WO1994002496A1 - Derivatives of 16-membered ring antibiotic macrolides - Google Patents
Derivatives of 16-membered ring antibiotic macrolides Download PDFInfo
- Publication number
- WO1994002496A1 WO1994002496A1 PCT/US1993/005210 US9305210W WO9402496A1 WO 1994002496 A1 WO1994002496 A1 WO 1994002496A1 US 9305210 W US9305210 W US 9305210W WO 9402496 A1 WO9402496 A1 WO 9402496A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carbons
- group
- alkyl
- pharmaceutically acceptable
- optionally substituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7048—Compounds having saccharide radicals and heterocyclic rings having oxygen as a ring hetero atom, e.g. leucoglucosan, hesperidin, erythromycin, nystatin, digitoxin or digoxin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Definitions
- This invention is concerned with new antibiotics.
- this invention relates to compounds which are derivatives of the macrolide antibiotics rosaramicin, repromicin, 5-mycaminosyltylonolide, desmycosin, lactenocin, O-demethyllactenocin, cirramycin A 1 , and 23-deoxymycaminosyltylonolide; to the pharmaceutically-acceptable acid addition salts of such derivatives; to a method of using such derivatives in the treatment of illnesses in animals caused by bacterial and mycoplasmic pathogens; and to pharmaceutical compositions useful therefor.
- the term "animals" includes mammals, fish and birds.
- agents known to combat bacterial infectious diseases in animals There are numerous agents known to combat bacterial infectious diseases in animals, but for many specific diseases the current agents of choice leave much to be desired. In some instances the agents may not persist long enough in the host and, therefore, require frequent dosing to maintain therapeutically effective blood and/or tissue levels. For meat producing animals (cattle, poultry, sheep and swine) this will require considerable labor intensive animal handling which is costly to the producer. In other cases, the agent may be poorly tolerated or even toxic to the host at therapeutically effective doses. Agents with increased potency, a longer half-life, an increased therapeutic index and a broader spectrum of antibacterial activity as well as agents with greater oral absorption would improve the scope of animal diseases that could be more effectively treated. Thus, the need for new antibacterial and anti-mycoplasmic agents with improved properties endures.
- bovine respiratory disease the principal causative bacterial pathogens of which are Pasteurella haemolytica. P. multocida and Haemophilus somnus; pasteurellosis in swine, goats, sheep and poultry (P. multocida): swine pleuropneumonia (Actinobacillus pleuropneumoniae); swine streptococcus infections (Streptococcus suis); and for all of the above mentioned hosts, infections by Mycoplasma spp.
- This invention is concerned with new antibiotics which are derivatives of the macrolides repromicin, rosaramicin, 5-mycaminosyltylonolide, desmycosin, lactenocin, O-demethyllactenocin, cirramycin A 1 , and 23-deoxymycaminosyltylonolide and to the acid addition salts of such derivatives.
- new antibiotics have enhanced potency against bacterial pathogens over the parent compounds and are active against mycoplasmic pathogens.
- m is 0 or 1 ;
- X 1 is H or CN
- Z is H or OH;
- Q is selected from the group consisting of H, OH, fluoro, chloro, bromo, iodo, OX 2 , SX 2 ,
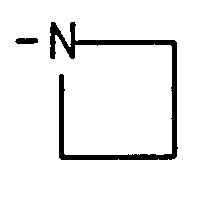
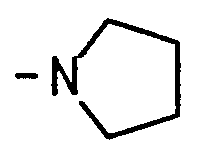
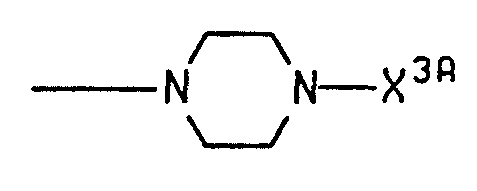
- azetidin-1-yl pyrrolidin-1-yl, piperidin-1-yl, 3,3-dimethylpiperidin-1-yl, hexahydroazepin-1-yl.octahydroazocin-1-yl.octahydroindol-1-yl,1 ,3,3a, 4,7,7a-hexahydroisoindol-2-yl, decahydroquinol-1-yl, decahydroisoquinol-2-yl, 1 ,2,3,4-tetrahydroisoquinol-2-yl, 1 ,2,3,6-tetrahydropyridin-1-yl, 4-alkylpiperazin-1-yl having 1 to 4 carbons in the alkyl portion, morpholino, 2,6-dimethylmorpholin-4-yl, thiomorpholino, and
- R 3 and R 4 are independently selected from the group consisting of H, alkyl having 1 to 4 carbons, hydroxyalkyl having 2 to 4 carbons, cycloalkyl having 3 to 8 carbons, alkenyl having 3 or 4 carbons, alkoxyalkyl having 1 to 4 carbons in the alkoxy portion and 2 to 4 carbons in the alkyl portion and alkoxyalkoxyalkyl having 1 to 4 carbons in each of the alkoxy portions and 2 to 4 carbons in the alkyl portion; and
- X 2 is selected from the group consisting of optionally substituted alkyl having 1 to 4 carbons, optionally substituted cycloalkyl having 4 to 8 carbon atoms, and an optionally substituted aryl, aralkyl or heteroaryl group selected from the group consisting of phenyl, benzyl, pyridinyl, quinolinyl, isoquinolinyl, quinazolinyl, pyrimidinyl, imidazolyl, oxazolyl, thiazolyl, benzimidazolyl, indolyl, benzoxazolyl and benzthiazolyl;
- optionally substituted alkyl and optionally substituted cycloalkyl can be substituted with 1 or 2 substituents independently selected from the group consisting of hydroxy, amino, N-alkylamino having 1 to 4 carbons, N,N-dia!kylamino having a total of 2 to 6 carbons and alkoxy having 1 to 4 carbons; and where the optionally substituted aryl, aralkyl and heteroaryl groups are optionally substituted with 1 or 2 substituents independently selected from the group consisting of alkyl having 1 to 4 carbons, fluoro, chloro, bromo, acetyl, amino, nitro, cyano, trifiuoromethyl, N-alkylamino having 1 to 4 carbons, N,N-dialkylamino having a total of 2 to 6 carbons, carboxyl, carboalkoxy having 1 to 4 carbons, carboxamido, sulfonamido, hydroxyalkyl having 1 to 4 carbons, aminoal
- R 1 is selected from the group consisting of H, alkyl having 1 to 4 carbons, aminoalkyl having 2 to 4 carbons, hydroxyalkyl having 2 to 4 carbons, N-alkylaminoalkyl having 1 to 4 carbons in the alkylamino portion and 2 to 4 carbons in the alkyl portion, benzyl, alkoxyalkyl having 2 to 4 carbons in the alkyl portion and 1 to 4 carbons in the alkoxy portion, N,N-dialkylaminoalkyl having a total of 2 to 6 carbons in the dialkylamino portion and 2 to 4 carbons in the alkyl portion, morpholino-(C 2 -C 4 )alkyl, piperidino-(C 2 - C 4 )alkyl, pyrrolidino-(C 2 -C 4 )alkyl, azetidinyl-(C 2 -C 4 )alkyl, and X 3 ;
- R 2 is selected from the group consisting of optionally substituted alkyl having 1 to 6 carbons,
- alkyl is optionally substituted with 1 or 2 substituents independently selected from the group consisting of hydroxy, cyano, amino, N-alkylamino having 1 to 4 carbons, N,N-dialkylamino having a total of 2 to 6 carbons, N-(hydroxyalkyl)amino having 2 to 4 carbons, N,N- bis(hydroxyalkyl)amino wherein each alkyl portion has 2 to 4 carbons, alkoxy having 1 to 4 carbons, alkoxycarbonyl having 1 to 4 carbons in the alkoxy portion, N.N-dialkylaminoalkoxy having a total of 2 to 6 carbons in the dialkylamino portion and 2 to 4 carbons in the alkoxy portion, alkoxyalkoxy having 1 to 4 carbons in each of the alkoxy portions, alkoxyalkoxyalkoxy having 1 to 4 carbons in each of the alkoxy portions, , , ,
- R 5 and R 6 are independently selected from hydrogen or alkyl having 1 to 4 carbons, or R 5 and R 6 are taken together with the nitrogen to which they are attached and form a saturated or unsaturated ring having 4 to 6 carbon atoms, morpholino or piperazino;
- A is CH 2 , NH, O, S or N-loweralkyl
- B 1 , B 2 , and B 3 are each independently selected from the group consisting of hydrogen and (C 1 -C 4 )alkyl;
- the optionally substituted cycloalkyl is optionally substituted with 1 to 5 substituents independently selected from the group consisting of hydroxy, fluoro, chloro, alkoxy having 1 to 4 carbons, hydroxyalkyl having 1 to 4 carbons, alkoxyalkyl having 1 to 4 carbons in each of the alkoxy and alkyl portions, amino, N-alkylamino having 1 to 4 carbons, N,N-dialkylamino having a total of
- G is (C 2 -C 4 )alkylene optionally substituted with (C 1 -C 4 )alkyl or hydroxyl;
- X 4 is selected from the group consisting of hydrogen, methyl and ethyl; X 3 and X 5 are independently selected from the group consisting of an optionally substituted hydroxyalkanoyl having 1 to 6 carbons, an amino acyl group, and dipeptidyl group,
- amino acyl group and the amino acyl groups of the dipeptidyl group are independently selected from the group consisting of the D- or L- form, when applicable, of alanyl, arginyl, asparagyl, aspartyl acid, cysteinyl, cystyl, glutamyl acid, glutamyl, glycyl, histidyl, hydroxyllysyl, hydroxyprolyl, isoleucyl, leucyl, lysyl, methionyl, phenylalanyl, prolyl, seryl, threonyl, tryptophyl, tyrosyl, valyl, ⁇ -alanyl, ⁇ - lysyl, N,N-dimethylglycyl, ⁇ , ⁇ -dimethylglycyl, ⁇ -aminobutyryl, 4- hydroxyphenylglycyl, phenylglycyl, ⁇ ,
- the optionally substituted hydroxyalkanoyl group is optionally substituted with an optionally substituted phenyl, wherein the optionally substituted phenyl is optionally substituted with 1 to 5 substituents selected from the group consisting of (C 1 -C 4 )alkyl, fluoro, chloro, bromo, iodo, (C 1 - C 4 )alkoxy, nitro, amino, cyano, hydroxy, trifluoromethyl and carboalkoxy having 1 to 4 carbons;
- X 4 and X 5 are taken together with the nitrogen to which they are attached and form , , , , , ,
- Y 1 is selected from the group consisting of C, CH, CH 2 , N and NH; Y 2 is O or S; n is 0, 1 or 2; R 7 is alkyl having 1 to 4 carbons
- R 8 is H or alkyl having 1 to 4 carbons
- R 9 is selected from the group consisting of H, alkyl having 1 to 4 carbons, hydroxy, alkoxy having 1 to
- R 8 and R 9 are taken together and form an oxo group
- X 3A is independently selected from the same group as X 3 ;or R 1 and R 2 are taken together with the nitrogen to which they are attached and form
- Y 3 is selected from the group consisting of C, CH, CH 2 , N and NH; Y 4 is O or S; b is 0, 1 or 2; R 10 is alkyl having 1 to 4 carbons
- R 11 is H or alkyl having 1 to 4 carbons
- R 12 is selected from the group consisting of H, alkyl having 1 to 4 carbons, hydroxy, alkoxy having 1 to 3 carbons, amino, N-alkylamino having 1 to 4 carbons and N,N-dialkylamino having a total of 2 to
- R 11 and R 12 are taken together and form an oxo group
- X 3 is as defined above;
- R 1 is H or alkyl having 1 to 4 carbons, R 2 cannot be unsubstituted alkyl.
- loweralkyl denotes an alkyl having 1 to 4 carbons.
- alkyl is meant to encompass both straight chain and branched alkyls.
- the amino acyl groups are derivatives of the corresponding amino acids and are well known in the art.
- the following D- or L- amino acids, where applicable, are used to derive the amino acyl groups of this invention: alanine, arginine, asparagine, aspartic acid, cysteine, cystine, glutamic acid, glutamine, glycine, histidine, hydroxyllysine, hydroxyproline, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophane, tyrosine, valine, ⁇ -alanine, ⁇ -lysine, N,N-dimethylglycine, ⁇ , ⁇ -dimethylglycine, ⁇ -aminobutyric acid, 4-hydroxyphenylglycine, phenylglycine, ⁇ , ⁇ -diaminobutyric acid, ornithine, homoserine, bi
- the dipeptidyl groups comprise derivatives of any possible combination of two of the amino acids listed hereinabove which have been coupled by conventional peptide synthesis methods well known to those skilled in the art.
- hydroxyalkanoyl groups are derivatives of the corresponding alkanoic acids and are well known in the art.
- a few examples of such groups, which are listed for illustration purposes and are not intended to limit the scope of the group, are glycolic acid, lactic acid and mandelic acid.
- a preferred group of compounds are those having the formula (I) or (II) where m is 0.
- Another preferred group of compounds are those having the formula (I) wherein m is 0; Z is H; R 1 is selected from the group consisting of H, alkyl having 1 to 4 carbons, hydroxyalkyl having 2 to 4 carbons and alkoxyalkyl having 2 to 4 carbons in the alkyl portion and 1 to 4 carbons in the alkoxy portion; R 2 is substituted alkyl having 2 to 5 carbons, wherein the substituents are selected from the group consisting of amino, N-alkylamino having 1 to 4 carbons, and N,N-dialkylamino having a total of 2 to 6 carbons; and Q is selected from the group consisting of hydrogen, homopiperidin-1-yl, piperidin-1-yl, pyrrolidin-1-yl, N,N-diethylamino, N,N-dimethylamino, N,N-dipropylamino,
- Yet still another group of preferred compounds are those having formula (II) wherein m is 0; Z is H; R 1 is selected from the group consisting of H, alkyl having 1 to 4 carbons, and alkoxyalkyl having 2 to 4 carbons in the alkyl portion and 1 to 4 carbons in the alkoxy portion; R 2 is selected from the group consisting of H, alkyl having 1 to 4 carbons and substituted alkyl having 2 to 5 carbons, wherein the substituents are selected from the group consisting of amino, N-alkylamino having 1 to 4 carbons, hydroxy, alkoxy having 1 to 4 carbons and N.N-dialkylamino having a total of 2 to 6 carbons; or R 1 and R 2 are taken together with the nitrogen to which they are attached and form a heterocyclic group selected from the group consisting of azetidinyl, pyrrolidino, piperidino, hexahydroazepinyl, and morpholino; and Q is hydrogen.
- Another preferred group of compounds are those compounds having the formula (I) wherein m is 0; R 1 is X 3 ; X 1 is H; R 2 is 3-(dimethylamino)propyl; and Q, Z and X 3 are as defined above for formula (I).
- a more preferred group of compounds within the above group of compounds are those wherein X 3 is an amino acyl group selected from the group consisting of L-alanyl, D-alanyl, glycyl, L-valyl, N,N-dimethylglycyl, N,N-dimethyl- ⁇ -aminobutyryl, N.N-diethyl- ⁇ -alanyl, sarcosyl, ⁇ . ⁇ -dimethylglycyl and ⁇ -aminobutyryl.
- the two most preferred compounds of this group of more preferred compounds are those wherein Q is H; Z is H and X 3 is glycyl, and wherein Q is H; Z is H and X 3 is L-alanyl.
- Q, Z, X 4 and X 5 are as defined above for formula (I).
- a more preferred group of compounds within this immediate group of compounds are those wherein G is propylene or 2,2-dimethylpropylene, X 4 is hydrogen or methyl and X 5 is an amino acyl group selected from the group consisting of L-alanyl, D-alanyl, glycyl, L-valyl, N,N-dimethylglycyl, sarcosyl, ⁇ , ⁇ -dimethylglycyl and ⁇ -aminobutyryl.
- the two most preferred compounds of this group of more preferred compounds are the compounds wherein Q is H; Z is H; X 4 is H; R 1 is H; G is 2,2-dimethylpropylene; and X 5 is L-alanyl; and wherein Q is H; Z is H; X 4 is methyl; R 1 is methyl; G is propylene and X 5 is glycyl.
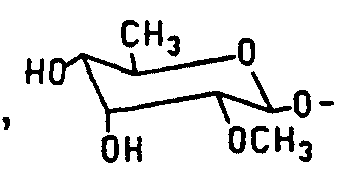
- the parent macrolides from which the compounds of this invention can be made are generally derivatized at the C-20 position with certain substituted amino groups. These derivatives are formed by reductive amination of the C-20 aldehyde group using reducing agents such as formic acid, sodium borohydride, sodium cyanoborohydride or sodium triacetoxyborohydride.
- reducing agents such as formic acid, sodium borohydride, sodium cyanoborohydride or sodium triacetoxyborohydride.
- the cyano derivative of the reductive amination products are inherently produced, in the reaction utilizing sodium cyanoborohydride, along with the non-cyano reductive amination product.
- Scheme I shows a method to obtain the intermediate for compounds of this invention where m is 1.
- the compounds of the present invention having the formula I or II, as defined above, are readily and generally prepared by reductive amination reactions of the appropriate macrolide, rosaramicin, repromicin, 5-mycaminosyltylonolide, desmycosin, lactenocin, O-demethyllactenosin, cirramycin A,, or 23-deoxymycaminosyltylonoIide, with an amine, optionally followed by conversion to the acid addition salt as detailed below.
- Derivatization of the parent macrolide at the C-23 position is carried out according to the method well known to those skilled in the art and as described in J. Antibiotics, 40(6), pp. 823-842, 1987, the contents of which are incorporated herein by reference.
- reaction conditions and reagents used to derivatize a compound of formula I or II at the C-20 position are dictated by the kind of amine that is used in the reaction.
- secondary amines are used in the reductive amination, the following procedure is utilized.
- a solution of a macrolide, such as repromicin is mixed with an excess, usually about 1.5 molar equivalent, of a secondary amine such as azetidine in a reaction inert solvent such as ethyl acetate.
- the reaction mixture is heated to about 60°C to 80°C, preferably about 70°C, with stirring.
- the type of amine to be used in the reductive amination is a primary amine
- a solution of sodium cyanoborohydride and the primary amine is made in a reaction-inert solvent such as methanol.
- the sodium cyanoborohydride is present at approximately four molar equivalents and the amine is present at about one molar equivalent of the macrolide.
- a methanol solution of the macrolide is added dropwise to the solution of sodium cyanoborohydride and the primary amine, and the mixture is stirred for about three to six hours, preferably about four hours.
- the desired C-20 amino derivative of the macrolide is isolated by standard techniques well known to those skilled in the art.
- the cyano derivative, where X 1 is CN is also produced in the reaction and can be isolated by standard techniques well known to those skilled in the art.
- the cyano derivatives can also be synthesized separately by the following method.
- a solution of zinc iodide and an appropriate macrolide is made in methanol.
- Trimethylsilylcyanide is added to the methanol solution and is stirred for about 15 minutes then the appropriate amine is added and the solution is heated at about 40° C for about 2 hours.
- the desired cyano derivative is isolated by standard methods well known in the art.
- a C-20 primary amino derivative of the macrolide, formed by the above method, can be further derivatized by N-methylating the secondary amino group at the C-20 position.
- This synthesis is carried out by suspending the C-20 secondary amino macrolide derivative in water and then adding formic acid. To the resulting solution, a 38% solution of aqueous formaldehyde is added and the reaction mixture is heated to reflux temperature. The reaction mixture is stirred at reflux for about four to six hours, preferably about five hours. It is then cooled to room temperature and the desired compound is isolated.
- a functionalized amine such as N,N-dimethyl-1 ,3-propanediamine
- the following is a method of effecting a reductive amination.
- a methanol solution of the macrolide is mixed with the appropriate amine and stirred at room temperature for approximately 30 minutes.
- the reaction mixture is then cooled to about 0°C and an equimolar amount of glacial acetic acid is added to the mixture and the reaction allowed to stir.
- a methanol solution of sodium cyanoborohydride is added to the reaction mixture, and the resulting solution is stirred for about one hour at about 0°C.
- the reaction is stopped by warming to room temperature and concentrating the reaction mixture, and then the desired macrolide derivative is isolated.
- a preferred method of accomplishing the same type of reaction is as follows. To a stirring solution of the macrolide in methanol is added the appropriate amine and stirred for about 30 minutes. The solution is then cooled to about 0 °C and sodium borohydride is added to it. After stirring for about 2 hours, the solution is concentrated to near dryness and the desired compound is isolated by conventional methods well known in the art. A secondary amine at the C-20 position can be further functionalized with an amino acyl group according to the following procedure.
- a dichloromethane solution of a N-protected amino acid or N-protected dipeptide (t-BOC is one of the preferred protecting groups), or an O-protected hydroxyalkanoic acid (acetate is one of the preferred protecting groups), dicyclohexylcarbodiimide and hydroxybenzotriazole (all of which are present in equimolar amounts) is cooled to about 0°C.
- t-BOC is one of the preferred protecting groups
- an O-protected hydroxyalkanoic acid acetate is one of the preferred protecting groups
- dicyclohexylcarbodiimide and hydroxybenzotriazole (all of which are present in equimolar amounts) is cooled to about 0°C.
- a C-20 secondary amino compound of formula I or II wherein R 1 is hydrogen and R 2 is as defined above.
- the solution is allowed to warm to room temperature and stirring is continued for about 48 to 72 hours.
- the crude product is isolated by conventional methods such
- a compound of formula I or II wherein the C-20 position is an aminoalkylamino can be further derivatized at the terminal amine by an amino acyl group according to the following procedure.
- a stirring solution of a compound of formula I or II having an aminoalkylamino group at the C-20 position in dimethylformamide is added a N-protected (t-BOC is preferred protecting group) amino acid hydroxysuccinimide ester, and the mixture is stirred for about 6 hours.
- the crude product is isolated by conventional methods such as silica gel chromatography.
- the N-protected amino acyl derivative is deprotected by conventional methods to yield the desired products.
- the pharmaceutically acceptable acid addition salts of the C-20 amino macrolide derivatives can be obtained by the following general procedure.
- the HCI salts can be isolated by dissolving the C-20 amino macrolide derivative in a methanolic HCI solution and then evaporating the volatile components to yield the desired salt.
- the methanolic HCI solution can be prepared by mixing acetyl chloride with methanol.
- other preferred pharmaceutically acceptable acid addition salts include citrate, phosphate, sulfate, methanesulfonate, palmitate, succinate, lactate, malate, maleate, tartrate, besylate, fumarate and stearate salts. All of such salts are prepared in a method analogous to the method used to form the HCI salt, that is, by adding equivalent amounts of the appropriate acid to the base.
- the starting macrolide rosaramicin is produced and isolated according to the method described by Wagman et al. in Journal of Antibiotics, Vol. XXV, No. 11 , pp. 641-646, November 1972.
- Repromicin is synthesized from rosaramicin using the method taught by Ganguly et al. in U.S. Patent 3,975,372.
- O-demethyllactenocin and 23-deoxymycaminosyltylonolide are produced and isolated according to the method described in Journal of Antibiotics, 35(12), pp. 1675-1682, 1982.
- Cirramycin A is produced and isolated according to the method described in Journal of Antibiotics, 22, p. 61 , 1969.
- the contents of the above references are incorporated herein by reference. All other starting materials and reagents required for the synthesis of the compounds of the present invention are readily available commercially or can be prepared according methods known in the literature.
- the antibacterial activity of the compounds of the present invention against bacterial pathogens is demonstrated by the compound's ability to inhibit growth of Pasteurella multocida and Pasteurella haemolytica.
- the following procedures are typical assays. Assay I is utilized to test for activity against Pasteurella multocida and Assay II is utilized to test for activity against Pasteurella haemolytica.
- This assay is based on the liquid dilution method in microliter format.
- a single colony of P. multocida (strain 59A067) is inoculated into 5 ml of brain heart infusion (BHl) broth.
- the test compounds are prepared by solubilizing 1 mg of the compound in 125 ⁇ of dimethylsulfoxide (DMSO). Dilutions of the test compound are prepared using uninoculated BHl broth.
- the concentrations of the test compound used range from 200//g/ml to 0.098 ⁇ g/ml by two-fold serial dilutions.
- the P. multocida inoculated BHl is diluted with uninoculated BHl broth to make a 10 4 cell suspension per 200 ⁇ l.
- the BHl cell suspensions are mixed with respective serial dilutions of the test compound, and incubated at 37° C for 18 hours.
- the minimum inhibitory concentration (MIC) is equal to the concentration of the compound exhibiting 100% inhibition of growth of P. multocida as determined by comparison with an uninoculated control.
- This assay is based on the agar dilution method using a Steers Replicator. Two to five colonies isolated from an agar plate are inoculated into BHl broth and incubated overnight at 37° C with shaking (200 rpm). The next morning, 300 //I of the fully grown P. haemolytica preculture is inoculated into 3 ml of fresh BHI broth and is incubated at 37 °C with shaking (200 rpm). The appropriate amounts of the test compounds are dissolved in ethanol and a series of two-fold serial dilutions are prepared. Two ml of the respective serial dilution is mixed with 18 ml of molten BHl agar and solidified. When the inoculated P.
- haemolytica culture reaches 0.5 McFarland standard density, about 5 ⁇ l of the P. haemolytica culture is inoculated onto BHl agar plates containing the various concentrations of the test compound using a Steers Replicator and incubated for 18 hours at 37° C. Initial concentrations of the test compound range from 100-200 A/g/ml. The MIC is equal to the concentration of the test compound exhibiting 100% inhibition of growth of P. haemolytica as determined by comparison with an uninoculated control.
- the in vivo activity of the compounds of formula I or II can be determined by conventional animal protection studies well known to those skilled in the art, usually carried out in mice.
- mice are allotted to cages (10 per cage) upon their arrival, and allowed to acclimate for a minimum of 48 hours before being used.
- Animals are inoculated with 0.5 ml of a 3 ⁇ 10 3 CFU/ml bacterial suspension (P. multocida strain 59A006) intraperitoneally.
- Each experiment has at least 3 non-medicated control groups including one infected with 0.1X challenge dose and two infected with 1X challenge dose; a 10X challenge data group may also be used.
- all mice in a given study can be challenged within 30-90 minutes, especially if a repeating syringe (such as a Cornwall ® syringe) is used to administer the challenge.
- a repeating syringe such as a Cornwall ® syringe
- the first compound treatment is given. It may be necessary for a second person to begin compound dosing if all of the animals have not been challenged at the end of 30 minutes.
- the routes of administration are subcutaneous or per os. Subcutaneous doses are administered into the loose skin in the back of the neck whereas oral doses are given by means of a feeding needle. In both cases, a volume of 0.2 ml is used per mouse. Compounds are administered 30 minutes, 4 hours, and 24 hours after challenge. A control compound of known efficacy administered by the same route is included in each test. Animals are observed daily, and the number of survivors in each group is recorded on the form provided. The P. multocida model monitoring continues for 96 hours (four days) post challenge. Surviving mice are asphyxiated with carbon dioxide at the end of the study.
- the PD 50 is a calculated dose at which the compound tested protects 50% of a group of mice from mortality due to the bacterial infection which would be lethal in the absence of drug treatment.
- an effective dose of a compound of formula I or II is administered to a susceptible or infected animal by parenteral (i.v., i.m. or s.c), oral or topical route.
- the effective dose will vary with the severity of the disease, and the age, weight and condition of the animal. However, the dose will usually range from about 0.25 to about 150 mg/kg, preferably from about 0.25 to about 25 mg/kg.
- a suitable vehicle for administering the dose parenterally is a solution of the compound in steril water, or a solution of the compound in a solvent comprising at least 50% water and a pharmaceutically acceptable cosolvent or cosolvents such as methanol, ethanol, isopropyl alcohol, propylene glycol, glycerol, carbonate esters like diethyl carbonate, dimethyl sulfoxide, N,N-dimethylformamide, N,N-dimethylacetamide, 1-methyl-2-pyrrolidinone, and the like.
- Suspensions are also suitable vehicles for administering the compounds of this invention.
- the suspending medium can be, for example, aqueous carboxymethyl cellulose, inert oils such as peanut oil, highly refined mineral oils, aqueous polyvinylpyrrolidone and so forth.
- Suitable physiologically acceptable adjuvants may be necessary to maintain the compound in suspension. These adjuvants may be chosen from among thickeners such as carboxymethyl cellulose, polyvinylpyrrolidone, gelatin, and the alginates.
- Surfactants are also useful as suspending agents. These surfactants include: lethicin, alkylphenol polyethylene oxide adducts, naphthalenesulfonates, alkylbenzenesulfonates and polyoxyethylene sorbitan esters. Agents affecting surface tension can also help in making useful suspensions. Such agents include silicone antifoams, sorbitol, and sugars. For intravenous use the total concentration of solutes should be controlled to render the preparation isotonic.
- compositions comprising a compound of the formula (I) or (II) or pharmaceutically acceptable salts thereof together with a pharmaceutically acceptable carrier or diluent.
- This invention also provides a method of treating a bacterial infection or a mycoplasmic infection in an animal in need thereof which method comprises administering to said animal a bacterial or mycoplasmic treating amount of a compound of the formula (I) or (II) or a pharmaceutically acceptable salt thereof.
- HPLC High Performance Liquid Chromatography
- a 45:55 (vol:vol) mixture of acetonitrile to aqueous 50 millimolar ammonium acetate is used as the eluant.
- the column temperature is maintained at 40° C and the flow rate is 1.0 ml per minute.
- Samples are dissolved in the eluant (2 mg/ml) and are injected (70 ⁇ l) into a Hewlett-Packard 1090 high performance liquid chromatography instrument; peaks corresponding to the sample input are detected by ultraviolet spectroscopy at either 254 or 280 nm.
- the filtrate from the initial product was combined with the insolubles that were saved, and the mixture was then chromatographed on 450 cc of silica gel. Elution with 1 :9 methanol/dichloromethane containing 1% ammonium hydroxide afforded 1.88 g (27%) of additional product for a total yield of 63% of the free base.
- Acetyl chloride (1.15 g, 14.66 mmol) was added dropwise to 75 ml of methanol, and the solution was allowed to stand at room temperature for 75 minutes.
- To this methanolic HCI solution was added 4.45 g (7.33 mmol) of 20-(azetidin-1-yl)-20-deoxorepromicin.
- the resulting pale yellow solution was allowed to stand at room temperature for two hours. The volatile components were evaporated and the residue was dried under reduced pressure to give 5.24 g of the title compound.
- 20-[3-(Morpholino)propyl]amino-20-deoxorepromicin was prepared from repromicin and 3-(morpholino)propylamine by Method B (omitting the HCI salt formation step).
- Method B To a suspension of 20-[3-(morpholino)propyl]amino-20-deoxorepromicin (200 mg, 0.290 mmol) in 1.25 ml of water was added 517 mg of formic acid.
- the resulting solution was treated with 84 ⁇ l (1.15 mmol) of 38% aqueous formaldehyde, and then heated under reflux with stirring for five hours. The reaction mixture was allowed to cool to room temperature, and then was stored at 5°C for overnight.
- Repromicin (18.00 g, 31.82 mmol) was dissolved in 300 ml of methanol. 3-(Dimethylamino)propylamine (8.00 ml, 63.63 mmol) was then added to the repromicin solution and the resulting reaction mixture was stirred at room temperature for 30 minutes. The reaction mixture was then cooled to 0°C and glacial acetic acid (1.80 ml, 31.82 mmol) was added. After 10 minutes, sodium cyanoborohydride (2.00 g, 31.82 mmol) was added at 0°C as a solution in 15 ml of methanol. The resulting reaction mixture was stirred at 0°C for 1 hour and then concentrated under reduced pressure.
- the BOC-protected material was dissolved in a mixture of trifluoroacetic acid and dichloromethane (1 :1 , 100 mL), and was stirred at about 0 °C for about 20 minutes. The solvent was removed with a rotary evaporator, and the residue was triturated with diethyl ether and was dried. This material was dissolved in water, and 1 N NaOH was added to bring the solution to pH 10. The resulting aqueous phase was extracted with chloroform, dried over sodium sulfate, and concentrated to afford 8.59 g (97%) of the desired product, m/e 724; HPLC RT: 2.16 minutes. EXAMPLE 6
- the BOC-protected intermediate (453 mg, 0.55 mmol) was dissolved in a mixture of dichloromethane and trifluoroacetic acid (1 :1 , 10 mL) and was stirred at about 0 °C for about 15 minutes. The solvent was removed with a rotary evaporator, and the residue was triturated with diethyl ether and dried to afford the desired product as the trifluoroacetate salt in quantitative yield, m/e 724.0.
- Rosaramicin 1.0 g, 1.72 mmol
- 54 mg of zinc iodide were added to a single neck round bottom flask. Enough anhydrous methanol was added to allow the mixture to stir easily. Trimethylsilylcyanide (0.3 mL, 2.15 mmol) was added in one portion; the resulting solution was stirred at room temperature for about 15 minutes. A 0.75 M solution of dimethylamine in methanol (12 mL, 9.0 mmol) was added. The resulting solution was heated at about 40° C for about two hours, and was then allowed to cool to room temperature. The volatile components were evaporated under reduced pressure to furnish 1.1 g of a yellow residue. This material was then purified by chromatography to give the free base of the title compound as a colorless solid: yield
- reaction mixture was quenched with 15 ml of saturated aqueous NaHCO 3 and extracted with ethyl acetate. The combined organic layers were washed with brine, dried (Na 2 SO 4 ), and concentrated under reduced pressure.
- the crude product was purified by flash chromatography (CHCl 3 /MEOH/NH 4 OH: 89: 10:1). The purified product was dissolved in 2 ml of CH 2 CI 2 and cooled to about 0°C. Anhydrous TFA (2 ml) was added. The resulting reaction mixture was stirred at about 0 °C for about 30 minutes. It was then concentrated under reduced pressure, triturated with Et 2 O, filtered, and dried to give 86 mg of the product as the TFA salt.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
- Radiation-Therapy Devices (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description









































































Claims
































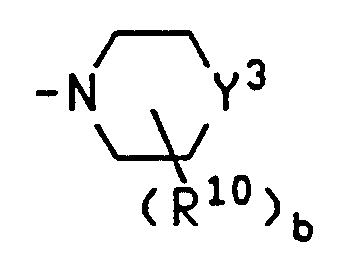








Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1019950700159A KR950702570A (en) | 1992-07-15 | 1993-06-07 | Derivatives of 16-membered macrolide antibiotics (DERIVATIVES OF 16-MEMBERED RING ANTIBIOTIC MACROLIDES) |
| DE69315839T DE69315839T2 (en) | 1992-07-15 | 1993-06-07 | DERIVATIVES OF 16-PIECE ANTIBIOTIC MACROLID |
| PL93307137A PL307137A1 (en) | 1992-07-15 | 1993-06-07 | Derivatives of 16-member cyclic antibiotic macrolydes |
| AU44010/93A AU4401093A (en) | 1992-07-15 | 1993-06-07 | Derivatives of 16-membered ring antibiotic macrolides |
| EP93914300A EP0649430B1 (en) | 1992-07-15 | 1993-06-07 | Derivatives of 16-membered ring antibiotic macrolides |
| US08/362,496 US5545624A (en) | 1992-07-15 | 1993-06-07 | Derivatives of 16-membered ring antibiotic macrolides |
| BR9306741A BR9306741A (en) | 1992-07-15 | 1993-06-07 | Derivatives of antibiotic macrolides with 16-membered ring |
| SK47-95A SK4795A3 (en) | 1992-07-15 | 1993-06-07 | Derivatives of macrolides antibiotics with 16-membered ring separated and used for treatment of illnesses and prarmaceutical agents on their base |
| NO950143A NO950143L (en) | 1992-07-15 | 1995-01-13 | Derivatives of antibiotic macrolides with 16-membered ring |
| GR980400232T GR3026064T3 (en) | 1992-07-15 | 1998-02-04 | Derivatives of 16-membered ring antibiotic macrolides. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US91424292A | 1992-07-15 | 1992-07-15 | |
| US07/914,242 | 1992-07-15 | ||
| US99624392A | 1992-12-23 | 1992-12-23 | |
| US07/996,243 | 1992-12-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994002496A1 true WO1994002496A1 (en) | 1994-02-03 |
Family
ID=27129638
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1993/005210 Ceased WO1994002496A1 (en) | 1992-07-15 | 1993-06-07 | Derivatives of 16-membered ring antibiotic macrolides |
Country Status (22)
| Country | Link |
|---|---|
| US (1) | US5545624A (en) |
| EP (1) | EP0649430B1 (en) |
| JP (1) | JP2836964B2 (en) |
| KR (1) | KR950702570A (en) |
| CN (1) | CN1083068A (en) |
| AT (1) | ATE161265T1 (en) |
| AU (1) | AU4401093A (en) |
| BR (1) | BR9306741A (en) |
| CA (1) | CA2140243A1 (en) |
| CZ (1) | CZ9995A3 (en) |
| DE (1) | DE69315839T2 (en) |
| DK (1) | DK0649430T3 (en) |
| FI (1) | FI933204A7 (en) |
| GR (1) | GR3026064T3 (en) |
| HU (1) | HUT65330A (en) |
| IL (1) | IL106263A0 (en) |
| MX (1) | MX9304301A (en) |
| PL (1) | PL307137A1 (en) |
| SK (1) | SK4795A3 (en) |
| TW (1) | TW226373B (en) |
| UY (1) | UY23617A1 (en) |
| WO (1) | WO1994002496A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2076107A1 (en) * | 1993-09-20 | 1995-10-16 | Pfizer | DERIVATIVES OF MACROLID ANTIBIOTICS OF RINGS OF 16 MEMBERS. |
| WO1996009312A1 (en) * | 1994-09-22 | 1996-03-28 | Pfizer Inc. | Antibiotic macrolides |
| EP0778283A3 (en) * | 1995-12-05 | 1998-01-28 | Pfizer Inc. | Antibiotic macrolides |
| WO2002032916A3 (en) * | 2000-09-25 | 2003-01-16 | Kosan Biosciences Inc | Sixteen-membered macrolide compounds |
| WO2008012343A3 (en) * | 2006-07-28 | 2008-04-17 | Intervet Int Bv | Macrolide synthesis process |
| CN101506220A (en) * | 2006-07-28 | 2009-08-12 | 英特威国际有限公司 | Macrolide synthesis method |
| WO2014187957A1 (en) * | 2013-05-23 | 2014-11-27 | Bayer Animal Health Gmbh | Tylosin derivatives and method for preparation thereof |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5677287A (en) * | 1993-03-18 | 1997-10-14 | Pfizer Inc. | Antibacterial 16-membered ring macrolides containing olefins at C-20 |
| CA2167201A1 (en) * | 1993-07-15 | 1995-01-26 | Pfizer Inc. | Amide derivatives of 16-membered ring antibiotic macrolides |
| US6605599B1 (en) * | 1997-07-08 | 2003-08-12 | Bristol-Myers Squibb Company | Epothilone derivatives |
| ES2213597T3 (en) * | 1999-08-30 | 2004-09-01 | Zaidan Hojin Biseibutsu Kagaku Kenkyu Kai | MACROLID ANTIBIOTICS AND TREATMENT OF PASTEURELOSIS. |
| US6664240B2 (en) | 2001-11-15 | 2003-12-16 | Enanta Pharmaceuticals, Inc. | Tylosin derivatives having antibacterial activity |
| SI1483251T1 (en) | 2002-03-12 | 2010-03-31 | Bristol Myers Squibb Co | C3-cyano epothilone derivatives |
| US7247617B2 (en) * | 2004-07-13 | 2007-07-24 | Kosan Biosciences Incorporated | Sixteen-member macrolide antiinfective agents |
| PT2782922T (en) | 2011-11-25 | 2018-02-05 | The Kitasato Inst | Antibacterial tylosin derivatives and methods for their preparation |
| FR2995605B1 (en) * | 2012-09-18 | 2014-09-19 | Sanofi Sa | MACROLIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION. |
| CN104788519B (en) * | 2014-01-22 | 2018-07-20 | 镇江威特药业有限责任公司 | Sixteen-membered ring triamine lactone derivatives and their applications |
| CN105777828A (en) * | 2014-12-22 | 2016-07-20 | 菏泽市方明制药有限公司 | Method for preparing high-quality tilmicosin through low-quality tylosin |
| US10287275B2 (en) | 2015-04-16 | 2019-05-14 | Victor Pharma Co., Ltd. Zhenjiang | 16-member triamilide derivatives and uses thereof |
| CN117304241B (en) * | 2023-11-30 | 2024-03-01 | 中国农业科学院饲料研究所 | Macrolide compound and preparation method and application thereof |
| CN117510561B (en) * | 2023-11-30 | 2024-04-02 | 中国农业科学院饲料研究所 | Tylosin derivative and preparation method and application thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0103465A1 (en) * | 1982-09-13 | 1984-03-21 | Eli Lilly And Company | 20-Amino macrolide derivatives |
| EP0104028A1 (en) * | 1982-09-13 | 1984-03-28 | Eli Lilly And Company | C-20-modified macrolide derivatives |
| GB2135670A (en) * | 1983-02-28 | 1984-09-05 | Lilly Co Eli | C-20- and c-23-modified macrolide derivatives |
| EP0240264A2 (en) * | 1986-03-31 | 1987-10-07 | Eli Lilly And Company | Improved process for preparing macrolide derivatives |
| EP0262903A2 (en) * | 1986-09-29 | 1988-04-06 | Eli Lilly And Company | New acyl derivatives of 20-modified tylosin and desmycosin compounds |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3975372A (en) * | 1975-02-28 | 1976-08-17 | Schering Corporation | Preparation of 12,13-desepoxy-12,13-dehydrorosamicin |
| JPS59225199A (en) * | 1983-02-10 | 1984-12-18 | Microbial Chem Res Found | 20,23-dideoxy-23-alkyleneimino-20-substituted amino mycaminosylleronolide |
| US4468511A (en) * | 1983-02-28 | 1984-08-28 | Eli Lilly And Company | C-20- And C-23-Modified macrolide derivatives |
| JPS59181299A (en) * | 1983-03-31 | 1984-10-15 | Microbial Chem Res Found | 20,23-dideoxy-20,23-bis-substituted amino-mycaminosylleronolide |
| US4820694A (en) * | 1986-09-29 | 1989-04-11 | Eli Lilly And Company | Modifications of 3-O-demethylmycinose in macrocin and lactenocin |
| US4920103A (en) * | 1986-09-29 | 1990-04-24 | Eli Lilly And Company | Modifications of mycinose and 3-O-demethylmycinose in tylosin-type macrolides |
| FR2638458A1 (en) * | 1988-10-27 | 1990-05-04 | Adir | NOVEL TYLOSIN DERIVATIVES, PROCESS FOR PREPARING THEM AND PHARMACEUTICAL COMPOSITIONS CONTAINING SAME |
-
1993
- 1993-05-25 TW TW082104137A patent/TW226373B/zh active
- 1993-06-07 JP JP6504438A patent/JP2836964B2/en not_active Expired - Lifetime
- 1993-06-07 CZ CZ9599A patent/CZ9995A3/en unknown
- 1993-06-07 DK DK93914300.4T patent/DK0649430T3/en active
- 1993-06-07 CA CA002140243A patent/CA2140243A1/en not_active Abandoned
- 1993-06-07 EP EP93914300A patent/EP0649430B1/en not_active Expired - Lifetime
- 1993-06-07 AT AT93914300T patent/ATE161265T1/en not_active IP Right Cessation
- 1993-06-07 PL PL93307137A patent/PL307137A1/en unknown
- 1993-06-07 SK SK47-95A patent/SK4795A3/en unknown
- 1993-06-07 DE DE69315839T patent/DE69315839T2/en not_active Expired - Fee Related
- 1993-06-07 AU AU44010/93A patent/AU4401093A/en not_active Abandoned
- 1993-06-07 WO PCT/US1993/005210 patent/WO1994002496A1/en not_active Ceased
- 1993-06-07 US US08/362,496 patent/US5545624A/en not_active Expired - Fee Related
- 1993-06-07 KR KR1019950700159A patent/KR950702570A/en not_active Ceased
- 1993-06-07 BR BR9306741A patent/BR9306741A/en not_active Application Discontinuation
- 1993-07-07 IL IL106263A patent/IL106263A0/en unknown
- 1993-07-14 HU HU9302016A patent/HUT65330A/en unknown
- 1993-07-14 UY UY23617A patent/UY23617A1/en unknown
- 1993-07-14 CN CN93108458A patent/CN1083068A/en active Pending
- 1993-07-14 FI FI933204A patent/FI933204A7/en unknown
- 1993-07-15 MX MX9304301A patent/MX9304301A/en unknown
-
1998
- 1998-02-04 GR GR980400232T patent/GR3026064T3/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0103465A1 (en) * | 1982-09-13 | 1984-03-21 | Eli Lilly And Company | 20-Amino macrolide derivatives |
| EP0104028A1 (en) * | 1982-09-13 | 1984-03-28 | Eli Lilly And Company | C-20-modified macrolide derivatives |
| GB2135670A (en) * | 1983-02-28 | 1984-09-05 | Lilly Co Eli | C-20- and c-23-modified macrolide derivatives |
| EP0240264A2 (en) * | 1986-03-31 | 1987-10-07 | Eli Lilly And Company | Improved process for preparing macrolide derivatives |
| EP0262903A2 (en) * | 1986-09-29 | 1988-04-06 | Eli Lilly And Company | New acyl derivatives of 20-modified tylosin and desmycosin compounds |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 102, 1985, Columbus, Ohio, US; abstract no. 185436n, '20,23-Dideoxy-20,23-bis(disubstituted amino)mikamycinosylrelonolide.' page 616 ;column 1 ; * |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2076107A1 (en) * | 1993-09-20 | 1995-10-16 | Pfizer | DERIVATIVES OF MACROLID ANTIBIOTICS OF RINGS OF 16 MEMBERS. |
| WO1996009312A1 (en) * | 1994-09-22 | 1996-03-28 | Pfizer Inc. | Antibiotic macrolides |
| EP0778283A3 (en) * | 1995-12-05 | 1998-01-28 | Pfizer Inc. | Antibiotic macrolides |
| US5760011A (en) * | 1995-12-05 | 1998-06-02 | Pfizer Inc. | Antibiotic macrolides |
| WO2002032916A3 (en) * | 2000-09-25 | 2003-01-16 | Kosan Biosciences Inc | Sixteen-membered macrolide compounds |
| WO2008012343A3 (en) * | 2006-07-28 | 2008-04-17 | Intervet Int Bv | Macrolide synthesis process |
| CN101506220A (en) * | 2006-07-28 | 2009-08-12 | 英特威国际有限公司 | Macrolide synthesis method |
| US8227429B2 (en) | 2006-07-28 | 2012-07-24 | Intervet International B.V. | Macrolide synthesis process and solid-state forms |
| US8263753B2 (en) | 2006-07-28 | 2012-09-11 | Intervet International B.V. | Macrolide synthesis process and solid-state forms |
| CN101506220B (en) * | 2006-07-28 | 2012-10-17 | 英特威国际有限公司 | Synthetic method of macrolide |
| US8461121B2 (en) | 2006-07-28 | 2013-06-11 | Intervet International B.V. | Macrolide synthesis process and solid-state forms |
| US8518900B2 (en) | 2006-07-28 | 2013-08-27 | Intervet International B.V. | Macrolide synthesis process and solid-state forms |
| WO2014187957A1 (en) * | 2013-05-23 | 2014-11-27 | Bayer Animal Health Gmbh | Tylosin derivatives and method for preparation thereof |
| KR20160014656A (en) * | 2013-05-23 | 2016-02-11 | 바이엘 애니멀 헬스 게엠베하 | Tylosin derivatives and method for preparation thereof |
| US9771389B2 (en) | 2013-05-23 | 2017-09-26 | The Kitasako Institute | Tylosin derivatives and method for preparation thereof |
| RU2714482C2 (en) * | 2013-05-23 | 2020-02-18 | Байер Энимэл Хельс ГмбХ | Tylosin derivatives and method for production thereof |
| KR102307218B1 (en) * | 2013-05-23 | 2021-10-05 | 바이엘 애니멀 헬스 게엠베하 | Tylosin derivatives and method for preparation thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| GR3026064T3 (en) | 1998-05-29 |
| US5545624A (en) | 1996-08-13 |
| FI933204L (en) | 1994-01-16 |
| CZ9995A3 (en) | 1995-07-12 |
| DE69315839D1 (en) | 1998-01-29 |
| UY23617A1 (en) | 1994-01-05 |
| FI933204A0 (en) | 1993-07-14 |
| EP0649430A1 (en) | 1995-04-26 |
| SK4795A3 (en) | 1995-07-11 |
| BR9306741A (en) | 1998-12-08 |
| FI933204A7 (en) | 1994-01-16 |
| HUT65330A (en) | 1994-05-02 |
| AU4401093A (en) | 1994-02-14 |
| DK0649430T3 (en) | 1998-02-09 |
| DE69315839T2 (en) | 1998-04-09 |
| PL307137A1 (en) | 1995-05-02 |
| KR950702570A (en) | 1995-07-29 |
| HU9302016D0 (en) | 1993-10-28 |
| JPH07504922A (en) | 1995-06-01 |
| ATE161265T1 (en) | 1998-01-15 |
| CN1083068A (en) | 1994-03-02 |
| MX9304301A (en) | 1994-02-28 |
| CA2140243A1 (en) | 1994-02-03 |
| EP0649430B1 (en) | 1997-12-17 |
| JP2836964B2 (en) | 1998-12-14 |
| IL106263A0 (en) | 1993-11-15 |
| TW226373B (en) | 1994-07-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0649430B1 (en) | Derivatives of 16-membered ring antibiotic macrolides | |
| US5591837A (en) | 5-O-desosaminylerythronolide a derivative | |
| KR100486053B1 (en) | Novel Erythromycin Derivatives, Method for Preparing Same, and Use Thereof as Drugs | |
| EP0708770B1 (en) | Amide derivatives of 16-membered ring antibiotic macrolides | |
| US4440759A (en) | 20-Amino tylosin derivatives and pharmaceutical compositions containing same | |
| US6440942B1 (en) | 14-membered macrolides derived from leucomycins | |
| US5677287A (en) | Antibacterial 16-membered ring macrolides containing olefins at C-20 | |
| EP0124216A1 (en) | C-20- and C-23-modified macrolide derivatives | |
| US4468511A (en) | C-20- And C-23-Modified macrolide derivatives | |
| AU704866B2 (en) | Antibiotic macrolides | |
| US20040235760A1 (en) | 5-O-mycaminosyltylonide derivatives | |
| EP0099747B1 (en) | C-23-modified derivatives of 5-0-mycaminosyl tylonolide | |
| WO1996009312A1 (en) | Antibiotic macrolides | |
| EP0099746B1 (en) | C-23-modified derivatives of macrolide antibiotics | |
| US4401660A (en) | Ester derivatives of 5-O-mycaminosyl tylonolide and method of using same | |
| MXPA96006111A (en) | Macroli antibiotics | |
| EP0262903A2 (en) | New acyl derivatives of 20-modified tylosin and desmycosin compounds | |
| USRE38520E1 (en) | Erythromycin derivatives, method for preparing same, and use thereof as drugs | |
| JPH06247996A (en) | 6-O-methylerythromycin A derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BR CA CZ JP KR NO NZ PL SK US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 253578 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993914300 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08362496 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4795 Country of ref document: SK Ref document number: 2140243 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1995-99 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993914300 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1995-99 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1993914300 Country of ref document: EP |
|
| WWR | Wipo information: refused in national office |
Ref document number: PV1995-99 Country of ref document: CZ |