詳細說明和特定實施態樣
為了簡明,本說明書引述之公開文獻(包括專利和專利申請案)所揭露的全部內容係倂入本文作為參考。
大多數化學名稱係使用IUPAC命名法則產生。某些化學名稱係使用不同之命名法則產生或使用此技術領域習知之替代名稱或商業名稱。若名稱與結構相衝突,結構勝出。
定義和一般術語
本發明之某些實施態樣將會詳細地加以說明且該等實施態樣之實例將會以附隨之結構和化學式說明。本發明欲涵蓋可包括於本發明之範圍內如申請專利範圍所定義者的所有替代、修飾及等同者。熟習此技術之人士將能確認與本說明書描述者相似或等同之許多可用於實施本發明的方法和材料。本發明不受限於本說明書描述之方法和材料。若倂入之文獻、專利及相似材料的一或多者與本說明書之揭露(包括但不限於定義之術語、術語使用、描述之技術或類似者)不同或相牴觸,將以本說明書為準。
另應瞭解的是,為清楚起見,描述於個別實施態樣之本發明的某些特徵亦可經組合而描述於單一實施態樣。相反地,為清楚起見,描述於單一實施態樣之本發明的多種不同之特徵亦可經個別描述或描述於任何適當之次組合中。
除非另有定義,本文使用之所有技術和科學術語具有如同熟習此技術之人士一般瞭解之相同意義。本說明書引述之所有專利和公開文獻的全部內容係倂入本文作為參考。
除非另有說明,適用所使用之下述定義。對於本發明之目的,依據元素周期表(CAS版)和Handbook of Chemistry and Physics,第75版,1994鑑別化學元素。此外,有機化學之一般原理係描述於"Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999和"March's Advanced Organic Chemistry", Michael B. Smith and Jerry March, John Wiley & Sons, New York: 2007,彼等之全部內容係倂入本文作為參考。
除非另有說明,上述且本文使用之術語具有下述之意義。若漏載定義,則參照熟習此技術之人士所瞭解之一般定義。若本文描述之定義與任何引述之公開文獻描述之定義相牴觸或不同,將以本說明書描述之定義為準。
本文使用之術語“包括”、“含有”及“包含”係以開放且不受限制之意思使用。
除非本文中另有清楚說明,本文使用之單數術語“一”和“該”包括複數意義。
為提供更清楚的描述,本文中某些定量表示不以“約”字定性。應瞭解的是,是否明確使用“約”字,所描述之每個量表示給定真實量且亦表示熟習此技術之人士所瞭解之合理的給定值之大約量,其包括該給定值因實驗及/或測量條件所產生之等同值和大約值。當產率係以%表示時,該產率係指實體之質量且係以相對於該實體於特定化學計量條件下可得之最大量的方式表示。除非另有不同說明,以%表示之濃度係指質量比。
化學定義
本文使用之“烷基”係指具有1至12個碳原子之飽和直鏈或支鏈烴基。代表性烷基包括但不限於甲基、乙基、正丙基、異丙基、2-甲基-1-丙基、2-甲基-2-丙基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、4-甲基-2-戊基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、丁基、異丁基、三級丁基、正戊基、異戊基、新戊基、正己基和類似基團及較長鏈之烷基,諸如庚基、辛基和類似基團。本文使用之“低碳烷基”表示具有1至6個碳原子之烷基。
本文使用之“烷基胺基”表示本文定義之胺基,其中該胺基之1個氫原子係經本文定義之烷基替代。烷基胺基可藉由下述通式-NH-烷基定義。該通式包括下述通式-NH-C
1
-C
10
烷基和-NH-C
1
-C
6
烷基之基團。烷基胺基之實例包括但不限於甲基胺基、乙基胺基、丙基胺基及丁基胺基。
本文使用之“二烷基胺基”表示本文定義之胺基,其中該胺基之2個氫原子係經本文定義之烷基替代。二烷基胺基可藉由下述通式-N(烷基)
2
定義,其中該等烷基可為相同或不同且可選自本文定義之烷基,例如C
1
-C
10
烷基或C
1
-C
6
烷基。
本文使用之“烷氧基”包括-O-(烷基),其中烷基係如上述定義者。
本文使用之“烷氧基烷基”表示-(伸烷基)-O-(烷基),其中每個“烷基”獨立地係上述定義之烷基。
本文使用之“胺基”係指-NH
2
。
“芳基”表示單環、雙環或三環芳香族基團,其中芳基之所有環係芳香族。對於雙環或三環系統,個別芳香族環彼此經稠合。芳基之實例包括但不限於苯基、萘基及蒽基。
本文使用之“芳氧基”係指-O-(芳基),其中芳基係如上述定義者。
本文使用之“芳基烷基”係指-(伸烷基)-(芳基),其中伸烷基和芳基係如上述定義者。芳基烷基的烷基之非限制性實例包含低碳烷基。適當的芳基烷基之非限制性實例包括苄基、2-苯乙基及萘基甲基。
本文使用之“芳基烷氧基”係指-O-(伸烷基)-芳基,其中伸烷基和芳基係如上述定義者。
本文使用之“氰基”表示具有碳原子與氮原子經由三鍵連接之取代基。
“氰基烷基”表示如上述定義之烷基,其中該烷基之1個氫原子係經氰基(-CN)替代。該氰基烷基之烷基部分提供與該分子之其餘部分的連接點。
本文使用之“氘”表示氫之安定同位素,其具有1個質子和1個中子。
本文使用之“鹵素”係指氟、氯、溴或碘。“鹵”表示氯、氟、溴或碘。
“鹵烷基”表示如上述定義之烷基,其中該烷基之1或多個(例如1、2或3個)氫原子係經鹵素原子(例如氟、溴或氯,特別是氟)替代。鹵烷基之實例包括但不限於單氟-、二氟-或三氟-甲基、-乙基或-丙基,例如3,3,3-三氟丙基、2-氟乙基、2,2,2-三氟乙基、氟甲基、二氟甲基或三氟甲基或溴乙基或氯乙基。同樣地,“氟烷基”係指如上述定義之烷基,其係經1或多個(例如1、2或3個)氟原子取代。
本文使用之“鹵烷氧基”係指-O-(鹵烷基),其中鹵烷基係如上述定義者。例示性鹵烷氧基係溴乙氧基、氯乙氧基、三氟甲氧基及2,2,2-三氟乙氧基。
“羥基”表示-OH。
“羥基烷基”表示經至少1個(例如1、2或3個)羥基取代之烷基。該羥基烷基之烷基部分提供與該分子之其餘部分的連接點。羥基烷基之實例包括但不限於羥基甲基、羥基乙基、1-羥基丙基、2-羥基異丙基、1,4-二羥基丁基及類似基團。
“側氧基”表示=O且可與碳原子或硫原子連接。“N-氧化物”係指氮原子之氧化形式。
本文使用之“環烷基”係指具有3至12個環碳原子的飽和或部分飽和之單環、稠合多環、橋連多環或螺多環的碳環。環烷基之非限制性範疇係具有3至6個碳原子的飽和或部分飽和之單環碳環。環烷基之說明性實例包括但不限於下述基團:

。
“環烷氧基”係指-O-(環烷基)。
本文使用之“雜芳基”係指具有3至15個選自碳、氧、氮、硒或硫的環原子之單環或稠合多環的芳香族雜環。適當之雜芳基不包括必須經荷電以呈芳香族性之環系統,諸如吡喃鎓。某些適當之5員雜芳基環(單環雜芳基或作為多環雜芳基之一部分)含有1個氧、硫或氮原子、或1個氮原子和1個氧或硫原子、或2、3或4個氮原子。某些適當之6員雜芳基環(單環雜芳基或作為多環雜芳基之一部分)含有1、2或3個氮原子。雜芳基之實例包括但不限於吡啶基、咪唑基、咪唑並吡啶基、嘧啶基、吡唑基、三唑基、吡嗪基、四唑基、呋喃基、噻吩基、異噁唑基、噻唑基、噁唑基、異噻唑基、吡咯基、喹啉基、異喹啉基、吲哚基、苯並咪唑基、苯並呋喃基、噌啉基、吲唑基、吲哚嗪基、呔嗪基、噠嗪基、三嗪基、異吲哚基、喋啶基、嘌呤基、噁二唑基、三唑基、噻二唑基、呋呫基、苯並呋呫基、苯並噻吩基、苯並噻唑基、苯並噁唑基、喹唑啉基、喹噁啉基、萘啶基及呋喃並吡啶基。
“雙環雜芳基”係指如上述定義之雜芳基,其含有2個構成之芳香族環,其中該2個環彼此經稠合且該2個環之至少一者係如上述定義之雜芳基。雙環雜芳基包括包含1、2、3或4個雜原子環員之雙環雜芳基且係未經取代或經1或多個選自胺基或鹵素之取代基取代,其中該雜芳基之1或多個N環員可選擇地係N-氧化物。
熟習此技術之人士當能瞭解上述列示或說明之雜芳基和環烷基的基團並非詳盡且亦可選擇屬於定義範圍之其他基團。
如本文所述,本文揭露之化合物可選擇地經1或多個取代基取代或可藉由本發明之特定類型、子類型及種類例示說明。
本文使用之“經取代”表示特定之基團或部分含有1或多個適當之取代基。本文使用之“未經取代”表示特定之基團不含有取代基。本文使用之“可選擇地經取代”表示特定之基團係未經取代或經特定數目之取代基取代。該“經取代”係用於描述結構系統且該取代表示發生於該系統上之任何允許之價位置。
本文使用之“1或多個取代基”表示可發生於該系統上之任何允許之價位置的1至最大可能數目之取代。於某些實施態樣中,1或多個取代基表示1、2、3、4或5個取代基。於另一實施態樣中,1或多個取代基表示1、2或3個取代基。
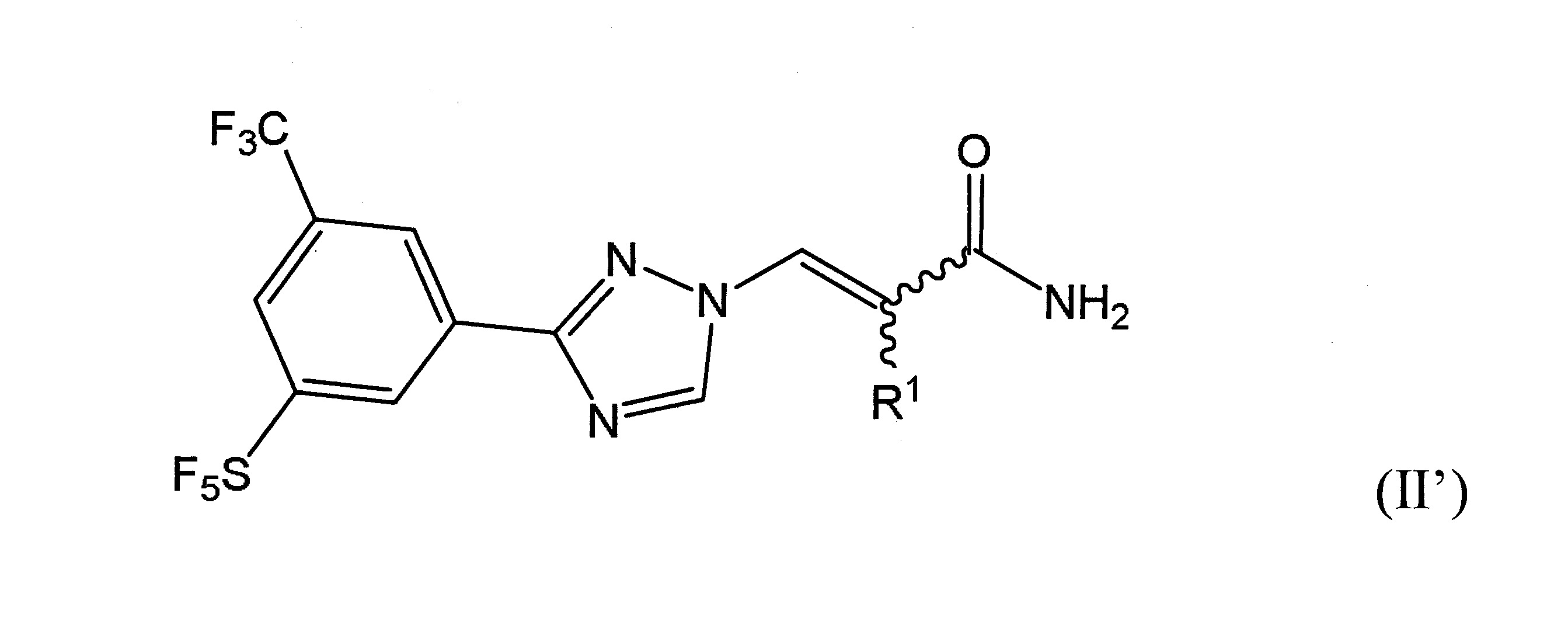
如本文所使用者,於含有經“
”鍵連接之取代基的式II’中,雙鍵表示E或Z構形。
以未滿足價表示之任何原子被假定為含有足夠數目之氫原子以滿足原子價。
當任何變異(例如烷基、伸烷基、雜芳基、R
1
及R
2
)於本文之任何化學式或描述中出現1次以上時,於每次出現時該變異之定義係獨立於該變異於每次其他出現時之定義。
本文使用之數目範圍欲包括依序全部數目。例如,“0至4”或“0-4”之範圍包括0、1、2、3及4且“10-20%”包括10%、11%、12%、13%、14%、15%、16%、17%、18%、19%及20%。同樣地,數目範圍亦欲包括依序小數。例如,“1-2%”包括1.0%、1.1%、1.2%、1.3%、1.4%、1.5%、1.6%、1.7%、1.8%、1.9%及2.0%。
當顯示多官能基時,與核連接之點係由線或連字號表示。例如,芳氧基-係指其中氧原子係與核連接之點且芳基係與該氧原子連接。
其他定義
本文使用之“個體”包含哺乳動物和非哺乳動物。哺乳動物之實例包括但不限於哺乳動物綱之任何成員:人類;非人類之靈長類動物,諸如黑猩猩和其他類人猿及猴子;農場動物,諸如牛、馬、綿羊、山羊及豬;家養動物,諸如兔、狗及貓;及,實驗室動物,其包括囓齒動物,諸如大鼠、小鼠及豚鼠,以及類似者。非哺乳動物之實例包括但不限於鳥、魚及類似者。於本發明之一實施態樣中,該哺乳動物係人類。
“病患”包括人類和動物。
“抑制劑”係指諸如化合物、藥物、酶活化劑或激素之分子,其阻斷或者干擾特定之生物活性。
“調節劑”係指諸如本發明之化合物的分子,其增加或降低或者影響給定之蛋白、受體及/或離子通道之活性。
“有效量”或“治療有效量”係指足以提供所欲之生物結果的試劑之量。該結果可為疾病或醫療條件之症狀、徵候或病因的減輕及/或緩和或生物系統之任何其他所欲之改變。例如,治療用途之“有效量”係指提供疾病狀態、徵候或醫療條件之臨床相關改變所需的化合物或包含該化合物之組成物的量。藉由使用慣常之實驗,熟習此技術之人士可決定用於任一個別案例之適當“有效”量。因此,“有效量”通常係指活性劑顯現所欲之治療功效的量。
本文使用之“處置”或“治療”包含“預防性”和“療效性”處置或治療。“預防性”處置表示延後疾病、疾病徵候或醫療條件的發展、抑制可能出現之徵候或降低疾病或徵候之發展或復發的風險。“療效性”治療包括減輕現有疾病、徵候或病況之嚴重程度或抑制現有疾病、徵候或病況之惡化。因此,治療包括改善或預防現有疾病徵候之惡化、預防其他徵候發生、改善或預防徵候之潛在的代謝病因、抑制病症或疾病,例如中止該病症或疾病之發展、緩解該病症或疾病、引起該病症或疾病之退化、緩解由該疾病或病症引起之病況或中止該疾病或病症之徵候。
應瞭解本文使用之“投予”化合物表示提供本發明之化合物或包含本發明之化合物或本發明之化合物的前藥之醫藥組成物給需要之個體。確定的是,熟習此非限制性技術之人士可使用有效量的本發明之化合物以治療正罹患神經系統和精神病症之病患或預防性治療罹患該等病症之病患。
本文使用之“組成物”欲包含產物,其包含特定量之特定成分,且包含任何產物,其係直接或間接由特定量之特定成分的組合產生。與醫藥組成物有關之組成物欲包含產物,其包含活性成分和作為載劑之惰性成分,且包含任何產物,其係直接或間接由該等成分之任何二或多者的組合、複合或聚積產生或由諸如引起該等成分之一或多者的解離之其他類型的反應或交互作用產生。於是,本發明之醫藥組成物包含由混合本發明之化合物和醫藥上可接受之載劑所製備之任何組成物。
其他化學描述
本文給予之任何化學式欲表示具有結構式所示之結構的化合物和某些變異體或型式。例如,本文給予之任何化學式的化合物可具有不對稱或手性中心並因此會以不同之立體異構形式存在。該通式之化合物的所有立體異構物(其包括光學異構物、鏡像異構物及非鏡像異構物)及彼等之混合物落入該通式之範圍。再者,某些結構可呈幾何異構物(即順式和反式異構物)、互變異構物或阻旋異構物。所有該等異構型式或彼等之混合物屬於本發明之一部分。因此,本文給予之任何化學式欲表示消旋物、一或多種鏡像異構形式、一或多種非鏡像異構形式、一或多種互變異構或阻旋異構形式及彼等之混合物。
“立體異構物”係指具有相同化學組成但原子或基團之空間配置不同的化合物。立體異構物包括鏡像異構物、非鏡像異構物、構形異構物(旋轉異構物)、幾何(順式/反式)異構物、阻旋異構物等。
“手性”係指具有與鏡像對非重疊性之分子且“非手性”係指與鏡像對重疊之分子。
“鏡像異構物”係指化合物之2個立體異構物,彼等彼此係非重疊之鏡像。
“非鏡像異構物”係指具有2或多個手性中心之立體異構物,該等異構物分子彼此不為鏡像。非鏡像異構物具有不同之物理性質,例如熔點、沸點、光譜特性或生物活性。藉由高解析分析方法,諸如電泳和層析(諸如HPLC),可分離非鏡像異構物之混合物。
本文使用之立體化學的定義和常規通常遵循文獻S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984)McGraw-Hill Book Company, New York和Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994。
許多有機化合物係以光學活性形式存在,即彼等能旋轉偏光面。於描述光學活性化合物時,使用字首D和L或R和S以表示分子之手性中心的絕對構形。使用字首d和l或(+)和(-)以表示化合物旋轉偏光面之符號,其中(-)或l表示該化合物呈左旋性。字首為(+)或d之化合物呈右旋性。特定之立體異構物可指鏡像異構物且該等立體異構物的混合物被稱為鏡像異構混合物。鏡像異構物之50:50混合物被稱為消旋混合物或消旋物,其可能發生於化學反應或製程中不具有立體選擇性或立體特異性的地方。
本文揭露之化合物的任何不對稱性原子(例如碳或類似者)可呈消旋性或富含鏡像異構物性,例如(R)-、(S)-或(R, S)-構形。於某些實施態樣中,於該(R)-或(S)-構形之每個不對稱性原子具有至少50%鏡像異構物過量、至少60%鏡像異構物過量、至少70%鏡像異構物過量、至少80%鏡像異構物過量、至少90%鏡像異構物過量、至少95%鏡像異構物過量或至少99%鏡像異構物過量。
取決於起始物和方法之選擇和不對稱性碳原子之數目,化合物可呈可能的立體異構物之一或彼等之混合物的形式,諸如消旋物和非鏡像異構混合物。使用手性合成組元或手性反應劑可製備光學活性(R)-和(S)-異構物,或使用慣用之技術可解析光學活性(R)-和(S)-異構物。若化合物含有雙鍵,則取代基可呈E或Z構形。若化合物含有經二取代之環烷基,則環烷基之取代基相對於該相同環烷基之另一取代基可呈順式或反式構形。
基於組分之理化性質的差異,藉由例如層析及/或分段結晶,立體異構物所形成之任何混合物可經分離成純或實質上純之幾何異構物、鏡像異構物或非鏡像異構物。藉由熟習此技術之人士所習知之方法,例如藉由分離終產物或中間體之非鏡像異構性鹽,該終產物或中間體所形成之任何消旋物可經解析成光學鏡像體。藉由手性層析,例如使用手性吸附劑之高效液相層析(HPLC),亦可解析消旋產物。藉由不對稱性合成,亦可製備較佳之鏡像異構物。參閱例如文獻Jacques, et al., Enantiomers, Racemates and Resolutions(Wiley Interscience, New York, 1981); Principles of Asymmetric Synthesis(2nd Ed. Robert E. Gawley, Jeffrey Aubé, Elsevier, Oxford, UK, 2012); Eliel, E. L. Stereochemistry of Carbon Compounds(McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions, p. 268(E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972); Chiral Separation Techniques: A Practical Approach (Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007)。
藉由熟習此技術之人士所習知之方法,諸如例如層析及/或分段結晶,基於個別非鏡像異構物之理化性質的差異,非鏡像異構混合物可經分離成個別非鏡像異構物。藉由與適當之光學活性化合物(例如手性輔助劑,諸如手性醇或Mosher氏醯基氯,或形成非鏡像異構性鹽之混合物)反應以轉化鏡像異構混合物成非鏡像異構混合物、分離該等非鏡像異構物及轉化(例如水解或脫鹽)個別非鏡像異構物為對應之純鏡像異構物,可分離鏡像異構物。藉由使用手性HPLC管柱,亦可分離鏡像異構物。
本發明之化合物可生成醫藥上可接受之鹽,其亦屬於本發明之範圍。“醫藥上可接受之鹽”係指式(I’)化合物之自由酸或鹼的鹽,其係非毒性、生理上可耐受、能與經調製之醫藥組成物互溶及另一方面適合調製及/或投予至個體。除非另有說明,應瞭解的是,本發明之化合物包括該化合物的醫藥上可接受之鹽。
化合物鹽包括與無機酸及/或有機酸所生成之酸性鹽及與無機鹼及/或有機鹼所生成之鹼性鹽。此外,當給予之化合物同時含有鹼性基團(諸如但不限於吡啶或咪唑)和酸性基團(諸如但不限於羧酸)時,熟習此技術之人士當能瞭解該化合物可呈兩性離子(“內鹽”),該鹽含括於本文使用之“鹽”的範圍。藉由例如令化合物與某一量(諸如當量)之適當酸或鹼於諸如使該鹽沉澱之介質或水溶性介質中反應並隨後經冷凍乾燥,可製備本發明之化合物的鹽。
例示性鹽包括但不限於硫酸鹽、檸檬酸鹽、乙酸鹽、草酸鹽、氯化物、溴化物、碘化物、硝酸鹽、硫酸氫鹽、磷酸鹽、酸式磷酸鹽、異菸鹼酸鹽、乳酸鹽、水楊酸鹽、酸式檸檬酸鹽、酒石酸鹽、油酸鹽、鞣酸鹽、泛酸鹽、酒石酸氫鹽、抗壞血酸鹽、丁二酸鹽、順丁烯二酸鹽、龍膽酸鹽、反丁烯二酸鹽、葡萄糖酸鹽、葡萄糖醛酸鹽、葡萄糖二酸鹽、甲酸鹽、苯甲酸鹽、麩胺酸鹽、甲磺酸鹽、乙磺酸鹽、苯磺酸鹽、對甲苯磺酸鹽及雙羥萘酸鹽(即1,1’-伸甲基‑雙(2-羥基-3-萘酸鹽))。醫藥上可接受之鹽可涉及包括其他分子,諸如乙酸離子、丁二酸離子或其他抗衡離子。該抗衡離子可為使母化合物上之電荷安定的任何有機或無機基團。再者,醫藥上可接受之鹽於結構上可含有超過1個帶電荷之原子。當多個帶電荷之原子係該醫藥上可接受之鹽的一部分時,實例可含有多個抗衡離子。因此,醫藥上可接受之鹽可含有1或多個帶電荷之原子及/或1或多個抗衡離子。
例示性酸加成鹽包括乙酸鹽、抗壞血酸鹽、苯甲酸鹽、苯磺酸鹽、硫酸氫鹽、硼酸鹽、丁酸鹽、檸檬酸鹽、樟腦酸鹽、樟腦磺酸鹽、反丁烯二酸鹽、氫氯酸鹽、氫溴酸鹽、氫碘酸鹽、乳酸鹽、順丁烯二酸鹽、甲磺酸鹽、萘磺酸鹽、硝酸鹽、草酸鹽、磷酸鹽、丙酸鹽、水楊酸鹽、丁二酸鹽、硫酸鹽、酒石酸鹽、硫氰酸鹽、甲苯磺酸鹽及類似者。
例示性鹼鹽包括銨鹽、鹼金屬鹽(諸如鈉鹽、鋰鹽及鉀鹽)、鹼土金屬鹽(諸如鈣鹽和鎂鹽)、有機鹼(例如有機胺,諸如二環己基胺和三級丁基胺)鹽及胺基酸(諸如精胺酸和離胺酸)鹽及類似者。含有氮之鹼性基團可經試劑季胺化,該試劑係諸如低碳烷基鹵化物(例如甲基氯、乙基氯、丁基氯、甲基溴、乙基溴、丁基溴、甲基碘、乙基碘及丁基碘)、硫酸二烷酯(例如硫酸二甲酯、硫酸二乙酯及硫酸二丁酯)、長鏈鹵化物(例如癸基氯、癸基溴、癸基碘、十二烷基氯、十二烷基溴、十二烷基碘、十八烷基氯、十八烷基溴及十八烷基碘)、芳烷基鹵化物(例如苄基溴和苯乙基溴)及類似者。
此外,通常被認為適合用於自醫藥化合物形成醫藥上有用之鹽的酸和鹼係見於文獻,例如P. Stahl et al, Camille G.(eds.)Handbook of Pharmaceutical Salts. Properties, Selection and Use.(2002)Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences(1977)66(1)1-19; P. Gould, International J. of Pharmaceutics(1986)33 201-217; Anderson et al, The Practice of Medicinal Chemistry(1996), Academic Press, New York;及The Orange Book(Food & Drug Administration, MD,可自FDA取得)。該等文獻併入本文作為參考。
此外,本文描述之任何化合物亦欲係指任何非溶劑化形式或該化合物之水合物、溶劑合物或多晶型及彼等之混合物,即使該等形式並未明確列示。“溶劑合物”表示本發明之化合物與一或多個溶劑分子之物理性結合。該物理性結合涉及不同程度之離子性和共價性鍵結,其包括氫鍵。於某些實例中,該溶劑合物將可被分離,例如當一或多個溶劑分子併入結晶固體之晶格時。“溶劑合物”包含溶液相和可被分離之溶劑合物二者。適當之溶劑合物包括與醫藥上可接受之溶劑(諸如水、乙醇及類似者)形成之溶劑合物。於某些實施態樣中,該溶劑係水且該溶劑合物係水合物。
本發明之一或多個化合物係可選擇地被轉化為溶劑合物。通常製備溶劑合物之方法係屬習知。因此,文獻例如M. Caira et al., J. Pharmaceutical Sci., 93(3), 601-611(2004)描述使用乙酸乙酯和水製備抗真菌劑“氟可那挫” (fluconazole)之溶劑合物。溶劑合物、半溶劑合物、水合物及類似者之相似製備係描述於文獻E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12(2004)和A. L. Bingham et al, Chem. Commun., 603-604(2001)。典型之非限制性方法涉及於高於周溫之溫度下溶解本發明之化合物於適量之溶劑(有機溶劑或水或彼等之混合物)中並於足以形成結晶之速率下冷卻溶液,隨後藉由標準方法分離該結晶。分析方法(諸如例如紅外光譜術)顯示於呈溶劑合物(或水合物)之結晶中存在溶劑(或水)。
本文給予之任何化學式亦欲表示化合物之未經標記和經同位素標記的形式。經同位素標記之化合物具有本文給予之化學式所示的結構,除了其中一或多個原子係經具有選擇之原子量或原子數的原子替代。可併入本發明之化合物的同位素之實例包括氫、碳、氮、氧、磷、氟、氯及碘之同位素,分別諸如
2
H、
3
H、
11
C、
13
C、
14
C、
15
N、
18
O、
17
O、
31
P、
32
P、
35
S、
18
F、
36
Cl及
125
I。該等經同位素標記之化合物可用於代謝研究(例如使用
14
C)、反應動力學研究(例如使用
2
H或
3
H)、偵測或顯像技術[諸如正電子發射電腦斷層掃描(PET)或單光子發射電腦斷層掃描攝影術(SPECT)],其包括藥物或受質組織分佈檢測或病患之放射治療。特定地,經
18
F或
11
C標記之化合物可特別適用於PET或SPECT研究。再者,經較重之同位素(諸如氘(
2
H))取代可提供因較佳之代謝安定性所導致的某些治療優異,例如增加之活體內半生期或降低之劑量需要。通常藉由實施反應圖或實施例描述之方法和下述之製備,經可方便取得之經同位素標記的反應劑替代未經同位素標記的反應劑,可製備本發明之經同位素標記之化合物。
對於本文描述之化合物,“鹽”、“溶劑合物”、“多晶型”及類似者將欲等同地應用於本發明之化合物的鏡像異構物、立體異構物、旋轉異構物、互變異構物、阻旋異構物及消旋物之鹽、溶劑合物及多晶型。
化學命名工具係軟體ChemDraw專業版16.0。
本發明之化合物的說明
本發明關於特定之分子和彼之醫藥上可接受之鹽或異構物。本發明另關於可用於調節功能失調性XPO1活性之分子和彼之醫藥上可接受之鹽、溶劑合物、酯或異構物。
本發明關於本文描述之化合物和彼之醫藥上可接受之鹽、溶劑合物、酯或異構物及包含本文描述之一或多種化合物和彼之醫藥上可接受之鹽或異構物之醫藥組成物。
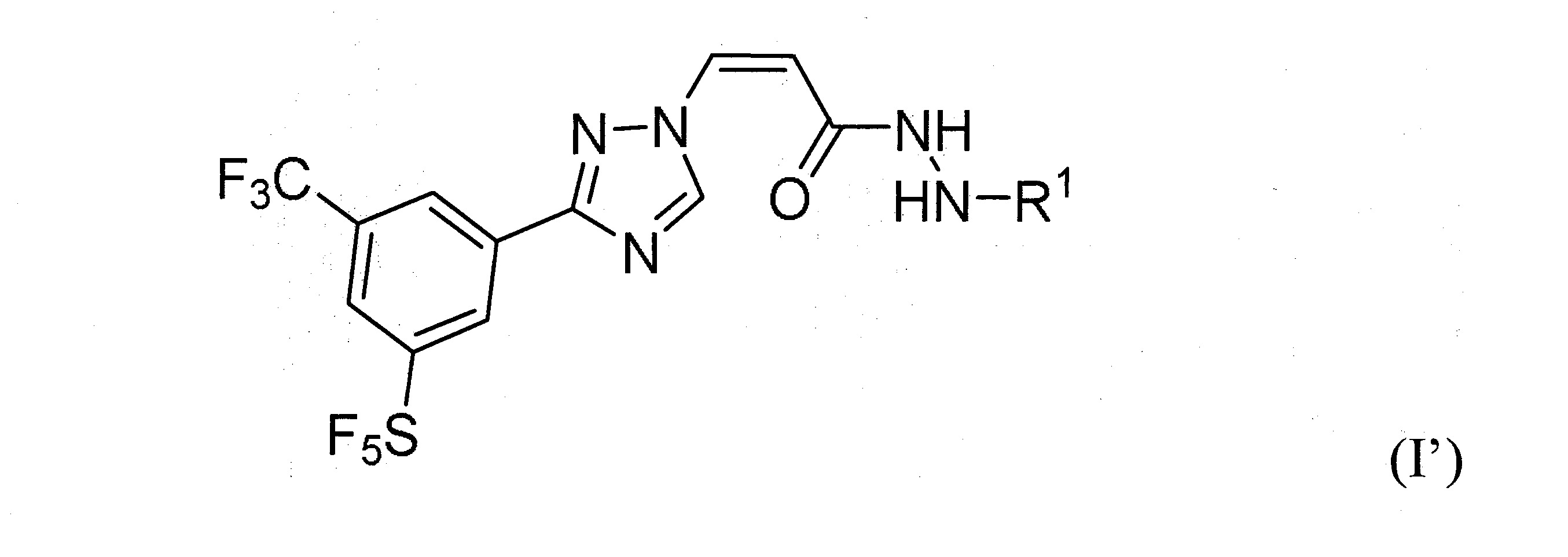
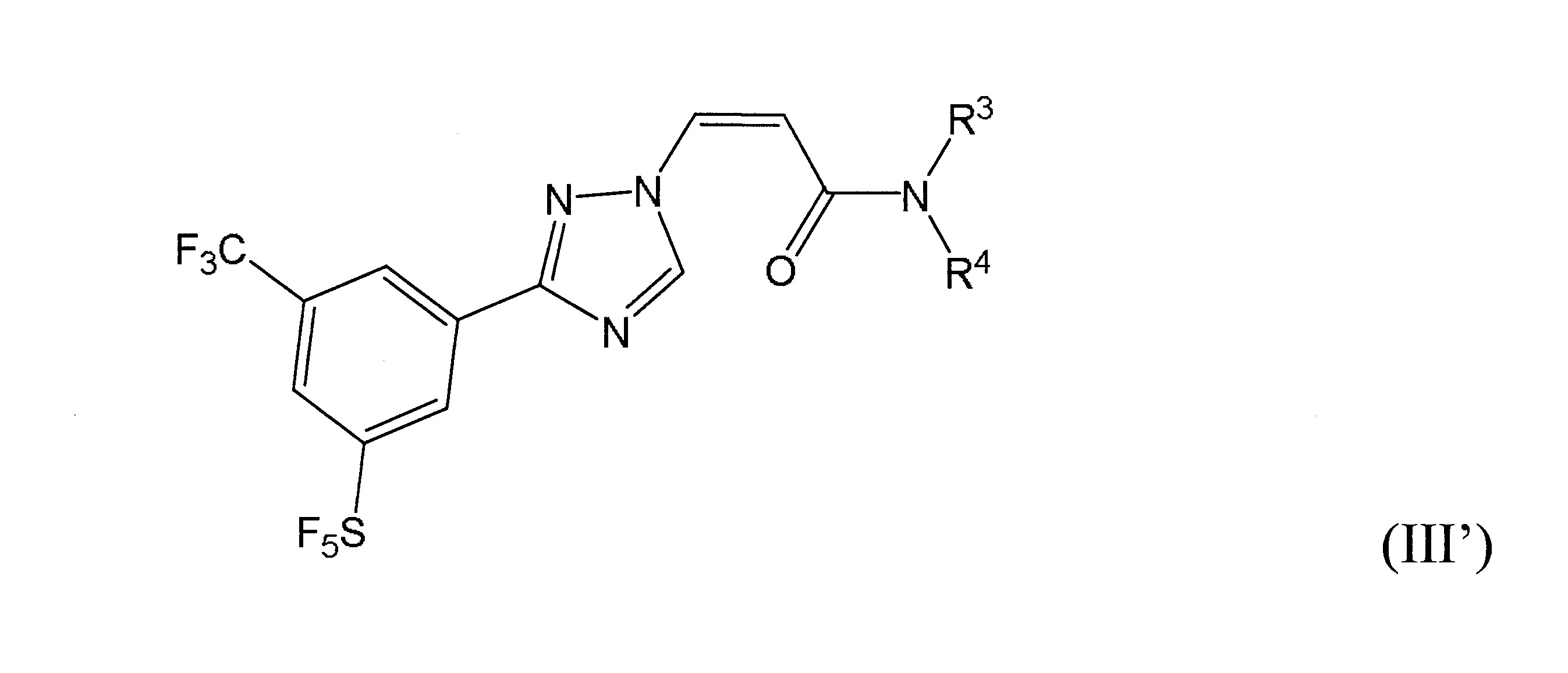
本發明之一方面提供用於調節哺乳動物之XPO1活性之化合物、組成物、套組及解毒劑,彼等含有式(I’)、式(II’)或式(III’)之結構的化合物:
或彼等之立體異構體、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中
R
1
係獨立地選自-C(=O)-R
2
、C
3-6
雜環烷基或C
5-10
雜芳基,R
1
之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、 -NO
2
、鹵素、胺基、氰基、C
1-12
烷基、C
2-12
烯基、C
2-12
炔基、C
1-12
烷氧基、C
1-12
鹵烷基、C
1-12
鹵烷氧基或C
1-12
烷硫基;且
R
2
係獨立地選自C
1-6
烷基、C
3-6
環烷基或C
3-6
雜環烷基,R
2
之任何烷基、環烷基及雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C
1-12
烷基、C
2-12
烯基、C
2-12
炔基、C
1-12
烷氧基、C
1-12
鹵烷基、C
1-12
鹵烷氧基或C
1-12
烷硫基;且
R
3
和R
4
係獨立地選自C
1-6
烷基、經取代之C
1-6
烷基、或R
3
和R
4
與彼等連接之N一起形成經取代或未經取代之C
4-10
環烷基胺基,R
3
和R
4
之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C
1-12
烷基、C
2-12
烯基、C
2-12
炔基、C
1-12
烷氧基、C
1-12
鹵烷基、C
1-12
鹵烷氧基或C
1-12
烷硫基。
於本發明之一實施態樣中,該化合物具有式(I’)結構。
於本發明之另一實施態樣中,該化合物具有式(II’)結構且含有R
1
之雙鍵的構形係E或Z。
再於本發明之另一實施態樣中,該化合物具有式(III’)結構。
再於另一實施態樣中,該化合物具有式(I’)結構且R
1
係-C(=O)-R
2
。
再於另一實施態樣中,R
2
係C
1-6
烷基。
再於另一實施態樣中,R
2
係C
3-6
環烷基。
再於另一實施態樣中,R
2
係經取代之C
1-6
烷基,其中取代基選自甲基、羥基或鹵素。
再於另一實施態樣中,R
2
係經取代之C
3-6
環烷基,其中取代基選自甲基、羥基或鹵素。
再於另一實施態樣中,R
2
係選自甲基、乙基、正丙基、異丙基、環丙基、正丁基、三級丁基、環丁基、異丁基、4-甲基-2-戊基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、環戊烯基、四氫呋喃基、
更佳地,R
2
係選自異丙基、環丙基、環丁基、4-甲基-2-戊基、三級丁基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、
最佳地,R
2
係選自三級丁基、2,2-二甲基-1-丁基、
;
可選擇地,R
2
係選自2-甲基環氧乙烷基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、正戊基、異戊基、新戊基、2,2-二甲基丁基或
。
再於另一實施態樣中,R
1
係C
3-6
雜環烷基,其中1或2個碳原子係經氮原子替代。
於某些實施態樣中,R
1
係C
5-6
雜芳基,其中1或2個碳原子係經氮原子或硫原子替代。
於某些實施態樣中,R
1
係C
6
雜芳基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自-NH
2
、-OH、鹵素或-CN。
於某些實施態樣中,R
1
係選自未經取代或經取代之
;
更佳地,R
1
係選自未經取代或經取代之
;
可選擇地,R
1
係選自未經取代或經取代之呋喃基、吡咯基、咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基或噻吩基。
於某些實施態樣中,R
1
係經取代之C
3-6
雜環烷基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自鹵素或-CN。
於某些實施態樣中,R
1
係未經取代或經取代之C
5-10
雜芳基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自鹵素或-CN。
於某些實施態樣中,R
1
係選自未經取代或經取代之;
更佳地,R
1
係未經取代或經取代之
;
可選擇地,R
1
係選自未經取代或經取代之苯基、萘基、呋喃基、苯並呋喃基、吡咯基、咪唑基、苯並咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基、噻吩基、苯並噻吩基、吲哚基、嘌呤基、喹啉基、異喹啉基或啡噁噻基。
於某些實施態樣中,該化合物係式(I’)化合物,且R
1
係-C(=O)-R
2
。
於某些實施態樣中,該化合物係式(I’)化合物,且R
1
係C
6
雜芳基,其中1或2個碳原子和相連接之氫原子係經氮原子替代。
於某些實施態樣中,R
3
和R
4
係獨立地C
1-4
烷基,或R
3
和R
4
與彼等連接之N一起形成C
4-10
環烷基胺基環。
於某些實施態樣中,R
3
和R
4
與彼等連接之N一起形成經取代之C
4-10
環烷基胺基,其中取代基係選自甲基、乙基、羥基或鹵素。
於某些實施態樣中,R
3
和R
4
結合一起形成環烷基胺基,其中C
4-10
環烷基胺基係選自
。
於某些實施態樣中,該化合物係式(III’)化合物,R
3
和R
4
與彼等連接之N一起形成經取代之環烷基胺基,其中取代基係選自甲基、羥基或鹵素,且該1或多個取代基係鹵素。
於某些實施態樣中,該化合物係選自式(I’)、式(II’)或式(III’)化合物。
於下述中,化學命名係基於ChemDraw專業版16.0。
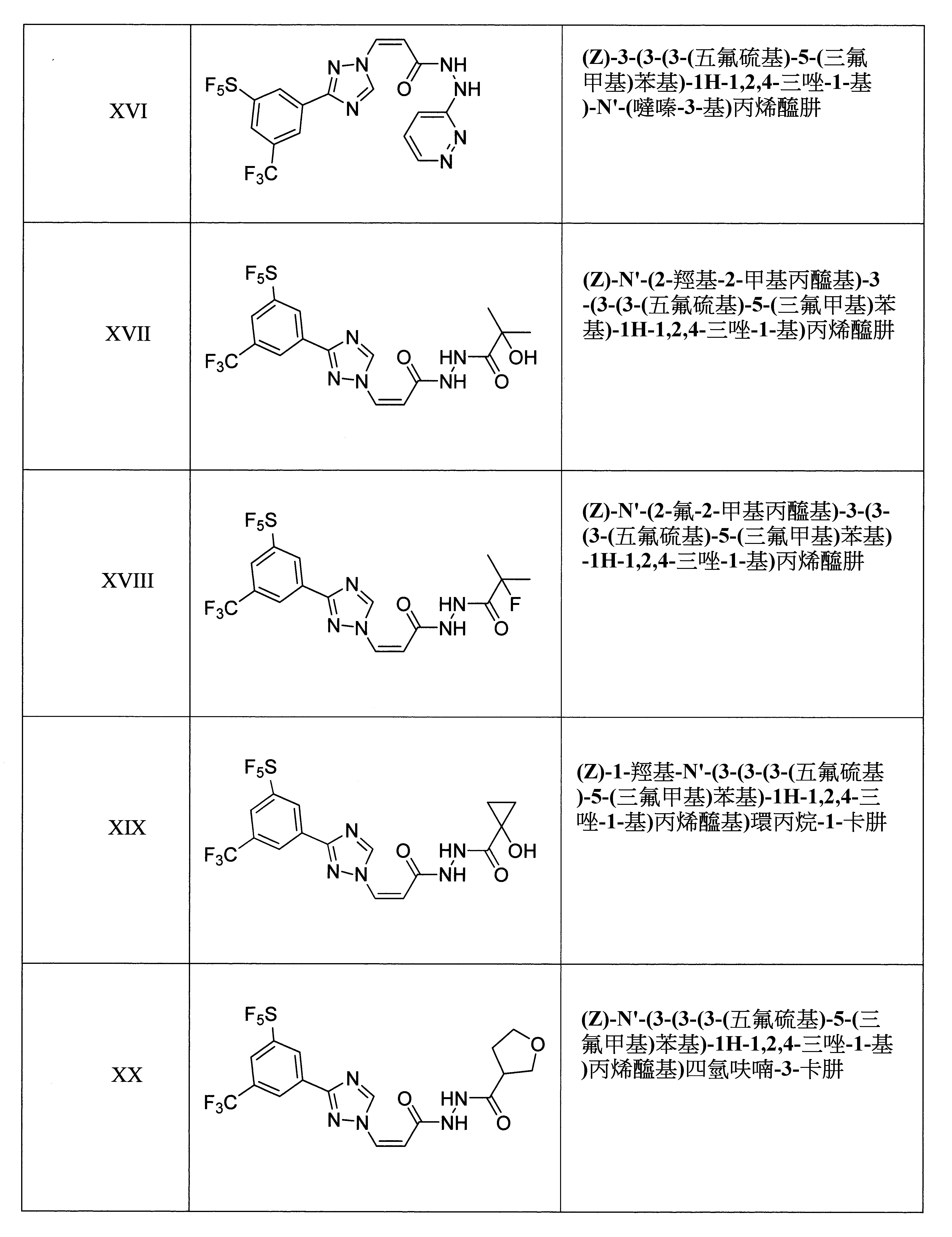
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(E)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼。
於某些實施態樣中,該化合物係(Z)-N'-異丁醯基-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼。
於某些實施態樣中,該化合物係(Z)-1-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼。
於某些實施態樣中,該化合物係(Z)-N'-乙醯基-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噠嗪-3-基)丙烯醯肼。
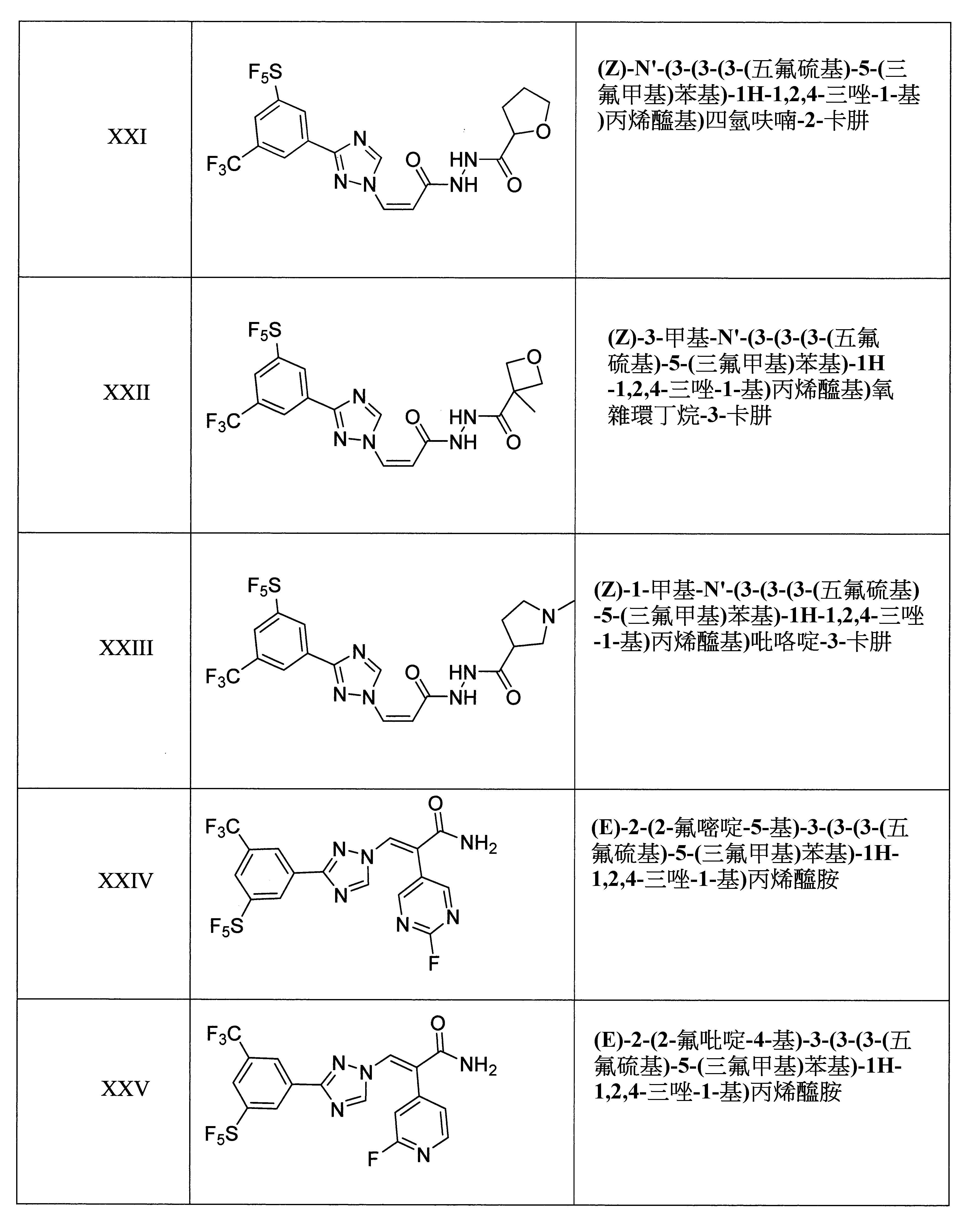
於某些實施態樣中,該化合物係(Z)-N'-(2-羥基-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-氟-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-1-羥基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)四氫呋喃-3-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)四氫呋喃-2-卡肼。
於某些實施態樣中,該化合物係(Z)-3-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)氧雜環丁烷-3-卡肼。
於某些實施態樣中,該化合物係(Z)-1-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)吡咯啶-3-卡肼。
於某些實施態樣中,該化合物係(E)-2-(2-氟嘧啶-5-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-2-(2-氟吡啶-4-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
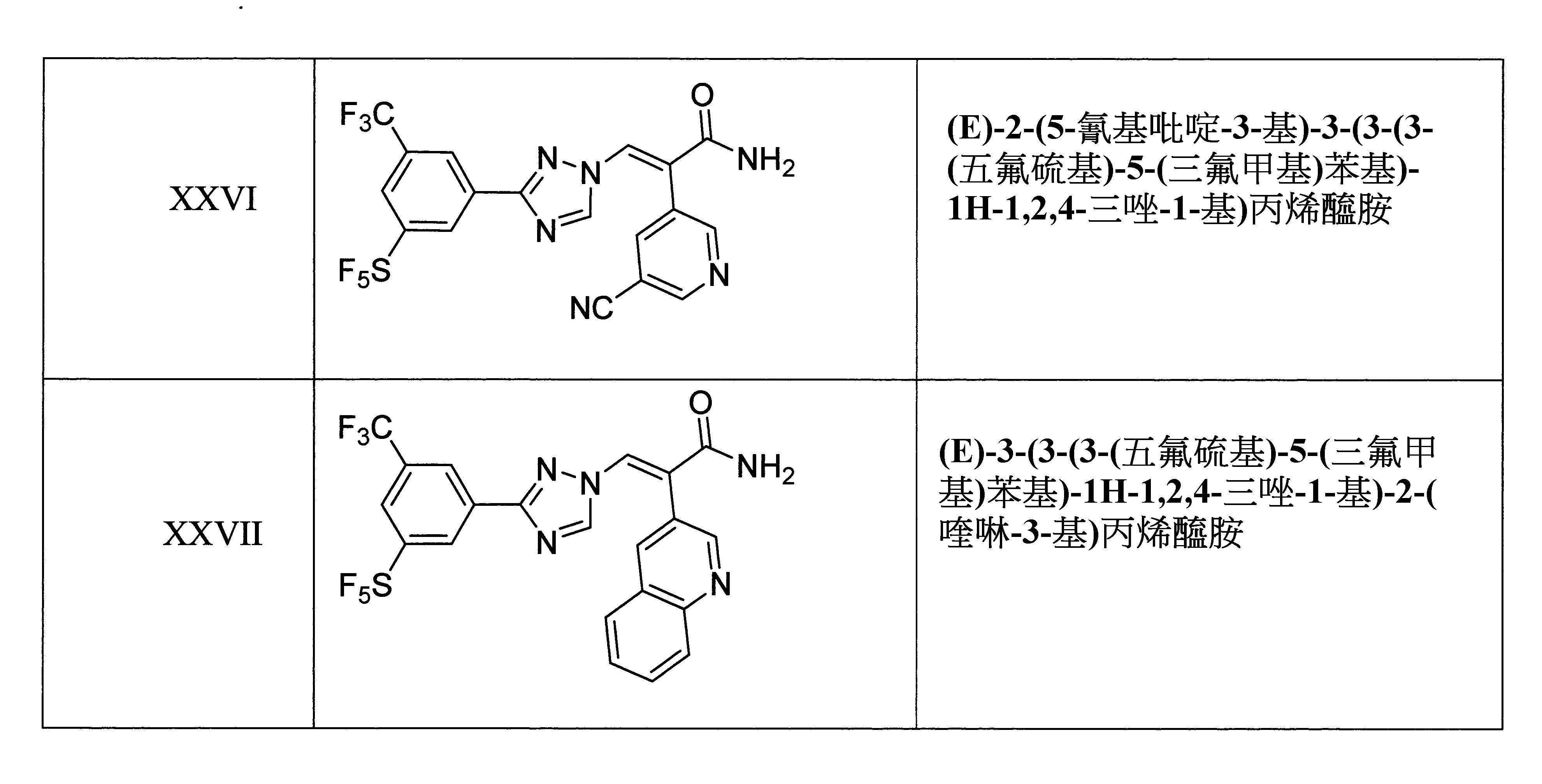
於某些實施態樣中,該化合物係(E)-2-(5-氰基吡啶-3-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(喹啉-3-基)丙烯醯胺。
本發明之另一方面提供用於調節哺乳動物之XPO1活性的化合物、組成物、套組及解毒劑,其中該化合物係選自由下列所組成之群組:




本發明之一方面關於本文揭露之化合物。
本發明之一方面關於本文揭露之化合物的製備方法。
於某些實施態樣中,提供具有通式(I’至III’)之某些化合物或上述某些化合物(I至XXVII)之製備方法,其包含下述步驟:
(a)提供式(A)化合物:
其中PG係鹵素(Cl、Br或I)、CN、N
3
、NH
2
、-COO-C
1-6
烷基、C
2-6
烯基或C
2-6
烯基-芳基;
(b)令該式(A)化合物與一或多種適當之反應劑反應以生成式(B)化合物:
(c)令步驟(b)之反應產物與式(C)化合物反應以生成式(D)化合物:
其中R
1
係如上述式(I’)、(II’)或(III’)化合物所定義者。
於某些實施態樣中,提供製備式(A)化合物之方法,其包含下述步驟:
(a)提供式(M)化合物:
(b)令該式(M)化合物與適當之反應劑反應以生成式(N)化合物:
(c)令步驟(b)之反應產物與適當之反應劑反應以生成式(J)化合物:
(d)令步驟(c)之反應產物與適當之反應劑反應以生成式(K)化合物:
。
本發明之一方面關於化合物,其係或可為功能失調性XPO1活性之調節劑。
本發明之一方面關於功能失調性XPO1活性之調節劑於製備藥物之用途,該藥物係用於治療、預防、抑制或除去腫瘤。
本發明之一方面關於功能失調性XPO1活性之調節劑於製備藥物之用途,該藥物係藉由調節病患體內之功能失調性XPO1活性以用於治療、預防、抑制或除去該病患之病症、疾病或醫療病況。
本發明亦描述一或多種合成本發明之化合物的方法。
本發明亦描述本發明之化合物的一或多種用途。
本發明亦描述本發明之化合物與輔助劑的一或多種用途,該輔助劑係諸如腫瘤壞死因子(TNF)、顆粒細胞集落刺激因子(GCSF)或其他化學治療劑。
本發明亦描述一或多種製備多種不同之醫藥組成物的方法,該等醫藥組成物包含本發明之化合物。
本發明亦描述本發明之多種不同之醫藥組成物於製備藥物之一或多種用途,該藥物係藉由調節病患體內之功能失調性XPO1活性以用於治療、預防、抑制或除去該病患之病症、疾病或醫療病況。
本發明之化合物的醫藥組成物及製備和投予
本發明提供一種包含本發明之化合物(例如實施例化合物)的醫藥組成物。依據本發明之特定實施例,該醫藥組成物可另包含醫藥上可接受之賦形劑、載體、佐劑、溶劑及彼等之組合。
本發明提供一種治療、預防或改善疾病或病症之方法,其包含投予安全且有效量之藥物的組合,該藥物含有本發明之化合物和一或多種治療活性劑。其中,該藥物的組合包含一或多種治療癌症之其他藥物。
本發明之醫藥組成物中該化合物之量係指可被有效地偵測以調節生物樣品和病患體內之功能失調性XPO1活性的量。該活性成分可以劑量投予需要該治療之個體,該劑量將於投藥途徑和治療期間提供不限於所欲之治療功效的最佳醫療功效。取決於疾病之本性和嚴重程度、病患之體重、病患依循之特定飲食、併行之藥物治療及熟習此技術之人士當能瞭解之其他因素,該劑量於不同病患之間將有所不同。依據特定之應用,製劑之單位劑量中活性化合物的量可於約1 mg至約1000 mg、較佳地約1 mg至約500 mg、更佳地約1 mg至約250 mg、甚佳地約1 mg至約50 mg之間加以變化或調整。
取決於病患之需要和欲治療之病況的嚴重程度,所使用之真實劑量可加以變化。決定對特定情況之適當劑量方案係屬此領域之技術。方便地,每日總劑量可如需要地區分為多個部分且於當天內投予該多個部分。依據主治臨床醫師之判斷,考量諸如病患之年齡、病況及大小以及欲治療之徵候的嚴重程度之諸多因素,調節投予本發明之化合物及/或彼之醫藥上可接受之鹽的量和頻率。經口服投予之典型建議的每日劑量方案的範圍可從約1 mg/天至約200 mg/天,較佳地介於10 mg/天至100 mg/天,該劑量可以1至多個劑量投予。再於另一實施態樣中,每日劑量範圍可從約1 mg/天至50 mg/病患/天。
亦將能瞭解的是,本發明之某些化合物於用於治療時可以游離形式存在,或適當地可以該等化合物的醫藥上可接受之衍生物或前藥形式存在。醫藥上可接受之衍生物包括醫藥上可接受之鹽、酯、該酯之鹽、或於投予需要治療之病患時能直接或間接提供本文描述之化合物的任何其他加成物或衍生物、或彼等之治療有效代謝物或殘基。
本發明之醫藥組成物可以批量形式製備和包裝,其中可抽取本文揭露之式(I至V)化合物的安全有效量並隨後以諸如粉末或糖漿之形式給予病患。通常,對病患投予介於0.0001至10 mg/kg體重/天之劑量以得到功能失調性XPO1活性之有效調節。可替代地,本發明之醫藥組成物可以單位劑型形式製備和包裝,其中每個形體上分開之單位含有本文揭露之式(I至V)化合物的安全有效量。當以單位劑型製備時,本發明之醫藥組成物通常含有約0.5 mg至1 g、或1 mg至700 mg、或5 mg至100 mg、或更佳地25 mg至60 mg之本發明之化合物。
當本發明之醫藥組成物亦含有除了本發明之化合物以外的一或多種其他活性成分時,本發明之化合物與第二活性成分的重量比可加以變化且取決於各別成分之有效劑量。因此,例如,當本發明之化合物與另一藥劑組合時,本發明之化合物與該另一藥劑的重量比範圍通常將從約1000:1至約1:1000,諸如約200:1至1:200。本發明之化合物與其他活性成分的組合通常亦將落入上述之範圍,但是於個別情況下,應使用該組合中每個活性成分之有效劑量。
本文使用之"醫藥上可接受之賦形劑"表示醫藥上可接受之材料、組成物或載體,其涉及賦予醫藥組成物之型式或一致性。當經混合時,每種賦形劑必須能與醫藥組成物之其他成分互溶,使得當投予病患時會實質上降低本發明之化合物的功效並將產生醫藥上不可接受之組成物的交互作用應予以避免。此外,每種賦形劑當然必須具有足夠高之純度以使呈醫藥上可接受者。
取決於所選擇之特定劑型,適當的醫藥上可接受之賦形劑可加以變化。此外,可選擇於組成物中具有特定功能之適當的醫藥上可接受之賦形劑。例如,可選擇使用某些醫藥上可接受之賦形劑以促進產製單一劑型。可選擇使用某些醫藥上可接受之賦形劑以促進產製安定劑型。可選擇使用某些醫藥上可接受之賦形劑以促進本發明之化合物當被投予給病患時自身體之一器官或部分被攜帶或轉運至身體之另一器官或部分。可選擇使用某些醫藥上可接受之賦形劑以增進病患之適應性。
適當的醫藥上可接受之賦形劑包括下述類型之賦形劑:稀釋劑、填料、結合劑、崩解劑、潤滑劑、助流劑、成粒劑、塗覆劑、潤濕劑、溶劑、共溶劑、懸浮劑、乳化劑、甜味劑、芳香劑、香味掩蓋劑、著色劑、抗結塊劑、保濕劑、螯合劑、塑化劑、黏度增加劑、抗氧化劑、防腐劑、安定劑、界面活性劑及緩衝劑。熟習此技術之人士當能瞭解某些醫藥上可接受之賦形劑可具有超過1種功能,且取決於該賦形劑存在於調製劑之量和存在於該調製劑之其他成分,該賦形劑可具有其他功能。
熟習此技術之人士所擁有之知識和技術使彼等能選擇可用於本發明之適量的適當醫藥上可接受之賦形劑。此外,熟習此技術之人士可取得許多資源,其描述醫藥上可接受之賦形劑且可用於選擇適當醫藥上可接受之賦形劑。實例包括Remington's Pharmaceutical Sciences(Mack Publishing Company)、The Handbook of Pharmaceutical Additives(Gower Publishing Limited)及The Handbook of Pharmaceutical Excipients(the American Pharmaceutical Association and the Pharmaceutical Press)。
文獻Remington: The Science and Practice of Pharmacy, 第21版,2005, D.B. Troy, Lippincott Williams及Wilkins, Philadelphia編輯、和Encyclopedia of Pharmaceutical Technology, J. Swarbrick和J. C. Boylan編輯,1988-1999, Marcel Dekker, New York揭露用於調製醫藥上可接受之組成物的多種不同之載體和製備該等載體之習知技術,該等文獻之內容倂入本文作為參考。除了當任何慣用之載體介質與本發明之化合物不相容(諸如產生任何不欲之生物功效或與該醫藥上可接受之組成物的任何其他成分進行有害之交互作用)以外,該等載體之用途屬於本發明之範疇。
使用熟習此技術之人士習知之技術和方法以製備本發明之醫藥組成物。某些此技術領域慣用之方法係描述於文獻Remington's Pharmaceutical Sciences(Mack出版公司)。
因此,本發明之另一方面關於一種製備醫藥組成物之方法。該醫藥組成物含有本發明描述之化合物和醫藥上可接受之賦形劑、載體、佐劑、載具或彼等之組合,且該方法包含混合多種不同之成分。可於例如正常周遭溫度和壓力下製備該含有本發明描述之化合物的醫藥組成物。
典型上,將本發明之化合物調製成適合藉由所欲之投藥途徑投予給病患的劑型。例如,劑型包括適合下述者:(1)口服投予,諸如片劑、膠囊、囊片、丸、錠劑、粉末、糖漿、酏劑、懸浮液、溶液、乳化液、香包及藥包;(2)非經腸投予,諸如無菌溶液、懸浮液及用於重構之粉末;(3)經皮投予,諸如經皮貼片;(4)直腸投予,諸如栓劑;(5)吸入投予,諸如氣溶膠、溶液及乾燥粉末;及(6)局部投予,諸如乳霜、軟膏、洗劑、溶液、糊、噴霧、泡沫及凝膠。
本發明之醫藥組成物可呈壓片劑、研製錠劑、咀嚼片、速溶片劑、多重壓片劑、腸溶衣片劑、糖衣片劑或膜衣片劑。腸溶衣片劑係塗覆層壓片劑,該塗覆層能抵抗胃酸之作用但可於小腸溶解或崩解並因此能於胃之酸性環境下保護活性成分。腸溶衣包括但不限於脂肪酸、脂質、水楊酸苯酯、蠟、蟲膠、氨化蟲膠及醋酸纖維素酞酸鹽。糖衣片劑係經糖塗覆之壓片劑,該糖塗覆可有益於掩蓋令人反感之味道或氣味且保護片劑免於遭受氧化。膜衣片劑係經水溶性材料之薄層或膜覆蓋的壓片劑。膜衣包括但不限於羥乙基纖維素、羧甲基纖維素鈉、聚乙二醇4000及醋酸纖維素酞酸鹽。膜衣賦予如同糖衣之一般特性。多重壓片劑係經超過一次壓縮循環所製成之壓片劑,其包括層狀片劑和壓塗或乾塗片劑。
自呈粉末、結晶或粒子形式之活性成分單獨或與一或多種本文描述之載體或賦形劑(其包括結合劑、崩解劑、控制釋出性聚合物、潤滑劑、稀釋劑及/或著色劑)組合,可製備該片劑劑型。芳香劑和甜味劑可特別用於生成咀嚼錠和片。
本發明之醫藥組成物可呈軟或硬膠囊,其可由明膠、甲基纖維素、澱粉或海藻酸鈣製備。硬明膠膠囊(亦稱為乾填充膠囊(DFC))係由兩部分組成,其中一部分滑置於另一部分上並因此完全包覆該活性成分。軟彈性膠囊(SEC)係軟球殼(諸如明膠殼),其係藉由加入甘油、山梨糖醇或類似多元醇經塑化。該軟明膠殼可含有防腐劑以防止微生物生長。適當之防腐劑係如本文描述者,其包括對羥苯甲酸甲酯和對羥苯甲酸丙酯及抗壞血酸。本文描述之液體、半固體及固體劑型可被封裝於膠囊內。適當之液體和半固體劑型包括於碳酸丙烯酯、植物油或三酸甘油酯之溶液和懸浮液。可製備含有該溶液之膠囊,如描述於美國專利號4,328,245、4,409,239及4,410,545。藉由熟習此技術之人士所習知之方法,亦可塗覆該膠囊以修改或維持該活性成分之溶解性。
本發明之醫藥組成物可呈液體和半固體劑型,其包括乳化液、溶液、懸浮液、酏劑及糖漿。乳化液係兩相系統,其中一液體係以小球形式分散於另一液體,該兩相系統可呈水包油或油包水。乳化液可包括醫藥上可接受之非水溶性液體或溶劑、乳化劑及防腐劑。懸浮液可包括醫藥上可接受之懸浮劑和防腐劑。水溶性醇溶液可包括醫藥上可接受之縮醛(諸如低碳烷基醛之二(低碳烷基)縮醛,例如乙醛二乙基縮醛)和含有1或多個羥基之與水互溶的溶劑,諸如丙二醇和乙醇。酏劑係呈澄清且經甜化之水醇溶液。糖漿係糖(例如蔗糖)之濃水溶液且可含有防腐劑。對於液體劑型,例如聚乙二醇溶液可經足量的醫藥上可接受之液體載體(例如水)稀釋以利投予時方便測量。
其他可使用之液體和半固體劑型包括但不限於含有本文描述之活性成分和二烷基化單或多烷二醇(其包括1,2-二甲氧基甲烷、二甘醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、聚乙二醇-350-二甲醚、聚乙二醇-550-二甲醚及聚乙二醇-750-二甲醚)的劑型,其中350、550及750係指該聚乙二醇之適當平均分子量。該等調製劑可進一步包含一或多種抗氧化劑,諸如2,6-二丁基羥基甲苯(BHT)、丁基羥基茴香醚(BHA)、沒食子酸丙酯、維生素E、對苯二酚、羥基香豆素、乙醇胺、卵磷脂、腦磷脂、抗壞血酸、蘋果酸、山梨糖醇、磷酸、亞硫酸氫鹽、間亞硫酸氫鈉、硫二丙酸和其酯及二硫代胺基甲酸酯。
適當地,口服投予之劑量單位調製劑可經微膠囊化。藉由例如塗覆或包埋於聚合物、蠟或類似者之粒子材料,該調製劑亦可經製備以延長或維持釋出。
供口服投予的本發明之醫藥組成物亦可呈脂質體、微胞、微球或奈米系統之型式。微胞劑型之製備可參閱美國專利號6,350,458。
本發明之醫藥組成物可呈將被重構成液體劑型之非發泡或發泡的粒子和粉末。用於該非發泡的粒子或粉末之醫藥上可接受之載體和賦形劑可包括稀釋劑、甜味劑及潤濕劑。用於該發泡的粒子或粉末之醫藥上可接受之載體和賦形劑可包括有機酸和二氧化碳來源。
著色劑和芳香劑可用於所有上述劑型。
本發明揭露之化合物亦可與作為標靶藥物載體之可溶性聚合物偶合。該等聚合物可包含經棕櫚醯基取代之聚乙烯吡咯啶酮、吡喃共聚物、聚羥基丙基甲基丙烯醯胺基酚、聚羥基乙基天冬醯胺基酚或聚乙二醇聚離胺酸。該等化合物可進一步與適合達成藥物之控制釋出的可生物降解之聚合物偶合,該等聚合物係例如聚乳酸、聚-ε-己內酯、聚羥基丁酸、聚原酸酯、聚縮醛、聚二羥基吡喃、聚氰基丙烯酸酯及水凝膠之交聯或兩性嵌段共聚物。
本發明之醫藥組成物可經調製成立即或修飾釋出之劑型,其包括延遲釋出、持續釋出、脈衝釋出、控制釋出、靶向釋出及程式化釋出之劑型。
本發明之醫藥組成物可與不會損害所欲之治療作用的其他活性成分或能補充所欲之作用的物質經共調製。
本發明之醫藥組成物可經由注射、輸注或植入之非經腸投予以供局部或系統性給藥。本文使用之非經腸投予包括靜脈內、動脈內、腹膜內、鞘內、腦室內、尿道內、胸骨內、顱內、肌內、滑膜內及皮下投予。
本發明之醫藥組成物可經調製成適合非經腸投予之任何劑型,其包括溶液、懸浮液、乳化液、微胞、脂質體、微球、奈米系統及於注射前適合液體之溶液或懸浮液的固體形式。依據熟習醫藥科學之人士習知之慣用方法(參閱如上述Remington: The Science and Practice of Pharmacy)可製備該等劑型。
用於非經腸投予之醫藥組成物可包括一或多種醫藥上可接受之載體和賦形劑,其包括但不限於水溶性載具、與水互溶之載具、非水溶性載具、抗微生物劑或抗微生物生長之防腐劑、安定劑、溶解促進劑、等張劑、緩衝劑、抗氧化劑、局部麻醉劑、懸浮劑和分散劑、潤濕劑或乳化劑、複合劑、隔離劑或螯合劑、防凍劑、凍乾保護劑、增稠劑、pH調節劑及惰性氣體。
適當之水溶性載具包括但不限於水、鹽水、生理食鹽水或磷酸鹽緩衝鹽水(PBS)、氯化鈉注射液、林格(Ringer)氏注射液、等滲葡萄糖注射液、無菌水注射液、葡萄糖和乳酸鹽之林格氏注射液。非水溶性載具包括但不限於植物來源之非揮發性油、蓖麻油、玉米油、棉籽油、橄欖油、花生油、薄荷油、紅花油、芝麻油、大豆油、氫化植物油、氫化大豆油、椰子油之中度鏈三酸甘油酯及棕櫚油。與水互溶之載具包括但不限於乙醇、1,3-丁二醇、液體聚乙二醇(例如聚乙二醇300和聚乙二醇400)、丙二醇、甘油、N-甲基-2-吡咯啶酮、N,N-二甲基乙醯胺及二甲亞碸。
適當之抗微生物劑或防腐劑包括但不限於酚、甲酚、汞劑、苄醇、氯丁醇、對羥基苯甲酸甲酯和丙酯、硫柳汞、氯化苄二烴銨(例如氯化本索寧)、對羥苯甲酸甲酯和丙酯及山梨酸。適當之等張劑包括但不限於氯化鈉、甘油及葡萄糖。適當之緩衝劑包括但不限於磷酸鹽和檸檬酸鹽。適當之抗氧化劑係如本文描述者,其包括亞硫酸氫鹽和間亞硫酸氫鈉。適當之局部麻醉劑包括但不限於普魯卡因(procaine)氫氯酸鹽。適當之懸浮劑和分散劑係如本文描述者,其包括羧甲基纖維素鈉、羥丙基甲基纖維素及聚乙烯吡咯啶酮。適當之乳化劑包括本文描述者,其包括聚氧乙烯山梨醇酐單月桂酸酯、聚氧乙烯山梨醇酐單油酸酯80及三乙醇胺油酸酯。適當之隔離劑或螯合劑包括但不限於EDTA。適當之pH調節劑包括但不限於氫氧化鈉、氫氯酸、檸檬酸及乳酸。適當之複合劑包括但不限於環糊精,其包括α-環糊精、β-環糊精、羥丙基-β-環糊精、β-環糊精磺丁醚及7-β-環糊精磺丁醚(CAPTISOL
®
, CyDex, Lenexa, Kans.)。
本發明之醫藥組成物可經調製以供單一或多重劑量投予。該單一劑量調製劑經包裝成安瓿、小瓶或針劑。該多重劑量非經腸調製劑必須含有呈抗細菌或抗真菌濃度之抗微生物劑。如此技術領域所習知且實施者,所有非經腸調製劑必須呈無菌。
於一實施態樣中,本發明之醫藥組成物係呈立即使用型無菌溶液。於另一實施態樣中,該醫藥組成物係呈無菌乾燥可溶性產物,其包括於使用前將與無菌載具重構之冷凍乾燥粉末和皮下片劑。再於另一實施態樣中,該醫藥組成物係呈立即使用型無菌懸浮液。再於另一實施態樣中,該醫藥組成物係呈於使用前將與載具重構之無菌乾燥可溶性產物。再於另一實施態樣中,該醫藥組成物係呈立即使用型無菌乳化液。
該醫藥組成物可經調製呈懸浮液、固體、半固體或觸變性液體以供植入儲存器投予。於一實施態樣中,本發明之醫藥組成物分散於固體內部基質,該固體內部基質被外部聚合性膜包圍,該外部聚合性膜不溶於體液但能允許該醫藥組成物之活性成分擴散通過。
適當之內部基質包括聚甲基丙烯酸甲酯、聚甲基丙烯酸丁酯、塑化的或未塑化的聚氯乙烯、塑化的尼龍、塑化的聚對苯二甲酸乙二酯、天然橡膠、聚異戊二烯、聚異丁烯、聚丁二烯、聚乙烯、乙烯-乙酸乙烯共聚物、矽氧橡膠、聚二甲基矽氧烷、碳酸矽氧共聚物、親水性聚合物(諸如丙烯酸和甲基丙烯酸之酯類的水凝膠)、膠原、經交聯之聚乙烯醇及經交聯之部分羥基化的聚乙酸乙烯酯。
適當之外部聚合性膜包括聚乙烯、聚丙烯、乙烯/丙烯共聚物、乙烯/丙烯酸乙酯共聚物、乙烯/乙酸乙烯共聚物、矽氧橡膠、聚二甲基矽氧烷、氯丁橡膠、氯化聚乙烯、聚氯乙烯、氯乙烯與乙酸乙烯之共聚物、二氯亞乙烯、乙烯與丙烯之離聚物、聚對苯二甲酸乙二酯、丁基橡膠、氧氯丙烷橡膠、乙烯/乙烯醇共聚物、乙烯/乙酸乙烯/乙烯醇三聚物及乙烯/乙烯氧基乙醇共聚物。
於其他方面,本發明之醫藥組成物經製備成適於藉由吸入以投予給病患之劑型,例如乾燥粉末、氣溶膠、懸浮液或溶液組成物。於一實施態樣中,本發明關於適於藉由吸入以投予給病患之呈乾燥粉末的劑型。藉由吸入以投予至肺之乾燥粉末組成物典型上包含呈微細分隔粉末的本文揭露之化合物或彼之醫藥上可接受之鹽和呈微細分隔粉末的一或多種醫藥上可接受之賦形劑。特別適用於乾燥粉末的醫藥上可接受之賦形劑係為熟習此技術之人士所習知且包括乳糖、澱粉、甘露醇及單糖、雙糖和多糖。藉由例如微粉化和碾磨可製備該微細分隔粉末。通常,大小減小(例如微粉化)之化合物可藉由約1至約10微米之D
50
值(藉由例如使用雷射粒徑分析測量)定義。
藉由懸浮或溶解本文揭露之化合物或彼之醫藥上可接受之鹽於液化推進劑可生成氣溶膠。適當之推進劑包括鹵代烴、烴及其他液化氣體。代表性推進劑包括三氯氟甲烷(推進劑11)、二氯氟甲烷(推進劑12)、二氯四氟乙烷(推進劑114)、四氟乙烷(HFA-134a)、1,1-二氟乙烷(HFA-152a)、二氟甲烷(HFA-32)、五氟乙烷(HFA-12)、七氟丙烷(HFA-227a)、全氟丙烷、全氟丁烷、全氟戊烷、丁烷、異丁烷及戊烷。典型上藉由定量噴霧劑(MDI)將包含式(A)化合物或彼之醫藥上可接受之鹽的氣溶膠投予給病患。該裝置係為熟習此技術之人士所習知。
該氣溶膠可含有典型上用於MDI之其他醫藥上可接受之賦形劑,諸如界面活性劑、潤滑劑、共溶劑及用於改善調製劑之物理安定性、改善閥性能、改善溶解性或改善氣味的其他賦形劑。
適於經皮投予之醫藥組成物可呈獨立貼片,該獨立貼片將長期維持與病患之表皮緊密接觸。例如,如文獻Pharmaceutical Research, 3(6), 318(1986)大抵描述者,藉由離子電滲自該貼片投予活性成分。
適於局部投予之醫藥組成物可經調製呈軟膏、乳霜、懸浮液、洗劑、粉末、溶液、糊、凝膠、噴霧、氣溶膠或油。使用水溶性或油性底質並添加適當之增稠劑及/或凝膠劑及/或溶劑可調製成例如軟膏、乳霜及凝膠。因此,該等適當之底質可例如包括水及/或油(諸如液體石蠟或植物油(諸如花生油或蓖麻油))或溶劑(諸如聚乙二醇)。依據該底質之本性可使用的增稠劑和凝膠劑包括軟石蠟、硬脂酸鋁、鯨蠟硬脂醇、聚乙二醇、羊毛脂、蜂蠟、聚羧乙烯和纖維素衍生物及/或單硬脂酸甘油酯及/或非離子性乳化劑。
可使用水溶性或油性底質以調製洗劑且通常洗劑亦將含有一或多種乳化劑、安定劑、分散劑、懸浮劑或增稠劑。
可藉助於任何適當之粉末底質(例如滑石、乳糖或澱粉)以形成供外部施用之粉末。可使用水溶性或非水溶性底質以調製滴劑且該滴劑亦可包含一或多種分散劑、溶解劑、懸浮劑或防腐劑。
經由每天一或多次施加至受影響之區域,可投予局部用製劑;對於皮膚表面,可方便地使用密封式敷料。經由連結性儲存系統可達成連續或延長給藥。
本發明之化合物和組成物的用途
本文描述之本發明之化合物或醫藥組成物可用於製造供治療、預防、改善或減輕個體之病症或疾病或癌症的藥物和供調節(例如阻斷)功能失調性XPO1活性的其他藥物,且本發明之化合物具有優異之藥物動力和藥效動力性質及較少之毒性副作用。
特定地,本發明之組成物的化合物的量可有效地且可偵測地調節功能失調性XPO1活性。本發明之化合物或醫藥組成物可用於預防、治療或減輕與功能失調性XPO1活性有關之疾病。
治療
於一實施態樣中,本文描述之治療包含對有需要之病患投予安全有效量的本發明之化合物或含有本發明之化合物的醫藥組成物。本文描述之每個實例包含治療上述疾病之方法,其包含對有需要之病患投予安全有效量的本發明之化合物或含有本發明之化合物的醫藥組成物。
於一實施態樣中,本發明之化合物或醫藥組成物可藉由任何適當之投予途徑投予,該投予途徑包括系統性投予和局部性投予。系統性投予包括口服投予、非經腸投予、經皮投予及直腸投予。非經腸投予係指非為經腸或經皮之投予途徑且典型上係藉由注射或輸注。非經腸投予包括靜脈內、肌內及皮下注射或輸注。局部性投予包括對皮膚投予和眼內、陰道內、吸入及鼻內投予。於一實施態樣中,本發明之化合物或醫藥組成物可經口投予。於另一實施態樣中,本發明之化合物或醫藥組成物可經吸入投予。於另一實施態樣中,本發明之化合物或醫藥組成物可經鼻內投予。
於一實施態樣中,本發明之化合物或醫藥組成物可經一次投予或依據給藥方案投予,其中於給定之期間內且於可變之時間間隔投予多個劑量。例如,每天可投予劑量1、2、3或4次。於一實施態樣中,每天投予劑量1次。於另一實施態樣中,每天投予劑量2次。可投予劑量直至達到所欲之治療功效或確定維持所欲之治療功效。本發明之化合物或醫藥組成物的適當給藥處方取決於該化合物之藥物動力性質,諸如吸收性、分佈性及代謝和去除之半生期,該等性質可由熟習此技術領域之人士測定。此外,對於本發明之化合物或醫藥組成物,包括給藥期間之適當給藥處方取決於欲治療之病症、欲治療之病症的嚴重程度、欲治療之病患的年齡和身體狀態、欲治療之病患的醫療史、同時治療之本性、所欲之治療功效及屬熟習此技術領域之人士的知識和專業之其他因素。熟習此技術領域之人士當能進一步瞭解的是:適當給藥處方可經調整以適於個別病患對該給藥處方之耐受性或個別病患歷時需要改變。
本發明之化合物可於投予一或多種其他治療劑之同時、之前或之後投予。本發明之化合物可分開投予、經相同或不同投予途徑投予或與其他藥劑之相同醫藥組成物一起投予。
對於約50至70 kg之個體,本發明之醫藥組成物或組合物可呈約1至1000 mg活性成分(較佳地約1至500 mg或約1至250 mg或約1至150 mg或約0.5至100 mg或約1至50 mg活性成分)之單位劑量。該化合物、醫藥組成物或組合物之治療有效劑量取決於個體之種類、體重、年齡、個別病況及欲治療之病症或疾病或彼之嚴重程度。熟習此技術領域之醫師、臨床醫師或獸醫當能輕易地決定用於預防、治療或抑制該病症或疾病之進展所需要的每一活性成分之有效量。
上述劑量性質可與有利地使用哺乳動物(例如小鼠、大鼠、狗及非人類之靈長類動物(諸如猴))或彼之經分離的器官、組織及製備物之活體外或活體內試驗相關連。本發明之化合物可以溶液(例如較佳地水溶液)之形式活體外投予且可以例如懸浮液或水溶液之形式活體內經局部、吸入、經腸或非經腸(有利地靜脈內)投予。
於一實施態樣中,本發明之化合物的治療有效劑量係每天約0.1 mg至約1000 mg。該醫藥組成物應提供約0.1 mg至約1000 mg該化合物之劑量。於一特定實施態樣中,製備醫藥單位劑型以提供每單位劑型約1 mg至約1000 mg、約10 mg至約500 mg、約20 mg至約200 mg、約25 mg至約100 mg或約30 mg至約60 mg之該活性成分或必要成分之組合。於一特定實施態樣中,製備醫藥單位劑型以提供約1 mg、5 mg、10 mg、20 mg、25 mg、50 mg、100 mg、250 mg、500 mg或1000 mg之該活性成分。
本發明之較佳實施態樣
一般合成方法
提供下述實施例以更完整地瞭解本發明。然而,應瞭解的是該等實施態樣僅提供實施本發明之方法,且本發明不限於該等實施態樣。
通常,本發明之化合物可藉由本文描述之方法製備,其中除非另有說明,取代基係如上述式(I’至III’)所定義者。下述非限制性反應圖和實施例係進一步例示說明本發明。
熟習此技術領域之人士當能瞭解,所描述之化學反應可方便地適於製備本文揭露之許多其他化合物,且製備本文揭露之化合物的其他方法係屬本文揭露之範圍。熟習此技術領域之人士當能瞭解,如下述實施例所揭露者,起始物可經變化且可使用額外之步驟以製備本發明所涵蓋之化合物。於某些情況下,為達成某些上述之轉化反應,保護某些反應官能基可能是必要的。通常,保護基團之需要及連接和除去該基團之必要條件係為熟習有機化學之人士所習知。例如,藉由熟習此技術之人士所習知之修改,例如藉由適當地保護干擾基團、使用非本文描述之此技術領域習知的其他適當反應劑及/或進行反應條件之慣常修改,可成功地進行本發明之非例示性化合物的合成。可替代地,習知之反應條件或本發明揭露之反應將可被應用於製備本發明揭露之其他化合物。
於下述實施例中,除非另有說明,所有溫度係為攝氏(℃)溫度。反應劑係購自於供應商,諸如Aldrich Chemical Company、Arco Chemical Company及Alfa Chemical Company,且除非另有說明,可未經進一步純化而使用。
化合物之製備
使用任何習知之有機合成技術可製備本發明之化合物(包括彼之鹽、酯、水合物或溶劑合物)且依據多種可能之合成途徑的任一者可合成本發明之化合物(包括彼之鹽、酯、水合物或溶劑合物)。
於適當之溶劑中可實施製備本發明之化合物的反應,且熟習有機化學之人士可輕易地選擇該溶劑。於實施該反應之溫度(例如可為介於溶劑之冷凍溫度至沸騰溫度之間的溫度)下,適當之溶劑可實質上不與起始物(反應物)、中間體或產物反應。給定之反應可於1種溶劑或超過1種溶劑之混合物中實施。取決於特定反應步驟,熟習此技術領域之人士當能選擇用於該特定反應步驟之適當溶劑。
依據此技術領域習知之任何適當方法可監測反應。例如,藉由光譜法(諸如核磁共振光譜術(例如
1
H或
13
C)、紅外光譜術、分光光譜術(例如UV-可見光)或質譜術)或藉由層析法(諸如高效液相層析(HPLC)、液相層析-質譜(LCMS)或薄層層析(TLC)),可監測產物生成。熟習此技術之人士可藉由多種不同之方法以純化化合物,該多種不同之方法包括高效液相層析(HPLC)(參閱文獻“Preparative LC-MS Purification: Improved Compound Specific Method Optimization” Karl F. Blom, Brian Glass, Richard Sparks, Andrew P. Combs J. Combi. Chem. 2004, 6(6), 874-883,彼之全部內容倂入本文作為參考)和正相矽膠層析。
使用下述之方法和合成有機化學領域所習知之合成方法或熟習此技術之人士能瞭解之變化/修改,可合成本發明之化合物。較佳之方法包括但不限於下述之方法。特定地,藉由依循如下所示之例示性一般合成反應圖所示的步驟,可合成本發明之式(A)化合物,且包括於該合成反應圖之反應物的簡稱或反應物的化學基團係定義於實施例。
反應圖1:使用下述反應順序以合成結構A1化合物
依據文獻(1. Chemistry-A European Journal,2012, 18, 10234-10238; 2. WO2013/19548; 3. Bioorganic & Medicinal Chemistry, 2008, 16, 9487-9497; 4. Organic Letter, 2014, 16, 4268-4271)揭露之相關方法但不限於該等揭露之方法可進行合成結構1A化合物之合成。其中PG係獨立地選自鹵素(Cl、Br、I)、CN、N
3
、NH
2
、-COO-C
1-6
烷基、C
2-6
烯基或C
2-6
烯基-芳基。硝基化合物1-1經NBS溴化並隨後經鐵還原以生成中間體1-2,該中間體1-2進一步經氰化和碘化以生成中間體1-3。隨後將三氟甲基加至芳基環上以生成重要之中間體化合物A1。
反應圖2:使用下述反應順序以合成結構A2化合物
另有可替代之方法以合成化合物A2。依據文獻(1. Chemistry-A European Journal,2012, 18, 10234-10238; 2. WO2013/19548; 3. Bioorganic & Medicinal Chemistry, 2008, 16, 9487-9497; 4. Organic Letter, 2014, 16, 4268-4271)揭露之相關方法但不限於該等揭露之方法,可進行合成結構A2化合物。硝基化合物2-1經NBS溴化並隨後經鐵還原以生成中間體2-2,該中間體2-2進一步經氰化和碘化以生成化合物2-3。該化合物2-3於酸條件下經環化以生成三唑化合物2-4,且使用三苯基甲基氯(TrtCl)以保護該三唑之胺基以生成安定之化合物2-5。使用經銅催化之三氟甲基化方法以導入三氟甲基以生成化合物2-6。隨後,令該化合物2-6與烯烴共軛並經醯胺化以得到終產物A2。
反應圖3:使用下述反應順序以合成結構A3化合物

依據文獻(1. WO2014205393; 2. Chemistry-A European Journal, 2012, 18, 1914-1917; 3. Journal of Medicinal Chemistry, 1995, 38, 3287-3296)揭露之相關方法但不限於該等揭露之方法,可進行合成結構A3化合物。自反應圖1合成化合物3-1,依循順序步驟以得到烯烴化合物3-2並隨後得到二溴化物化合物3-3,且隨後經除去1個溴以得到化合物3-4。於鈴木(Suzuki)反應條件下,可合成多種化合物且經
醯胺化以生成終產物A3。
例示之化合物的製備和特性
經由不同之反應圖可製備本發明之化合物。經由不同之反應圖合成10個例示化合物的詳細製備方法係說明如下且亦列示特性結果。
除非另有說明,所有反應劑係購自供應商且未經進一步純化。如為必要,使用藉由標準方法進行乾燥之溶劑。用於薄層層析(TLC)之板係預先經E. Merck矽膠60F254(厚度0.24 nm)塗覆之鋁板,其隨後可見於UV光(365 nm和254 nm)下或可見於經5%十二鉬磷酸(dodecamolybdophosphoric acid)之乙醇溶液染色且隨後經加熱。使用購自供應商之矽膠(200至400篩孔)進行管柱層析。室溫下使用Agilent 400-MR NMR光譜儀(400.00 MHz;1小時)記錄
1
H NMR光譜。使用溶劑訊號作為
1
H NMR參考值(CDCl
3
,7.26 ppm;CD
3
OD,3.31 ppm;DMSO-d6,2.50 ppm;D
2
O,4.79 ppm)。使用下列簡稱以說明多重性:s=單線;d=雙線;t=三線;q=四線;br. s.=寬單線;dd=雙重雙線;td=三重雙線;dt=重雙三線;dq=重雙四線;m=多重線。實驗細節使用之其他簡稱說明如下:δ=低磁場下自四甲基矽烷之化學位移(每百萬分);Ar=芳基;Ac=醯基;Boc=三級丁氧羰基;Bn=苄基;DCM=二氯甲烷;DMF=N,N’-二甲基甲醯胺;DIPEA=二異丙基乙胺;DMAP=4-(二甲基胺基)吡啶;DMSO=二甲亞碸;EA=乙酸乙酯;Et=乙基;Me=甲基;Hz=赫茲(hertz);HPLC=高效液相層析;J=偶合常數(於NMR中);min=分鐘;h=小時;NMR=核磁共振;prep=製備性;t-Bu=三級丁基;iPr=異丙基;TBAF=氟化四丁基銨;tert=三級芳基;TFA=三氟乙酸;THF=四氫呋喃;TLC=薄層層析。
實施例
應瞭解下述詳細說明之本發明之實施態樣僅為例示性說明本發明且不應解釋為限制本發明。未有特殊技術或條件之實施例可依據此技術領域之文件描述的技術或條件或依據產品之指示說明加以實施。藉由例行性購置以取得反應劑或無製造商之儀器。熟習此技術之人士當能瞭解:如示於下述實施例,起始物可經變化且可使用額外之步驟以產製本發明之化合物。
實施例1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼(I)
步驟1:合成丙炔酸異丙酯(2)
於25℃下對丙炔酸1(30 g,0.43莫耳)於異丙醇(100 ml)之混合物加入BF
3
乙醚絡合物(122.1 g,0.86莫耳)。經攪拌10分鐘後,令反應混合物經加熱至90℃並經攪拌2小時。藉由TLC監測反應完全。令反應混合物降低至25至30℃並經碎冰驟冷且隨後經二氯甲烷(2×150 ml)萃取。令有機層先後經水和鹽水溶液沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以生成該丙炔酸異丙酯(35.2 g,73%產率)。
步驟2:合成(Z)-3-碘丙烯酸異丙酯(3)
於25℃下將丙炔酸異丙酯2(35.2 g,0.31毫莫耳)加入至乙酸(200 ml)並令反應混合物經攪拌10分鐘。於攪拌下加入碘化鈉(70.0 g,0.47毫莫耳)(觀察到暗褐色)。提高溫度至110℃並維持反應物於該溫度下達2小時。藉由TLC監測反應完全。令反應混合物冷卻至室溫,經冰冷水(200 ml)驟冷且經攪拌30分鐘。將甲基三級丁基醚(MTBE)(200 ml)加入至該反應混合物並再經攪拌30分鐘。分離有機層並令水層再經MTBE(200 ml)萃取。令結合之有機層經NaHCO
3
(2×100 ml)、NaHSO
3
(2×100 ml)及鹽水(100 ml)沖洗,置於硫酸鈉上乾燥並於35℃下經真空下濃縮以生成呈淡黃色油之(Z)-3-碘丙烯酸異丙酯(48.5 g,61%產率)。
1
H NMR(400 MHz, CDC1
3
)δ 7.38(d, J=8.0 Hz, 1 H), 6.83(d, J=8.0 Hz, 1 H), 5.07-5.13(m, 1 H), 1.27(d, J=8.0 Hz, 6 H)。
步驟3:合成(3-溴-5-硝基苯基)五氟硫烷(5)
將化合物4(9.25 g,37.1毫莫耳)、TFA(20 ml)及濃H
2
SO
4
(100 ml)加入至圓底燒瓶(250 ml),隨後令混合物經劇烈攪拌並經30分鐘分批加入NBS(9.92 g,55.7毫莫耳)且令反應物於25℃下經攪拌12小時。將混合物倒入至冰水並經EA(3×100 ml)萃取。令結合之有機層經飽和NaHCO
3
(3×100 ml)和水(3×100 ml)沖洗,置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=25:1)純化以得到呈淡黃色油之標的化合物5(12.04 g,99%產率)。
1
H NMR(400 MHz, CDC1
3
)δ 8.57(s, 1 H), 8.55(s, 1 H), 8.22(s, 1 H)。
步驟4:合成3-溴-5-(五氟硫基)苯胺(6)
於25℃下對化合物5(5.9 g,18.0毫莫耳)於MeOH(40 ml)和水(10 ml)之溶液加入Fe(5.0 g,90毫莫耳)和NH
4
Cl(4.8 g,90毫莫耳)。令混合物於90℃下經攪拌2小時並隨後經過濾,經MeOH(20 ml)沖洗且除去溶劑。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機相置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=10:1)純化以得到呈淡黃色油之標的化合物6(5.1 g,96%產率)。
1
H NMR(400 MHz, CDC1
3
)δ 7.35(s, 1 H), 7.20(s, 1 H), 6.99(s, 1 H)。
步驟5:合成3-胺基-5-(五氟硫基)苄腈(7)
對圓底燒瓶(250 ml)加入化合物6(8.68 g,29.2毫莫耳)和NMP(100 ml)並隨後加入CuCN(5.26 g,58.5毫莫耳)且令反應物於180℃和N
2
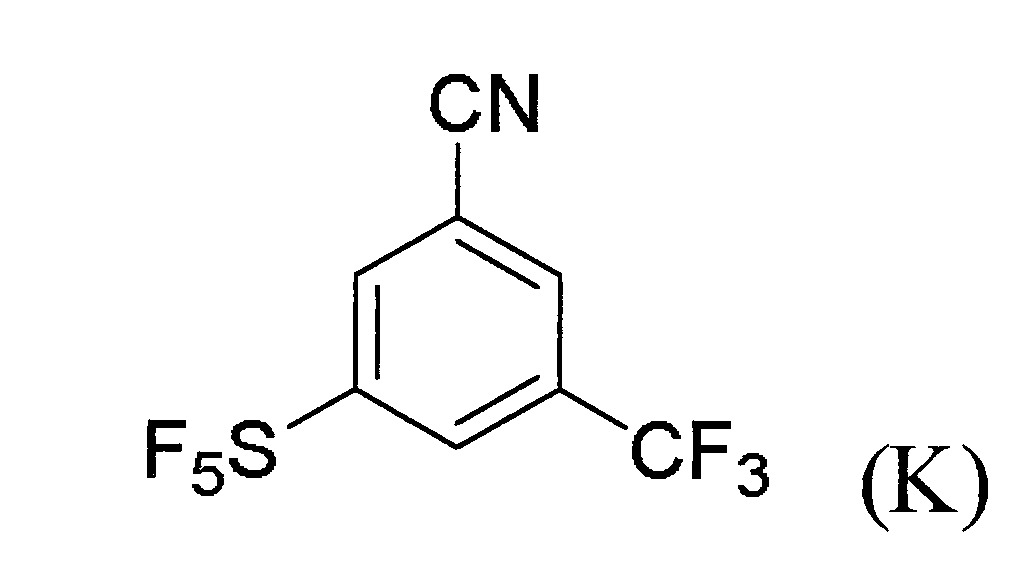
氛圍下經攪拌6小時。令混合物經冷卻至25℃並經過濾。對濾液加入EA(200 ml)並經水(3×200 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=10:1)純化以得到呈淡黃色固體之標的化合物7(3.3 g,46%產率)。
步驟6:合成3-碘-5-(五氟硫基)苄腈(8)
令化合物7(4.23 g,17.3毫莫耳)懸浮於濃H
2
SO
4
(8.7 ml)與水(17.3 ml)之混合物並經冷卻至0℃。經1小時加入NaNO
2
(1.23 g,17.9毫莫耳)之水(3.5 ml)溶液並令所生成之混合物於0℃下再經攪拌1小時。隨後經1小時逐滴加入CuI(173 mg,0.87毫莫耳)和KI(3.05 g,18毫莫耳)之水(3.5 ml)溶液並隨後令混合物於25℃下經攪拌10小時。對該混合物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈淡黃色固體之標的化合物8(3.65 g,59%產率)。
1
H NMR(400 MHz, CDC1
3
)δ 8.28(s, 1 H), 8.13(s, 1 H), 8.01(s, 1 H)。
步驟7:合成3-碘-5-(五氟硫基)苯硫代甲醯胺(benzothioamide) (9)
對化合物8(3.6 g,10毫莫耳)之DMF(50 ml)溶液加入NaHS(1.1 g,20毫莫耳)和MgCl
2 .
6H
2
O(2 g,10毫莫耳)並令混合物於25℃下經攪拌2小時。對該混合物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以得到粗製產物9(3.8 g,98%產率),其未經任何純化而用於下一個步驟。
MS(ESI): [M+H
+
]=389.9。
步驟8:合成3-(3-碘-5-(五氟硫基)苯基)-1H-1,2,4-三唑(10)
對化合物9(3.32 g,8.53毫莫耳)之DMF(15 ml)溶液加入N
2
H
4 .
H
2
O(853 mg,17.1毫莫耳)並令混合物於25℃下經攪拌3小時。隨後加入甲酸(10 ml)並令混合物於90℃下經攪拌3小時。令反應物經冷卻至25℃並經飽和NaHCO
3
(50 ml)驟冷且經EA(50 ml)萃取。令有機層經鹽水(50 ml)沖洗,置於硫酸鈉上乾燥並經真空下濃縮以得到粗製產物10(3.36 g,99%產率),其未經進一步純化而用於下一個步驟。
MS(ESI): [M+H
+
]=397.9。
步驟9:合成3-(3-碘-5-(五氟硫基)苯基)-1-三苯甲基-1H-1,2,4-三唑(11)
對化合物10(3.37 g,8.53毫莫耳)之DCM(50 ml)溶液加入Et
3
N(1.29 g,12.8毫莫耳)和TrtCl(3.57 g,12.8毫莫耳)並令混合物於25℃下經攪拌2小時且隨後經水(3×50 ml)沖洗。令有機層經乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為DCM:MeOH=50:1)純化以得到呈淡黃色固體之標的化合物11(5.36 g,98%產率)。
1
H NMR(400 MHz, CDCl
3
)δ 8.57(s, 1 H), 8.46(s, 1 H), 8.05(s, 1 H), 8.01(s, 1 H), 7.37-7.15(m, 15 H)。
步驟10:合成3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1-三苯甲基-1H-1,2,4-三唑(12)
對三頸燒瓶(100 ml)載入化合物11(5.36 g,8.4毫莫耳)、KF(1.46 g,25.2毫莫耳)、CuI(320 mg,1.7毫莫耳)及1,10-啡啉(306 mg,1.7毫莫耳)並隨後於氮氛圍下對該燒瓶內粗製材料抽真空3次。隨後將DMSO(20 ml)與DMF(10 ml)之混合溶液、TMSCF
3
(4.76 g,33.6毫莫耳)及B(OMe)
3
(2.55 g,25.2毫莫耳)加入至上述反應溶液並隨後令反應物於70℃和N
2
氛圍下經攪拌24小時。令混合物經冷卻至25℃並經過濾。對濾液加入EA(100 ml)並經水(3×50 ml)和鹽水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以得到呈黑色固體之標的化合物12(4.88 g,99%產率),其未經任何純化而用於下一個步驟。
19
F NMR(400 MHz, CDCl
3
)δ 62.45 ppm, -62.79 ppm。
步驟11:合成3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑(13)
對化合物12(4.88 g,8.4毫莫耳)之DCM(50 ml)溶液加入TFA(1.67 g,14.3毫莫耳)和Et
3
SiH(1.95 g,16.8毫莫耳),令混合物於25℃下經攪拌1小時且隨後於真空下經濃縮以除去該有機溶液。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=1:1)純化以得到呈淡黃色固體之標的化合物13(2.1 g,74%產率)。
MS(ESI): [M+H
+
]=340.1。
步驟12:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(14)
對化合物13(2.1 g,6.2毫莫耳)之DMF(15 ml)溶液加入DABCO(1.39 g,12.4毫莫耳)和化合物3(2.08 g,8.7毫莫耳),令混合物於25℃下經攪拌3小時且隨後經飽和NH
4
Cl (50 ml)驟冷並經EA(100 ml)萃取。令有機層經鹽水(3×50 ml)沖洗,置於硫酸鈉上乾燥且經過濾和真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=30:1)純化以得到呈白色固體之標的化合物14(2.09 g,75%產率)。
MS(ESI): [M+H
+
]=452.1。
1
H NMR(400 MHz, CDCl
3
)δ 8.64(s, 1 H), 8.51(s, 1 H), 8.34(s, 1 H), 8.11(s, 1 H), 7.96(d, J=14.1 Hz, 1 H), 7.69(d, J=13.7 Hz, 1 H), 5.15(m, 1 H), 1.32(d, J=6.3 Hz, 6 H)。
步驟13:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸(15)
對化合物14(2.09 g,4.63毫莫耳)之THF(20 ml)溶液加入LiOH
.
H
2
O(3.34 g,55.6毫莫耳,1N,56 ml H
2
O),令混合物於25℃下經攪拌3小時且隨後經1N HCl酸化至pH=3並經EA(3×100 ml)萃取。令有機層經飽和NaHCO
3
(50 ml)沖洗,置於硫酸鈉上乾燥且經真空下濃縮以得到呈白色固體之標的化合物15(1.89 g,99%產率)。
MS(ESI): [M+H
+
]=410.0。
步驟14:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼(I)
對化合物15(1.01 g,2.5毫莫耳)、化合物16(826 mg,7.5毫莫耳)、DIEA(1.29 g,10毫莫耳)之EA/DCM(10 ml/10 ml,v/v)溶液加入T
3
P(6.36 g,10毫莫耳,50% EA溶液)。令混合物於-40℃下經攪拌3小時且隨後令有機溶液經真空下濃縮。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機相經濃縮並經製備性TLC(DCM:MeOH:NH
3 .
H
2
O= 15:1:0.1)純化以得到呈黃色固體之標的化合物I(594 mg,48%產率)。
MS(ESI): [M+H
+
]=502.1。
1
H NMR(400 MHz, DMSO-d6)δ 10.51(s, 1 H), 9.56(s, 1 H), 9.11(s, 1 H), 8.61(s, 1 H), 8.48(s, 1 H), 8.39(s, 1 H),8.07(s, 2 H), 7.90(s, 1 H), 7.50(d, J=10 Hz, 1 H), 6.06(d, J=10.4 Hz, 1 H)。
實施例2:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼(II)
步驟1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼(II)
對化合物15(40 mg,0.1毫莫耳)、2-肼基吡啶(32 mg,0.3毫莫耳)及DIEA(52 mg,0.4毫莫耳)於EA:DCM(1 ml:1 ml)之溶液加入T
3
P(190 mg,0.3毫莫耳,50% EA溶液)。令混合物於-40℃下經攪拌3小時並隨後令有機溶液經真空下濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機相經濃縮並經製備性TLC(DCM:MeOH:NH
3 .
H
2
O= 15:1:0.1)純化以得到呈黃色固體之標的化合物II(23 mg,46%產率)。隨後與HCl/二噁烷溶液生成產物以得到固體產物。
MS(ESI): [M+H
+
]=501.1。
1
H NMR(400 MHz, DMSO-d6)δ 11.17(s, 1 H), 9.52(s, 1 H), 8.62(s, 1 H), 8.49(s, 1 H), 8.41(s, 1 H), 8.02(m, 2 H), 7.63(d, J=10.4 Hz, 1 H), 7.20(m, 1 H), 7.04(m, 1 H), 6.12(d, J=10.5 Hz, 1 H)。
實施例3:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼(III)
步驟1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸(15)
於0℃下對化合物16(150 mg,0.33毫莫耳)之乾燥THF(15 ml)攪拌溶液逐滴加入LiOH(2.6 ml,2.64毫莫耳,1M)溶液。令反應物於25℃下經攪拌3小時。隨後令反應混合物經HCl(1M)驟冷直至pH=2至3並經EA(3×20 ml)萃取。令有機層經鹽水(40 ml)沖洗,置於硫酸鈉上乾燥且經濃縮以生成呈白色固體之化合物15(125 mg,92%產率)。
MS(ESI): [M+H]
+
=409.9。
步驟2:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼(III)
令化合物15(125 mg,0.31毫莫耳)之DCM:EA(5 ml:5 ml,v/v)攪拌溶液冷卻至-78℃並於0℃下加入三甲基乙醯基醯肼(54.0 mg,0.46毫莫耳)、DIEA(80.0 mg,0.62毫莫耳)及T
3
P(395 mg,0.62毫莫耳,50重量% EA溶液)。令反應物於25℃下經攪拌3小時並隨後經飽和氯化銨(20 ml)驟冷。令混合物經DCM萃取並令有機層經鹽水沖洗,置於硫酸鈉上乾燥且經真空下濃縮以生成粗製白色固體。將該固體加入至MeCN(5 ml)並於25℃下經攪拌2小時且隨後經過濾,經MeCN(10 ml)沖洗以生成呈白色固體之標的產物III(110 mg,69%產率)。
MS(ESI): [M+H]
+
=506.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.41(s, 1 H), 9.66(d, J=8 Hz, 2 H), 8.66(s, 1 H), 8.56(s, 1 H), 8.43(s, 1 H), 7.49(d, J=10.8 Hz, 1 H), 6.04(d, J=10.4 Hz, 1 H), 1.17(s, 9 H)。
實施例4:(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(IV)
步驟1:合成2,3-二溴-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙酸異丙酯(20)
於0℃下對化合物16(9.0 g,20毫莫耳)之DCM(60 ml)溶液加入Br
2
(6.31 g,40毫莫耳)並隨後令混合物於25℃下經攪拌10小時。令該混合物經水(3×60 ml)和飽和Na
2
S
2
O
3
(60 ml)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈黃色固體之化合物20(12.2 g,99%產率)。
MS(ESI): [M+H]
+
=611.9。
步驟2:合成(Z)-2-溴-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(21)
於-20℃下對化合物20(12.2 g,20毫莫耳)之THF(120 ml)攪拌溶液逐滴加入氫氧化鋰水合物(13 ml,40毫莫耳)水溶液(3M)。令反應物於-20℃下經攪拌2.5小時。隨後令反應混合物經HCl(1M)驟冷直至pH=4並經EA(200 ml)萃取。令有機層經水(3×100 ml)和鹽水(3×100 ml)沖洗,置於硫酸鈉上乾燥且經過濾和濃縮以生成呈黃色固體之粗製產物21(10.6 g,99%產率)。
MS(ESI): [M+H]
+
=529.9。
1
H NMR(400 MHz, CDCl
3
)δ 9.47(s, 1 H), 8.76(s, 1 H), 8.75(s, 1 H), 8.59(s, 1 H), 8.07(s, 1 H), 5.25-5.19(m, 1 H), 1.39(d, J=8 Hz, 6 H)。
步驟3:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(22)
於25℃和氮氛圍下對化合物21(1.06 g,2毫莫耳)和嘧啶-5-基亞硼酸(boronic acid)(397 mg,3.2毫莫耳)之二噁烷:水(10 ml:3 ml)溶液加入Pd(PPh
3
)
4
(231 mg,0.2毫莫耳)和Cs
2
CO
3
(1.3 g,4毫莫耳)。令反應混合物於85℃和氮氛圍下經攪拌4小時。冷卻至25℃並令有機溶液經濃縮。對殘餘物加入EA(200 ml)並隨後經水(3×100 ml)沖洗,令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=2:1)純化以得到呈白色固體之化合物22(603 mg,57%產率)。
MS(ESI): [M+H]
+
=530.1。
1
H NMR(400MHz, DMSO-d6)δ 9.26(s, 1 H), 9.19(s, 1 H), 8.78(s, 2 H), 8.68(s, 1 H), 8.38(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 5.13-5.07(m, 1 H), 1.27(d, J=8 Hz, 6 H)。
步驟4:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸(23)
於0℃下對化合物22(1.16 g,2.2毫莫耳)之DCM(100 ml)溶液加入AlCl
3(
1.2 g,8.8毫莫耳)。令反應混合物於25℃下經攪拌2.5小時並隨後回溫至30℃且於該溫度下經攪拌0.5小時。令反應物經水(15 ml)驟冷並令有機溶液經濃縮。對殘餘物加入EA(100 ml)並隨後經水(3×100 ml)和HCl溶液(30 ml,1N)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(DCM:MeOH:AcOH=10:1:0.1,v/v)純化以得到呈白色固體之化合物23(684 mg,64%產率)。
MS(ESI): [M+H]
+
=487.9。
步驟5:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(IV)
於0℃下對化合物23(684 mg,1.4毫莫耳)之乾燥THF(30 ml)攪拌溶液逐滴加入NMM(283 mg,2.8毫莫耳)和氯甲酸異丙酯(346 mg,2.8毫莫耳)。令反應物於0℃下經攪拌20分鐘。隨後於0℃下對混合物加入NH
4
OH(294 mg,8.4毫莫耳)溶液,令該混合物於25℃下再經攪拌5分鐘,隨後經水(50 ml)驟冷且令有機溶液經濃縮。對殘餘物加入EA(100 ml)並隨後經水(3×100 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為EA)純化以得到呈白色固體之化合物IV(480 mg,71%產率)。
MS(ESI): [M+H]
+
=486.9。
1
H NMR(400 MHz, DMSO-d6)δ 9.19(s, 1 H), 9.14(s, 1 H), 8.72(s, 2 H), 8.42(s, 1 H), 8.36(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 7.63(s, 1 H), 7.39(s, 1 H)。
實施例5:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(V)
步驟1:合成(E)-2,3-二碘丙烯酸異丙酯(26)
於25℃下對化合物25(336 mg,3毫莫耳)之CH
3
CN(5 ml)攪拌溶液加入CuI(29 mg,0.15毫莫耳)和I
2
(1.14 g,4.5毫莫耳)。令反應物於80℃下經攪拌8小時。隨後將MTBE (20 ml)加入至混合物並令該混合物經水(3×20 ml)和飽和Na
2
S
2
O
3
溶液(3×20 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈淡黃色油之化合物26(453 mg,41%產率)。
1
H NMR(400 MHz, CDCl
3
)δ 7.61(s, 1 H), 5.19-5.10(m, 1 H), 1.36(d, J=6.4 Hz, 6 H)。
步驟2:合成(E)-2-碘-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(27)和(Z)-2-碘-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(28)
於0℃下對化合物13(780 mg,2.3毫莫耳)之DMF(10 ml)溶液加入DABCO(515 mg,4.6毫莫耳)和化合物26(1.68 g,4.6毫莫耳),令混合物於80℃下經攪拌0.5小時,隨後經飽和NH
4
Cl(50 ml)驟冷且經EA(100 ml)萃取。令有機層經鹽水(3×50 ml)沖洗並經乾燥、過濾及濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈白色固體之標的化合物27(1.1 g,83%產率):
MS(ESI): [M+H]
+
=577.9;
1
H NMR(400 MHz, CDCl
3
)δ 8.67(s, 1 H), 8.52(s, 2 H), 8.04(s, 1 H), 7.57(s, 1 H), 5.22-5.16(m, 1 H), 1.34(d, J=8 Hz, 6 H);和
呈白色固體之化合物28(210 mg,16%產率):
MS(ESI): [M+H]
+
=577.9;
1
H NMR(400 MHz, CDCl
3
)δ 9.52(s, 1 H), 8.81(s, 1 H), 8.75(s, 1 H), 8.59(s, 1 H), 8.07(s, 1 H), 5.23-5.17(m, 1 H), 1.38(d, J=8 Hz, 6 H)。
步驟3:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(29)和(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(30)
於25℃和氮氛圍下對化合物27(58 mg,0.1毫莫耳)和嘧啶-5-基亞硼酸(20 mg,0.16毫莫耳)之二噁烷:水(3 ml:1 ml)溶液加入Pd(dppf)Cl
2
(8 mg,0.01毫莫耳)和K
2
CO
3
(28 mg,0.2毫莫耳)。令反應混合物於50℃和氮氛圍下經攪拌2小時。冷卻至25℃並令有機溶液經濃縮。對殘餘物加入EA(50 ml)並隨後經水(3×50 ml)沖洗,令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=2:1)純化以得到呈白色固體之標的化合物29(24 mg,46%產率):
MS(ESI): [M+H]
+
=530.1;
1
H NMR(400 MHz, DMSO-d6)δ 9.21(s, 1 H), 9.02(s, 1 H), 8.92(s, 2 H), 8.59(s, 1 H), 8.47(s, 1 H), 8.43(s, 1 H), 8.19(s, 1 H), 5.21-5.16(m, 1 H), 1.25(d, J=8 Hz, 6 H);和
呈白色固體之化合物30(12 mg,23%產率):
MS(ESI): [M+H]
+
=530.1;
1
H NMR(400 MHz, DMSO-d6)δ 9.26(s, 1 H), 9.19(s, 1 H), 8.78(s, 2 H), 8.68(s, 1 H), 8.38(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 5.13-5.07(m, 1 H), 1.27(d, J=8 Hz, 6 H)。
步驟4:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸(31)
於0℃下對化合物28(20 mg,2.2毫莫耳)之DCM(3 ml)溶液加入AlCl
3
(21 mg,0.16毫莫耳)。隨後令反應混合物於35℃下經攪拌4小時。令反應物經水(5 ml)驟冷並令有機溶液經濃縮。對殘餘物加入EA(10 ml)並隨後經水(3×10 ml)和HCl溶液(10 ml,1N)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(DCM:MeOH:AcOH=10:1:0.1,v/v)純化以得到呈白色固體之化合物31(10 mg,56%產率)。
MS(ESI): [M+H]
+
=487.9。
步驟5:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(V)
對化合物31(10 mg,0.02毫莫耳)之乾燥THF(2 ml)攪拌溶液加入NMM(4 mg,0.04毫莫耳)並於0℃下逐滴加入氯甲酸異丙酯(5 mg,0.04毫莫耳)之THF(1 ml)溶液。令反應物於0℃下經攪拌20分鐘。隨後於0℃下對混合物加入NH
4
OH(2 mg,0.04毫莫耳)溶液,令該混合物於0℃下再經攪拌5分鐘,隨後經水(5 ml)驟冷且令有機溶液經濃縮。對殘餘物加入EA(10 ml)並隨後經水(3×10 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為EA)純化以得到呈白色固體之化合物V(5 mg,50%產率)。
MS(ESI): [M+H]
+
=486.9。
1
H NMR(400 MHz, DMSO-d6)δ 9.22(s, 1 H), 8.96(s, 3 H), 8.66(s, 1 H), 8.52(s, 1 H), 8.44(s, 1 H), 8.10(s, 1 H), 8.00(s, 1 H), 7.93(s, 1 H)。
實施例6:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼(VI)
步驟1:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼(VI)
於0℃下對化合物15(100 mg,0.24毫莫耳)和2-肼基噻唑氫氯酸鹽34(45 mg,0.29毫莫耳)之CH
3
CN:EA(4 ml:2 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(228 mg,0.36毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌10小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈灰色固體之化合物VI(8 mg,7%產率)。
MS(ESI): [M+H]
+
=506.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.92(s, 1 H), 9.58(s, 1 H), 8.65(s, 1 H), 8.53(s, 1 H), 8.43(s, 1 H), 7.57(d, J=12 Hz, 1 H), 7.22(d, J=4 Hz, 1 H), 6.89(d, J=4 Hz, 1 H), 6.04(d, J=8 Hz, 1 H)。
實施例7:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼(VII)
步驟1:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼(VII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和環丙烷卡肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物VII(40 mg,68%產率)。
MS(ESI): [M+H]
+
=492.8。
1
H NMR(400 MHz, DMSO-d6)δ 10.54(s, 1 H), 10.31(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 1.71-1.67(m, 1 H), 0.79-0.74(m, 4 H)。
實施例8:(Z)-N'-異丁醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(VIII)
步驟1:(Z)-N'-異丁醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(VIII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和異丁醯肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物VIII(40 mg,68%產率)。
MS(ESI): [M+H]
+
=494.8。
1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.59(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 2.55-2.48(m, 1 H), 1.06(d, J=8 Hz, 6 H)。
實施例9:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼(IX)
步驟1:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼(IX)
於0℃下對化合物15(50 mg,0.12毫莫耳)和丁醯肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物IX(40 mg,68%產率)。
MS(ESI): [M+H]
+
=494.8。
1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.04(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.16(t, J=8 Hz, 2 H), 1.61-1.52(m, 2 H), 0.90(t, J=8 Hz, 3 H)。
實施例10:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼(X)
步驟1:合成2-(環丁烷羰基)肼-1-羧酸三級丁酯(43)
於0℃下對化合物41(500 mg,5毫莫耳)和化合物42(726 mg,5.5毫莫耳)之DCM(20 ml)溶液加入DIEA(1.29 g,10毫莫耳)和T
3
P(6.36 g,10毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物43(1 g,93%產率)。
MS(ESI): [M+H]
+
=215.4。
步驟2:合成環丁烷卡肼氫氯酸鹽(44)
對化合物43(1 g,4.7毫莫耳)之DCM(20 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物44(0.7 g,99%產率)。
MS(ESI): [M+H]
+
=115.3。
步驟3:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼(X)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物44(28 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物X(11 mg,18%產率)。
MS(ESI): [M+H]
+
=506.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.49(s, 1 H), 9.93(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 3.18-3.10(m, 1 H), 2.23-1.90(m, 6 H)。
實施例11:(Z)-1-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼(XI)
步驟1:(Z)-1-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼(XI)
於0℃下對化合物15(40 mg,0.1毫莫耳)和1-甲基環丙烷卡肼(17 mg,0.15毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(52 mg,0.4毫莫耳)和T
3
P(254 mg,0.4毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XI(21 mg,41%產率)。
MS(ESI): [M+H]
+
=506.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.35(s, 1 H), 9.65(s, 1 H), 9.62(s, 1 H), 8.65(s, 1 H), 8.54(s, 1 H), 8.40(s, 1 H), 7.49(d, J=8 Hz, 1 H), 6.02(d, J=8 Hz, 1 H), 1.31(s, 3 H), 1.02(s, 2 H), 0.62(s, 2 H)。
實施例12:(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XII)
步驟1:合成2-(3-甲基氧雜環丁烷-3-羰基)肼-1-羧酸三級丁酯(47)
於0℃下對化合物46(116 mg,1毫莫耳)和化合物42 (145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T
3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×15 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物47(230 mg,99%產率)。
MS(ESI): [M+H]
+
=231.4。
步驟2:合成3-氯-2-(羥基甲基)-2-甲基丙烷肼氫氯酸鹽(48)
對化合物47(230 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令混合物經濃縮和真空下乾燥以得到呈白色固體之化合物48(200 mg,99%產率)。
MS(ESI): [M+H]
+
=166.3。
步驟3:合成(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XII)
於0℃下對化合物15(59 mg,0.14毫莫耳)和化合物48(45 mg,0.22毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(72 mg,0.56毫莫耳)和T
3
P(356 mg,0.56毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XII(10 mg,13%產率)。
MS(ESI): [M+H]
+
=559.0。
1
H NMR(400 MHz, DMSO-d6)δ 10.65(s, 1 H), 9.88(s, 1 H), 9.63(s, 1 H), 8.66(s, 1 H), 8.55(s, 1 H), 8.40(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 5.16(s, 1 H), 3.81(s, 2 H), 3.59(s, 2 H), 1.22(s, 3 H)。
實施例13:(Z)-3-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼(XIII)之一般合成
步驟1:合成(Z)-3-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼(XIII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和3-甲基丁烷肼(21 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XIII(37 mg,61%產率)。
MS(ESI): [M+H]
+
=508.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.07-1.95(m, 3 H), 0.93(d, J=8 Hz, 6 H)。
實施例14:(Z)-N'-乙醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XIV)之一般合成
步驟1:合成乙醯肼(52)
對化合物51(2.4 g,27.3毫莫耳)之EtOH(5 ml)溶液加入肼水合物(1.03 g,20.6毫莫耳)並隨後令混合物於80℃下經攪拌12小時。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物52(1.3 g,86%產率)。
步驟2:合成(Z)-N'-乙醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XIV)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物52(14 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XIV(20 mg,36%產率)。
MS(ESI): [M+H]
+
=466.8。
1
H NMR(400 MHz, DMSO-d6)δ 10.51(s, 1 H), 10.09(s, 1 H), 9.62(s, 1 H), 8.66(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 1.91(s, 3 H)。
實施例15:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼(XV)
步驟1:合成2-丙醯基肼-1-羧酸三級丁酯(57)
於25℃下對化合物56(74 mg,1毫莫耳)和化合物42(145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T
3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物57(190 mg,99%產率)。
MS(ESI): [M+H]
+
=192.3。
步驟2:合成丙醯肼氫氯酸鹽(58)
對化合物57(190 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物58(120 mg,94%產率)。
步驟3:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼(XV)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物58(23 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XV(24 mg,42%產率)。
MS(ESI): [M+H]
+
=480.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.03(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.20(q, J=8 Hz, 2 H), 1.05(t, J=8 Hz, 3 H)。
實施例16:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼(XVI)
步驟1:合成2-(環戊烷羰基)肼-1-羧酸三級丁酯(62)
於25℃下對化合物61(114 mg,1毫莫耳)和化合物42 (145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T
3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物62(220 mg,96%產率)。
MS(ESI): [M+H]
+
=229.3。
步驟2:合成環戊烷卡肼氫氯酸鹽(63)
對化合物62(220 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物63(160 mg,97%產率)。
MS(ESI): [M+H]
+
=129.3。
步驟3:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼(XVI)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物63(30 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T
3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XVI(30 mg,48%產率)。
MS(ESI): [M+H]
+
=520.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.65(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 2.72-2.65(m, 1 H), 1.84-1.52(m, 8 H)。
實施例17:(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XVII)
步驟1:合成2-甲基丙-3,3,3-d3酸(66)
於0℃和氮氛圍下對LDA(50 ml,74.3毫莫耳)之THF (150 ml)溶液加入化合物65(2.2 g,29.7毫莫耳)並隨後令混合物於80℃下經攪拌2小時。冷卻至0℃,逐滴加入CD
3
I (4.8 g,32.7毫莫耳)且隨後於80℃下經攪拌10小時。對混合物加入水(50 ml)並隨後經EA(50 ml)萃取。令無機層經1N HCl酸化至pH=4並隨後經EA(50 ml)萃取。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈淡黃色油之化合物66(1.68 g,62%產率)。
1
H NMR(400 MHz, CDCl
3
)δ 2.57(q, J=8 Hz, 1 H), 1.19(d, J=8 Hz, 3 H)。
步驟2:合成2-甲基-2-(甲基-d3)丙-3,3,3-d3酸(67)
於0℃和氮氛圍下對LDA(31 ml,46.3毫莫耳)之THF (90 ml)溶液加入化合物66(1.68 g,18.5毫莫耳)並隨後令混合物於80℃下經攪拌2小時。冷卻至0℃,逐滴加入CD
3
I(3 g,20.8毫莫耳)且隨後於80℃下經攪拌10小時。對混合物加入水(50 ml)並隨後經EA(50 ml)萃取。令無機層經1N HCl酸化至pH=4並隨後經EA(50 ml)萃取。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈黃色油之化合物67(1.17 g,59%產率)。
1
H NMR(400 MHz, CDCl
3
)δ 1.22(s, 3 H)。
步驟3:合成2-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)肼-1-羧酸三級丁酯(68)
於25℃下對化合物67(540 mg,5毫莫耳)、肼羧酸三級丁酯(660 mg,5毫莫耳)及Et
3
N(1.01 g,10毫莫耳)之DCM (50 ml)溶液加入EDCI(1.05 g,5.5毫莫耳)和HOBT(740 mg,5.5毫莫耳)並隨後令混合物於25℃下經攪拌12小時。隨後令該混合物經水(3×50 ml)沖洗。令有機溶液經濃縮並經矽膠管柱(流洗溶劑為PE:EA=5:1至3:1)純化以得到呈白色固體之化合物68(412 mg,37%產率)。
MS(ESI): [M+H]
+
=223.5。
1
H NMR(400 MHz, CDCl
3
)δ 7.37(s, 1 H), 6.48(s, 1 H), 1.47(s, 9 H), 1.24(s, 3 H)。
步驟4:合成2-甲基-2-(甲基-d3)丙烷肼-3,3,3-d3氫氯酸鹽(69)
對單頸燒瓶加入化合物68(412 mg,1.9毫莫耳)和HCl之二噁烷(10 ml,4M)溶液。令混合物於25℃下經攪拌1小時。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物69(300 mg,99%產率)。
MS(ESI): [M+H]
+
=123.3。
步驟5:合成(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XVII)
於-78℃下對化合物15(700 mg,1.7毫莫耳)和化合物69(300 mg,1.88毫莫耳)之DCM(50 ml)溶液加入DIEA(877 mg,6.8毫莫耳)和T
3
P(4.3 g,6.8毫莫耳,50% EA溶液),令混合物於-78℃下經攪拌30分鐘,隨後回溫至0℃且經攪拌1小時。令該混合物於35℃和真空下經濃縮。對殘餘物加入EA(100 ml)並經水(3×100 ml)和鹽水(3×100 ml)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到粗製產物,令該粗製產物經CH
3
CN(15 ml)碾製12小時且經過濾。收集濾餅並經DCM(15 ml)碾製12小時且經過濾。令濾餅經DCM(15 ml)沖洗並經收集且經真空下乾燥達5小時以得到呈白色固體之化合物XVII(670 mg,77%產率)。
MS(ESI): [M+H]
+
=514.9。
1
H NMR(400 MHz, DMSO-d6)δ 10.36(s, 1 H), 9.67(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.44(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 1.16(s, 3 H)。
實施例18:抑制細胞增生檢測
使用CellTiter-Glo發光細胞存活檢測(其使用Promega CellTiter 96®發光CellTiter Glo套組)以研究測試化合物之增生抑制作用。令化合物再懸浮於DMSO以製備25 mM儲存液。令癌細胞株或正常組織之細胞接種於96孔槽盤(1.5至5×10
3
個細胞/孔槽)隔夜。令化合物經2或3倍系列稀釋以得到10個濃度。隨後藉由Bravo(Agilent)將藥物加入至該等細胞孔槽並經培育72小時。藉由加入CellTiter-Glo試劑(依據製造商之指示)使細胞裂解並隨後使用EnVision (PerkinElmer)讀取發光值。
依據下述方法計算生長%:
經DMSO處理之細胞作為載具對照組(高度對照組;HC)且培養基單獨作為背景組(低度對照組;LC)。
生長%=100×(樣品發光值-LC發光值)/(HC發光值-LC發光值)。
使用Graphpad Prism藉由濃度反應曲線擬合計算IC
50
值。
測試之血液癌細胞株包括OCI-AML3、MV-4-11、KG-1、霞-1、K562及THP-1;測試之實體癌細胞株包括U87MG、U251、T98G、LN229、A172、H460、H2009及A549;測試之正常細胞株包括HCN2和3T3L1。結果示於表1,其中該等測試化合物顯示對腦、肺及血液之癌細胞株的生長抑制作用。然而,該等測試化合物對活體外培養之正常細胞未顯現毒性。
實施例19:REV-GFP移位檢測
先前研究已報告REV貨載之細胞核堆積係CRM1抑制作用之標記。細胞核轉出之選擇性抑制劑(SINE)處理引起以劑量依賴性之方式自細胞質部位至細胞核之REV的清楚且迅速之轉移。為評估測試化合物對XPO1之抑制作用,藉由位於中國之HD Biosciences公司產製REV-GFP U2OS選殖株並令細胞經生長培養基培養。隨後於37℃和5% CO
2
培養器中將細胞接種於96孔槽盤(100 μl生長培養基;7,000個細胞/孔槽)隔夜。來普黴素B(leptomycin B;LMB)之最高濃度設定為50 nM並經系列稀釋以作為陽性對照組,同時其他化合物之最高濃度設定為10 µM。於37℃和5% CO
2
下培育經不同化合物或DMSO處理之細胞達1小時。隨後於室溫下令細胞固定於4%甲醛達15分鐘。
令細胞經沖洗並於室溫和黑暗下經Hoechst 33342作業溶液(100 μl)染色10分鐘,隨後藉由高量顯像系統Image Xpress(Molecular Devices)且使用20倍接物鏡使細胞螢光顯像。針對Hoechst(350/461 nm)和GFP/FITC(488/509 nm) (激動和最大發射波長)設定濾鏡。依據下式計算REV細胞核移位之功效%:
功效%=(I
測試組
-I
對照組
)/(I
LMB
-I
對照組
)×100
I
測試組
-源自測試化合物之訊號;I
LMB
-50 nM LMB組之訊號;I
對照組
-載具對照組之訊號。
分別經LMB和化合物III處理1小時後,REV之細胞核再分佈存有劑量依賴性,且所有測試濃度之化合物III未降低細胞存活。數據顯示化合物III藉由抑制CRM1以增加REV細胞核堆積且對細胞存活無作用。結果示於圖1和2。其中圖1顯示LMB對Rev-GFP-U2OS細胞之REV再分佈的功效(A. LMB之1小時劑量反應功效的代表性影像,綠色表示與Rev共軛之EGFP;B.經LMB處理之REV再分佈的劑量反應曲線,載具組經設定為0且使用50 nM LMB組以代表100%功效,數值表示平均值±SEM(n=2),EC50=0.11 nM;C. LMB對細胞數之功效,每個組之經分析的相對比經載具組校正,所有值表示平均值±SEM(n=2))。其中圖2顯示化合物III對REV貨載抑制作用之EC50且化合物III未影響細胞存活。如示於表2,使用Rev-GFP以評估其他測試化合物。
實施例20:顯示持續但可逆之XPO1抑制作用的化合物洗除分析
為評估經洗除後測試化合物對XPO1蛋白之抑制作用的期間,使用REV-GFP細胞進行洗除分析。將測試化合物和陽性對照組加入至細胞孔槽並令盤於37℃和5% CO
2
下經培育1小時。除去含有藥物之培養基,令細胞經沖洗且加入新鮮培養基。令細胞於特定時間點0、4、24、48、72及96小時下經顯像以評估REV細胞核轉出之抑制作用。使用50 nM LMB,直至洗除後24小時,REV蛋白持續保留於細胞核。於72小時,功效逐漸降低至30%。然而,如細胞數降低所顯示者,相對於對照組,LMB顯示毒性。化合物III (EC90)亦抑制REV細胞核轉出。起始自洗除後4小時,功效逐漸減少且於24小時喪失功效。細胞對化合物III處理顯現良好之耐受性且未觀察到細胞數減少。結論為相對於陽性對照組LMB,亦與XPO1蛋白共價結合之化合物III於增加REV於細胞核之再分佈上顯現顯著且可逆之功效且於24小時內未對細胞生長產生任何副作用。結果示於圖3和4。其中圖3顯示於清洗研究中LMB之持續的XPO1抑制作用(A.於洗除後LMB對REV再分佈之作用的代表性影像;B.於洗除後LMB對REV再分佈之作用的摘要;C. LMB對細胞存活之作用,每個組之經分析的相對比經每個組於0小時校正,所有值表示平均值±SEM(n=3))。圖4顯示於Rev-GFP-U2OS細胞,化合物III對REV再分佈之洗除功效。於此洗除REV-GFP移位研究,於EC90和EC50(400 nM和120 nM)評估化合物III。經1小時處理後,化合物III(EC50)對REV之細胞核保留顯示約40%功效。此功效於洗除後4小時逐漸減少至20%且於24小時完全喪失。然而,化合物III(400 nM)於4小時顯現持續之XPO1抑制作用且於24小時逐漸減少。未觀察到對細胞存活有顯著作用。結論為化合物III顯現對XPO1蛋白之時間和劑量依賴性抑制作用且該作用持續4小時且呈可逆性。A.於洗除後化合物III對REV再分佈之作用的代表性影像;B.於洗除後化合物III對REV再分佈之作用的摘要;C.化合物III對細胞存活之作用。
實施例21:藥物動力(PK)和腦:血漿之比
自小鼠(n=3或5)收集AUC血液以助於總計10個時間點(給藥前、5分鐘、15分鐘、30分鐘、1小時、2小時、4小時、8小時、12小時及24小時)。於指定時間點,使用異氟醚麻醉動物且藉由眼眶後穿刺於每個時間點收集血液(約110 µl)至經預先冷卻之K
2
EDTA管。將血液樣品置於濕冰上並經離心(2000 x g,5分鐘,4℃)以於30分鐘得到收集之樣品的血漿。所有樣品經冷凍儲存於約-80℃下直至分析使用。於進行分析前,令樣品與溶於乙腈之內部標準品混合,經渦旋振盪和離心且注入上清液以進行分析。使用LC-MS-MS儀器(API 4000, Triple Quadrupole LC/MS/MS質譜儀)測量血漿中化合物之濃度。使用Phoenix Win Nonlin 6.3套裝軟體PO-Noncompartmental模式200(血管外輸入)計算AUC值。結果示於表3。
腦:血漿(B:P)之比
對另一組小鼠或大鼠(n=3或5)給藥(口服10 mg/kg)並隨後於最高血漿濃度之時間點(T
max
,給藥後1小時)處死且於該時間點收集末端血漿和腦組織。經收集後,使用冰鹽水輕洗腦組織,置於濾紙上乾燥,經稱重且置於乾冰上急速冷凍。所有樣品經冷凍儲存於約-80℃下直至分析使用。於分析時,令腦組織經均質化(均質化溶液PBS,pH 7.4),與溶於乙腈之內部標準品混合,經渦旋振盪和離心且注入上清液以進行分析。使用LC-MS-MS儀器(API 4000,Triple Quadrupole LC/MS/MS質譜儀)測量血漿中化合物之濃度。使用相同方法(除了均質化步驟)處理血漿樣品並依據所產生之標準曲線計算每個基質中化合物之濃度。PK檢測和B:P比測定之結果示於表4和圖5,其中圖5顯示於腦通透之匣PK中化合物III與KPT-350的腦行為之比較。
實施例22:最大耐受劑量(MTD)研究
於研究前令雌性BALB/c裸鼠(6至8週大/18至22 g;Beijing AniKeeper Biotech Co., Ltd.提供)經檢疫7天。經由獸醫評估動物之一般健康狀態。於進行研究前排除顯現異常之動物。依據Pharmaron, Inc.之標準作業程序(SOP)進行動物照顧和使用之一般程序。
每天對所有動物之體重稱重。例行性監測一般行為,諸如活動力、食物和水消耗(僅藉由籠邊檢查)、眼遮蓋/毛糾結及任何其他異常現象。記錄任何死亡及/或異常臨床徵候。藉由二氧化碳和隨後頸脫位使顯現嚴重痛苦及/或疼痛之明顯徵候的動物經人道處死以確認死亡。若有下述情況使動物安樂死:明顯體重減少>20%或動物不能獲取足夠之食物或水。結果示於圖6。其中圖6顯示MTD研究中小鼠體重變化%(對於KPT-330組,經給藥1週後,所有小鼠死亡或因體重減少達20%而經麻醉;然而,化合物I組之所有小鼠(口服25 mg/kg;隔天每週3次達2週)存活。投予調製劑:10%NMP/10%表面活性劑Solutol/80%(0.5%泊咯沙姆(Poloxamer)188+0.5% PVP))。
實施例23:評估化合物III對BALB/c裸鼠之U87MG-luc人神經膠質母細胞瘤原位模式的抗腫瘤功效
23.1. 研究設計
動物:使用雌性BALB/c裸鼠(6至8週大/18至22 g;Beijing AniKeeper Biotech Co., Ltd.提供)。依據Pharmaron, Inc.之標準作業程序(SOP)進行動物照顧和使用之一般程序。
分組和處置:於腫瘤細胞接種後第7天開始分組和處置。令小鼠顯像以監測腫瘤生長並隨後藉由電腦產生之隨機方式將該等小鼠隨機指定至各別組。於腫瘤移植後,所有測試藥物經口服投予(20 mg/kg;3次/週)達4週(n=12/組)。
23.2. 實驗方法和測量參數
細胞培養:活體外維持人神經膠質母細胞瘤U87-luc腫瘤細胞株呈單層培養並用於腫瘤接種。
腫瘤接種和隨機化:藉由置入針(AP: 2.0 mm;ML:0.5至1.0 mm;DV:3.0 mm),將懸浮於MEM培養基(2 µl)的2.5×10
5
個表現螢光素酶之U87MG-luc腫瘤細胞自前囟注射至右前腦。經1分鐘緩慢進行該注射。經完成注射後,維持該針於另一分鐘。記錄測量參數:腫瘤生長(經顯像分析監測)、體重及存活天數。
中止標準:當動物體重減少>20%或動物不能獲取足夠之食物或水時,藉由二氧化碳使各別動物經人道處死。
統計分析:如研究設計者,對所有測量參數值以值±標準誤差記錄數據。藉由SPSS 17.0統計軟體進行所有統計試驗且設定顯著水準為p<0.05。
23.3. 結果
整體上,於治療期間終了時,與載具對照組相比較,化合物III(20 mg/kg)顯現呈生物發光訊號減少99%之顯著抗腫瘤活性(p<0.05)。相較於化合物III處理組動物之中間存活時間(MST)為47天(p<0.0007),載具組動物之MST為39.5天。
關於安全性輪廓,大多數動物對化合物III(20 mg/kg)耐受良好,雖然個別動物顯現低於10%之體重減少,該個別動物需要跳躍劑量。於治療期間未觀察到其他顯著之臨床異常。結論為化合物III(20 mg/kg口服投予;3次/週達4週)能顯著延長帶有GBM腫瘤之小鼠的存活。同時,藉由抑制GBM腫瘤生長且延長帶有U87MG-luc原位腫瘤之動物組的存活,化合物III證實穿透BBB之能力。
結果示於圖7至9。其中圖7顯示於U87MG-luc原位模式中化合物III之腫瘤生長抑制作用(Y軸為對數刻度;p<0.05)。其中圖8顯示於20 mg/kg之每週3次經4週治療後,載具組對化合物III處理組之存活曲線。其中圖9顯示於治療期間和之後(第7至65天)的體重變化。
實施例24:藉由IHC於U87MG-luc原位小鼠模式中評估化合物III之PD功效
基於腫瘤之生物發光訊號,藉由電腦產生之隨機方式將小鼠隨機指定至各別組,該各別組分別包括每組5隻動物之載具組和化合物III處理組。口服給予化合物III(20 mg/kg;3次/週於第1、3及5天)達2週。IHC研究之動物於最後給藥和正常鹽水灌流且隨後經4%三聚甲醛固定後6小時被安樂死。收集含有腫瘤之完整腦並於石蠟密封程序前保持於固定劑中達24小時。
IHC定量之數據示於圖9。與載具對照組(*p<0.05;**p<0.01) 相比較,口服投予(3次/週達2週)化合物III或陽性對照組(KPT-330)顯著降低腫瘤之CRM1和Ki67的表現,其表示化合物III藉由抑制CRM1之表現顯現抗腫瘤功效。如示於腫瘤之降低的Ki67量,化合物III抑制腫瘤增生。
結果示於圖10。其中圖10說明藉由XPO1化合物III之Ki67和CRM1的降低。










 彼等之立體異構物、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中
R1
係獨立地選自-C(=O)-R2
、C3-6
雜環烷基或C5-10
雜芳基;R1
之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、 -NO2
、鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基(alkylsulfanyl);且
R2
係獨立地選自C1-6
烷基、C3-6
環烷基或C3-6
雜環烷基;R2
之任何烷基、環烷基或雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基;且
R3
和R4
係獨立地選自C1-6
烷基或經取代之C1-6
烷基,或R3
和R4
與彼等連接之N一起形成經取代或未經取代之C4-10
環烷基胺基;R3
和R4
之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基。
於另一方面,本發明關於醫藥組成物,其各別包含有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。本發明之醫藥組成物可進一步包含至少一種醫藥上可接受之賦形劑、載劑、佐劑、溶劑、撐體或彼等之組合。
於另一方面,本發明關於一種藉由調節外輸蛋白-1活性以治療罹患病症或疾病的個體之方法,該病症或疾病包括特別是某些神經系統病症或癌症,該方法包含對需要該治療之個體投予有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽,或包含對需要該治療之個體投予有效量之醫藥組成物,該醫藥組成物包含有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。
再於另一方面,本發明關於一種治療罹患某些神經系統病症或疾病的個體之方法,該等病症或疾病包含肌萎縮性脊髓側索硬化症、癲癇、創傷性腦損傷、亨丁頓舞蹈症(Huntington’s disease)、帕金森氏症(Parkinson’s disease)、類風濕性關節炎及全身性紅斑狼瘡。
再於另一方面,本發明關於一種治療罹患癌症的個體之方法,該癌症包含淋巴瘤、脂肪肉瘤、多發性骨髓瘤、骨髓增生異常綜合症、前列腺癌、大腸直腸癌、子宮內膜癌、胰臟癌、胃癌、瀰漫性大B細胞淋巴瘤、非小細胞肺癌、卵巢瘤、乳癌、急性骨髓性白血病、胸腺瘤、食道癌、神經膠質母細胞瘤及其他實體腫瘤。
本發明之一方面關於式(I’-III’)化合物於製備藥物之用途,該藥物係用於治療、預防、抑制或除去某些神經系統病症或癌症,該藥物進一步包含附加性治療,諸如輻射或治療有效量之一或多種可選擇地附加性活性成分,該等附加性活性成分包含化學治療劑、TK或RTK抑制劑、BCL2抑制劑、FLT3抑制劑、EGFR抑制劑、促細胞凋亡藥物、抗體-藥物共軛體(ADC)、免疫檢查點抑制劑、CAR-T、個人化癌症疫苗及化學激活素/細胞激素。
於本發明之另一方面,該式(I’-III’)化合物和彼等之醫藥上可接受之鹽係作為XPO-1活性之調節劑。因此,本發明關於一種調節個體的XPO-1活性之方法,其包含暴露該個體於有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。
再於另一方面,本發明關於製造式(I’-III’)化合物和彼等之醫藥上可接受之鹽之方法。
於本發明之該等化合物、醫藥組成物及方法的某些實施態樣中,該式(I’-III’)化合物係選自下述之詳細說明所描述或例示的下位概念之化合物或該化合物之醫藥上可接受之鹽。
於另一較佳實施態樣中,本發明關於製備醫藥組成物之方法,各該醫藥組成物包含有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。本發明之醫藥組成物可進一步包含至少一種醫藥上可接受之賦形劑、載劑、佐劑、溶劑、撐體或彼等之組合。
若經調製呈固定劑量,該等組合產物使用本說明書所描述(或熟習此技術之人士所習知)之劑量範圍內的本發明之化合物和各劑量範圍內的其他醫藥活性劑或處置。例如,CDC2抑制劑“奧羅莫(olomucine)”業已經發現於引起細胞凋亡上能與習知之細胞毒性劑協同增效地作用(J. Cell Sci.,(1995)108, 2897)。當組合調製劑係不適當時,本發明之化合物亦可與習知之抗癌劑或細胞毒性劑依序投予。於任何組合治療時,本發明不受限於投藥順序;於投予習知之抗癌劑或細胞毒性劑之前或之後,可投予式(I’-III’)化合物。例如,經依序投予抗癌劑,周期蛋白依賴性激酶抑制劑“黃酮吡醇(flavopiridol)”之細胞毒性活性受到影響(Cancer Research,(1997)57, 3375)。該等技術係熟習此技術之人士和主治醫師所習知。
藉由投予流體(諸如水)、環利尿劑、一或多種化學治療劑或抗腫瘤劑(諸如甲醯四氫葉酸(leucovorin)和氟脲嘧啶)、附加性化學治療劑(諸如惠爾血添(filgrastim)和紅血球生成素)或上述之任何組合可增強任何上述方法。
另一實施態樣係一種投予本發明之化合物至需要該化合物的個體(例如人)之方法,其係藉由投予該個體本發明之醫藥調製劑。
另一實施態樣係一種製備本發明之醫藥調製劑之方法,其係藉由混合至少一種本發明之醫藥上可接受之化合物和可選擇地一或多種醫藥上可接受之添加劑或賦形劑。
為自本發明描述之化合物製備醫藥組成物,惰性醫藥上可接受之載劑可為固體或液體。固體型式製劑包括粉末、片劑、可分散性粒子、膠囊、珠、藥包及栓劑。該粉末和片劑可包含約5%至約95%活性成分。適當之固體載劑係此技術所習知,例如碳酸鎂、硬脂酸鎂、滑石、糖或乳糖。可使用片劑、粉末、藥包及膠囊作為適合口服投予之固體劑型。醫藥上可接受之載劑的實例和製造多種不同組成物之方法可見於文獻A. Gennaro(ed.), Remington's Pharmaceutical Sciences,第18版,(1990), Mack Publishing Co., Easton, Pa。
液體型式製劑包括溶液、懸浮液及乳化液。可提及之實例係用於非經腸注射或添加甜味劑之水或水-丙二醇溶液和用於口服溶液、懸浮液及乳化液之乳白劑。液體型式製劑亦可包括用於鼻內投予之溶液。
適合吸入之氣溶膠製劑可包括溶液和呈粉末形式之固體,彼等可與醫藥上可接受之載劑(諸如惰性壓縮氣體,例如氮)組合。
亦包括欲於使用前即時轉化為用於口服或非經腸投予之液體型式製劑的固體型式製劑。該液體型式包括溶液、懸浮液及乳化液。
本發明之化合物亦可經皮投遞。經皮組成物之型式可為乳霜、洗劑、氣溶膠及/或乳化液且可包括於如此技術為此目的慣用之基質型或儲備型經皮貼片。
本發明之化合物亦可經皮下投遞。
較佳地,該化合物經口服或靜脈內投予。
較佳地,該醫藥製劑係呈單位劑型。於該劑型中,製劑被細分為大小適當之單位劑量,該單位劑量含有適量之活性成分,例如達到所欲目的的有效量之活性成分。
依據特定應用,該製劑之單位劑量中活性成分之量可於約1 mg至約1000 mg、較佳地約1 mg至約500 mg、更佳地約1 mg至約250 mg、甚佳地約1 mg至約200 mg之範圍內變化或調整。
取決於病患之需要和欲治療之病況的嚴重程度,所使用之真實劑量可加以變化。決定對特定情況之適當劑量方案係屬此領域之技術。方便地,每日總劑量可如需要地區分為多個部分且於當天內投予該多個部分。依據主治臨床醫師之判斷,考量諸如病患之年齡、病況及大小以及欲治療之徵候的嚴重程度之諸多因素,調節投予本發明之化合物及/或彼之醫藥上可接受之鹽的量和頻率。經口服投予之典型建議的每日劑量方案的範圍可從約1 mg/天至約200 mg/天,該劑量可呈1至2個分開之劑量。
本發明揭露之任何實施態樣可與其他實施態樣組合,只要該等實施態樣彼此不對立,即使該等實施態樣係描述於本發明之不同方面。此外,一實施態樣之任何技術特徵可被施加至其他實施態樣之對應技術特徵,只要該等實施態樣彼此不對立,即使該等實施態樣係描述於本發明之不同方面。
上述僅概要本發明之某些方面且本質上不欲作為限制。本發明之該等方面和其他方面及額外之實施態樣、特徵及優點將由下述之詳細說明和本發明之實施方式顯現。
彼等之立體異構物、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中
R1
係獨立地選自-C(=O)-R2
、C3-6
雜環烷基或C5-10
雜芳基;R1
之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、 -NO2
、鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基(alkylsulfanyl);且
R2
係獨立地選自C1-6
烷基、C3-6
環烷基或C3-6
雜環烷基;R2
之任何烷基、環烷基或雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基;且
R3
和R4
係獨立地選自C1-6
烷基或經取代之C1-6
烷基,或R3
和R4
與彼等連接之N一起形成經取代或未經取代之C4-10
環烷基胺基;R3
和R4
之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基。
於另一方面,本發明關於醫藥組成物,其各別包含有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。本發明之醫藥組成物可進一步包含至少一種醫藥上可接受之賦形劑、載劑、佐劑、溶劑、撐體或彼等之組合。
於另一方面,本發明關於一種藉由調節外輸蛋白-1活性以治療罹患病症或疾病的個體之方法,該病症或疾病包括特別是某些神經系統病症或癌症,該方法包含對需要該治療之個體投予有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽,或包含對需要該治療之個體投予有效量之醫藥組成物,該醫藥組成物包含有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。
再於另一方面,本發明關於一種治療罹患某些神經系統病症或疾病的個體之方法,該等病症或疾病包含肌萎縮性脊髓側索硬化症、癲癇、創傷性腦損傷、亨丁頓舞蹈症(Huntington’s disease)、帕金森氏症(Parkinson’s disease)、類風濕性關節炎及全身性紅斑狼瘡。
再於另一方面,本發明關於一種治療罹患癌症的個體之方法,該癌症包含淋巴瘤、脂肪肉瘤、多發性骨髓瘤、骨髓增生異常綜合症、前列腺癌、大腸直腸癌、子宮內膜癌、胰臟癌、胃癌、瀰漫性大B細胞淋巴瘤、非小細胞肺癌、卵巢瘤、乳癌、急性骨髓性白血病、胸腺瘤、食道癌、神經膠質母細胞瘤及其他實體腫瘤。
本發明之一方面關於式(I’-III’)化合物於製備藥物之用途,該藥物係用於治療、預防、抑制或除去某些神經系統病症或癌症,該藥物進一步包含附加性治療,諸如輻射或治療有效量之一或多種可選擇地附加性活性成分,該等附加性活性成分包含化學治療劑、TK或RTK抑制劑、BCL2抑制劑、FLT3抑制劑、EGFR抑制劑、促細胞凋亡藥物、抗體-藥物共軛體(ADC)、免疫檢查點抑制劑、CAR-T、個人化癌症疫苗及化學激活素/細胞激素。
於本發明之另一方面,該式(I’-III’)化合物和彼等之醫藥上可接受之鹽係作為XPO-1活性之調節劑。因此,本發明關於一種調節個體的XPO-1活性之方法,其包含暴露該個體於有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。
再於另一方面,本發明關於製造式(I’-III’)化合物和彼等之醫藥上可接受之鹽之方法。
於本發明之該等化合物、醫藥組成物及方法的某些實施態樣中,該式(I’-III’)化合物係選自下述之詳細說明所描述或例示的下位概念之化合物或該化合物之醫藥上可接受之鹽。
於另一較佳實施態樣中,本發明關於製備醫藥組成物之方法,各該醫藥組成物包含有效量之至少一種式(I’-III’)化合物或式(I’-III’)化合物之醫藥上可接受之鹽。本發明之醫藥組成物可進一步包含至少一種醫藥上可接受之賦形劑、載劑、佐劑、溶劑、撐體或彼等之組合。
若經調製呈固定劑量,該等組合產物使用本說明書所描述(或熟習此技術之人士所習知)之劑量範圍內的本發明之化合物和各劑量範圍內的其他醫藥活性劑或處置。例如,CDC2抑制劑“奧羅莫(olomucine)”業已經發現於引起細胞凋亡上能與習知之細胞毒性劑協同增效地作用(J. Cell Sci.,(1995)108, 2897)。當組合調製劑係不適當時,本發明之化合物亦可與習知之抗癌劑或細胞毒性劑依序投予。於任何組合治療時,本發明不受限於投藥順序;於投予習知之抗癌劑或細胞毒性劑之前或之後,可投予式(I’-III’)化合物。例如,經依序投予抗癌劑,周期蛋白依賴性激酶抑制劑“黃酮吡醇(flavopiridol)”之細胞毒性活性受到影響(Cancer Research,(1997)57, 3375)。該等技術係熟習此技術之人士和主治醫師所習知。
藉由投予流體(諸如水)、環利尿劑、一或多種化學治療劑或抗腫瘤劑(諸如甲醯四氫葉酸(leucovorin)和氟脲嘧啶)、附加性化學治療劑(諸如惠爾血添(filgrastim)和紅血球生成素)或上述之任何組合可增強任何上述方法。
另一實施態樣係一種投予本發明之化合物至需要該化合物的個體(例如人)之方法,其係藉由投予該個體本發明之醫藥調製劑。
另一實施態樣係一種製備本發明之醫藥調製劑之方法,其係藉由混合至少一種本發明之醫藥上可接受之化合物和可選擇地一或多種醫藥上可接受之添加劑或賦形劑。
為自本發明描述之化合物製備醫藥組成物,惰性醫藥上可接受之載劑可為固體或液體。固體型式製劑包括粉末、片劑、可分散性粒子、膠囊、珠、藥包及栓劑。該粉末和片劑可包含約5%至約95%活性成分。適當之固體載劑係此技術所習知,例如碳酸鎂、硬脂酸鎂、滑石、糖或乳糖。可使用片劑、粉末、藥包及膠囊作為適合口服投予之固體劑型。醫藥上可接受之載劑的實例和製造多種不同組成物之方法可見於文獻A. Gennaro(ed.), Remington's Pharmaceutical Sciences,第18版,(1990), Mack Publishing Co., Easton, Pa。
液體型式製劑包括溶液、懸浮液及乳化液。可提及之實例係用於非經腸注射或添加甜味劑之水或水-丙二醇溶液和用於口服溶液、懸浮液及乳化液之乳白劑。液體型式製劑亦可包括用於鼻內投予之溶液。
適合吸入之氣溶膠製劑可包括溶液和呈粉末形式之固體,彼等可與醫藥上可接受之載劑(諸如惰性壓縮氣體,例如氮)組合。
亦包括欲於使用前即時轉化為用於口服或非經腸投予之液體型式製劑的固體型式製劑。該液體型式包括溶液、懸浮液及乳化液。
本發明之化合物亦可經皮投遞。經皮組成物之型式可為乳霜、洗劑、氣溶膠及/或乳化液且可包括於如此技術為此目的慣用之基質型或儲備型經皮貼片。
本發明之化合物亦可經皮下投遞。
較佳地,該化合物經口服或靜脈內投予。
較佳地,該醫藥製劑係呈單位劑型。於該劑型中,製劑被細分為大小適當之單位劑量,該單位劑量含有適量之活性成分,例如達到所欲目的的有效量之活性成分。
依據特定應用,該製劑之單位劑量中活性成分之量可於約1 mg至約1000 mg、較佳地約1 mg至約500 mg、更佳地約1 mg至約250 mg、甚佳地約1 mg至約200 mg之範圍內變化或調整。
取決於病患之需要和欲治療之病況的嚴重程度,所使用之真實劑量可加以變化。決定對特定情況之適當劑量方案係屬此領域之技術。方便地,每日總劑量可如需要地區分為多個部分且於當天內投予該多個部分。依據主治臨床醫師之判斷,考量諸如病患之年齡、病況及大小以及欲治療之徵候的嚴重程度之諸多因素,調節投予本發明之化合物及/或彼之醫藥上可接受之鹽的量和頻率。經口服投予之典型建議的每日劑量方案的範圍可從約1 mg/天至約200 mg/天,該劑量可呈1至2個分開之劑量。
本發明揭露之任何實施態樣可與其他實施態樣組合,只要該等實施態樣彼此不對立,即使該等實施態樣係描述於本發明之不同方面。此外,一實施態樣之任何技術特徵可被施加至其他實施態樣之對應技術特徵,只要該等實施態樣彼此不對立,即使該等實施態樣係描述於本發明之不同方面。
上述僅概要本發明之某些方面且本質上不欲作為限制。本發明之該等方面和其他方面及額外之實施態樣、特徵及優點將由下述之詳細說明和本發明之實施方式顯現。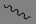 ”鍵連接之取代基的式II’中,雙鍵表示E或Z構形。
以未滿足價表示之任何原子被假定為含有足夠數目之氫原子以滿足原子價。
當任何變異(例如烷基、伸烷基、雜芳基、R1
及R2
)於本文之任何化學式或描述中出現1次以上時,於每次出現時該變異之定義係獨立於該變異於每次其他出現時之定義。
本文使用之數目範圍欲包括依序全部數目。例如,“0至4”或“0-4”之範圍包括0、1、2、3及4且“10-20%”包括10%、11%、12%、13%、14%、15%、16%、17%、18%、19%及20%。同樣地,數目範圍亦欲包括依序小數。例如,“1-2%”包括1.0%、1.1%、1.2%、1.3%、1.4%、1.5%、1.6%、1.7%、1.8%、1.9%及2.0%。
當顯示多官能基時,與核連接之點係由線或連字號表示。例如,芳氧基-係指其中氧原子係與核連接之點且芳基係與該氧原子連接。
其他定義
本文使用之“個體”包含哺乳動物和非哺乳動物。哺乳動物之實例包括但不限於哺乳動物綱之任何成員:人類;非人類之靈長類動物,諸如黑猩猩和其他類人猿及猴子;農場動物,諸如牛、馬、綿羊、山羊及豬;家養動物,諸如兔、狗及貓;及,實驗室動物,其包括囓齒動物,諸如大鼠、小鼠及豚鼠,以及類似者。非哺乳動物之實例包括但不限於鳥、魚及類似者。於本發明之一實施態樣中,該哺乳動物係人類。
“病患”包括人類和動物。
“抑制劑”係指諸如化合物、藥物、酶活化劑或激素之分子,其阻斷或者干擾特定之生物活性。
“調節劑”係指諸如本發明之化合物的分子,其增加或降低或者影響給定之蛋白、受體及/或離子通道之活性。
“有效量”或“治療有效量”係指足以提供所欲之生物結果的試劑之量。該結果可為疾病或醫療條件之症狀、徵候或病因的減輕及/或緩和或生物系統之任何其他所欲之改變。例如,治療用途之“有效量”係指提供疾病狀態、徵候或醫療條件之臨床相關改變所需的化合物或包含該化合物之組成物的量。藉由使用慣常之實驗,熟習此技術之人士可決定用於任一個別案例之適當“有效”量。因此,“有效量”通常係指活性劑顯現所欲之治療功效的量。
本文使用之“處置”或“治療”包含“預防性”和“療效性”處置或治療。“預防性”處置表示延後疾病、疾病徵候或醫療條件的發展、抑制可能出現之徵候或降低疾病或徵候之發展或復發的風險。“療效性”治療包括減輕現有疾病、徵候或病況之嚴重程度或抑制現有疾病、徵候或病況之惡化。因此,治療包括改善或預防現有疾病徵候之惡化、預防其他徵候發生、改善或預防徵候之潛在的代謝病因、抑制病症或疾病,例如中止該病症或疾病之發展、緩解該病症或疾病、引起該病症或疾病之退化、緩解由該疾病或病症引起之病況或中止該疾病或病症之徵候。
應瞭解本文使用之“投予”化合物表示提供本發明之化合物或包含本發明之化合物或本發明之化合物的前藥之醫藥組成物給需要之個體。確定的是,熟習此非限制性技術之人士可使用有效量的本發明之化合物以治療正罹患神經系統和精神病症之病患或預防性治療罹患該等病症之病患。
本文使用之“組成物”欲包含產物,其包含特定量之特定成分,且包含任何產物,其係直接或間接由特定量之特定成分的組合產生。與醫藥組成物有關之組成物欲包含產物,其包含活性成分和作為載劑之惰性成分,且包含任何產物,其係直接或間接由該等成分之任何二或多者的組合、複合或聚積產生或由諸如引起該等成分之一或多者的解離之其他類型的反應或交互作用產生。於是,本發明之醫藥組成物包含由混合本發明之化合物和醫藥上可接受之載劑所製備之任何組成物。
其他化學描述
本文給予之任何化學式欲表示具有結構式所示之結構的化合物和某些變異體或型式。例如,本文給予之任何化學式的化合物可具有不對稱或手性中心並因此會以不同之立體異構形式存在。該通式之化合物的所有立體異構物(其包括光學異構物、鏡像異構物及非鏡像異構物)及彼等之混合物落入該通式之範圍。再者,某些結構可呈幾何異構物(即順式和反式異構物)、互變異構物或阻旋異構物。所有該等異構型式或彼等之混合物屬於本發明之一部分。因此,本文給予之任何化學式欲表示消旋物、一或多種鏡像異構形式、一或多種非鏡像異構形式、一或多種互變異構或阻旋異構形式及彼等之混合物。
“立體異構物”係指具有相同化學組成但原子或基團之空間配置不同的化合物。立體異構物包括鏡像異構物、非鏡像異構物、構形異構物(旋轉異構物)、幾何(順式/反式)異構物、阻旋異構物等。
“手性”係指具有與鏡像對非重疊性之分子且“非手性”係指與鏡像對重疊之分子。
“鏡像異構物”係指化合物之2個立體異構物,彼等彼此係非重疊之鏡像。
“非鏡像異構物”係指具有2或多個手性中心之立體異構物,該等異構物分子彼此不為鏡像。非鏡像異構物具有不同之物理性質,例如熔點、沸點、光譜特性或生物活性。藉由高解析分析方法,諸如電泳和層析(諸如HPLC),可分離非鏡像異構物之混合物。
本文使用之立體化學的定義和常規通常遵循文獻S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984)McGraw-Hill Book Company, New York和Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994。
許多有機化合物係以光學活性形式存在,即彼等能旋轉偏光面。於描述光學活性化合物時,使用字首D和L或R和S以表示分子之手性中心的絕對構形。使用字首d和l或(+)和(-)以表示化合物旋轉偏光面之符號,其中(-)或l表示該化合物呈左旋性。字首為(+)或d之化合物呈右旋性。特定之立體異構物可指鏡像異構物且該等立體異構物的混合物被稱為鏡像異構混合物。鏡像異構物之50:50混合物被稱為消旋混合物或消旋物,其可能發生於化學反應或製程中不具有立體選擇性或立體特異性的地方。
本文揭露之化合物的任何不對稱性原子(例如碳或類似者)可呈消旋性或富含鏡像異構物性,例如(R)-、(S)-或(R, S)-構形。於某些實施態樣中,於該(R)-或(S)-構形之每個不對稱性原子具有至少50%鏡像異構物過量、至少60%鏡像異構物過量、至少70%鏡像異構物過量、至少80%鏡像異構物過量、至少90%鏡像異構物過量、至少95%鏡像異構物過量或至少99%鏡像異構物過量。
取決於起始物和方法之選擇和不對稱性碳原子之數目,化合物可呈可能的立體異構物之一或彼等之混合物的形式,諸如消旋物和非鏡像異構混合物。使用手性合成組元或手性反應劑可製備光學活性(R)-和(S)-異構物,或使用慣用之技術可解析光學活性(R)-和(S)-異構物。若化合物含有雙鍵,則取代基可呈E或Z構形。若化合物含有經二取代之環烷基,則環烷基之取代基相對於該相同環烷基之另一取代基可呈順式或反式構形。
基於組分之理化性質的差異,藉由例如層析及/或分段結晶,立體異構物所形成之任何混合物可經分離成純或實質上純之幾何異構物、鏡像異構物或非鏡像異構物。藉由熟習此技術之人士所習知之方法,例如藉由分離終產物或中間體之非鏡像異構性鹽,該終產物或中間體所形成之任何消旋物可經解析成光學鏡像體。藉由手性層析,例如使用手性吸附劑之高效液相層析(HPLC),亦可解析消旋產物。藉由不對稱性合成,亦可製備較佳之鏡像異構物。參閱例如文獻Jacques, et al., Enantiomers, Racemates and Resolutions(Wiley Interscience, New York, 1981); Principles of Asymmetric Synthesis(2nd Ed. Robert E. Gawley, Jeffrey Aubé, Elsevier, Oxford, UK, 2012); Eliel, E. L. Stereochemistry of Carbon Compounds(McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions, p. 268(E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972); Chiral Separation Techniques: A Practical Approach (Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007)。
藉由熟習此技術之人士所習知之方法,諸如例如層析及/或分段結晶,基於個別非鏡像異構物之理化性質的差異,非鏡像異構混合物可經分離成個別非鏡像異構物。藉由與適當之光學活性化合物(例如手性輔助劑,諸如手性醇或Mosher氏醯基氯,或形成非鏡像異構性鹽之混合物)反應以轉化鏡像異構混合物成非鏡像異構混合物、分離該等非鏡像異構物及轉化(例如水解或脫鹽)個別非鏡像異構物為對應之純鏡像異構物,可分離鏡像異構物。藉由使用手性HPLC管柱,亦可分離鏡像異構物。
本發明之化合物可生成醫藥上可接受之鹽,其亦屬於本發明之範圍。“醫藥上可接受之鹽”係指式(I’)化合物之自由酸或鹼的鹽,其係非毒性、生理上可耐受、能與經調製之醫藥組成物互溶及另一方面適合調製及/或投予至個體。除非另有說明,應瞭解的是,本發明之化合物包括該化合物的醫藥上可接受之鹽。
化合物鹽包括與無機酸及/或有機酸所生成之酸性鹽及與無機鹼及/或有機鹼所生成之鹼性鹽。此外,當給予之化合物同時含有鹼性基團(諸如但不限於吡啶或咪唑)和酸性基團(諸如但不限於羧酸)時,熟習此技術之人士當能瞭解該化合物可呈兩性離子(“內鹽”),該鹽含括於本文使用之“鹽”的範圍。藉由例如令化合物與某一量(諸如當量)之適當酸或鹼於諸如使該鹽沉澱之介質或水溶性介質中反應並隨後經冷凍乾燥,可製備本發明之化合物的鹽。
例示性鹽包括但不限於硫酸鹽、檸檬酸鹽、乙酸鹽、草酸鹽、氯化物、溴化物、碘化物、硝酸鹽、硫酸氫鹽、磷酸鹽、酸式磷酸鹽、異菸鹼酸鹽、乳酸鹽、水楊酸鹽、酸式檸檬酸鹽、酒石酸鹽、油酸鹽、鞣酸鹽、泛酸鹽、酒石酸氫鹽、抗壞血酸鹽、丁二酸鹽、順丁烯二酸鹽、龍膽酸鹽、反丁烯二酸鹽、葡萄糖酸鹽、葡萄糖醛酸鹽、葡萄糖二酸鹽、甲酸鹽、苯甲酸鹽、麩胺酸鹽、甲磺酸鹽、乙磺酸鹽、苯磺酸鹽、對甲苯磺酸鹽及雙羥萘酸鹽(即1,1’-伸甲基‑雙(2-羥基-3-萘酸鹽))。醫藥上可接受之鹽可涉及包括其他分子,諸如乙酸離子、丁二酸離子或其他抗衡離子。該抗衡離子可為使母化合物上之電荷安定的任何有機或無機基團。再者,醫藥上可接受之鹽於結構上可含有超過1個帶電荷之原子。當多個帶電荷之原子係該醫藥上可接受之鹽的一部分時,實例可含有多個抗衡離子。因此,醫藥上可接受之鹽可含有1或多個帶電荷之原子及/或1或多個抗衡離子。
例示性酸加成鹽包括乙酸鹽、抗壞血酸鹽、苯甲酸鹽、苯磺酸鹽、硫酸氫鹽、硼酸鹽、丁酸鹽、檸檬酸鹽、樟腦酸鹽、樟腦磺酸鹽、反丁烯二酸鹽、氫氯酸鹽、氫溴酸鹽、氫碘酸鹽、乳酸鹽、順丁烯二酸鹽、甲磺酸鹽、萘磺酸鹽、硝酸鹽、草酸鹽、磷酸鹽、丙酸鹽、水楊酸鹽、丁二酸鹽、硫酸鹽、酒石酸鹽、硫氰酸鹽、甲苯磺酸鹽及類似者。
例示性鹼鹽包括銨鹽、鹼金屬鹽(諸如鈉鹽、鋰鹽及鉀鹽)、鹼土金屬鹽(諸如鈣鹽和鎂鹽)、有機鹼(例如有機胺,諸如二環己基胺和三級丁基胺)鹽及胺基酸(諸如精胺酸和離胺酸)鹽及類似者。含有氮之鹼性基團可經試劑季胺化,該試劑係諸如低碳烷基鹵化物(例如甲基氯、乙基氯、丁基氯、甲基溴、乙基溴、丁基溴、甲基碘、乙基碘及丁基碘)、硫酸二烷酯(例如硫酸二甲酯、硫酸二乙酯及硫酸二丁酯)、長鏈鹵化物(例如癸基氯、癸基溴、癸基碘、十二烷基氯、十二烷基溴、十二烷基碘、十八烷基氯、十八烷基溴及十八烷基碘)、芳烷基鹵化物(例如苄基溴和苯乙基溴)及類似者。
此外,通常被認為適合用於自醫藥化合物形成醫藥上有用之鹽的酸和鹼係見於文獻,例如P. Stahl et al, Camille G.(eds.)Handbook of Pharmaceutical Salts. Properties, Selection and Use.(2002)Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences(1977)66(1)1-19; P. Gould, International J. of Pharmaceutics(1986)33 201-217; Anderson et al, The Practice of Medicinal Chemistry(1996), Academic Press, New York;及The Orange Book(Food & Drug Administration, MD,可自FDA取得)。該等文獻併入本文作為參考。
此外,本文描述之任何化合物亦欲係指任何非溶劑化形式或該化合物之水合物、溶劑合物或多晶型及彼等之混合物,即使該等形式並未明確列示。“溶劑合物”表示本發明之化合物與一或多個溶劑分子之物理性結合。該物理性結合涉及不同程度之離子性和共價性鍵結,其包括氫鍵。於某些實例中,該溶劑合物將可被分離,例如當一或多個溶劑分子併入結晶固體之晶格時。“溶劑合物”包含溶液相和可被分離之溶劑合物二者。適當之溶劑合物包括與醫藥上可接受之溶劑(諸如水、乙醇及類似者)形成之溶劑合物。於某些實施態樣中,該溶劑係水且該溶劑合物係水合物。
本發明之一或多個化合物係可選擇地被轉化為溶劑合物。通常製備溶劑合物之方法係屬習知。因此,文獻例如M. Caira et al., J. Pharmaceutical Sci., 93(3), 601-611(2004)描述使用乙酸乙酯和水製備抗真菌劑“氟可那挫” (fluconazole)之溶劑合物。溶劑合物、半溶劑合物、水合物及類似者之相似製備係描述於文獻E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12(2004)和A. L. Bingham et al, Chem. Commun., 603-604(2001)。典型之非限制性方法涉及於高於周溫之溫度下溶解本發明之化合物於適量之溶劑(有機溶劑或水或彼等之混合物)中並於足以形成結晶之速率下冷卻溶液,隨後藉由標準方法分離該結晶。分析方法(諸如例如紅外光譜術)顯示於呈溶劑合物(或水合物)之結晶中存在溶劑(或水)。
本文給予之任何化學式亦欲表示化合物之未經標記和經同位素標記的形式。經同位素標記之化合物具有本文給予之化學式所示的結構,除了其中一或多個原子係經具有選擇之原子量或原子數的原子替代。可併入本發明之化合物的同位素之實例包括氫、碳、氮、氧、磷、氟、氯及碘之同位素,分別諸如2
H、3
H、11
C、13
C、14
C、15
N、18
O、17
O、31
P、32
P、35
S、18
F、36
Cl及125
I。該等經同位素標記之化合物可用於代謝研究(例如使用14
C)、反應動力學研究(例如使用2
H或3
H)、偵測或顯像技術[諸如正電子發射電腦斷層掃描(PET)或單光子發射電腦斷層掃描攝影術(SPECT)],其包括藥物或受質組織分佈檢測或病患之放射治療。特定地,經18
F或11
C標記之化合物可特別適用於PET或SPECT研究。再者,經較重之同位素(諸如氘(2
H))取代可提供因較佳之代謝安定性所導致的某些治療優異,例如增加之活體內半生期或降低之劑量需要。通常藉由實施反應圖或實施例描述之方法和下述之製備,經可方便取得之經同位素標記的反應劑替代未經同位素標記的反應劑,可製備本發明之經同位素標記之化合物。
對於本文描述之化合物,“鹽”、“溶劑合物”、“多晶型”及類似者將欲等同地應用於本發明之化合物的鏡像異構物、立體異構物、旋轉異構物、互變異構物、阻旋異構物及消旋物之鹽、溶劑合物及多晶型。
化學命名工具係軟體ChemDraw專業版16.0。
本發明之化合物的說明
本發明關於特定之分子和彼之醫藥上可接受之鹽或異構物。本發明另關於可用於調節功能失調性XPO1活性之分子和彼之醫藥上可接受之鹽、溶劑合物、酯或異構物。
本發明關於本文描述之化合物和彼之醫藥上可接受之鹽、溶劑合物、酯或異構物及包含本文描述之一或多種化合物和彼之醫藥上可接受之鹽或異構物之醫藥組成物。
本發明之一方面提供用於調節哺乳動物之XPO1活性之化合物、組成物、套組及解毒劑,彼等含有式(I’)、式(II’)或式(III’)之結構的化合物:
”鍵連接之取代基的式II’中,雙鍵表示E或Z構形。
以未滿足價表示之任何原子被假定為含有足夠數目之氫原子以滿足原子價。
當任何變異(例如烷基、伸烷基、雜芳基、R1
及R2
)於本文之任何化學式或描述中出現1次以上時,於每次出現時該變異之定義係獨立於該變異於每次其他出現時之定義。
本文使用之數目範圍欲包括依序全部數目。例如,“0至4”或“0-4”之範圍包括0、1、2、3及4且“10-20%”包括10%、11%、12%、13%、14%、15%、16%、17%、18%、19%及20%。同樣地,數目範圍亦欲包括依序小數。例如,“1-2%”包括1.0%、1.1%、1.2%、1.3%、1.4%、1.5%、1.6%、1.7%、1.8%、1.9%及2.0%。
當顯示多官能基時,與核連接之點係由線或連字號表示。例如,芳氧基-係指其中氧原子係與核連接之點且芳基係與該氧原子連接。
其他定義
本文使用之“個體”包含哺乳動物和非哺乳動物。哺乳動物之實例包括但不限於哺乳動物綱之任何成員:人類;非人類之靈長類動物,諸如黑猩猩和其他類人猿及猴子;農場動物,諸如牛、馬、綿羊、山羊及豬;家養動物,諸如兔、狗及貓;及,實驗室動物,其包括囓齒動物,諸如大鼠、小鼠及豚鼠,以及類似者。非哺乳動物之實例包括但不限於鳥、魚及類似者。於本發明之一實施態樣中,該哺乳動物係人類。
“病患”包括人類和動物。
“抑制劑”係指諸如化合物、藥物、酶活化劑或激素之分子,其阻斷或者干擾特定之生物活性。
“調節劑”係指諸如本發明之化合物的分子,其增加或降低或者影響給定之蛋白、受體及/或離子通道之活性。
“有效量”或“治療有效量”係指足以提供所欲之生物結果的試劑之量。該結果可為疾病或醫療條件之症狀、徵候或病因的減輕及/或緩和或生物系統之任何其他所欲之改變。例如,治療用途之“有效量”係指提供疾病狀態、徵候或醫療條件之臨床相關改變所需的化合物或包含該化合物之組成物的量。藉由使用慣常之實驗,熟習此技術之人士可決定用於任一個別案例之適當“有效”量。因此,“有效量”通常係指活性劑顯現所欲之治療功效的量。
本文使用之“處置”或“治療”包含“預防性”和“療效性”處置或治療。“預防性”處置表示延後疾病、疾病徵候或醫療條件的發展、抑制可能出現之徵候或降低疾病或徵候之發展或復發的風險。“療效性”治療包括減輕現有疾病、徵候或病況之嚴重程度或抑制現有疾病、徵候或病況之惡化。因此,治療包括改善或預防現有疾病徵候之惡化、預防其他徵候發生、改善或預防徵候之潛在的代謝病因、抑制病症或疾病,例如中止該病症或疾病之發展、緩解該病症或疾病、引起該病症或疾病之退化、緩解由該疾病或病症引起之病況或中止該疾病或病症之徵候。
應瞭解本文使用之“投予”化合物表示提供本發明之化合物或包含本發明之化合物或本發明之化合物的前藥之醫藥組成物給需要之個體。確定的是,熟習此非限制性技術之人士可使用有效量的本發明之化合物以治療正罹患神經系統和精神病症之病患或預防性治療罹患該等病症之病患。
本文使用之“組成物”欲包含產物,其包含特定量之特定成分,且包含任何產物,其係直接或間接由特定量之特定成分的組合產生。與醫藥組成物有關之組成物欲包含產物,其包含活性成分和作為載劑之惰性成分,且包含任何產物,其係直接或間接由該等成分之任何二或多者的組合、複合或聚積產生或由諸如引起該等成分之一或多者的解離之其他類型的反應或交互作用產生。於是,本發明之醫藥組成物包含由混合本發明之化合物和醫藥上可接受之載劑所製備之任何組成物。
其他化學描述
本文給予之任何化學式欲表示具有結構式所示之結構的化合物和某些變異體或型式。例如,本文給予之任何化學式的化合物可具有不對稱或手性中心並因此會以不同之立體異構形式存在。該通式之化合物的所有立體異構物(其包括光學異構物、鏡像異構物及非鏡像異構物)及彼等之混合物落入該通式之範圍。再者,某些結構可呈幾何異構物(即順式和反式異構物)、互變異構物或阻旋異構物。所有該等異構型式或彼等之混合物屬於本發明之一部分。因此,本文給予之任何化學式欲表示消旋物、一或多種鏡像異構形式、一或多種非鏡像異構形式、一或多種互變異構或阻旋異構形式及彼等之混合物。
“立體異構物”係指具有相同化學組成但原子或基團之空間配置不同的化合物。立體異構物包括鏡像異構物、非鏡像異構物、構形異構物(旋轉異構物)、幾何(順式/反式)異構物、阻旋異構物等。
“手性”係指具有與鏡像對非重疊性之分子且“非手性”係指與鏡像對重疊之分子。
“鏡像異構物”係指化合物之2個立體異構物,彼等彼此係非重疊之鏡像。
“非鏡像異構物”係指具有2或多個手性中心之立體異構物,該等異構物分子彼此不為鏡像。非鏡像異構物具有不同之物理性質,例如熔點、沸點、光譜特性或生物活性。藉由高解析分析方法,諸如電泳和層析(諸如HPLC),可分離非鏡像異構物之混合物。
本文使用之立體化學的定義和常規通常遵循文獻S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984)McGraw-Hill Book Company, New York和Eliel, E. and Wilen, S., "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., New York, 1994。
許多有機化合物係以光學活性形式存在,即彼等能旋轉偏光面。於描述光學活性化合物時,使用字首D和L或R和S以表示分子之手性中心的絕對構形。使用字首d和l或(+)和(-)以表示化合物旋轉偏光面之符號,其中(-)或l表示該化合物呈左旋性。字首為(+)或d之化合物呈右旋性。特定之立體異構物可指鏡像異構物且該等立體異構物的混合物被稱為鏡像異構混合物。鏡像異構物之50:50混合物被稱為消旋混合物或消旋物,其可能發生於化學反應或製程中不具有立體選擇性或立體特異性的地方。
本文揭露之化合物的任何不對稱性原子(例如碳或類似者)可呈消旋性或富含鏡像異構物性,例如(R)-、(S)-或(R, S)-構形。於某些實施態樣中,於該(R)-或(S)-構形之每個不對稱性原子具有至少50%鏡像異構物過量、至少60%鏡像異構物過量、至少70%鏡像異構物過量、至少80%鏡像異構物過量、至少90%鏡像異構物過量、至少95%鏡像異構物過量或至少99%鏡像異構物過量。
取決於起始物和方法之選擇和不對稱性碳原子之數目,化合物可呈可能的立體異構物之一或彼等之混合物的形式,諸如消旋物和非鏡像異構混合物。使用手性合成組元或手性反應劑可製備光學活性(R)-和(S)-異構物,或使用慣用之技術可解析光學活性(R)-和(S)-異構物。若化合物含有雙鍵,則取代基可呈E或Z構形。若化合物含有經二取代之環烷基,則環烷基之取代基相對於該相同環烷基之另一取代基可呈順式或反式構形。
基於組分之理化性質的差異,藉由例如層析及/或分段結晶,立體異構物所形成之任何混合物可經分離成純或實質上純之幾何異構物、鏡像異構物或非鏡像異構物。藉由熟習此技術之人士所習知之方法,例如藉由分離終產物或中間體之非鏡像異構性鹽,該終產物或中間體所形成之任何消旋物可經解析成光學鏡像體。藉由手性層析,例如使用手性吸附劑之高效液相層析(HPLC),亦可解析消旋產物。藉由不對稱性合成,亦可製備較佳之鏡像異構物。參閱例如文獻Jacques, et al., Enantiomers, Racemates and Resolutions(Wiley Interscience, New York, 1981); Principles of Asymmetric Synthesis(2nd Ed. Robert E. Gawley, Jeffrey Aubé, Elsevier, Oxford, UK, 2012); Eliel, E. L. Stereochemistry of Carbon Compounds(McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions, p. 268(E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972); Chiral Separation Techniques: A Practical Approach (Subramanian, G. Ed., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2007)。
藉由熟習此技術之人士所習知之方法,諸如例如層析及/或分段結晶,基於個別非鏡像異構物之理化性質的差異,非鏡像異構混合物可經分離成個別非鏡像異構物。藉由與適當之光學活性化合物(例如手性輔助劑,諸如手性醇或Mosher氏醯基氯,或形成非鏡像異構性鹽之混合物)反應以轉化鏡像異構混合物成非鏡像異構混合物、分離該等非鏡像異構物及轉化(例如水解或脫鹽)個別非鏡像異構物為對應之純鏡像異構物,可分離鏡像異構物。藉由使用手性HPLC管柱,亦可分離鏡像異構物。
本發明之化合物可生成醫藥上可接受之鹽,其亦屬於本發明之範圍。“醫藥上可接受之鹽”係指式(I’)化合物之自由酸或鹼的鹽,其係非毒性、生理上可耐受、能與經調製之醫藥組成物互溶及另一方面適合調製及/或投予至個體。除非另有說明,應瞭解的是,本發明之化合物包括該化合物的醫藥上可接受之鹽。
化合物鹽包括與無機酸及/或有機酸所生成之酸性鹽及與無機鹼及/或有機鹼所生成之鹼性鹽。此外,當給予之化合物同時含有鹼性基團(諸如但不限於吡啶或咪唑)和酸性基團(諸如但不限於羧酸)時,熟習此技術之人士當能瞭解該化合物可呈兩性離子(“內鹽”),該鹽含括於本文使用之“鹽”的範圍。藉由例如令化合物與某一量(諸如當量)之適當酸或鹼於諸如使該鹽沉澱之介質或水溶性介質中反應並隨後經冷凍乾燥,可製備本發明之化合物的鹽。
例示性鹽包括但不限於硫酸鹽、檸檬酸鹽、乙酸鹽、草酸鹽、氯化物、溴化物、碘化物、硝酸鹽、硫酸氫鹽、磷酸鹽、酸式磷酸鹽、異菸鹼酸鹽、乳酸鹽、水楊酸鹽、酸式檸檬酸鹽、酒石酸鹽、油酸鹽、鞣酸鹽、泛酸鹽、酒石酸氫鹽、抗壞血酸鹽、丁二酸鹽、順丁烯二酸鹽、龍膽酸鹽、反丁烯二酸鹽、葡萄糖酸鹽、葡萄糖醛酸鹽、葡萄糖二酸鹽、甲酸鹽、苯甲酸鹽、麩胺酸鹽、甲磺酸鹽、乙磺酸鹽、苯磺酸鹽、對甲苯磺酸鹽及雙羥萘酸鹽(即1,1’-伸甲基‑雙(2-羥基-3-萘酸鹽))。醫藥上可接受之鹽可涉及包括其他分子,諸如乙酸離子、丁二酸離子或其他抗衡離子。該抗衡離子可為使母化合物上之電荷安定的任何有機或無機基團。再者,醫藥上可接受之鹽於結構上可含有超過1個帶電荷之原子。當多個帶電荷之原子係該醫藥上可接受之鹽的一部分時,實例可含有多個抗衡離子。因此,醫藥上可接受之鹽可含有1或多個帶電荷之原子及/或1或多個抗衡離子。
例示性酸加成鹽包括乙酸鹽、抗壞血酸鹽、苯甲酸鹽、苯磺酸鹽、硫酸氫鹽、硼酸鹽、丁酸鹽、檸檬酸鹽、樟腦酸鹽、樟腦磺酸鹽、反丁烯二酸鹽、氫氯酸鹽、氫溴酸鹽、氫碘酸鹽、乳酸鹽、順丁烯二酸鹽、甲磺酸鹽、萘磺酸鹽、硝酸鹽、草酸鹽、磷酸鹽、丙酸鹽、水楊酸鹽、丁二酸鹽、硫酸鹽、酒石酸鹽、硫氰酸鹽、甲苯磺酸鹽及類似者。
例示性鹼鹽包括銨鹽、鹼金屬鹽(諸如鈉鹽、鋰鹽及鉀鹽)、鹼土金屬鹽(諸如鈣鹽和鎂鹽)、有機鹼(例如有機胺,諸如二環己基胺和三級丁基胺)鹽及胺基酸(諸如精胺酸和離胺酸)鹽及類似者。含有氮之鹼性基團可經試劑季胺化,該試劑係諸如低碳烷基鹵化物(例如甲基氯、乙基氯、丁基氯、甲基溴、乙基溴、丁基溴、甲基碘、乙基碘及丁基碘)、硫酸二烷酯(例如硫酸二甲酯、硫酸二乙酯及硫酸二丁酯)、長鏈鹵化物(例如癸基氯、癸基溴、癸基碘、十二烷基氯、十二烷基溴、十二烷基碘、十八烷基氯、十八烷基溴及十八烷基碘)、芳烷基鹵化物(例如苄基溴和苯乙基溴)及類似者。
此外,通常被認為適合用於自醫藥化合物形成醫藥上有用之鹽的酸和鹼係見於文獻,例如P. Stahl et al, Camille G.(eds.)Handbook of Pharmaceutical Salts. Properties, Selection and Use.(2002)Zurich: Wiley-VCH; S. Berge et al, Journal of Pharmaceutical Sciences(1977)66(1)1-19; P. Gould, International J. of Pharmaceutics(1986)33 201-217; Anderson et al, The Practice of Medicinal Chemistry(1996), Academic Press, New York;及The Orange Book(Food & Drug Administration, MD,可自FDA取得)。該等文獻併入本文作為參考。
此外,本文描述之任何化合物亦欲係指任何非溶劑化形式或該化合物之水合物、溶劑合物或多晶型及彼等之混合物,即使該等形式並未明確列示。“溶劑合物”表示本發明之化合物與一或多個溶劑分子之物理性結合。該物理性結合涉及不同程度之離子性和共價性鍵結,其包括氫鍵。於某些實例中,該溶劑合物將可被分離,例如當一或多個溶劑分子併入結晶固體之晶格時。“溶劑合物”包含溶液相和可被分離之溶劑合物二者。適當之溶劑合物包括與醫藥上可接受之溶劑(諸如水、乙醇及類似者)形成之溶劑合物。於某些實施態樣中,該溶劑係水且該溶劑合物係水合物。
本發明之一或多個化合物係可選擇地被轉化為溶劑合物。通常製備溶劑合物之方法係屬習知。因此,文獻例如M. Caira et al., J. Pharmaceutical Sci., 93(3), 601-611(2004)描述使用乙酸乙酯和水製備抗真菌劑“氟可那挫” (fluconazole)之溶劑合物。溶劑合物、半溶劑合物、水合物及類似者之相似製備係描述於文獻E. C. van Tonder et al, AAPS PharmSciTech., 5(1), article 12(2004)和A. L. Bingham et al, Chem. Commun., 603-604(2001)。典型之非限制性方法涉及於高於周溫之溫度下溶解本發明之化合物於適量之溶劑(有機溶劑或水或彼等之混合物)中並於足以形成結晶之速率下冷卻溶液,隨後藉由標準方法分離該結晶。分析方法(諸如例如紅外光譜術)顯示於呈溶劑合物(或水合物)之結晶中存在溶劑(或水)。
本文給予之任何化學式亦欲表示化合物之未經標記和經同位素標記的形式。經同位素標記之化合物具有本文給予之化學式所示的結構,除了其中一或多個原子係經具有選擇之原子量或原子數的原子替代。可併入本發明之化合物的同位素之實例包括氫、碳、氮、氧、磷、氟、氯及碘之同位素,分別諸如2
H、3
H、11
C、13
C、14
C、15
N、18
O、17
O、31
P、32
P、35
S、18
F、36
Cl及125
I。該等經同位素標記之化合物可用於代謝研究(例如使用14
C)、反應動力學研究(例如使用2
H或3
H)、偵測或顯像技術[諸如正電子發射電腦斷層掃描(PET)或單光子發射電腦斷層掃描攝影術(SPECT)],其包括藥物或受質組織分佈檢測或病患之放射治療。特定地,經18
F或11
C標記之化合物可特別適用於PET或SPECT研究。再者,經較重之同位素(諸如氘(2
H))取代可提供因較佳之代謝安定性所導致的某些治療優異,例如增加之活體內半生期或降低之劑量需要。通常藉由實施反應圖或實施例描述之方法和下述之製備,經可方便取得之經同位素標記的反應劑替代未經同位素標記的反應劑,可製備本發明之經同位素標記之化合物。
對於本文描述之化合物,“鹽”、“溶劑合物”、“多晶型”及類似者將欲等同地應用於本發明之化合物的鏡像異構物、立體異構物、旋轉異構物、互變異構物、阻旋異構物及消旋物之鹽、溶劑合物及多晶型。
化學命名工具係軟體ChemDraw專業版16.0。
本發明之化合物的說明
本發明關於特定之分子和彼之醫藥上可接受之鹽或異構物。本發明另關於可用於調節功能失調性XPO1活性之分子和彼之醫藥上可接受之鹽、溶劑合物、酯或異構物。
本發明關於本文描述之化合物和彼之醫藥上可接受之鹽、溶劑合物、酯或異構物及包含本文描述之一或多種化合物和彼之醫藥上可接受之鹽或異構物之醫藥組成物。
本發明之一方面提供用於調節哺乳動物之XPO1活性之化合物、組成物、套組及解毒劑,彼等含有式(I’)、式(II’)或式(III’)之結構的化合物:

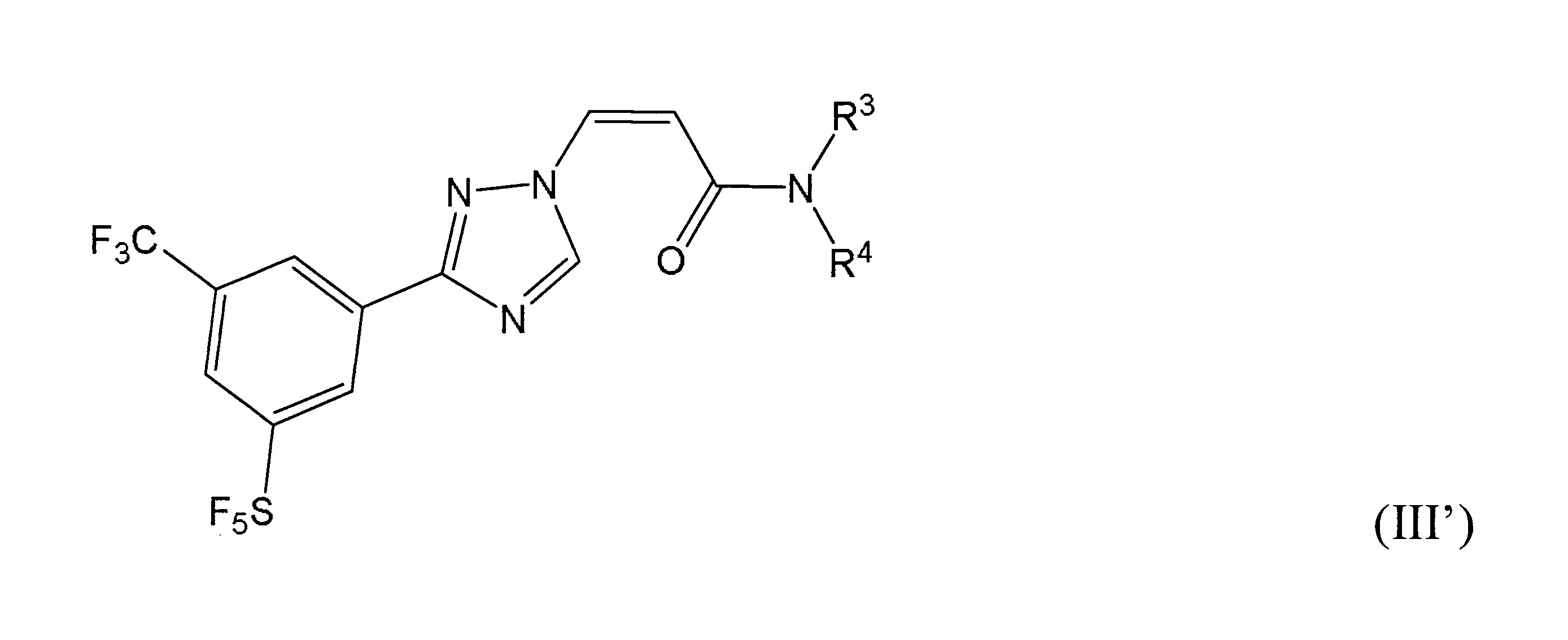 或彼等之立體異構體、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中
R1
係獨立地選自-C(=O)-R2
、C3-6
雜環烷基或C5-10
雜芳基,R1
之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、 -NO2
、鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基;且
R2
係獨立地選自C1-6
烷基、C3-6
環烷基或C3-6
雜環烷基,R2
之任何烷基、環烷基及雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基;且
R3
和R4
係獨立地選自C1-6
烷基、經取代之C1-6
烷基、或R3
和R4
與彼等連接之N一起形成經取代或未經取代之C4-10
環烷基胺基,R3
和R4
之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基。
於本發明之一實施態樣中,該化合物具有式(I’)結構。
於本發明之另一實施態樣中,該化合物具有式(II’)結構且含有R1
之雙鍵的構形係E或Z。
再於本發明之另一實施態樣中,該化合物具有式(III’)結構。
再於另一實施態樣中,該化合物具有式(I’)結構且R1
係-C(=O)-R2
。
再於另一實施態樣中,R2
係C1-6
烷基。
再於另一實施態樣中,R2
係C3-6
環烷基。
再於另一實施態樣中,R2
係經取代之C1-6
烷基,其中取代基選自甲基、羥基或鹵素。
再於另一實施態樣中,R2
係經取代之C3-6
環烷基,其中取代基選自甲基、羥基或鹵素。
再於另一實施態樣中,R2
係選自甲基、乙基、正丙基、異丙基、環丙基、正丁基、三級丁基、環丁基、異丁基、4-甲基-2-戊基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、環戊烯基、四氫呋喃基、
或彼等之立體異構體、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中
R1
係獨立地選自-C(=O)-R2
、C3-6
雜環烷基或C5-10
雜芳基,R1
之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、 -NO2
、鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基;且
R2
係獨立地選自C1-6
烷基、C3-6
環烷基或C3-6
雜環烷基,R2
之任何烷基、環烷基及雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基;且
R3
和R4
係獨立地選自C1-6
烷基、經取代之C1-6
烷基、或R3
和R4
與彼等連接之N一起形成經取代或未經取代之C4-10
環烷基胺基,R3
和R4
之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12
烷基、C2-12
烯基、C2-12
炔基、C1-12
烷氧基、C1-12
鹵烷基、C1-12
鹵烷氧基或C1-12
烷硫基。
於本發明之一實施態樣中,該化合物具有式(I’)結構。
於本發明之另一實施態樣中,該化合物具有式(II’)結構且含有R1
之雙鍵的構形係E或Z。
再於本發明之另一實施態樣中,該化合物具有式(III’)結構。
再於另一實施態樣中,該化合物具有式(I’)結構且R1
係-C(=O)-R2
。
再於另一實施態樣中,R2
係C1-6
烷基。
再於另一實施態樣中,R2
係C3-6
環烷基。
再於另一實施態樣中,R2
係經取代之C1-6
烷基,其中取代基選自甲基、羥基或鹵素。
再於另一實施態樣中,R2
係經取代之C3-6
環烷基,其中取代基選自甲基、羥基或鹵素。
再於另一實施態樣中,R2
係選自甲基、乙基、正丙基、異丙基、環丙基、正丁基、三級丁基、環丁基、異丁基、4-甲基-2-戊基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、環戊烯基、四氫呋喃基、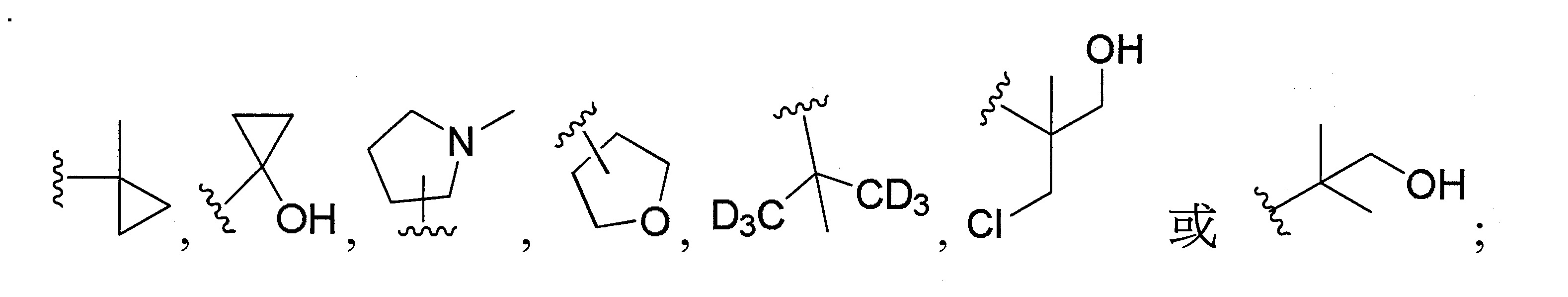 更佳地,R2
係選自異丙基、環丙基、環丁基、4-甲基-2-戊基、三級丁基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、
更佳地,R2
係選自異丙基、環丙基、環丁基、4-甲基-2-戊基、三級丁基、2,2-二甲基-1-丁基、3,3-二甲基-1-丁基、2-乙基-1-丁基、 最佳地,R2
係選自三級丁基、2,2-二甲基-1-丁基、
最佳地,R2
係選自三級丁基、2,2-二甲基-1-丁基、 ;
可選擇地,R2
係選自2-甲基環氧乙烷基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、正戊基、異戊基、新戊基、2,2-二甲基丁基或
;
可選擇地,R2
係選自2-甲基環氧乙烷基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、正戊基、異戊基、新戊基、2,2-二甲基丁基或 。
再於另一實施態樣中,R1
係C3-6
雜環烷基,其中1或2個碳原子係經氮原子替代。
於某些實施態樣中,R1
係C5-6
雜芳基,其中1或2個碳原子係經氮原子或硫原子替代。
於某些實施態樣中,R1
係C6
雜芳基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自-NH2
、-OH、鹵素或-CN。
於某些實施態樣中,R1
係選自未經取代或經取代之
。
再於另一實施態樣中,R1
係C3-6
雜環烷基,其中1或2個碳原子係經氮原子替代。
於某些實施態樣中,R1
係C5-6
雜芳基,其中1或2個碳原子係經氮原子或硫原子替代。
於某些實施態樣中,R1
係C6
雜芳基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自-NH2
、-OH、鹵素或-CN。
於某些實施態樣中,R1
係選自未經取代或經取代之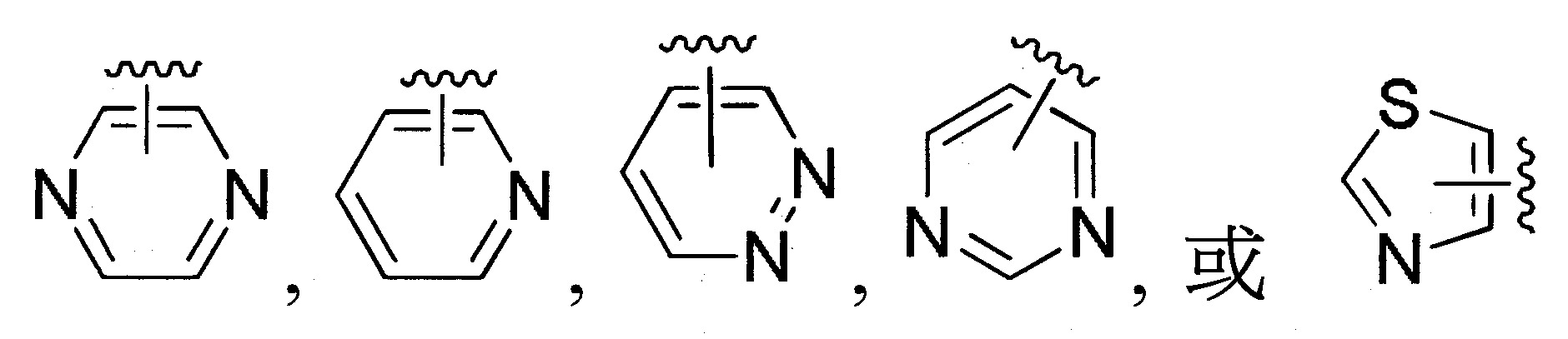 ;
更佳地,R1
係選自未經取代或經取代之
;
更佳地,R1
係選自未經取代或經取代之 ;
可選擇地,R1
係選自未經取代或經取代之呋喃基、吡咯基、咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基或噻吩基。
於某些實施態樣中,R1
係經取代之C3-6
雜環烷基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自鹵素或-CN。
於某些實施態樣中,R1
係未經取代或經取代之C5-10
雜芳基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自鹵素或-CN。
於某些實施態樣中,R1
係選自未經取代或經取代之;
;
可選擇地,R1
係選自未經取代或經取代之呋喃基、吡咯基、咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基或噻吩基。
於某些實施態樣中,R1
係經取代之C3-6
雜環烷基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自鹵素或-CN。
於某些實施態樣中,R1
係未經取代或經取代之C5-10
雜芳基,其中1或2個碳原子係經氮原子替代,且該1或多個取代基係選自鹵素或-CN。
於某些實施態樣中,R1
係選自未經取代或經取代之; 更佳地,R1
係未經取代或經取代之
更佳地,R1
係未經取代或經取代之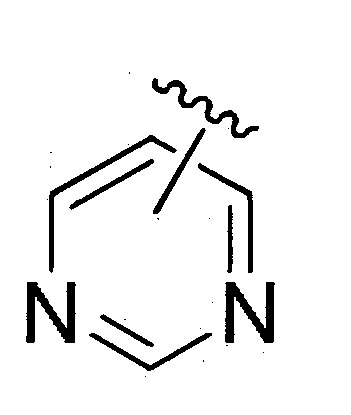 ;
可選擇地,R1
係選自未經取代或經取代之苯基、萘基、呋喃基、苯並呋喃基、吡咯基、咪唑基、苯並咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基、噻吩基、苯並噻吩基、吲哚基、嘌呤基、喹啉基、異喹啉基或啡噁噻基。
於某些實施態樣中,該化合物係式(I’)化合物,且R1
係-C(=O)-R2
。
於某些實施態樣中,該化合物係式(I’)化合物,且R1
係C6
雜芳基,其中1或2個碳原子和相連接之氫原子係經氮原子替代。
於某些實施態樣中,R3
和R4
係獨立地C1-4
烷基,或R3
和R4
與彼等連接之N一起形成C4-10
環烷基胺基環。
於某些實施態樣中,R3
和R4
與彼等連接之N一起形成經取代之C4-10
環烷基胺基,其中取代基係選自甲基、乙基、羥基或鹵素。
於某些實施態樣中,R3
和R4
結合一起形成環烷基胺基,其中C4-10
環烷基胺基係選自
;
可選擇地,R1
係選自未經取代或經取代之苯基、萘基、呋喃基、苯並呋喃基、吡咯基、咪唑基、苯並咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基、噻吩基、苯並噻吩基、吲哚基、嘌呤基、喹啉基、異喹啉基或啡噁噻基。
於某些實施態樣中,該化合物係式(I’)化合物,且R1
係-C(=O)-R2
。
於某些實施態樣中,該化合物係式(I’)化合物,且R1
係C6
雜芳基,其中1或2個碳原子和相連接之氫原子係經氮原子替代。
於某些實施態樣中,R3
和R4
係獨立地C1-4
烷基,或R3
和R4
與彼等連接之N一起形成C4-10
環烷基胺基環。
於某些實施態樣中,R3
和R4
與彼等連接之N一起形成經取代之C4-10
環烷基胺基,其中取代基係選自甲基、乙基、羥基或鹵素。
於某些實施態樣中,R3
和R4
結合一起形成環烷基胺基,其中C4-10
環烷基胺基係選自 。
於某些實施態樣中,該化合物係式(III’)化合物,R3
和R4
與彼等連接之N一起形成經取代之環烷基胺基,其中取代基係選自甲基、羥基或鹵素,且該1或多個取代基係鹵素。
於某些實施態樣中,該化合物係選自式(I’)、式(II’)或式(III’)化合物。
於下述中,化學命名係基於ChemDraw專業版16.0。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(E)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼。
於某些實施態樣中,該化合物係(Z)-N'-異丁醯基-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼。
於某些實施態樣中,該化合物係(Z)-1-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼。
於某些實施態樣中,該化合物係(Z)-N'-乙醯基-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噠嗪-3-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-羥基-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-氟-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-1-羥基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)四氫呋喃-3-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)四氫呋喃-2-卡肼。
於某些實施態樣中,該化合物係(Z)-3-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)氧雜環丁烷-3-卡肼。
於某些實施態樣中,該化合物係(Z)-1-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)吡咯啶-3-卡肼。
於某些實施態樣中,該化合物係(E)-2-(2-氟嘧啶-5-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-2-(2-氟吡啶-4-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-2-(5-氰基吡啶-3-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(喹啉-3-基)丙烯醯胺。
本發明之另一方面提供用於調節哺乳動物之XPO1活性的化合物、組成物、套組及解毒劑,其中該化合物係選自由下列所組成之群組:
。
於某些實施態樣中,該化合物係式(III’)化合物,R3
和R4
與彼等連接之N一起形成經取代之環烷基胺基,其中取代基係選自甲基、羥基或鹵素,且該1或多個取代基係鹵素。
於某些實施態樣中,該化合物係選自式(I’)、式(II’)或式(III’)化合物。
於下述中,化學命名係基於ChemDraw專業版16.0。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(E)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼。
於某些實施態樣中,該化合物係(Z)-N'-異丁醯基-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼。
於某些實施態樣中,該化合物係(Z)-1-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼。
於某些實施態樣中,該化合物係(Z)-N'-乙醯基-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(二氟甲基)-5-(五氟硫基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噠嗪-3-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-羥基-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-N'-(2-氟-2-甲基丙醯基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼。
於某些實施態樣中,該化合物係(Z)-1-羥基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)四氫呋喃-3-卡肼。
於某些實施態樣中,該化合物係(Z)-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)四氫呋喃-2-卡肼。
於某些實施態樣中,該化合物係(Z)-3-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)氧雜環丁烷-3-卡肼。
於某些實施態樣中,該化合物係(Z)-1-甲基-N'-(3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)吡咯啶-3-卡肼。
於某些實施態樣中,該化合物係(E)-2-(2-氟嘧啶-5-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-2-(2-氟吡啶-4-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-2-(5-氰基吡啶-3-基)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯胺。
於某些實施態樣中,該化合物係(E)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(喹啉-3-基)丙烯醯胺。
本發明之另一方面提供用於調節哺乳動物之XPO1活性的化合物、組成物、套組及解毒劑,其中該化合物係選自由下列所組成之群組: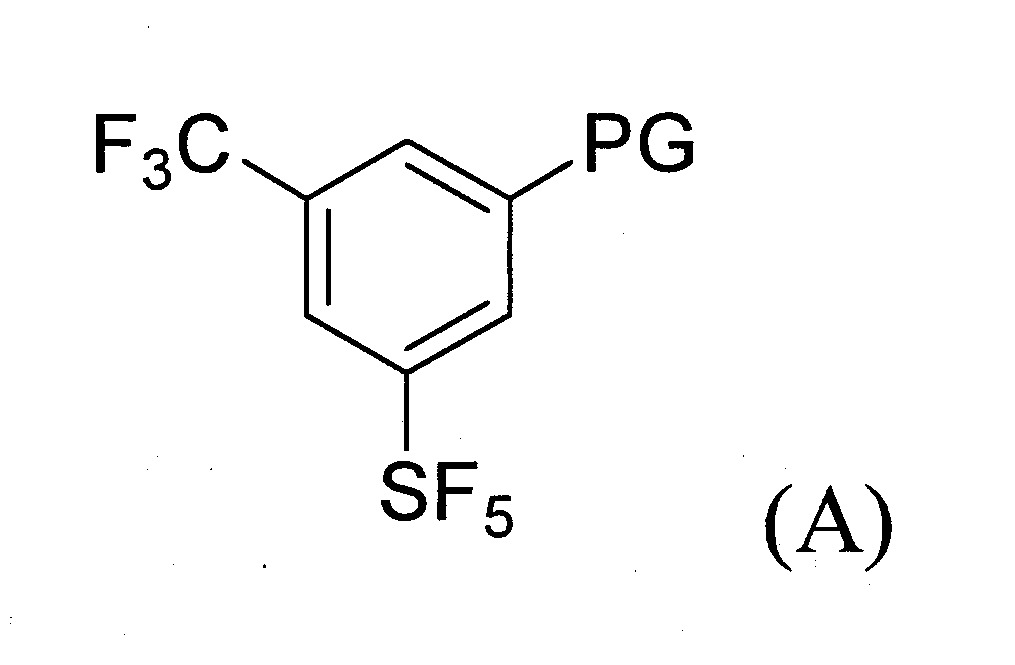 其中PG係鹵素(Cl、Br或I)、CN、N3
、NH2
、-COO-C1-6
烷基、C2-6
烯基或C2-6
烯基-芳基;
(b)令該式(A)化合物與一或多種適當之反應劑反應以生成式(B)化合物:
其中PG係鹵素(Cl、Br或I)、CN、N3
、NH2
、-COO-C1-6
烷基、C2-6
烯基或C2-6
烯基-芳基;
(b)令該式(A)化合物與一或多種適當之反應劑反應以生成式(B)化合物: (c)令步驟(b)之反應產物與式(C)化合物反應以生成式(D)化合物:
(c)令步驟(b)之反應產物與式(C)化合物反應以生成式(D)化合物: 其中R1
係如上述式(I’)、(II’)或(III’)化合物所定義者。
於某些實施態樣中,提供製備式(A)化合物之方法,其包含下述步驟:
(a)提供式(M)化合物:
其中R1
係如上述式(I’)、(II’)或(III’)化合物所定義者。
於某些實施態樣中,提供製備式(A)化合物之方法,其包含下述步驟:
(a)提供式(M)化合物: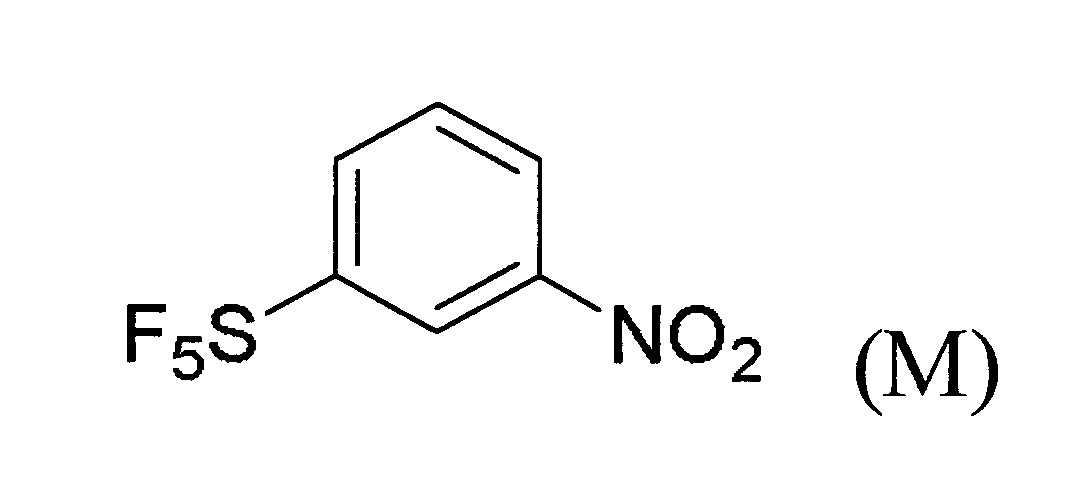 (b)令該式(M)化合物與適當之反應劑反應以生成式(N)化合物:
(b)令該式(M)化合物與適當之反應劑反應以生成式(N)化合物: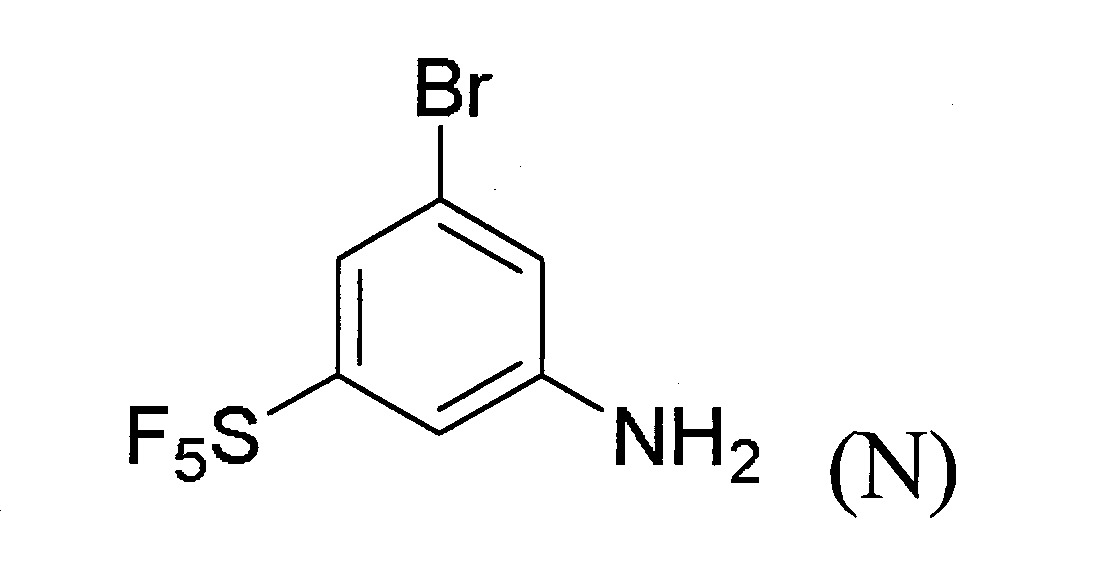 (c)令步驟(b)之反應產物與適當之反應劑反應以生成式(J)化合物:
(c)令步驟(b)之反應產物與適當之反應劑反應以生成式(J)化合物: (d)令步驟(c)之反應產物與適當之反應劑反應以生成式(K)化合物:
(d)令步驟(c)之反應產物與適當之反應劑反應以生成式(K)化合物: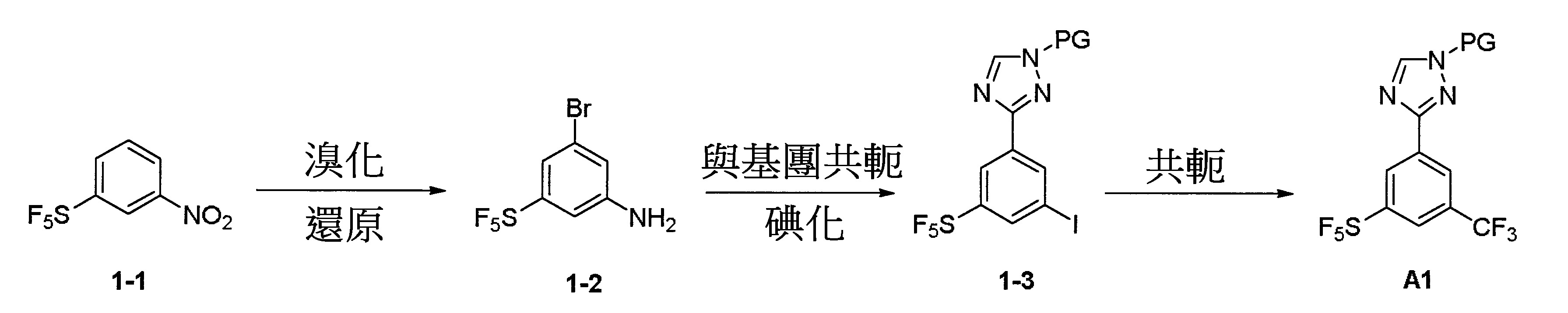 依據文獻(1. Chemistry-A European Journal,2012, 18, 10234-10238; 2. WO2013/19548; 3. Bioorganic & Medicinal Chemistry, 2008, 16, 9487-9497; 4. Organic Letter, 2014, 16, 4268-4271)揭露之相關方法但不限於該等揭露之方法可進行合成結構1A化合物之合成。其中PG係獨立地選自鹵素(Cl、Br、I)、CN、N3
、NH2
、-COO-C1-6
烷基、C2-6
烯基或C2-6
烯基-芳基。硝基化合物1-1經NBS溴化並隨後經鐵還原以生成中間體1-2,該中間體1-2進一步經氰化和碘化以生成中間體1-3。隨後將三氟甲基加至芳基環上以生成重要之中間體化合物A1。
反應圖2:使用下述反應順序以合成結構A2化合物
依據文獻(1. Chemistry-A European Journal,2012, 18, 10234-10238; 2. WO2013/19548; 3. Bioorganic & Medicinal Chemistry, 2008, 16, 9487-9497; 4. Organic Letter, 2014, 16, 4268-4271)揭露之相關方法但不限於該等揭露之方法可進行合成結構1A化合物之合成。其中PG係獨立地選自鹵素(Cl、Br、I)、CN、N3
、NH2
、-COO-C1-6
烷基、C2-6
烯基或C2-6
烯基-芳基。硝基化合物1-1經NBS溴化並隨後經鐵還原以生成中間體1-2,該中間體1-2進一步經氰化和碘化以生成中間體1-3。隨後將三氟甲基加至芳基環上以生成重要之中間體化合物A1。
反應圖2:使用下述反應順序以合成結構A2化合物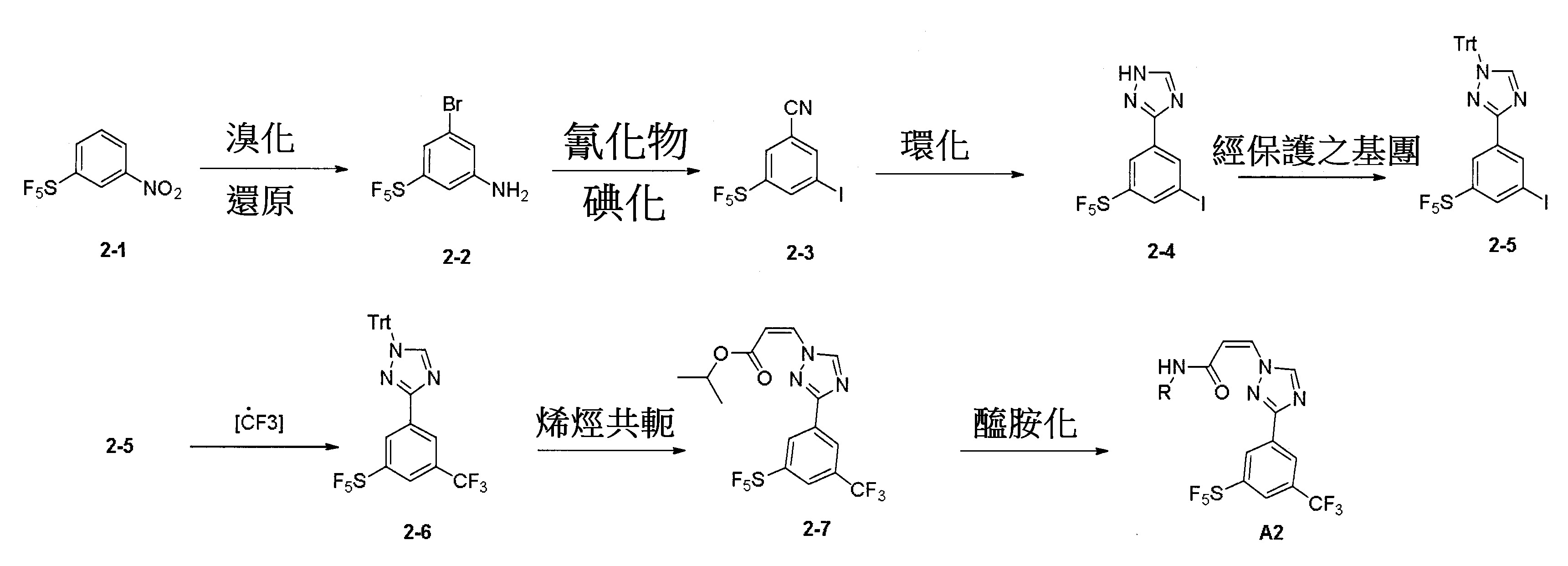 另有可替代之方法以合成化合物A2。依據文獻(1. Chemistry-A European Journal,2012, 18, 10234-10238; 2. WO2013/19548; 3. Bioorganic & Medicinal Chemistry, 2008, 16, 9487-9497; 4. Organic Letter, 2014, 16, 4268-4271)揭露之相關方法但不限於該等揭露之方法,可進行合成結構A2化合物。硝基化合物2-1經NBS溴化並隨後經鐵還原以生成中間體2-2,該中間體2-2進一步經氰化和碘化以生成化合物2-3。該化合物2-3於酸條件下經環化以生成三唑化合物2-4,且使用三苯基甲基氯(TrtCl)以保護該三唑之胺基以生成安定之化合物2-5。使用經銅催化之三氟甲基化方法以導入三氟甲基以生成化合物2-6。隨後,令該化合物2-6與烯烴共軛並經醯胺化以得到終產物A2。
反應圖3:使用下述反應順序以合成結構A3化合物
另有可替代之方法以合成化合物A2。依據文獻(1. Chemistry-A European Journal,2012, 18, 10234-10238; 2. WO2013/19548; 3. Bioorganic & Medicinal Chemistry, 2008, 16, 9487-9497; 4. Organic Letter, 2014, 16, 4268-4271)揭露之相關方法但不限於該等揭露之方法,可進行合成結構A2化合物。硝基化合物2-1經NBS溴化並隨後經鐵還原以生成中間體2-2,該中間體2-2進一步經氰化和碘化以生成化合物2-3。該化合物2-3於酸條件下經環化以生成三唑化合物2-4,且使用三苯基甲基氯(TrtCl)以保護該三唑之胺基以生成安定之化合物2-5。使用經銅催化之三氟甲基化方法以導入三氟甲基以生成化合物2-6。隨後,令該化合物2-6與烯烴共軛並經醯胺化以得到終產物A2。
反應圖3:使用下述反應順序以合成結構A3化合物 實施例1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼(I)
實施例1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼(I) 步驟1:合成丙炔酸異丙酯(2)
於25℃下對丙炔酸1(30 g,0.43莫耳)於異丙醇(100 ml)之混合物加入BF3
乙醚絡合物(122.1 g,0.86莫耳)。經攪拌10分鐘後,令反應混合物經加熱至90℃並經攪拌2小時。藉由TLC監測反應完全。令反應混合物降低至25至30℃並經碎冰驟冷且隨後經二氯甲烷(2×150 ml)萃取。令有機層先後經水和鹽水溶液沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以生成該丙炔酸異丙酯(35.2 g,73%產率)。
步驟2:合成(Z)-3-碘丙烯酸異丙酯(3)
於25℃下將丙炔酸異丙酯2(35.2 g,0.31毫莫耳)加入至乙酸(200 ml)並令反應混合物經攪拌10分鐘。於攪拌下加入碘化鈉(70.0 g,0.47毫莫耳)(觀察到暗褐色)。提高溫度至110℃並維持反應物於該溫度下達2小時。藉由TLC監測反應完全。令反應混合物冷卻至室溫,經冰冷水(200 ml)驟冷且經攪拌30分鐘。將甲基三級丁基醚(MTBE)(200 ml)加入至該反應混合物並再經攪拌30分鐘。分離有機層並令水層再經MTBE(200 ml)萃取。令結合之有機層經NaHCO3
(2×100 ml)、NaHSO3
(2×100 ml)及鹽水(100 ml)沖洗,置於硫酸鈉上乾燥並於35℃下經真空下濃縮以生成呈淡黃色油之(Z)-3-碘丙烯酸異丙酯(48.5 g,61%產率)。1
H NMR(400 MHz, CDC13
)δ 7.38(d, J=8.0 Hz, 1 H), 6.83(d, J=8.0 Hz, 1 H), 5.07-5.13(m, 1 H), 1.27(d, J=8.0 Hz, 6 H)。
步驟3:合成(3-溴-5-硝基苯基)五氟硫烷(5)
將化合物4(9.25 g,37.1毫莫耳)、TFA(20 ml)及濃H2
SO4
(100 ml)加入至圓底燒瓶(250 ml),隨後令混合物經劇烈攪拌並經30分鐘分批加入NBS(9.92 g,55.7毫莫耳)且令反應物於25℃下經攪拌12小時。將混合物倒入至冰水並經EA(3×100 ml)萃取。令結合之有機層經飽和NaHCO3
(3×100 ml)和水(3×100 ml)沖洗,置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=25:1)純化以得到呈淡黃色油之標的化合物5(12.04 g,99%產率)。1
H NMR(400 MHz, CDC13
)δ 8.57(s, 1 H), 8.55(s, 1 H), 8.22(s, 1 H)。
步驟4:合成3-溴-5-(五氟硫基)苯胺(6)
於25℃下對化合物5(5.9 g,18.0毫莫耳)於MeOH(40 ml)和水(10 ml)之溶液加入Fe(5.0 g,90毫莫耳)和NH4
Cl(4.8 g,90毫莫耳)。令混合物於90℃下經攪拌2小時並隨後經過濾,經MeOH(20 ml)沖洗且除去溶劑。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機相置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=10:1)純化以得到呈淡黃色油之標的化合物6(5.1 g,96%產率)。1
H NMR(400 MHz, CDC13
)δ 7.35(s, 1 H), 7.20(s, 1 H), 6.99(s, 1 H)。
步驟5:合成3-胺基-5-(五氟硫基)苄腈(7)
對圓底燒瓶(250 ml)加入化合物6(8.68 g,29.2毫莫耳)和NMP(100 ml)並隨後加入CuCN(5.26 g,58.5毫莫耳)且令反應物於180℃和N2
氛圍下經攪拌6小時。令混合物經冷卻至25℃並經過濾。對濾液加入EA(200 ml)並經水(3×200 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=10:1)純化以得到呈淡黃色固體之標的化合物7(3.3 g,46%產率)。
步驟6:合成3-碘-5-(五氟硫基)苄腈(8)
令化合物7(4.23 g,17.3毫莫耳)懸浮於濃H2
SO4
(8.7 ml)與水(17.3 ml)之混合物並經冷卻至0℃。經1小時加入NaNO2
(1.23 g,17.9毫莫耳)之水(3.5 ml)溶液並令所生成之混合物於0℃下再經攪拌1小時。隨後經1小時逐滴加入CuI(173 mg,0.87毫莫耳)和KI(3.05 g,18毫莫耳)之水(3.5 ml)溶液並隨後令混合物於25℃下經攪拌10小時。對該混合物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈淡黃色固體之標的化合物8(3.65 g,59%產率)。1
H NMR(400 MHz, CDC13
)δ 8.28(s, 1 H), 8.13(s, 1 H), 8.01(s, 1 H)。
步驟7:合成3-碘-5-(五氟硫基)苯硫代甲醯胺(benzothioamide) (9)
對化合物8(3.6 g,10毫莫耳)之DMF(50 ml)溶液加入NaHS(1.1 g,20毫莫耳)和MgCl2 .
6H2
O(2 g,10毫莫耳)並令混合物於25℃下經攪拌2小時。對該混合物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以得到粗製產物9(3.8 g,98%產率),其未經任何純化而用於下一個步驟。
MS(ESI): [M+H+
]=389.9。
步驟8:合成3-(3-碘-5-(五氟硫基)苯基)-1H-1,2,4-三唑(10)
對化合物9(3.32 g,8.53毫莫耳)之DMF(15 ml)溶液加入N2
H4 .
H2
O(853 mg,17.1毫莫耳)並令混合物於25℃下經攪拌3小時。隨後加入甲酸(10 ml)並令混合物於90℃下經攪拌3小時。令反應物經冷卻至25℃並經飽和NaHCO3
(50 ml)驟冷且經EA(50 ml)萃取。令有機層經鹽水(50 ml)沖洗,置於硫酸鈉上乾燥並經真空下濃縮以得到粗製產物10(3.36 g,99%產率),其未經進一步純化而用於下一個步驟。
MS(ESI): [M+H+
]=397.9。
步驟9:合成3-(3-碘-5-(五氟硫基)苯基)-1-三苯甲基-1H-1,2,4-三唑(11)
對化合物10(3.37 g,8.53毫莫耳)之DCM(50 ml)溶液加入Et3
N(1.29 g,12.8毫莫耳)和TrtCl(3.57 g,12.8毫莫耳)並令混合物於25℃下經攪拌2小時且隨後經水(3×50 ml)沖洗。令有機層經乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為DCM:MeOH=50:1)純化以得到呈淡黃色固體之標的化合物11(5.36 g,98%產率)。1
H NMR(400 MHz, CDCl3
)δ 8.57(s, 1 H), 8.46(s, 1 H), 8.05(s, 1 H), 8.01(s, 1 H), 7.37-7.15(m, 15 H)。
步驟10:合成3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1-三苯甲基-1H-1,2,4-三唑(12)
對三頸燒瓶(100 ml)載入化合物11(5.36 g,8.4毫莫耳)、KF(1.46 g,25.2毫莫耳)、CuI(320 mg,1.7毫莫耳)及1,10-啡啉(306 mg,1.7毫莫耳)並隨後於氮氛圍下對該燒瓶內粗製材料抽真空3次。隨後將DMSO(20 ml)與DMF(10 ml)之混合溶液、TMSCF3
(4.76 g,33.6毫莫耳)及B(OMe)3
(2.55 g,25.2毫莫耳)加入至上述反應溶液並隨後令反應物於70℃和N2
氛圍下經攪拌24小時。令混合物經冷卻至25℃並經過濾。對濾液加入EA(100 ml)並經水(3×50 ml)和鹽水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以得到呈黑色固體之標的化合物12(4.88 g,99%產率),其未經任何純化而用於下一個步驟。19
F NMR(400 MHz, CDCl3
)δ 62.45 ppm, -62.79 ppm。
步驟11:合成3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑(13)
對化合物12(4.88 g,8.4毫莫耳)之DCM(50 ml)溶液加入TFA(1.67 g,14.3毫莫耳)和Et3
SiH(1.95 g,16.8毫莫耳),令混合物於25℃下經攪拌1小時且隨後於真空下經濃縮以除去該有機溶液。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=1:1)純化以得到呈淡黃色固體之標的化合物13(2.1 g,74%產率)。
MS(ESI): [M+H+
]=340.1。
步驟12:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(14)
對化合物13(2.1 g,6.2毫莫耳)之DMF(15 ml)溶液加入DABCO(1.39 g,12.4毫莫耳)和化合物3(2.08 g,8.7毫莫耳),令混合物於25℃下經攪拌3小時且隨後經飽和NH4
Cl (50 ml)驟冷並經EA(100 ml)萃取。令有機層經鹽水(3×50 ml)沖洗,置於硫酸鈉上乾燥且經過濾和真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=30:1)純化以得到呈白色固體之標的化合物14(2.09 g,75%產率)。
MS(ESI): [M+H+
]=452.1。1
H NMR(400 MHz, CDCl3
)δ 8.64(s, 1 H), 8.51(s, 1 H), 8.34(s, 1 H), 8.11(s, 1 H), 7.96(d, J=14.1 Hz, 1 H), 7.69(d, J=13.7 Hz, 1 H), 5.15(m, 1 H), 1.32(d, J=6.3 Hz, 6 H)。
步驟13:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸(15)
對化合物14(2.09 g,4.63毫莫耳)之THF(20 ml)溶液加入LiOH.
H2
O(3.34 g,55.6毫莫耳,1N,56 ml H2
O),令混合物於25℃下經攪拌3小時且隨後經1N HCl酸化至pH=3並經EA(3×100 ml)萃取。令有機層經飽和NaHCO3
(50 ml)沖洗,置於硫酸鈉上乾燥且經真空下濃縮以得到呈白色固體之標的化合物15(1.89 g,99%產率)。
MS(ESI): [M+H+
]=410.0。
步驟14:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼(I)
對化合物15(1.01 g,2.5毫莫耳)、化合物16(826 mg,7.5毫莫耳)、DIEA(1.29 g,10毫莫耳)之EA/DCM(10 ml/10 ml,v/v)溶液加入T3
P(6.36 g,10毫莫耳,50% EA溶液)。令混合物於-40℃下經攪拌3小時且隨後令有機溶液經真空下濃縮。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機相經濃縮並經製備性TLC(DCM:MeOH:NH3 .
H2
O= 15:1:0.1)純化以得到呈黃色固體之標的化合物I(594 mg,48%產率)。
MS(ESI): [M+H+
]=502.1。1
H NMR(400 MHz, DMSO-d6)δ 10.51(s, 1 H), 9.56(s, 1 H), 9.11(s, 1 H), 8.61(s, 1 H), 8.48(s, 1 H), 8.39(s, 1 H),8.07(s, 2 H), 7.90(s, 1 H), 7.50(d, J=10 Hz, 1 H), 6.06(d, J=10.4 Hz, 1 H)。
實施例2:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼(II)
步驟1:合成丙炔酸異丙酯(2)
於25℃下對丙炔酸1(30 g,0.43莫耳)於異丙醇(100 ml)之混合物加入BF3
乙醚絡合物(122.1 g,0.86莫耳)。經攪拌10分鐘後,令反應混合物經加熱至90℃並經攪拌2小時。藉由TLC監測反應完全。令反應混合物降低至25至30℃並經碎冰驟冷且隨後經二氯甲烷(2×150 ml)萃取。令有機層先後經水和鹽水溶液沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以生成該丙炔酸異丙酯(35.2 g,73%產率)。
步驟2:合成(Z)-3-碘丙烯酸異丙酯(3)
於25℃下將丙炔酸異丙酯2(35.2 g,0.31毫莫耳)加入至乙酸(200 ml)並令反應混合物經攪拌10分鐘。於攪拌下加入碘化鈉(70.0 g,0.47毫莫耳)(觀察到暗褐色)。提高溫度至110℃並維持反應物於該溫度下達2小時。藉由TLC監測反應完全。令反應混合物冷卻至室溫,經冰冷水(200 ml)驟冷且經攪拌30分鐘。將甲基三級丁基醚(MTBE)(200 ml)加入至該反應混合物並再經攪拌30分鐘。分離有機層並令水層再經MTBE(200 ml)萃取。令結合之有機層經NaHCO3
(2×100 ml)、NaHSO3
(2×100 ml)及鹽水(100 ml)沖洗,置於硫酸鈉上乾燥並於35℃下經真空下濃縮以生成呈淡黃色油之(Z)-3-碘丙烯酸異丙酯(48.5 g,61%產率)。1
H NMR(400 MHz, CDC13
)δ 7.38(d, J=8.0 Hz, 1 H), 6.83(d, J=8.0 Hz, 1 H), 5.07-5.13(m, 1 H), 1.27(d, J=8.0 Hz, 6 H)。
步驟3:合成(3-溴-5-硝基苯基)五氟硫烷(5)
將化合物4(9.25 g,37.1毫莫耳)、TFA(20 ml)及濃H2
SO4
(100 ml)加入至圓底燒瓶(250 ml),隨後令混合物經劇烈攪拌並經30分鐘分批加入NBS(9.92 g,55.7毫莫耳)且令反應物於25℃下經攪拌12小時。將混合物倒入至冰水並經EA(3×100 ml)萃取。令結合之有機層經飽和NaHCO3
(3×100 ml)和水(3×100 ml)沖洗,置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=25:1)純化以得到呈淡黃色油之標的化合物5(12.04 g,99%產率)。1
H NMR(400 MHz, CDC13
)δ 8.57(s, 1 H), 8.55(s, 1 H), 8.22(s, 1 H)。
步驟4:合成3-溴-5-(五氟硫基)苯胺(6)
於25℃下對化合物5(5.9 g,18.0毫莫耳)於MeOH(40 ml)和水(10 ml)之溶液加入Fe(5.0 g,90毫莫耳)和NH4
Cl(4.8 g,90毫莫耳)。令混合物於90℃下經攪拌2小時並隨後經過濾,經MeOH(20 ml)沖洗且除去溶劑。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機相置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=10:1)純化以得到呈淡黃色油之標的化合物6(5.1 g,96%產率)。1
H NMR(400 MHz, CDC13
)δ 7.35(s, 1 H), 7.20(s, 1 H), 6.99(s, 1 H)。
步驟5:合成3-胺基-5-(五氟硫基)苄腈(7)
對圓底燒瓶(250 ml)加入化合物6(8.68 g,29.2毫莫耳)和NMP(100 ml)並隨後加入CuCN(5.26 g,58.5毫莫耳)且令反應物於180℃和N2
氛圍下經攪拌6小時。令混合物經冷卻至25℃並經過濾。對濾液加入EA(200 ml)並經水(3×200 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=10:1)純化以得到呈淡黃色固體之標的化合物7(3.3 g,46%產率)。
步驟6:合成3-碘-5-(五氟硫基)苄腈(8)
令化合物7(4.23 g,17.3毫莫耳)懸浮於濃H2
SO4
(8.7 ml)與水(17.3 ml)之混合物並經冷卻至0℃。經1小時加入NaNO2
(1.23 g,17.9毫莫耳)之水(3.5 ml)溶液並令所生成之混合物於0℃下再經攪拌1小時。隨後經1小時逐滴加入CuI(173 mg,0.87毫莫耳)和KI(3.05 g,18毫莫耳)之水(3.5 ml)溶液並隨後令混合物於25℃下經攪拌10小時。對該混合物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈淡黃色固體之標的化合物8(3.65 g,59%產率)。1
H NMR(400 MHz, CDC13
)δ 8.28(s, 1 H), 8.13(s, 1 H), 8.01(s, 1 H)。
步驟7:合成3-碘-5-(五氟硫基)苯硫代甲醯胺(benzothioamide) (9)
對化合物8(3.6 g,10毫莫耳)之DMF(50 ml)溶液加入NaHS(1.1 g,20毫莫耳)和MgCl2 .
6H2
O(2 g,10毫莫耳)並令混合物於25℃下經攪拌2小時。對該混合物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以得到粗製產物9(3.8 g,98%產率),其未經任何純化而用於下一個步驟。
MS(ESI): [M+H+
]=389.9。
步驟8:合成3-(3-碘-5-(五氟硫基)苯基)-1H-1,2,4-三唑(10)
對化合物9(3.32 g,8.53毫莫耳)之DMF(15 ml)溶液加入N2
H4 .
H2
O(853 mg,17.1毫莫耳)並令混合物於25℃下經攪拌3小時。隨後加入甲酸(10 ml)並令混合物於90℃下經攪拌3小時。令反應物經冷卻至25℃並經飽和NaHCO3
(50 ml)驟冷且經EA(50 ml)萃取。令有機層經鹽水(50 ml)沖洗,置於硫酸鈉上乾燥並經真空下濃縮以得到粗製產物10(3.36 g,99%產率),其未經進一步純化而用於下一個步驟。
MS(ESI): [M+H+
]=397.9。
步驟9:合成3-(3-碘-5-(五氟硫基)苯基)-1-三苯甲基-1H-1,2,4-三唑(11)
對化合物10(3.37 g,8.53毫莫耳)之DCM(50 ml)溶液加入Et3
N(1.29 g,12.8毫莫耳)和TrtCl(3.57 g,12.8毫莫耳)並令混合物於25℃下經攪拌2小時且隨後經水(3×50 ml)沖洗。令有機層經乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為DCM:MeOH=50:1)純化以得到呈淡黃色固體之標的化合物11(5.36 g,98%產率)。1
H NMR(400 MHz, CDCl3
)δ 8.57(s, 1 H), 8.46(s, 1 H), 8.05(s, 1 H), 8.01(s, 1 H), 7.37-7.15(m, 15 H)。
步驟10:合成3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1-三苯甲基-1H-1,2,4-三唑(12)
對三頸燒瓶(100 ml)載入化合物11(5.36 g,8.4毫莫耳)、KF(1.46 g,25.2毫莫耳)、CuI(320 mg,1.7毫莫耳)及1,10-啡啉(306 mg,1.7毫莫耳)並隨後於氮氛圍下對該燒瓶內粗製材料抽真空3次。隨後將DMSO(20 ml)與DMF(10 ml)之混合溶液、TMSCF3
(4.76 g,33.6毫莫耳)及B(OMe)3
(2.55 g,25.2毫莫耳)加入至上述反應溶液並隨後令反應物於70℃和N2
氛圍下經攪拌24小時。令混合物經冷卻至25℃並經過濾。對濾液加入EA(100 ml)並經水(3×50 ml)和鹽水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮以得到呈黑色固體之標的化合物12(4.88 g,99%產率),其未經任何純化而用於下一個步驟。19
F NMR(400 MHz, CDCl3
)δ 62.45 ppm, -62.79 ppm。
步驟11:合成3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑(13)
對化合物12(4.88 g,8.4毫莫耳)之DCM(50 ml)溶液加入TFA(1.67 g,14.3毫莫耳)和Et3
SiH(1.95 g,16.8毫莫耳),令混合物於25℃下經攪拌1小時且隨後於真空下經濃縮以除去該有機溶液。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機層置於硫酸鈉上乾燥並經真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=1:1)純化以得到呈淡黃色固體之標的化合物13(2.1 g,74%產率)。
MS(ESI): [M+H+
]=340.1。
步驟12:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(14)
對化合物13(2.1 g,6.2毫莫耳)之DMF(15 ml)溶液加入DABCO(1.39 g,12.4毫莫耳)和化合物3(2.08 g,8.7毫莫耳),令混合物於25℃下經攪拌3小時且隨後經飽和NH4
Cl (50 ml)驟冷並經EA(100 ml)萃取。令有機層經鹽水(3×50 ml)沖洗,置於硫酸鈉上乾燥且經過濾和真空下濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=30:1)純化以得到呈白色固體之標的化合物14(2.09 g,75%產率)。
MS(ESI): [M+H+
]=452.1。1
H NMR(400 MHz, CDCl3
)δ 8.64(s, 1 H), 8.51(s, 1 H), 8.34(s, 1 H), 8.11(s, 1 H), 7.96(d, J=14.1 Hz, 1 H), 7.69(d, J=13.7 Hz, 1 H), 5.15(m, 1 H), 1.32(d, J=6.3 Hz, 6 H)。
步驟13:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸(15)
對化合物14(2.09 g,4.63毫莫耳)之THF(20 ml)溶液加入LiOH.
H2
O(3.34 g,55.6毫莫耳,1N,56 ml H2
O),令混合物於25℃下經攪拌3小時且隨後經1N HCl酸化至pH=3並經EA(3×100 ml)萃取。令有機層經飽和NaHCO3
(50 ml)沖洗,置於硫酸鈉上乾燥且經真空下濃縮以得到呈白色固體之標的化合物15(1.89 g,99%產率)。
MS(ESI): [M+H+
]=410.0。
步驟14:合成(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡嗪-2-基)丙烯醯肼(I)
對化合物15(1.01 g,2.5毫莫耳)、化合物16(826 mg,7.5毫莫耳)、DIEA(1.29 g,10毫莫耳)之EA/DCM(10 ml/10 ml,v/v)溶液加入T3
P(6.36 g,10毫莫耳,50% EA溶液)。令混合物於-40℃下經攪拌3小時且隨後令有機溶液經真空下濃縮。對殘餘物加入EA(100 ml)並經水(3×50 ml)沖洗。令有機相經濃縮並經製備性TLC(DCM:MeOH:NH3 .
H2
O= 15:1:0.1)純化以得到呈黃色固體之標的化合物I(594 mg,48%產率)。
MS(ESI): [M+H+
]=502.1。1
H NMR(400 MHz, DMSO-d6)δ 10.51(s, 1 H), 9.56(s, 1 H), 9.11(s, 1 H), 8.61(s, 1 H), 8.48(s, 1 H), 8.39(s, 1 H),8.07(s, 2 H), 7.90(s, 1 H), 7.50(d, J=10 Hz, 1 H), 6.06(d, J=10.4 Hz, 1 H)。
實施例2:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼(II) 步驟1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼(II)
對化合物15(40 mg,0.1毫莫耳)、2-肼基吡啶(32 mg,0.3毫莫耳)及DIEA(52 mg,0.4毫莫耳)於EA:DCM(1 ml:1 ml)之溶液加入T3
P(190 mg,0.3毫莫耳,50% EA溶液)。令混合物於-40℃下經攪拌3小時並隨後令有機溶液經真空下濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機相經濃縮並經製備性TLC(DCM:MeOH:NH3 .
H2
O= 15:1:0.1)純化以得到呈黃色固體之標的化合物II(23 mg,46%產率)。隨後與HCl/二噁烷溶液生成產物以得到固體產物。
MS(ESI): [M+H+
]=501.1。1
H NMR(400 MHz, DMSO-d6)δ 11.17(s, 1 H), 9.52(s, 1 H), 8.62(s, 1 H), 8.49(s, 1 H), 8.41(s, 1 H), 8.02(m, 2 H), 7.63(d, J=10.4 Hz, 1 H), 7.20(m, 1 H), 7.04(m, 1 H), 6.12(d, J=10.5 Hz, 1 H)。
實施例3:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼(III)
步驟1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(吡啶-2-基)丙烯醯肼(II)
對化合物15(40 mg,0.1毫莫耳)、2-肼基吡啶(32 mg,0.3毫莫耳)及DIEA(52 mg,0.4毫莫耳)於EA:DCM(1 ml:1 ml)之溶液加入T3
P(190 mg,0.3毫莫耳,50% EA溶液)。令混合物於-40℃下經攪拌3小時並隨後令有機溶液經真空下濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機相經濃縮並經製備性TLC(DCM:MeOH:NH3 .
H2
O= 15:1:0.1)純化以得到呈黃色固體之標的化合物II(23 mg,46%產率)。隨後與HCl/二噁烷溶液生成產物以得到固體產物。
MS(ESI): [M+H+
]=501.1。1
H NMR(400 MHz, DMSO-d6)δ 11.17(s, 1 H), 9.52(s, 1 H), 8.62(s, 1 H), 8.49(s, 1 H), 8.41(s, 1 H), 8.02(m, 2 H), 7.63(d, J=10.4 Hz, 1 H), 7.20(m, 1 H), 7.04(m, 1 H), 6.12(d, J=10.5 Hz, 1 H)。
實施例3:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼(III) 步驟1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸(15)
於0℃下對化合物16(150 mg,0.33毫莫耳)之乾燥THF(15 ml)攪拌溶液逐滴加入LiOH(2.6 ml,2.64毫莫耳,1M)溶液。令反應物於25℃下經攪拌3小時。隨後令反應混合物經HCl(1M)驟冷直至pH=2至3並經EA(3×20 ml)萃取。令有機層經鹽水(40 ml)沖洗,置於硫酸鈉上乾燥且經濃縮以生成呈白色固體之化合物15(125 mg,92%產率)。
MS(ESI): [M+H]+
=409.9。
步驟2:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼(III)
令化合物15(125 mg,0.31毫莫耳)之DCM:EA(5 ml:5 ml,v/v)攪拌溶液冷卻至-78℃並於0℃下加入三甲基乙醯基醯肼(54.0 mg,0.46毫莫耳)、DIEA(80.0 mg,0.62毫莫耳)及T3
P(395 mg,0.62毫莫耳,50重量% EA溶液)。令反應物於25℃下經攪拌3小時並隨後經飽和氯化銨(20 ml)驟冷。令混合物經DCM萃取並令有機層經鹽水沖洗,置於硫酸鈉上乾燥且經真空下濃縮以生成粗製白色固體。將該固體加入至MeCN(5 ml)並於25℃下經攪拌2小時且隨後經過濾,經MeCN(10 ml)沖洗以生成呈白色固體之標的產物III(110 mg,69%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.41(s, 1 H), 9.66(d, J=8 Hz, 2 H), 8.66(s, 1 H), 8.56(s, 1 H), 8.43(s, 1 H), 7.49(d, J=10.8 Hz, 1 H), 6.04(d, J=10.4 Hz, 1 H), 1.17(s, 9 H)。
實施例4:(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(IV)
步驟1:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸(15)
於0℃下對化合物16(150 mg,0.33毫莫耳)之乾燥THF(15 ml)攪拌溶液逐滴加入LiOH(2.6 ml,2.64毫莫耳,1M)溶液。令反應物於25℃下經攪拌3小時。隨後令反應混合物經HCl(1M)驟冷直至pH=2至3並經EA(3×20 ml)萃取。令有機層經鹽水(40 ml)沖洗,置於硫酸鈉上乾燥且經濃縮以生成呈白色固體之化合物15(125 mg,92%產率)。
MS(ESI): [M+H]+
=409.9。
步驟2:(Z)-3-(3-(3-(五氟硫基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-三甲基乙醯基丙烯醯肼(III)
令化合物15(125 mg,0.31毫莫耳)之DCM:EA(5 ml:5 ml,v/v)攪拌溶液冷卻至-78℃並於0℃下加入三甲基乙醯基醯肼(54.0 mg,0.46毫莫耳)、DIEA(80.0 mg,0.62毫莫耳)及T3
P(395 mg,0.62毫莫耳,50重量% EA溶液)。令反應物於25℃下經攪拌3小時並隨後經飽和氯化銨(20 ml)驟冷。令混合物經DCM萃取並令有機層經鹽水沖洗,置於硫酸鈉上乾燥且經真空下濃縮以生成粗製白色固體。將該固體加入至MeCN(5 ml)並於25℃下經攪拌2小時且隨後經過濾,經MeCN(10 ml)沖洗以生成呈白色固體之標的產物III(110 mg,69%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.41(s, 1 H), 9.66(d, J=8 Hz, 2 H), 8.66(s, 1 H), 8.56(s, 1 H), 8.43(s, 1 H), 7.49(d, J=10.8 Hz, 1 H), 6.04(d, J=10.4 Hz, 1 H), 1.17(s, 9 H)。
實施例4:(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(IV) 步驟1:合成2,3-二溴-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙酸異丙酯(20)
於0℃下對化合物16(9.0 g,20毫莫耳)之DCM(60 ml)溶液加入Br2
(6.31 g,40毫莫耳)並隨後令混合物於25℃下經攪拌10小時。令該混合物經水(3×60 ml)和飽和Na2
S2
O3
(60 ml)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈黃色固體之化合物20(12.2 g,99%產率)。
MS(ESI): [M+H]+
=611.9。
步驟2:合成(Z)-2-溴-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(21)
於-20℃下對化合物20(12.2 g,20毫莫耳)之THF(120 ml)攪拌溶液逐滴加入氫氧化鋰水合物(13 ml,40毫莫耳)水溶液(3M)。令反應物於-20℃下經攪拌2.5小時。隨後令反應混合物經HCl(1M)驟冷直至pH=4並經EA(200 ml)萃取。令有機層經水(3×100 ml)和鹽水(3×100 ml)沖洗,置於硫酸鈉上乾燥且經過濾和濃縮以生成呈黃色固體之粗製產物21(10.6 g,99%產率)。
MS(ESI): [M+H]+
=529.9。1
H NMR(400 MHz, CDCl3
)δ 9.47(s, 1 H), 8.76(s, 1 H), 8.75(s, 1 H), 8.59(s, 1 H), 8.07(s, 1 H), 5.25-5.19(m, 1 H), 1.39(d, J=8 Hz, 6 H)。
步驟3:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(22)
於25℃和氮氛圍下對化合物21(1.06 g,2毫莫耳)和嘧啶-5-基亞硼酸(boronic acid)(397 mg,3.2毫莫耳)之二噁烷:水(10 ml:3 ml)溶液加入Pd(PPh3
)4
(231 mg,0.2毫莫耳)和Cs2
CO3
(1.3 g,4毫莫耳)。令反應混合物於85℃和氮氛圍下經攪拌4小時。冷卻至25℃並令有機溶液經濃縮。對殘餘物加入EA(200 ml)並隨後經水(3×100 ml)沖洗,令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=2:1)純化以得到呈白色固體之化合物22(603 mg,57%產率)。
MS(ESI): [M+H]+
=530.1。1
H NMR(400MHz, DMSO-d6)δ 9.26(s, 1 H), 9.19(s, 1 H), 8.78(s, 2 H), 8.68(s, 1 H), 8.38(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 5.13-5.07(m, 1 H), 1.27(d, J=8 Hz, 6 H)。
步驟4:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸(23)
於0℃下對化合物22(1.16 g,2.2毫莫耳)之DCM(100 ml)溶液加入AlCl3(
1.2 g,8.8毫莫耳)。令反應混合物於25℃下經攪拌2.5小時並隨後回溫至30℃且於該溫度下經攪拌0.5小時。令反應物經水(15 ml)驟冷並令有機溶液經濃縮。對殘餘物加入EA(100 ml)並隨後經水(3×100 ml)和HCl溶液(30 ml,1N)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(DCM:MeOH:AcOH=10:1:0.1,v/v)純化以得到呈白色固體之化合物23(684 mg,64%產率)。
MS(ESI): [M+H]+
=487.9。
步驟5:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(IV)
於0℃下對化合物23(684 mg,1.4毫莫耳)之乾燥THF(30 ml)攪拌溶液逐滴加入NMM(283 mg,2.8毫莫耳)和氯甲酸異丙酯(346 mg,2.8毫莫耳)。令反應物於0℃下經攪拌20分鐘。隨後於0℃下對混合物加入NH4
OH(294 mg,8.4毫莫耳)溶液,令該混合物於25℃下再經攪拌5分鐘,隨後經水(50 ml)驟冷且令有機溶液經濃縮。對殘餘物加入EA(100 ml)並隨後經水(3×100 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為EA)純化以得到呈白色固體之化合物IV(480 mg,71%產率)。
MS(ESI): [M+H]+
=486.9。1
H NMR(400 MHz, DMSO-d6)δ 9.19(s, 1 H), 9.14(s, 1 H), 8.72(s, 2 H), 8.42(s, 1 H), 8.36(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 7.63(s, 1 H), 7.39(s, 1 H)。
實施例5:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(V)
步驟1:合成2,3-二溴-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙酸異丙酯(20)
於0℃下對化合物16(9.0 g,20毫莫耳)之DCM(60 ml)溶液加入Br2
(6.31 g,40毫莫耳)並隨後令混合物於25℃下經攪拌10小時。令該混合物經水(3×60 ml)和飽和Na2
S2
O3
(60 ml)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈黃色固體之化合物20(12.2 g,99%產率)。
MS(ESI): [M+H]+
=611.9。
步驟2:合成(Z)-2-溴-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(21)
於-20℃下對化合物20(12.2 g,20毫莫耳)之THF(120 ml)攪拌溶液逐滴加入氫氧化鋰水合物(13 ml,40毫莫耳)水溶液(3M)。令反應物於-20℃下經攪拌2.5小時。隨後令反應混合物經HCl(1M)驟冷直至pH=4並經EA(200 ml)萃取。令有機層經水(3×100 ml)和鹽水(3×100 ml)沖洗,置於硫酸鈉上乾燥且經過濾和濃縮以生成呈黃色固體之粗製產物21(10.6 g,99%產率)。
MS(ESI): [M+H]+
=529.9。1
H NMR(400 MHz, CDCl3
)δ 9.47(s, 1 H), 8.76(s, 1 H), 8.75(s, 1 H), 8.59(s, 1 H), 8.07(s, 1 H), 5.25-5.19(m, 1 H), 1.39(d, J=8 Hz, 6 H)。
步驟3:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(22)
於25℃和氮氛圍下對化合物21(1.06 g,2毫莫耳)和嘧啶-5-基亞硼酸(boronic acid)(397 mg,3.2毫莫耳)之二噁烷:水(10 ml:3 ml)溶液加入Pd(PPh3
)4
(231 mg,0.2毫莫耳)和Cs2
CO3
(1.3 g,4毫莫耳)。令反應混合物於85℃和氮氛圍下經攪拌4小時。冷卻至25℃並令有機溶液經濃縮。對殘餘物加入EA(200 ml)並隨後經水(3×100 ml)沖洗,令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=2:1)純化以得到呈白色固體之化合物22(603 mg,57%產率)。
MS(ESI): [M+H]+
=530.1。1
H NMR(400MHz, DMSO-d6)δ 9.26(s, 1 H), 9.19(s, 1 H), 8.78(s, 2 H), 8.68(s, 1 H), 8.38(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 5.13-5.07(m, 1 H), 1.27(d, J=8 Hz, 6 H)。
步驟4:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸(23)
於0℃下對化合物22(1.16 g,2.2毫莫耳)之DCM(100 ml)溶液加入AlCl3(
1.2 g,8.8毫莫耳)。令反應混合物於25℃下經攪拌2.5小時並隨後回溫至30℃且於該溫度下經攪拌0.5小時。令反應物經水(15 ml)驟冷並令有機溶液經濃縮。對殘餘物加入EA(100 ml)並隨後經水(3×100 ml)和HCl溶液(30 ml,1N)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(DCM:MeOH:AcOH=10:1:0.1,v/v)純化以得到呈白色固體之化合物23(684 mg,64%產率)。
MS(ESI): [M+H]+
=487.9。
步驟5:合成(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(IV)
於0℃下對化合物23(684 mg,1.4毫莫耳)之乾燥THF(30 ml)攪拌溶液逐滴加入NMM(283 mg,2.8毫莫耳)和氯甲酸異丙酯(346 mg,2.8毫莫耳)。令反應物於0℃下經攪拌20分鐘。隨後於0℃下對混合物加入NH4
OH(294 mg,8.4毫莫耳)溶液,令該混合物於25℃下再經攪拌5分鐘,隨後經水(50 ml)驟冷且令有機溶液經濃縮。對殘餘物加入EA(100 ml)並隨後經水(3×100 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為EA)純化以得到呈白色固體之化合物IV(480 mg,71%產率)。
MS(ESI): [M+H]+
=486.9。1
H NMR(400 MHz, DMSO-d6)δ 9.19(s, 1 H), 9.14(s, 1 H), 8.72(s, 2 H), 8.42(s, 1 H), 8.36(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 7.63(s, 1 H), 7.39(s, 1 H)。
實施例5:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(V)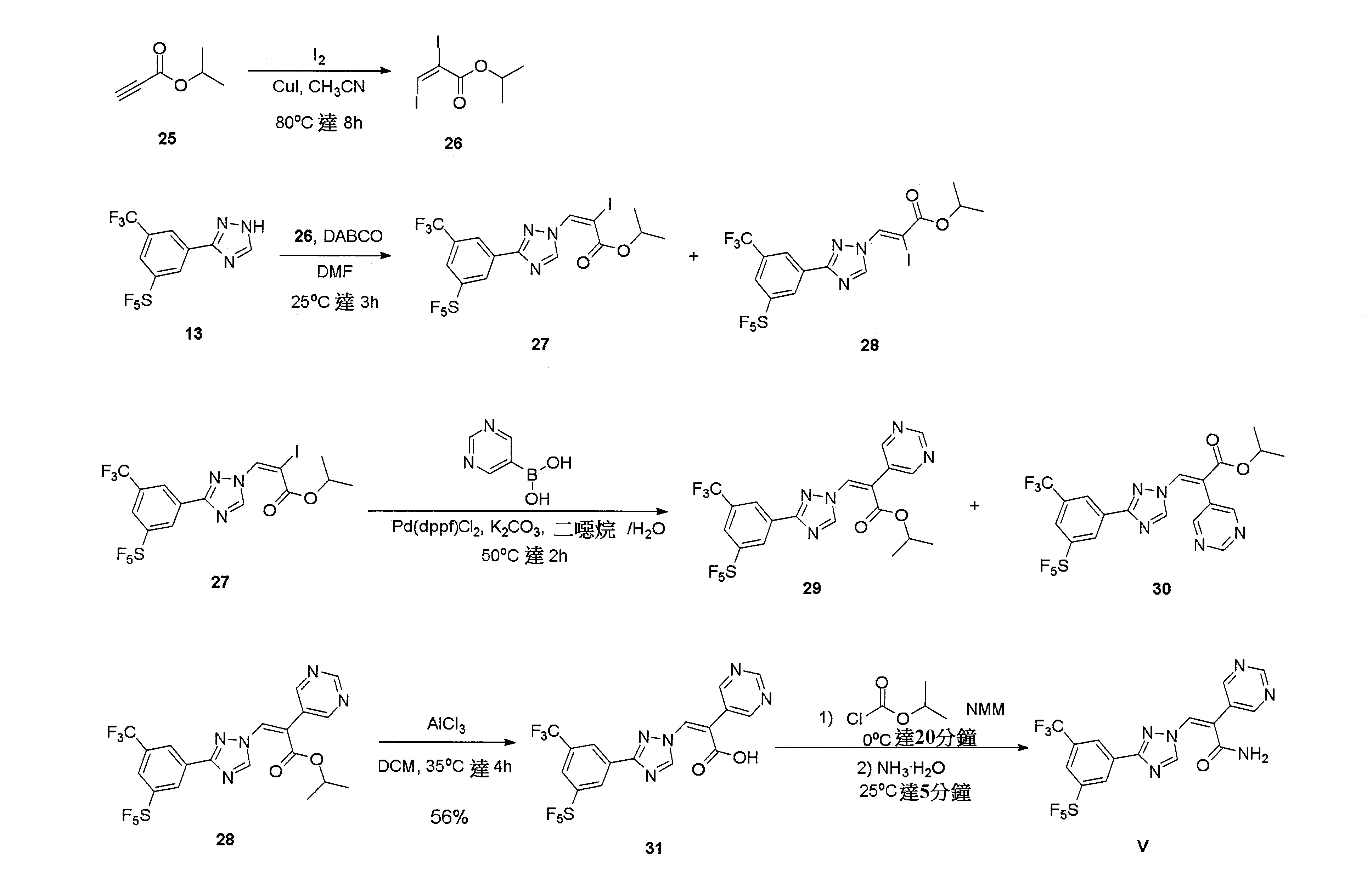 步驟1:合成(E)-2,3-二碘丙烯酸異丙酯(26)
於25℃下對化合物25(336 mg,3毫莫耳)之CH3
CN(5 ml)攪拌溶液加入CuI(29 mg,0.15毫莫耳)和I2
(1.14 g,4.5毫莫耳)。令反應物於80℃下經攪拌8小時。隨後將MTBE (20 ml)加入至混合物並令該混合物經水(3×20 ml)和飽和Na2
S2
O3
溶液(3×20 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈淡黃色油之化合物26(453 mg,41%產率)。1
H NMR(400 MHz, CDCl3
)δ 7.61(s, 1 H), 5.19-5.10(m, 1 H), 1.36(d, J=6.4 Hz, 6 H)。
步驟2:合成(E)-2-碘-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(27)和(Z)-2-碘-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(28)
於0℃下對化合物13(780 mg,2.3毫莫耳)之DMF(10 ml)溶液加入DABCO(515 mg,4.6毫莫耳)和化合物26(1.68 g,4.6毫莫耳),令混合物於80℃下經攪拌0.5小時,隨後經飽和NH4
Cl(50 ml)驟冷且經EA(100 ml)萃取。令有機層經鹽水(3×50 ml)沖洗並經乾燥、過濾及濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈白色固體之標的化合物27(1.1 g,83%產率):
MS(ESI): [M+H]+
=577.9;1
H NMR(400 MHz, CDCl3
)δ 8.67(s, 1 H), 8.52(s, 2 H), 8.04(s, 1 H), 7.57(s, 1 H), 5.22-5.16(m, 1 H), 1.34(d, J=8 Hz, 6 H);和
呈白色固體之化合物28(210 mg,16%產率):
MS(ESI): [M+H]+
=577.9;1
H NMR(400 MHz, CDCl3
)δ 9.52(s, 1 H), 8.81(s, 1 H), 8.75(s, 1 H), 8.59(s, 1 H), 8.07(s, 1 H), 5.23-5.17(m, 1 H), 1.38(d, J=8 Hz, 6 H)。
步驟3:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(29)和(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(30)
於25℃和氮氛圍下對化合物27(58 mg,0.1毫莫耳)和嘧啶-5-基亞硼酸(20 mg,0.16毫莫耳)之二噁烷:水(3 ml:1 ml)溶液加入Pd(dppf)Cl2
(8 mg,0.01毫莫耳)和K2
CO3
(28 mg,0.2毫莫耳)。令反應混合物於50℃和氮氛圍下經攪拌2小時。冷卻至25℃並令有機溶液經濃縮。對殘餘物加入EA(50 ml)並隨後經水(3×50 ml)沖洗,令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=2:1)純化以得到呈白色固體之標的化合物29(24 mg,46%產率):
MS(ESI): [M+H]+
=530.1;1
H NMR(400 MHz, DMSO-d6)δ 9.21(s, 1 H), 9.02(s, 1 H), 8.92(s, 2 H), 8.59(s, 1 H), 8.47(s, 1 H), 8.43(s, 1 H), 8.19(s, 1 H), 5.21-5.16(m, 1 H), 1.25(d, J=8 Hz, 6 H);和
呈白色固體之化合物30(12 mg,23%產率):
MS(ESI): [M+H]+
=530.1;1
H NMR(400 MHz, DMSO-d6)δ 9.26(s, 1 H), 9.19(s, 1 H), 8.78(s, 2 H), 8.68(s, 1 H), 8.38(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 5.13-5.07(m, 1 H), 1.27(d, J=8 Hz, 6 H)。
步驟4:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸(31)
於0℃下對化合物28(20 mg,2.2毫莫耳)之DCM(3 ml)溶液加入AlCl3
(21 mg,0.16毫莫耳)。隨後令反應混合物於35℃下經攪拌4小時。令反應物經水(5 ml)驟冷並令有機溶液經濃縮。對殘餘物加入EA(10 ml)並隨後經水(3×10 ml)和HCl溶液(10 ml,1N)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(DCM:MeOH:AcOH=10:1:0.1,v/v)純化以得到呈白色固體之化合物31(10 mg,56%產率)。
MS(ESI): [M+H]+
=487.9。
步驟5:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(V)
對化合物31(10 mg,0.02毫莫耳)之乾燥THF(2 ml)攪拌溶液加入NMM(4 mg,0.04毫莫耳)並於0℃下逐滴加入氯甲酸異丙酯(5 mg,0.04毫莫耳)之THF(1 ml)溶液。令反應物於0℃下經攪拌20分鐘。隨後於0℃下對混合物加入NH4
OH(2 mg,0.04毫莫耳)溶液,令該混合物於0℃下再經攪拌5分鐘,隨後經水(5 ml)驟冷且令有機溶液經濃縮。對殘餘物加入EA(10 ml)並隨後經水(3×10 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為EA)純化以得到呈白色固體之化合物V(5 mg,50%產率)。
MS(ESI): [M+H]+
=486.9。1
H NMR(400 MHz, DMSO-d6)δ 9.22(s, 1 H), 8.96(s, 3 H), 8.66(s, 1 H), 8.52(s, 1 H), 8.44(s, 1 H), 8.10(s, 1 H), 8.00(s, 1 H), 7.93(s, 1 H)。
實施例6:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼(VI)
步驟1:合成(E)-2,3-二碘丙烯酸異丙酯(26)
於25℃下對化合物25(336 mg,3毫莫耳)之CH3
CN(5 ml)攪拌溶液加入CuI(29 mg,0.15毫莫耳)和I2
(1.14 g,4.5毫莫耳)。令反應物於80℃下經攪拌8小時。隨後將MTBE (20 ml)加入至混合物並令該混合物經水(3×20 ml)和飽和Na2
S2
O3
溶液(3×20 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈淡黃色油之化合物26(453 mg,41%產率)。1
H NMR(400 MHz, CDCl3
)δ 7.61(s, 1 H), 5.19-5.10(m, 1 H), 1.36(d, J=6.4 Hz, 6 H)。
步驟2:合成(E)-2-碘-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(27)和(Z)-2-碘-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯酸異丙酯(28)
於0℃下對化合物13(780 mg,2.3毫莫耳)之DMF(10 ml)溶液加入DABCO(515 mg,4.6毫莫耳)和化合物26(1.68 g,4.6毫莫耳),令混合物於80℃下經攪拌0.5小時,隨後經飽和NH4
Cl(50 ml)驟冷且經EA(100 ml)萃取。令有機層經鹽水(3×50 ml)沖洗並經乾燥、過濾及濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=5:1)純化以得到呈白色固體之標的化合物27(1.1 g,83%產率):
MS(ESI): [M+H]+
=577.9;1
H NMR(400 MHz, CDCl3
)δ 8.67(s, 1 H), 8.52(s, 2 H), 8.04(s, 1 H), 7.57(s, 1 H), 5.22-5.16(m, 1 H), 1.34(d, J=8 Hz, 6 H);和
呈白色固體之化合物28(210 mg,16%產率):
MS(ESI): [M+H]+
=577.9;1
H NMR(400 MHz, CDCl3
)δ 9.52(s, 1 H), 8.81(s, 1 H), 8.75(s, 1 H), 8.59(s, 1 H), 8.07(s, 1 H), 5.23-5.17(m, 1 H), 1.38(d, J=8 Hz, 6 H)。
步驟3:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(29)和(E)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸異丙酯(30)
於25℃和氮氛圍下對化合物27(58 mg,0.1毫莫耳)和嘧啶-5-基亞硼酸(20 mg,0.16毫莫耳)之二噁烷:水(3 ml:1 ml)溶液加入Pd(dppf)Cl2
(8 mg,0.01毫莫耳)和K2
CO3
(28 mg,0.2毫莫耳)。令反應混合物於50℃和氮氛圍下經攪拌2小時。冷卻至25℃並令有機溶液經濃縮。對殘餘物加入EA(50 ml)並隨後經水(3×50 ml)沖洗,令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(流洗溶劑為PE:EA=2:1)純化以得到呈白色固體之標的化合物29(24 mg,46%產率):
MS(ESI): [M+H]+
=530.1;1
H NMR(400 MHz, DMSO-d6)δ 9.21(s, 1 H), 9.02(s, 1 H), 8.92(s, 2 H), 8.59(s, 1 H), 8.47(s, 1 H), 8.43(s, 1 H), 8.19(s, 1 H), 5.21-5.16(m, 1 H), 1.25(d, J=8 Hz, 6 H);和
呈白色固體之化合物30(12 mg,23%產率):
MS(ESI): [M+H]+
=530.1;1
H NMR(400 MHz, DMSO-d6)δ 9.26(s, 1 H), 9.19(s, 1 H), 8.78(s, 2 H), 8.68(s, 1 H), 8.38(s, 1 H), 8.13(s, 1 H), 8.09(s, 1 H), 5.13-5.07(m, 1 H), 1.27(d, J=8 Hz, 6 H)。
步驟4:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯酸(31)
於0℃下對化合物28(20 mg,2.2毫莫耳)之DCM(3 ml)溶液加入AlCl3
(21 mg,0.16毫莫耳)。隨後令反應混合物於35℃下經攪拌4小時。令反應物經水(5 ml)驟冷並令有機溶液經濃縮。對殘餘物加入EA(10 ml)並隨後經水(3×10 ml)和HCl溶液(10 ml,1N)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮。令粗製產物經矽膠管柱(DCM:MeOH:AcOH=10:1:0.1,v/v)純化以得到呈白色固體之化合物31(10 mg,56%產率)。
MS(ESI): [M+H]+
=487.9。
步驟5:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-2-(嘧啶-5-基)丙烯醯胺(V)
對化合物31(10 mg,0.02毫莫耳)之乾燥THF(2 ml)攪拌溶液加入NMM(4 mg,0.04毫莫耳)並於0℃下逐滴加入氯甲酸異丙酯(5 mg,0.04毫莫耳)之THF(1 ml)溶液。令反應物於0℃下經攪拌20分鐘。隨後於0℃下對混合物加入NH4
OH(2 mg,0.04毫莫耳)溶液,令該混合物於0℃下再經攪拌5分鐘,隨後經水(5 ml)驟冷且令有機溶液經濃縮。對殘餘物加入EA(10 ml)並隨後經水(3×10 ml)沖洗。令有機層置於硫酸鈉上乾燥且經濃縮。令殘餘物經矽膠管柱(流洗溶劑為EA)純化以得到呈白色固體之化合物V(5 mg,50%產率)。
MS(ESI): [M+H]+
=486.9。1
H NMR(400 MHz, DMSO-d6)δ 9.22(s, 1 H), 8.96(s, 3 H), 8.66(s, 1 H), 8.52(s, 1 H), 8.44(s, 1 H), 8.10(s, 1 H), 8.00(s, 1 H), 7.93(s, 1 H)。
實施例6:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼(VI) 步驟1:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼(VI)
於0℃下對化合物15(100 mg,0.24毫莫耳)和2-肼基噻唑氫氯酸鹽34(45 mg,0.29毫莫耳)之CH3
CN:EA(4 ml:2 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(228 mg,0.36毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌10小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈灰色固體之化合物VI(8 mg,7%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.92(s, 1 H), 9.58(s, 1 H), 8.65(s, 1 H), 8.53(s, 1 H), 8.43(s, 1 H), 7.57(d, J=12 Hz, 1 H), 7.22(d, J=4 Hz, 1 H), 6.89(d, J=4 Hz, 1 H), 6.04(d, J=8 Hz, 1 H)。
實施例7:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼(VII)
步驟1:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-(噻唑-2-基)丙烯醯肼(VI)
於0℃下對化合物15(100 mg,0.24毫莫耳)和2-肼基噻唑氫氯酸鹽34(45 mg,0.29毫莫耳)之CH3
CN:EA(4 ml:2 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(228 mg,0.36毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌10小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈灰色固體之化合物VI(8 mg,7%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.92(s, 1 H), 9.58(s, 1 H), 8.65(s, 1 H), 8.53(s, 1 H), 8.43(s, 1 H), 7.57(d, J=12 Hz, 1 H), 7.22(d, J=4 Hz, 1 H), 6.89(d, J=4 Hz, 1 H), 6.04(d, J=8 Hz, 1 H)。
實施例7:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼(VII) 步驟1:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼(VII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和環丙烷卡肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物VII(40 mg,68%產率)。
MS(ESI): [M+H]+
=492.8。1
H NMR(400 MHz, DMSO-d6)δ 10.54(s, 1 H), 10.31(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 1.71-1.67(m, 1 H), 0.79-0.74(m, 4 H)。
實施例8:(Z)-N'-異丁醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(VIII)
步驟1:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷卡肼(VII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和環丙烷卡肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物VII(40 mg,68%產率)。
MS(ESI): [M+H]+
=492.8。1
H NMR(400 MHz, DMSO-d6)δ 10.54(s, 1 H), 10.31(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 1.71-1.67(m, 1 H), 0.79-0.74(m, 4 H)。
實施例8:(Z)-N'-異丁醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(VIII) 步驟1:(Z)-N'-異丁醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(VIII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和異丁醯肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物VIII(40 mg,68%產率)。
MS(ESI): [M+H]+
=494.8。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.59(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 2.55-2.48(m, 1 H), 1.06(d, J=8 Hz, 6 H)。
實施例9:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼(IX)
步驟1:(Z)-N'-異丁醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(VIII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和異丁醯肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物VIII(40 mg,68%產率)。
MS(ESI): [M+H]+
=494.8。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.59(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 2.55-2.48(m, 1 H), 1.06(d, J=8 Hz, 6 H)。
實施例9:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼(IX) 步驟1:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼(IX)
於0℃下對化合物15(50 mg,0.12毫莫耳)和丁醯肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物IX(40 mg,68%產率)。
MS(ESI): [M+H]+
=494.8。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.04(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.16(t, J=8 Hz, 2 H), 1.61-1.52(m, 2 H), 0.90(t, J=8 Hz, 3 H)。
實施例10:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼(X)
步驟1:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁醯肼(IX)
於0℃下對化合物15(50 mg,0.12毫莫耳)和丁醯肼(18 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物IX(40 mg,68%產率)。
MS(ESI): [M+H]+
=494.8。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.04(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.16(t, J=8 Hz, 2 H), 1.61-1.52(m, 2 H), 0.90(t, J=8 Hz, 3 H)。
實施例10:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼(X) 步驟1:合成2-(環丁烷羰基)肼-1-羧酸三級丁酯(43)
於0℃下對化合物41(500 mg,5毫莫耳)和化合物42(726 mg,5.5毫莫耳)之DCM(20 ml)溶液加入DIEA(1.29 g,10毫莫耳)和T3
P(6.36 g,10毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物43(1 g,93%產率)。
MS(ESI): [M+H]+
=215.4。
步驟2:合成環丁烷卡肼氫氯酸鹽(44)
對化合物43(1 g,4.7毫莫耳)之DCM(20 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物44(0.7 g,99%產率)。
MS(ESI): [M+H]+
=115.3。
步驟3:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼(X)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物44(28 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物X(11 mg,18%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.49(s, 1 H), 9.93(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 3.18-3.10(m, 1 H), 2.23-1.90(m, 6 H)。
實施例11:(Z)-1-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼(XI)
步驟1:合成2-(環丁烷羰基)肼-1-羧酸三級丁酯(43)
於0℃下對化合物41(500 mg,5毫莫耳)和化合物42(726 mg,5.5毫莫耳)之DCM(20 ml)溶液加入DIEA(1.29 g,10毫莫耳)和T3
P(6.36 g,10毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物43(1 g,93%產率)。
MS(ESI): [M+H]+
=215.4。
步驟2:合成環丁烷卡肼氫氯酸鹽(44)
對化合物43(1 g,4.7毫莫耳)之DCM(20 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物44(0.7 g,99%產率)。
MS(ESI): [M+H]+
=115.3。
步驟3:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丁烷卡肼(X)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物44(28 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物X(11 mg,18%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.49(s, 1 H), 9.93(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 3.18-3.10(m, 1 H), 2.23-1.90(m, 6 H)。
實施例11:(Z)-1-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼(XI) 步驟1:(Z)-1-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼(XI)
於0℃下對化合物15(40 mg,0.1毫莫耳)和1-甲基環丙烷卡肼(17 mg,0.15毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(52 mg,0.4毫莫耳)和T3
P(254 mg,0.4毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XI(21 mg,41%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.35(s, 1 H), 9.65(s, 1 H), 9.62(s, 1 H), 8.65(s, 1 H), 8.54(s, 1 H), 8.40(s, 1 H), 7.49(d, J=8 Hz, 1 H), 6.02(d, J=8 Hz, 1 H), 1.31(s, 3 H), 1.02(s, 2 H), 0.62(s, 2 H)。
實施例12:(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XII)
步驟1:(Z)-1-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環丙烷-1-卡肼(XI)
於0℃下對化合物15(40 mg,0.1毫莫耳)和1-甲基環丙烷卡肼(17 mg,0.15毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(52 mg,0.4毫莫耳)和T3
P(254 mg,0.4毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XI(21 mg,41%產率)。
MS(ESI): [M+H]+
=506.9。1
H NMR(400 MHz, DMSO-d6)δ 10.35(s, 1 H), 9.65(s, 1 H), 9.62(s, 1 H), 8.65(s, 1 H), 8.54(s, 1 H), 8.40(s, 1 H), 7.49(d, J=8 Hz, 1 H), 6.02(d, J=8 Hz, 1 H), 1.31(s, 3 H), 1.02(s, 2 H), 0.62(s, 2 H)。
實施例12:(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XII) 步驟1:合成2-(3-甲基氧雜環丁烷-3-羰基)肼-1-羧酸三級丁酯(47)
於0℃下對化合物46(116 mg,1毫莫耳)和化合物42 (145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×15 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物47(230 mg,99%產率)。
MS(ESI): [M+H]+
=231.4。
步驟2:合成3-氯-2-(羥基甲基)-2-甲基丙烷肼氫氯酸鹽(48)
對化合物47(230 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令混合物經濃縮和真空下乾燥以得到呈白色固體之化合物48(200 mg,99%產率)。
MS(ESI): [M+H]+
=166.3。
步驟3:合成(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XII)
於0℃下對化合物15(59 mg,0.14毫莫耳)和化合物48(45 mg,0.22毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(72 mg,0.56毫莫耳)和T3
P(356 mg,0.56毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XII(10 mg,13%產率)。
MS(ESI): [M+H]+
=559.0。1
H NMR(400 MHz, DMSO-d6)δ 10.65(s, 1 H), 9.88(s, 1 H), 9.63(s, 1 H), 8.66(s, 1 H), 8.55(s, 1 H), 8.40(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 5.16(s, 1 H), 3.81(s, 2 H), 3.59(s, 2 H), 1.22(s, 3 H)。
實施例13:(Z)-3-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼(XIII)之一般合成
步驟1:合成2-(3-甲基氧雜環丁烷-3-羰基)肼-1-羧酸三級丁酯(47)
於0℃下對化合物46(116 mg,1毫莫耳)和化合物42 (145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×15 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物47(230 mg,99%產率)。
MS(ESI): [M+H]+
=231.4。
步驟2:合成3-氯-2-(羥基甲基)-2-甲基丙烷肼氫氯酸鹽(48)
對化合物47(230 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令混合物經濃縮和真空下乾燥以得到呈白色固體之化合物48(200 mg,99%產率)。
MS(ESI): [M+H]+
=166.3。
步驟3:合成(Z)-N'-(3-氯-2-(羥基甲基)-2-甲基丙醯基)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XII)
於0℃下對化合物15(59 mg,0.14毫莫耳)和化合物48(45 mg,0.22毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(72 mg,0.56毫莫耳)和T3
P(356 mg,0.56毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XII(10 mg,13%產率)。
MS(ESI): [M+H]+
=559.0。1
H NMR(400 MHz, DMSO-d6)δ 10.65(s, 1 H), 9.88(s, 1 H), 9.63(s, 1 H), 8.66(s, 1 H), 8.55(s, 1 H), 8.40(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 5.16(s, 1 H), 3.81(s, 2 H), 3.59(s, 2 H), 1.22(s, 3 H)。
實施例13:(Z)-3-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼(XIII)之一般合成 步驟1:合成(Z)-3-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼(XIII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和3-甲基丁烷肼(21 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XIII(37 mg,61%產率)。
MS(ESI): [M+H]+
=508.9。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.07-1.95(m, 3 H), 0.93(d, J=8 Hz, 6 H)。
實施例14:(Z)-N'-乙醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XIV)之一般合成
步驟1:合成(Z)-3-甲基-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)丁烷肼(XIII)
於0℃下對化合物15(50 mg,0.12毫莫耳)和3-甲基丁烷肼(21 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XIII(37 mg,61%產率)。
MS(ESI): [M+H]+
=508.9。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.07-1.95(m, 3 H), 0.93(d, J=8 Hz, 6 H)。
實施例14:(Z)-N'-乙醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XIV)之一般合成 步驟1:合成乙醯肼(52)
對化合物51(2.4 g,27.3毫莫耳)之EtOH(5 ml)溶液加入肼水合物(1.03 g,20.6毫莫耳)並隨後令混合物於80℃下經攪拌12小時。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物52(1.3 g,86%產率)。
步驟2:合成(Z)-N'-乙醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XIV)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物52(14 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XIV(20 mg,36%產率)。
MS(ESI): [M+H]+
=466.8。1
H NMR(400 MHz, DMSO-d6)δ 10.51(s, 1 H), 10.09(s, 1 H), 9.62(s, 1 H), 8.66(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 1.91(s, 3 H)。
實施例15:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼(XV)
步驟1:合成乙醯肼(52)
對化合物51(2.4 g,27.3毫莫耳)之EtOH(5 ml)溶液加入肼水合物(1.03 g,20.6毫莫耳)並隨後令混合物於80℃下經攪拌12小時。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物52(1.3 g,86%產率)。
步驟2:合成(Z)-N'-乙醯基-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XIV)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物52(14 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XIV(20 mg,36%產率)。
MS(ESI): [M+H]+
=466.8。1
H NMR(400 MHz, DMSO-d6)δ 10.51(s, 1 H), 10.09(s, 1 H), 9.62(s, 1 H), 8.66(s, 1 H), 8.55(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 1.91(s, 3 H)。
實施例15:(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼(XV) 步驟1:合成2-丙醯基肼-1-羧酸三級丁酯(57)
於25℃下對化合物56(74 mg,1毫莫耳)和化合物42(145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物57(190 mg,99%產率)。
MS(ESI): [M+H]+
=192.3。
步驟2:合成丙醯肼氫氯酸鹽(58)
對化合物57(190 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物58(120 mg,94%產率)。
步驟3:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼(XV)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物58(23 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XV(24 mg,42%產率)。
MS(ESI): [M+H]+
=480.9。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.03(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.20(q, J=8 Hz, 2 H), 1.05(t, J=8 Hz, 3 H)。
實施例16:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼(XVI)
步驟1:合成2-丙醯基肼-1-羧酸三級丁酯(57)
於25℃下對化合物56(74 mg,1毫莫耳)和化合物42(145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物57(190 mg,99%產率)。
MS(ESI): [M+H]+
=192.3。
步驟2:合成丙醯肼氫氯酸鹽(58)
對化合物57(190 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物58(120 mg,94%產率)。
步驟3:合成(Z)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)-N'-丙醯基丙烯醯肼(XV)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物58(23 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XV(24 mg,42%產率)。
MS(ESI): [M+H]+
=480.9。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.03(s, 1 H), 9.63(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.03(d, J=12 Hz, 1 H), 2.20(q, J=8 Hz, 2 H), 1.05(t, J=8 Hz, 3 H)。
實施例16:(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼(XVI)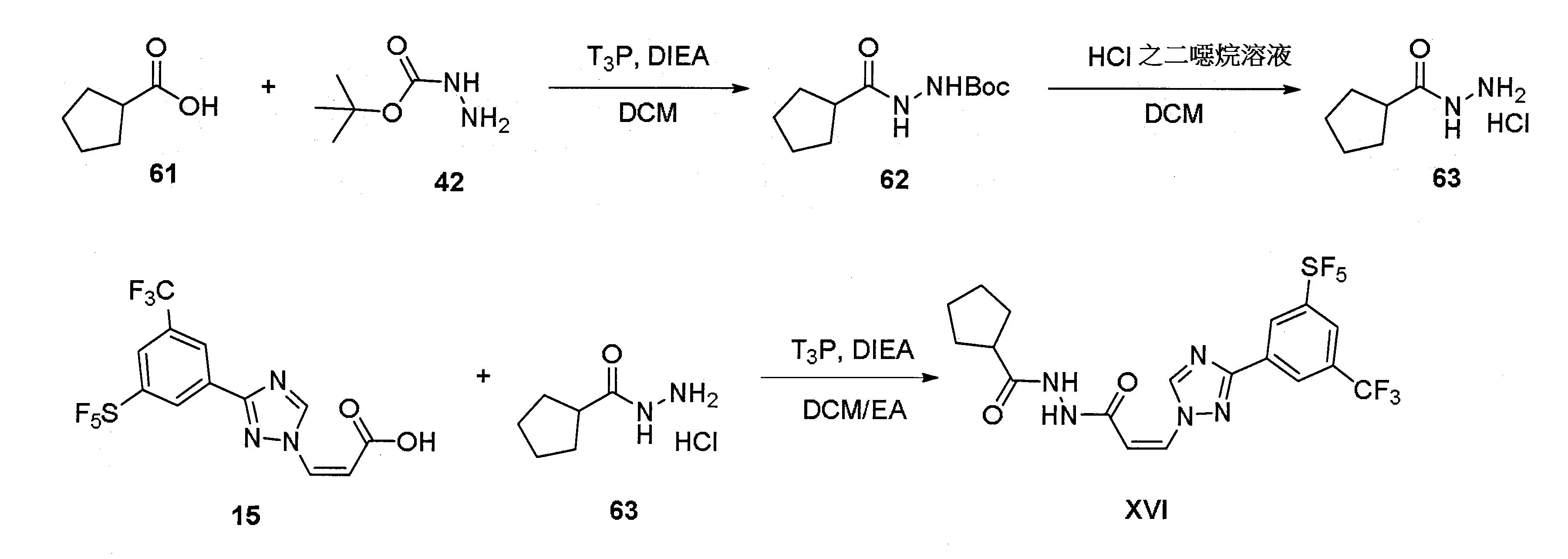 步驟1:合成2-(環戊烷羰基)肼-1-羧酸三級丁酯(62)
於25℃下對化合物61(114 mg,1毫莫耳)和化合物42 (145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物62(220 mg,96%產率)。
MS(ESI): [M+H]+
=229.3。
步驟2:合成環戊烷卡肼氫氯酸鹽(63)
對化合物62(220 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物63(160 mg,97%產率)。
MS(ESI): [M+H]+
=129.3。
步驟3:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼(XVI)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物63(30 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XVI(30 mg,48%產率)。
MS(ESI): [M+H]+
=520.9。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.65(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 2.72-2.65(m, 1 H), 1.84-1.52(m, 8 H)。
實施例17:(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XVII)
步驟1:合成2-(環戊烷羰基)肼-1-羧酸三級丁酯(62)
於25℃下對化合物61(114 mg,1毫莫耳)和化合物42 (145 mg,1.1毫莫耳)之DCM(10 ml)溶液加入DIEA(258 mg,2毫莫耳)和T3
P(1.27 g,2毫莫耳,50% EA溶液)並隨後令混合物於25℃下經攪拌1小時。令該混合物經水(3×20 ml)沖洗。令有機溶液經濃縮和真空下乾燥以得到呈白色固體之化合物62(220 mg,96%產率)。
MS(ESI): [M+H]+
=229.3。
步驟2:合成環戊烷卡肼氫氯酸鹽(63)
對化合物62(220 mg,1毫莫耳)之DCM(10 ml)溶液加入HCl之二噁烷(10 ml,4M)溶液並隨後令混合物於25℃下經攪拌30分鐘。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物63(160 mg,97%產率)。
MS(ESI): [M+H]+
=129.3。
步驟3:合成(Z)-N'-(3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯基)環戊烷卡肼(XVI)
於0℃下對化合物15(50 mg,0.12毫莫耳)和化合物63(30 mg,0.18毫莫耳)之DCM:EA(3 ml:3 ml)溶液加入DIEA(62 mg,0.48毫莫耳)和T3
P(305 mg,0.48毫莫耳,50% EA溶液)並隨後令混合物於0℃下經攪拌1小時。令該混合物經濃縮。對殘餘物加入EA(50 ml)並經水(3×50 ml)沖洗。令有機溶液經濃縮並經製備性TLC(EA)純化以得到呈白色固體之化合物XVI(30 mg,48%產率)。
MS(ESI): [M+H]+
=520.9。1
H NMR(400 MHz, DMSO-d6)δ 10.50(s, 1 H), 10.02(s, 1 H), 9.65(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.42(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 2.72-2.65(m, 1 H), 1.84-1.52(m, 8 H)。
實施例17:(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XVII) 步驟1:合成2-甲基丙-3,3,3-d3酸(66)
於0℃和氮氛圍下對LDA(50 ml,74.3毫莫耳)之THF (150 ml)溶液加入化合物65(2.2 g,29.7毫莫耳)並隨後令混合物於80℃下經攪拌2小時。冷卻至0℃,逐滴加入CD3
I (4.8 g,32.7毫莫耳)且隨後於80℃下經攪拌10小時。對混合物加入水(50 ml)並隨後經EA(50 ml)萃取。令無機層經1N HCl酸化至pH=4並隨後經EA(50 ml)萃取。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈淡黃色油之化合物66(1.68 g,62%產率)。1
H NMR(400 MHz, CDCl3
)δ 2.57(q, J=8 Hz, 1 H), 1.19(d, J=8 Hz, 3 H)。
步驟2:合成2-甲基-2-(甲基-d3)丙-3,3,3-d3酸(67)
於0℃和氮氛圍下對LDA(31 ml,46.3毫莫耳)之THF (90 ml)溶液加入化合物66(1.68 g,18.5毫莫耳)並隨後令混合物於80℃下經攪拌2小時。冷卻至0℃,逐滴加入CD3
I(3 g,20.8毫莫耳)且隨後於80℃下經攪拌10小時。對混合物加入水(50 ml)並隨後經EA(50 ml)萃取。令無機層經1N HCl酸化至pH=4並隨後經EA(50 ml)萃取。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈黃色油之化合物67(1.17 g,59%產率)。1
H NMR(400 MHz, CDCl3
)δ 1.22(s, 3 H)。
步驟3:合成2-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)肼-1-羧酸三級丁酯(68)
於25℃下對化合物67(540 mg,5毫莫耳)、肼羧酸三級丁酯(660 mg,5毫莫耳)及Et3
N(1.01 g,10毫莫耳)之DCM (50 ml)溶液加入EDCI(1.05 g,5.5毫莫耳)和HOBT(740 mg,5.5毫莫耳)並隨後令混合物於25℃下經攪拌12小時。隨後令該混合物經水(3×50 ml)沖洗。令有機溶液經濃縮並經矽膠管柱(流洗溶劑為PE:EA=5:1至3:1)純化以得到呈白色固體之化合物68(412 mg,37%產率)。
MS(ESI): [M+H]+
=223.5。1
H NMR(400 MHz, CDCl3
)δ 7.37(s, 1 H), 6.48(s, 1 H), 1.47(s, 9 H), 1.24(s, 3 H)。
步驟4:合成2-甲基-2-(甲基-d3)丙烷肼-3,3,3-d3氫氯酸鹽(69)
對單頸燒瓶加入化合物68(412 mg,1.9毫莫耳)和HCl之二噁烷(10 ml,4M)溶液。令混合物於25℃下經攪拌1小時。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物69(300 mg,99%產率)。
MS(ESI): [M+H]+
=123.3。
步驟5:合成(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XVII)
於-78℃下對化合物15(700 mg,1.7毫莫耳)和化合物69(300 mg,1.88毫莫耳)之DCM(50 ml)溶液加入DIEA(877 mg,6.8毫莫耳)和T3
P(4.3 g,6.8毫莫耳,50% EA溶液),令混合物於-78℃下經攪拌30分鐘,隨後回溫至0℃且經攪拌1小時。令該混合物於35℃和真空下經濃縮。對殘餘物加入EA(100 ml)並經水(3×100 ml)和鹽水(3×100 ml)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到粗製產物,令該粗製產物經CH3
CN(15 ml)碾製12小時且經過濾。收集濾餅並經DCM(15 ml)碾製12小時且經過濾。令濾餅經DCM(15 ml)沖洗並經收集且經真空下乾燥達5小時以得到呈白色固體之化合物XVII(670 mg,77%產率)。
MS(ESI): [M+H]+
=514.9。1
H NMR(400 MHz, DMSO-d6)δ 10.36(s, 1 H), 9.67(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.44(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 1.16(s, 3 H)。
實施例18:抑制細胞增生檢測
使用CellTiter-Glo發光細胞存活檢測(其使用Promega CellTiter 96®發光CellTiter Glo套組)以研究測試化合物之增生抑制作用。令化合物再懸浮於DMSO以製備25 mM儲存液。令癌細胞株或正常組織之細胞接種於96孔槽盤(1.5至5×103
個細胞/孔槽)隔夜。令化合物經2或3倍系列稀釋以得到10個濃度。隨後藉由Bravo(Agilent)將藥物加入至該等細胞孔槽並經培育72小時。藉由加入CellTiter-Glo試劑(依據製造商之指示)使細胞裂解並隨後使用EnVision (PerkinElmer)讀取發光值。
依據下述方法計算生長%:
經DMSO處理之細胞作為載具對照組(高度對照組;HC)且培養基單獨作為背景組(低度對照組;LC)。
生長%=100×(樣品發光值-LC發光值)/(HC發光值-LC發光值)。
使用Graphpad Prism藉由濃度反應曲線擬合計算IC50
值。
測試之血液癌細胞株包括OCI-AML3、MV-4-11、KG-1、霞-1、K562及THP-1;測試之實體癌細胞株包括U87MG、U251、T98G、LN229、A172、H460、H2009及A549;測試之正常細胞株包括HCN2和3T3L1。結果示於表1,其中該等測試化合物顯示對腦、肺及血液之癌細胞株的生長抑制作用。然而,該等測試化合物對活體外培養之正常細胞未顯現毒性。
步驟1:合成2-甲基丙-3,3,3-d3酸(66)
於0℃和氮氛圍下對LDA(50 ml,74.3毫莫耳)之THF (150 ml)溶液加入化合物65(2.2 g,29.7毫莫耳)並隨後令混合物於80℃下經攪拌2小時。冷卻至0℃,逐滴加入CD3
I (4.8 g,32.7毫莫耳)且隨後於80℃下經攪拌10小時。對混合物加入水(50 ml)並隨後經EA(50 ml)萃取。令無機層經1N HCl酸化至pH=4並隨後經EA(50 ml)萃取。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈淡黃色油之化合物66(1.68 g,62%產率)。1
H NMR(400 MHz, CDCl3
)δ 2.57(q, J=8 Hz, 1 H), 1.19(d, J=8 Hz, 3 H)。
步驟2:合成2-甲基-2-(甲基-d3)丙-3,3,3-d3酸(67)
於0℃和氮氛圍下對LDA(31 ml,46.3毫莫耳)之THF (90 ml)溶液加入化合物66(1.68 g,18.5毫莫耳)並隨後令混合物於80℃下經攪拌2小時。冷卻至0℃,逐滴加入CD3
I(3 g,20.8毫莫耳)且隨後於80℃下經攪拌10小時。對混合物加入水(50 ml)並隨後經EA(50 ml)萃取。令無機層經1N HCl酸化至pH=4並隨後經EA(50 ml)萃取。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到呈黃色油之化合物67(1.17 g,59%產率)。1
H NMR(400 MHz, CDCl3
)δ 1.22(s, 3 H)。
步驟3:合成2-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)肼-1-羧酸三級丁酯(68)
於25℃下對化合物67(540 mg,5毫莫耳)、肼羧酸三級丁酯(660 mg,5毫莫耳)及Et3
N(1.01 g,10毫莫耳)之DCM (50 ml)溶液加入EDCI(1.05 g,5.5毫莫耳)和HOBT(740 mg,5.5毫莫耳)並隨後令混合物於25℃下經攪拌12小時。隨後令該混合物經水(3×50 ml)沖洗。令有機溶液經濃縮並經矽膠管柱(流洗溶劑為PE:EA=5:1至3:1)純化以得到呈白色固體之化合物68(412 mg,37%產率)。
MS(ESI): [M+H]+
=223.5。1
H NMR(400 MHz, CDCl3
)δ 7.37(s, 1 H), 6.48(s, 1 H), 1.47(s, 9 H), 1.24(s, 3 H)。
步驟4:合成2-甲基-2-(甲基-d3)丙烷肼-3,3,3-d3氫氯酸鹽(69)
對單頸燒瓶加入化合物68(412 mg,1.9毫莫耳)和HCl之二噁烷(10 ml,4M)溶液。令混合物於25℃下經攪拌1小時。令該混合物經濃縮和真空下乾燥以得到呈白色固體之化合物69(300 mg,99%產率)。
MS(ESI): [M+H]+
=123.3。
步驟5:合成(Z)-N'-(2-甲基-2-(甲基-d3)丙醯基-3,3,3-d3)-3-(3-(3-(五氟硫烷基)-5-(三氟甲基)苯基)-1H-1,2,4-三唑-1-基)丙烯醯肼(XVII)
於-78℃下對化合物15(700 mg,1.7毫莫耳)和化合物69(300 mg,1.88毫莫耳)之DCM(50 ml)溶液加入DIEA(877 mg,6.8毫莫耳)和T3
P(4.3 g,6.8毫莫耳,50% EA溶液),令混合物於-78℃下經攪拌30分鐘,隨後回溫至0℃且經攪拌1小時。令該混合物於35℃和真空下經濃縮。對殘餘物加入EA(100 ml)並經水(3×100 ml)和鹽水(3×100 ml)沖洗。令有機溶液置於硫酸鈉上乾燥且經過濾和濃縮以得到粗製產物,令該粗製產物經CH3
CN(15 ml)碾製12小時且經過濾。收集濾餅並經DCM(15 ml)碾製12小時且經過濾。令濾餅經DCM(15 ml)沖洗並經收集且經真空下乾燥達5小時以得到呈白色固體之化合物XVII(670 mg,77%產率)。
MS(ESI): [M+H]+
=514.9。1
H NMR(400 MHz, DMSO-d6)δ 10.36(s, 1 H), 9.67(s, 1 H), 9.64(s, 1 H), 8.67(s, 1 H), 8.56(s, 1 H), 8.44(s, 1 H), 7.49(d, J=12 Hz, 1 H), 6.02(d, J=12 Hz, 1 H), 1.16(s, 3 H)。
實施例18:抑制細胞增生檢測
使用CellTiter-Glo發光細胞存活檢測(其使用Promega CellTiter 96®發光CellTiter Glo套組)以研究測試化合物之增生抑制作用。令化合物再懸浮於DMSO以製備25 mM儲存液。令癌細胞株或正常組織之細胞接種於96孔槽盤(1.5至5×103
個細胞/孔槽)隔夜。令化合物經2或3倍系列稀釋以得到10個濃度。隨後藉由Bravo(Agilent)將藥物加入至該等細胞孔槽並經培育72小時。藉由加入CellTiter-Glo試劑(依據製造商之指示)使細胞裂解並隨後使用EnVision (PerkinElmer)讀取發光值。
依據下述方法計算生長%:
經DMSO處理之細胞作為載具對照組(高度對照組;HC)且培養基單獨作為背景組(低度對照組;LC)。
生長%=100×(樣品發光值-LC發光值)/(HC發光值-LC發光值)。
使用Graphpad Prism藉由濃度反應曲線擬合計算IC50
值。
測試之血液癌細胞株包括OCI-AML3、MV-4-11、KG-1、霞-1、K562及THP-1;測試之實體癌細胞株包括U87MG、U251、T98G、LN229、A172、H460、H2009及A549;測試之正常細胞株包括HCN2和3T3L1。結果示於表1,其中該等測試化合物顯示對腦、肺及血液之癌細胞株的生長抑制作用。然而,該等測試化合物對活體外培養之正常細胞未顯現毒性。 實施例19:REV-GFP移位檢測
先前研究已報告REV貨載之細胞核堆積係CRM1抑制作用之標記。細胞核轉出之選擇性抑制劑(SINE)處理引起以劑量依賴性之方式自細胞質部位至細胞核之REV的清楚且迅速之轉移。為評估測試化合物對XPO1之抑制作用,藉由位於中國之HD Biosciences公司產製REV-GFP U2OS選殖株並令細胞經生長培養基培養。隨後於37℃和5% CO2
培養器中將細胞接種於96孔槽盤(100 μl生長培養基;7,000個細胞/孔槽)隔夜。來普黴素B(leptomycin B;LMB)之最高濃度設定為50 nM並經系列稀釋以作為陽性對照組,同時其他化合物之最高濃度設定為10 µM。於37℃和5% CO2
下培育經不同化合物或DMSO處理之細胞達1小時。隨後於室溫下令細胞固定於4%甲醛達15分鐘。
令細胞經沖洗並於室溫和黑暗下經Hoechst 33342作業溶液(100 μl)染色10分鐘,隨後藉由高量顯像系統Image Xpress(Molecular Devices)且使用20倍接物鏡使細胞螢光顯像。針對Hoechst(350/461 nm)和GFP/FITC(488/509 nm) (激動和最大發射波長)設定濾鏡。依據下式計算REV細胞核移位之功效%:
功效%=(I測試組
-I對照組
)/(ILMB
-I對照組
)×100
I測試組
-源自測試化合物之訊號;ILMB
-50 nM LMB組之訊號;I對照組
-載具對照組之訊號。
分別經LMB和化合物III處理1小時後,REV之細胞核再分佈存有劑量依賴性,且所有測試濃度之化合物III未降低細胞存活。數據顯示化合物III藉由抑制CRM1以增加REV細胞核堆積且對細胞存活無作用。結果示於圖1和2。其中圖1顯示LMB對Rev-GFP-U2OS細胞之REV再分佈的功效(A. LMB之1小時劑量反應功效的代表性影像,綠色表示與Rev共軛之EGFP;B.經LMB處理之REV再分佈的劑量反應曲線,載具組經設定為0且使用50 nM LMB組以代表100%功效,數值表示平均值±SEM(n=2),EC50=0.11 nM;C. LMB對細胞數之功效,每個組之經分析的相對比經載具組校正,所有值表示平均值±SEM(n=2))。其中圖2顯示化合物III對REV貨載抑制作用之EC50且化合物III未影響細胞存活。如示於表2,使用Rev-GFP以評估其他測試化合物。
實施例19:REV-GFP移位檢測
先前研究已報告REV貨載之細胞核堆積係CRM1抑制作用之標記。細胞核轉出之選擇性抑制劑(SINE)處理引起以劑量依賴性之方式自細胞質部位至細胞核之REV的清楚且迅速之轉移。為評估測試化合物對XPO1之抑制作用,藉由位於中國之HD Biosciences公司產製REV-GFP U2OS選殖株並令細胞經生長培養基培養。隨後於37℃和5% CO2
培養器中將細胞接種於96孔槽盤(100 μl生長培養基;7,000個細胞/孔槽)隔夜。來普黴素B(leptomycin B;LMB)之最高濃度設定為50 nM並經系列稀釋以作為陽性對照組,同時其他化合物之最高濃度設定為10 µM。於37℃和5% CO2
下培育經不同化合物或DMSO處理之細胞達1小時。隨後於室溫下令細胞固定於4%甲醛達15分鐘。
令細胞經沖洗並於室溫和黑暗下經Hoechst 33342作業溶液(100 μl)染色10分鐘,隨後藉由高量顯像系統Image Xpress(Molecular Devices)且使用20倍接物鏡使細胞螢光顯像。針對Hoechst(350/461 nm)和GFP/FITC(488/509 nm) (激動和最大發射波長)設定濾鏡。依據下式計算REV細胞核移位之功效%:
功效%=(I測試組
-I對照組
)/(ILMB
-I對照組
)×100
I測試組
-源自測試化合物之訊號;ILMB
-50 nM LMB組之訊號;I對照組
-載具對照組之訊號。
分別經LMB和化合物III處理1小時後,REV之細胞核再分佈存有劑量依賴性,且所有測試濃度之化合物III未降低細胞存活。數據顯示化合物III藉由抑制CRM1以增加REV細胞核堆積且對細胞存活無作用。結果示於圖1和2。其中圖1顯示LMB對Rev-GFP-U2OS細胞之REV再分佈的功效(A. LMB之1小時劑量反應功效的代表性影像,綠色表示與Rev共軛之EGFP;B.經LMB處理之REV再分佈的劑量反應曲線,載具組經設定為0且使用50 nM LMB組以代表100%功效,數值表示平均值±SEM(n=2),EC50=0.11 nM;C. LMB對細胞數之功效,每個組之經分析的相對比經載具組校正,所有值表示平均值±SEM(n=2))。其中圖2顯示化合物III對REV貨載抑制作用之EC50且化合物III未影響細胞存活。如示於表2,使用Rev-GFP以評估其他測試化合物。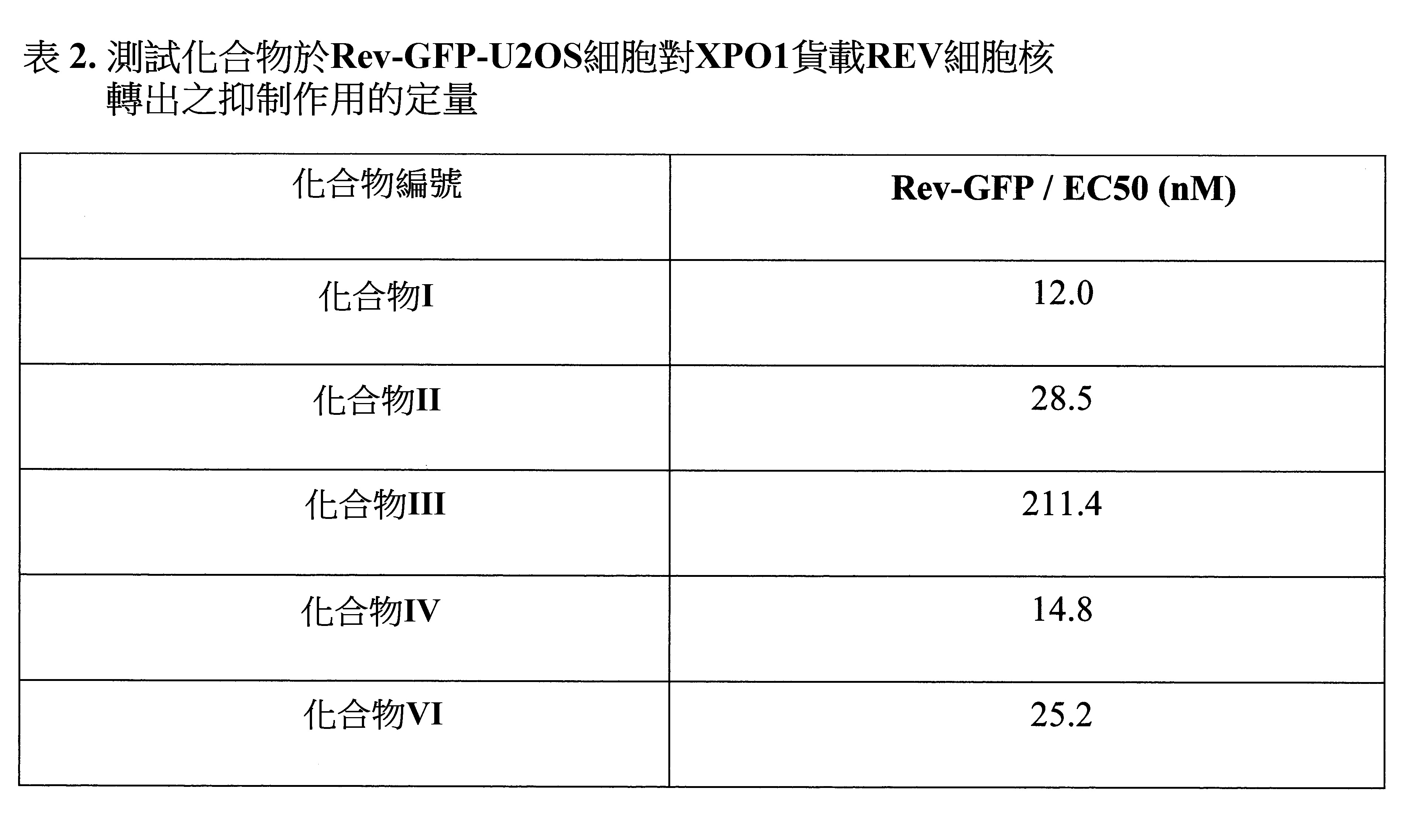 實施例20:顯示持續但可逆之XPO1抑制作用的化合物洗除分析
為評估經洗除後測試化合物對XPO1蛋白之抑制作用的期間,使用REV-GFP細胞進行洗除分析。將測試化合物和陽性對照組加入至細胞孔槽並令盤於37℃和5% CO2
下經培育1小時。除去含有藥物之培養基,令細胞經沖洗且加入新鮮培養基。令細胞於特定時間點0、4、24、48、72及96小時下經顯像以評估REV細胞核轉出之抑制作用。使用50 nM LMB,直至洗除後24小時,REV蛋白持續保留於細胞核。於72小時,功效逐漸降低至30%。然而,如細胞數降低所顯示者,相對於對照組,LMB顯示毒性。化合物III (EC90)亦抑制REV細胞核轉出。起始自洗除後4小時,功效逐漸減少且於24小時喪失功效。細胞對化合物III處理顯現良好之耐受性且未觀察到細胞數減少。結論為相對於陽性對照組LMB,亦與XPO1蛋白共價結合之化合物III於增加REV於細胞核之再分佈上顯現顯著且可逆之功效且於24小時內未對細胞生長產生任何副作用。結果示於圖3和4。其中圖3顯示於清洗研究中LMB之持續的XPO1抑制作用(A.於洗除後LMB對REV再分佈之作用的代表性影像;B.於洗除後LMB對REV再分佈之作用的摘要;C. LMB對細胞存活之作用,每個組之經分析的相對比經每個組於0小時校正,所有值表示平均值±SEM(n=3))。圖4顯示於Rev-GFP-U2OS細胞,化合物III對REV再分佈之洗除功效。於此洗除REV-GFP移位研究,於EC90和EC50(400 nM和120 nM)評估化合物III。經1小時處理後,化合物III(EC50)對REV之細胞核保留顯示約40%功效。此功效於洗除後4小時逐漸減少至20%且於24小時完全喪失。然而,化合物III(400 nM)於4小時顯現持續之XPO1抑制作用且於24小時逐漸減少。未觀察到對細胞存活有顯著作用。結論為化合物III顯現對XPO1蛋白之時間和劑量依賴性抑制作用且該作用持續4小時且呈可逆性。A.於洗除後化合物III對REV再分佈之作用的代表性影像;B.於洗除後化合物III對REV再分佈之作用的摘要;C.化合物III對細胞存活之作用。
實施例21:藥物動力(PK)和腦:血漿之比
自小鼠(n=3或5)收集AUC血液以助於總計10個時間點(給藥前、5分鐘、15分鐘、30分鐘、1小時、2小時、4小時、8小時、12小時及24小時)。於指定時間點,使用異氟醚麻醉動物且藉由眼眶後穿刺於每個時間點收集血液(約110 µl)至經預先冷卻之K2
EDTA管。將血液樣品置於濕冰上並經離心(2000 x g,5分鐘,4℃)以於30分鐘得到收集之樣品的血漿。所有樣品經冷凍儲存於約-80℃下直至分析使用。於進行分析前,令樣品與溶於乙腈之內部標準品混合,經渦旋振盪和離心且注入上清液以進行分析。使用LC-MS-MS儀器(API 4000, Triple Quadrupole LC/MS/MS質譜儀)測量血漿中化合物之濃度。使用Phoenix Win Nonlin 6.3套裝軟體PO-Noncompartmental模式200(血管外輸入)計算AUC值。結果示於表3。
實施例20:顯示持續但可逆之XPO1抑制作用的化合物洗除分析
為評估經洗除後測試化合物對XPO1蛋白之抑制作用的期間,使用REV-GFP細胞進行洗除分析。將測試化合物和陽性對照組加入至細胞孔槽並令盤於37℃和5% CO2
下經培育1小時。除去含有藥物之培養基,令細胞經沖洗且加入新鮮培養基。令細胞於特定時間點0、4、24、48、72及96小時下經顯像以評估REV細胞核轉出之抑制作用。使用50 nM LMB,直至洗除後24小時,REV蛋白持續保留於細胞核。於72小時,功效逐漸降低至30%。然而,如細胞數降低所顯示者,相對於對照組,LMB顯示毒性。化合物III (EC90)亦抑制REV細胞核轉出。起始自洗除後4小時,功效逐漸減少且於24小時喪失功效。細胞對化合物III處理顯現良好之耐受性且未觀察到細胞數減少。結論為相對於陽性對照組LMB,亦與XPO1蛋白共價結合之化合物III於增加REV於細胞核之再分佈上顯現顯著且可逆之功效且於24小時內未對細胞生長產生任何副作用。結果示於圖3和4。其中圖3顯示於清洗研究中LMB之持續的XPO1抑制作用(A.於洗除後LMB對REV再分佈之作用的代表性影像;B.於洗除後LMB對REV再分佈之作用的摘要;C. LMB對細胞存活之作用,每個組之經分析的相對比經每個組於0小時校正,所有值表示平均值±SEM(n=3))。圖4顯示於Rev-GFP-U2OS細胞,化合物III對REV再分佈之洗除功效。於此洗除REV-GFP移位研究,於EC90和EC50(400 nM和120 nM)評估化合物III。經1小時處理後,化合物III(EC50)對REV之細胞核保留顯示約40%功效。此功效於洗除後4小時逐漸減少至20%且於24小時完全喪失。然而,化合物III(400 nM)於4小時顯現持續之XPO1抑制作用且於24小時逐漸減少。未觀察到對細胞存活有顯著作用。結論為化合物III顯現對XPO1蛋白之時間和劑量依賴性抑制作用且該作用持續4小時且呈可逆性。A.於洗除後化合物III對REV再分佈之作用的代表性影像;B.於洗除後化合物III對REV再分佈之作用的摘要;C.化合物III對細胞存活之作用。
實施例21:藥物動力(PK)和腦:血漿之比
自小鼠(n=3或5)收集AUC血液以助於總計10個時間點(給藥前、5分鐘、15分鐘、30分鐘、1小時、2小時、4小時、8小時、12小時及24小時)。於指定時間點,使用異氟醚麻醉動物且藉由眼眶後穿刺於每個時間點收集血液(約110 µl)至經預先冷卻之K2
EDTA管。將血液樣品置於濕冰上並經離心(2000 x g,5分鐘,4℃)以於30分鐘得到收集之樣品的血漿。所有樣品經冷凍儲存於約-80℃下直至分析使用。於進行分析前,令樣品與溶於乙腈之內部標準品混合,經渦旋振盪和離心且注入上清液以進行分析。使用LC-MS-MS儀器(API 4000, Triple Quadrupole LC/MS/MS質譜儀)測量血漿中化合物之濃度。使用Phoenix Win Nonlin 6.3套裝軟體PO-Noncompartmental模式200(血管外輸入)計算AUC值。結果示於表3。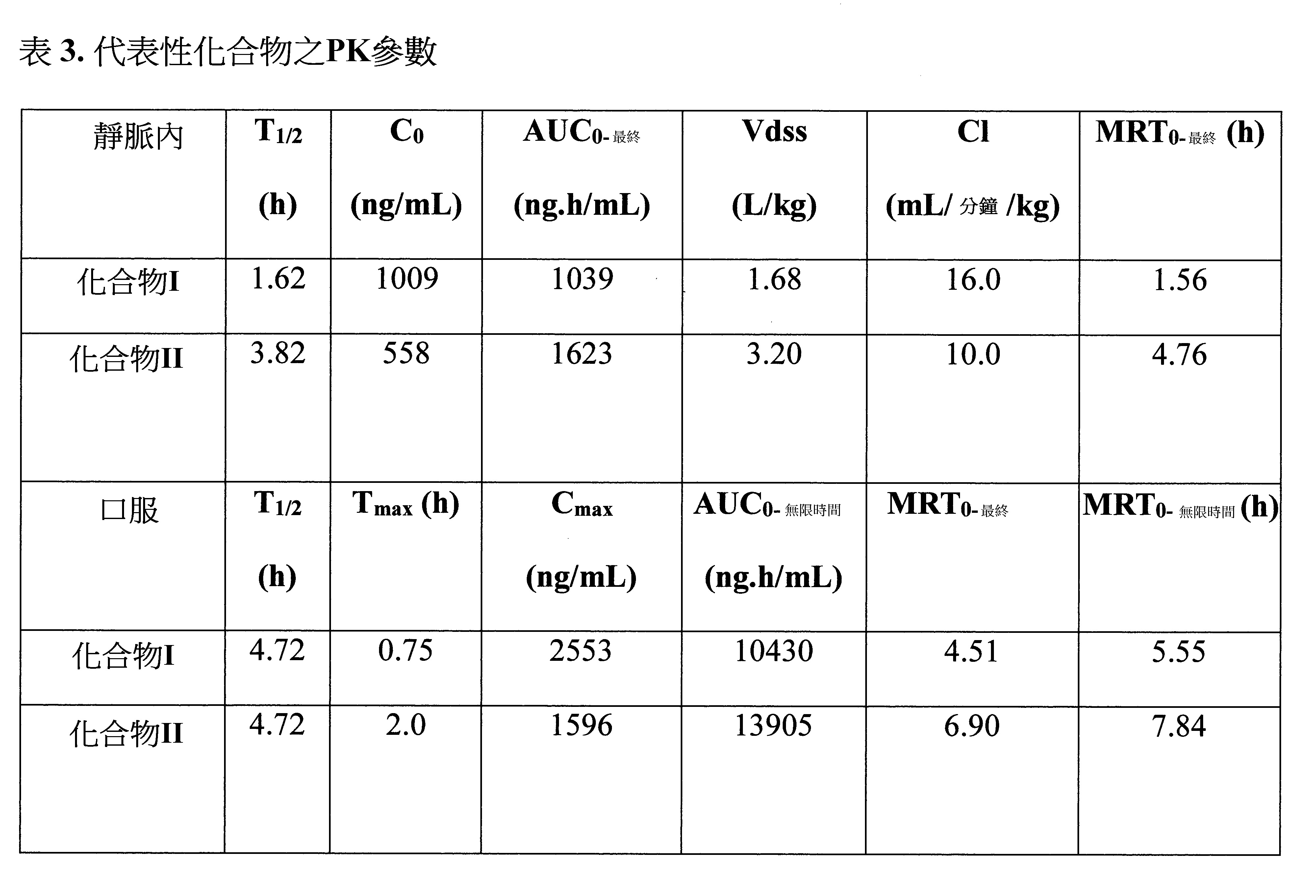 腦:血漿(B:P)之比
對另一組小鼠或大鼠(n=3或5)給藥(口服10 mg/kg)並隨後於最高血漿濃度之時間點(Tmax
,給藥後1小時)處死且於該時間點收集末端血漿和腦組織。經收集後,使用冰鹽水輕洗腦組織,置於濾紙上乾燥,經稱重且置於乾冰上急速冷凍。所有樣品經冷凍儲存於約-80℃下直至分析使用。於分析時,令腦組織經均質化(均質化溶液PBS,pH 7.4),與溶於乙腈之內部標準品混合,經渦旋振盪和離心且注入上清液以進行分析。使用LC-MS-MS儀器(API 4000,Triple Quadrupole LC/MS/MS質譜儀)測量血漿中化合物之濃度。使用相同方法(除了均質化步驟)處理血漿樣品並依據所產生之標準曲線計算每個基質中化合物之濃度。PK檢測和B:P比測定之結果示於表4和圖5,其中圖5顯示於腦通透之匣PK中化合物III與KPT-350的腦行為之比較。
腦:血漿(B:P)之比
對另一組小鼠或大鼠(n=3或5)給藥(口服10 mg/kg)並隨後於最高血漿濃度之時間點(Tmax
,給藥後1小時)處死且於該時間點收集末端血漿和腦組織。經收集後,使用冰鹽水輕洗腦組織,置於濾紙上乾燥,經稱重且置於乾冰上急速冷凍。所有樣品經冷凍儲存於約-80℃下直至分析使用。於分析時,令腦組織經均質化(均質化溶液PBS,pH 7.4),與溶於乙腈之內部標準品混合,經渦旋振盪和離心且注入上清液以進行分析。使用LC-MS-MS儀器(API 4000,Triple Quadrupole LC/MS/MS質譜儀)測量血漿中化合物之濃度。使用相同方法(除了均質化步驟)處理血漿樣品並依據所產生之標準曲線計算每個基質中化合物之濃度。PK檢測和B:P比測定之結果示於表4和圖5,其中圖5顯示於腦通透之匣PK中化合物III與KPT-350的腦行為之比較。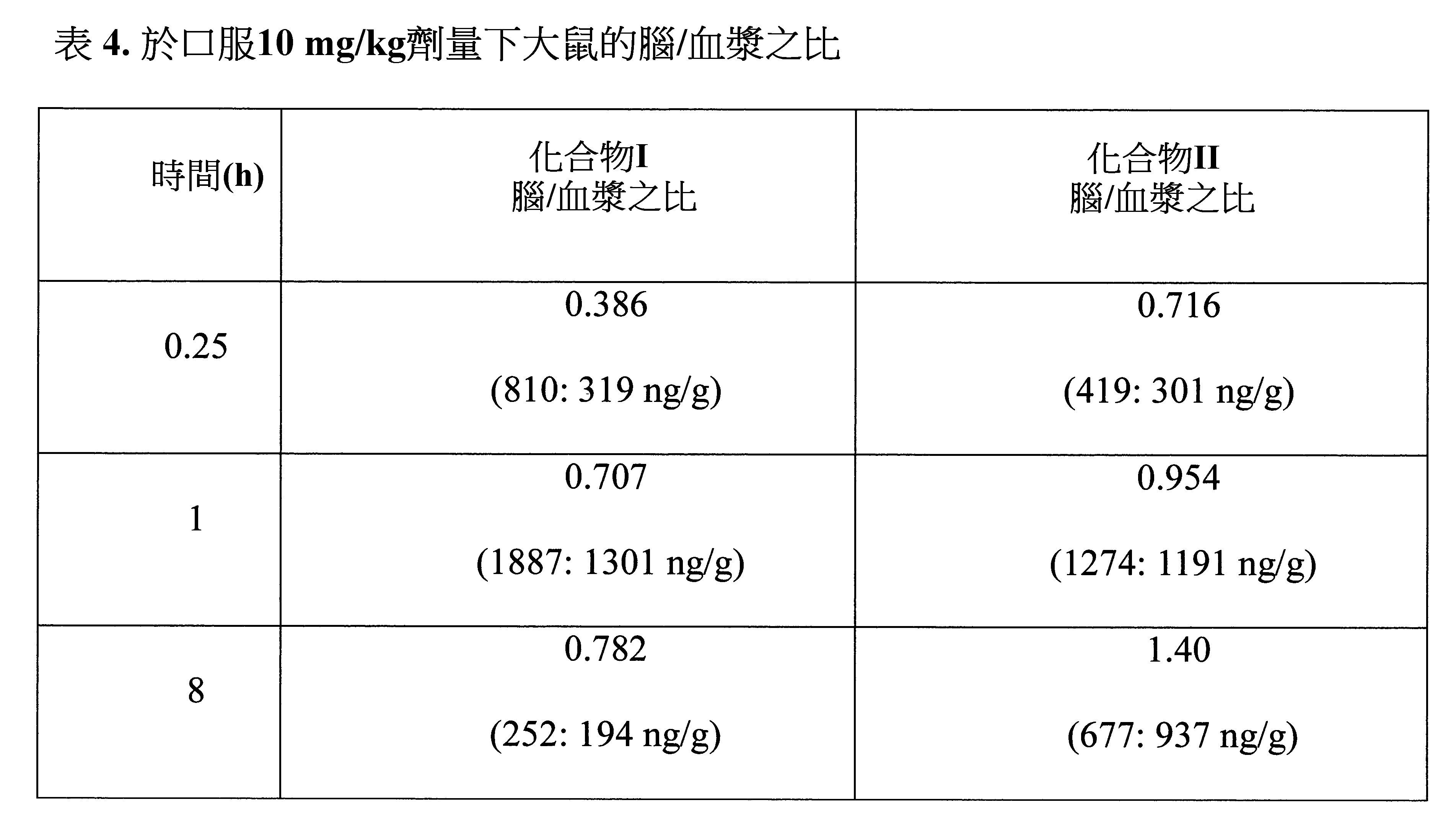 實施例22:最大耐受劑量(MTD)研究
於研究前令雌性BALB/c裸鼠(6至8週大/18至22 g;Beijing AniKeeper Biotech Co., Ltd.提供)經檢疫7天。經由獸醫評估動物之一般健康狀態。於進行研究前排除顯現異常之動物。依據Pharmaron, Inc.之標準作業程序(SOP)進行動物照顧和使用之一般程序。
實施例22:最大耐受劑量(MTD)研究
於研究前令雌性BALB/c裸鼠(6至8週大/18至22 g;Beijing AniKeeper Biotech Co., Ltd.提供)經檢疫7天。經由獸醫評估動物之一般健康狀態。於進行研究前排除顯現異常之動物。依據Pharmaron, Inc.之標準作業程序(SOP)進行動物照顧和使用之一般程序。 每天對所有動物之體重稱重。例行性監測一般行為,諸如活動力、食物和水消耗(僅藉由籠邊檢查)、眼遮蓋/毛糾結及任何其他異常現象。記錄任何死亡及/或異常臨床徵候。藉由二氧化碳和隨後頸脫位使顯現嚴重痛苦及/或疼痛之明顯徵候的動物經人道處死以確認死亡。若有下述情況使動物安樂死:明顯體重減少>20%或動物不能獲取足夠之食物或水。結果示於圖6。其中圖6顯示MTD研究中小鼠體重變化%(對於KPT-330組,經給藥1週後,所有小鼠死亡或因體重減少達20%而經麻醉;然而,化合物I組之所有小鼠(口服25 mg/kg;隔天每週3次達2週)存活。投予調製劑:10%NMP/10%表面活性劑Solutol/80%(0.5%泊咯沙姆(Poloxamer)188+0.5% PVP))。
實施例23:評估化合物III對BALB/c裸鼠之U87MG-luc人神經膠質母細胞瘤原位模式的抗腫瘤功效
23.1. 研究設計
動物:使用雌性BALB/c裸鼠(6至8週大/18至22 g;Beijing AniKeeper Biotech Co., Ltd.提供)。依據Pharmaron, Inc.之標準作業程序(SOP)進行動物照顧和使用之一般程序。
分組和處置:於腫瘤細胞接種後第7天開始分組和處置。令小鼠顯像以監測腫瘤生長並隨後藉由電腦產生之隨機方式將該等小鼠隨機指定至各別組。於腫瘤移植後,所有測試藥物經口服投予(20 mg/kg;3次/週)達4週(n=12/組)。
23.2. 實驗方法和測量參數
細胞培養:活體外維持人神經膠質母細胞瘤U87-luc腫瘤細胞株呈單層培養並用於腫瘤接種。
腫瘤接種和隨機化:藉由置入針(AP: 2.0 mm;ML:0.5至1.0 mm;DV:3.0 mm),將懸浮於MEM培養基(2 µl)的2.5×105
個表現螢光素酶之U87MG-luc腫瘤細胞自前囟注射至右前腦。經1分鐘緩慢進行該注射。經完成注射後,維持該針於另一分鐘。記錄測量參數:腫瘤生長(經顯像分析監測)、體重及存活天數。
中止標準:當動物體重減少>20%或動物不能獲取足夠之食物或水時,藉由二氧化碳使各別動物經人道處死。
統計分析:如研究設計者,對所有測量參數值以值±標準誤差記錄數據。藉由SPSS 17.0統計軟體進行所有統計試驗且設定顯著水準為p<0.05。
23.3. 結果
整體上,於治療期間終了時,與載具對照組相比較,化合物III(20 mg/kg)顯現呈生物發光訊號減少99%之顯著抗腫瘤活性(p<0.05)。相較於化合物III處理組動物之中間存活時間(MST)為47天(p<0.0007),載具組動物之MST為39.5天。
關於安全性輪廓,大多數動物對化合物III(20 mg/kg)耐受良好,雖然個別動物顯現低於10%之體重減少,該個別動物需要跳躍劑量。於治療期間未觀察到其他顯著之臨床異常。結論為化合物III(20 mg/kg口服投予;3次/週達4週)能顯著延長帶有GBM腫瘤之小鼠的存活。同時,藉由抑制GBM腫瘤生長且延長帶有U87MG-luc原位腫瘤之動物組的存活,化合物III證實穿透BBB之能力。
結果示於圖7至9。其中圖7顯示於U87MG-luc原位模式中化合物III之腫瘤生長抑制作用(Y軸為對數刻度;p<0.05)。其中圖8顯示於20 mg/kg之每週3次經4週治療後,載具組對化合物III處理組之存活曲線。其中圖9顯示於治療期間和之後(第7至65天)的體重變化。
實施例24:藉由IHC於U87MG-luc原位小鼠模式中評估化合物III之PD功效
基於腫瘤之生物發光訊號,藉由電腦產生之隨機方式將小鼠隨機指定至各別組,該各別組分別包括每組5隻動物之載具組和化合物III處理組。口服給予化合物III(20 mg/kg;3次/週於第1、3及5天)達2週。IHC研究之動物於最後給藥和正常鹽水灌流且隨後經4%三聚甲醛固定後6小時被安樂死。收集含有腫瘤之完整腦並於石蠟密封程序前保持於固定劑中達24小時。
IHC定量之數據示於圖9。與載具對照組(*p<0.05;**p<0.01) 相比較,口服投予(3次/週達2週)化合物III或陽性對照組(KPT-330)顯著降低腫瘤之CRM1和Ki67的表現,其表示化合物III藉由抑制CRM1之表現顯現抗腫瘤功效。如示於腫瘤之降低的Ki67量,化合物III抑制腫瘤增生。
結果示於圖10。其中圖10說明藉由XPO1化合物III之Ki67和CRM1的降低。
每天對所有動物之體重稱重。例行性監測一般行為,諸如活動力、食物和水消耗(僅藉由籠邊檢查)、眼遮蓋/毛糾結及任何其他異常現象。記錄任何死亡及/或異常臨床徵候。藉由二氧化碳和隨後頸脫位使顯現嚴重痛苦及/或疼痛之明顯徵候的動物經人道處死以確認死亡。若有下述情況使動物安樂死:明顯體重減少>20%或動物不能獲取足夠之食物或水。結果示於圖6。其中圖6顯示MTD研究中小鼠體重變化%(對於KPT-330組,經給藥1週後,所有小鼠死亡或因體重減少達20%而經麻醉;然而,化合物I組之所有小鼠(口服25 mg/kg;隔天每週3次達2週)存活。投予調製劑:10%NMP/10%表面活性劑Solutol/80%(0.5%泊咯沙姆(Poloxamer)188+0.5% PVP))。
實施例23:評估化合物III對BALB/c裸鼠之U87MG-luc人神經膠質母細胞瘤原位模式的抗腫瘤功效
23.1. 研究設計
動物:使用雌性BALB/c裸鼠(6至8週大/18至22 g;Beijing AniKeeper Biotech Co., Ltd.提供)。依據Pharmaron, Inc.之標準作業程序(SOP)進行動物照顧和使用之一般程序。
分組和處置:於腫瘤細胞接種後第7天開始分組和處置。令小鼠顯像以監測腫瘤生長並隨後藉由電腦產生之隨機方式將該等小鼠隨機指定至各別組。於腫瘤移植後,所有測試藥物經口服投予(20 mg/kg;3次/週)達4週(n=12/組)。
23.2. 實驗方法和測量參數
細胞培養:活體外維持人神經膠質母細胞瘤U87-luc腫瘤細胞株呈單層培養並用於腫瘤接種。
腫瘤接種和隨機化:藉由置入針(AP: 2.0 mm;ML:0.5至1.0 mm;DV:3.0 mm),將懸浮於MEM培養基(2 µl)的2.5×105
個表現螢光素酶之U87MG-luc腫瘤細胞自前囟注射至右前腦。經1分鐘緩慢進行該注射。經完成注射後,維持該針於另一分鐘。記錄測量參數:腫瘤生長(經顯像分析監測)、體重及存活天數。
中止標準:當動物體重減少>20%或動物不能獲取足夠之食物或水時,藉由二氧化碳使各別動物經人道處死。
統計分析:如研究設計者,對所有測量參數值以值±標準誤差記錄數據。藉由SPSS 17.0統計軟體進行所有統計試驗且設定顯著水準為p<0.05。
23.3. 結果
整體上,於治療期間終了時,與載具對照組相比較,化合物III(20 mg/kg)顯現呈生物發光訊號減少99%之顯著抗腫瘤活性(p<0.05)。相較於化合物III處理組動物之中間存活時間(MST)為47天(p<0.0007),載具組動物之MST為39.5天。
關於安全性輪廓,大多數動物對化合物III(20 mg/kg)耐受良好,雖然個別動物顯現低於10%之體重減少,該個別動物需要跳躍劑量。於治療期間未觀察到其他顯著之臨床異常。結論為化合物III(20 mg/kg口服投予;3次/週達4週)能顯著延長帶有GBM腫瘤之小鼠的存活。同時,藉由抑制GBM腫瘤生長且延長帶有U87MG-luc原位腫瘤之動物組的存活,化合物III證實穿透BBB之能力。
結果示於圖7至9。其中圖7顯示於U87MG-luc原位模式中化合物III之腫瘤生長抑制作用(Y軸為對數刻度;p<0.05)。其中圖8顯示於20 mg/kg之每週3次經4週治療後,載具組對化合物III處理組之存活曲線。其中圖9顯示於治療期間和之後(第7至65天)的體重變化。
實施例24:藉由IHC於U87MG-luc原位小鼠模式中評估化合物III之PD功效
基於腫瘤之生物發光訊號,藉由電腦產生之隨機方式將小鼠隨機指定至各別組,該各別組分別包括每組5隻動物之載具組和化合物III處理組。口服給予化合物III(20 mg/kg;3次/週於第1、3及5天)達2週。IHC研究之動物於最後給藥和正常鹽水灌流且隨後經4%三聚甲醛固定後6小時被安樂死。收集含有腫瘤之完整腦並於石蠟密封程序前保持於固定劑中達24小時。
IHC定量之數據示於圖9。與載具對照組(*p<0.05;**p<0.01) 相比較,口服投予(3次/週達2週)化合物III或陽性對照組(KPT-330)顯著降低腫瘤之CRM1和Ki67的表現,其表示化合物III藉由抑制CRM1之表現顯現抗腫瘤功效。如示於腫瘤之降低的Ki67量,化合物III抑制腫瘤增生。
結果示於圖10。其中圖10說明藉由XPO1化合物III之Ki67和CRM1的降低。

 彼等之立體異構物、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中 R1 係選自-C(=O)-R2 、C3-6 雜環烷基或C5-10 雜芳基;R1 之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、-NO2 、 鹵素、胺基、氰基、C1-12 烷基、C2-12 烯基、C2-12 炔基、C1-12 烷氧基、C1-12 鹵烷基、C1-12 鹵烷氧基或C1-12 烷硫基(alkylsulfanyl);且 R2 係選自C1-6 烷基、C3-6 環烷基或C3-6 雜環烷基;R2 之任何烷基、環烷基或雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12 烷基、C2-12 烯基、C2-12 炔基、C1-12 烷氧基、C1-12 鹵烷基、C1-12 鹵烷氧基或C1-12 烷硫基;且 R3 和R4 係獨立地選自C1-6 烷基,或R3 和R4 與彼等連接之N一起形成經取代或未經取代之C4-10 環烷基胺基;R3 和R4 之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12 烷基、C2-12 烯基、C2-12 炔基、C1-12 烷氧基、C1-12 鹵烷基、C1-12 鹵烷氧基或C1-12 烷硫基。
彼等之立體異構物、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥,其中 R1 係選自-C(=O)-R2 、C3-6 雜環烷基或C5-10 雜芳基;R1 之任何雜環烷基或雜芳基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自氘、-OH、-SH、-NO2 、 鹵素、胺基、氰基、C1-12 烷基、C2-12 烯基、C2-12 炔基、C1-12 烷氧基、C1-12 鹵烷基、C1-12 鹵烷氧基或C1-12 烷硫基(alkylsulfanyl);且 R2 係選自C1-6 烷基、C3-6 環烷基或C3-6 雜環烷基;R2 之任何烷基、環烷基或雜環烷基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12 烷基、C2-12 烯基、C2-12 炔基、C1-12 烷氧基、C1-12 鹵烷基、C1-12 鹵烷氧基或C1-12 烷硫基;且 R3 和R4 係獨立地選自C1-6 烷基,或R3 和R4 與彼等連接之N一起形成經取代或未經取代之C4-10 環烷基胺基;R3 和R4 之任何烷基或環烷基胺基係可選擇地獨立地經一或多個取代基取代,該一或多個取代基選自鹵素、胺基、氰基、C1-12 烷基、C2-12 烯基、C2-12 炔基、C1-12 烷氧基、C1-12 鹵烷基、C1-12 鹵烷氧基或C1-12 烷硫基。
 ; 最佳地,R2 係選自三級丁基、2,2-二甲基-1-丁基、
; 最佳地,R2 係選自三級丁基、2,2-二甲基-1-丁基、 ; 可選擇地,R2 係選自2-甲基環氧乙烷基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、正戊基、異戊基、新戊基、2,2-二甲基丁基或
; 可選擇地,R2 係選自2-甲基環氧乙烷基、2-甲基-1-丁基、3-甲基-1-丁基、2-甲基-3-丁基、2,2-二甲基-1-丙基、2-甲基-1-戊基、3-甲基-1-戊基、4-甲基-1-戊基、2-甲基-2-戊基、3-甲基-2-戊基、正戊基、異戊基、新戊基、2,2-二甲基丁基或 。
。
 ; 可選擇地,R1 係選自未經取代或經取代之呋喃基、吡咯基、咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基或噻吩基。
; 可選擇地,R1 係選自未經取代或經取代之呋喃基、吡咯基、咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基或噻吩基。
 ; 可選擇地,R1 係選自未經取代或經取代之苯基、萘基、呋喃基、苯並呋喃基、吡咯基、咪唑基、苯並咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基、噻吩基、苯並噻吩基、吲哚基、嘌呤基、喹啉基、異喹啉基或啡噁噻基。
; 可選擇地,R1 係選自未經取代或經取代之苯基、萘基、呋喃基、苯並呋喃基、吡咯基、咪唑基、苯並咪唑基、三唑基、四唑基、噁唑基、噁二唑基、1,3,5-三嗪基、噻唑基、噻吩基、苯並噻吩基、吲哚基、嘌呤基、喹啉基、異喹啉基或啡噁噻基。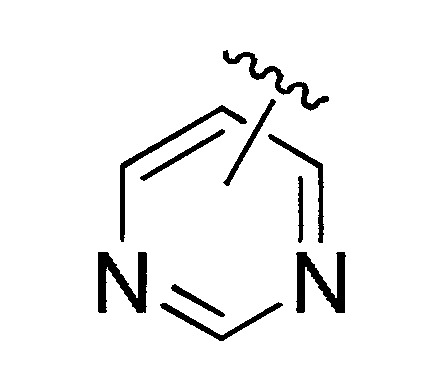
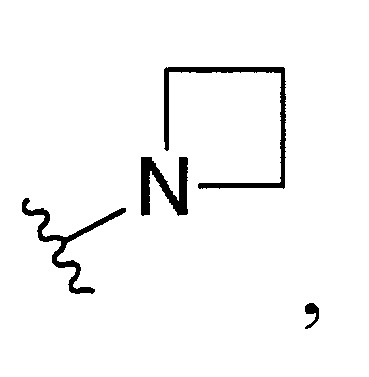 。
。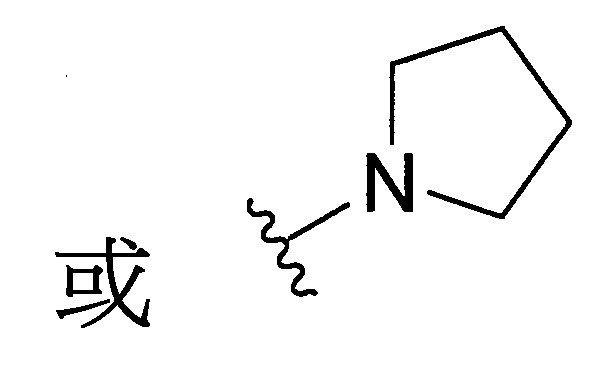




 彼等之立體異構物、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥。
彼等之立體異構物、N-氧化物、溶劑合物、代謝物、醫藥上可接受之鹽或前藥。