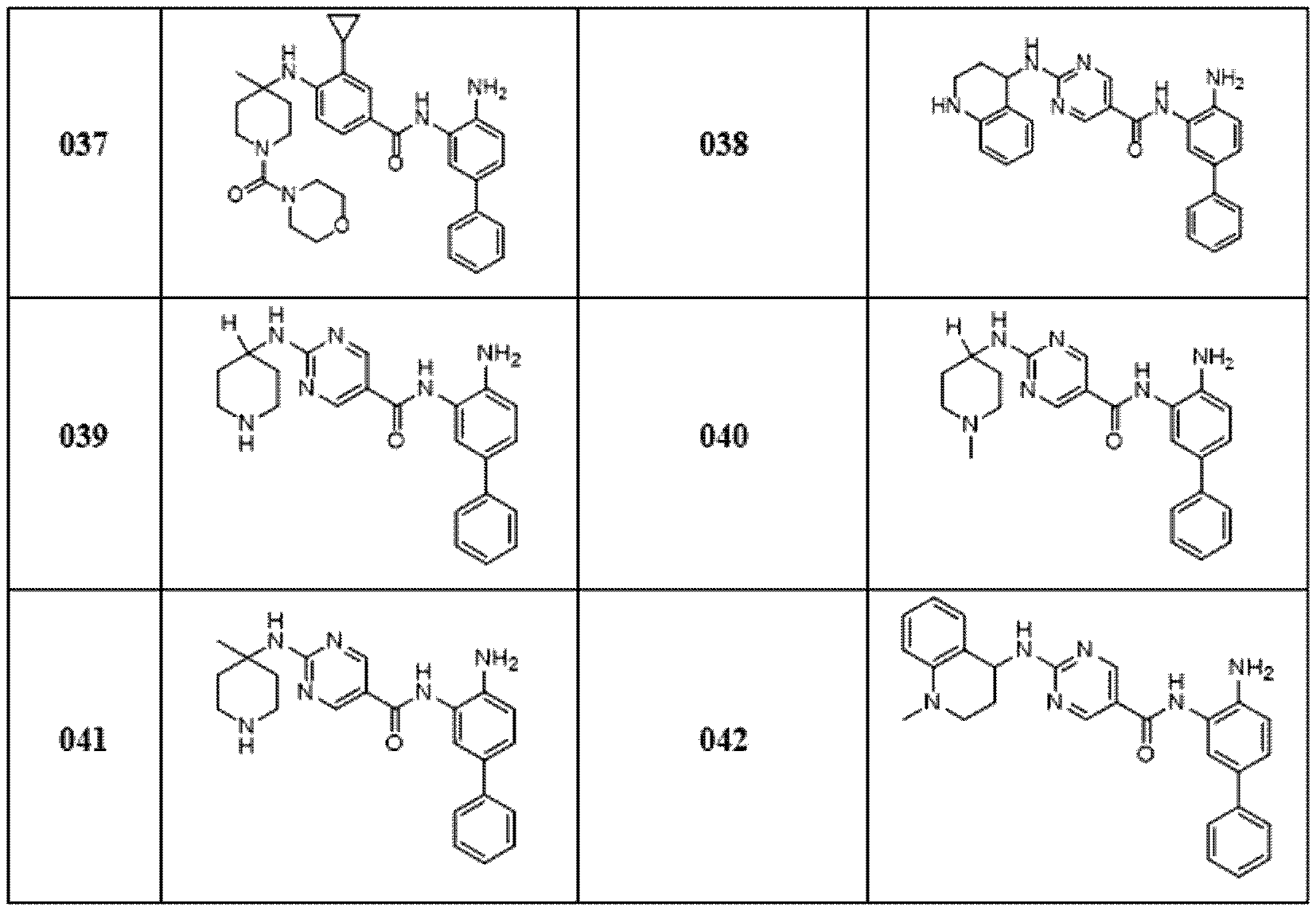
KR20170095964A - Hdac1/2 억제제로서 피페리딘 유도체 - Google Patents
Hdac1/2 억제제로서 피페리딘 유도체 Download PDFInfo
- Publication number
- KR20170095964A KR20170095964A KR1020177019370A KR20177019370A KR20170095964A KR 20170095964 A KR20170095964 A KR 20170095964A KR 1020177019370 A KR1020177019370 A KR 1020177019370A KR 20177019370 A KR20177019370 A KR 20177019370A KR 20170095964 A KR20170095964 A KR 20170095964A
- Authority
- KR
- South Korea
- Prior art keywords
- compound
- mmol
- mixture
- alkyl
- stirred
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Withdrawn
Links
Images






Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Diabetes (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
화합물, 이러한 화합물을 포함하는 약제학적 조성물, HDAC1 및/또는 HDAC2 활성과 연관된 질환 또는 장애를 치료하기 위해 이러한 화합물을 사용하는 방법이 본원에 제공된다.
Description
[관련 출원]
본 출원은 그 전체가 참조로서 본원에 포함되는, 2014년 12월 12일자로 출원된 미국 가특허 출원 제62/091,221호, 및 2015년 10월 8일자로 출원된 미국 가특허 출원 제62/238,931호의 우선권을 주장한다.
[기술분야]
본 발명은 화합물, 이러한 화합물을 포함하는 약제학적 조성물, HDAC1 및/또는 HDAC2 활성과 연관된 질환 또는 장애를 치료하기 위해 이러한 화합물을 사용하는 방법에 관한 것이다.
현재 관심있는 생물학적 표적은 히스톤 디아세틸라제 (HDAC)이다 (예를 들어, 암의 치료를 위한 히스톤 디아세틸라제의 억제제의 사용에 관한 논의: Marks et al. Nature Reviews Cancer 2001, 7,194; Johnstone et al. Nature Reviews Drug Discovery 2002, 287 참조). 리신 잔기의 아세틸화 및 탈아세틸화를 통한 단백질의 번역-후 변형은 이들의 세포 기능을 조절하는데 결정적인 역할을 한다. HDAC는 히스톤 단백질 및 다른 전사 조절자의 N-아세틸-리신 잔기의 탈아세틸화를 통해 유전자 발현을 조절하는 아연 히드롤라제이다 (Hassig et al. Curr . Opin . Chem. Biol. 1997, 1, 300-308). HDAC는 세포 모양 및 분화를 제어하는 세포 경로에 참여하고, HDAC 억제제는 다른 난치성 암을 치료하는데 효과적인 것으로 나타난바 있다 (Warrell et al. J. Natl. Cancer Inst. 1998, 90, 1621-1625).
Zn을 보조인자로 사용하는, 11종의 인간 HDACs가 동정되어 있고 (Taunton et al. Science 1996, 272, 408-411; Yang et al. J. Biol. Chem. 1997, 272, 28001- 28007. Grozinger et al. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 4868-4873; Kao et al. Genes Dev. 2000, 14, 55-66. Hu et al. J. Biol. Chem. 2000, 275, 15254-15264; Zhou et al. Proc. Natl. Acad. Sci U.S.A. 2001, 98, 10572-10577; Venter et al. Science 2001, 291, 1304-1351) 이들 멤버는 이들의 효모 상동체 (orthologues)에 대한 서열 상동성을 기초로 3개 부류 (클래스 I, II, 및 IV)로 구분된다 (O. Witt et al.Cancer Letters, 2009, 277, 8-21). 클래스 I HDAC는 HDAC1, HDAC2, HDAC3, 및 HDAC8을 포함하고, "고전적 (classical)" HDAC로 지칭되며, 이는 그의 염기에 Zn2+ 이온을 갖는 촉매 포켓을 내포한다.
구조적으로 다양한 HDAC 억제제, 특히 HDAC의 특정 부류 및 개별적 HDAC의 강력하고/하거나 선택적인 억제제인 것들을 제조할 필요가 여전히 존재한다.
발명의 개요
화합물, 이러한 화합물을 포함하는 약제학적 조성물, HDAC와 연관된 질환 또는 장애, 특히 HDAC1 및/또는 HDAC2 발현을 포함하는 질환 또는 장애를 치료하기 위해 이러한 화합물을 사용하는 방법이 본원에 제공된다. HDAC1 및/또는 HDAC2 발현을 포함하는 질환에는, 이로 제한되는 것은 아니지만, 다양한 유형의 암 및 이상혈색소, 예컨대, 겸상적혈구성 빈혈 및 베타-지중해성 빈혈이 포함된다.
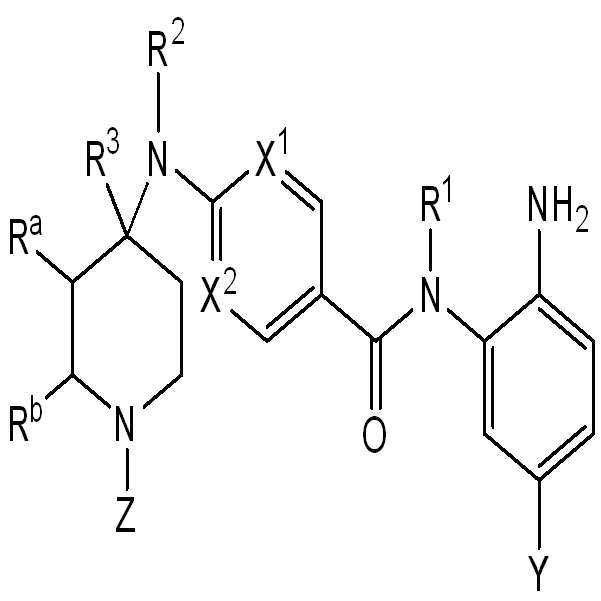
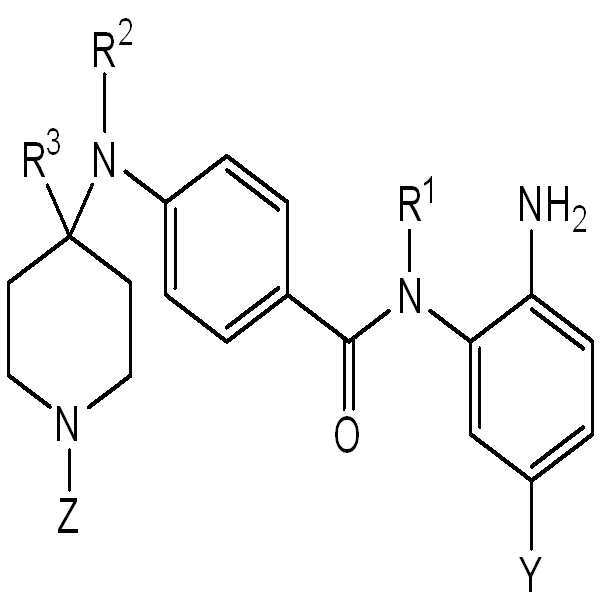
따라서, 일 측면에서, 화학식 I의 화합물 또는 이의 약제학적으로 허용가능한 염이 본원에 제공된다:

I
실시양태에서, 화학식 II의 화합물 또는 이의 약제학적으로 허용가능한 염이 본원에 제공된다:

II
특정 실시양태에서, 화학식 III의 화합물 또는 이의 약제학적으로 허용가능한 염이 본원에 제공된다:

III
다른 실시양태에서, 표 1의 화합물 또는 이의 약제학적으로 허용가능한 염이 본원에 제공된다.
다른 측면에서, 약제학적으로 허용가능한 담체와 함께, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 포함하는 약제학적 조성물이 본원에 제공된다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2의 활성을 억제하는 방법이 본원에 제공된다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 개체에 투여하는 것을 포함하는, 개체에서 다른 HDACs에 비해 HDAC1 및/또는 HDAC2 각각의 활성을 선택적으로 억제하는 방법이 본원에 제공된다. 일부 실시양태에서, 화합물은 HDAC1 및/또는 HDAC2 각각에 대해서 다른 HDACs에 비해 5 내지 1000배 더 높은 선택성을 갖는다. 다른 실시양태에서, 화합물은 HDAC 효소 어세이에서 시험될 때 HDAC1 및/또는 HDAC2 각각에 대해서 다른 HDACs에 비해 5 내지 1000배 더 높은 선택성을 갖는다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 이를 필요로 하는 개체에 투여하는 것을 포함하는, 개체에서 하나 이상의 HDACs에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다. 일부 실시양태에서, 질환은 HDAC1 및 HDAC2에 의해 매개된다. 다른 실시양태에서, 질환은 HDAC1에 의해 매개된다. 또 다른 실시양태에서, 질환은 HDAC2에 의해 매개된다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 개체에 투여하는 것을 포함하는, 개체에서 질환을 치료하는 방법이 본원에 제공된다. 실시양태에서, 질환은 골수형성이상 증후군이다. 실시양태에서, 질환은 이상혈색소이다. 다른 실시양태에서, 질환은 겸상적혈구병이다. 또 다른 실시양태에서, 질환은 베타-지중해성 빈혈이다.
추가의 실시양태에서, 질환은 암이다. 암은 폐암, 결장암, 유방암, 신경모세포종, 백혈병, 또는 림프종으로부터 선택될 수 있다. 다른 추가의 실시양태에서, 암은 신경모세포종이다. 백혈병은 급성 골수성 백혈병 또는 급성 거대핵세포성 백혈병일 수 있다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 이를 필요로 하는 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 급성 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
본원에 기술된 치료 방법의 추가의 실시양태에서, 치료되는 개체는 인간이다.
또 다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염의 치료학적 유효량을 이를 필요로 하는 개체에 투여하는 것을 포함하는, GATA 결합 단백질 2 (Gata2) 결핍과 연관된 질환 또는 장애를 치료하는 방법이 본원에 제공된다.
여전히 다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염과 세포를 접촉시키는 것을 포함하는, 세포에서 GATA 결합 단백질 2 (Gata2) 발현을 증가시키는 방법이 본원에 제공된다.
추가의 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 개체에 투여하는 것을 포함하는, 개체에서 HbG (감마 글로빈) 발현을 유도하는 방법이 본원에 제공된다.
본 출원은, 일반적으로, 화합물, 이러한 화합물을 포함하는 약제학적 조성물, 및 HDAC 활성과 연관된 질환 또는 장애, 특히 임의의 유형의 HDAC1 및/또는HDAC2 발현을 포함하는 질환 또는 장애를 치료하기 위해 이러한 화합물을 사용하는 방법에 관한 것이다.
정의
본 발명을 기술하는데 사용된 다양한 용어의 정의가 하기에 열거된다. 이들 정의는, 특별한 경우로 달리 제한되지 않는 한, 개별적으로 또는 더 큰 그룹의 일부로서, 본 명세서 및 청구범위 전체에 걸쳐서 사용되는 바와 같은 용어에 적용된다.
용어 "약"은 일반적으로 값의 10%, 5%, 또는 1% 이하의 가능한 변형을 지시한다. 예를 들어, "약 25 mg/kg"은 일반적으로, 그의 가장 넓은 의미에서, 22.5 - 27.5 mg/kg의 값, 즉 25 ± 2.5 mg/kg을 나타낼 것이다.
용어 "알킬"은, 특정 실시양태에서, 각각 1개 내지 6개, 또는 1개 내지 8개 사이 탄소 원자를 함유하는 포화된, 직쇄 또는 분지쇄 탄화수소 모이어티를 지칭한다. 알킬 치환기에서 탄소 원자의 수는 접두사 "Cx-Cy" (여기서 x는 치환기 내 탄소 원자의 최소 수이고 y는 이의 최대 수임)로 표시낼 수 있다. 유사하게, Cx 쇄 서술은 x 탄소 원자를 함유하는 기 (즉, 헤테로원자의 수는 포함하지 않음)를 나타낸다. C1-C6-알킬 모이어티의 예시에는, 이로 제한되는 것은 아니지만, 메틸, 에틸, 프로필, 이소프로필, n-부틸, tert-부틸, 네오펜틸, n-헥실 모이어티가 포함되고; C1-C8-알킬 모이어티의 예시에는, 이로 제한되는 것은 아니지만, 메틸, 에틸, 프로필, 이소프로필, n-부틸, tert-부틸, 네오펜틸, n-헥실, 헵틸, 및 옥틸 모이어티가 포함된다.
용어 "알콕시"는 -O-알킬 모이어티를 지칭한다. C1-C6-알콕시의 비-제한적인 예시에는 메톡시, 에톡시, 1-프로폭시, 2-프로폭시, n-부톡시, t-부톡시, 펜톡시, 헥속시 등이 포함된다. 알콕시의 알킬 부분은 직쇄 또는 분지쇄일 수 있다.
용어 "아릴"은, 이로 제한되는 것은 아니지만, 페닐 (즉, C6-아릴), 나프틸, 테트라히드로나프틸, 인다닐, 이데닐 등을 포함하는, 융합 또는 비-융합된, 하나 이상의 방향족 고리를 갖는 단환 또는 다환의 탄소환식 고리를 지칭한다. 일부 실시양태에서, 아릴 기는 6개 탄소 원자 (예컨대, C6-아릴)를 갖는다. 일부 실시양태에서, 아릴 기는 6개 내지 10개 탄소 원자 (예컨대, C6-C10-아릴)를 갖는다. 일부 실시양태에서, 아릴 기는 6개 내지 16개 탄소 원자를 갖는다.
용어 "사이클로알킬"은 단환 또는 다환의 포화된 또는 부분적으로 불포화된 탄소환식 고리 화합물을 지칭한다. C3-C6-사이클로알킬의 예시에는, 이로 제한되는 것은 아니지만, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 및 사이클로헥실이 포함되고; C3-C8-사이클로알킬에는, 이로 제한되는 것은 아니지만, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 사이클로펜틸 및 사이클로옥틸이 포함되고; C3-C12-사이클로알킬의 예시에는, 이로 제한되는 것은 아니지만, 사이클로프로필, 사이클로부틸, 사이클로펜틸, 사이클로헥실, 비사이클로[2.2.1]헵틸, 및 비사이클로[2.2.2]옥틸이 포함된다. 단일 수소 원자의 제거에 의해 탄소-탄소 이중 결합을 갖는 단환 또는 다환의 탄소환식 고리 화합물로부터 유래된 일가 기가 또한 고려된다. 이러한 기의 예시에는, 이로 제한되는 것은 아니지만, 사이클로프로페닐, 사이클로부테닐, 사이클로펜테닐, 사이클로헥세닐, 사이클로헵테닐, 사이클로옥테닐 등이 포함된다.
용어 "헤테로아릴"은 적어도 하나의 방향족 고리를 갖는 단환 또는 다환의 (예컨대, 이환 또는 삼환 또는 그 이상의) 융합 또는 비-융합된 모이어티 또는 고리 시스템을 지칭하고, 여기서 고리-형성 원자의 하나 이상은 산소, 황, 또는 질소와 같은 헤테로원자이다. 일부 실시양태에서, 헤테로아릴 기는 1개 내지 8개 탄소 원자, 1개 내지 6개 탄소 원자, 2개 내지 6개 탄소 원자 (예컨대, C1-C8-헤테로아릴, C1-C6-헤테로아릴, 또는 C2-C6-헤테로아릴)를 갖는다. 추가의 실시양태에서, 헤테로아릴 기는 1개 내지 15개 탄소 원자를 갖는다. 일부 실시양태에서, 헤테로아릴 기는 그의 하나의 고리 원자가 산소, 황 및 질소로부터 선택되는 5개 내지 16개 고리 원자를 함유하고; 0, 1, 2, 또는 3개 고리 원자가 산소, 황 및 질소로부터 독립적으로 선택된 추가의 헤테로원자이고; 잔여 고리 원자는 탄소이다. 헤테로아릴에는, 이로 제한되는 것은 아니지만, 피리디닐, 피라지닐, 피리미디닐, 피롤릴, 피라졸릴, 이미다졸릴, 티아졸릴, 옥사졸릴, 이소옥사졸릴, 티아졸릴, 티아디아졸릴, 옥사디아졸릴, 티오페닐, 퓨라닐, 인돌릴, 퀴놀리닐, 이소퀴놀리닐, 벤즈이미다졸릴, 벤조옥사졸릴, 퀸옥살리닐, 아크리디닐 등이 포함된다.
용어 "헤테로사이클릴"은 비-방향족 3-, 4-, 5-, 6- 또는 7-원 고리 또는 이환기 또는 삼환기 융합 또는 비-융합 시스템을 지칭하는데, 여기서 (i) 각 고리는 산소, 황 및 질소로부터 독립적으로 선택된 1개 내지 3개 사이의 헤테로원자를 함유하고 잔여 원자는 탄소이고 (예컨대, C2-C6-헤테로사이클릴, C3-C6-헤테로사이클릴,또는 C3-C5-헤테로사이클릴), (ii) 각 5-원 고리는 0 내지 1개 이중 결합을 갖고 각 6-원 고리는 0 내지 2개 이중 결합을 가지며, (iii) 질소 및 황 헤테로원자는 선택적으로 산화될 수 있고, (iv) 질소 헤테로원자는 선택적으로 사차화될 수 있으며, (iv) 임의의 상기 고리는 벤젠 고리에 융합될 수 있다. 용어 "헤테로사이클릴"에는, 이로 제한되는 것은 아니지만, [1,3]디옥솔란, 피롤리디닐, 피라졸리닐, 피라졸리디닐, 이미다졸리닐, 이미다졸리디닐, 피페리디닐, 피페라지닐, 옥사졸리디닐, 이속사졸리디닐, 모르폴리닐, 티아졸리디닐, 이소티아졸리디닐, 및 테트라히드로퓨릴이 포함된다.
용어 "할로" 및 "할로겐"은 불소, 염소, 브롬 및 요오드로부터 선택되는 원자를 지칭한다.
용어 "할로알킬"은 임의의 하나 이상의 알킬 탄소 원자가 상기에 정의된 바와 같은 할로로 치환된 알킬 라디칼을 지칭한다. 할로알킬은 모노할로알킬, 디할로알킬, 및 폴리할로알킬 라디칼을 포함한다. 용어 "할로알킬"에는, 이로 제한되는 것은 아니지만, 플루오로메틸, 디플루오로메틸, 트리플루오로메틸, 클로로메틸, 디클로로메틸, 트리클로로메틸, 및 펜타플루오로에틸이 포함된다.
용어 "히드록시"는 -OH 라디칼을 지칭한다.
용어 "HDAC"는 히스톤 디아세틸라제를 지칭하는데, 이는 코어 히스톤 내 리신 잔기로부터 아세틸기를 제거하고, 그로써 응축되고 전사적으로 침묵화된 (transcriptionally silenced) 크로마틴의 형성을 초래하는 효소이다. 현재 18종의 공지된 히스톤 디아세틸라제가 있는데, 이들은 4개 그룹으로 분류된다. HDAC1, HDAC2, HDAC3, 및 HDAC8을 포함하는 클래스 I HDAC는 효모 RPD3 유전자와 관련된다. HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, 및 HDAC10을 포함하는 클래스 II HDAC는 효모 Hda1 유전자와 관련된다. 서투인 (sirtuins)으로도 알려진 클래스 III HDAC는 Sir2 유전자와 관련되며 SIRT1-7을 포함한다. 단지 HDAC11만을 포함하는 클래스 IV HDAC는 클래스 I 및 II HDACs 둘 다의 특징을 갖는다. 용어 "HDAC"는, 달리 특정되지 않는 한, 공지된 18종의 히스톤 디아세틸라제의 임의의 하나 이상을 지칭한다.
용어 "억제제"는 용어 길항제와 동의어이다.
용어 "약제학적으로 허용가능한 염"은 적절한 의학적 판단의 범주 내에서, 과도한 독성, 자극, 알러지 반응 등이 없이 인간 및 하등 동물의 조직과 접촉 사용하기에 적합한, 본 발명의 방법에 의해 형성된 화합물의 그러한 염을 지칭하고, 합리적 이익/위험 비율에 상응한다. 또한, "약제학적으로 허용가능한 염"은 모체 화합물이 기존의 산 또는 염기 모이어티를 그의 염 형태로 전환시킴으로써 변형된, 기술된 화합물의 유도체를 지칭한다. 약제학적으로 허용가능한 염의 예시에는, 이로 제한되는 것은 아니지만, 아민과 같은 염기성 잔기의 무기질 또는 유기산; 카르복실산과 같은 산성 잔기의 알칼리 또는 유기산; 등이 포함된다. 본 발명의 약제학적으로 허용가능한 염은, 예를 들어, 비-독성 무기산 또는 유기산으로부터 형성된 모체 화합물의 통상적인 비-독성 염을 포함한다. 본 발명의 약제학적으로 허용가능한 염은 통상적인 화학적 방법에 의해 염기성 또는 산성 모이어티를 함유하는, 모체 화합물로부터 합성될 수 있다. 일반적으로, 이러한 염은 이들 화합물의 유리산 또는 염기 형태를 물 중 또는 유기 용매 중, 또는 이 둘의 혼합물 중에서 적합한 염기 또는 산의 화학량론적 양과 반응시켜 제조될 수 있고; 일반적으로, 에테르, 에틸 아세테이트, 에탄올, 이소프로판올, 또는 아세토니트릴과 같은 비수성 매질이 바람직하다. 적합한 염의 목록이, 그 전체가 참조로서 본원에 포함되는, 문헌 [Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, Pa., 1985, p. 1418 and Journal of Pharmaceutical Science, 66, 2 (1977)]에서 확인된다.
본 발명에 의해 구상되는 치환기 및 변수의 조합은 오로지 안정한 화합물의 형성을 초래하는 것들이다. 용어 "안정한"은 제조를 허용하기에 충분한 안정성을 보유하고 본원에 기술된 목적 (예컨대, 개체에의 치료적 또는 예방적 투여)을 위해 유용하기에 충분한 시간 동안에 화합물의 온전성 (integrity)을 유지하는 화합물을 지칭한다.
용어 "개체"는 포유동물을 지칭한다. 따라서, 개체는, 예를 들어, 개, 고양이, 말, 소, 돼지, 기니아 피그 등을 지칭한다. 바람직하게는, 개체는 인간이다. 개체가 인간인 경우, 그러면 개체는 본원에서 환자로 언급될 수 있다.
본 발명의 화합물
일 측면에서, 화학식 I의 화합물 또는 이의 약제학적으로 허용가능한 염이 본원에 제공된다:
I
상기 식에서,
X1은 CR7 또는 N이고;
X2는 CH 또는 N이고;
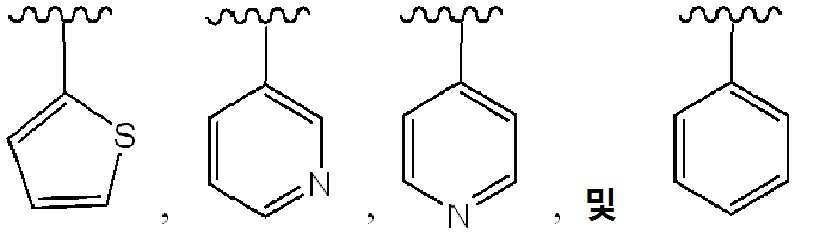
Y는 하기로 구성된 군으로부터 선택되고:
Z는 H, C1-C6-알킬, C6-아릴, C(O)NR4R5, C(O)OR6, C(O)C1-C6-알킬, C(O)C0-C6-알킬-C6-아릴, C(O)-C3-C6-사이클로알킬, C(O)-C2-C6-헤테로사이클릴, 및 C(O)C0-6-알킬-헤테로아릴로 구성된 군으로부터 선택되고, 여기서 아릴, 헤테로아릴, 사이클로알킬, 및 헤테로사이클릴 기는 C1-C6-알킬, 할로, C1-C6-할로알킬, 히드록시, 또는 C1-C6-알콕시의 1개 또는 2개로 선택적으로 치환되고;
Ra 및 Rb는 H이거나, 또는 Ra 및 Rb는 함께 융합된 C6-아릴을 형성하고;
R1은 H 및 C1-C6-알킬로 구성된 군으로부터 선택되고;
R2는 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택되고;
R3은 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택되거나;
또는 R2 및 R3은 함께 C3-C6-헤테로사이클릴을 형성하고;
R4는 H, C1-C6-알킬, C1-C6-알킬-OH, 및 C1-C6-NH2로 구성된 군으로부터 선택되고;
R5는 C1-C6-알킬이거나;
또는 R4 및 R5는 함께 C2-C6-헤테로사이클릴을 형성하고, 여기서 헤테로사이클릴은 C1-C6-알킬, 할로, C1-C6-할로알킬, 히드록시, 또는 C1-C6-알콕시의 1개 또는 2개로 선택적으로 치환되고;
R6은 C1-C6-알킬 및 C0-C6-알킬-C6-아릴로 구성된 군으로부터 선택되고, 여기서 아릴은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되고;
R7은 H, C1-C6-알킬, 및 C3-C6-사이클로알킬로 구성된 군으로부터 선택된다.
화학식 I의 화합물의 실시양태에서, Ra 및 Rb는 H이고 R3은 H 및 C6-아릴로 구성된 군으로부터 선택된다.
화학식 I의 화합물의 실시양태에서, Y는 하기로 구성된 군으로부터 선택된다:

다른 화학식 I의 화합물의 실시양태에서, Z는 H, C1-C6-알킬, C6-아릴, C(O)OR6, C(O)C1-C6-알킬, C(O)C0-C6-알킬-C6-아릴, C(O)-C3-C6-사이클로알킬, 및 C(O)C0-6-알킬-헤테로아릴로 구성된 군으로부터 선택되고, 여기서 아릴, 헤테로아릴, 및 사이클로알킬 기는 C1-C6-알킬, 할로, C1-C6-할로알킬, 히드록시, 또는 C1-C6-알콕시의 1개 또는 2개로 선택적으로 치환된다.
실시양태에서, 화학식 I의 화합물은 화학식 II의 화합물 또는 이의 약제학적으로 허용가능한 염이다:
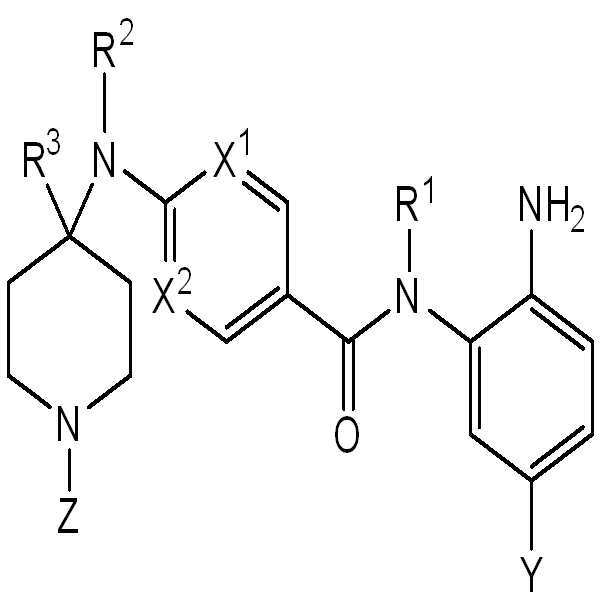
II
화학식 I 또는 화학식 II의 화합물의 실시양태에서, X1 및 X2는 각각 N이거나, 또는 X1 및 X2는 각각 CH이다.
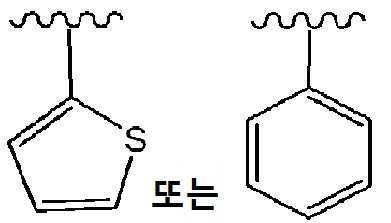
화학식 I 또는 화학식 II의 화합물의 다른 실시양태에서, Y는 하기이다:
화학식 I 또는 화학식 II의 화합물의 다른 실시양태에서, Z는 C(O)NR4R5, C(O)OR6, C(O)-C3-C6-사이클로알킬, C(O)-C2-C6-헤테로사이클릴, 및 C(O)C0-6-알킬-헤테로아릴로 구성된 군으로부터 선택되고, 여기서 헤테로아릴, 사이클로알킬, 또는 헤테로사이클릴은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되고;
R6은 C6-아릴이다.
화학식 I 또는 화학식 II의 화합물의 또 다른 실시양태에서, Z는 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택된다.
화학식 I 또는 화학식 II의 화합물의 실시양태에서, R1은 H이다.
화학식 I 또는 화학식 II의 화합물의 다른 실시양태에서, R2는 H이다.
화학식 I 또는 화학식 II의 화합물의 다른 실시양태에서, R3은 H, 메틸, 에틸, 이소프로필, 또는 페닐이다. 화학식 I 또는 화학식 II의 화합물의 다른 실시양태에서, R3은 H 또는 C6-아릴로 구성된 군으로부터 선택된다. 화학식 I 또는 화학식 II의 화합물의 다른 추가의 실시양태에서, R3은 H이다.
실시양태에서, 화학식 I의 화합물은 화학식 IIa의 화합물 또는 이의 약제학적으로 허용가능한 염이다:
IIa
화학식 IIa의 화합물의 실시양태에서, Y는 하기이다:

화학식 IIa의 화합물의 다른 실시양태에서, Z는 C(O)NR4R5, C(O)OR6, C(O)-C3-C6-사이클로알킬, C(O)-C2-C6-헤테로사이클릴, 및 C(O)C0-6-알킬-헤테로아릴로 구성된 군으로부터 선택되고, 여기서 헤테로아릴, 사이클로알킬, 또는 헤테로사이클릴은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되고;
R6은 C6-아릴이다.
화학식 IIa의 화합물의 또 다른 실시양태에서, Z는 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택된다.
화학식 IIa의 화합물의 실시양태에서, R1은 H이다.
화학식 IIa의 화합물의 다른 실시양태에서, R2는 H이다.
화학식 IIa의 화합물의 다른 실시양태에서, R3은 H, 메틸, 에틸, 이소프로필, 또는 페닐이다. 화학식 IIa의 화합물의 다른 추가의 실시양태에서, R3은 H이다.
특정 실시양태에서, R1은 H이고; R2는 H이고; R3은 H이다.
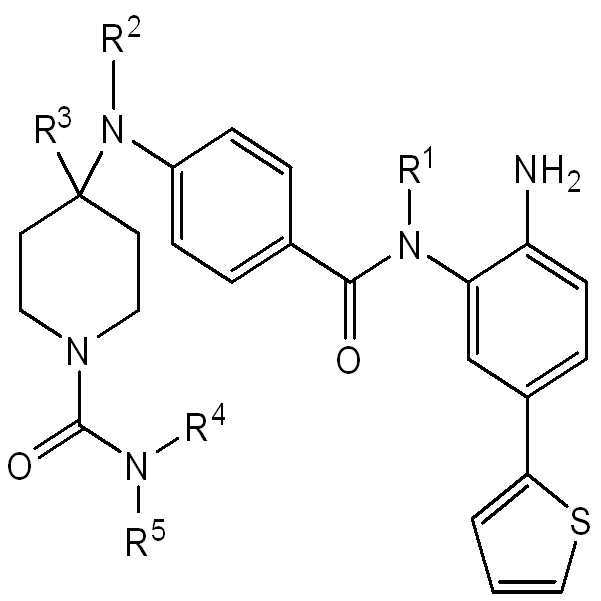
추가의 실시양태에서, 화학식 I의 화합물은 화학식 III의 화합물 또는 이의 약제학적으로 허용가능한 염이다:

III
화학식 III의 화합물의 실시양태에서, X1 및 X2는 각각 N이거나, 또는 X1 및 X2는 각각 CH이다.
화학식 III의 화합물의 실시양태에서, R2는 H이다.
화학식 III의 화합물의 실시양태에서, R3은 H, 메틸, 또는 이소프로필이다.
화학식 III의 화합물의 실시양태에서, R4는 H이고 R5는 C1-C6-알킬이다.
화학식 III의 화합물의 다른 실시양태에서, R4 및 R5는 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환된다.
화학식 III의 화합물의 다른 실시양태에서, X1 및 X2는 N이고;
R1은 H이고;
R2는 H이고;
R3은 H 또는 C1-C4-알킬이고;
R4 및 R5는 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환된다.
화학식 III의 화합물의 다른 실시양태에서, X1 및 X2는 N이고;
R1은 H이고;
R2는 H이고;
R3은 H이고;
R4 및 R5는 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환된다.
추가의 실시양태에서, 화학식 I의 화합물은 화학식 IIIa의 화합물 또는 이의 약제학적으로 허용가능한 염이다:
IIIa
화학식 IIIa의 화합물의 실시양태에서, R2는 H이다.
화학식 IIIa의 화합물의 실시양태에서, R3은 H, 메틸, 또는 이소프로필이다. 화학식 IIIa의 화합물의 추가의 실시양태에서, R3은 H이다.
화학식 III의 화합물의 실시양태에서, R4는 H이고 R5는 C1-C6-알킬이다.
화학식 IIIa의 화합물의 실시양태에서, R4 및 R5는 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환된다.
화학식 IIIa의 화합물의 다른 실시양태에서,
R1은 H이고;
R2는 H이고;
R3은 H이다.
화학식 IIIa의 화합물의 다른 실시양태에서,
R1은 H이고;
R2는 H이고;
R3은 H이고;
R4 및 R5는 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환된다.
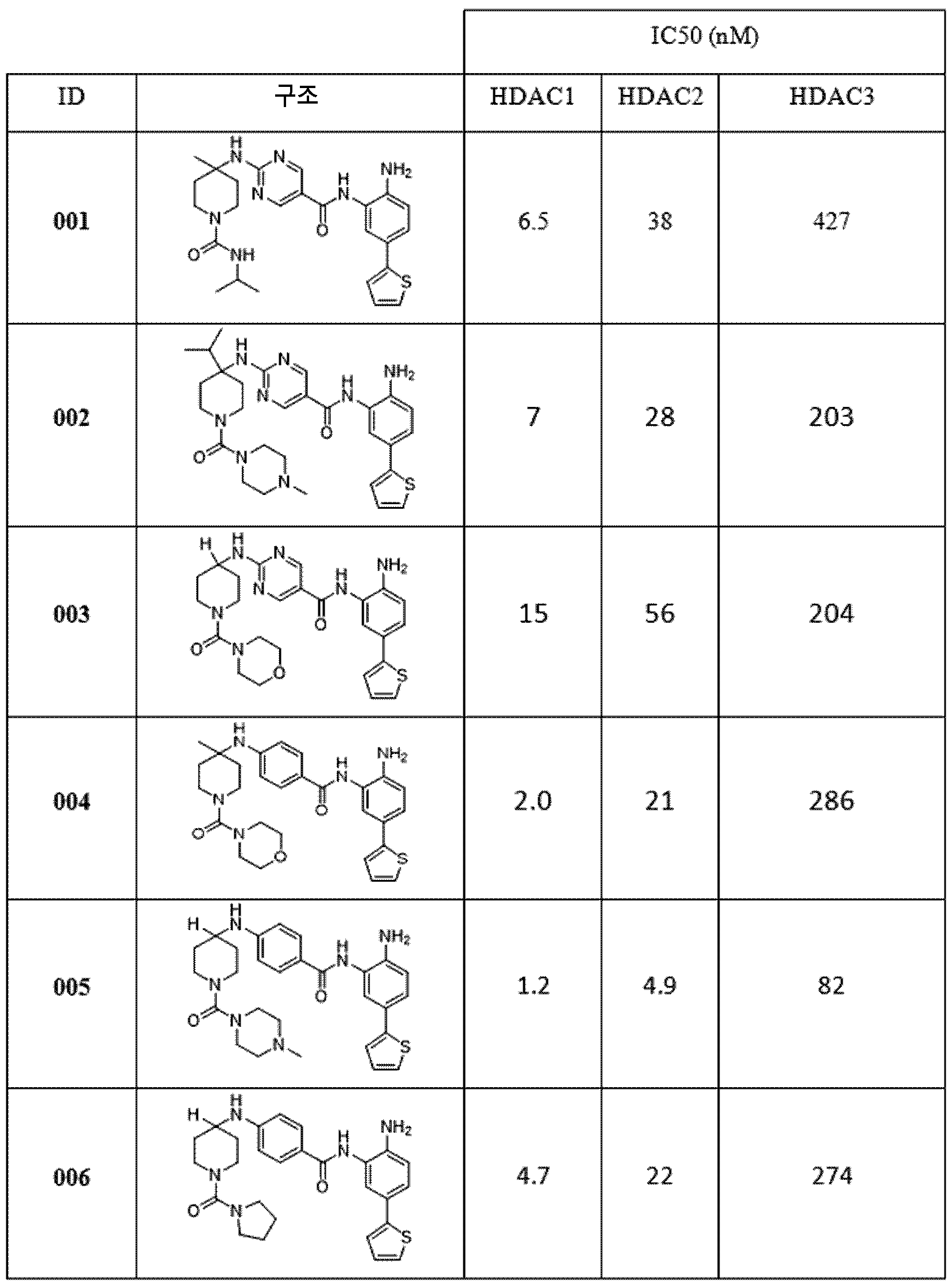
다른 측면에서, 표 1에 제시된 임의의 화합물로부터 선택된 화합물 및 이의 약제학적으로 허용가능한 염이 본원에 제공된다:





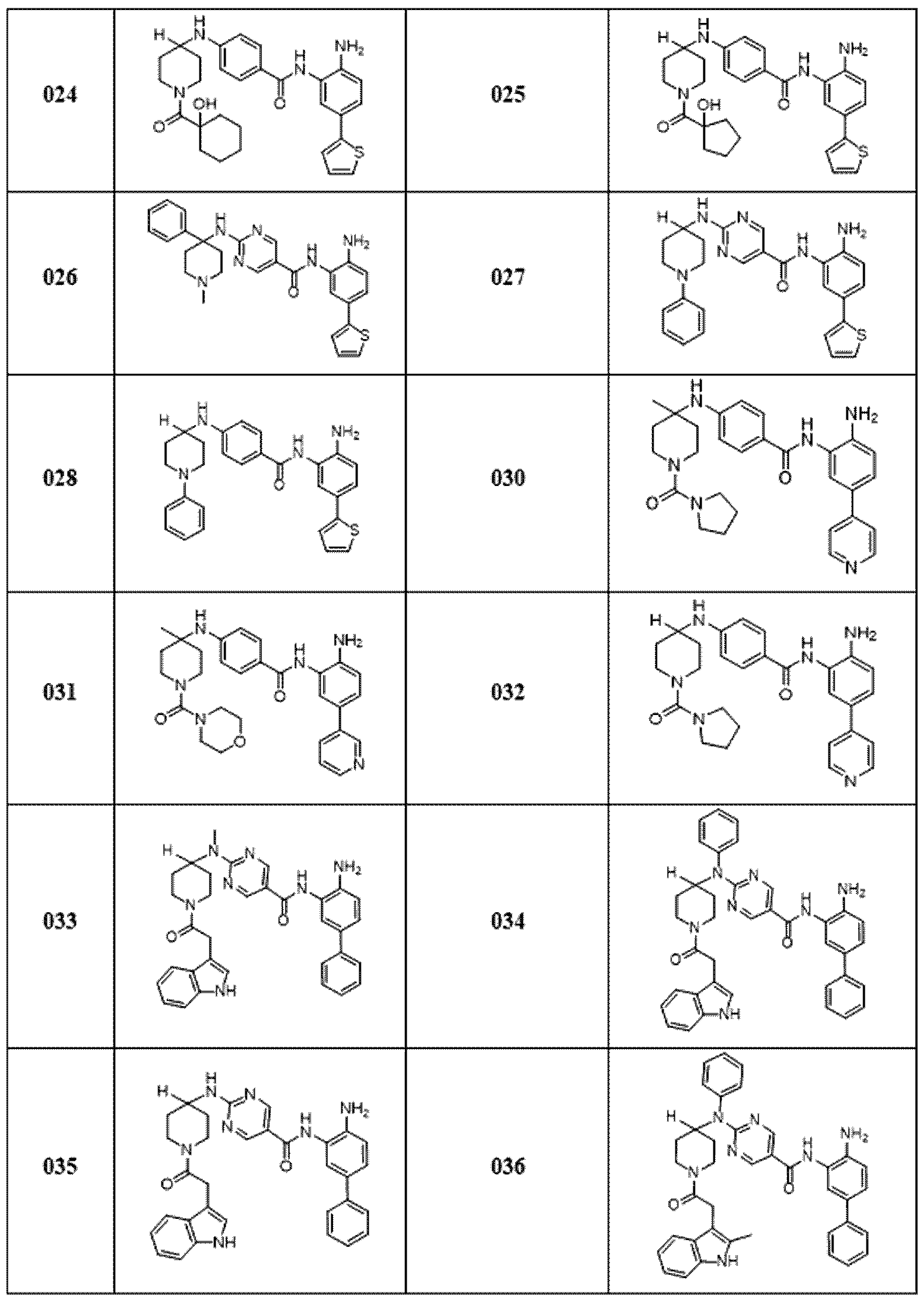
다른 측면에서, 표 1a에 제시된 임의의 화합물로부터 선택된 화합물 및 이의 약제학적으로 허용가능한 염이 본원에 제공된다:
[표 1a]


바람직한 실시양태에서, 본 발명의 화합물은 하기 특성 중 하나 이상을 갖는다: 화합물은 적어도 하나의 히스톤 디아세틸라제 (HDAC)를 억제할 수 있고; 화합물은 HDAC1 및/또는 HDAC2를 억제할 수 있고; 화합물은 다른 HDACs에 비해 HDAC1 및/또는 HDAC2를 선택적으로 억제한다.
본 발명의 다른 목적은 본원에 기술된 장애 또는 질환의 치료에 사용하기 위한 약제의 제조에 있어서 본원에 기술된 바와 같은 (예컨대, 본원의 임의의 화학식의) 화합물의 용도이다. 본 발명의 다른 목적은 본원에서 장애 또는 질환의 치료에 사용하기 위한 본원에 기술된 바와 같은 (예컨대, 본원의 임의의 화학식의) 화합물의 용도이다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 합성하는 방법이 본원에 제공된다. 본 발명의 화합물의 합성은 하기 실시예에서 확인할 수 있다. 따라서, 실시양태는 본원에 서술된 반응의 임의의 하나 또는 이들의 조합을 이용하여 본원의 임의의 화학식의 화합물을 제조하는 방법이다. 방법은 본원에 서술된 하나 이상의 중간체 또는 화학적 시약의 사용을 포함할 수 있다.
다른 측면에서, 본원에 서술된 임의의 화학식의 동위원소 표지된 화합물이 본원에 제공된다. 이러한 화합물은 화합물 내로 도입된 방사능일 수 있는 하나 이상의 동위원소 (예컨대, 3H, 2H, 14C, 13C, 35S, 32P, 125I, 및 131I)를 갖는다. 이러한 화합물은 치료적 적용뿐만 아니라 약물 대사 연구 및 진단학에 유용하다.
본 발명의 화합물의 보호된 유도체가 당업자에게 공지된 수단에 의해 제조될 수 있다. 보호기의 생성 및 이들의 제거에 적용가능한 기법의 상세한 설명은 문헌 [T. W. Greene, "Protecting Groups in Organic Chemistry," 3rd edition, John Wiley and Sons, Inc., 1999] 및 이의 후속 버전에서 확인할 수 있다.
본 발명의 화합물은 용매화물 (예컨대, 수화물)로서, 본 발명의 방법 동안에 용이하게 제조되거나 형성될 수 있다. 본 발명의 화합물의 수화물은 디옥산, 테트라히드로푸란 또는 메탄올과 같은 유기 용매를 사용하여 수성/유기 용매 혼합물로부터의 재결정화에 의해 용이하게 제조될 수 있다.
또한, 본 발명의 화합물은 하나 이상의 이중 결합, 또는 하나 이상의 비대칭 중심을 가질 수 있다. 이러한 화합물은 라세미체, 라세미 혼합물, 단일 거울상이성질체, 개별 부분입체이성질체, 부분입체이성질체 혼합물, 및 시스- 또는 트랜스- 또는 E- 또는 Z- 이중 이성질체 형태, 및 절대 입체화학 측면에서, 아미노산에 대해 (R)- 또는 (S)-, 또는 (D)- 또는 (L)-로서 정의될 수 있는 다른 입체이성질체 형태로서 야기된다. 이들 화합물의 이러한 모든 이성질체 형태는 본 발명에 명백히 포함된다. 광학 이성질체는 상기에 기술된 절차에 의해, 또는 라세미 혼합물의 분해에 의해 이들 각각의 광학 활성 전구체로부터 제조될 수 있다. 분해는 분해제의 존재하에서 크로마토그래피에 의해 또는 반복 재결정화에 의해 또는 당업자에게 공지된 이들 기법의 일부 조합에 의해 수행될 수 있다. 분해와 관련한 추가의 설명은 문헌 [Jacques, et al., "Enantiomers, Racemates, and Resolutions" (John Wiley & Sons, 1981)]에서 확인할 수 있다. 본 발명의 화합물은 또한 다중 호변이성질체 형태로 나타낼 수 있는데, 일부 경우에, 본 발명은 본원에 기술된 화합물의 모든 호변이성질체 형태를 명백히 포함한다. 본원에 기술된 화합물이 올레핀 이중 결합 또는 기하학적 비대칭의 다른 중심을 함유하는 경우, 달리 특정되지 않는 한, 화합물은 E 및 Z 기하학적 이성질체 둘 다를 포함하는 것으로 의도된다. 유사하게, 모든 호변이성질체 형태가 또한 포함되는 것으로 의도된다. 본원에 나타나는 임의의 탄소-탄소 이중 결합의 형상은 단지 편의를 위해 선택된 것이고, 문맥이 그렇게 언급하지 않는 한 특정 형상을 지정하는 것으로 의도되지 않으며; 따라서 트랜스로서 본원에 임의로 묘사된 탄소-탄소 이중 결합은 시스, 트랜스, 또는 임의의 비율로 이 둘의 혼합물일 수 있다. 이들 화합물의 모든 이러한 이성질체 형태는 본 발명에 명백히 포함된다.
합성된 화합물은 반응 혼합물로부터 분리되고 칼럼 크로마토그래피, 고압 액체 크로마토그래피, 또는 재결정화와 같은 방법에 의해 추가로 정제될 수 있다. 당업자에게 인식될 수 있는 바와 같이, 본원의 화학식의 화합물을 합성하는 추가의 방법이 당업자에게 명백할 것이다. 부가적으로, 다양한 합성 단계가 목적하는 화합물을 제공하기 위해 교대 순서 또는 차례로 수행될 수 있다. 또한, 본원에 서술된 용매, 온도, 반응 기간 등은 단지 설명을 위한 것이고 당업자는 반응 조건의 변화가 본 발명의 목적하는 화합물을 생산할 수 있음을 인식할 것이다. 본원에 기술된 화합물을 합성하는데 유용한 합성 화학 변형 및 보호기 방법론 (보호 및 탈보호)은 당해 분야에 공지되어 있다.
본원에서 변수의 임의의 정의에 있어서 화학적 기의 열거의 인용은 임의의 단일 기 또는 열거된 기들의 조합으로서의 변수의 정의를 포함한다. 본원에서 변수에 대한 실시양태의 인용은 임의의 단일 실시양태로서 또는 임의의 다른 실시양태 또는 그의 일부와 조합하여 그 실시양태를 포함한다.
약제학적 조성물
약제학적으로 허용가능한 담체와 함께, 본 발명의 화합물, 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 조성물이 또한 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 본 발명의 임의의 화합물 (즉, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염)을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 화학식 I의 화합물, 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 화학식 II의 화합물, 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 화학식 IIa의 화합물, 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 화학식 III의 화합물, 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 화학식 IIIa의 화합물, 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 표 1의 화합물 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 표 1a의 화합물 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
일 측면에서, 약제학적으로 허용가능한 담체와 함께, 화합물 005 또는 이의 약제학적으로 허용가능한 염을 포함하는 약제학적 화합물이 본원에 제공된다.
이들 약제학적 조성물은 하나 이상의 약제학적으로 허용가능한 담체와 함께 조제된 본 발명의 화합물의 치료적 유효량을 포함한다. 용어 "약제학적으로 허용가능한 담체"는 비-독성, 불활성 고체 또는 액체 충전제, 희석제, 캡슐화 재료 (encapsulating material) 또는 임의의 유형의 제제 보조제를 의미한다. 약제학적 조성물은 인간 및 다른 동물에 경구로, 직장으로, 비경구로, 수조내로 (intracisternally), 질내로, 복강내로, 국소적으로 (분말, 연고, 또는 점적약제에 의해서와 같이), 볼내로 (buccally), 또는 구강 또는 비강 스프레이로서 투여될 수 있다.
본 발명의 화합물은 임의의 통상적인 경로, 특히 소화관내로, 예를 들어, 경구로, 예컨대, 정제 또는 캡슐의 형태로, 또는 비경구로, 예컨대, 주사가능 용액 또는 현탁액의 형태로, 국소적으로, 예컨대, 로숀, 젤, 연고 또는 크림의 형태로, 또는 비강 또는 좌약 형태로 약제학적 조성물로서 투여될 수 있다.
적어도 하나의 약제학적으로 허용가능한 담체 또는 희석제와 연합하여 유리 형태 또는 약제학적으로 허용가능한 염 형태로 본 발명의 화합물을 포함하는 약제학적 조성물은 혼합, 과립화 또는 코팅 방법에 의해 통상적인 방식으로 제조될 수 있다. 예를 들어, 경구 조성물은 a) 희석제, 예컨대, 락토스, 덱스트로스, 수크로스, 만니톨, 소르비톨, 셀룰로스 및/또는 글리신; b) 윤활제, 예컨대, 실리카, 탈크, 스테아르산, 그의 마그네슘 또는 칼슘 염 및/또는 폴리에틸렌글리콜; 정제의 경우 또한 c) 결합제, 예컨대, 마그네슘 알루미늄 실리케이트, 전분 페이스트, 젤라틴, 트라가칸스, 메틸셀룰로스, 소디움 카르복시메틸셀룰로스 및 폴리비닐피롤리돈; 바람직하다면 d) 붕해제, 예컨대, 전분, 아가, 알긴산 또는 그의 나트륨 염, 또는 발포성 혼합물; 및/또는 e) 흡수제, 척색제, 향미제 및 감미제와 함께 활성 성분을 포함하는 정제 또는 젤라틴 캡슐일 수 있다. 주사가능 조성물은 수성 등장 용액 또는 현탁액일 수 있고, 좌약은 지방 유탁액 또는 현탁액으로부터 제조될 수 있다. 조성물은 안정화되고/되거나 애주번트, 예컨대, 보존제, 안정화제, 습윤제 또는 유화제, 용액 프로모터, 삼투압 조절을 위한 염 및/또는 완충제를 함유할 수 있다. 나아가, 이들은 또한 다른 치료학적으로 유용한 물질을 함유할 수 있다. 경피 적용에 적합한 제제는 담체와 함께 본 발명의 화합물의 유효량을 포함한다. 담체는 숙주의 피부를 통과하는 것을 보조하는 흡수가능한 약제학적으로 허용가능한 용매를 포함할 수 있다. 예를 들어, 경피 장치는 백킹 부재 (backing member), 선택적으로 담체와 함께 화합물을 함유하는 저장소, 선택적으로 연장된 기간에 걸쳐서 제어되고 미리 지정된 속도로 숙주의 피부에 화합물을 전달하기 위한 속도 제어 장벽, 및 피부에 장치를 고정하기 위한 수단을 포함하는 밴드 (bandage)의 형태이다. 매트릭스 경피 제제가 또한 사용될 수 있다. 예컨대, 피부 및 눈에의 국소 적용에 적합한 제제는 바람직하게는 당해 분야에 잘 알려진 수성 용액, 연고, 크림 또는 젤이다. 이들은 용해제, 안정화제, 장력 증강제, 완충제 및 보존제를 함유할 수 있다.
정제, 당제, 캡슐, 알약, 및 과립의 고체 투약 형태는 장용 코팅, 방출 제어 코팅 및 약제학적 제제 분야에 잘 알려진 다른 코팅과 같은 코팅 및 쉘 (shell)과 함께 제조될 수 있다. 이러한 고체 투약 형태에서, 활성 화합물은 수크로스, 락토스 또는 전분과 같은 적어도 하나의 불활성 희석제와 혼합될 수 있다. 이러한 투약 형태는, 또한 통상적인 관례에서와 같이, 불활성 희석제 이외의 추가적 물질, 예컨대, 정제화 윤활제 및 마그네슘 스테아레이트 및 미결정 셀룰로스와 같은 다른 정제화 보조제를 또한 포함할 수 있다. 캡슐, 정제 및 알약의 경우에, 투약 형태는 또한 완충제를 포함할 수 있다.
치료 방법
목적하는 결과를 달성하는데 필요한 바와 같은 그러한 양으로 그러한 시간 동안 본 발명의 화합물의 치료학적 유효량을 개체에 투여함으로써, 인간 또는 다른 동물과 같은 개체에서 장애를 치료 또는 예방하는 방법이 본원에 제공된다. 본 발명의 화합물의 "치료학적 유효량"이라는 용어는 개체에서 장애의 증상을 감소시키기에 충분한 화합물의 양을 의미한다. 의학적 분야에서 잘 이해되는 바와 같이, 본 발명의 화합물의 치료학적 유효량은 임의의 의학적 처치에 적용가능한 합리적인 이익/위험 비율에서일 것이다.
본원에 사용된 바와 같이, 용어 "치료하는" 또는 "치료"는 개체에서 적어도 하나의 증상을 완화, 감소 또는 경감하거나 질환의 진행 지연을 초래하는 것을 포함한다. 예를 들어, 치료는 암과 같은 장애의 여러 증상 중 하나의 약화 또는 장애의 완전한 퇴치일 수 있다. 본 명세서의 의미 내에서, 용어 "치료하다"는 또한 질환의 정지, 질환 개시 (즉, 질환의 임상 증상으로 나타나기 전의 기간)의 지연 및/또는 질환의 발병 또는 악화의 위험을 감소시키는 것을 나타낸다. 용어 "보호하다"는 본원에서 개체, 예컨대, 포유동물 또는 인간에서 질환의 발생, 지속 또는 악화를, 적절하게, 예방, 지연, 또는 치료, 또는 이들 모두를 의미하는데 사용된다. 본원에 사용된 바와 같이, 용어 "예방하다", "예방하는" 또는 "예방"은 예방되는 상태, 질환 또는 장애와 연관되거나 그에 의해 야기된 적어도 하나의 증상의 예방을 포함한다.
일반적으로, 본 발명의 화합물은, 단독으로 또는 하나 이상의 치료제와 조합하여, 당해 분야에 공지된 통상의 허용가능한 임의의 방식을 통해 치료학적 유효량으로 투여될 것이다. 치료학적 유효량은 질환의 중증도, 개체의 연령 및 상대적 건강상태, 사용된 화합물의 효능 및 다른 인자에 따라서 광범위하게 달라질 수 있다.
특정 실시양태에서, 본 발명의 화합물의 치료적 양 또는 용량은 약 0.1 mg/kg 내지 약 500 mg/kg (약 0.18 mg/m2 내지 약 900 mg/m2), 대안적으로 약 1 내지 약 50 mg/kg (약 1.8 내지 약 90 mg/m2)의 범위일 수 있다. 일반적으로, 본 발명에 따른 치료 레지멘 (regimen)은 이러한 치료를 필요로 하는 환자에 단일 또는 다중 용량으로 하루에 본 발명의 화합물(들)의 약 10 mg 내지 약 1000 mg의 투여를 포함한다. 치료적 양 및 용량은 또한 다른 제제와의 공동-사용의 가능성뿐만 아니라, 투여 경로에 따라서 달라질 것이다.
개체 상태의 개선 시, 필요하다면, 본 발명의 화합물, 조성물 또는 조합의 유지 용량이 투여될 수 있다. 이어서, 투여의 복용량 또는 빈도, 또는 둘 다는, 증상의 함수로서, 개선된 상태가 유지되는 수준으로 감소될 수 있고, 증상이 목적하는 수준으로 경감되었다면 치료는 중단되어야 한다. 그러나, 개체는 질환 증상의 임의의 재발 시 장기적으로 간헐적 치료를 요구할 수 있다.
그러나, 본 발명의 화합물 및 조성물의 총 일일 사용량은 건전한 의학적 판단의 범위 내에서 주치의에 의해 결정될 것으로 이해될 것이다. 임의의 특정 환자를 위한 특정 억제 용량은 치료되는 장애, 장애의 중증도; 사용되는 특정 화합물의 활성; 사용되는 특정 조성물; 환자의 연령, 체중, 일반 건강상태, 성별 및 식이; 사용된 특정 화합물의 투여 시간, 투여 경로, 및 배설률; 치료의 기간; 사용된 특정 화합물과 조합하여 또는 동시에 사용되는 약물; 및 의학 분야에 잘 알려진 관련 인자를 비롯한 다양한 인자에 따라 좌우될 것이다.
일 측면에서, 본 발명은 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 투여하는 것을 포함하는, 개체에서 다른 HDACs에 비해 HDAC 및/또는 HDAC2 각각의 활성을 선택적으로 억제하는 방법을 제공한다.
실시양태에서, 화합물은 다른 HDACs에 비해 약 2 내지 1000배 (5 내지 1000배, 10 내지 1000배, 5 내지 100배 등과 같은 범위를 포함함) 더 우수한 HDAC1 및/또는 HDAC2 각각에 대한 선택성을 갖는다. 다른 실시양태에서, 화합물은 HDAC 효소 어세이에서 시험될 때, 다른 HDACs에 비해 약 2 내지 1000배 (5 내지 1000배, 10 내지 1000배, 5 내지 100배 등과 같은 범위를 포함함) 더 우수한 HDAC1 및/또는 HDAC2 각각에 대한 선택성을 갖는다.
다른 측면에서, 본 발명은 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염을 개체에 투여하는 것을 포함하는, 개체에서 HDAC, 특히 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법을 제공한다. 본 발명의 선택적 HDAC1 및 HDAC2 억제제는 유리한 약물동태학 프로파일을 갖는다 (예컨대, 실시예 44 참조).
일 측면에서, 화학식 I의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 II의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 IIa의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 III의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 IIIa의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 표 1의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 표 1a의 화합물 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화합물 005 또는 이의 약제학적으로 허용가능한 염의 의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개되는 질환을 치료하는 방법이 본원에 제공된다.
HDAC1 및 HDAC2의 억제는 태아 글로빈을 저해하기에 충분하다. 적혈구 분화를 거치는 배양된 인간 CD34+ 골수 세포에서, 이들 화합물은 태아 헤모글로빈 발현의 용량 의존적 증가를 유도할 수 있다 (예컨대, 실시예 45 참조).
따라서, 화합물은 HDAC 억제를 통해 태아 글로빈을 저해할 수 있다. 따라서, 실시양태에서, 화합물은 이상혈색소를 앓고 있거나 이에 걸리기 쉬운 개체를 치료할 수 있다. 바람직한 실시양태에서, 화합물은 겸상적혈구병 또는 베타-지중해성 빈혈을 치료할 수 있다.
다른 실시양태에서, 본 발명의 화합물은 골수형성이상 증후군의 치료에 유용하다.
특정 실시양태에서, 본 발명의 화합물은 항암제로 유용하다. 본 발명의 화합물은 암세포에서 아폽토시스를 유도할 수 있고, 그로써 암 또는 증식 질환과 같은 질환을 치료할 수 있다. 실시양태에서, 본 발명의 화합물은 종양 세포 사멸을 초래하거나 종양 세포의 성장을 억제함으로써 암의 치료에 유용할 수 있다.
특정 실시양태에서, 암은 폐암, 직장결장암, 유방암, 전립선암, 간암, 췌장암, 뇌암, 신장암, 난소암, 위암 (stomach cancer), 피부암, 골암, 위암 (gastric cancer), 유방암, 신경교종, 교아종, 신경모세포종, 간세포암종, 유두상 신세포암종, 두경부 편평세포암종, 백혈병, 림프종, 골수종, 망막아세포종, 자궁경부암, 흑색종 및/또는 피부암, 방광암, 자궁암, 정소암, 식도암, 및 고형 종양이다. 일부 실시양태에서, 암은 폐암, 결장암, 유방암, 신경모세포종, 백혈병, 또는 림프종이다. 다른 실시양태에서, 암은 폐암, 결장암, 유방암, 신경모세포종, 백혈병, 또는 림프종이다. 추가의 실시양태에서, 암은 비-소세포 폐암 (NSCLC) 또는 소세포 폐암이다. 다른 실시양태에서, 암은 신경모세포종이다.
추가의 실시양태에서, 암은 백혈병 또는 림프종과 같은 혈액암이다. 특정 실시양태에서, 림프종은 호지킨 림프종 또는 비-호지킨 림프종이다. 특정 실시양태에서, 백혈병은 골수, 림프구, 골수성 (myelocytic), 림프구성 (lymphoblastic), 또는 거핵세포성 백혈병이다. 특정 실시양태에서, 백혈병은 급성 골수성 백혈병 및 거대핵세포성 백혈병이다.
다른 측면에서, 화학식 I, 화학식 II, 화학식 IIa, 화학식 III, 또는 화학식 IIIa의 화합물, 표 1에 제시된 화합물, 표 1a에 제시된 화합물, 또는 이들의 약제학적으로 허용가능한 염의 치료학적 유효량을 이를 필요로 하는 개체에 투여하는 것을 포함하는, 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 I의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 II의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 IIa의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 III의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
일 측면에서, 화학식 IIIa의 화합물, 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 개체에 투여하는 것을 포함하는, 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 거대핵세포성 백혈병을 치료하는 방법이 본원에 제공된다.
본원에 서술된 방법은 개체가 특별히 명시된 치료를 필요로 하는 것으로 확인된 방법을 포함한다. 이러한 치료를 필요로 하는 개체를 확인하는 것은 개체 또는 건강 관리 전문가의 판단 내일 수 있고 주관적 (예컨대, 의견) 또는 객관적 (예컨대, 검사 또는 진단 방법에 의해 측정가능함)일 수 있다.
또한, 상기에 논의된 바와 같이, 본 발명의 화합물은 HDAC1 및/또는 HDAC2의 선택적 억제제이고, 따라서 이들 히스톤 디아세틸라제 (HDAC)에 의해 조절되는 장애의 치료에 유용하다. 예를 들어, 본 발명의 화합물은 암 (예컨대, 폐암, 결장암, 유방암, 신경모세포종, 백혈병, 또는 림프종 등)의 치료에 유용할 수 있다. 따라서, 또 다른 측면에서, 본 발명의 치료 방법에 따르면, 종양 세포는 사멸되거나, 또는 상기 종양 세포를 본원에 기술된 바와 같은 본 발명의 화합물 또는 조성물과 접촉시킴으로써 이들의 성장이 억제된다.
따라서, 본 발명의 다른 측면에서, 본원에 기술된 바와 같은 본 발명의 화합물 (즉, 본원의 임의의 화학식)의 치료학적 유효량을 이를 필요로 하는 개체에 투여하는 것을 포함하는, 암의 치료 방법이 제공된다. 특정 실시양태에서, 개체는 이러한 치료를 필요로 하는 것으로 확인된다. 특정 실시양태에서, 목적하는 결과를 달성하는데 필요한 바와 같은 그러한 양으로 그러한 시간 동안, 이를 필요로 하는 개체에 본 발명의 화합물, 또는 본 발명의 화합물을 포함하는 약제학적 조성물의 치료학적 유효량을 투여하는 것을 포함하는, 암의 치료 방법이 제공된다. 본 발명의 특정 실시양태에서, 본 발명의 화합물 또는 약제학적 조성물의 "치료학적 유효량"은 종양 세포를 사멸하거나 이들의 성장을 억제하는데 효과적인 양이다. 본 발명의 방법에 따르면, 화합물 및 조성물은 종양 세포를 사멸하거나 이들의 성장을 억제하는데 효과적인 임의의 양 및 임의의 투여 경로를 사용하여 투여될 수 있다. 따라서, 본원에 사용된 바와 같이, 표현 "종양 세포를 사멸하거나 이들의 성장을 억제하는데 효과적인 양"은 종양 세포를 사멸하거나 이들의 성장을 억제하는데 충분한 제제의 양을 지칭한다. 요구되는 정확한 양은 개체의 종, 연령 및 일반 건강상태, 감염의 중증도, 특정 항암제, 그의 투여 방식 등에 따라서 개체마다 달라질 것이다.
특정 실시양태에서, 방법은 이를 필요로 하는 개체 (이로 제한되는 것은 아니지만, 인간 또는 동물을 포함함)에 화합물 또는 이의 약제학적으로 허용가능한 유도체의 치료학적 유효량의 투여를 포함한다. 특정 실시양태에서, 본 발명의 화합물은, 이로 제한되는 것은 아니지만, 폐암 (예컨대, 비-소세포 폐암), 직장결장암, 유방암, 전립선암, 간암, 췌장암, 뇌암, 신장암, 난소암, 위암, 골암, 피부암, 위암, 유방암, 신경교종, 교아종, 신경모세포종, 간세포암종, 유두상 신세포암종, 두경부 편평세포암종, 백혈병 (예컨대, CML, AML, CLL, ALL), 림프종 (비-호지킨 및 호지킨), 골수종, 망막아세포종, 자궁경부암, 흑색종 및/또는 피부암, 방광암, 자궁암, 정소암, 식도암, 및 고형 종양을 포함하는 암 및 기타 증식성 장애의 치료에 유용하다.
특정 실시양태에서, 본 발명은 개체가 인간인, 본원에 기술된 임의의 장애를 치료하기 위한 방법을 제공한다.
전술한 바에 따르면, 본 발명은 이러한 치료를 필요로 하는 개체에서 상기에 기술된 임의의 질환 또는 장애를 예방 또는 치료하는 방법을 추가로 제공하는데, 이 방법은 본 발명의 화합물 또는 이의 약제학적으로 허용가능한 염의 치료학적 유효량을 상기 개체에 투여하는 것을 포함한다. 임의의 상기 용도를 위해, 요구되는 복용량은 투여 방식, 치료되는 특정 상태 및 목적하는 효과에 따라서 달라질 것이다.
도 1은 HDAC1, HDAC2, 및 HDAC3에 대한 화합물 003의 HDAC 억제 프로파일을 보여주는 그래프이다 (실시예 43 참조).
도 2는 40 mg/kg의 화합물 003의 경구 투여 시 시간의 함수로서 래트에서의 혈장 농도를 나타낸다 (실시예 44 참조).
도 3은 또 다른 공지의 HDAC1/2 억제제인, 화합물 A와 비교하여 화합물 003의 시험관 내 태아 글로빈 유도를 나타낸다 (실시예 45 참조).
도 4는 HDAC1, HDAC2, 및 HDAC3에 대한 화합물 005의 HDAC 억제 프로파일을 보여주는 그래프이다 (실시예 43 참조).
도 5는 20 mg/kg의 화합물 005의 경구 투여 시 시간의 함수로서 래트에서의 혈장 농도를 나타낸다 (실시예 44 참조).
도 6은 또 다른 공지의 HDAC1/2 억제제인, 화합물 A와 비교하여 화합물 005의 시험관 내 태아 글로빈 유도를 나타낸다 (실시예 45 참조).
도 7은 다양한 HDAC1/2 억제제 (화합물 005 및 A)를 이용한 적혈구 전구세포의 처리가 Gata2 mRNA의 유도를 초래함을 나타낸다.
도 2는 40 mg/kg의 화합물 003의 경구 투여 시 시간의 함수로서 래트에서의 혈장 농도를 나타낸다 (실시예 44 참조).
도 3은 또 다른 공지의 HDAC1/2 억제제인, 화합물 A와 비교하여 화합물 003의 시험관 내 태아 글로빈 유도를 나타낸다 (실시예 45 참조).
도 4는 HDAC1, HDAC2, 및 HDAC3에 대한 화합물 005의 HDAC 억제 프로파일을 보여주는 그래프이다 (실시예 43 참조).
도 5는 20 mg/kg의 화합물 005의 경구 투여 시 시간의 함수로서 래트에서의 혈장 농도를 나타낸다 (실시예 44 참조).
도 6은 또 다른 공지의 HDAC1/2 억제제인, 화합물 A와 비교하여 화합물 005의 시험관 내 태아 글로빈 유도를 나타낸다 (실시예 45 참조).
도 7은 다양한 HDAC1/2 억제제 (화합물 005 및 A)를 이용한 적혈구 전구세포의 처리가 Gata2 mRNA의 유도를 초래함을 나타낸다.
실시예가 설명을 목적으로 하기에 기술되었고 본 발명의 특별한 특정 실시양태를 기술하기 위해 개시되었다. 그러나, 청구범위의 범주가 본원에 개시된 실시예에 의해 어떠한 방식으로도 제한되는 것은 아니다. 개시된 실시양태에 대한 다양한 변화 및 변형이 당업자에게 명백할 것이며, 제한 없이, 본 발명의 화학적 구조, 치환체, 유도체, 제제 및/또는 방법에 관한 것들을 포함하는, 이러한 변화 및 변형이 본 발명의 요지 및 첨부된 청구범위의 범주에서 벗어남 없이 이루어질 수 있다. 본원의 반응식에서 구조의 변수의 정의는 본원에서 제시된 화학식의 상응하는 위치의 변수에 부합한다.
실시예
1: 화합물 001의 합성
단계 1: 리튬 비스(트리메틸실릴)아미드 (THF 중의 1.0 M 용액, 240 ml, 240 mmol)를 N2 하의 -76℃에서 화합물 1 (25 g, 120 mmol) 함유 둥근-바닥 플라스크에 서서히 첨가하였다. 반응을 -76℃에서 4시간 동안 교반한 후, 이오도메탄 (15 ml, 240 mmol)을 시스템 내로 주입하였다. 반응 혼합물을 -76℃에서 30분간 교반한 후, 실온으로 가온하고 밤새 교반하였다. 반응 혼합물을 150 ml 포화 수성 NH4Cl로 ??칭하고 (quenched), 물로 희석하고 EtOAc (에틸 아세테이트, 또는 EA)로 추출하였다. 유기층을 물 및 소금물로 세척하고 황산나트륨 상에서 건조시키고, 여과하고 농축하여 연한 황색 고체로서 표적 화합물 2 (25g, 93%)를 수득하였다.
단계 2: K2CO3 (31 g, 224 mmol)를 DMSO (120 ml) 중의 화합물 2 (25g, 111 mmol)의 용액에 첨가하였다. H2O2 (100 ml)를 60℃에서 서서히 시스템에 첨가하고, 반응을 60℃에서 밤새 교반하였다. 그 후에 시스템을 냉수 내로 도입하고 EA로 추출하였다. 유기층을 물 및 소금물로 세척하고 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 3 (26 g, 96%)을 수득하였다.
단계 3: 화합물 3 (26 g, 107 mmol)을 ACN (아세토니트릴) (200 ml) 및 5 N KOH (100 ml)로 용해시켰다. 그 후에 1,3-디브로모-5,5-디메틸이미다졸리딘-2,4-디온 (15 g, 54 mmol)을 시스템 내로 첨가하였다. 혼합물을 밤새 교반하고 농축하여 ACN을 제거하였다. 수상의 pH를 빙조 (ice bath)에서 2 N HCl을 이용해 약 5로 조정하고, EA로 추출하고 분리하였다. 그 후에 수상의 pH를 10으로 조정하였다. 침전물을 수집하여 백색 고체 (16 g, 69%)로서 화합물 4를 수득하였다.
단계 4: 1,4-디옥산 (25 ml) 중의 화합물 4 (2 g, 9.34 mmol), 2-클로로피리미딘 (2.6 g, 14.02 mmol) 및 DIPEA (5.3 g, 28.03 mmol)의 용액을 밤새 95℃에서 가열하였다. 그 후에 반응 혼합물을 농축하고 EA/PE = 1/5를 이용한 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 5 (1.8 g, 53%)를 수득하였다.
단계 5: 화합물 5 (465 mg, 1.28 mmol), THF (10 ml) 중의 2 N NaOH (10 ml, 20 mmol), 및 EtOH (10 ml)의 용액을 55℃에서 2시간 동안 가열하였다. 반응액을 농축하고 수상의 pH를 약 5-6 사이로 조정하였다. 결과 용액을 EA로 추출하고, 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 6 (400 mg, 93%)을 수득하였다.
단계 6: DMF (10 mL) 중의 화합물 6 (400 mg, 1.19 mmol), 아민 (345 mg, 1.19 mmol), EDCI (307 mg, 2.38 mmol) 및 DMAP (290 mg, 2.38 mmol)의 혼합물을 55℃에서 밤새 가열하였다. 혼합물을 물과 혼합하고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 결과 조성물을 EA/PE=1/2를 이용한 실리카 겔 컬럼으로 정제하여 자주색 고체로서 화합물 7 (400 mg, 55%)을 수득하였다.
단계 7: 1,4-디옥산 (10 ml) 중의 화합물 7 (400 mg, 0.65 mmol)과 HCl/1,4-디옥산 (5 ml, 20 mmol)의 용액을 실온에서 밤새 교반하였다. 반응액을 농축하고 PE로 세척하여 회색 고체로서 표적 화합물 8 (350 mg, 100%)을 수득하였다.
단계 8: 화합물 8 (162 mg, 0.4 mmol) 및 Et3N (80 mg, 0.8 mmol)을 THF (5 ml) 중에 용해시켰다. 이소프로필 이소시아네이트 (CAS: 1795-48-8, 1.2 eq)를 시스템 내에 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하고 Pre-HPLC로 정제하여 화합물 001 (35 mg, 16%)을 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.51 (s, 1H), 8.84 (s, 2H), 7.50 (s, 1H), 7.44 (d, J = 2.0 Hz, 1H), 7.35 (d, J = 5.1 Hz, 1H), 7.29 (dd, J = 8.3, 2.1 Hz, 1H), 7.23 (d, J = 2.7 Hz, 1H), 7.05 (dd, J = 5.0, 3.6 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 6.09 (d, J = 7.6 Hz, 1H), 5.20 (s, 2H), 3.53 (d, J = 13.6 Hz, 2H), 3.05 (t, J = 10.7 Hz, 2H), 2.25 (d, J = 13.8 Hz, 2H), 1.53 - 1.44 (m, 2H), 1.42 (s, 3H), 1.08 (d, J = 6.6 Hz, 1H), 1.04 (d, J = 6.6 Hz, 6H). LCMS: m/z = 494 (M+H).
실시예 2: 화합물 002의 합성

단계 1: N2로 세정된 (flushed) 비등의 3-목 플라스크 내 1 (3 g, 14.28 mmol)의 용액에 -78℃에서 LHDMS (1 M, 21.4 ml)를 첨가하였다. 반응을 3시간 동안 교반한 후 2-이오도프로판 (3.6 g, 21.43 mmol)을 서서히 첨가하였다. 반응액을 -78℃에서 교반하고, 밤새 실온으로 가온하였다. 혼합물을 H2O (2 ml)로 ??칭하고 농축하고, EA (200 ml)에 용해시키고, 물 (100 ml×2)에 이어서 포화 NaCl (수성, 100 ml)로 세척하였다. 유기층을 농축하여 갈색 고체로서 화합물 2 (4 g, 100%)를 수득하였다.
단계 2: DMSO (30 ml) 중의 2 (1 g, 3.97 mmol)의 용액에 K2CO3 (1.6 g, 11.9 mmol)을 첨가하였다. 결과 반응 혼합물을 60℃에서 교반하였다. 그 후에 H2O2 (30% aq, 5 ml)를 한 방울씩 첨가하고 2시간 동안 교반하였다. EA (100 ml)를 혼합물에 첨가한 후 이를 물 (50 ml×2) 및 포화 NaCl (aq, 50 ml)로 세척하였다. 무수 Na2SO4로 건조시키고 농축하여 백색 고체로서 화합물 3 (1 g, 90%)을 수득하였다.
단계 3: ACN (50 ml) 중의 화합물 3 (2.7 g, 10 mmol)의 용액에 KOH (4 N, aq, 50 ml) 및 DBDMH (2.81 g, 5 mmol)를 0℃에서 첨가하였다. 결과 반응 혼합물을 밤새 실온에서 교반하였다. 혼합물을 농축하고 1 N HCl을 첨가하여 pH를 약 6으로 조정하였다. 그 후에 혼합물을 EA (50 ml)로 추출하고, KOH를 첨가하여 수상을 약 pH 9로 조정하였다. 결과 수상을 EA (50 ml×3)로 추출하였다. EA 상을 무수 Na2SO4로 건조시키고 농축하여 무색 액체로서 화합물 4 (1 g, 40%)를 수득하였다.
단계 4: NMP (10 ml) 중의 화합물 4 (500 mg, 2.06 mmol) 및 에틸 2-클로로피리미딘-5-카르복실레이트 (384 mg, 2.06 mmol)의 용액을 N2로 세정하고 140℃에서 1시간 동안 교반하였다. EA (100 ml)를 혼합물에 첨가하고, 이를 그 후에 물 (50 ml×2) 및 포화 NaCl (aq, 50 ml)로 세척하였다. 농축 및 실리카 겔 컬럼 크로마토그래피 (PE/EA=5/1)에 의한 정제로 백색 고체로서 화합물 5 (120 mg 15%)를 수득하였다.
단계 5: EtOH (aq, 95%, 5 ml) 중의 화합물 5 (400 mg)의 혼합물에 NaOH (2 M, 5 ml)를 첨가하고 반응을 55℃에서 2시간 동안 교반하였다. 물 (50 ml)을 혼합물에 첨가하고 pH를 시트르산을 이용해 약 7로 조정하였다. 결과 수성 혼합물을 EA (50 ml×3)로 추출하였다. EA 상을 무수 Na2SO4로 건조시키고 농축하여 백색 고체로서 화합물 6 (340 mg, 조생성물)을 수득하였다.
단계 6: DMF (5 ml) 중의 6 (100 mg, 조생성물), 아민 (79.6 mg, 0.274 mmol), EDCI (71 mg, 0.549 mmol), HOAT (75 mg, 0.549 mmol), DMAP (3.4 mg, 0.027 mmol), DIPEA (142 mg, 1.1 mmol)의 용액을 55℃에서 밤새 교반하였다. EA (100 ml)를 혼합물에 첨가한 후 이를 물 (50 ml×3)로 세척하고 농축하여 갈색 오일로서 화합물 7 (200 mg, 조생성물)을 수득하였다.
단계 7: DCM (2 ml) 중의 7 (200 mg, 조생성물)의 용액에 TFA (2 ml)를 첨가하고 결과 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 농축하여 갈색 오일로서 화합물 8 (200 mg, 20%)을 수득하였다.
단계 8: DCM (5 ml) 중의 화합물 8 (100 mg, 조생성물)의 용액에 DIPEA (88.8 mg, 0.688 mmol) 및 (44.6 mg, 0.275 mmol)를 첨가하였다. 반응을 0℃에서 1시간 동안 교반하였다. 혼합물을 농축하고 Pre-HPLC로 정제하여 백색 고체로서 화합물 002 (34 mg, 35%)를 수득하였다.
1H NMR (400 MHz, DMSO) δ 9.75 (s, 1H), 9.64 (s, 1H), 8.84 (s, 2H), 7.58 (s, 1H), 7.47 (d, J = 1.9 Hz, 1H), 7.39 (d, J = 4.9 Hz, 1H), 7.35 (dd, J = 8.3, 2.1 Hz, 1H), 7.28 (d, J = 3.3 Hz, 1H), 7.09 - 7.04 (m, 1H), 6.88 (d, J = 8.4 Hz, 1H), 3.64 (d, J = 10.5 Hz, 2H), 3.50 (d, J = 11.9 Hz, 2H), 3.37 (d, J = 8.4 Hz, 2H), 2.98 (dd, J = 30.8, 11.3 Hz, 6H), 2.81 (s, 3H), 2.61 - 2.55 (m, 1H), 2.38 (d, J = 11.7 Hz, 2H), 1.51 (t, J = 11.1 Hz, 2H), 0.86 (d, J = 6.9 Hz, 6H). LCMS: m/z = 563 (M+H).
실시예 3: 화합물 003의 합성

단계 1: 1,4-디옥산 (20 ml) 중의 2-Cl-피리미딘 (1.86 g, 10 mmol), 아민 (3.00, 15 mmol), 및 NEt3 (3.0 g, 30 mmol)의 혼합물을 95℃에서 밤새 교반하였다. 혼합물을 농축하고, EA (60 ml) 및 수성 시트르산 (60 mL)을 첨가하고, 결과 혼합물을 30분간 교반하였다. 유기층을 분리하고, 건조시키고, 농축하여 연한 황색 고체로서 화합물 2 (3.4 g, 수율: 97%)를 수득하였다.
단계 2: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 2 (3.5 g, 10 mmol) 및 NaOH (2 M, 15 ml)의 혼합물을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축하고, pH <7이 될 때까지 수성 시트르산을 첨가하였다. 결과 혼합물을 30분간 교반하고 여과하여 연한 황색 고체로서 화합물 3 (2.8 g, 수율: 90%)을 수득하였다.
단계 3: DMF (25 ml) 중의 화합물 3 (3.2 g, 10 mmol), 화합물 아민 (2.9 g, 10 mmol), HOAT (2.0 g, 15 mmol), EDCI (3.8 g, 20 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA (100 ml) 및 수성 포화 NaCl (100 ml)을 혼합물에 첨가하고 결과 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)을 세척하고, 건조 및 농축하여 잔사를 생성하고, 이를 CH3CN (10~20 ml)으로 세척하여 회색 고체로서 화합물 4 (2.9 g, 50%)를 수득하였다.
단계 4: DCM (30 ml) 중의 화합물 4 (2.9 g, 5 mmol)의 용액을 실온에서 2시간 동안 TFA (5 ml)에 첨가하였다. 혼합물을 농축하여 추가의 정제 없이 화합물 5 (2.9 g, 100%)를 수득하였다.
단계 5: DCM (5 ml) 중의 화합물 4 (197 mg, 0.5 mmol) 및 NEt3 (250 mg, 2.5 mmol)의 용액에 모르폴린-4-카르보닐 클로라이드 (194 mg, 0.65 mmol)를 0℃에서 첨가하였다. LCMS를 사용하여 반응의 종결을 모니터링하였다. NH3-H2O (0.5 ml)를 반응 혼합물에 첨가한 후 이를 30분간 교반하고 잔사를 농축하였다. 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 003 (114 mg, 45%)을 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.54 (s, 1H), 8.85 (s, 2H), 7.88 (d, J = 7.9 Hz, 1H), 7.44 (d, J = 1.9 Hz, 1H), 7.35 (d, J = 5.0 Hz, 1H), 7.29 (dd, J = 8.3, 2.1 Hz, 1H), 7.24 (d, J = 3.4 Hz, 1H), 7.05 (dd, J = 4.9, 3.7 Hz, 1H), 6.79 (d, J = 8.3 Hz, 1H), 5.21 (s, 2H), 4.01 (dd, J = 7.2, 3.3 Hz, 1H), 3.68 - 3.51 (m, 7H), 3.18 - 3.04 (m, 5H), 2.88 (t, J = 11.7 Hz, 2H), 1.86 (d, J = 10.0 Hz, 2H), 1.45 (dd, J = 20.5, 11.3 Hz, 2H). LCMS: m/z = 508 (M+H)
실시예 4: 화합물 004의 합성
단계 1: Tol (50 ml) 중의 아민 1 (1 g, 4.67 mmol), 메틸 4-브로모벤조에이트 (1 g, 4.67 mmol), Pd2(dba)3 (428 mg, 0.47 mmol), Ruphos (218 mg, 0.47 mmol), Cs2CO3 (4.5 g, 14.0 mmol)의 용액을 N2로 세정하고 98℃에서 밤새 교반하였다. 혼합물을 여과, 농축하고, 실리카 겔 크로마토그래피 (PE:EA=5:1-1:1)로 정제하여 황색 고체로서 화합물 2 (1.1 g, 65%)를 수득하였다.
단계 2: EtOH (수성 95%, 5 ml) 중의 화합물 2 (1.1 g)의 혼합물에 NaOH (2 M, 5 ml)를 첨가하고 결과 용액을 55℃에서 2시간 동안 교반하였다. 물 (50 ml)을 혼합물에 첨가하고 시트르산을 이용해 pH를 7로 조정하였다. 결과 혼합물을 EA (50 ml×3)로 추출하였다. EA 상을 무수 Na2SO4를 사용해 건조시키고 농축하여 백색 고체로서 화합물 3 (1 g, 조생성물)을 수득하였다.
단계 3: DMF (20 ml) 중의 화합물 3 (800 mg, 조생성물), 아민 (694 mg, 2.395 mmol), EDCI (618 mg, 4.790 mmol), HOAT (651 mg, 4.790 mmol), DMAP (29 mg, 0.239 mmol), 및 DIPEA (927 mg, 7.186 mmol)의 용액을 55℃에서 3일간 교반하였다. EA (100 ml)를 혼합물에 첨가하고 결과 용액을 물 (50 ml×3)로 세척하고, 농축하고, 실리카 겔 크로마토그래피 (PE:EA=6:1-1:1)로 정제하여 갈색 고체로서 화합물 4 (650 mg,45%)를 수득하였다.
단계 4: DCM (2 ml) 중의 화합물 4 (300 mg)의 용액에 TFA (2 ml)를 첨가하고 결과 용액을 실온에서 2시간 동안 교반하였다. 혼합물을 농축하여 갈색 오일로서 화합물 5 (400 mg, 조생성물)를 수득하였다.
단계 5: DCM (20 ml) 중의 화합물 5 (200 mg, 조생성물)의 용액에 DIPEA (190 mg, 1.478 mmol) 및 모르폴린-4-카르보닐 클로라이드 (110 mg, 0.739 mmol)를 첨가하였다. 결과 반응 혼합물을 0℃에서 1시간 동안 교반하였다. 혼합물을 농축하고 Pre-HPLC로 정제하여 백색 고체로서 화합물 004 (67.4 mg)를 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.85 (s, 1H), 7.81 (t, J = 21.9 Hz, 2H), 7.58 (d, J = 1.7 Hz, 1H), 7.53 - 7.42 (m, 2H), 7.40 (d, J = 3.1 Hz, 1H), 7.11 (dd, J = 5.0, 3.6 Hz, 2H), 6.92 (s, 2H), 3.63 - 3.47 (m, 4H), 3.31 - 3.01 (m, 8H), 1.98 (d, J = 14.0 Hz, 2H), 1.63 (d, J = 12.9 Hz, 2H), 1.37 (s, 3H). LCMS: m/z = 520 (M+H).
실시예 5: 화합물 005의 합성
단계 1: 톨루엔 (20 ml) 중의 화합물 1 (1 g, 5 mmol) 및 메틸 4-브로모벤조에이트 (1.1 g, 5 mmol)의 용액에 Pd2(dba)3 (230 mg, 0.25 mmol), Ruphos (290 mg, 0.5 mmol) 및 Cs2CO3 (4.9 g, 15 mmol)를 첨가하였다. 반응을 N2 대기 하에서 95℃에서 밤새 교반하였다. 출발 물질이 완전히 소비된 후, 이종 혼합물을 규조토를 통해 여과하고 진공 농축하여 점성 오일을 수득하고, 이를 실리카 겔 컬럼으로 정제하여 화합물 2 (1 g, 60%)를 수득하였다.
단계 2: 화합물 2 (1 g, 3 mmol)를 MeOH (10 ml) 및 THF (10 ml)에 용해시켰다. 그 후에 2 N NaOH (15 ml)를 용액 내에 첨가하였다. 반응을 55℃에서 1시간 동안 교반하였다. 반응 혼합물을 농축하여 용매를 제거하고 pH를 약 4~5 사이로 조정하고 EA로 추출하였다. 유기상을 소금물로 세척하고 Na2SO4 상에서 건조시켰다. 유기상의 농축은 백색 고체로서 화합물 3 (800 mg, 82%)을 생성하였다.
단계 3: DMF (10 ml) 중의 화합물 3 (500 mg, 1.56 mmol) 및 아민 (453 mg, 1.56 mmol)의 용액에 HOAT (421 mg, 3.1 mmol), EDCI (592 mg, 3.1 mmol) 및 DIPEA (400 mg, 3.1 mmol)를 첨가하였다. 반응을 55℃에서 밤새 교반하였다. 반응을 물로 ??칭하고 EA로 추출하였다. 유기상을 소금물로 세척하고 Na2SO4 상에서 건조시켰다. 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 4 (800 mg, 80%)를 수득하였다.
단계 4: DCM (10 ml) 중의 화합물 4 (800 mg, 1.35 mmol)의 용액에 TFA (5 ml)를 첨가하였다. 용액을 실온에서 30분간 교반하였다. 용액을 농축하여 회색 고체로서 화합물 5 (600 mg, 100%)를 수득하였다.
단계 5: 화합물 5 (80 mg, 0.20 mmol) 및 4-메틸피페라진-1-카르보닐 클로라이드 히드로클로라이드 (40 mg, 0.20 mmol)의 용액에 트리에틸아민 (100 mg, 1 mmol)을 0℃에서 첨가하였다. 반응을 0℃에서 2시간 동안 교반한 후 실리카 겔을 통해 여과하였다. 농축 및 Pre-HPLC로 정제하여 백색 고체로서 화합물 004 (15 mg, 15%)를 수득하였다.
1H NMR (400 MHz, DMSO) δ 9.37 (s, 1H), 7.78 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 2.1 Hz, 1H), 7.36 (dd, J = 5.1, 1.0 Hz, 1H), 7.28 (d, J = 2.1 Hz, 1H), 7.26 (d, J = 2.2 Hz, 1H), 7.24 (dd, J = 3.5, 1.1 Hz, 1H), 7.05 (dd, J = 5.1, 3.6 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.65 (d, J = 8.8 Hz, 2H), 6.20 (d, J = 8.1 Hz, 1H), 5.08 (s, 2H), 3.57 (d, J = 13.0 Hz, 3H), 3.15 (s, 4H), 2.91 (t, J = 11.3 Hz, 2H), 2.33 (s, 3H), 2.20 (s, 3H), 1.91 (d, J = 10.2 Hz, 2H), 1.41 - 1.24 (m, 2H). LCMS: m/z = 519 (M+H).
실시예 6: 화합물 006의 합성

단계 1-4: 화합물 5를 수득하기 위해 실시예 5의 단계 1-4를 참조한다.
단계 5: 화합물 5 (100 mg, 0.25 mmol) 및 피롤리딘-1-카르보닐 클로라이드 (34 mg, 0.25 mmol)의 용액에 트리에틸아민 (100 mg, 1 mmol)을 0℃에서 첨가하였다. 반응을 0℃에서 2시간 동안 교반한 후 실리카 겔을 통해 여과하였다. 농축 및 Pre-HPLC로 정제하여 백색 고체로서 화합물 006 (10 mg, 8%)을 수득하였다.
1H NMR (400 MHz, DMSO) δ 9.50 (s, 1H), 7.79 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 2.1 Hz, 1H), 7.40 (d, J = 4.2 Hz, 1H), 7.33 (dd, J = 8.3, 2.1 Hz, 1H), 7.29 (d, J = 2.7 Hz, 1H), 7.07 (dd, J = 5.1, 3.6 Hz, 1H), 6.91 (d, J = 8.3 Hz, 1H), 6.66 (d, J = 8.8 Hz, 2H), 3.63 (d, J = 13.2 Hz, 2H), 3.53 (s, 1H), 3.26 (t, J = 6.5 Hz, 4H), 2.87 (t, J = 11.2 Hz, 2H), 1.91 (d, J = 10.8 Hz, 2H), 1.75 (t, J = 6.5 Hz, 5H), 1.34 (dd, J = 20.6, 10.1 Hz, 2H). LCMS: m/z = 490 (M+H).
실시예 7: 화합물 007의 합성
단계 1: 리튬 비스(트리메틸실릴)아미드 (THF 중의 1.0 M 용액, 240 ml, 240 mmol)의 용액을 화합물 1 (25 g, 120 mmol) 함유 둥근-바닥 플라스크 내로 질소 대기 하 -76℃에서 서서히 첨가하였다. 혼합물을 -76℃에서 4시간 동안 교반하였다. 그 후에, 이오도메탄 (15 ml, 240 mmol)을 시스템 내에 첨가하였다. 반응 혼합물을 -76℃에서 30분간 교반한 후, 실온으로 가온하고 밤새 교반하였다. 반응 혼합물을 150 ml 포화 수성 NH4Cl로 ??칭하고, 물로 희석하고 EtOAc로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 연한 황색 고체로서 표적 화합물 2 (25 g, 93%)을 제공하였다.
단계 2: DMSO (120 ml) 중의 화합물 2 (25 g, 111 mmol)의 용액 내로 K2CO3 (31 g, 224 mmol)을 첨가하였다. H2O2 (100 ml)를 60℃에서 서서히 시스템 내로 첨가하고 반응을 60℃에서 밤새 교반하였다. 종결 후, 시스템을 냉수로 ??칭하고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 3 (26 g, 96%)을 수득하였다.
단계 3: 200 ml CH3CN 중의 화합물 3 (26 g, 107 mmol)의 용액에 5 N KOH (100 ml)를 첨가하였다. 그 후에 1,3-디브로모-5,5-디메틸이미다졸리딘-2,4-디온 (15 g, 54 mmol)을 시스템 내로 첨가하였다. 반응을 밤새 교반하였다. 종결 후, 혼합물을 농축하여 CH3CN을 제거하고, 수상의 pH를 빙조에서 2 N HCl을 이용해 5로 조정하고, EA로 추출하고 분리하였다. 그 후에 수상의 pH를 10으로 조정하였다. 침전물을 수집하여 백색 고체로서 화합물 4 (16.1 g, 69%)를 제공하였다.
단계 4: 1,4-디옥산 (25 ml) 중의 화합물 4 (2 g, 9.34 mmol)의 용액에 에틸 2-클로로피리미딘-5-카르복실레이트 (2.6 g, 14.02 mmol) 및 DIPEA (5.3 g, 28.03 mmol)를 첨가하였다. 반응을 95℃에서 밤새 교반하였다. 농축 및 EA/PE = 1/5를 이용한 실리카 겔 컬럼의 정제에 의해 연한 황색 고체로서 화합물 5 (1.8 g, 53%)를 제공하였다.
단계 5: THF (10 ml) 및 EtOH (10 ml) 중의 화합물 5 (465 mg, 1.28 mmol) 및 2 N NaOH (10 ml, 20 mmol)의 용액을 55℃에서 2시간 동안 교반하였다. 농축하고 수상의 pH를 5-6으로 조정한 후 이어서 EA (2×15 ml)로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 화합물 6 (400 mg, 93%)을 제공하였다.
단계 6: DMF (10 ml) 중의 화합물 6 (400 mg, 1.19 mmol), tert-부틸 2-아미노-4-(티오펜-2-일)페닐카르바메이트 (345 mg, 1.19 mmol), EDCI (307 mg, 2.38 mmol) 및 DMAP (290 mg, 2.38 mmol)의 용액을 형성하였다. 반응을 55℃에서 밤새 교반하였다. 종결 후, 혼합물을 물 내로 첨가하고 EA (2×15 ml)로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 그 후에 EA/PE=1/2를 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 7 (400 mg, 55%)을 제공하였다.
단계 7: 1,4-디옥산 (10 ml) 중의 화합물 7 (400 mg, 0.65 mmol)의 용액에 HCl/1,4-디옥산 (5 ml, 20 mmol)을 실온에서 첨가하였다. 농축 및 PE로 세척하여 회색 고체로서 화합물 8 (350 mg, 100%)을 제공하였다.
단계 8: THF (10 ml) 중의 화합물 8 (200 mg, 0.45 mmol) 및 Et3N (101 mg, 2.2 eq)의 용액에 페닐 카르보노클로리데이트 (78 mg, 0.5 mmol)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 종결 후, 혼합물을 농축하고 Prep-HPLC로 정제하여 화합물 007 (66 mg, 28%)을 제공하였다.
1H NMR (500 MHz, DMSO) δ 9.71 (s, 1H), 8.88 (s, 2H), 7.68 (s, 1H), 7.50 (s, 1H), 7.42-7.35 (m, 4H), 7.30 (d, J = 2.9 Hz, 1H), 7.22 (t, J = 7.3 Hz, 1H), 7.12 (d, J = 7.9 Hz, 2H), 7.10-7.06 (m, 1H), 6.91 (d, J = 8.2 Hz, 1H), 3.76 (d, J = 60.0 Hz, 2H), 3.32 (d, J = 79.9 Hz, 2H), 2.40 (s, 2H), 1.65 (s, 2H), 1.48 (s, 3H). LCMS: m/z = 529 (M+H)+.
실시예 8: 화합물 008의 합성
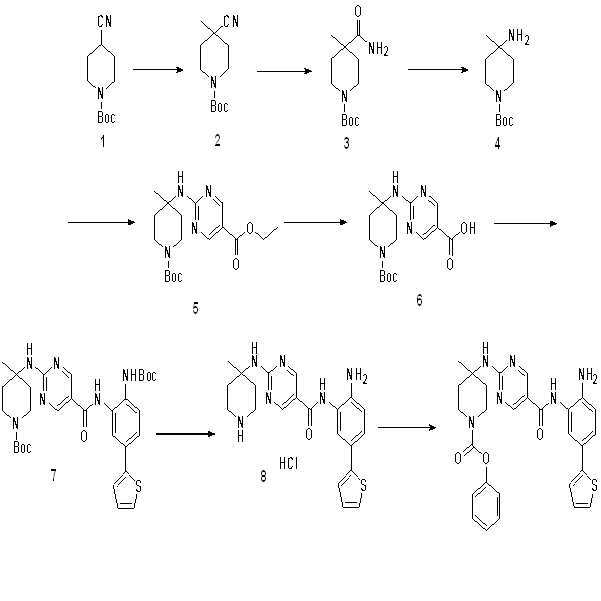
단계 1-7: 화합물 8을 수득하기 위해 실시예 7의 단계 1-7을 참조한다.
단계 8: THF (5 ml) 중의 화합물 8 (100 mg, 0.22 mmol) 및 Et3N (44 mg, 0.44 mmol)의 용액에 모르핀-1-카르보닐 클로라이드 (36 mg, 1.1 eq)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 종결 후, 혼합물을 농축하고 Prep-HPLC로 정제하여 화합물 008 (50 mg, 44%)을 제공하였다.
1H NMR (500 MHz, DMSO) δ 9.52 (s, 1H), 8.85 (s, 2H), 7.56 (s, 1H), 7.44 (s, 1H), 7.35 (d, J = 5.0 Hz, 1H), 7.29 (d, J = 8.3 Hz, 1H), 7.23 (d, J = 3.1 Hz, 1H), 7.07 - 7.03 (m, 1H), 6.79 (d, J = 8.4 Hz, 1H), 5.22 (s, 2H), 3.56 (s, 4H), 3.08 (d, J = 22.9 Hz, 6H), 2.31 (d, J = 12.9 Hz, 2H), 1.56 (t, J = 10.1 Hz, 2H), 1.42 (s, 3H). LCMS: m/z = 522 (M+H)+
실시예 9: 화합물 009의 합성

단계 1: 1,4-디옥산 (20 ml) 중의 화합물 1 (1.86 g, 10 mmol), 화합물 Boc-아민 (3.0 g, 15 mmol), 및 NEt3 (3.0 g, 30 mmol)의 혼합물을 95℃에서 밤새 교반하였다. 혼합물을 농축하고, EA (60 ml) 및 수성 시트르산 (60 ml)을 첨가한 후, 혼합물을 30분간 교반하였다. 유기층을 분리하고, 건조시키고 농축하여 연한 황색 고체로서 화합물 2 (3.4 g, 97%)를 수득하였다.
단계 2: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 2 (3.5 g, 10 mmol) 및 NaOH (2 M, 15 ml)의 혼합물을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축하고, 수성 시트르산을 첨가하여 pH <7로 조정한 후, 혼합물을 30분간 교반하고, 여과하여 연한 황색 고체로서 화합물 3 (2.8 g, 90%)을 제공하였다.
단계 3: DMF (25 ml) 중의 화합물 3 (3.2 g, 10 mmol), 화합물 Boc-아민 (2.9 g, 10 mmol), HOAT (2.0 g, 15 mmol), 및 EDCI (3.8 g, 20 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 수득하고, 이를 CH3CN (10-20 ml)으로 세척하여 회색 고체로서 화합물 4 (2.9 g, 50%)를 수득하였다.
단계 3: DCM (30 ml) 중의 화합물 4 (2.9 g, 5 mmol)의 용액에 TFA (5 ml)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 농축하여 추가의 정제 없이 화합물 5 (2.9 g, 100%)를 제공하였다.
단계 4: DCM (5 ml) 중의 화합물 5 (197 mg, 0.5 mmol) 및 NEt3 (250 mg, 2.5 mmol)의 용액에 피페리딘-1-카르보닐 클로라이드 (96 mg, 0.65 mmol)를 0℃에서 첨가하였다. 반응이 종결될 때까지 LCMS로 모니터링하였다. 혼합물에 NH3H2O (0.5 ml)를 첨가하고, 혼합물을 30분간 교반하고 농축하여 잔사를 수득하고, 이를 실리카 겔 컬럼으로 정제하여 화합물 009 (114 mg, 45%)를 제공하였다.
1H NMR (500 MHz, DMSO) δ 9.69 (s, 1H), 8.86 (s, 2H), 7.89 (d, J = 8.0 Hz, 1H), 7.49 (s, 1H), 7.40 (d, J = 5.1 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.30 (d, J = 3.3 Hz, 1H), 7.07 (dd, J = 5.0, 3.7 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 3.99 (s, 1H), 3.56 (d, J = 11.5 Hz, 2H), 3.10 (s, 4H), 2.84 (t, J = 11.6 Hz, 2H), 1.85 (d, J = 10.7 Hz, 2H), 1.52 (s, 2H), 1.46 (d, J = 8.3 Hz, 6H). LCMS: m/z = 506 (M+H)+.
실시예 10: 화합물 010의 합성

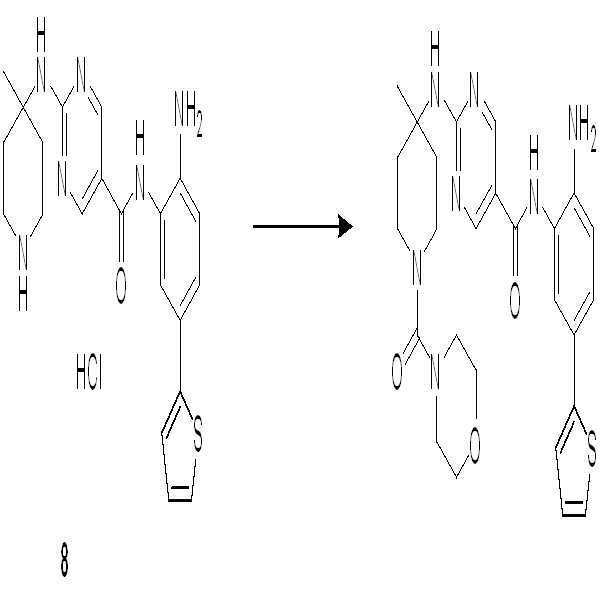
단계 1-7: 화합물 8을 수득하기 위해 실시예 7의 단계 1-7을 참조한다.
단계 8: THF (5 ml) 중의 화합물 8 (100 mg, 0.22 mmol) 및 Et3N (44 mg, 0.45 mmol)의 용액에 피페라진-1-카르보닐 클로라이드 (60 mg, 0.24 mmol)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 종결 후, 혼합물을 농축하여 오일로서 조 화합물 9 (110 mg, 조생성물)를 제공하였다.
단계 9: DCM (5 ml) 중의 화합물 9 (100 mg)의 용액에 TFA (1 ml)를 실온에서 40분간 첨가하였다. 종결 후, 혼합물을 농축하고 Prep-HPLC로 정제하여 황색 고체로서 화합물 010 (35 mg, 30%, 2 단계)을 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.70 (s, 1H), 8.86 (s, 2H), 8.79 (s, 2H), 7.63 (s, 1H), 7.49 (d, J = 1.8 Hz, 1H), 7.40 (d, J = 5.0 Hz, 1H), 7.36 (dd, J = 8.3, 1.9 Hz, 1H), 7.29 (d, J = 3.2 Hz, 1H), 7.10-7.04 (m, 1H), 6.90 (d, J = 8.3 Hz, 1H), 3.37 (s, 1H), 3.29 (s, 4H), 3.09 (s, 6H), 2.32 (d, J = 13.6 Hz, 2H), 1.56 (t, J = 10.2 Hz, 2H), 1.43 (s, 3H). LCMS: m/z = 521 (M+H)+.
실시예 11: 화합물 011의 합성
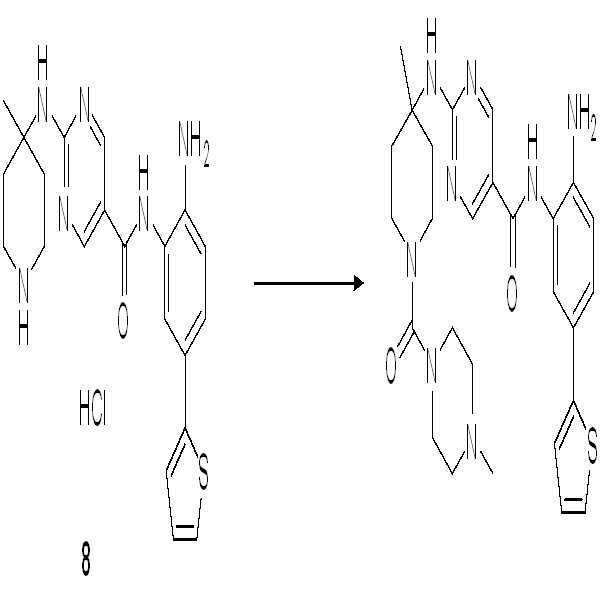
단계 1-7: 화합물 8을 수득하기 위해 실시예 7의 단계 1-7을 참조한다.
단계 8: THF (5 ml) 중의 화합물 8 (100 mg, 0.22 mmol) 및 Et3N (45 mg, 0.45 mmol)의 용액에 4-메틸피페라진-1-카르보닐 클로라이드 (40 mg, 0.24 mmol)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 실리카 겔을 통해 여과하고 EA로 세척하였다. 농축하고 Prep-HPLC로 정제하여 화합물 011 (42 mg, 36%)을 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.88 (s, 1H), 9.72 (s, 1H), 8.86 (s, 2H), 7.64 (s, 1H), 7.49 (d, J = 1.8 Hz, 1H), 7.41 (d, J = 5.0 Hz, 1H), 7.37 (dd, J = 8.3, 2.0 Hz, 1H), 7.30 (d, J = 2.9 Hz, 1H), 7.08 (dd, J = 4.9, 3.7 Hz, 1H), 6.92 (d, J = 8.4 Hz, 1H), 3.63 (d, J = 11.2 Hz, 2H), 3.38 (d, J = 8.2 Hz, 3H), 3.15-2.97 (m, 6H), 2.81 (s, 3H), 2.33 (d, J = 13.5 Hz, 2H), 1.57 (t, J = 10.1 Hz, 2H), 1.43 (s, 3H). LCMS: m/z = 535 (M+H)+.
실시예 12: 화합물 012의 합성

단계 1: THF (5 ml) 중의 화합물 6 (95 mg, 0.79 mmol) 및 Et3N (159 mg, 1.6 mmol)의 용액에 비스(트리클로로메틸)카보네이트 (119 mg, 0.4 mmol)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하고 다음 단계 (단계 2)에서 바로 사용하였다.
단계 2: 화합물 8을 수득하기 위해 실시예 7의 단계 1-7을 참조한다. THF (5 ml) 중의 화합물 8 (162 mg, 0.4 mmol) 및 Et3N (80 mg, 0.8 mmol)의 용액을 화합물 7을 함유하는 단계 1의 반응 혼합물의 용액에 첨가하였다. 반응을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하고 Pre-HPLC로 정제하여 화합물 012 (35 mg, 16%)를 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.73 (s, 1H), 8.86 (s, 2H), 7.62 (s, 1H), 7.50 (s, 1H), 7.42 (d, J = 5.0 Hz, 1H), 7.38 (d, J = 8.4 Hz, 1H), 7.31 (d, J = 3.1 Hz, 1H), 7.08 (dd, J = 5.0, 3.7 Hz, 1H), 6.93 (d, J = 8.1 Hz, 1H), 3.34 (s, 1H), 3.31 (s, 1H), 3.23 (d, J = 5.6 Hz, 4H), 3.08 (t, J = 10.8 Hz, 2H), 2.31 (d, J = 13.7 Hz, 2H), 1.94 (d, J = 13.9 Hz, 4H), 1.57 (t, J = 10.0 Hz, 2H), 1.43 (s, 3H).LCMS: m/z = 556 (M+H)+.
실시예 13: 화합물 013의 합성

단계 1: tol (30 ml) 중의 메틸 4-이오도벤조에이트 (2.6 g, 10 mmol), 화합물 4 (6.4 g, 30 mmol), Pd2(dba)3 (915 mg, 1 mmol), Ruphos (467 mg, 1 mmol) 및 Cs2CO3 (9.75g, 30 mmol, 3eq)의 혼합물을 질소 대기 하 95℃에서 밤새 교반하였다. 혼합물을 냉각시키고, EA (100 ml)를 첨가하였다. 혼합물을 여과하고 농축하여 잔사를 수득하고, 이를 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 5 (2.2 g, 60%)를 수득하였다.
단계 2: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 5 (500 mg, 1.4 mmol)의 용액에 수성 NaOH (2 M, 15 ml)를 첨가하고, 혼합물을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하고, 물 (100 ml)을 첨가한 후 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정하였고, 그 후에 여과하여 백색 고체로서 화합물 6 (369 mg, 80%)을 제공하였다.
단계 3: DMF (5 ml) 중의 화합물 6 (150 mg, 0.45 mmol), 화합물 Boc-아민 (130 mg, 0.45 mmol), HOAT (103 mg, 0.76 mmol), EDCI (145 mg, 0.76 mmol) 및 NEt3 (154 mg, 1.52 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 수득하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 7 (123 mg, 45%)을 제공하였다.
단계 4: DCM (5 ml) 중의 화합물 7 (120 mg, 0.20 mmol)의 용액에 TFA (3 ml)를 실온에서 2시간 동안 첨가하였다. 혼합물을 농축하여 화합물 8 (80 mg, 100%)을 제공하고, 이를 추가 정제 없이 다음 단계에 사용하였다.
단계 5: THF (5 ml) 중의 화합물 8 (80 mg, 0.20 mmol) 및 Et3N (106 mg, 1.05 mmol)의 용액에 화합물 피롤리딘-1-카르보닐 클로라이드 (74 mg, 0.30 mmol)를 0℃에서 첨가하고 2시간 동안 교반하였다. 혼합물에 EA (50 ml) 및 수성 포화 NaCl (50 ml)을 첨가하고, 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 제공하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 013 (99 mg, 80%)을 수득하였다.
1H NMR (500 MHz, DMSO) δ 9.35 (s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 2.0 Hz, 1H), 7.36 (d, J = 5.1 Hz, 1H), 7.27 (dd, J = 8.3, 2.1 Hz, 1H), 7.24 (d, J = 3.5 Hz, 1H), 7.05 (dd, J = 5.0, 3.6 Hz, 1H), 6.79 (dd, J = 8.5, 3.5 Hz, 3H), 5.89 (s, 1H), 5.07 (d, J = 12.4 Hz, 2H), 3.31 (d, J = 7.8 Hz, 1H), 3.25 (t, J = 6.4 Hz, 5H), 3.10 (t, J = 10.5 Hz, 2H), 1.97 (d, J = 13.9 Hz, 2H), 1.74 (s, 4H), 1.59 (t, J = 9.9 Hz, 2H), 1.37 (s, 3H).LCMS: m/z = 504 (M+H)+.
실시예 14: 화합물 014의 합성
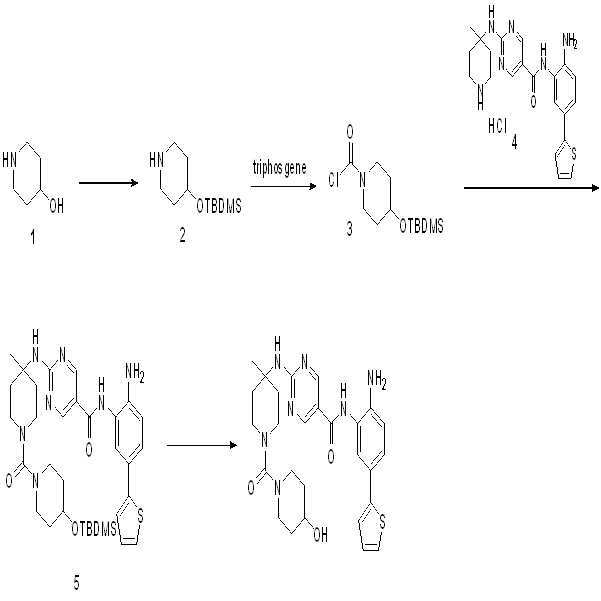
단계 1: CH2Cl2 (25 ml) 중의 4-히드록시피페리딘 (화합물 1, 0.8 g, 7.9 mmol), 피리딘 (1.3 g, 16 mmol) 및 tert-부틸디메틸실릴 클로라이드 (1.4 g, 9.5 mmol)의 용액을 0℃에서 형성하였다. 혼합물을 3시간 동안 교반하였다. 종결 후, 혼합물을 농축하고 EA:PE = 1:5를 이용한 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 2 (1.5 g, 88%)를 수득하였다.
단계 2: THF (5 ml) 중의 화합물 2 (95 mg, 0.44 mmol) 및 Et3N (89 mg, 0.9 mmol)의 용액에 트리포스젠 (74 mg, 0.25 mmol)을 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하고 다음 단계에서 바로 사용하였다.
단계 3: THF (5 ml) 중의 화합물 4 (162 mg, 0.4 mmol) 및 Et3N (80 mg, 0.8 mmol)의 용액에 화합물 3을 함유하는 단계 2로부터의 상기 용액을 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하고 임의의 추가 정제 없이 다음 단계에 사용하였다.
단계 4: THF (5 ml) 중의 화합물 5 (조 혼합물)의 용액에 TBAF (5 방울)를 0℃에서 첨가하였다. 혼합물을 실온에서 2시간 동안 교반한 후, 농축하고 Prep-HPLC로 정제하여 화합물 014 (29 mg, 15%, 3 단계)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.67 (s, 1H), 8.86 (s, 2H), 7.56 (s, 1H), 7.48 (s, 1H), 7.39 (d, J = 4.9 Hz, 1H), 7.37-7.31 (m, 1H), 7.28 (d, J = 3.3 Hz, 1H), 7.10-7.03 (m, 1H), 6.88 (d, J = 8.3 Hz, 1H), 3.59 (s, 3H), 3.42 (s, 1H), 3.25 (d, J = 13.6 Hz, 2H), 3.03 (t, J = 11.0 Hz, 2H), 2.83 (t, J = 10.6 Hz, 2H), 2.29 (d, J = 13.5 Hz, 2H), 1.70 (d, J = 9.9 Hz, 2H), 1.57 (t, J = 10.3 Hz, 2H), 1.44 (d, J = 10.4 Hz, 3H), 1.34-1.27 (m, 2H).LCMS: m/z = 536 (M+H)+.
실시예 15: 화합물 015의 합성

단계 1-4: 화합물 8을 수득하기 위해 실시예 13의 단계 1-4를 참조한다.
단계 5: THF (5 ml) 중의 화합물 8 (80 mg, 0.20 mmol) 및 Et3N (106 mg, 1.05 mmol)의 용액에 tert-부틸 4-(클로로카르보닐)피페라진-1-카르복실레이트 (74 mg, 0.30 mmol)를 0℃에서 첨가하고, 혼합물을 2시간 동안 교반하였다. 혼합물에 EA (20 ml) 및 수성 포화 NaCl (20 ml)을 첨가하고, 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 수득하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 9 (99 mg, 80%)를 수득하였다.
단계 6: DCM (5 ml) 중의 화합물 9 (90 mg, 0.15 mmol)의 용액에 TFA (3 ml)를 서서히 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하여 잔사를 수득하고, 이를 Prep-HPLC로 정제하여 백색 고체로서 화합물 015 (45 mg, 60%)를 수득하였다.
1H NMR (400 MHz, DMSO) δ 9.58 (s, 1H), 8.78 (s, 2H), 7.78 (t, J = 9.6 Hz, 2H), 7.52 (d, J = 2.1 Hz, 1H), 7.42 (d, J = 5.1 Hz, 1H), 7.36 (dd, J = 8.3, 2.1 Hz, 1H), 7.31 (d, J = 3.5 Hz, 1H), 7.08 (dd, J = 5.0, 3.6 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 8.4 Hz, 2H), 5.97 (s, 6H), 3.18 (t, J = 10.6 Hz, 2H), 3.10 (s, 4H), 2.02-1.93 (m, 2H), 1.60 (t, J = 9.7 Hz, 2H), 1.37 (s, 3H).LCMS: m/z = 519 (M+H)+.
실시예 16: 화합물 016의 합성

단계 1-4: 화합물 8을 수득하기 위해 실시예 13의 단계 1-4를 참조한다.
단계 5: THF (5 ml) 중의 화합물 8 (80 mg, 0.20 mmol) 및 Et3N (106 mg, 1.05 mmol)의 용액에 4-메틸피페라진-1-카르보닐 클로라이드 (49 mg, 0.30 mmol)를 0℃에서 첨가하고, 혼합물을 2시간 동안 교반하였다. 혼합물을 Prep-TLC로 정제하여 황색 고체로서 화합물 016 (49 mg, 46%)을 수득하였다.
1H NMR (400 MHz, DMSO) δ 9.79 (s, 1H), 9.58 (s, 1H), 7.79 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 2.1 Hz, 1H), 7.42 (d, J = 4.2 Hz, 1H), 7.36 (dd, J = 8.3, 2.1 Hz, 1H), 7.31 (d, J = 3.5 Hz, 1H), 7.08 (dd, J = 5.0, 3.6 Hz, 1H), 6.95 (d, J = 8.3 Hz, 1H), 6.84 (d, J = 8.3 Hz, 2H), 3.64 (d, J = 11.6 Hz, 2H), 3.43-3.28 (m, 4H), 3.18 (t, J = 10.3 Hz, 2H), 3.10-2.96 (m, 4H), 2.81 (s, 3H), 1.99 (d, J = 13.4 Hz, 2H), 1.60 (t, J = 9.7 Hz, 2H), 1.37 (s, 3H).LCMS: m/z = 533 (M+H)+.
실시예 17: 화합물 017의 합성

단계 1: 둥근-바닥 플라스크 내 리튬 비스(트리메틸실릴)아미드 (THF 중의 1.0 M, 240 ml, 240 mmol)의 용액에 화합물 1 (25 g, 120 mmol)을 질소 대기 하 -76℃에서 서서히 첨가하였다. 혼합물을 4시간 동안 -76℃에서 교반한 후, 이오도에탄 (17 ml, 240 mmol)을 시스템 내로 한 방울씩 첨가하였다. 반응 혼합물을 추가 30분간 교반한 후 실온으로 가온하고 밤새 교반하였다. 잔사를 150 ml 포화 수성 NH4Cl로 ??칭하고, 물로 희석하고 EtOAc로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 연한 황색 고체로서 표적 화합물 2 (20 g, 70%)를 제공하였다.
단계 2: 화합물 2 (20 g, 84 mmol)의 용액에 DMSO (120 ml) 중의 K2CO3 (23 g, 168 mmol)를 첨가하였다. H2O2 (100 ml)를 60℃에서 매우 서서히 시스템에 첨가한 후 반응을 60℃에서 밤새 교반하였다. 냉수를 첨가하고, 혼합물을 EA (3*100 ml)로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 3 (21 g, 99%)을 수득하였다.
단계 3: CH3CN (200 ml) 및 5 N KOH (100 ml) 중의 화합물 3 (20.5 g, 80 mmol)의 용액에 1,3-디브로모-5,5-디메틸이미다졸리딘-2,4-디온 (11.1 g, 40 mmol)을 첨가하였다. 혼합물을 실온에서 밤새 교반하였다. 종결 후, 혼합물을 농축하여 CH3CN을 제거하였다. 2 N HCl을 사용하여 빙조에서 수상의 pH를 5로 조정하고, 혼합물을 EA로 추출하고 분리하였다. 그 후에 수상의 pH를 10으로 조정하였다. 침전물을 수집하여 백색 고체로서 화합물 4 (10 g, 55%)를 제공하였다.
단계 4: 톨루엔 (25 ml) 중의 화합물 4 (1.0 g, 4.4 mmol), 에틸 4-브로모벤조에이트 (943 mg, 4.4 mmol), Pd2(dba)3 (202 mg, 0.22 mmol), Ruphos (103 mg, 0.22 mmol) 및 Cs2CO3 (2.9 g, 8.8 mmol)의 용액을 100℃에서 밤새 교반하였다. 혼합물을 농축하고 EA:PE = 1:5를 이용한 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 5 (1.0 g, 59%)를 제공하였다.
단계 5: THF (10 ml) 및 EtOH (10 ml) 중의 화합물 5 (233 mg, 0.65 mmol) 및 2 N NaOH (10 ml, 20 mmol)의 용액을 55℃에서 2시간 동안 교반하였다. 농축하고 수상의 pH를 5-6으로 조정한 후, 이어서 EA (3×15 ml)로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 화합물 6 (200 mg, 93%)을 제공하였다.
단계 6: DMF (10 ml) 중의 화합물 6 (200 mg, 0.6 mmol), tert-부틸 2-아미노-4-(티오펜-2-일)페닐카르바메이트 (173 mg, 0.6 mmol), EDCI (154 mg, 1.2 mmol) 및 DMAP (145 mg, 1.2 mmol)의 혼합물을 55℃에서 밤새 교반하였다. 혼합물을 물로 희석하고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 그 후에 EA:PE=1:2를 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 7 (200 mg, 55%)을 수득하였다.
단계 7: 1,4-디옥산 (10 ml) 중의 화합물 7 (200 mg, 0.32 mmol)의 용액에 HCl/1,4-디옥산 (5 ml, 20 mmol)을 첨가하고, 혼합물을 실온에서 밤새 교반하였다. 농축하고 PE로 세척하여 회색 고체로서 화합물 8 (175 mg, 100%)을 제공하였다.
단계 8: THF (5 ml) 중의 화합물 8 (95 mg, 0.21 mmol) 및 Et3N (106 mg, 1.05 mmol)의 용액에 모르폴린-4-카르보닐 클로라이드 (50 mg, 0.3 mmol)를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 실리카 겔을 통해 여과하고 EA로 세척하였다. 농축하고 Prep-HPLC로 정제하여 화합물 017 (10 mg, 9%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.56 (s, 1H), 7.76 (d, J = 8.7 Hz, 2H), 7.52 (s, 1H), 7.42 (d, J = 4.8 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.31 (d, J = 3.0 Hz, 1H), 7.11-7.04 (m, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.81 (d, J = 8.5 Hz, 2H), 5.86 (s, 1H), 3.55 (s, 8H), 3.11 (s, 4H), 2.02 (d, J = 13.3 Hz, 2H), 1.76 (d, J = 6.6 Hz, 2H), 1.50 (t, J = 10.9 Hz, 2H), 0.75 (t, J = 7.3 Hz, 3H).LCMS: m/z = 534 (M+H)+.
실시예 18: 화합물 018의 합성

단계 1: 디옥산 (20 ml) 중의 화합물 1 (1.2 g, 5 mmol)의 용액에 DIPEA (1.3 g, 10 mmol) 및 에틸 2-클로로피리미딘-5-카르복실레이트 (5.5 mmol, 1.0 g)를 첨가하였다. 혼합물을 100℃에서 밤새 교반하였다. 종결 후, 농축하고 PE:EA=2:1를 이용한 Prep-TLC로 직접 정제하여 백색 고체로서 화합물 2 (1.37 g, 70%)를 제공하였다.
단계 2: EtOH (5 ml) 및 THF (5 ml) 중의 화합물 2 (220 mg, 0.56 mmol)의 용액에 수성 NaOH (2 M, 2 ml)를 첨가하였다. 반응을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하였다. 잔사에 물 (10 ml)을 첨가하고, 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정하고, 혼합물을 여과하여 백색 고체로서 화합물 3 (200 mg, 조생성물)을 제공하였다.
단계 3: DMF (10 ml) 중의 화합물 3 (200 mg, 조생성물), 화합물 tert-부틸 2-아미노-4-(티오펜-2-일)페닐카르바메이트 (160 mg, 0.55 mmol), EDCI (154 mg, 1.2 mmol) 및 DMAP (145 mg, 1.2 mmol)의 혼합물을 67℃에서 밤새 교반하였다. 혼합물을 물로 희석하고 EA로 추출하여 황색 오일로서 화합물 4 (200 mg, 55%)를 제공하였다.
단계 4: DCM (10 ml) 중의 화합물 4 (200 mg, 0.32 mmol)의 용액에 TFA (2 ml)를 첨가하고, 혼합물을 실온에서 30분간 교반하였다. 농축하고 PE로 세척하여 갈색 오일로서 화합물 5 (250 mg, 조생성물)를 제공하였다.
단계 5: THF (5 ml) 중의 화합물 5 (250 mg, 조생성물) 및 Et3N (106 mg, 1.05 mmol)의 용액에 피롤리딘-1-카르보닐 클로라이드 (70 mg, 0.5 mmol)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 실리카 겔을 통해 여과하고 EA로 세척하였다. 농축하고 Prep-HPLC로 정제하여 화합물 018 (61 mg, 60%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.72 (s, 1H), 8.91 (s, 2H), 7.53 (s, 1H), 7.42 (d, J = 4.8 Hz, 1H), 7.39 (d, J = 8.3 Hz, 1H), 7.32 (d, J = 3.2 Hz, 1H), 7.12- 7.04 (m, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.85-6.13 (m, 1H), 3.70 (dd, J = 13.5, 7.5 Hz, 4H), 3.29 (s, 4H), 3.07 (t, J = 10.5 Hz, 2H), 2.81 (t, J = 12.8 Hz, 2H), 2.11 (dd, J = 14.5, 7.9 Hz, 2H), 1.93-1.84 (m, 2H), 1.76 (s, 4H), 1.32 (d, J = 12.1 Hz, 2H). LCMS: m/z = 532 (M+H)+.
실시예 19: 화합물 019의 합성

단계 1: 톨루엔 (30 ml) 중의 메틸 4-브로모벤조에이트 (2.1 g, 10 mmol, 1 eq), 화합물 1 (6.0 g, 30 mmol, 3 eq), Pd2(dba)3 (915 mg, 1 mmol, 0.1 eq), Xantphos (478 mg, 1 mmol, 0.1 eq) 및 Cs2CO3 (9.75 g, 30 mmol, 3 eq)의 혼합물을 질소 대기 하 95℃에서 밤새 교반하였다. 혼합물을 냉각시키고, EA (100 ml)를 첨가하고, 여과하고 농축하여 잔사를 수득하고, 이를 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 2 (1.97 g, 59%)를 수득하였다.
단계 2: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 2 (3.34 g, 10 mmol)의 용액에 수성 NaOH (2 M, 15 ml)를 첨가하고 60℃에서 5시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하였다. 혼합물을 물 (100 ml)로 희석하고, 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정하고 여과하여 백색 고체로서 화합물 3 (3.07 g, 96%)을 제공하였다.
단계 3: Py (15 ml) 중의 화합물 3 (3.2 g, 10 mmol, 1 eq), Boc-아민 (2.6 g, 9 mmol, 0.9 eq), EDCI (5.7 g, 30 mmol, 3 eq)의 혼합물을 25℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 시트르산 (50 ml)을 첨가하고, 혼합물을 30분간 교반하였다. 유기층을 분리하고, 건조시키고 농축하여 잔사를 수득하고, 이를 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 4 (3.7 g, 70%)를 제공하였다.
단계 4: DCM (20 ml) 중의 화합물 4 (2.96 g, 5 mmol)의 용액에 TFA (20 ml)를 첨가하고, 혼합물을 실온에서 1시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하였다. 혼합물에 물 (100 ml) 및 NaOH (2 M 용액)를 첨가하여 pH >7로 조정하고, 혼합물을 여과하여 연한 황색 고체로서 화합물 5 (1.86 g, 95%)를 제공하였다.
단계 5: THF (5 ml) 중의 화합물 5 (100 mg, 0.25 mmol) 및 Et3N (50 mg, 0.5 mmol)의 용액에 tert-부틸 4-(클로로카르보닐)피페라진-1-카르복실레이트 (68 mg, 0.275 mmol)를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 종결 후, 혼합물을 농축하여 다음 단계를 위한 조 화합물 6 (110 mg)을 제공하였다.
단계 6: DCM (5 ml) 중의 화합물 6 (110 mg, 조생성물)의 용액에 TFA (2 ml)를 서서히 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하여 잔사를 수득하고, 이를 Prep-HPLC로 정제하여 백색 고체로서 화합물 019 (40 mg, 32%, 2 단계)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.37 (s, 1H), 8.71 (s, 2H), 7.79 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 2.1 Hz, 1H), 7.36 (d, J = 5.1 Hz, 1H), 7.30-7.20 (m, 3H), 7.05 (dd, J = 5.1, 3.6 Hz, 1H), 6.81 (d, J = 8.3 Hz, 1H), 6.66 (d, J = 8.8 Hz, 2H), 3.63 (d, J = 13.5 Hz, 2H), 3.48-3.42 (m, 1H), 3.30 (s, 4H), 3.11 (s, 4H), 2.96 (t, J = 11.9 Hz, 2H), 1.92 (d, J = 9.8 Hz, 2H), 1.34 (d, J = 10.8 Hz, 2H), 1.05 (t, J = 7.0 Hz, 1H). LCMS: m/z = 505 (M+H)+.
실시예 20: 화합물 020의 합성

단계 1-4: 화합물 5를 수득하기 위해 실시예 19의 단계 1-4를 참조한다.
단계 5: THF (5 ml) 중의 화합물 5 (100 mg, 0.25 mmol) 및 Et3N (50 mg, 0.5 mmol)의 용액에 tert-부틸 4-(클로로카르보닐)피페라진-1-카르복실레이트 (41 mg, 0.275 mmol)를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하여 잔사를 수득하고, 이를 Prep-HPLC로 정제하여 백색 고체로서 화합물 020 (32 mg, 25%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.37 (s, 1H), 7.78 (d, J = 8.6 Hz, 2H), 7.45 (d, J = 2.0 Hz, 1H), 7.36 (d, J = 4.4 Hz, 1H), 7.30 - 7.20 (m, 2H), 7.05 (dd, J = 5.0, 3.7 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.65 (d, J = 8.8 Hz, 2H), 6.20 (d, J = 7.9 Hz, 1H), 5.07 (s, 2H), 3.60-3.52 (m, 6H), 3.18 - 3.06 (m, 4H), 2.92 (t, J = 11.6 Hz, 2H), 2.08 (s, 1H), 1.91 (d, J = 10.2 Hz, 2H), 1.33 (dd, J = 20.7, 9.8 Hz, 2H). LCMS: m/z = 506 (M+H)+.
실시예 21: 화합물 021의 합성

단계 1-4: 화합물 5를 수득하기 위해 실시예 19의 단계 1-4를 참조한다.
단계 5: THF (5 ml) 중의 화합물 5 (100 mg, 0.25 mmol) 및 Et3N (50 mg, 0.5 mmol)의 용액에 사이클로헥산카르보닐 클로라이드 (41 mg, 0.275 mmol)를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하여 잔사를 수득하고, 이를 Prep-HPLC로 정제하여 백색 고체로서 화합물 021 (30 mg, 24%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.61 (s, 1H), 7.81 (d, J = 8.6 Hz, 2H), 7.53 (d, J = 1.8 Hz, 1H), 7.43 (d, J = 4.8 Hz, 1H), 7.38 (d, J = 8.3 Hz, 1H), 7.33 (d, J = 3.3 Hz, 1H), 7.13-7.05 (m, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.68 (d, J = 8.7 Hz, 2H), 4.29 (d, J = 11.3 Hz, 1H), 3.92 (d, J = 12.5 Hz, 1H), 3.66-3.57 (m, 1H), 3.27-3.13 (m, 1H), 2.85-2.73 (m, 1H), 2.67-2.56 (m, 1H), 2.03-1.88 (m, 2H), 1.75-1.58 (m, 5H), 1.37-1.12 (m, 7H). LCMS: m/z = 503 (M+H)+.
실시예 22: 화합물 022의 합성

단계 1-4: 화합물 5를 수득하기 위해 실시예 19의 단계 1-4를 참조한다.
단계 5: THF (5 ml) 중의 화합물 5 (100 mg, 0.25 mmol) 및 Et3N (50 mg, 0.5 mmol)의 용액에 테트라히드로-2H-피란-4-카르보닐 클로라이드 (41 mg, 0.275 mmol)를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. 그 후에 혼합물을 농축하여 잔사를 수득하고, 이를 Prep-HPLC로 정제하여 백색 고체로서 화합물 022 (20 mg, 16%)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.57 (s, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.52 (s, 1H), 7.46-7.29 (m, 3H), 7.1-7.06 (m, 1H), 6.96 (s, 1H), 6.68 (d, J = 8.6 Hz, 2H), 4.29 (d, J = 14.4 Hz, 1H), 3.97 (d, J = 13.9 Hz, 1H), 3.85 (d, J = 9.7 Hz, 2H), 3.71-3.55 (m, 1H), 3.39 (t, J = 11.5 Hz, 2H), 3.22 (t, J = 11.6 Hz, 1H), 2.98-2.87 (m, 1H), 2.81 (t, J = 12.1 Hz, 1H), 2.08-1.86 (m, 2H), 1.68-1.43 (m, 4H), 1.39-1.13 (m, 2H). LCMS: m/z = 505 (M+H)+.
실시예 23: 화합물 023의 합성

단계 1-7: 화합물 8을 수득하기 위해 실시예 17의 단계 1-7을 참조한다.
단계 8: THF (5 ml) 중의 화합물 8 (175 mg, 0.4 mmol) 및 Et3N (106 mg, 1.05 mmol)의 혼합물에 tert-부틸 4-(클로로카르보닐) 피페라진-1-카르복실레이트 (125 mg, 0.5 mmol)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 실리카 겔을 통해 여과하고 EA로 세척하여 황색 오일로서 화합물 9 (175 mg, 67%)를 수득하였다.
단계 9: 1,4-디옥산 (10 ml) 중의 화합물 9 (175 mg, 0.28 mmol)의 용액에 실온에서 HCl/1,4-디옥산 (5 ml, 20 mmol)을 첨가하고 이어서 밤새 교반하였다. 농축하고 PE로 세척하여 황색 고체로서 표적 화합물 023 (92 mg, 63%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 10.21 (s, 1H), 9.30 (s, 3H), 7.91 (d, J = 8.1 Hz, 2H), 7.81 (s, 1H), 7.59 (d, J = 5.9 Hz, 2H), 7.51 (d, J = 3.3 Hz, 1H), 7.45 (d, J = 8.4 Hz, 1H), 7.19-7.12 (m, 1H), 6.90 (s, 2H), 3.48-3.41 (m, 1H), 3.33 (s, 4H), 3.18-3.10 (m, 2H), 3.06 (s, 4H), 2.04 (d, J = 13.0 Hz, 2H), 1.77 (d, J = 7.0 Hz, 2H), 1.59-1.48 (m, 2H), 1.08-1.02 (m, 2H), 0.78 (t, J = 7.0 Hz, 3H). LCMS: m/z = 533 (M+H)+.
실시예 24: 화합물 024의 합성

단계 1: TFA (5 ml) 중의 화합물 2 (300 mg, 0.90 mmol)의 용액을 실온에서 밤새 교반하였다. 그 후에 농축하고 PE로 세척하여 회색 고체로서 표적 화합물 3 (200 mg, 95%)을 수득하였다.
단계 2: DMF (10 ml) 중의 화합물 3 (180 mg, 0.77 mmol), 1-히드록시사이클로헥산-카르복실산 (111 mg, 0.77 mmol), HATU (351 mg, 0.92 mmol) 및 DIPEA (199 mg, 1.5 mmol)의 혼합물을 형성하였다. 반응을 실온에서 2시간 동안 교반하였다. 혼합물을 물에 용해시키고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 그 후에 EA:PE=1:2를 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 4 (150 mg, 54%)를 수득하였다.
단계 3: THF (10 ml) 및 EtOH (10 ml) 중의 화합물 4 (150 mg, 0.42 mmol) 및 2 N NaOH (10 ml, 20 mmol)의 용액을 60℃에서 6시간 동안 교반하였다. 그 후에 혼합물을 농축하고, 수상의 pH를 5-6로 조정하고 이어서 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 5 (130 mg, 90%)을 수득하였다.
단계 4: DMF (10 ml) 중의 화합물 5 (70 mg, 0.2 mmol), tert-부틸 2-아미노-4-(티오펜-2-일)페닐카르바메이트 (59.0 mg, 0.2 mmol), EDCI (39.0 mg, 0.3 mmol), HOAT (41 mg, 0.3 mmol) 및 DIPEA (52 mg, 0.4 mmol)의 혼합물을 형성하였다. 혼합물을 65℃에서 밤새 교반하였다. 그 후에 혼합물을 물에 용해시키고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 그 후에 EA:PE=1:2를 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 6 (20 mg, 16%)을 수득하였다.
단계 5: DCM (10 ml) 중의 화합물 6 (20 mg, 0.03 mmol)의 용액에 HCl/1,4-디옥산 (2 ml, 8.0 mmol)을 첨가하고 이어서 실온에서 밤새 교반하였다. 농축하고 PE로 세척하여 백색 고체로서 표적 화합물 024 (12 mg, 71%)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.41 (s, 1H), 7.80 (d, J = 8.5 Hz, 2H), 7.46 (s, 1H), 7.36 (d, J = 4.9 Hz, 1H), 7.30-7.22 (m, 2H), 7.09-7.03 (m, 1H), 6.80 (d, J = 8.4 Hz, 1H), 6.66 (d, J = 8.6 Hz, 2H), 6.26 (d, J = 7.9 Hz, 1H), 5.24 (s, 1H), 5.09 (s, 2H), 2.04-1.87 (m, 3H), 1.76-1.39 (m, 10H), 1.35-1.14 (m, 6H). LCMS: m/z = 519 (M+H)+.
실시예 25: 화합물 025의 합성

단계 1: 화합물 3을 수득하기 위해 실시예 24의 단계 1을 참조한다.
단계 2: DMF (10 ml) 중의 화합물 3 (400 mg, 1.7 mmol), 1-히드록시사이클로펜탄-카르복실산 (222 mg, 1.7 mmol), HATU (969 mg, 2.6 mmol) 및 DIPEA (439 mg, 3.4 mmol)의 혼합물을 형성하였다. 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 물에 용해시키고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 그 후에 EA:PE=1:2를 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 4 (300 mg, 51%)를 제공하였다.
단계 3: THF (10 ml) 및 EtOH (10 ml) 중의 화합물 4 (300 mg, 0.87 mmol)의 용액에 2 N NaOH (10 ml, 20 mmol)를 60℃에서 첨가하고, 결과 반응 혼합물을 6시간 동안 교반하였다. 농축하고 수상의 pH를 5-6으로 조정한 후 이어서 EA (2×15 ml)로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 5 (250 mg, 87%)를 수득하였다.
단계 4: DMF (10 ml) 중의 화합물 5 (250 mg, 0.75 mmol), tert-부틸 2-아미노-4-(티오펜-2-일)페닐카르바메이트 (218 mg, 0.75 mmol), EDCI (143.0 mg, 1.1 mmol), HOAT (147 mg, 1.1 mmol) 및 DIPEA (194 mg, 1.5 mmol)의 혼합물을 형성하였다. 반응을 65℃에서 밤새 교반하였다. 그 후에 혼합물을 물에 용해시키고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. 그 후에 EA:PE=1:2를 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 6 (50 mg, 11%)을 수득하였다.
단계 5: DCM 중의 화합물 6 (50 mg, 0.08 mmol)의 용액에 HCl/1,4-디옥산 (2 ml, 8.0 mmol)을 첨가하였다. 혼합물을 실온에서 밤새 교반하였다. 농축하고 PE로 세척하여 백색 고체로서 표적 화합물 025 (13 mg, 32%)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.61 (s, 1H), 7.81 (d, J = 8.6 Hz, 2H), 7.53 (s, 1H), 7.46-7.30 (m, 3H), 7.11-7.07 (m, 1H), 6.98 (d, J = 8.0 Hz, 1H), 6.68 (d, J = 8.7 Hz, 2H), 4.57-4.44 (m, 1H), 4.36-4.19 (m, 1H), 3.24-3.17 (m, 1H), 2.90-2.79 (m, 1H), 2.39-2.25 (m, 1H), 2.08-1.91 (m, 4H), 1.74-1.63 (m, 4H), 1.57-1.52 (m, 2H), 1.38-1.22 (m, 2H).LCMS: m/z = 505 (M+H)+.
실시예 26: 화합물 026의 합성

단계 1: THF (5 ml) 및 EtOH (5 ml) 중의 화합물 1 (300 mg, 0.88 mmol)의 혼합물에 2 M NaOH를 (5 m에 걸쳐) 첨가하고, 결과 반응 혼합물을 60℃에서 5시간 동안 교반하였다. 혼합물을 농축하여 잔사를 제공하고, 이를 플래시 칼럼으로 정제하여 황색 고체로서 화합물 2 (180 mg, 수율: 65%)를 수득하였다.
단계 2: DMF (4 ml) 중의 화합물 2 (150 mg, 0.48 mmol), 화합물 Boc-아민 (139 mg, 0.48 mmol), HOAT (131 mg, 096 mmol), 및 EDCI (183 mg, 0.96 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (30 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 제공하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 3 (100 mg, 35%)을 수득하였다.
단계 3: DCM (3 ml) 중의 화합물 3 (100 mg, 0.17 mmol)의 용액에 TFA (3 ml)를 첨가하고, 결과 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하고, 이를 Prep-HPLC로 정제하여 백색 고체로서 화합물 026 (20 mg, 24%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.53 (s, 1H), 8.76 (d, J = 86.5 Hz, 2H), 8.41 (d, J = 13.8 Hz, 1H), 7.43-7.38 (m, 3H), 7.37-7.31 (m, 3H), 7.29 (dd, J = 8.3, 2.2 Hz, 1H), 7.22 (dd, J = 7.7, 4.2 Hz, 2H), 7.04 (dd, J = 5.1, 3.6 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 3.46 (d, J = 11.2 Hz, 2H), 3.34-3.20 (m, 2H), 2.96-2.90 (m, 2H), 2.86 (d, J = 4.5 Hz, 3H), 2.26-2.08 (m, 2H). LCMS: m/z = 485 (M+H)+.
실시예 27: 화합물 027의 합성

단계 1: 톨루엔 (100 ml) 중의 화합물 1 (3.0 g, 15 mmol), 브로모벤젠 (7.0 g, 45 mmol), Pd2(dba)3 (1.4 g, 1.5 mmol), Ruphos (700 mg, 1.5 mmol) 및 Cs2CO3 (14.6 g, 45 mmol)의 혼합물을 질소 대기 하 95℃에서 밤새 교반하였다. 혼합물을 여과하고 농축한 후 이어서 PE (30 ml)로 세척하여 연한 황색 고체로서 화합물 2 (3.5 g, 수율: 85%)를 제공하였다.
단계 2: 1,4-디옥산 (50 ml) 중의 화합물 2 (3.5 g, 12.6 mmol)의 용액에 1,4-디옥산 (9.45 ml, 37.8 mmol) 중의 HCl을 실온에서 첨가하고, 혼합물을 밤새 교반하였다. 혼합물을 여과하여 백색 고체로서 화합물 3 (2.0 g, 90%)을 제공하였다.
단계 3: 1,4-디옥산 (15 ml) 중의 화합물 3 (500 mg, 2.8 mmol), 에틸 2-클로로피리미딘-5-카르복실레이트 (352 mg, 1.9 mmol), 및 NEt3 (576 mg, 5.7 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 시트르산 (30 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하고, PE (30 ml)로 세척하여 황색 고체로서 화합물 4 (500 mg, 81%)를 수득하였다.
단계 4: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 4 (500 mg, 1.5 mmol)의 용액에 수성 NaOH (2 M, 15 ml)를 첨가하고, 결과 반응 혼합물을 60℃에서 5시간 동안 교반하였다. 혼합물에 수성 포화 시트르산을 첨가하여 pH <7로 조정한 후 이어서 여과하여 백색 고체로서 화합물 5 (400 mg, 87%)를 수득하였다.
단계 5: DMF (5 ml) 중의 화합물 5 (150 mg, 0.5mmol), 화합물 Boc-아민 (145 mg, 0.5 mmol), HOAT (136 mg, 1 mmol), EDCI (191 mg, 1 mmol) 및 NEt3 (202 mg, 2 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하고, CH3CN (10-20 mL)으로 세척하여 황색 고체로서 화합물 6 (140 mg, 49%)을 제공하였다.
단계 6: DCM (5 ml) 중의 화합물 6 (140 mg, 0.25 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하고 Prep-HPLC로 정제하여 백색 고체로서 화합물 027 (110 mg, 95%)을 제공하였다.
1H NMR (500 MHz, DMSO) δ 9.52 (s, 1H), 8.86 (s, 2H), 7.87 (d, J = 7.8 Hz, 1H), 7.45 (s, 1H), 7.35 (d, J = 4.9 Hz, 1H), 7.29 (d, J = 8.3 Hz, 1H), 7.25-7.18 (m, 3H), 7.07-7.02 (m, 1H), 6.96 (d, J = 8.2 Hz, 2H), 6.83-6.73 (m, 2H), 5.20 (s, 2H), 3.72 (d, J = 12.2 Hz, 2H), 2.82 (t, J = 11.7 Hz, 2H), 1.99 (s, 1H), 1.96 (d, J = 10.5 Hz, 2H), 1.69-1.58 (m, 2H). LCMS: m/z = 471 (M+H)+.
실시예 28: 화합물 028의 합성

단계 1-2: 화합물 3을 수득하기 위해 실시예 27의 단계 1-2를 참조한다.
단계 3: DMSO (10 ml) 중의 화합물 3 (300 mg, 1.7 mmol), 4-플루오로벤조니트릴 (181 mg, 1.5 mmol), 및 K2CO3 (414 mg, 3 mmol)의 혼합물을 100℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 시트르산 (30 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하고, 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 4 (300 mg, 72%)를 제공하였다.
단계 4: 6 M HCl (20 ml) 중의 화합물 4 (300 mg, 1.1 mmol)의 용액을 80℃에서 3일간 교반하였다. 혼합물을 여과하여 백색 고체로서 화합물 5 (250 mg, 78%)를 제공하였다.
단계 5: DMF (5 ml) 중의 화합물 5 (150 mg, 0.5mmol), 화합물 Boc-아민 (145 mg, 0.5 mmol), HOAT (136 mg, 1 mmol), EDCI (191 mg, 1 mmol) 및 NEt3 (202 mg, 2 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (80 ml) 및 수성 포화 NaCl (80 ml)를 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (30 ml×2)로 세척하고, 건조시키고 농축하고, Prep-TLC로 정제하여 황색 고체로서 화합물 6 (50 mg, 18%)을 제공하였다.
단계 6: DCM (5 ml) 중의 화합물 6 (50 mg, 0.09 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하고 Prep-HPLC로 정제하여 백색 고체로서 화합물 028 (5 mg, 12%)을 제공하였다.
1H NMR (500 MHz, DMSO) δ 9.36 (s, 1H), 7.79 (d, J = 8.6 Hz, 2H), 7.46 (d, J = 1.9 Hz, 1H), 7.35 (d, J = 5.0 Hz, 1H), 7.27 (dd, J = 8.3, 2.0 Hz, 1H), 7.25-7.18 (m, 3H), 7.08-7.03 (m, 1H), 6.96 (d, J = 8.2 Hz, 2H), 6.80 (d, J = 8.3 Hz, 1H), 6.75 (t, J = 7.2 Hz, 1H), 6.67 (d, J = 8.7 Hz, 2H), 6.22 (d, J = 8.0 Hz, 1H), 5.07 (s, 2H), 3.70 (d, J = 12.8 Hz, 2H), 3.53 (s, 1H), 2.89 (t, J = 11.0 Hz, 2H), 2.01 (d, J = 10.8 Hz, 2H), 1.51 (dd, J = 20.6, 10.5 Hz, 2H). LCMS: m/z = 469 (M+H)+.
실시예 29: 화합물 029의 합성
단계 1- 2: 화합물 6을 수득하기 위해 실시예 13의 단계 1-2를 참조한다.
단계 3: DMF (25 ml) 중의 화합물 6 (630 mg, 1.89 mmol), 화합물 Boc-아민 (484 mg, 1.7 mmol), HOAT (510 mg, 3.78 mmol), EDCI (721 mg, 3.78 mmol), DIPEA (487 mg, 3.78 mmol) 및 DMAP (461 mg, 3.78 mmol)의 혼합물을 67℃에서 3일간 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하고, 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 7 (473 mg, 46%)을 제공하였다.
단계 4: DCM (10 ml) 중의 화합물 7 (250 mg, 0.42 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하여 화합물 8 (300 mg, 조생성물)을 제공하고 추가의 정제 없이 다음 단계에서 사용하였다.
단계 5: THF (5 ml) 중의 화합물 8 (150 mg, 조생성물) 및 Et3N (50 mg, 0.5 mmol)의 용액에 화합물 모르폴린-4-카르보닐 클로라이드 (37 mg, 0.25 mmol)를 0℃에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물에 EA (10 ml) 및 수성 포화 NaCl (10 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (10 ml×2)로 세척하고, 건조시키고 농축하고, Prep-HPLC로 정제하여 백색 고체로서 화합물 029 (44 mg, 35%)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.36 (s, 1H), 8.51 (dd, J = 4.7, 1.5 Hz, 2H), 7.76 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 2.1 Hz, 1H), 7.57 (dd, J = 4.7, 1.6 Hz, 2H), 7.47 (dd, J = 8.4, 2.2 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 6.79 (d, J = 8.8 Hz, 2H), 5.87 (s, 1H), 5.29 (s, 2H), 3.60-3.51 (m, 4H), 3.32-3.24 (m, 2H), 3.17-3.08 (m, 6H), 1.98 (d, J = 13.8 Hz, 2H), 1.64-1.55 (m, 2H), 1.36 (s, 3H). LCMS: m/z = 515 (M+H)+.
실시예 30: 화합물 030의 합성
단계 1-3: 화합물 8을 수득하기 위해 실시예 29의 단계 1-3를 참조한다.
단계 3: THF (5 ml) 중의 화합물 8 (150 mg, 조생성물) 및 Et3N (50 mg, 0.5 mmol)의 용액에 화합물 피롤리딘-1-카르보닐 클로라이드 (33 mg, 0.25 mmol)를 0℃에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물에 EA (10 ml) 및 수성 포화 NaCl (10 ml)를 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (10 ml×2)로 세척하고, 건조시키고 농축하고, Prep-HPLC로 정제하여 백색 고체로서 화합물 030 (17 mg, 14%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.34 (d, J = 10.2 Hz, 1H), 8.51 (d, J = 5.5 Hz, 2H), 7.76 (d, J = 8.6 Hz, 2H), 7.67 (d, J = 1.8 Hz, 1H), 7.58 (d, J = 5.9 Hz, 2H), 7.48 (dd, J = 8.3, 1.9 Hz, 1H), 6.88 (d, J = 8.4 Hz, 1H), 6.79 (d, J = 8.6 Hz, 2H), 5.87 (s, 1H), 5.31 (s, 2H), 3.31-3.20 (m, 6H), 3.10 (t, J = 10.5 Hz, 2H), 1.98 (d, J = 13.5 Hz, 2H), 1.74 (s, 4H), 1.59 (t, J = 10.1 Hz, 2H), 1.37 (s, 3H).LCMS: m/z = 499 (M+H)+.
실시예 31: 화합물 031의 합성

단계 1: DMF (25ml) 중의 화합물 6 (270 mg, 0.8 mmol), 화합물 Boc-아민 (205 mg, 0.72 mmol), HOAT (216 mg, 1.6 mmol), EDCI (305 mg, 1.6 mmol) DIPEA (206 mg, 1.6 mmol) 및 DMAP (195 mg, 1.6 mmol)의 혼합물을 67℃에서 3일간 교반하였다. 혼합물에 EA (60 ml) 및 수성 포화 NaCl (60 ml)를 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (20 ml×2)로 세척하고, 건조시키고 농축하고, 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 7 (400 mg, 83%)을 제공하였다.
단계 2: DCM (10 ml) 중의 화합물 7 (200 mg, 0.33 mmol)의 용액에 TFA (3 ml)를 첨가하고, 결과 반응 혼합물을 실온에서 2시간 동안 교반하였다. 혼합물을 농축하여 화합물 8 (250 mg, 조생성물)을 제공하고 추가의 정제 없이 다음 단계에 사용하였다.
단계 3: THF (5 ml) 중의 화합물 8 (150 mg, 조생성물) 및 Et3N (50 mg, 0.5 mmol)의 용액에 화합물 모르폴린-4-카르보닐 클로라이드 (37 mg, 0.25 mmol)를 0℃에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물에 EA (10 ml) 및 수성 포화 NaCl (10 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (10 ml×2)로 세척하고, 건조시키고 농축하고, Prep-HPLC로 정제하여 백색 고체로서 화합물 031 (55 mg, 60%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.57 (s, 1H), 9.09 (s, 1H), 8.76-8.58 (m, 2H), 8.01-7.69 (m, 4H), 7.56 (d, J = 7.2 Hz, 1H), 7.05-6.87 (m, 3H), 3.56 (s, 4H), 3.17 (d, J = 39.8 Hz, 8H), 1.96 (s, 2H), 1.61 (s, 2H), 1.37 (s, 3H).LCMS: m/z = 515 (M+H)+.
실시예 32: 화합물 032의 합성

단계 1- 2: 화합물 3을 수득하기 위해 실시예 19의 단계 1-2를 참조한다.
단계 3: CH3CN (5 ml) 중의 화합물 3 (150 mg, 0.45mmol), 화합물 Boc-아민 (130 mg, 0.45 mmol), HOAT (103 mg, 0.76 mmol), EDCI (145 mg, 0.76 mmol) 및 NEt3 (154 mg, 1.52 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (10 ml) 및 수성 포화 NaCl (10 ml)를 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (10 ml×2)로 세척하고, 건조시키고 농축하고, 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 4 (90 mg, 30%)를 제공하였다.
단계 4: DCM (5 ml) 중의 화합물 4 (90 mg, 0.15 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하여 화합물 5 (58 mg, 조생성물)를 제공하고 추가의 정제 없이 다음 단계에 사용하였다.
단계 5: THF (5 ml) 중의 화합물 5 (58 mg, 0.15 mmol) 및 Et3N (33 mg, 0.33 mmol)의 용액에 화합물 피롤리딘-1-카르보닐 클로라이드 (20 mg, 015 mmol)를 0℃에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 반응 혼합물을 Prep-HPLC로 정제하여 백색 고체로서 화합물 032 (12 mg, 17%)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.37 (s, 1H), 8.58 (d, J = 5.1 Hz, 2H), 7.79 (d, J = 8.8 Hz, 4H), 7.76 (d, J = 2.1 Hz, 1H), 7.59 (dd, J = 8.5, 2.2 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 6.66 (d, J = 8.8 Hz, 2H), 6.22 (d, J = 7.4 Hz, 1H), 3.63 (d, J = 13.2 Hz, 2H), 3.26 (t, J = 6.4 Hz, 4H), 2.87 (t, J = 11.3 Hz, 2H), 1.91 (d, J = 10.0 Hz, 2H), 1.75 (t, J = 6.5 Hz, 4H), 1.34 (dd, J = 20.2, 10.4 Hz, 2H).LCMS: m/z = 485 (M+H)+.
실시예 33: 화합물 033의 합성
단계 1: 1,4-디옥산 (20 ml) 중의 화합물 1 (2.1 g, 9.6 mmol), 에틸 2-클로로피리미딘-5-카르복실레이트 (600 mg, 3.2 mmol) 및 NEt3 (970 mg, 9.6 mmol)의 혼합물을 95℃에서 밤새 교반하였다. 혼합물을 농축한 후 이어서 EA (60 ml) 및 수성 시트르산 (60 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 건조시키고 농축하여 연한 황색 고체로서 화합물 2 (880 mg, 수율: 75%)를 제공하였다.
단계 2: DCM (10 ml) 중의 화합물 2 (880 mg, 2.4 mmol)의 용액에 TFA (5 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하여 황색 고체로서 화합물 3 (630 mg, 99%)을 제공하였다.
단계 3: DMF (15 ml) 중의 화합물 3 (630 mg, 2.4 mmol), 2-인돌아세트산 (531 mg, 3 mmol), HOAT (816 mg, 6 mmol), EDCI (1.1 g, 6 mmol) 및 NEt3 (1.2 g, 12 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 수득하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 4 (600 mg, 60%)를 수득하였다.
단계 4: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 4 (600 mg, 1.4 mmol)의 용액에 수성 NaOH (2 M, 15 ml)를 첨가하고, 결과 반응 혼합물을 60℃에서 5시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하였다. 혼합물에 물 (100 ml) 및 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정한 후, 이어서 여과하여 백색 고체로서 화합물 5 (400 mg, 72%)를 수득하였다.
단계 5: DMF (5 ml) 중의 화합물 5 (150 mg, 0.38 mmol), 화합물 Boc-아민 (108 mg, 0.38 mmol), HOAT (103 mg, 0.76 mmol), EDCI (145 mg, 0.76 mmol) 및 NEt3 (154 mg, 1.52 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (20 ml) 및 수성 포화 NaCl (20 mL)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (20 ml×2)로 세척하고, 건조시키고 농축하고, Prep-TLC로 정제하여 백색 고체로서 화합물 6 (150 mg, 59%)을 제공하였다.
단계 6: DCM (5 ml) 중의 화합물 6 (150 mg, 0.23 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하고 Prep-HPLC로 정제하여 백색 고체로서 화합물 033 (28 mg, 22%)을 제공하였다.
1H NMR (500 MHz, DMSO) δ 10.94 (s, 1H), 9.75 (s, 1H), 8.91 (s, 2H), 7.58 (dd, J = 15.8, 7.9 Hz, 5H), 7.41 (t, J = 7.6 Hz, 4H), 7.36 (d, J = 8.1 Hz, 1H), 7.31-7.24 (m, 3H), 7.08 (t, J = 7.5 Hz, 1H), 7.01-6.97 (m, 2H), 4.84 (d, J = 11.4 Hz, 2H), 4.58 (d, J = 14.0 Hz, 2H), 4.14 (d, J = 11.9 Hz, 1H), 3.82 (q, J = 15.1 Hz, 3H), 3.09 (t, J = 12.1 Hz, 1H), 2.82 (s, 3H), 2.63 (t, J = 12.0 Hz, 1H), 1.63-1.44 (m, 5H), 1.31 (d, J = 8.2 Hz, 2H).LCMS: m/z = 562 (M+H)+.
실시예 34: 화합물 034의 합성

단계 1: 화합물 2를 수득하기 위해 실시예 3의 단계 1을 참조한다.
단계 2: DMSO (50 ml) 중의 화합물 2 (5.25 g, 15 mmol)의 용액에 K2CO3 (4.14 g, 30 mmol) 및 Cu (960 mg, 15 mmol)를 첨가하였다. 혼합물을 질소 대기 하 145℃에서 밤새 교반하였다. 혼합물을 여과하고, 여과물을 수집한 후 이어서 칼럼 상에서 정제하여 백색 고체로서 화합물 3 (2.4, 38%)을 제공하였다.
단계 3: 1,4-디옥산 (15 ml) 중의 화합물 3 (600 mg, 1.5 mmol)의 용액에 1,4-디옥산 (10 ml) 중의 4 M HCl을 실온에서 밤새 첨가하였다. 혼합물을 여과하여 황색 고체로서 화합물 4 (437 mg, 95%)를 수득하였다.
단계 4: 5 ml DMF 중의 화합물 4 (437 mg, 1.75 mmol), EDCI (543 mg, 3.5 mmol), HOAT (476 mg, 3.5 mmol), DIPEA (903 mg, 7 mmol), 및 2-인돌아세트산 (307 mg, 1.75 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA로 추출 후, EA를 이용한 컬럼으로 정제하여 표적 화합물 5 (600 mg, 84 %)를 제공하였다.
단계 5: EtOH/THF 중의 화합물 5 (600 mg, 1.47 mmol)의 용액에 2 N NaOH (5 ml)를 첨가한 후, 결과 반응 혼합물을 60℃에서 밤새 교반하였다. 용매를 증발시키고, 혼합물을 EA로 추출한 후, 병합된 유기층을 건조시켜 백색 고체로서 목적하는 화합물 6 (140 mg, 25%)을 수득하였다.
단계 6: 5 ml DMF 중의 화합물 6 (200 mg, 0.33 mmol), EDCI (102 mg, 0.66 mmol), HOAT (88 mg, 0.66 mmol), DIPEA (170 mg, 1.32 mmol), 및 아민 (102 mg, 0.33 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA로 추출 후, 백색 고체로서 표적 화합물 7 (200 mg, 20 %)을 제공하였다.
단계 7: 5 ml CH2Cl2 중의 화합물 7 (200 mg, 0.28 mmol)의 혼합물에 2 ml TFA를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. EA로 추출 후, 표적 화합물 034 (10 mg, 6 %)를 Prep-HPLC로 정제하였다.
1H NMR (500 MHz, DMSO) δ 10.86 (s, 1H), 9.67 (s, 1H), 8.85 (s, 1H), 7.57-7.52 (m, 2H), 7.47.41 (m, 3H), 7.40-7.35 (m, 3H), 7.33 (d, J = 8.1 Hz, 1H), 7.26 (t, J = 7.4 Hz, 1H), 7.12 (s, 1H), 7.09 (d, J = 8.8 Hz, 1H), 7.07-7.04 (m, 2H), 6.92 (t, J = 7.0 Hz, 1H), 4.96 (s, 1H), 4.49 (d, J = 10.4 Hz, 1H), 4.12 (d, J = 10.9 Hz, 1H), 3.71 (q, J = 15.0 Hz, 2H), 3.44 (q, J = 7.0 Hz, 2H), 3.10 (t, J = 12.3 Hz, 1H), 2.64 (t, J = 12.2 Hz, 1H), 2.09 (s, 2H), 1.87 (dd, J = 26.7, 12.8 Hz, 2H), 1.16-1.08 (m, 1H), 1.05 (t, J = 7.0 Hz, 3H). LCMS: m/z = 622 (M+H)+.
실시예 35: 화합물 035의 합성

단계 1: 1,4-디옥산 (20 ml) 중의 에틸 2-클로로피리미딘-5-카르복실레이트 (1.86 g, 10 mmol), 화합물 1 (3.00, 15 mmol), 및 NEt3 (3.0 g, 30 mmol)의 혼합물을 95℃에서 밤새 교반하였다. 혼합물을 농축하고, EA (60 ml) 및 수성 시트르산 (60 ml)을 첨가하였다. 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 건조시키고 농축하여 연한 황색 고체로서 화합물 2 (3.4 g, 수율: 97%)를 제공하였다.
단계 2: 1,4-디옥산 (15 ml) 중의 화합물 2 (600 mg, 1.7 mmol)의 용액에 1,4-디옥산 (10 ml) 중의 4 M HCl을 실온에서 첨가하고, 결과 반응 혼합물을 밤새 교반하였다. 혼합물을 여과하여 황색 고체로서 화합물 3 (427 mg, 100%)을 수득하였다.
단계 3: DMF (15 ml) 중의 화합물 3 (427 mg, 1.7 mmol), 화합물 2-인돌아세트산 (301 mg, 1.7 mmol), HOAT (462 mg, 3.4 mmol), EDCI (649 mg, 3.4 mmol) 및 NEt3 (687 mg, 6.8 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 수득하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 4 (584 mg, 84%)를 제공하였다.
단계 4: EtOH (15 ml) 및 THF (15 ml) 중의 화합물 4 (500 mg, 1.2 mmol)의 용액에 수성 NaOH (2 M, 15 mL)를 첨가하고, 결과 반응 혼합물을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축하여 잔사를 제공하였다. 혼합물에 물 (100 mL) 및 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정한 후 이어서 여과하여 백색 고체로서 화합물 5 (342 mg, 75%)를 수득하였다.
단계 5: DMF (5 ml) 중의 화합물 5 (150 mg, 0.39 mmol), 화합물 Boc-아민 (113 mg, 0.39 mmol), HOAT (103 mg, 0.76 mmol), EDCI (145 mg, 0.76 mmol) 및 NEt3 (154 mg, 1.52 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 잔사를 수득하고, 이를 Prep-TLC로 정제하여 황색 고체로서 화합물 6 (150 mg, 59%)을 제공하였다.
단계 6: DCM (5 ml) 중의 화합물 6 (150 mg, 0.23 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하고 Prep-HPLC로 정제하여 백색 고체로서 화합물 035 (2 mg, 1.5%)를 제공하였다.
1H NMR (400 MHz, DMSO) δ 10.92 (s, 1H), 9.57 (s, 1H), 8.86 (s, 2H), 7.84 (d, J = 7.8 Hz, 1H), 7.56 (dd, J = 12.6, 7.9 Hz, 4H), 7.49 (s, 1H), 7.36 (dt, J = 17.7, 8.5 Hz, 5H), 7.27 - 7.19 (m, 3H), 7.07 (t, J = 7.4 Hz, 1H), 6.97 (t, J = 7.3 Hz, 1H), 6.84 (d, J = 8.3 Hz, 1H), 5.15 (s, 2H), 4.33 (d, J = 14.0 Hz, 1H), 3.78 (d, J = 2.4 Hz, 2H), 3.15 (t, J = 12.5 Hz, 1H), 2.77 (t, J = 11.7 Hz, 1H), 1.84 (s, 2H), 1.32 (d, J = 10.9 Hz, 2H). LCMS: m/z = 548 (M+H)+.
실시예 36: 화합물 036의 합성

단계 1- 3: 화합물 4를 수득하기 위해 실시예 34의 단계 1-3을 참조한다.
단계 4: 10 ml DMF 중의 화합물 4 (500 mg, 1.53 mmol), EDCI (573 mg, 3 mmol), HOAT (405 mg, 3 mmol), DIPEA (390 mg, 3 mmol), 및 2-메틸-3-인돌아세트산 (289 mg, 1.53 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 얼음물로 ??칭한 후, 표적 화합물 5 (508 mg, 69 %)를 침전시키고 백색 고체로서 수집하였다.
단계 5: EtOH/THF (10 ml) 중의 화합물 5 (508 mg, 1 mmol)의 용액에 2 N NaOH (5 ml)를 첨가한 후, 결과 반응 혼합물을 60℃에서 밤새 교반하였다. 용매를 증발시킨 후, 혼합물의 pH를 4-5로 조정하였다. 침전물을 수집하여 백색 고체로서 목적하는 화합물 6 (460 mg, 96%)을 수득하였다.
단계 6: 5 ml DMF 중의 화합물 6 (200 mg, 0.42 mmol), EDCI (160 mg, 0.84 mmol), HOAT (113 mg, 0.84 mmol), DIPEA (108 mg, 0.84 mmol), 및 아민 (120 mg, 0.42 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA로 추출 후, 표적 화합물 7 (200 mg, 조생성물)을 오일로서 제공하였다.
단계 7: 5 ml CH2Cl2 중의 화합물 7 (200 mg, 조생성물)의 혼합물에 2 ml TFA를 첨가하고, 혼합물을 실온에서 2시간 동안 교반하였다. EA로 추출 후, 표적 화합물 036 (94 mg, 46 %)을 Prep-HPLC로 정제하였다.
1H NMR (500 MHz, DMSO) δ 10.74 (s, 1H), 9.78 (s, 1H), 8.84 (s, 2H), 7.61-7.54 (m, 3H), 7.48- 7.36 (m, 6H), 7.34-7.25 (m, 2H), 7.22 (d, J = 8.0 Hz, 1H), 7.04-6.94 (m, 4H), 6.86 (t, J = 7.4 Hz, 1H), 4.93 (t, J = 11.9 Hz, 1H), 4.48 (d, J = 13.2 Hz, 1H), 3.98 (d, J = 12.1 Hz, 1H), 3.64 (dd, J = 45.6, 15.4 Hz, 2H), 3.07 (t, J = 12.4 Hz, 1H), 2.62 (t, J = 12.2 Hz, 1H), 2.22 (s, 3H), 1.85 (d, J = 11.3 Hz, 1H), 1.75 (d, J = 10.8 Hz, 1H), 1.08 (d, J = 8.4 Hz, 1H), 0.94 (d, J = 8.5 Hz, 1H). LCMS: m/z = 636 (M+H)+.
실시예
37: 화합물 037의 합성

단계 1: 화합물 5를 수득하기 위해 실시예 13의 단계 1을 참조한다.
단계 2: DCM (10 ml) 중의 화합물 5 (250 mg, 0.69 mmol) 및 NBS (123 mg, 069 mmol)의 혼합물을 0℃에서 30분간 교반하였다. 혼합물에 EA (20 ml) 및 수성 포화 Na2SO3 (20 ml)를 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (20 ml×2)로 세척하고, 건조시키고 농축하고, Prep-TLC로 정제하여 황색 고체로서 화합물 6 (258 mg, 85%)을 수득하였다.
단계 3: 톨루엔 (30 ml) 및 H2O (5 ml) 중의 화합물 6 (240 mg, 0.55 mmol), 사이클로프로필보론산 (232 mg, 2.7 mmol), Pd(OAc)2 (11 mg, 0.05 mmol), 트리사이클로헥실포스핀 (14 mg, 0.05 mmol) 및 K3PO4 (360 mg, 1.7 mmol)의 혼합물을 질소 대기 하 95℃에서 밤새 교반하였다. 혼합물을 냉각시키고, 혼합물에 EA (100 ml)를 첨가하였다. 여과, 농축, 및 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 7 (200 mg, 90%)을 수득하였다.
단계 4: EtOH (15 mL) 및 THF (15 ml) 중의 화합물 7 (200 mg, 0.5 mmol)의 용액에 수성 NaOH (2 M, 15 mL)를 첨가하고, 결과 반응 혼합물을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축한 후, 혼합물에 물 (20 ml) 및 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정한 후 이어서 여과하여 백색 고체로서 화합물 8 (168 mg, 90%)을 수득하였다.
단계 5: DMF (5 ml) 중의 화합물 8 (150 mg, 0.40 mmol), 화합물 Boc-아민 (114 mg, 0.40 mmol), HOAT (103 mg, 0.76 mmol), EDCI (145 mg, 0.76 mmol) 및 NEt3 (154 mg, 1.52 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물을 EA (10 ml×2)로 추출하고, 건조시키고 농축하고, Prep-TLC로 정제하여 황색 고체로서 화합물 9 (120 mg, 50%)를 수득하였다.
단계 6: DCM (5 ml) 중의 화합물 9 (120 mg, 0.20 mmol)의 용액에 TFA (3 ml)를 실온에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물을 농축하여 화합물 10 (80 mg, 100%)을 제공하고 추가의 정제 없이 다음 단계에 사용하였다.
단계 7: THF (5 ml) 중의 화합물 10 (80 mg, 0.20 mmol) 및 Et3N (106 mg, 1.05 mmol)의 용액에 모르폴린-4-카르보닐 클로라이드 (45 mg, 0.30 mmol)를 0℃에서 첨가하고, 결과 반응 혼합물을 2시간 동안 교반하였다. 혼합물에 EA (10 ml) 및 수성 포화 NaCl (10 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 농축하고, Prep-HPLC로 정제하여 황색 고체로서 화합물 037 (3 mg, 3%)을 제공하였다.
1H NMR (400 MHz, MeOD) δ 7.78 (d, J = 7.1 Hz, 2H), 7.58 (d, J = 7.6 Hz, 2H), 7.48 (s, 1H), 7.39 (t, J = 7.9 Hz, 3H), 7.26 (t, J = 7.4 Hz, 1H), 6.99 (d, J = 8.3 Hz, 1H), 6.93 (d, J = 9.2 Hz, 1H), 3.71-3.66 (m, 4H), 3.50 (d, J = 13.6 Hz, 2H), 3.30-3.20 (m, 6H), 2.19 (d, J = 13.9 Hz, 2H), 2.05 (s, 1H), 1.84-1.67 (m, 3H), 1.53 (s, 3H), 1.33 (d, J = 17.9 Hz, 1H), 1.06-0.99 (m, 2H), 0.67 (d, J = 4.4 Hz, 2H). LCMS: m/z = 554 (M+H)+.
실시예 38: 화합물 038의 합성
단계 1: CH2Cl2 (50 ml) 중의 화합물 1 (1.5 g, 10.2 mmol), (Boc)2O (2.2 g, 10.2 mmol) 및 TEA (2.06 g, 20.4 mmol)의 혼합물을 실온에서 밤새 교반하였다. 농축 및 EA:PE = 1:5를 이용한 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 2 (1.5 g, 60%)를 수득하였다.
단계 2: MeOH (20 ml) 중의 화합물 2 (1.4 g, 5.7 mmol), NaBH3CN (539 mg, 8.6 mmol) 및 암모늄 아세테이트 (3.1 g, 40.0 mmol)의 혼합물을 70℃에서 2시간 동안 교반하였다. 농축 및 EA:PE = 1:1을 이용한 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 3 (1.2 g, 85%)을 제공하였다.
단계 3: 1,4-디옥산 (25 ml) 중의 화합물 3 (800 mg, 3.2 mmol), 에틸 2-클로로피리미딘-5-카르복실레이트 (603 mg, 3.2 mmol) 및 DIPEA (826 mg, 6.4 mmol)의 용액을 형성하였다. 혼합물을 95℃로 가열하고 밤새 교반하였다. 농축 및 EA:PE = 1:10을 이용한 실리카 겔 컬럼으로 정제하여 연한 황색 고체로서 화합물 4 (1.0 g, 79%)를 제공하였다.
단계 4: THF (10 ml) 및 EtOH (10 ml) 중의 화합물 4 (1.0 g, 2.5 mmol) 및 2 N NaOH (10 ml, 20 mmol)의 용액을 형성하였다. 혼합물을 60℃로 가열하고 6시간 동안 교반하였다. 농축 및 수상의 pH를 5-6으로 조정한 후 이어서 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하여 백색 고체로서 표적 화합물 5 (600 mg, 65%)를 수득하였다.
단계 5: DMF (10 ml) 중의 화합물 5 (100 mg, 0.27 mmol), tert-부틸 3-아미노비페닐-4-일카르바메이트 (77 mg, 0.27 mmol), EDCI (53.0 mg, 0.41 mmol), HOAT (55.0 mg, 0.41 mmol) 및 DIEA (70 mg, 0.54 mmol)의 혼합물을 형성하였다. 혼합물을 65℃로 가열하고 밤새 교반하였다. 그 후에 혼합물을 물에 용해시키고 EA로 추출하였다. 유기층을 물 및 소금물로 세척한 후 황산나트륨 상에서 건조시키고, 여과하고 농축하였다. EA:PE=1:1을 이용한 실리카 겔 컬럼으로 정제하여 자색 고체로서 화합물 6 (130 mg, 76%)을 수득하였다.
단계 6: CH2Cl2 (10 ml) 중의 화합물 6 (130 mg, 0.2 mmol)의 교반된 용액에 HCl/1,4-디옥산 (2 ml, 8.0 mmol)을 첨가하고, 결과 반응 혼합물을 실온에서 밤새 교반하였다. 농축 및 PE로 세척하여 백색 고체로서 표적 화합물 038 (55 mg, 62%)을 제공하였다.
1H NMR (400 MHz, DMSO) δ 9.58 (s, 1H), 8.92 (s, 2H), 8.10 (d, J = 8.8 Hz, 1H), 7.6-7.47 (m, 3H), 7.45-7.29 (m, 3H), 7.24 (t, J = 7.3 Hz, 1H), 6.98-6.81 (m, 3H), 6.54-6.40 (m, 2H), 5.85 (s, 1H), 5.29 (s, 1H), 3.13-3.04 (m, 1H), 1.94 (d, J = 5.1 Hz, 2H), 1.18 (t, J = 7.2 Hz, 2H), 0.85 (s, 1H).LCMS: m/z = 437 (M+H)+.
실시예 39: 화합물 039의 합성
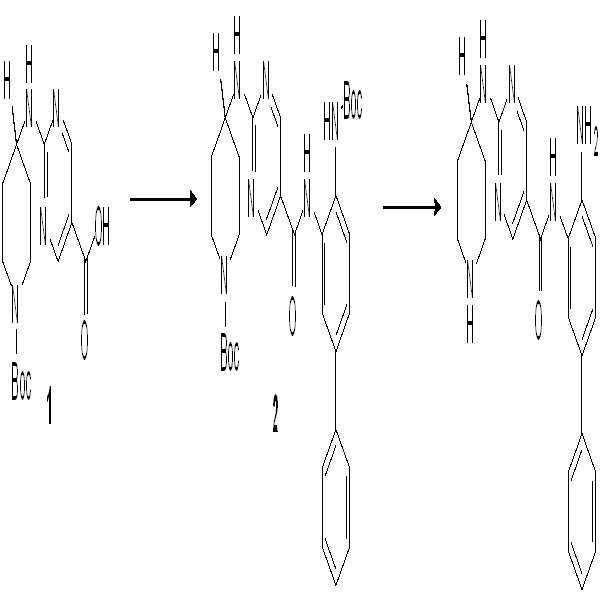
단계 1: 5 ml DMF 중의 화합물 1 (200 mg, 0.62 mmol), EDCI (192 mg, 1.24 mmol), HOAT (170 mg, 1.24 mmol), DIPEA (400 mg, 2.5 mmol), 및 아민 (177 mg, 0.62 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA로 추출 후, 표적 화합물 2 (220 mg, 59 %)를 조 오일로서 제공하였다.
단계 2: 5 ml CH2Cl2 중의 화합물 2 (220 mg, 0.4 mmol)의 혼합물에 1 ml TFA를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. EA (10 ml) 및 물 (10 ml×2)로 추출 후, 표적 화합물 039 (120 mg, 67%)를 Prep-HPLC로 정제하였다.
1H NMR (400 MHz, DMSO) δ 9.65 (s, 1H), 8.89 (s, 2H), 8.58 (s, 1H), 8.32 (s, 1H), 8.07 (d, J = 7.3 Hz, 1H), 7.62-7.49 (m, 3H), 7.39 (dd, J = 17.1, 9.2 Hz, 3H), 7.26 (t, J = 7.4 Hz, 1H), 6.92 (s, 1H), 4.13 (s, 1H), 3.33 (d, J = 12.8 Hz, 2H), 3.05 (d, J = 9.9 Hz, 2H), 2.05 (d, J = 11.4 Hz, 2H), 1.77-1.63 (m, 2H). LCMS: m/z = 389 (M+H)+.
실시예 40: 화합물 040의 합성
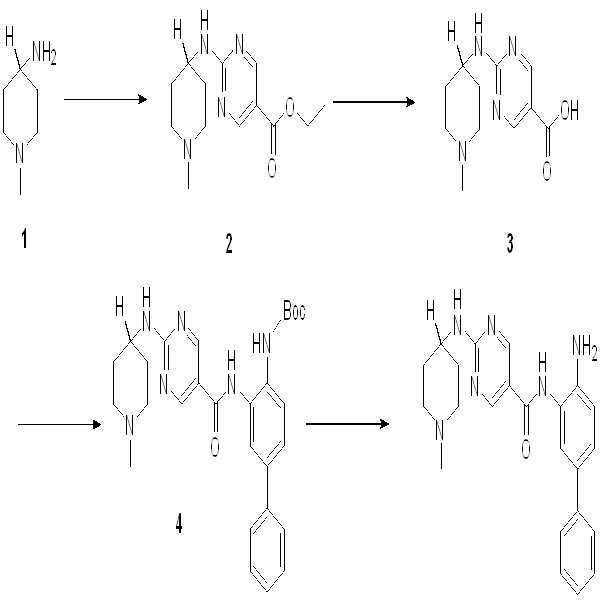
단계 1: 5 ml CH2Cl2 중의 화합물 1 (2.0 g, 17.5 mmol), 에틸 2-클로로피리미딘-5-카르복실레이트 (3.2 g, 17.5mmol), 및 DIPEA (4.5 mg, 35.0 mmol)의 혼합물을 실온에서 2시간 동안 교반하였다. EA (2×50 ml)로 추출 후, 병합된 유기층을 건조시켜 백색 고체로서 표적 화합물 2 (2.0 g, 43%)를 제공하였다.
단계 2: 6 M HCl 중의 화합물 2 (400 mg, 1.5 mmol)의 용액을 100℃에서 밤새 교반하고, 목적하는 화합물 3 (300 mg, 85%)을 동결 건조기로 건조시켰다.
단계 3: 5 ml DMF 중의 화합물 3 (150 mg, 0.63 mmol), EDCI (197 mg, 1.26 mmol), HOAT (171 mg, 1.26 mmol), DIPEA (325 mg, 2.5 mmol), 및 아민 (179 mg, 0.63 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA로 추출 후, 표적 화합물 (200 mg, 63 %)을 CH2Cl2:CH3OH (10:1)을 이용한 칼럼으로 정제하였다.
단계 4: 5 ml CH2Cl2 중의 화합물 4 (100 mg, 0.2 mmol)의 혼합물에 TFA (1 ml)를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. EA (10 ml) 및 물 (10 ml×2)로 추출 후, 표적 화합물 040 (20 mg, 25%)을 Prep-HPLC로 정제하였다.
1H NMR (400 MHz, DMSO) δ 9.52 (s, 1H), 8.84 (s, 2H), 7.78 (d, J = 7.5 Hz, 1H), 7.59 - 7.46 (m, 3H), 7.39 (t, J = 7.5 Hz, 2H), 7.32 (d, J = 7.5 Hz, 1H), 7.24 (t, J = 7.0 Hz, 1H), 6.85 (d, J = 8.3 Hz, 1H), 5.14 (s, 2H), 3.77 (s, 1H), 2.75 (d, J = 11.5 Hz, 2H), 2.16 (s, 3H), 1.95 (t, J = 10.9 Hz, 2H), 1.83 (d, J = 10.4 Hz, 2H), 1.55 (d, J = 9.5 Hz, 2H).LCMS: m/z = 403 (M+H)+.
실시예 41: 화합물 041의 합성
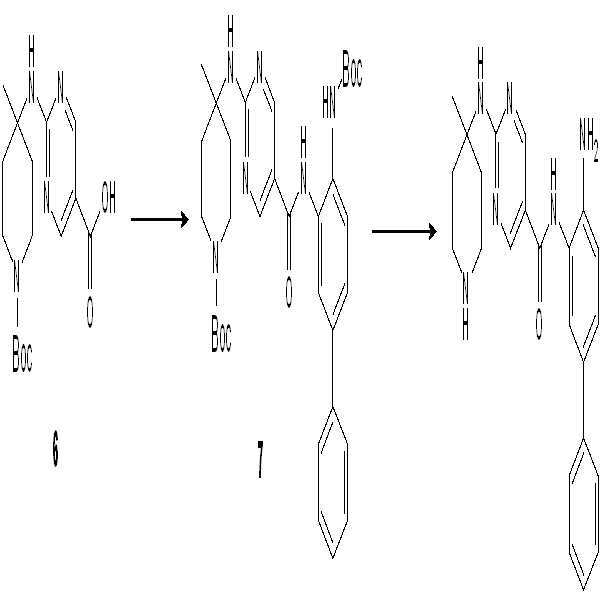
단계 1- 5: 화합물 6을 수득하기 위해 실시예 7의 단계 1-5를 참조한다.
단계 6: 5 ml DMF 중의 화합물 6 (200 mg, 0.62 mmol), EDCI (192 mg, 1.24 mmol), HOAT (170 mg, 1.24 mmol), DIPEA (400 mg, 2.5 mmol), 및 아민 (177 mg, 0.62 mmol)의 혼합물을 60℃에서 밤새 교반하였다. EA로 추출 후, 표적 화합물 7 (187 mg, 50 %)을 조 오일로서 제공하였다.
단계 7: 5 ml CH2Cl2 중의 화합물 7 (186 mg, 0.31 mmol)의 혼합물에 1 ml TFA를 첨가하였다. 혼합물을 실온에서 2시간 동안 교반하였다. EA (10 ml) 및 물 (10 ml×2)로 추출 후, 표적 화합물 041 (90 mg, 70%)을 Prep-HPLC로 정제하였다.
1H NMR (400 MHz, DMSO) δ 9.57 (s, 1H), 8.88 (s, 2H), 7.63 (s, 1H), 7.54 (d, J = 7.4 Hz, 2H), 7.49 (s, 1H), 7.39 (t, J = 7.7 Hz, 2H), 7.35-7.29 (m, 1H), 7.24 (t, J = 7.3 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 5.16 (s, 2H), 3.04 (d, J = 13.0 Hz, 2H), 2.96 (t, J = 10.8 Hz, 2H), 2.43 (s, 2H), 1.68 (t, J = 10.7 Hz, 2H), 1.44 (s, 3H). LCMS: m/z = 402 (M+H)+.
실시예 42: 화합물 042의 합성
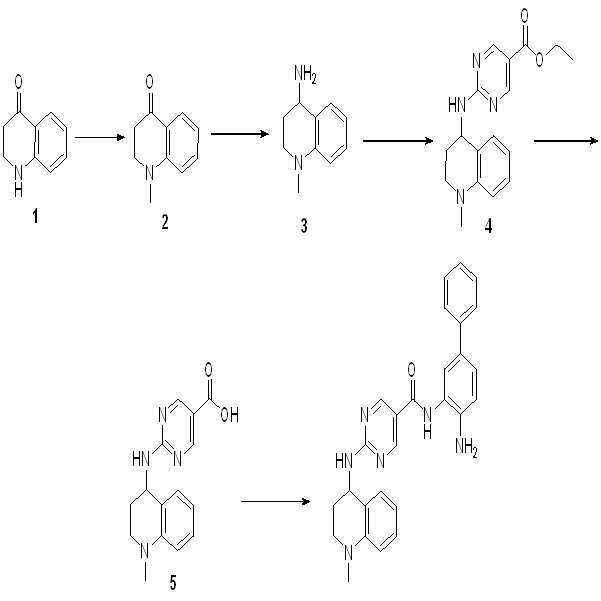
단계 1: DMF (30 ml) 중의 화합물 1 (2.0 g, 13.6 mmol), 이오도메탄 (5.8 g, 40.8 mmol), 및 K2CO3 (5.5 g, 40.8 mmol)의 혼합물을 80℃에서 밤새 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하고, 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 2 (1.5 g, 68%)를 수득하였다.
단계 2: MeOH (30 ml) 중의 화합물 2 (1.5 g, 9.3 mmol), NaBH3CN (1.8 g, 27.9 mmol), 및 AcONH4 (3.6 g, 46.5 mmol)의 혼합물을 70℃에서 5시간 동안 교반하였다. 혼합물에 EA (100 ml) 및 수성 포화 NaCl (100 ml)을 첨가하고, 결과 반응 혼합물을 30분간 교반하였다. 유기층을 분리하고, 수성 포화 NaCl (50 ml×2)로 세척하고, 건조시키고 농축하여 백색 고체로서 화합물 3 (1.4 g, 80%)을 제공하였다.
단계 3: DCM (30 ml) 중의 화합물 3 (1.6 g, 10 mmol), 에틸 2-클로로피리미딘-5-카르복실레이트 (1.7 g, 9 mmol), 및 DIPEA (3.9 g, 30 mmol)의 혼합물을 실온에서 밤새 교반하였다. 혼합물을 실리카 겔 컬럼으로 정제하여 황색 고체로서 화합물 4 (800 mg, 29%)를 제공하였다.
단계 4: EtOH (20 ml) 및 THF (20 ml) 중의 화합물 4 (800 mg, 2.6 mmol)의 용액에 수성 NaOH (2 M, 20 ml)를 첨가하였다. 혼합물을 60℃에서 2시간 동안 교반하였다. 혼합물을 농축하여 잔사를 수득하고, 혼합물에 물 (100 ml) 및 수성 시트르산을 첨가하여 0℃에서 pH <7로 조정한 후 이어서 여과하여 백색 고체로서 화합물 5 (600 mg, 80%)를 제공하였다.
단계 5: DMF (5 mL) 중의 화합물 5 (150 mg, 0.53mmol), 화합물 Boc-아민 (152 mg, 0.53 mmol), HOAT (103 mg, 0.76 mmol), EDCI (145 mg, 0.76 mmol) 및 NEt3 (154 mg, 1.52 mmol)의 혼합물을 60℃에서 밤새 교반하였다. 혼합물을 EA (10 ml) 및 물 (10 ml×2)로 추출하였다. 유기층을 분리하고, 수성 포화 NaCl (20 ml×2)로 세척하고, 건조시키고 농축하고, Prep-TLC로 정제하여 황색 고체로서 화합물 6 (45 mg, 15%)을 제공하였다.
단계 6: 1,4-디옥산 (5 ml) 중의 화합물 6 (45 mg, 0.08 mmol)의 용액에 1,4-디옥산 (0.5 ml) 중의 HCl을 실온에서 첨가하고, 결과 반응 혼합물을 밤새 교반하였다. 혼합물을 여과하여 화합물 042 (15 mg, 41%)를 제공하였다.
1H NMR (500 MHz, DMSO) δ 10.43 (s, 1H), 9.00 (d, J = 29.0 Hz, 2H), 8.30 (d, J = 8.6 Hz, 1H), 7.80 (d, J = 1.9 Hz, 1H), 7.67 (d, J = 7.4 Hz, 2H), 7.61 (dd, J = 8.3, 1.9 Hz, 1H), 7.49 (t, J = 7.7 Hz, 2H), 7.45-7.37 (m, 2H), 7.12 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.3 Hz, 1H), 6.72 (d, J = 8.2 Hz, 1H), 6.63 (t, J = 7.3 Hz, 1H), 5.32 (d, J = 6.8 Hz, 1H), 3.39 (s, 2H), 2.90 (s, 3H), 2.07 (dd, J = 22.1, 4.2 Hz, 2H). LCMS: m/z = 451 (M+H)+.
실시예 43: HDAC 효소 어세이
시험용 화합물을 DMSO 중에서 50배 최종 농도로 희석하고 10.3배 희석액 시리즈 (ten point three fold dilution series)를 제조하였다. 화합물을 어세이 버퍼 (50 mM HEPES, pH 7.4, 100 mM KCl, 0.001% 트윈-20, 0.05% BSA, 20 μM 트리스(2-카르복시에틸)포스핀) 중에서 이의 6배 최종 농도로 희석하였다. HDAC 효소 (BPS Biosciences로부터 구입함)를 어세이 버퍼 중에서 이의 1.5배 최종 농도로 희석하고 기질의 첨가 전에 24시간 동안 화합물과 사전-인큐베이션하였다.
각 효소에 대한 기질 트리펩티드 기질 3 (인 하우스 합성)은 기질 적정 곡성에 의해 결정되는 바와 같은 Km과 동일하였다. 사용된 효소 및 기질 농도를 표 2에 나타내었다. 기질을 0.3 μM 시퀀싱 등급 트립신 (Sigma)과 함께 어세이 버퍼 중에서 이의 6배 최종 농도로 희석하였다. 기질/트립신 믹스 (mix)를 효소/혼합물 믹스에 첨가하고, 플레이트를 60초간 흔들어주고 Spectramax M5 미세적정 플레이트 판독기 (microtiter plate reader) 내에 넣어 두었다. 형광의 발색을 30분간 모니터링하고 반응의 선행 속도를 계산하였다. IC50을 4개의 파라미터 곡선 적합화 (four parameter curve fit)에 의한 그래프 패드 프리즘 (Graph Pad Prism)을 사용하여 결정하였다. 본 발명의 화합물에 대해 수득된 IC50 값을 표 1에 나타내었다. 곡선의 예시는 도 1 및 4에서 확인된다.

실시예
44:
약물동태학
웅성 SD 래트를 밤새 절식시켰다. 본 발명의 화합물을 디메틸 아세트아미드에 10배 최종 농도로 용해시킨 후, 솔루톨 (Solutol) HS 15 (BASF)를 10%의 최종 농도로 첨가하였다. 마지막으로, 80% 식염수를 첨가하고 볼텍싱하여 투명한 용액을 달성하였다. IV 투약의 경우, 3마리 동물의 발등에 정맥을 통해 1 mg/kg 화합물을 주입하였다. PO 투약의 경우, 5 mg/kg의 화합물을 경구 위관영양 (oral gavage)에 의해 전달하였다. 꼬리 정맥을 통해 투약한 후 5분, 15분, 30분, 1시간, 2시간, 4시간, 8시간 및 24시간째에 K2EDTA 튜브 내로 혈액을 수집하였다. 혈액을 4℃에서 5분간 2000 g로 원심분리하여 혈장을 수득하였다. 혈장을 아세토니트릴로 추출하고 화합물의 수준을 LC/MS/MS로 분석하였다. 혈장 내 화합물의 수준을 래트 혈장의 표준 곡선으로부터 계산하였다. IV 소거율 및 면적하 곡선을 WinNonLin 소프트웨어를 사용하여 계산하였다. IV 및 경구 투약에 대한 용량 조정된 면적하 곡선을 사용하여 경구 생체이용률을 계산하였다.
결과의 요약을 표 2뿐만 아니라 표 3, 표 4에 나타내었다.
게잡이 원숭이 (Cynomolgus monkey) 약물동태학을 화합물 005에 대해 결정하였다. 화합물 005를 0.5% 히드록시프로필 메틸셀룰로스에 용해시키고 20 mg/kg 체중의 게잡이 원숭이에게 먹이로 공급하였다. 혈장 샘플을 시간의 경과에 따라 수집하고 상기에 기술된 바와 같이 분석하였다. 각 시점에서의 혈장 수준을 도 5에 나타내었다.
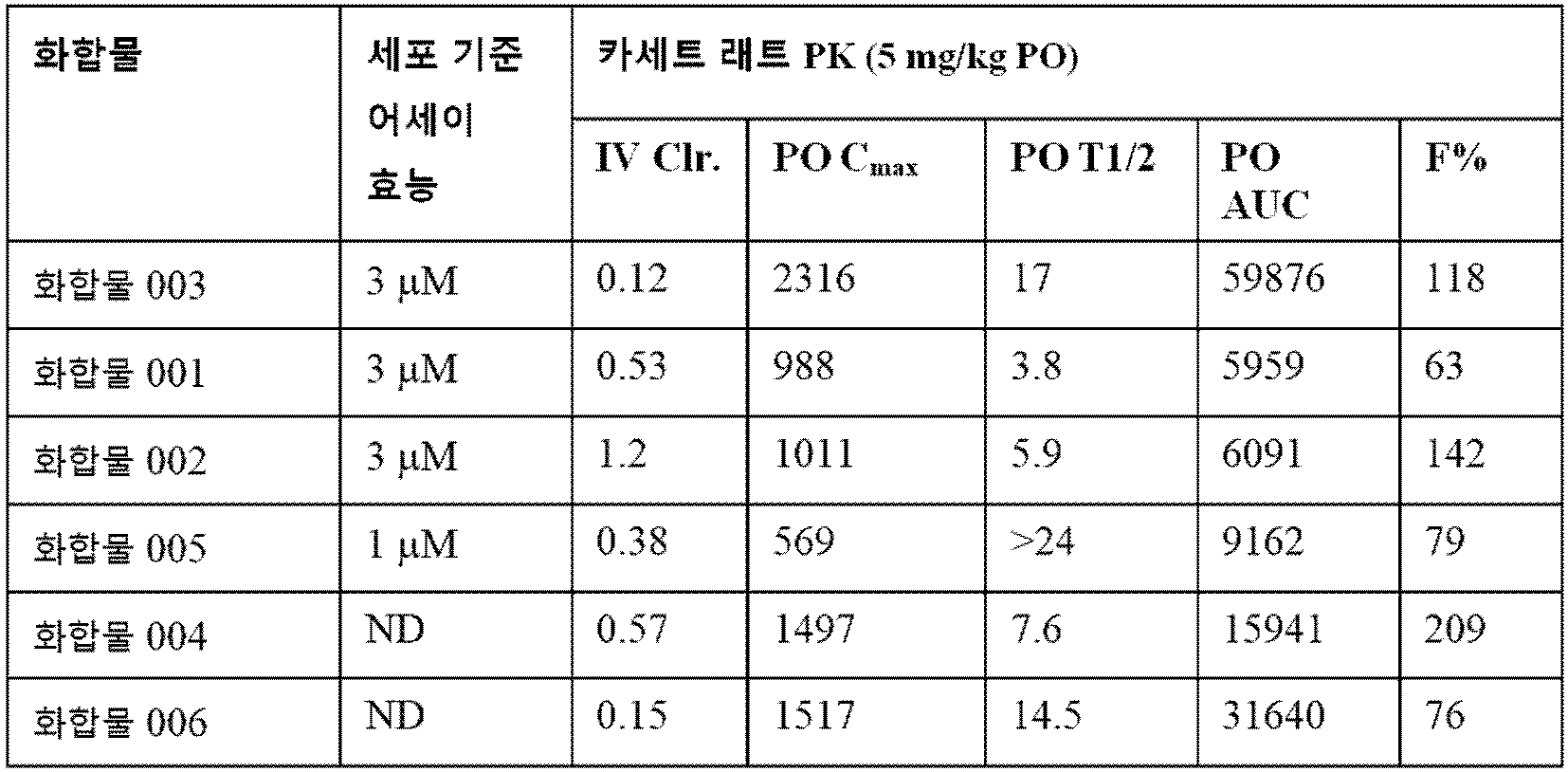
표 3: 태아 글로빈 유도 어세이에서 화합물을 시험하였고 기준선 이상의 태아 글로빈 유전자 발현에서의 2배 증가가 달성되는 가장 낮은 농도를 "세포 기준 어세이 효능 (Cell based assay potency)" 칼럼에 나타내었다. 약물동태학 특성을 래트 카세트 투약 실험에서 평가하였다. IV 소거율 (IV Clr.)은 L/hr/kg의 단위이다. 경구 최대 혈장 농도 (PO Cmax)는 ng/ml의 단위이다. 경구 혈장 반감기 (PO T1/2)는 시간의 단위이다. 경구 면적하 곡선 (PO AUC)은 시간×ng/ml의 단위이다. 경구 경로로 흡수된 부분 (F%)은 용량 조정된, IV 곡선하 면적에 대한 경구 면적하 곡선의 백분율이다.

실시예
45: 태아 글로빈 유도
인간 골수로부터 분리된 CD34+ 세포를 Bradner JE (Proc Natl Acad Sci USA. 2010 Jul 13; 107(28): 12617-22)에 의해 기술된 방법을 이용하여 시험관 내 배양하였는데, 이는 세포의 적혈구 계통으로의 분화를 지지하는 배지 중에서 7일간의 증폭기에 이어서 적혈구 세포 발생이 지속되는 3일간의 분화기로 이루어진다. 분화기의 말미에, 이들 세포는 원발성 후기 적혈모구 (primarily late erythroblasts)이다. mRNA 수준을 성체 대 (major) β-글로빈 (β), 성체 소 (minor) β-글로빈 (δ), 태아 β-유사 글로빈 (HbG, γ), 및 배아 β-유사 글로빈 (ε)에 대해 고안된 프라이머/프로브 세트를 사용한 정량적 실시간 PCR에 의해 결정하였다. 단백질 수준을 태아 헤모글로빈 (HbF) 또는 성체 헤모글로빈 (HbA)에 대한 형광 항체를 사용한 유세포분석에 의해 결정하였다.
도 3에 나타난 실험에서, 세포를 비히클 (DMSO) 0.3, 1 및 3 μM의 화합물 A, 및 0.3, 1, 및 3 μM의 화합물 003의 존재하에서 분화시켰다. 도 6에 나타난 실험에서, 세포를 비히클 (DMSO), 0.3, 1 및 3 μM의 화합물 A, 및 0.3, 1, 및 3 μM의 화합물 005의 존재하에서 분화시켰다. 글로빈 mRNA 수준을 분화 3일째에 결정하였다.
화합물 A는 하기에 나타낸 구조를 갖는 HDAC1/2 억제제 (IC50은 HDAC1, HDAC2, 및 HDAC3에 대해 각각 약 4, 15, 및 114 nM임)이다. 화합물 A의 특성화 및 합성은 미국 공개특허 제2014-0128391호에서 확인된다.

화합물 A
실시예 46: 추가 연구
화합물 003이 hERG, CYP 억제, 및 유전독성을 나타내지 않음을 보여주는 추가적인 실험을 수행하였다.
hERG 어세이를 위해, hERG cDNA로 안정하게 형질감염되고 hERG 채널을 발현하는 CHO 세포주를 3-8×106 세포/ml의 밀도로 QPatch 플레이트 (Sophion) 내에 접종하였다. 세포를 -80 mV의 보유 전위에서 전압 클래핑하였다 (clamped). hERG 전류를 5초간 +20 mV에서 탈분극시켜 활성화키고, 그 후에 전류를 5초간 -50 mV로 되돌려서 불활성화를 제거하고 비활성화 꼬리 전류를 관찰하였다. 꼬리 전류 크기의 최대량을 사용하여 hERG 전류 진폭을 결정하였다. 화합물 003의 6개 용량 (30, 10, 3, 1, 0.3 및 0.1 μM)을 선택하여 적합화 곡선 및 IC50을 수득하였다. 데이터를 Sophion 및 Graphpad Prism에 의해 제공된 어세이 소프트웨어를 사용하여 분석하였다. 화합물 003은 30 μM의 최대 농도에서 hERG 채널의 활성을 억제하지 않았다.
Cyp 억제를 위해, BD Gentest로부터의 인간 간 마이크로솜을 화합물 003 (10, 3.33, 1.11, 0.37, 0.12, 0.04, 0.01 μM) 및 기질 (CYP1A2: 30 μM의 페나세틴 (Phenacetin); CYP2C9: 10 μM의 디클로페낙; CYP2C19: 35 μM의 S-메페니토인 (S-Mephenytoin); CYP3A4: 5 μM의 미다졸람 및 80 μM의 테스토스테론; CYP2D6: 10 μM의 부푸랄롤 (Bufuralol))과 하기 인큐베이션 시간 동안 인큐베이션하였다: CYP1A2, 2C9, 2D6: 10분, 37℃; CYP2C19: 45분, 37℃; CYP3A4: 5분, 37℃. 기질 전환을 액체 크로마토그래피/질량 분광법/질량 분광법 (LC/MS/MS)으로 측정하였다. 그래프 패드 프리즘 내 곡선 적합화에 의해 억제를 측정하였다. 화합물 003은 최대 10 μM에서 임의의 Cyp의 활성을 억제하지 않았다.
유전독성의 경우, Ames et al. (1975)에 의해 기술된 바와 같은 세균 시험 균주 TA98, TA100, TA1535 및 TA97와 Green and Muriel (1976)에 의해 기술된 바와 같은 E. coli 시험 균주 WP2를 희생 5일 전에 500 mg/kg의 용량으로, Aroclor 1254 (옥수수유 중의 200 mg/mL)가 복강내로 주입되었던 웅성 스프라그 다우리 래트로부터 제조된 상업적으로 구매한 (MolTox; Boone, NC) 간 균질액 (S9)이 존재 또는 부재하는 24 웰 플레이트에서 250, 75, 25, 7.5, 2.5, 0.75, 0.25, 및 0.075 μg/웰로 화합물 003과 인큐베이션하였다. 돌연변이 유발성을
비-허용 배지 상에서 형성된 콜로니의 수를 계수함으로써 평가하였다. 화합물 003은 S9 활성화의 존재 또는 부재인 임의의 균주의 관련 콜로니의 수를 증가시키지 않았다.
인간 말초 혈액 림프구 (HPBL)에서의 소핵 (micronucleus) 형성 어세이에서 유전독성을 추가로 평가하였다. HPBL을 건강한 공여자로부터 수득하고 S9 활성화 존재 또는 부재 하에서 4시간 동안 3500, 2450, 1715, 1200, 840, 588, 412, 288, 202, 141, 98.8, 69.2, 48,4, 및 33.9 μg/ml에서 화합물 003에 노출시켰다. 세포를 PBS로 세척하고 6 μg/ml의 사이토콜라신 (cytocholasin) B를 함유하는 완전 배지에서 24시간 동안 인큐베이션하였다. 그 후에 세포를 용해시키고, 고정하고, 현미경 슬라이드 위에 올려 놓았다. 단핵, 이핵, 및 소핵 세포의 수를 맹검 (blinded) 조건하에서 계수하였다. 화합물 003은 임의의 농도에서 소핵 세포의 수를 증가시키지 않았다.
실시예 47: 다양한 HDAC1/2 억제제의 적혈구 전구체의 처리는 Gata2 mRNA의 유도를 초래함
인간 골수 유래 CD34+ 세포를 문헌 [Sankaran et al., Science, vol. 322(5909), pp. 1839-42 (2008)]에 기술된 바와 같이 7일간 증폭시켰다. 그 후에 세포를 지시된 농도의 화합물 005, 화합물 A (또 다른 공지의 HDAC1/2 억제제), 또는 비히클 대조군 (DMSO)의 지시된 농도의 존재하에서 적혈구형성 (erythropoiesis)을 지지하는 배지에서 3일간 분화시켰다 (Hu et al., "Isolation and functional characterization of human erythroblasts at distinct stages: implications for understanding of normal and disordered erythropoiesis in vivo", Blood, vol. 121(16), pp. 3246-53 (2005)). Gata2 mRNA를 정량적 실시간 PCR을 사용하여 결정하고 베타-액틴 mRNA 대조군 수준에 대비하여 나타내었다. 일차 적혈구 전구체의 화합물 005 또는 화합물 A 처리는 %HbG (도 6) 및 Gata2 mRNA (도 7)의 등가 용량 및 시간-의존적 유도를 초래한다.
참조로 통합
본원의 전반에 걸쳐 인용된 모든 참조(참고 문헌, 발행된 특허, 공개된 특허출원 및 계류중인 특허 출원을 포함함)의 내용은 본원에 전체적으로 명시적으로 포함된다. 달리 정의되지 않는 한, 본원에서 사용되는 모든 기술 및 과학 용어는 당업자에게 일반적으로 공지된 의미를 따른다.
균등물
당업자는 통상적인 실험만을 사용하여 본원에 기술된 특정 실시양태의 다수 균등물을 인식하거나 확인할 수 있을 것이다. 이러한 균등물은 하기 청구범위에 포함되는 것으로 의도된다.
Claims (34)
- 화학식 I의 화합물 또는 이의 약제학적으로 허용가능한 염:

I
상기 식에서,
X1은 CR7 또는 N이고;
X2는 CH 또는 N이고;
Y는 하기로 구성된 군으로부터 선택되고:

Z는 H, C1-C6-알킬, C6-아릴, C(O)NR4R5, C(O)OR6, C(O)C1-C6-알킬, C(O)C0-C6-알킬-C6-아릴, C(O)-C3-C6-사이클로알킬, C(O)-C2-C6-헤테로사이클릴, 및 C(O)C0-6-알킬-헤테로아릴로 구성된 군으로부터 선택되고, 여기서 아릴, 헤테로아릴, 사이클로알킬, 및 헤테로사이클릴 기는 C1-C6-알킬, 할로, C1-C6-할로알킬, 히드록시, 또는 C1-C6-알콕시의 1개 또는 2개로 선택적으로 치환되고;
Ra 및 Rb는 H이거나, 또는 Ra 및 Rb는 함께 융합된 C6-아릴을 형성하고;
R1은 H 및 C1-C6-알킬로 구성된 군으로부터 선택되고;
R2는 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택되고;
R3은 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택되거나;
또는 R2 및 R3은 함께 C2-C6-헤테로사이클릴을 형성하고;
R4는 H, C1-C6-알킬, C1-C6-알킬-OH, 및 C1-C6-NH2로 구성된 군으로부터 선택되고;
R5는 C1-C6-알킬이거나;
또는 R4 및R5는 함께 C2-C6-헤테로사이클릴을 형성하고, 여기서 헤테로사이클릴은 C1-C6-알킬, 할로, C1-C6-할로알킬, 히드록시, 또는 C1-C6-알콕시의 1개 또는 2개로 선택적으로 치환되고;
R6은 C1-C6-알킬 및 C0-C6-알킬-C6-아릴로 구성된 군으로부터 선택되고, 여기서 아릴은 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되고;
R7은 H, C1-C6-알킬, 및 C3-C6-사이클로알킬로 구성된 군으로부터 선택된다. - 제1항에 있어서, 화학식 II의 구조를 갖는 화합물 또는 이의 약제학적으로 허용가능한 염:
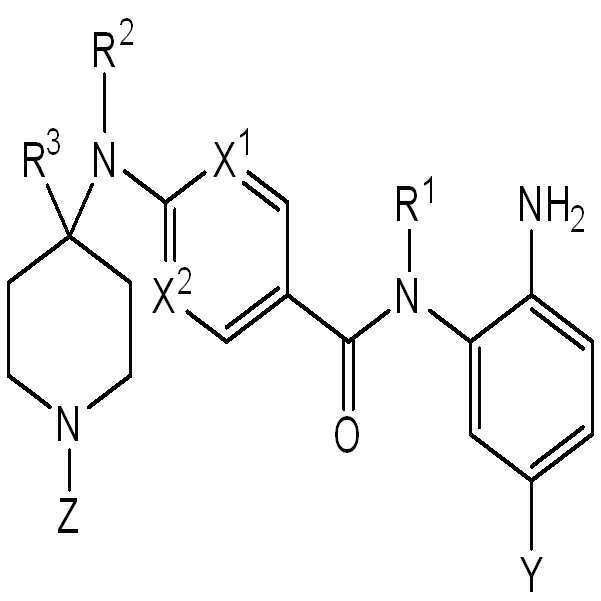
II - 제1항 또는 제2항에 있어서, X1 및 X2가 각각 N이거나, 또는 X1 및 X2가 각각 CH인 것인, 화합물.
- 제1항 내지 제3항 중 어느 한 항에 있어서, Y가 하기인 것인, 화합물:

- 제1항 내지 제4항 중 어느 한 항에 있어서, Z가 C(O)NR4R5, C(O)OR6, C(O)-C3-C6-사이클로알킬, C(O)-C2-C6-헤테로사이클릴, 및 C(O)C0-6-알킬-헤테로아릴로 구성된 군으로부터 선택되고,여기서 헤테로아릴, 사이클로알킬, 또는 헤테로사이클릴이 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되고;
R6이 C6-아릴인 것인, 화합물. - 제1항 내지 제4항 중 어느 한 항에 있어서, Z가 H, C1-C6-알킬, 및 C6-아릴로 구성된 군으로부터 선택되는 것인, 화합물.
- 제1항 내지 제6항 중 어느 한 항에 있어서, R1이 H인 것인, 화합물.
- 제1항 내지 제7항 중 어느 한 항에 있어서, R2가 H인 것인, 화합물.
- 제1항 내지 제8항 중 어느 한 항에 있어서, R3이 H, 메틸, 에틸, 이소프로필, 또는 페닐인 것인, 화합물.
- 제1항에 있어서, 화학식 III의 구조를 갖는 화합물 또는 이의 약제학적으로 허용가능한 염:

III - 제10항에 있어서, X1 및 X2가 각각 N이거나, 또는 X1 및 X2가 각각 CH인 것인, 화합물.
- 제10항 또는 제11항에 있어서, R2가 H인 것인, 화합물.
- 제10항 내지 제12항 중 어느 한 항에 있어서, R3이 H, 메틸, 또는 이소프로필인 것인, 화합물.
- 제10항 내지 제13항 중 어느 한 항에 있어서, R4가 H이고 R5가 C1-C6-알킬인 것인, 화합물.
- 제10항 내지 제13항 중 어느 한 항에 있어서, R4 및 R5가 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐이 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되는 것인, 화합물.
- 제10항 내지 제15항 중 어느 한 항에 있어서,
X1 및 X2가 N이고;
R1이 H이고;
R2가 H이고;
R3이 H 또는 C1-C4-알킬이고;
R4 및 R5가 함께 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐로 구성된 군으로부터 선택되는 헤테로사이클릴을 형성하고, 여기서 모르폴리닐, 피페리디닐, 피페라지닐, 및 피롤리디닐이 C1-C6-알킬, 할로, 또는 히드록시의 1개 또는 2개로 선택적으로 치환되는 것인, 화합물. - 제1항 내지 제16항 중 어느 한 항에 있어서, 하기로부터 선택되는 화합물 또는 이의 약제학적으로 허용가능한 염:
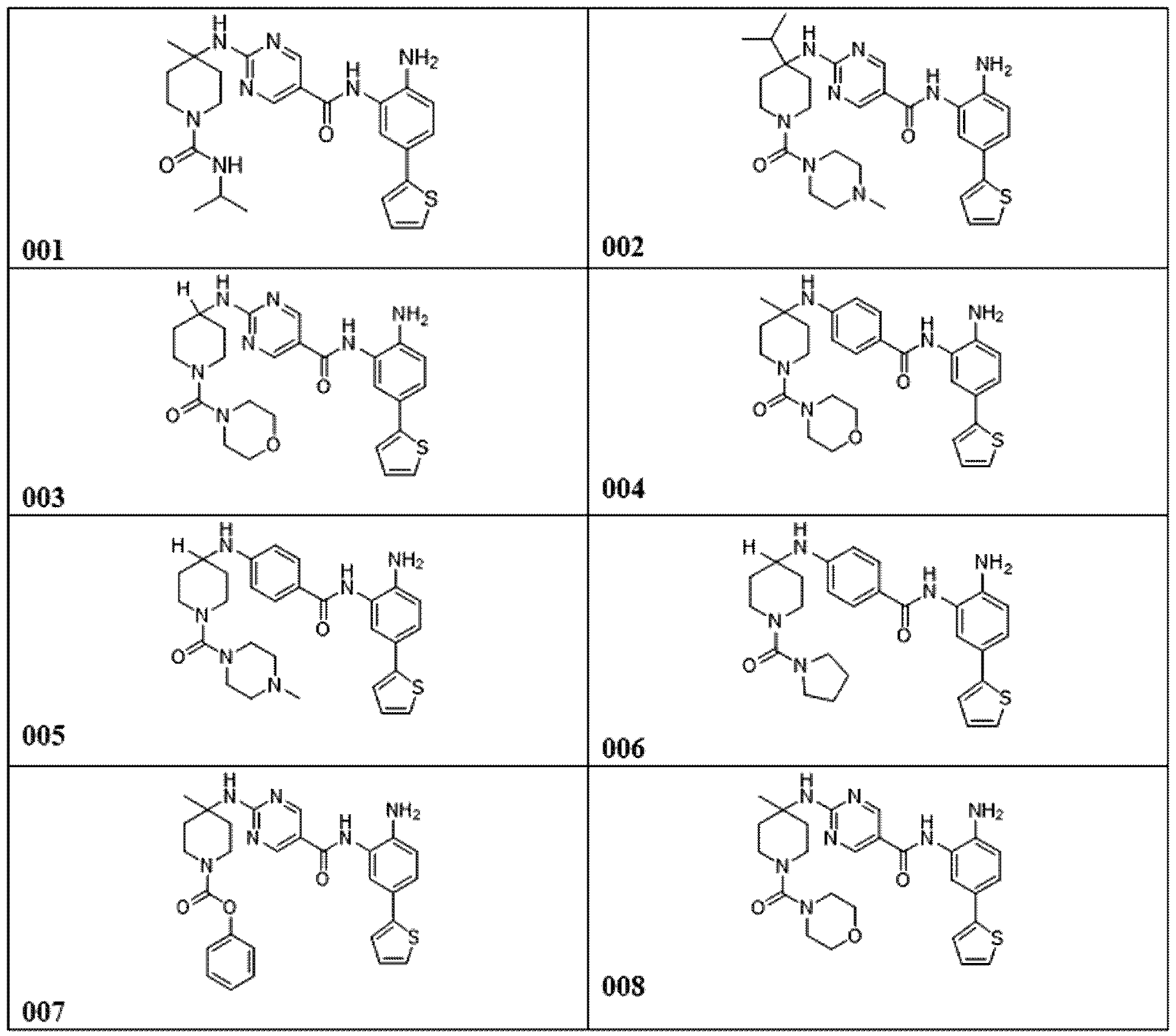
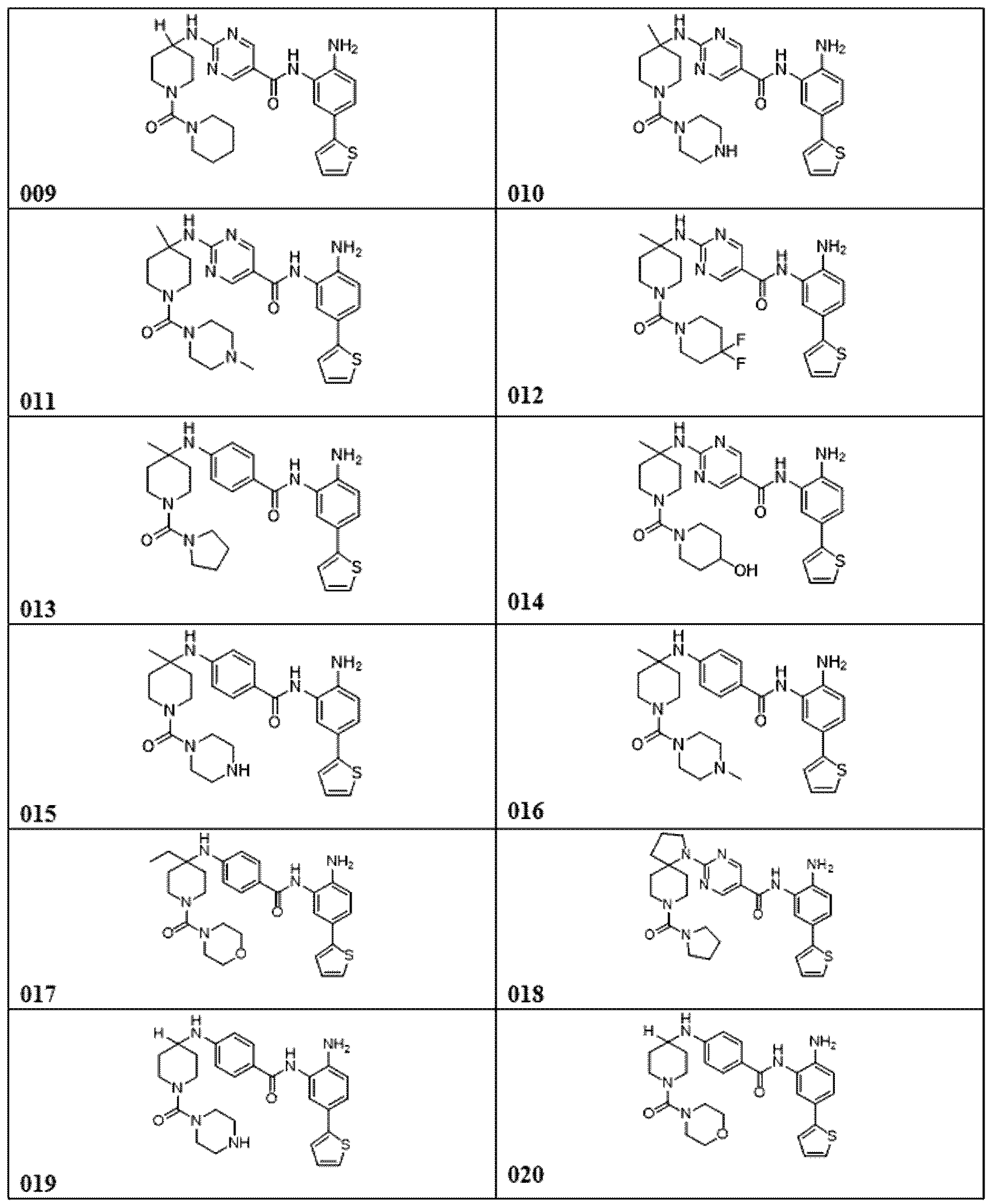

- 제10항 내지 제16항 중 어느 한 항에 있어서, 하기로부터 선택되는 화합물 또는 이의 약제학적으로 허용가능한 염:

- 제1항 내지 제18항 중 어느 한 항의 화합물 및 약제학적으로 허용가능한 담체를 포함하는 약제학적 조성물.
- 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염을 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2의 활성을 억제하는 방법.
- 개체에 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염을 투여하는 것을 포함하는, 개체에서 HDAC1 및/또는 HDAC2에 의해 매개된 질환을 치료하는 방법.
- 제21항에 있어서, 질환이 골수형성이상 증후군 (myelodysplastic syndrome)인 것인, 방법.
- 제21항에 있어서, 질환이 이상혈색소증 (hemoglobinopathy)인 것인, 방법.
- 제23항에 있어서, 이상혈색소증이 겸상적혈구병 (sickle-cell disease) 또는 베타-지중해성 빈혈 (beta-thalassemia)인 것인, 방법
- 제21항에 있어서, 질환이 폐암, 결장암, 유방암, 신경모세포종 (neuroblastoma), 백혈병, 또는 림프종인 것인, 방법
- 제21항에 있어서, 질환이 급성 골수성 백혈병 또는 급성 거대핵세포성 백혈병인 것인, 방법.
- 제21항에 있어서, 질환이 신경모세포종인 것인, 방법.
- 개체에 치료학적 유효량의 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염을 투여하는 것을 포함하는, 이를 필요로 하는 개체에서 겸상적혈구병, 베타-지중해성 빈혈, 골수형성이상 증후군, 급성 골수성 백혈병, 신경모세포종, 또는 급성 거대핵세포성 백혈병을 치료하는 방법.
- 제20항 내지 제28항 중 어느 한 항에 있어서, 개체가 인간인 것인, 방법.
- GATA 결합 단백질 2 (Gate2)결핍과 연관된 질환 또는 장애의 치료를 필요로 하는 개체에 치료학적 유효량의 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염을 투여하는 것을 포함하는, GATA 결합 단백질 2 (Gata2) 결핍과 연관된 질환 또는 장애를 치료하는 방법.
- 세포를 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염과 접촉시키는 것을 포함하는, 세포에서 GATA 결합 단백질 2 (Gata2) 발현을 증가시키는 방법.
- 제31항에 있어서, Gata2 과발현이 HbG (감마 글로빈)을 유도하는 것인, 방법.
- 개체에 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염을 투여하는 것을 포함하는, 개체에서 HbG (감마 글로빈) 발현을 유도하는 방법.
- 제33항에 있어서, 제1항 내지 제18항 중 어느 한 항의 화합물 또는 이의 약제학적으로 허용가능한 염이 개체에서 HbG가 약 2배 증가 내지 약 20배 증가되는 용량으로 투여되는 것인, 방법.
Applications Claiming Priority (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462091221P | 2014-12-12 | 2014-12-12 | |
| US62/091,221 | 2014-12-12 | ||
| US201562238931P | 2015-10-08 | 2015-10-08 | |
| US62/238,931 | 2015-10-08 | ||
| PCT/US2015/065289 WO2016094824A1 (en) | 2014-12-12 | 2015-12-11 | Piperidine derivatives as hdac1/2 inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20170095964A true KR20170095964A (ko) | 2017-08-23 |
Family
ID=56108272
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020177019370A Withdrawn KR20170095964A (ko) | 2014-12-12 | 2015-12-11 | Hdac1/2 억제제로서 피페리딘 유도체 |
Country Status (17)
| Country | Link |
|---|---|
| US (6) | US9790180B2 (ko) |
| EP (2) | EP3725787B1 (ko) |
| JP (1) | JP6693957B2 (ko) |
| KR (1) | KR20170095964A (ko) |
| CN (1) | CN107835810A (ko) |
| AU (2) | AU2015360270B9 (ko) |
| BR (1) | BR112017012504A2 (ko) |
| CA (1) | CA2970500C (ko) |
| CL (1) | CL2017001511A1 (ko) |
| EA (1) | EA035120B1 (ko) |
| ES (1) | ES2816641T3 (ko) |
| HK (1) | HK1251566A1 (ko) |
| IL (1) | IL252828A0 (ko) |
| MX (2) | MX2017007623A (ko) |
| PH (1) | PH12017501095A1 (ko) |
| SG (1) | SG11201704759QA (ko) |
| WO (1) | WO2016094824A1 (ko) |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2839037B1 (en) | 2012-04-19 | 2018-12-26 | Acetylon Pharmaceuticals, Inc. | Biomarkers to identify patients that will respond to treatment and treating such patients |
| US9145412B2 (en) | 2012-11-02 | 2015-09-29 | Acetylon Pharmaceuticals, Inc. | Selective HDAC1 and HDAC2 inhibitors |
| ES2929576T3 (es) | 2013-10-08 | 2022-11-30 | Acetylon Pharmaceuticals Inc | Combinaciones de inhibidores de histona deacetilasa 6 y el inhibidor de Her2 lapatinib para el uso en el tratamiento del cáncer de mama |
| CR20160308A (es) | 2013-12-03 | 2016-11-08 | Acetylon Pharmaceuticals Inc | Combinaciones de inhibidores de histona deacetilasa y farmacos inmunomoduladores |
| AU2015360270B9 (en) * | 2014-12-12 | 2019-12-05 | Regenacy Pharmaceuticals, Llc | Piperidine derivatives as HDAC1/2 inhibitors |
| AU2015362394C1 (en) | 2014-12-12 | 2021-01-28 | Japan Tobacco Inc. | Dihydropyrimidine-2-one compounds and medicinal uses thereof |
| AR105812A1 (es) | 2015-06-08 | 2017-11-15 | Acetylon Pharmaceuticals Inc | Métodos para la preparación de inhibidores de proteína deacetilasa |
| AR104935A1 (es) | 2015-06-08 | 2017-08-23 | Acetylon Pharmaceuticals Inc | Formas cristalinas de un inhibidor de histona deacetilasa |
| US11324744B2 (en) | 2016-08-08 | 2022-05-10 | Acetylon Pharmaceuticals Inc. | Methods of use and pharmaceutical combinations of histone deacetylase inhibitors and CD20 inhibitory antibodies |
| WO2019167981A1 (ja) | 2018-02-28 | 2019-09-06 | 日本たばこ産業株式会社 | 4-メチルジヒドロピリミジノン化合物及びその医薬用途 |
| CN110372572A (zh) * | 2019-08-23 | 2019-10-25 | 苏州汉德创宏生化科技有限公司 | 一种4,4-二氟哌啶-1-甲酰氯的合成方法 |
| EP4565221A1 (en) | 2022-08-05 | 2025-06-11 | Tango Therapeutics, Inc. | An hdac inhibitor for treating cancer with a modified stk11 activity or expression |
| WO2024167423A1 (en) | 2023-02-07 | 2024-08-15 | Captor Therapeutics S.A. | Gspt1 degrader compounds |
| WO2025240627A1 (en) * | 2024-05-14 | 2025-11-20 | Cedars-Sinai Medical Center | Methods and compositions for treating pancreatic cancer |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60326436D1 (en) * | 2002-03-13 | 2009-04-16 | Janssen Pharmaceutica Nv | Aminoderivate als histone-deacetylase-inhibitoren |
| ES2306859T3 (es) * | 2002-03-13 | 2008-11-16 | Janssen Pharmaceutica Nv | Derivados de sulfonil como nuevos inhibidores de histona deacetilasa. |
| KR101153335B1 (ko) * | 2003-09-24 | 2012-07-05 | 메틸진 인코포레이티드 | 히스톤 데아세틸라제의 억제제 |
| AR057579A1 (es) * | 2005-11-23 | 2007-12-05 | Merck & Co Inc | Compuestos espirociclicos como inhibidores de histona de acetilasa (hdac) |
| KR101495611B1 (ko) | 2006-04-07 | 2015-02-25 | 메틸진 인코포레이티드 | 히스톤 데아세틸라아제의 억제제 |
| CA2649861A1 (en) | 2006-04-26 | 2007-11-08 | Merck & Co., Inc. | Disubstituted aniline compounds |
| CA2651681A1 (en) * | 2006-05-18 | 2007-11-29 | Merck & Co., Inc. | Aryl-fused spirocyclic compounds |
| EP2174938A1 (en) * | 2006-10-12 | 2010-04-14 | SuperGen, Inc. | Quinoline derivatives for modulating DNA methylation |
| JP2010120852A (ja) | 2007-03-09 | 2010-06-03 | Daiichi Sankyo Co Ltd | 新規なジアミド誘導体 |
| WO2009033281A1 (en) | 2007-09-14 | 2009-03-19 | Methylgene Inc. | Cancer combination therapy with a selective inhibitor of histone deacetylase hdac1, hdac2 and/or hdac3 and a microtubule stabilizer |
| US7790746B2 (en) * | 2007-10-12 | 2010-09-07 | Supergen, Inc. | Quinoline derivatives for modulating DNA methylation |
| US7939546B2 (en) * | 2007-10-12 | 2011-05-10 | Supergen, Inc. | Quinoline derivatives for modulating DNA methylation |
| TWI600638B (zh) | 2010-01-22 | 2017-10-01 | 艾斯特隆製藥公司 | 作爲蛋白質去乙醯酶抑制劑之反式醯胺化合物及其使用方法 |
| WO2012018499A2 (en) | 2010-08-05 | 2012-02-09 | Acetylon Pharmaceuticals | Specific regulation of cytokine levels by hdac6 inhibitors |
| BR112013011868A2 (pt) | 2010-11-16 | 2016-08-23 | Acetylon Pharmaceuticals Inc | compostos de pirimidina hidróxi amida como inibidores da proteína desacetilase, composição farmacêutica e uso dos referidos compostos |
| WO2012106343A2 (en) | 2011-02-01 | 2012-08-09 | The Board Of Trustees Of The University Of Illinois | Hdac inhibitors and therapeutic methods using the same |
| CN102775356B (zh) * | 2011-05-13 | 2015-07-22 | 江苏恒谊药业有限公司 | 4-氨基喹唑啉衍生物及其应用 |
| KR20130077390A (ko) * | 2011-12-29 | 2013-07-09 | 제이더블유중외제약 주식회사 | 단백질 키나아제 저해활성을 가지는 6-아미노-3-카복스아미도인다졸 유도체 |
| EP2839037B1 (en) | 2012-04-19 | 2018-12-26 | Acetylon Pharmaceuticals, Inc. | Biomarkers to identify patients that will respond to treatment and treating such patients |
| US9145412B2 (en) | 2012-11-02 | 2015-09-29 | Acetylon Pharmaceuticals, Inc. | Selective HDAC1 and HDAC2 inhibitors |
| US9139583B2 (en) | 2013-02-01 | 2015-09-22 | Acetylon Pharmaceuticals, Inc. | Selective HDAC3 inhibitors |
| JP2016511237A (ja) | 2013-02-01 | 2016-04-14 | アセチロン ファーマシューティカルズ インコーポレイテッドAcetylon Pharmaceuticals,Inc. | 選択的hdac3阻害剤 |
| EP3004141A4 (en) | 2013-06-03 | 2017-05-31 | Acetylon Pharmaceuticals, Inc. | Histone deacetylase ( hdac) biomarkers in multiple myeloma |
| ES2929576T3 (es) | 2013-10-08 | 2022-11-30 | Acetylon Pharmaceuticals Inc | Combinaciones de inhibidores de histona deacetilasa 6 y el inhibidor de Her2 lapatinib para el uso en el tratamiento del cáncer de mama |
| US20150105383A1 (en) | 2013-10-10 | 2015-04-16 | Acetylon Pharmaceuticals, Inc. | HDAC Inhibitors, Alone Or In Combination With PI3K Inhibitors, For Treating Non-Hodgkin's Lymphoma |
| WO2015054474A1 (en) | 2013-10-10 | 2015-04-16 | Acetylon Pharmaceuticals, Inc. | Pyrimidine hydroxy amide compounds as histone deacetylase inhibitors |
| EP3054954A4 (en) | 2013-10-10 | 2017-12-13 | Acetylon Pharmaceuticals, Inc. | Hdac inhibitors, alone or in combination with btk inhibitors, for treating non-hodgkin's lymphoma |
| CA2926808A1 (en) | 2013-10-11 | 2015-04-16 | Acetylon Pharmaceuticals, Inc. | Combinations of histone deacetylase inhibitors and immunomodulatory drugs |
| CR20160308A (es) | 2013-12-03 | 2016-11-08 | Acetylon Pharmaceuticals Inc | Combinaciones de inhibidores de histona deacetilasa y farmacos inmunomoduladores |
| ES2936812T3 (es) | 2013-12-20 | 2023-03-22 | Acetylon Pharmaceuticals Inc | Biomarcadores de histona desacetilasa 6 (HDAC6) en mieloma múltiple |
| US9464073B2 (en) | 2014-02-26 | 2016-10-11 | Acetylon Pharmaceuticals, Inc. | Pyrimidine hydroxy amide compounds as HDAC6 selective inhibitors |
| SG11201700094TA (en) | 2014-07-07 | 2017-02-27 | Acetylon Pharmaceuticals Inc | Treatment of leukemia with histone deacetylase inhibitors |
| US20160137630A1 (en) | 2014-10-08 | 2016-05-19 | Acetylon Pharmaceuticals, Inc. | Induction of gata2 by hdac1 and hdac2 inhibitors |
| AU2015360270B9 (en) | 2014-12-12 | 2019-12-05 | Regenacy Pharmaceuticals, Llc | Piperidine derivatives as HDAC1/2 inhibitors |
-
2015
- 2015-12-11 AU AU2015360270A patent/AU2015360270B9/en not_active Ceased
- 2015-12-11 SG SG11201704759QA patent/SG11201704759QA/en unknown
- 2015-12-11 JP JP2017531260A patent/JP6693957B2/ja not_active Expired - Fee Related
- 2015-12-11 MX MX2017007623A patent/MX2017007623A/es active IP Right Grant
- 2015-12-11 EP EP20160447.7A patent/EP3725787B1/en not_active Withdrawn - After Issue
- 2015-12-11 US US14/966,556 patent/US9790180B2/en not_active Expired - Fee Related
- 2015-12-11 HK HK18111028.3A patent/HK1251566A1/zh unknown
- 2015-12-11 ES ES15868544T patent/ES2816641T3/es active Active
- 2015-12-11 BR BR112017012504-8A patent/BR112017012504A2/pt not_active Application Discontinuation
- 2015-12-11 CN CN201580076059.1A patent/CN107835810A/zh active Pending
- 2015-12-11 WO PCT/US2015/065289 patent/WO2016094824A1/en not_active Ceased
- 2015-12-11 CA CA2970500A patent/CA2970500C/en active Active
- 2015-12-11 EA EA201791259A patent/EA035120B1/ru not_active IP Right Cessation
- 2015-12-11 KR KR1020177019370A patent/KR20170095964A/ko not_active Withdrawn
- 2015-12-11 EP EP15868544.6A patent/EP3292113B1/en active Active
-
2017
- 2017-06-09 PH PH12017501095A patent/PH12017501095A1/en unknown
- 2017-06-09 MX MX2020002487A patent/MX2020002487A/es unknown
- 2017-06-11 IL IL252828A patent/IL252828A0/en unknown
- 2017-06-13 CL CL2017001511A patent/CL2017001511A1/es unknown
- 2017-09-11 US US15/700,998 patent/US10358421B2/en active Active
- 2017-09-29 US US15/719,748 patent/US10239837B2/en active Active
-
2019
- 2019-06-06 US US16/433,386 patent/US10968180B2/en not_active Expired - Fee Related
-
2020
- 2020-02-06 AU AU2020200846A patent/AU2020200846A1/en not_active Abandoned
-
2021
- 2021-02-24 US US17/184,118 patent/US11702389B2/en active Active
-
2023
- 2023-05-05 US US18/313,016 patent/US20230348389A1/en not_active Abandoned
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20230348389A1 (en) | Piperidine derivatives as hdac1/2 inhibitors | |
| CA2994688C (en) | 1,3,4-oxadiazole derivative compounds as histone deacetylase 6 inhibitor, and the pharmaceutical composition comprising the same | |
| EP2951153B1 (en) | Selective hdac3 inhibitors | |
| EP3055299B1 (en) | Pyrimidine hydroxy amide compounds as histone deacetylase inhibitors | |
| KR102885432B1 (ko) | CCR8 억제제로 Tregs를 표적화하는 방법 및 조성물 | |
| EP3562810B1 (en) | Benzhydroxamic acid derivatives as selective hdac6 inhibitors | |
| CA2754613C (en) | Piperazine compound capable of inhibiting prostaglandin d synthase | |
| CA2684037A1 (en) | Compounds with anti-cancer activity | |
| US20180215743A1 (en) | 1,3,4-oxadiazole amide derivative compound as histone deacetylase 6 inhibitor, and pharmaceutical composition containing same | |
| JP6906449B2 (ja) | 複素環スルホンアミド誘導体及びそれを含有する医薬 | |
| EP4460492A1 (en) | Compounds containing a hydroxyphenyl moiety and their use | |
| JP5330377B2 (ja) | 3,4−ジヒドロキナゾリン誘導体 | |
| JP2025540298A (ja) | Hdac阻害剤およびその使用 | |
| HK40012833A (en) | Benzhydroxamic acid derivatives as selective hdac6 inhibitors | |
| HK40012833B (en) | Benzhydroxamic acid derivatives as selective hdac6 inhibitors | |
| HK1214818B (en) | Selective hdac3 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20170712 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| N231 | Notification of change of applicant | ||
| PN2301 | Change of applicant |
Patent event date: 20171228 Comment text: Notification of Change of Applicant Patent event code: PN23011R01D |
|
| PC1203 | Withdrawal of no request for examination | ||
| WITN | Application deemed withdrawn, e.g. because no request for examination was filed or no examination fee was paid |