]+.
(9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (377 mg, 1.272 mmol)을 28℃에서 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (450 mg, 1.272 mmol), 및 TEA (0.886 mL, 6.36 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 30분 동안 교반하고, (R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (571 mg, 2.54 mmol)을 첨가하였다. 반응 혼합물을 70℃에서 10시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (5 mL)과 EtOAc (15 mL) 사이에 분배하였다. EtOAc 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하였다. 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 그레이스에 의해 C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: MeOH를 사용하여 정제하고, 생성물을 물 중 0.1% 포름산 중 91% MeOH로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 수성 층을 DCM으로 추출하고, DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 황색 고체로서 수득하였다 (250 mg, 0.391 mmol, 30.8% 수율), LCMS (m/z): 604.14 [M+H]+.
(9S)-2-클로로-3-아이오도-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

NIS (2.58 g, 11.45 mmol)를 클로로포름 (40 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (2.0 g, 9.54 mmol)의 교반 용액에 질소 하에 0℃에서 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 용매를 감압 하에 완전히 증발시켜 조 생성물을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 20% EtOAc (구배 시스템)로 용리시켜 목적 생성물 (9S)-2-클로로-3-아이오도-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 연황색 고체로서 수득하였다 (1.65 g, 4.53 mmol, 47.5% 수율), LCMS (m/z): 335.90 [M+H]+.
(9S)-2-클로로-3-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

1,4-디옥산 (15 mL) & 물 (1.5 mL) 중 (9S)-2-클로로-3-아이오도-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (1.65 g, 4.92 mmol), 트리메틸보록신 (1.639 mL, 4.92 mmol) 및 탄산칼륨 (2.039 g, 14.75 mmol)의 현탁액을 교반하고, 아르곤으로 실온에서 15분 동안 탈기하고, 테트라키스(트리페닐포스핀)팔라듐(0) (0.568 g, 0.492 mmol)을 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 90℃에서 48시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 셀라이트를 통해 여과하고, EtOAc (30 ml)로 세척하였다. 여과물을 취하고, 농축시키고, EtOAc (50 ml) 중에 용해시켰다. EtOAc 층을 물 (15 ml)에 이어서 염수 용액 (15 ml)으로 세척하고, Na2SO4로 건조시키고, 여과하고, 농축시켜 조 생성물을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 헥산 중 20% EtOAc (구배 시스템)로 용리시키면서 정제하여 목적 생성물 (9S)-2-클로로-3-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 연황색 고체로서 수득하였다 (0.800 g, 3.55 mmol, 72.1% 수율), (TLC 용리액: 헥산 중 50% EtOAc: Rf-0.3.; UV 활성). LCMS (m/z): 224.90 [M+H]+.
(9S)-3-메틸-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

1,4-디옥산 (20 mL) & 물 (4 mL) 중 (9S)-2-클로로-3-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (800 mg, 3.58 mmol), (2-메틸피리딘-4-일)보론산 (612 mg, 4.47 mmol) 및 인산삼칼륨 (2277 mg, 10.73 mmol)의 현탁액을 교반하고, 아르곤으로 실온에서 15분 동안 탈기하였다. Pd2(dba)3 (164 mg, 0.179 mmol) 및 X-Phos (170 mg, 0.358 mmol)를 반응 혼합물에 첨가하였다. 이어서, 반응 혼합물을 90℃에서 16시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 셀라이트를 통해 여과하고, EtOAc로 세척하였다. 여과물을 취하고, 농축시키고, EtOAc로 용해시켰다. EtOAc 층을 물에 이어서 염수 용액으로 세척하고, Na2SO4로 건조시키고, 여과하고, 농축시켜 조 생성물을 수득하였다. 조 생성물을 에테르 (20 ml) 세척에 의해 정제하여 목적 생성물 (9S)-3-메틸-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 회백색 고체로서 수득하였다 (600 mg, 2.121 mmol, 59.3% 수율), LCMS (m/z): 281.16 [M+H]+.
2,6-디클로로-4-메틸-3-니트로피리딘의 합성

0℃로 냉각시킨 트리플루오로아세트산 무수물 (24.98 mL, 177 mmol) 중 2,6-디클로로-4-메틸피리딘 (5g, 30.9 mmol)의 현탁액에 질산 (2.90 mL, 64.8 mmol)을 적가하였다. 생성된 용액을 실온에서 18시간 동안 교반하였다. 반응 혼합물을 물 (40 mL) 중 메타중아황산나트륨 (5.87 g, 30.9 mmol)의 냉각 용액에 천천히 첨가하고, 실온에서 2시간 동안 교반하였다. 반응 혼합물을 8N NaOH (25 mL) 용액을 사용하여 pH 7로 중화시키고, DCM (100 mL)으로 2회 추출하였다. 합한 유기 층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 2,6-디클로로-4-메틸-3-니트로피리딘을 회백색 고체로서 수득하였다 (6.2g, 29.4 mmol, 95% 수율), LCMS (m/z): 206.98 [M+H]+.
(S)-디메틸 2-((6-클로로-4-메틸-3-니트로피리딘-2-일)아미노)펜탄디오에이트의 합성

절차: (S)-디메틸 2-아미노펜탄디오에이트 히드로클로라이드 (147 g, 696 mmol)를 테트라히드로푸란 (THF) (2.5 L) 중 2,6-디클로로-4-메틸-3-니트로피리딘 (120 g, 580 mmol) 및 중탄산나트륨 (146 g, 1739 mmol)의 현탁액에 질소 하에 0℃에서 첨가하였다. 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 여과하고, EtOAc (2 x 30 mL)로 세척하였다. 여과물을 진공 하에 농축시켜 조 물질을 수득하였다. 조 물질을 칼럼 크로마토그래피에 의해 실리카 겔 (100-200 메쉬)을 사용하여 정제하고, 생성물을 석유 에테르 중 10% EtOAc로 용리시켜 (S)-디메틸 2-((6-클로로-4-메틸-3-니트로피리딘-2-일)아미노)펜탄디오에이트를 황색 고체로서 수득하였다 (50g, 135 mmol, 23.31% 수율), LCMS (m/z): 345.1 [M+H]+.
(S)-메틸 3-(6-클로로-8-메틸-2-옥소-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로파노에이트의 합성

철 (40.4 g, 723 mmol)을 실온에서 이소프로판올 (450 mL) 및 물 (90 mL) 중 (S)-디메틸 2-((6-클로로-4-메틸-3-니트로피리딘-2-일)아미노)펜탄디오에이트 (50 g, 145 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 40℃로 가열하고, 아세트산 (12.42 mL, 217 mmol)을 첨가하였다. 반응 혼합물을 80℃에서 2시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 포화 NaHCO3로 염기성화시키고, 셀라이트의 패드를 통해 여과하고, DCM (3 X 50mL)으로 세척하였다. 여과물을 분리하고, 염수 용액으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 감압 하에 농축시켜 조 생성물을 수득하였다. 조 생성물을 회백색 고체로서 에테르 (20 ml)로 세척함으로써 정제하여 목적 생성물 (S)-메틸 3-(6-클로로-8-메틸-2-옥소-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로파노에이트를 수득하였다 (29g, 101 mmol, 69.8% 수율), LCMS (m/z): 284.06 [M+H]+.
(S)-3-(6-클로로-8-메틸-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로판-1-올의 합성

절차: 2M 수소화알루미늄리튬 (256 mL, 511 mmol)을 테트라히드로푸란 (THF) (290 mL) 중 염화알루미늄 (19.08 g, 143 mmol)의 교반 용액에 질소 하에 기체 발생을 제어하는 속도로 적가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물 (알란 AlH3)을 테트라히드로푸란 (THF) (450 mL) 중 (S)-메틸 3-(6-클로로-8-메틸-2-옥소-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로파노에이트 (29 g, 102 mmol)의 교반 용액에 질소 하에 30분에 걸쳐 -78℃에서 적가하였다. 반응물을 16시간 동안 주위 온도로 가온되도록 하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 0℃에서 10% NaOH 용액으로 켄칭하고, 16시간 동안 교반하고, 셀라이트의 패드를 통해 여과하고, (300 ml) DCM으로 세척하였다. DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 생성물을 에테르 (2 X 50 ml)로 세척함으로써 정제하여 (S)-3-(6-클로로-8-메틸-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로판-1-올을 회백색 황색 고체로서 수득하였다 (22 g, 69.8 mmol, 68.3% 수율), LCMS (m/z): 242.0 [M+H]+.
(9S)-2-클로로-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

(S)-3-(6-클로로-8-메틸-1,2,3,4-테트라히드로피리도[2,3-b]피라진-3-일)프로판-1-올 (21 g, 87 mmol)의 교반 용액에 HBr (11.79 ml, 217 mmol)을 28℃에서 첨가하고, 100℃에서 12시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 28℃로 냉각시키고, 빙수 (50 mL)에 부었다. 수성 층을 포화 NaHCO3 용액 (120 ml)으로 중화시켰다. 수성 층을 EtOAc (2X 50 ml)로 추출하였다. EtOAc 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 생성물을 석유 에테르 중 50% EtOAc로 용리시켜 화합물을 수득하였다. 화합물을 n-펜탄으로 세척하여 순수한 (9S)-2-클로로-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 백색 고체로서 수득하였다 (13 g, 57.8 mmol, 66.5% 수율), LCMS (m/z): 224.09 [M+H]+.
(9S)-4-메틸-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

1,4-디옥산 (40 mL);물 (10 mL) 중 (9S)-2-클로로-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 2.235 mmol), (2-메틸피리딘-4-일)보론산 (459 mg, 3.35 mmol) 및 K3PO4 (1423 mg, 6.71 mmol)의 탈기된 용액에 x-phos (213 mg, 0.447 mmol),Pd2(dba)3 (205 mg, 0.224 mmol)을 첨가하였다. 반응 혼합물을 110℃에서 12시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (15 mL)과 EtOAc (2 X15 mL) 사이에 분배하였다. EtOAc 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하였다. 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 디에틸 에테르 (15 mL)로 세척함으로써 정제하여 순수한 (9S)-4-메틸-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 황색 고체로서 수득하였다 (550 mg, 1.920 mmol, 86% 수율), LCMS (m/z): 281.05 [M+H]+.
(9S)-3-클로로-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신의 합성

0℃에서 질소 하에 교반하는 클로로포름 (20 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (2.0 g, 7.51 mmol)의 용액에 NCS (1.504 g, 11.26 mmol)를 10분 동안 조금씩 첨가하였다. 반응 혼합물을에 0℃에서서 4시간 동안 교반하였다. 반응물을 빙냉수 (20 ml)로 켄칭하고, DCM (3x30 ml)으로 추출하였다. 유기 상을 분리하고, 물 (2x20 ml) 및 염수 용액 (2x20 ml)으로 세척한 다음, 유기 상을 무수 황산나트륨 상에서 건조시키고, 이를 여과하고, 감압 하에 증발시켜 조 생성물을 수득하였다. 조 생성물 (N35964-5-A1 & N35964-7-A1)을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50% EtOAc (구배 시스템)로 용리시켜 목적 생성물 (9S)-3-클로로-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신을 연황색 고체로서 수득하였다 (1.2 g, 3.60 mmol, 47.9% 수율), LCMS (m/z): 300.17 [M+H]+.
(9S)-페닐 3-클로로-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복실레이트의 합성

질소 하에 0℃에서 교반하는 디클로로메탄 (DCM) (35 mL) 중 페닐 카르보노클로리데이트 (0.195 mL, 1.555 mmol) 및 피리딘 (0.149 mL, 1.837 mmol)의 용액에 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.413 mmol)을 첨가하였다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 포화 중탄산나트륨 용액으로 켄칭하고, DCM (200 mL)으로 2회 추출하였다. 합한 DCM 층을 물 (80mL)로 세척하고, Na2SO4로 건조시키고, 여과하고, 고진공 하에 농축시켜 조 생성물을 수득하였다. 조 생성물을 실리카 겔 (100-200 메쉬) 칼럼에 첨가하고, 30%Hex/EtOAc로 용리시키고, 수집된 분획을 감압 하에 증류시켜 화합물을 수득하였다. 화합물을 펜탄으로 세척하여 (9S)-페닐 3-클로로-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복실레이트의 순수한 화합물을 백색 고체로서 수득하였다 (500 mg, 1.044 mmol, 73.9% 수율), LCMS (m/z): 474.30 [M+H]+.
(9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (377 mg, 1.272 mmol)을 28℃에서 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (450 mg, 1.272 mmol), 및 TEA (0.886 mL, 6.36 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 30분 동안 교반하고, (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (571 mg, 2.54 mmol)을 첨가하였다. 반응 혼합물을 70℃에서 10시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (15 mL)과 EtOAc (25 mL) 사이에 분배하였다. EtOAc 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하였다. 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 그레이스에 의해 C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: MeOH를 사용하여 정제하고, 생성물을 물 중 0.1% 포름산 중 91% MeOH로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 수성 층을 DCM으로 추출하고, DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 황색 고체로서 수득하였다 (270 mg, 0.430 mmol, 33.8% 수율), LCMS (m/z): 606.11 [M+H]+.
(9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

TEA (1.064 mL, 7.63 mmol) 및 트리포스겐 (377 mg, 1.272 mmol)을 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (450 mg, 1.272 mmol)의 교반 용액에 질소 하에 실온에서 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민 (573 mg, 2.54 mmol)을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 용매를 감압 하에 완전히 증발시키고, 물 (10 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50% EtOAc (구배 시스템)로 용리시켜 목적 생성물 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (200 mg, 0.271 mmol, 21.27% 수율), LCMS (m/z): 605.2 [M+H]+.
(9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

TEA (1.064 mL, 7.63 mmol) 및 트리포스겐 (377 mg, 1.272 mmol)을 테트라히드로푸란 (THF) (45 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (450 mg, 1.272 mmol)의 교반 용액에 질소 하에 실온에서 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. (S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-아민 (860 mg, 3.82 mmol)을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 용매를 감압 하에 완전히 증발시키고, 물 (30 mL)과 EtOAc (100 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 20% EtOAc (구배 시스템)로 용리시켜 목적 생성물 (9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (200 mg, 0.317 mmol, 24.91% 수율), LCMS (m/z): 605.19 [M+H]+.
(9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
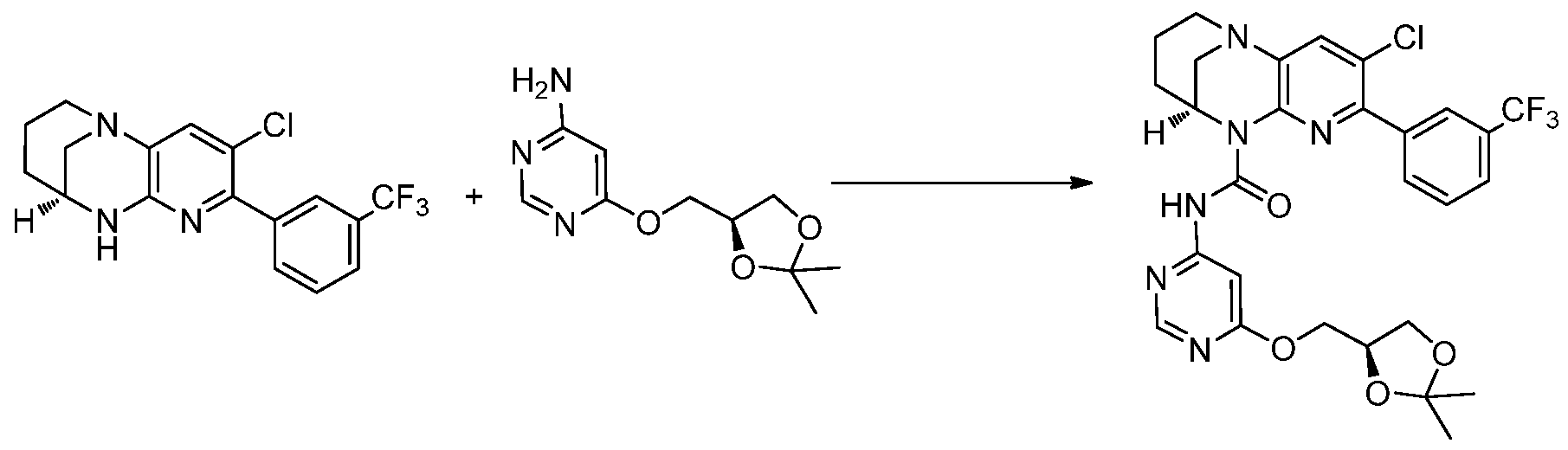
테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.4 g, 1.131 mmol) 및 TEA (0.788 mL, 5.65 mmol)의 용액에 트리포스겐 (0.336 g, 1.131 mmol)을 첨가하고, 1시간 동안 질소 하에 실온에서 교반하였다. 이 반응 혼합물에 (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민 (0.382 g, 1.696 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하고, 반응의 진행을 TLC 및 LCMS에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (50 mL)에 붓고, EtOAc (2 X 50mL)로 추출하였다. 유기 층을 물 및 염수로 세척하였다. 유기 층을 Na2SO4을 통해 여과하고, 농축시켜 조 화합물을 수득하였다. 조 화합물을 중성 알루미나에 의해 정제하고, 헥산 중 50% 에틸 아세테이트로 용리시켜 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.3 g, 0.486 mmol, 43.0% 수율), LCMS (m/z): 605.55 [M+H]+.
(9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (417 mg, 1.405 mmol)을 실온에서 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-6,7,8,9,10,10a-헥사히드로-4aH-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.405 mmol) 및 TEA (1.175 mL, 8.43 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 1시간 동안 교반하고, (R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-아민 (791 mg, 3.51 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 감압 하에 증발시키고, 물 (50 mL)로 희석하고, 에틸 아세테이트 (2 X 75 mL)에 이어서 염수 용액 (50 mL)으로 추출하고, 층을 분리하고, 무수 Na2SO4로 건조시키고, 여과하고, 농축시켜 조 생성물을 수득하였다. 조 생성물을 중성 알루미나 칼럼에 적용하고, 헥산 중 50-60% 에틸 아세테이트로 용리시켰다. 수집된 분획을 감압 하에 증발시켜 (9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 순수한 화합물을 회백색 고체로서 수득하였다 (350 mg, 0.544 mmol, 38.7% 수율), LCMS (m/z): 605.23 [M+H]+.
(9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (252 mg, 0.848 mmol)에 이어서 트리에틸아민 (1.182 mL, 8.48 mmol)을 테트라히드로푸란 (THF) (15 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.413 mmol)의 교반 용액에 실온에서 첨가하고, 30분 동안 교반하였다. 이어서, (R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민 (637 mg, 2.83 mmol)을 반응 혼합물에 실온에서 첨가하고, 80℃에서 15.5시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (40mL)로 희석하고, 에틸 아세테이트 (2X60 mL)로 추출하고, 염수 용액 (30 mL)으로 세척하였다. 유기 층을 분리하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 정제하고, 실리카 겔 (100-200 메쉬) 및 용리액으로서 석유 에테르 중 30% 에틸 아세테이트를 사용하여 (9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 갈색 점착성 고체로서 수득하였다 (200 mg, 0.324 mmol, 22.92% 수율), LCMS (m/z): 606.99 [M+H]+.
(9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (419 mg, 1.413 mmol)을 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.413 mmol), 및 TEA (1.182 mL, 8.48 mmol)의 교반 용액에 질소 하에 28℃에서 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하고, (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민 (955 mg, 4.24 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 반응 혼합물을 물 (20 mL)과 EtOAc (2x 25mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 그레이스에 의해, C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: ACN을 사용하여 정제하고, 생성물을 물 중 0.1% 포름산 50% 중 ACN으로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 침전된 고체를 여과하고, 건조시켜 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (120 mg, 0.191 mmol, 13.48% 수율), LCMS (m/z): 605.23 [M+H]+.
(9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (0.419 g, 1.413 mmol)을 테트라히드로푸란 (THF) (15 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5 g, 1.413 mmol), TEA (0.985 mL, 7.07 mmol))의 용액에 첨가하고, 질소 하에 실온에서 1시간 동안 교반하였다. 이 반응 혼합물에 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민 (0.637 g, 2.83 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하고, 반응의 진행을 TLC 및 LCMS에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 빙수 (50 mL)에 붓고, EtOAc (2 X 100 mL)로 추출하였다. 합한 유기 층을 물 (50 mL), 염수 용액 (50 mL)으로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50% EtOAc로 용리시켜 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (0.52g, 0.387 mmol, 27.4% 수율), LCMS (m/z): 605.20 [M+H]+.
(9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

(9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (600 mg, 1.696 mmol)에 이어서 트리포스겐 (302 mg, 1.018 mmol)을 25℃에서 테트라히드로푸란 (THF) (20 mL) 중 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민 (458 mg, 2.035 mmol) 및 TEA (1.182 mL, 8.48 mmol)의 용액에 첨가하고, 70℃에서 16시간 동안 교반하였다. 반응의 진행을 LCMS 및 TLC에 의해 모니터링하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (20 mL)과 EtOAc (2X50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 샘플을 디클로로메탄에 로딩하고, 실리카 5g 상에서 0-15% 메탄올-디클로로메탄을 80분에 걸쳐 사용하여 정제하였다. 적절한 분획을 합하고, 진공 하에 증발시켜 목적 생성물 220 mg을 회백색 고체로서 수득하였다, LCMS (m/z): 605.17 [M+H]+.
(9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (20 mL) 중 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민 (637 mg, 2.83 mmol)의 용액에 트리포스겐 (419 mg, 1.413 mmol) 및 트리에틸아민 (1.182 mL, 8.48 mmol)을 첨가하고, 이에 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민 (637 mg, 2.83 mmol)을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 빙수로 켄칭하고, 에틸 아세테이트 2x 25 ml로 추출하고, 합한 유기 층을 Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (100-200 실리카 겔)에 의해 DCM 중 2% 메탄올로 용리시키면서 정제하여 순수한 화합물 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연갈색 고체로서 수득하였다 (200 mg, 0.308 mmol, 21.82% 수율), LCMS (m/z): 605.1 [M+H]+.
(9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

(9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (1 g, 2.83 mmol)을 테트라히드로푸란 (THF) (30 mL) 중에 용해시키고, 0℃에서 질소 하에 교반하고, 트리포스겐 (0.839 g, 2.83 mmol), DIPEA (2.468 mL, 14.13 mmol)를 첨가하였다. 반응 혼합물을 실온에서 16 동안 교반하였다. 이에 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (1.268 g, 5.65 mmol)을 첨가하고, 밀봉된 튜브 중에서 16시간 동안 80℃에서 교반하였다. 반응 혼합물을 실온으로 되도록 하고, 물 200 ml로 켄칭하고, 에틸 아세테이트 3x200 ml로 추출하고, 합한 유기 층을 Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피에 의해 정제하여 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (300 mg, 0.486 mmol, 17.18% 수율). LCMS (m/z): 604.20 [M+H]+.
(9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

TEA (1.970 mL, 14.13 mmol)에 이어서 트리포스겐 (0.839 g, 2.83 mmol)을 테트라히드로푸란 (THF) (20 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (1 g, 2.83 mmol)의 용액에 실온에서 첨가하고, 6시간 동안 실온에서 교반하고, (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (0.697 g, 3.11 mmol)을 첨가하고, 80℃에서 10시간 동안 가열하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (25 mL)과 EtOAc (40 mL x 2) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득한 다음, 이를 칼럼 크로마토그래피 (100-200 실리카 겔 사용, 칼럼을 헥산 중 80% 에틸 아세테이트로 용리시키미)에 의해 정제하여 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (350 mg, 0.562 mmol, 19.88% 수율), LCMS (m/z): 604.24 [M+H]+.
(9S)-3-클로로-N-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

DIPEA (2.52 mL, 14.13 mmol)에 이어서 트리포스겐 (0.503 g, 1.696 mmol)을 70℃에서 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (1.0 g, 2.83 mmol) 및 (S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민 (0.828 g, 3.67 mmol)의 용액에 첨가하고, 25℃에서 16시간 동안 교반하였다. 반응의 진행을 LCMS 및 TLC에 의해 모니터링하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (100 mL)과 EtOAc (2X100 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 샘플을 디클로로메탄에 로딩하고, 실리카 (Si) 5g 상에서 0-15% 메탄올-디클로로메탄을 80분에 걸쳐 사용하여 정제하였다. 적절한 분획을 합하고, 진공 하에 증발시켜 목적 생성물 300 mg을 회백색 고체로서 수득하였다, LCMS (m/z): 605.21 [M+H]+.
(9S)-3-클로로-N-(4-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

TEA (1.970 mL, 14.13 mmol)에 이어서 트리포스겐 (0.839 g, 2.83 mmol)을 테트라히드로푸란 (THF) (30 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (1 g, 2.83 mmol)의 용액에 실온에서 첨가하고, 6시간 동안 실온에서 교반하고, (R)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민 (0.700 g, 3.11 mmol)을 첨가하고, 80℃에서 10시간 동안 가열하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (25 mL)과 EtOAc (40 mL x 2) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-3-클로로-N-(4-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 검으로서 수득하였다 (300 mg, 0.411 mmol, 14.52% 수율), LCMS (m/z): 605.26 [M+H]+.
(9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
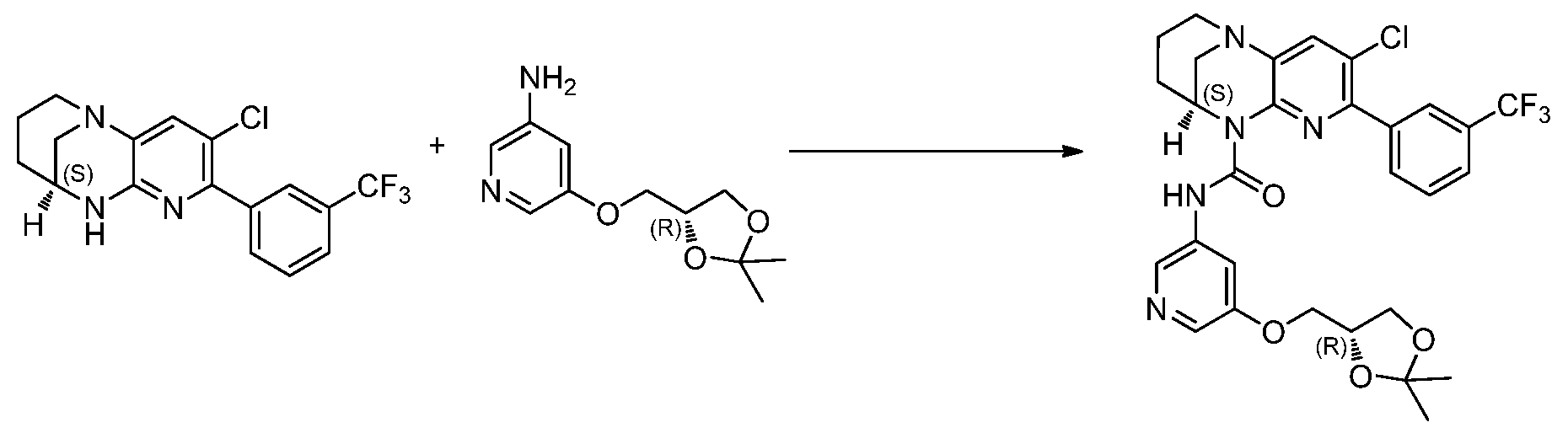
DIPEA (329 mg, 2.54 mmol)에 이어서 트리포스겐 (252 mg, 0.848 mmol)을 테트라히드로푸란 (THF) (50 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (300 mg, 0.848 mmol)의 용액에 첨가하고, 25℃에서 3시간 동안 교반하고, (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민 (380 mg, 1.696 mmol)을 첨가하고, 70℃에서 16시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (20 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 칼럼 크로마토그래피 (100-200) 메쉬에 의해 정제하고, 석유 에테르 중 30% EtOAc로 용리시켜 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (250 mg, 0.414 mmol, 48.8% 수율), LCMS (m/z): 604.00[M+H]+.
(9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (419 mg, 1.413 mmol)을 28℃에서 테트라히드로푸란 (THF) (20 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.413 mmol), 및 TEA (0.985 mL, 7.07 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 30분 동안 교반하고, (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민 (634 mg, 2.83 mmol)을 첨가하였다. 반응 혼합물을 70℃에서 15.5시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (30 mL)과 EtOAc (100 mL) 사이에 분배하였다. EtOAc 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하였다. 여과물을 증발시켜 조 물질을 수득하였다. 샘플을 디클로로메탄에 로딩하고, 실리카 (Si) 5g 상에서 0-50% 에틸 아세테이트/석유 에테르를 80분에 걸쳐 사용하여 정제하였다. 적절한 분획을 합하고, 진공 하에 증발시켜 목적 생성물 250 mg을 회백색 고체로서 수득하였다. LCMS (m/z): 604.22 [M+H]+.
(9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (20 mL) 중 고체 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (600 mg, 1.696 mmol)에 TEA (1.182 mL, 8.48 mmol)를 첨가하고, 고체 트리포스겐 (302 mg, 1.018 mmol)을 6시간 동안 교반하고, (R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민 (571 mg, 2.54 mmol)을 첨가하였다. 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 반응 혼합물을 TLC에 의해 모니터링하였다. 유기 상을 증발시키고, 물 50 mL를 첨가하고, 에틸 아세테이트로 추출하고, 포화 염수 100 mL로 세척하고, Na2SO4 상에서 건조시키고, 진공 하에 증발시켜 조 생성물을 수득하였다. 잔류물을 콤비플래쉬 (100% ACN 역상 칼럼)를 통해 정제하였다. 수집된 분획을 증발시켜 순수한 화합물 (9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 수득하였다 (180 mg, 0.273 mmol, 16.12% 수율), LCMS (m/z): 604.09 [M+H]+.
(9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

DIPEA (877 mg, 6.78 mmol)에 이어서 트리포스겐 (671 mg, 2.261 mmol)을 테트라히드로푸란 (THF) (25 mL) 중 (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민 (1014 mg, 4.52 mmol)의 용액에 첨가하고, 25℃에서 1시간 동안 교반하고, (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (800 mg, 2.261 mmol)을 첨가하고, 70℃에서 18시간 동안 가열하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (20 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다 (TLC 용리액: DCM 중 5% 메탄올 Rf: 0.3; UV 활성). 조 화합물을 칼럼 크로마토그래피 (C-18: 용리, 1% 수성 포름산 중 80% ACN으로 용리시킴)에 의해 정제하여 LCMS: 73%로 460 mg을 수득하였다. 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬)에 의해 헥산 중 50% EtOAc로 용리시켜 추가로 정제하여 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 갈색빛 점착성 물질로서 수득하였다 (300 mg, 0.490 mmol, 21.65% 수율), LCMS (m/z): 604.0 (M+H)+.
(9S)-3-클로로-N-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
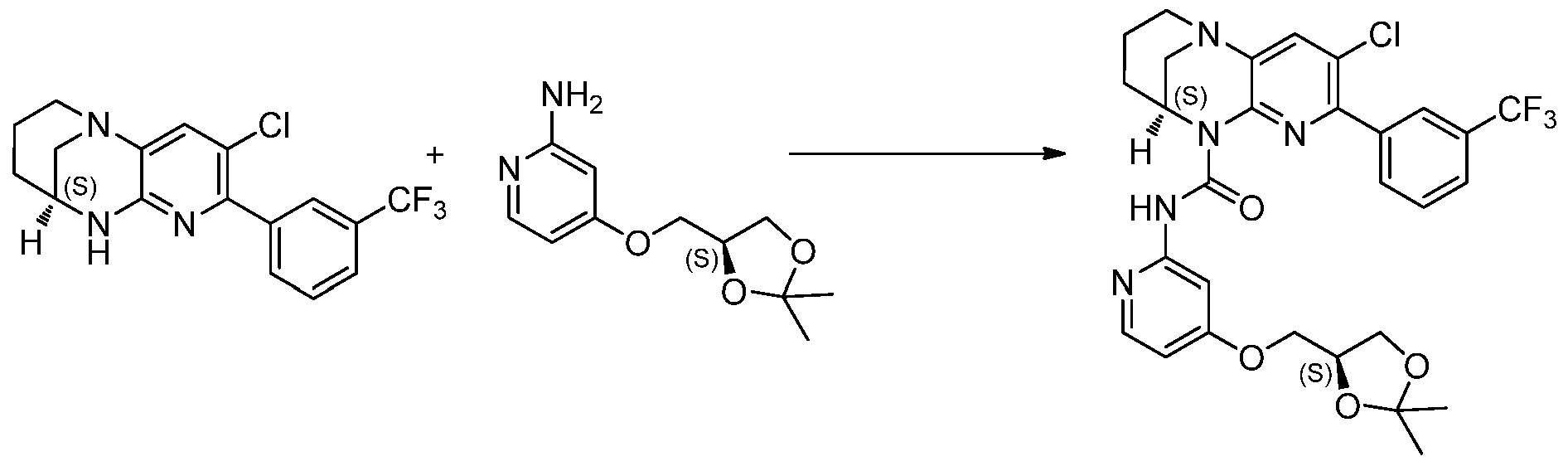
실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (20 mL) 중 고체 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (600 mg, 1.696 mmol)에 TEA (1.182 mL, 8.48 mmol)를 첨가하고, 고체 트리포스겐 (302 mg, 1.018 mmol)을 16시간 동안 교반하고, (S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (571 mg, 2.54 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 TLC에 의해 모니터링하였다. 유기 상을 증발시키고, 물 50 mL를 첨가하고, 에틸 아세테이트로 추출하고, 포화 염수 100 mL로 세척하고, Na2SO4 상에서 건조시키고, 진공 하에 증발시켜 조 생성물을 수득하였다. 조 생성물을 플래쉬 크로마토그래피에 의해 정제하고, 분획을 수집하여 화합물을 수득하고, 디에틸 에테르 및 펜탄으로 세척하고, 여과하고, 펜탄으로 세척하여 순수한 화합물 (9S)-3-클로로-N-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 수득하였다 (216 mg, 0.319 mmol, 18.79% 수율), LCMS (m/z): 604.11 [M+H]+.
(9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

절차: 트리포스겐 (377 mg, 1.272 mmol)을 28℃에서 테트라히드로푸란 (THF) (10.0 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (450 mg, 1.272 mmol), 및 TEA (0.886 mL, 6.36 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 30분 동안 교반하고, (S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-아민 (571 mg, 2.54 mmol)을 첨가하였다. 반응 혼합물을 70℃에서 10시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (5 mL)과 EtOAc (15 mL) 사이에 분배하였다. EtOAc 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하였다. 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 크로마토그래피 (그레이스, C-18 역 칼럼 사용, 이동상 A: 물 중 0.1% 포름산; B: MeOH, 용리액 A 중 91% B)에 의해 정제하였다. 합한 분획을 농축시키고, 포화 NaHCO3을 사용하여 염기성화시켰다. 수성 층을 DCM으로 추출하고, DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (300.0 mg, 0.489 mmol, 38.5% 수율), LCMS (m/z): 604.13[M+H]+.
(9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (50 mL) 중 고체 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (600 mg, 1.696 mmol)에 TEA (0.236 mL, 1.696 mmol)를 첨가하고, 고체 트리포스겐 (503 mg, 1.696 mmol)을 30분 동안 교반하고, (R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-아민 (380 mg, 1.696 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 TLC에 의해 모니터링하였다. 유기 상을 증발시키고, 물 50 mL를 첨가하고, 에틸 아세테이트로 추출하고, 포화 염수 100 mL로 세척하고, Na2SO4 상에서 건조시키고, 진공 하에 증발시켜 조 생성물을 수득하였다. 조 생성물을 실리카 겔 칼럼에 첨가하고, DCM/MeOH로 용리시켰다. 분획을 수집하여 일부 순수한 화합물을 수득하고, 다시 콤비플래쉬 (100% ACN; 120g 역상 칼럼)에 의해 정제하였다. 분획을 수집하여 순수한 화합물 (9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 수득하였다 (450 mg, 0.732 mmol, 43.2% 수율), LCMS (m/z): 604.02 [M+H]+.
(9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

트리포스겐 (377 mg, 1.272 mmol)을 28℃에서 테트라히드로푸란 (THF) (30 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (450 mg, 1.272 mmol) 및 TEA (1.064 mL, 7.63 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 2시간 동안 교반하고, (R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민 (573 mg, 2.54 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. TLC 용리액: 100% 에틸 아세테이트 Rf: 0.3, UV 활성. 반응 혼합물을 실온으로 냉각시키고, 용매를 감압 하에 완전히 증발시키고, 물 (10 mL)과 EtOAc (2 X 50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다. 조 물질을 DCM으로 희석하고, 중성 알루미나로 흡수시키고, 석유 에테르 중 35-40% EtOAc로 용리시키고, 분획을 수집하고, 농축시켜 (9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 갈색 고체로서 수득하였다 (220 mg, 0.244 mmol, 19.18% 수율), LCMS (m/z): 605.00 [M+H]+.
(9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

테트라히드로푸란 (THF) (20 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (400 mg, 1.131 mmol), 트리포스겐 (336 mg, 1.131 mmol) 및 트리에틸아민 (0.788 mL, 5.65 mmol)의 용액을 질소 하에 실온에서 30분 동안 교반하였다. 이 반응 혼합물에 (S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민 (509 mg, 2.261 mmol)을 첨가하였다. 반응 혼합물을 70℃에서 16시간 동안 교반하고, 반응의 진행을 TLC에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (10 mL)에 붓고, EtOAc (3 X 20 mL)로 추출하였다. 합한 유기 층을 물 (20 mL), 염수 용액 (20 mL)으로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. TLC 용리액: 100% EtOAc/헥산, Rf: 0.3, UV 활성. 조 화합물을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 석유 에테르 중 20% EtOAc로 용리시켜 순수한 (9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (300 mg, 0.395 mmol, 34.9% 수율), LCMS (m/z): 605.23 [M+H
(9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

실온에서 메탄올 (500 mL) 중 (9S)-2-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (50 g, 238 mmol)의 현탁액에 TEA (100 mL, 715 mmol) 및 PdCl2(dppf)-CH2Cl2 부가물 (9.74 g, 11.92 mmol)을 오토클레이브 내에서 첨가하고, CO(100psi) 기체로 충전하였다. 이를 탈기하고, 다시 CO(300-350 psi) 기체로 충전하였다. 반응 혼합물을 130℃로 16시간 동안 가열하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 출발 물질이 소모되었음을 나타내었다. 반응물을 실온으로 냉각시키고, 셀라이트를 통해 여과하고, 셀라이트 층을 메탄올(500mL)로 세척하였다. 여과물을 농축시켜 흑색 점착성 화합물을 수득하였다. 이 회백색 고체에 메탄올(200mL)을 첨가하고, 15분 동안 교반하고, 고체를 여과하고, 건조시켜 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 수득하였다 (30 g, 108 mmol, 45.1% 수율))(N36489-30-A2), LCMS (m/z): 234.07 (M+H)+.
(9S)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (20 mL), 물 (20 mL) 중 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (3 g, 12.86 mmol)의 용액에. 반응 혼합물을 0℃로 냉각시키고, 물 (10 mL) 중 LiOH (0.462 g, 19.29 mmol)의 용액을 첨가하였다. 반응물을 28℃에서 2시간 동안 교반하고, 반응의 진행을 TLC에 의해 모니터링하였다. 용매를 감압 하에 증발시키고, 물로 희석하고, 1N HCl (pH;4-5)을 사용하여 0℃에서 산성화시켰다. 고체를 여과하고, 물로 세척하고, 진공 하에 건조시켜 (9S)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 회백색 고체로서 수득하였다 (2.0 g, 8.54 mmol, 66.4% 수율), LCMS (m/z): 220.00 [M+H]+.
(9S)-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드의 합성

N,N-디메틸포름아미드 (DMF) (20 mL) 중 (9S)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (2.0 g, 9.12 mmol), HATU (5.20 g, 13.68 mmol)의 교반 용액에 DIPEA (6.37 mL, 36.5 mmol)를 첨가하였다. (R)-1,1,1-트리플루오로프로판-2-아민 (1.032 g, 9.12 mmol)을 0℃에서 반응 혼합물에 첨가하였다. 반응 혼합물을 28℃에서 16시간 동안 교반하고, 반응의 진행을 TLC에 의해 모니터링하였다. 반응 혼합물을 물 (40 mL)에 붓고, EtOAc(3X30 mL)로 추출하였다. 합한 유기 층을 염수 용액으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 중성 알루미나 및 용리액으로서 석유 에테르 중 30% EtOAc를 사용하여 정제하여 (9S)-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드를 회백색 고체로서 수득하였다 (1.8 g, 5.59 mmol, 61.3% 수율), LCMS (m/z): 314.85 [M+H]+.
(9S)-N10-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성
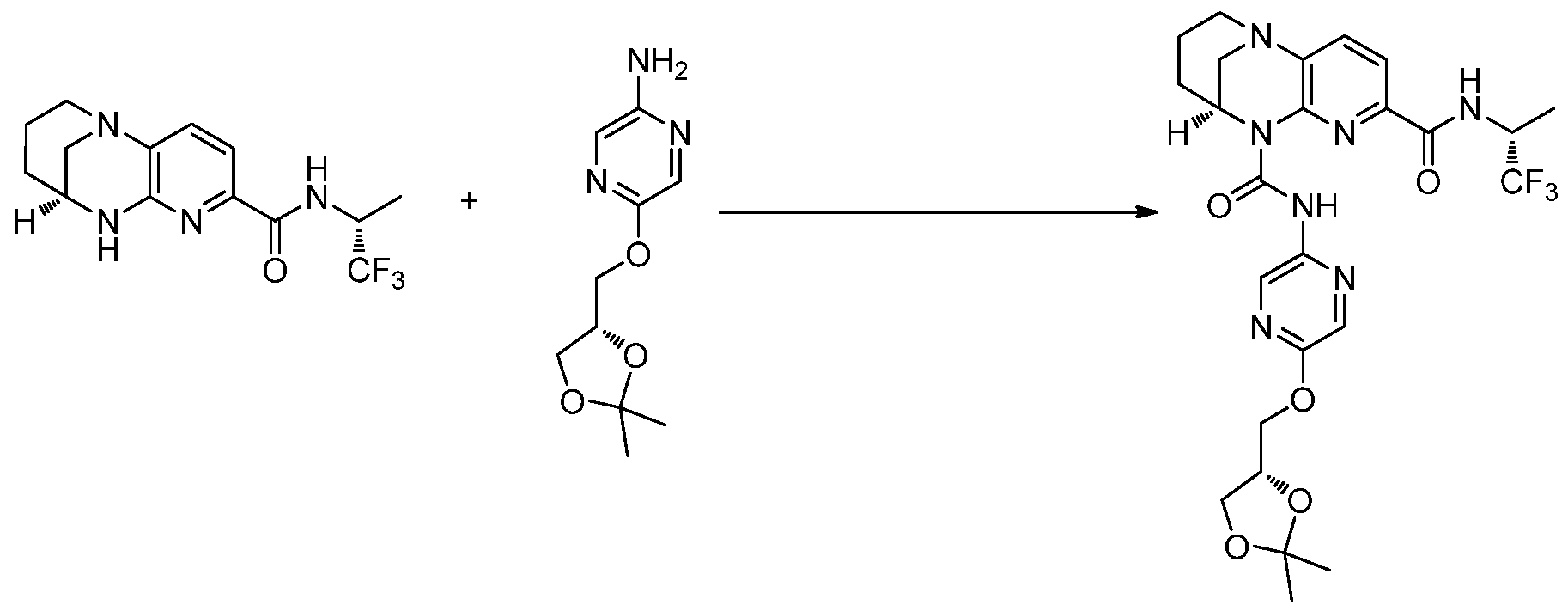
28℃에서 질소 하에 테트라히드로푸란 (THF) (20 mL) 중 (9S)-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드 (300 mg, 0.954 mmol), 트리포스겐 (283 mg, 0.954 mmol) 및 트리에틸아민 (0.665 mL, 4.77 mmol)의 교반 용액에 첨가하고, 반응 혼합물을 실온에서 30분 동안 교반하였다. (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민 (322 mg, 1.432 mmol)을 반응 혼합물에 첨가하였다. 반응 혼합물을 70℃에서 16시간 동안 교반하고, 반응의 진행을 TLC에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (30 mL)과 EtOAc (3X30 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 조 물질을 그레이스에 의해, C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: MeOH를 사용하여 정제하고, 생성물을 물 중 MeOH 및 0.1% 포름산 65%로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 침전된 고체를 여과하고, 건조시켜 (9S)-N10-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (200 mg, 0.349 mmol, 36.6% 수율), LCMS (m/z): 566.35 [M+H]+.
(9S)-10-((5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성
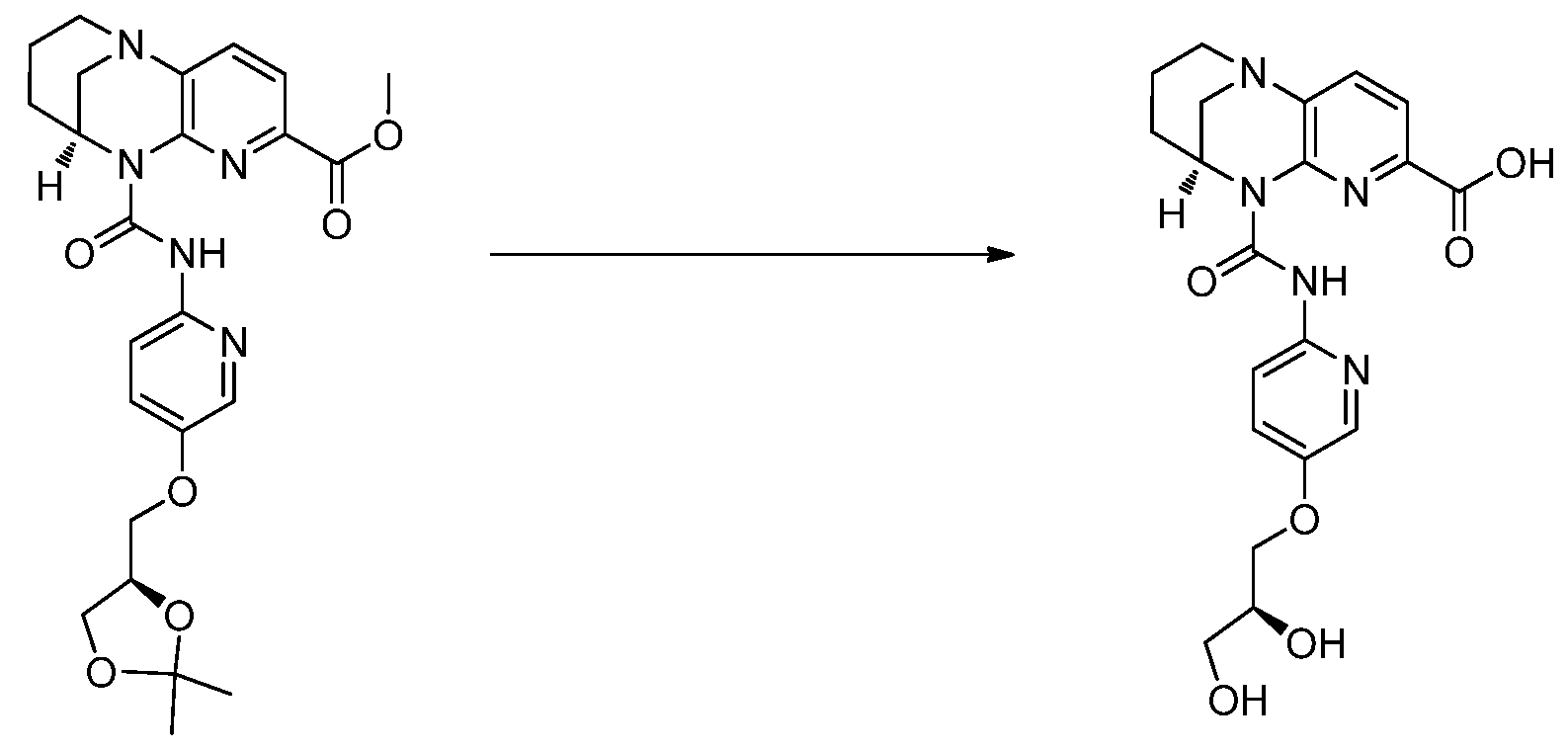
테트라히드로푸란 (THF) (10 mL), 물 (10 mL) 중 (9S)-메틸 10-((5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (400 mg, 0.827 mmol)의 교반 용액에 0℃에서 물 (1 mL) 중 LiOH (29.7 mg, 1.241 mmol)의 용액을 첨가하였다. 반응 혼합물을 28℃에서 2시간 동안 교반하고, 반응의 진행을 TLC에 의해 모니터링하였다. 반응 용매를 감압 하에 증발시키고, 물로 희석하고, 1N HCl (pH;2-3)로 0℃에서 산성화시키고, DCM (3x40 mL)으로 추출하였다. 합한 유기 층을 물, 염수 용액으로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 물질을 그레이스에 의해, C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: MeOH를 사용하여 정제하고, 생성물을 물 중 MeOH 및 0.1% 포름산 50%로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 침전된 고체를 여과하고, 건조시켜 (9S)-10-((5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 회백색 고체로서 수득하였다 (350 mg, 0.802 mmol, 97% 수율), LCMS (m/z): 430.46 [M+H]+.
(9S)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

28℃에서 질소 하에 교반하는 테트라히드로푸란 (THF) (10 mL) 중 (9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (500 mg, 1.065 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (241 mg, 2.130 mmol)의 용액에 HATU (486 mg, 1.278 mmol) 및 DIPEA (0.372 mL, 2.130 mmol)를 첨가하고, 반응 혼합물을 28℃에서 16시간 동안 교반하였다. 반응 혼합물을 빙수로 켄칭하고, 에틸 아세테이트 3x50 ml로 추출하고, 합한 유기 층을 염수 용액 100 ml로 세척하고, Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피 (100-200 실리카 겔)에 의해 용리액으로서 석유 에테르중 80% EtOAc의 구배 혼합물을 사용하여 정제하여 (9S)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (200 mg, 0.341 mmol, 32.0% 수율), LCMS (m/z): 565.15 [M+H]+.
(9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (35 mL) 및 물 (35.0 mL) 중 (9S)-메틸 10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (1.0 g, 2.068 mmol)의 용액에 LiOH (0.074 g, 3.10 mmol)를 첨가하였다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 감압 하에 농축시켜 화합물 (9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 회백색 고체로서 수득하였다 (900 mg, 1.876 mmol, 91% 수율), LCMS (m/z): 470.16 (M+H)+.
(9S)-메틸 10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

0℃에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (1.0 g, 4.29 mmol), 트리포스겐 (0.763 g, 2.57 mmol)의 용액에 DIPEA (3.74 mL, 21.43 mmol)를 첨가하였다. 이어서, 반응 혼합물을 28℃에서 30분 동안 교반하고, (S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (1.442 g, 6.43 mmol)을 첨가한 다음, 반응 혼합물을 80℃에서 15.5시간 동안 교반하였다. 반응물을 LCMS 및 TLC에 의해 모니터링하였다. 반응 혼합물을 냉수 (50 mL)에 붓고, 에틸 아세테이트 (2x 100mL)로 추출하였다. 유기 층을 무수 Na2SO4 상에서 건조시키고, 진공 하에 농축시켜 조 생성물을 수득하였다, LCMS (m/z): 484.14 (M+H)+.
(9S)-메틸 10-((4-브로모피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

0℃에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (1.0 g, 4.29 mmol), TEA (3.59 mL, 25.7 mmol)의 용액에 트리포스겐 (1.272 g, 4.29 mmol)을 첨가하였다. 이어서, 반응 혼합물을 30℃에서 30분 동안 교반하고, 4-브로모피리딘-2-아민 (2.225 g, 12.86 mmol)을 첨가한 다음, 반응 혼합물을 80℃에서 15.5시간 동안 교반하였다. 반응물을 LCMS 및 TLC에 의해 모니터링하였다. 반응 혼합물을 냉수 (20 mL)에 붓고, 에틸 아세테이트 (2x50mL)로 추출하였다. 유기 층을 무수 Na2SO4 상에서 건조시키고, 진공 하에 농축시켜 조 생성물을 수득하였다, LCMS (m/z): 431.9 (M+H)+.
(9S)-메틸 10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트; 및 (9S)-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성
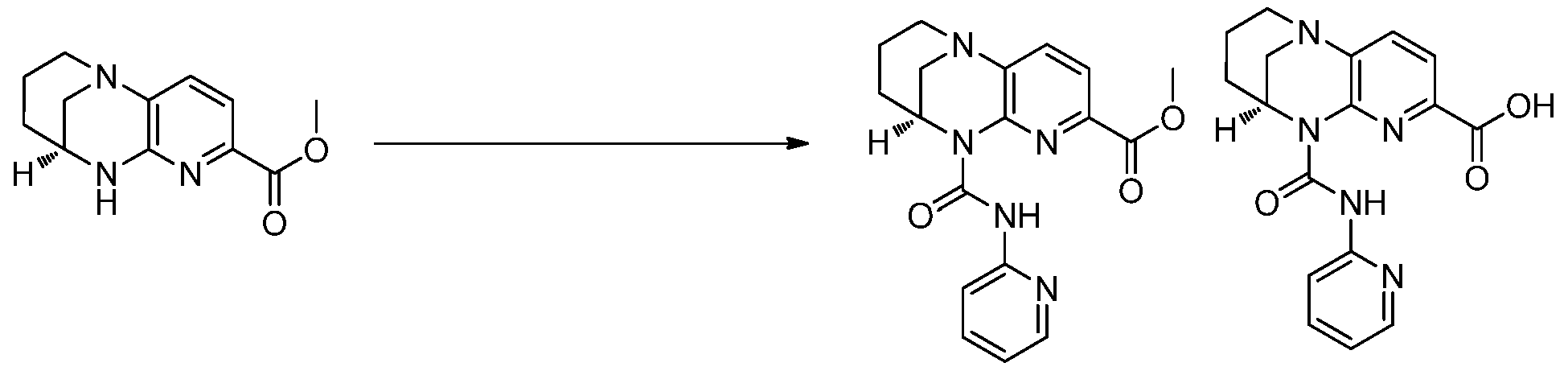
실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (100 mL) 중 NaH (8.57 g, 214 mmol)의 현탁액에 고체 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (10 g, 42.9 mmol)를 첨가하고, 실온에서 15분 후에 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (15.45 g, 64.3 mmol)을 첨가하였다. 생성된 반응 혼합물을 80℃에서 4시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 출발 물질이 소모되었음을 나타내었다. 반응물을 실온으로 냉각시키고, 에틸 아세테이트(100mL)로 희석하고, 빙냉수(250mL)로 켄칭하였다. 유기 층을 분리하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 다 (9S)-메틸 10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 갈색 고체 화합물로서 수득하였 (10 g, 11.35 mmol, 26.5% 수율). LCMS (m/z): 354.16 (M+H)+. 수성 층을 2N HCl을 사용하여 pH 5 내지 6으로 산성화시켰다. 수성 층을 진공 하에 농축시켜 회백색 고체 화합물을 수득하였다. 이를 DCM 중 10%메탄올 (1.0L) 중에 용해시키고, 무기물을 여과하였다. 여과물을 농축시키고, 건조시켜 (9S)-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 담갈색 고체로서 수득하였다 (3 g, 5.46 mmol, 12.75% 수율). LCMS (m/z): 340.0 (M+H)+.
(9S)-메틸 3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성
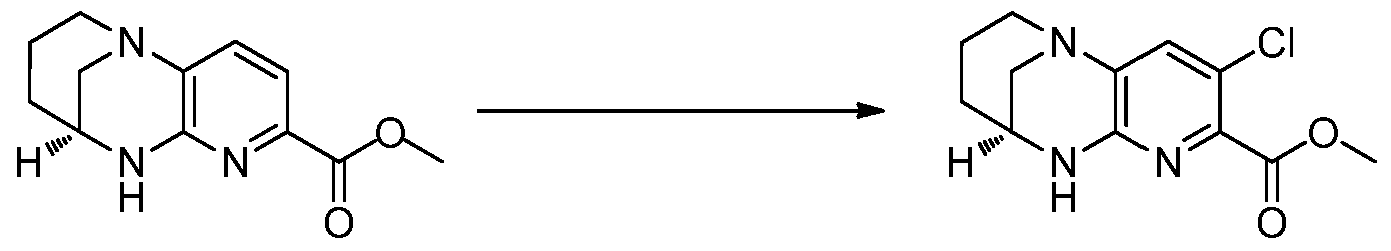
질소 하에 교반하는 0℃에서 클로로포름 (25 mL) 중 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (2.2 g, 9.43 mmol)의 현탁액에 1-클로로피롤리딘-2,5-디온 (1.637 g, 12.26 mmol)을 2분에 걸쳐 0℃에서 로트별로 첨가하였다. 0℃에서 30분 후, 반응 혼합물을 실온에서 3시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 2개의 비 극성 스폿 및 소량의 미반응 SM을 나타내었다. 반응물을 물 50 ml로 희석하고, DCM (2X100 ml)으로 추출하였다. 합한 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 수득하였다. 조 물질을 콤비플래쉬에 의해 실리카 겔 칼럼 (40 g, 헥산 중 50% EtOAc)을 사용하여 정제하였다. 순수한 화합물을 함유하는 분획을 합하고, 농축시켜 목적 화합물 (9S)-메틸 3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 연갈색 점성 액체로서 수득하였다 (1g, 3.29 mmol, 34.9% 수율), LCMS (m/z): 267.96 (M+H)+.
(9S)-메틸 10-(이속사졸-3-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

0℃에서 질소 하에 교반하는 테트라히드로푸란 (THF) (10 mL) 중 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (600 mg, 2.57 mmol), 트리포스겐 (458 mg, 1.543 mmol)의 용액에 DIPEA (2.246 mL, 12.86 mmol)를 첨가하였다. 이어서, 반응 혼합물을 30℃에서 30분 동안 교반하고, 이속사졸-3-아민 (324 mg, 3.86 mmol)을 첨가한 다음, 반응 혼합물을 80℃에서 15.5시간 동안 교반하였다. 반응물을 LCMS 및 TLC에 의해 모니터링하였다. 반응 혼합물을 냉수 (50 mL)에 붓고, 에틸 아세테이트 (2x 50mL)로 추출하였다. 유기 층을 무수 Na2SO4 상에서 건조시키고, 진공 하에 농축시켜 조 생성물을 수득하였다, LCMS (m/z): 344.17 (M+H)+.
(9S)-10-(이속사졸-3-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (25 mL) 및 물 (25.00 mL) 중 (9S)-메틸 10-(이속사졸-3-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (0.5 g, 1.456 mmol)의 용액에 LiOH (0.052 g, 2.184 mmol)를 첨가하였다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 감압 하에 농축시켜 모든 THF를 제거한 다음, 2N.HCl로 pH=4까지 산성화시켰다. 이어서 침전된 고체를 여과하고, 건조시켜 화합물 (9S)-10-(이속사졸-3-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 회백색 고체로서 수득하였다 (450 mg, 1.346 mmol, 92% 수율), LCMS (m/z): 330.05 (M+H)+.
(9S)-메틸 10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

0℃에서 질소 하에 교반하는 (9S)-메틸 7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (2 g, 8.57 mmol)의 용액에 트리포스겐 (2.54 g, 8.57 mmol) 및 TEA (7.17 mL, 51.4 mmol)를 첨가하였다. 이어서, 반응 혼합물을 30℃에서 30분 동안 교반하고, 6-메틸-1H-피라졸로[3,4-b]피리딘-3-아민 (507 mg, 3.42 mmol)을 첨가한 다음, 반응 혼합물을 90℃에서 16시간 동안 교반하였다. 반응물을 LCMS 및 TLC에 의해 모니터링하였다. 반응 혼합물을 냉수 (30 mL)에 붓고, 에틸 아세테이트 (2x 50 mL)로 추출하였다. 유기 층을 무수 Na2SO4 상에서 건조시키고, 진공 하에 농축시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피 (100-200 실리카 겔)에 의해 용리액으로서 DCM 중 10% 메탄올의 구배 혼합물을 사용하여 정제하여 화합물 (9S)-메틸 10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 연갈색 고체로서 수득하였다 (1.5 g, 2.338 mmol, 27.3% 수율), LCMS (m/z): 408.00 (M+H)+.
(9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (10 mL) 및 물 (10.00 mL) 중 (9S)-메틸 10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (1.5 g, 3.68 mmol)의 용액에 LiOH (0.132 g, 5.52 mmol)를 첨가하였다. 반응 혼합물을 실온에서 2시간 동안 교반하였다. 반응 혼합물을 감압 하에 농축시켜 Li 염으로서의 화합물 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 연갈색 고체로서 수득하였다 (1.2 g, 3.03 mmol, 82% 수율), LCMS (m/z): 394.1 (M+H)+.
(9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

실온에서 테트라히드로푸란 (THF) (80 mL) 중 NaH (0.448 g, 18.68 mmol)의 교반 용액에 조금씩 1분 동안 (9S)-메틸 3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (1 g, 3.74 mmol)를 첨가하였다. 실온에서 15분 후, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (1.077 g, 4.48 mmol)을 첨가하였다. 생성된 반응 혼합물을 실온에서 1시간 동안에 이어서 70℃에서 3시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 비 극성 스폿, 극성 스폿의 형성 및 SM의 완전한 소모를 나타내었다. 반응물을 빙냉수 100 ml로 희석하고, 수성 층을 EtOAc (100 ml)로 세척하고, 수성 층의 pH를 1N HCl로 4로 조정하고, 수성 층을 감압 하에 농축시켜 조 물질을 수득하고, 생성된 조 물질을 DCM 중 10% MeOH로 추출하고, 유기 층을 농축시켜 (9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 수득하였다 (500 mg, 0.744 mmol, 19.91% 수율), LCMS (m/z): 374.20 (M+H)+.
(9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성
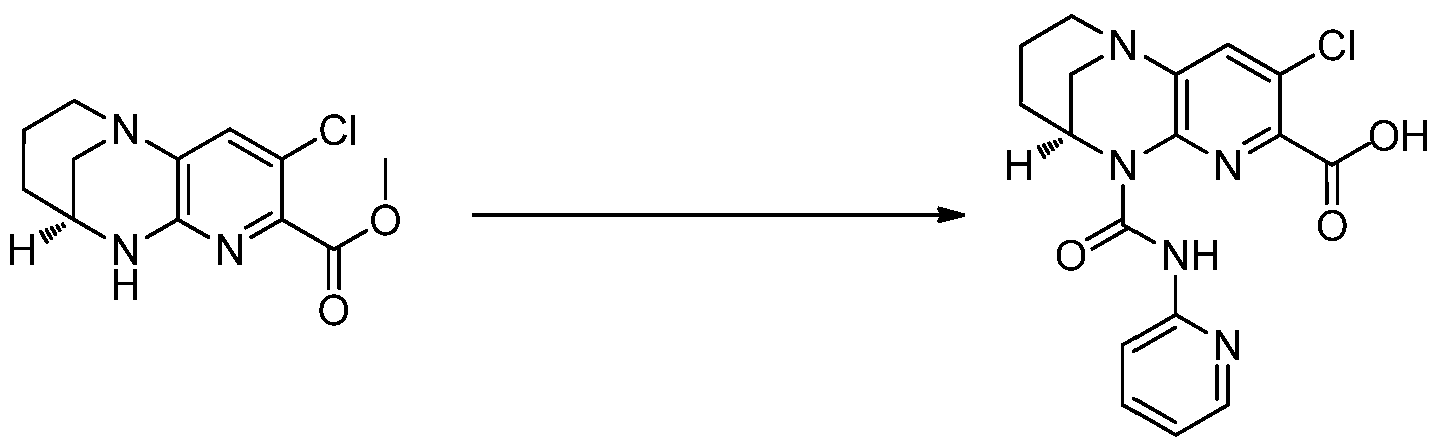
실온에서 테트라히드로푸란 (THF) (30 mL) 중 NaH (224 mg, 5.60 mmol)의 교반 용액에 조금씩 1분 동안 (9S)-메틸 3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (500mg, 1.868 mmol)를 첨가하였다. 실온에서 15분 후, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (538 mg, 2.241 mmol)을 첨가하였다. 생성된 반응 혼합물을 실온에서 1시간 동안에 이어서 70℃에서 3시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 비 극성 스폿, 극성 스폿의 형성 및 SM의 완전한 소모를 나타내었다. 반응물을 빙냉수 100 ml로 희석하고, 수성 층을 EtOAc (100 ml)로 세척하고, 수성 층의 pH를 1N HCl로 4로 조정하고, 수성 층을 감압 하에 농축시켜 조 물질을 수득하고, 생성된 조 물질을 DCM 중 10% MeOH로 추출하고, 유기 층을 농축시켜 (9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 갈색 N35384-85-A1로서 수득하였다 (300 mg, 0.525 mmol, 28.1% 수율), LCMS (m/z): 374.08 (M+H)+.
(9S)-3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (35 mL) 및 물 (35.0 mL) 중 (9S)-메틸 3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (2.0 g, 7.47 mmol)의 용액에 LiOH (0.268 g, 11.21 mmol)를 실온에서 첨가하고, 16시간 동안 교반하였다 (TLC 용리액: 에틸 아세테이트 중 10% MeOH Rf: 0.2; UV 활성). 반응 혼합물을 물 (100 mL)로 희석하고, 에틸 아세테이트 (2X 100 mL)로 추출하여 모든 불순물을 제거하였다. 수성 층을 수성 HCl (5 mL)을 사용하여 산성화시키고, 감압 하에 농축시켜 (9S)-3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 담갈색 고체로서 수득하였다 (1.02 g, 3.32 mmol, 44.4% 수율). LCMS (m/z): 253.91 [M+H]+.
(9S)-3-클로로-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드의 합성

실온에서 질소 하에 N,N-디메틸포름아미드 (DMF) (5 mL) 중 (9S)-3-클로로-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (1.02 g, 4.02 mmol)의 용액에 HATU (1.835 g, 4.82 mmol), DIPEA (1.404 mL, 8.04 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (0.682 g, 6.03 mmol)을 첨가하고, 실온에서 16시간 동안 교반하였다. 반응 혼합물을 빙수로 희석하고, 에틸 아세테이트 2x 100 mL로 추출하였다. 합한 유기 층을 염수로 세척하고, Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (100-200 실리카 겔, 5% CH2Cl2/MeOH로 용리시킴)에 의해 정제하여 (9S)-3-클로로-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드를 회백색 고체로서 수득하였다 (800 mg, 1.991 mmol, 49.5% 수율), LCMS (m/z): 348.96 [M+H]+.
(9S)-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

실온에서 테트라히드로푸란 (THF) (100 mL), 물 (100 mL) 중 (9S)-메틸 10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (10 g, 28.3 mmol)의 용액에 수산화리튬 (1.355 g, 56.6 mmol)을 첨가하였다. 반응 혼합물을 실온에서 3시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 출발 물질이 소모되었음을 나타내었다. 반응물을 에틸 아세테이트(100mL)로 희석하고, 유기 층을 분리하였다. 수성 층을 1N HCl을 사용하여 PH- 5-6으로 산성화시켰다. 수성 층을 농축시켜 담갈색 고체를 수득하였다. 이에 DCM(500mL) 중 10%메탄올을 첨가하고, 15분 동안 교반하고, 무기물을 여과하였다. 여과물물을 농축시켜 (9S)-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 담갈색 고체로서 수득하였다 (6 g, 17.47 mmol, 61.7% 수율). LCMS (m/z): 340.15 (M+H)+.
(9S)-10-((4-브로모피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (35 mL) 및 물 (35.0 mL) 중 (9S)-메틸 10-((4-브로모피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (0.800 g, 1.851 mmol)의 용액에 LiOH (0.066 g, 2.78 mmol)를 첨가하였다. 반응 혼합물을 실온에서 1시간 동안 교반하였다. 반응 혼합물을 감압 하에 농축시켜 생성물 Li 염을 수득하였다. 이어서, 염을 물 (20 mL)로 희석하고, 에틸 아세테이트 (2X 50 mL)로 완전히 세척한 다음, 수성 층을 수성 HCl로 산성화시켜 (9S)-10-((4-브로모피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 회백색 고체로서 수득하였다 (600 mg, 1.357 mmol, 73.3% 수율), LCMS (m/z): 419.90 [M+H]+.
(9S)-N10-(4-브로모피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성
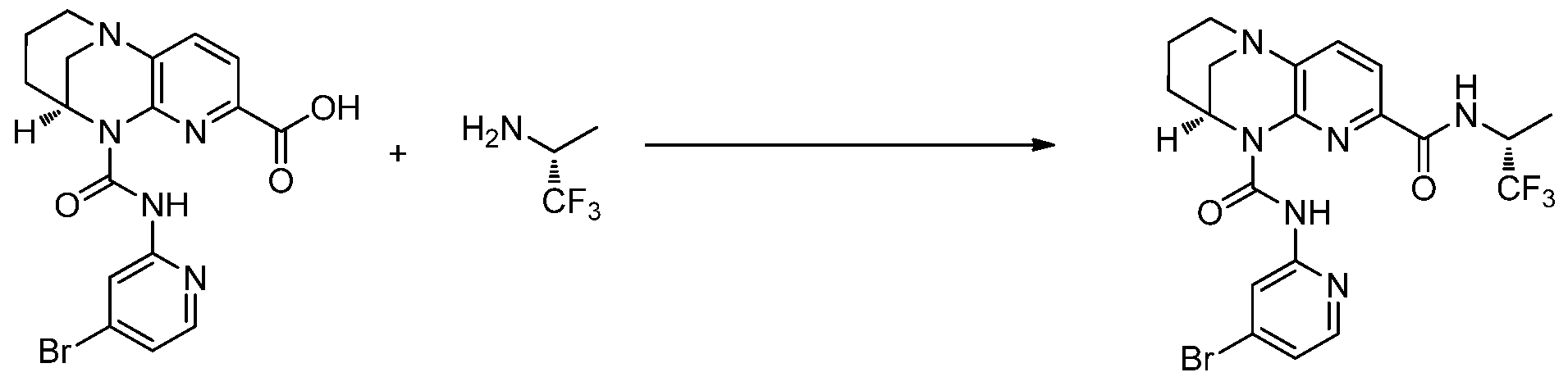
28℃에서 질소 하에 교반하는 N,N-디메틸포름아미드 (DMF) (15 mL) 중 (9S)-10-((4-브로모피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (480 mg, 1.148 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (195 mg, 1.721 mmol)의 용액에 HATU (524 mg, 1.377 mmol) 및 DIPEA (0.401 mL, 2.295 mmol)를 첨가하고, 반응 혼합물을 28℃에서 16시간 동안 교반하였다. 반응 혼합물을 빙수로 켄칭하고, 에틸 아세테이트 3x20 ml로 추출하고, 합한 유기 층을 염수 용액 20 ml로 세척하고, Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 100-200 실리카 겔 및 용리액으로서 80-100% 에틸 아세테이트/헥산을 사용하여 정제하였다. 수집된 분획을 증류시켜 (9S)-N10-(4-브로모피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (500 mg, 0.960 mmol, 84% 수율), LCMS (m/z): 565.9 [M+H]+.
(9S)-N2-(2,2-디플루오로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

20℃에서 질소 하에 교반하는 테트라히드로푸란 (THF) (10 mL) 중 (9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (500 mg, 1.065 mmol)의 용액에 HATU (486 mg, 1.278 mmol) 및 DIPEA (0.372 mL, 2.130 mmol)를 첨가하고, 반응 혼합물을 28℃에서 16시간 동안 교반하였다. 반응 혼합물을 빙수로 희석하고, 에틸 아세테이트 3x50 ml로 추출하고, 합한 유기 층을 염수 용액 100 ml로 세척하고, Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 100-200 메쉬 실리카겔을 사용하고, 80%에서 순수한 에틸 아세테이트로 용리시켜 정제하였다, LCMS (m/z): 547.35 [M+H]+.
하기의 합성:
(9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드(피크-1); 및
(9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (피크-2)

ACN (10 mL) 중 (9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (2.5 g, 5.32 mmol)의 교반 용액에 HATU (4.05 g, 10.65 mmol) 및 DMAP (1.301 g, 10.65 mmol)를 첨가하였다. 반응 혼합물을 실온에서 15분 동안 교반한 다음, 밀봉된 튜브 중에서 2,2-디플루오로시클로프로판아민, 히드로클로라이드 (1.380 g, 10.65 mmol)를 첨가하고, 반응 혼합물을 90℃에서 16시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. 반응 혼합물을 실온으로 냉각시키고, 물(50 mL)로 희석하고, 에틸 아세테이트(2x50 mL)로 추출하고, 유기 층을 분리하고, Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 물질을 수득하였다. 수득된 조 물질을 칼럼에 의해 실리카 (100-200 메쉬/ 용리액으로서 석유 에테르 중 1- 80% EtOH)를 사용하여 정제하고, 수집된 분획을 감압 하에 농축시켜 순수한 화합물 700mg을 수득하였다. 수득된 순수한 화합물을 SFC에 의해 정제하였다.
SFC 정제하여 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (피크-1) (230 mg, 0.414 mmol, 7.77% 수율) 및 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (피크-2) (250 mg, 0.454 mmol, 8.53% 수율)를 수득하였다.
칼럼/치수: 키랄팩 IC(250 X30)mm,5μ
% CO2: 50.0%
% 공용매: 50.0% (100% 메탄올)
총 유량: 100.0 g/분
배압: 100.0 bar
UV: 210 nm
스택 시간: 6.5분
로드/주입: 9.5 mg
용해도: 메탄올
총 주입 횟수: 70
기기 세부사항: 제조/모델: 타르 SFC- 200-뉴-2
LCMS (m/z): 545.02 [M+H]+. (피크 1)
LCMS (m/z): 545.89 [M+H]+. (피크 2)
(9S)-N10-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

N,N-디메틸포름아미드 (DMF) (8 mL) 중 (9S)-10-((5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (400 mg, 0.827 mmol)의 교반 용액에 0℃에서 DIPEA (0.722 mL, 4.14 mmol), HATU (472 mg, 1.241 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (140 mg, 1.241 mmol)을 첨가하였다. 반응 혼합물을 27℃에서 3시간 동안 교반하였다. 반응 혼합물을 냉수 (100 mL)로 희석하고, 15분 동안 교반하였다. 침전된 고체를 부흐너 깔때기를 통해 여과하고, 물로 세척하고, 감압 하에 건조시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피 (중성 알루미나, 용리액: 헥산 중 65-70% 에틸 아세테이트)에 의해 정제하였다. 수집된 분획을 감압 하에 농축시켜 목적 생성물 (9S)-N10-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 갈색 고체로서 수득하였다 (350 mg, 0.580 mmol, 70.1% 수율), LCMS (m/z): 579.22 [M+H]+.
(9S)-10-((5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (20 mL) & 물 (3.0 mL) 중 (9S)-메틸 10-((5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (450 mg, 0.904 mmol)의 교반 용액에 LiOH.H2O (21.66 mg, 0.904 mmol)를 0℃에서 첨가하였다. 반응 혼합물을 27℃에서 6시간 동안 교반하였다. 반응 혼합물 용매를 감압 하에 완전히 증발시키고, 시트르산 용액을 사용하여 0℃에서 산성화시켰다. 수성 층을 디클로로메탄 (2x 50 mL)으로 추출하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다. 조 화합물을 디에틸 에테르 및 펜탄으로 연화처리하였다, LCMS (m/z): 484.28 [M+H]+.
(9S)-메틸 10-((5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

테트라히드로푸란 (THF) (45 mL) 중 (9S)-메틸 4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (600 mg, 2.426 mmol)의 교반 용액에 질소 하에 28℃에서 TEA (2.029 mL, 14.56 mmol) 및 트리포스겐 (720 mg, 2.426 mmol)을 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반한 다음, (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (1088 mg, 4.85 mmol)을 반응 혼합물에 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 용매를 감압 하에 완전히 증발시키고, 물 (30 mL)과 EtOAc (2 X 40 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다. TLC 용리액: 100% EtOAc/헥산, Rf : 0.3, UV 활성. 조 화합물을 그레이스에 의해, C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: ACN을 사용하여 정제하고, 생성물을 물 중 70-75% ACN/0.1% 포름산으로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 수성 층을 DCM으로 추출하였다. DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 순수한 (9S)-메틸 10-((5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 회백색 고체로서 수득하였다 (500 mg, 0.936 mmol, 38.6% 수율), LCMS (m/z): 498.48 [M+H]+.
(9S)-메틸 4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성
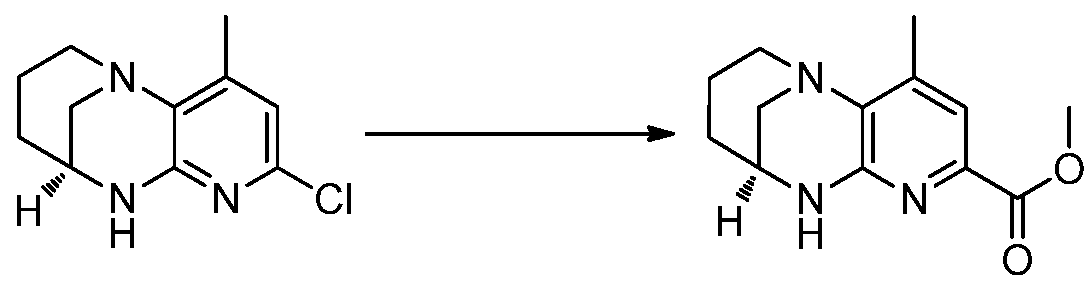
메탄올 (100 mL) 중 (9S)-2-클로로-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (6 g, 26.8 mmol)의 교반 용액에 탈기된 30분 동안에 이어서 트리에틸아민 (18.69 mL, 134 mmol) 및 PdCl2(dppf)-CH2Cl2 부가물 (1.095 g, 1.341 mmol)을 첨가하고, 300 psi CO 기체로 충전하였다. 반응 혼합물을 강철 용기 중에서 115℃에서 16시간 동안 교반하였다. 반응물을 TLC에 의해 모니터링하였다. 반응 혼합물을 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 DCM (50 mL) 중에 용해시키고, 물 (10 mL)로 세척하였다. DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-메틸 4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 갈색 고체로서 수득하였다 (7g, 23.76 mmol, 89% 수율), LCMS (m/z): 248.11 [M+H]+.
(9S)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

N,N-디메틸포름아미드 (DMF) (8 mL) 중 (9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (450 mg, 0.931 mmol)의 교반 용액에 0℃에서 DIPEA (0.813 mL, 4.65 mmol), HATU (531 mg, 1.396 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (158 mg, 1.396 mmol)을 첨가하였다. 반응 혼합물을 27℃에서 3시간 동안 교반하였다. 반응 혼합물을 냉수 (100 mL)로 희석하고, 15분 동안 교반하였다. 침전된 고체를 부흐너 깔때기를 통해 여과하고, 물로 세척하고, 감압 하에 건조시켜 조 화합물을 수득하였다. 화합물을 에테르로 연화처리하여 (9S)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (330 mg, 0.481 mmol, 51.7% 수율), LCMS (m/z): 579.32 [M+H]+.
(9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산의 합성

테트라히드로푸란 (THF) (20.0 mL) & 물 (5.00 mL)중 (9S)-메틸 10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (550 mg, 1.105 mmol)의 교반 용액에 LiOH.H2O (39.7 mg, 1.658 mmol)를 0℃에서 첨가하였다. 반응 혼합물을 27℃에서 6시간 동안 교반하였다. 반응 혼합물 용매를 감압 하에 완전히 증발시키고, 시트르산 용액을 사용하여 0℃에서 산성화시켰다. 수성 층을 디클로로메탄(2x 50 mL)으로 추출하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다. 조 화합물을 디에틸 에테르 및 펜탄으로 연화처리하여 (9S)-10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산을 회백색 고체로서 수득하였다 (450 mg, 0.892 mmol, 81% 수율), LCMS (m/z):484.30 [M+H]+.
(9S)-메틸 10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트의 합성

테트라히드로푸란 (THF) (55 mL) 중 (9S)-메틸 4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트 (800 mg, 3.24 mmol)의 교반 용액에 질소 하에 28℃에서 TEA (2.71 mL, 19.41 mmol) 및 트리포스겐 (960 mg, 3.24 mmol)을 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반한 다음, (S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민 (2176 mg, 9.71 mmol)을 반응 혼합물에 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 용매를 감압 하에 완전히 증발시키고, 물 (30 mL)과 EtOAc (2 X 40 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다. TLC 용리액: 100% EtOAc/헥산, Rf : 0.2, UV 활성. 조 화합물을 그레이스에 의해, C-18 역 칼럼, 이동상 A: 물 중 0.1% 포름산; B: ACN을 사용하여 정제하고, 생성물을 물 중 65-70% ACN/0.1% 포름산으로 용리시켰다. 용매를 증발시키고, 포화 NaHCO3으로 염기성화시켰다. 수성 층을 DCM으로 추출하였다. DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 순수한 (9S)-메틸 10-((4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)카르바모일)-4-메틸-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실레이트를 회백색 고체로서 수득하였다 (650mg, 1.125 mmol, 34.8% 수율), LCMS (m/z): 498.48 [M+H]+.
(R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민의 합성

0℃에서 질소 하에 N-메틸-2-피롤리돈 (NMP) (100 mL) 중 NaH (11.67 g, 292 mmol)의 교반 현탁액에 N-메틸-2-피롤리돈 (NMP) (100 mL) 중 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (25.7 g, 194 mmol)의 용액을 0℃에서 10분 동안 적가하였다. 10분 후, N-메틸-2-피롤리돈 (NMP) (100 mL) 중 6-클로로피리딘-2-아민 (25 g, 194 mmol)의 용액을 10분 동안 0℃에서 적가하였다. 반응 혼합물을 100℃에서 36시간 동안 가열하였다. TLC는 생성물과 함께 소량의 출발 물질을 나타내었다.
반응 혼합물을 빙냉수 (600 mL)에 붓고, 수성 층을 EtOAc (2 x 500 mL)로 추출하였다. 유기 층을 물 (3 x 300 mL)로 세척하여 과량의 NMP를 제거하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 100-200 실리카 겔 및 용리액으로서 석유 에테르 중 12-15% EtOAc를 사용하여 정제하여 (R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민을 황색 농후한 액체로서 수득하였다 (10 g, 44.6 mmol, 22.93% 수율).
(S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민의 합성
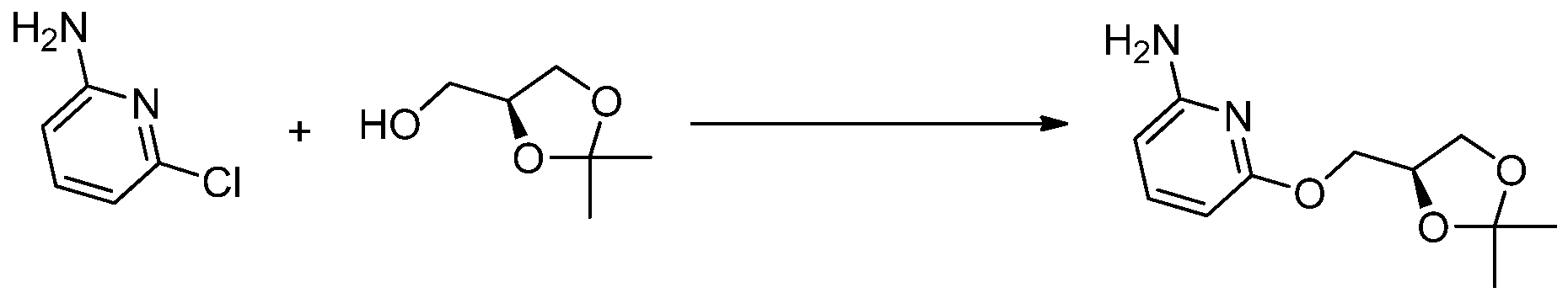
0℃에서 질소 하에 N-메틸-2-피롤리돈 (NMP) (800 mL) 중 NaH (62.2 g, 1556 mmol)의 교반 현탁액에 N-메틸-2-피롤리돈 (NMP) (300 mL) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (206 g, 1556 mmol)의 용액을 2시간 동안 적가하였다. 추가로 10분 동안 교반한 후 N-메틸-2-피롤리돈 (NMP) (300 mL) 중 6-클로로피리딘-2-아민 (200 g, 1556 mmol)의 용액을 30분 동안 0℃에서 적가하였다. 반응 혼합물을 120℃에서 48시간 동안 교반하였다. TLC는 출발 물질이 있었음을 나타내었다. 반응 혼합물을 빙냉수 (2000 mL)에 붓고, 수성 층을 EtOAc (3 x 1000 mL)로 추출하였다. 합한 유기 층을 물 (3 x 1000 mL)로 세척하여 과량의 NMP를 제거하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 100-200 실리카 겔 (용리액 석유 에테르 중 12-15% EtOAc)을 사용하여 정제하여 목적 순수한 생성물 (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민을 황색 점성 액체로서 수득하였다 (75 g, 325 mmol, 20.92% 수율). LCMS (m/z): 225 [M+H]+.
(R)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민의 합성
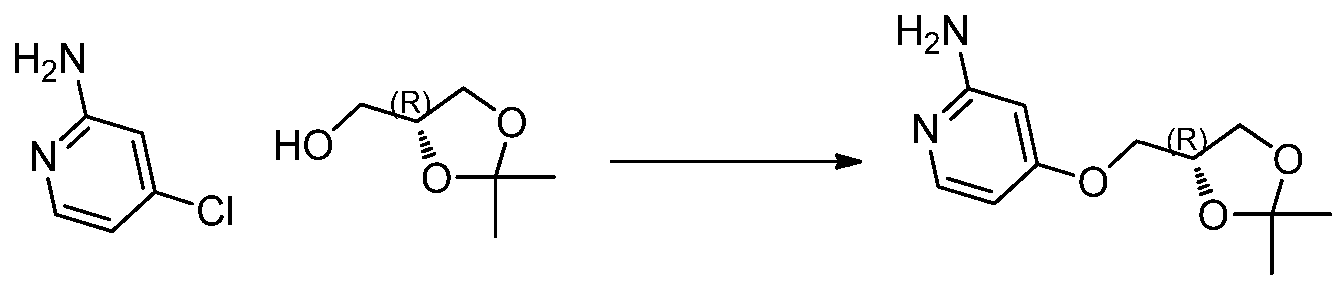
밀봉된 튜브 중에서 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (3.000 g, 22.70 mmol), 4-클로로피리딘-2-아민 (1.459 g, 11.35 mmol) 및 나트륨 (0.522 g, 22.70 mmol)의 현탁액에. 반응 혼합물을 140℃에서 16시간 동안 교반하였다. 다음에, 반응 혼합물을 실온으로 냉각시키고, MeOH 중에 용해시키고, 빙수에 붓고, EtOAc로 추출하였다. 유기 상을 염수 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 실리카 겔을 사용하여 정제하고, 2-3% MeOH/DCM으로 용리시켜 순수한 화합물을 수득하였다 (1.1g, 21%), LCMS (m/z) 225.2 [M+H]+.
(S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민의 합성
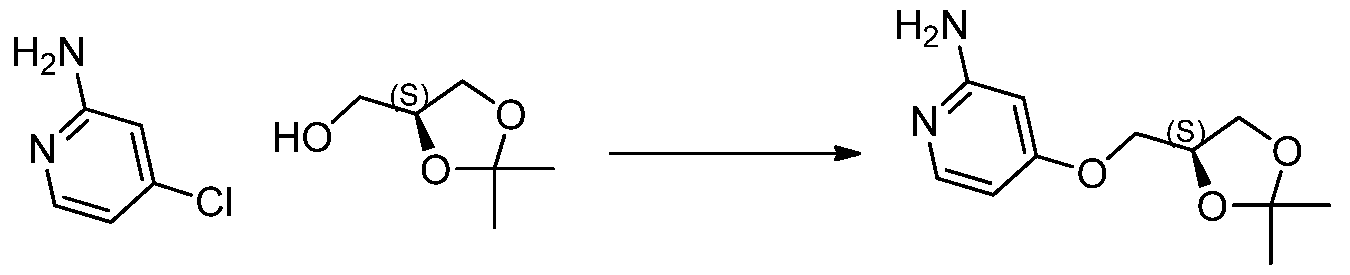
밀봉된 튜브 중에서 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (3.000 g, 22.70 mmol), 4-클로로피리딘-2-아민 (1.459 g, 11.35 mmol) 및 나트륨 (0.522 g, 22.70 mmol)의 현탁액에. 반응 혼합물을 140℃에서 16시간 동안 교반한 다음, 실온으로 냉각시키고, MeOH 중에 용해시키고, 빙수에 붓고, EtOAc로 추출하였다. 유기 상을 염수 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 여과하고, 증발시켰다. 조 물질을 실리카 겔 칼럼 크로마토그래피에 의해 정제하고, 2-3% MeOH/DCM로 용리시켜 목적 생성물을 수득하였다 (1.2g, 22%), LCMS (m/z) 225.2 [M+H]+.
(R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민의 합성
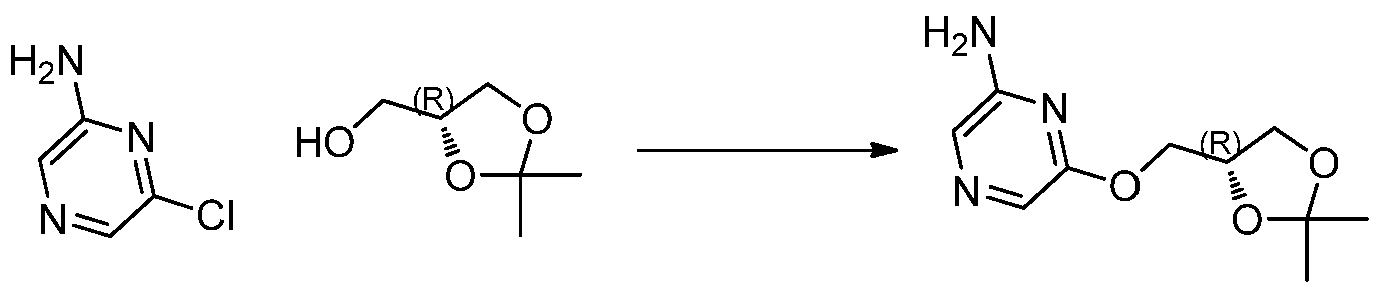
0℃에서 질소 하에 교반하는 테트라히드로푸란 (THF) (50 mL) 중 6-클로로피라진-2-아민 (5 g, 38.6 mmol), 수소화나트륨 (2.316 g, 57.9 mmol) 및 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (5.61 g, 42.5 mmol)의 용액에 첨가하고, 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 반응 혼합물을 빙냉수로 켄칭하고, 에틸 아세테이트로 추출하였다. 유기 층을 Na2SO4 상에서 건조시켰다. 용매를 감압 하에 증발시켜 조 생성물을 수득하였다. 조 생성물을 실리카 겔 칼럼에 첨가하고, DCM/MeOH로 용리시켰다. 생성물을 포함하는 분획을 합하고, 감압 하에 증발시켜 목적 생성물을 수득하였다 (2.8g, 11.9 mmol, 31%), LCMS (m/z) 225.9 [M+H]+.
(S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민의 합성

6-클로로피라진-2-아민 (0.980 g, 7.57 mmol),(S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (2 g, 15.13 mmol) 및 나트륨 (0.348 g, 15.13 mmol)을 밀봉 튜브 중에서 녹이고, 130℃에서 16시간 동안 가열한 다음, 반응 혼합물을 메탄올 및 빙냉수 (100 mL)로 켄칭하고, 에틸 아세테이트 (5 x 50 mL)로 추출하였다. 합한 유기 층을 물, 포화 염수 용액으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 농축시켜 생성물을 수득하였다 (1 g, 4.26 mmol, 28.2% 수율), LCMS (m/z) 265.1 [M+H]+.
(S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민의 합성
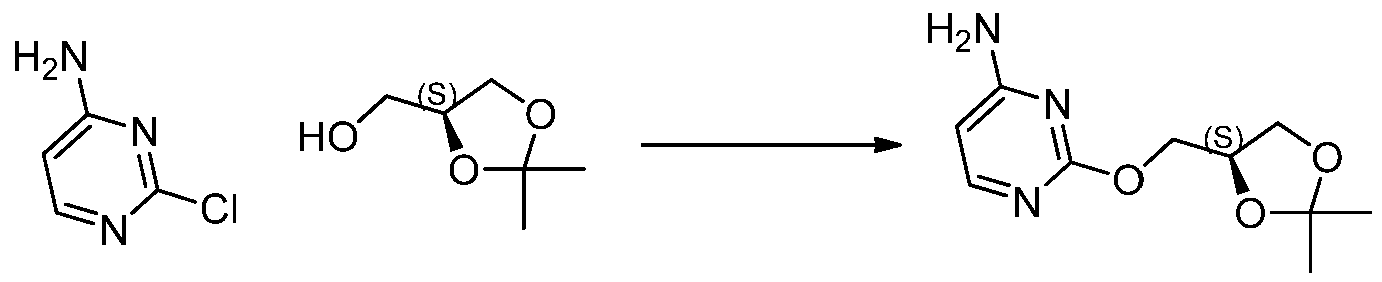
실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (50 mL) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (10.20 g, 77 mmol), 및 NaH (4.63 g, 116 mmol)의 현탁액에 2-클로로피리미딘-4-아민 (5 g, 38.6 mmol)을 15분에 걸쳐 조금씩 첨가하였다. 반응 혼합물을 70℃에서 16시간 동안 교반하였다. 다음에, 반응 혼합물을 수성 NaHCO3의 용액으로 켄칭한 다음, EtOAc로 추출하고, Na2SO4 상에서 건조시키고, 증발시켰다. 조 생성물을 실리카 겔 칼럼에 첨가하고, 50% Hex/EtOAc로 용리시켰다. 수집된 분획을 증발시켜 목적 생성물을 회백색 고체로서 수득하였다 (3 g, 11.84 mmol, 30.7% 수율), LCMS (m/z) 226.2 [M+H]+.
(R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민의 합성

실온에서 테트라히드로푸란 (THF) (30 mL) 중 수소화나트륨 (0.817 g, 34.1 mmol)의 용액에 THF (5 mL) 중 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (3 g, 22.70 mmol)의 용액을 1분에 걸쳐 첨가하고, 실온에서 15분 동안 교반한 다음, 2-클로로피리미딘-4-아민 (2.059 g, 15.89 mmol)을 실온에서 조금씩 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 물에 붓고, EtOAc (3 X 100mL)로 추출하였다. 이어서, 합한 유기 층을 물, 염수 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 증발시켜 조 화합물 4.0 g을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 100-200 실리카 겔 메쉬를 사용하여 정제하고, 2-3% MeOH/DCM으로 용리시켜 순수한 화합물을 수득하였다 (2.5g, 10.42 mmol, 46%), LCMS (m/z) 226.2 [M+H]+.
(S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-아민의 합성
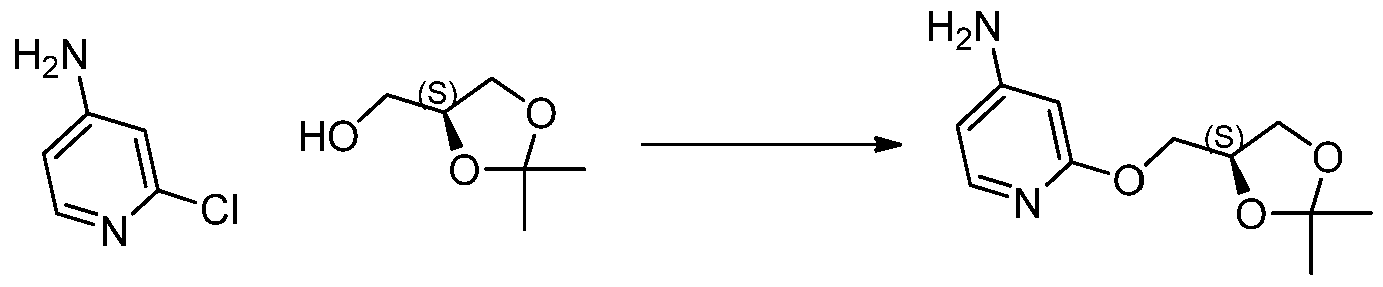
2-클로로피리딘-4-아민 (1.459 g, 11.35 mmol), (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (3.0 g, 22.70 mmol)의 현탁액에 나트륨 (0.522 g, 22.70 mmol)을 첨가하였다. 반응 혼합물을 140℃에서 16시간 동안 교반하고, 반응의 진행을 TLC에 의해 모니터링하였다. 반응 혼합물을 MeOH 중에 용해시키고, 빙수에 붓고, EtOAc (3 X 100mL)로 추출하였다. 이어서, 합한 유기 층을 물, 염수 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 증발시켜 조 화합물 4.0 g을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 100-200 실리카 겔 메쉬를 사용하여 정제하고, 2-3% MeOH/DCM으로 용리시켜 (S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-아민을 수득하였다 (2.5 g, 10.73 mmol, 47.3% 수율), LCMS (m/z) 225.3 [M+H]+.
(R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-아민의 합성

밀봉된 튜브 중에서 실온에서 2-클로로피리딘-4-아민 (4 g, 31.1 mmol), (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (2.056 g, 15.56 mmol) 및 나트륨 (0.715 g, 31.1 mmol)의 용액에. 반응 혼합물을 140℃에서 48시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, MeOH에 이어서 물로 켄칭하였다. 이어서, 반응물을 EtOAc로 추출하였다. 이어서, 유기 층을 물에 이어서 염수 용액으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 완전히 증류시켰다. 조 생성물을 실리카 겔 칼럼에 첨가하고, Hex/EtOAc (1:1)로 용리시켰다. 수집된 분획을 증발시켜 목적 생성물을 수득하였다 (2.250 g, 9.93 mmol, 31.9% 수율), LCMS (m/z) 225.0 [M+H]+.
페닐 (1-메틸-1H-피라졸-4-일)카르바메이트의 합성

페닐 카르보노클로리데이트 (2.90 g, 18.53 mmol)를 디클로로메탄 (DCM) (50 mL) 중 피리딘 (3.12 mL, 38.6 mmol)의 교반 용액에 0℃에서 첨가하고, 15분 동안 교반한 다음, 1-메틸-1H-피라졸-4-아민 (1.5g, 15.45 mmol)을 동일한 온도에서 첨가하였다. 반응 혼합물을 실온에서 4시간 동안 교반하였다. 출발 물질의 소모 후 (TLC에 의해 모니터링함), 빙냉수를 첨가하고, 분리된 유기 층을 물 및 염수로 세척하였다. 유기 층을 황산나트륨을 통해 여과하고, 농축시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피에 의해 60-120(실리카 겔)을 사용하여 정제하고, 헥산 중 50% 에틸 아세테이트로 용리시켜 목적 생성물을 담갈색 고체로서 수득하였다 (1.6g, 6.41 mmol, 42% 수율), LCMS (m/z) 218.1 (M+H)+.
페닐 피리딘-3-일카르바메이트의 합성

실온에서 질소 하에 교반하는 디클로로메탄 (DCM) (30 mL) 중 페닐 카르보노클로리데이트 (2.163 g, 13.81 mmol), 및 피리딘 (1.375 mL, 17.00 mmol)의 용액에 피리딘-3-아민 (1.0 g, 10.63 mmol)을 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물을 포화 중탄산나트륨 용액으로 켄칭하였다. 유기 층을 분리하고, 수성 층을 DCM (50 ml)으로 추출하였다. 합한 DCM 층을 물로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 고진공 하에 농축시켜 조 생성물을 수득하였다. 조 생성물을 실리카 겔 칼럼에 첨가하고, 20% EtOAc/헥산으로 용리시켰다. 수집된 분획을 증발시켜 목적 생성물을 백색 고체로서 수득하였다 (1.3 g, 6.01 mmol, 57%), LCMS (m/z) 215.1 (M+H)+.
페닐 피리미딘-2-일카르바메이트의 합성

실온에서 질소 하에 교반하는 디클로로메탄 (DCM) (10 mL) 중 페닐 카르보노클로리데이트 (2.140 g, 13.67 mmol), 및 피리딘 (1.361 mL, 16.82 mmol)의 용액에 피리미딘-2-아민 (1.0 g, 10.51 mmol)을 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물을 포화 중탄산나트륨 용액으로 켄칭하였다. 유기 층을 분리하고, 수성 층을 DCM (50 ml)으로 추출하였다. 합한 DCM 층을 물로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 고진공 하에 농축시켜 조 생성물을 수득하였다. 이를 실리카 겔 칼럼에 첨가하고, 20% EtOAc/헥산으로 용리시켰다. 수집된 분획을 증발시켜 목적 생성물을 수득하였다 (1.6 g, 6.49 mmol, 61.7%), LCMS (m/z) 216.3 (M+H)+.
페닐 (5-플루오로피리딘-2-일)카르바메이트의 합성

실온에서 질소 하에 교반하는 디클로로메탄 (DCM) (40 mL) 중 페닐 카르보노클로리데이트 (1.397 g, 8.92 mmol), 및 피리딘 (0.721 mL, 8.92 mmol)의 용액에 5-플루오로피리딘-2-아민 (1.0 g, 8.92 mmol)을 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물을 포화 중탄산나트륨 용액으로 켄칭하였다. 유기 층을 분리하고, 수성 층을 DCM (20 ml)으로 추출하였다. 합한 유기 층을 물에 이어서 염수 용액으로 세척하고, 황산나트륨으로 건조시키고, 여과하고, 진공 하에 농축시켜 목적 생성물을 수득하였다 (1.4 g, 5.94 mmol, 67%), LCMS (m/z) 233.2 (M+H)+.
페닐 (2-메틸-2H-인다졸-5-일)카르바메이트의 합성

실온에서 질소 하에 교반하는 디클로로메탄 (DCM) (40 mL) 중 페닐 카르보노클로리데이트 (1.064 g, 6.79 mmol), 및 피리딘 (0.550 mL, 6.79 mmol)의 용액에 2-메틸-2H-인다졸-5-아민 (1 g, 6.79 mmol)을 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 반응 혼합물을 포화 중탄산나트륨 용액으로 켄칭하였다. 유기 층을 분리하고, 수성 층을 DCM (20 ml)으로 추출하였다. 합한 유기 층을 물에 이어서 염수 용액으로 세척하고, 황산나트륨으로 건조시키고, 진공 하에 농축시켜 페닐 (2-메틸-2H-인다졸-5-일)카르바메이트를 수득하였다 (1.3 g, 4.82 mmol, 70.9% 수율), LCMS (m/z) 268.1 (M+H)+.
페닐 피리다진-3-일카르바메이트의 합성

25℃에서 질소 하에 5분 동안 교반하는 디클로로메탄 (10 ml) 중 페닐 카르보노클로리데이트 (1.070 g, 6.83 mmol), 피리딘 (0.665 g, 8.41 mmol)의 용액에 디클로로메탄 (5ml) 중 피리다진-3-아민 (0.5 g, 5.26 mmol)의 현탁액을 첨가하였다. 반응 혼합물을 25℃에서 1시간 동안 교반하였다. 다음에, 유기 상을 물 3 mL, 포화 염수 3 mL로 세척하고, 황산나트륨 상에서 건조시키고, 진공 하에 농축시켜 조 생성물을 백색 고체로서 수득하였다. 화합물을 헥산으로 세척하고, 감압 하에 건조시켰다, LCMS (m/z) 216.2 (M+H)+.
페닐 피리미딘-4-일카르바메이트의 합성

25℃에서 질소 하에 교반하는 DCM (15 ml) 중 페닐 카르보노클로리데이트 (1.070 g, 6.83 mmol), 피리딘 (0.665 g, 8.41 mmol)의 용액에 DCM (5 ml) 중 피리미딘-4-아민 (0.5 g, 5.26 mmol)의 현탁액을 5분 동안 적가하였다. 반응 혼합물을 25℃에서 1시간 동안 교반하였다. 유기 상을 물 3 mL, 염수 3 mL로 세척하고, 황산나트륨 상에서 건조시키고, 진공 하에 농축시켜 조 생성물을 회백색 고체로서 수득하였다. 조 화합물을 헥산으로 세척한 다음, 감압 하에 건조시켜 목적 생성물을 수득하였다 (500 mg, 1.95 mmol, 37%), LCMS (m/z) 215.9 (M+H)+.
3-((6-아미노피리딘-2-일)옥시)프로판-1-올의 합성

프로판-1,3-디올 (1.358 g, 17.84 mmol)을 N-메틸-2-피롤리돈 (NMP) (5 mL) 중 NaH (1.070 g, 44.6 mmol)의 교반 용액에 0℃에서 첨가하고, 1시간 동안 교반하고, 이어서 6-플루오로피리딘-2-아민 (1.0 g, 8.92 mmol)을 첨가하고, 2시간 동안 80℃에서 교반하였다. 반응물을 실온으로 냉각시키고, 천천히 빙냉수에 첨가하고, 에틸 아세테이트로 희석하였다. 분리된 유기 층을 물 및 염수로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 화합물을 수득하였다. 조 화합물을 100-200 실리카 겔을 사용하여 정제하고, 100% 에틸 아세테이트로 용리시켜 3-((6-아미노피리딘-2-일)옥시)프로판-1-올을 갈색 점성 물질로서 수득하였다 (0.4g, 1.760 mmol, 19.73% 수율), LCMS (m/z): 169.22 [M+H]+.
6-(3-((테트라히드로-2H-피란-2-일)옥시)프로폭시)피리딘-2-아민의 합성

1,4-디옥산 (20 mL) 중 3-((테트라히드로-2H-피란-2-일)옥시)프로판-1-올 (4.2 g, 26.2 mmol)을 1,4-디옥산 (20 mL) 중 NaH (1.307 g, 32.7 mmol)의 용액에 0℃에서 첨가하고, 반응 혼합물을 28℃에서 30분 동안 교반하였다. 1,4-디옥산 (20 mL) 중 6-클로로피리딘-2-아민 (2.8g, 21.78 mmol)을 반응 혼합물에 0℃에서 첨가하고, 반응 혼합물을 100℃에서 10시간 동안 교반하였다. 반응 혼합물을 물 (20 mL)과 DCM (2 X 25 mL) 사이에 분배하였다. DCM 층을 포화 NaHCO3 용액으로 세척하고, 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 6-(3-((테트라히드로-2H-피란-2-일)옥시)프로폭시)피리딘-2-아민을 갈색 오일로서 수득하였다 (5.5 g, 18.12 mmol, 83% 수율), LCMS (m/z): 253.2 [M+H]+.
3-((테트라히드로-2H-피란-2-일)옥시)프로판-1-올의 합성
p-톨루엔술폰산 1수화물 (0.678 g, 3.57 mmol)을 0℃에서 디클로로메탄 (DCM) (50 mL) 중 프로판-1,3-디올 (5.43 g, 71.3 mmol), 및 3,4-디히드로-2H-피란 (3g, 35.7 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 28℃에서 2시간 동안 교반하였다. 반응 혼합물을 물 (20 mL)과 DCM (2 X 25 mL) 사이에 분배하였다. DCM 층을 포화 NaHCO3 용액으로 세척하고, 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 3-((테트라히드로-2H-피란-2-일)옥시)프로판-1-올을 무색 오일로서 수득하였다 (4.2g, 26.2 mmol, 73.5% 수율).
(S)-3-브로모-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘의 합성
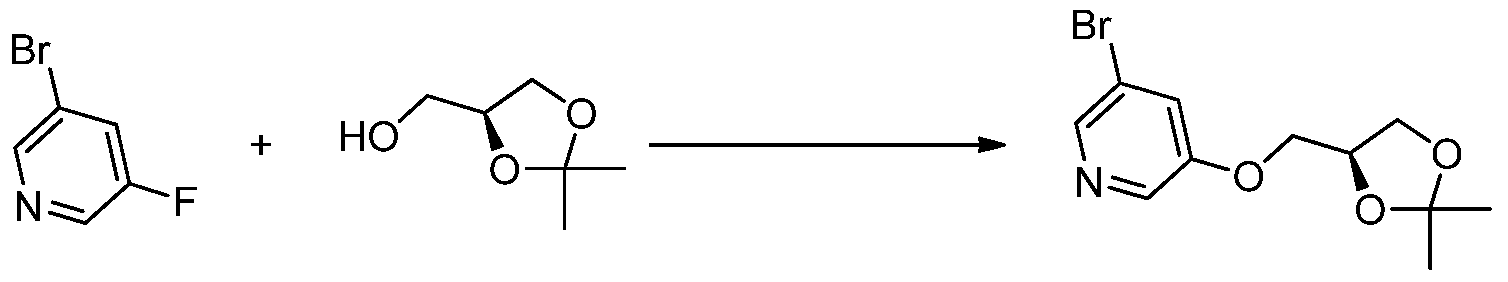
탄산세슘 (37.0 g, 114 mmol)을 다중구 RB에 녹였다. 이어서, 플라스크를 0℃로 냉각시키고, N-메틸-2-피롤리돈 (NMP) (100 mL)을 3분의 기간에 걸쳐 천천히 첨가하였다. 생성된 반응 혼합물을 질소 하에 15분 동안 교반하였다. 이어서, (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (10 g, 76 mmol)을 0℃에서 5분의 기간에 걸쳐 적가하였다. 이 현탁액을 실온에서 1시간 동안 교반하였다. 현탁액을 연황색 용액이 되었으며, 그 후에 3-브로모-5-플루오로피리딘 (7.62 mL, 73.9 mmol)을 첨가하였다. 생성된 용액을 75℃에서 24시간 동안 교반하였다. 반응 진행을 TLC 헥산 중 40% EtOAc에 의해 모니터링하였다. TLC는 24시간 후에 SM의 소모 및 새로운 스폿의 형성을 나타내었다. 반응물을 실온으로 냉각시키고, 물 (500 mL)로 희석하였다. 수성 층을 에틸 아세테이트 (2X300 mL)로 추출하였다. 유기 층을 염수 (250 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 갈색 오일을 수득하였다. 조 생성물을 칼럼 크로마토그래피에 의해 100-200 메쉬 크기 실리카 겔을 거쳐 정제하였다. 칼럼을 구배 EtOAc/헥산으로 용리시켰다. 목적 화합물을 헥산 중 20% EtOAc로 용리시켰다. 순수한 화합물을 함유하는 화합물 분획을 감압 하에 농축시켜 (S)-3-브로모-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘을 연황색 점성 오일로서 수득하였다 (10 g, 34.0 mmol, 44.9% 수율), LCMS (m/z): 289.99 [M+H]+.
(R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민의 합성

(R)-3-브로모-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘 (50g, 174 mmol), 액체 암모니아 (25 mL, 1155 mmol)를 밀봉된 튜브 중에서 녹였다. 이어서, 황산구리(II) (5.54 g, 34.7 mmol)를 0℃에서 첨가하였다. 생성된 청색 용액을 120℃로 2시간 동안 가열하였다. 반응 진행을 TLC DCM 중 10% MeOH에 의해 모니터링하였으며, TLC는 24시간 후에 새로운 스폿의 형성 및 SM의 소모를 나타내었다. 완결된 후, 반응물을 실온으로 냉각시켰다. 반응물을 20% NaOH를 사용하여 pH 10으로 만들고, NaCl로 포화시키고, 에틸 아세테이트 (30 mL* 2)로 추출하였다. 합한 유기 층을 염수 (20 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 갈색 고체를 수득하였으며, 이를 디에틸 에테르로 연화처리하고, 4시간 동안 교반한 다음, 여과하여 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민을 연갈색 고체로서 수득하였다 (35.4 g, 146 mmol, 84% 수율), LCMS (m/z): 225.29 [M+H]+.
(S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민의 합성

(S)-3-브로모-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘 (10 g, 34.7 mmol), 액체 암모니아 (100 mL, 4621 mmol)를 밀봉된 튜브 중에서 녹였다. 생성된 갈색 용액을 120℃로 24시간 동안 가열하였다. 반응 진행을 TLC DCM 중 10% MeOH에 의해 모니터링하였으며, TLC는 24시간 후에 새로운 스폿의 형성 및 SM의 소모를 나타내었다. 완결된 후, 반응물을 실온으로 냉각시켰다. 반응물을 20% NaOH를 사용하여 pH 10으로 만들고, NaCl로 포화시키고, 에틸 아세테이트 (30 mL* 2)로 추출하였다. 합한 유기 층을 염수 (20 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민을 연갈색 고체로서 수득하였다 (6 g, 25.8 mmol, 74.2% 수율), LCMS (m/z): 225.10 [M+H]+.
(R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민의 합성

THF (100 mL) 중 NaH (11.35 g, 473 mmol)의 현탁액에 THF (150 mL) 중 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (25 g, 189 mmol)의 용액을 질소 하에 0℃에서 적가하였다. 생성된 현탁액을 실온에서 1시간 동안 교반하였다. 6-클로로피리미딘-4-아민 (19.61 g, 151 mmol)을 반응 혼합물에 조금씩 실온에서 첨가하고, 생성된 현탁액을 90℃로 48시간 동안 가열하였다. 반응이 완결된 후 (TLC에 의해 모니터링함, 이는 아주 소량의 출발 물질 및 새로운 스폿이 극성에서 관찰되었음을 나타냄), 반응 혼합물을 빙수 (500 mL)에 붓고, 수성 층을 EtOAc (2 X 1000 mL)로 추출하였다. 합한 유기부를 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 담갈색 고체 (조 물질)를 수득하였다. 조 물질을 실리카 겔 칼럼 (100-200, DCM 중 3%MeOH)에 의해 정제하였다. 순수한 화합물을 함유하는 분획을 합하고, 농축시켜 목적 생성물 (R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민 (13 g, 53.9 mmol, 28.5% 수율)을 회백색 고체로서 수득하고, 또한 불순한 화합물 (10 g)로서 수득하였다. LCMS (m/z):226.17 (M+H)+.
(S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민의 합성

THF (150 mL) 중 NaH (9.08 g, 378 mmol)의 현탁액에 THF (200 mL) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (20 g, 151 mmol)의 용액을 질소 하에 0℃에서 적가하고, 생성된 현탁액을 실온에서 1시간 동안 교반하였다. 6-클로로피리미딘-4-아민 (15.68 g, 121 mmol)을 반응물에 조금씩 실온에서 첨가하고, 생성된 현탁액을 90℃로 48시간 동안 가열하였다. 반응이 완결된 후 (TLC에 의해 모니터링함, 출발 물질은 완전히 소모되고 새로운 스폿이 극성에서 관찰되었음), 반응물을 빙수 (200 mL)에 붓고, 에틸 아세테이트 (2X400 ml)로 추출하였다. 합한 유기부를 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 담갈색 고체를 수득하였다. 수득된 고체를 디에틸 에테르 (200 ml) 중에서 30분 동안 교반하고, 여과하고, 진공 하에 건조시켜 (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-아민을 담갈색 고체로서 수득하였다 (13 g, 57.3 mmol, 37.9% 수율), LCMS (m/z): 225.96 [M+H]+.
(R)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민의 합성

THF (150 mL) 중 NaH (9.08 g, 378 mmol)의 현탁액에 THF (250 mL) 중 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (20 g, 151 mmol)의 용액을 질소 하에 0℃에서 적가하였다. 생성된 현탁액을 실온에서 1시간 동안 교반하였다. 4-클로로피리미딘-2-아민 (15.68 g, 121 mmol)을 반응 혼합물에 조금씩 실온에서 첨가하고, 생성된 현탁액을 90℃로 48시간 동안 가열하였다. 반응이 완결된 후 (TLC에 의해 모니터링함, 출발 물질이 완전히 소모되고 새로운 스폿이 극성에서 관찰되었음), 반응 혼합물을 빙수 (250 mL)에 붓고, 수성 층을 EtOAc (2 X 300 mL)로 추출하였다. 합한 유기부를 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 연황색 액체 (조 물질)를 수득하였다. 수득된 조 물질을 칼럼 (100-200 실리카 겔)에 의해 0-50% EtOAc-석유 에테르를 사용하여 정제하여 (R)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민을 연황색 고체로서 수득하였다 (13 g, 57.0 mmol, 37.7% 수율), LCMS (m/z): 226.20 [M+H]+.
(S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민의 합성

1,4-디옥산 (200 mL) 중 NaH (8.25 g, 189 mmol)의 현탁액에 1,4-디옥산 (50 mL) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (10 g, 76 mmol)의 용액을 질소 하에 0℃에서 적가하였다. 생성된 현탁액을 실온에서 1시간 동안 교반하였다. 4-클로로피리미딘-2-아민 (7.84 g, 60.5 mmol)을 반응 혼합물에 조금씩 실온에서 첨가하고, 생성된 현탁액을 90℃로 48시간 동안 가열하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (200 mL)과 EtOAc (200 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하였다 (TLC 용리액: 순수한 에틸 아세테이트 Rf 0.3; UV 활성). 조 화합물을 칼럼 크로마토그래피 (100-200 메쉬 실리카 겔, 헥산 중 60% 에틸 아세테이트로 용리시킴)에 의해 정제하여 (S)-4-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민을 연황색 고체로서 수득하였다 (8.0 g, 35.4 mmol, 46.8% 수율), LCMS (m/z) 226.30 (M+H)+.
(R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민의 합성

테트라히드로푸란 (THF) (20 mL) 중 (R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진 (12 g, 49.0 mmol)의 교반 용액에 수산화암모늄 (300 mL, 1926 mmol) 및 황산구리(II) (1.566 g, 9.81 mmol)를 밀봉된 튜브 중에서 첨가하였다. 반응 혼합물을 120℃에서 18시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였으며, TLC는 미반응 SM과 함께 극성 스폿의 형성을 나타내었다. 반응 혼합물을 물 (300 mL)로 희석하고, EtOAc(3x 200mL)로 추출하고, 유기 층을 합하고, 물 (100 mL), 염수 용액 (100 mL)으로 세척하고, 유기 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민을 황색 유성 조 화합물로서 수득하였다 (10 g, 3.97 mmol, 8.09% 수율), LCMS (m/z): 226.13 (M+H)+.
(S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진의 합성

0℃에서 질소 하에 교반하는 N,N-디메틸포름아미드 (DMF) (50 mL) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (8.87 g, 67.1 mmol)의 현탁액에 탄산세슘 (32.8 g, 101 mmol)을 첨가하고, 생성된 반응 혼합물을 0℃에서 1시간 동안 교반하였다. 이에 2,5-디클로로피라진 (10 g, 67.1 mmol)을 첨가하였다. 생성된 반응 혼합물을 100℃에서 6시간 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 출발 물질이 소모되어 0.3 Rf의 새로운 극성 스폿을 형성하였음을 나타내었다. 반응물을 실온으로 냉각시키고, 물(100mL)을 첨가하고, 에틸 아세테이트(100mL)로 추출하였다. 유기 층을 물(100mLX2)로 세척하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 담갈색 액체로서 수득하였다. 조 생성물을 실리카 겔 (60-120) 칼럼에 첨가하고, Hex/EtOAc로 용리시켰다. 수집된 분획: 헥산 중 30% EtOAc로 생성물을 용리시켰다. 생성물 분획을 농축시켜 (S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진을 담갈색 액체로서 수득하였다 (12 g, 47.7 mmol, 71.0% 수율), LCMS (m/z): 244.90 [M+H]+.
(S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민의 합성

실온에서 교반하는 테트라히드로푸란 (THF) (10 mL) 중 (S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진 (10 g, 40.9 mmol)의 용액에 실온에서 수산화암모늄 (63.7 mL, 409 mmol) 및 황산구리(II) (3.26 g, 20.44 mmol)를 첨가하였다. 반응 혼합물을 밀봉된 튜브 중에서 130℃에서 2일 동안 교반하였다. 반응의 진행을 TLC에 의해 모니터링하였다. TLC는 출발 물질이 소모되었음을 나타내었다. 반응물을 실온으로 냉각시키고, 물(100mL)로 희석하고, 에틸 아세테이트 (250mLX2)로 추출하였다. 유기 층을 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 화합물을 갈색 점착성 화합물로서 수득하였다. 조 생성물을 실리카 겔 칼럼에 첨가하고, DCM/EtOAc로 용리시켰다. 수집된 분획: 석유 에테르 중 50% EtOAc로 생성물을 용리시켰다. 생성물 분획을 농축시켜 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민을 담갈색 고체로서 수득하였다 (2 g, 8.77 mmol, 21.46% 수율)(N35119-51-A2). NMR: CDCl3 중에서 일치, LCMS (m/z): 226.09 [M+H]+.
(R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민의 합성

(R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (27.8 g, 210 mmol)을 NMP (200 mL) 중 KOtBu (45.8 g, 408 mmol)의 교반 용액에 0℃에서 첨가한 다음, 실온에서 1시간 동안 교반하고, 0℃로 냉각시키고, 6-클로로피리딘-3-아민 (15 g, 117 mmol)을 첨가하고, 110℃로 144시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (500 mLX2)과 EtOAc (200 mL x 4) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 수득하고, 칼럼 크로마토그래피에 의해 정제하여 (100-200 실리카 겔 사용, 칼럼을 헥산 중 50% 에틸 아세테이트로 용리시킴)에 의해 정제하여 (R)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민을 갈색 오일로서 수득하였다 (8 g, 35.1 mmol, 30.1% 수율), LCMS (m/z): 225.16 [M+H]+.
(S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민의 합성
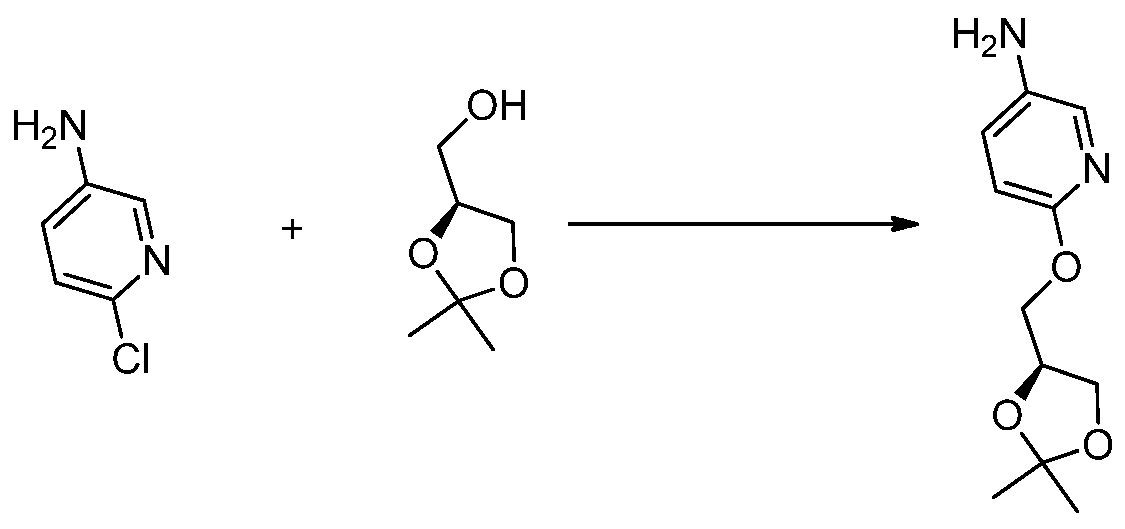
(S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (18.50 g, 140 mmol)을 NMP (600 mL) 중 KOtBu (30.5 g, 272 mmol)의 교반 용액에 0℃에서 첨가한 다음, 실온에서 1시간 동안 교반하고, 0℃로 냉각시키고, 6-클로로피리딘-3-아민 (10.0 g, 78 mmol)을 첨가하고, 110℃로 88시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (50 mLX2) 및 EtOAc (100 mL x 2) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 검으로서 수득하였다 (TLC: 용리액: 100% 에틸 아세테이트, Rf 0.5; UV 활성:). 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬)에 의해 정제하고, 헥산 중 50% EtOAc로 용리시키면서 (S)-6-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-아민을 암색 점착성 물질로서 수득하였다 (10.0 g, 41.7 mmol, 53.6% 수율), LCMS (m/z) 225.0 (M+H)+.
(R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민의 합성

(R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (30.7 g, 232 mmol)을 NMP (800 mL) 중 KOtBu (70.1 g, 624 mmol)의 교반 용액에 0℃에서 첨가한 다음, 실온에서 1시간 동안 교반하고, 0℃로 냉각시킨 다음, 5-플루오로피리딘-2-아민 (20 g, 178 mmol)을 첨가하고, 110℃로 114시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (500 mLX2) 및 EtOAc (500 mL x 4) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득한 다음, 이를 칼럼 크로마토그래피 (100-200 실리카 겔 사용, 칼럼을 헥산 중 80% 에틸 아세테이트로 용리시킴)에 의해 정제하여 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민을 갈색 오일로서 수득하였다 (10 g, 40.1 mmol, 22.50% 수율), LCMS: 225.0 (M+H).
(S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민의 합성
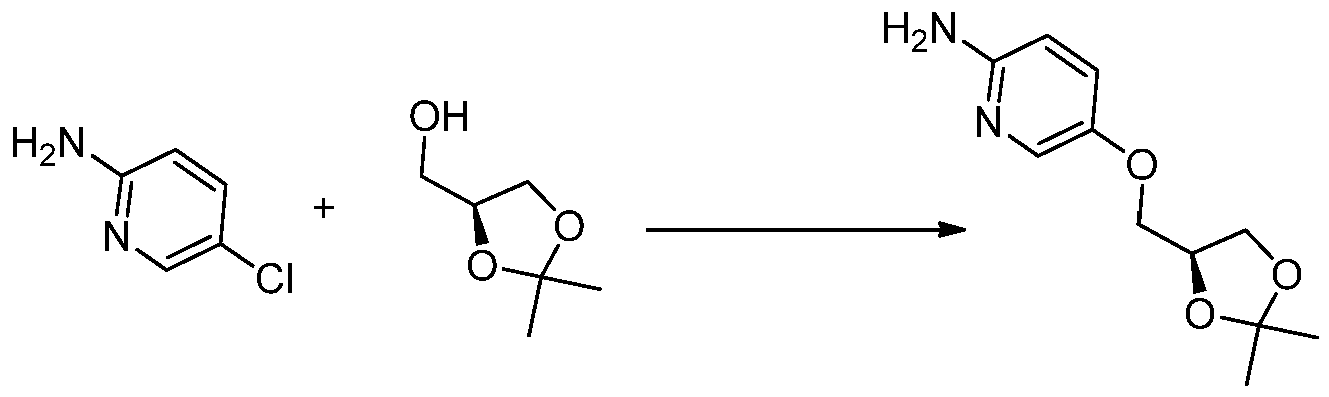
NaH (12.84 g, 268 mmol)를 디메틸 술폭시드 (DMSO) (100 mL) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (31.8 g, 241 mmol)의 교반 용액에 0℃에서 첨가한 다음, 실온에서 1시간 동안 교반하고, 0℃로 냉각시키고, 5-플루오로피리딘-2-아민 (15.0 g, 134 mmol)을 첨가하고, 110℃로 60시간 동안 가열하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (50 mL)과 EtOAc (100 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다 (TLC: 용리액: 100% 에틸 아세테이트, Rf 0.5; UV 활성). 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬)에 의해 정제하고, 헥산 중 50% EtOAc로 용리시켜 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-아민을 연황색 점착성 물질로서 수득하였다 (7.2 g, 32.1 mmol, 23.99% 수율), LCMS (m/z): 225.1 (M+H)+.
(R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-아민의 합성

테트라히드로푸란 (75 mL)을 NaH (5.56 g, 0℃, (R)-(2,2-디메틸-1,3-디옥솔란-4-일에서 232 mmol))메탄올 (12.46 mL에 첨가하고, 테트라히드로푸란 (50 mL 중 100 mmol)을 반응 혼합물에 0℃에서 첨가하고, 반응 혼합물을 28℃에서 1시간 동안 교반하였다. 테트라히드로푸란 (25 mL) 중 2-클로로피리미딘-5-아민 (10 g, 77 mmol)을 첨가하고, 70℃에서 16시간 동안 교반하였다. 반응 혼합물을 냉수 (30 mL)로 켄칭하고, 에틸 아세테이트(3 x 80 mL)로 추출하였다. 유기 층을 물 (2 X 50 mL) 및 포화 염수 용액 (50 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켰다. 조 화합물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하고, 생성물을 헥산 중 40-45% 에틸 아세테이트로 용리시켜 (R)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-아민을 연황색 고체로서 수득하였다 (6.5 g, 28.3 mmol, 36.6% 수율), LCMS (m/z): 226.0 [M+H]+.
(S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-아민의 합성
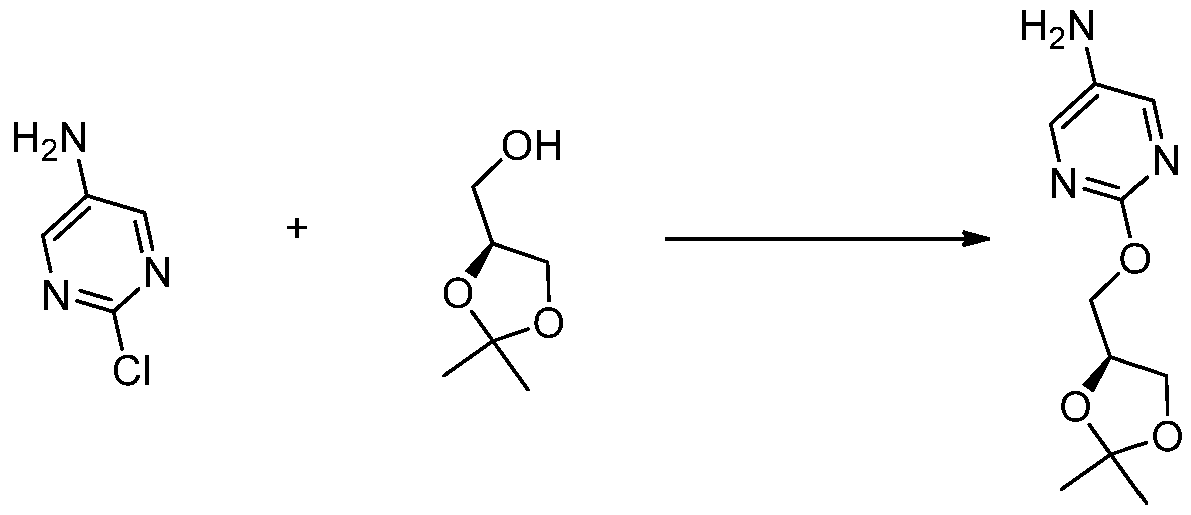
THF (100 ml) 중 NaH (6.17 g, 154 mmol)의 현탁액에 THF (50 ml) 중 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (13.26 g, 100 mmol)을 0℃에서 첨가하고, 반응 혼합물을 25℃에서 1시간 동안 교반하였다. 이에 THF (50 ml) 중 2-클로로피리미딘-5-아민 (10 g, 77 mmol)을 0℃에서 첨가하고, 80℃로 천천히 가열하고, 80℃에서 16시간 동안 교반하였다. 반응이 완결된 후, 반응 혼합물을 염화암모늄 (10 ml)으로 켄칭하고, 에틸 아세테이트 (3x20 ml)로 추출하였다. 유기 층을 분리하고, 염수로 세척하고, Na2SO4 상에서 건조시키고, 이를 여과하고, 감압 하에 농축시켜 조 물질을 수득하였다. 이 조 물질을 디에틸 에테르로 연화처리하여 (S)-2-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-아민을 갈색 고체로서 수득하였다 (5.0 g, 19.77 mmol, 25.6% 수율), LCMS (m/z): 226.1 [M+H]+.
(S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일) 메틸) 피라진의 합성

DMF (1000 mL) 중 탄산세슘 (492 g, 1510 mmol)의 교반 용액에 0℃에서 (S)-(2,2-디메틸-1,3-디옥솔란-4-일) 메탄올 (133 g, 1007 mmol)을 첨가하였다. 생성된 반응 혼합물을 실온에서 30분 동안 교반하였다. 이어서, DMF (500 mL) 중 2,5-디클로로피라진 (150 g, 1007 mmol)의 용액을 0℃에서 첨가하고, 생성된 반응 혼합물을 100℃에서 4시간 동안 교반하였다 (TLC 시스템: 석유 에테르 중 20% 에틸 아세테이트, Rf: 0.5, UV 활성). 반응 혼합물을 빙냉수 (500 mL)로 희석하고, EtOAc (3 x 300 mL)로 추출하였다. 합한 유기 층을 물 (2 x 200 mL) 및 염수 용액 (100 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 화합물을 플래쉬 칼럼 크로마토그래피 (실리카 겔: 100-200 메쉬, 용리액: 헥산 중 10% EtOAc)에 의해 정제하여 목적 생성물 (S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일) 메톡시) 피라진을 황색 액체로서 수득하였다 (200 g, 768 mmol, 76% 수율). LCMS (m/z): 245.1 [M+H]+.
(S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민의 합성

THF (30 mL) 중 (S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진 (120 g, 490 mmol)의 교반 용액에 밀봉된 튜브 중에서 수산화암모늄 (1000 mL, 6420 mmol) 및 황산구리(II) (15.66 g, 98 mmol)를 첨가하고, 생성된 반응 혼합물을 120℃에서 48시간 동안 교반하였다 (TLC 시스템: 석유 에테르 중 50% 에틸 아세테이트, Rf: 0.4, UV 활성). 반응 혼합물을 물 (300 mL)로 희석하고, EtOAc (3x 500 mL)로 추출하였다. 합한 유기 층을 물 (200 mL) 및 염수 용액 (200 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 물질을 플래쉬 칼럼 크로마토그래피 (100-200 메쉬 실리카겔 사용, 화합물을 헥산 중 40% EtOAc로 용리시킴)에 의해 정제하여 목적 생성물 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민을 황색 결정 고체로서 수득하였다 (65 g, 280 mmol, 57.2% 수율). LCMS (m/z): 226.13 [M+H]+.
(R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일) 메톡시)피라진의 합성
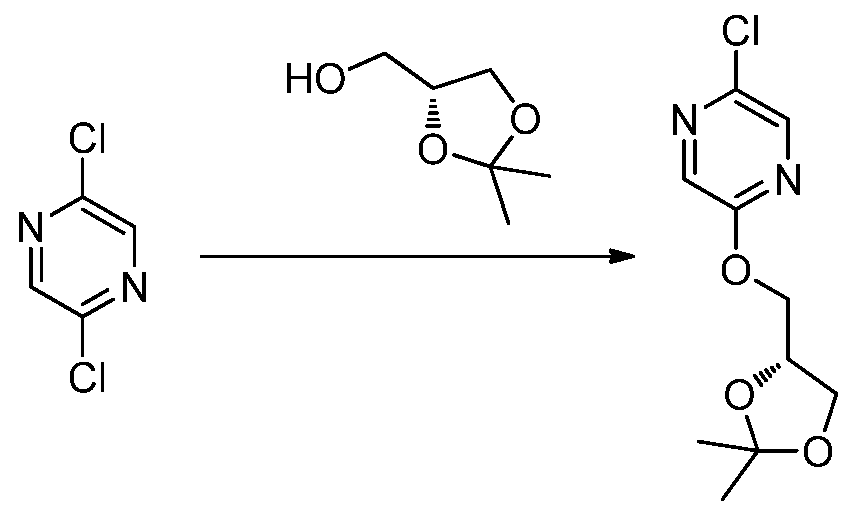
DMF (100 mL) 중 탄산세슘 (32.8 g, 101 mmol)의 교반 현탁액에 (R)-(2,2-디메틸-1,3-디옥솔란-4-일) 메탄올 (8.87 g, 67.1 mmol)을 0℃에서 첨가하고, 실온에서 30분 동안 교반하였다. 이어서, 2,5-디클로로피라진 (10 g, 67.1 mmol)을 첨가하고, 생성된 반응 혼합물을 100℃에서 4시간 동안 교반하였다 (TLC 시스템: 헥산 중 20% 에틸 아세테이트, Rf: 0.5, UV 활성). 반응 혼합물을 빙냉수 (200 mL)로 희석하고, EtOAc (3 x 100 mL)로 추출하였다. 합한 유기 층을 물 (2x50 mL) 및 염수 용액 (50 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 (R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진을 황색 유성 화합물로서 수득하였다 (12 g, 43.8 mmol, 65.3% 수율). LCMS (m/z): 244.99 [M+H]+.
(R)-5-((2,2-디메틸-1,3-디옥솔란-4-일) 메톡시) 피라진-2-아민의 합성

테트라히드로푸란 (10 mL) 중 (R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진 (8 g, 32.7 mmol)의 교반 용액에 밀봉된 튜브 중에서 수산화암모늄 (400 mL, 2568 mmol) 및 황산구리(II) (1.044 g, 6.54 mmol)를 첨가하고, 반응 혼합물을 120℃에서 48시간 동안 교반하였다 (TLC 시스템: 헥산 중 50% 에틸 아세테이트, Rf: 0.4, UV 활성). 반응 혼합물을 물 (200 mL)로 희석하고, EtOAc (3x 50 mL)로 추출하였다. 합한 유기 층을 물 (50 mL), 염수 용액 (50 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 물질을 플래쉬 칼럼 크로마토그래피 (100-200 메쉬 실리카겔 사용, 화합물을 헥산 중 40% EtOAc로 용리시킴)에 의해 정제하고, 순수한 분획을 수집하고, 감압 하에 농축시켜 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-아민을 황색 결정 고체로서 수득하였다 (2 g, 8.65 mmol, 26.4% 수율). LCMS (m/z): 226.10 [M+H]+.
(S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘의 합성

0℃에서 THF (100 mL) 중 2-클로로피리미딘-5-올 (13 g, 100 mmol)의 교반 용액에 (S)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (13.16 g, 100 mmol), 트리페닐포스핀 (32.7 g, 124 mmol)에 이어서 DEAD (19.71 mL, 124 mmol)를 첨가하고, 반응을 실온에서 4시간 동안 교반하였다 (TLC 용리 시스템: 석유 에테르 중 30% EtOAc; Rf-0.5; UV 활성). 반응 혼합물을 물 (50 mL)로 켄칭하고, EtOAc (2x75 mL)로 추출하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 물질을 크로마토그래피 (실리카겔, 용리액: 헥산 중 20% EtOAc)에 의해 정제하여 (S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘을 회백색 고체로서 수득하였다 (20g, 79 mmol, 79% 수율). LCMS (m/z): 245.10; [M+H]+.
(S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민의 합성

밀봉된 튜브에 들은 (S)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘 (10 g, 40.9 mmol) 및 수성 암모니아 (66.3 ml, 1226 mmol)의 혼합물을 120℃에서 24시간 동안 가열하였다 (TLC 용리 시스템: 100% EtOAc; Rf-0.2; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 물 (50 mL)로 켄칭하고, EtOAc (2x75 mL)로 추출하였다. 유기 층을 분리하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 황색 고체로서 수득하였다. 조 화합물을 n-펜탄 (50 mL)으로 연화처리하여 (S)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민을 회백색 고체로서 수득하였다 (6.6g, 28.6 mmol, 70.0% 수율). LCMS (m/z): 226.17; [M+H]+.
(R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘의 합성

0℃에서 THF (100 mL) 중 2-클로로피리미딘-5-올 (20 g, 153 mmol)의 교반 용액에 (R)-(2,2-디메틸-1,3-디옥솔란-4-일)메탄올 (24.30 g, 184 mmol), 트리페닐포스핀 (50.2 g, 192 mmol)에 이어서 DEAD (30.3 mL, 192 mmol)를 첨가하고, 반응물을 실온에서 12시간 동안 교반하였다 (TLC 용리 시스템: 석유 에테르 중 70% EtOAc; Rf-0.5; UV 활성). 반응 혼합물을 물 (100 mL)로 켄칭하고, EtOAc (200 mL)로 추출하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 물질을 크로마토그래피 (실리카겔, 용리액: 헥산 중 35% EtOAc)에 의해 정제하여 (R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘을 백색 고체로서 수득하였다 (23 g, 91 mmol, 59.5% 수율). LCMS (m/z): 245.06; [M+H]+.
(R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민의 합성

밀봉된 튜브에 들은 (R)-2-클로로-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘 (5 g, 20.44 mmol) 및 수성 암모니아 (50 ml, 924 mmol)의 혼합물을 48시간 동안 120℃로 가열하였다 (TLC 용리 시스템: 100% EtOAc; Rf-0.2; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 물 (50 mL)로 켄칭하고, DCM (2x75 mL)으로 추출하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (R)-5-((2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-아민을 연황색 고체로서 수득하였다 (2.7 g, 11.5 mmol, 57.5% 수율). LCMS (m/z): 226.02; [M+H]+.
화합물 실시예
실시예 1
(9S)-N-(피리딘-2-일)-2-((S)-2-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

NaH (0.254 g, 10.57 mmol)를 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-((S)-2-(트리플루오로메틸)피롤리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.550 g, 1.761 mmol)의 교반 용액에 질소 하에 실온에서 교반하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 이어서, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (0.635 g, 2.64 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (10 mL)과 EtOAc (25 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 헥산 중 75-80% EtOAc로 용리시켜 순수한 (9S)-N-(피리딘-2-일)-2-((S)-2-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.275 g, 0.631 mmol, 35.8% 수율), LCMS (m/z): 433.2 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.60 (s, 1 H), 8.20 - 8.29 (m, 1 H), 8.09 (dt, J=8.33, 0.88 Hz, 1 H), 7.72 - 7.81 (m, 1 H), 7.37 (d, J=8.55 Hz, 1 H), 7.05 (ddd, J=7.29, 4.88, 0.99 Hz, 1 H), 6.45 (d, J=8.55 Hz, 1 H), 5.11 - 5.19 (m, 1 H), 4.74 (br s, 1 H), 3.91 (dt, J=10.19, 5.21 Hz, 1 H), 3.50 - 3.62 (m, 1 H), 3.27 (d, J=1.75 Hz, 1 H), 3.11 - 3.22 (m, 1 H), 3.01 - 3.08 (m, 1 H), 2.81 (br d, J=13.37 Hz, 1 H), 2.07 - 2.28 (m, 4 H), 1.94 - 2.02 (m, 1 H), 1.81 - 1.92 (m, 1 H), 1.21 - 1.40 (m, 2 H)
실시예 2
(9S)-N-(5-플루오로피리딘-3-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (15 mL) 중 (9S)-2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.561 mmol)의 교반 용액에 트리에틸아민 (0.653 mL, 4.68 mmol) 및 트리포스겐 (232 mg, 0.780 mmol)을 30℃에서 첨가하고, 실온에서 1시간 동안 교반하였다. 이어서, 5-플루오로피리딘-3-아민 (525 mg, 4.68 mmol)을 30℃에서 첨가하고, 반응을 70℃에서 16시간 동안 가열하였다. THF를 감압 하에 증발시키고, 수득된 잔류물을 물로 희석하고, CH2Cl2 (3x50 mL)로 추출하였다. 합한 유기 층을 물, 염수로 세척하고, Na2SO4 상에서 건조시키고, 용매를 감압 하에 증발시켜 조 화합물을 수득하였으며, 이를 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: DCM 중 3% MeOH)에 의해 정제하여 (9S)-N-(5-플루오로피리딘-3-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (200 mg, 0.4366 mmol, 35% 수율), (TLC: Rf = 0.3, EtOAc 중 10% MeOH), LCMS (m/z): 459.25[M+H]+.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.19 (s, 1 H), 8.93 (d, J=5.04 Hz, 1 H), 8.47 (s, 1 H), 8.37 (s, 1 H), 8.33 - 8.21 (m, 2 H), 8.03 (d, J=11.18 Hz, 1 H), 7.88 (d, J=8.11 Hz, 1 H), 7.78 (d, J=8.11 Hz, 1 H), 4.83 (s, 1 H), 3.43 (d, J=12.06 Hz, 1 H), 3.25 (s, 2H), 2.90 (d, J=13.81 Hz, 1 H), 2.11 - 1.84 (m, 2 H), 1.33 (s, 2 H).
실시예 3
(9S)-N-(피리미딘-5-일)-2-(5-(트리플루오로메틸)피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (12 mL) 중 (9S)-2-(5-(트리플루오로메틸)피리딘-3-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.6 g, 1.873 mmol), 트리에틸아민 (0.783 mL, 5.62 mmol)의 용액에 트리포스겐 (0.278 g, 0.937 mmol)을 0℃에서 첨가하고, 실온에서 30분 동안 교반하였다. 이어서, 피리미딘-5-아민을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 용매를 감압 하에 제거하여 조 물질을 수득하고, 디클로로메탄으로 희석하고, 물 및 염수 용액으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 유기 층을 감압 하에 증발시켜 조 생성물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, CH2Cl2 중 2% MeOH로 용리시킴)에 의해 정제하여 (9S)-N-(피리미딘-5-일)-2-(5-(트리플루오로메틸)피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (459 mg, 1.016 mmol, 54.2% 수율), (TLC: 헥산 중 80% EtOAc, Rf = 0.5), LCMS (m/z): 442.2 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.19 (s, 1 H), 9.48 (d, J=2.19 Hz, 1 H), 9.08 (s, 1H), 8.96 (s, 1 H), 8.88 (s, 1 H), 8.71 (s, 1 H), 7.85 (d, J=8.11 Hz, 1 H), 7.77 (d, J=7.89 Hz, 1 H), 4.84 (s, 1 H), 3.43 (dd, J=13.59, 1.97 Hz, 1 H), 3.33 (s, 2 H), 2.90 (d, J=13.81 Hz, 2 H), 1.95 (d, J=10.30 Hz, 2 H), 1.33 (m, 2 H).
실시예 4
(9S)-2-(2-메틸피리딘-4-일)-N-(피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
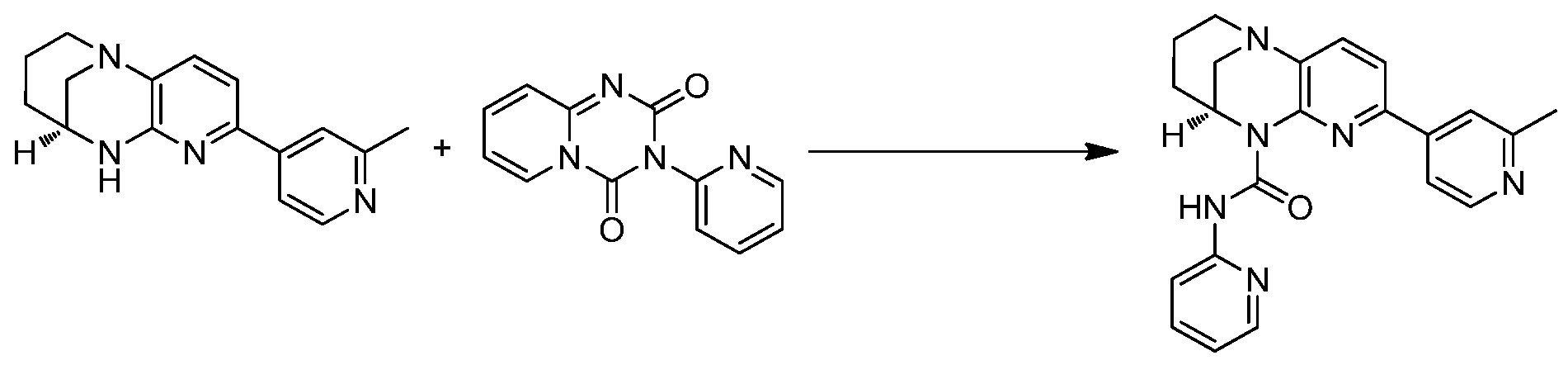
NaH (0.297 g, 12.39 mmol)을 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.550 g, 2.065 mmol)의 교반 용액에 첨가하고, 질소 하에 실온에서 교반하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반하였다. 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (0.744 g, 3.10 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (20 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 헥산 중 70% EtOAc: Rf = 0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 헥산 중 50-60% EtOAc로 용리시켜 순수한 (9S)-2-(2-메틸피리딘-4-일)-N-(피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (0.292 g, 0.751 mmol, 36.4% 수율), LCMS (m/z): 387 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.99 (s, 1 H), 8.59 (d, J=5.48 Hz, 1 H), 8.41 - 8.45 (m, 1 H), 8.27 (s, 1 H), 8.18 (d, J=8.33 Hz, 1 H), 7.97 (br d, J=3.73 Hz, 1 H), 7.82 - 7.86 (m, 2 H), 7.71 (d, J=7.89 Hz, 1 H), 7.11 - 7.16 (m, 1 H), 4.86 (br s, 1 H), 3.40 (br d, J=13.37 Hz, 1 H), 3.29 (s, 2 H), 2.91 (br d, J=13.15 Hz, 1 H), 2.64 (s, 3 H), 1.93 - 2.05 (m, 2 H), 1.32 (br s, 2 H)
실시예 5
(9S)-N-(5-플루오로피리딘-3-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.6 g, 2.253 mmol)의 용액에 트리에틸아민 (1.884 mL, 13.52 mmol) 및 트리포스겐 (0.669 g, 2.253 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반하였다. 다음에, 5-플루오로피리딘-3-아민 (0.758 g, 6.76 mmol)을 실온에서 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시켰다. 이어서, 반응 혼합물을 감압 하에 농축시키고, 물 (20 mL)과 디클로로메탄 (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.4; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 100% EtOAc로 용리시켜 순수한 (9S)-N-(5-플루오로피리딘-3-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (0.314 g, 0.770 mmol, 34.2% 수율), LCMS (m/z): 405.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 13.71 (s, 1H), 8.74 - 8.55 (m, 1H), 8.44 (d, J = 1.7 Hz, 1H), 8.29 (d, J = 2.6 Hz, 1H), 8.18 - 7.99 (m, 1H), 7.81 (d, J = 1.7 Hz, 1H), 7.73 (d, J = 2.8 Hz, 3H), 4.82 (s, 1H), 3.39 (d, J = 1.9 Hz, 1H), 3.30 (s, 2H), 2.92 (s, 1H), 2.57 (s, 3H), 2.01 (s, 2H), 1.32 (s, 2H).
실시예 6
(9S)-N-(피리다진-3-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (8 mL) 중 (9S)-2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.4 g, 1.249 mmol), 페닐 피리다진-3-일카르바메이트 (0.806 g, 3.75 mmol)의 교반 용액에 질소 하에 25℃에서 DMAP (0.458 g, 3.75 mmol)를 첨가하였다. 반응 혼합물을 80℃에서 16시간 동안 가열하였다. 실온으로 냉각되도록 하고, 용매를 진공 하에 제거하여 조 물질을 수득하고, CH2Cl2로 희석하고, 물, 염수 용액으로 세척하고, 무수 Na2SO4 상에서 건조시켰다. 유기 층을 진공 하에 농축시켜 조 화합물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: CH2Cl2 중 4% MeOH)에 의해 정제하여 (9S)-N-(피리다진-3-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (320 mg, 0.711 mmol, 56.9% 수율), (TLC: 헥산 중 80% EtOAc Rf: 0.6), LCMS (m/z): 442.2 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 14.27 (s, 1H), 8.98 (dd, J = 4.7, 1.5 Hz, 1H), 8.90 (d, J = 5.1 Hz, 1H), 8.59 - 8.48 (m, 2H), 8.38 (dd, J = 9.0, 1.5 Hz, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.71 (dd, J = 9.0, 4.7 Hz, 1H), 5.25 - 4.49 (m, 1H), 3.42 (dd, J = 13.7, 1.9 Hz, 1H), 3.30 (s, 2H), 2.94 (dt, J = 13.8, 2.5 Hz, 1H), 2.02 (m, 2H), 1.34 (q, J = 5.2, 4.4 Hz, 2H).
실시예 7
(9S)-N-(피리딘-2-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (5 mL) 중 (9S)-2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.250 g, 0.780 mmol)의 용액에 수소화나트륨 (0.156 g, 3.90 mmol)을 실온에서 1 부분으로 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 이어서, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (0.562 g, 2.341 mmol)을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 실온으로 냉각되도록 하고, 이를 빙냉수로 켄칭하고, 에틸 아세테이트 (3x50 mL)로 추출하였다. 합한 유기 층을 물, 포화 염수 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: CH2Cl2 중 2% MeOH)에 의해 정제하여 (9S)-N-(피리딘-2-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (302 mg, 0.681 mmol, 87% 수율), (TLC: 헥산 중 70% EtOAc, Rf: 0.5), LCMS (m/z): 441.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 13.85 (s, 1H), 8.94 (d, J = 5.2 Hz, 1H), 8.64 (dd, J = 1.7, 0.9 Hz, 1H), 8.55 - 8.50 (m, 1H), 8.34 (ddd, J = 4.8, 2.0, 0.9 Hz, 1H), 8.15 (dt, J = 8.3, 1.0 Hz, 1H), 7.98 (d, J = 8.1 Hz, 1H), 7.86 - 7.79 (m, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.12 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 4.87 (t, J = 2.6 Hz, 1H), 3.41 (dd, J = 13.6, 1.9 Hz, 1H), 3.30 (s, 2H), 2.92 (d, J = 14.0 Hz, 1H), 2.09 - 1.86 (m, 2H), 1.34 (d, J = 8.5 Hz, 2H).
실시예 8
(9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

NaH (0.277 g, 11.53 mmol)를 테트라히드로푸란 (THF) (20 mL) 중 (9S)-2-(3)피롤리딘-1-일)- 7,8,9,10-테트라히드로-6H-5,9-메타-(트리플루오로메틸노피리도[2,3-b][1,4]디아조신 (0.6 g, 1.921 mmol)의 용액에 첨가하고, 30분 동안 교반하였다. 이어서, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (0.692 g, 2.88 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 물 (20 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 헥산 중 70% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 헥산 중 50-60% EtOAc로 용리시켜 입체이성질체의 혼합물 (9:1)을 수득하였으며, 추가로 SFC 정제하여 순수한 (9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서로서 수득하였다 (0.422 g, 0.973 mmol, 50.6% 수율), LCMS (m/z): 433.3 [M+H]+.
분석용 SFC 조건: 피크-I 칼럼/치수: 키랄셀 OX-H (250 X 4.6)mm,5u
% CO2: 60.0%
% 공용매: 40.0% (MeOH 중 0.5% DEA)
총 유량: 4.0g/분
배압: 100 bar
온도: 26.8℃
UV: 263nm
1H NMR (400 MHz, DMSO-d6): δ 13.81 (s, 1H), 8.23 - 8.18 (m, 1H), 8.11 (dt, J = 8.3, 1.0 Hz, 1H), 7.76 (ddd, J = 8.5, 7.4, 1.9 Hz, 1H), 7.32 (d, J = 8.5 Hz, 1H), 7.04 (ddd, J = 7.3, 4.9, 1.0 Hz, 1H), 6.24 (d, J = 8.5 Hz, 1H), 4.71 (s, 1H), 3.96 (d, J = 8.5 Hz, 1H), 3.78 (d, J = 7.0 Hz, 1H), 3.74 (s, 1H), 3.66 (d, J = 4.1 Hz, 2H), 3.59 - 3.38 (m, 1H), 3.26 (d, J = 1.9 Hz, 1H), 3.12 (dd, J = 12.7, 3.5 Hz, 1H), 3.05 (s, 1H), 2.80 (d, J = 13.4 Hz, 1H), 2.41 - 2.29 (m, 1H), 2.15 (dd, J = 12.7, 8.1 Hz, 1H), 1.96 (s, 1H), 1.83 (s, 2H), 1.31 (s, 2H).
실시예 9
(9S)-N-(피리미딘-4-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
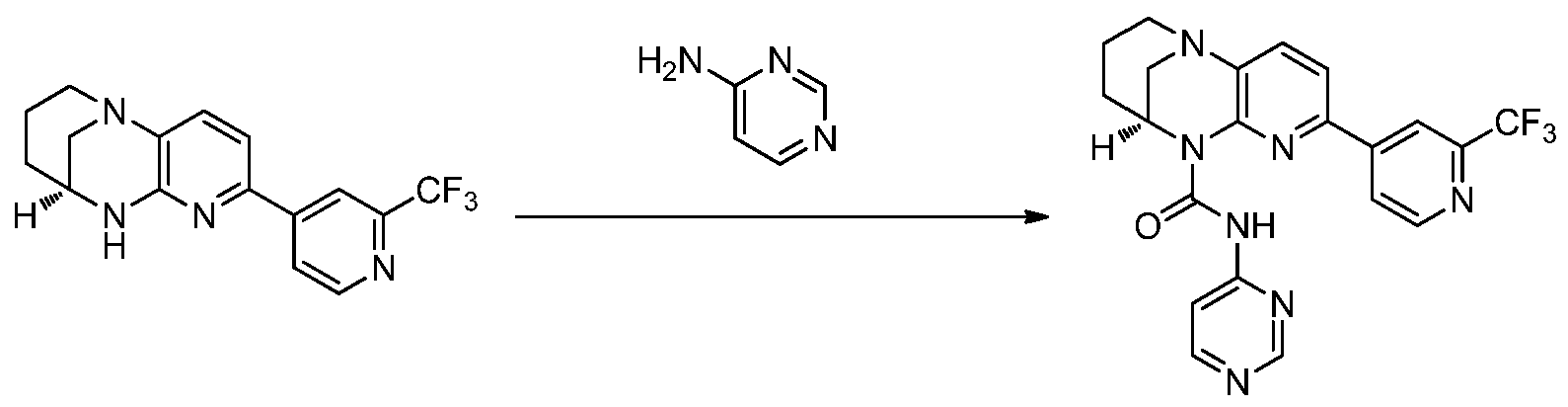
질소 하에 교반하는 25℃에서 THF (10 mL) 중 (9S)-2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5 g, 1.561 mmol), 트리에틸아민 (0.653 mL, 4.68 mmol)에 비스(트리클로로메틸) 카르보네이트 (0.232 g, 0.780 mmol)를 1회 충전량으로 첨가하였다. 반응 혼합물을 25℃에서 30분 동안 교반하였다. 이어서, 피리미딘-4-아민 (0.445 g, 4.68 mmol)을 한 부분으로 첨가하고, 반응물을 65℃에서 16시간 동안 교반하였다. 실온으로 냉각되도록 하고, 용매를 진공 하에 제거하고, 조 물질을 DCM으로 희석하고, 물, 염수 용액으로 세척하고, 황산나트륨 상에서 건조시키고, 진공 하에 농축시켜 조 화합물을 수득하였다. 조 혼합물을 정제용 HPLC에 의해 정제하여 (9S)-N-(피리미딘-4-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (199 mg, 0.446 mmol, 28.6% 수율), (TLC: 헥산 중 70% EtOAc, Rf: 0.6), LCMS (m/z): 442.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 14.18 (s, 1H), 8.97 (d, J = 5.1 Hz, 1H), 8.85 (d, J = 1.2 Hz, 1H), 8.69 (d, J = 5.8 Hz, 1H), 8.60 (dd, J = 1.9, 0.8 Hz, 1H), 8.50 (dd, J = 5.1, 1.7 Hz, 1H), 8.12 (dd, J = 5.8, 1.3 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.80 (d, J = 8.1 Hz, 1H), 4.93 - 4.73 (m, 1H), 3.42 (dd, J = 13.7, 1.9 Hz, 1H), 3.32 (d, J = 8.2 Hz, 2H), 2.93 (dt, J = 13.9, 2.5 Hz, 1H), 2.12 - 1.83 (m, 2H), 1.33 (d, J = 7.8 Hz, 2H).
실시예 10
(9S)-N-(피리미딘-5-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (10 mL) 중 (9S)-2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5 g, 1.561 mmol), 트리에틸아민 (0.653 mL, 4.68 mmol)의 용액에 질소 분위기 하에 비스(트리클로로메틸) 카르보네이트 (0.232 g, 0.780 mmol)를 25℃에서 첨가하고, 실온에서 30분 동안 교반하였다. 이어서, 피리미딘-5-아민 (0.445 g, 4.68 mmol)을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 가열하였다. 실온으로 냉각되도록 하고, 용매를 회전증발기 상에서 제거하고, 조 물질을 CH2Cl2 (20ml)로 희석하고, 물 (5ml), 염수 용액 (5ml)으로 세척한 다음, 황산나트륨 상에서 건조시켰다. 유기 용매를 감압 하에 증발시켜 조 생성물을 수득하였다. 조 혼합물을 정제용 HPLC (포름산, ACN 25%)에 의해 정제하여 (9S)-N-(피리미딘-5-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (250 mg, 0.56 mmol, 45% 수율), (TLC: 헥산 중 80% EtOAc, Rf: 0.5), LCMS (m/z): 442.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 13.07 (s, 1H), 9.08 - 8.72 (m, 4H), 8.38 (dd, J = 1.7, 0.9 Hz, 1H), 8.28 (dd, J = 5.2, 1.6 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 4.85 (t, J = 2.7 Hz, 1H), 3.43 (dd, J = 13.7, 1.9 Hz, 1H), 3.33 (d, J = 10.1 Hz, 2H), 2.91 (d, J = 13.7 Hz, 1H), 2.15 - 1.79 (m, 2H), 1.33 (s, 2H).
실시예 11
(9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

NaH (0.110 g, 4.60 mmol)를 질소 하에 실온에서 교반하는 테트라히드로푸란 (THF) (20 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-II, 0.250 g, 0.766 mmol)의 교반 용액에 첨가하였다. 반응 혼합물을 실온에서 30분 동안 교반하였다. 이어서, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (0.276 g, 1.149 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응물을 실온으로 냉각시키고, 물 (10 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 헥산 중 70% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하고, 헥산 중 50-60% EtOAc로 용리시켜 순수한 (9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.2 g, 0.439 mmol, 57.3% 수율), LCMS (m/z): 447.2 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 13.50 (s, 1H), 8.32 - 8.18 (m, 1H), 8.16 - 8.03 (m, 1H), 7.76 (ddd, J = 8.8, 7.4, 2.0 Hz, 1H), 7.35 (d, J = 8.6 Hz, 1H), 7.05 (dd, J = 7.8, 4.5 Hz, 1H), 6.60 (d, J = 8.7 Hz, 1H), 4.72 (d, J = 2.7 Hz, 1H), 4.27 (s, 2H), 3.31 - 3.24 (m, 1H), 3.22 - 2.89 (m, 4H), 2.80 (d, J = 13.3 Hz, 1H), 2.51 (s, 1H), 1.99 (t, J = 12.7 Hz, 2H), 1.85 (d, J = 11.6 Hz, 2H), 1.75 - 1.43 (m, 2H), 1.25 (s, 2H).
실시예 12
(9S)-2-(2-메틸피리딘-4-일)-N-(피리미딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (7 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.350 g, 1.314 mmol), DMAP (0.482 g, 3.94 mmol)의 교반 용액에 25℃에서 페닐 피리미딘-4-일카르바메이트 (0.848 g, 3.94 mmol)를 첨가하였다. 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 용매를 진공 하에 제거하고, 조 물질을 CH2Cl2 (20ml)로 희석하고, 물 (5 mL), 포화 염수 용액 (5 mL)으로 세척하고, Na2SO4 상에서 건조시켰다. 유기 용매를 감압 하에 증발시켜 조 생성물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, CH2Cl2 중 3% MeOH로 용리시킴)에 의해 정제하여 (9S)-2-(2-메틸피리딘-4-일)-N-(피리미딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (207 mg, 0.529 mmol, 40.3% 수율), (TLC: DCM 중 10% MeOH, Rf: 0.7), LCMS (m/z): 388.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 14.32 (s, 1H), 8.95 (d, J = 1.3 Hz, 1H), 8.69 (d, J = 5.8 Hz, 1H), 8.61 (d, J = 5.3 Hz, 1H), 8.17 (d, J = 1.7 Hz, 1H), 8.15 (dd, J = 5.8, 1.3 Hz, 1H), 7.95 (dd, J = 5.3, 1.8 Hz, 1H), 7.86 (d, J = 8.1 Hz, 1H), 7.74 (d, J = 8.1 Hz, 1H), 4.90 - 4.67 (m, 1H), 3.40 (dd, J = 13.7, 1.9 Hz, 1H), 3.28 (s, 2H), 3.00 - 2.81 (m, 1H), 2.64 (s, 3H), 2.13 - 1.71 (m, 2H), 1.41 - 1.14 (m, 2H).
실시예 13
(9S)-2-(2-메틸피리딘-4-일)-N-(피라진-2-일)-8,9-디히드로-6H-5, 9-메타노피리도 [2, 3-b] [1, 4] 디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.700 g, 2.63 mmol)의 용액에 트리에틸아민 (2.198 mL, 15.77 mmol) 및 트리포스겐 (0.780 g, 2.63 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반하였다. 피라진-2-아민 (0.750 g, 7.88 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응 혼합물을 농축 건조시킨 다음, 물 (20 mL)과 디클로로메탄 (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다. 조 물질을 칼럼 크로마토그래피에 의해 (100-200 메쉬) 실리카 겔을 사용하여 정제하여 순수한 (9S)-2-(2-메틸피리딘-4-일)-N-(피라진-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다, LCMS (m/z): 388.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 14.27 (s, 1H), 9.59 (d, J = 0.9 Hz, 1H), 8.67 - 8.60 (m, 1H), 8.32 (d, J = 1.0 Hz, 2H), 8.10 (dt, J = 1.9, 0.7 Hz, 1H), 7.74 - 7.67 (m, 1H), 7.62 - 7.50 (m, 2H), 5.06 - 5.00 (m, 1H), 3.51 - 3.27 (m, 3H), 3.03 (d, J = 13.6 Hz, 1H), 2.74 (s, 3H), 2.26 (s, 1H), 2.08 - 1.84 (m, 1H), 1.43 (d, J = 6.2 Hz, 2H).
실시예 14
(9S)-2-(2-메틸피리딘-4-일)-N-(피리다진-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (3 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.150 g, 0.563 mmol), 페닐 피리다진-3-일카르바메이트 (0.364 g, 1.690 mmol)의 교반 용액에 25℃에서 DMAP (0.206 g, 1.690 mmol)를 첨가하였다. 반응 혼합물을 80℃에서 16시간 동안 교반하였다. 용매를 감압 하에 제거하고, 조 화합물을 DCM (10ml)으로 희석하고, 물(3ml), 포화 염수 용액(3ml)으로 세척하고, Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: CH2Cl2 중 2% MeOH)에 의해 정제하여 (9S)-2-(2-메틸피리딘-4-일)-N-(피리다진-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (271 mg, 0.692 mmol, 123% 수율), (TLC: CH2Cl2 중 10% MeOH, Rf: 0.6), LCMS (m/z): 388.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 14.66 (s, 1H), 8.94 (dd, J = 4.7, 1.5 Hz, 1H), 8.66 (dd, J = 5.2, 0.8 Hz, 1H), 8.50 (dd, J = 9.1, 1.5 Hz, 1H), 8.29 (d, J = 1.8 Hz, 1H), 7.74 (dd, J = 5.4, 1.8 Hz, 1H), 7.57 (d, J = 4.3 Hz, 2H), 7.46 (dd, J = 9.0, 4.7 Hz, 1H), 5.00 (p, J = 2.6 Hz, 1H), 3.50 - 3.22 (m, 3H), 3.12 - 2.96 (m, 1H), 2.82 (s, 3H), 2.39 - 2.10 (m, 1H), 1.95 (tdd, J = 14.2, 5.5, 3.0 Hz, 1H), 1.58 - 1.33 (m, 2H).
실시예 15
(9S)-N-(2-메틸-2H-인다졸-5-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

DMAP (0.550 g, 4.51 mmol)를 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.4 g, 1.502 mmol) 및 페닐 (2-메틸-2H-인다졸-5-일)카르바메이트 (0.803 g, 3.00 mmol)의 교반 용액에 질소 분위기 하에 실온에서 첨가하였다. 반응물을 70℃에서 48시간 동안 교반하였다. 반응물을 실온으로 냉각시키고, 감압 하에 농축시켰다. EtOAc를 반응물에 첨가하고, 10분 동안 교반한 다음, 여과하고, 고체를 EtOAc로 세척하였다. 여과물을 취하고, 농축시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 80% EtOAc: Rf-0.4; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 60% EtOAc로 용리시켜 순수한 (9S)-N-(2-메틸-2H-인다졸-5-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.18 g, 0.406 mmol, 27.1% 수율), LCMS (m/z): 440.4 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.44 (s, 1H), 8.66 (d, J = 5.2 Hz, 1H), 8.18 (dd, J = 1.9, 0.8 Hz, 1H), 7.82 (d, J = 0.9 Hz, 1H), 7.69 (d, J = 1.6 Hz, 1H), 7.63 (d, J = 9.1 Hz, 1H), 7.60 - 7.50 (m, 2H), 7.40 (d, J = 7.9 Hz, 1H), 7.27 - 7.19 (m, 1H), 5.05 (t, J = 2.7 Hz, 1H), 4.20 (s, 3H), 3.50 - 3.23 (m, 3H), 3.01 (d, J = 13.5 Hz, 1H), 2.67 (s, 3H), 2.28 (s, 1H), 1.93 (s, 1H), 1.58 (s, 2H).
실시예 16
(9S)-2-(2-메틸피리딘-4-일)-N-(피리미딘-5-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (12 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.6 g, 2.253 mmol), 트리에틸아민 (0.942 mL, 6.76 mmol) 및 비스(트리클로로메틸) 카르보네이트 (0.334 g, 1.126 mmol)를 질소 하에 실온에서 30분 동안 교반한 다음, 1 부분으로 피리미딘-5-아민 (0.643 g, 6.76 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 용매를 감압 하에 제거하고, 수득된 조 물질을 CH2Cl2 (35ml)로 희석하고, 물 (5 mL), 포화 염수 용액 (5 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: CH2Cl2 중 3% MeOH)에 의해 정제하여 반 순수한 화합물을 수득하였다. 반 순수한 화합물을 다시 정제용 HPLC (30% ACN/ 물)에 의해 정제하여 (9S)-2-(2-메틸피리딘-4-일)-N-(피리미딘-5-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (261 mg, 0.667 mmol, 29.6% 수율), (TLC: CH2Cl2 중 10% MeOH, Rf: 0.4), LCMS (m/z): 388 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.81 (s, 1H), 9.03 (s, 2H), 8.94 (s, 1H), 8.69 (dd, J = 5.2, 0.9 Hz, 1H), 7.75 - 7.55 (m, 2H), 7.55 - 7.46 (m, 1H), 7.43 (d, J = 8.0 Hz, 1H), 5.08 - 4.89 (m, 1H), 3.49 - 3.17 (m, 3H), 3.02 (d, J = 13.7 Hz, 1H), 2.69 (s, 3H), 2.36 - 2.13 (m, 1H), 2.06 - 1.79 (m, 1H), 1.59 - 1.32 (m, 2H).
실시예 17
(9S)-N-(피라진-2-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (18 mL) 중 (9S)-2-(2-(트리플루오로메틸)피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.9 g, 2.81 mmol)의 교반 용액에 비스(트리클로로메틸) 카르보네이트 (0.417 g, 1.405 mmol)를 실온에서 첨가하고, 30분 동안 교반하였다. 이어서, 피라진-2-아민 및 Et3N (2.94 ml, 21.8 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 유기 용매를 감압 및 CH2Cl2 (30 ml) 하에 제거하였다. 유기 층을 물 (5ml), 포화 염수 용액 (5ml)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: DCM 중 4% MeOH)에 의해 정제하여 (9S)-N-(피라진-2-일)-2-(2-(트리플루오로메틸)피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (476 mg, 1.059 mmol, 37.7% 수율), (TLC: 헥산 중 80% EtOAc, Rf: 0.5), LCMS (m/z): 442.2 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 14.06 (s, 1H), 9.56 (d, J = 1.4 Hz, 1H), 8.87 (d, J = 5.1 Hz, 1H), 8.46 (dd, J = 1.8, 0.9 Hz, 1H), 8.37 - 8.23 (m, 2H), 8.14 (dd, J = 5.1, 1.7 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.56 (s, 1H), 5.15 - 4.92 (m, 1H), 3.52 - 3.26 (m, 3H), 3.03 (d, J = 14.0 Hz, 1H), 2.26 (s, 1H), 2.05 - 1.88 (m, 1H), 1.53 - 1.29 (m, 2H).
실시예 18
(9S)-N-(피리미딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

테트라히드로푸란 (500 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크 1 500 mg, 1.532 mmol)의 용액에 DMAP (562 mg, 4.60 mmol) 및 페닐 2-(피리미딘-2-일)아세테이트 (985 mg, 4.60 mmol)를 첨가하였다. 반응 혼합물을 65℃에서 28시간 동안 교반하였다. 반응 혼합물을 물 (50 mL)로 희석하고, EtOAc (2 X 75 mL)로 추출하였다. 합한 유기 층을 물로 세척하고, 염수 용액으로 포화시키고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 백색 고체로서 수득하였다 (TLC 용리액: DCM 중 10% MeOH: Rf-0.4; UV 활성). 조 생성물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하고, 생성물을 헥산 중 25% 에틸 아세테이트로 용리시켜 (9S)-N-(피리미딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노-피리도 [2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (320 mg, 0.711 mmol, 46.4% 수율). LCMS (m/z) 448.3 [M+H]+.
1H NMR (400 MHz, DMSO-d6): δ 13.80 (s, 1H), 8.62 (d, J = 4.8 Hz, 2H), 7.36 (d, J = 8.6 Hz, 1H), 7.15 (t, J = 4.8 Hz, 1H), 6.61 (d, J = 8.6 Hz, 1H), 4.69 (s, 1H), 4.30 (d, J = 12.7 Hz, 1H), 4.20 (d, J = 13.0 Hz, 1H), 3.26 (d, J = 1.9 Hz, 1H), 3.20 - 2.89 (m, 4H), 2.79 (d, J = 13.4 Hz, 1H), 2.55 (d, J = 10.8 Hz, 1H), 1.99 (d, J = 12.6 Hz, 2H), 1.82 (h, J = 6.1 Hz, 2H), 1.72 - 1.44 (m, 2H), 1.42 - 1.15 (m, 2H).
실시예 19
(9S)-N-(4,5-디메틸티아졸-2-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (15 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (600 mg, 2.253 mmol)의 용액에 트리에틸아민 (0.942 mL, 6.76 mmol) 및 트리포스겐 (334 mg, 1.126 mmol)을 30℃에서 첨가하고, 1시간 동안 교반하였다. 이어서, 4,5-디메틸티아졸-2-아민 히드로클로라이드 (556 mg, 3.38 mmol)를 30℃에서 첨가하고, 반응을 70℃에서 16시간 동안 가열하였다. 용매를 감압 하에 증발시키고, 잔류물을 물 (40 ml)로 희석하고, DCM (2x 40 ml)으로 추출하였다. 합한 유기 층을 물, 염수로 세척하고, 무수 황산나트륨 상에서 건조시키고, 용매를 감압 하에 증발시켜 조 화합물을 수득하였다. 조 혼합물을 플래쉬 칼럼 크로마토그래피 및 정제용 HPLC에 의해 정제하여 (9S)-N-(4,5-디메틸티아졸-2-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (275 mg, 0.655 mmol, 42% 수율), (TLC: EtOAc 중 10% MeOH, Rf: 0.3), LCMS (m/z): 421.27 [M+H]+.
1H NMR (400 MHz, CDCl3): δ ppm 14.79 (s, 2 H), 8.64 (d, J=5.26 Hz, 2 H), 8.02 (s, 1 H), 7.64 (dd, J=5.26, 1.53 Hz, 1 H), 7.54 (q, J=8.11 Hz, 1 H), 4.99 (s, 1 H), 3.42 - 3.18 (m, 3 H), 3.12 - 2.89 (m, 1 H), 2.79 (s, 3 H), 2.28 (s, 3 H), 2.22 (s, 3 H), 2.00 - 1.75 (m, 1 H), 1.52 - 1.35 (m, 2 H).
실시예 20
(9S)-N-(6-메톡시피라진-2-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.6 g, 2.253 mmol)의 용액에 트리에틸아민 (1.884 mL, 13.52 mmol) 및 트리포스겐 (0.669 g, 2.253 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반하였다. 6-메톡시피라진-2-아민 (0.846 g, 6.76 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 감압 하에 농축시키고, 물 (20 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50% EtOAc로 용리시켜 순수한 (9S)-N-(6-메톡시피라진-2-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.220 g, 0.526 mmol, 23.34% 수율), LCMS (m/z): 418.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.64 (s, 1H), 9.45 - 8.78 (m, 1H), 8.60 (dd, J = 5.3, 0.8 Hz, 1H), 7.96 (d, J = 0.5 Hz, 1H), 7.80 (ddd, J = 5.3, 1.7, 0.7 Hz, 1H), 7.62 (dt, J = 1.8, 0.7 Hz, 1H), 7.59 (s, 1H, 7.57 (s, 1H), 7.46 (d, J = 8.0 Hz, 1H), 5.04 (t, J = 2.6 Hz, 1H), 3.84 (s, 3H), 3.57 - 3.26 (m, 3H), 3.15 - 2.91 (m, 1H), 2.64 (s, 3H), 2.28 (d, J = 14.3 Hz, 1H), 2.12 - 1.85 (m, 1H), 1.43 (s, 2H)
실시예 21
((9S)-N-(6-메틸피라진-2-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성:

트리포스겐 (0.279 g, 0.939 mmol)을 25℃에서 테트라히드로푸란 (50 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.25g, 0.939 mmol)의 교반 용액에 첨가하였다. 트리에틸아민 (0.654 mL, 4.69 mmol)을 첨가하고, 이어서 6-메틸피라진-2-아민 (0.410 g, 3.75 mmol)의 첨가를 첨가하였다. 반응 혼합물을 70℃에서 15시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 물 (50 mL)과 디클로로메탄 (50 mL) 사이에 분배하였다. 분리된 유기 층을 물 및 염수로 세척하였다. 유기 층을 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다 (TLC 용리액: EtOAc 중 10% MeOH; Rf = 0.4; UV 활성). 조 화합물을 중성 알루미나를 사용하여 정제하고, 100% 에틸 아세테이트로 용리시켜 (9S)-N-(6-메틸피라진-2-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 황색 고체로서 수득하였다 (0.11g, 0.272 mmol, 29.0% 수율), LCMS (m/z): 402.26 [M+H]+.
1H NMR (CDCl3,400 MHz): δ 14.02 (s, 1 H), 9.37 (s, 1 H), 8.68 (d, J=5.26 Hz, 1 H), 8.13 - 8.26 (m, 1 H), 7.98 (dd, J=5.26, 1.75 Hz, 1 H), 7.69 - 7.80 (m, 1 H), 7.49 - 7.63 (m, 2 H), 5.03 (t, J=2.19 Hz, 1 H), 3.30 - 3.47 (m, 2 H), 3.03 (br d, J=13.81 Hz, 1 H), 2.69 (s, 2 H), 2.49 - 2.59 (m, 2 H), 2.15 - 2.30 (m, 1 H), 1.84 - 1.99 (m, 1 H), 1.24 - 1.66 (m, 3 H).
실시예 22
(9S)-N-(피리미딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크 II 0.600 g, 1.838 mmol)의 용액에 DMAP (0.674 g, 5.52 mmol) 및 페닐 피리미딘-2-일카르바메이트 (1.187 g, 5.52 mmol)를 첨가하고, 65℃에서 36시간 동안 교반하였다. 반응물을 실온으로 냉각시킨 다음, 감압 하에 농축시켰다. 잔류물을 물 (10 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 헥산 중 80% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 35-40% EtOAc로 용리시켜 순수한 (9S)-N-(피리미딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.375 g, 0.834 mmol, 45.4% 수율), LCMS (m/z): 448.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.97 (s, 1H), 8.59 (d, J = 4.8 Hz, 2H), 7.31 (d, J = 8.6 Hz, 1H), 6.93 (t, J = 4.8 Hz, 1H), 6.40 (d, J = 8.6 Hz, 1H), 4.93 (q, J = 2.6 Hz, 1H), 4.44 - 4.07 (m, 2H), 3.31 (d, J = 1.9 Hz, 1H), 3.19 (d, J = 3.4 Hz, 2H), 3.10 - 2.81 (m, 3H), 2.44 (dtt, J = 15.6, 7.7, 3.9 Hz, 1H), 2.29 (d, J = 13.4 Hz, 1H), 2.17 - 2.07 (m, 1H), 1.93 (dt, J = 13.2, 3.3 Hz, 1H), 1.85 (ddd, J = 13.8, 5.3, 3.1 Hz, 2H), 1.77 - 1.68 (m, 1H), 1.55 - 1.43 (m, 1H), 1.31 (d, J = 14.0 Hz, 1H).
실시예 23
(9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

NaH (0.154 g, 6.43 mmol)를 테트라히드로푸란 (25 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-I, 0.350 g, 1.072 mmol)의 용액에 첨가하고, 질소 하에 0℃에서 교반하였다. 반응 혼합물을 30℃에서 30분 동안 교반하고, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (0.386 g, 1.609 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 24시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 메탄올 (5 mL)로 켄칭하였다. 반응 혼합물을 물로 희석하고, EtOAc (2 X 100mL)로 추출하였다. 유기 층을 물에 이어서 염수 용액으로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 물질을 갈색 고체로서 수득하였다. 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 60% 에틸 아세테이트로 용리시켜 순수한 (9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.320 g, 0.708 mmol, 66.0% 수율), LCMS (m/z): 447.34 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.58 (s, 1H), 8.46 - 8.04 (m, 2H), 7.65 (ddd, J = 8.9, 7.3, 1.9 Hz, 1H), 7.35 - 7.10 (m, 1H), 6.94 (ddd, J = 7.3, 4.9, 1.1 Hz, 1H), 6.38 (d, J = 8.6 Hz, 1H), 5.10 - 4.73 (m, 1H), 4.59 - 4.37 (m, 1H), 4.31 - 4.19 (m, 1H), 3.30 (dd, J = 13.4, 1.9 Hz, 1H), 3.26 - 3.13 (m, 2H), 3.06 - 2.88 (m, 3H), 2.48-2.36 (m, J = 16.3, 8.3, 4.1 Hz, 1H), 2.21 (d, J = 14.4 Hz, 1H), 2.12 (d, J = 12.1 Hz, 1H), 1.94 (s, 1H), 1.88 - 1.68 (m, 2H), 1.67 - 1.57 (m, 1H), 1.50 (s, 1H), 1.31 (d, J = 13.6 Hz, 1H).
실시예 24
(9S)-N-(5-플루오로피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리의 피크-II, 0.600 g, 1.838 mmol)의 용액에 DMAP (0.674 g, 5.52 mmol) 및 페닐 (5-플루오로피리딘-2-일)카르바메이트 (1.281 g, 5.52 mmol)를 첨가하였다. 반응 혼합물을 65℃에서 36시간 동안 교반하였다. 반응물을 실온으로 냉각시키고, 감압 하에 농축시켰다. 잔류물을 물 (10 mL)과 EtOAc (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 헥산 중 80% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 25-30% EtOAc로 용리시켜 순수한 (9S)-N-(5-플루오로피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (0.288 g, 0.608 mmol, 33.1% 수율), LCMS (m/z): 465.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.62 (s, 1H), 8.20 (ddd, J = 9.2, 4.1, 0.6 Hz, 1H), 8.09 (s, 1H), 7.39 (ddd, J = 9.1, 7.8, 3.0 Hz, 1H), 7.30 (d, J = 8.5 Hz, 1H), 6.39 (d, J = 8.6 Hz, 1H), 4.87 (p, J = 2.8 Hz, 1H), 4.30 (dtt, J = 12.5, 3.7, 1.8 Hz, 2H), 3.31 (dd, J = 13.4, 1.9 Hz, 1H), 3.25 - 3.13 (m, 2H), 3.07 - 2.85 (m, 3H), 2.44 (dt, J = 7.4, 3.6 Hz, 1H), 2.26 - 2.05 (m, 2H), 1.99 - 1.83 (m, 2H), 1.78 - 1.66 (m, 1H), 1.62 (dd, J = 12.4, 3.8 Hz, 1H), 1.53 - 1.44 (m, 1H), 1.37 - 1.26 (m, 1H).
실시예 25
(9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
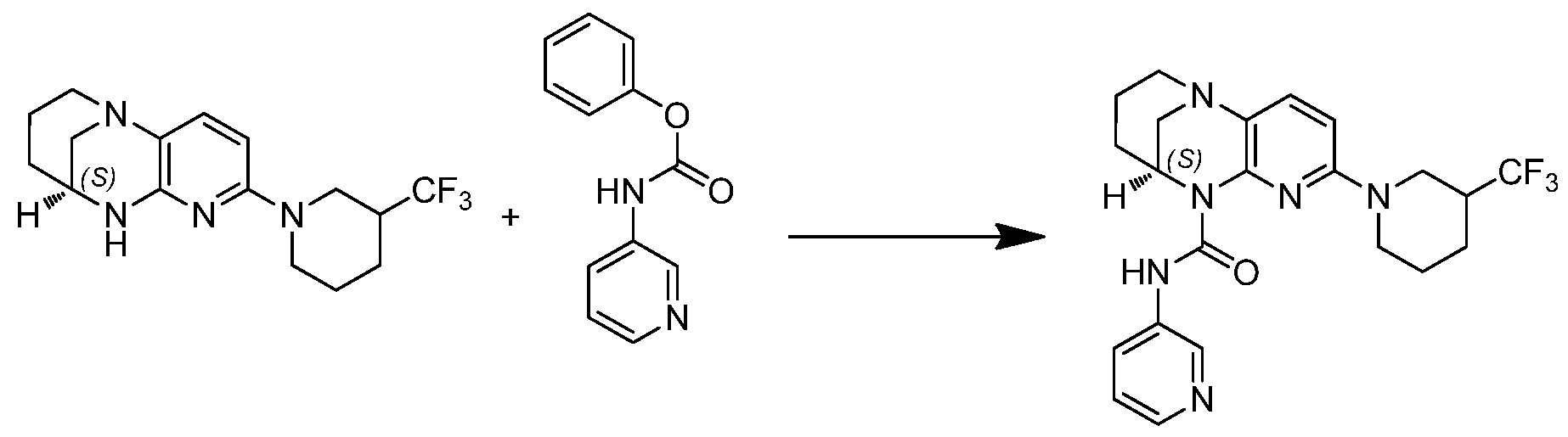
테트라히드로푸란 (30 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리의 피크-I, 600 mg, 1.838 mmol)의 용액에, DMAP (674 mg, 5.52 mmol) 및 페닐 피리딘-3-일카르바메이트 (1182 mg, 5.52 mmol)를 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 물 (50 mL)로 희석하고, 에틸 아세테이트 (2x75 mL)로 추출하였다. 합한 유기 층을 물로 세척하고, 염수 용액으로 포화시키고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 백색 고체로서 수득하였다 (TLC 용리액: 100% 순도 에틸 아세테이트; Rf 값: 0.4; UV 활성). 조 생성물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하여 생성물을 헥산 중 25% 에틸 아세테이트로 용리시켜 (9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (350 mg, 0.782 mmol, 42.5% 수율), LCMS (m/z): 447.26 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 12.94 (s, 1H), 8.62 (dd, J = 2.6, 0.7 Hz, 1H), 8.32 (dd, J = 4.7, 1.5 Hz, 1H), 8.15 (ddd, J = 8.4, 2.7, 1.5 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.29 - 7.24 (m, 1H), 6.41 (d, J = 8.6 Hz, 1H), 4.90 (t, J = 2.6 Hz, 1H), 4.22 - 4.04 (m, 2H), 3.31 (dd, J = 13.5, 1.9 Hz, 1H), 3.25 - 3.14 (m, 2H), 2.99 - 2.88 (m, 3H), 2.48-2.36 (m, 1H), 2.25 - 2.18 (m, 1H), 2.13 (dd, J = 13.2, 4.0 Hz, 1H), 1.98 - 1.92 (m, 1H), 1.85 (tdd, J = 13.9, 5.2, 2.9 Hz, 1H), 1.71 (dtd, J = 16.5, 8.3, 7.7, 4.3 Hz, 1H), 1.63 (d, J = 3.8 Hz, 1H), 1.57 - 1.44 (m, 1H), 1.33 (d, J = 14.2 Hz, 1H).
실시예 26
(9S)-N-(1-메틸-1H-피라졸-4-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

DMAP (0.573 g, 4.69 mmol)을 테트라히드로푸란 (10 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (0.5g, 1.877 mmol), 및 페닐 (1-메틸-1H-피라졸-4-일)카르바메이트 (1.01g, 4.692 mmol)의 교반 용액에 실온에서 첨가하고, 밀봉된 튜브 중에서 24시간 동안 80℃로 냉각시켰다. 반응 혼합물을 28℃로 냉각시키고, 에틸 아세테이트 및 물로 희석하였다. 유기 층을 분리하고, 물 및 염수로 세척하였다. 유기 층을 황산나트륨 상에서 건조시키고, 감압 하에 증발시켜 조 화합물을 수득하였다 (TLC 용리액: EtOAc 중 10% MeOH; Rf-0.4; UV 활성). 조 화합물을 중성 알루미나를 사용하여 정제하고, 헥산 중 75% 에틸 아세테이트로 용리시켜 (9S)-N-(1-메틸-1H-피라졸-4-일)-2-(2-메틸피리딘-4-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.17g, 0.430 mmol, 22.89% 수율), LCMS (m/z): 390.28 [M+H]+.
1H NMR (CDCl3, 400 MHz): δ 13.27 (s, 1 H), 8.67 (d, J=5.26 Hz, 1 H), 7.87 (s, 1 H), 7.63 (d, J=1.53 Hz, 1 H), 7.53 - 7.57 (m, 2 H), 7.43 (d, J=0.66 Hz, 1 H), 7.40 (s, 1 H), 4.99 (t, J=2.30 Hz, 1 H), 3.89 (s, 3 H), 3.22 - 3.42 (m, 3 H), 2.98 (br d, J=13.59 Hz, 1 H), 2.70 (s, 3 H), 2.20 - 2.28 (m, 1 H), 1.92 (tdd, J=13.67, 13.67, 5.54, 3.07 Hz, 1 H), 1.32 - 1.50 (m, 2 H).
실시예 27
(9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
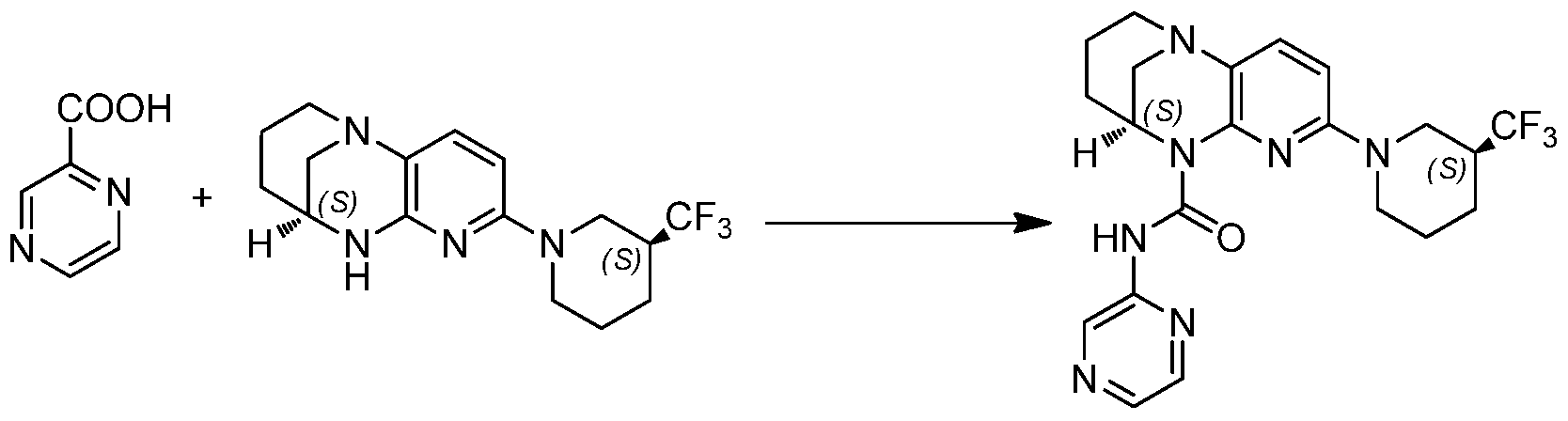
디페닐 포스포르아지데이트 (1.996 g, 7.25 mmol)를 테트라히드로푸란 (THF) (60 mL) 중 피라진-2-카르복실산 (중간체 SFC 분리의 피크-II, 0.600 g, 4.83 mmol)의 교반 용액에 첨가하고, DIPEA (4.22 mL, 24.17 mmol)를 아르곤과 함께 실온에서 교반하였다. 반응 혼합물을 실온에서 2시간 동안 교반하고, (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (1.105 g, 3.38 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 실온으로 냉각시키고, 감압 하에 농축시켰다. 잔류물을 물 (30 mL)과 EtOAc (100 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50% EtOAc로 용리시켜 순수한 (9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (0.367 g, 0.815 mmol, 16.86% 수율), LCMS (m/z): 448.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 13.79 (s, 1H), 9.53 (d, J = 1.5 Hz, 1H), 8.25 (d, J = 2.5 Hz, 1H), 8.20 (d, J = 1.6 Hz, 1H), 7.32 (d, J = 8.6 Hz, 1H), 6.42 (d, J = 8.6 Hz, 1H), 4.90 (s, 1H), 4.28 (s, 2H), 3.32 (dd, J = 13.5, 1.9 Hz, 1H), 3.21 (dd, J = 12.5, 3.3 Hz, 2H), 3.10 - 2.86 (m, 3H), 2.44 (dt, J = 7.6, 3.6 Hz, 1H), 2.28 - 2.18 (m, 1H), 2.11 (d, J = 3.6 Hz, 1H), 1.99 - 1.80 (m, 2H), 1.78 - 1.65 (m, 1H), 1.65 - 1.60 (m, 1H), 1.49 (s, 1H), 1.35 (s, 1H).
실시예 28
(9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
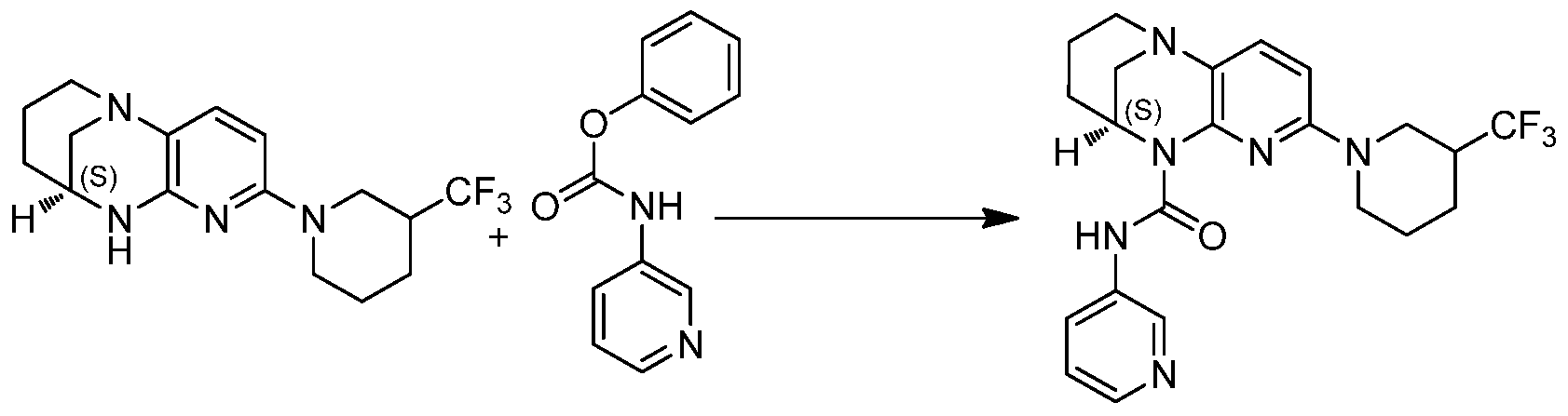
테트라히드로푸란 (THF) (70 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리의 피크-II, (중간체 SFC 분리로부터의 피크-II, 0.700 g, 2.145 mmol)의 용액에 DMAP (0.786 g, 6.43 mmol)를 첨가하고, 페닐 피리딘-3-일카르바메이트 (1.378 g, 6.43 mmol)를 65℃에서 36시간 동안 교반한 후, 반응물을 실온으로 냉각시키고, 감압 하에 농축시켰다. 잔류물을 물 (20 mL)과 EtOAc (75 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 헥산 중 80% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50% EtOAc로 용리시켜 순수한 (9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (0.355 g, 0.794 mmol, 37.0% 수율), LCMS (m/z): 447.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ ppm 12.92 (s, 1 H), 8.62 (d, J=2.63 Hz, 1 H), 8.32 (dd, J=4.71, 1.43 Hz, 1 H), 8.08 - 8.21 (m, 1 H), 7.33 (d, J=8.55 Hz, 1 H), 7.20 - 7.29 (m, 1 H), 6.40 (d, J=8.55 Hz, 1 H), 4.90 (br s, 1 H), 4.00 - 4.21 (m, 2 H), 3.31 (dd, J=13.48, 1.86 Hz, 1 H), 3.17 - 3.26 (m, 2 H), 2.88 - 3.03 (m, 3 H), 2.36 - 2.49 (m, 1 H), 2.19 - 2.25 (m, 1 H), 2.10 - 2.16 (m, 1 H), 1.92 - 1.99 (m, 1 H), 1.86 (tdd, J=13.84, 13.84, 5.10, 2.96 Hz, 1 H), 1.65 - 1.76 (m, 1 H), 1.46 - 1.58 (m, 2 H), 1.29 - 1.38 (m, 1 H)
실시예 29
(9S)-N-(5-플루오로피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

테트라히드로푸란 (30 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (SFC 분리로부터의 피크-I, 450 mg, 1.379 mmol)의 용액에 DMAP (505 mg, 4.14 mmol) 및 페닐 (5-플루오로피리딘-2-일)카르바메이트 (961 mg, 4.14 mmol)를 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 물 (50 mL)로 희석하고, 에틸 아세테이트 (2x75 mL)로 추출하였다. 합한 유기 층을 물로 세척하고, 염수 용액으로 포화시키고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 회백색 고체로서 수득하였다 (TLC 용리액: 100% 에틸 아세테이트; Rf 값: 0.4; UV 활성). 조 생성물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하여 생성물을 헥산 중 25% 에틸 아세테이트로 용리시켜 (9S)-N-(5-플루오로피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (140 mg, 0.296 mmol, 21.46% 수율), LCMS (m/z): 465.21 [M+H]+.
1H NMR (400 MHz, CDCl3): δ ppm 1.15 - 1.43 (m, 1 H) 1.43 - 1.68 (m, 2 H) 1.68 - 1.89 (m, 3 H) 2.04 - 2.28 (m, 2 H) 2.42 (dtd, J=11.67, 7.92, 7.92, 4.06 Hz, 1 H) 2.86 - 3.09 (m, 3 H) 3.10 - 3.36 (m, 3 H) 4.23 - 4.47 (m, 2 H) 4.87 (br s, 1 H) 6.39 (d, J=8.55 Hz, 1 H) 7.3 (d, J=8.8 Hz, 1 H) 7.34 - 7.59 (m, 1 H) 8.09 (d, J=2.8 Hz, 1 H) 8.20 (dd, J=9.21, 4.17 Hz, 1 H) 13.65 (s, 1 H).
실시예 30
(9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

테트라히드로푸란 (50 mL) 디페닐포스포릴 아지드 (1.996 g, 7.25 mmol) 및 DIPEA (4.22 mL, 24.17 mmol) 중 피라진-2-카르복실산 (0.6 g, 4.83 mmol)의 현탁액을 반응 혼합물에 첨가하였다. 반응 혼합물을 2시간 동안 28℃에서 교반하고, (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-I 1.105 g, 3.38 mmol)을 반응 혼합물에 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 물 (50mL)로 희석하고, 에틸 아세테이트 (2 X 75 mL)로 추출하였다. 합한 유기 층을 물로 세척하고, 염수 용액으로 포화시키고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 회백색 고체로서 수득하였다 (TLC 용리액: 100% 에틸 아세테이트; Rf 값: 0.3; UV 활성). 조 생성물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하여 생성물을 헥산 중 30% 에틸 아세테이트로 용리시켜 (9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)-피페리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (0.330 g, 0.728 mmol, 15.05% 수율), LCMS (m/z): 448.25 [M+H]+.
1H NMR (400 MHz, CDCl3): δ ppm 1.31 - 1.34 (m, 1H)1.45 - 1.67 (m, 2 H) 1.68 - 1.79 (m, 1 H) 1.82 - 1.99 (m, 2 H) 2.05 - 2.18 (m, 2 H) 2.42 (dddt, J=15.65, 11.78, 8.03, 3.97, 3.97 Hz, 1 H) 2.86 - 3.09 (m, 3 H) 3.11 - 3.35 (m, 3 H) 4.21 - 4.39 (m, 2 H) 4.89 (br s, 1 H) 6.42 (d, J=8.55 Hz, 1 H) 7.32 (d, J=8.4 Hz, 1 H) 8.14 - 8.20 (m, 1 H) 8.24 (d, J=2.8 Hz, 1 H) 9.53 (d, J=1.53 Hz, 1 H) 13.81 (s, 1 H)
실시예 31
(9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 히드로클로라이드의 합성

디에틸 에테르 중 2.0 M 염산 (2 mL, 4.00 mmol)을 0℃에서 (9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (180 mg, 0.416 mmol)에 첨가하였다. 반응 혼합물을 28℃에서 4시간 동안 교반하고, 농축시켜 조 화합물을 수득하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.1; UV 활성). 조 화합물을 디에틸 에테르 (2x5 mL)로 세척하여 순수한 (9S)-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)피롤리딘-1-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 히드로클로라이드를 연갈색 고체로서 수득하였다 (83mg, 0.177 mmol, 42.78% 수율), LCMS (m/z): 433.28 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 15.02 (br s, 1 H), 13.34 (br s, 1 H), 8.25 (br d, J=3.73 Hz, 1 H), 8.13 (br d, J=8.33 Hz, 1 H), 8.06 (d, J=8.77 Hz, 1 H), 7.75- 7.68 (m, 1 H), 7.04- 6.98 (m, 1 H), 6.15 (s, 1 H), 5.30 -5.24 (b, s, 1 H), 4.12 -3.94 (m, 2 H), 3.73 -3.59 (m, 4 H), 3.49 -3.37 (m, 2 H), 3.18-3.04 (m, 1 H), 2.45 - 2.25 (m, 3 H), 1.86 (d, J=12.50 Hz, 2 H).
실시예 32
(9S)-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 분위기 하에 THF (30 mL) 중 (9S)-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (300 mg, 1.126 mmol)의 교반 용액에 페닐 피리딘-3-일카르바메이트 (724 mg, 3.38 mmol), DMAP (413 mg, 3.38 mmol)를 첨가하고, 65℃에서 48시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.4; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 농축시키고, 잔류물을 물 (20 mL)과 EtOAc (100 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 물질을 칼럼 크로마토그래피 (중성 알루미나, 용리액: DCM 중 2% MeOH)에 의해 정제하여 목적 생성물 (9S)-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (200 mg, 0.516 mmol, 45.8% 수율). LCMS (m/z): 387.09 [M+H]+, Rt = 1.21분.
1H NMR (400 MHz, CDCl3): δppm 13.61 (s, 1 H), 8.84 - 8.62 (m, 2 H), 8.33 (dd, J=4.71, 1.21 Hz, 1 H), 8.17 (dd, J=8.33, 1.54 Hz, 1 H), 7.63 (s, 1 H), 7.58 (d, J=7.89 Hz, 1 H), 7.52 (br d, J=5.04 Hz, 1 H), 7.42 (d, J=8.11 Hz, 1 H), 7.28 (br d, J=4.60 Hz, 1 H), 5.02 (br s, 1 H), 3.45 - 3.31 (m, 3 H), 3.01 (br d, J=13.59 Hz, 1 H), 2.69 (s, 3 H), 2.27 (br d, J=12.93 Hz, 1 H), 3.01 - 1.85 (m, 1 H), 1.54 - 1.32 (m, 2 H).
실시예 33
(9S)-3-클로로-N2-시클로프로필-N10-(피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성.

실온에서 질소 하에 DMF (6 mL) 중 (9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (650 mg, 1.739 mmol)의 교반 용액에 DIPEA (0.911 mL, 5.22 mmol), HATU (1322 mg, 3.48 mmol) 및 시클로프로판아민 (119 mg, 2.087 mmol)을 첨가한 다음, 반응 혼합물을 16시간 동안 교반하였다 (TLC 시스템: DCM 중 5% 메탄올. Rf: 0.30). 반응물을 빙냉수 50 mL로 희석하고, EtOAc (3 x 100 mL)로 추출하였다. 합한 유기 층을 염수 (100 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 화합물을 수득하였다. 조 물질을 콤비플래쉬 (실리카 겔 칼럼 사용, 헥산 중 75% EtOAc)에 의해 정제하였다. 순수한 화합물을 함유하는 분획을 합하고, 농축시켜 목적 화합물 (9S)-3-클로로-N2-시클로프로필-N10-(피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-H)-디카르복스아미드를 회백색 고체로서 수득하였다 (97 mg, 0.229 mmol, 13.15% 수율). LCMS (m/z): 413.10 [M+H]+, Rt = 1.84분.
1H NMR (400 MHz, CDCl3): δ ppm 13.12 (br s, 1 H), 8.33 (dd, J=4.93, 0.99 Hz, 1 H), 8.14 (d, J=8.33 Hz, 1 H), 7.51 - 7.76 (m, 2 H), 7.60 (m, 1 H), 7.09 (br, 1 H), 4.97 (br s, 1 H), 3.23 - 3.44 (m, 3 H), 3.04 (ddt, J=10.91, 7.18, 3.89, 3.89 Hz, 1 H), 2.93 (br d, J=13.81 Hz, 1 H), 2.10 - 2.32 (m, 1 H), 1.80 - 2.07 (m, 1 H), 1.22 - 1.46 (m, 2 H), 0.81 - 0.98 (m, 4 H)
실시예 34
(9S)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성:

메탄올 (10 mL) 중 (9S)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (200 mg, 0.354 mmol)의 용액에 수성 HCl (0.308 mL, 3.54 mmol)을 5분의 기간에 걸쳐 0℃에서 적가하고, 실온에서 1시간 동안 교반하였다 (TLC 시스템: 100% 에틸 아세테이트. Rf 값: 0.3). 이어서, 반응 혼합물을 포화 NaHCO3 용액 (20 mL)으로 켄칭하고, DCM (2x30 mL)으로 추출하였다. 합한 유기 층을 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 생성물을 수득하였다. 조 생성물을 펜탄: 디에틸 에테르 (1:1)로 연화처리하여 요구되는 순도의 목적 생성물 (9S)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (100 mg, 0.187 mmol, 52.7% 수율). LCMS (m/z): 525.14 [M+H]+, Rt =1.53분.
1H NMR (400 MHz, CDCl3): δ ppm 13.84 (s, 1 H), 8.16 (br d, J=9.21 Hz, 1 H), 8.09 (d, J=5.92 Hz, 1 H), 8.00 (d, J=7.89 Hz, 1 H), 7.80 (d, J=2.19 Hz, 1 H), 7.61 (d, J=7.89 Hz, 1 H), 6.60 (dd, J=5.81, 2.30 Hz, 1 H), 5.17 - 5.04 (m, 1 H), 4.96 (br s, 1 H), 4.25 - 4.03 (m, 3 H), 3.89 - 3.71 (m, 2 H), 3.44 - 3.18 (m, 3 H), 2.96 (br d, J=14.03 Hz, 1 H), 2.59 (br s, 1 H), 2.22 (br d, J=14.47 Hz, 1 H), 2.06 - 1.76 (m, 2 H), 1.60 (d, J=7.02 Hz, 3 H), 1.45 - 1.33 (m, 2 H).
실시예 35
(9S)-N2-(2,2-디플루오로프로필)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-N2-(2,2-디플루오로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (210 mg, 0.384 mmol)의 용액에 수성 HCl (0.667 mL, 7.68 mmol)을 5분의 기간에 걸쳐 0℃에서 적가하고, 실온에서 1시간 동안 교반하였다 (TLC 시스템: 100% 에틸 아세테이트. Rf 값: 0.3). 이어서, 반응 혼합물을 포화 NaHCO3 용액 (20 mL)으로 켄칭하고, DCM (2x30 mL)으로 추출하였다. 합한 유기 층을 무수 황산나트륨 및 농축 감압 하에 상에서 건조시키고, 펜탄: 디에틸 에테르 (1:1)로 연화처리하여 목적 생성물 (9S)-N2-(2,2-디플루오로프로필)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (95 mg, 0.186 mmol, 48.5% 수율). LCMS (m/z): 507.14 [M+H]+, Rt =1.40분.
1H NMR (400 MHz, CDCl3): δ ppm 14.25 (s, 1 H), 8.69 (br t, J=6.47 Hz, 1 H), 8.12 (d, J=5.92 Hz, 1 H), 7.95 (d, J=8.11 Hz, 1 H), 7.71 (d, J=2.19 Hz, 1 H), 7.60 (d, J=7.89 Hz, 1 H), 6.58 (dd, J=5.92, 2.41 Hz, 1 H), 4.94 (br s, 1 H), 4.24 - 4.08 (m, 3 H), 4.04 - 3.90 (m, 2 H), 3.87 - 3.73 (m, 2 H), 3.43 - 3.24 (m, 3 H), 2.97 (br d, J=14.03 Hz, 1 H), 2.62 - 2.50 (m, 1 H), 2.27 - 2.16 (m, 1 H), 2.08 - 1.89 (m, 2 H), 1.69 (t, J=18.64 Hz, 3 H), 1.465 - 1.35 (m, 2 H).
실시예 36
(9S)-N2-시클로프로필-N10-(피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성
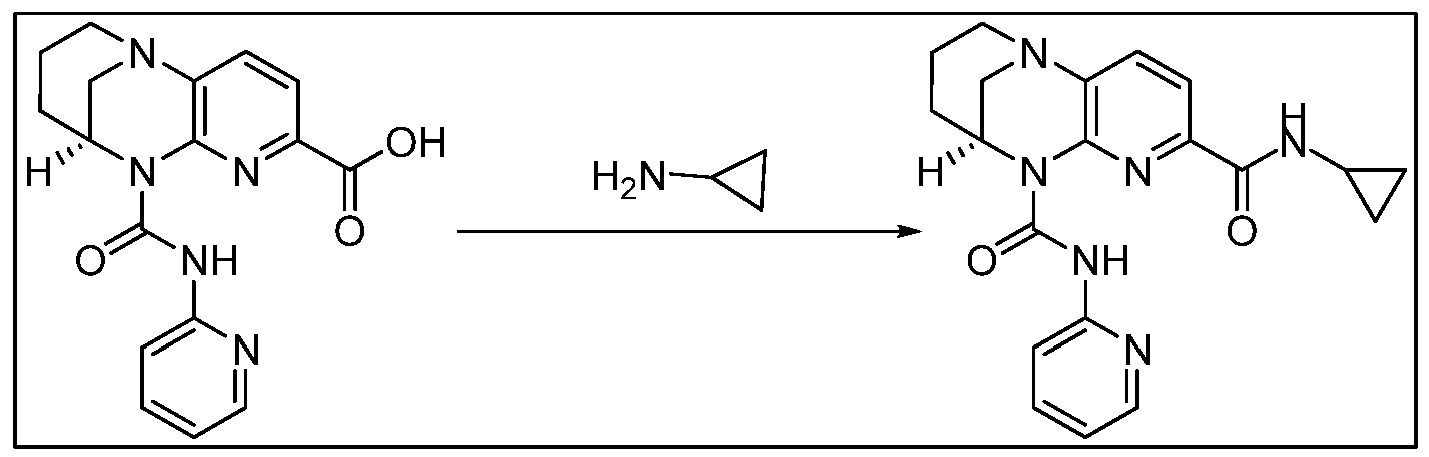
0℃에서 질소 하에 DMF (60 mL) 중 (9S)-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산(6g,17.68mmol)의 교반 용액에 DIPEA (9.26 mL, 53.0 mmol), HATU (13.45 g, 35.4 mmol) 및 시클로프로판아민 (1.514 g, 26.5 mmol)을 첨가하고, 반응 혼합물을 실온에서 16시간 동안 교반하였다 (TLC 시스템: 헥산 중 70% 에틸아세테이트, Rf 값: 0.3). 반응 혼합물에 빙냉수 (100 mL)를 첨가하고, 15분 동안 교반하였다. 생성된 고체를 여과하고, 건조시키고, 콤비플래쉬 크로마토그래피 (실리카겔 칼럼 사용, DCM 중 3% 메탄올로 용리시킴)에 의해 정제하여 (9S)-N2-시클로프로필-N10-(피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (4.05 g, 10.70 mmol, 60.5% 수율). LCMS (m/z): 379.16 [M+H]+, Rt=1.97분.
1H NMR (400 MHz, CDCl3): δ ppm 13.70 - 14.14 (m, 1 H), 8.29 - 8.42 (m, 1 H), 8.16 (dt, J=8.33, 0.88 Hz, 2 H), 7.97 (d, J=7.89 Hz, 1 H), 7.72 (td, J=7.84, 1.64 Hz, 1 H), 7.58 (d, J=7.89 Hz, 1 H), 7.04 (ddd, J=7.29, 4.99, 1.10 Hz, 1 H), 4.97 (t, J=2.19 Hz, 1 H), 3.29 - 3.42 (m, 3 H), 3.10 (ddt, J=10.93, 7.32, 3.92, 3.92 Hz, 1 H), 2.96 (br d, J=13.59 Hz, 1 H), 2.16 - 2.29 (m, 1 H), 1.86 - 1.99 (m, 1 H), 1.59 (s, 1 H), 1.32 - 1.45 (m, 2 H), 0.81 - 1.02 (m, 4 H).
실시예 37
(9S)-N10-(피리딘-2-일)-N2-(2,2,2-트리플루오로에틸)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

0℃에서 질소 하에 DMF (50 mL) 중 (9S)-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (7g,20.63mmol)의 교반 용액에 DIPEA (10.81 mL, 61.9 mmol), HATU (15.69 g, 41.3 mmol) 및 2,2,2-트리플루오로에탄아민 (2.248 g, 22.69 mmol)을 첨가한 다음, 반응물을 실온에서 16시간 동안 교반하였다 (TLC 시스템: 헥산 중 70% 에틸아세테이트, Rf 값: 0.3). 반응 혼합물에 빙냉수 (200 mL)를 첨가하고, 30분 동안 교반하였다. 생성된 고체를 여과하고, 건조시키고, 콤비플래쉬 크로마토그래피 (실리카겔 칼럼 사용, DCM 중 3% 메탄올로 용리시킴)에 의해 정제하여 생성물을 회백색 고체로서 수득하였다. 고체 생성물을 80℃에서 에탄올 (500 mL) 중에 용해시키고, 팔라듐 스캐빈저 (실리사이클 브랜드, 8 g)을 첨가하고, 5시간 동안 계속 가열하였다. 반응 혼합물을 셀라이트를 통해 여과하고, 여과물을 농축시켜 (9S)-N10-(피리딘-2-일)-N2-(2,2,2-트리플루오로에틸)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (8.1 g, 19.17 mmol, 93% 수율). LCMS (m/z): 421.1 [M+H]+, Rt=2.23분.
1H NMR (400 MHz, CDCl3): δ ppm 14.13 - 14.37 (m, 1 H), 8.71 (br t, J=5.92 Hz, 1 H), 8.25 (dd, J=4.93, 0.99 Hz, 1 H), 8.11 (d, J=8.33 Hz, 1 H), 7.96 (d, J=7.89 Hz, 1 H), 7.70 - 7.77 (m, 1 H), 7.61 (d, J=7.89 Hz, 1 H), 7.00 - 7.05 (m, 1 H), 4.97 (t, J=2.30 Hz, 1 H), 4.13 - 4.37 (m, 2 H), 3.27 - 3.45 (m, 3 H), 2.97 (br d, J=13.59 Hz, 1 H), 2.18 - 2.27 (m, 1 H), 1.87 - 2.00 (m, 1 H), 1.34 - 1.46 (m, 2 H).
실시예 38
(9S)-3-클로로-N10-(피리딘-2-일)-N2-(2,2,2-트리플루오로에틸)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성.

실온에서 질소 하에 아세토니트릴 (15 mL) 중 (9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (900 mg, 2.408 mmol)의 현탁액에 DIPEA (1.262 mL, 7.22 mmol), 2-클로로-1-메틸피리딘-1-윰 아이오다이드 (615 mg, 2.408 mmol) 및 2,2,2-트리플루오로에탄아민 (238 mg, 2.408 mmol)을 첨가한 다음, 생성된 반응 혼합물을 16시간 동안 교반하였다 (TLC 시스템: 헥산 중 75% EtOAc, Rf 값: 0.30). 반응물을 물 100 mL)로 희석하고, EtOAc (2 x 150 mL)로 추출하였다. 합한 유기 층을 염수 (80 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 수득하였다. 조 물질을 콤비플래쉬 (실리카 겔 칼럼 사용, 헥산 중 70% EtOAc)에 의해 정제하였다. 화합물을 함유하는 분획을 합하고, 농축시켜 목적 화합물 (9S)-3-클로로-N10-(피리딘-2-일)-N2-(2,2,2-트리플루오로에틸)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (52 mg, 0.114 mmol, 4.75% 수율). LCMS (m/z): 455.05 [M+H]+, Rt = 2.17분.
1H NMR (400 MHz, CDCl3): δ ppm 13.43 (s, 1 H), 8.29 - 8.49 (m, 1 H), 8.25 (d, J=4.17 Hz, 1 H), 8.11 (d, J=8.33 Hz, 1 H), 7.67 - 7.84 (m, 1 H), 7.59 (s, 1 H), 6.95 - 7.14 (m, 1 H), 4.97 (br s, 1 H), 4.12 - 4.37 (m, 2 H), 3.28 - 3.44 (m, 3 H), 2.95 (br d, J=13.81 Hz, 1 H), 2.10 - 2.33 (m, 1 H), 1.86 - 2.07 (m, 1 H), 1.25 - 1.50 (m, 2 H).
실시예 39
(9S)-3-클로로-N10-(피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성.

실온에서 질소 하에 DMF (6 mL) 중 (9S)-3-클로로-10-(피리딘-2-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (500 mg, 1.338 mmol)의 교반 용액에 DIPEA (0.701 mL, 4.01 mmol), HATU (1017 mg, 2.68 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (182 mg, 1.605 mmol)을 첨가한 다음, 생성된 반응 혼합물을 실온에서 16시간 동안 교반하였다 (TLC 시스템: DCM 중 5% MeOH, Rf 값: 0.30). 반응물을 빙냉수 50 mL로 희석하고, EtOAc (2x100 mL)로 추출하였다. 합한 유기 층을 염수 (80 mL)로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 화합물을 수득하였다. 조 물질을 콤비플래쉬 (실리카 겔 칼럼 사용, 헥산 중 80% EtOAc)에 의해 정제하였다. 화합물을 함유하는 분획을 합하고, LCMS에 의해 농축시켜 목적 화합물을 60% 순도로 수득하였다. 이를 추가로 정제용 HPLC (칼럼: 크로마실 페닐 (150x25) mm 10μ; MP-A: 10mM 중탄산암모늄 (수성), MP-B: 아세토니트릴; 방법: 50:50; 유량: 20 ml/분 용해도: ACN+THF)에 의해 정제하여 목적 생성물 (9S)-3-클로로-N10-(피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (50 mg, 0.106 mmol, 7.93% 수율). LCMS (m/z): 469.16 [M+H]+, Rt = 2.22분.
1H NMR (400 MHz, CDCl3): δ ppm 12.89 (s, 1 H), 8.27 (d, J=4.17 Hz, 1 H), 8.15 (d, J=8.33 Hz, 1 H), 7.64 - 7.80 (m, 2 H), 7.59 (s, 1 H), 6.90 - 7.15 (m, 1 H), 5.09 (dq, J=16.83, 7.55 Hz, 1 H), 4.89 - 5.01 (m, 1 H), 3.26 - 3.44 (m, 3 H), 2.94 (br d, J=13.81 Hz, 1 H), 2.14 - 2.35 (m, 1 H), 1.85 - 2.09 (m, 1 H), 1.39 - 1.63 (m, 3 H), 1.40-1.32 (s, 2 H).
실시예 40
(9S)-N10-(이속사졸-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

실온에서 질소 하에 DMF (15 mL) 중 (9S)-10-(이속사졸-3-일카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (400 mg, 1.215 mmol) 및 (R)-1,1,1-트리플루오로프로판-2-아민 (206 mg, 1.822 mmol)의 교반 용액에 HATU (554 mg, 1.458 mmol), DIPEA (0.424 mL, 2.429 mmol)를 첨가하고, 16시간 동안 교반하였다 (TLC 시스템: 에틸아세테이트. Rf 값: 0.6). 반응 혼합물을 빙냉수 (30 mL)로 켄칭하고, 에틸 아세테이트 (3x50 mL)로 추출하였다. 합한 유기 층을 염수 용액 (30 mL)으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카 겔, 석유 에테르 중 60% 에틸아세테이트 사용)에 의해 정제하여 목적 생성물 (9S)-N10-(이속사졸-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (160 mg, 0.373 mmol, 30.7% 수율). LCMS (m/z): 425.09 [M+H]+, Rt =2.14분.
1H NMR (400 MHz, CDCl3): δ ppm 13.55 (s, 1 H), 8.30 (d, J=1.32 Hz, 1 H), 7.96 (d, J=7.89 Hz, 1 H), 7.62 (d, J=7.89 Hz, 1 H), 7.23 (br s, 1 H), 6.99 (d, J=1.75 Hz, 1 H), 4.89 - 5.05 (m, 2 H), 3.32 - 3.47 (m, 3 H), 2.96 (br d, J=13.81 Hz, 1 H), 2.22 (dt, J=14.69, 2.96 Hz, 1 H), 1.90 - 2.02 (m, 1 H), 1.64 (d, J=7.02 Hz, 3 H), 1.33 - 1.45 (m, 2 H).
실시예 41
(9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-(2,2,2-트리플루오로에틸)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성.

N,N-디메틸포름아미드 (5 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (200 mg, 0.508 mmol)의 교반 용액에 HATU (193 mg, 0.508 mmol) 및 DIPEA (0.089 mL, 0.508 mmol)를 질소 분위기 하에 28℃에서 첨가하고, 30분 동안 실온에서 교반한 다음, 2,2,2-트리플루오로에탄아민 히드로클로라이드 (103 mg, 0.763 mmol)를 첨가하고, 반응 혼합물을 28℃에서 16시간 동안 교반하였다 (TLC: 10% MeOH/CH2Cl2, Rf 값: 0.4). 반응 혼합물을 물 (40 mL)로 희석하고, EtOAc (2x40 mL)로 추출하였다. 합한 유기 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카겔:100-200 메쉬, 용리액: 5% 메탄올 /DCM)에 의해 정제하여 목적 생성물 (9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-(2,2,2-트리플루오로에틸)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (100 mg, 0.211 mmol, 41.5% 수율). LCMS (m/z): 475.10 [M+H]+, Rt = 1.96분.
1H NMR (400 MHz, CDCl3-d): δ ppm 13.85 (s, 1 H), 10.39 (s, 1 H), 8.75 (d, J=8.33 Hz, 1 H), 8.04 -7.90 (m, 2 H), 7.63 (d, J=7.89 Hz, 1 H), 7.03 (d, J=8.55 Hz, 1 H), 5.00 (d, J=2.19 Hz, 1 H), 4.37- 4.12 (m, 2 H), 3.48 -3.30 (m, 3 H), 3.00 (d, J=13.59 Hz, 1 H), 2.69 (s, 3 H), 2.37 -2.25 (m, 1 H), 2.07 -1.88 (m, 1 H), 1.48 -1.39 (m, 2 H).
실시예 42
(9S)-N2-시클로프로필-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

DMF (15 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (300 mg, 0.763 mmol), HATU (435 mg, 1.144 mmol) 및 DIPEA (0.400 mL, 2.288 mmol)의 교반 용액에 시클로프로판아민 (65.3 mg, 1.144 mmol)을 실온에서 첨가하고, 반응 혼합물을 실온에서 16시간 동안 교반하였다 (TLC: 순수한 에틸 아세테이트, Rf 값: 0.3, UV 활성). 반응 혼합물을 물 (100 mL)로 희석하고, EtOAc (2x100 mL)로 추출하였다. 합한 유기 층을 포화 염수 용액 (50 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카 겔: 100-200 메쉬, 용리액: n-헥산 중 80% 에틸 아세테이트)에 의해 정제하여 목적 생성물 (9S)-N2-시클로프로필-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (90 mg, 0.202 mmol, 26.5% 수율). LCMS (m/z): 433.16 [M+H]+, Rt =1.76분.
1H NMR (400 MHz, CDCl3): δ ppm 13.67 -13.63 (m, 1 H), 13.65 (s, 1 H), 11.16 (s, 1 H), 8.71 (d, J=8.33 Hz, 1 H), 7.93 (d, J=7.89 Hz, 1 H), 7.60 (d, J=8.11 Hz, 1 H), 7.52 (d, J=2.85 Hz, 1 H), 7.03 (d, J=8.55 Hz, 1 H), 5.01 (s, 1 H), 3.42- 3.31 (m, 3 H), 3.12 -3.05 (m, 1 H), 3.00 (d, J=13.81 Hz, 1 H), 2.73 (s, 3 H), 2.27 (d, J=14.47 Hz, 1 H), 2.01- 1.88 (m, 1 H), 1.45- 1.36 (m, 2 H), 0.91- 0.85 (m, 3 H).
실시예 43
(9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

DMF (15 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (200 mg, 0.508 mmol), HOBT (97 mg, 0.635 mmol), EDC (122 mg, 0.635 mmol) 및 DIPEA (0.266 mL, 1.525 mmol)의 교반 용액에 (R)-1,1,1-트리플루오로프로판-2-아민 (69.0 mg, 0.610 mmol)을 실온에서 첨가하고, 반응 혼합물을 실온에서 16시간 동안 교반하였다 (TLC: 순수한 에틸 아세테이트, Rf 값: 0.3, UV 활성). 반응 혼합물을 물 (100 mL)로 희석하고, EtOAc (2x100 mL)로 추출하였다. 합한 유기 층을 염수 용액 (50 mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (100-200 실리카 겔 사용, 화합물을 n-헥산 중 80% 에틸 아세테이트로 용리시킴)에 의해 정제하여 목적 생성물 (9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (119 mg, 0.237 mmol, 46.6% 수율). LCMS: (m/z): 489.2 [M+H] +, Rt = 2.06분.
1H NMR (400 MHz, CDCl3): δ ppm 13.63 (s, 1 H), 10.14 (s, 1 H), 8.69 (d, J=8.33 Hz, 1 H), 7.96 (d, J=7.89 Hz, 1 H), 7.69 (d, J=9.21 Hz, 1 H), 7.62 (d, J=7.89 Hz, 1 H), 7.03 (d, J=8.55 Hz, 1 H), 5.12- 4.95 (m, 2 H), 3.51-3.18 (m, 3 H), 2.93 - 3.05 (m, 1 H), 2.68 (s, 3 H), 2.29 (d, J=14.47 Hz, 1 H), 2.04- 1.87 (m, 1 H), 1.57 (s, 3 H), 1.49 - 1.25 (m, 2 H).
실시예 44
(9S)-N10-(4-(2-메틸티아졸-5-일)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

실온에서 질소 하에 1,4-디옥산 (20 mL) 및 물 (1.0 mL) 중 (9S)-N10-(4-브로모피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (350 mg, 0.682 mmol) 및 2-메틸-5-(4,4,5,5-테트라메틸-1,3,2-디옥사보롤란-2-일)티아졸 (230 mg, 1.023 mmol)의 용액에 K3PO4 (434 mg, 2.046 mmol)를 첨가하고, 아르곤으로 20분 동안 퍼징함으로써 탈기하였다. 이어서, PdCl2(dppf)-CH2Cl2 부가물 (55.7 mg, 0.068 mmol)을 첨가하고, 혼합물을 110℃에서 3.5시간 동안 두었다 (TLC 시스템: 100% 에틸 아세테이트. Rf 값: 0.3). 반응 혼합물을 실온으로 냉각시키고, 물 (30 mL)로 희석하고, 에틸 아세테이트 (2x50 mL)로 추출하였다. 합한 유기 층을 염수 용액 (50 mL)으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (100-200 실리카겔, 석유 에테르 중 50% EtOAc로 용리시킴)에 의해 정제하여 목적 생성물(9S)-N10-(4-(2-메틸티아졸-5-일)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (320 mg, 0.594 mmol, 87% 수율). LCMS (m/z): 532.20 [M+H]+, Rt =2.60분.
1H NMR (400 MHz, CDCl3): δ ppm 13.88 (s, 1 H), 8.39 (s, 1 H), 8.26 (d, J=5.26 Hz, 1 H), 8.13 - 7.89 (m, 3 H), 7.62 (d, J=8.11 Hz, 1 H), 7.15 (dd, J=5.26, 1.53 Hz, 1 H), 5.20 - 5.06 (m, 1 H), 5.00 (br s, 1 H), 3.44 - 3.21 (m, 3 H), 3.03 - 2.90 (m, 1 H), 2.76 (s, 3 H), 2.25 (br d, J=14.47 Hz, 1 H), 2.05 -1.89 (m, 1 H), 1.62 (d, J=7.02 Hz, 3 H), 1.49 - 1.36 (m, 2 H).
실시예 45
(9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-(1,1,1-트리플루오로-3-히드록시프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성.

0℃에서 질소 하에 피리딘 (40 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (4.5 g, 11.44 mmol)의 교반 현탁액에 EDC (4.39 g, 22.88 mmol)에 이어서 2-아미노-3,3,3-트리플루오로프로판-1-올 (2.215 g, 17.16 mmol)을 첨가하고, 반응 혼합물을 실온에서 16시간 동안 교반하였다 (TLC 시스템 DCM 중 5% 메탄올. Rf 값 0.3). 반응 혼합물을 농축시키고, 잔류물을 EtOAc (200mL) 중에 용해시키고, 물 (2 x 100 mL)로 세척하였다. 합한 유기 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 갈색 고체로서 수득하였다. 고체를 디에틸에테르 (50mL)로 연화처리하고, 여과하고, 건조시켜 목적 화합물을 부분입체이성질체 혼합물로서 수득하였다. 부분입체이성질체를 정제용 키랄 SFC (칼럼/치수: 키랄팩 AD-H (250 x 30)mm,5μ; % CO2: 50.0; % 공용매: 50.0% (MeOH); 총 유량: 100.0 g/분,배압: 100.0 bar; UV: 213 nm, 스택 시간: 6.7분, 로드/주입: 95.0mg, 용해도: MeOH, 총 주입 횟수: 60, 기기 세부사항: 제조/모델: 타르 SFC-200 뉴-1)에 의해 분리하였다.
피크-1 SFC로부터 수집된 분획을 농축시키고, 디에틸에테르(20 mL)로 연화처리하고, 건조시키고, 모터에서 분쇄하여 연화처리하여 (9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-(1,1,1-트리플루오로-3-히드록시프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (920 mg, 1.821 mmol, 15.92% 수율). LCMS (m/z): 505.23 [M+H]+. Rt =1.85분.
실시예 46
(9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-(1,1,1-트리플루오로-3-히드록시프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성.
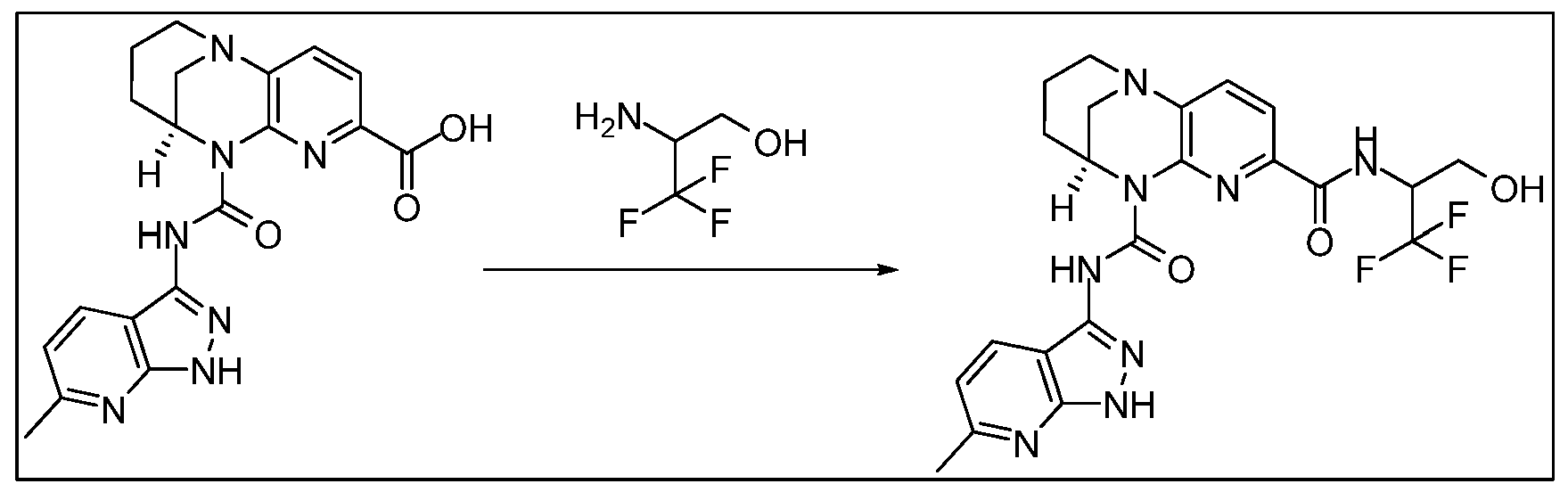
0℃에서 질소 하에 피리딘 (40 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (4.5 g, 11.44 mmol)의 교반 현탁액에 EDC (4.39 g, 22.88 mmol)에 이어서 2-아미노-3,3,3-트리플루오로프로판-1-올 (2.215 g, 17.16 mmol)을 첨가하고, 반응 혼합물을 실온에서 16시간 동안 교반하였다 (TLC 시스템 DCM 중 5% 메탄올. Rf 값 0.3). 반응 혼합물을 농축시키고, 잔류물을 EtOAc (200mL) 중에 용해시키고, 물 (2 x 100 mL)로 세척하였다. 합한 유기 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 갈색 고체로서 수득하였다. 고체를 디에틸에테르 (50mL)로 연화처리하고, 여과하고, 건조시켜 목적 화합물을 부분입체이성질체 혼합물로서 수득하였다. 부분입체이성질체를 정제용 키랄 SFC (칼럼/치수: 키랄팩 AD-H (250 x 30)mm,5μ; % CO2: 50.0; % 공용매: 50.0% (MeOH); 총 유량: 100.0 g/분,배압: 100.0 bar; UV: 213 nm, 스택 시간: 6.7분, 로드/주입: 95.0mg, 용해도: MeOH, 총 주입 횟수: 60, 기기 세부사항: 제조/모델: 타르 SFC-200 뉴-1)에 의해 분리하였다.
피크-2 SFC로부터 수집된 분획을 농축시키고, 디에틸에테르(20 mL)로 세척하고, 건조시키고, 모터에서 분쇄하여 (9S)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-(1,1,1-트리플루오로-3-히드록시프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (1 g, 1.966 mmol, 17.19% 수율). LCMS (m/z):505.2323 [M+H]+. Rt=1.85분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.15 (s, 1 H), 12.92 - 13.08 (m, 1 H), 8.56 (d, J=8.99 Hz, 1 H), 8.39 (d, J=8.33 Hz, 1 H), 7.60 - 7.85 (m, 2 H), 7.03 (d, J=8.55 Hz, 1 H), 5.24 (t, J=6.36 Hz, 1 H), 4.74 - 4.91 (m, 2 H), 3.85 (t, J=6.25 Hz, 2 H), 3.43 (dd, J=13.48, 1.64 Hz, 3 H), 2.81 - 3.04 (m, 1 H), 2.57 (s, 3 H), 2.05 (br d, J=18.42 Hz, 2 H), 1.33 (br d, J=8.33 Hz, 2 H).
실시예 47
(9S)-4-메틸-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성:

실온에서 THF (30 mL) 중 (9S)-4-메틸-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드 (300 mg, 0.914 mmol)의 교반 용액에 TEA (0.764 mL, 5.48 mmol), 트리포스겐 (271 mg, 0.914 mmol)를 첨가하고, 30분 동안 교반한 다음, 6-메틸-1H-피라졸로[3,4-b]피리딘-3-아민 (271 mg, 1.827 mmol)을 첨가하고, 반응 혼합물을 65℃에서 16시간 동안 가열하였다 (TLC 용리 시스템: 100% EtOAc: Rf-0.4; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 물 (20 mL)로 켄칭하고, EtOAc (2x35 mL)로 추출하였다. 유기 층을 분리하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 물질을 크로마토그래피 (그레이스, C-18 역 칼럼 사용, 이동상 A: 물 중 0.1% 포름산; B: ACN, 용리액 A 중 44% B)에 의해 정제하고, 합한 분획을 농축시키고, 포화 NaHCO3으로 염기성화시켰다. 수성 층을 DCM으로 추출하고, 합한 DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-4-메틸-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 황색 고체로서 수득하였다 (105 mg, 0.204 mmol, 22.34% 수율). LCMS (m/z): 503.25 [M+H]+, Rt = 2.38분.
1H NMR (400MHz, CDCl3): δ ppm 13.87 (s, 1 H), 9.81 (br s, 1 H), 8.70 (d, J=8.1 Hz, 1 H), 7.84 (s, 1 H), 7.68 (br d, J=8.8 Hz, 1 H), 7.02 (d, J=8.3 Hz, 1 H), 5.11 - 4.90 (m, 2 H), 3.39 (br d, J=13.4 Hz, 1 H), 3.21 (br d, J=6.8 Hz, 2 H), 2.97 (br d, J=14.0 Hz, 1 H), 2.66 (s, 3 H), 2.43 (s, 3 H), 2.38 - 2.13 (m, 1 H), 2.13 - 1.86 (m, 1 H), 1.63 - 1.49 (m, 3 H), 1.42 (br d, J=6.1 Hz, 2 H).
실시예 48
(9S)-N2-(2,2-디플루오로시클로프로필)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

실온에서 질소 하에 교반하는 DMF (10 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (500 mg, 2.54 mmol)의 교반 용액에 HATU (2.9 g, 7.63 mmol) 및 DMAP (0.621 g, 5.08 mmol)를 첨가하였다. 이에 2,2-디플루오로시클로프로판아민, 히드로클로라이드 (0.823 g, 6.35 mmol)를 첨가하고, 반응 혼합물을 50℃에서 16시간 동안 교반하였다 (TLC 용리액: DCM 중 10% MeOH: Rf-0.4; UV 활성). 반응 혼합물을 실온으로 냉각되도록 하고, 빙수로 희석하고, 에틸아세테이트 (2x50 mL)로 추출하였다. 합한 유기 층을 염수 용액으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 칼럼 크로마토그래피 (실리카겔:100-200 메쉬, DCM 중 2% 메탄올)에 의해 정제하여 부분입체이성질체 혼합물을 수득하고, SFC 분리 (조건:(칼럼/치수): 키랄팩 IC (250 X30)mm, 5μ,% CO2: 50.0% 공용매: 50.0% (용리액: 100% M 에탄올),총 유량: 100.0 g/분, 배압: 100.0 bar, UV: 217 nm, 스택 시간:16.5분, 로드/주입: 46.0 mg, 용해도: 메탄올, 총 주입 횟수: 7, 기기 세부사항: 제조/모델: 타르 SFC- 200 (OLD))에 의해 정제하여 2개의 피크를 피크-I 및 피크-II로서 수득하였다
피크-II: 연갈색 고체로서 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (73 mg, 0.155 mmol, 6.08% 수율). LCMS (m/z): 469.2 [M+H]+, Rt = 1.94분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.2 (s, 1 H), 12.8 (s, 1 H), 8.64 (br s, 1 H), 8.41 (d, J=8.55 Hz, 1 H), 7.74 - 7.6 (m, 1 H), 7.64 - 7.62 (m, 1 H), 7.05 (d, J=8.55 Hz, 1 H), 4.87 (s, 1 H), 3.56 - 3.39 (m, 2 H), 3.35 (br s, 2 H), 2.87 (d, J=13.37 Hz, 1 H), 2.55 (s, 3 H), 2.07 - 1.9 (m, 4 H), 1.35 - 1.22 (m, 2 H).
실시예 49
(9S)-N10-(5-((R)-2,3-디히드록시프로폭시)피라진-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

질소 분위기 하에 메탄올 (10 mL) 중 (9S)-N10-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (200 mg, 0.354 mmol)의 교반 용액에 0℃에서 수성 HCl (4 ml, 16.00 mmol)을 첨가하였다. 생성된 반응 혼합물을 28℃에서 1시간 동안 교반하였다 (TLC 시스템: DCM 중 5% 메탄올, Rf: 0.3 중, UV 활성). 용매를 감압 하에 증발시키고, 포화 NaHCO3 용액으로 염기성화시키고 (pH:8-9까지), 침전된 고체를 여과하여 목적 생성물 (9S)-N10-(5-((R)-2,3-디히드록시프로폭시)피라진-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (110 mg, 0.207 mmol, 58.4% 수율). LCMS (m/z): 526.26 [M+H]+, Rt = 1.87분.
1H NMR (400 MHz, CDCl3): δ 13.64 (s, 1 H), 8.99 (s, 1 H), 8.96-7.96 (m, 2 H), 7.71 (d, J=9.43 Hz, 1 H), 7.62 (d, J=7.89 Hz, 1 H), 5.15-4.93 (m, 2 H), 4.54-4.39 (m, 2 H), 4.16-4.03 (m, 1 H), 3.85-3.66 (m, 2 H), 3.46-3.27 (m, 3 H), 3.08 (d, J=3.95 Hz, 1 H), 2.96 (d, J=14.03 Hz, 1 H), 2.33-2.18 (m, 2 H), 2.02-1.87 (m, 1 H), 1.57 (d, J=7.02 Hz, 3 H), 1.42 (s, 2 H).
실시예 50
(9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

0℃에서 메탄올 (5 mL) 중 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (270 mg, 0.496 mmol)의 교반 용액에 염산 (0.030 mL, 0.992 mmol)을 5분의 기간에 걸쳐 적가하였다. 이어서, 반응 혼합물을 실온에서 2시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.1; UV 활성). 반응 혼합물을 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 디에틸 에테르 (2x 30 ml), n-펜탄 (2x 30 ml)으로 세척하여 목적 생성물 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (150 mg, 0.297 mmol, 59.8% 수율). LCMS (m/z): 505.23 [M+H]+, Rt = 1.50분.
1H NMR (400 MHz, TFA-d): δ ppm 8.58- 8.49 (m, 1 H), 8.28 (d, J=7.7 Hz, 1 H), 8.01- 7.98 (m, 1 H), 7.39 - 7.20 (m, 2 H), 5.40-5.36 (m, 1 H), 4.68- 4.39 (m, 3 H), 4.45 - 3.80 (m, 6 H), 3.62 - 3.49 (m, 1 H), 2.39 - 2.29 (m, 1 H), 2.30 - 1.80 (m, 5 H).
실시예 51
(9S)-3-클로로-N10-(4-(2-메틸옥사졸-5-일)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

실온에서 THF (20 mL) 중 (9S)-3-클로로-N-((R)-1,1,1-트리플루오로프로판-2-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복스아미드 (700mg, 2.007mmol)의 용액에 TEA (1.399 mL, 10.04 mmol)에 이어서 트리포스겐 (357 mg, 1.204 mmol)를 첨가하고, 30분 동안 교반한 다음, 4-(2-메틸옥사졸-5-일)피리딘-2-아민 (527 mg, 3.01 mmol)을 첨가하고, 반응 혼합물을 80℃로 16시간 동안 가열하였다 (TLC 시스템:100% 에틸아세테이트, Rf 값: 0.4). 반응 혼합물을 냉수 (50 mL)로 희석하고, EtOAc (2x50 mL)로 추출하였다. 합한 유기 층을 염수 용액 (30 mL)으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 정제용 HPLC (칼럼: 크로모실 페닐 C18 (150*25)mm 10u, 이동상-A: 10 mM 중탄산암모늄 (수성), 이동상-B: 아세토니트릴, 구배: 시간 T/%B = 0/45, 12/45,12.5/100, 15/100, 15.5/45, 칼럼 온도: 주위, 유량: 30 ml/분, 희석제: 아세토니트릴+THF+물)에 의해 정제하여 (9S)-3-클로로-N10-(4-(2-메틸옥사졸-5-일)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도-[2,3-b][1,4]-디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (95 mg, 0.171 mmol, 8.53% 수율). LCMS (m/z): 550.20 [M+H]+, Rt = 2.36분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.04 (s, 1 H), 9.22 (d, J=8.77 Hz, 1 H), 8.36 (s, 1 H), 8.30 (d, J=5.26 Hz, 1 H), 7.81 (d, J=12.50 Hz, 2 H), 7.39 (dd, J=5.26, 1.53 Hz, 1 H), 4.93 - 4.71 (m, 2 H), 3.48 - 3.39 (m, 1 H), 3.32 - 3.30 (m, 2 H), 2.88 (br d, J=13.81 Hz, 1 H), 2.53 (s, 3 H), 2.02 - 1.88 (m, 2 H), 1.46 (d, J=7.02 Hz, 3 H), 1.38 - 1.22 (m, 2 H).
실시예 52
(9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

0℃에서 메탄올 (5 mL) 중 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (230 mg, 0.422 mmol)의 교반 용액에 염산 (0.026 mL, 0.845 mmol)을 5분의 기간에 걸쳐 적가하였다. 이어서, 반응 혼합물을 실온에서 2시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf- 0.1; UV 활성). 이어서, 반응 혼합물을 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 에테르 (2x 30 ml), 펜탄 (2x 30 ml)으로 세척하여 목적 생성물 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (175 mg, 0.346 mmol, 82% 수율). LCMS (m/z): 505.23 [M+H]+, Rt = 1.50분.
1H NMR (400 MHz, TFA-d): δ ppm 8.56 - 8.50 (m, 1 H), 8.39 (d, J=7.7 Hz, 1 H), 8.02 (m, 1 H), 7.24 - 7.10 (m, 2 H), 5.41-5.39 (m, 1 H), 4.64- 4.41 (m, 3 H), 4.39 - 3.89 (m, 6 H), 3.68 - 3.51-3.39 (m, 1 H), 2.41 - 2.29 (m, 1 H), 2.22 - 1.76 (m, 5 H).
실시예 53
(9S)-N2-(2,2-디플루오로시클로프로필)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

실온에서 질소 하에 교반하는 DMF (10 mL) 중 (9S)-10-((6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (500 mg, 2.54 mmol)의 교반 용액에 HATU (2.9 g, 7.63 mmol) 및 DMAP (0.621 g, 5.08 mmol)를 첨가하였다. 이에 2,2-디플루오로시클로프로판아민, 히드로클로라이드 (0.823 g, 6.35 mmol)를 첨가하고, 반응 혼합물을 50℃에서 16시간 동안 교반하였다 (TLC 용리액: DCM 중 10% MeOH: Rf-0.4; UV 활성). 반응 혼합물을 실온으로 냉각되도록 하고, 빙수로 희석하고, 에틸아세테이트 (2x50 mL)로 추출하였다. 합한 유기 층을 염수 용액으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 칼럼 크로마토그래피 (실리카겔:100-200 메쉬, DCM 중 2% 메탄올)에 의해 정제하여 부분입체이성질체 혼합물을 수득하고, SFC 분리 (조건:(칼럼/치수): 키랄팩 IC (250 X30)mm, 5μ,% CO2: 50.0% 공용매: 50.0% (용리액: 100% M 에탄올),총 유량: 100.0 g/분, 배압: 100.0 bar, UV: 217 nm, 스택 시간:16.5분, 로드/주입: 46.0 mg, 용해도: 메탄올, 총 주입 횟수: 7, 기기 세부사항: 제조/모델: 타르 SFC- 200 (OLD))에 의해 정제하여 2개의 피크를 피크-I 및 피크-II로서 수득하였다.
피크-I: 회백색 고체로서 (9S)-N2-(2,2-디플루오로시클로프로필)-N10-(6-메틸-1H-피라졸로[3,4-b]피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (70 mg, 0.147 mmol, 5.80% 수율). LCMS (m/z): 469.2 [M+H]+, Rt =1.92분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.26 (s, 1 H), 12.99 (s, 1 H), 8.64 (br s, 1 H), 8.41 (d, J=8.55 Hz, 1 H), 7.74 - 7.69 (m, 1 H), 7.66 - 7.62 (m, 1 H), 7.03 (d, J=8.55 Hz, 1 H), 4.87 (br s, 1 H), 3.54 - 3.39 (m, 2 H), 3.32 (br s, 2 H), 2.88 (d, J=13.37 Hz, 1 H), 2.57 (s, 3 H), 2.07 - 1.88 (m, 4 H), 1.35 - 1.24 (m, 2 H).
실시예 54
(9S)-N10-(5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

N,N-디메틸포름아미드 (10 mL) 중 (9S)-10-((5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)카르바모일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2-카르복실산 (300 mg, 0.699 mmol) 및 HATU (398 mg, 1.048 mmol)의 교반 용액에 DIPEA (0.488 mL, 2.79 mmol)를 첨가하고, 이어서 (R)-1,1,1-트리플루오로프로판-2-아민 히드로클로라이드 (104 mg, 0.699 mmol)를 실온에서 반응 혼합물에 첨가하였다. 생성된 반응 혼합물을 28℃에서 16시간 동안 교반하였다 (TLC 용리액: DCM 중 10% MeOH: Rf-0.3; UV 활성). 반응 혼합물을 물 (30 mL)에 붓고, EtOAc (2X20 mL)로 추출하였다. 합한 유기 층을 염수 용액으로 세척하고, Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 화합물을 그레이스 (C-18 역 칼럼, 용리액: 물 중 60% MeOH 및 0.1% 포름산)에 의해 정제하여 목적 생성물 (9S)-N10-(5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 연황색 고체로서 수득하였다 (169 mg, 0.310 mmol, 44.4% 수율). LCMS (m/z): 525.25 [M+H]+, Rt = 1.86분.
1H NMR (400 MHz, CDCl3): δ ppm 13.62 (s, 1 H), 8.14 (d, J=8.99 Hz, 1 H), 8.06 - 7.90 (m, 3 H), 7.71 - 7.51 (m, 1 H), 7.41 7.24 (m, 1 H), 5.02 - 5.21 (m, 1 H), 4.98 (br s, 1 H), 4.24 - 4.03 (m, 3 H), 3.95 - 3.83 (m, 1 H), 3.76 - 3.82 (m, 1 H), 3.41 - 3.27 (m, 3 H), 2.95 (d, J=14.03 Hz, 1 H), 2.55 (br s, 1 H), 2.23 (d, J=15.13 Hz, 1 H), 1.87 - 2.01 (m, 2 H), 1.61 (d, J=7.02 Hz, 3 H), 1.48 - 1.34 (m, 2 H).
실시예 55
(9S)-N10-(6-((R)-2,3-디히드록시프로폭시)피리다진-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성

질소 하에 메탄올 (10 mL) 중 (9S)-N10-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리다진-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (220 mg, 0.389 mmol)의 교반 용액에 0℃에서 수성 HCl (0.8 ml, 3.20 mmol)을 첨가하였다. 반응 혼합물을 28℃에서 1시간 동안 교반하였다 (TLC 시스템: DCM 중 5% 메탄올, Rf: 0.3, UV 활성). 용매를 감압 하에 증발시키고, 포화 NaHCO3 용액으로 염기성화시키고 (pH: 8-9까지), DCM (3X30 mL)으로 추출하였다. 합한 유기 층을 물, 염수 용액으로 세척하고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 화합물을 정제용 HPLC (조건: MP-A: 10 Mm 아세트산암모늄(수성) MP-B: 아세토니트릴 칼럼: 크로마실 C18(250*21.2) mm,10 μ 방법 (T/%B): 65:35 유량: 20 ml/분 용해도: 아세토니트릴 + MeOH + THF)에 의해 정제하여 목적 생성물 (9S)-N10-(6-((R)-2,3-디히드록시프로폭시)피리다진-3-일)-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (130 mg, 0.234 mmol, 60.1% 수율). LCMS (m/z): 524.2 [M+H]+, Rt = 5.98분.
1H NMR (400 MHz, CDCl3): δ ppm 14.34 (s, 1 H), 8.42 (d, J=9.43 Hz, 1 H), 8.07 (d, J=9.21 Hz, 1 H), 8.01 (d, J=7.89 Hz, 1 H), 7.63 (d, J=7.89 Hz, 1 H), 7.10 (d, J=9.43 Hz, 1 H), 5.12 - 5.00 (m, 1 H), 4.96 (br s, 1 H), 4.65 - 4.54 (m, 2 H), 4.16 - 4.08 (m, 1 H), 3.82 - 3.66 (m, 2 H), 3.49 (br s, 1 H), 3.43 - 3.30 (m, 3 H), 2.97 (d, J=13.81 Hz, 1 H), 2.34 (br s, 1 H), 2.26 - 2.17 (m, 1 H), 2.01 - 1.89 (m, 1 H), 1.71 (d, J=7.02 Hz, 3 H), 1.47 - 1.36 (m, 2 H).
실시예 56
(9S)-N10-(5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성:

0℃에서 메탄올 (15 mL) 중 (9S)-N10-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (300 mg, 0.519 mmol)의 교반 용액에 수성 HCl (1.5 ml, 49.4 mmol)을 첨가하고, 질소 하에 실온에서 1시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.2; UV 활성). 반응 혼합물을 0℃에서 포화 중탄산나트륨 용액으로 염기성화시키고 (pH-8-9까지), 용매를 감압 하에 증발시켰다. 잔류물을 물로 희석하고, 생성된 고체를 부흐너 깔때기를 통해 여과하고, 감압 하에 건조시켜 조 화합물을 수득하였다. 조 물질을 크로마토그래피 (그레이스, C-18 역 칼럼 사용, 이동상 A: 물 중 0.1% 포름산; B: ACN, 용리액 A 중 45-50% B)에 의해 정제하고, 합한 분획을 농축시킨 다음, 포화 NaHCO3으로 염기성화시켰다. 침전된 고체를 여과하고, 건조시켜 (9S)-N10-(5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (214 mg, 0.395 mmol, 76% 수율). LCMS (m/z): 539.29 [M+H]+, Rt = 2.14분.
1H NMR (400 MHz, CDCl3): δ ppm 13.83 (s, 1 H), 8.14 (d, J=8.99 Hz, 1 H), 7.93 - 8.01 (m, 2 H), 7.87 (s, 1 H), 7.32 (dd, J=9.10, 2.96 Hz, 1 H), 5.04 - 5.17 (m, 1 H), 4.95 (br s, 1 H), 4.04 - 4.19 (m, 3 H), 3.72 - 3.92 (m, 2 H), 3.37 (dd, J=13.59, 1.75 Hz, 1 H), 3.14 - 3.25 (m, 2 H), 2.92 (br d, J=13.37 Hz, 1 H), 2.58 (d, J=4.60 Hz, 1 H), 2.41 (s, 3 H), 2.26 (br d, J=14.47 Hz, 1 H), 1.86 - 2.02 (m, 2 H), 1.53 - 1.64 (m, 3 H), 1.33 - 1.47 (m, 2 H).
실시예 57
(9S)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드의 합성:

0℃에서 메탄올 (15 mL) 중 (9S)-N10-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드 (300mg, 0.519 mmol)의 교반 용액에 수성 HCl (1.5 mL, 18.00 mmol)을 첨가하고, 질소 하에 실온에서 1시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH Rf-0.3; UV 활성). 반응 혼합물을 0℃에서 포화 중탄산나트륨 용액으로 염기성화시키고 (pH-8-9까지), 용매를 감압 하에 증발시켰다. 잔류물을 물로 희석하고, 침전된 고체를 여과하고, 감압 하에 건조시켜 조 물질을 수득하였다. 조 물질을 크로마토그래피 (그레이스, C-18 역 칼럼 사용, 이동상 A: 물 중 0.1% 포름산; B: ACN, 용리액 A 중 50-55% B)에 의해 정제하고, 합한 분획을 농축시킨 다음, 포화 NaHCO3 용액으로 염기성화시켰다. 침전된 고체를 여과하고, 건조시켜 (9S)-N10-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-4-메틸-N2-((R)-1,1,1-트리플루오로프로판-2-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-2,10(7H)-디카르복스아미드를 회백색 고체로서 수득하였다 (230 mg, 0.424 mmol, 82% 수율). LCMS (m/z): 539.32 [M+H]+, Rt = 1.78분.
1H NMR (400 MHz, CDCl3): δ ppm 14.06 (s, 1 H), 8.15 (br d, J=9.21 Hz, 1 H), 8.08 (d, J=5.92 Hz, 1 H), 7.89 (s, 1 H), 7.81 (d, J=2.41 Hz, 1 H), 6.59 (dd, J=5.70, 2.41 Hz, 1 H), 5.00 - 5.15 (m, 1 H), 4.93 (br s, 1 H), 4.10 - 4.22 (m, 3 H), 3.69 - 3.90 (m, 2 H), 3.37 (dd, J=13.48, 1.64 Hz, 1 H), 3.13 - 3.23 (m, 2 H), 2.93 (br d, J=13.37 Hz, 1 H), 2.57 (d, J=3.73 Hz, 1 H), 2.41 (s, 3 H), 2.26 (br d, J=14.25 Hz, 1 H), 1.86 - 2.07 (m, 2 H), 1.50 - 1.63 (m, 3 H), 1.32 - 1.45 (m, 2 H).
실시예 58
(9S)-N-(5-플루오로피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드의 합성
실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (40 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-II 0.700 g, 2.145 mmol)의 용액에 트리에틸아민 (1.794 mL, 12.87 mmol) 및 트리포스겐 (0.636 g, 2.145 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반하였다. 5-플루오로피리딘-2-아민 (0.721 g, 6.43 mmol)을 실온에서 첨가한 다음, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 농축 건조시켰다. 잔류물을 물 (10 mL)과 디클로로메탄 (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 35-40% EtOAc로 용리시켜 순수한 (9S)-N-(5-플루오로피리딘-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드를 회백색 고체로서 수득하였다 (0.220 g, 0.468 mmol, 21.82% 수율), LCMS (m/z): 4653 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 12.14 (s, 1H), 8.41 (dd, J = 9.2, 4.2 Hz, 1H), 8.15 (d, J = 3.0 Hz, 1H), 8.02 (s, 1H), 7.42 (ddd, J = 9.2, 7.7, 3.0 Hz, 1H), 5.51 (d, J = 4.6 Hz, 1H), 3.76 (s, 1H), 3.58 - 3.46 (m, 1H), 3.41 - 3.17 (m, 4H), 3.03 (s, 1H), 2.88 - 2.72 (m, 3H), 2.14 (d, J = 13.7 Hz, 1H), 2.08 (s, 1H), 2.00 - 1.77 (m, 3H), 1.53 - 1.42 (m, 2H), 1.37 - 1.21 (m, 2H).
실시예 59
(9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드의 합성

실온에서 10분 동안 교반하는 테트라히드로푸란 (50mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-I, 500 mg, 1.532 mmol)의 현탁액에 트리에틸아민 (0.641 mL, 4.60 mmol) 및 트리포스겐 (227 mg, 0.766 mmol)을 첨가하였다. 반응 혼합물을 30분 동안 교반하고, 피라진-2-아민 (437 mg, 4.60 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응물을 농축시키고, 잔류물을 물 (25 mL)로 희석하고, EtOAc (2x75 mL)로 추출하였다. 합한 유기 층을 물 및 포화 염수 용액으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 백색 고체로서 수득하였다 (TLC 용리액: 100%에틸 아세테이트: Rf-0.4; UV 활성). 조 생성물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하여 생성물을 헥산 중 60% 에틸 아세테이트로 용리시켜 (9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드를 회백색 고체로서 수득하였다 (150 mg, 0.319 mmol, 20.82% 수율), LCMS (m/z): 448.35 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 12.31 (s, 1H), 9.70 (d, J = 1.5 Hz, 1H), 8.30 (dd, J = 2.5, 0.4 Hz, 1H), 8.26 (dd, J = 2.6, 1.5 Hz, 1H), 8.03 (d, J = 0.7 Hz, 1H), 5.55 (d, J = 4.6 Hz, 1H), 3.76 (s, 1H), 3.56 - 3.39 (m, 1H), 3.29 - 3.20 (m, 4H), 3.02 (t, J = 11.7 Hz, 1H), 2.93 - 2.78 (m, 3H), 2.15 (d, J = 13.1 Hz, 1H), 2.06 - 1.82 (m, 4H), 1.54 - 1.42 (m, 2H), 1.36 (s, 1H).
실시예 60
(9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-II 0.550 g, 1.685 mmol)의 용액에 트리에틸아민 (1.409 mL, 10.11 mmol) 및 트리포스겐 (0.500 g, 1.685 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반한 다음, 피리딘-3-아민 (0.476 g, 5.06 mmol)을 실온에서 첨가하였다. 이어서, 반응 혼합물을 65℃에서 16시간 동안 교반한 다음, 28℃로 냉각시키고, 감압 하에 농축시켰다. 잔류물을 물 (10 mL)과 디클로로메탄 (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50-60% EtOAc로 용리시켜 순수한 (9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드를 회백색 고체로서 수득하였다 (0.264 g, 0.587 mmol, 34.8% 수율), LCMS (m/z): 447.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 11.74 (s, 1H), 8.59 (d, J = 2.6 Hz, 1H), 8.44 (ddd, J = 8.4, 2.7, 1.5 Hz, 1H), 8.32 (dd, J = 4.7, 1.6 Hz, 1H), 8.02 (d, J = 0.8 Hz, 1H), 7.30 (dd, J = 8.4, 4.7 Hz, 1H), 5.53 (d, J = 5.0 Hz, 1H), 3.76 (s, 1H), 3.58 - 3.41 (m, 1H), 3.24 (ddt, J = 10.1, 7.3, 2.2 Hz, 4H), 3.12 (t, J = 11.7 Hz, 1H), 2.90 - 2.70 (m, 2H), 2.55 (dtt, J = 15.5, 7.5, 3.9 Hz, 1H), 2.12 (d, J = 13.0 Hz, 1H), 2.01 - 1.84 (m, 3H), 1.81 - 1.66 (m, 1H), 1.53 (td, J = 12.7, 4.3 Hz, 2H), 1.35 (s, 1H), 1.26 (s, 1H).
실시예 61
(9S)-N-(피리딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드의 합성

실온에서 10분 동안 교반하는 테트라히드로푸란 (50mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리의 피크-I, 600 mg, 1.840 mmol)의 현탁액에 트리에틸아민 (0.796 mL, 5.521 mmol) 및 트리포스겐 (279 mg, 0.9202 mmol)을 첨가하였다. 반응 혼합물을 30분 동안 교반하고, 피리딘-2-아민 (519 mg, 5.521 mmol)을 첨가하였다. 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응물을 농축시키고, 잔류물을 물 (25 mL)로 희석하고, EtOAc (2x75 mL)로 추출하였다. 합한 유기 층을 물 및 포화 염수 용액으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 농축시켜 조 물질을 백색 고체로서 수득하였다 (TLC 용리액: 100%에틸 아세테이트: Rf-0.4; UV 활성). 조 생성물을 칼럼 크로마토그래피 (중성 알루미나)에 의해 정제하여 생성물을 헥산 중 60% 에틸 아세테이트로 용리시켜 (9S)-N-(피라딘-3-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드를 회백색 고체로서 수득하였다 (354 mg, 0.791mmol, 43% 수율), LCMS (m/z): 447.28 [M+H]+.
1H NMR (400 MHz, CDCl3): δ 11.66 (s, 1 H), 8.60 (s, 1 H), 8.47 - 8.40 (m, 1 H), 8.33 (s, 1 H), 8.01 (s, J=0.66 Hz, 1 H), 7.34 - 7.28 (m, 1 H), 5.53 (br d, J=4.17 Hz, 1 H), 3.76 (s, 1 H), 3.47 (dt, J=12.17, 1.70 Hz, 1 H), 3.30 - 3.19 (m, 4 H), 3.09 (t, J=11.62 Hz, 1 H), 2.95 - 2.76 (m, 2 H), 2.65 - 2.48 (m, 1 H), 2.18 - 2.07 (m, 1 H), 1.98 -1.85 (m, 3 H),1.78 - 1.68 (m, 2 H),1.62 - 1.44 (m, 2 H),1.39 - 1.30 (m, 1 H).
실시예 62
(9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드의 합성

실온에서 질소 하에 교반하는 테트라히드로푸란 (THF) (30 mL) 중 (9S)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (중간체 SFC 분리로부터의 피크-II 0.700 g, 2.145 mmol)의 용액에 트리에틸아민 (1.794 mL, 12.87 mmol) 및 트리포스겐 (0.636 g, 2.145 mmol)을 첨가하였다. 이어서, 반응 혼합물을 실온에서 30분 동안 교반한 후, 피라진-2-아민 (0.612 g, 6.43 mmol)을 첨가하였다. 이어서, 반응 혼합물을 65℃에서 16시간 동안 교반하였다. 반응 혼합물을 28℃로 냉각시키고, 감압 하에 농축시켰다. 생성된 잔류물을 물 (10 mL)과 디클로로메탄 (50 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 갈색 고체로서 수득하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 조 물질을 칼럼 크로마토그래피에 의해 중성 알루미나를 사용하여 정제하고, 헥산 중 50-60% EtOAc로 용리시켜 순수한 (9S)-N-(피라진-2-일)-2-(3-(트리플루오로메틸)피페리딘-1-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-3-카르복스아미드를 연황색 고체로서 수득하였다 (105 mg, 0.227 mmol, 10.58% 수율), LCMS (m/z): 448.3 [M+H]+.
1H NMR (400 MHz, CDCl3): δ ppm 12.36 (s, 1 H), 9.69 (d, J=1.53 Hz, 1 H), 8.28 (d, J=2.41 Hz, 1 H), 8.23 - 8.25 (m, 1 H), 8.03 (s, 1 H), 5.54 (br d, J=4.17 Hz, 1 H), 3.75 (br s, 1 H), 3.48 (br d, J=10.30 Hz, 1 H), 3.18 - 3.27 (m, 4 H), 3.05 (t, J=11.84 Hz, 1 H), 2.74 - 2.85 (m, 3 H), 2.14 (br d, J=12.93 Hz, 1 H), 2.02 (dt, J=13.21, 4.03 Hz, 1 H), 1.81 - 1.92 (m, 3 H), 1.39 - 1.52 (m, 2 H), 1.27 - 1.36 (m, 1 H).
실시예 63
(9S)-3-클로로-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

THF (10 mL) 중 (9S)-3-클로로-2-(3-(트리플루오로메틸)페닐)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.413 mmol)의 교반 용액에 NaH (33.9 mg, 1.413 mmol)를 0℃에서 첨가하고, 질소 분위기 하에 30분 동안 교반하였다. 이어서, 3-(피리딘-2-일)-2H-피리도[1,2-a][1,3,5]트리아진-2,4(3H)-디온 (340 mg, 1.413 mmol)을 이 반응 혼합물에 첨가하고, 80℃에서 16시간 동안 교반하였다 (TLC 용리액: 순수한 에틸 아세테이트; Rf: 0.25). 반응 혼합물을 빙수로 켄칭하고, 에틸 아세테이트 (2x15 mL)로 추출하였다. 합한 유기 층을 염수 용액 (20 mL)으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (실리카-겔: 100-200 메쉬, 용리액: 석유 에테르 중 70% 에틸 아세테이트)에 의해 정제하여 목적 생성물 (9S)-3-클로로-N-(피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (260 mg, 0.548 mmol, 38.7% 수율). LCMS (m/z): 474.05 [M+H]+, Rt = 3.0분.
1H NMR (400 MHz, CDCl3): δ ppm 13.40 (s, 1 H), 8.30 - 8.23 (m, 2 H), 8.20 - 8.26 (m, 1 H), 8.20 - 8.13 (m, 1 H), 7.78 - 7.69 (m, 1 H), 7.59 - 7.57 (m, 2 H), 7.52 - 7.48 (m, 1 H), 6.97 - 6.91 (m, 1 H), 5.02 - 4.97 (m, 1 H), 3.41 - 3.37 (m, 3 H), 2.96 - 2.95 (m, 1 H), 2.92 (d, J=14.03 Hz, 1 H), 2.41 - 2.06 (m, 1 H), 1.63 - 1.21 (m, 2 H).
실시예 64
(9S)-3-클로로-N-(6-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성:
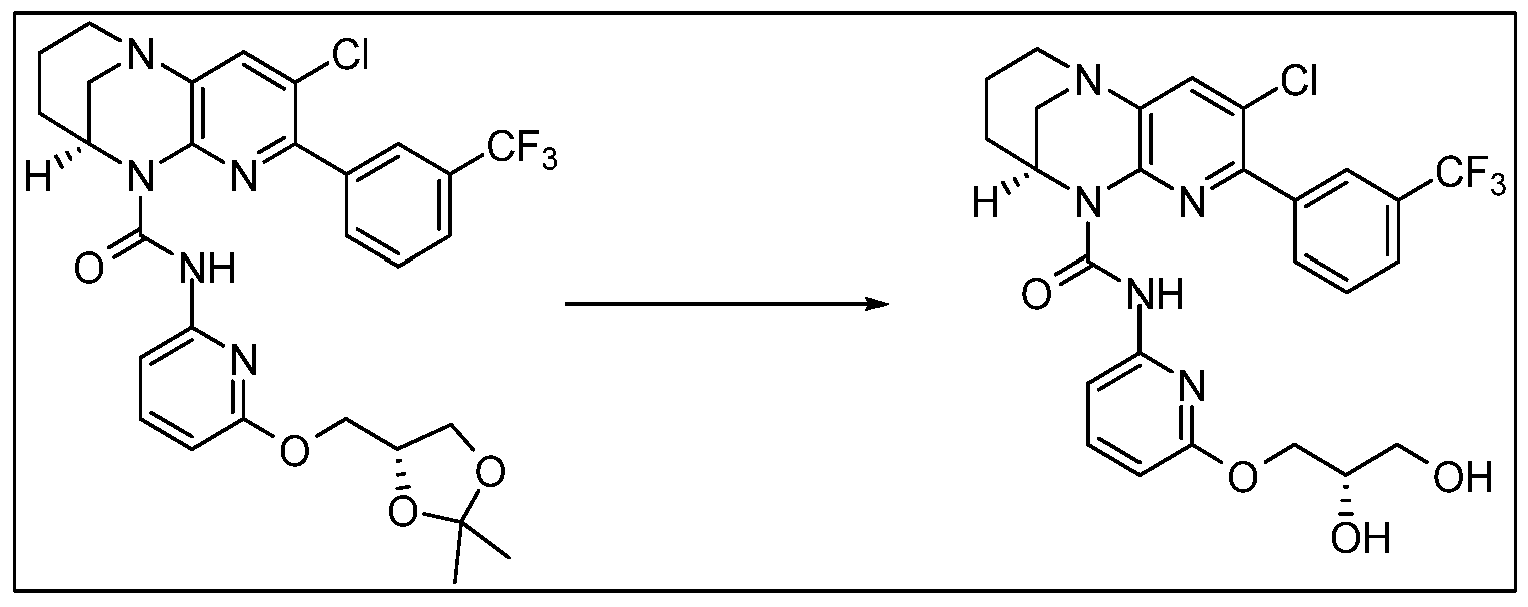
메탄올 (10 mL) 중 (9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (250 mg, 0.414 mmol)의 교반 용액에 수성 HCl (0.345 mL, 4.14 mmol, 36%)을 0℃에서 첨가하고, 실온에서 4시간 동안 교반하였다. TLC에 의해 반응이 완결된 후, 휘발성 물질을 감압 하에 증발시켜 조 물질을 수득하였다 (TLC 용리액 시스템: 100% EtOAc,Rf-0.5, UV 활성). 조 물질을 물 (5 ml)로 희석하고, 10% 중탄산나트륨 용액으로 염기성화시키고 (pH 8까지), 침전된 고체를 여과하고, 물로 세척하고, 진공 하에 건조시켜 (9S)-3-클로로-N-(6-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (162 mg, 0.285 mmol, 68.8% 수율). LCMS (m/z): 564.13 [M+H]+, Rt =2.50분.
1H NMR (400 MHz, CDCl3): δ ppm 13.10 (br s, 1 H), 7.89 - 8.16 (m, 2 H), 7.72 - 7.80 (m, 1 H), 7.63 - 7.70 (m, 1 H), 7.47 - 7.61 (m, 2 H), 6.42 (d, J=7.89 Hz, 1 H), 5.00 (br s, 1 H), 3.92 (qd, J=11.91, 4.60 Hz, 2 H), 3.67 - 3.81 (m, 1 H), 3.48 (br s, 2 H), 3.27 - 3.42 (m, 2 H), 2.98 (br d, J=12.93 Hz, 2 H), 2.80 (br d, J=5.48 Hz, 1 H), 2.25 (br d, J=15.13 Hz, 2 H), 1.94 (br d, J=12.50 Hz, 2 H), 1.22 - 1.49 (m, 2 H).
실시예 65
(9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성:

메탄올 (10 mL) 중 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (270 mg, 0.447 mmol)의 교반 용액에 수성 HCl (1 mL, 32.9 mmol, 36%)을 0℃에서 첨가하고, 실온에서 4시간 동안 교반하였다. 반응이 완결된 후, 휘발성 물질을 감압 하에 증발시켜 조 물질을 수득하였다 (TLC 용리액 시스템: 100% EtOAc,Rf-0.5, UV 활성). 조 물질을 물 (5 ml)로 희석하고, 10% 중탄산나트륨 용액으로 염기성화시키고, 침전된 고체를 여과하고, 물로 세척하고, 진공 하에 건조시켜 (9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (142 mg, 0.248 mmol, 55.5% 수율). LCMS (m/z): 564.13 [M+H]+, Rt =2.51분.
1H NMR (400 MHz, CDCl3): δ ppm 13.11 (s, 1 H), 7.89 - 8.14 (m, 2 H), 7.61 - 7.82 (m, 2 H), 7.44 - 7.61 (m, 3 H), 6.42 (d, J=7.89 Hz, 1 H), 5.00 (br s, 1 H), 3.83 - 4.04 (m, 2 H), 3.63 - 3.83 (m, 1 H), 3.42 - 3.57 (m, 2 H), 3.28 - 3.41 (m, 3 H), 2.98 (br d, J=14.03 Hz, 1 H), 2.60 - 2.87 (m, 1 H), 2.17 - 2.39 (m, 2 H), 1.86 - 1.98 (m, 1 H), 1.24 - 1.52 (m, 2 H).
실시예 66
(9S)-3-클로로-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 하에 THF (30 mL) 중 (9S)-3-클로로-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (300 mg, 0.997 mmol)의 교반 용액에 트리에틸아민 (0.834 mL, 5.98 mmol), 트리포스겐 (296 mg, 0.997 mmol)을 첨가하고, 4시간 동안 교반한 다음, 피리딘-3-아민 (282 mg, 2.99 mmol)을 첨가하고, 반응물을 65℃에서 16시간 동안 가열하였다 (TLC 용리액: 100% EtOAc: Rf-0.3; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 농축시키고, 잔류물을 물 (5 mL)과 EtOAc (20 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 물질을 칼럼 크로마토그래피 (중성 알루미나, 용리액: 헥산 중 60% 에틸 아세테이트)에 의해 정제하여 목적 생성물 (9S)-3-클로로-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (135 mg, 0.319 mmol, 31.9% 수율). LCMS (m/z): 550.13 [M+H]+, Rt = 1.95분.
1H NMR (400 MHz, CDCl3): δ ppm 13.11 (s, 1 H), 8.71 (d, J=5.04 Hz, 1 H), 8.49 (d, J=2.19 Hz, 1 H), 8.29 (dd, J=4.60, 1.32 Hz, 1 H), 8.05 - 7.97 (m, 1 H), 7.59 (s, 1 H), 7.54 (s, 1 H), 7.51 (d, J=5.04 Hz, 1 H), 7.22 (dd, J=8.33, 4.82 Hz, 1 H), 5.00 (br s, 1 H), 3.42 - 3.32 (m, 3 H), 2.98 (br d, J=14.03 Hz, 1 H), 2.70 (s, 3 H), 2.25 (br d, J=14.25 Hz, 1 H), 2.00 - 1.87 (m, 1 H), 1.51 - 1.40 (m, 2 H)
실시예 67
(9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

0℃에서 메탄올 (10 mL) 중 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (200 mg, 0.331 mmol)의 교반 용액에 수성 HCl (0.201 mL, 6.61 mmol, 36%)을 첨가하고, 2시간 동안 교반하였다 (TLC 용리액: 100% EtOAc: Rf-0.2; UV 활성). 반응 혼합물을 포화 중탄산나트륨 용액으로 염기성화시키고 (pH-8-9까지), 용매를 감압 하에 증발시켰다. 잔류물을 물 (10 mL)로 희석하고, 디클로로메탄 (2x15 mL)으로 추출하였다. 합한 유기 추출물을 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 진공 하에 증발시키고, 조 물질을 펜탄 (10 mL)으로 연화처리하여 목적 생성물 (9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (123 mg, 0.214 mmol, 64.6% 수율). LCMS (m/z): 565.14 [M+H]+, Rt = 2.33분.
1H NMR (400 MHz, CDCl3): δ ppm 13.27 (s, 1 H), 8.88 (s, 1 H), 8.03 (s, 1 H), 7.96 (br d, J=7.67 Hz, 1 H), 7.92 (s, 1 H), 7.78 - 7.74 (m, 1 H), 7.72 - 7.66 (m, 1 H), 7.61 (s, 1 H), 5.01 (br s, 1 H), 3.99 - 3.92 (m, 1 H), 3.91 - 3.83 (m, 2 H), 3.66 - 3.59 (m, 2 H), 3.42 - 3.31 (m, 3 H), 2.99 (br d, J=13.59 Hz, 1 H), 2.56 (d, J=4.82 Hz, 1 H), 2.26 (br d, J=15.35 Hz, 1 H), 2.07 (br t, J=6.03 Hz, 1 H), 2.00 - 1.86 (m, 1 H), 1.52 - 1.39 (m, 2 H)
실시예 68
(9S)-3-메틸-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 질소 분위기 하에 THF (25 mL) 중 (9S)-3-메틸-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (250 mg, 0.892 mmol)의 교반 용액에 페닐 피리딘-3-일카르바메이트 (573 mg, 2.68 mmol), DMAP (327 mg, 2.68 mmol)를 첨가하고, 65℃에서 48시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.4; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 농축시키고, 잔류물을 물 (10 mL)과 EtOAc (30 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 물질을 칼럼 크로마토그래피 (중성 알루미나, 용리액: 헥산 중 70% EtOAc)에 의해 정제하여 (9S)-3-메틸-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 연황색 고체로서 수득하였다 (178 mg, 0.437 mmol, 49.0% 수율). LCMS (m/z): 401.1 [M+H]+, Rt = 3.342분.
1H NMR (400 MHz, CDCl3): δ ppm 13.57 (s, 1 H), 8.68 (d, J=5.26 Hz, 1 H), 8.48 (d, J=2.41 Hz, 1 H), 8.26 (dd, J=4.71, 1.43 Hz, 1 H), 8.08 - 7.99 (m, 1 H), 7.42 - 7.36 (m, 2 H), 7.30 (dd, J=5.15, 1.21 Hz, 1 H), 7.20 (dd, J=8.33, 4.60 Hz, 1 H), 4.97 (br s, 1 H), 3.42 - 3.28 (m, 3 H), 3.00 (br d, J=13.59 Hz, 1 H), 2.69 (s, 3 H), 2.36 (s, 3 H), 2.30 - 2.20 (m, 1 H), 2.01 - 1.87 (m, 1 H), 1.54 - 1.35 (m, 2 H)
실시예 69
(4S)-8-클로로-N-(2-((R)-2,3-디히드록시프로폭시)피리미딘-5-일)-7-(3-(트리플루오로메틸)페닐)-3,4-디히드로-1,4-메타노피리도[2,3-b][1,4]디아제핀-5(2H)-카르복스아미드의 합성

실온에서 메탄올 (10 mL) 중 (9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (150 mg, 0.248 mmol)의 교반 용액에 수성 HCl (1.240 mL, 2.479 mmol, 36%)을 첨가하고, 2시간 동안 교반하였다 (TLC 용리액: 100% EtOAc: Rf-0.2; UV 활성). 반응 혼합물을 포화 중탄산나트륨 용액으로 염기성화시키고 (pH 8-9까지), 용매를 감압 하에 증발시켰다. 잔류물을 물 (5 mL)로 희석하고, 디클로로메탄 (2x10 mL)으로 추출하였다. 합한 유기 추출물을 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 진공 하에 증발시키고, 조 물질을 디에틸에테르 (10 mL) 및 펜탄 (10 mL)으로 연화처리하여 목적 생성물 (9S)-3-클로로-N-(2-((R)-2,3-디히드록시프로폭시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (89 mg, 0.153 mmol, 61.6% 수율). LCMS (m/z): 565.17 [M+H]+, Rt = 2.30분.
1H NMR (400 MHz, CDCl3): δ ppm 13.10 (s, 1 H), 8.58 (s, 2 H), 8.02 (s, 1 H), 7.94 (d, J=7.89 Hz, 1 H), 7.77 (d, J=7.89 Hz, 1 H), 7.71 - 7.65 (m, 1 H), 7.61 (s, 1 H), 4.98 (br s, 1 H), 4.51 - 4.41 (m, 2 H), 4.15 - 4.06 (m, 1 H), 3.82 - 3.67 (m, 2 H), 3.44 - 3.31 (m, 3 H), 3.14 (d, J=5.26 Hz, 1 H), 2.98 (br d, J=13.59 Hz, 1 H), 2.34 - 2.18 (m, 2 H), 2.00 - 1.88 (m, 1 H), 1.54 - 1.42 (m, 2 H)
실시예 70
(9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
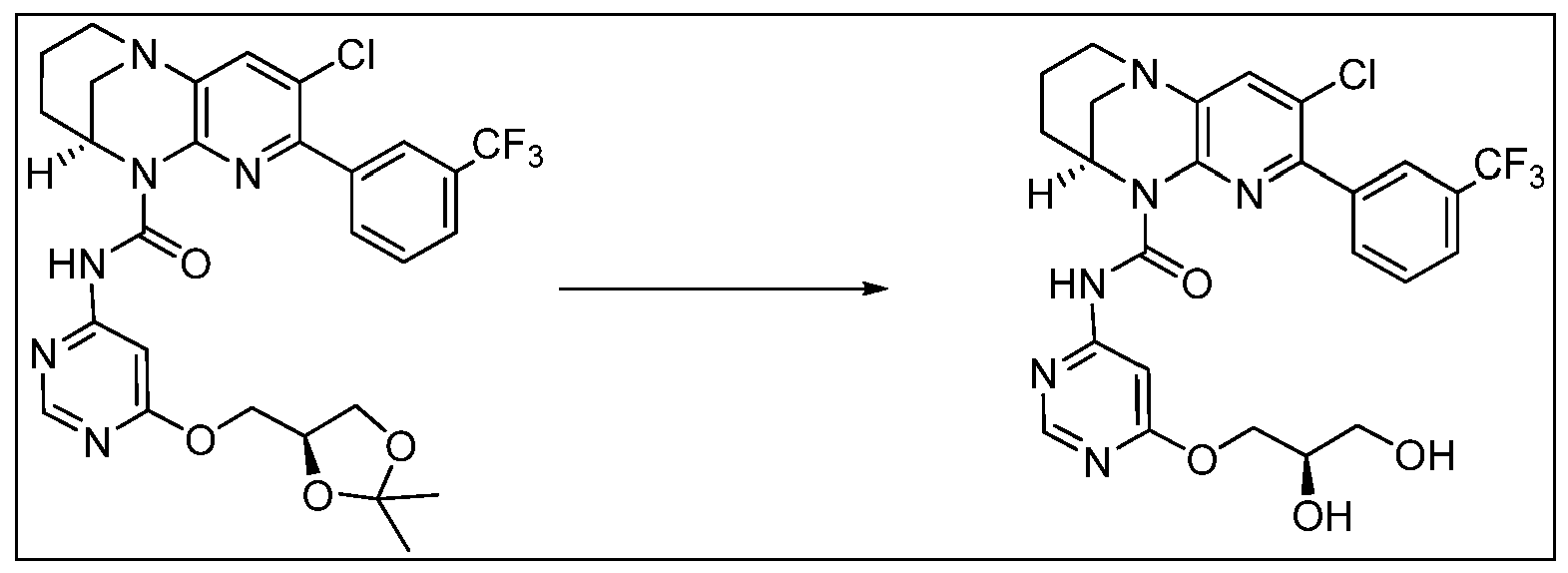
0℃에서 메탄올 (10 mL) 중 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일) 메톡시) 피리미딘-4-일)-2-(3-(트리플루오로메틸) 페닐)-8,9-디히드로-6H-5,9 메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (0.25 g, 0.413 mmol)의 교반 용액에 수성 HCl (0.517 mL, 6.20 mmol, 36%)을 첨가하고, 실온에서 1시간 동안 교반하였다 (TLC 용리액: 100% EtOAc: Rf-0.2; UV 활성). 반응 혼합물을 포화 중탄산나트륨 용액을 첨가하여 염기성화시킨 다음 (pH-8-9까지), 농축시켰다. 잔류물을 물 (10 mL)로 희석하고, EtOAc (2x25 mL)로 추출하였다. 합한 유기 추출물을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 물질을 디에틸 에테르 (10 mL)로 연화처리하여 목적 생성물 (9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.18g, 0.311 mmol, 75% 수율). LCMS (m/z): 565.14 [M+H]+, Rt = 2.51분.
1H NMR (400 MHz, CDCl3): δ ppm 13.64 (s, 1 H), 8.35 (s, 1 H), 8.19 (s, 1 H), 8.11 (br d, J = 7.89 Hz, 1 H), 7.75 (br d, J = 8.11 Hz, 1 H), 7.69 - 7.60 (m, 2 H), 7.55 (s, 1 H), 4.95 (br s, 1 H), 4.51 - 4.46 (m, 2 H), 4.06 - 3.98 (m, 1 H), 3.75 - 3.63 (m, 2 H), 3.48 - 3.31 (m, 4 H), 2.99 (br d, J = 13.59 Hz, 1 H), 2.51 (br t, J = 6.14 Hz, 1 H), 2.23 (br d, J = 12.28 Hz, 1 H), 1.98 - 1.88 (m, 1 H), 1.49 - 1.40 (m, 2 H).
실시예 71
(9S)-3-클로로-N-시클로프로필-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

실온에서 THF (30 mL) 중 (9S)-페닐 3-클로로-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복실레이트 (400 mg, 0.844 mmol)의 교반 용액에 DMAP (516 mg, 4.22 mmol), 시클로프로판아민 (0.301 mL, 4.22 mmol)을 첨가하고, 65℃에서 16시간 동안 교반하였다 (TLC 용리액: 석유 에테르 중 70% 에틸 아세테이트, Rf : 0.4). 반응 혼합물을 실온으로 냉각시키고, 진공 하에 농축시키고, 잔류물을 물 (40 mL)과 EtOAc (80 mL) 사이에 분배하였다. 유기 층을 분리하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 화합물을 칼럼 크로마토그래피 (실리카 겔 사용, 용리액 헥산 중 50% 에틸아세테이트)에 이어서 정제용 HPLC (칼럼: 엑스브리지 C 18(150x4.6mm, 3.5μ), 이동상: A: 0.01 M 중탄산암모늄 B: ACN,구배: 시간/%B: 0/5,0.8/5,5/50,8/95,12/95,12.5/5,15/5, 온도: 주위, 유량: 0.8ml/분, 희석제: ACN)에 의해 정제하여 (9S)-3-클로로-N-시클로프로필-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (118 mg, 0.269 mmol, 31.9% 수율). LCMS (m/z): 437.05 [M+H]+; Rt = 2.03분.
1H NMR (400 MHz, CDCl3): δ ppm 10.51 (br s, 1 H), 7.97 (s, 1 H), 7.92 (d, J=7.67 Hz, 1 H), 7.71 (d, J=7.67 Hz, 1 H), 7.64-7.59 (m, 1 H), 7.50 (s, 1 H), 4.95 (t, J=2.19 Hz, 1 H), 3.35 - 3.26 (m, 3 H), 2.89 (br d, J=13.59 Hz, 1 H), 2.79 (tq, J=7.19, 3.68 Hz, 1 H), 2.24 - 2.16 (m, 1 H), 1.92 - 1.81 (m, 1 H), 1.52 - 1.33 (m, 2 H), 0.75 - 0.67 (m, 2 H), 0.46 - 0.40 (m, 2 H).
실시예 72
(9S)-3-클로로-N-(2-((S)-2,3-디히드록시프로폭시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

0℃에서 메탄올 (20 mL) 중 (9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9 메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (300 mg, 0.496 mmol)의 교반 용액에 수성 HCl (1.2 mL, 14.40 mmol)을 첨가하고, 4시간 동안 교반하였다 (TLC 용리액: 100% EtOAc, Rf: 0.2). 반응 혼합물을 진공 하에 농축시키고, 잔류물을 포화 NaHCO3 용액 (20 mL)으로 염기성화시켰다. 생성된 고체를 여과하고, 키랄 (칼럼/치수: 키랄팩 AD-H (250 X30)mm,5μ % CO2: 50.0% % 공용매: 50.0% (MeOH 중 0.5% DEA), 총 유량: 70.0 g/분, 배압: 100.0 bar, UV: 260 nm, 스택 시간: 14분, 로드/주입: 38.0 mg, 용해도: 메탄올, 총 주입 횟수: 8 기기 세부사항: 제조/모델: 타르 SFC-80)에 의해 정제하여 (9S)-3-클로로-N-(2-((S)-2,3-디히드록시프로폭시)피리미딘-5-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (139 mg, 0.239 mmol, 48.1% 수율). LCMS (m/z): 565.17 [M+H]+, Rt = 2.30분.
1H NMR (400 MHz, CDCl3): δ ppm 13.10 (s, 1 H), 8.58 (s, 2 H), 8.08 - 7.91 (m, 2 H), 7.78 (s, 1 H), 7.71 - 7.57 (m, 2 H), 4.98 (br s, 1 H),4.59 - 4.38 (m, 2 H), 4.17 - 4.05 (m, 1 H), 3.87 - 3.63 (m, 2 H), 3.47 - 3.26 (m, 3 H), 2.98 (d, J=13.81 Hz, 1 H), 2.23 (d, J=14.25 Hz, 2 H), 2.04 - 1.83 (m, 2 H),1.69 - 1.38 (m, 2 H)
실시예 73
(9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피라진-2-일)-2-(3-(트리플루오로 메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (2 mL) 중 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (200 mg, 0.331 mmol)의 교반 용액에 염산 (0.5 mL, 16.46 mmol)을 0℃에서 5분의 기간에 걸쳐 적가하였다. 이어서, 반응 혼합물을 실온에서 2시간 동안 교반하였다 (TLC 용리액: DCM 중 10% MeOH: Rf-0.3). 용매를 증발시키고, 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 물로 세척하여 목적 생성물 (9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (130 mg, 0.229 mmol, 69.2% 수율). LCMS (m/z): 565.17 [M+H]+, Rt =2.51분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.35 (s, 1 H), 8.83 (d, J=1.32 Hz, 1 H), 8.18 - 8.12 (m, 2 H), 7.91 - 7.85 (m, 3 H), 7.83 - 7.76 (m, 1 H), 4.92 (d, J=5.26 Hz, 1 H), 4.94 - 4.90 (m, 1 H), 4.83 (brs, 1 H), 4.63 (t, J=5.70 Hz, 1 H), 4.30 (dd, J=10.85, 4.06 Hz, 1 H), 4.15 (dd, J=10.74, 6.58 Hz, 1 H), 3.85 - 3.76 (m, 1 H), 3.48 - 3.37 (m, 3 H), 3.32 (d, J=3.07 Hz, 1 H), 2.91 (d, J=14.03 Hz, 1 H), 2.11 - 1.76 (m, 2 H), 1.50 - 1.27 (m, 2 H).
실시예 74
(9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (8 mL) 및 테트라히드로푸란 (5 mL) 중 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도 [2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (250 mg, 0.414 mmol)의 교반 용액에 HCl (0.5 mL, 16.46 mmol)을 0℃에서 첨가한 다음, 실온에서 2시간 동안 교반하였다 (TLC 용리액: 순수한 에틸 아세테이트, Rf: 0.2). 반응 혼합물을 진공 하에 농축시키고, 잔류물을 포화 NaHCO3 용액으로 중화시키고, 수득된 고체를 여과하고, n-펜탄 (10 mLx3)으로 세척하여 목적 생성물 (9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (201 mg, 0.354 mmol, 86% 수율). LCMS (m/z): 564.17 [M+H]+, Rt = 2.44분.
1H NMR (400 MHz, CDCl3): δ ppm 13.34 (s, 1 H), 8.22 (s, 1 H), 8.14 (d, J=7.89 Hz, 1 H), 8.04 (d, J=8.99 Hz, 1 H), 7.95 (d, J=3.07 Hz, 1 H), 7.73 (d, J=7.89 Hz, 1 H), 7.67 - 7.61(m, 1 H), 7.58 (s, 1 H), 7.25 - 7.22 (m, 1 H), 4.98 (br s, 1 H), 4.14 - 4.03 (m, 3 H), 3.89 - 3.82 (m, 1 H), 3.79 - 3.72 (m, 1 H), 3.40 - 3.31 (m, 3 H), 2.98 (d, J=13.81 Hz, 1 H), 2.54 (d, J=4.38 Hz, 1 H), 2.25 (d, J=12.06 Hz, 1 H), 1.97 - 1.87 (m, 2 H), 1.52 - 1.35 (m, 2 H).
실시예 75
(9S)-3-클로로-N-(4-((S)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (5 mL) 및 테트라히드로푸란 (5 mL) 중 (9S)-3-클로로-N-(4-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (200 mg, 0.331 mmol)의 교반 용액에 HCl (0.3 mL, 9.87 mmol)을 0℃에서 첨가한 다음, 실온에서 1시간 동안 교반하였다 (TLC 시스템: 순수한 에틸 아세테이트, Rf: 0.1). 반응 혼합물을 진공 하에 농축시키고, 잔류물을 포화 NaHCO3 용액으로 중화시키고, 수득된 고체를 여과하고, n-펜탄 (10 mLx2)으로 세척하여 목적 생성물 (9S)-3-클로로-N-(4-((S)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (114 mg, 0.200 mmol, 60.4% 수율). LCMS (m/z): 565.17 [M+H]+, Rt = 2.05분.
1H NMR (400 MHz, CDCl3): δ ppm 13.90 (s, 1 H), 8.21 - 8.16 (m, 2 H), 8.10 (d, J=7.67 Hz, 1 H), 7.73 (d, J=7.89 Hz, 1 H), 7.66 - 7.59 (m, 2 H), 6.42 (d, J=5.70 Hz, 1 H), 5.03 (br s, 1 H), 4.64 (dd, J=11.95, 5.37 Hz, 1 H), 4.46 (dd, J=11.95, 4.49 Hz, 1 H), 4.11 (d, J=6.36 Hz, 1 H), 3.94 - 3.88 (m, 1 H), 3.64 - 3.57 (m, 2 H), 3.46 - 3.30 (m, 4 H), 2.97 (d, J=14.03 Hz, 1 H), 2.27 (d, J=12.50 Hz, 1 H), 1.97 -1.86 (m, 1 H), 1.52 - 1.39 (m, 2 H).
실시예 76
(9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
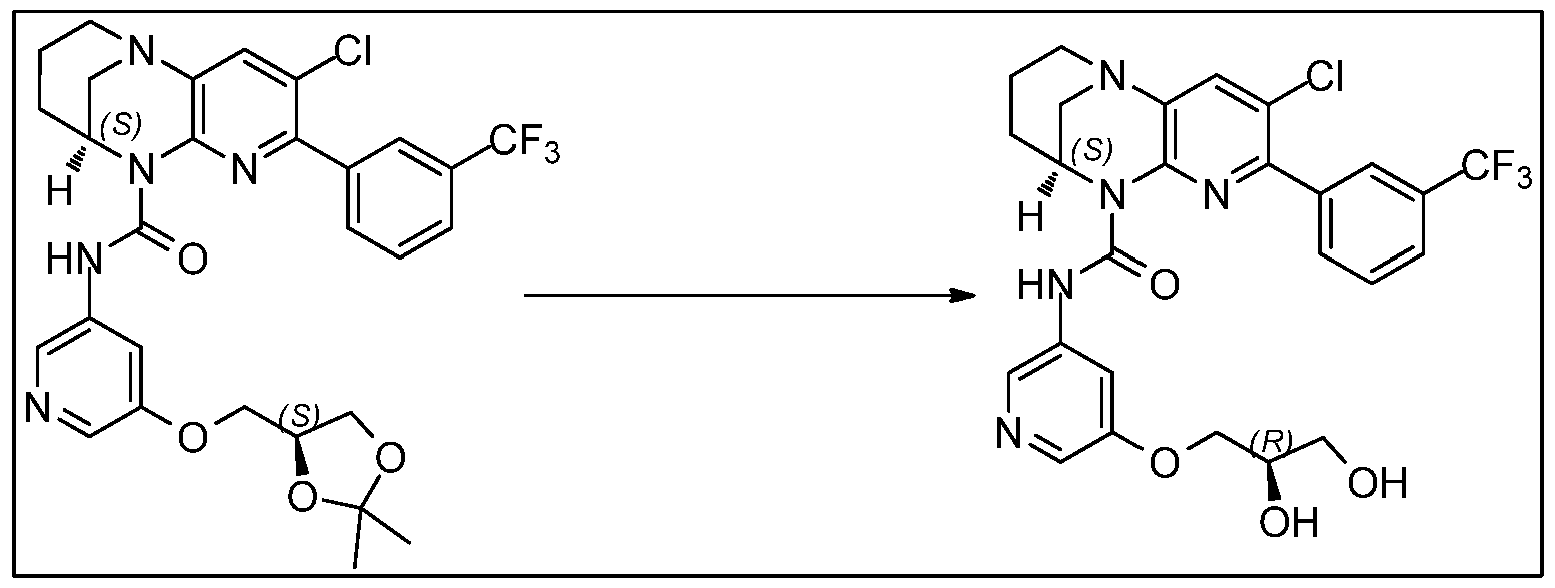
메탄올 (10 mL) 중 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (240 mg, 0.397 mmol)의 교반 용액에 수성 HCl (0.345 mL, 3.97 mmol)을 2분의 기간에 걸쳐 0℃에서 적가하고, 실온에서 1시간 동안 교반하였다 (TLC 시스템: 100% 에틸 아세테이트. Rf 값: 0.3). 이어서, 반응 혼합물을 포화 NaHCO3 용액 (20 mL)과 DCM (30 mL) 사이에 분배하고, 분리된 유기 층을 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 생성물을 플래쉬 칼럼 크로마토그래피 (100-200 실리카겔, 에틸 아세테이트 중 0-2% MeOH로 용리시킴)에 의해 정제하여 목적 생성물 (9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (170 mg, 0.299 mmol, 75% 수율). LCMS (m/z): 564.20 [M+H]+, Rt =2.17분.
1H NMR (400 MHz, CDCl3): δ ppm 13.21 (s, 1 H), 8.04 (s, 1 H), 7.99 (d, J=2.41 Hz, 1 H), 7.94 (d, J=7.45 Hz, 1 H), 7.88 (t, J=2.30 Hz, 1 H), 7.83 (d, J=1.97 Hz, 1 H), 7.76 - 7.80 (m, 1 H), 7.67 - 7.74 (m, 1 H), 7.61 (s, 1 H), 4.97 (brs, 1 H), 4.01 - 4.13 (m, 3 H), 3.79 - 3.86 (m, 2 H), 3.71 - 3.77 (m, 1 H), 3.30 - 3.42 (m, 3 H), 2.99 (br d, J=13.81 Hz, 1 H), 2.63 (brs, 1 H), 2.24 (brd, J=14.69 Hz, 1 H), 1.88 - 1.99 (m, 1 H), 1.39 - 1.48 (m, 1 H), 1.26 (s, 1 H).
실시예 77
(9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (200 mg, 0.331 mmol)의 교반 용액에 수성 HCl (0.287 mL, 3.31 mmol)을 2분의 기간에 걸쳐 0℃에서 적가하고, 실온에서 1시간 동안 교반하였다 (TLC 시스템: 100% 에틸 아세테이트. Rf 값: 0.4). 이어서, 반응 혼합물을 포화 NaHCO3 용액 (20 mL)과 DCM (30 mL) 사이에 분배하고, 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 목적 생성물 (9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피라진-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (171 mg, 0.299 mmol, 91% 수율). LCMS (m/z): 565.17 [M+H]+, Rt =2.51분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.35 (s, 1 H), 8.83 (s, 1 H), 8.15 (s, 2 H), 7.26 - 7.96 (m, 4 H), 4.92 (d, J=5.26 Hz, 1 H), 4.83 (br s, 1 H), 4.63 (br t, J=5.81 Hz, 1 H), 4.30 (br dd, J=10.85, 3.84 Hz, 1 H), 4.15 (br dd, J=10.63, 6.69 Hz, 1 H), 3.81 (br dd, J=10.19, 5.15 Hz, 1 H), 3.30 - 3.63 (m, 5 H), 2.91 (br d, J=13.37 Hz, 1 H), 1.88 - 2.11 (m, 2 H), 1.34 (br d, J=4.38 Hz, 2 H).
실시예 78
(9S)-3-클로로-N-(4-((R)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-3-클로로-N-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (290 mg, 0.479 mmol)의 용액에 수성 HCl (0.416 mL, 4.79 mmol)을 5분의 기간에 걸쳐 0℃에서 적가하고, 실온에서 1시간 동안 교반하였다 (TLC 시스템: 100% 에틸 아세테이트. Rf 값: 0.3). 이어서, 반응 혼합물을 포화 수성 NaHCO3 용액 (20 mL)과 DCM (30 mL) 사이에 분배하였다. 분리된 유기 층을 무수 황산나트륨 상에서 건조시키고, 감압 하에 농축시켜 목적 생성물 (9S)-3-클로로-N-(4-((R)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (100 mg, 0.173 mmol, 36.1% 수율). LCMS (m/z): 565.10 [M+H]+, Rt =2.07분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.42 (s, 1 H), 8.31 - 8.18 (m, 1 H), 8.17 - 7.97 (m, 2 H), 7.95 - 7.80 (m, 1 H), 6.58 - 6.41 (m, 1 H), 4.92 (br d, J=5.04 Hz, 1 H), 4.80 (br s, 1 H), 4.62 (br t, J=5.70 Hz, 1 H), 4.24 - 4.13 (m, 2 H), 4.08 (br dd, J=10.85, 5.81 Hz, 1 H), 3.79 - 3.67 (m, 2 H), 3.52 - 3.31 (m, 3 H), 3.34 - 3.30 (m, 2 H), 2.90 (br d, J=12.72 Hz, 1 H), 2.04 - 1.78 (m, 2 H), 1.42 - 1.19 (m, 2 H).
실시예 79
(9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-3-클로로-N-(6-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (300 mg, 0.497 mmol)의 교반 용액에 HCl (0.151 mL, 4.97 mmol)을 0℃에서 첨가하고, 25℃에서 2시간 동안 교반하였다 (TLC 시스템: 100% 에틸아세테이트, Rf 값: 0.2). 반응 혼합물을 포화 NaHCO3 용액 (10 mL)으로 켄칭하고, DCM (2x30 mL)으로 추출하였다. 합한 유기 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 화합물을 n-펜탄 (3 x 10mL)으로 연화처리하여 목적 생성물 (9S)-3-클로로-N-(6-((R)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 백색 고체로서 수득하였다 (140 mg, 0.248 mmol, 49.8% 수율). LCMS (m/z): 564.17 [M+H]+, Rt = 2.42분.
1H NMR (400 MHz, CDCl3): δ ppm 12.92 (s, 1 H), 8.09 (d, J=2.85 Hz, 1 H), 8.03 (s, 1 H), 7.96 (d, J=7.67 Hz, 1 H), 7.77 (dd, J=8.88, 2.74 Hz, 2 H), 7.71 - 7.64 (m, 1 H), 7.59 (s, 1 H), 6.73 (d, J=8.99 Hz, 1 H), 4.98 (brs, 1 H), 4.43 - 4.39 (m, 2 H), 4.01 - 3.95 (m, 2 H), 3.70 - 3.61 (m, 2 H), 3.42 - 3.30 (m, 3 H), 2.98 (d, J=13.81 Hz, 1 H), 2.69 (t, J=6.47 Hz, 1 H), 2.24 (d, J=12.06 Hz, 1 H), 1.98 - 1.87 (m, 1 H), 1.53 - 1.40 (m, 2 H).
실시예 80
(9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리 플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (5 mL) 중 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도 [2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (200 mg, 0.331 mmol)의 교반 용액에 수성 HCl (1.0 mL, 32.9 mmol)을 0℃에서 5분의 기간에 걸쳐 첨가하였다. 이어서, 반응 혼합물을 실온에서 1시간 동안 교반하고 (TLC 용리액: DCM 중 10% MeOH Rf: 0.3; UV 활성), 반응 혼합물을 빙냉수 (10 mL)에 붓고, 반응 혼합물의 PH를 포화 NaHCO3 용액을 사용하여 중성으로 조정하고, EtOAc (2x25 mL)로 추출하였다. 합한 유기 층을 물 (10 mL), 염수 용액 (10mL)으로 세척하고, 무수 Na2SO4 상에서 건조시키고, 여과하고, 감압 하에 농축시켜 조 화합물을 수득하였다. 조 화합물을 플래쉬 칼럼 크로마토그래피 (실리카겔: 100-200 메쉬, 용리액: DCM 중 5% MeOH)에 의해 정제하여 목적 생성물 (9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸) 페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (110 mg, 0.190 mmol, 57.4% 수율). LCMS (m/z): 564.38 [M+H]+, Rt = 2.17분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.07 (s, 1 H), 8.18 - 8.10 (m, 2 H), 7.95 - 7.81 (m, 5 H), 7.64 (br s, 1 H), 4.96 (d, J=5.04 Hz, 1 H), 4.82 (br s, 1 H), 4.67 (t, J=5.59 Hz, 1 H), 3.99 (dd, J=9.32, 3.84 Hz, 1 H), 3.87 - 3.75 (m, 2 H), 3.45 (t, J=5.59 Hz, 3 H), 3.40 (s, 1 H), 2.89 (d, J=13.59 Hz, 1 H), 2.04 - 1.90 (m, 2 H), 1.41- 1.29 (m, 3 H).
실시예 81
(9S)-3-클로로-N-(2-((S)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

0℃에서 질소 하에 메탄올 (10 mL) 중 (9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (170 mg, 0.281 mmol)의 용액에 수성 HCl (1 mL, 32.9 mmol, 36%)을 첨가하고, 실온에서 2시간 동안 교반하였다 (TLC 용리액: 100% 에틸 아세테이트: Rf-0.2; UV 활성). 반응 혼합물을 0℃에서 포화 중탄산나트륨 용액으로 염기성화시키고 (pH-8-9까지), 용매를 감압 하에 증발시켰다. 잔류물을 물 (30 mL)로 희석하고, DCM (2x100 mL)으로 추출하였다. 합한 유기 추출물을 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 물질을 백색 고체로서 수득하였다. 조 물질을 에테르로 연화처리하고, 여과하여 (9S)-3-클로로-N-(2-((S)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (70 mg, 0.121 mmol, 43.0% 수율). LCMS (m/z): 565.17 [M+H]+, Rt = 2.32분.
1H NMR (400 MHz, CDCl3): δ ppm 13.57 (s, 1 H), 8.32 (d, J=5.70 Hz, 1 H), 8.15 - 7.97 (m, 2 H), 7.87 - 7.67 (m, 3 H), 7.62 (s, 1 H), 4.95 (br s, 1 H), 4.31 - 4.07 (m, 2 H), 4.00 - 3.85 (m, 1 H), 3.77 - 3.52 (m, 2 H), 3.43 - 3.21 (m, 3 H), 3.16 (br d, J=5.26 Hz, 1 H), 3.00 (br d, J=13.81 Hz, 1 H), 2.41 (br t, J=6.25 Hz, 1 H), 2.24 (br d, J=14.69 Hz, 1 H), 2.03 - 1.85 (m, 1 H), 1.54 - 1.37 (m, 2 H).
실시예 82
(9S)-3-클로로-N-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-3-클로로-N-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도 [2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (180 mg, 0.298 mmol)의 교반 용액에 염산 (5 mL, 165 mmol)을 0℃에서 5분의 기간에 걸쳐 첨가하였다. 이어서, 반응 혼합물을 30℃에서 2시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.5; UV 활성). 용매를 증발시키고, 반응 혼합물을 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 물 (10 mL) 및, n-펜탄 (2 x 20 mL)으로 세척하여 목적 생성물 (9S)-3-클로로-N-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (120 mg, 0.212 mmol, 35.5% 수율). LCMS (m/z): 564 [M+H]+, Rt = 6.0분.
1H NMR (400 MHz, CDCl3): δ ppm 13.43 (s, 1 H), 8.23 (s, 1 H), 8.16 (d, J=8.11 Hz, 1 H), 8.04 (d, J=5.70 Hz, 1 H), 7.71 - 7.79 (m, 2 H), 7.60 - 7.67 (m, 1 H), 7.58 (s, 1 H), 6.53 (dd, J=5.70, 2.19 Hz, 1 H), 4.96 (s, 1 H), 4.08 - 4.18 (m, 2 H), 3.79 - 3.87 (m, 1 H), 3.70 - 3.77 (m, 1 H), 3.31 - 3.40 (m, 3 H), 3.00 (d, J=13.59 Hz, 1 H), 2.60 (s, 3 H), 2.24 (d, J=12.93 Hz, 1 H), 2.05 (d, J=7.67 Hz, 1 H), 1.86 - 1.98 (m, 1 H), 1.38 - 1.52 (m, 1 H).
실시예 83
(9S)-3-클로로-N-(2-((S)-2,3-디히드록시프로폭시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-3-클로로-N-(2-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도 [2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (420 mg, 0.695 mmol)의 교반 용액에 염산 (5 mL, 165 mmol)을 0℃에서 5분의 기간에 걸쳐 첨가하였다. 이어서, 반응 혼합물을 30℃에서 30분 동안 교반하였다 (TLC 시스템: DCM 중 5% MeOH: Rf-0.5; UV 활성). 이어서, 용매를 증발시키고, 반응 혼합물을 중탄산나트륨 용액으로 중화시키고, 여과하고, 물 및 n-펜탄 (2x 20 mL)으로 세척하여 목적 생성물 (9S)-3-클로로-N-(2-((S)-2,3-디히드록시프로폭시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (299 mg, 0.517 mmol, 37.1% 수율). LCMS (m/z): 564.20 [M+H]+, Rt = 2.35분.
1H NMR (400 MHz, CDCl3): δ ppm 13.29 (s, 1 H), 8.02 (s, 1 H), 7.97 (d, J=7.67 Hz, 1 H), 7.85 (d, J=5.70 Hz, 1 H), 7.81 (d, J=7.45 Hz, 1 H), 7.73-7.65 (m, 1 H), 7.61 (s, 1 H), 7.06 (d, J=1.53 Hz, 1 H), 6.69 (dd, J=5.70, 1.75 Hz, 1 H), 4.97 (s, 1 H), 4.46- 4.42 (m, 2 H), 4.28 (d, J=4.82 Hz, 1 H), 3.96 (d, J=4.38 Hz, 1 H), 3.69 -3.60 (m, 2 H), 3.41 -3.32 (m, 3 H), 2.98 (d, J=13.81 Hz, 1 H), 2.85 (t, J=6.36 Hz, 1 H), 2.23 (d, J=14.03 Hz, 1 H), 2.01-1.87 (m, 1 H), 1.49 -1.38 (m, 2 H).
실시예 84
(9S)-3-클로로-N-(6-((S)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
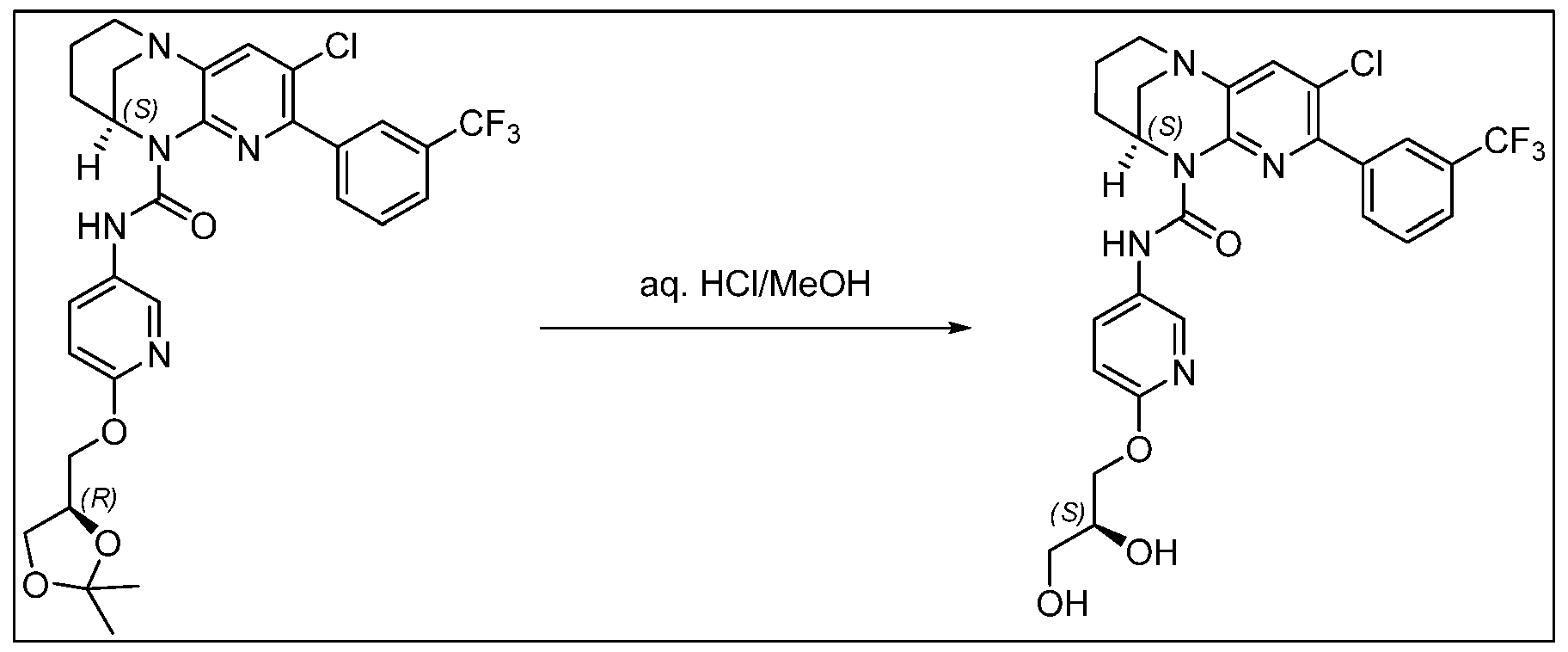
메탄올 (10mL) 중 (9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (150 mg, 0.248 mmol)의 교반 용액에 염산 (4 mL, 132 mmol)을 0℃에서 5분의 기간에 걸쳐 첨가하였다. 이어서, 반응 혼합물을 30℃에서 30분 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.5; UV 활성). 용매를 증발시키고, 반응 혼합물을 중탄산나트륨 용액으로 중화시키고, DCM (2 x 50.mL)으로 추출하였다. 합한 유기 층을 염수 용액으로 세척하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 증발시켜 조 화합물을 수득하였다. 조 생성물을 콤비-플래쉬 크로마토그래피 (120g 역상 칼럼: 용리액: 100% 아세토니트릴)에 의해 정제하고, 수득된 생성물을 n-펜탄 (2 x 20 mL)으로 세척하여 목적 생성물 (9S)-3-클로로-N-(6-((S)-2,3-디히드록시프로폭시)피리딘-3-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (108 mg, 0.190 mmol, 77% 수율). LC-MS (m/z): 564.17 [M+H]+, Rt = 2.42분.
1H NMR (400 MHz, CDCl3): δ ppm 12.92 (s, 1 H), 8.09 (d, J=2.85 Hz, 1 H), 8.03 (s, 1 H), 7.96 (d, J=7.67 Hz, 1 H), 7.77 (dd, J=8.99, 2.63 Hz, 2 H), 7.71- 7.63 (m, 1 H), 7.59 (s, 1 H), 6.73 (d, J=8.77 Hz, 1 H), 4.99 (s, 1 H), 4.42 (d, J=4.17 Hz, 2 H), 3.97 (s, 2 H), 3.73- 3.61 (m, 2 H), 3.40- 3.31 (m, 3 H), 2.98 (d, J=13.59 Hz, 1 H), 2.67 (t, J=6.14 Hz, 1 H), 2.23 (d, J=14.03 Hz, 1 H), 1.99- 1.87 (m, 1 H), 1.49- 1.38 (m, 2 H).
실시예 85
(9S)-3-클로로-N-(2-((R)-2,3-디히드록시프로폭시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (5.0 mL) 중 (9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (250 mg, 0.414 mmol)의 교반 현탁액에 수성 HCl (0.349 mL, 4.14 mmol, 36%)을 0℃에서 첨가하고, 실온에서 5시간 동안 교반하였다. 반응이 완결된 후, 휘발성 물질을 감압 하에 증발시켜 조 물질 (TLC 용리액 시스템: DCM 중 5% 메탄올,Rf-0.2, UV 활성)을 수득하였다. 조 물질을 물 (5 ml)로 희석하고, 수성 중탄산나트륨 용액으로 염기성화시켰다 (pH 8까지). 침전된 고체를 여과하고, 물로 세척하고, 진공 하에 건조시켜 (9S)-3-클로로-N-(2-((R)-2,3-디히드록시프로폭시)피리딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (149.0 mg, 0.262 mmol, 63.4% 수율). LCMS (m/z): 564.20 [M+H]+, Rt =2.37분.
1H NMR (400 MHz, CDCl3): δ ppm 13.30 (s, 1 H), 8.02 (s, 1 H), 7.97 (br d, J=7.67 Hz, 1 H) 7.78 - 7.88 (m, 2 H), 7.66 - 7.73 (m, 1 H), 7.61 (s, 1 H), 7.06 (d, J=1.32 Hz, 1 H), 6.69 (dd, J=5.70, 1.53 Hz, 1 H), 4.96 (br s, 1 H), 4.43 (d, J=4.60 Hz, 2 H), 4.30 (br s, 1 H), 3.97 (br d, J=3.73 Hz, 1 H), 3.65 (br d, J=4.38 Hz, 2 H), 3.29 - 3.42 (m, 3 H), 2.87 - 3.01 (m, 2 H), 2.23 (br d, J=14.47 Hz, 1 H), 1.87 - 2.00 (m, 1 H), 1.39 - 1.55 (m, 2 H).
실시예 86
(9S)-3-클로로-N-(6-((S)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

0℃에서 메탄올 (5 mL) 중 (9S)-3-클로로-N-(6-(((R)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메톡시피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (200 mg, 0.331 mmol)의 교반 용액에 염산 (1 mL, 32.9 mmol)을 5분의 기간에 걸쳐 적가하였다. 이어서, 반응 혼합물을 28℃에서 30분 동안 교반하였다 (TLC 용리액: DCM 중 10% MeOH: Rf-0.2; UV 활성). 반응 혼합물을 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 디에틸에테르로 연화처리하여 목적 생성물 (9S)-3-클로로-N-(6-((S)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (110 mg, 0.189 mmol, 57.1% 수율). LCMS (m/z): 565.10 [M+H]+, Rt: 2.52분.
1H NMR (400 MHz, CDCl3): δ ppm 13.64 (s, 1 H), 8.35 (d, J=0.66 Hz, 1 H), 8.19 (s, 1 H), 8.11 (d, J=7.89 Hz, 1 H), 7.75 (d, J=8.11 Hz, 1 H), 7.67 - 7.62 (m, 1 H), 7.61 (s, 1 H), 7.55 (d, J=0.88 Hz, 1 H), 4.95 (br s, 1 H), 4.55 - 4.42 (m, 2 H), 3.98 - 4.08 (m, 1 H), 3.75 - 3.62 (m, 2 H), 3.50 - 3.31 (m, 4 H), 2.99 (d, J=13.59 Hz, 1 H), 2.52 (t, J=6.25 Hz, 1 H), 2.22 (br d, J=14.69 Hz, 1 H), 2.02 - 1.82 (m, 1 H), 1.53 - 1.37 (m, 2 H).
실시예 87
(9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로 메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
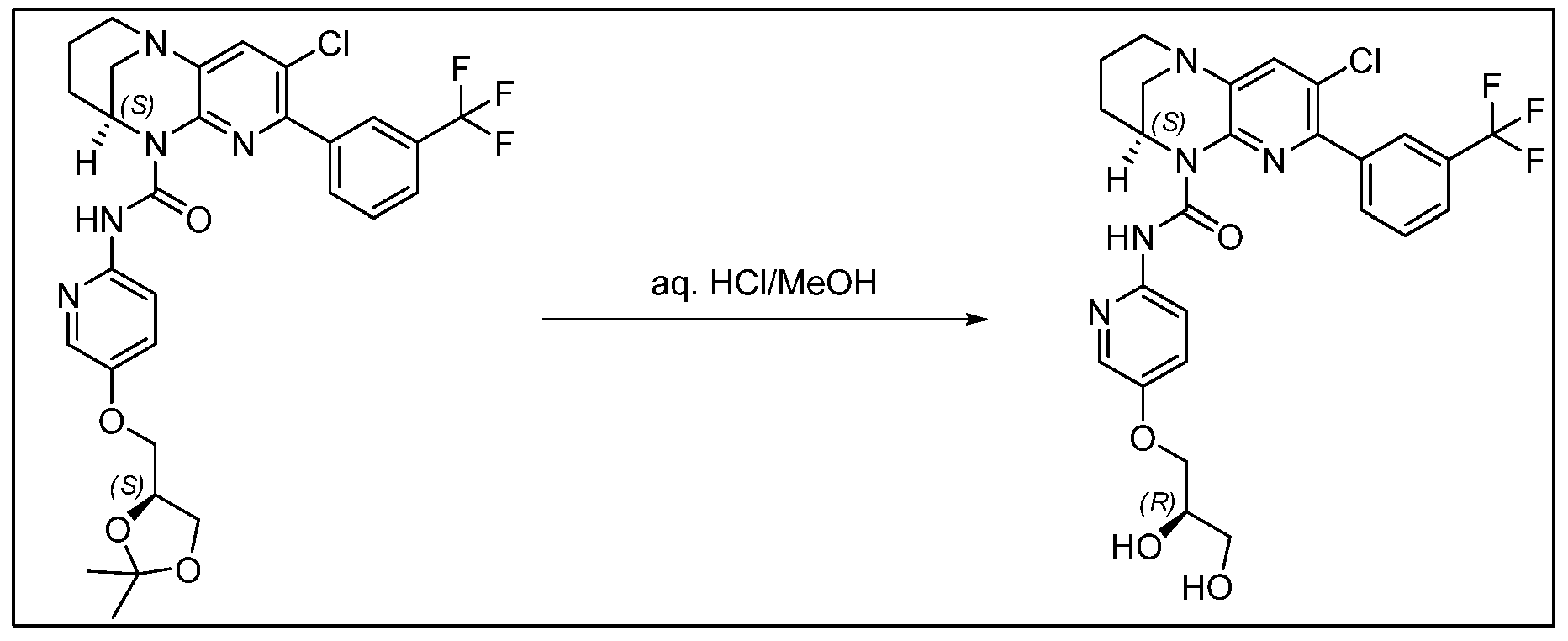
메탄올 (5 mL) 중 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (250 mg, 0.414 mmol)의 교반 용액에 0℃에서 수성 HCl (0.126 mL, 4.14 mmol)을 첨가하였다. 반응 혼합물을 1시간 동안 교반하였다 (TLC 시스템: Rf: 0.4, EtOAc). 실온에서 및 감압 하에 농축시켜 잔류물을 수득하였다. 잔류물을 포화 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 건조시켜 목적 생성물 (9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (160 mg, 0.274 mmol, 66.1% 수율). LCMS (m/z): 564.1 [M+H]+, Rt = 2.44분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.22 (s, 1 H), 8.23- 8.12 (m, 2 H), 7.99 (d, J=9.21 Hz, 1 H), 7.90- 7.84 (m, 3 H), 7.83 -7.78 (m, 1 H), 7.43 (dd, J=9.21, 3.07 Hz, 1 H), 4.95 (br s, 1 H), 4.83 (br s, 1 H), 4.65 (br s, 1 H), 4.02 (dd, J=9.87, 3.95 Hz, 1 H), 3.92- 3.85 (m, 1 H), 3.82 -3.74 (m, 1 H), 3.48- 3.31 (m, 5 H), 2.89 (d, J=13.81 Hz, 1 H), 2.06- 1.89 (m, 2 H), 1.42 -1.30 (m, 2 H).
실시예 88
(9S)-3-클로로-N-(2-((R)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

0℃에서 질소 하에 메탄올 (10 mL) 중 (9S)-3-클로로-N-(2-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (300 mg, 0.496 mmol)의 교반 용액에 수성 HCl (1 ml, 4.00 mmol, 36%)을 첨가하고, 실온에서 1시간 동안 교반하였다 (TLC 용리액: DCM 중 5% 메탄올, Rf: 0.3, UV 활성). 반응 혼합물에 포화 NaHCO3 용액을 첨가하고 (pH-8-9까지), EtOAc (3x10 mL)로 추출하였다. 합한 유기 추출물을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 생성물을 수득하였다. 조 화합물을 크로마토그래피 (그레이스 기기, C-18 칼럼 사용, 이동상 A: 물 중 0.1% 포름산; B: ACN, 생성물을 90% ACN/물 중 0.1% 포름산으로 용리시킴)에 의해 정제하였다. 후속적으로 필요한 분획을 농축시키고, 포화 NaHCO3으로 염기성화시키고, DCM으로 추출하였다. 합한 추출물을 무수 Na2SO4 상에서 건조시키고, 여과하고, 증발시켜 (9S)-3-클로로-N-(2-((R)-2,3-디히드록시프로폭시)피리미딘-4-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (170 mg, 0.292 mmol, 58.9% 수율). LCMS (m/z): 565.2 [M+H]+, Rt = 2.30분.
1H NMR (400 MHz, CDCl3): δ ppm 13.57 (s, 1 H), 8.32 (d, J=5.70 Hz, 1 H), 8.10 - 8.05 (m, 2 H), 7.79 - 7.69 (m, 3 H), 7.62 (s, 1 H), 4.95 (br s, 1 H), 4.24 - 4.12 (m, 2 H), 3.97 (dq, J=10.06, 5.05 Hz, 1 H), 3.72 - 3.58 (m, 2 H), 3.41 - 3.32 (m, 3 H), 3.11 (d, J=5.26 Hz, 1 H), 3.00 (br d, J=14.03 Hz, 1 H), 2.37 (t, J=6.36 Hz, 1 H), 2.24 (br d, J=14.03 Hz, 1 H), 2.00 - 1.88 (m, 1 H), 1.53 - 1.39 (m, 2 H)
실시예 89
(9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (15 mL) 중 (9S)-3-클로로-N-(5-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (120 mg, 0.198 mmol)의 교반 용액에 수성 HCl (6.03 μL, 0.198 mmol, 36%)을 0℃에서 첨가하고, 실온에서 1시간 동안 교반하였다 (TLC 용리액: EtOAc 중 10%MeOH: Rf-0.1; UV 활성). 반응 혼합물을 0℃에서 포화 중탄산나트륨 용액으로 염기성화시키고 (pH-8-9까지), 농축시켰다. 잔류물을 물 (8 mL)로 희석하고, DCM (2x25 mL)으로 추출하였다. 합한 유기 추출물을 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 감압 하에 증발시켜 (9S)-3-클로로-N-(5-((R)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 갈색 고체로서 수득하였다 (40 mg, 0.068 mmol, 34.4% 수율). LCMS (m/z): 565.17 [M+H]+, Rt =2.25분.
1H NMR (400 MHz, DMSO-d6): δ ppm 13.43 (s, 1 H), 8.33 (s, 2 H), 8.00 - 8.24 (m, 2 H), 7.83 - 7.91 (m, 2 H), 7.83 - 7.91 (m, 1 H), 5.01 (br d, J=3.95 Hz, 1 H), 4.80 (br s, 1 H), 4.47 - 4.74 (m, 1 H), 4.12 (dd, J=10.08, 3.73 Hz, 2 H), 3.89 - 4.06 (m, 1 H), 3.36 - 3.48 (m, 3 H), 3.30 - 3.34 (m, 2 H), 2.89 (br d, J=13.59 Hz, 1H), 1.98 (br s, 2 H), 1.35 (br s, 2 H).
실시예 90
(9S)-4-메틸-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성.

실온에서 THF (30 mL) 중 (9S)-4-메틸-2-(2-메틸피리딘-4-일)-7,8,9,10-테트라히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신 (500 mg, 1.783 mmol)의 교반 용액에 DIPEA (0.934 mL, 5.35 mmol), 트리포스겐 (318 mg, 1.070 mmol)를 첨가하고, 1시간 동안 교반한 다음, 피리딘-3-아민 (252 mg, 2.68 mmol)을 첨가하고, 반응 혼합물을 75℃로 16시간 동안 가열하였다 (TLC 용리 시스템: DCM 중 10% MeOH; Rf-0.3; UV 활성). 반응 혼합물을 실온으로 냉각시키고, 물 (15 mL)로 켄칭하고, EtOAc (2x15 mL)로 추출하였다. 유기 층을 분리하고, 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 증발시켜 조 화합물을 수득하였다. 조 물질을 크로마토그래피 (그레이스, C-18 역 칼럼 사용, 이동상 A: 물 중 0.1% 포름산; B: ACN, 용리액 A 중 56% B)에 의해 정제하였다. 합한 분획을 증발시키고, 포화 NaHCO3 용액으로 염기성화시켰다. 수성 층을 DCM으로 추출하고, DCM 층을 무수 Na2SO4 상에서 건조시키고, 여과하고, 여과물을 증발시켜 (9S)-4-메틸-2-(2-메틸피리딘-4-일)-N-(피리딘-3-일)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 황색 고체로서 수득하였다 (160 mg, 0.395 mmol, 22.17% 수율). LCMS (m/z): 400.9 [M+H]+, Rt =4.45분.
1H NMR (400 MHz, CDCl3): δ ppm 13.83 (s, 1 H), 8.57 - 8.76 (m, 2 H), 8.31 (dd, J=4.71, 1.43 Hz, 1 H), 8.02 - 8.23 (m, 1 H), 7.61 (s, 1 H), 7.51 (dd, J=5.04, 1.53 Hz, 1 H), 7.20 - 7.33 (m, 2 H), 4.99 (br s, 1 H), 3.38 (dd, J=13.59, 1.75 Hz, 1 H), 3.12 - 3.28 (m, 2 H), 2.98 (br d, J=13.59 Hz, 1 H), 2.68 (s, 3 H), 2.44 (s, 3 H), 2.13 - 2.41 (m, 1 H), 1.85 - 2.00 (m, 1 H), 1.44 (dt, J=6.08, 3.21 Hz, 2 H).
실시예 91
(9S)-N-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-3-메틸-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성

메탄올 (10 mL) 중 (9S)-N-(4-(((S)-2,2-디메틸-1,3-디옥솔란-4-일)메톡시)피리딘-2-일)-3-메틸-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (300 mg, 0.514 mmol)의 교반 용액에 HCl (1 mL, 32.9 mmol)을 0℃에서 5분의 기간에 걸쳐 적가하였다. 이어서, 반응 혼합물을 30℃에서 1시간 동안 교반하였다 (TLC 용리액: DCM 중 5% MeOH: Rf-0.3; UV 활성). 용매를 감압 하에 증발시키고, 중탄산나트륨 용액으로 중화시키고, 수득된 고체를 여과하고, 1:1 비율의 디에틸에테르 (50 ml) 및 펜탄 (50 ml)으로 연화처리하여 순수한 화합물 (9S)-N-(4-((R)-2,3-디히드록시프로폭시)피리딘-2-일)-3-메틸-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (250 mg, 0.453 mmol, 88% 수율). LCMS (m/z): 544.26 [M+H] +, Rt = 2.23분.
1H NMR (400 MHz, CDCl3): δ: 13.76 (s, 1 H), 8.08 - 8.00 (m, 2 H), 7.97 - 7.88 (m, 2 H), 7.73 - 7.57 (m, 2 H), 7.38 (d, J = 0.7 Hz, 1 H), 7.23 (dd, J = 9.0, 3.1 Hz, 1 H), 4.95 (d, J = 3.1 Hz, 1 H), 4.16 - 4.00 (m, 3 H), 3.85 (dd, J = 11.4, 3.8 Hz, 2 H), 3.76 (dd, J = 11.4, 5.4 Hz, 3 H), 3.01 (d, J = 13.6 Hz, 1 H), 2.55 - 2.4 (m, 1 H), 2.39 (s, 3 H), 2.25 (d, J = 14.4 Hz, 1 H), 1.98 - 1.84 (m, 2 H), 1.42 - 1.33 (m, 1 H).
실시예 92
(9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드의 합성
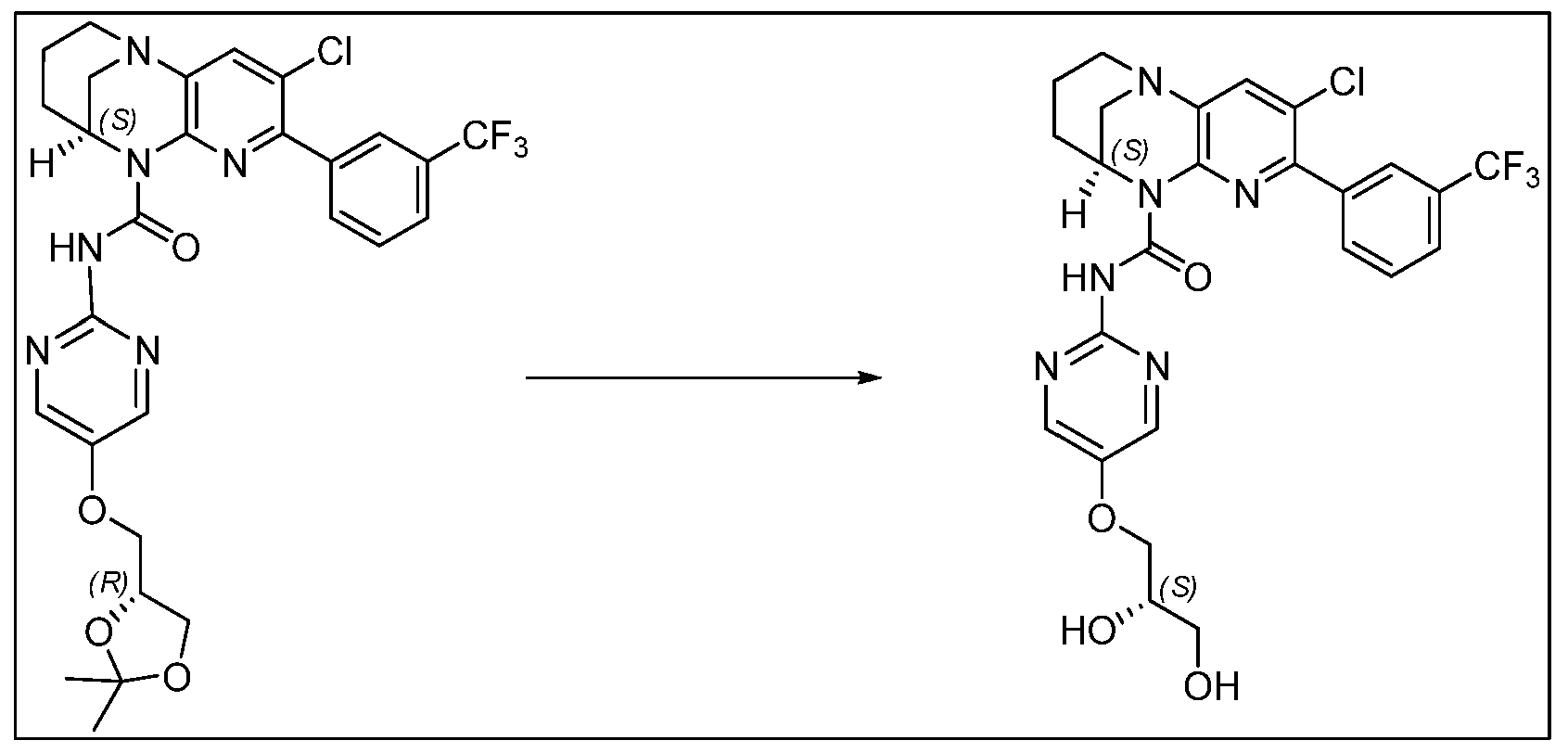
메탄올 (5 mL) 중 (9S)-3-클로로-N-(5-(((R)-2,2-디메틸-1,3-디옥솔란-4- 일)메톡시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드 (0.420g, 0.694 mmol)의 교반 용액에 수성 HCl (0.5 mL, 6.00 mmol, 12M)을 0℃에서 첨가하고, 실온에서 1시간 동안 교반하였다 (TLC 용리액 100% 에틸아세테이트: Rf = 0.1; UV 활성). 반응 혼합물을 포화 중탄산나트륨 용액을 첨가하여 염기성화시킨 다음 (pH-8-9까지), 농축시켰다. 잔류물을 물 (10 mL)로 희석하고, 에틸 아세테이트 (20 mL)로 추출하였다. 합한 유기 추출물을 무수 황산나트륨 상에서 건조시키고, 여과하고, 여과물을 진공 하에 증발시키고, 조 물질을 디에틸에테르 (10 mL)로 연화처리하여 목적 생성물 (9S)-3-클로로-N-(5-((S)-2,3-디히드록시프로폭시)피리미딘-2-일)-2-(3-(트리플루오로메틸)페닐)-8,9-디히드로-6H-5,9-메타노피리도[2,3-b][1,4]디아조신-10(7H)-카르복스아미드를 회백색 고체로서 수득하였다 (0.083g, 0.145 mmol, 20.90% 수율). LCMS (m/z): 565.14 [M+H] +, Rt = 2.25분.
1H NMR (400 MHz, CDCl3): δ 13.74 (s, 1 H), 8.29 (s, 2 H), 8.18 (s, 1 H), 8.10 (d, J=7.89 Hz, 1 H), 7.77-7.75 (m, 1 H), 7.65 -7.56 (m, 2 H), 5.03 (br s, 1 H), 4.21 -4.00 (m, 3 H), 3.86 (br dd, J=11.18, 2.63 Hz, 1 H), 3.76 (br dd, J=11.18, 4.82 Hz, 1 H), 3.44 -3.24 (m, 3 H), 2.98 (br d, J=13.59 Hz, 1 H), 2.62 (br s, 1 H), 2.38 - 2.21 (m, 1 H), 2.01 (br s, 2 H), 1.98 -1.75 (m, 2 H).
실시예 93. 전장 SIRT1 생산
C-말단 His6 태그를 갖는 전장 인간 SIRT1 (hSIRT1) 단백질을 발현시키고, 문헌 [Hubbard. et al. (2013) Science 339, 1216]에 기재된 바와 같이 정제하였다. 각각의 세포 페이스트를 얼음 상에서 EDTA-무함유 프로테아제 억제제 칵테일 정제인 컴플리트 (로슈)로 보충된 1,000 U 벤조나제 뉴클레아제 (시그마 알드리치) 함유 완충제 A (50 mM 트리스-HCl pH 7.5, 250 mM NaCl, 25 mM 이미다졸, 및 0.1 mM TCEP) 중에 재현탁시켰다. 40 W에서 총 12분 동안 50% 온 및 50% 오프로의 펄스 초음파처리에 의해 세포를 파괴하였다. 불용성 파편을 원심분리에 의해 제거하였다. 정화된 상청액을 1 mL His트랩 FF 크루드 칼럼 (지이 라이프사이언시스) 상에 직접 로딩하였다. 완충제 A로 세척한 후, SIRT1을 완충제 B (50 mM 트리스-HCl pH 7.5, 250 mM NaCl, 500 mM 이미다졸 및 0.1 mM TCEP)로 용리시켰다. 단백질을 하이-로드 슈퍼덱스 200 16/60 칼럼 (지이 라이프사이언시스)을 사용하여 완충제 C (50 mM 트리스-HCl pH 7.5, 300 mM NaCl, 0.1 mM TCEP) 로 크기 배제 크로마토그래피에 의해 추가로 정제하였다. 표준물로서 BSA를 사용하여 브래드포드 검정에 의해 효소 농도를 결정하였다. 최종 단백질 순도를 겔 밀도측정법에 의해 평가하였다. 단백질을 LC/MS에 의해 확인하였다. 모든 단백질은 90% 초과의 순도였다.
실시예 94. SIRT1 탈아세틸화 반응
SIRT1 탈아세틸화 반응을 반응 완충제 (50 mM HEPES-NaOH, pH 7.5, 150 mM NaCl, 1 mM DTT, 및 1% DMSO) 중에서 25℃에서 수행하며, 연속 PNC1/GDH 커플링 검정을 사용하여 니코틴아미드 생산을 모니터링하거나 (Smith, B. C. et al. (2009) Anal Biochem 394, 101) 또는 질량 분광측정법에 의해 O-아세틸 ADP 리보스 (OAcADPr) 생산을 모니터링하였다 (Hubbard. et al. (2013) Science 339, 1216). 사용된 PNC1/GDH 커플링 시스템 성분의 최종 농도는 20 단위/mL 소 GDH (시그마-알드리치), 1 uM 효모 PNC1, 3.4 mM α-케토글루타레이트, 및 220 μM NADH 또는 NADPH였다. 6.22 mM-1cm-1의 흡광 계수 및 0.81 cm의 경로길이를 사용하여 340 nm에서의 흡광도를 사용된 150 uL 반응물에 대한 생성물 농도로 전환시켰다. OAcADPr 생산을 모니터링하는 검정을 0.05% BSA 함유 반응 완충제 중에서 수행하고, 탈아세틸화 반응물을 정지 용액으로 켄칭함으로써 1% 포름산 및 5 mM 니코틴아미드의 최종 농도를 제공하는 시점을 정하였다. 켄칭된 반응물을 1:1 아세토니트릴:메탄올로 5배 희석하고, 5,000 x g에서 10분 동안 회전시켜 단백질을 침전시킨 후, 전기분무 이온화 공급원이 피팅된 ABSciex API 4000 질량 분광계에 커플링된 애질런트 래피드파이어 200 고처리량 질량 분광측정법 시스템 (애질런트, 매사추세츠주 웨이크필드)으로 분석하였다. p53-기반 Ac-p53(W5) (Ac-RHKKAcW-NH2) 및 TAMRA (Ac-EE-K(비오틴)-GQSTSSHSK(Ac)NleSTEG-K(5TMR)-EE-NH2) 펩티드를 각각 아메리칸 센츄리 펩티드 및 바이오펩티드, 인크로부터 입수하였다. 기질 KM 결정은 1종의 기질 농도를 제2 기질의 고정된 포화 농도에서 변동시킴으로써 수행하였다. SIRT1 활성화 및 억제 검정은 25℃에서 0.05% BSA 함유 반응 완충제 중에서 실행하고, OAcADPr 검정을 사용하여 분석하였다. 효소 및 화합물을 20분 동안 예비-인큐베이션한 후, 기질을 첨가하였다. 전장 hSIRT1의 활성화 스크린을 위해, 화합물을 용량 반응으로 이중 시험하였다. KM-조정 활성화제에 대해 감수성이도록 하기 위해, 그의 KM 값의 대략 1/10의 기질 농도를 사용하였다. 5종의 화합물의 용량-의존성을 시험하고, 배수-활성화 데이터를 수학식 1에 의해 기재하였다.
<수학식 1>

여기서 vx/v0은 활성화제 (X)의 존재 (vx) 대 부재 (v0) 하의 반응 속도의 비이고, RVmax는 무한 활성화제 농도에서의 상대 속도이고, EC50은 RVmax의 1/2를 생성하는데 필요한 활성화제의 농도이고, b는 vx/v0의 최소 값이다.
실시예 95. 생화학적 활성
질량 분광측정법 기반 검정은 SIRT1 활성의 조정제를 확인하는데 사용하였다. TAMRA 기반 검정은 하기와 같은 20개 아미노산 잔기를 갖는 펩티드: Ac-EE-K(비오틴)-GQSTSSHSK(Ac)NleSTEG-K(5TMR)-EE-NH2 (서열식별번호: 1)를 이용하였으며, 여기서 K(Ac)는 아세틸화 리신 잔기이고, Nle는 노르류신이다. 펩티드를 C-말단에서 형광단 5TMR (여기 540 nm/방출 580 nm)로 표지하였다. 펩티드 기질의 서열은 여러 변형을 갖는 p53을 기반으로 하였다. 추가로, 서열에 자연적으로 존재하는 메티오닌 잔기를 노르류신으로 대체하였으며, 이는 메티오닌이 합성 및 정제 동안 산화에 대해 감수성일 수 있기 때문이다. Trp 기반 검정은 하기와 같은 아미노산 잔기를 갖는 펩티드: Ac-R-H-K-K(Ac)-W-NH2 (서열식별번호: 2)를 이용하였다.
TAMRA 기반 질량 분광측정법 검정은 하기와 같이 수행하였다: 0.5 μM 펩티드 기질 및 120 μM βNAD+를 반응 완충제 (50 mM 트리스-아세테이트 pH 8, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 5 mM DTT, 0.05% BSA) 중에서 25℃에서 25분 동안 10 nM SIRT1과 함께 인큐베이션하였다. SIRT1 단백질을 SirT1 유전자를 T7-프로모터 함유 벡터 내로 클로닝함으로써 수득한 다음, 이를 형질전환시키고, BL21(DE3) 박테리아 세포에서 발현시켰다. 시험 화합물을 상기 반응 혼합물에 다양한 농도로 첨가하고, 생성된 반응물을 모니터링하였다. SIRT1과 함께 25분 동안 인큐베이션한 후, 10 μL의 10% 포름산를 첨가하여 반응을 정지시켰다. 생성된 반응물을 추후 질량 스펙트럼 분석을 위해 밀봉 및 동결시켰다. 시르투인-매개 NAD-의존성 탈아세틸화 반응에 의해 형성된 탈아세틸화 기질 펩티드의 양 (또는 대안적으로 생성된 O-아세틸-ADP-리보스 (OAADPR)의 양)의 결정은 시험 화합물 결여 대조군 반응에 대비하여 다양한 농도의 시험 화합물의 존재 하의 상대적 SIRT1 활성의 정밀 측정을 가능하게 하였다.
Trp 질량 분광측정법 검정은 하기와 같이 수행하였다. 0.5 μM 펩티드 기질 및 120 μM βNAD+를 반응 완충제 (50 mM HEPES pH 7.5, 1500 mM NaCl, 1 mM DTT, 0.05% BSA) 중에서 25℃에서 25분 동안 10 nM SIRT1과 함께 인큐베이션하였다. SIRT1 단백질을 SirT1 유전자를 T7-프로모터 함유 벡터 내로 클로닝함으로써 수득한 다음, 이를 BL21(DE3) 박테리아 세포에서 발현시키고, 하기에 더욱 상세하게 기재된 바와 같이 정제하였다. 시험 화합물을 상기 반응 혼합물에 다양한 농도로 첨가하고, 생성된 반응물을 모니터링하였다. SIRT1과 함께 25분 동안 인큐베이션한 후, 10 μL의 10% 포름산을 첨가하여 반응을 정지시켰다. 생성된 반응물을 추후 질량 스펙트럼 분석을 위해 밀봉 및 동결시켰다. 이어서, 상대적 SIRT1 활성은 시험 화합물 결여 대조군 반응에 대비하여 다양한 농도의 시험 화합물의 존재 하의 NAD-의존성 시르투인 탈아세틸화 반응에 의해 형성된 O-아세틸-ADP-리보스 (OAADPR)의 양 (또는, 대안적으로, 생성된 탈아세틸화 Trp 펩티드의 양)을 측정함으로써 결정하였다. 시험 작용제가 SIRT1에 의한 탈아세틸화를 활성화시킨 정도는 EC1.5 (즉, SIRT1 활성을 시험 화합물 결여 대조군에 비해 50% 증가시키는데 필요한 화합물의 농도), 및 퍼센트 최대 활성화 (즉, 시험 화합물에 대해 수득된 대조군 (100%) 대비 최대 활성)로서 표현하였다.
시르투인 활성의 억제에 대한 대조군은 반응 개시 시에 음성 대조군으로서 1 μL의 500 mM 니코틴아미드를 첨가함 (예를 들어, 최대 시르투인 억제의 결정을 허용함)으로써 수행하였다. 시르투인 활성의 활성화에 대한 대조군은 화합물 대신에 1 μL의 DMSO 함유 10 nM 시르투인 단백질을 사용하여 수행하여, 검정의 선형 범위 내의 주어진 시점에서의 기질의 탈아세틸화의 양을 결정하였다. 상기 시점은 시험 화합물에 대해 사용된 것과 동일하였으며, 선형 범위 내에서 종점은 속도의 변화를 나타낸다.
상기 검정을 위해, SIRT1 단백질을 하기와 같이 발현 및 정제하였다. SirT1 유전자를 T7-프로모터 함유 벡터 내로 클로닝하고, BL21(DE3)로 형질전환시켰다. 단백질을 18℃에서 밤새 N-말단 His-태그 융합 단백질로서 1 mM IPTG로의 유도에 의해 발현시키고, 30,000 x g에서 수확하였다. 세포를 용해 완충제 (50 mM 트리스-HCl, 2 mM 트리스[2-카르복시에틸] 포스핀 (TCEP), 10 μM ZnCl2, 200 mM NaCl) 중에서 리소자임을 사용하여 용해시키고, 완전 용해를 위해 초음파로 10분 동안 추가로 처리하였다. 단백질을 Ni-NTA 칼럼 (아머샴) 상에서 정제하고, 순수한 단백질을 함유하는 분획을 모으고, 농축시키고, 크기결정 칼럼 (세파덱스 S200 26/60 글로벌) 상에서 구동시켰다. 가용성 단백질을 함유하는 피크를 수집하고, 이온-교환 칼럼 (모노큐) 상에서 구동시켰다. 구배 용리 (200 mM - 500 mM NaCl)시켜 순수한 단백질을 수득하였다. 상기 단백질을 농축시키고, 투석 완충제 (20 mM 트리스-HCl, 2 mM TCEP)에 대해 밤새 투석하였다. 단백질을 분취하고, 추가 사용 시까지 -80℃에서 동결시켰다.
SIRT1을 활성화시키는 화학식 I의 시르투인-조정 화합물을 상기 기재된 검정을 사용하여 확인하였으며, 하기 표 1에 제시되어 있다. EC1 .5 값은 SIRT1의 150% 활성화를 유발하는 시험 화합물의 농도를 나타낸다. 화학식 I의 활성화 화합물에 대한 EC1.5 값은 A (EC1.5 <1 μM), B (EC1.5 1-25 μM), C (EC1.5 >25 μM)에 의해 나타내어진다. 퍼센트 최대 배수 활성화는 A (배수 활성화 ≥150%) 또는 B (배수 활성화 <150%)에 의해 나타내어진다. "NT"는 시험되지 않음을 의미하고; "ND"는 결정가능하지 않음을 의미한다. 표 내의 화합물 넘버링은 화합물 번호 10, 및 도 4 및 실시예 90-106에서의 STAC 넘버링 시스템에 상응하는 괄호 넘버링 (#)으로 시작한다 (즉, 화합물 번호 68은 또한 STAC 1이므로, 이는 68(1)로서 제시됨, 추가의 STAC: 546(3), 444(4), 314(5), 816(7), 76(8), 및 81(9)).
본 발명의 화합물은 2종의 상이한 화학 도면 및/또는 화학 명명 컴퓨터 프로그램에 의해 생성 시의, 즉 각각의 회사인 켐 액손 (제이켐-엑셀) 및 캠브리지 소프트 (켐드로우(ChemDraw)®)에 의해 생성 시의 2종의 상이한 화학 명명법 규정에 의해 명명된 것임에 유의한다.
표 1



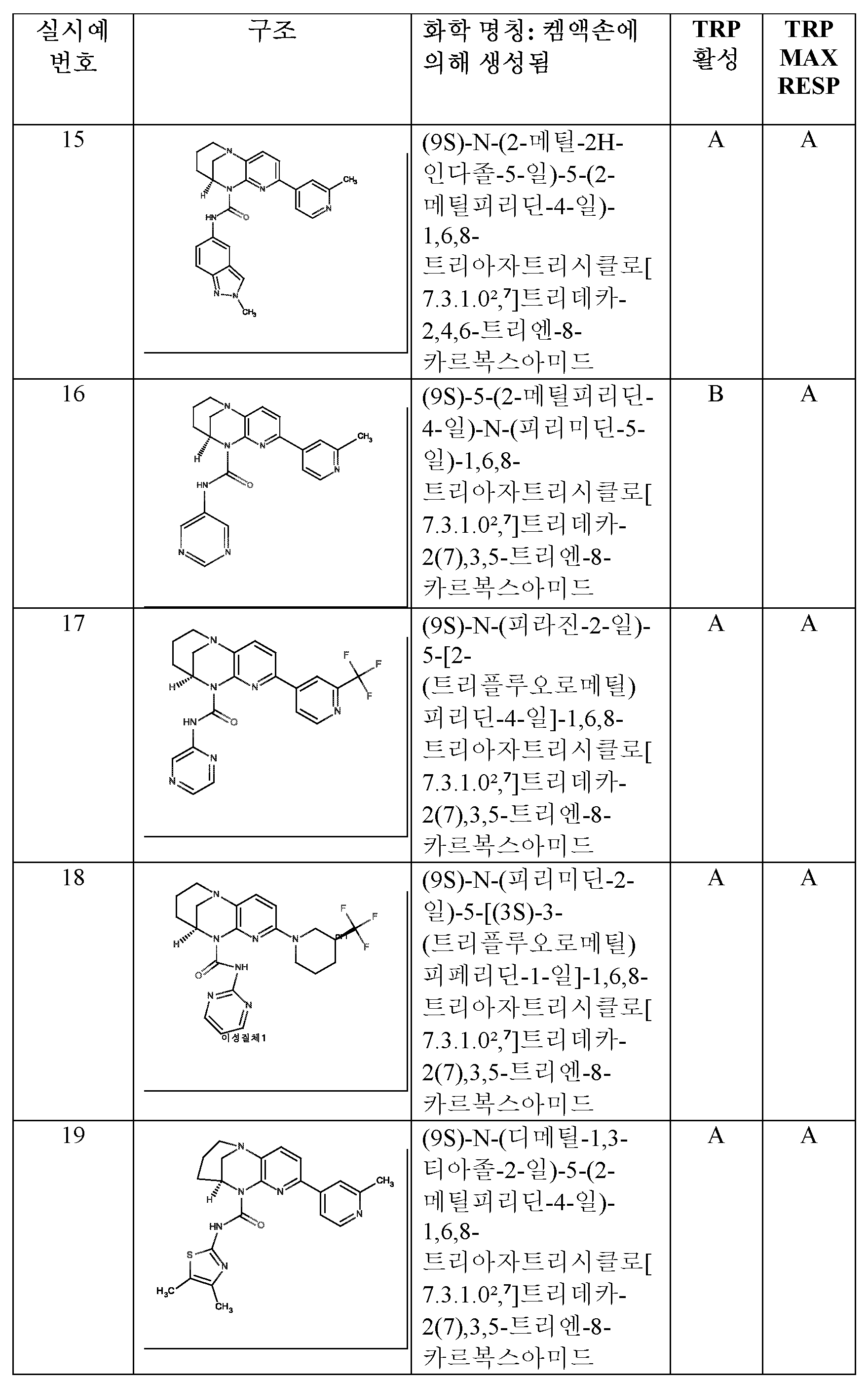






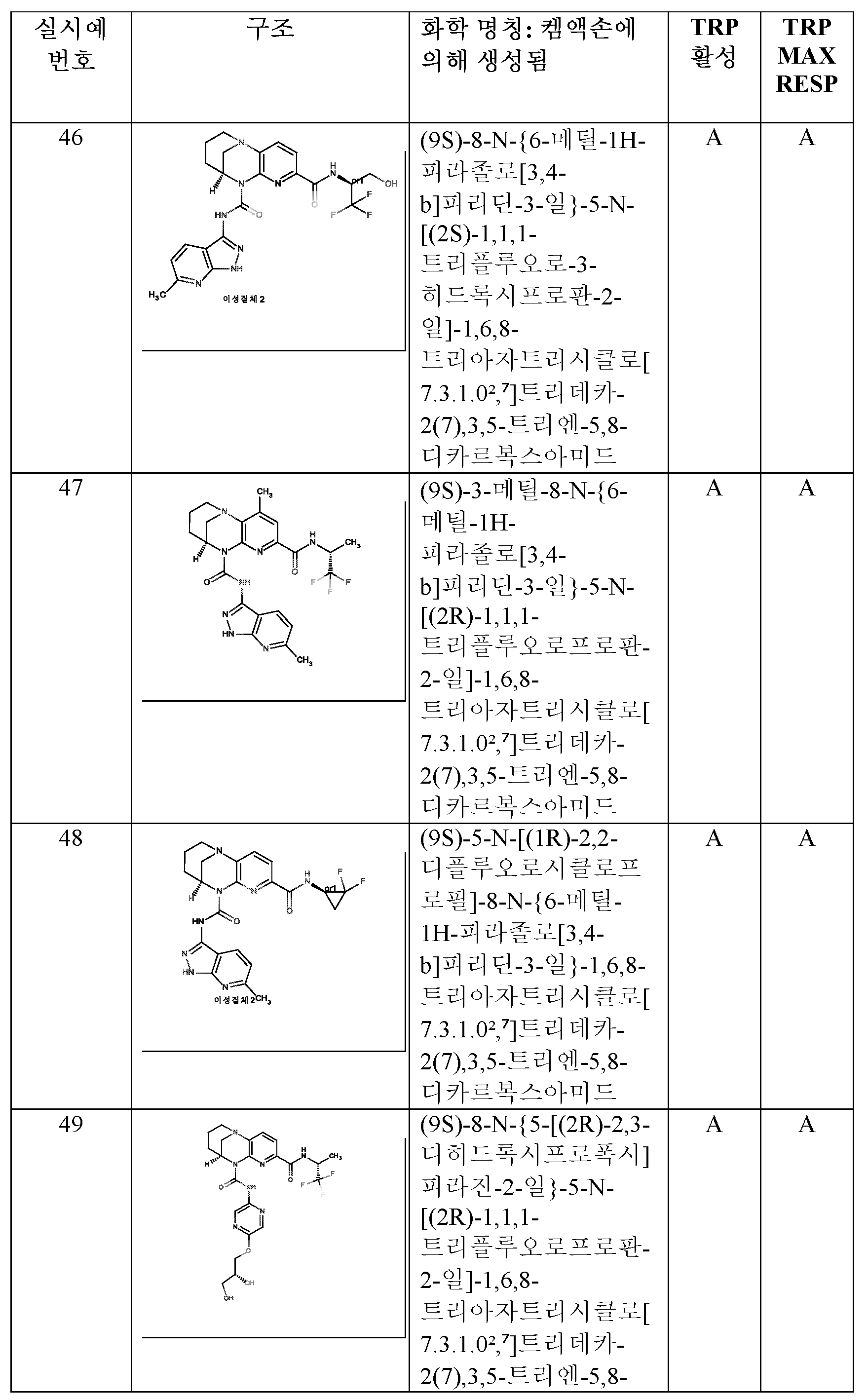










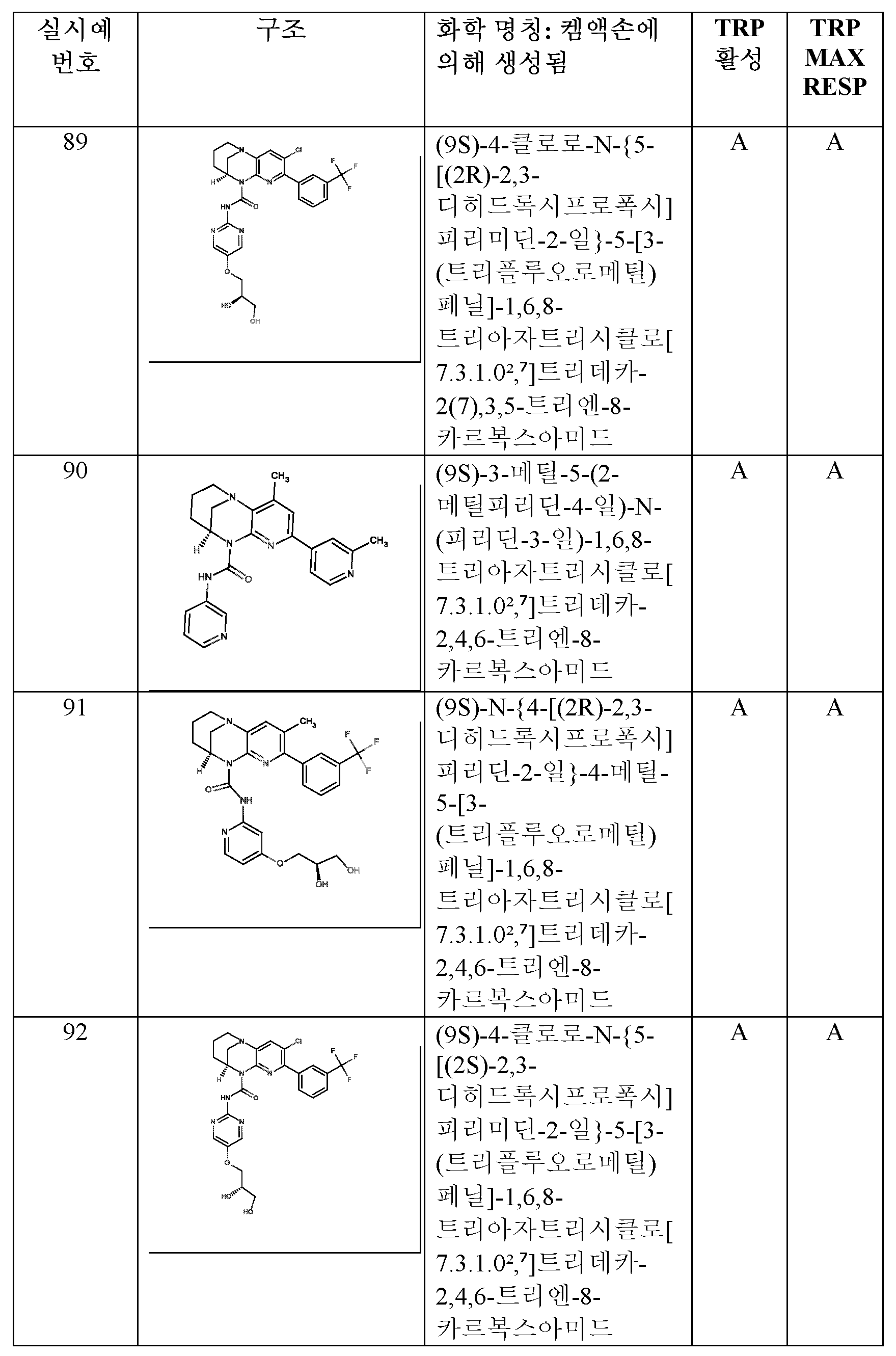
실시예 96
본 발명은 세포의 수명을 증가시키며, 예를 들어 노화 또는 스트레스와 관련된 질환 또는 장애, 당뇨병, 비만, 신경변성 질환, 심혈관 질환, 혈액 응고 장애, 염증, 암 및/또는 홍조 뿐만 아니라 증가된 미토콘드리아 활성으로부터 이익을 얻을 질환 또는 장애를 포함하나, 이에 제한되지는 않는 매우 다양한 질환 및 장애를 치료 및/또는 예방하기에 유용한 것으로 과학 문헌에 공지된 시르투인 조정제에 관한 것이다.
치료 잠재력 이외에도, SIRT1 활성 및 소분자 시르투인 조정제에 의한 활성화의 구조적 및 생물물리학적 연구는 시르투인의 생물학적 기능, 시르투인 활성화의 작용 메카니즘의 이해를 발전시키고, 신규 시르투인 조정제를 확인하는 검정의 개발을 보조하기에 유용할 것이다.
상기에 기초하여, 하기 참고 문헌 각각이 시르투인 조정제로서의 본 발명의 화합물의 유용성 및 그와 하기 참고문헌에 예시 또는 개시된 바와 같은 다양한 질환의 상호연관성을 증명하기 위해 인용된다:
1. Marcia C. Haigis and David A. Sinclair, Mammalian Sirtuins: Biological Insights and Disease Relevance, Annu Rev Pathol. 2010 ; 5: 253-295.
하이기스 및 싱클레어는 하기를 교시하고 있다:
"노화는 질환 예컨대 제II형 당뇨병, 신경변성 질환, 암, 및 심혈관 질환으로부터의 증가된 발생률 및 사망률로 이어지는 다발성 기관계의 건강한 기능의 저하를 동반한다. 역사적으로, 연구원은 더 우수한 약물을 설계하기를 희망하여, 질환의 근본 원인을 확인하기 위한 전략으로서 단리된 기관에서의 개별 경로를 조사하는데 집중해 왔다. 효모에서의 노화의 연구는 유기체의 수명 및 전반적 건강을 증가시키는 다중 경로에 영향을 미치는 시르투인으로서 공지된 보존된 효소의 패밀리의 발견으로 이어졌다. 10년 전에 가장 먼저 공지된 포유동물 시르투인인 SIRT1의 발견 이래로, 시르투인, 그의 조절, 및 포유동물 생리학 및 건강 수명을 광범위하게 개선시키는 것에 대한 그의 능력의 우리의 효소학적 이해에 있어서 주요한 발전이 이루어져 왔다. 본 종설논문은 지난 10년의 발견 발전사항 및 내년도에 본 분야가 직면할 과제를 요약 및 논의한다 (그의 요약 및 참고문헌 참조)."
2. Gizem Donmez1 et al., SIRT1 and SIRT2: emerging targets in neurodegeneration, EMBO Mol Med (2013) 5, 344-352.
기젬 돈메츠1 등은 하기를 교시하고 있다:
"시르투인은 연령-관련 질환 예컨대 암, 당뇨병, 심혈관 및 신경변성 질환에 대해 보호 효과를 갖는 것으로 공지된 NAD-의존성 단백질 데아세틸라제이다. 포유동물에서, 7종의 시르투인 (SIRT1-7)이 존재하며, 이는 세포하 국재화 및 기능에 있어서 다양성을 나타낸다. SIRT1은 그와 수명 연장의 초기 연관성 및 칼로리 제한에서의 수반으로 인해 광범위하게 조사되어 왔으며, 다른 시르투인의 중요한 생물학적 및 치료적 역할은 단지 최근에 인식된 바 있다. 여기서, 우리는 신경변성 질환에서의 SIRT1 및 SIRT2의 잠재적 역할 및 효과를 검토한다. 우리는 알츠하이머병 (AD), 파킨슨병 (PD) 및 헌팅톤병 (HD)을 포함한 다양한 신경변성 질환에서의 SIRT1 및 SIRT2의 상이한 기능 및 표적을 논의한다. 우리는 또한 신경변성 상태에서 가능한 연관성으로 인해 뉴런 분화에서의 SIRT1의 역할을 커버하며, 이들 장애에서의 SIRT1 및 SIRT2의 잠재적 치료 가치에 대한 전망으로 결론짓는다 (그의 요약 및 참고문헌 참조)."
3. Bracke et al., Targeted silencing of DEFB4 in a bioengineered skin-humanized mouse model for psoriasis: development of siRNA SECosome-based novel therapies; Exp Dermatol. 2014 Mar;23(3):199-201. doi: 10.1111/exd.12321.
특히, 브라케 등은 하기를 교시하고 있다:
"건선은 매우 다양한 임상 징후를 제시하는 복합 염증성 피부 질환이다. 인간 β 데펜신-2 (hBD-2)는 건선성 병변에서 고도로 상향조절되며, 질환 활성에 대한 바이오마커로서 정의된 바 있다. 우리는 건선에 대한 생체공학적 피부-인간화 마우스 모델에서 DEFB4-siRNA-함유 SECosome의 국소 적용에 의해 hBD-2를 표적화하는 것의 잠재적 이익을 연구하였다. 건선성 표현형에서의 유의한 개선이 조직학적 검사에 의해, 피부 아키텍처의 정상화 및 피부 구획 내 혈관의 수 및 크기에서의 감소와 함께 관찰되었다. 치료는 정상 재생 인간 피부에서 발견되는 것들과 유사한 수준으로의 트랜스글루타미나제 활성, 필라그린 발현 및 각질층 외관의 회복으로 이어진다. SECosome 기술의 사용과 함께 건선에 대한 신뢰성있는 피부-인간화 마우스 모델의 생체이용률은 이러한 질환에 대한 잠재적 치료 표적을 확인하기 위한 가치있는 전임상 도구를 제공할 수 있다."
4. Karline Guilloteau et al., Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22 Recapitulates Some Features of Psoriasis Oncostatin M, IL-1a, and TNF-a,J Immunol 2010; 184:5263-5270.
길로토 등은 하기를 교시하고 있다:
"각질세포는 환경 및 면역 세포 자극에 반응하는 피부 염증의 조절에서 결정적인 역할을 한다. 이들은 면역 세포 상에서 또는 직접 공격자 상에서 자가분비 또는 주변분비 방식으로 작용할 수 있는 가용성 인자를 생산한다. 각질세포 유전자 발현에 대한 36종의 시토카인의 활성의 스크리닝은 IL-17A, IL-22, 온코스타틴 M, TNF-a, 및 IL-1a가 피부 염증을 유발함에 있어서 강력한 시토카인인 것으로 확인시켰다. 이들 5종의 염증유발 시토카인은 CXCL8 및 b-데펜신 2 (BD2)의 생산을 상승작용적으로 증가시켰다. 추가로, 인간 피부 외식편에 대한 생체외 연구는 시토카인의 동일한 조합에 반응하는 BD2, S100A7, 및 CXCL8 발현의 상향조절을 증명하였다. 마우스에서의 이들 5종의 시토카인의 생체내 피내 주사는 호중구 침윤과 연관된 CXCL1, CXCL2, CXCL3, S100A9, 및 BD3 발현을 증가시켰다. 우리는 정량적 실시간 PCR 분석을 사용하여 이러한 상승작용적 효과를 확인 및 확장시켰으며, 9종의 케모카인 및 12종의 항미생물 펩티드의 증가된 발현을 관찰하였다. 이러한 시토카인 조합의 존재 하에 자극된 각질세포에 의한 CXCL, CXCL5, 및 CXCL8의 생산은 증가된 호중구 화학주성 활성과 연관되어 있었다. 유사하게, BD2, BD3, 및 S100A7의 높은 생산은 증가된 항미생물 활성과 연관되어 있었다. 최종적으로, 염증성 각질세포의 이러한 시험관내 모델에서 관찰된 전사 프로파일은 병변 건선성 피부 중 1종과 상관관계가 있었다. 우리의 결과는 각질세포에 대한 IL-17A, IL-22, 온코스타틴 M, TNF-a, 및 IL-1a의 중요한 강화 활성을 증명한다. 이는 이들 시토카인이 과다발현되고 상승작용하여 케모카인 및 항미생물 펩티드 생산의 상향조절에 있어서 중요한 역할을 할 수 있는 건선의 맥락에서 특히 흥미롭다. The Journal of Immunology, 2010, 184: 5263-5270 (그의 요약 및 참고문헌 참조)".
실시예 97
검정의 설명:
PBMC 검정
시르투인 1 (Sirt1)은 침묵 정보 조절인자 2 (Sir2)의 상동체 및 NAD 의존성 클래스 III 히스톤 데아세틸라제의 구성원이다. Sirt1은 히스톤, 전사 인자 및 비히스톤 단백질 상의 리신 잔기를 탈아세틸화시킨다. Sirt1은 노화, 세포 주기 조절, 아폽토시스, 대사 조정 및 염증에 수반되는 것으로 제시된 바 있다. Sirt1의 활성화는, NF-kB 전사를 억제하고 TNF알파의 수준을 하향조절하는 핵 인자 kB (NF-kB) 전사 인자의 RelA/p65 서브유닛의 리신 310에서의 탈아세틸화를 유발한다. TNF알파는 대식세포 및 단핵구에 의해 주로 생산되는 다면발현성 시토카인이다. TNF알파는 건선을 포함한 면역 방어 및 만성 염증에 밀접하게 수반된다. 제1형 시토카인 예컨대 TNFa의 발현은 건선성 피부에서 증가되는 것으로 공지되었으며, 이는 건선의 병인에 있어서 중요한 역할을 한다 (Uyemura K et al., 1993, J. Invest Dermatol, 101, p701). 중요하게, 항-TNF 작용제는 건선에 대해 임상적으로 사용되어 왔다. 따라서, 염증 세포에서 TNFa 발현의 감소를 유발하는 Sirt1 활성화제는 중등도 내지 중증 건선 환자에서 치료 효과를 가져야 한다.
PBMC/TNF알파 세포 기반 검정은 말초 혈액 단핵 세포 (PBMC)의 리포폴리사카라이드 (LPS) 자극에 반응하는 TNF알파의 방출을 억제하는 Sirt1의 활성화제를 확인하기 위해 개발되었다. 간략하게, PBMC는 LPS에 의해 자극되어, TNF알파 분비의 생산의 증가로 이어졌다. TNF알파 단백질 수준은 TNF알파 HTRF (균질 시간 분해 형광) 키트 (시스바이오, 인크.)에 의해 측정하였다. 세포 용해 및 TNF알파 검출은 제조업체의 지침에 따라 수행하였다. Sirt1 활성화제를 TNFa 방출에 대한 그의 억제 효과를 평가하기 위해 LPS의 존재 하에 시험하였으며, IC50은 용량-반응 실험에서 결정하였다.
베타-데펜신 2 (bD2) 검정
시르투인은 광범위한 생리학적 기능을 갖는 NAD-의존성 데아세틸라제의 패밀리이며, 류마티스 관절염 및 제I형 당뇨병을 포함한 다수의 자가면역 및 대사 장애에 연루된 바 있다. SIRT1의 기질은 다양하며, 선천성 및 적응성 면역 반응에 있어서 잘 확립된 역할을 갖는 염증 요인 예컨대 NFκB, AP-1, FOXO, 및 p53을 포함한다.
건선은 유전, 자가면역 및 환경 요인에 의해 유발된 만성 염증성 피부 장애이다. 병변은 표피에서의 각질세포의 과다증식 및 백색 인편에 의해 덮인 만성 홍반성 플라크를 유발하는 염증 세포의 침윤을 특징으로 한다. 이전의 연구는 SIRT1이 STAT3 아세틸화의 직접 억제를 통해, 건선에서의 주요 시토카인인 IL-22의 효과를 방해할 수 있다는 것을 제시한 바 있다 (Sestito et al., 2011). 추가로, SIRT1 과다발현 및 레스베라트롤 처리 (SIRT1 활성화) 둘 다는 각질세포 분화를 유발할 수 있다 (Blander et al., 2009).
베타-데펜신 2 (bD2)는 상피로부터 분비될 수 있는 항미생물 펩티드이며, 여기서 이는 기억 T-세포, 미성숙 수지상 세포, 및 호중구에 대한 화학유인물질로서 작용한다. 이와 같이, bD2는 피부에서 염증 반응의 주요 부분이다. bD2는 정상 피부와 비교하여 건선 환자의 병변 표피 세포에서 유도될 뿐만 아니라 건선 환자에서의 질환 중증도에 대한 혈청 바이오마커이다 (Jansen et al., 2009; Kamsteeg et al. 2009). 추가로, bD2는 최근 연구가 증가된 베타-데펜신 유전자 카피수와 건선 위험 사이의 유의한 연관성을 밝혀냄에 따라 유전학적으로 건선과 연관될 수 있다 (Hollox et al., 2008). 주요하게, bD2 siRNA의 국소 전달은 건선에 대한 생체공학적 피부-인간화 마우스 모델에서 정상 피부 아키텍처 및 단백질 발현의 회복을 유발하였다 (Bracke et al., 2014).
건선성 염증을 모방하도록 생성된 시험관내 각질세포 염증 검정은 이전에 기재되었다 (Guilloteau et al., 2010; Teng et al. 2014). 이들 연구에서, IL-1알파, IL-17A, IL-22, OSM, 및 TNF알파 ("M5"로 지칭됨)의 시토카인 칵테일은 시험관내 1차 인간 각질세포에서 "건선형" 전사 프로파일을 생성하도록 상승작용하는 것으로 밝혀졌다. 이들 연구에서, bD2는 각질세포 염증의 유발에 대한 가장 강한 반응인자 중 1종이었다.
따라서, 본 검정은 추가로 국소 건선 프로그램에 대한 SIRT1 활성화제 화합물의 효능을 평가하기 위해 개발되었다. 구체적으로, 조건은 건선성 염증을 유발하도록 M5 시토카인 조합물로 시험관내 처리된 불멸화 인간 각질세포 세포주 (HaCaT)에 대해 최적화되었다 (상기 참고문헌에서와 같음). 48시간의 시간 프레임에서, bD2 분비는 bD2 ELISA 검정 (알파 다이아그노스틱스)에 의해 측정 시에 비자극된 각질세포와 비교하여 현저하게 증가된다. 이러한 bD2 유발은 건선성 염증을 억제하는 것으로 공지된 화합물의 처리로, 또는 중요하게는 SIRT1 활성화제의 하위세트로 억제될 수 있다. 병행하여, 48시간의 검정의 길이에 걸친 세포독성을 셀타이터-글로 발광 세포 생존율 검정 (프로메가)에 의해 확인하여 독성이 bD2 반응에 있어서 역할을 할 수 있는지의 여부를 결정하였다.
참고문헌:
Blander G, Bhimavarapu A, Mammone T, Maes D, Elliston K, Reich C, Matsui MS, Guarente L, Loureiro JJ. SIRT1 promotes differentiation of normal human keratinocytes. J Invest Dermatol. 2009 Jan;129(1):41-9.
Bracke S, Carretero M, Guerrero-Aspizua S, Desmet E, Illera N, Navarro M, Lambert J, Del Rio M. Targeted silencing of DEFB4 in a bioengineered skin-humanized mouse model for psoriasis: development of siRNA SECosome-based novel therapies. Exp Dermatol. 2014 Mar;23(3):199-201.
Guilloteau K, Paris I, Pedretti N, Boniface K, Juchaux F, Huguier V, Guillet G, Bernard FX, Lecron JC, Morel F. Skin Inflammation Induced by the Synergistic Action of IL-17A, IL-22, Oncostatin M, IL-1alpha, and TNFalpha Recapitulates Some Features of Psoriasis. J Immunol. 2010 Mar 24.
Jansen PA, Rodijk-Olthuis D, Hollox EJ, Kamsteeg M, Tjabringa GS, de Jongh GJ, van Vlijmen-Willems IM, Bergboer JG, van Rossum MM, de Jong EM, den Heijer M, Evers AW, Bergers M, Armour JA, Zeeuwen PL, Schalkwijk J. Beta-defensin-2 protein is a serum biomarker for disease activity in psoriasis and reaches biologically relevant concentrations in lesional skin. PLoS One. 2009;4(3):e4725.
Kamsteeg M, Jansen PA, van Vlijmen-Willems IM, van Erp PE, Rodijk-Olthuis D, van der Valk PG, Feuth T, Zeeuwen PL, Schalkwijk J. Molecular diagnostics of psoriasis, atopic dermatitis, allergic contact dermatitis and irritant contact dermatitis. Br J Dermatol. 2010 Mar;162(3):568-78.
Sestito R, Madonna S, Scarponi C, Cianfarani F, Failla CM, Cavani A, Girolomoni G, Albanesi C. STAT3-dependent effects of IL-22 in human keratinocytes are counterregulated by sirtuin 1 through a direct inhibition of STAT3 acetylation. FASEB J. 2011 Mar;25(3):916-27.
Teng X, Hu Z, Wei X, Wang Z, Guan T, Liu N, Liu X, Ye N, Deng G, Luo C, Huang N, Sun C, Xu M, Zhou X, Deng H, Edwards CK 3rd, Chen X, Wang X, Cui K, Wei Y, Li J. IL-37 ameliorates the inflammatory process in psoriasis by suppressing proinflammatory cytokine production. J Immunol. 2014 Feb 15;192(4):1815-23.
건선 & IL-17
건선은 유전, 환경 및 면역병리학적 요인에 의해 영향을 받는 다인성 발병기전을 갖는 만성 재발성 염증성 자가면역 피부 장애이다 (Griffiths CE et al., Lancet 2007;370:263-71). 건선은 점착성 은색 인편을 갖는 상승된 잘 구획화된 홍반성 타원형 플라크의 재발 에피소드를 특징으로 한다. 조직학적으로, 건선의 특징은 활성화된 T 세포, 호중구 및 수지상 세포에 의한 각질세포의 과다증식 및 피부 침윤에 의해 유발된 망상 돌기의 과장을 갖는 비후화된 핵형성 각질세포 층의 존재이다 (Schon MP N. Engl. J. Med. 352: 1899-1912).
증거의 축적물은 그의 서명 시토카인 IL-17 A, IL-17 F 및 IL-22에 의해 유도된 Th17-매개 질환으로서의 건선을 시사한다. IL-22는 각질세포의 증식을 유발하는 반면에, IL-17A는 호중구, 수지상 세포 및 선청성 림프성 세포를 포함한 추가의 염증 세포를 동원하는 케모카인 및 다른 염증유발 매개체를 분비하도록 각질세포를 자극한다 (Martin DA et al., J Invest Dermatol 2013; 133:17-26).
건선을 매개함에 있어서의 IL-17 경로의 임상 검증은 IL-17을 표적화하는 모노클로날 항체 요법을 사용하여 질환의 유의한 개선을 나타내는 성공적 Ph3 연구에 의해 증명된다 (Langley et al., NEJM 2014). 추가로, IL-17 억제 후 건선 병변에서의 전신 전사 프로파일링은 각질세포 및 백혈구 하위세트로부터의 다중 염증성 인자를 비-병변 피부에서 관찰된 것과 유사한 수준으로 억제하였다 (Russell et al., J Immunol 2014, 192: 3828-3836). 이와 함께, 이들 발견은 건선 발병기전을 매개함에 있어서의 IL-17의 역할을 지지한다.
방법 (생체외 피부 검정)
Th17 시토카인 칵테일을 사용하는 생체외 인간 피부 외식편에서의 피부-상주 면역 세포의 자극은 Th17 관련 시토카인 (IL-17A, IL-17F 및 IL-22)의 현저한 상향조절을 유발하며, 이는 이러한 시스템을 건선에 대한 인간 조직-기반 모델로서 확립한다. IL-17A, IL-17 F 및 IL-22의 발현을 조정하는 것에 대한 시험 화합물의 능력은 Th17 시토카인 칵테일로의 자극후 생체외 피부 배양 방법을 사용하여 평가하였다.
간략하게, 복부성형술 수술로부터 수득된 생체외 인간 피부를 프로세싱하여 지방을 제거하고, 조직을 ~750 마이크로미터로 피부분절화하였다. 이어서, 피부분절 피부를 각각 항생제/항진균제 용액을 함유하는 실온 PBS 중에서 5-10분의 2회 연속 린스로 세정하였다. 이어서, 피부 절편을 일회용 단일-사용 생검 펀치를 사용하여 10 mm 직경의 원형 절편으로 절단한 다음, 이를 각질화 배지를 사용하여 제조된 30 μl의 64% 소 콜라겐 용액 (오르가노제네시스, #200-055)을 함유하는 0.4 μm PCF 막 트랜스웰 (밀리셀 #PIHP01250)의 상부 챔버에 넣었다. 피부 샘플을 가습 챔버 내에서 37℃에서 30분 동안 콜라겐 용액에 정치되도록 하였다. 트랜스웰 상의 피부 샘플을 6-웰 플레이트 (웰당 1개의 샘플)로 옮기고, 하부 챔버를 1 ml 완전 배지 (각질화 배지)로 채웠다.
복부성형술 수술 후 제1 일에, 피부 외식편을 각질화 배지 중에서 배양하고, 37℃에서 밤새 인큐베이션되도록 하였다. 구체적으로, 인간 피부 외식편 (조건당 N=3)을 Th17 칵테일 (CD3, 1 μg/ml, CD28, 2 μg/ml, IL-1b, 10 ng/ml, IL-6, 5 ng/ml, TGFb, 1 ng/ml, IL-21, 10 ng/ml, 항-IL-4, 1 μg/ml 및 항-INFg, 1 μg/ml)로 자극하였다. 1, 3 및 10uM의 시험 화합물을 Th17 칵테일로서 동시에 첨가하였다. 조직을 Th17 활성화 후 24시간에 수확하고, RNA를 qPCR을 사용하여 전사체 정량화 (IL-17A, IL-17F,IL-22)를 위해 단리하였다.
전체 RNA를 퀴아젠 미니 RNA 단리 키트 (Cat # 74106)를 사용하여 ~40 mg의 조직으로부터 단리하였다. 간략하게, 조직을 2-분 휴지기를 갖는 10회 사이클 동안 6300 rpm에서 30초 동안 1% 2-베타-메르캅토-에탄올로 보충된 300 μl의 RLT 완충제을 사용하여 프리셀리스-24 기계에서 세절 및 균질화하였다. 10 μl 프로테이나제 K를 함유하는 490 μl의 물을 상기 균질물에 첨가하고, 55℃에서 15분 동안 소화시켰다. 소화된 조직을 10,000 G에서 3분 동안 회전시켜 세포 파편으로 펠릿화시키고, 상청액을 제조업체의 프로토콜에 따라 퀴아젠 RN이지 미니 칼럼을 사용하는 RNA 단리에 사용하였다. 전체 RNA를 나노드롭 2000을 사용하여 정량화하고, 애질런트 생물분석기 상에서 분석하였다 (첨부된 파일). 1.4 μg의 RNA를 인비트로젠 슈퍼스크립트 VILO cDNA 합성 키트 (# 11754-050)를 사용하여 20 μl PCR 부피로 주형으로서 사용하여 cDNA 주형을 생성하였다. 이어서, 정량화될 각각의 유전자에 대한 특이적 택맨 프로브로의 후속 qPCR을 위해, cDNA를 1:25 희석하였다. 관심 유전자의 상대 발현의 RNA 수준을 델타 델타 CT 공식을 사용하여 계산하였다.
등가물
본 발명은 특히 시르투인-조정 화합물 및 그의 방법 또는 용도를 제공한다. 본 발명의 구체적 실시양태가 논의되었지만, 상기 명세서는 예시적이며, 제한적인 것은 아니다. 본 발명의 많은 변형은 본 명세서의 검토 시에 관련 기술분야의 통상의 기술자에게 명백해질 것이다. 본 발명의 전체 범주는 청구범위를 그의 전체 등가물 범주와 함께 참조하고, 본 명세서를 이러한 변형과 함께 참조함으로써 결정되어야 한다.
Claims (18)
- 화학식 I의 화합물 또는 그의 제약상 염.
<화학식 I>
여기서
X1 또는 X2는 독립적으로 -N 또는 -C로부터 선택되고;
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
R4는 수소 또는 -C(O)NRbRc이고;
여기서
X2가 -N인 경우에, R2는 존재하지 않거나; 또는
X2가 -C인 경우에, R2는 상기 정의된 바와 같고;
상기 정의된 바와 같은 각각의 R1, R2, R3 및 R4는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, 또는 Rf는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
Rg는 -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬이고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, 또는 Rf는 수소, 할로겐, -OH, -C≡N, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, 카르보시클릴, -(CH2)x-카르보시클릴, -헤테로시클릴, -O-헤테로시클릴 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)-OH로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
m은 1 내지 3의 정수이고;
n은 1 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다. - 제1항에 있어서,
m은 1이고;
n은 3이고;
R4는 수소 또는 -C(O)NRbRc이고, 여기서 Rb 및 Rc는 상기 제1항에 정의된 바와 같은 것인
화학식 I의 화합물. - 제1항에 있어서, R1은 하기로부터 선택된 것인 화학식 I의 화합물.



- 제1항에 있어서, R1은 하기로부터 선택된 것인 화학식 I의 화합물.

- 제1항에 있어서, R4는 하기로부터 선택된 것인 화학식 I의 화합물.


- 제1항에 있어서, R4는 하기인 화학식 I의 화합물.
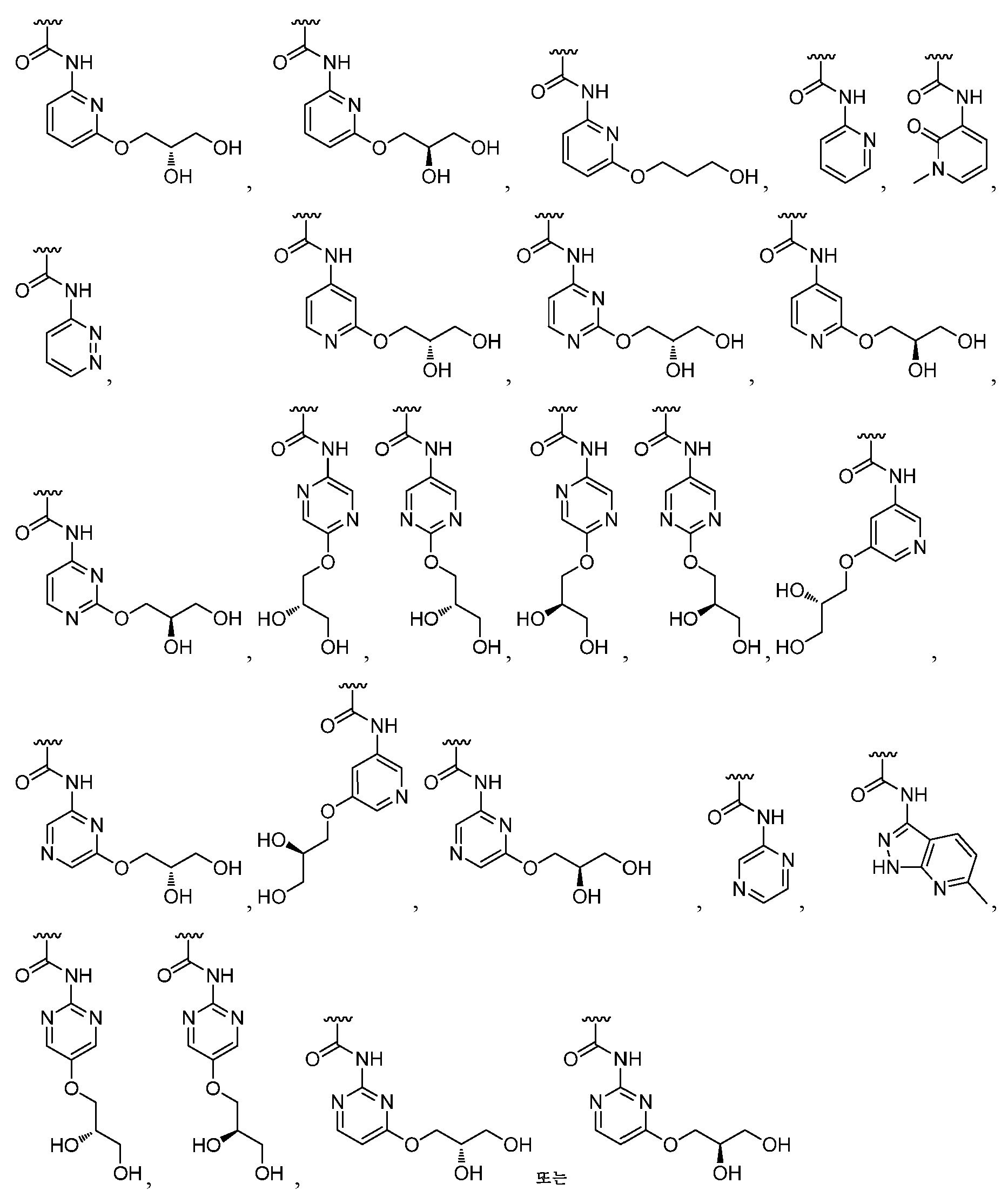
- 화학식 V의 화합물 또는 그의 제약상 염.
<화학식 V>

여기서
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
R4는 수소 또는 -C(O)-NRbRc이고;
여기서
상기 정의된 바와 같은 각각의 R1, R2, R3 또는 R4는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, 또는 Rf는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, 또는 Rf는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRgRh, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형-C1-C6 할로알콕시, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, -헤테로시클릴, -O-헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)ORi로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
여기서
상기 정의된 바와 같은 각각의 Rg, Rh 및 Ri는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬 또는 -직쇄형 또는 분지형-C1-C6 할로알킬로부터 선택되고;
m은 1 내지 3의 정수이고;
n은 2 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다. - 화학식 VI의 화합물 또는 그의 제약상 염.
<화학식 VI>

여기서
R1은 수소, 할로겐, -CN, 카르보시클릴, 헤테로시클릴, -N-치환된 헤테로시클릴, 아릴, 헤테로아릴, -C(O)Ra 또는 -C(O)-NRbRc이고;
R2는 할로겐, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, 또는 -C(O)-NRbRc이고;
R3은 수소, 할로겐, -히드록시, -직쇄형 또는 분지형 C1-C6 알킬, 또는 -직쇄형 또는 분지형-C1-C6 할로알킬이고;
각각의 R5 및 R6은 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6시클로알킬, -(CH2)xC1-C6시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴, -(CHRg)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 R1, R2, R3, R5 및 R6은 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRdRe, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형 C1-C6 할로알콕시, -O-직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), 또는 -C(O)ORf로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
상기 정의된 바와 같은 각각의 Ra, Rb, Rc, Rd, Re, Rf 또는 Rg는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -C1-C6-시클로알킬, -(CH2)xC1-C6-시클로알킬, 헤테로시클릴, -N-헤테로시클릴, 아릴, 헤테로아릴, 또는 -(CH2)x헤테로아릴로부터 선택되고;
여기서
상기 정의된 바와 같은 각각의 Ra, Rb, Rc Rd, Re, Rf 또는 Rg는 수소, 할로겐, -OH, -(CH2)xOH, -C≡N, -NRhRi, -직쇄형 또는 분지형 C1-C6 알킬, -직쇄형 또는 분지형-C1-C6 할로알킬, -직쇄형 또는 분지형 C1-C6 알콕시, -직쇄형 또는 분지형-C1-C6 할로알콕시, -C1-C6 시클로알킬, -(CH2)x- 시클로알킬, 헤테로시클릴, -헤테로시클릴, -O-헤테로시클릴, 아릴, -헤테로아릴, -(CH2)x-헤테로아릴, -O-(CH2)xCH(OH)CH2(OH), -(CH2)x-OH, 또는 -C(O)ORj로부터 선택된 1개 이상의 치환기로 임의로 추가로 치환되고;
여기서
상기 정의된 바와 같은 각각의 Rh, Ri 및 Rj는 독립적으로 수소, -직쇄형 또는 분지형 C1-C6 알킬 또는 -직쇄형 또는 분지형-C1-C6 할로알킬로부터 선택되고;
m은 1 내지 3의 정수이고;
n은 2 내지 3으로부터 선택된 정수이고;
x는 0, 또는 1 내지 6의 정수이다. - 표 I에 제시된 바와 같은 화합물 또는 그의 제약상 허용되는 염.
- 제1항 내지 제9항 중 어느 한 항의 화합물 및 제약상 허용되는 담체를 포함하는 제약 조성물.
- 제13항에 있어서, 추가의 활성제를 추가로 포함하는 제약 조성물.
- 인슐린 저항성, 대사 증후군, 대사 기능장애, 당뇨병 또는 그의 합병증의 치료 또는 인슐린 감수성의 증가를 필요로 하는 대상체에게 제1항 내지 제9항 중 어느 한 항에 따른 화합물 또는 제10항 또는 제11항에 따른 제약 조성물을 투여하는 것을 포함하는, 인슐린 저항성, 대사 증후군, 대사 기능장애, 당뇨병 또는 그의 합병증을 치료하거나 또는 인슐린 감수성을 증가시키는 방법.
- 감소된 SIRT1 발현 또는 활성으로부터 유발된 질환 또는 장애의 치료를 필요로 하는 대상체에게 제1항 내지 제9항 중 어느 한 항에 따른 화합물 또는 그의 제약상 허용되는 염 또는 제11항 또는 제12항에 따른 제약 조성물을 투여하는 것을 포함하는, 상기 질환 또는 장애를 치료하는 방법.
- 제13항에 있어서, 감소된 SIRT1 발현 또는 활성으로부터 유발된 질환 또는 장애가 노화 또는 스트레스, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환으로부터 선택되나, 이에 제한되지는 않는 것인 방법.
- 제14항에 있어서, 노화 또는 스트레스와 관련된 질환, 당뇨병, 대사 기능장애, 신경변성 질환, 심혈관 질환, 암 또는 염증성 질환이 건선, 아토피성 피부염, 여드름, 장미증, 염증성 장 질환, 골다공증, 패혈증, 관절염, COPD, 전신 홍반성 루푸스 및 안부 염증으로부터 선택된 것인 방법.
- 건선의 치료를 필요로 하는 대상체에게 제1항 내지 제9항 중 어느 한 항에 따른 화합물 또는 제11항 또는 제12항에 따른 제약 조성물을 투여하는 것을 포함하는, 건선을 치료하는 방법.
- 제1항 내지 제9항 중 어느 한 항에 있어서, 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증을 앓고 있거나 또는 그에 걸리기 쉬운 대상체의 치료에서의 또는 대상체에서 인슐린 감수성을 증가시키기 위한 요법에 사용하기 위한 화합물.
- 인슐린 저항성, 대사 증후군, 당뇨병 또는 그의 합병증의 치료에 사용하기 위한 또는 대상체에서 인슐린 감수성을 증가시키기 위한 의약의 제조에서의, 제1항 내지 제9항 중 어느 한 항에 정의된 바와 같은 화합물의 용도.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462081916P | 2014-11-19 | 2014-11-19 | |
| US62/081,916 | 2014-11-19 | ||
| PCT/IB2015/058980 WO2016079711A1 (en) | 2014-11-19 | 2015-11-19 | Substituted bridged urea analogs as sirtuin modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20170087907A true KR20170087907A (ko) | 2017-07-31 |
Family
ID=54705247
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020177016278A Withdrawn KR20170083134A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
| KR1020177016280A Withdrawn KR20170083135A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
| KR1020177016279A Withdrawn KR20170087907A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
| KR1020177016277A Withdrawn KR20170083133A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020177016278A Withdrawn KR20170083134A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
| KR1020177016280A Withdrawn KR20170083135A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020177016277A Withdrawn KR20170083133A (ko) | 2014-11-19 | 2015-11-19 | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 |
Country Status (21)
| Country | Link |
|---|---|
| US (4) | US10072011B2 (ko) |
| EP (4) | EP3221317B1 (ko) |
| JP (4) | JP6806679B2 (ko) |
| KR (4) | KR20170083134A (ko) |
| CN (4) | CN107207521A (ko) |
| AU (4) | AU2015348941A1 (ko) |
| BR (4) | BR112017010595A2 (ko) |
| CA (4) | CA2968029A1 (ko) |
| CL (1) | CL2017001275A1 (ko) |
| CO (1) | CO2017004994A2 (ko) |
| CR (1) | CR20170209A (ko) |
| DO (1) | DOP2017000123A (ko) |
| EA (1) | EA201791074A1 (ko) |
| ES (1) | ES2823748T3 (ko) |
| IL (1) | IL252252A0 (ko) |
| MX (1) | MX2017006658A (ko) |
| PE (1) | PE20171257A1 (ko) |
| PH (1) | PH12017500913A1 (ko) |
| RU (3) | RU2017120858A (ko) |
| SG (1) | SG11201703824QA (ko) |
| WO (4) | WO2016079709A1 (ko) |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016086088A2 (en) | 2014-11-25 | 2016-06-02 | Northwestern University | Wound healing through sirt1 overexpression |
| CN108329388A (zh) * | 2018-01-18 | 2018-07-27 | 天津市湖滨盘古基因科学发展有限公司 | 一种人的沉默配型信息调节蛋白突变蛋白及其应用 |
| CN108395436B (zh) * | 2018-05-14 | 2021-01-12 | 鹤壁市人民医院 | 一种咪唑并吡嗪医药中间体的制备方法 |
| CN108707150B (zh) * | 2018-05-14 | 2021-04-16 | 江苏惠利生物科技有限公司 | 一种咪唑并吡嗪医药中间体的制备方法 |
| CN110478339A (zh) * | 2019-08-12 | 2019-11-22 | 昆明理工大学 | 紫铆因在制备靶向恢复突变p53构象药物中的应用 |
| KR20220056223A (ko) | 2019-09-02 | 2022-05-04 | 메르크 파텐트 게엠베하 | 유기 전계발광 디바이스용 재료 |
| WO2021100059A1 (en) * | 2019-11-19 | 2021-05-27 | Lupin Limited | Process for preparing chroman compounds |
| PT4090654T (pt) * | 2020-01-17 | 2025-10-07 | Lupin Ltd | Métodos, processos e intermediários para a preparação de compostos de cromano |
| CN114890955A (zh) * | 2022-06-23 | 2022-08-12 | 江西瑞威尔生物科技有限公司 | 一种n取代吗啉类有机化合物的制备工艺 |
| WO2025194094A1 (en) * | 2024-03-15 | 2025-09-18 | The Regents Of The University Of Michigan | Small molecule modulators of sirt5 and uses thereof |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4727064A (en) | 1984-04-25 | 1988-02-23 | The United States Of America As Represented By The Department Of Health And Human Services | Pharmaceutical preparations containing cyclodextrin derivatives |
| US4683195A (en) | 1986-01-30 | 1987-07-28 | Cetus Corporation | Process for amplifying, detecting, and/or-cloning nucleic acid sequences |
| WO2006094210A2 (en) * | 2005-03-03 | 2006-09-08 | Sirtris Pharmaceuticals, Inc. | Tetrahydroquinoxalinone sirtuin modulators |
| UA108596C2 (xx) | 2007-11-09 | 2015-05-25 | Інгібітори пептиддеформілази | |
| US9556201B2 (en) * | 2009-10-29 | 2017-01-31 | Glaxosmithkline Llc | Bicyclic pyridines and analogs as sirtuin modulators |
| AU2012352442A1 (en) * | 2011-12-12 | 2014-07-03 | Smb Innovation Aps | Novel heterocyclic compounds useful in sirtuin binding and modulation |
| CN105392788B (zh) * | 2013-05-13 | 2018-10-23 | 葛兰素史密斯克莱有限责任公司 | 用作沉默调节蛋白调节剂的取代的桥连脲类似物 |
-
2015
- 2015-11-19 JP JP2017526852A patent/JP6806679B2/ja not_active Expired - Fee Related
- 2015-11-19 CN CN201580073782.4A patent/CN107207521A/zh active Pending
- 2015-11-19 BR BR112017010595-0A patent/BR112017010595A2/pt not_active Application Discontinuation
- 2015-11-19 CA CA2968029A patent/CA2968029A1/en not_active Abandoned
- 2015-11-19 KR KR1020177016278A patent/KR20170083134A/ko not_active Withdrawn
- 2015-11-19 EP EP15801271.6A patent/EP3221317B1/en active Active
- 2015-11-19 EP EP15804601.1A patent/EP3220918A1/en not_active Withdrawn
- 2015-11-19 WO PCT/IB2015/058977 patent/WO2016079709A1/en not_active Ceased
- 2015-11-19 AU AU2015348941A patent/AU2015348941A1/en not_active Abandoned
- 2015-11-19 JP JP2017526965A patent/JP2017534665A/ja active Pending
- 2015-11-19 RU RU2017120858A patent/RU2017120858A/ru unknown
- 2015-11-19 CN CN201580073769.9A patent/CN107207520A/zh active Pending
- 2015-11-19 US US15/527,581 patent/US10072011B2/en not_active Expired - Fee Related
- 2015-11-19 WO PCT/IB2015/058980 patent/WO2016079711A1/en not_active Ceased
- 2015-11-19 EP EP15816522.5A patent/EP3221322A1/en not_active Withdrawn
- 2015-11-19 CR CR20170209A patent/CR20170209A/es unknown
- 2015-11-19 KR KR1020177016280A patent/KR20170083135A/ko not_active Withdrawn
- 2015-11-19 KR KR1020177016279A patent/KR20170087907A/ko not_active Withdrawn
- 2015-11-19 CN CN201580073311.3A patent/CN107108627A/zh active Pending
- 2015-11-19 US US15/527,941 patent/US20180319810A1/en not_active Abandoned
- 2015-11-19 MX MX2017006658A patent/MX2017006658A/es unknown
- 2015-11-19 AU AU2015348944A patent/AU2015348944A1/en not_active Abandoned
- 2015-11-19 RU RU2017120855A patent/RU2017120855A/ru unknown
- 2015-11-19 ES ES15801271T patent/ES2823748T3/es active Active
- 2015-11-19 US US15/527,851 patent/US20190048016A1/en not_active Abandoned
- 2015-11-19 WO PCT/IB2015/058981 patent/WO2016079712A1/en not_active Ceased
- 2015-11-19 KR KR1020177016277A patent/KR20170083133A/ko not_active Withdrawn
- 2015-11-19 JP JP2017526950A patent/JP2017534664A/ja active Pending
- 2015-11-19 BR BR112017010599A patent/BR112017010599A2/pt not_active Application Discontinuation
- 2015-11-19 PE PE2017000883A patent/PE20171257A1/es not_active Application Discontinuation
- 2015-11-19 AU AU2015348942A patent/AU2015348942B2/en not_active Ceased
- 2015-11-19 US US15/527,440 patent/US20170355705A1/en not_active Abandoned
- 2015-11-19 SG SG11201703824QA patent/SG11201703824QA/en unknown
- 2015-11-19 JP JP2017526875A patent/JP2017534663A/ja not_active Withdrawn
- 2015-11-19 CA CA2968027A patent/CA2968027A1/en not_active Abandoned
- 2015-11-19 AU AU2015348943A patent/AU2015348943A1/en not_active Abandoned
- 2015-11-19 CA CA2968030A patent/CA2968030A1/en not_active Abandoned
- 2015-11-19 EP EP15801270.8A patent/EP3221316A1/en not_active Withdrawn
- 2015-11-19 RU RU2017120857A patent/RU2017120857A/ru unknown
- 2015-11-19 WO PCT/IB2015/058978 patent/WO2016079710A1/en not_active Ceased
- 2015-11-19 EA EA201791074A patent/EA201791074A1/ru unknown
- 2015-11-19 CN CN201580073897.3A patent/CN107207509B/zh not_active Expired - Fee Related
- 2015-11-19 BR BR112017010601A patent/BR112017010601A2/pt not_active Application Discontinuation
- 2015-11-19 CA CA2968032A patent/CA2968032A1/en not_active Abandoned
- 2015-11-19 BR BR112017010602A patent/BR112017010602A2/pt not_active Application Discontinuation
-
2017
- 2017-05-11 IL IL252252A patent/IL252252A0/en unknown
- 2017-05-17 PH PH12017500913A patent/PH12017500913A1/en unknown
- 2017-05-18 DO DO2017000123A patent/DOP2017000123A/es unknown
- 2017-05-18 CL CL2017001275A patent/CL2017001275A1/es unknown
- 2017-05-19 CO CONC2017/0004994A patent/CO2017004994A2/es unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20170087907A (ko) | 시르투인 조정제로서의 치환된 가교된 우레아 유사체 | |
| US10590135B2 (en) | Substituted bridged urea analogs as sirtuin modulators | |
| KR20140077965A (ko) | 시르투인 조절제로서의 치환된 비시클릭 아자-헤테로사이클 및 유사체 | |
| WO2016081692A2 (en) | Substituted bridged urea analogs as sirtuin modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20170614 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| PC1203 | Withdrawal of no request for examination |