KR20200032127A - 메틸롬 분석을 이용한 암 검출 및 분류 - Google Patents
메틸롬 분석을 이용한 암 검출 및 분류 Download PDFInfo
- Publication number
- KR20200032127A KR20200032127A KR1020207004066A KR20207004066A KR20200032127A KR 20200032127 A KR20200032127 A KR 20200032127A KR 1020207004066 A KR1020207004066 A KR 1020207004066A KR 20207004066 A KR20207004066 A KR 20207004066A KR 20200032127 A KR20200032127 A KR 20200032127A
- Authority
- KR
- South Korea
- Prior art keywords
- dna
- methylated
- acellular
- cancer
- derived
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Images





















Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6804—Nucleic acid analysis using immunogens
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- G—PHYSICS
- G06—COMPUTING OR CALCULATING; COUNTING
- G06F—ELECTRIC DIGITAL DATA PROCESSING
- G06F18/00—Pattern recognition
- G06F18/20—Analysing
- G06F18/24—Classification techniques
- G06F18/241—Classification techniques relating to the classification model, e.g. parametric or non-parametric approaches
-
- G—PHYSICS
- G06—COMPUTING OR CALCULATING; COUNTING
- G06N—COMPUTING ARRANGEMENTS BASED ON SPECIFIC COMPUTATIONAL MODELS
- G06N20/00—Machine learning
-
- G—PHYSICS
- G06—COMPUTING OR CALCULATING; COUNTING
- G06N—COMPUTING ARRANGEMENTS BASED ON SPECIFIC COMPUTATIONAL MODELS
- G06N3/00—Computing arrangements based on biological models
- G06N3/02—Neural networks
- G06N3/08—Learning methods
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/20—Allele or variant detection, e.g. single nucleotide polymorphism [SNP] detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G16B40/20—Supervised data analysis
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B5/00—ICT specially adapted for modelling or simulations in systems biology, e.g. gene-regulatory networks, protein interaction networks or metabolic networks
- G16B5/20—Probabilistic models
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2522/00—Reaction characterised by the use of non-enzymatic proteins
- C12Q2522/10—Nucleic acid binding proteins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2537/00—Reactions characterised by the reaction format or use of a specific feature
- C12Q2537/10—Reactions characterised by the reaction format or use of a specific feature the purpose or use of
- C12Q2537/164—Methylation detection other then bisulfite or methylation sensitive restriction endonucleases
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
- G16B30/20—Sequence assembly
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Physics & Mathematics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Analytical Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Biotechnology (AREA)
- Immunology (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- General Engineering & Computer Science (AREA)
- Theoretical Computer Science (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- Medical Informatics (AREA)
- Pathology (AREA)
- Data Mining & Analysis (AREA)
- Bioinformatics & Computational Biology (AREA)
- Evolutionary Biology (AREA)
- Software Systems (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Evolutionary Computation (AREA)
- Artificial Intelligence (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Hospice & Palliative Care (AREA)
- Oncology (AREA)
- General Physics & Mathematics (AREA)
- Bioethics (AREA)
- Databases & Information Systems (AREA)
- Epidemiology (AREA)
- Public Health (AREA)
- Computing Systems (AREA)
- Mathematical Physics (AREA)
Abstract
Description
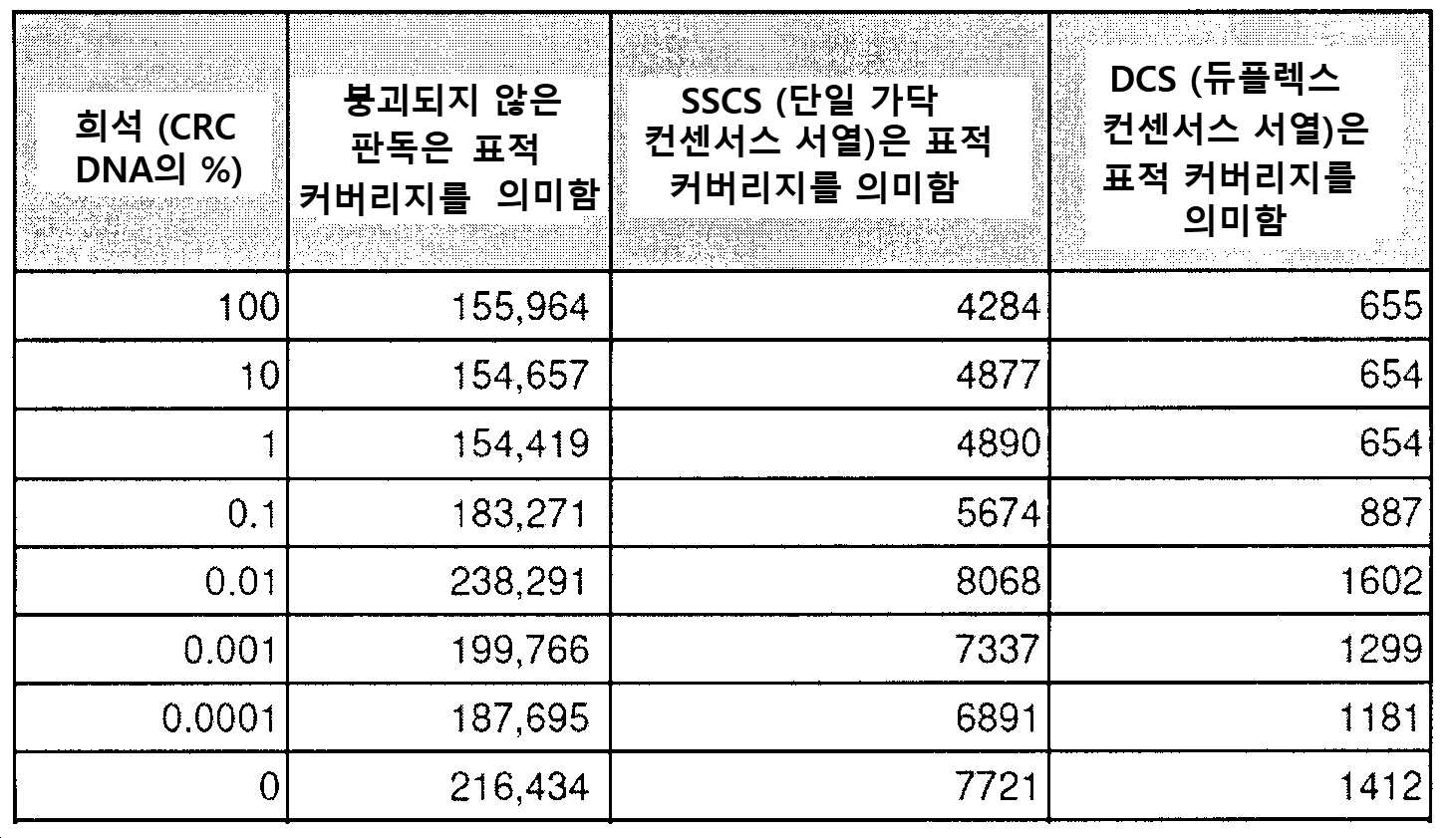
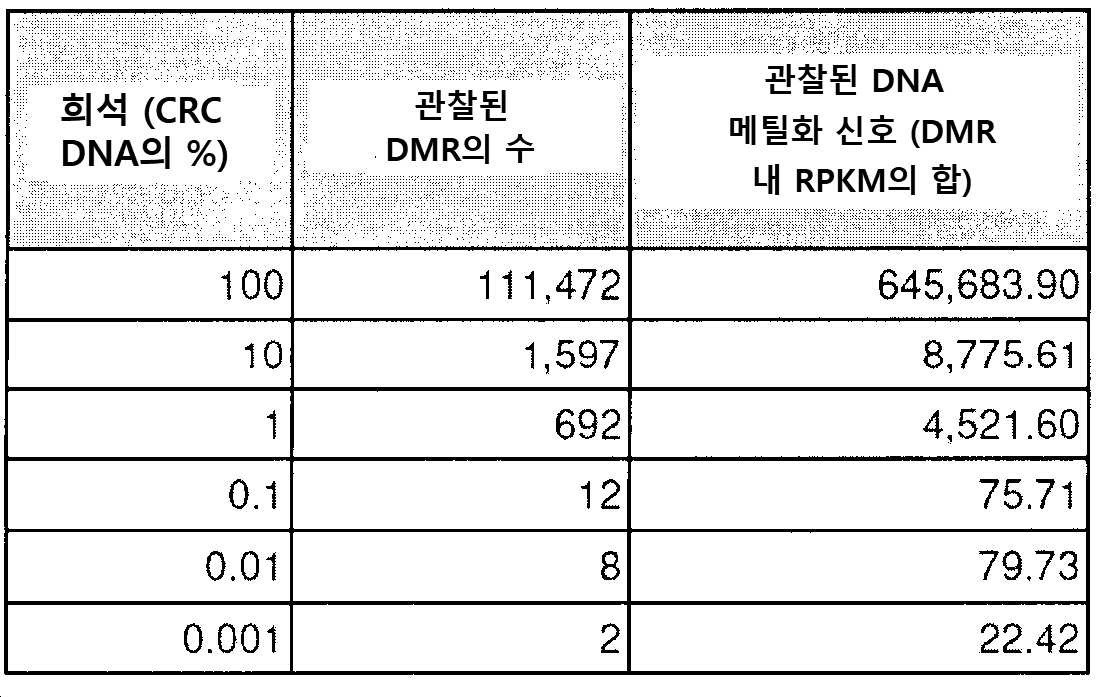
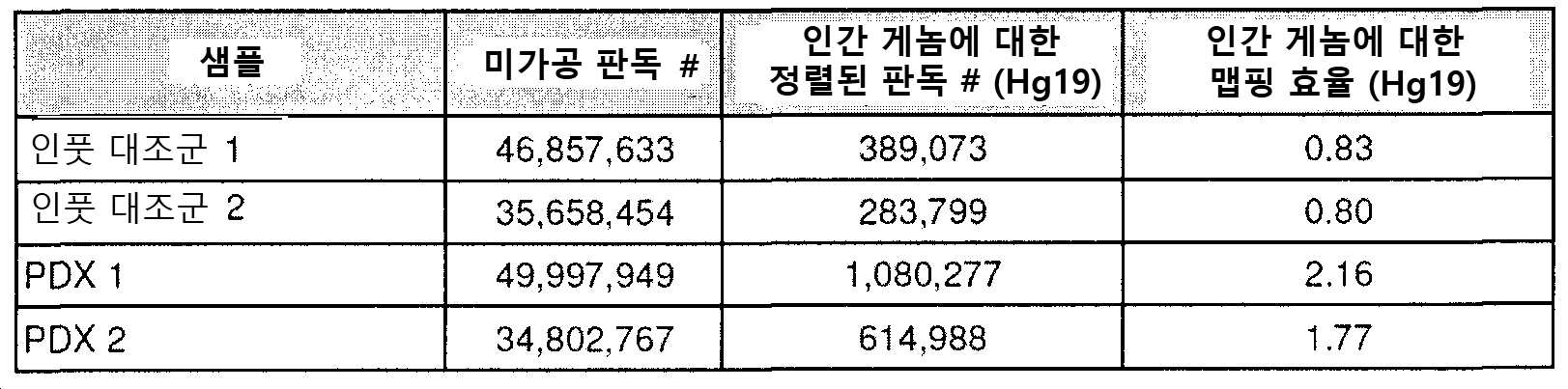
도 1은 cfDNA의 메틸롬 분석이 낮은 양의 인풋 DNA에서 ctDNA를 집적(enrich)하고 검출하기 위한 고도의 민감성 접근법이다. a) ctDNA의 농도 (컬럼), 조사되는 DMR의 수(열) 및 서열분석 깊이(x-축)의 함수로서 적어도 하나의 후생변이를 검출할 확률의 컴퓨터 시뮬레이션. b) 혈장 cfDNA를 모방하기 위해 단편화된 HCT116 세포주 기원의 1 내지 100 ng의 인풋 DNA에 대한 DNA 메틸화 신호 간의 게놈-전체 피어슨 상호관계. 각각의 농도는 2개의 생물학적 레플리케이트를 갖는다. c) ENCODE(ENCSR000DFS)로부터 수득된 HCT116(Green Tracks) + PRBS(감소된 표현 바이설파이트 서열분석(Reduced Representation Bisulfite Sequencing)) HCT116 데이터 및 GEO(GSM1465024)로부터 수득된 WGBS(전체-게놈 바이설파이트 서열분석(Whole-Genome Bisulfite Sequencing)) HCT116 데이터로부터 상이한 농도의 인풋 DNA 기원의 cfMeDIP-seq로부터 수득된 DNA 메틸화 프로파일. 히트맵(heatmap) (RRBS 트랙)에 대해, 황색은 메틸화된 것을 의미하고 청색은 비메틸화된 것을 의미하며 회색은 적용 범위가 없음을 의미한다. d-e) CRC 세포주 HCT116의 다중 골수종(MM) 세포주 MM1.S. cfMeDIP-seq로의 연속 희석은 순수 HCT116 DNA(100% CRC), 순수 MM1.S DNA(100% MM) 및 MM DNA로 희석된 10%, 1%, 0.1%, 0.01%, 및 0.001% CRC DNA에서 수행하였다. 모든 DNA는 혈장 cfDNA로 보망하기 위해 단편화시켰다. 본원 발명자들은 DMR의 관찰된 것 대 예상된(D) 수 및 (E) 상기 DMR 내 DNA 메틸화 신호(RPKM 중) 간의 거의 완벽한 선형 상호관계(r2=0.99, p<0.0001)를 관찰하였다. f) 동일한 연속 희석에서, 공지된 체세포 돌연변이는 백그라운드 서열분석기 및 폴리머라제 오류율 상에서 초심도(>10,000X) 표적화된 서열분석에 의해 1/100 대립형질유전자 분획에서만 검출 가능하다. CRC 세포주에서 각각의 돌연변이의 부위에서 각각의 염기 또는 삽입/결실을 함유하는 판독 분획을 나타낸다. g) 2명의 결장직장 암 환자 기원의 환자-유래된 이종이식체(PDX)를 보유한 마우스의 혈장에서 총 cfDNA(인간 + 마우스)의 백분율로서 ctDNA(인간)의 빈도.
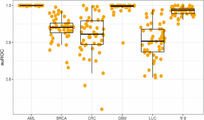
도 2는 종양 분류를 가능하게 하는 혈장 cfDNA의 메틸롬 분석을 보여준다. a) 암 분류를 위한 기계 학습 분류기 구성의 접근법을 입증하는 도식. b) 다중-분류 탄력적 네트 머신 학습 분류기 내 함유된 DMR의 히트맵. 분류기는 건강한 공여자(n=24), 폐암(n=25), 유방암(n=25), 결장직장암(n=23), 급성 골수성 백혈병(AML) (n=28), 및 신경모세포종 다형태(GBM) (n=71) 기원의 혈장 DNA 샘플에 대해 트레이닝하였다. 계층 클러스터링 방법: 워드(Ward). c) 모델의 10% 또는 25%에서 동정된 암-유형 관련된 DMR의 tSNE(t-분포 확률적 임베딩(t-Distributed Stochastic Neighbor Embedding))에 의한 2D 가시화. d) 혈장 cfDNA 메틸화-기반 다중암 분류기를 위한 수행능 이점. 탄력적 네트 머신 학습 분류기의 50배 생성 후 각각의 암 타입 및 건강한 공여자를 위해 y축 상에 나타낸 수용자 작동자 곡선 이하 면적(auROC).
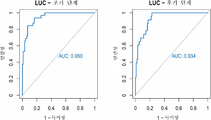
도 3은 독립적 코호트 상에서 다중암 분류기의 확증을 보여준다. a) ROC 곡선은 폐암(LUC) (n=55 LUC 대 n=97 기타), AML(n=35 AML 대 n=117 기타), 및 건강한 공여자(n=62 건강한 공여자 대 n=90 기타)의 코호트 상에서 다중암 분류기의 독립적 확장을 위해 나타낸다. b) ROC 곡선은 초기 단계 LUC(n=32 단계 I-II LUC 대 n=97 기타) 및 후기 단계 LUC(n=23 단계 III-IV LUC 대 n=97 기타) 상에 다중암 분류기의 독립적 확증을 위해 나타낸다.
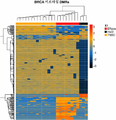
도 4는 종양 서브타입 분류를 가능하게 하는 혈장 cfDNA의 메틸롬 분석을 보여준다. a) 암 서브타입 관련 DMR의 tSNE(t-분포 확률적 임베딩)에 의한 2D 가시화. 유방암 서브타입은 특유한 종양 카피수 비정상(HER2 상태) 뿐만 아니라 특유한 유전자 발현 패턴 및 전사 인자 활성(ER 상태)을 갖는 종양을 보유한 환자들을 구분하는 능력을 보여준다. AML 서브타입은 특유한 재배열(FLT3 상태)을 갖는 종양을 보유한 환자들을 구분하는 능력을 보여준다. 신경모세포종 다형태(GBM) 서브타입은 특유한 점 돌연변이(IDH 유전자 돌연변이 상태)를 갖는 종양을 보유한 환자들을 구분하는 능력을 보여준다. 폐암 서브타입은 예후적 및 치료학적 관련성(선암 종 대 편평 암종 대 소세포 암종)을 갖는 특유한 조직학을 사용한 종양을 보유한 환자들을 구분하는 능력을 보여준다. b) 유방암 혈장 샘플에서 3개의 유방암 서브타입의 정확한 식별을 가능하게 하는 상부 DMR을 보여주는 히트맵. c) AML 환자 혈장 샘플 중에서 FLT3-ITD 상태의 정확한 식별을 가능하게 하는 상부 DMR을 보여주는 히트맵. d) 신경모세포종 다형태(GBM) 환자 혈장 샘플에서 IDH 유전자 돌연변이 상태의 정확한 식별을 가능하게 하는 상부 DMR을 보여주는 히트맵. e) 폐암 혈장 샘플에서 3개의 폐암 조직학의 정확한 식별을 가능하게 하는 상부 DMR을 보여주는 히트맵.
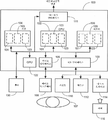
도 5는 본원에 기재된 바와 같이 하나 이상의 구현예을 가능하게 하기 위한 플랫폼을 제공하기 위해 적합하게 구성된 컴퓨터 장치, 및 관련된 통신 네트워크, 장치, 소프트웨어 및 펌웨어를 보여준다.
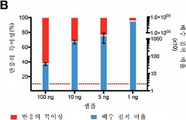
도 6은 서열분석 포화 분석 및 품질 관리를 보여준다. a) 상기 도면은 혈장 cfDNA를 모방하기 위해 단편화된 HCT116 DNA로부터의 각각의 인풋 농도에 대한 각각의 레플리케이트로부터 cfMeDIP-seq 데이터를 분석하는 바이오컨덕터 패키지(Bioconductor package) MEDIPS로부터의 포화 분석 결과를 보여준다. b) 프로토콜은 HCT116 세포주의 4개의 출발 DNA 농도(100, 10, 5, 및 1 ng)의 2개의 레플리케이에서 시험하였다. 반응의 특이성은 메틸화되고 비메틸화된 스파이크드-인(spiked-in) 에이. 탈리아나(A. thaliana) DNA를 사용하여 계산하였다. 배수 집적 비율은 단편화된 HCT116 DNA(메틸화된 고환-특이적 H2B, TSH2B0 및 비메틸화된 인간 DNA 영역(GAPDH 프로모터)에 대한 프라이머)의 게놈 영역을 사용하여 계산하였다. 수평 점선은 25의 배수-집적 비율 역치를 나타낸다. 오류 막대는 ± 1 s.e.m을 나타낸다. c) 서열분석된 샘플의 CpG 집적 스코어는 인풋 대조군과 비교하여 면역침전된 샘플로부터의 게놈 영역 내 CpG의 강한 집적을 보여준다. CpG 집적 스코어는 상기 영역의 CpG의 상대적 빈도를 인간 게놈의 CpG의 상대적 빈도로 나누어 수득하였다. 오류 막대는 ± 1 s.e.m을 나타낸다.

Claims (38)
- 대상체에서 암 세포 유래 DNA의 존재를 검출하는 방법으로서,
상기 방법이:
(a) 대상체로부터 무세포성 DNA(cell-free DNA) 샘플을 제공하는 단계;
(b) 메틸화된 무세포성 DNA의 후속적 서열분석을 허용하기 위해, 상기 샘플에 대해 라이브러리를 제조하는 단계;
(c) 제1 소정량의 충전제 DNA를 상기 샘플에 첨가한 다음 선택적으로 상기 샘플을 변성시키는 단계로서, 상기 충전제 DNA는 적어도 일부가 메틸화된 것인, 단계;
(d) 메틸화된 폴리뉴클레오타이드에 대한 선택적인 결합제를 사용하여 메틸화된 무세포성 DNA를 포획하는 단계;
(e) 상기 포획된 메틸화된 무세포성 DNA를 서열분석하는 단계;
(f) 상기 포획된 메틸화된 무세포성 DNA의 서열과, 건강한 개체 및 암성 개체로부터의 메틸화된 무세포성 대조군 DNA 서열을, 비교하는 단계;
(g) 상기 포획된 메틸화된 무세포성 DNA의 하나 이상의 서열과 암성 개체로부터 유래된 메틸화된 무세포성 DNA 서열 간에 통계학적으로 유의한 유사성이 존재하는 경우, 암 세포 유래 DNA의 존재를 동정하는 단계
를 포함하는, 방법. - 제1항에 있어서,
상기 샘플이 대상체의 혈액 또는 혈장으로부터 유래되는, 방법. - 제1항 또는 제2항에 있어서,
비교 단계 (f)가 통계적 분류기(statistical classifier)를 이용한 피트(fit)를 토대로 하는, 방법. - 제3항에 있어서,
상기 분류기가 기계 학습으로 파생되는(machine learning-derived), 방법. - 제4항에 있어서,
상기 분류기가 탄력적 네트 분류기(elastic net classifier), 라소(lasso), 서포트 벡터 머신(support vector machine), 랜덤 포레스트(random forest) 또는 신경 네트워크인, 방법. - 제1항 내지 제5항 중 어느 한 항에 있어서,
상기 건강한 개체 및 암성 개체로부터의 메틸화된 무세포성 대조군 DNA 서열이 건강한 개체와 암성 개체 간에 차별적으로 메틸화된 영역 (DMR)의 데이터베이스에 포함된, 방법. - 제1항 내지 제6항 중 어느 한 항에 있어서,
상기 건강한 개체 및 암성 개체로부터의 메틸화된 무세포성 대조군 DNA 서열이, 무세포성 DNA로부터 유래된 DNA에서 건강한 개체와 암성 개체 간에 차별적으로 메틸화되는, 메틸화된 무세포성 대조군 DNA 서열로 한정되는, 방법. - 제7항에 있어서,
상기 메틸화된 무세포성 대조군 DNA 서열이 혈장 유래 DNA에서 건강한 개체와 암성 개체 간에 차별적으로 메틸화되는, 방법. - 제1항 내지 제8항 중 어느 한 항에 있어서,
상기 샘플이 무세포성 DNA를 100 ng, 75 ng, 또는 50 ng 미만으로 포함하는, 방법. - 제1항 내지 제9항 중 어느 한 항에 있어서,
상기 제1 소정량의 충전제 DNA가 메틸화된 충전제 DNA를 약 5%, 10%, 15%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 또는 100%로 포함하고, 나머지는 비-메틸화된 충전제 DNA이고, 바람직하게 메틸화된 충전제 DNA를 5% 내지 50%, 10% 내지 40%, 또는 15% 내지 30%로 포함하는, 방법. - 제1항 내지 제9항 중 어느 한 항에 있어서,
상기 제1 소정량의 충전제 DNA가 20 ng 내지 100 ng, 바람직하게 30 ng 내지 100 ng, 보다 바람직하게 50 ng 내지 100 ng인, 방법. - 제1항 내지 제11항 중 어느 한 항에 있어서,
상기 샘플의 무세포성 DNA와 상기 제1 소정량의 충전제 DNA를 합하여 총 DNA의 50 ng 이상, 바람직하게 총 DNA의 100 ng 이상을 차지하는, 방법. - 제1항 내지 제12항 중 어느 한 항에 있어서,
상기 충전제 DNA가 50 bp 내지 800 bp 길이이고, 바람직하게 100 bp 내지 600 bp 길이이고, 보다 바람직하게 200 bp 내지 600 bp 길이인, 방법. - 제1항 내지 제13항 중 어느 한 항에 있어서,
상기 충전제 DNA가 이중 가닥인, 방법. - 제1항 또는 제2항에 있어서,
상기 충전제 DNA가 정크(junk) DNA인, 방법. - 제1항 내지 제3항 중 어느 한 항에 있어서,
상기 충전제 DNA가 내인성 또는 외인성 DNA인, 방법. - 제16항에 있어서,
상기 충전제 DNA가 비-인간 DNA, 바람직하게 λDNA인, 방법. - 제1항 내지 제17항 중 어느 한 항에 있어서,
상기 충전제 DNA가 인간 DNA에 대한 얼라인먼트(alignment)가 없는, 방법. - 제1항 내지 제18항 중 어느 한 항에 있어서,
상기 결합제가 메틸-CpG-결합 도메인을 포함하는 단백질인, 방법. - 제1항 내지 제19항 중 어느 한 항에 있어서,
상기 단백질이 MBD2 단백질인, 방법. - 제1항 내지 제20항 중 어느 한 항에 있어서,
단계 (d)가 메틸화된 무세포성 DNA를 항체를 사용해 면역침전시키는 것을 포함하는, 방법. - 제21항에 있어서,
면역침전을 위해 상기 샘플에 항체를 0.05 μg 이상, 바람직하게 0.16 μg 이상으로 첨가하는 것을 포함하는, 방법. - 제21항에 있어서,
상기 항체가 5-MeC 항체인, 방법. - 제21항에 있어서,
면역침전 반응을 확인하기 위해 단계 (c) 후 제2 소정량의 대조군 DNA를 상기 샘플에 첨가하는 단계를 더 포함하는, 방법. - 제1항 내지 제23항 중 어느 한 항에 있어서,
메틸화된 무세포성 DNA의 포획을 확인하기 위해 단계 (c) 후 제2 소정량의 대조군 DNA를 상기 샘플에 첨가하는 단계를 더 포함하는, 방법. - 제1항 내지 제25항 중 어느 한 항에 있어서,
암 세포 유래 DNA의 존재를 동정하는 단계가 암 세포의 기원 조직을 동정하는 단계를 더 포함하는, 방법. - 제26항에 있어서,
상기 암 세포의 기원 조직을 동정하는 단계가 암 서브타입을 동정하는 단계를 더 포함하는, 방법. - 제27항에 있어서,
상기 암 서브타입이 병기(stage), 조직학, 유전자 발현 패턴, 카피수 이상, 재배열 또는 점 돌연변이 상태를 기반으로 암을 구별하는, 방법. - 제1항 내지 제28항 중 어느 한 항에 있어서,
단계 (f)에서 비교가 게놈-전체(genome-wide)에서 수행되는, 방법. - 제1항 내지 제28항 중 어느 한 항에 있어서,
단계 (f)에서 비교가 게놈-전체에서 특정 조절 영역으로 제한되는, 방법. - 제30항에 있어서,
상기 조절 영역이 FANTOM5 인핸서, CpG 아일랜드, CpG 쇼어(shores), CpG 셸프(Shelves) 또는 이들의 조합인, 방법. - 제1항 내지 제31항 중 어느 한 항에 있어서,
단계 (f) 및 (g)가 컴퓨터 프로세서에 의해 수행되는, 방법. - 암 세포 유래 DNA의 존재를 검출하고 암 서브타입을 동정하는 방법으로서, 상기 방법이:
a. 대상체 샘플로부터 메틸화된 무세포성 DNA의 서열분석 데이터를 입수하는 단계;
b. 포획한 메틸화된 무세포성 DNA의 서열을, 건강한 개체 및 암성 개체로부터의 메틸화된 무세포성 대조군 DNA 서열과, 비교하는 단계;
c. 상기 포획한 메틸화된 무세포성 DNA의 하나 이상의 서열과 암성 개체로부터 유래된 메틸화된 무세포성 DNA 서열 간에 통계학적으로 유의한 유사성이 존재하는 경우, 암 세포 유래 DNA의 존재를 동정하는 단계; 및
d. 추가적으로, 단계 c에서 암 세포 유래 DNA가 동정된 경우, 단계 b에서의 비교에 기초하여 암 세포의 기원 조직 및 암 서브타입을 동정하는 단계
를 포함하는, 방법. - 암 세포 유래 DNA의 존재를 검출하고 암 서브타입을 동정하기 위한 컴퓨터-구현 방법(computer-implemented method)으로서,
상기 방법이:
a. 적어도 하나의 프로세서에서 대상체 샘플로부터 메틸화된 무세포성 DNA의 서열분석 데이터를 입수하는 단계;
b. 적어도 하나의 프로세서에서 포획한 메틸화된 무세포성 DNA의 서열을, 건강한 개체 및 암성 개체로부터의 메틸화된 무세포성 대조군 DNA 서열과, 비교하는 단계;
c. 적어도 하나의 프로세서에서 상기 포획한 메틸화된 무세포성 DNA의 하나 이상의 서열과 암성 개체로부터의 메틸화된 무세포성 DNA 서열 간에 통계학적으로 유의한 유사성이 존재하는 경우, 암 세포 유래 DNA의 존재를 동정하는 단계, 및 추가적으로, 단계 c로부터의 암 세포 기원의 DNA가 동정되는 경우, 단계 b에서의 비교에 기초하여 암 세포의 기원 조직 및 암 서브타입을 동정하는 단계
를 포함하는, 방법. - 프로세서 및 상기 프로세서에 연결된 메모리가 구비된 범용 컴퓨터와 연계하여 사용하기 위한 컴퓨터 프로그램 제품으로서,
상기 컴퓨터 프로그램 제품이 암호화된 컴퓨터 메카니즘을 가진 컴퓨터 판독 가능한 저장 매체를 포함하고,
상기 컴퓨터 프로그램 메카니즘이 컴퓨터의 메모리에 로딩되어, 컬퓨터에 의해 제34항의 방법을 수행하게 할 수 있는, 컴퓨터 프로그램 제품. - 제35항에 따른 컴퓨터 프로그램 제품을 저장하기 위한 저장된 데이터 구조를 가진 컴퓨터 판독 가능한 매체.
- 암 세포 유래 DNA의 존재를 검출하고 암 서브타입을 동정하기 위한 장치로서,
상기 장치가:
적어도 하나의 프로세서; 및
적어도 하나의 프로세서와 통신하는 전자 메모리를 포함하고,
상기 전자 메모리는, 상기 적어도 하나의 프로세서에서 실행되는 경우, 상기 적어도 하나의 프로세서가:
a. 대상체 샘플로부터 메틸화된 무세포성 DNA의 서열분석 데이터를 입수하고;
b. 포획한 메틸화된 무세포성 DNA의 서열과 건강한 개체 및 암성 개체로부터 유래된 메틸화된 무세포성 대조군 DNA 서열을 비교하고;
c. 포획한 메틸화된 무세포성 DNA의 하나 이상의 서열과 암성 개체로부터 유래된 메틸화된 무세포성 DNA 서열 간에 통계학적으로 유의한 유사성이 존재하는 경우, 암 세포 유래 DNA의 존재를 동정하고, 암 세포 유래 DNA가 동정되는 경우, 추가적으로, 단계 b에서의 비교에 기초하여 암 세포의 기원 조직 및 암 서브타입을 동정하게 하는, 프로세서-실행가능한 코드(processor-executable code)를 저장한, 장치. - 암 세포 유래 DNA의 존재를 검출하고 2 이상의 가능성있는 장기로부터 암 세포가 발생하는 암의 위치를 결정하는 방법으로서,
상기 방법이:
(a) 대상체로부터 무세포성 DNA의 샘플을 제공하는 단계;
(b) 메틸화된 폴리뉴클레오타이드에 대한 선택적인 결합제를 사용하여 상기 샘플로부터 메틸화된 무세포성 DNA를 포획하는 단계;
(c) 상기 포획된 메틸화된 무세포성 DNA를 서열분석하는 단계;
(d) 상기 포획된 메틸화된 무세포성 DNA의 서열 패턴을 2 이상의 대조군 개체 집단(들)의 DNA 서열 패턴과 비교하는 단계로서, 상기 2 이상의 집단은 각각 서로 다른 장기에 암이 위치하는, 단계; 및
(e) 상기 무세포성 DNA와 상기 2 이상의 집단 간의 메틸화 패턴에 대한 통계학적으로 유의한 유사성에 기초하여 암 세포가 기원한 장기를 결정하는 단계
를 포함하는, 방법.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020247002257A KR102930572B1 (ko) | 2017-07-12 | 2018-07-11 | 메틸롬 분석을 이용한 암 검출 및 분류 |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762531527P | 2017-07-12 | 2017-07-12 | |
| US62/531,527 | 2017-07-12 | ||
| PCT/CA2018/000141 WO2019010564A1 (en) | 2017-07-12 | 2018-07-11 | DETECTION AND CLASSIFICATION OF CANCER USING METHYLOME ANALYSIS |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020247002257A Division KR102930572B1 (ko) | 2017-07-12 | 2018-07-11 | 메틸롬 분석을 이용한 암 검출 및 분류 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| KR20200032127A true KR20200032127A (ko) | 2020-03-25 |
| KR102628878B1 KR102628878B1 (ko) | 2024-01-23 |
Family
ID=65000926
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020207004066A Active KR102628878B1 (ko) | 2017-07-12 | 2018-07-11 | 메틸롬 분석을 이용한 암 검출 및 분류 |
| KR1020247002257A Active KR102930572B1 (ko) | 2017-07-12 | 2018-07-11 | 메틸롬 분석을 이용한 암 검출 및 분류 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020247002257A Active KR102930572B1 (ko) | 2017-07-12 | 2018-07-11 | 메틸롬 분석을 이용한 암 검출 및 분류 |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US12031184B2 (ko) |
| EP (1) | EP3652741A4 (ko) |
| JP (2) | JP2020537487A (ko) |
| KR (2) | KR102628878B1 (ko) |
| CN (2) | CN118600004A (ko) |
| BR (1) | BR112020000681A2 (ko) |
| CA (1) | CA3069754A1 (ko) |
| WO (1) | WO2019010564A1 (ko) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023191197A1 (ko) * | 2022-03-29 | 2023-10-05 | 주식회사 아이엠비디엑스 | 암 진단을 위한 다중 분석 예측 모델의 제조 방법 |
| WO2024091028A1 (ko) * | 2022-10-28 | 2024-05-02 | 주식회사 클리노믹스 | Cell-free dna를 이용한 건강 및 질병관리 시스템 및 방법 |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102522067B1 (ko) * | 2016-05-03 | 2023-04-13 | 유니버시티 헬스 네트워크 | 무-세포 메틸화된 dna의 포획 방법 및 이의 이용 |
| MX2019007444A (es) | 2016-12-22 | 2019-08-16 | Guardant Health Inc | Metodos y sistemas para analisis de moleculas de acido nucleico. |
| US12227737B2 (en) * | 2017-07-12 | 2025-02-18 | University Health Network | Cancer detection and classification using methylome analysis |
| BR112020000681A2 (pt) | 2017-07-12 | 2020-07-14 | University Health Network | detecção e classificação de cancro utilizando análise de metilome |
| US12428684B2 (en) * | 2018-07-05 | 2025-09-30 | Active Genomes Expressed Diagnostics Corp | Methods for detecting and treating a tumorigenic phenotype of the liver |
| CN109097439A (zh) * | 2018-09-04 | 2018-12-28 | 上海交通大学 | 一种检测少量样品全基因组dna甲基化的方法 |
| US11891653B2 (en) | 2019-09-30 | 2024-02-06 | Guardant Health, Inc. | Compositions and methods for analyzing cell-free DNA in methylation partitioning assays |
| CN114868191A (zh) * | 2019-10-11 | 2022-08-05 | 格瑞尔有限责任公司 | 利用起源组织阈值的癌症分类 |
| CN115087744A (zh) * | 2019-11-06 | 2022-09-20 | 大学健康网络 | 用于无细胞MeDIP测序的合成加标对照及其使用方法 |
| EP4617375A3 (en) * | 2019-11-26 | 2025-10-29 | Guardant Health, Inc. | Methods, compositions and systems for improving the binding of methylated polynucleotides |
| WO2021119471A1 (en) * | 2019-12-13 | 2021-06-17 | Grail, Inc. | Cancer classification using patch convolutional neural networks |
| CN111154846A (zh) * | 2020-01-13 | 2020-05-15 | 四川大学华西医院 | 一种甲基化核酸的检测方法 |
| CN119193839A (zh) * | 2020-05-09 | 2024-12-27 | 广州燃石医学检验所有限公司 | 癌症预后方法 |
| US12592321B2 (en) * | 2020-06-19 | 2026-03-31 | University Health Network | Cancer detection and classification using methylome analysis |
| CA3182321A1 (en) * | 2020-06-19 | 2021-12-23 | Scott BRATMAN | Multimodal analysis of circulating tumor nucleic acid molecules |
| AU2021292311A1 (en) * | 2020-06-20 | 2023-02-16 | Grail, Llc | Detection and classification of human papillomavirus associated cancers |
| JP2023547620A (ja) | 2020-10-23 | 2023-11-13 | ガーダント ヘルス, インコーポレイテッド | 分配および塩基変換を使用してdnaを解析するための組成物および方法 |
| CN112382342A (zh) * | 2020-11-24 | 2021-02-19 | 山西三友和智慧信息技术股份有限公司 | 一种基于集成特征选择的癌症甲基化数据分类方法 |
| CN112820407B (zh) * | 2021-01-08 | 2022-06-17 | 清华大学 | 利用血浆游离核酸检测癌症的深度学习方法和系统 |
| CN116762132B (zh) * | 2021-01-14 | 2026-03-31 | 深圳华大生命科学研究院 | 基于游离dna的疾病预测模型及其构建方法和应用 |
| CN114507737A (zh) * | 2022-03-22 | 2022-05-17 | 杭州医学院 | 一种石棉相关疾病甲基化标记物的检测引物组合、试剂盒及扩增子测序文库的构建方法 |
| CN119604623A (zh) * | 2022-05-25 | 2025-03-11 | 阿黛拉公司 | 用于无细胞核酸处理的方法和系统 |
| CN115274124B (zh) * | 2022-07-22 | 2023-11-14 | 江苏先声医学诊断有限公司 | 一种基于数据驱动的肿瘤早筛靶向Panel和分类模型的动态优化方法 |
| WO2024025831A1 (en) * | 2022-07-25 | 2024-02-01 | Grail, Llc | Sample contamination detection of contaminated fragments with cpg-snp contamination markers |
| CN115132273B (zh) * | 2022-08-01 | 2023-07-28 | 广州燃石医学检验所有限公司 | 一种肿瘤形成风险与肿瘤组织来源的评估方法及系统 |
| CN117766028B (zh) * | 2022-09-16 | 2025-03-11 | 深圳吉因加医学检验实验室 | 一种基于甲基化差异预测样本来源的方法及装置 |
| CN115376616B (zh) * | 2022-10-24 | 2023-04-28 | 臻和(北京)生物科技有限公司 | 一种基于cfDNA多组学的多分类方法及装置 |
| CN121569344A (zh) * | 2023-07-14 | 2026-02-24 | 夸登特健康公司 | 使用来自液体活检的dna甲基化对结肠直肠肿瘤进行分类 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015536639A (ja) * | 2012-09-20 | 2015-12-24 | ザ チャイニーズ ユニバーシティ オブ ホンコン | 血漿による胎児または腫瘍のメチロームの非侵襲的決定 |
| WO2016115530A1 (en) * | 2015-01-18 | 2016-07-21 | The Regents Of The University Of California | Method and system for determining cancer status |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004524030A (ja) | 2001-02-23 | 2004-08-12 | ザ ジョンズ ホプキンス ユニバーシティー スクール オブ メディシン | 膵臓がんにおけるメチル化の異なる配列 |
| US6818762B2 (en) | 2001-05-25 | 2004-11-16 | Maine Molecular Quality Controls, Inc. | Compositions and methods relating to nucleic acid reference standards |
| CN102308212A (zh) * | 2008-12-04 | 2012-01-04 | 加利福尼亚大学董事会 | 用于确定前列腺癌诊断和预后的材料和方法 |
| EP2514836B1 (en) | 2011-04-19 | 2016-06-29 | Max-Planck-Gesellschaft zur Förderung der Wissenschaften e.V. | Prostate cancer markers |
| US9732390B2 (en) | 2012-09-20 | 2017-08-15 | The Chinese University Of Hong Kong | Non-invasive determination of methylome of fetus or tumor from plasma |
| JP6681841B2 (ja) | 2014-05-09 | 2020-04-15 | ライフコーデックス アーゲー | 特別な細胞タイプに由来するdnaの検出とそれに関連する方法 |
| TWI813141B (zh) * | 2014-07-18 | 2023-08-21 | 香港中文大學 | Dna混合物中之組織甲基化模式分析 |
| WO2016094330A2 (en) * | 2014-12-08 | 2016-06-16 | 20/20 Genesystems, Inc | Methods and machine learning systems for predicting the liklihood or risk of having cancer |
| DE102015009187B3 (de) | 2015-07-16 | 2016-10-13 | Dimo Dietrich | Verfahren zur Bestimmung einer Mutation in genomischer DNA, Verwendung des Verfahrens und Kit zur Durchführung des Verfahrens |
| WO2017100356A1 (en) | 2015-12-07 | 2017-06-15 | Data4Cure, Inc. | A method and system for ontology-based dynamic learning and knowledge integration from measurement data and text |
| KR102522067B1 (ko) | 2016-05-03 | 2023-04-13 | 유니버시티 헬스 네트워크 | 무-세포 메틸화된 dna의 포획 방법 및 이의 이용 |
| CN110168099B (zh) | 2016-06-07 | 2024-06-07 | 加利福尼亚大学董事会 | 用于疾病和病症分析的无细胞dna甲基化模式 |
| US12227737B2 (en) | 2017-07-12 | 2025-02-18 | University Health Network | Cancer detection and classification using methylome analysis |
| BR112020000681A2 (pt) | 2017-07-12 | 2020-07-14 | University Health Network | detecção e classificação de cancro utilizando análise de metilome |
| US20210156863A1 (en) | 2017-11-03 | 2021-05-27 | University Health Network | Cancer detection, classification, prognostication, therapy prediction and therapy monitoring using methylome analysis |
-
2018
- 2018-07-11 BR BR112020000681-5A patent/BR112020000681A2/pt not_active Application Discontinuation
- 2018-07-11 CN CN202410710233.4A patent/CN118600004A/zh active Pending
- 2018-07-11 CN CN201880059089.5A patent/CN111094590A/zh active Pending
- 2018-07-11 KR KR1020207004066A patent/KR102628878B1/ko active Active
- 2018-07-11 US US16/630,299 patent/US12031184B2/en active Active
- 2018-07-11 JP JP2020501564A patent/JP2020537487A/ja active Pending
- 2018-07-11 WO PCT/CA2018/000141 patent/WO2019010564A1/en not_active Ceased
- 2018-07-11 KR KR1020247002257A patent/KR102930572B1/ko active Active
- 2018-07-11 EP EP18832886.8A patent/EP3652741A4/en active Pending
- 2018-07-11 CA CA3069754A patent/CA3069754A1/en active Pending
-
2022
- 2022-02-09 US US17/668,314 patent/US20220251665A1/en not_active Abandoned
-
2023
- 2023-07-21 JP JP2023119203A patent/JP2023139162A/ja active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2015536639A (ja) * | 2012-09-20 | 2015-12-24 | ザ チャイニーズ ユニバーシティ オブ ホンコン | 血漿による胎児または腫瘍のメチロームの非侵襲的決定 |
| WO2016115530A1 (en) * | 2015-01-18 | 2016-07-21 | The Regents Of The University Of California | Method and system for determining cancer status |
Non-Patent Citations (28)
| Title |
|---|
| Aravanis, A.M., M. Lee, and R.D. Klausner, Next-Generation Sequencing of Circulating Tumor DNA for Early Cancer Detection. Cell, 2017. 168(4): p. 571-574. |
| Beltran, H., et al., Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. 2016. 22(3): p. 298-305. |
| Chakravarthy, A., et al., Human Papillomavirus Drives Tumor Development Throughout the Head and Neck: Improved Prognosis Is Associated With an Immune Response Largely Restricted to the Oropharynx. J Clin Oncol, 2016. 34(34): p. 4132-4141. |
| Chan, K.C., et al., Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc Natl Acad Sci U S A, 2013. 110(47): p. 18761-8. |
| Diaz, L.A., Jr. and A. Bardelli, Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol, 2014. 32(6): p. 579-86. |
| Fang, F., et al., Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med, 2011. 3(75): p. 75ra25. |
| Fleischhacker, M. and B. Schmidt, Circulating nucleic acids (CNAs) and cancer--a survey. Biochim Biophys Acta, 2007. 1775(1): p. 181-232. |
| Hinoue, T., et al., Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res, 2012. 22(2): p. 271-82. |
| Hoadley, K.A., et al., Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell, 2014. 158(4): p. 929-44. |
| Kandoth, C., et al., Mutational landscape and significance across 12 major cancer types. Nature, 2013. 502(7471): p. 333-9. |
| Laurens van der Maaten, G.H., Visualizing Data using t-SNE. Journal of Machine Learning Research, 2008. 9: p. 2579-2605. |
| Law, C.W., et al., voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol, 2014. 15(2): p. R29. |
| Legendre, C., et al., Whole-genome bisulfite sequencing of cell-free DNA identifies signature associated with metastatic breast cancer. Clin Epigenetics, 2015. 7: p. 100. |
| Lehmann-Werman, R., et al., Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A, 2016. 113(13): p. E1826-34. |
| Lienhard, M., et al., MEDIPS: genome-wide differential coverage analysis of sequencing data derived from DNA enrichment experiments. Bioinformatics, 2014. 30(2): p. 284-6. |
| Mack, S.C., et al., Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature, 2014. 506(7489): p. 445-50. |
| Martincorena, I., et al., Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science, 2015. 348(6237): p. 880-6. |
| McGranahan, N., et al., Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci Transl Med, 2015. 7(283): p. 283ra54. |
| Nat Protoc, 7(4): 617-636. (2012.03.08.) * |
| Newman, A.M., et al., An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med, 2014. 20(5): p. 548-54. |
| Potter, N.T., et al., Validation of a real-time PCR-based qualitative assay for the detection of methylated SEPT9 DNA in human plasma. Clin Chem, 2014. 60(9): p. 1183-91. |
| Sharma, S., T.K. Kelly, and P.A. Jones, Epigenetics in cancer. Carcinogenesis, 2010. 31(1): p. 27-36. |
| Stirzaker, C., et al., Methylome sequencing in triple-negative breast cancer reveals distinct methylation clusters with prognostic value. Nat Commun, 2015. 6: p. 5899. |
| Sturm, D., et al., Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell, 2012. 22(4): p. 425-37. |
| Sun, K., et al., Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. Proc Natl Acad Sci U S A, 2015. 112(40): p. E5503-12. |
| Taiwo, O., et al., Methylome analysis using MeDIP-seq with low DNA concentrations. Nat Protoc, 2012. 7(4): p. 617-36. |
| Visvanathan, K., et al., Monitoring of Serum DNA Methylation as an Early Independent Marker of Response and Survival in Metastatic Breast Cancer: TBCRC 005 Prospective Biomarker Study. J Clin Oncol, 2016: p. JCO2015662080. |
| Zauber, P., S. Marotta, and M. Sabbath-Solitare, KRAS gene mutations are more common in colorectal villous adenomas and in situ carcinomas than in carcinomas. Int J Mol Epidemiol Genet, 2013. 4(1): p. 1-10. |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023191197A1 (ko) * | 2022-03-29 | 2023-10-05 | 주식회사 아이엠비디엑스 | 암 진단을 위한 다중 분석 예측 모델의 제조 방법 |
| WO2024091028A1 (ko) * | 2022-10-28 | 2024-05-02 | 주식회사 클리노믹스 | Cell-free dna를 이용한 건강 및 질병관리 시스템 및 방법 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3652741A1 (en) | 2020-05-20 |
| KR102628878B1 (ko) | 2024-01-23 |
| US20200308651A1 (en) | 2020-10-01 |
| US20220251665A1 (en) | 2022-08-11 |
| CN118600004A (zh) | 2024-09-06 |
| JP2020537487A (ja) | 2020-12-24 |
| EP3652741A4 (en) | 2021-04-21 |
| CA3069754A1 (en) | 2019-01-17 |
| WO2019010564A1 (en) | 2019-01-17 |
| JP2023139162A (ja) | 2023-10-03 |
| KR102930572B1 (ko) | 2026-02-24 |
| US12031184B2 (en) | 2024-07-09 |
| BR112020000681A2 (pt) | 2020-07-14 |
| KR20240018667A (ko) | 2024-02-13 |
| CN111094590A (zh) | 2020-05-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR102628878B1 (ko) | 메틸롬 분석을 이용한 암 검출 및 분류 | |
| US11560558B2 (en) | Methods of capturing cell-free methylated DNA and uses of same | |
| US20210156863A1 (en) | Cancer detection, classification, prognostication, therapy prediction and therapy monitoring using methylome analysis | |
| US12227737B2 (en) | Cancer detection and classification using methylome analysis | |
| JP7665659B2 (ja) | 循環腫瘍核酸分子のマルチモーダル分析 | |
| US20250277270A1 (en) | Heatrich-bs: heat enrichment of cpg-rich regions for bisulfite sequencing | |
| KR20240046525A (ko) | 세포-유리 dna에 대한 tet-보조 피리딘 보란 시퀀싱과 관련된 조성물 및 방법 | |
| AU2024341199A1 (en) | Methods and systems for methylation sequencing | |
| US20230416841A1 (en) | Inferring transcription factor activity from dna methylation and its application as a biomarker | |
| CN121464223A (zh) | 用于无细胞核酸处理的方法和系统 | |
| US12592321B2 (en) | Cancer detection and classification using methylome analysis | |
| HK40112829A (zh) | 使用甲基化组分析进行癌症检测和分类 | |
| CA3022606C (en) | Methods of capturing cell-free methylated dna and uses of same | |
| HK40092784A (zh) | 循环肿瘤核酸分子的多模态分析 | |
| HK40116163A (en) | Methods of capturing cell-free methylated dna and uses of same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
Patent event date: 20200211 Patent event code: PA01051R01D Comment text: International Patent Application |
|
| PG1501 | Laying open of application | ||
| A201 | Request for examination | ||
| PA0201 | Request for examination |
Patent event code: PA02012R01D Patent event date: 20210114 Comment text: Request for Examination of Application |
|
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection |
Comment text: Notification of reason for refusal Patent event date: 20230316 Patent event code: PE09021S01D |
|
| E701 | Decision to grant or registration of patent right | ||
| PE0701 | Decision of registration |
Patent event code: PE07011S01D Comment text: Decision to Grant Registration Patent event date: 20231019 |
|
| GRNT | Written decision to grant | ||
| PR0701 | Registration of establishment |
Comment text: Registration of Establishment Patent event date: 20240119 Patent event code: PR07011E01D |
|
| PR1002 | Payment of registration fee |
Payment date: 20240119 End annual number: 3 Start annual number: 1 |
|
| PG1601 | Publication of registration |