RU2249266C2 - Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива аэс - Google Patents
Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива аэс Download PDFInfo
- Publication number
- RU2249266C2 RU2249266C2 RU2003100275A RU2003100275A RU2249266C2 RU 2249266 C2 RU2249266 C2 RU 2249266C2 RU 2003100275 A RU2003100275 A RU 2003100275A RU 2003100275 A RU2003100275 A RU 2003100275A RU 2249266 C2 RU2249266 C2 RU 2249266C2
- Authority
- RU
- Russia
- Prior art keywords
- extractant
- mol
- solution
- extraction
- nitric acid
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 37
- 239000002915 spent fuel radioactive waste Substances 0.000 title claims abstract description 6
- 238000000605 extraction Methods 0.000 title claims description 47
- 238000011084 recovery Methods 0.000 title abstract 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 claims abstract description 51
- 229910052761 rare earth metal Inorganic materials 0.000 claims abstract description 48
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims abstract description 33
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 claims abstract description 23
- 229910052750 molybdenum Inorganic materials 0.000 claims abstract description 23
- 229910017604 nitric acid Inorganic materials 0.000 claims abstract description 23
- 229910052727 yttrium Inorganic materials 0.000 claims abstract description 23
- VWQVUPCCIRVNHF-UHFFFAOYSA-N yttrium atom Chemical compound [Y] VWQVUPCCIRVNHF-UHFFFAOYSA-N 0.000 claims abstract description 22
- 229910052726 zirconium Inorganic materials 0.000 claims abstract description 21
- 229910052742 iron Inorganic materials 0.000 claims abstract description 17
- 235000006408 oxalic acid Nutrition 0.000 claims abstract description 16
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 claims abstract description 15
- QCWXUUIWCKQGHC-UHFFFAOYSA-N Zirconium Chemical group [Zr] QCWXUUIWCKQGHC-UHFFFAOYSA-N 0.000 claims abstract description 15
- 239000011733 molybdenum Substances 0.000 claims abstract description 15
- 238000005406 washing Methods 0.000 claims abstract description 14
- 239000012634 fragment Substances 0.000 claims abstract description 10
- 239000002253 acid Substances 0.000 claims abstract description 9
- 230000002441 reversible effect Effects 0.000 claims abstract description 8
- GWXLDORMOJMVQZ-UHFFFAOYSA-N cerium Chemical group [Ce] GWXLDORMOJMVQZ-UHFFFAOYSA-N 0.000 claims abstract description 6
- VBIXEXWLHSRNKB-UHFFFAOYSA-N ammonium oxalate Chemical compound [NH4+].[NH4+].[O-]C(=O)C([O-])=O VBIXEXWLHSRNKB-UHFFFAOYSA-N 0.000 claims abstract description 5
- 150000003754 zirconium Chemical class 0.000 claims abstract description 4
- 229930195734 saturated hydrocarbon Natural products 0.000 claims abstract description 3
- JYFHYPJRHGVZDY-UHFFFAOYSA-N Dibutyl phosphate Chemical compound CCCCOP(O)(=O)OCCCC JYFHYPJRHGVZDY-UHFFFAOYSA-N 0.000 claims abstract 3
- STCOOQWBFONSKY-UHFFFAOYSA-N tributyl phosphate Chemical compound CCCCOP(=O)(OCCCC)OCCCC STCOOQWBFONSKY-UHFFFAOYSA-N 0.000 claims abstract 3
- 239000000243 solution Substances 0.000 claims description 41
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 claims description 12
- 238000013467 fragmentation Methods 0.000 claims description 9
- 238000006062 fragmentation reaction Methods 0.000 claims description 9
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 7
- 229910052684 Cerium Inorganic materials 0.000 claims description 6
- 239000000203 mixture Substances 0.000 claims description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 claims description 5
- 239000012074 organic phase Substances 0.000 claims description 5
- 238000012545 processing Methods 0.000 claims description 5
- 230000008929 regeneration Effects 0.000 claims description 5
- 238000011069 regeneration method Methods 0.000 claims description 5
- 239000007864 aqueous solution Substances 0.000 claims description 2
- -1 ammonium ions Chemical class 0.000 claims 1
- 239000007924 injection Substances 0.000 claims 1
- 238000002347 injection Methods 0.000 claims 1
- DBRMBYFUMAFZOB-UHFFFAOYSA-N molybdenum nitric acid Chemical compound [Mo].[N+](=O)(O)[O-] DBRMBYFUMAFZOB-UHFFFAOYSA-N 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 10
- 238000005260 corrosion Methods 0.000 abstract description 2
- 230000007797 corrosion Effects 0.000 abstract description 2
- 239000000126 substance Substances 0.000 abstract description 2
- 239000003795 chemical substances by application Substances 0.000 abstract 4
- 238000000658 coextraction Methods 0.000 abstract 1
- 238000000926 separation method Methods 0.000 description 11
- ZUROCNHARMFRKA-UHFFFAOYSA-N 4,5-dibromo-1h-pyrrole-2-carboxylic acid Chemical compound OC(=O)C1=CC(Br)=C(Br)N1 ZUROCNHARMFRKA-UHFFFAOYSA-N 0.000 description 9
- NLZUEZXRPGMBCV-UHFFFAOYSA-N Butylhydroxytoluene Chemical compound CC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 NLZUEZXRPGMBCV-UHFFFAOYSA-N 0.000 description 9
- 101000823778 Homo sapiens Y-box-binding protein 2 Proteins 0.000 description 9
- 101100431668 Homo sapiens YBX3 gene Proteins 0.000 description 9
- 102100022221 Y-box-binding protein 3 Human genes 0.000 description 9
- DIOQZVSQGTUSAI-UHFFFAOYSA-N decane Chemical compound CCCCCCCCCC DIOQZVSQGTUSAI-UHFFFAOYSA-N 0.000 description 6
- 238000009826 distribution Methods 0.000 description 6
- 230000007423 decrease Effects 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 4
- 150000007513 acids Chemical class 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 238000011160 research Methods 0.000 description 3
- 229910052693 Europium Inorganic materials 0.000 description 2
- 244000309464 bull Species 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000004140 cleaning Methods 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 229920002725 thermoplastic elastomer Polymers 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-L Oxalate Chemical compound [O-]C(=O)C([O-])=O MUBZPKHOEPUJKR-UHFFFAOYSA-L 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 230000000536 complexating effect Effects 0.000 description 1
- 238000010668 complexation reaction Methods 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- OGPBJKLSAFTDLK-UHFFFAOYSA-N europium atom Chemical compound [Eu] OGPBJKLSAFTDLK-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000011824 nuclear material Substances 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 230000010349 pulsation Effects 0.000 description 1
- 238000012958 reprocessing Methods 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- 239000002699 waste material Substances 0.000 description 1
Classifications
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02E—REDUCTION OF GREENHOUSE GAS [GHG] EMISSIONS, RELATED TO ENERGY GENERATION, TRANSMISSION OR DISTRIBUTION
- Y02E30/00—Energy generation of nuclear origin
- Y02E30/30—Nuclear fission reactors
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02W—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO WASTEWATER TREATMENT OR WASTE MANAGEMENT
- Y02W30/00—Technologies for solid waste management
- Y02W30/50—Reuse, recycling or recovery technologies
Landscapes
- Manufacture And Refinement Of Metals (AREA)
- Extraction Or Liquid Replacement (AREA)
Abstract
Изобретение относится к области переработки отработавшего ядерного топлива. Сущность изобретения: способ экстракционной переработки высокоактивного рафината Пурекс-процесса включает совместное экстрагирование трансплутониевых, редкоземельных элементов, осколочных молибдена и циркония, а также коррозионного железа с помощью раствора 0,1-0,4 моль/л циркониевой соли дибутилфосфорной кислоты в 30% (об.) трибутилфосфате с предельными углеводородами. Затем производят реэкстракцию трансплутониевых элементов с цериевой подгруппой редкоземельных элементов в раствор азотной кислоты при ее концентрации 3,5-5 моль/л в реэкстракте, промытом от молибдена оборотным экстрагентом. Далее, проводят совместную или раздельную реэкстракцию осколочного молибдена с иттрием и иттриевой подгруппы редкоземельных элементов. Регенерируют экстрагент путем промывки в противотоке части экстрагента, выведенной, исходя из поступления циркония с исходным раствором, щавелевой кислотой при добавлении оксалата аммония в ступень ввода экстрагента при температуре процесса 35-40°С. Преимущество изобретения заключается в повышении качества переработки. 6 з.п. ф-лы, 7 табл., 1 ил.
Description
Изобретение относится к технологии переработки отработанного ядерного топлива атомных электростанций (ОЯТ АЭС) и, в частности, к экстракционной переработке высокоактивного рафината от Пурекс-процесса. Изобретение может быть использовано в комплексных технологических схемах экстракционной переработки облученных ядерных материалов при подготовке их к захоронению, а также в технологии извлечения и концентрирования редкоземельных элементов (РЗЭ).
Известно групповое разделение трансплутониевых элементов (ТПЭ) и РЗЭ с помощью процесса, обратного "Талспик-процессу" (Del Сul G.D., Toth L.M., Bond W.D. et al. // Separ. Sci. & Techn. 1997. V.32, No.1-4, P.431-446). Сущность процесса состоит в совместной экстракции трехвалентных РЗЭ и ТПЭ органическими растворами длинноцепочечных диалкилфосфорной (или диалкилфосфоновой) кислот и разделении этих элементов на стадии реэкстракции смесью кислот оксикарбоновой и полиаминополиуксусной (комплексона). Основное количество ТПЭ, образующих более прочные комплексы с кислотами, чем РЗЭ, переходят в водную фазу, тогда как большее количество РЗЭ (цериевая подгруппа) остается в органическом растворе. После разделения групп ТПЭ остаются в водной фазе в присутствии иттрия, а также РЗЭ иттриевой подгруппы в присутствии большого количества комплексообразующих веществ. При этом значительные количества осколочных циркония и молибдена, а также коррозионного железа остаются в органической фазе и требуют дополнительных операций для их локализации, а сам процесс проводится с использованием экстрагента, несовместимого с Пурекс-процессом. Таким образом, решение задачи очистки ТПЭ от РЗЭ иттриевой подгруппы при обеспечении локализации осадкообразующих элементов с использованием ТБФ-совместимого экстрагента представляется весьма актуальным.
Известен также способ (Патент РФ №2106030, Бюл. №6, 1998 г., МПК G 21 С 19/46), который может быть принят за прототип. Он состоит в экстракционном извлечении и разделении ТПЭ и РЗЭ из азотнокислых растворов с помощью циркониевой соли дибутилфосфорный кислоты (ЦС ДБФК), растворенной в экстрагенте Пурекс-процесса, т.е. 30% ТБФ с углеводородным разбавителем. Сущность метода заключается в том, что извлечение ТПЭ и РЗЭ проводят из растворов, содержащих 0,5-3,0 моль/л HNO3, а реэкстракцию ТПЭ и РЗЭ из органической фазы проводят раствором азотной кислоты концентрацией 3-12 моль/л. При этом мольное соотношение Zr:ДБФК=1:50-1:4.
Основным недостатком прототипа является отсутствие конкретных приемов разделения ТПЭ и РЗЭ экстрагентом на основе ЦС ДБФК. Необходимость предварительного удаления осколочных Мо и Zr также является неудобством данного метода, так как либо снижает его универсальность, либо вызывает определенные трудности при очистке экстракта осколочных Мо, Zr и коррозионного Fe от ТПЭ и РЗЭ. Кроме того, не предложены пути локализации железа, образующегося в результате коррозии аппаратуры.
Предлагаемым способом решается задача создания непрерывного процесса переработки высокоактивного рафината от регенерации ОЯТ АЭС при его фракционировании с извлечением долгоживущих α-нуклидов.
Для достижения указанного технического результата предлагается способ, состоящий в последовательном проведении следующих операций:
- экстракция РЗЭ и ТПЭ, а также осколочных Zr, Мо и коррозионного Fe раствором ЦС ДБФК в 30% ТБФ при соотношении Zr:ДБФК=1:9-1:15 в предельных углеводородах из азотнокислого раствора с последующей промывкой экстракта;
- реэкстракция ТПЭ в смеси с РЗЭ цериевой подгруппы азотной кислотой с концентрацией ее в реэкстракте 3,5-5 моль/л с отмывкой реэкстракта оборотным экстрагентом от РЗЭ иттриевой подгруппы и осколочного Мо;
- реэкстракция РЗЭ иттриевой подгруппы и осколочного молибдена (совместно или раздельно) с помощью раствора 0,3-1 моль/л перекиси водорода в 4-7 моль/л азотной кислоте с удалением обоих компонентов из экстрагента промывкой его водой;
- отделение части экстрагента от его рециклируемого основного потока по балансу с цирконием, поступившим с исходным раствором, для проведения его регенерации путем реэкстракции циркония и коррозионного железа раствором 0,5-0,8 моль/л щавелевой кислоты при добавлении оксалата аммония в протоке экстрактора (с целью нейтрализации следов азотной кислоты) и последующая промывка регенерированного экстрагента от экстрагированной щавелевой кислоты.
Предлагаемый способ иллюстрируется схемой (см. чертеж), на которой изображены блоки многоступенчатых экстракторов. Водные потоки изображены нечетными цифрами, органические - четными. В блоке 1 осуществляется экстракция компонентов из исходного раствора и промывка экстракта, в блоке 2 - разделение ТПЭ и иттриевой подгруппы РЗЭ, в блоках 3 и 4 - совместная или раздельная реэкстракция осколочного Мо и РЗЭ (Y-подгруппы) и в блоках 5 и 6 - регенерация экстрагента, из которых в блоке 5 производится реэкстракция Zr и сопутствующего коррозионного Fe щавелевой кислотой, а в блоке 6 - отмывка экстрагента от щавелевой кислоты.
Данная технологическая схема позволяет максимально облегчить разделение компонентов на проводимых последовательно операциях реэкстракции и осуществить выбор состава экстрагента и условий экстракции исходя из этого принципа, что легче понять на конкретных примерах, приведенных ниже.
Заявляемая схема совмещает в себе несколько технических приемов, каждый из которых позволяет оптимизировать отдельную операцию фракционирования элементов, а в совокупности - схему в целом. К таким приемам относится, прежде всего, переменное соотношение ДБФК:Zr по стадиям процесса. Отношение ДБФК:Zr=9, при котором экстракция ТПЭ и РЗЭ является максимальной (Патент РФ №2106030, Бюл. №6, 1998 г., МПК G 21 С 19/46), создается только в ступени ввода исходного раствора за счет экстракции из него осколочного Zr оборотным экстрагентом с соотношением ДБФК:Zr=12,5, обеспечивающим максимальное извлечение осколочного Мо на этой операции, а также на операции отмывки реэкстракта Am от РЗЭ иттриевой подгруппы и осколочного Мо; при этом коэффициенты распределения РЗЭ снижаются незначительно. На разделительных операциях соотношение ДБФК:Zr=10. На реэкстракцию циркония (вместе с коррозионным железом) выводится доля экстрагента по балансу с поступающим в схему осколочным цирконием, причем “пустой” экстрагент смешивается с оборотным экстрагентом, создавая тем самым в нем вышеупомянутое соотношение ДБФК:Zr=12,5.
Такая оптимизация процесса возможна лишь при содержании ДБФК в 30% ТБФ на уровне 0,1-0,4 моль/л, поскольку при более низкой и более высокой концентрации экстрагента ухудшается разделение ТПЭ и РЗЭ иттриевой подгруппы.
Вторым отличительным признаком предлагаемого способа является использование перекиси водорода в сильнокислой среде для усиления реэкстракции иттрия и иттриевых земель. Попутно обеспечивается известная реэкстракция осколочного молибдена. Вместе с тем, варьированием соотношения потоков фаз в выбранной области концентраций компонентов реэкстракцию осколочного Мо можно проводить как совместно с иттрием и другими РЗЭ, так и селективно до них.
Третьим отличительным признаком схемы является выбор области концентраций компонентов для реэкстракции циркония и коррозионного железа. При более высокой концентрации ДБФК и более низкой концентрации щавелевой кислоты процесс неэффективен из-за слабого комплексования циркония. Для реэкстракции коррозионного железа надо гарантированно убирать из экстрагента следы азотной кислоты, для чего в первую ступень реэкстракционного блока вводится буферная добавка оксалата аммония.
Совокупность технологических операций в перечисленной последовательности позволяет создать замкнутый экстракционный цикл первичного фракционирования ТПЭ и достичь очистки Am от осколочного Мо и Y в 10 раз, а от Eu-Sm в 1,5-3 раза.
ПРИМЕРЫ
Пример 1. Приготовлены порции экстрагента, содержащего 0,1-0,4 моль/л дибутилфосфорной кислоты (ДБФК), растворенной в 30% ТБФ с деканом или изопаром-L (смесь разветвленных изопарофинов с длиной углеродной цепи 11-15) и цирконий в соотношении Zr:ДБФК=1:9. После уравновешивания органической фазы с азотной кислотой произведено ее контактирование с исходным водным раствором. Соотношение объемов фаз при экстракции 1:1; время контакта 5 минут. Результаты сведены в табл.1.
Из приведенных данных следует, что рабочая область концентраций ДБФК находится в пределах 0,1-0,4 моль/л, причем с увеличением концентрации НNО3 в растворе коэффициенты распределения Y, РЗЭ и ТПЭ уменьшаются. Однако у Y снижение экстракции менее выражено, что затрудняет его кислотную реэкстракцию. Для удовлетворительной экстракции Мо концентрация ДБФК должна быть не ниже 0,15 моль/л, тогда как при реэкстракции иттрия азотной кислотой необходимо использовать экстрагент с концентрацией ДБФК не выше 0,2 моль/л. Кроме того, в указанном пределе концентраций при кислотности около 5 моль/л обеспечивается наилучшее разделение ТПЭ и РЗЭ иттриевой подгруппы. Таким образом, оптимальная концентрация ДБФК в 30% ТБФ составляет 0,15-0,2 моль/л.

Пример 2. Было изучено влияние соотношения Zr:ДБФК на экстракцию ТПЭ, РЗЭ и Мо. Методика проведения экспериментов аналогична примеру 1. Результаты исследований приведены в табл.2.
В результате проведенных исследований было обнаружено, что максимальная экстракция ТПЭ и РЗЭ, включая Y, из 1,5 моль/л НNО3 наблюдается при Zr:ДБФК=1:9, а из 5 моль/л НNО3 оптимальное соотношение Zr:ДБФК для иттрия сохраняется равной 1:9, тогда как для остальных РЗЭ и Am изменение соотношения Zr:ДБФК в экстрагенте в пределах 1:8-1:15 практически не влияет на экстракцию элементов; кроме того, еще раз подтверждается возможность эффективного отделения Am и Се (по-видимому, и других цериевых земель) от европия и прочих иттриевых земель. Максимальная экстракция Мо из 1,5 моль/л HNO3 приходится на соотношение Zr:ДБФК=1:13,5; а в растворах 5 моль/л HNO3 его экстракция несколько усиливается с понижением содержания Zr в органической фазе в области соотношений Zr:ДБФК>9.
| Таблица 2 Влияние соотношения Zr:ДБФК на экстракцию ТПЭ, РЗЭ и Мо с помощью ЦС ДБФК (0,2 моль/л ДБФК) в 30% ТБФ с деканом из растворов HNO3. |
||||||||||
| Zr:ДБФК | 1,5 моль/л HNO3 | 5 моль/л HNO3 | ||||||||
| Мо | Y | Се | Eu | Am | Мо | Y | Се | Eu | Am | |
| 1:6 | 10,7 | 2,4 | 5,9 | 4,8 | 2,4 | 2,2 | - | - | - | - |
| 1:8 | 13,3 | 9,8 | 13,1 | 19 | 3,7 | 2,4 | 1,1 | - | 0,15 | - |
| 1:9 | 21 | 12,5 | 16 | 23 | 4,5 | 3,4 | 1,7 | 0,07 | 0,28 | 0,06 |
| 1:10 | 23 | 12,5 | 15,7 | 21 | 4,5 | - | - | 0,06 | 0,17 | 0,05 |
| 1:12 | 24 | - | 11,8 | 15 | 4,2 | 3,5 | - | - | - | - |
| 1:14 | 23 | 8 | 10,2 | 12,2 | 4,1 | - | - | - | - | - |
| 1:15 | 21,5 | - | - | - | - | 3,8 | 1,2 | 0,06 | 0,16 | 0,05 |
| 1:18 | 19,7 | 2,9 | 8,8 | 9,3 | 3,9 | 4 | - | - | - | - |
Пример 3. В этом примере были изучены условия реэкстракции Y, РЗЭ, Мо и Fe в присутствии Н2O2 (табл.3). Методика проведения экспериментов аналогична примеру 1.
Коэффициент распределения Мо резко снижается с повышением концентраций H2O2 и HNO3. Коэффициенты распределения РЗЭ и Y в ЦС ДБФК также неожиданно понижаются в присутствии Н2O2, причем этот эффект сохраняется с ростом концентрации до 4-5 моль/л HNO3, чем можно воспользоваться для реэкстракции Y совместно с Мо или после него. Эффект, вероятно, связан с обратимым взаимодействием H2O2 с циркониевым ядром ЦС ДБФК.
| Таблица 3 Зависимость коэффициентов распределения ряда РЗЭ и Y при их реэкстракции из раствора ЦС ДБФК в 30% ТБФ с деканом от концентрации HNO3 и Н2О2. Концентрации: ДБФК- 0,2 моль/л, Zr-0,02 2 моль/л; концентрации элементов – 100 мг/л. |
||||||||||||||||||
| HNO3, моль/л | Коэффициент распределения элемента (D) при концентрации | |||||||||||||||||
| перекиси водорода в водной фазе, моль/л | ||||||||||||||||||
| Am | Се | Eu | Y | Мо | ||||||||||||||
| 0 | 0,5 | 1,0 | 0 | 0,5 | 1,0 | 0 | 0,5 | 1,0 | 3,0 | 0 | 0,5 | 1,5 | 3,0 | 0 | 0,5 | 1,0 | 2,0 | |
| 1,5 | 4,1 | - | 0,19 | 16 | - | 0,4 | 23 | - | 0,28 | - | 12,5 | 3,2 | 2,8 | 2,5 | 22 | 0,41 | - | - |
| 2,5 | - | - | - | - | - | - | 2,2 | - | 0,17 | - | 5,2 | 1,2 | 0,97 | 1,1 | 7,3 | 0,25 | - | - |
| 4,0 | 0,1 | 0,07 | 0,05 | 0,25 | 0,1 | 0,08 | 0,4 | 0,15 | 0,14 | 0,12 | 2,4 | 1,1 | 0,84 | 0,72 | - | 0,01 | 0,01 | 0,01 |
| 6,0 | - | - | - | - | - | - | 0,1 | - | 0,12 | 0,1 | 1,15 | 1,1 | 0,59 | 0,8 | - | 0,02 | 0,07 | 0,08 |
| 8,0 | 0,02 | - | - | 0,03 | - | - | 0,09 | - | - | - | 0,75 | - | 1,2 | - | 11,4 | 0,07 | 0,08 | 0,1 |
Пример 4. В этом примере были изучены условия реэкстракции Мо, Fe и Zr раствором щавелевой кислоты. Результаты исследований приведены в табл.4.
Из приведенных данных по реэкстракции Fe, Мо и Zr щавелевой кислотой следует, что железо реэкстрагируется труднее Zr, причем исходная концентрация Н2С2O4 должна составлять 0,4-0,5 моль/л; при этом опасны самые малые количества азотной кислоты. Для нейтрализации HNO3, поступающей с экстрактом, полезно использовать оксалатный буферный раствор с равновесной концентрацией оксалат-иона 0,33-0,35 моль/л и только на одной ступени, так как при иной обработке выпадает осадок и/или ухудшается реэкстракция компонентов.

Пример 5. Была изучена также экстракция самой щавелевой кислоты ЦС ДБФК. Было установлено, что ее экстракция в значительной мере зависит от температуры проведения процесса. Результаты исследований приведены в табл.5.
Поэтому при проведении процесса в многоступенчатом каскаде при соотношении потоков Vopг:Vвод=2 и при температуре 20°С концентрация щавелевой кислоты в водной фазе не обеспечивает реэкстракцию Fe и Zr вследствие ее высокой экстракции. Для избежания этого явления предлагается повышать температуру операции на этом узле технологической схемы до 35-40°С.

Пример 6. Стенд состоял из трех блоков лабораторных пульсационных экстракторов типа "смеситель-отстойник". Опыты проводили при температуре 20±1°С. Был приготовлен имитатор следующего состава, мг/л: Сr-80, Мn-85, Со-120, Ni-60, Sr-130, Mo-560, Cs-200, Ca-60, Ba-70, Al-60, Na-290, Y-50, Ce-500, Nd-500, Eu-200 в 1,5 моль/л HNO3. Раствор был помечен реальным производственным рафинатом; при этом фиксировались удельные активности гамма-излучающих Еu152 и Am241, а также суммарная бета-активность, создаваемая, главным образом, Y90. Технологическая схема стенда в опыте соответствовала представленной на чертеже, но без блока реэкстракции Мо. Следует отметить, что Zr и Fe в опыте отсутствовали, и доля экстрагента, поступавшего на расциколовку Zr и Fe, была незначительной. Условия проведения и конечные результаты отражены в табл.6.
Рафинат первого экстракционного блока содержал более 99% общей γ-активности, 50% общей β-активности (Cs, Sr) и ~0,1% общей α-активности. Am и Еu в рафинате обнаружены не были, но в нем оказалось около 10% Мо по балансу.
На втором блоке удалось достичь полноты реэкстракции Am (99%) с очисткой от Еu в 3 раза; при этом содержание Мо в реэкстракте РЗЭ и ТПЭ оказалось около 1 г/л (около 48% по балансу), хотя этого не следовало ожидать, исходя из данных примера 2. Реэкстракция Мо и оставшейся части РЗЭ, а также Y в 3 блоке протекала достаточно эффективно.
| Таблица 6 Показатели технологического процесса. Экстрагент - 0,15 моль/л ДБФК (Zr:ДБФК=1:9) в 30% ТБФ с изопаром L. |
|||||||||
| Код потока | Характеристика потока | Состав растворов | |||||||
| HNO3, моль/л | Реагент | РЗЭ г/л | Мо г/л | Eu152 Бк/л | Am241 Бк/л | β общ. Бк/л | |||
| Наменован. | Конц. моль/л | ||||||||
| 11 | Исходный раствор | 1,46 | 1,1 | 0,56 | 9,1 Е6 | 6,8 Е6 | 3,2 Е7 | ||
| 12 | Экстрагент | ||||||||
| 13 | Рафинат | 1,0 | 0,054 | - | - | 1,5 Е7 | |||
| 14 | Экстракт Мо, РЗЭ, ТПЭ | ||||||||
| 15 | Промывной раствор | 0,1 | |||||||
| 20 | Подпитка HNO3 | 10 | |||||||
| 22 | Экстрагент | ||||||||
| 24 | Экстракт Мо, Y | ||||||||
| 25 | Реэкстрагент РЗЭ, ТПЭ | 10 | |||||||
| 27 | Промывной раствор | 7 | |||||||
| 29 | Реэкстракт РЗЭ, ТПЭ | 3,9 | 0,9 | 1 Е7 | 4,4 Е7 | 4,5 Е7 | |||
| 45 | Промывной раствор | 0,1 | |||||||
| 47 | Реэкстрагент Мо, Y | 4 | Н2O2 | 7 | |||||
| 49 | Реэкстракт Мо и РЗЭ | 5,6 | Н2O2 | 0,8 | 0,44 | 9 Е6 | 1,3 Е5 | 1,8 Е7 | |
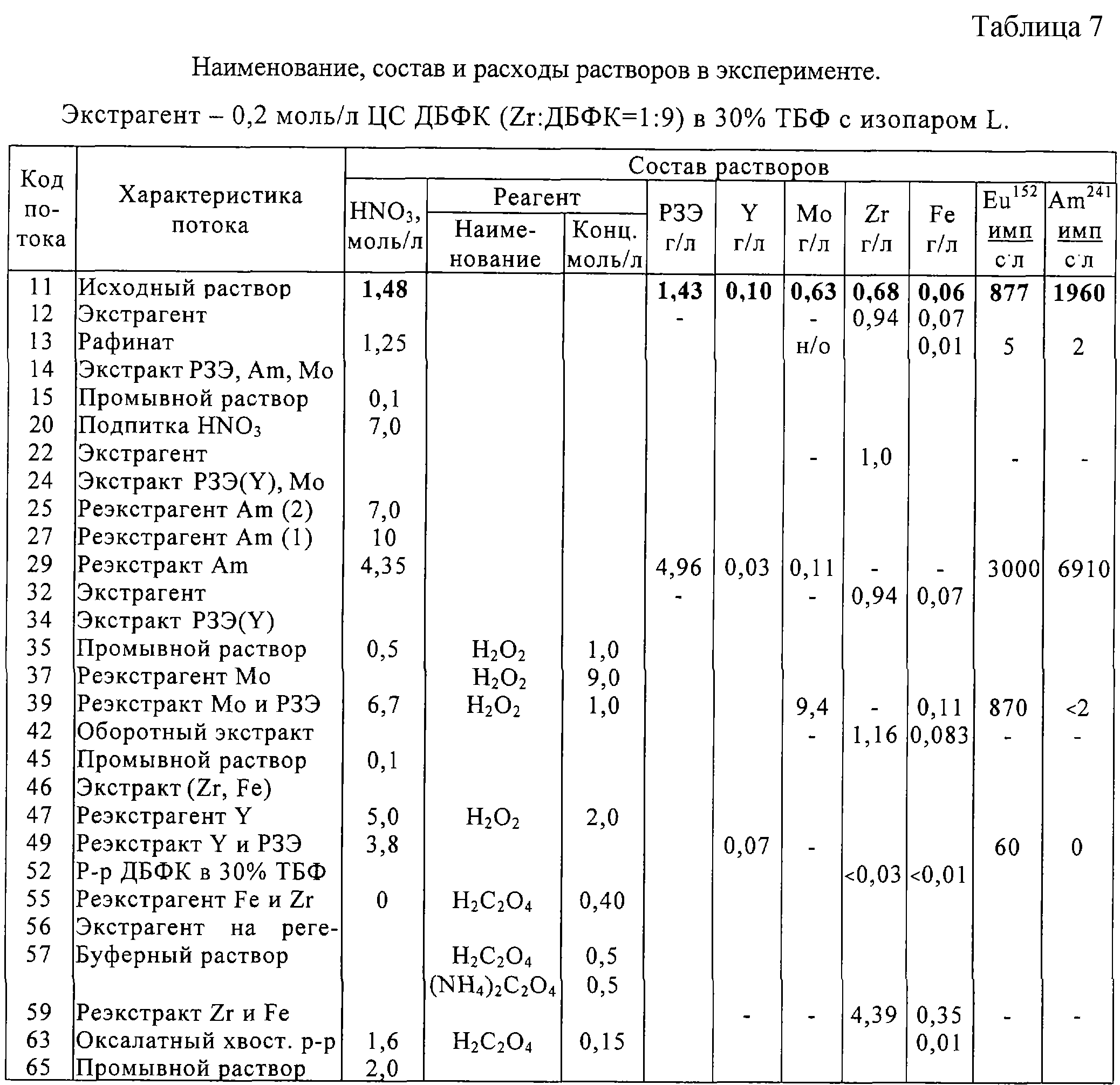
Пример 7. Испытания процесса на стенде центробежных экстракторов были проведены по полной технологической схеме, приведенной на чертеже. Характеристика технологических потоков и сводные результаты представлены в табл.7. Температура процесса была на уровне 32-43°С.
Экстракция осуществлялась из модельного рафината от переработки ОЯТ ВВЭР-1000 с выгоранием 40 ГВт·сут/т после его разбавления вдвое для достижения концентрации азотной кислоты 1,5 моль/л. Исходный раствор содержал 1,4 г/л суммы РЗЭ (а также метки Am241 и Eu152). В качестве оборотного экстрагента использовали раствор 0,2 моль/л ЦС ДБФК в 30% ТБФ с изопаром-L при пониженном соотношении Zr:ДБФК, равном 1:12,5 (1:10 после экстракции Zr) в целях улучшения экстракции Мо при реэкстракции Am совместно с цериевыми РЗЭ, благодаря чему концентрацию молибдена в данном реэкстракте удалось снизить до 0,11 г/л (5% по балансу).
На блоке реэкстракции молибдена совместно с ним смывается 7% экстрагированного Еu и заметная часть железа (10-15%). На долю операции расцикловки Zr (совместно с Fe) приходится 70% нагрузки по реэкстракции Fe. В результате был получен реэкстракт Мо с концентрированием в ~8 раз относительно его содержания в исходном (неразбавленном) рафинате Пурекс-процесса. В блоке реэкстракции Am было достигнуто полное его выведение с очисткой от Мо в 10 раз, а от Еu-Sm в 1,5-2 раза.
Пример 8. Технологическая схема отличается от схемы, представленной на чертеже и описанной в примере 6, отсутствием блока для отмывки экстрагента от оксалата и двух потоков: промывной раствор (65) и выходящий оксалатный раствор (63). При этом первый экстракционный блок должен быть увеличен на соответствующее число ступеней ликвидированного блока. Этот технологический прием позволяет сократить общий объем отходов и число подаваемых реагентов.
Claims (7)
1. Способ экстракционной переработки высокоактивного рафината Пурекс-процесса для отработанного ядерного топлива атомных электростанций с использованием в качестве экстрагента циркониевой соли дибутилфосфорной кислоты, растворенной в разбавленном трибутилфосфате для извлечения трансплутониевых, редкоземельных элементов и осколочного молибдена, отличающийся тем, что совместно экстрагируют трансплутониевые, редкоземельные элементы, осколочные молибден и цирконий, а также коррозионное железо с помощью раствора 0,1-0,4 моль/л циркониевой соли дибутилфосфорной кислоты в 30% (об.) трибутилфосфате с предельными углеводородами, причем соотношение циркония к дибутилфосфорной кислоте составляет 1:12-1:15 в оборотном экстрагенте и 1:9-1:12 в ступени ввода исходного раствора и на последующей операции кислотной промывки, затем производят реэкстракцию трансплутониевых элементов с цериевой подгруппой редкоземельных элементов в раствор азотной кислоты при ее концентрации 3,5-5 моль/л в реэкстракте, промытом от молибдена оборотным экстрагентом, проводят реэкстракцию иттрия и иттриевой подгруппы редкоземельных элементов вместе или раздельно с осколочным молибденом с получением их раствора в 3,5-5 моль/л азотной кислоты при добавлении 0,3-1 моль/л перекиси водорода в реэкстрагирующий раствор с последующей промывкой экстрагента водой и регенерируют экстрагент путем промывки в противотоке части экстрагента, выведенной, исходя из поступления циркония с исходным раствором, щавелевой кислотой при добавлении оксалата аммония в ступень ввода экстрагента при температуре процесса 35-40°С, причем экстрагированную щавелевую кислоту удаляют путем последующей промывки экстрагента азотной кислотой и водой.
2. Способ по п.1, отличающийся тем, что исходный водный раствор содержит 1-2 моль/л азотной кислоты при составе промывного раствора 0,1 моль/л азотной кислоты.
3. Способ по п.1, отличающийся тем, что реэкстракцию трансплутониевых элементов проводят с тремя вводами раствора азотной кислоты следующих концентраций: 7 моль/л - в точку вывода экстракта, 10 моль/л - в середину реэкстракционной зоны, а также в середину зоны промывки реэкстракта от осколочного молибдена оборотным экстрагентом, причем соотношение потоков выбирают с учетом концентрации экстрагента и содержащихся в нем элементов.
4. Способ по п.1, отличающийся тем, что для обеспечения концентрации 3,5-5 моль/л азотной кислоты при реэкстракции иттрия и его подгруппы редкоземельных элементов совместно или раздельно с осколочным молибденом вводят в среднюю часть экстракционного блока раствор 10 моль/л азотной кислоты и раствор перекиси водорода в 4-7 моль/л азотной кислоте при подаче воды в ступень вывода экстрагента с реэкстракции, при этом расчетная концентрация перекиси водорода в выходящем реэкстракте должна составлять 0,3-1 моль/л, а соотношение органического и водного потоков на стадии реэкстракции находится в пределах 0,8-2 в зависимости от концентрации экстрагента.
5. Способ по п.1 или 4, отличающийся тем, что раздельную реэкстракцию осколочного молибдена проводят до реэкстракции иттрия и редкоземельных элементов его подгруппы и промывают реэкстракт осколочного молибдена оборотным экстрагентом для удаления из него иттрия и редкоземельных элементов его подгруппы.
6. Способ по п.1, отличающийся тем, что регенерацию части экстрагента осуществляют путем реэкстракции циркония и сопутствующего коррозионного железа для удаления их в количествах, поступивших с исходным раствором, проводят раствором 0,5 моль/л щавелевой кислоты при подаче в ступень ввода экстрагента раствора оксалата аммония, исходя из количества азотной кислоты в органической фазе, поступающей на регенерацию, и при соотношении ионов аммония к общему количеству вводимой щавелевой кислоты не выше чем 1:4.
7. Способ по п.1 или 6, отличающийся тем, что промывку экстрагента от экстрагированной щавелевой кислоты осуществляют раствором 3 моль/л азотной кислоты при ее подаче 5 моль/л азотной кислоты в середину экстракционного блока и подаче воды в крайнюю ступень блока по ходу экстрагента, либо совмещают операцию с экстракцией трансплутониевых и редкоземельных элементов путем увеличения числа ступеней в экстракционном блоке с выведением щавелевой кислоты в рафинат.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2003100275A RU2249266C2 (ru) | 2003-01-04 | 2003-01-04 | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива аэс |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2003100275A RU2249266C2 (ru) | 2003-01-04 | 2003-01-04 | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива аэс |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| RU2003100275A RU2003100275A (ru) | 2004-08-10 |
| RU2249266C2 true RU2249266C2 (ru) | 2005-03-27 |
Family
ID=35560799
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| RU2003100275A RU2249266C2 (ru) | 2003-01-04 | 2003-01-04 | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива аэс |
Country Status (1)
| Country | Link |
|---|---|
| RU (1) | RU2249266C2 (ru) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2295166C1 (ru) * | 2005-07-19 | 2007-03-10 | Российская Федерация в лице Федерального агентства по атомной энергии | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива атомных электростанций |
| RU2384902C1 (ru) * | 2009-02-09 | 2010-03-20 | Открытое акционерное общество "Сибирский химический комбинат" | Способ очистки оксидов урана от примесей |
| RU2447523C2 (ru) * | 2010-06-11 | 2012-04-10 | Открытое акционерное общество "Сибирский химический комбинат" | Способ очистки регенерированного урана |
| RU2502142C1 (ru) * | 2012-04-19 | 2013-12-20 | Федеральное государственное унитарное предприятие "Научно-исследовательский институт Научно-производственное объединение "ЛУЧ" (ФГУП "НИИ НПО "ЛУЧ") | Способ переработки уран-молибденовой композиции |
| RU2522544C2 (ru) * | 2012-06-15 | 2014-07-20 | Федеральное государственное унитарное предприятие "Научно-производственное объединение "Радиевый институт им. В.Г. Хлопина" | Способ селективного извлечения радионуклидов из радиоактивных азотнокислых растворов (варианты) |
| RU2663882C1 (ru) * | 2013-12-20 | 2018-08-13 | Коммиссариат А Л' Энержи Атомик Э Оз Энержи Альтернатив | Способ переработки отработанного ядерного топлива, включающий стадию очистки урана (vi) от по меньшей мере одного актинида (iv) путем получения комплекса данного актинида (iv) |
| RU2709826C1 (ru) * | 2019-02-18 | 2019-12-23 | Российская Федерация, от имени которой выступает Государственная корпорация по атомной энергии "Росатом" | Способ переработки высокоактивных отходов с фракционированием радионуклидов |
| CN112680609A (zh) * | 2020-12-14 | 2021-04-20 | 中国人民解放军63653部队 | 一种钚回收离子液体萃取剂及其从含钚废液中萃取分离钚的方法 |
| US11091819B2 (en) | 2013-01-18 | 2021-08-17 | Rare Element Resources Ltd. | Extraction of metals from metallic compounds |
| CN114350984A (zh) * | 2020-10-14 | 2022-04-15 | 厦门稀土材料研究所 | 磷酸类萃取沉淀剂分离回收稀土的方法 |
| RU2774155C1 (ru) * | 2021-07-27 | 2022-06-15 | Частное Учреждение По Обеспечению Научного Развития Атомной Отрасли "Наука И Инновации" (Частное Учреждение "Наука И Инновации") | Способ экстракционного выделения трансплутониевых и редкоземельных элементов |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2960690B1 (fr) * | 2010-05-27 | 2012-06-29 | Commissariat Energie Atomique | Procede de traitement de combustibles nucleaires uses ne necessitant pas d'operation de desextraction reductrice du plutonium |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1330535A (en) * | 1970-07-07 | 1973-09-19 | Atomic Energy Authority Uk | Processing of irradiated nuclear reactor fuel |
| RU2080666C1 (ru) * | 1993-07-27 | 1997-05-27 | Научно-производственное объединение "Радиевый институт им.В.Г.Хлопина" | Способ обработки высокоактивных азотнокислых рафинатов от регенерации топлива аэс |
| RU2106030C1 (ru) * | 1994-10-11 | 1998-02-27 | Научно-производственное объединение "Радиевый институт им.В.Г.Хлопина" | Способ экстракционного извлечения и разделения тпэ и рзэ из азотнокислых растворов |
-
2003
- 2003-01-04 RU RU2003100275A patent/RU2249266C2/ru not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1330535A (en) * | 1970-07-07 | 1973-09-19 | Atomic Energy Authority Uk | Processing of irradiated nuclear reactor fuel |
| RU2080666C1 (ru) * | 1993-07-27 | 1997-05-27 | Научно-производственное объединение "Радиевый институт им.В.Г.Хлопина" | Способ обработки высокоактивных азотнокислых рафинатов от регенерации топлива аэс |
| RU2106030C1 (ru) * | 1994-10-11 | 1998-02-27 | Научно-производственное объединение "Радиевый институт им.В.Г.Хлопина" | Способ экстракционного извлечения и разделения тпэ и рзэ из азотнокислых растворов |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2295166C1 (ru) * | 2005-07-19 | 2007-03-10 | Российская Федерация в лице Федерального агентства по атомной энергии | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива атомных электростанций |
| RU2384902C1 (ru) * | 2009-02-09 | 2010-03-20 | Открытое акционерное общество "Сибирский химический комбинат" | Способ очистки оксидов урана от примесей |
| RU2447523C2 (ru) * | 2010-06-11 | 2012-04-10 | Открытое акционерное общество "Сибирский химический комбинат" | Способ очистки регенерированного урана |
| RU2502142C1 (ru) * | 2012-04-19 | 2013-12-20 | Федеральное государственное унитарное предприятие "Научно-исследовательский институт Научно-производственное объединение "ЛУЧ" (ФГУП "НИИ НПО "ЛУЧ") | Способ переработки уран-молибденовой композиции |
| RU2522544C2 (ru) * | 2012-06-15 | 2014-07-20 | Федеральное государственное унитарное предприятие "Научно-производственное объединение "Радиевый институт им. В.Г. Хлопина" | Способ селективного извлечения радионуклидов из радиоактивных азотнокислых растворов (варианты) |
| US11091819B2 (en) | 2013-01-18 | 2021-08-17 | Rare Element Resources Ltd. | Extraction of metals from metallic compounds |
| RU2663882C1 (ru) * | 2013-12-20 | 2018-08-13 | Коммиссариат А Л' Энержи Атомик Э Оз Энержи Альтернатив | Способ переработки отработанного ядерного топлива, включающий стадию очистки урана (vi) от по меньшей мере одного актинида (iv) путем получения комплекса данного актинида (iv) |
| RU2709826C1 (ru) * | 2019-02-18 | 2019-12-23 | Российская Федерация, от имени которой выступает Государственная корпорация по атомной энергии "Росатом" | Способ переработки высокоактивных отходов с фракционированием радионуклидов |
| CN114350984A (zh) * | 2020-10-14 | 2022-04-15 | 厦门稀土材料研究所 | 磷酸类萃取沉淀剂分离回收稀土的方法 |
| CN112680609A (zh) * | 2020-12-14 | 2021-04-20 | 中国人民解放军63653部队 | 一种钚回收离子液体萃取剂及其从含钚废液中萃取分离钚的方法 |
| RU2774155C1 (ru) * | 2021-07-27 | 2022-06-15 | Частное Учреждение По Обеспечению Научного Развития Атомной Отрасли "Наука И Инновации" (Частное Учреждение "Наука И Инновации") | Способ экстракционного выделения трансплутониевых и редкоземельных элементов |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN102549176B (zh) | 液-液萃取操作中在镅和锔之间和/或在镧系元素之间分离因子的提高 | |
| RU2438200C2 (ru) | Групповое разделение актинидов из сильнокислой водной фазы | |
| Nasab et al. | Determination of optimum process conditions for the separation of thorium and rare earth elements by solvent extraction | |
| El-Nadi | Lanthanum and neodymium from Egyptian monazite: Synergistic extractive separation using organophosphorus reagents | |
| RU2249266C2 (ru) | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива аэс | |
| CN102471824B (zh) | 用于从硝酸水相中选择性地回收镅的方法 | |
| GB2305291A (en) | A method of separating trivalent actinides and rare earth elements | |
| CN112458319A (zh) | 一种基于离子液体萃取体系分离重稀土元素的方法 | |
| US2824783A (en) | Separation of scandium from aqueous solutions | |
| US7157003B2 (en) | Cyclic method for separating chemical elements present in an aqueous solution | |
| RU2106030C1 (ru) | Способ экстракционного извлечения и разделения тпэ и рзэ из азотнокислых растворов | |
| Shaohua et al. | Study on separation technology of Pr and Nd in D2EHPA-HCl-LA coordination extraction system | |
| CN1037914C (zh) | 从生产堆浓缩高放废液中分离锕系元素的方法 | |
| JPS63198897A (ja) | ジルコニウムとウラン又はプルトニウムのごとき1種類以上の別の金属と共に有機溶媒中に存在するテクネチウムを分離すべく、特に照射済核燃料の再処理に使用し得る方法 | |
| US3640678A (en) | Yttrium purification process | |
| JP2007503526A (ja) | 三価のキュリウムから三価のアメリシウムを分離する方法 | |
| US2780518A (en) | Process for recovery of uranium from aqueous solutions | |
| CN1032765C (zh) | 镅和锔与裂变产物稀土分离的方法 | |
| RU2727140C1 (ru) | Способ экстракционной переработки облученного ядерного топлива | |
| US3615171A (en) | Process of separating yttrium from lanthanide rare earths | |
| RU2454740C1 (ru) | Способ выведения нептуния при фракционировании долгоживущих радионуклидов | |
| RU2623943C1 (ru) | Экстракционная смесь для извлечения тпэ и рзэ из высокоактивного рафината переработки оят аэс и способ её применения (варианты) | |
| Hérès et al. | The separation of extractants implemented in the DIAMEX-SANEX process | |
| RU2773142C2 (ru) | Способ экстракционного извлечения и разделения РЗЭ | |
| RU2295166C1 (ru) | Способ экстракционной переработки высокоактивного рафината пурекс-процесса для отработанного ядерного топлива атомных электростанций |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| MM4A | The patent is invalid due to non-payment of fees |
Effective date: 20140105 |