WO1984001155A1 - Hydroxyvitamin d2 compounds and process for preparing them - Google Patents
Hydroxyvitamin d2 compounds and process for preparing them Download PDFInfo
- Publication number
- WO1984001155A1 WO1984001155A1 PCT/US1983/001393 US8301393W WO8401155A1 WO 1984001155 A1 WO1984001155 A1 WO 1984001155A1 US 8301393 W US8301393 W US 8301393W WO 8401155 A1 WO8401155 A1 WO 8401155A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compounds
- hydrogen
- configuration
- carbon
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC1=*CCC1 Chemical compound CC1=*CCC1 0.000 description 6
- WUDWACWWYNEPAH-UHFFFAOYSA-N CC(C)(COCCO)C(CS(c1ccccc1)(=[O-2])=O)I Chemical compound CC(C)(COCCO)C(CS(c1ccccc1)(=[O-2])=O)I WUDWACWWYNEPAH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J53/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/02—Nutrients, e.g. vitamins, minerals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/18—Radicals substituted by singly bound oxygen or sulfur atoms
- C07D317/20—Free hydroxyl or mercaptan
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/10—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings
- C07D317/14—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 not condensed with other rings with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D317/18—Radicals substituted by singly bound oxygen or sulfur atoms
- C07D317/22—Radicals substituted by singly bound oxygen or sulfur atoms etherified
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J17/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, having an oxygen-containing hetero ring not condensed with the cyclopenta(a)hydrophenanthrene skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J53/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class
- C07J53/002—Carbocyclic rings fused
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Definitions
- This invention relates to biologically active vitamin D compounds.
- this invention relates to a process for the preparation of hydroxylated derivatives of vitamin D 2 , and to novel intermediates used in this process.
- this invention relates to the synthesis of 25-hydroxyvitamin D 2 and the 24-epimer thereof, to certain alkyl and aryl-analogs and to 5,6-trans-, and the acyl derivatives of these compounds.
- the D vitamins are very iinportant agents for the control of calcium and phosphate metabolism in animals and humans, and have long been used as dietary supplements and in clinical practice to assure proper bone growth and development. It is now known that the in vivo activity of these vitamins, specifically of vitamin D 2 and D 3 , is dependent on metabolism to hydroxylated forms. Thus, vitamin D 3 undergoes two successive hydroxylation reactions in vivo, leading first to 25-hydroxyvita ⁇ t ⁇ in D 3 and then to 1,25-dihydroxyvitamin D 3 and the latter is indeed thought to be the c ⁇ tpound responsible for the well-known beneficial effects of vitamin D 3 .
- vitamin D 2 which is commonly used as a dietary supplement, undergoes an analogous hydroxylation sequence to its active forms, being first converted to 25-hydroxyvitamin D 2 (25-OH-D 2 .) and then to 1,25-dihydroxyvitamin D 2 (1,25-(OH) 2 D 3 ).
- the hydroxylated forms of vitamin D 2 named above are, because of their potency and other beneficial properties, highly desirable dietary supplements, or pharmaceutical agents, for the cure or prevention of bone or related diseases, and their value and possible use is recognized in patents relating to these corripounds [U.S. Letters Patents 3,585,221 and 3,880,894].
- ltfhereas all metabolites of vitamin D 3 have been prepared by chemical synthesis, there has been but little work on the preparation of vitamin D 2 metabolites.
- the present process provides the 5,6-trans-isomers of the above compounds, and novel intermediates that are of utility for the preparation of 25-OH- D 2 -.analogs and/or isotopically-labeled products.
- acyl signifies an aliphatic acyl group of from 1 to about 6 carbons, in all possible isomeric forms (e.g. formyl, acetyl, propionyl, butyryl, isobutyryl, valeryl, etc.), or an aromatic acyl group (aroyl group) such as benzoyl, the isomeric methyl-benzoyls, the isomeric nitro- or halo-benzoyIs, etc., or a dicarboxylic acyl group of from 2 to 6 atoms chain length, i.e.
- alkyl refers to a lower alkyl group of 1 to 6 carbons in all possible isomeric forms, e.g.
- aryl signifies a phenyl or substituted phenyl group, e.g. alkylphenyl, methoxyphenyl, etc.
- the overall process developed for the preparation of the above c ⁇ ipounds may be divided into two general phases, namely (a) addition of a side chain fragment to a suitable steroidal precursor to produce a 5,7-diene steroid as the central intermediate, and (b) conversion of this 5,7-diene to the vitamin D structure with, as required, further modification of the side chain to produce the desired 25-hydroxylated co ⁇ pounds.
- This general scheme allows for sore flexibility in the choice of specific starting material and in the exact order of individual process steps, two features that are of considerable practical advantage and convenience.
- Process Scheme I presents an embodiment of the overall process, whereas Process Scheme II illustrates sane of the options available for executing the last four steps of the synthesis.
- Starting materials for the present process are steroidal 22-aldehydes in which the ring B double bond(s) is (are) protected in an appropriate manner.
- suitable compounds are for example, the PTAD-diene-protected-22-aldehyde (1) (where PTAD refers to the phenyltriazoline-3,5-dione-protecting group shown) or the 3,5-cyclo-22-aldehyde (4) wherein the ⁇ 5 -double bond is protected via i-ether formation.
- PTAD refers to the phenyltriazoline-3,5-dione-protecting group shown
- 3,5-cyclo-22-aldehyde (4) wherein the ⁇ 5 -double bond is protected via i-ether formation.
- Both of these compounds are known products (see for example Barton et al. J. Chem. See. (C) 1968 (1971); and Heyl et al. U.S. Patent 2,623,052) and both can be carried through the steps of the present
- the first step of this process comprises the addition of a suitable side chain fragment.
- a suitable side chain fragment For example, condensation of aldehyde (1) with a sulfonyl-side chain fragment as shown in the Scheme (sulfone A, further described below) present in the form of its anion, in an ether or hydrocarbon solvent, provides the hydroxy-sulfone intermediate (2) .
- the anion of the sulfone A side chain fragment is generated by treatment of the sulfone with a strong base, such as lithium diethylamide, n-butyl lithium or ethyl magnesium bromide (or similar Grignard reagent) in an ether or hydrocarbon solvent, and to this solution of sulfone anion the steroid aldehyde (compound1) as an ether or hydrocarbon solution is then added.
- a strong base such as lithium diethylamide, n-butyl lithium or ethyl magnesium bromide (or similar Grignard reagent) in an ether or hydrocarbon solvent
- steroid aldehyde compound1
- the reaction may be conducted advantageously at about room temperature, and is best effected under an inert atmosphere.
- the next step comprises the removal of the hydroxy- and phenylsulfonyl groups in the side chain with formation of the 22 (23) -trans-double bond.
- the analogous treatment of compound (5) gives the 22-olefinic ccmpound (6).
- the 22-hydroxy group in compounds (2) or (5) can also be acylated or sulfonylated (e.g. mesylated) prior to the Na/Hg-reduction step, but this is not generally required.
- the third operation of the process involves conversion of these ring B-protected steroids to the desired 5,7-diene intermediate (7).
- this conversion is accomplished in a single step, namely treatment of (3) with a strong hydride reducing agent (e.g. LiAlH 4 )in an ether solvent at reflux temperature gives the diene (7) .
- a strong hydride reducing agent e.g. LiAlH 4
- This same material, compound (7) is produced from the i-ether derivative (6), in several steps, all of which are conventional and well-known in the art.
- the i-ether (6) is first solvolyzed in glacial acetic acid at reflux for ca. 2 hours to yield the corresponding 5-ene-3-acetate derivative (6a).
- This compound in a hydrocarbon solution (e.g. hexane) at reflux temperature preferably under an inert atmosphere, is then treated with a brominating reagent (e.g. 1,3-dibromo-5,5-dimethylhydantoin) over a period of about 20 minutes, and the resulting C-7-bromo-intermediate is directly dehydrobrcminated by dissolution in xylene and treatment with a base (e.g. s-collidine) at reflux temperatures under an inert atmosphere for about 90 minutes.
- a base e.g. s-collidine
- Conversion of 5,7-diene (7) to the final vitamin D products comprises a sequence of four steps, the precise order of which may be altered as convenient.
- the sequence shown in Process Scheme I involves first the irradiation of an ether or hydrocarbon solution of the 5,7-diene (7) with ultraviolet light to yield the previtamin analog (8) which by warming (50-90°C) in a suitable solvent (e.g. ethanol, hexane) undergoes iscmerization to the vitamin D 2 -analog (9) .
- a suitable solvent e.g. ethanol, hexane
- ketal removal is achieved by heating ketal (9) in hydroxylic solvent under acid catalysis for 1-2 hours. (It is desirable to monitor the progress of reaction by periodic chromatographic analysis of the crude reaction mixture.
- the product, ketone (10) thereby obtained, is then alkylated in the final step by means of a Grignard reagent (an alkyl-, or aryl-magnesium halide, e.g. methyl magnesium branide in this case) to give the 25-hydroxyvitamin D 2 compound (11) .
- a Grignard reagent an alkyl-, or aryl-magnesium halide, e.g. methyl magnesium branide in this case
- Alkylation via an alkyllithium reagent, e.g. methyl lithium is also effective and convenient. If the side chain fragment, sulfone A, as used in the first step, is racemic, i.e.
- compound (11) will be obtained as a mixture of two C-24-epimers, i.e. (24S)-epimer (11a) which corresponds to the natural product, and the (24R) -epimer (11b) which is 25-OH-24-epi-D 2 .
- C-24-epimers are conveniently separated by high performance liquid chromatography (HPLC) on an efficient microparticulate silica gel column to obtain 25-OH-D 2 (11a) and 25-OH-24-epi-D 2 (11b) in pure form.
- racemic sulfone A as the side Chain starting material, the early synthetic intermediates, e.g. (3) [or (6), and (6a)] as well as the 5,7-diene (7) and subsequent intermediates (8) , (9) , and (10) also occur as the two C-24-epimers.
- separation of epimers can be conducted at any of these intermediate stages, and the (24R) and (24S) epimers may then be processed separately through the remaining steps to yield 25-OH-D 2 (11a) or 25-OH-24-epi-D 2 (11b) as desired. It is generally convenient to effect separation at the stage of the final products.
- the ketal in diene (7A) may be hydrolyzed first (see sequence B in Process Scheme II) , to yield the diene-ketone identified as (7B) in the scheme, which after irradiation gives the previtamin ketone (8B) , and thermal iscmerization then leads to vitamin D 2 -ketone (10) , which via a final Grignard reaction yields the 25-OH-D 2 -epimers (11) .
- the 5,7-diene-ketone (7B) is first reacted with Grignard reagent to yield the 25-hydroxy intermediate (7C) , which after irradiation gives the corresponding 25-OH-previtamin D 2 (8C) , and a final thermal iscmerization provides the 25-OH-D 2 products (11).
- sequence A is generally preferred, because of the utility of intermediates such as (9A) and (10) for the preparation of other vitamin D 2 -analogs and/or labeled derivatives (see also below).
- the 5,7-diene (7) may be used as the free hydroxy compound or as its C-3-acylate.
- the final 25-OH-D 2 products will be obtained as the free hydroxy coirpounds or, if desired, as the C-3-, or C-25-acylates, or 3,25-diacylates.
- synthesis according to sequence A or B would normally provide the 25-OH-D 2 products as the free hydroxy compounds since the final Grignard reaction co ⁇ mon to both sequences removes any acyl groups.
- Sequence C can be used to produce the 25-OH- D 2 epimers (11) as the free hydroxy compounds, or as the 3- or 25-monoacylate, or 3,25-diacylate, depending on the intermediate used.
- the 5,7-diene intermediate (7C) shown in Process Scheme II may be used as the 3-acyl, or
- 25-OH-D 2 -epimers 25-OH-D 2 (11a) or 25-OH-24-epi-D 2 (11b) when obtained in the free hydroxy forms, are also conveniently acylated at the C-3 or C-25, or at both positions, by reaction with acid anhydrides or acyl chlorides using conventional conditions.
- 25-OH-D 2 (11a) nay be acylated to yield, for example, the 25-OH-D 2 -3--acetate, or the corresponding 3,25-diacetate.
- the 3-moncacetate in a like fashion, may be further acylated at C-25 by treatment with a different acylating reagent, or, alternatively, the 3,25-diacetate may be selectively hydrolyzed by mild base (KOH/MeOH) to give the 25-monoacetate, which if desired can be reacylated with a different acyl group at C-3.
- KOH/MeOH mild base
- suitable acylating agents are propionic, butyric, pentanoic or hexanoic anhydrides or the corresponding acid chlorides, or aromatic acylating agents such as the acid chlorides of benzoic or substituted benzoic acids, or the anhydrides of dicarboxylic acids, such as succinic, glutaric, adipic, diglycolic anhydrides, or the acyl chlorides of these dicarboxylic acid monoesters.
- the 5 ,6-trans-isomers of 25-OH-D 2 and 25-OH-24-epi-D 2 are compounds of potential utility in medical applications because of their considerable vitamin D-like activity.
- These 5 ,6-trans-compounds are prepared from the 5,6-cis-iscmers (i.e. 11a or 11b) by iodine catalyzed iscmerization according to the procedures of Verlcop et al. Rec. Trav. Chim.
- 25-keto-intermediate i.e. compound (10) in Process Scheme I
- keto-vitamin D compound (10) also serves as a convenient intermediate for the synthesis of 25-OH-D 2 -analogs, of the formula (12) shown below.
- X 1 and X 2 are selected from hydrogen and acyl, and where Y is an alkyl group other than methyl or an aryl group.
- ketone (10) is prepared by reaction of ketone (10) with the appropriate alkyl- or aryl- Grignard or alkyl- or aryl-lithium reagent.
- sulfone A is itself prepared according to the process shown in Process Scheme III.
- This synthesis is straightforward and involves as a first step the reaction of commercially available 4-hydroxy-3-methyl-butan-2-one with p-toluenesulfonylchloride to form the corresponding toluenesulfonyl ester.
- This product is then treated with thiophenol in the presence of base (e.g. potassium t-butylate) whereby the toluenesulfonyl group is displaced and the corresponding phenylthioether is formed.
- base e.g. potassium t-butylate
- the ketone group is protected as the ethylene ketal by reaction with ethylene glycol under acid catalysis, using conventional conditions well established in the art.
- Oxidation of this product with a peracid (e.g. perbenzoic acid, or m-chloroperbenzoic acid) in halocarbon solution (e.g. CH 2 Cl 2 ) then provides the desired sulfone, labeled sulfone A, as shown in Process Scheme III.
- a peracid e.g. perbenzoic acid, or m-chloroperbenzoic acid
- halocarbon solution e.g. CH 2 Cl 2
- optically active starting materials for example, the ethylene ketal of (3R)-4-hydroxy-3-methylbutan-2-one or the ethylene ketal of (3s)-4-hydroxy-3-methylbutan-2-one.
- Each of these ethylene ketals is then processed through the appropriate steps of Process Scherre III, namely a) tosylation, b) phenylsulfide formation, and c) peracid oxidation, to yield from the (R) ketal starting material the (S)-enantiomer of sulfone A, and from the (S) -ketal the (R)-enanti ⁇ ner of sulfone A.
- the (R) and (S) ketal starting materials are themselves conveniently obtained from commercially available recemic ⁇ -methylaceto- acetate ethyl ester (ethyl 2-methyl-3-oxo-butonate) as follows:
- the keto ester is converted to the ethylene ketal ester by reaction with ethylene glycol under acid catalysis using conventional procedures, and the ester function is then reduced (LiAlH 4 in ether) to yield the racemic ketal-alcohol (2,2-ethylenedioxy-3-methylbutan-4-ol).
- Resolution of the racemic mixture is accomplished by conversion to a mixture of diastereomers (by reaction of the alcohol function with an optically active acylating agent) which are then separated.
- the alcohol can be converted to the corresponding ⁇ -methoxy- ⁇ -trifluoromethylphenylacetyl derivative (or similar optically active acylate) by reaction in pyridine solution with the chloride of optically active (+) ⁇ -methoxy- ⁇ - trifluorcmethyl-phenylacetic acid (according to the procedures of, for example, Dale et al. , J. Org. Chem. 34, 2543 (1961); Eguchi et al. , Proc. Natl. Acad. Sci.
- (3R)-4-hydroxy-3-methylbutan-2-one and (3S)-4-hydroxy-3-methylbutaan-2-one can also be prepared from naturally occurring optically active substrates.
- (3S)-3-hydroxy-2-methylpropanoic acid ⁇ -hydroxyisobutyric acid) with methyl lithium there is obtained (3S)-4-hydroxy-3-methylbutan-2-one; and the corresponding (3R)-hydroxybutanone can be prepared from the same (S)-hydroxy-iisobutyric acid, by transposition of functionalities, i.e. elaboration of the hydroxymethyl group to a methyl ketone function, and reduction of the acid to alcohol, according to obvious and conventional procedures.
- the C-22 aldehyde (1) is obtained by degradation of ergosterol acetate (in which the ring B diene system has been protected by Diels-Alder addition of 4-phenyl-1,2,4-triazoline-3,5-dione) according to the procedure of Barton et al. (supra).
- the i-ether aldehyde (4) is obtained from stigmasterol by the method of U.S. Patent 2,623,052.
- Grignard reagent is prepared from Mg (535 mg; 22.22 mmol) and ethyl bromide in ether (10 ml) , and the vigorously stirred solution is treated with sulfone A (6 g;
- Grignard reagent is prepared from Mg (75 mg, 3.1 mmol) and ethyl bromide in ether (10 ml) .
- sulfone A 891 mg; 3.3 mmol
- benzene 5 ml
- a solution of aldehyde (4) 290 mg
- the reaction is continued for 2.5 h, then quenched with saturated (NH 4 ) 3 SO 4 solution (5 ml) and diluted with ether.
- the separated organic layer is washed with water, dried, and evaporated.
- the oily residue containing (5) is treated with acetic anhydride (2 ml) and pyridine (2 ml).
- the reaction mixture is allowed to stand for 24 h, poured into water and extracted with benzene.
- the benzene extract is washed with an aqueous solution of CuSO 4 , water, dried, and evaporated.
- the crude product [the acetate of (5)] is dissolved in meth-anol saturated with Na 2 HPO 4 and sodium amalgam (5.65%, 8 g) is added.
- the reaction mixture is stirred at 4°C for 16 h. After the reaction, mercury is removed by filtration, methanol is evaporated, and water and benzene are added to dissolve the residue.
- the benzene layer is dried and evaporated.
- the reaction mixture is irradiated under argon atmosphere for 18 min with a mercury arc lamp (Hanovia SA-1) fitted with a Vycor filter.
- Grignard reagent is prepared from magnesium (240 mg) and methyl iodide in anhydrous ether (20 ml). To one-tenth of this solution (2 ml; 0.5 M solution of CH 3 Mgl) ketone (10) (16 mg; 0.04 mmol) in ether (2 ml) is added. The reaction mixture is stirred at room temperature for 2 h under an inert atmosphere, then quenched with squeous solution of NH 4 Cl, diluted with benzene and washed with water. The organic layer is separated, dried and evaporated.
- the crude product is first purified by silica gel column chromatography (elution with 20% ether in benzene) and the mixture of (11a) and (11b) (16 mg; 96%) thereby obtained is then repeatedly Chr ⁇ ratographed on HPLC column using 2% 2- ⁇ ropanol in hexane as an eluent to separate the 24-stereoisomers, 24-epi-25-OH-D 2 (11b) and 25-OH-D 2 (11a) .
- optically active (S)-isulfone A having the structure
- 25-OH-D 2 (compound 11a) is dissolved in ether containing a drop of pyridine and treated with a solution of iodine in hexane (ca. 0.5 mg/ml) for 15 min. Addition of an aqueous solution of sodium thiosulfate, separation of the organic phase, and evaporation of solvents yields a residue, from which the desired 25-hydroxy-5,6-trans-vitamin D 2 is isolated by HPLC using a microparticulate silica gel column and 2% of 2-propanol in hexane as eluent.
- Mg is reacted with the following halides, ethyl iodide; propyl iodide; isopropyl bromide; butyl bromide; sec.-butyl iodide; isobutyl iodide; pentyl iodide; and phenyl bromide, to obtain the corresponding Grign.ard reagents.
- halides ethyl iodide
- propyl iodide isopropyl bromide
- butyl bromide butyl bromide
- sec.-butyl iodide isobutyl iodide
- pentyl iodide and phenyl bromide
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Nutrition Science (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
Abstract
Hydroxylated derivatives of vitamin D2., processes for preparing such compounds, intermediates utilized in such processes and certain isotopically labelled vitamin D2 compounds. The vitamin D2 derivatives would find application in the treatment of or prophylaxsis for various disease states characterized by calcium and phosphorous imbalances and as a substitute for vitamin D3 and its metabolites in their various applications.
Description
Description
Hydroxyvitamin D2 Compounds Process for Preparing Them
Technical Field
This invention relates to biologically active vitamin D compounds.
More specifically, this invention relates to a process for the preparation of hydroxylated derivatives of vitamin D2, and to novel intermediates used in this process.
Still more specifically, this invention relates to the synthesis of 25-hydroxyvitamin D2 and the 24-epimer thereof, to certain alkyl and aryl-analogs and to 5,6-trans-, and the acyl derivatives of these compounds.
Background
The D vitamins are very iinportant agents for the control of calcium and phosphate metabolism in animals and humans, and have long been used as dietary supplements and in clinical practice to assure proper bone growth and development. It is now known that the in vivo activity of these vitamins, specifically of vitamin D2 and D3, is dependent on metabolism to hydroxylated forms. Thus, vitamin D3 undergoes two successive hydroxylation reactions in vivo, leading first to 25-hydroxyvitaιtιin D3 and then to 1,25-dihydroxyvitamin D3 and the latter is indeed thought to be the cαtpound responsible for the well-known beneficial effects of vitamin D3. Likewise, vitamin D2, which is commonly used as a dietary supplement, undergoes an analogous hydroxylation sequence to its active forms, being first converted to 25-hydroxyvitamin D2 (25-OH-D2.) and then to 1,25-dihydroxyvitamin D2 (1,25-(OH)2D3). These facts are well established and well known in the art [see, for example, Suda et al. Biochemistry 8, 3515 (1969) and Jones et al. Biochemistry 14, 1250 (1975)].
Like the metabolites of the vitamin D3 series, the hydroxylated forms of vitamin D2 named above are, because of their potency and other beneficial properties, highly desirable dietary supplements, or pharmaceutical agents, for the cure or prevention of bone or related diseases, and their value and possible use is recognized in patents relating to these corripounds [U.S. Letters Patents 3,585,221 and 3,880,894]. ltfhereas all metabolites of vitamin D3 have been prepared by chemical synthesis, there has been but little work on the preparation of vitamin D2 metabolites. The known synthetic processes for the metabolites of the D3-series (especially as far as they relate to the preparation of side chain hydroxylated compounds) are, of course, in general not suitable for the preparation of the corresponding vitamin D2 metabolites, since the latter are characterized by a side chain structure (i.e. presence of a double bond and an extra methyl group) which requires a different synthetic approach from that applicable to side chain hydroxylated D3 compounds.
Two compovinds structurally related to 25-OH-D2 have been prepared, namely 22-dehydro-25-hydroxycholecalciferol, which may be considered a 24-desmethyl analog of 25-OH-D2 (see U.S. Patent 3,786,062), and 24,25-dihydroxyvitamin D2, the 24-hydroxy-analog of 25-OH-D2 [Jones et al. Biochemistry 18, 1094 (1979) ] . However, the synthetic methods proposed in these reports are not applicable to the preparation of 25-OH-D2 itself. No synthesis of the latter compound has appeared in the literature, and although there is the mention in the paper by Salmond et al. (Tetrahedron Letters, 1695-1698 (1977), see p. 1697 and footnote 11) of the successful preparation of 25-OH-D2, no information on the overall process has been published to date.
Disclosure of Invention
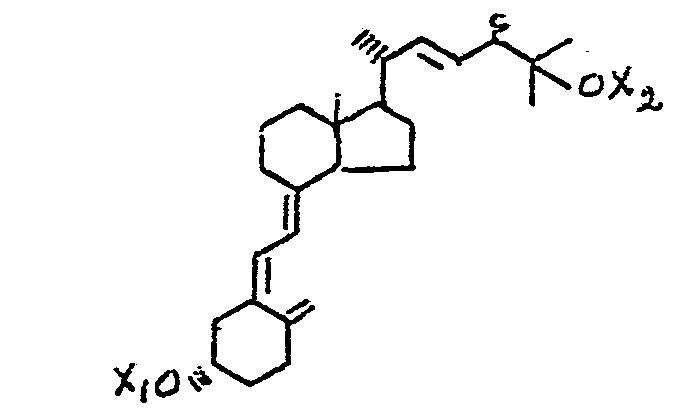
A novel and convenient synthesis of 25-hydroxylated vitamin D2 compounds has now been developed and is fully described herein. This synthesis provides 25-hydroxyvitamin D2 (25-CΗ-D2) and its 24-epimer, 25-hydroxy-24-epi-vitamin D2 (25-OH-24-epi D2) , characterized by the structures shown below (where X1 and X2 are hydrogen and Y is methyl) ,


as well as the corresponding alkyl or aryl analogs, characterized by the structures above where Y is alkyl or aryl, and the hydroxy-protected derivatives of these compounds characterized by the structures above, where either of X1 and X2, or both of X1 and X2 are acyl.
In addition, the present process provides the 5,6-trans-isomers of the above compounds, and novel intermediates that are of utility for the preparation of 25-OH- D2-.analogs and/or isotopically-labeled products.
The term "acyl", as used in this specification or in the claims, signifies an aliphatic acyl group of from 1 to about 6 carbons, in all possible isomeric forms (e.g. formyl, acetyl, propionyl, butyryl, isobutyryl, valeryl, etc.), or an aromatic acyl group (aroyl group) such as benzoyl, the isomeric methyl-benzoyls, the isomeric nitro- or halo-benzoyIs, etc., or a dicarboxylic acyl group of from 2 to 6 atoms chain length, i.e. acyl groups of the type ROOC(CH2)nCO-, or ROOCCH2-O-CH2CO-, where n has values between 0 and 4 inclusive and R is hydrogen or an alkyl radical, such as oxalyl, malonyl, succinoyl, glutaryl, adipyl, diglycolyl. The term
"alkyl" refers to a lower alkyl group of 1 to 6 carbons in all possible isomeric forms, e.g. methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec.-butyl, pentyl, etc., and the word "aryl" signifies a phenyl or substituted phenyl group, e.g. alkylphenyl, methoxyphenyl, etc.
'The overall process developed for the preparation of the above cαipounds may be divided into two general phases, namely (a) addition of a side chain fragment to a suitable steroidal precursor to produce a 5,7-diene steroid as the central intermediate, and (b) conversion of this 5,7-diene to the vitamin D structure with, as required, further modification of the side chain to produce the desired 25-hydroxylated coπpounds. This general scheme allows for sore flexibility in the choice of specific starting material and in the exact order of individual process steps, two features that are of considerable practical advantage and convenience.
The reaction sequence illustrated by Process Scheme I presents an embodiment of the overall process, whereas Process Scheme II illustrates sane of the options available for executing the last four steps of the synthesis.
Starting materials for the present process are steroidal 22-aldehydes in which the ring B double bond(s) is (are) protected in an appropriate manner. As shown in Process Scheme I, suitable compounds are for example, the PTAD-diene-protected-22-aldehyde (1) (where PTAD refers to the phenyltriazoline-3,5-dione-protecting group shown) or the 3,5-cyclo-22-aldehyde (4) wherein the Δ 5-double bond is protected via i-ether formation. Both of these compounds are known products (see for example Barton et al. J. Chem. See. (C) 1968 (1971); and Heyl et al. U.S. Patent 2,623,052) and both can be carried through the steps of the present process in a basically analogous fashion.
The first step of this process comprises the addition of a suitable side chain fragment. Thus, condensation of aldehyde (1) with a sulfonyl-side chain fragment as shown in
 the Scheme (sulfone A, further described below) present in the form of its anion, in an ether or hydrocarbon solvent, provides the hydroxy-sulfone intermediate (2) . The anion of the sulfone A side chain fragment is generated by treatment of the sulfone with a strong base, such as lithium diethylamide, n-butyl lithium or ethyl magnesium bromide (or similar Grignard reagent) in an ether or hydrocarbon solvent, and to this solution of sulfone anion the steroid aldehyde (compound1) as an ether or hydrocarbon solution is then added. The reaction may be conducted advantageously at about room temperature, and is best effected under an inert atmosphere.
the Scheme (sulfone A, further described below) present in the form of its anion, in an ether or hydrocarbon solvent, provides the hydroxy-sulfone intermediate (2) . The anion of the sulfone A side chain fragment is generated by treatment of the sulfone with a strong base, such as lithium diethylamide, n-butyl lithium or ethyl magnesium bromide (or similar Grignard reagent) in an ether or hydrocarbon solvent, and to this solution of sulfone anion the steroid aldehyde (compound1) as an ether or hydrocarbon solution is then added. The reaction may be conducted advantageously at about room temperature, and is best effected under an inert atmosphere.
The analogous addition of sulfone A to aldehyde (4) provides the hydroxy-sulfone intermediate characterized by structure (5) in Process Scheme I.
The next step comprises the removal of the hydroxy- and phenylsulfonyl groups in the side chain with formation of the 22 (23) -trans-double bond. Thus, treatment of compound (2), in methanol solution saturated with NaHPO4, with sodium amalgam under an inert atmosphere, gives cαnpound (3) featuring the desired trans-22-double bond in the side chain. The analogous treatment of compound (5) gives the 22-olefinic ccmpound (6). If desired, the 22-hydroxy group in compounds (2) or (5) can also be acylated or sulfonylated (e.g. mesylated) prior to the Na/Hg-reduction step, but this is not generally required.
It is to be noted that, as shown in Process Scheme I, addition of the side chain fragment, sulfone A, to the aldehydes (1) or (4) , does not cause epimerization at the asymetric center of carbon 20, i.e. the stereochemistry at that center is retained, as is required. If desired, retention of stereochemistry at carbon 20 may be checked at this stage of the synthesis by the conversion of intermediates of type (3) or (6) back to the original aldehyde starting materials. For example, subjecting compound (6) to ozonolysis with reductive work-up, using fully conventional and standard conditions, yields the corresponding C-22-aldehyde, i.e. the
aldehyde of structure (4) . Spectroscopic and chrcmatographic comparison of the aldehyde obtained from ozonolysis with the original starting material confirms retention of C-20 stereochemistry.
The third operation of the process involves conversion of these ring B-protected steroids to the desired 5,7-diene intermediate (7). In the case of the PTAD-diene-protected compound (3) , this conversion is accomplished in a single step, namely treatment of (3) with a strong hydride reducing agent (e.g. LiAlH4)in an ether solvent at reflux temperature gives the diene (7) . This same material, compound (7), is produced from the i-ether derivative (6), in several steps, all of which are conventional and well-known in the art. The i-ether (6) is first solvolyzed in glacial acetic acid at reflux for ca. 2 hours to yield the corresponding 5-ene-3-acetate derivative (6a). This compound, in a hydrocarbon solution (e.g. hexane) at reflux temperature preferably under an inert atmosphere, is then treated with a brominating reagent (e.g. 1,3-dibromo-5,5-dimethylhydantoin) over a period of about 20 minutes, and the resulting C-7-bromo-intermediate is directly dehydrobrcminated by dissolution in xylene and treatment with a base (e.g. s-collidine) at reflux temperatures under an inert atmosphere for about 90 minutes. The resulting product, the 5,7-diene-3-acetate is then isolated in the usual way and purified by high performance liquid chromatography or thin layer chromatography on silica gel plates. Simple hydrolysis of the acetate (5% KOH in MeOH) then provides 5,7-diene (7). This hydrolysis step may, however, also be αnitted since both the 5,7-diene-3-ol (7) or the corresponding 3-O-acylates can be used for the subsequent steps of the process. Any such 3-O-acylates are, of course, also readily accessible by simple acylation of (7) according to conventional procedures.
Conversion of 5,7-diene (7) to the final vitamin D products comprises a sequence of four steps, the precise order
of which may be altered as convenient. The sequence shown in Process Scheme I involves first the irradiation of an ether or hydrocarbon solution of the 5,7-diene (7) with ultraviolet light to yield the previtamin analog (8) which by warming (50-90°C) in a suitable solvent (e.g. ethanol, hexane) undergoes iscmerization to the vitamin D2-analog (9) . The next step, removal of the ketal protecting group, is a critical one, since ketal removal by hydrolysis to give the corresponding keto-derivative (10) must be accomplished without iscmerization of the 22(23)-double bond to the conjugated 23 (24)-position. Isomerization of a β, δ-unsaturated ketone to the conjugated αβ-unsaturated ketone can readily occur under conditions of ketal hydrolysis, but must be avoided in this case, since it would defeat the purpose of the entire synthetic sequence. In the present process, ketal removal is achieved by heating ketal (9) in hydroxylic solvent under acid catalysis for 1-2 hours. (It is desirable to monitor the progress of reaction by periodic chromatographic analysis of the crude reaction mixture. Analysis by HPLC is suitable and convenient.) The product, ketone (10) thereby obtained, is then alkylated in the final step by means of a Grignard reagent (an alkyl-, or aryl-magnesium halide, e.g. methyl magnesium branide in this case) to give the 25-hydroxyvitamin D2 compound (11) . Alkylation via an alkyllithium reagent, e.g. methyl lithium, is also effective and convenient. If the side chain fragment, sulfone A, as used in the first step, is racemic, i.e. exists as a mixture of its (R) and (S)-enantioners, then compound (11) will be obtained as a mixture of two C-24-epimers, i.e. (24S)-epimer (11a) which corresponds to the natural product, and the (24R) -epimer (11b) which is 25-OH-24-epi-D2. These C-24-epimers are conveniently separated by high performance liquid chromatography (HPLC) on an efficient microparticulate silica gel column to obtain 25-OH-D2 (11a) and 25-OH-24-epi-D2 (11b) in pure form.
It is to be noted that with racemic sulfone A as the side Chain starting material, the early synthetic intermediates, e.g. (3) [or (6), and (6a)] as well as the 5,7-diene (7) and subsequent intermediates (8) , (9) , and (10) also occur as the two C-24-epimers. If desired and convenient, separation of epimers can be conducted at any of these intermediate stages, and the (24R) and (24S) epimers may then be processed separately through the remaining steps to yield 25-OH-D2 (11a) or 25-OH-24-epi-D2 (11b) as desired. It is generally convenient to effect separation at the stage of the final products.
Since the mixtures of (24R)- and (24S)-epimers arise when the side chain fragment, sulfone A, as used in the above process is itself a racemic compound, i.e. is present as an enantiomeric mixture of the (R) and (S) forms, it is also possible, if desired, to circumvent the need for epimer separation by the use of optically active sulfone A. Thus, use of the (R) -epimer of sulfone A in the present process yields specifically 25-OH- D2 (11a) , while use of the corresponding (S)-epimer of sulfone A provides 25-OH-24-epi-D2 (11b) as well as, of course, the respective intermediates in the pure (24R) or (24S) forms; the use of such optically pure sulfone starting material requires no other modification of the process steps described.
It is also important to note that the exact sequence of steps between the 5,7-diene (7) and the final products may be altered. Indeed, there are three convenient synthetic sequences, all involving the same steps, but in different order. These alternate sequences are shown in Process Scheme II, where X1 and X2 in the structural drawings signify hydrogen or an acyl group such as for example acetyl, propionyl, butyryl, benzoyl or substituted benzoyl.
The first sequence (identified by the letter A in Process Scheme II), leading from diene (7A) (which, when X1=H, corresponds to diene (7) of Process Scheme I) to intermediates
(8A) , (9A) , and to the final product (11) presents the orderof reactions as discussed above.
Alternatively, the ketal in diene (7A) may be hydrolyzed first (see sequence B in Process Scheme II) , to yield the diene-ketone identified as (7B) in the scheme, which after irradiation gives the previtamin ketone (8B) , and thermal iscmerization then leads to vitamin D2-ketone (10) , which via a final Grignard reaction yields the 25-OH-D2-epimers (11) .
In the third sequence (C) , the 5,7-diene-ketone (7B) is first reacted with Grignard reagent to yield the 25-hydroxy intermediate (7C) , which after irradiation gives the corresponding 25-OH-previtamin D2 (8C) , and a final thermal iscmerization provides the 25-OH-D2 products (11).
Thus, these three sequences differ only in the exact order in which specific steps are carried out, but the experimental conditions for the individual steps are analogous to the procedures described earlier, and as described in detail by the Examples. Among the three alternate sequences, sequence A is generally preferred, because of the utility of intermediates such as (9A) and (10) for the preparation of other vitamin D2-analogs and/or labeled derivatives (see also below).
For any of these sequences, the 5,7-diene (7) may be used as the free hydroxy compound or as its C-3-acylate. Depending on the sub.sequent reaction sequence, the final 25-OH-D2 products will be obtained as the free hydroxy coirpounds or, if desired, as the C-3-, or C-25-acylates, or 3,25-diacylates. Thus, synthesis according to sequence A or B would normally provide the 25-OH-D2 products as the free hydroxy compounds since the final Grignard reaction coπmon to both sequences removes any acyl groups. Sequence C can be used to produce the 25-OH- D2 epimers (11) as the free hydroxy compounds, or as the 3- or 25-monoacylate, or 3,25-diacylate, depending on the intermediate used. For example, the 5,7-diene intermediate (7C) shown in Process Scheme II, may be used as the 3-acyl, or


25-acyl, or 3,25-diacyl derivatives, which are available from the 3,25-diol by reaction with acyl chloride or acid anhydride reagents according to conventional procedures. Thus, reaction of 3,25-diol intermediate (7C) with acetic anhydride is pyridine at room temperature gives the 3-acetate, and the corresponding 3,25-diacεtate is obtained by further acylation at elevated teπperature; the latter may be selectively hydrolyzed with dilute KOH/MeOH at room temperature to give the 25-mono-acetate. Further conversion of any of such acyl intermediates through the remaining steps [to (8C) and (11)] in Process Scheme II, then yields the 25-OH-D2-epimers (11) in any desired acylated form.
The individual 25-OH-D2-epimers, 25-OH-D2 (11a) or 25-OH-24-epi-D2 (11b) when obtained in the free hydroxy forms, are also conveniently acylated at the C-3 or C-25, or at both positions, by reaction with acid anhydrides or acyl chlorides using conventional conditions. Thus, 25-OH-D2 (11a) nay be acylated to yield, for example, the 25-OH-D2-3--acetate, or the corresponding 3,25-diacetate. The 3-moncacetate, in a like fashion, may be further acylated at C-25 by treatment with a different acylating reagent, or, alternatively, the 3,25-diacetate may be selectively hydrolyzed by mild base (KOH/MeOH) to give the 25-monoacetate, which if desired can be reacylated with a different acyl group at C-3. In addition to acetic anhydride, suitable acylating agents are propionic, butyric, pentanoic or hexanoic anhydrides or the corresponding acid chlorides, or aromatic acylating agents such as the acid chlorides of benzoic or substituted benzoic acids, or the anhydrides of dicarboxylic acids, such as succinic, glutaric, adipic, diglycolic anhydrides, or the acyl chlorides of these dicarboxylic acid monoesters.
In addition to the acylates, the 5 ,6-trans-isomers of 25-OH-D2 and 25-OH-24-epi-D2 are compounds of potential utility in medical applications because of their considerable vitamin D-like activity. These 5 ,6-trans-compounds are
prepared from the 5,6-cis-iscmers (i.e. 11a or 11b) by iodine catalyzed iscmerization according to the procedures of Verlcop et al. Rec. Trav. Chim. Pays Bas 78, 1004 (1969) , and the corresponding 3- and/or 25-acylates are likewise obtained by analogous iscmerization of the corresponding 5,6-cis-acylates, or by acylation of the 5,6-trans-25-OH-D compounds.
It is to be noted also that the 25-keto-intermediate (i.e. compound (10) in Process Scheme I) can serve as a substrate for the convenient preparation of 25-OH-D2 or its 24-epimer in isotopically labeled form, namely by reaction of the ketone with commercially available isotopically labeled
Grignard or methyl lithium reagents to provide 25-OH-D2 compounds labeled at carbon 26 with 13C, 14C, 2H or 3H.
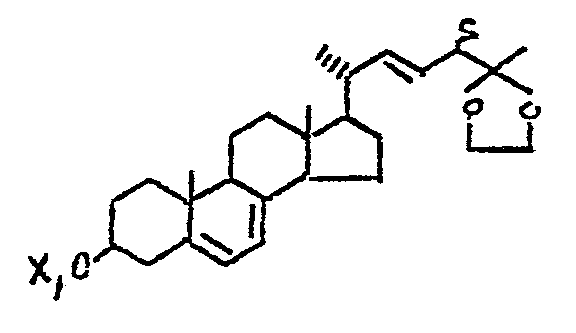
Furthermore, the keto-vitamin D compound (10) also serves as a convenient intermediate for the synthesis of 25-OH-D2-analogs, of the formula (12) shown below.

Since the coπpounds, where Y is a higher homolog of methyl, are generally more lipophilic, the alkyl- or aryl-analogues represented by structure (12) above or their 5,6-trans-isomers, are expected to have utility in applications where a greater degree of lipcphilicity is desired.
The required side chain fragment, sulfone A, is itself prepared according to the process shown in Process Scheme III. This synthesis is straightforward and involves as a first step the reaction of commercially available 4-hydroxy-3-methyl-butan-2-one with p-toluenesulfonylchloride to form the corresponding toluenesulfonyl ester. This product is then treated with thiophenol in the presence of base (e.g. potassium t-butylate) whereby the toluenesulfonyl group is displaced and the corresponding phenylthioether is formed. In the next step, the ketone group is protected as the ethylene ketal by reaction with ethylene glycol under acid catalysis, using conventional conditions well established in the art. Oxidation of this product with a peracid (e.g. perbenzoic acid, or m-chloroperbenzoic acid) in halocarbon solution (e.g. CH2Cl2) then provides the desired sulfone, labeled sulfone A, as shown in Process Scheme III.
If sulfone A is desired in optically active form, i.e. as the pure (R) or (S) -epimer, it is appropriate to use optically active starting materials , for example, the ethylene ketal of (3R)-4-hydroxy-3-methylbutan-2-one or the ethylene ketal of (3s)-4-hydroxy-3-methylbutan-2-one. Each of these ethylene ketals is then processed through the appropriate steps of Process Scherre III, namely a) tosylation, b) phenylsulfide formation, and c) peracid oxidation, to yield from the (R) ketal starting material the (S)-enantiomer of sulfone A, and
from the (S) -ketal the (R)-enantiαner of sulfone A. The (R) and (S) ketal starting materials are themselves conveniently obtained from commercially available recemic α-methylaceto- acetate ethyl ester (ethyl 2-methyl-3-oxo-butonate) as follows: The keto ester is converted to the ethylene ketal ester by reaction with ethylene glycol under acid catalysis using conventional procedures, and the ester function is then reduced (LiAlH4 in ether) to yield the racemic ketal-alcohol (2,2-ethylenedioxy-3-methylbutan-4-ol). Resolution of the racemic mixture is accomplished by conversion to a mixture of diastereomers (by reaction of the alcohol function with an optically active acylating agent) which are then separated. For exartple, the alcohol can be converted to the corresponding α-methoxy-α-trifluoromethylphenylacetyl derivative (or similar optically active acylate) by reaction in pyridine solution with the chloride of optically active (+) α-methoxy-α- trifluorcmethyl-phenylacetic acid (according to the procedures of, for example, Dale et al. , J. Org. Chem. 34, 2543 (1961); Eguchi et al. , Proc. Natl. Acad. Sci. USA 78, 6579 (1981)); this disastereαrteric acyl-derivative mixture is now separable by HPLC or similar chromatographic πethods into its two components, namely the acylate of the (R) -enantiσmer and the acylate of the (S)-enantiomer. Removal of the acyl group in each compound by base hydrolysis under standard conditions then provides the ethylene ketal of (3R)-4-hydroxy-3-methylbutan-2-one, and the ethylene ketal of (3S)-4-hydroxy-3-methylbutan-2-one, which are then separately processed to the respective sulfone A enantiomer as described above. If desired, optically active hydroxybutanone intermediates, i.e. (3R)-4-hydroxy-3-methylbutan-2-one and (3S)-4-hydroxy-3-methylbutaan-2-one, can also be prepared from naturally occurring optically active substrates. Thus, by reaction of the known (S)-3-hydroxy-2-methylpropanoic acid β-hydroxyisobutyric acid) with methyl lithium there is obtained (3S)-4-hydroxy-3-methylbutan-2-one; and the
corresponding (3R)-hydroxybutanone can be prepared from the same (S)-hydroxy-iisobutyric acid, by transposition of functionalities, i.e. elaboration of the hydroxymethyl group to a methyl ketone function, and reduction of the acid to alcohol, according to obvious and conventional procedures.
The present invention is further described by means of the following illustrative examples. In these examples, numerals designating specific products (e.g. cαrpσunds (1) , (2) , (3) , etc. in Examples 1 through 12, or compounds (7A), (8B) , (8C) , etc. in Examples 13 and 14) refer to the structures so numbered in Process Schemes I or II. Example 1
The C-22 aldehyde (1) is obtained by degradation of ergosterol acetate (in which the ring B diene system has been protected by Diels-Alder addition of 4-phenyl-1,2,4-triazoline-3,5-dione) according to the procedure of Barton et al. (supra). The i-ether aldehyde (4) is obtained from stigmasterol by the method of U.S. Patent 2,623,052. Example 2
Synthesis of the Side Chain Fragment (Sulfone A)
To a stirred solution of 4-hydroxy-3-methylbutan-2-one (12.75 g; 0.125 mol) in pyridine (100 ml) is added p-toluenesulfonyl chloride (p-TsCl) (33.25 g, 0.175 mol) in portions, and after standing for 14 h at room temperature, the reaction mixture is poured into water and extracted with CH2Cl2. The extract is washed several times with aqueous CuSO4 solution and water and then dried over anhydrous sodium sulfate. Removal of solvent under reduced pressure gives the crude tosylate which is used directly for the next reaction.
Thiophenol (14 g) dissolved in DMF (100 ml) is treated with t-BuOK (14 g) . To this reagent, the tosylate is added and after 12 h at room temperature, the reaction mixture is poured into water and extracted with CH2Cl2. The extract is washed with aqueous Na2CO3 solution and water, then dried. Evaporation of solvent gives an oily residue which is purified
by silica gel column chromatography. Pure phenyl sulfide is eluted with benzene (yield 15 g).
To this phenyl sulfide derivative (15 g) , in benzene (100 ml) , ethylene glycol (6 g) and p-TsOH (20 mg) is added and the reaction mixture is heated under a Dean-Stark trap for 3 h. After cooling, it is extracted with Na2CO3 solution and water, then dried and the solvent is evaporated. The product, the desired ketal, is chrαnatographically homogenous and can be used in the next step without further purification.
Crude ketal in dichloromethane (250 ml) solution is treated with m-chloroperbenzoic acid (m-CPBA) (80-85%, 27 g, added in portions) while maintaining the temperature of the reaction mixture below 30°C. After the addition of reagent, the reaction is allowed to stand at room temperature with occasional shaking. When the reaction reaches completion (about 1.5 h) , the aromatic acids are removed by extraction with aqueous NH3, and the organic layer is washed with water and dried. Evaporation of solvent gives the oily sulfone (sulfone A) in essentially quantitative yield (19 g) . The product is substantially pure (homogenous by TIC) and can be used without any further purification; 1H-NMR; δ; 1.18 (d, J= 7 Hz, 3H, CH3--CH-), 1.19 (s, 3H, CH3-C-), 3.84 (m, 4H, ketal-H), 7.3-7.6 and 7.6-7.9 (m, 3H + 2H, aromatic protons);

IR, 1305,1147,1082 cm-1; mass spectrum, m/z (rel.
 intensity): 255 (M+-Me, 21), 184 (66), 87 (92), 43 (100).
intensity): 255 (M+-Me, 21), 184 (66), 87 (92), 43 (100).
Example 3
Coupling of Sulfone A to Aldehyde (1) : Hydroxysulfone
(2) and Olefin (3). Grignard reagent is prepared from Mg (535 mg; 22.22 mmol) and ethyl bromide in ether (10 ml) , and the vigorously stirred solution is treated with sulfone A (6 g;
2.22 irtnol) in benzene (6 ml) . The precipitate formed is ground with a spatula, stirring is continued, and after 15 min the aldehyde (1) (2.0 g) is added in benzene (10 ml). The reaction mixture is stirred at room temperature for 24 h, then poured into aqueous (NH4)2SO4 solution and extracted with
benzene. The organic layer, after washing with water, drying and evaporation gives an oily residue which is chromatographed on silica gel. In the benzene-ether fractions (8:2) , excess sulfone is recovered (4.5 g); elution with benzene-ether (3:1) affords unreacted aldehyde (1) (1.0 g); the reaction products
(2) are eluted with ethyl acetate.
The crude mixture of steroidal α-hydroxysulfones (2) is dissolved in methanol (200 ml) saturated with Na2HPO4. Sodium amalgam (5.65%, 15 g) is added and the reaction mixture is stirred at 4°C for 15 h.
After completion of the Na/Hg reduction, mercury is removed by filtration, and methanol by evaporation under reduced pressure, water is added and the organic material is
'extracted with benzene. After drying and evaporation of solvent, the oily residue is chromatographed on a silica gel column. Elution with benzene-ether (1:4) gives compound (3) a colorless foam; 1H-NMR, δ : 0.80 (s, 18-H), 0.97 (s, 19-H),
1.22 (s, 26-H), 3.93 (m, 4H, ketal-H), 4.44 (m, 1H, 3 -H) ,
5.25-5.45 (m, 2H, 22-H and 23-H) , 6.23 and 6.39 (doublets, J =
8 Hz, 2 x 1H, 7-H and 6-H) , 7.25-7.45 (m, 5H, -C6H5);
IR,
 3603 (0-H) , 1749, 1692 (C=0) , 1406,1038 cm-1; mass spectrum, m/z: 440 (M+ - triazoline, 24) , 87 (100) .
3603 (0-H) , 1749, 1692 (C=0) , 1406,1038 cm-1; mass spectrum, m/z: 440 (M+ - triazoline, 24) , 87 (100) .
(To increase yield, unreacted aldehyde (1) , as recovered above, can be recycled through the sulfone addition, and the resulting α-hydrσxy sulfcnes (2) are then, as above, traeated with sodium .amalgam in buffered methanol to provide additional olefin (3) . The above reactions are preferably conducted under an inert atmosphere, such as argon.) Example 4
Coupling of Sulfone A to Aldehyde (4) Hydroxysulfone (5) and Olefin (6) .
Grignard reagent is prepared from Mg (75 mg, 3.1 mmol) and ethyl bromide in ether (10 ml) . To the stirred solution of ethyl magnesium bromide, sulfone A (891 mg; 3.3 mmol) in
benzene (5 ml) is added. After stirring the resulting suspension at room temperature for 15 min, a solution of aldehyde (4) (290 mg) in benzene (5 ml) is added. The reaction is continued for 2.5 h, then quenched with saturated (NH4)3SO4 solution (5 ml) and diluted with ether. The separated organic layer is washed with water, dried, and evaporated. The oily residue containing (5) is treated with acetic anhydride (2 ml) and pyridine (2 ml). The reaction mixture is allowed to stand for 24 h, poured into water and extracted with benzene. The benzene extract is washed with an aqueous solution of CuSO4, water, dried, and evaporated. The crude product [the acetate of (5)] is dissolved in meth-anol saturated with Na2HPO4 and sodium amalgam (5.65%, 8 g) is added. The reaction mixture is stirred at 4°C for 16 h. After the reaction, mercury is removed by filtration, methanol is evaporated, and water and benzene are added to dissolve the residue. The benzene layer is dried and evaporated. The oily residue is chromatographed over silica gel. Elution with benzene-ether mixture (93:7) affords compound (6) (206 mg; 54%), 1H-NMR, δ: 0.74 (s, 18-H), 1.04 (s, 19-H) , 1.25 (s, 26-H), 2.78 (m, 1H, 6 -H), 3.34 (s, 3H, -OCH3), 3.97 (m, 4H, ketal-H), 5.25-5.45 (m, 2H, 22-H and 23-H) , IR, 3470
 (O-H) , 1095 cm-1; mass spectrum, m/z (rel. intensity): 456 (M+, 1) , 441 (M+-Me, 45), 87 (100). It should be noted that the acetylation step described above is not essential and may be omitted if desired; i.e. the hydroxysulfone (5) may be submitted directly to Na/Hg-reduction, as in Example 3. The above reactions are preferably conducted under an inert atmosphere, e.g. argon. Example 5
(O-H) , 1095 cm-1; mass spectrum, m/z (rel. intensity): 456 (M+, 1) , 441 (M+-Me, 45), 87 (100). It should be noted that the acetylation step described above is not essential and may be omitted if desired; i.e. the hydroxysulfone (5) may be submitted directly to Na/Hg-reduction, as in Example 3. The above reactions are preferably conducted under an inert atmosphere, e.g. argon. Example 5
Removal of PTAD-protecting Group: 5,7-Diene (7).
A mixture of the compound (3) (1 g) and lithium aluminum hydride (1.8 g) in THF (120 ml) is heated under reflux for 10 h. After cooling, excess reagent is destroyed with a few drops of water, and the mixture is dried over anhydrous MgSO4,
filtered, and solvent is evaporated to give colorless crystalline material. Crude diene 7 is repeatedly crystallized from ethanol; first and second crops combined give 415 mg of (7) . The mother liquor is chromatographed on silica, gel column, to give with benzene-ether (7:3) , an additional 120 mg of (7) ; total yield 535 mg (79%) ; m.p. 132-134°C (from ethanol) , 1HI-NMR, δ:0.63 (s, 18-H) , 0.95 (s, 19-H) , 1.23 (s, 26-H) , 3.63 (m, 1H, 3 -H) , 3.95 (m, 4H, ketal-H), 5.20-5.50 (m, 3H, 22-H, 23-H and 7-H) , 5.57 (m, IH,
6-H); IR, 3430 (O-H) , 1063, 1038 cm-1; mass spectrum, m/z rel.
 int.): 440 (M+, 50), 407 (M+-H2O-Me, 11), 87 (100);UV, 282 nm (ε= 11,000).
int.): 440 (M+, 50), 407 (M+-H2O-Me, 11), 87 (100);UV, 282 nm (ε= 11,000).

Example 6
Irradiation of Cortpound (7) : Previtamin Analog (8) .
A solution of diene (7) (50 mg) in 150 ml of benzene-ether (1:4) is cooled on ice and deoxygenated with argon for 20 min. The reaction mixture is irradiated under argon atmosphere for 18 min with a mercury arc lamp (Hanovia SA-1) fitted with a Vycor filter. The solvent is evaporated and the residue is ctocimatographed on HPLC (6.2 mm x 25 cm microparticulate silica gel, 4 ml/min, 1400 psi) and eluted with 2% 2-propanol in hexane to yield 22 mg (44%) of previtamin (8); 1H-NMR; δ: 0.73 (s, 18-H) , 1.24 (s, 26-H), 1.64 (s, 19-H) , 3.96 (m, 5H, ketal-H and 3 -H) , 5.35 (m, 2H, 22-H and 23-H) , 5.50 (m, 1H, 9-H), 569 and 5.94 (doublets, J
= 11.5 Hz, 2 x 1H, 6-H and 7-H) ; UV,

(ε= 8,900). Example 7
Iscmerization of (8) to the Vitamin-Analog (9). Previtamin 8 (22 mg) is dissolved in ethanol (40 ml) and heated under reflux for 150 min (argon atmosphere). The product is purified by HPLC to yield 18 mg (82%) of the pure vitamin (9); 1H-NMR, δ: 0.75 (s, 18-H) , 1.24 (s, 26-H) , 3.94 (m, 5H, ketal-H and 3 -H) , 4.81 and 5.04 (2 narrow m, 2 x IH, 19 (Z)- and 19(E)-H), 5.33 (m, 2H, 22-H and 23-H) , 6.03 (d, J =
11 Hz, 1H, 7-H) , 6.22 (d, J = 11 Hz, IH, 6- spectrum, m/z (rel. int.): 440 (M+, 17), 87 (100), UV 265 nm
 (ε= 17,000). Exaπple 8
(ε= 17,000). Exaπple 8
Hydrolysis of the ketal: Keto-Vitamin D2-Analog (10).
To the solution of compound (9) (18 mg) in ethanol (35 ml) , p-toluenesulfonic acid (7.5 mg) in water (1 ml) is added and the reaction mixture is heated under reflux for 90 min
(the reaction course is monitored by HPLC) . The solvent is evaporated, the residue is dissolved in benzene and extracted with water. The benzene solution is dried (anhydrous MgSO4) , and evaporated to yield product (10) (16 mg; 99%). 1H-NMR, δ:
0.57 (s, 18-H) , 1.04 (d, J = 7 Hz, 21-H) , 1.13 (d, J = 7 Hz,
28-H) , 2.12 (s, 3H, 26-H) , 3.10 (m, 1H, 24-H) , 3.96 (m, 1H, 3
-H) , 4.82 and 5.05 (2 narrow m, 2 x 1 H, 19(Z)- and 19(E)-H),
5.2-5.5 (m, 2H, 22-H and 23-H) , 6.03 (d, J = 11.5 Hz, 1H,
7-H) , 6.22 (d, J = 11.5 Hz, 1H, 6-H) , IR,
 3596 (O-H) ,
3596 (O-H) ,
1709 cm (C=0) ; mass spectrum, m/z (rel. int.): 396 (M+, 41),
363 (M+-H2O-Me, 13) , 271 (M+-side chain, 16) , 253 (m+-side chain-H2O, 23), 136 (100), 118 (95); UV, 265 nm (ε =

17,900). Example 9
Reaction of Ketone (10) with Methylmagnesium Iodide: 25-OH-D2, (11a), and its Epimer (11b) .
Grignard reagent is prepared from magnesium (240 mg) and methyl iodide in anhydrous ether (20 ml). To one-tenth of this solution (2 ml; 0.5 M solution of CH3Mgl) ketone (10) (16 mg; 0.04 mmol) in ether (2 ml) is added. The reaction mixture is stirred at room temperature for 2 h under an inert atmosphere, then quenched with squeous solution of NH4Cl, diluted with benzene and washed with water. The organic layer is separated, dried and evaporated. The crude product is first purified by silica gel column chromatography (elution with 20% ether in benzene) and the mixture of (11a) and (11b)
(16 mg; 96%) thereby obtained is then repeatedly Chrαratographed on HPLC column using 2% 2-ρropanol in hexane as an eluent to separate the 24-stereoisomers, 24-epi-25-OH-D2 (11b) and 25-OH-D2 (11a) . Chrαnatography and rec±romatography of each stereoisomer yields 4 mg of (11b) (collected at 68 ml) , 4 mg of (11a) (collected at 74 ml) and 7 mg of the mixture of both epimers. Treatment of 2 mg of the epimer mixture with excess acetic anhydride in pyridine solution at room temperature overnight followed by standard work-up yields the corresponding 3-O-acetates.
25-OH-D2 (11a) : + 56.8° (C = 0.2 in EtOH) ; 1H-NMR,
 δ: 0.57 (s, 18-H) , 1.00 (d, J = 7 Hz, 28-H) , 1.04 (d, J = 7 Hz, 21-H), 1.15 and 1.17 (2 singlets, 26-H and 27-H) , 3.95 (m, 1H, 3 -H) , 4.82 and 5.05 (2 narrow m, 2 x 1H, 19 (Z)- and 19(E)-H), 5.23-5.43 (m, 2H, 22-H and 23-H) , 6.05 and 6.22 (2 doublets, J = 11 Hz, 2 x 1H, 7-H and 6-H) ; IR,
δ: 0.57 (s, 18-H) , 1.00 (d, J = 7 Hz, 28-H) , 1.04 (d, J = 7 Hz, 21-H), 1.15 and 1.17 (2 singlets, 26-H and 27-H) , 3.95 (m, 1H, 3 -H) , 4.82 and 5.05 (2 narrow m, 2 x 1H, 19 (Z)- and 19(E)-H), 5.23-5.43 (m, 2H, 22-H and 23-H) , 6.05 and 6.22 (2 doublets, J = 11 Hz, 2 x 1H, 7-H and 6-H) ; IR,

(O-H) , 1645, 1631 (C=C) , 971 cm-1 (trans C=C) ; mass spectrum, m/z (rel. int. ) : 412 (M+, 63) , 394 (M+-H2O, 10) , 379 (M+-H2O-Me, 23) , 271 (M+-side chain, 37) , 253 (M+-side chain-H2O, 43) , 136 (100) , 118 (86) , 59 (99) , UV,
 nm ( ε = 17,950) .
nm ( ε = 17,950) .
24-epi-25-OH-D2 (11b) :
 + 50.7° (C = 0.2 in EtOH) , 1H-NMR, δ: 0.57 (s, 18-H) , 0 99 (d, J = 7 Hz, 28-H) , 1.03 (d, J = 7 Hz, 21-H) , 1.14 and 1.16 (2 singlets, 26-H and 27-H) , 3.94 (m, 1H, 3 -H) , 4.82 and 5.03 (2 narrow m, 2 x 1H, 19 (Z) - and 19 (E) -H) , 5.20-5.40 (m, 2H, 22-H and 23-H) , 6.04 and 6.22
+ 50.7° (C = 0.2 in EtOH) , 1H-NMR, δ: 0.57 (s, 18-H) , 0 99 (d, J = 7 Hz, 28-H) , 1.03 (d, J = 7 Hz, 21-H) , 1.14 and 1.16 (2 singlets, 26-H and 27-H) , 3.94 (m, 1H, 3 -H) , 4.82 and 5.03 (2 narrow m, 2 x 1H, 19 (Z) - and 19 (E) -H) , 5.20-5.40 (m, 2H, 22-H and 23-H) , 6.04 and 6.22
(2 doublets, J = 11 Hz, 2 x 1H, 7-H and 6-H) , IR,
 (OH), 1643, 1630 (C=C) , 971 cm-1 (trans C=C) ; mass spectrum, m/z (rel. int.): 412 (M+, 62) 394 (M+-H2O; 12), 379 (M+-H2O-Me, 31) , 271 (M+-side chain, 44) , 253 (M+-side chain-H2O, 55), 136 (100), 118 (67), 59 (38); UV,
(OH), 1643, 1630 (C=C) , 971 cm-1 (trans C=C) ; mass spectrum, m/z (rel. int.): 412 (M+, 62) 394 (M+-H2O; 12), 379 (M+-H2O-Me, 31) , 271 (M+-side chain, 44) , 253 (M+-side chain-H2O, 55), 136 (100), 118 (67), 59 (38); UV,
 nm (ε= 17,300).
nm (ε= 17,300).
It should be noted that from pure provitamin (7) further synthesis (i.e. the irradiation, isomerization, deketalization and Grignard reaction steps) may be accomplished without
chraomatographic purification of any intermediate. Careful column chrcmatography on silica gel before the final separation on HPLC removes all by-products.
By reaction of 25-OH-D2 (11a) with each of the following acylating reagents, acetic anhydride propionic anhydride, benzoyl chloride and .succinic anhydride, under conventional conditions there is obtained, respectively:
25-OH-D2-3-acetate
25-OH-D2-3,25-diacetate
25-OH-D2-3-propionate
25-OH-D2-3,25-diprcpionate
25-OH-D2-3-benzoate
25-OH- D2-3,25-dibenzoate
25-OH- D2-3-hemisuccinate.
By reaction of 25-OH-24-epi-D2 (11b) with, respectively, acetic anhydride, benzoyl chloride and diglycolic anhydride, tender conventional conditions, there is obtained, respectively:
25-OH-24-epi-D2-3,25-diacetate
25-OH-24-epi-D2-3-benzoate
25-OH-24-epi-D2-3-hemidiglycolate. Example 10
By coupling of aldehyde (1) with optically active (R) -sulfone A having the structure


Using optically active (S)-isulfone A, having the structure

in the reactions described in Example 3, there is obtained compound (3) , having the (24R) -side chain structure as shown below

and reduction of this product according to the conditions of Exartple 5 provides the 5,7-diene (7) having the (24R) configuration. Irradiation of (24R)-(7) according to Example 6 gives the previtamin D analog (8) with the (24R) configuration, and by subsequent thermal iscmerization, according to the conditions of Example 7, there is obtained the vitamin D compound (9) having the (24R)-side chain configuration. Ketal hydrolysis, according to the conditions of Exarrple 8, then yields the (24R) -25-ketovitamin D (10) , and by a reaction of this product with a methyl Grignard reagent
according to the conditions of Exarrple 9, there is obtained 25-hydroxy-24-epi-vitamin D2 (structure 11b, in Process Scheme I). Example 12
Preparations of 5,6-trans-Compounds
25-OH-D2 (compound 11a) is dissolved in ether containing a drop of pyridine and treated with a solution of iodine in hexane (ca. 0.5 mg/ml) for 15 min. Addition of an aqueous solution of sodium thiosulfate, separation of the organic phase, and evaporation of solvents yields a residue, from which the desired 25-hydroxy-5,6-trans-vitamin D2 is isolated by HPLC using a microparticulate silica gel column and 2% of 2-propanol in hexane as eluent.
By the same procedure, there is obtained from 25-hydroxy-24-epi-D2 the corresponding trans-isomer, namely 25-hydroxy-5,6-trans-24-epi-D2.
From 25-OH-D23-acetate, there is obtained 25-OH-5,6-trans-D23-acetate, and from 25-OH-24-epi-D2 3-acetate there is obtained 25-OH-5,6-trans-24-epi-D2 3-acetate by the application of the above isorrerization procedure.
Acylation of 25-OH-5,6-trans-D2 or 25-OH-5,6-trans-24-epi-D2 under conventional conditions provides the respective acylates, such as:
25-OH-5,6-trans-D2-3-acetate
25-OH-5,6-trans-D2-3,25-diacetate
25-OH-5,6-trans-D2-S-benzoate
25-OH-5,6-trans-D2-3-acetate-25-benzoate
25-OH-5,6-trans-24-epi-D2-3-acetate
25-OH-5,6-trans-24-epi-D2-3,25-dibenzoate. Example 13
Hydrolysis of 5,7-diene-25-ketal (ccmpound (7A) , where X1=H) using the conditions described in Example 8 gives 3β-hydroxy-24-methyl 27-norcholesta-5,7,22-trien-25-one {compound 7B, where X1=H) . Irradiation of this product under
conditions analogous to those of Example 6 gives the 25-keto previtamin D2 analog characterized by structure (8B) , where X1=H. Heating of (8B) in an ethanol .solution according to the conditions of Exarrple 7 provides the 25-keto vitamin D2 product (compound 10, where X1=H).
Example 14
Reaction of 3β-hydroxy-24-methyl-27-norccholesta-5,7,22-trien-25-one (compound (7B) , where X1=H) as obtained in Exarrple 13 with methyl magnesium bromide in accordance with the conditions of Example 9 gives 24-methylcholesta-5,7,22-triene-3β,25-diol (compound (7C) , where X1=X2=H). Irradiation of this product, according to the conditiαis of Exarrple 6, gives 25-hydroxy previtamin D2 product characterized by structure (8C, where X1=X2=H) . Thermal isαnzerlation of this previtamin using the conditions of Example 7 provides the 25-hydroxyvitamin D2 compound (11; where X1=X2=H) .
Processing of 24-methylcholesta-5,7,22-triene-3β,25-diol 3,25-diacetate (cαipound 7C, X1=X2=acetyl) through the reaction steps involving irradiation and thermal iscmerization according to the conditions of Examples 6 and 7 respectively gives the 25-OH-D23,25-diacetate epimers (compound (11) , where X1=X2=acetyl). Exarrple 15
Using the conditions analogous to those of Example 9, Mg is reacted with the following halides, ethyl iodide; propyl iodide; isopropyl bromide; butyl bromide; sec.-butyl iodide; isobutyl iodide; pentyl iodide; and phenyl bromide, to obtain the corresponding Grign.ard reagents.
By reaction of each of these reagents with ketone (10) by procedures analogous to that of Example 9, there are obtained, respectively, the following products: compound (12) where X1=X2H, Y=ethyl compound (12) where X1=X2=H, Y=propyl compound (12) where X1=X2=H, Y=isopropyl
compound (12) where X1=X2=H, Y=butyl compound (12) where X1=X2=H, Y=sec.-butyl compound (12) where X1=X2=H, Y=isobutyl compound (12) where X1=X2=H, Y=pentyl compound (12) where X1=X2=H, Y=phenyl
By reaction of ketone (10) with i-sotopically labeled methyl Grign-ard reagents, namely 13CH3Mgl, 14CH3Mgl, C2H3Mgl, C3H3Mgl, under conditions analogous to those of Example 9, there are obtained, respectively, the following products: compound (12) where Y=13CH3, X1=X2=H cαrpound (12) where Y=14CH3, X1=X2=H compound (12) where Y=C2H3, X1=X3=H compound (12) where Y=C3H3, X1=X2=H characterized by isotopic substitution in the methyl group of carbon 26 of the molecule.
Claims
Claims
1. A process for preparing a compounds having the formula

2. The process of Claim 1 wherein X1 and X2 are hydrogen and
Y is methyl.
The process according to Claims 1 or 2, wherein the ketal hydrolysis step precedes the irradiation step, and where the resulting 25-ketone is then subjected sequentially to the irradiation, isomerization and alkylation steps.
The process according to Claim 3, wherein the alkylation reaction precedes the irradiation step, and where the resulting 25-hydroxy-5,7-diene resulting from the alkylation is subjected sequentially to the irradiation and isomerization reactions.
5. The process of Claims 1 or 2 wherein the 5,7-diene steroid has the 24(R) configuration. 6. The process of Claims 1 or 2 wherein the 5,7-diene steroid has the 24 (S) configuration. 7. The process of Claims 3 or 4, wherein the 5,7-diene steroid has the (24R) or the (24S) configuration. 8. Coπpounds having the formula
 where N is a steroid nucleus selected from the group consisting of
where N is a steroid nucleus selected from the group consisting of

wherein X1 is hydrogen or acyl.
9. The compounds of Claim 8 wherein the asymetric center at carbon 24 has the (R) configuration.
10. The compounds of Claim 8 wherein the asymetric center at carbon 24 has the (S) configuration.
11. Compounds having th


12. The compounds of Claim 11 wherein X1 and X2 are hydrogen or acetyl and Y is methyl.
13. The compounds of Claims 11 or 12 wherein the asymetric center at carbon 24 has the (R) configuration.
14. The cαrpounds of Claims 11 or 12 wherein the asymetric center at carbon 24 has the (S) configuration.
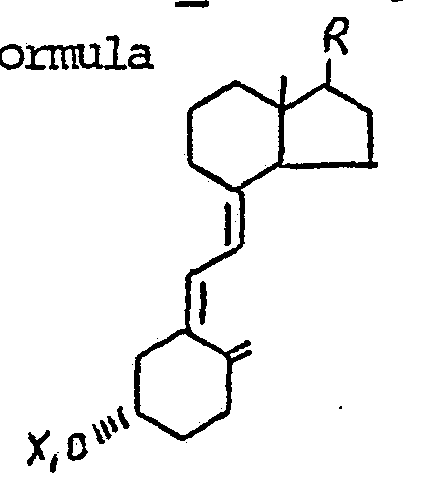
15. Coπpounds having the formula

wherein X1 is hydrogen or acyl, and where R is selected from the group consisting of

16. The compounds of Claim 15 wherein X1 and X2 are hydrogen or acetyl and Y is methyl.
17. The compounds of Claims 15 or 16 wherein the asymetric center at carbon 24 has the (R) configuration.
18. The compounds of Claims 15 or 16 wherein the asymetric center at carbon 24 has the (S) configuration.
19. Compounds having the f

wherein X~2 is hydrogen or acyl and Y is alkyl or aryl, with the proviso that when Y is methyl, X1 and X2 cannot both be hydrogen. 20. The coπpounds of Claim 19, wherein R is the group
 and X1 is hydrogen, acetyl or benzoyl.
and X1 is hydrogen, acetyl or benzoyl.
21. The compounds of Claim 20 wherein the asymetric center at carbon 24 has the (R) configuration.
22. The cαrpounds of Claim 20 wherein the asymetric center at carbon 24 has the (S) configuration.
23. The compounds of Claim 19 wherein R is the group
 and X1 is hydrogen, acetyl or benzoyl.
and X1 is hydrogen, acetyl or benzoyl.
24. The compounds of Claim 23 wherein the asymetric center at carbon 24 has the (R) configuration.
25. The compounds of Claim 23 wherein the asymetric center at carbon 24 has the (S) configuration.
26. The compounds of Claim 19 wherein R is the group

27. The ccttpounds of Claim 26 wherein X1 is selected from the group consisting of acetyl, benzoyl, succinyl, and diglycolyl, and X2 is selected from the group consisting of hydrogen, acetyl, benzoyl, succinyl and diglycolyl and Y is methyl.
28. The compounds of Claims 26 or 27 wherein the asymetric center at carbon 24 has the (R) configuration.
29. The compounds of Claims 26 r 27 wherein the asymetric center at carbon 24 has the (S) configuration.
30. Cαrpounds having the formula

31. The compounds of Claim 30 wherein the asymetric center at carbon 24 has the (R) configuration.
32. The compounds of Claim 30 wherein the asymetric center at carbon 24 has the (S) configuration.
33. The compound of Claim 31 wherein X1 and X2 are hydrogen and Y is methyl.
34. The compound of Claim 32 wherein X1 and X2 are hydrogen and Y is methyl.
35. The compounds of Claim 26 wherein X1 and X2 are hydrogen and Y is an isotopically labelled methyl group.
36. The compounds of Claim 35 wherein Y is selected from 13CH3, 14CH3, C2H3 and C3H3.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19833390212 DE3390212T1 (en) | 1982-09-20 | 1983-09-12 | Hydroxyvitamin D? 2? Compounds and processes for their preparation |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US06/420,191 US4448721A (en) | 1982-09-20 | 1982-09-20 | Hydroxyvitamin D2 compounds and process for preparing same |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1984001155A1 true WO1984001155A1 (en) | 1984-03-29 |
Family
ID=23665448
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1983/001393 Ceased WO1984001155A1 (en) | 1982-09-20 | 1983-09-12 | Hydroxyvitamin d2 compounds and process for preparing them |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US4448721A (en) |
| EP (1) | EP0119251A1 (en) |
| JP (6) | JPS59501746A (en) |
| CA (1) | CA1283422C (en) |
| CH (1) | CH671222A5 (en) |
| DE (2) | DE3390212T1 (en) |
| FR (3) | FR2540869B1 (en) |
| GB (2) | GB2127023B (en) |
| IE (1) | IE56174B1 (en) |
| IL (1) | IL69763A (en) |
| WO (1) | WO1984001155A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991012240A1 (en) * | 1990-02-14 | 1991-08-22 | Wisconsin Alumni Research Foundation | Process for preparing vitamin d2 compounds and the corresponding 1 alpha-hydroxylated derivatives |
Families Citing this family (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5036061A (en) * | 1983-05-09 | 1991-07-30 | Deluca Hector F | Process for the preparation of 1 alpha,25-dihydroxylated vitamin D2 and related compounds |
| US4505906A (en) * | 1984-01-30 | 1985-03-19 | Wisconsin Alumni Research Foundation | Hydroxyvitamin D2 isomers |
| US5120722A (en) * | 1984-02-08 | 1992-06-09 | Hoffmann-La Roche Inc. | Trihydroxy-cholecacliferol and trihydroxy-ergocalciferol for treating leukemia |
| US4588716A (en) * | 1984-05-04 | 1986-05-13 | Wisconsin Alumni Research Foundation | Method for treating metabolic bone disease in mammals |
| US4588528A (en) | 1984-05-31 | 1986-05-13 | Wisconsin Alumni Research Foundation | 1,24-dihydroxy-Δ22 -vitamin D3 and process for preparing same |
| US4857518A (en) * | 1984-10-04 | 1989-08-15 | Wisconsin Alumni Research Foundation | Hydroxylated 24-homo-vitamin D derivatives and methods for preparing same |
| EP0197949A1 (en) * | 1984-10-04 | 1986-10-22 | Wisconsin Alumni Research Foundation | Vitamin d derivatives and methods for preparing same |
| US5225579A (en) * | 1988-03-16 | 1993-07-06 | Hoxan Corporation | Method of manufacturing vitamin D2, Vitamin D3, activated type vitamin D2, activated type vitamin D3, and their derivatives |
| US5030772A (en) * | 1990-02-14 | 1991-07-09 | Deluca Hector F | Process for preparing vitamin D2 compounds and the corresponding 1 α-hydroxylated derivatives |
| US5260290A (en) * | 1990-02-14 | 1993-11-09 | Wisconsin Alumni Research Foundation | Homologated vitamin D2 compounds and the corresponding 1α-hydroxylated derivatives |
| US5063234A (en) * | 1990-05-25 | 1991-11-05 | Eli Lilly And Company | Method of inhibiting demineralization of bone |
| DE69233074T2 (en) * | 1991-01-08 | 2004-03-18 | Bone Care International Inc., Madison | Process for the preparation of 1-alpha-24-dihydroxy vitamin D2 |
| US20040009958A1 (en) * | 1991-01-08 | 2004-01-15 | Bone Care International, Inc. | Methods for preparation and use of 1alpha,24(S)-dihydroxyvitamin D2 |
| EP0631500B1 (en) * | 1992-01-29 | 1998-04-22 | Bone Care International, Inc. | 1 alpha-HYDROXY-24-(EPI)-VITAMIN D4 |
| TW267161B (en) * | 1992-11-20 | 1996-01-01 | Hoffmann La Roche | |
| US5716946A (en) * | 1996-02-13 | 1998-02-10 | Wisconsin Alumni Research Foundation | Multiple sclerosis treatment |
| US6479474B2 (en) * | 1999-07-08 | 2002-11-12 | Wisconsin Alumni Research Foundation | Dietary calcium as a supplement to vitamin D compound treatment of multiple sclerosis |
| EP1434782A2 (en) * | 2001-10-01 | 2004-07-07 | University of Virginia Patent Foundation | 2-propynyl adenosine analogs having a2a agonist activity and compositions thereof |
| US7094775B2 (en) * | 2004-06-30 | 2006-08-22 | Bone Care International, Llc | Method of treating breast cancer using a combination of vitamin D analogues and other agents |
| US20060003950A1 (en) * | 2004-06-30 | 2006-01-05 | Bone Care International, Inc. | Method of treating prostatic diseases using a combination of vitamin D analogues and other agents |
| US7947682B2 (en) * | 2004-12-29 | 2011-05-24 | University Of Southern California | Substituted N′-pyrrolo[1,2-a]quinoxalin-4-yl-hydrazides as anti-cancer agents |
| SI3095447T1 (en) | 2006-02-03 | 2022-02-28 | Opko Renal, Llc | Treatment shortages vitamin D s 25-hydroxyvitamin D2 in 25-hydroxyvitamin D3 |
| WO2007120972A2 (en) * | 2006-02-10 | 2007-10-25 | University Of Virginia Patent Foundation | Method to treat sickle cell disease |
| US8188063B2 (en) * | 2006-06-19 | 2012-05-29 | University Of Virginia Patent Foundation | Use of adenosine A2A modulators to treat spinal cord injury |
| HUE037309T2 (en) | 2006-06-21 | 2018-08-28 | Opko Ireland Global Holdings Ltd | Therapy using vitamin D repletion agent and vitamin D hormone replacement agent |
| PL2148661T3 (en) | 2007-04-25 | 2013-07-31 | Cytochroma Inc | Oral controlled release compositions comprising vitamin d compound and waxy carrier |
| DK2148684T3 (en) | 2007-04-25 | 2013-04-22 | Cytochroma Inc | Method of treating vitamin D insufficiency and deficiency |
| US8058259B2 (en) * | 2007-12-20 | 2011-11-15 | University Of Virginia Patent Foundation | Substituted 4-{3-[6-amino-9-(3,4-dihydroxy-tetrahydro-furan-2-yl)-9H-purin-2-yl]-prop-2-ynyl}-piperidine-1-carboxylic acid esters as A2AR agonists |
| CN102046812A (en) | 2008-04-02 | 2011-05-04 | 赛特克罗公司 | Methods, compositions, uses and kits for vitamin D deficiency and related disorders |
| US9212136B2 (en) * | 2008-07-22 | 2015-12-15 | Azad Pharmaceuticals Ingredients Ag | Methods for producing paricalcitol |
| EA201691980A1 (en) | 2009-01-27 | 2017-07-31 | БЕРГ ЭлЭлСи | WAYS TO REDUCE SIDE EFFECTS ASSOCIATED WITH CHEMOTHERAPY |
| CN102655869B (en) | 2009-08-14 | 2016-08-10 | 博格有限责任公司 | Vitamin D3 and its analogs for hair loss |
| PT2552484T (en) | 2010-03-29 | 2020-04-03 | Opko Ireland Global Holdings Ltd | Methods and compositions for reducing parathyroid levels |
| CA2698160C (en) * | 2010-03-30 | 2017-07-25 | Alphora Research Inc. | Stabilized doxercalciferol and process for manufacturing the same |
| KR101847947B1 (en) | 2013-03-15 | 2018-05-28 | 옵코 아이피 홀딩스 Ⅱ 인코포레이티드 | Stabilized modified release vitamin d formulation |
| WO2014194133A1 (en) | 2013-05-29 | 2014-12-04 | Berg Llc | Preventing or mitigating chemotherapy induced alopecia using vitamin d |
| US10220047B2 (en) | 2014-08-07 | 2019-03-05 | Opko Ireland Global Holdings, Ltd. | Adjunctive therapy with 25-hydroxyvitamin D and articles therefor |
| MY198547A (en) | 2016-03-28 | 2023-09-04 | Opko Ireland Global Holdings Ltd | Methods of vitamin d treatment |
| CN115947678B (en) * | 2023-03-10 | 2024-01-16 | 成都诺森医学检验有限公司 | Synthesis method of stable isotope 2H (D) labeled 25-hydroxyvitamin D2 |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4229358A (en) * | 1978-07-26 | 1980-10-21 | Wisconsin Alumni Research Foundation | Fluorovitamin D compounds and processes for their preparation |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL150682B (en) * | 1968-07-01 | 1976-09-15 | Wisconsin Alumni Res Found | PROCESS FOR PREPARING A PHARMACOLOGICALLY ACTIVE PREPARATION CONTAINING A VITAMIN D DERIVATIVE AND FORMED FOOD PRODUCTS. |
| US3880894A (en) * | 1974-05-24 | 1975-04-29 | Wisconsin Alumni Res Found | 1,25-Dihydroxyergocalciferol |
| US4105660A (en) * | 1977-06-29 | 1978-08-08 | Merck & Co., Inc. | Preparation of 3β-hydroxy-27-norcholest-5,7-dien-25-one |
| US4145346A (en) * | 1977-10-03 | 1979-03-20 | Merck & Co., Inc. | Preparation of 3β-hydroxy-27-norcholest-5-ene-25-one and intermediates thereof |
| US4188345A (en) * | 1978-07-26 | 1980-02-12 | Wisconsin Alumni Research Foundation | Fluorovitamin D compounds and processes for their preparation |
| US4265822A (en) * | 1979-09-10 | 1981-05-05 | Wisconsin Alumni Research Foundation | Process for preparing 1-hydroxylated vitamin D compounds from 5,6-trans-vitamin D compounds |
| DE3048698A1 (en) * | 1980-12-23 | 1982-07-15 | Wisconsin Alumni Research Foundation, 53707 Madison, Wis. | 1-Alpha-hydroxy-vitamin=D prepn. - by allylic oxidn. of 5,6-trans-vitamin=D followed by irradiation |
-
1982
- 1982-09-20 US US06/420,191 patent/US4448721A/en not_active Expired - Lifetime
-
1983
- 1983-09-12 CH CH2538/84A patent/CH671222A5/de not_active IP Right Cessation
- 1983-09-12 DE DE19833390212 patent/DE3390212T1/en active Granted
- 1983-09-12 DE DE3348322A patent/DE3348322C2/de not_active Expired - Lifetime
- 1983-09-12 EP EP83903074A patent/EP0119251A1/en not_active Withdrawn
- 1983-09-12 JP JP58503103A patent/JPS59501746A/en active Granted
- 1983-09-12 WO PCT/US1983/001393 patent/WO1984001155A1/en not_active Ceased
- 1983-09-18 IL IL69763A patent/IL69763A/en not_active IP Right Cessation
- 1983-09-19 CA CA000436979A patent/CA1283422C/en not_active Expired - Lifetime
- 1983-09-20 GB GB08325131A patent/GB2127023B/en not_active Expired
- 1983-09-20 FR FR838314942A patent/FR2540869B1/en not_active Expired
- 1983-09-20 IE IE2203/83A patent/IE56174B1/en not_active IP Right Cessation
-
1984
- 1984-07-09 GB GB08417477A patent/GB2142922B/en not_active Expired
- 1984-10-15 FR FR8415789A patent/FR2555172B1/en not_active Expired
- 1984-10-15 FR FR848415790A patent/FR2555173B1/en not_active Expired
-
1989
- 1989-12-27 JP JP1336823A patent/JPH02209862A/en active Granted
- 1989-12-27 JP JP1336821A patent/JPH02209887A/en active Granted
- 1989-12-27 JP JP1336822A patent/JPH02209888A/en active Granted
- 1989-12-27 JP JP1336824A patent/JPH02209863A/en active Granted
- 1989-12-27 JP JP1336825A patent/JPH0610187B2/en not_active Expired - Lifetime
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4229358A (en) * | 1978-07-26 | 1980-10-21 | Wisconsin Alumni Research Foundation | Fluorovitamin D compounds and processes for their preparation |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1991012240A1 (en) * | 1990-02-14 | 1991-08-22 | Wisconsin Alumni Research Foundation | Process for preparing vitamin d2 compounds and the corresponding 1 alpha-hydroxylated derivatives |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4448721A (en) | Hydroxyvitamin D2 compounds and process for preparing same | |
| US5750746A (en) | Homologated vitamin D2 compounds and the corresponding 1α-hydroxylated derivatives | |
| US4769181A (en) | 1,25-dihydroxyvitamin D2 compounds | |
| US5030772A (en) | Process for preparing vitamin D2 compounds and the corresponding 1 α-hydroxylated derivatives | |
| US5036061A (en) | Process for the preparation of 1 alpha,25-dihydroxylated vitamin D2 and related compounds | |
| EP0157781B1 (en) | 1,23-dihydroxyvitamin d compounds | |
| US5030626A (en) | Fluorine derivatives of vitamin D3 and process for producing the same | |
| EP0250755A2 (en) | Fluorine derivatives of vitamin D3 and process for producing the same | |
| EP0154627A1 (en) | 23,23-difluoro-25-hydroxy-vitamin d 3? and process for preparing same | |
| JP2818493B2 (en) | Method for producing 25-hydroxyvitamin D2 compounds and corresponding 1α-hydroxylated derivatives | |
| JP2818494B2 (en) | Process for producing vitamin D2 compounds and corresponding 1α-hydroxylated derivatives | |
| AU7307791A (en) | Process for preparing vitamin d2 compounds and the corresponding 1 alpha-hydroxylated derivatives | |
| JPH05339230A (en) | Activated type vitamin d2 and the production of its derivative | |
| JPH0455424B2 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Designated state(s): CH DE JP |
|
| AL | Designated countries for regional patents |
Designated state(s): FR |
|
| RET | De translation (de og part 6b) |
Ref document number: 3390212 Country of ref document: DE Date of ref document: 19841129 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3390212 Country of ref document: DE |