KR20200080272A - 비침습적 산전 검사 및 암 검출을 위한 핵산 크기 범위의 용도 - Google Patents
비침습적 산전 검사 및 암 검출을 위한 핵산 크기 범위의 용도 Download PDFInfo
- Publication number
- KR20200080272A KR20200080272A KR1020207014693A KR20207014693A KR20200080272A KR 20200080272 A KR20200080272 A KR 20200080272A KR 1020207014693 A KR1020207014693 A KR 1020207014693A KR 20207014693 A KR20207014693 A KR 20207014693A KR 20200080272 A KR20200080272 A KR 20200080272A
- Authority
- KR
- South Korea
- Prior art keywords
- size
- cancer
- pattern
- comparing
- dna
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images



































Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
- C12Q1/6886—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material for cancer
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
- C12Q1/6883—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes for diseases caused by alterations of genetic material
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B15/00—ICT specially adapted for analysing two-dimensional [2D] or three-dimensional [3D] molecular structures, e.g. structural or functional relations or structure alignment
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B20/00—ICT specially adapted for functional genomics or proteomics, e.g. genotype-phenotype associations
- G16B20/10—Ploidy or copy number detection
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B30/00—ICT specially adapted for sequence analysis involving nucleotides or amino acids
-
- G—PHYSICS
- G16—INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR SPECIFIC APPLICATION FIELDS
- G16B—BIOINFORMATICS, i.e. INFORMATION AND COMMUNICATION TECHNOLOGY [ICT] SPECIALLY ADAPTED FOR GENETIC OR PROTEIN-RELATED DATA PROCESSING IN COMPUTATIONAL MOLECULAR BIOLOGY
- G16B40/00—ICT specially adapted for biostatistics; ICT specially adapted for bioinformatics-related machine learning or data mining, e.g. knowledge discovery or pattern finding
- G16B40/20—Supervised data analysis
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6809—Methods for determination or identification of nucleic acids involving differential detection
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2537/00—Reactions characterised by the reaction format or use of a specific feature
- C12Q2537/10—Reactions characterised by the reaction format or use of a specific feature the purpose or use of
- C12Q2537/16—Assays for determining copy number or wherein the copy number is of special importance
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2537/00—Reactions characterised by the reaction format or use of a specific feature
- C12Q2537/10—Reactions characterised by the reaction format or use of a specific feature the purpose or use of
- C12Q2537/165—Mathematical modelling, e.g. logarithm, ratio
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2545/00—Reactions characterised by their quantitative nature
- C12Q2545/10—Reactions characterised by their quantitative nature the purpose being quantitative analysis
- C12Q2545/113—Reactions characterised by their quantitative nature the purpose being quantitative analysis with an external standard/control, i.e. control reaction is separated from the test/target reaction
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/154—Methylation markers
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2600/00—Oligonucleotides characterized by their use
- C12Q2600/156—Polymorphic or mutational markers
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Organic Chemistry (AREA)
- Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Molecular Biology (AREA)
- Immunology (AREA)
- Medical Informatics (AREA)
- Pathology (AREA)
- Spectroscopy & Molecular Physics (AREA)
- Biochemistry (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Computational Biology (AREA)
- Theoretical Computer Science (AREA)
- Evolutionary Biology (AREA)
- Data Mining & Analysis (AREA)
- Oncology (AREA)
- Hospice & Palliative Care (AREA)
- Artificial Intelligence (AREA)
- Evolutionary Computation (AREA)
- Computer Vision & Pattern Recognition (AREA)
- Bioethics (AREA)
- Software Systems (AREA)
- Epidemiology (AREA)
- Public Health (AREA)
- Databases & Information Systems (AREA)
- Crystallography & Structural Chemistry (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Chemical Kinetics & Catalysis (AREA)
Abstract
Description
도 2a는 본 발명의 구현예에 따른 혈장 DNA 단편의 크기에 대한 이수성 염색체에 대한 측정된 태아 DNA 분율을 보여준다.
도 2b는 본 발명의 구현예에 따른 정배수성(euploidy) 및 21번 삼염색체성 태아로부터의 DNA를 포함한 시료에 대한 크기 밴드에 대한 z-점수를 보여준다.
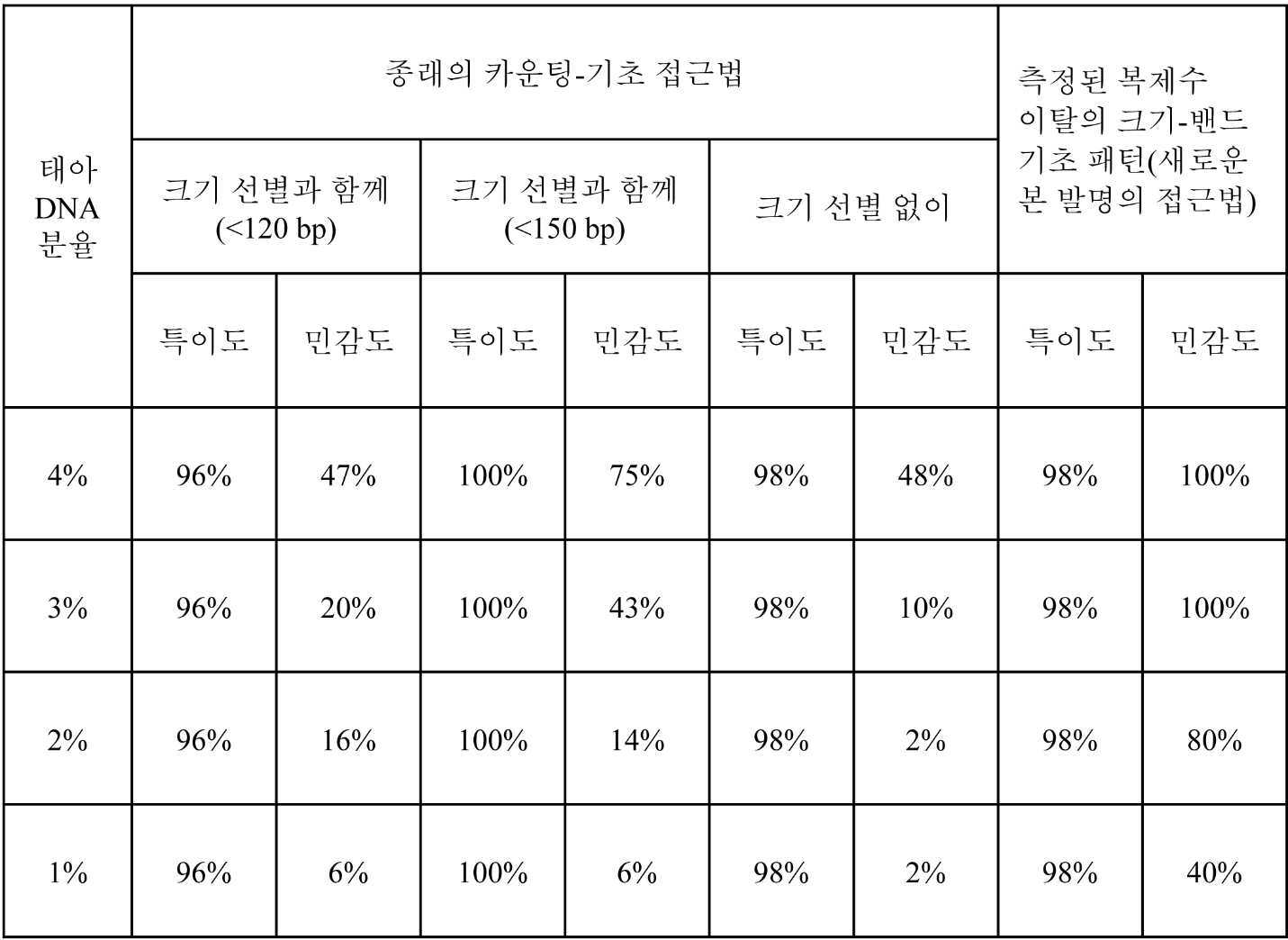
도 3은 본 발명의 구현예에 따라 4%의 태아 DNA 분율을 갖는 상이한 개별 임신에 걸쳐 이수성 염색체에 대한 측정된 게놈 표현(genomic representation)의 크기-밴드 기초의 변화 패턴을 보여준다.
도 4a는 본 발명의 구현예에 따라 정배수성 및 21번 삼염색체성 태아를 갖는 임신 사이에서 크기-밴드 기초의 변화 패턴의 히트맵 플롯을 보여준다.
도 4b는 본 발명의 구현예에 따라 정배수성 및 21번 삼염색체성 태아를 갖는 임신 사이에서 크기-밴드 기초의 변화 패턴의 t-SNE(t-분포 확률적 임베딩; t-distributed stochastic neighbor embedding) 플롯을 보여준다.
도 4c는 본 발명의 구현예에 따라 정배수성 및 21번 삼염색체성 태아를 갖는 임신 사이에서 종래의 z-점수 접근법을 사용한 z-점수 분포를 보여준다.
도 5a 및 도 5b는 본 발명의 구현예에 따라 상이한 크기 밴드 중에서 z-점수 패턴을 학습함으로써 신경망 기초 모델에 대한 성능 평가를 보여준다.
도 6은 염색체 영역이 본 발명의 구현예에 따라 대상체로부터의 생물학적 시료에서 복제수 이상을 나타내는지를 결정하는 방법을 보여준다.
도 7은 본 발명의 구현예에 따라 간세포암종(HCC) 환자의 혈장 DNA에서 측정된 메틸화의 크기-밴드 기초의 변화 패턴을 보여준다.
도 8은 본 발명의 구현예에 따라 대상체로부터 생물학적 시료에서 암 분류를 결정하는 방법을 보여준다.
도 9는 본 발명의 구현예에 따라 간세포암종(HCC) 환자의 혈장 DNA에서 측정된 복제수 이상의 크기-밴드 기초의 변화 패턴을 보여준다.
도 10은 본 발명의 구현예에 따라 암 검출을 위한 크기-밴드 게놈 표현(GR) 접근법에 대한 작업흐름(workflow)을 예시한다.
도 11a, 도 11b 및 도 11c는 본 발명의 구현예에 따라 크기-밴드 GR 접근법과 종래의 z-점수 접근법 사이의 비교를 보여준다.
도 12는 본 발명의 구현예에 따른 암 분류를 결정하는 방법을 보여준다.
도 13은 본 발명의 구현예에 따른 암 검출을 위한 크기-밴드 메틸화 밀도(MD) 접근법에 대한 작업흐름을 예시한다.
도 14a, 14b 및 14c는 본 발명의 구현예에 따라 크기-밴드 MD 접근법과 종래의 z-점수 접근법 사이의 비교를 보여준다.
도 15는 본 발명의 구현예에 따른 시스템을 예시한다.
도 16은 본 발명의 구현예에 따른 컴퓨터 시스템을 보여준다.
용어
용어 "시료", "생물학적 시료" 또는 "환자 시료"는 살아 있는 또는 죽은 대상체로부터 유래되는 임의의 조직 또는 물질을 포함하는 것으로 의미된다. 생물학적 시료는 세포-무함유 시료일 수 있으며, 이는 대상체로부터의 핵산 분자와 잠재적으로는 병원체, 예를 들어, 바이러스로부터의 핵산 분자의 혼합물을 포함할 수 있다. 생물학적 시료는 일반적으로, 핵산(예를 들어, DNA 또는 RNA) 또는 이의 단편을 포함한다. 용어 "핵산"은 일반적으로, 데옥시리보핵산(DNA), 리보핵산(RNA) 또는 이들의 임의의 하이브리드 또는 단편을 지칭한다. 시료 내의 핵산은 세포-무함유 핵산일 수 있다. 시료는 액체 시료 또는 고체 시료(예를 들어, 세포 또는 조직 시료)일 수 있다. 생물학적 시료는 체액, 예컨대 혈액, 혈장, 혈청, 소변, 질액, 수류(예를 들어, 고환의)로부터의 유체, 질 플러싱 유체(vaginal flushing fluid), 흉수(pleural fluid), 복수(ascitic fluid), 뇌척수액, 침, 땀, 눈물, 가래, 기관지폐포 세척액, 유두로부터의 배출액, 신체(예를 들어, 갑상선, 유방)의 상이한 부분들로부터의 흡인액 등일 수 있다. 대변 시료도 또한 사용될 수 있다. 다양한 구현예에서, 세포-무함유 DNA에 대해 농화되었던 생물학적 시료(예를 들어, 원심분리 프로토콜을 통해 수득된 혈장 시료) 내의 대부분의 DNA는 세포-무함유일 수 있다(예를 들어, 50%, 60%, 70%, 80%, 90%, 95% 또는 99% 초과의 DNA가 세포-무함유일 수 있음). 원심분리 프로토콜은 예를 들어, 3,000 g x 10분, 유체 부분을 수득하고 잔여 세포를 제거하기 위해 예를 들어, 30,000 g에서 또 다른 10분 동안 재-원심분리할 수 있다.
본원에 사용된 바와 같이, 용어 "좌위(locus)" 또는 이의 복수형 "좌위들"은 게놈에 걸쳐 변동을 갖는 임의의 길이의 뉴클레오타이드(또는 염기쌍)의 장소 또는 주소(address)이다. 용어 "서열 판독"은 핵산 분자의 모두 또는 일부, 예를 들어, DNA 단편으로부터 수득되는 서열을 지칭한다. 일 구현예에서, 단편의 단지 하나의 말단이 시퀀싱된다. 대안적으로, 단편의 두 말단 모두(예를 들어, 각각의 말단으로부터 약 30 bp)가 시퀀싱되어, 2개의 서열 판독을 발생시킬 수 있다. 그 후에, 짝형성된(paired) 서열 판독은 기준 게놈에 정렬될 수 있으며, 이는 단편의 길이를 제공할 수 있다. 보다 다른 구현예에서, 선형 DNA 단편은 예를 들어, 결찰에 의해 고리화될 수 있고, 결찰 부위를 포괄하는(spanning) 부분(part)이 시퀀싱될 수 있다.
본원에 사용된 바와 같이, 용어 "단편"(예를 들어, DNA 단편)은 적어도 3개의 연속 뉴클레오타이드를 포함하는 폴리뉴클레오타이드 또는 폴리펩타이드 서열의 일부를 지칭할 수 있다. 핵산 단편은 부모 폴리펩타이드의 생물학적 활성 및/또는 일부 특징을 보유할 수 있다. 핵산 단편은 이중 가닥 또는 단일 가닥이거나, 메틸화 또는 비메틸화되거나, 온전하거나 또는 닉킹되거나(nicked), 다른 거대분자, 예를 들어, 지질 입자, 단백질과 복합체화될 수 있거나 복합체화되지 않을 수 있다. 종양-유래 핵산은 종양 세포 내의 병원체로부터의 병원체 핵산을 포함하여 종양 세포로부터 방출되는 임의의 핵산을 지칭할 수 있다.
용어 "검정법"은 일반적으로, 핵산의 특성을 결정하는 기술을 지칭한다. 검정법(예를 들어, 제1 검정법 또는 제2 검정법)은 일반적으로, 시료 내 핵산의 양, 시료 내 핵산의 유전적 동일성, 시료 내 핵산의 복제수 변동(복제수 변동), 시료 내 핵산의 메틸화 상태, 시료 내 핵산의 단편 크기 분포, 시료 내 핵산의 돌연변이 상태, 또는 시료 내 핵산의 단편화 패턴을 결정하는 기술을 지칭한다. 당업자에게 알려진 임의의 검정법이 본원에서 언급된 핵산의 임의의 특성을 검출하는 데 사용될 수 있다. 핵산의 특성은 핵산의 서열, 양, 유전적 동일성, 복제수, 하나 이상의 뉴클레오타이드 위치에서의 메틸화 상태, 크기, 하나 이상의 뉴클레오타이드 위치에서 핵산 내 돌연변이, 및 핵산의 단편화 패턴(예를 들어, 핵산이 단편화하는 뉴클레오타이드 위치(들))을 포함한다. 용어 "검정법"은 용어 "방법"과 상호교환적으로 사용될 수 있다. 검정법 또는 방법은 특정 민감도 및/또는 특이도를 가질 수 있고, 진단 툴로서의 이들의 상대적인 유용성은 ROC-AUC 통계를 사용하여 측정될 수 있다.
본원에 사용된 바와 같이, 용어 "무작위 시퀀싱"은 일반적으로, 시퀀싱된 핵산 단편이 시퀀싱 절차 전에 구체적으로 식별되거나 또는 사전-결정되지 않은 시퀀싱을 지칭한다. 특이적인 유전자 좌위를 표적화하는 서열-특이적인 프라이머가 요구되지 않는다. 일부 구현예에서, 어댑터(adapter)는 단편의 말단에 첨가되고, 시퀀싱을 위한 프라이머가 상기 어댑터에 부착된다. 따라서, 임의의 단편은 동일한 유니버셜 어댑터에 부착하는 동일한 프라이머를 이용하여 시퀀싱될 수 있고, 따라서 시퀀싱은 무작위일 수 있다. 대규모 병렬 시퀀싱은 무작위 시퀀싱을 사용하여 수행될 수 있다.
"핵산"은 데옥시리보뉴클레오타이드 또는 리보뉴클레오타이드 및 단일 가닥 또는 이중 가닥 형태의 이들의 중합체를 지칭할 수 있다. 용어는 합성, 천연 발생 및 비-천연 발생이며 기준 핵산과 유사한 결합 특성을 갖고 기준 뉴클레오타이드와 유사한 방식으로 대사되는 기지의(known) 뉴클레오타이드 유사체 또는 변형된 백본 잔기 또는 연결(linkage)을 함유하는 핵산을 포괄할 수 있다. 이러한 유사체의 예는 비제한적으로, 포스포로티오에이트, 포스포라미다이트, 메틸 포스포네이트, 카이랄-메틸 포스포네이트, 2-O-메틸 리보뉴클레오타이드, 펩타이드-핵산(PNA)을 포함할 수 있다.
다르게 나타내지 않는 한, 특정 핵산 서열은 또한, 이의 보존적으로 변형된 변이체(예를 들어, 축퇴성(degenerate) 코돈 치환) 및 상보적 서열, 뿐만 아니라 명쾌하게 나타낸 서열을 내재적으로 포괄한다. 구체적으로, 축퇴성 코돈 치환은, 하나 이상의 선택된(또는 모든) 코돈의 제3 위치가 혼합-염기 및/또는 데옥시이노신 잔기로 치환된 서열을 발생시킴으로써 달성될 수 있다(문헌[Batzer et al., Nucleic Acid Res. 19:5081 (1991); Ohtsuka et al., J. Biol. Chem. 260:2605-2608 (1985); Rossolini et al., Mol. Cell. Probes 8:91-98 (1994)]). 용어 핵산은 유전자, cDNA, mRNA, 올리고뉴클레오타이드 및 폴리뉴클레오타이드와 상호교환적으로 사용된다.
용어 "뉴클레오타이드"는 천연 발생 리보뉴클레오타이드 또는 데옥시리보뉴클레오타이드 단량체를 지칭하는 것 외에도, 문맥상 분명하게 다르게 나타내지 않는 한, 뉴클레오타이드가 사용되는 특정 맥락(예를 들어, 상보적 염기에의 혼성화)에 관하여 기능적으로 동등한 이의 유도체 및 유사체를 포함하여 관련된 구조적 변이체를 지칭하는 것으로 이해될 수 있다.
"서열 판독"은 핵산 분자 중 임의의 부분 또는 모두로부터 시퀀싱된 뉴클레오타이드 열(string)을 지칭한다. 예를 들어, 서열 판독은 생물학적 시료에 존재하는 전체 핵산 단편일 수 있다. 또한 예로서, 서열 판독은 생물학적 시료에 존재하는 핵산 단편으로부터 시퀀싱된 뉴클레오타이드(예를 들어, 20 내지 150개 염기)의 짧은 열, 핵산 단편 중 하나의 말단 또는 두 말단 모두에서 뉴클레오타이드의 짧은 열, 또는 전체 핵산 단편의 시퀀싱일 수 있다. 서열 판독은 여러 가지 방식으로, 예를 들어, 혼성화 프로브 또는 포착 프로브에서 시퀀싱 기술을 사용하거나 프로브를 사용하여, 또는 증폭 기술, 예컨대 중합효소 연쇄 반응(PCR) 또는 단일 프라이머를 사용하는 선형 증폭 또는 등온 증폭에서, 또는 생물물리학적 측정, 예컨대 질량 분광분석법을 기초로 하여 수득될 수 있다. 서열 판독은 단일-분자 시퀀싱으로부터 수득될 수 있다. "단일-분자 시퀀싱"은 주형 DNA 분자의 클론 복사체로부터 염기 서열 정보를 해석할 필요 없이, 단일 주형 DNA 분자를 시퀀싱하여 서열 판독을 수득하는 것을 지칭한다. 단일-분자 시퀀싱은 전체 분자 또는 DNA 분자의 단지 일부를 시퀀싱할 수 있다. 대부분의 DNA 분자, 예를 들어, 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95% 또는 99% 초과가 시퀀싱될 수 있다.
용어 "유니버셜 시퀀싱"은, 어댑터가 단편의 말단에 첨가되고, 시퀀싱용 프라이머가 상기 어댑터에 부착되는 시퀀싱을 지칭한다. 따라서, 임의의 단편이 동일한 프라이머로 시퀀싱될 수 있으므로, 시퀀싱은 무작위일 수 있다.
"임상적-관련(clinically-relevant)" DNA의 예는 모체 혈장 내 태아 DNA 및 환자의 혈장 내 종양 DNA를 포함한다. 또 다른 예는 이식 환자의 혈장 내 이식-연관 DNA의 양의 측정을 포함한다. 추가의 예는 대상체의 혈장 내 조혈모 DNA 및 비-조혈모 DNA의 상대량의 측정을 포함한다. 이러한 후자의 구현예는 조혈모 및 비-조혈모 조직을 수반하는 병리학적 과정 또는 손상을 검출하거나 모니터링하거나 예후화하는 데 사용될 수 있다.
용어 "암의 수준"(또는 보다 일반적으로 "질병의 수준" 또는 "질환의 수준")은, 암이 존재하는지의 여부(즉, 존재 또는 부재), 암의 병기, 종양의 크기, 전이가 존재하는지의 여부, 신체의 총 종양 부담, 치료에 대한 암의 반응, 및/또는 암의 중증도의 다른 측정치(예를 들어, 암의 재발)를 지칭할 수 있다. 암의 수준은 수치(예를 들어, 확률) 또는 다른 지표, 예컨대 부호, 알파벳 글자 및 색상일 수 있다. 수준은 제로(0)일 수 있다. 암의 수준은 또한, 전악성(premalignant) 또는 전암성(precancerous) 질환(상태)을 포함한다. 암의 수준은 다양한 방식으로 사용될 수 있다. 예를 들어, 스크리닝은, 암을 갖고 있는 것으로 이전에는 알려지지 않은 개체에 암이 존재하는지 체크할 수 있다. 평가는 암을 진단받은 개체를 조사하여, 시간 경과에 따른 암의 진전을 모니터링하거나, 치료법의 효능을 연구하거나, 예후를 결정할 수 있다. 일 구현예에서, 예후는 환자가 암으로 사망할 가능성, 특정한 기간 또는 시간 후에 암이 진전되는 가능성, 또는 암이 전이될 가능성으로서 표현될 수 있다. 검출은 '스크리닝'을 의미할 수 있거나, 암의 제안적인 특색(예를 들어, 증상 또는 다른 양성 시험)을 갖는 개체가 암을 갖고 있는지 체크하는 것을 의미할 수 있다. "병상(pathology)의 수준"은 병원체와 연관된 병상의 수준을 지칭할 수 있으며, 이때 상기 수준은 암에 대해 상기 기재된 바와 같을 수 있다. 질병/질환의 수준 또한, 암에 대해 상기 기재된 바와 같을 수 있다. 암이 병원체와 연관되어 있는 경우, 암의 수준은 병상의 수준의 유형일 수 있다.
본원에 사용된 바와 같이, 용어 "염색체 이수성"은 2배체 게놈으로부터 염색체의 정량적 양의 변동을 의미한다. 상기 변동은 획득(gain) 또는 소실(loss)일 수 있다. 상기 변동은 하나의 염색체의 전체 또는 염색체의 영역을 수반할 수 있다.
본원에 사용된 바와 같이, 용어 "서열 불균형" 또는 "이상"은 기준 양으로부터 임상적으로 관련된 염색체 영역의 양에서 적어도 하나의 컷오프(cutoff) 값에 의해 정의되는 바와 같은 임의의 유의한 편차를 의미한다. 서열 불균형은 염색체 투여량 불균형, 대립유전자 불균형, 돌연변이 투약량 불균형, 복제수 불균형, 반수체 투약량 불균형, 및 다른 유사한 불균형을 포함할 수 있다. 일례로, 대립유전자 불균형은, 종양이 결실된 유전자의 하나의 대립유전자 또는 증폭된 유전자의 하나의 대립유전자, 또는 이의 게놈에서 2개 대립유전자의 차별적인 증폭을 가짐으로써 시료 내 특정 좌위에서 불균형을 생성할 때 발생할 수 있다. 또 다른 예로서, 환자는 종양 억제자 유전자에서 유전받은(inherited) 돌연변이를 가질 수 있을 것이다. 그 후에, 상기 환자는 종양 억제자 유전자의 비-돌연변이화된 대립유전자가 결실되는 종양을 발병시키게 될 수 있을 것이다. 따라서, 종양 내에서, 돌연변이 투여량 불균형이 존재한다. 종양이 이의 DNA를 환자의 혈장 내로 방출할 때, 종양 DNA는 혈장 내 환자의 (정상 세포로부터의) 구성적(constitutional) DNA와 혼합될 것이다. 본원에 기재된 방법의 사용을 통해, 혈장에서 이러한 DNA 혼합물의 돌연변이 투여량 불균형이 검출될 수 있다. 이상은 염색체 영역의 결실 또는 증폭을 포함할 수 있다.
포유류 게놈에서 "DNA 메틸화"는 전형적으로, CpG 디뉴클레오타이드 중에서 시토신 잔기의 5' 탄소에 메틸기의 첨가(즉, 5-메틸시토신)를 지칭한다. DNA 메틸화는 다른 맥락, 예를 들어, CHG 및 CHH의 시토신에서 발생할 수 있으며, 이때 H는 아데닌, 시토신 또는 티민이다. 시토신 메틸화는 또한, 5-하이드록시메틸시토신의 형태일 수 있다. 비-시토신 메틸화, 예컨대 N6-메틸아데닌이 또한, 보고되었다.
"분류"는 시료의 특정한 특성과 연관된 임의의 수치(들) 또는 다른 특징(들)을 지칭한다. 예를 들어, "+" 부호(또는 단어 "양성")는, 시료가 결실 또는 증폭을 갖고 있는 것으로 분류됨을 의미할 수 있을 것이다. 분류는 2진(binary)(예를 들어, 양성 또는 음성)일 수 있거나, 더 많은 수준의 분류(예를 들어, 1 내지 10, 또는 0 내지 1의 규모)를 가질 수 있다.
용어 "컷오프" 및 "역치"는 작동에 사용되는 사전-결정된 수치를 지칭할 수 있다. 역치 또는 기준 값은, 이 값의 초과 또는 미만에서 특정 분류, 예를 들어, 질환의 분류, 예컨대 대상체가 질환을 갖고 있는지의 여부 또는 질환의 중증도가 적용되는 값일 수 있다. 컷오프는 시료 또는 대상체의 특징을 참조하거나 참조하지 않으면서 사전-결정될 수 있다. 예를 들어, 컷오프는 시험되는 대상체의 연령 또는 성별에 기초하여 선택될 수 있다. 컷오프는 시험 데이터의 출력 후 그리고 이에 기초하여 선택될 수 있다. 예를 들어, 소정의 컷오프는, 시료의 시퀀싱이 소정의 깊이에 도달할 때 사용될 수 있다. 또 다른 예로서, 하나 이상의 질환의 기지의 분류 및 측정된 특징적인 값(예를 들어, 메틸화 수준, 통계학적 크기 값, 또는 카운트(count))을 갖는 기준 대상체는 상이한 질환들 및/또는 질환의 분류(예를 들어, 대상체가 질환을 갖고 있는지 여부)를 분간하기 위해 기준 수준을 결정하는 데 사용될 수 있다. 이들 용어 중 임의의 용어는 이들 맥락 중 임의의 맥락에서 사용될 수 있다. 당업자가 이해하는 바와 같이, 컷오프는 요망되는 민감도 및 특이도를 달성하도록 선택될 수 있다.
"부위"("게놈 부위"로도 지칭됨)는 단일 부위에 상응하며, 이는 단일 염기 위치 또는 상관(correlated) 염기 위치의 그룹, 예를 들어, 상관 염기 위치의 CpG 부위 또는 더 큰 그룹일 수 있다. "좌위"는 다수의 부위들을 포함하는 영역에 상응할 수 있다. 좌위는 단지 하나의 부위를 포함할 수 있으며, 이는 상기 좌위를 해당 맥락에서 부위에 동등하게 만들 것이다.
각각의 게놈 부위(예를 들어, CpG 부위)에 대한 "메틸화 지수"는 (예를 들어, 시퀀스 판독 또는 프로브로부터 결정된 바와 같은) DNA 단편의 비율을 지칭할 수 있으며, 부위를 망라하는 판독의 총 수에 걸쳐 해당 부위에서의 메틸화를 보여준다. "판독"은 DNA 단편으로부터 수득된 정보(예를 들어, 부위에서의 메틸화 상태)에 상응할 수 있다. 판독은 특정 메틸화 상태의 DNA 단편에 우선적으로 혼성화하는 시약(예를 들어, 프라이머 또는 프로브)을 사용하여 수득될 수 있다. 전형적으로, 이러한 시약은 DNA 분자의 메틸화 상태에 따라 이들 분자를 차별적으로 변형시키거나 차별적으로 인지하는 과정, 예를 들어, 비설파이트 전환, 또는 메틸화-민감성 제한 효소, 메틸화 결합 단백질, 또는 항-메틸시토신 항체로 처리한 후 적용된다. 또 다른 구현예에서, 메틸시토신 및 하이드록시메틸시토신을 인지하는 단일 분자 시퀀싱 기술은 메틸화 상태를 명시하고 메틸화 지수를 결정하는 데 사용될 수 있다.
영역의 "메틸화 밀도"는 영역 내의 부위를 망라하는 판독의 총 수로 나눈, 메틸화를 보여주는 영역 내의 부위에서의 판독의 수를 지칭할 수 있다. 상기 부위는 특이적인 특징, 예를 들어, CpG 부위라는 특징을 가질 수 있다. 따라서, 영역의 "CpG 메틸화 밀도"는 영역(예를 들어, 특정 CpG 부위, CpG 섬(island) 내의 CpG 부위, 또는 더 큰 영역) 내의 CpG 부위를 망라하는 판독의 총 수로 나눈, CpG 메틸화를 보여주는 판독의 수를 지칭할 수 있다. 예를 들어, 인간 게놈에서 각각의 100-kb 빈(bin)에 대한 메틸화 밀도는, 100-kb 영역으로 맵핑된(mapped) 시퀀스 판독에 의해 망라된 모든 CpG 부위의 비율로서 CpG 부위에서 비설파이트 처리(메틸화된 시토신에 상응함) 후 전환되지 않은 시토신의 총 수로부터 결정될 수 있다. 이 분석은 다른 빈 크기, 예를 들어, 500 bp, 5 kb, 10 kb, 50-kb 또는 1-Mb 등에 대해서도 수행될 수 있다. 영역은 전체 게놈, 염색체 또는 염색체의 일부(예를 들어, 염색체 아암(arm))일 수 있을 것이다. CpG 부위의 메틸화 지수는, 영역이 해당 CpG 부위만 포함할 때, 상기 영역에 대한 메틸화 밀도와 동일하다. "메틸화된 시토신의 비율"은 영역 내의 분석된 시토신 잔기, 즉, CpG 맥락 외부의 시토신을 포함하여 이들의 총 수에 걸쳐, 메틸화된(예를 들어, 비설파이트 전환 후 전환되지 않는) 것으로 보이는 시토신 부위, "C"의 수를 지칭할 수 있다. 메틸화된 시토신의 메틸화 지수, 메틸화 밀도 및 비율은, "메틸화 수준"의 예이며, 이는 부위에서 메틸화된 판독의 카운트를 수반하는 다른 비를 포함할 수 있다. 비설파이트 전환 외에도, 비제한적으로 메틸화 상태에 민감한 효소(예를 들어, 메틸화-민감성 제한 효소), 메틸화 결합 단백질, 메틸화 상태에 민감한 플랫폼을 사용하는 단일 분자 시퀀싱(예를 들어, 나노포어 시퀀싱(문헌[Schreiber et al. Proc Natl Acad Sci 2013; 110: 18910-18915]) 및 Pacific Biosciences 단일 분자 실시간 분석(문헌[Flusberg et al. Nat Methods 2010; 7: 461-465]))을 포함하여 당업자에게 알려진 다른 과정이 DNA 분자의 메틸화 상태에 대한 정보를 얻는 데 사용될 수 있다.
"메틸화-인식 시퀀싱"은 비제한적으로 비설파이트 시퀀싱, 또는 메틸화-민감성 제한 효소 절단에 뒤이은 시퀀싱, 항-메틸시토신 항체 또는 메틸화 결합 단백질을 사용한 면역침전, 또는 메틸화 상태의 명시(elucidation)를 가능하게 하는 단일 분자 시퀀싱을 포함하여 당업자가 시퀀싱 과정 동안 DNA 분자의 메틸화 상태를 확인할 수 있게 하는 임의의 시퀀싱 방법을 지칭한다. "메틸화-인식 검정법" 또는 "메틸화-민감성 검정법"은 시퀀싱 기초 방법과 비-시퀀싱 기초 방법 둘 모두, 예컨대 MSP, 프로브 기초 조사(interrogation), 혼성화, 제한 효소 절단 및 뒤이은 밀도 측정, 항-메틸시토신 면역검정법, 메틸화된 시토신 또는 하이드록시메틸시토신의 비율의 질량 분광분석법 조사, 시퀀싱이 후속하지 않는 면역침전 등을 포함할 수 있다.
"분리 값"(또는, 상대 존재비(relative abundance))은 2개의 값, 예를 들어, DNA 분자의 2개 양, 2개의 분획 기여도(fractional contribution), 또는 2개의 메틸화 수준, 예컨대 시료(혼합물) 메틸화 수준 및 기준 메틸화 수준을 수반하는 차이 또는 비에 상응한다. 분리 값은 단순한 차이 또는 비일 수 있을 것이다. 예로서, x/y의 정비(direct ratio)는 분리 값, 뿐만 아니라 x/(x+y)이다. 분리 값은 다른 인자, 예를 들어, 곱셈 인자(multiplicative factor)를 포함할 수 있다. 다른 예로서, 값들의 함수의 차이 또는 비, 예를 들어, 2개 값의 자연 로그(ln)의 차이 또는 비가 사용될 수 있다. 분리 값은 차이 및/또는 비를 포함할 수 있다. 메틸화 수준은 예를 들어, (예를 들어, 특정 부위에서) 메틸화된 DNA 분자와 다른 DNA 분자(예를 들어, 특정 부위에서의 모든 다른 DNA 분자 또는 단지 비메틸화된 DNA 분자) 사이의 상대 존재비의 일례이다. 다른 DNA 분자의 양은 정규화 인자(normalization factor)로서 작용할 수 있다. 또 다른 예로서, 모든 또는 비메틸화된 DNA 분자의 강도에 대한 메틸화된 DNA 분자의 강도(예를 들어, 형광 또는 전기적 강도)가 결정될 수 있다. 상대 존재비는 또한, 1 부피 당 강도를 포함할 수 있다.
용어 "대조군", "대조군 시료", "기준", "기준 시료", "정상" 및 "정상 시료"는 일반적으로 특정 질환을 갖고 있지 않는, 또는 그렇지 않다면 건강한 시료를 설명하기 위해 상호교환적으로 사용될 수 있다. 일례에서, 본원에 개시된 바와 같은 방법은 종양을 갖고 있는 대상체 상에서 수행될 수 있으며, 이때, 기준 시료는 대상체의 건강한 조직으로부터 가져온 시료이다. 또 다른 예에서, 기준 시료는 질병, 예를 들어, 암 또는 특정 병기(stage)의 암을 갖는 대상체로부터 가져온 시료이다. 기준 시료는 대상체로부터, 또는 데이터베이스로부터 수득될 수 있다. 기준은 일반적으로, 대상체로부터 시료를 시퀀싱함으로써 수득되는 시퀀스 판독을 맵핑하는 데 사용되는 기준 게놈을 지칭한다. 기준 게놈은 일반적으로, 생물학적 시료 및 구성적(constitutional) 시료로부터의 시퀀스 판독이 정렬되고 비교될 수 있는 반수체 또는 2배체 게놈을 지칭한다. 반수체 게놈의 경우, 각각의 좌위에 오직 1개의 뉴클레오타이드가 존재한다. 2배체 게놈의 경우, 이형접합체성(heterozygous) 좌위가 식별될 수 있으며, 이때, 이러한 좌위는 2개의 대립유전자를 가지며, 여기서 대립유전자는 상기 좌위로의 정렬을 위한 매치를 가능하게 할 수 있다. 기준 게놈은 예를 들어, 하나 이상의 바이러스 게놈을 포함함으로써 바이러스에 상응할 수 있다.
본원에 사용된 바와 같이 어구 "건강한"은 일반적으로, 양호한 건강을 소유한 대상체를 지칭한다. 이러한 대상체는 임의의 악성 또는 비-악성 질병의 부재를 실증한다. "건강한 개체"는 검정되는 질환과 관련이 없는 다른 질병 또는 질환을 갖고 있을 수 있으며, 통상적으로 "건강한" 것으로 여겨지지 않을 수 있다.
용어 "암" 또는 "종양"은 상호교환적으로 사용될 수 있고, 일반적으로, 조직의 비정상적인 덩어리(mass)를 지칭하며, 상기 덩어리의 성장은 정상 조직의 성장을 능가하고 이와 조화되지 않는다. 암 또는 종양은 하기 특징에 따라 "양성" 또는 "악성"으로서 정의될 수 있다: 형태 및 기능성을 포함하여 세포 분화의 정도, 성장 속도, 국소 침습, 및 전이. "양성" 종양은 일반적으로 잘 분화되어 있으며, 악성 종양보다 특징적으로 더 느린 성장을 갖고, 기원 부위로 국소화된 채로 남아 있다. 또한, 양성 종양은 원위부로 침윤하거나, 침습하거나 전이하는 능력을 갖지 않는다. "악성" 종양은 일반적으로 불량하게 분화되어 있으며(퇴화(anaplasia)), 주변 조직의 점진적인 침윤, 침습 및 파괴가 동반되는 특징적으로 신속한 성장을 가진다. 더욱이, 악성 종양은 원위부로 전이하는 능력을 가진다. "병기"는, 악성 종양이 얼마나 진행되어 있는지 설명하는 데 사용될 수 있다. 초기 암 또는 악성물은 말기 악성물보다 신체에서 더 적은 종양 부담(burden), 일반적으로 더 적은 증상, 더 양호한 예후, 및 더 양호한 치료 결과와 연관이 있다. 말기 또는 진행 병기(advanced stage) 암 또는 악성물은 종종, 원위부 전이 및/또는 림프 확산(lymphatic spread)과 연관이 있다.
용어 "위양성"(FP)은 질환을 갖지 않는 대상체를 지칭할 수 있다. 위양성은 일반적으로, 종양, 암, 전암성 질환(예를 들어, 전암성 병변), 국소화된 또는 전이된 암, 비-악성 질병을 갖지 않거나 그렇지 않다면 건강한 대상체를 지칭한다. 용어 위양성은 일반적으로, 질환을 갖지 않지만 본 개시내용의 검정법 또는 방법에 의해 상기 질환을 갖는 것으로 식별되는 대상체를 지칭한다.
용어 "민감도" 또는 "진양성률"(TPR; true positive rate)은 진양성 및 위음성의 수의 합계로 나눈, 진양성의 수를 지칭할 수 있다. 민감도는 실제로 질환을 갖고 있는 집단의 비율을 올바르게 식별하는 검정법 또는 방법의 능력을 특징화할 수 있다. 예를 들어, 민감도는 암을 갖고 있는 집단 내의 대상체의 수를 올바르게 식별하는 방법의 능력을 특징화할 수 있다. 또 다른 예에서, 민감도는 암을 시사하는 하나 이상의 마커를 올바르게 식별하는 방법의 능력을 특징화할 수 있다.
용어 "특이도" 또는 "진음성률"(TNR; true negative rate)은 진음성 및 위양성의 수의 합계로 나눈 진음성의 수를 지칭할 수 있다. 특이도는 실제로 질환을 갖고 있지 않는 집단의 비율을 올바르게 식별하는 검정법 또는 방법의 능력을 특징화할 수 있다. 예를 들어, 특이도는 암을 갖고 있지 않는 집단 내의 대상체의 수를 올바르게 식별하는 방법의 능력을 특징화할 수 있다. 또 다른 예에서, 특이도는 암을 시사하는 하나 이상의 마커를 올바르게 식별하는 방법의 능력을 특징화할 수 있다.
용어 "ROC" 또는 "ROC 곡선"은 수신자 조작자 특성 곡선(receiver operator characteristic curve)을 지칭할 수 있다. ROC 곡선은 2진 분류기 시스템의 성능의 그래프 표현일 수 있다. 임의의 주어진 방법에 대해, ROC 곡선은 다양한 역치 설정에서 민감도를 특이도에 대해 도시함으로써 발생될 수 있다. 대상체에서 종양의 존재를 검출하는 방법의 민감도 및 특이도는 상기 대상체의 혈장 시료 내 다양한 농도의 종양-유래 핵산에서 결정될 수 있다. 더욱이, 3개 매개변수(예를 들어, 민감도, 특이도, 및 역치 설정) 중 적어도 하나를 고려하면, ROC 곡선은 임의의 미지의 매개변수에 대한 값 또는 예상 값을 결정할 수 있다. 미지의 매개변수는 ROC 곡선에 피팅된(fitted) 곡선을 사용하여 결정될 수 있다. 용어 "AUC" 또는 "ROC-AUC"는 일반적으로, 수신자 조작자 특성 곡선 아래 면적을 지칭한다. 이러한 계측(metric)은 방법의 민감도와 특이도 둘 모두를 고려하여, 상기 방법의 진단 유용성의 측정치를 제공할 수 있다. 일반적으로, ROC-AUC는 0.5 내지 1.0의 범위이며, 이때, 0.5에 더 근접한 값은 상기 방법이 제한된 진단 유용성(예를 들어, 더 낮은 민감도 및/또는 특이도)을 가짐을 시사하고, 1.0에 더 근접한 값은 더 큰 진단 유용성(예를 들어, 더 높은 민감도 및/또는 특이도)을 가짐을 시사한다. 예를 들어, 문헌[Pepe et al, " Limitations of the Odds Ratio in Gauging the Performance of a Diagnostic, Prognostic, or Screening Marker," Am. J. Epidemiol 2004, 159 (9): 882-890]을 참조하고, 이는 참조에 의해 본 명세서에 포함된다. 확률 함수, 교차비(odds ratio), 정보 이론, 예측 값, 보정(적합도(goodness-of-fit)를 포함함), 및 재분류 측정을 사용하여 진단 유용성을 특징화하는 또 다른 접근법은 문헌[Cook, "Use and Misuse of the Receiver Operating Characteristic Curve in Risk Prediction," Circulation 2007, 115: 928-935]에 따라 요약되어 있으며, 이는 참조에 의해 본 명세서에 포함된다.
용어 "약" 또는 "대략"은 당업자에 의해 결정된 바와 같은 특정 값에 대한 허용 가능한 오차 범위 내를 의미할 수 있으며, 이는 부분적으로는 값이 어떻게 측정되거나 결정되는가, 즉, 측정 시스템의 한계에 의존할 수 있다. 예를 들어, "약"은 당업의 관행에 따라, 1 이내 또는 1 초과의 표준 편차를 의미할 수 있다. 대안적으로, "약"은 주어진 값의 20% 이하, 10% 이하, 5% 이하, 또는 1% 이하의 범위를 의미할 수 있다. 대안적으로, 특히 생물학적 시스템 또는 과정에 관하여, 용어 "약" 또는 "대략"은 값의 승수(order of magnitude) 이내, 5-배 이내, 보다 바람직하게는 2-배 이내를 의미할 수 있다. 특정 값이 출원 및 청구항에 기재되어 있는 경우, 다르게 언급되지 않는 한, 특정 값에 대한 허용 가능한 오차 범위 내를 의미하는 용어 "약"이 추정되어야 한다. 용어 "약"은 당업자에 의해 보편적으로 이해되는 바와 같은 의미를 가질 수 있다. 용어 "약"은 ±10%를 지칭할 수 있다. 용어 "약"은 ±5%를 지칭할 수 있다.
본원에서 사용되는 용어는 특정 사례만 설명하기 위한 것이고, 제한하려는 것이 아니다. 본원에 사용된 바와 같은, 단수형("a", "an" 및 "the")은 맥락상 다르게 명확하게 시사하지 않는 한, 복수형도 포함시키려는 것이다. "또는"의 사용은 "포함하거나 또는"을 의미하고, 구체적으로 다르게 시사되지 않는 한 "배제하거나 또는"을 의미하도록 의도되지 않는다. 용어 "~에 기초하는"은 "적어도 부분적으로 ~에 기초하는"을 의미하도록 의도된다. 더욱이, 용어 "포함하는", "포함한다", "갖는", "가진다", "있는" 또는 이의 변형이 상세한 설명 및/또는 청구범위에서 사용되는 정도까지, 이러한 용어는 용어 "포함하는"과 유사한 방식으로 포함하도록 의도된다.

Claims (43)
- 염색체 영역이 대상체로부터의 생물학적 시료에서 복제수 이상(copy number aberration)을 나타내는지를 결정하는 방법으로서, 상기 생물학적 시료는 임상적-관련(clinically-relevant) DNA 분자 및 다른 DNA 분자를 포함하는 세포-무함유 DNA 분자의 혼합물을 포함하고, 상기 방법은 하기 단계를 포함하는, 방법:
복수의 크기 범위의 각각의 크기 범위에 대해:
크기 범위에 상응하는 생물학적 시료로부터 세포-무함유 DNA 분자의 제1 양을 측정하는 단계, 및
크기 범위에 상응하는 세포-무함유 DNA 분자의 상기 제1 양, 및 크기 범위 내의 것이 아닌 크기를 포함하는 제2 크기 범위 내의 DNA 분자의 제2 양을 사용하여 크기 비를 컴퓨터 시스템에 의해 계산하는 단계;
복수의 크기 범위에 대한 복수의 기준 크기 비를 포함하는 기준 크기 패턴을 수득하는 단계로서, 상기 기준 크기 패턴은 염색체 영역에 복제수 이상을 갖는 대상체로부터의 또는 복제수 이상을 갖지 않는 대상체로부터의 복수의 기준 시료로부터 결정되는 단계;
복수의 크기 비를 상기 기준 패턴과 비교하는 단계;
상기 비교에 기초하여 염색체 영역이 복제수 이상을 나타내는지를 결정하는 단계. - 제1항에 있어서, 상기 임상적-관련 DNA 분자는 태아 DNA 또는 모체 DNA를 포함하는, 방법.
- 제1항에 있어서, 상기 임상적-관련 DNA 분자는 종양 DNA를 포함하고, 다른 DNA 분자는 비-종양 DNA를 포함하는, 방법.
- 제2항에 있어서, 상기 복제수 이상은 이수성(aneuploidy)인, 방법.
- 제3항에 있어서, 상기 복제수 이상은 암의 지표(indication)인, 방법.
- 제1항에 있어서, 복수의 크기 범위의 각각의 크기 범위는 밴드폭을 특징으로 하는, 방법.
- 제6항에 있어서, 상기 밴드폭은 50 bp 내지 200 bp 범위인, 방법.
- 제1항에 있어서, 상기 각각의 크기 범위는 복수의 크기 범위의 임의의 다른 크기 범위와 비-중첩되는, 방법.
- 제1항에 있어서, 상기 각각의 크기 범위는 복수의 크기 범위의 적어도 하나의 다른 크기 범위와 중첩되는, 방법.
- 제1항에 있어서, 상기 크기 비는 z-점수를 포함하는, 방법.
- 제1항에 있어서, 상기 제2 크기 범위는 복수의 크기 범위의 각각의 크기 범위보다 큰 범위인, 방법.
- 제1항에 있어서, 상기 제2 크기 범위는 생물학적 시료 내의 세포-무함유 DNA 분자의 모든 크기 또는 염색체 영역 내 세포-무함유 DNA 분자의 모든 크기를 포함하는, 방법.
- 제1항에 있어서, 상기 세포-무함유 DNA 분자는 게놈 영역으로부터의 것인, 방법.
- 제13항에 있어서, 상기 게놈 영역은 염색체인, 방법.
- 제13항에 있어서, 상기 게놈 영역은 염색체 아암(arm)인, 방법.
- 제1항에 있어서,
복수의 크기 비를 기준 크기 패턴과 비교하는 단계는 하기 단계를 포함하는, 방법:
복수의 크기 비의 각각의 크기 비를 상응하는 크기 범위에서 기준 크기 비와 비교하는 단계,
각각의 크기 비가 상응하는 크기 범위에서 기준 크기 비와 통계학적으로 유사한지 결정하는 단계. - 제1항에 있어서,
복수의 크기 비를 기준 크기 패턴과 비교하는 단계는 하기 단계를 포함하는, 방법:
복수의 크기 범위에 대한 복수의 크기 비를 포함하는 크기 패턴을 결정하는 단계;
상기 크기 패턴을 기준 크기 패턴과 비교하는 단계,
크기 패턴이 기준 크기 패턴과 유사한 모양을 갖는지를 결정하는 단계. - 제16항에 있어서,
상기 기준 크기 패턴은 복제수 이상을 갖는 대상체로부터의 복수의 기준 시료로부터 결정되고,
상기 방법은 추가로 하기 단계를 포함하는, 방법:
비교에 기초하여 염색체 영역이 복제수 이상을 나타내는지를 결정하는 단계. - 제1항에 있어서,
기준 크기 패턴을 수득하고 복수의 크기 비를 상기 기준 크기 패턴과 비교하는 단계는 복수의 크기 비를 기계 학습(machine learning) 모델 내로 입력하는 단계를 포함하고,
상기 기계 학습 모델은 복수의 기준 시료로부터의 복수의 트레이닝(training) 크기 패턴을 사용하여 트레이닝된, 방법. - 제1항에 있어서, 복수의 크기 비를 기준 크기 패턴과 비교하는 단계는, 상기 복수의 크기 비를, 복수의 기준 시료로부터 결정되는 복수의 역치 값과 비교하는 단계를 포함하는, 방법.
- 대상체로부터의 생물학적 시료에서 암 분류를 결정하는 방법으로서, 상기 생물학적 시료는 종양 DNA 분자 및 비-종양 DNA 분자를 포함하는 세포-무함유 DNA 분자의 혼합물을 포함하고, 상기 방법은 하기 단계를 포함하는, 방법:
복수의 크기 범위의 각각의 크기 범위에 대해:
크기 범위에 상응하는 생물학적 시료로부터 메틸화된 세포-무함유 DNA 분자의 제1 양을 측정하는 단계, 및
크기 범위에 상응하는 메틸화된 세포-무함유 DNA 분자의 상기 제1 양, 및 크기 범위 내의 것이 아닌 크기를 포함하는 제2 크기 범위 내의 DNA 분자의 제2 양을 사용하여 메틸화 수준을 컴퓨터 시스템에 의해 계산하는 단계;
복수의 크기 범위에 대한 복수의 기준 메틸화 수준을 포함하는 기준 크기 패턴을 수득하는 단계로서, 상기 기준 크기 패턴은 암을 갖는 대상체로부터의 또는 암을 갖지 않는 대상체로부터의 복수의 기준 시료로부터 결정되는 단계;
복수의 메틸화 수준을 상기 기준 패턴과 비교하는 단계; 및
상기 비교에 기초하여 암의 수준을 결정하는 단계. - 제21항에 있어서, 상기 제2 양은 메틸화된 세포-무함유 DNA 분자인, 방법.
- 제21항에 있어서, 상기 메틸화된 세포-무함유 DNA 분자는 염색체 아암으로부터의 것인, 방법.
- 제21항에 있어서,
복수의 메틸화 수준을 기준 크기 패턴과 비교하는 단계는 하기 단계를 포함하는, 방법:
복수의 크기 범위의 각각의 메틸화 수준을 상응하는 크기 범위에서 기준 메틸화 수준과 비교하는 단계,
각각의 메틸화 수준이 상응하는 크기 범위에서 기준 메틸화 수준과 통계학적으로 유사한지 결정하는 단계. - 제21항에 있어서,
복수의 메틸화 수준을 기준 크기 패턴과 비교하는 단계는 하기 단계를 포함하는, 방법:
복수의 크기 범위에 대한 복수의 메틸화 수준을 포함하는 크기 패턴을 결정하는 단계;
상기 크기 패턴을 기준 크기 패턴과 비교하는 단계,
크기 패턴이 기준 크기 패턴과 유사한 모양을 갖는지를 결정하는 단계. - 제24항에 있어서,
상기 기준 크기 패턴은 암을 갖는 대상체로부터의 복수의 기준 시료로부터 결정되고,
상기 방법은 추가로 하기 단계를 포함하는, 방법:
대상체가 암을 갖는지를 결정하는 단계. - 제21항에 있어서, 메틸화된 세포-무함유 DNA 분자의 상기 제1 양은 게놈 영역으로부터의 것인, 방법.
- 제27항에 있어서, 상기 게놈 영역은 염색체 아암이고, 상기 염색체 아암은 1p, 1q, 8p, 8q, 13q 및 14q로 이루어진 군으로부터 선택되는, 방법.
- 제21항에 있어서, 복수의 메틸화 수준을 기준 크기 패턴과 비교하는 단계는, 복수의 메틸화 수준을 복수의 기준 시료로부터 결정되는 복수의 역치 값과 비교하는 단계를 포함하는, 방법.
- 제21항에 있어서,
복수의 크기 범위는 M개의 크기 범위를 포함하며,
메틸화된 세포-무함유 DNA 분자의 제1 양을 측정하는 단계는 크기 범위에 상응하고 N개의 게놈 영역에 대한 각각의 게놈 영역에 상응하는 메틸화된 세포-무함유 DNA 분자의 제1 양을 측정하는 단계를 포함하며,
크기 범위에 상응하고 게놈 영역에 상응하는 메틸화된 세포-무함유 DNA의 상기 제1 양 및 제2 양을 사용하여 메틸화 수준을 계산하는 것은 NХM개의 메틸화 수준의 측정 벡터를 발생시키며, N은 1 이상의 정수이고 M은 1 초과의 정수이고,
기준 크기 패턴은 N개의 게놈 영역 및 M개의 크기 범위에 대한 기준 메틸화 수준의 기준 벡터를 포함하고, 상기 기준 크기 패턴은 암을 갖는 대상체로부터의 또는 암을 갖지 않는 대상체로부터의 복수의 기준 시료로부터 결정되고,
복수의 메틸화 수준을 기준 크기 패턴과 비교하는 단계는 상기 측정 벡터를 상기 기준 벡터와 비교하는 단계를 포함하는, 방법. - 대상체로부터의 생물학적 시료에서 암 분류를 결정하는 방법으로서, 상기 생물학적 시료는 종양 DNA 분자 및 비-종양 DNA 분자를 포함하는 세포-무함유 DNA 분자의 혼합물을 포함하고, 상기 방법은 하기 단계를 포함하는, 방법:
N개의 게놈 영역의 각각의 게놈 영역에 대해:
M개의 크기 범위의 각각의 크기 범위에 대해:
크기 범위에 상응하고 게놈 영역에 상응하는 생물학적 시료로부터 세포-무함유 DNA 분자의 제1 양을 측정하는 단계, 및
크기 범위에 상응하고 게놈 영역에 상응하는 세포-무함유 DNA 분자의 상기 제1 양, 및 크기 범위 내의 것이 아닌 크기를 포함하는 제2 크기 범위 내의 DNA 분자의 제2 양을 사용하여 크기 비를 컴퓨터 시스템에 의해 계산하여, NХM개의 크기 비의 측정 벡터를 발생시키는 단계로서, N은 1 이상의 정수이고 M은 1 초과의 정수인, 단계;
N개의 게놈 영역 및 M개의 크기 범위에 대한 기준 크기 비의 기준 벡터를 포함하는 기준 크기 패턴을 수득하는 단계로서, 상기 기준 크기 패턴은 암을 갖는 대상체로부터의 또는 암을 갖지 않는 대상체로부터의 복수의 기준 시료로부터 결정되는 단계;
측정 벡터를 상기 기준 패턴과 비교하는 단계; 및
상기 비교에 기초하여 암의 수준을 결정하는 단계. - 제31항에 있어서, 각각의 게놈 영역은 염색체 아암인, 방법.
- 제31항에 있어서,
기준 크기 패턴은 기계 학습 모델을 사용하여 결정되고, 상기 기계 학습 모델은 서포트 벡터 머신(support vector machine), 결정 트리(decision tree), 나이브 베이즈 분류(naive Bayes classification), 로지스틱 회귀(logistic regression), 클러스터링 알고리즘(clustering algorithm), 주성분 분석(principal component analysis), 특이값 분해(singular value decomposition), t-분포 확률적 임베딩(t-distributed stochastic neighbor embedding), 및 인공 신경망(artificial neural network)으로 이루어진 군으로부터 선택되는 하나 이상을 포함하는, 방법. - 제31항에 있어서, 측정 벡터를 기준 벡터와 비교하는 단계는, 암을 갖는 것으로 결정된 개체에 대한 그리고 암을 갖지 않는 것으로 결정된 개체에 대한 상이한 게놈 영역에 대한 크기 비를 포함하는 트레이닝 벡터의 트레이닝 세트로 트레이닝된 기계 학습 모델을 사용하는 단계를 포함하는, 방법.
- 제31항에 있어서, 상기 암은 간세포암종을 포함하는, 방법.
- 제31항에 있어서, 상기 암의 수준은 암의 확률을 포함하는, 방법.
- 제31항에 있어서,
기준 크기 패턴을 수득하고 측정 벡터를 기준 벡터와 비교하는 단계는 기계 학습 모델을 사용하는 단계를 포함하며,
상기 기계 학습 모델은 복수의 기준 크기 패턴을 사용하여 트레이닝되었으며,
측정 벡터를 기준 벡터와 비교하는 단계는 상기 기준 벡터에 대한 상기 측정 벡터의 유사성을 특징화하는 컷오프 값을 결정하는 단계를 포함하고,
암의 수준을 결정하는 단계는 상기 컷오프 값을 사용하는, 방법. - 제31항에 있어서, 측정 벡터를 기준 벡터와 비교하는 단계는 NХM개의 크기 비를 복수의 기준 시료로부터 결정되는 복수의 역치 값과 비교하는 단계를 포함하는, 방법.
- 제1항의 동작을 수행하도록 컴퓨터 시스템을 제어하기 위한 복수의 명령을 저장하는 비-일시성(non-transitory) 컴퓨터 판독 가능 매체를 포함하는 컴퓨터 제품.
- 하기를 포함하는 시스템:
제39항의 컴퓨터 제품; 및
비-일시성 컴퓨터 판독 가능 매체 상에 저장된 명령을 실행하기 위한 하나 이상의 프로세서. - 제1항 내지 제39항의 방법 중 임의의 방법을 수행하기 위한 수단을 포함하는 시스템.
- 제1항 내지 제39항의 방법 중 임의의 방법을 수행하도록 구성된 시스템.
- 제1항 내지 제39항의 방법 중 임의의 방법의 단계를 각각 수행하는 모듈을 포함하는 시스템.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762580906P | 2017-11-02 | 2017-11-02 | |
| US62/580,906 | 2017-11-02 | ||
| PCT/CN2018/113640 WO2019085988A1 (en) | 2017-11-02 | 2018-11-02 | Using nucleic acid size range for noninvasive prenatal testing and cancer detection |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| KR20200080272A true KR20200080272A (ko) | 2020-07-06 |
Family
ID=66243987
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| KR1020207014693A Ceased KR20200080272A (ko) | 2017-11-02 | 2018-11-02 | 비침습적 산전 검사 및 암 검출을 위한 핵산 크기 범위의 용도 |
Country Status (11)
| Country | Link |
|---|---|
| US (3) | US11168356B2 (ko) |
| EP (2) | EP3704264B1 (ko) |
| JP (2) | JP2021501609A (ko) |
| KR (1) | KR20200080272A (ko) |
| CN (2) | CN117079713A (ko) |
| AU (1) | AU2018359944A1 (ko) |
| CA (1) | CA3081538A1 (ko) |
| DK (1) | DK3704264T3 (ko) |
| IL (1) | IL274097A (ko) |
| TW (2) | TWI828637B (ko) |
| WO (1) | WO2019085988A1 (ko) |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11168356B2 (en) * | 2017-11-02 | 2021-11-09 | The Chinese University Of Hong Kong | Using nucleic acid size range for noninvasive cancer detection |
| CN112805563B (zh) * | 2018-05-18 | 2025-06-13 | 约翰·霍普金斯大学 | 用于评估和/或治疗癌症的无细胞dna |
| WO2020232109A1 (en) * | 2019-05-13 | 2020-11-19 | Grail, Inc. | Model-based featurization and classification |
| US11869661B2 (en) | 2019-05-22 | 2024-01-09 | Grail, Llc | Systems and methods for determining whether a subject has a cancer condition using transfer learning |
| AU2020391556B2 (en) * | 2019-11-29 | 2024-01-04 | GC Genome Corporation | Artificial intelligence-based chromosomal abnormality detection method |
| IL303888B2 (en) * | 2020-02-05 | 2025-09-01 | Univ Hong Kong Chinese | Molecular testing using long cell-free fragments in pregnancy |
| CA3169488A1 (en) * | 2020-02-28 | 2021-09-02 | Collin MELTON | Identifying methylation patterns that discriminate or indicate a cancer condition |
| CN113362893A (zh) * | 2020-03-06 | 2021-09-07 | 福建和瑞基因科技有限公司 | 肿瘤筛查模型的构建方法及应用 |
| CA3200221A1 (en) * | 2020-11-27 | 2022-06-02 | Yong Bai | Method and system of detecting fetal chromosomal abnormalities |
| CN114634982A (zh) * | 2020-12-15 | 2022-06-17 | 广州市基准医疗有限责任公司 | 一种检测多核苷酸变异的方法 |
| TW202305142A (zh) * | 2021-04-08 | 2023-02-01 | 香港中文大學 | 游離dna甲基化及核酸酶介導之片段化 |
| CN116246704B (zh) * | 2023-05-10 | 2023-08-15 | 广州精科生物技术有限公司 | 用于胎儿无创产前检测的系统 |
| WO2024249175A1 (en) * | 2023-05-26 | 2024-12-05 | Illumina, Inc. | Methods for discriminating between fetal and maternal events in non-invasive prenatal test samples |
| TW202530420A (zh) * | 2023-10-13 | 2025-08-01 | 創新診斷科技中心 | 長游離dna之基因體來源、片段組學及轉錄相關性 |
| US20260085358A1 (en) * | 2024-07-09 | 2026-03-26 | Centre For Novostics | Relative and absolute cell-free dna concentrations for clinical utilities |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL2557520T3 (pl) | 2007-07-23 | 2021-10-11 | The Chinese University Of Hong Kong | Określanie zaburzenia równowagi sekwencji kwasu nukleinowego |
| WO2009051842A2 (en) | 2007-10-18 | 2009-04-23 | The Johns Hopkins University | Detection of cancer by measuring genomic copy number and strand length in cell-free dna |
| US20120208711A1 (en) * | 2009-10-02 | 2012-08-16 | Centre For Addiction And Mental Health | Method for Analysis of DNA Methylation Profiles of Cell-Free Circulating DNA in Bodily Fluids |
| CA2780016C (en) | 2009-11-06 | 2017-09-19 | The Chinese University Of Hong Kong | Size-based genomic analysis |
| CN105243295B (zh) | 2010-11-30 | 2018-08-17 | 香港中文大学 | 与癌症相关的遗传或分子畸变的检测 |
| US9892230B2 (en) * | 2012-03-08 | 2018-02-13 | The Chinese University Of Hong Kong | Size-based analysis of fetal or tumor DNA fraction in plasma |
| US9732390B2 (en) | 2012-09-20 | 2017-08-15 | The Chinese University Of Hong Kong | Non-invasive determination of methylome of fetus or tumor from plasma |
| SI4056712T1 (sl) * | 2012-09-20 | 2024-10-30 | The Chinese University Of Hong Kong | Neinvazivno določanje metiloma tumorja iz plazme |
| WO2014204991A1 (en) * | 2013-06-17 | 2014-12-24 | Verinata Health, Inc. | Method for determining copy number variations in sex chromosomes |
| US10174375B2 (en) * | 2013-09-20 | 2019-01-08 | The Chinese University Of Hong Kong | Sequencing analysis of circulating DNA to detect and monitor autoimmune diseases |
| TWI813141B (zh) | 2014-07-18 | 2023-08-21 | 香港中文大學 | Dna混合物中之組織甲基化模式分析 |
| US10364467B2 (en) | 2015-01-13 | 2019-07-30 | The Chinese University Of Hong Kong | Using size and number aberrations in plasma DNA for detecting cancer |
| US10319463B2 (en) | 2015-01-23 | 2019-06-11 | The Chinese University Of Hong Kong | Combined size- and count-based analysis of maternal plasma for detection of fetal subchromosomal aberrations |
| WO2018209125A1 (en) * | 2017-05-10 | 2018-11-15 | Fred Hutchinson Cancer Research Center | Epstein barr virus antibodies, vaccines, and uses of the same |
| WO2019075251A2 (en) * | 2017-10-12 | 2019-04-18 | Nantomics, Llc | CANCER SCORE FOR EVALUATION AND PREDICTION OF RESPONSE FROM BIOLOGICAL FLUIDS |
| US11168356B2 (en) * | 2017-11-02 | 2021-11-09 | The Chinese University Of Hong Kong | Using nucleic acid size range for noninvasive cancer detection |
-
2018
- 2018-11-01 US US16/178,378 patent/US11168356B2/en active Active
- 2018-11-02 CN CN202311065158.2A patent/CN117079713A/zh active Pending
- 2018-11-02 JP JP2020544091A patent/JP2021501609A/ja active Pending
- 2018-11-02 WO PCT/CN2018/113640 patent/WO2019085988A1/en not_active Ceased
- 2018-11-02 KR KR1020207014693A patent/KR20200080272A/ko not_active Ceased
- 2018-11-02 TW TW107139084A patent/TWI828637B/zh active
- 2018-11-02 AU AU2018359944A patent/AU2018359944A1/en not_active Abandoned
- 2018-11-02 CA CA3081538A patent/CA3081538A1/en active Pending
- 2018-11-02 EP EP18874285.2A patent/EP3704264B1/en active Active
- 2018-11-02 DK DK18874285.2T patent/DK3704264T3/da active
- 2018-11-02 TW TW112148593A patent/TWI874039B/zh active
- 2018-11-02 CN CN201880083024.4A patent/CN111712582B/zh active Active
- 2018-11-02 EP EP23190219.8A patent/EP4254417B1/en active Active
-
2020
- 2020-04-21 IL IL274097A patent/IL274097A/en unknown
-
2021
- 2021-11-08 US US17/521,333 patent/US12247259B2/en active Active
-
2023
- 2023-08-04 JP JP2023127899A patent/JP2023139321A/ja active Pending
-
2025
- 2025-02-14 US US19/054,355 patent/US20260015673A1/en active Pending
Also Published As
| Publication number | Publication date |
|---|---|
| TWI828637B (zh) | 2024-01-11 |
| CA3081538A1 (en) | 2019-05-09 |
| AU2018359944A1 (en) | 2020-05-07 |
| EP3704264A1 (en) | 2020-09-09 |
| WO2019085988A1 (en) | 2019-05-09 |
| CN111712582B (zh) | 2023-09-12 |
| IL274097A (en) | 2020-06-30 |
| EP3704264A4 (en) | 2021-08-11 |
| EP4254417B1 (en) | 2025-04-02 |
| US20190130065A1 (en) | 2019-05-02 |
| TWI874039B (zh) | 2025-02-21 |
| CN117079713A (zh) | 2023-11-17 |
| CN111712582A (zh) | 2020-09-25 |
| JP2023139321A (ja) | 2023-10-03 |
| US11168356B2 (en) | 2021-11-09 |
| EP3704264B1 (en) | 2023-09-27 |
| DK3704264T3 (da) | 2023-10-30 |
| TW201928065A (zh) | 2019-07-16 |
| US20220064714A1 (en) | 2022-03-03 |
| TW202428883A (zh) | 2024-07-16 |
| JP2021501609A (ja) | 2021-01-21 |
| EP4254417A3 (en) | 2023-12-20 |
| US20260015673A1 (en) | 2026-01-15 |
| US12247259B2 (en) | 2025-03-11 |
| EP4254417A2 (en) | 2023-10-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12247259B2 (en) | Using nucleic acid size range for noninvasive cancer detection | |
| JP7607264B2 (ja) | 無細胞dna末端特性 | |
| JP7781441B2 (ja) | 無細胞ウイルス核酸を用いる癌スクリーニングの強化 | |
| CN108026572B (zh) | 游离dna的片段化模式的分析 | |
| CN113710818A (zh) | 病毒相关联的癌症风险分层 | |
| HK40102019A (en) | Using nucleic acid size range for noninvasive prenatal testing and cancer detection | |
| HK40102019B (en) | Using nucleic acid size range for noninvasive prenatal testing and cancer detection | |
| HK40101694A (zh) | 使用核酸大小范围进行非侵入性产前检查和癌症检测 | |
| US20250125051A1 (en) | Genomic origin, fragmentomics, and transcriptional correlation of long cell-free dna | |
| HK40031026B (zh) | 使用核酸大小范围进行非侵入性产前检查和癌症检测 | |
| HK40030257B (en) | Using nucleic acid size range for noninvasive prenatal testing and cancer detection | |
| HK40030257A (en) | Using nucleic acid size range for noninvasive prenatal testing and cancer detection | |
| HK40031026A (en) | Using nucleic acid size range for noninvasive prenatal testing and cancer detection | |
| HK40079331A (en) | Analysis of fragmentation patterns of cell-free dna | |
| TW202519666A (zh) | 與表觀遺傳修飾相關的游離dna片段化模式的用途 | |
| HK40023330A (en) | Enhancement of cancer screening using cell-free viral nucleic acids | |
| HK1251264B (en) | Analysis of fragmentation patterns of cell-free dna |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PA0105 | International application |
St.27 status event code: A-0-1-A10-A15-nap-PA0105 |
|
| P11-X000 | Amendment of application requested |
St.27 status event code: A-2-2-P10-P11-nap-X000 |
|
| P13-X000 | Application amended |
St.27 status event code: A-2-2-P10-P13-nap-X000 |
|
| PG1501 | Laying open of application |
St.27 status event code: A-1-1-Q10-Q12-nap-PG1501 |
|
| A201 | Request for examination | ||
| PA0201 | Request for examination |
St.27 status event code: A-1-2-D10-D11-exm-PA0201 |
|
| P22-X000 | Classification modified |
St.27 status event code: A-2-2-P10-P22-nap-X000 |
|
| D13-X000 | Search requested |
St.27 status event code: A-1-2-D10-D13-srh-X000 |
|
| R18-X000 | Changes to party contact information recorded |
St.27 status event code: A-3-3-R10-R18-oth-X000 |
|
| D14-X000 | Search report completed |
St.27 status event code: A-1-2-D10-D14-srh-X000 |
|
| N231 | Notification of change of applicant | ||
| PN2301 | Change of applicant |
St.27 status event code: A-3-3-R10-R13-asn-PN2301 St.27 status event code: A-3-3-R10-R11-asn-PN2301 |
|
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection |
St.27 status event code: A-1-2-D10-D21-exm-PE0902 |
|
| T11-X000 | Administrative time limit extension requested |
St.27 status event code: U-3-3-T10-T11-oth-X000 |
|
| T11-X000 | Administrative time limit extension requested |
St.27 status event code: U-3-3-T10-T11-oth-X000 |
|
| P11-X000 | Amendment of application requested |
St.27 status event code: A-2-2-P10-P11-nap-X000 |
|
| P13-X000 | Application amended |
St.27 status event code: A-2-2-P10-P13-nap-X000 |
|
| E601 | Decision to refuse application | ||
| PE0601 | Decision on rejection of patent |
St.27 status event code: N-2-6-B10-B15-exm-PE0601 |
|
| PN2301 | Change of applicant |
St.27 status event code: A-3-3-R10-R13-asn-PN2301 St.27 status event code: A-3-3-R10-R11-asn-PN2301 |
|
| P11-X000 | Amendment of application requested |
St.27 status event code: A-2-2-P10-P11-nap-X000 |
|
| P13-X000 | Application amended |
St.27 status event code: A-2-2-P10-P13-nap-X000 |
|
| PX0901 | Re-examination |
St.27 status event code: A-2-3-E10-E12-rex-PX0901 |
|
| E902 | Notification of reason for refusal | ||
| PE0902 | Notice of grounds for rejection |
St.27 status event code: A-1-2-D10-D21-exm-PE0902 |
|
| T11-X000 | Administrative time limit extension requested |
St.27 status event code: U-3-3-T10-T11-oth-X000 |
|
| PX0601 | Decision of rejection after re-examination |
St.27 status event code: N-2-6-B10-B17-rex-PX0601 |
|
| X601 | Decision of rejection after re-examination |