WO1992019604A1 - Novel isoxazole derivative and salt thereof - Google Patents
Novel isoxazole derivative and salt thereof Download PDFInfo
- Publication number
- WO1992019604A1 WO1992019604A1 PCT/JP1992/000571 JP9200571W WO9219604A1 WO 1992019604 A1 WO1992019604 A1 WO 1992019604A1 JP 9200571 W JP9200571 W JP 9200571W WO 9219604 A1 WO9219604 A1 WO 9219604A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- atom
- reaction
- acid
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6527—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having nitrogen and oxygen atoms as the only ring hetero atoms
- C07F9/653—Five-membered rings
Definitions
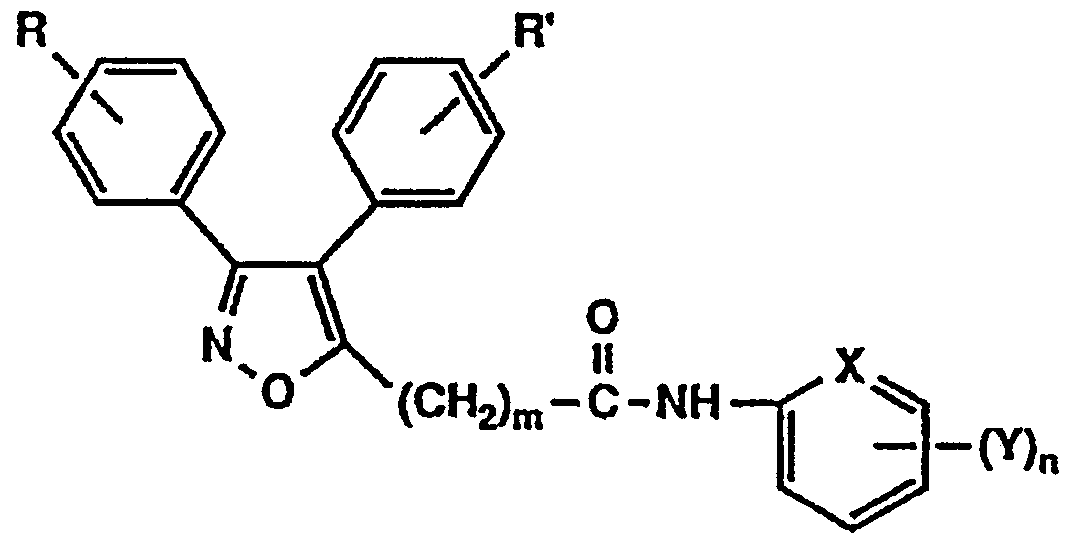
- the present invention relates to a novel isoxazole derivative having a lipoxygenase inhibitory activity and a cyclooxygenase inhibitory activity, a salt thereof, and a pharmaceutical use thereof.
- Leukotrienes which are 5-lipoxygenase products of arachidonic acid
- prosulin glandins which are cyclooxygenase products
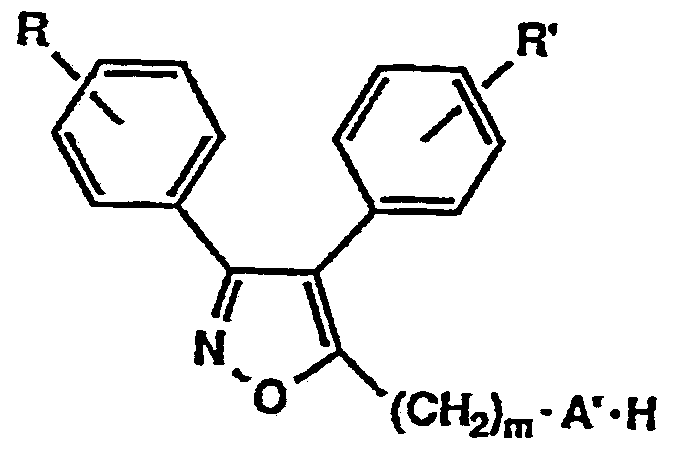
- the present inventors have conducted intensive studies in view of the problems of the background art, and as a result, have found that the novel isooxoazole derivative represented by the following general formula (1) has excellent lipoxygenase inhibitory activity and cyclooxygenase inhibitory activity. Have been found to be useful as pharmaceuticals, and have completed the present invention. That is, the present invention relates to the general formula (1)
- R and R ' are the same or different and each represent a hydrogen atom, a lower alkyl group, a lower alkoxy group, or a halogen atom.
- m represents 0 to 5
- A represents 1 NH—, 10—, or a direct bond.
- X represents a nitrogen atom or a carbon atom.
- n 0 to 3
- Y has a hydroxy group, a lower alkoxy group, a lower alkyl group, a lower alkoxycarbonyloxy group, a lower alkoxycarbonylmethyloxy group, a carboxymethyloxy group, and a protecting group.
- n is 2 or 3
- Y represents the same or different and represents these groups, and n is 2 or more. In this case, Y may form a methylenedioxy group.
- Y may form a methylenedioxy group.
- the compound of the present invention represented by the general formula (1) has excellent lipoxygenase inhibitory activity and cyclooxygenase inhibitory activity.
- examples of the lipoxygenase include 5-lipoxygenase, 12-lipoxygenase, and 15-lipoxygenase.
- the compound of the present invention has an excellent inhibitory effect on 5-lipoxygenase. ing.
- the compound of the present invention has excellent lipoxygenase inhibitory activity and cyclooxygenase inhibitory activity, and is an anti-asthmatic agent, an anti-allergic agent, an agent for brain diseases, a cardiovascular agent, a therapeutic agent for nephritis, an anti-inflammatory analgesic agent, an anti-rheumatic agent It is useful as a therapeutic agent for skin diseases typified by psoriasis, etc. and an agent for liver diseases.
- the present invention provides an anti-asthmatic agent, an anti-allergic agent, an agent for brain diseases, an agent for cardiovascular diseases, a therapeutic agent for nephritis, an anti-inflammatory analgesic, comprising an effective amount of the compound of the above general formula (1) and a pharmaceutical carrier.
- An object of the present invention is to provide an antirheumatic agent, a therapeutic agent for skin diseases represented by psoriasis and the like, and an agent for liver diseases.
- the present invention provides an asthma, allergy, or brain disease, which comprises administering an effective amount of the compound represented by the general formula (1) to a patient.
- the present invention provides a method for treating skin diseases and liver diseases typified by disease, cardiovascular disease, kidney disease, inflammation, rheumatism, psoriasis, etc.
- the present invention provides a compound of the above general formula (1) for treating skin diseases and liver diseases represented by breathing, allergy, brain disease, circulatory disease, renal disease, inflammation, rheumatism, psoriasis and the like. Concerning the use of things.
- the present invention relates to the use of the compound of the above general formula (1) for producing a lipoxygenase inhibitor.
- the present invention relates to the use of the compound represented by the above general formula (1) for the production of a 5-lipoxygenase inhibitor.
- the present invention relates to the use of the compound of the above general formula (1) for producing a cyclooxygenase inhibitor.
- the present invention relates to the use of the compound of the above general formula (1) for inhibiting lipoxygenase.
- the present invention relates to the use of the compound of the above general formula (1) for inhibiting 5-lipoxygenase.
- the present invention relates to the use of the compound of the above general formula (1) for cyclooxygenase inhibition.
- examples of the halogen atom represented by R include a fluorine atom, a chlorine atom, a bromine atom, an iodine atom and the like.
- Lower alues represented by R, R 'and Y The kill group includes, for example, methyl group, ethyl group, n-propyl group, isopropyl group, n-butyl group, isobutyl group, sec-butyl group, t-butyl group, etc.
- lower alkoxy groups represented by R, R 'and Y include, for example, methoxy, ethoxy, n-propoxy, isopropoxy, n —A straight-chain or branched alkoxy group having 1 to 4 carbon atoms such as —butoxy group, isobutoxy group, sec—butoxy group and t—butoxy group.
- Examples of the lower alkoxycarbonyloxy group represented by Y include a methoxycarbonyloxy group, an ethoxycarbonyloxy group, an n-propoxycarbonyloxy group, an isopropoxycarbonyloxy group, and an n-butoxycarbonyloxy group , Isobutoxycarbonyloxy, sec-butoxycarbonyloxy, t-butoxycarbonyloxy, pentyloxycarbonyloxy, isopentyloxycarbonyloxy, hexyloxycarbonyloxy
- Examples of the linear or branched alkoxycarbonyloxy group having 2 to 7 carbon atoms such as a lower alkoxycarbonylmethyloxy group include a methoxycarbonylmethyloxy group and an ethoxycarbonylmethyl group.
- the amino acid residue optionally having a protecting group represented by Y indicates a group formed by removing a hydrogen atom from a carboxyl group of an amino acid, and the amino acid includes, for example, glycine Natural or synthetic amino acids such as, alanine, methionine, barin, serine, proline, leucine, isoleucine, glutamine, histidine, phenylalanine, and fuunylglycine; and amino acids.
- the protecting group for an amino group include a lower alkyl group having 1 to 6 carbon atoms, a lower acyl group having 2 to 5 carbon atoms, a lower alkoxycarbonyl group having 2 to 5 carbon atoms, and a benzyloxycarbonyl group.
- amino acids having a protecting group examples include N, N-dimethylglycine, N-acetylglycine, Nt-butoxycarbonylglycine, and N-benzene.
- examples include ziroxycarbonylglycine, N-acetylvinyl, N-t-butoxycarbonylparine, etc. Wear.
- Examples of the lower alkylcarbonyloxy group represented by Y include a methylcarbonyloxy group, an ethylcarbonyloxy group, an ⁇ -propylcarbonyloxy group, an isopropylpropylcarbonyloxy group, an ⁇ -butyl A straight-chain or branched alkylcarbonyloxy group having 2 to 5 carbon atoms such as a carbonyloxy group, a dibutylcarbonyloxy group, and a sec-butylcarbonyloxy group or a t-butylcarbonyloxy group;
- Examples of the di-lower alkyl phosphate residue include a dimethyl phosphate ester residue, a getyl phosphate ester residue, a dipropyl phosphate ester residue, a dibutyl phosphate ester residue, etc.
- the salt of the isoxazole compound of the present invention for example, as the salt of the basic group, there may be mentioned inorganic salts such as hydrochloride, sulfate, nitrate and phosphate, maleate, succinate and the like.
- Organic acid salts such as sulfonic acid salt, fumarate salt, p-toluenesulfonate salt and methansulfonate salt.
- the acid group salt include sodium salt and potassium salt.
- R and R ′ are the same Or different hydrogen atoms, lower alkyl groups, lower alkoxy groups or halogen atoms, and R and R 'are the same or different and are lower alkoxy groups or halogen atoms. More preferred.
- m is 0 to 3
- A is one NH— or a direct bond
- Y is a hydroxy group, a lower alkoxy group, a lower alkyl group or a lower alkoxycarbonyloxy group
- n is 2 or 3 or more
- X is preferably a carbon atom.
- m is more preferably 1 to 3.
- preferred compounds are those in which R and R ′ are the same or different and are a hydrogen atom, a lower alkyl group, a lower alkoxy group or a halogen atom, and m is 0-3.
- A is one NH— or a direct bond
- ⁇ represents a hydroxy group, a lower alkoxy group, a lower alkyl group or a lower alkoxycarbonyloxy group
- ⁇ is 2 or 3
- X is a compound showing a carbon atom. It is.
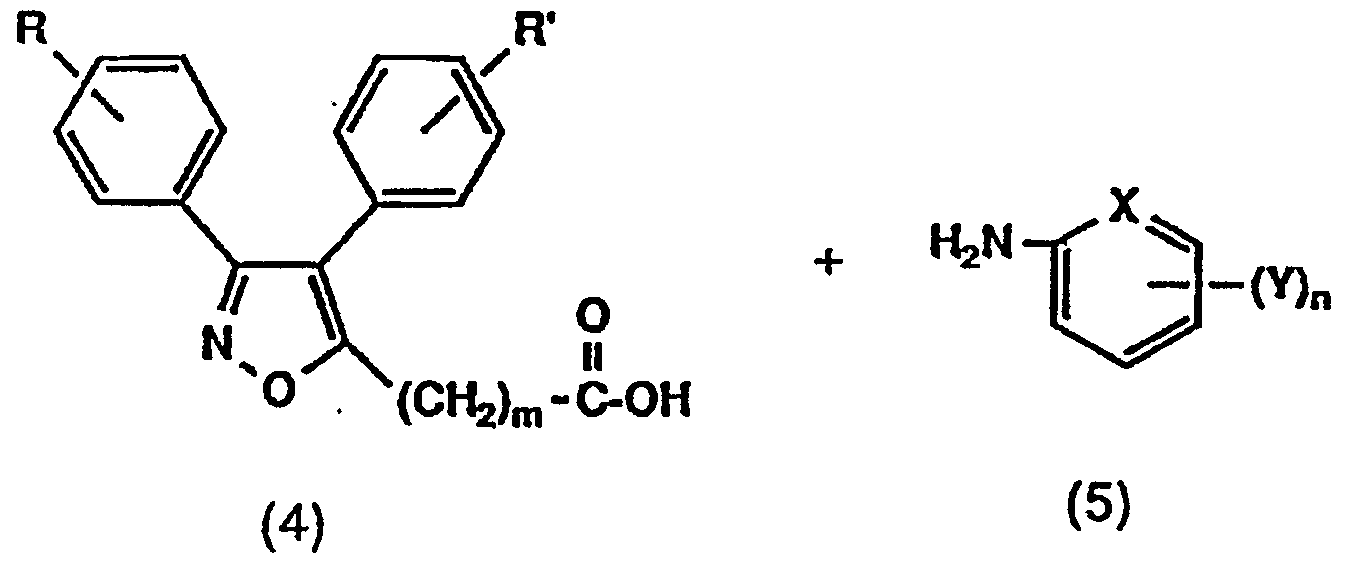
- the compound of the present invention represented by the general formula (1) is produced by the following reaction process formulas (i) to (vii). Reaction scheme (i)>
- R, R ", m, Y, X and n are the same as above. Represents 10— or 1 NH—, and H represents 1 or 2. ]
- Tetrahydrovinylil group and the like can be used, and as a method for introducing these protecting groups, Journal of American Chemical Society, Journal of American Chemical Society 100, 801 The method can be performed according to the method described in 9 7 8).
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include ethers such as ether and tetrahydrofuran, halogenated hydrocarbons such as methylene chloride and chloroform, and benzene. , Toluene and other aromatic hydrocarbons, N, N-dimethylformamide, dimethylsulfoxide, acetonitrile and other non-protonic polar solvents can be used.
- the condensing agent for example, N, N-dicyclohexyl
- the condensing agent for example, N, N-dicyclohexyl
- the catalyst include, for example, 4-dimethylaminopyridine, 11-hydroxybenzotriazole, pyridin, triethylamine and the like.
- the compound of the general formula (3) is about 1 to 2 equivalents
- the condensing agent is about 1 to 3 equivalents
- the catalyst is 0.1 to 2 equivalents to the compound of the general formula (2). It is preferable to use it.
- the reaction temperature is from about ice-cooling to about room temperature, and the reaction proceeds advantageously in about 1 to 48 hours.
- An isoxazole derivative represented by (lb) is obtained.
- Y of the compound represented by the general formula (5) is a hydroxy group
- the compound can be protected and then condensed similarly to the method of the above reaction scheme (i).
- the solvent and the catalyst for example, those exemplified in the reaction step formula (i) can be used.
- the condensing agent include, for example, 1,3-thiazolidin-12-thione and the like in addition to the condensing agent exemplified in the reaction step formula (i).
- the reaction proceeds advantageously by carrying out the reaction under the same reaction conditions as in the reaction scheme (i).
- the dialkylmethylphosphonate and n-butyllithium are stirred in a solvent under a nitrogen atmosphere at 178 ° C or lower for 10 to 30 minutes, and the ester represented by the general formula (6) is added to the solution. A compound represented by the general formula (7) is obtained.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include ethers such as ether and tetrahydrofuran. Can be exemplified.
- the reaction temperature is about 78 to about ice-cooling, and the reaction time is about 30 minutes to about 2 hours.
- examples of the dialkylmethylphosphonate include dimethylmethylphosphonate and getylmethylphosphonate.
- the obtained compound represented by the general formula (7) is reacted with sodium hydride in a solvent under water cooling for 30 minutes to 1 hour, and then the aldehyde represented by the general formula (8) is reacted. Then, the mixture is reacted under ice-cooling to about room temperature for 2 to 6 hours to obtain the desired isoxazole derivative represented by the general formula (1c).
- the solvent for example, the solvent used for synthesizing the compound (7) can be used.
- Y in the compound represented by the general formula (8) is a hydroxy group, the compound can be protected and then condensed in the same manner as in the reaction step formula (i). In the reaction, it is preferable to use about 1 to 2 equivalents each of the sodium hydride and the compound of the general formula (8) with respect to the compound of the general formula (7). 5 Reaction scheme (iv)>
- reaction it is preferable to use about 1 to 1.5 equivalents of the compound of the general formula (8) and about 1 to 2 equivalents of the base for the compound of the general formula (4).
- the reaction temperature is a temperature at which the solvent is refluxed, and is about 100 to 150 when no solvent is used, and the reaction proceeds advantageously in about 2 to 5 hours.
- the reaction of the reaction scheme (iv) 6 The conditions are not limited to the above, but are usually performed
- the isoxazole derivative represented by (1d) can be obtained.
- the compound represented by the general formula (9) After reacting the compound represented by the general formula (9) with alkyl or phenyl lithium in a solvent, the compound is reacted with a well-known 1,3-dioxolan-1-methyl-ethyl ester, Subsequently, the compound is reacted with an acid to obtain a compound represented by the general formula (10).
- the solvent is not particularly limited as long as it does not participate in the reaction.
- ethers such as ethyl ether and tetrahydrofuran
- saturated alkyls such as hexane and cyclohexan can be used.
- the alkyllithium examples include methyllithium, n-butyllithium, sec-butyllithium, t-butyllithium, and the like.
- the acid for example, inorganic acids such as hydrochloric acid and sulfuric acid, and organic acids such as p-toluenesulfonic acid can be used.
- the alkyllithium compound is added to the compound of the formula (9) in an amount of 2 to 3 equivalents of phenyllithium and an acid until the reaction solution becomes acidic.
- an inert dry gas such as nitrogen or argon is used.
- the reaction proceeds advantageously in an atmosphere.
- the reaction temperature is about 178 ° C to room temperature, and the reaction time is about 6 to 12 hours, and the reaction proceeds advantageously.
- R, R ′, m, A, Y, X and n are the same as above.
- the desired isoxazole derivative represented by the general formula (1f) is obtained.
- the solvent is not particularly limited as long as it does not participate in the reaction. Ethers such as ethyl ether and tetrahydrofuran, aromatic hydrocarbons such as benzene and toluene, and halogenated hydrocarbons such as black form and methylene chloride can be used. In the reaction, it is preferable to use 1 to 2 equivalents of Lawson reagent relative to the compound of the general formula (12).
- the reaction temperature is from room temperature to about the boiling point of the solvent.
- the reaction time is about 1 to 6 hours, and the reaction proceeds advantageously.
- Y' is a lower alkoxycarbonyloxy group A lower alkoxycarbonylmethyloxy group, an amino acid residue which may have a protecting group, a lower alkyl carboxy group, a pyridyl carboxy group, a dimethylaminophenyl carboxy group or a linyl group Shows an acid di-lower alkyl residue.
- the compound represented by the general formula (13) is dissolved in a suitable solvent in a lower alkoxycarbonyl chloride (lower alkyl carbonate lower alkyl ester), ⁇ -halogenated acetic acid lower alkyl ester, amino acid or amino acid.
- a lower alkoxycarbonyl chloride lower alkyl carbonate lower alkyl ester
- ⁇ -halogenated acetic acid lower alkyl ester amino acid or amino acid.
- ⁇ using protected amino acids, lower fatty acids or their acid chlorides, nicotinic acid, isonicotinic acid, dimethylaminobenzoic acid or di-lower alkyl chlorides and condensing agents By reacting, the desired isoxoxazole derivative represented by the general formula (1 g) is obtained.
- lower alkoxycarbonyl chloride examples include methoxycarbonyl chloride, ethoxycarbonyl chloride, n-propoxycarbonyl chloride, isopropoxycarbonyl chloride, and n-butoxycanolebonyl.
- alkoxycarbonyl chlorides having 2 to 5 carbon atoms such as chloride, isobutoxycarbonyl chloride, sec-butoxycarbonyl chloride, t-butoxycarbonyl chloride.
- lower alkyl esters of carboxylic acid examples include, for example, methyl acetic acid methyl ester and methyl bromoperate And chloroacetic acid ethyl ester, bromoacetic acid ethyl ester, bromoacetic acid propyl ester, bromoacetic acid butyl ester and the like.
- Amino acids include glycine, alanine, methionine, valine, serine, proline, leucine, isoleucine, gurumin, histidine, and phenine.
- Examples thereof include natural or synthetic amino acids such as dilulanine and phenylglycine, and the above-mentioned N-protected amino acids, in which the amino group is usually protected, are preferred.
- the protecting group any of the above protecting groups for amino acid can be used.
- Examples of the lower fatty acid include linear or branched fatty acids having 2 to 5 carbon atoms, such as acetic acid, propionic acid, butyric acid, isobutyric acid, valeric acid, isovaleric acid, and bivalic acid.
- Examples of chlorides include acetic acid chloride, propionic acid chloride, butyric acid chloride, isobutyric acid chloride, valeric acid chloride, and isovaleric acid chloride.
- chlorides of straight-chain or branched fatty acids having 2 to 5 carbon atoms such as amides and bivalic acid chlorides.
- the di-lower alkyl chlorides include:
- Di (Ci-C4 alkyl) phosphoric acid chlorides such as dimethylchlorophosphate, getylchlorophosphate, dipropizolechlorophosphate, dibutylchlorophosphate.
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include ethers such as ether and tetrahydrofuran, methylene chloride, chloroform and the like, hydrogenated hydrocarbons, and benzene. And aromatic hydrocarbons such as toluene, and non-polar polar solvents such as N, N-dimethylformamide and dimethylsulfoxide.
- the condensing agent usually used for peptide synthesis is used.
- an additive may be used if necessary, and the reaction proceeds advantageously when an organic amine such as N, N-dimethylaminopyridine or 1-hydroxybenzotriazole is used as the additive. In some cases.
- the reaction with lower alkoxycarbonyl chloride, lower alkyl ester of carboxylic acid, acid chloride of lower fatty acid or di-lower alkyl chloride generally uses a base as a condensing agent.
- the base include organic bases such as pyridine and triethylamine, sodium hydrogen carbonate, sodium hydrogen carbonate, sodium carbonate, sodium carbonate, hydrogenation and the like.
- examples include inorganic bases such as sodium.
- the use ratio of the reaction raw material is represented by the general formula (13)
- the organic amine is used as an additive, the amount of the organic amine to be used may be 1 to 2.5 equivalents S to the compound of the general formula (13). .
- the reaction time is about 1 to 15 hours, and the reaction is completed under ice cooling to about room temperature.
- an N-protected amino acid it may be deprotected according to a conventional method, if necessary.
- the deprotecting agent those usually used, for example, inorganic acids such as hydrochloric acid and sulfuric acid, p-toluene Organic acids such as sulfonic acid, trifluoroacetic acid, and formic acid can be used.
- the conditions for deprotection may be the same as those in a known and commonly used method used for ordinary peptide synthesis.
- R and R ′ are the same as above.
- R represents a lower alkyl group, and z-represents a lower alkoxycarbol group or a ditolyl group.
- Examples of the lower alkyl group represented by are the lower alkyl groups described above.
- Examples of the lower alkoxycarbonyl group represented by ⁇ ′ include a methoxycarbonyl group, an ethoxycarbonyl group, an ⁇ -propoxycarbonyl group, an isopropoxycarbonyl group, an ⁇ -butoxycarbonyl group, and an isobutoxycarbonyl group.
- the solvent examples include alcohols such as methanol, ethanol and tert-butanol, ethers such as ether and tetrahydrofuran, and aromatic hydrocarbons such as benzene and toluene. And non-protonic polar solvents such as N, N-dimethylformamide and dimethylsulfoxide.
- the base for example, sodium hydroxide, potassium hydroxide, sodium hydride, sodium amide, sodium methoxide And strong bases such as potassium, tert-butoxide, and butyllithium; and organic bases such as triethylamine, getylaminopyridine, and pyridin.
- the reaction ratio is preferably about 1 to 3 equivalents of the compound of the formula (15) and about 0.1 to 3 equivalents of the base to the compound of the formula (14).
- the reaction temperature is about the boiling point of the solvent under ice cooling, and the reaction proceeds advantageously in about 0.5 to 20 hours.o
- the compound represented by the general formula (17) is obtained by reacting the compound represented by the general formula (16) obtained in the step A with a hydroxylamine or a salt thereof in a suitable solvent. obtain.
- the hydroxylamine salt used in the reaction is not particularly limited, and examples thereof include generally commercially available hydrochlorides and sulfates.
- the solvent is not particularly limited as long as it does not participate in the reaction.
- the reaction ratio is preferably about 1 to 10 equivalents of hydroxylamin or a salt thereof to the compound of general formula (16).
- the reaction temperature is from room temperature to about the boiling point of the solvent, and the reaction proceeds advantageously in about 1 to 30 hours. In this reaction, add an acid or base as necessary, or in a mixed solvent such as a buffer. The reaction may be carried out.
- the compound represented by the general formula (17) is cyclized in a suitable solvent using a halogenating agent or the like, or is reacted with an oxidizing agent in a suitable solvent or without a solvent.
- a compound represented by the general formula (18) is obtained.
- the solvent is not particularly limited as long as it does not participate in the reaction.
- the solvent exemplified in the step A can be used, and acetic acid or the like can be used.
- the halogenating agent used in the cyclization reaction include, for example, chlorine, bromine, iodine, N-chlorosuccinic acid imide, N-bromosuccinic acid imide and the like.
- the ratio of the reaction is preferably 1 to 3 equivalents of the halogenating agent to the compound of the general formula (17).
- the reaction temperature is about -70 to 150, and the reaction time is about 1 to 24 hours, and the reaction proceeds advantageously.
- the oxidizing agent for example, oxides such as permanganate potassium, manganese dioxide, and potassium periodate, metal salts such as lead tetraacetate and mercury acetate, and peroxides such as hydrogen peroxide and peracetic acid Objects and the like.
- oxides such as permanganate potassium, manganese dioxide, and potassium periodate
- metal salts such as lead tetraacetate and mercury acetate
- peroxides such as hydrogen peroxide and peracetic acid Objects and the like.
- the compound represented by the general formula (18) can also be obtained by an organic electrolytic oxidation method using oxygen oxidation using air or oxygen and anodization.
- the general formula (17) It is preferable to use about 0.2 to 10 equivalents of the oxidizing agent to the compound.
- the reaction temperature is about 100 from ice-cooling, and the reaction proceeds advantageously in about 5 minutes to 10 hours.
- the reaction temperature is about ⁇ 0 ° C. to 100, and the reaction time is about 5 minutes to 10 hours, and the reaction proceeds advantageously.
- these reactions proceed efficiently in the presence of a catalyst, and the use of a catalyst of the general formula (17) in an amount of about 1 ⁇ 10 ′′ to: L0 times the equivalent of the compound of general formula (17)
- the catalyst include metals such as cobalt, rhodium, palladium, chromium, cerium and ruthenium, and metal compounds such as metal salts, metal oxides and metal complexes.
- a compound represented by the general formula (18) in which is represented by a ditolyl group the compound is converted into a carboxylic acid by solvolysis or hydrolysis in the presence of an acid or a base, followed by esterification and further reduction.
- a compound represented by the general formula (19) is obtained.
- the solvolysis or hydrolysis is carried out by the solvolysis method described in JP-A-60-75471 or a hydrolysis method commonly used in the art.
- Acids used in solvolysis or hydrolysis reactions include inorganic acids such as hydrochloric acid, sulfuric acid, and nitric acid, and salts Examples of the group include inorganic bases such as sodium hydroxide, potassium hydroxide, and sodium carbonate.
- Esterification can also be carried out by a method usually used in the art, for example, by using an acid as a catalyst in an alcohol solvent such as methanol or ethanol.
- an acid include inorganic acids such as hydrochloric acid and sulfuric acid, and organic acids such as p-toluenesulfonic acid.
- the ester can be reduced by using a reducing agent in a suitable solvent.
- a reducing agent for example, the solvent exemplified in Step A can be used.
- the reducing agent include lithium aluminum hydride and sodium borohydride.
- the ratio of the reaction is preferably such that the reducing agent is used in an amount of about 1 to 10 times equivalent to the ester compound.
- the reaction temperature ranges from ice-cooling to room temperature, and the reaction proceeds advantageously in about 10 minutes to 24 hours.
- the intermediate ( ⁇ ′ is a carboxylic acid) can also be obtained by the method described in JP-A-56-59764.
- the compound represented by the general formula (20) is obtained by reacting de, triphenylphosphine and ethyl azodicarboxylate in a suitable solvent.
- suitable solvent include ethers such as ether and tetrahydrofuran, halogenated hydrocarbons such as methylene chloride, and culoform, and aromatic hydrocarbons such as benzene and toluene. .
- the reaction ratio is preferably such that phthalimid, triphenylphosphine and getyl azodicarbonate are each used in an amount of about 1 to 2 equivalents to the alcohol of the general formula (19).
- the reaction temperature is about ice-cold to about room temperature, and the reaction time is about 1 to 48 hours, and the reaction proceeds efficiently.
- the compound represented by the general formula (21) is obtained by reacting the compound represented by the general formula (20) under the conditions of a Gabriel reaction generally performed.
- the reaction is carried out, for example, in an ethanol solvent, using about 1 to 1.1 equivalents of hydrazine hydrate with respect to the compound of the general formula (20), and at room temperature to near the boiling point of ethanol, 1 to 2 times.
- the reaction proceeds advantageously by reacting for about 4 hours.
- the desired amine can be obtained by hydrolysis of an acid or alkali which is usually performed.
- R and R ′ are the same as above.
- a solvent for example, bis (chloroacetic acid) anhydride, bis (chloropropionic acid) anhydride, etc.
- the solvent is not particularly limited as long as it does not participate in the reaction.
- examples include ethers such as ethyl ether and tetrahydrofuran, and saturated alkyls such as hexane and cyclohexane. .
- alkyl lithium examples include methyl lithium, Examples thereof include n-butyllithium, sec-butyllithium, and tert-butyllithium.
- the reaction is carried out using about 2 to 3 equivalents of alkyllithium or phenylarthium and about 1 to 2 equivalents of 0> -chloroanhydro lower fatty acid, preferably nitrogen, argon, etc. with respect to the compound of the general formula (9).
- the reaction proceeds advantageously by performing the reaction in an inert dry gas atmosphere.
- the reaction temperature is about 120 to about room temperature
- the reaction time is about 1 to 2 hours for the reaction with alkyllithium or phenyllithium, and about 0.5 to 2 hours for the reaction with The reaction proceeds advantageously.
- the compound represented by the general formula (23) is obtained by reacting the obtained compound represented by the general formula (22) with ammonia in a solvent.
- the solvent is not particularly limited as long as it does not participate in the reaction.
- alcohols such as methanol and ethanol, water, and the like can be used.
- Ammonia can be passed through the above solvent through ammonia gas or used as ammonia water.
- the reaction rate is such that an excess amount of ammonia is used relative to the compound of the general formula (22), the reaction temperature is from room temperature to the boiling point of the solvent, and the reaction proceeds advantageously in about 2 to 12 hours. . ⁇ Reaction process formula (X)>
- the compound represented by the general formula (26) is obtained by reacting the compound represented by the general formula (22) with an inorganic base in a solvent.
- a solvent for example, alcohols such as methanol and ethanol, or water can be used alone or as a mixture, and a solvent such as tetrahydrofuran can be used as an auxiliary solvent.
- the inorganic bases include sodium hydroxide, sodium hydroxide, sodium carbonate, sodium carbonate, calcium oxide and the like.
- the reaction temperature is from room temperature to about the boiling point of the solvent, and the reaction time is about 1 to 6 hours, and the reaction proceeds advantageously.
- the compound represented by the general formula (9) is obtained by carrying out the oximation process in the same manner as in the step B of the above reaction scheme (viii).
- the compound represented by the general formula (9) After reacting with dimethyl or phenyllithium, it is further reacted with an acid anhydride to obtain a carboxylic acid represented by the general formula (24).
- the solvent is not particularly limited as long as it does not participate in the reaction, and examples thereof include ethers such as ethyl ether and tetrahydrofuran, saturated alkyls such as hexane and cyclohexane, and chloroform. Halogenated hydrocarbons such as mouth form and methylene chloride can be used.
- the alkyllithium include methyllithium, n-butyllithium, sec-butyllithium, tert-butyllithium, and the like.
- Examples of the acid anhydride include succinic anhydride, glutaric anhydride, adipic anhydride, heptandioic anhydride and the like.
- the reaction uses about 2 to 3 equivalents of alkyllithium or phenyllithium and about 1 to 2 equivalents of an acid anhydride with respect to the compound of the general formula (9), and is preferably inert such as nitrogen or argon.
- the reaction proceeds advantageously in a dry gas atmosphere.
- the reaction temperature is about 120 ° C. to room temperature, and the reaction time is preferably about 2 to 2 hours for reaction with alkyllithium or phenyllithium, and about 0.5 to 2 hours for reaction with acid anhydride. proceed.
- a compound represented by the general formula (25) is obtained in the same manner as in the esterification method used in the step D of the above reaction scheme (vi ii).
- the compound represented by the general formulas (24) and (25) obtained by the reaction scheme (xi) is reacted according to the method of the reaction scheme (viii) to obtain a compound of the general formula (2) can also be converted to the compound represented by '.
- the compound of the present invention having a basic group obtained by the above reaction can be obtained by mixing the above-mentioned inorganic acid with a solvent such as ethers, lower alcohols, ethyl ethyl drone, and hexane at a temperature of about room temperature.
- a salt of a basic group can be obtained by a conventionally known method such as a reaction with an organic acid.
- the compound of the present invention having an acidic group obtained by the above reaction can be obtained by replacing the above compound with an inorganic acid or an organic acid in a solvent such as sodium hydroxide, Reacts with alkali metals such as lithium and calcium hydroxide or alkaline earth metal hydroxides or strong bases such as sodium methoxide, potassium ethoxide and sodium hydride.
- the salt can be converted into a salt form of an acidic group by a conventionally known method such as a salting method.
- various administration forms can be adopted depending on the purpose of prevention or treatment.
- the form include oral preparations, injections, suppositories, plasters, It may be any of a patch and the like, and these administration forms can be respectively manufactured by a commonly used formulation method known to those skilled in the art.
- an excipient if necessary, a binder, a disintegrant, a lubricant, a coloring agent, a flavoring agent, a flavoring agent, and the like are added to the compound of the present invention, and the mixture is treated in a usual manner.
- Tablets, coated tablets, granules, powders, capsules and the like can be manufactured.
- additives may be those commonly used in the art, such as lactose, sucrose, sodium chloride, sodium glucose, starch, starch, and the like. Carbonated calcium carbonate, cellulose, microcrystalline cellulose, silicic acid, etc.
- binders are water, ethanol, propanol, single syrup, pudose solution, starch solution, gelatin solution, carboxy Disintegrants such as methylcellulose, hydroxypropylcellulose, hydroxypropyl starch, methylcellulose, ethylcellulose, shellac, calcium phosphate, polyvinylidene, etc .; dry starch, alginic acid Sodium, agar powder, sodium bicarbonate, calcium carbonate, sodium raurylsulfate, monoglyceryl stearate , Lactose, etc., lubricating agents such as purified talc, stearate, borax, polyethylene glycol, etc., and flavoring agents such as sucrose, orange peel, citric acid, tartaric acid, etc. .
- the compound of the present invention is flavored.
- Oral liquids, syrups, elixirs, etc. can be manufactured in a conventional manner by adding agents, buffers, stabilizers, and flavoring agents.
- the flavoring agents mentioned above may be used, and sodium citrate or the like may be used as a buffer, and tragacanth, arabic gum, gelatin or the like may be used as a stabilizer.
- a PH regulator, a buffer, a stabilizer, an isotonicity agent, a local anesthetic, etc. are added to the compound of the present invention, and a subcutaneous, intramuscular and intravenous injection is prepared by a conventional method.
- a pH regulator and the buffer include sodium citrate, sodium phosphate, and sodium phosphate.
- the stabilizer include sodium pyrosulfite, EDTA, thioglycolic acid, and thiolactic acid.
- local anesthetics include proforce hydrochloride and lidocaine hydrochloride.
- the compound of the present invention may contain a pharmaceutical carrier known in the art, such as polyethylene glycol, lanolin, cocoa butter, or fatty acid triglyceride, and, if necessary, After adding a surfactant such as (registered trademark), it can be produced by an ordinary method.
- a pharmaceutical carrier known in the art, such as polyethylene glycol, lanolin, cocoa butter, or fatty acid triglyceride, and, if necessary, After adding a surfactant such as (registered trademark), it can be produced by an ordinary method.
- bases, stabilizers, wetting agents, preservatives, etc. commonly used for the compound of the present invention may be added as necessary. It is mixed and formulated in a usual manner.
- the base include fluid paraffin, white petrolatum, beeswax, octyl dodecyl alcohol, paraffin and the like.
- the preservative include methyl paraoxybenzoate, ethyl ethyl paraoxybenzoate, propyl paraoxybenzoate, and the like.
- the above-mentioned plaster, cream, gel, paste or the like may be applied to a usual support in a usual manner.
- a woven or non-woven fabric made of cotton, fabric, or synthetic fiber, a film such as soft vinyl chloride, polyethylene, or polyurethane, or a foam sheet is suitable. .
- the amount of the compound of the present invention to be incorporated in each of the above-mentioned dosage unit forms is not fixed depending on the condition of the patient to which the present invention is to be applied or on its dosage form. ⁇ 100 mg, about 0.1-500 mg for injections, about 5 ⁇ : LOOO mg for suppositories.
- the daily dose of the drug having the above dosage form depends on the patient's condition, weight, age, sex, etc., and cannot be determined unconditionally, but is usually about 0.1 to 500 mg per adult per day. , Preferably about 1 to: LOOO mg should be used, and it is preferable to administer it once or in 2 to 4 divided doses.
- Step E 5- (2-Hydroxityl) -13,4-bis (4-methoxyphenyl) isoxazole obtained in Example 4 1.3 g, triphenylphosphine 1. ⁇ g and phthalimide 600 mg of dry tetrahydrofuran solution 15 ml in a nitrogen atmosphere and water-cooled getyl azodicarboxylate
- Step F 1.5 g of this phthalimid form was suspended in 15 ml of ethanol, 165 mg of hydrazine hydrate was added, and the mixture was stirred at room temperature for 40.5 hours. The precipitated crystals are removed by filtration, washed with 10 ml of ethanol, combined with the mother liquor, concentrated under reduced pressure, and purified by silica gel column chromatography (20% methanol / close-up form). (2-Aminoethyl) 1-3,4-bis (4-methoxyphenyl) isoxazole
- Step A In 40 ml of tert-butanol, 128 g of deoxyanisoyin, 67.3 g of potassium tert-butoxide and 116 g of methyl 3-methoxyacrylate In addition, the mixture was stirred at 70 for 3 hours. After completion of the reaction, n-hexane was added to the reaction mixture, and the mixture was left at room temperature. The precipitate was collected by filtration, dissolved by adding 100 ml of ethyl acetate and 300 ml of 3N-sulfuric acid, and then the organic layer was separated. The extract was washed successively with a saline solution and dried over anhydrous magnesium sulfate. The organic layer was concentrated under reduced pressure to obtain 1553 g (yield: 90%) of methyl 4,5-bis (4-methoxyphenyl) -15-oxo-13-pentenoate as an oil.
- Step B Methyl 4,5-bis (4-methoxyphenyl) — 5-year-old 3-pentenoate 24.5 g and hydroxylaminate hydrochloride 51.5 g in methanol
- a mixed solvent of 65 ml of water and 72 ml of water the mixture was heated under reflux for 23 hours. At this time, 0.9 equivalent of sodium hydrogencarbonate was added to the reaction mixture to allow the reaction to proceed. 4 2 Added separately.
- methanol was distilled off under reduced pressure. Water and ethyl acetate were added to the residue to dissolve it, and the organic layer was separated, washed with saturated saline, and dried over anhydrous magnesium sulfate.
- the aqueous layer was acidified with concentrated hydrochloric acid, and then extracted with 100 ml of ethyl ethyl phosphate.
- the extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- the residue was dissolved in methanol (80 ml), a few drops of concentrated sulfuric acid were added, the mixture was stirred at room temperature for 12 hours, and the solvent was distilled off under reduced pressure. Dissolve the residue in 100 ml of ethyl acetate and add saturated After washing with 50 ml of an aqueous sodium hydrogen solution and 50 ml of a saturated saline solution, the extract was dried over anhydrous magnesium sulfate and concentrated under reduced pressure.
- Example 13 300 mg of the compound 13 obtained in Example 13 was dissolved in 5 ml of methylene chloride, and 94 mg of N-t-butoxycarbonylglycine, 4 mg of 4-dimethylaminoaminopyridine was added. Then, under water cooling, 132 mg of N, N'-dicyclohexylcarbodiimide was added, followed by stirring for 2 days. The precipitated crystals were collected by filtration, washed with ethyl acetate, the mother liquor and the washing solution were combined, diluted with 70 ml of ethyl acetate, washed with 15 ml of water, and dried over anhydrous magnesium sulfate.
- Example 4 3 to 4 4> Using the compound 38 obtained in Example 38, compounds 43 and 44 shown in Table 1 were synthesized in the same manner as in Example 22.
- Compound 46 shown in Table 1 was synthesized in the same manner as in Example 45.
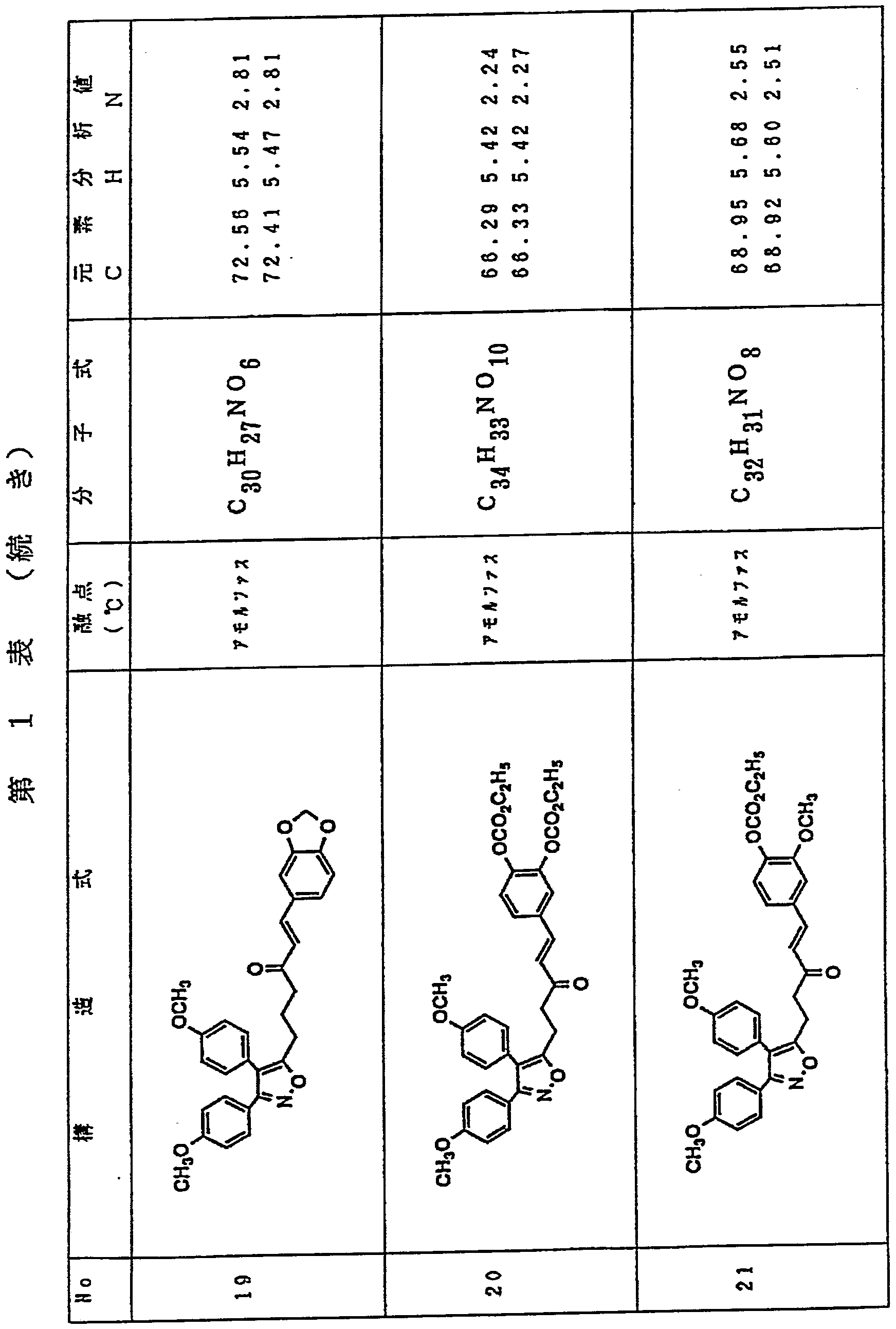
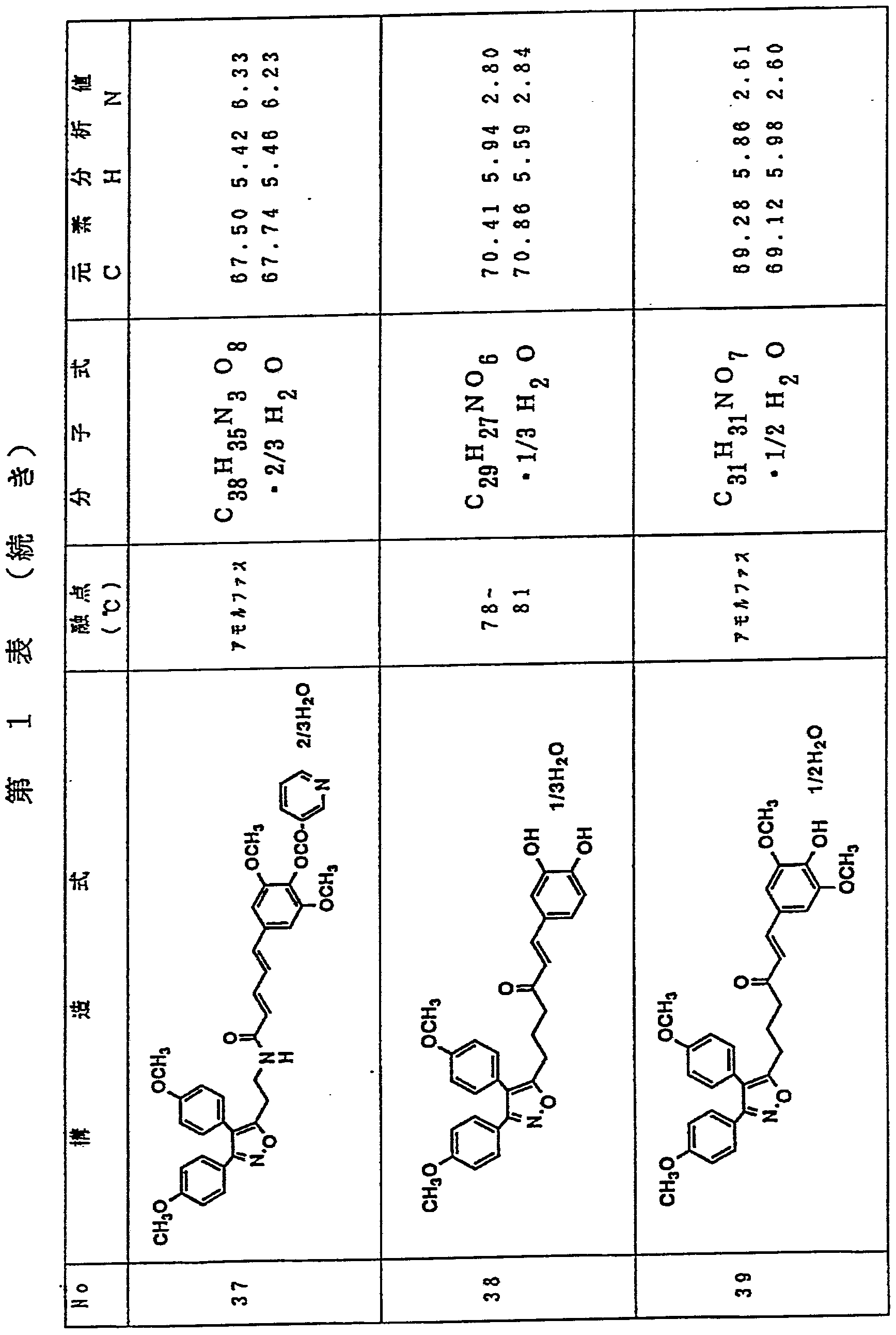
- Table 1 shows the structures, melting points, molecular formulas, and elemental analysis values of the compounds of the present invention produced in Examples 1 to 48 described above.
- the upper row shows the analytical values and the lower row shows the theoretical values.
- Tablets were prepared in the following proportions according to a conventional method.
- Granules were prepared at the following mixing ratios according to a conventional method.
- Fine granules were prepared according to the usual method at the following mixing ratio
- Suppositories were prepared at the following compounding ratios according to a conventional method.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Description




























Claims











Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/140,020 US5399577A (en) | 1991-05-01 | 1992-05-01 | Isoxazole derivatives and salts thereof |
| EP92909853A EP0633254A1 (en) | 1991-05-01 | 1992-05-01 | Novel isoxazole derivative and salt thereof |
| AU17405/92A AU658629B2 (en) | 1991-05-01 | 1992-05-01 | Novel isoxazole derivative and salt thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP10013691 | 1991-05-01 | ||
| JP3/100136 | 1991-05-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1992019604A1 true WO1992019604A1 (en) | 1992-11-12 |
Family
ID=14265903
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1992/000571 Ceased WO1992019604A1 (en) | 1991-05-01 | 1992-05-01 | Novel isoxazole derivative and salt thereof |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US5399577A (ja) |
| EP (1) | EP0633254A1 (ja) |
| KR (1) | KR0138498B1 (ja) |
| AU (1) | AU658629B2 (ja) |
| CA (1) | CA2102209C (ja) |
| WO (1) | WO1992019604A1 (ja) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0623603A4 (en) * | 1992-10-30 | 1995-03-08 | Taiho Pharmaceutical Co Ltd | STYRENE DERIVATIVES AND SALTS THEREOF. |
| WO1995011888A1 (de) * | 1993-10-28 | 1995-05-04 | F. Joh. Kwizda Gesellschaft M.B.H. | Verfahren zur herstellung von 3,4-diaryl-5-isoxazolylessigsäure derivaten |
| US5633272A (en) * | 1995-02-13 | 1997-05-27 | Talley; John J. | Substituted isoxazoles for the treatment of inflammation |
| US5859257A (en) * | 1995-02-13 | 1999-01-12 | G. D. Searle & Co. | Isoxazole compounds as cyclooxygenase inhibitors |
| US6432999B2 (en) | 1995-06-02 | 2002-08-13 | Pharmacia Corporation | Pyrazole substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6512121B2 (en) | 1998-09-14 | 2003-01-28 | G.D. Searle & Co. | Heterocyclo substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6515014B2 (en) | 1995-06-02 | 2003-02-04 | G. D. Searle & Co. | Thiophene substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6613790B2 (en) | 2001-04-17 | 2003-09-02 | Pharmacia Corporation | Prodrugs of COX-2 inhibitors |
| US6673818B2 (en) | 2001-04-20 | 2004-01-06 | Pharmacia Corporation | Fluoro-substituted benzenesulfonyl compounds for the treatment of inflammation |
| US6677364B2 (en) | 1998-04-20 | 2004-01-13 | G.D. Searle & Co. | Substituted sulfonylphenylheterocycles as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6809204B2 (en) | 2001-10-12 | 2004-10-26 | Onconova Therapeutics, Inc. | Processes for the preparation of substituted isoxazoles and 2-isoxazolines |
| US7329401B2 (en) | 2002-04-15 | 2008-02-12 | The Regents Of The University Of California | Cyclooxygenase-2 selective agents useful as imaging probes and related methods |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04134077A (ja) * | 1990-09-21 | 1992-05-07 | Taiho Yakuhin Kogyo Kk | イソオキサゾール化合物 |
| KR100330609B1 (ko) * | 1994-02-16 | 2002-11-22 | 시오노기세이야쿠가부시키가이샤 | 3-이속사졸카복실산의 제조방법 |
| EP0833622B8 (en) * | 1995-06-12 | 2005-10-12 | G.D. Searle & Co. | Compositions comprising a cyclooxygenase-2 inhibitor and a 5-lipoxygenase inhibitor |
| DE19953024A1 (de) | 1999-11-04 | 2001-05-10 | Merck Patent Gmbh | Isoxazolderivate als Phosphodiesterase VII-Hemmer |
| DE10237883A1 (de) * | 2002-08-19 | 2004-03-04 | Merckle Gmbh Chem.-Pharm. Fabrik | Substituierte Isoxazolderivate und ihre Verwendung in der Pharmazie |
| JP4921162B2 (ja) * | 2003-02-11 | 2012-04-25 | ヴァーナリス(ケンブリッジ)リミテッド | 熱ショックタンパク質の阻害剤としてのイソオキサゾール化合物類 |
| ITMI20040019A1 (it) * | 2004-01-12 | 2004-04-12 | Univ Bari | Derivati isossazolici e loro impiego come inibitori della ciclossigenasi |
| US7989450B2 (en) | 2008-01-11 | 2011-08-02 | Universita' Degli Studi Di Bari | Functionalized diarylisoxazoles inhibitors of ciclooxygenase |
| US9359346B2 (en) * | 2011-11-29 | 2016-06-07 | Vivozon, Inc. | Benzamide derivative and use thereof |
| EP2949653A1 (en) * | 2014-05-27 | 2015-12-02 | Banoglu, Erden | Isoxazole derivatives as leukotriene biosynthesis inhibitors |
| WO2015191636A2 (en) * | 2014-06-09 | 2015-12-17 | Baylor College Of Medicine | Xanthine oxidase inhibitors and methods of use |
| EP3782990B1 (en) * | 2016-04-27 | 2023-06-07 | Università degli Studi di Bari "Aldo Moro" | Mofezolac derivative as imaging agent in a diagnostic method of cancer |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1128526A (en) * | 1979-10-05 | 1982-07-27 | Cdc Life Sciences Inc. | 3,4-diarylisoxazol-5-acetic acids |
| JPS6075471A (ja) * | 1983-10-03 | 1985-04-27 | Sankyo Kasei Kogyo Kk | 3,4−ジフエニルイソオキサゾ−ル−5−酢酸類の製造方法 |
| JP2622887B2 (ja) * | 1988-11-21 | 1997-06-25 | 大鵬薬品工業株式会社 | イソキサゾール誘導体及びその製法 |
| JP2796749B2 (ja) * | 1990-01-24 | 1998-09-10 | 大鵬薬品工業株式会社 | イソキサゾール誘導体の製造方法 |
| JPH04134077A (ja) * | 1990-09-21 | 1992-05-07 | Taiho Yakuhin Kogyo Kk | イソオキサゾール化合物 |
-
1992
- 1992-05-01 US US08/140,020 patent/US5399577A/en not_active Expired - Fee Related
- 1992-05-01 CA CA002102209A patent/CA2102209C/en not_active Expired - Fee Related
- 1992-05-01 WO PCT/JP1992/000571 patent/WO1992019604A1/ja not_active Ceased
- 1992-05-01 EP EP92909853A patent/EP0633254A1/en not_active Withdrawn
- 1992-05-01 AU AU17405/92A patent/AU658629B2/en not_active Ceased
-
1993
- 1993-11-01 KR KR1019930703302A patent/KR0138498B1/ko not_active Expired - Fee Related
Non-Patent Citations (1)
| Title |
|---|
| Tetrahedron Letters, No. 47, (1964), G. LOVOECCHIO et al., "Synthesis of Biisoxazolinic Spiro Compounds", p. 3495-7. * |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0623603A4 (en) * | 1992-10-30 | 1995-03-08 | Taiho Pharmaceutical Co Ltd | STYRENE DERIVATIVES AND SALTS THEREOF. |
| WO1995011888A1 (de) * | 1993-10-28 | 1995-05-04 | F. Joh. Kwizda Gesellschaft M.B.H. | Verfahren zur herstellung von 3,4-diaryl-5-isoxazolylessigsäure derivaten |
| US5633272A (en) * | 1995-02-13 | 1997-05-27 | Talley; John J. | Substituted isoxazoles for the treatment of inflammation |
| US5859257A (en) * | 1995-02-13 | 1999-01-12 | G. D. Searle & Co. | Isoxazole compounds as cyclooxygenase inhibitors |
| US5985902A (en) * | 1995-02-13 | 1999-11-16 | G.D. Searle & Co. | Substituted isoxazole for the treatment of inflammation |
| US6753344B2 (en) | 1995-06-02 | 2004-06-22 | Pharmacia Corporation | Thiophene substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6515014B2 (en) | 1995-06-02 | 2003-02-04 | G. D. Searle & Co. | Thiophene substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6432999B2 (en) | 1995-06-02 | 2002-08-13 | Pharmacia Corporation | Pyrazole substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6875785B2 (en) | 1995-06-02 | 2005-04-05 | Pharmacia Corporation | Heterocyclo substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6998415B2 (en) | 1996-05-31 | 2006-02-14 | Pharmacia Corporation | Substituted sulfonylphenylheterocycles as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6677364B2 (en) | 1998-04-20 | 2004-01-13 | G.D. Searle & Co. | Substituted sulfonylphenylheterocycles as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6512121B2 (en) | 1998-09-14 | 2003-01-28 | G.D. Searle & Co. | Heterocyclo substituted hydroxamic acid derivatives as cyclooxygenase-2 and 5-lipoxygenase inhibitors |
| US6613790B2 (en) | 2001-04-17 | 2003-09-02 | Pharmacia Corporation | Prodrugs of COX-2 inhibitors |
| US6673818B2 (en) | 2001-04-20 | 2004-01-06 | Pharmacia Corporation | Fluoro-substituted benzenesulfonyl compounds for the treatment of inflammation |
| US6699884B2 (en) | 2001-04-20 | 2004-03-02 | Pharmacia Corporation | Fluoro-substituted benzenesulfonyl compounds for the treatment of inflammation |
| US6809204B2 (en) | 2001-10-12 | 2004-10-26 | Onconova Therapeutics, Inc. | Processes for the preparation of substituted isoxazoles and 2-isoxazolines |
| US7094903B2 (en) | 2001-10-12 | 2006-08-22 | Onconova Therapeutics, Inc. | Processes for the preparation of substituted isoxazoles and 2-isoxazolines |
| US7329401B2 (en) | 2002-04-15 | 2008-02-12 | The Regents Of The University Of California | Cyclooxygenase-2 selective agents useful as imaging probes and related methods |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2102209A1 (en) | 1992-11-02 |
| CA2102209C (en) | 1997-11-18 |
| EP0633254A1 (en) | 1995-01-11 |
| KR0138498B1 (ko) | 1998-05-15 |
| US5399577A (en) | 1995-03-21 |
| AU1740592A (en) | 1992-12-21 |
| AU658629B2 (en) | 1995-04-27 |
| EP0633254A4 (en) | 1994-02-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1992019604A1 (en) | Novel isoxazole derivative and salt thereof | |
| JP6832946B2 (ja) | キナーゼ阻害剤およびその中間体の調製方法 | |
| US5478856A (en) | Styrene derivatives and salts thereof | |
| MA27660A1 (fr) | Derive heteroarylcarbamoylbenzene | |
| WO1992005162A1 (en) | Isoxazole compound, pharmaceutically acceptable salt thereof, and medicinal use thereof | |
| CA2481742C (fr) | Procedes de preparation de combretastatines | |
| JP3507124B2 (ja) | ベンジリデン誘導体の製造法 | |
| JPWO1992019604A1 (ja) | 新規イソオキサゾール誘導体及びその塩 | |
| WO2003076374A1 (en) | PROCESS FOR PRODUCING trans-4-AMINO-1-CYCLOHEXANECARBOXYLIC ACID DERIVATIVE | |
| WO2011116490A1 (zh) | 制备消旋卡多曲的方法 | |
| JP2018090551A (ja) | L−カルノシン誘導体またはその塩、及びl−カルノシンまたはその塩の製造方法 | |
| JP2008542415A (ja) | レニン阻害剤の製造方法 | |
| CN104788353B (zh) | 一种合成4‑氧代‑l‑脯氨酸衍生物的方法 | |
| JPH0629229B2 (ja) | システインプロティナーゼ阻害剤 | |
| JPH06220029A (ja) | 1,4−ベンズオキサジン誘導体 | |
| WO1991011444A1 (en) | Piperazine compound, production thereof, and medicinal use thereof | |
| CH616659A5 (ja) | ||
| JPH04217650A (ja) | δ−アミノレブリン酸の酸付加塩の製造方法 | |
| KR950003333B1 (ko) | α,β-불포화케톤 및 케토옥심유도체 | |
| JPH069553A (ja) | 1−[|2s|−メチル−3−メルカプトプロピオニル−ピロリジン−|2s|−カルボン酸の製法 | |
| JP4013772B2 (ja) | 2−ヒドロキシイミノ−3−オキソプロピオニトリル及びその製法 | |
| DE3914227A1 (de) | Verfahren zur herstellung von 2,7-difluor-9-fluorenon und neue zwischenprodukte | |
| CA3214107A1 (en) | New process for the synthesis of 5-{5-chloro-2-[(3s)-3- [(morpholin-4-yl)methyl]-3,4-dihydroisoquinoline-2(1h)- carbonyl]phenyl}-1,2-dimethyl-1h-pyrrole-3-carboxylic acid derivatives and its application for the production of pharmaceutical compounds | |
| JPS61246176A (ja) | アミノラクトンの調製方法 | |
| KR100468314B1 (ko) | 레보설피리드 제조용 중간체 및 그 제조방법 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IT LU MC NL SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1992909853 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2102209 Country of ref document: CA Ref document number: 08140020 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1992909853 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1992909853 Country of ref document: EP |