WO1994007498A1 - Composition pharmaceutique utilisee pour inhiber la production des facteurs de necrose tumorale - Google Patents
Composition pharmaceutique utilisee pour inhiber la production des facteurs de necrose tumorale Download PDFInfo
- Publication number
- WO1994007498A1 WO1994007498A1 PCT/JP1993/001443 JP9301443W WO9407498A1 WO 1994007498 A1 WO1994007498 A1 WO 1994007498A1 JP 9301443 W JP9301443 W JP 9301443W WO 9407498 A1 WO9407498 A1 WO 9407498A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- amino
- tumor necrosis
- general formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/78—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 2
- C07D239/80—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/78—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 2
- C07D239/80—Oxygen atoms
- C07D239/82—Oxygen atoms with an aryl radical attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present invention relates to a pharmaceutical composition for inhibiting tumor necrosis factor production or secretion. More specifically, as a therapeutic agent containing a quinazoline compound or a salt thereof as an active ingredient, and in which tumor necrosis factor is considered to be involved in the pathogenesis, for example, cachexia, sepsis, and multiple organ failure. And an effective pharmaceutical composition for inhibiting tumor necrosis factor production or secretion.
- Tumor necrosis factor is a peptide consisting of 157 amino acids and having a molecular weight of about 17,000, including macrophages. Is one of the site cytokines produced from various cells.
- TNF was initially found to be a cytokine with tumor-damaging effects, but subsequent studies have shown that its effects extend to many normal cells in addition to tumor cells. It is clear that For example, inhibition of lipoprotein lipase activity of fat globules, expression of HLA antigens on vascular endothelial cells and fibroblasts, production of interleukin-1 in fibroblasts or macrophages, tumors Activation of injured macrophages, suppression of CFU, fibroblasts, endothelial cells, Production of colony stimulating factor in certain types of tumor cells, inhibition of cartilage synthesis and absorption of proteoglycan, activation of neutrophils and generation of superoxide, production of procoagulant factor in vascular endothelial cells , proliferation of fibroblasts, changes in membrane potential of the skeletal muscle, fibroblasts Lee printer monounsaturated Russia down of - production of / 5 2, etc.
- TNF has been reported to be the same substance as cachectin, a trigger of cachexia in cancer and infectious diseases, which promotes catabolic metabolism in the whole body and leads to extreme depletion.
- anti-TNF antibodies have also been shown to inhibit sepsis (Starnes, HF Jr., Pearce, K., Tewari, Yim, JH, Zou, J-, Abrams, JS, J. Immunol. ⁇ 45, 4 18 5-4 19 1 (199 0), Beutler, B., Mi 1 sark, IW, Cerami, A., Science, 229, 86 9-8 7 1 (1 9 8 5),
- TNF inhibitors that can be used as therapeutic agents for such pathological conditions are desired.
- pentoxifylline having a methylxanthine skeleton As a compound having a TNF inhibitory action, for example, pentoxifylline having a methylxanthine skeleton is known.
- This compound has lethal protective activity in endotoxin shock model mice, improves mood and suppresses weight loss in patients with severe pulmonary tuberculosis, and inhibits cancer patients.
- Have been reported to improve mood and suppress weight loss Zaabel, P., Schade, FU, Schlaak, M., Immunobiol., 187, 447-463 ( 1 9 9
- TNF inhibitory activity is a factor, the conventional I Ri glucoside Koruchikoi de, protease inhibitors, off O Suho lipase A 2 inhibitors, Li Pokishigenaze inhibitors, PAF (platelet aggregating factor) antagonists, La Jikarusu force base emissions suicide over, prostaglandin Grad down di emissions F 2 or I 2, etc. anti-TNF antibody is known. It is expected that the relationship between TNF and disease state will be further clarified using such small molecules or antibodies. However, these compounds have side effects due to their wide variety of pharmacological effects. Therefore, there is a demand for the development of a safer low molecular weight compound by a new mechanism of action.
- the present invention provides a disease in which TNF is considered to be involved in the onset of TNF through its TNF production or secretion inhibitory action, such as cachexia, sepsis, multiple organ failure, and rheumatoid arthritis. Ulcerative colitis, Behcet's disease, systemic lupus erythematosus (SLE), bone marrow transplant rejection (GvHD), multiple organ failure, malaria, acquired immunodeficiency syndrome (AIDS), It provides remedies for meningitis, fulminant hepatitis, Bowl's disease, etc.
- the compound used as an active ingredient in the present invention is a known compound, for example, as described in the literature Chem. Pharm. Bull., 29
- the present inventors have found that a quinazoline compound has strong TNF production or secretion inhibitory activity, and have completed the present invention.
- the object of the present invention is to provide a compound represented by the general formula (1)
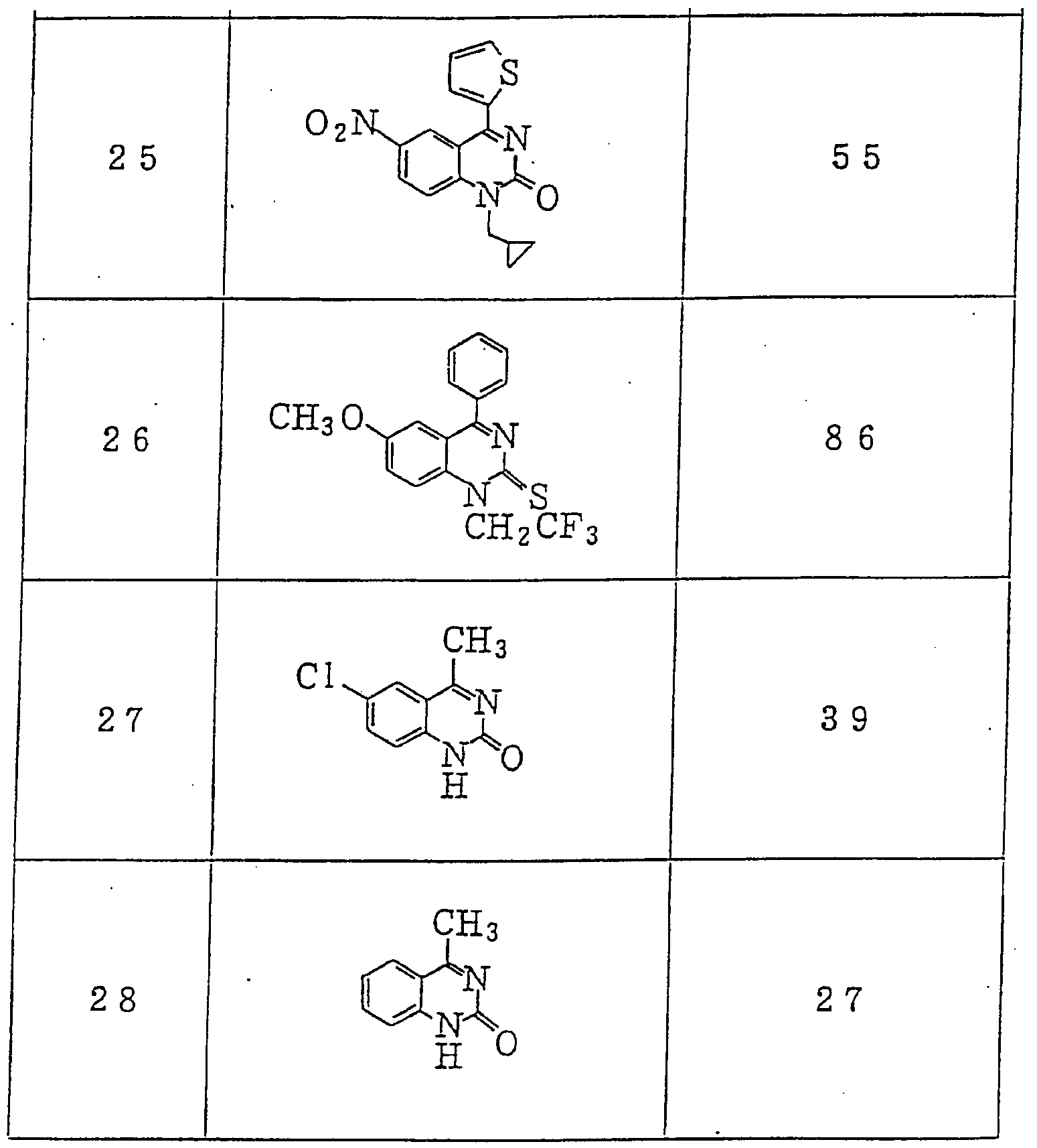
- R 1 is a hydrogen atom, a halogen atom, a hydroxy group, an amino group, a substituted amino group, a nitro group, a cyano group, an alkyl group, an alkoxy group, an alkylthio group.
- R 2 is a phenyl group, a substituted phenyl group, a phenyl group, a furyl group, a cycloalkyl group, a cycloalkenyl group, an alkyl group, an amino group or a substituted amino group
- R 3 represents a hydrogen atom
- R 4 represents a hydrogen atom or an alkyl group
- R 5 , R 6 R 7 represents an oxo group or a thioxo group when R 5 and R 6 are taken together, and R 7 is a hydrogen atom, an alkyl group, a peralkyl group, or a phenyl group.
- a substituted phenyl group or a formula—X—Y (wherein X represents an alkylene group, and Y represents a cycloalkyl group, a phenyl group, a substituted phenyl group, a carboxyl group, Represents a droxy group, an alkoxy group, an aminocarbonyl group, a substituted aminocarbonyl group, an alkoxycarbonyl group, an amino group or a substituted amino group.) other will table Wa binding R 6 and R 7 are One Do together, R.
- R 1 R 2 RR 4 has the same meaning as described above.
- An object of the present invention is to provide a pharmaceutical composition for inhibiting the production or secretion of tumor necrosis factor, comprising a compound represented by the formula or a salt thereof as an active ingredient.
- Still another object of the present invention is to produce a tumor necrosis factor containing a compound represented by the above general formula (1) or the above general formula (2) or a salt thereof as an active ingredient.
- An object of the present invention is to provide a use of the compound or a salt thereof for producing a secretion inhibitory pharmaceutical composition.
- FIG. 1 is a graph showing the results of a galactosamine-loaded mouse-end toxic shock model lethality inhibition test in Example 2 of the present invention.
- FIG. 2 is a graph showing the characteristics of the third embodiment of the present invention.
- 7 is a graph showing the results of a TNF production inhibition test in a load mouse toxin shock model.
- the horizontal axis is LPS, D-ga1N and 5% dimethylsulfoxide 10% nickel solution (control) or LPS, D-ga1N and 3 types 3 shows the elapsed time after administration of the solution of the compound of the compound number 33 in Example 1.
- the vertical axis indicates the TNF activity in the serum of the mouse (3 mice each) collected at each elapsed time. TNF activity (U / ml) is the average value of the measured values of the serum of 3 mice. And standard deviation (s.d.).
- Examples of the alkyl group include a lower alkyl group
- examples of the alkoxy group include a lower alkoxy group
- examples of the alkylthio group include a lower alkylthio group
- examples of the acyl group include
- a lower alkanol or aryl group is an alkoxycarbonyl group
- a lower alkoxycarbonyl group is, for example, a cycloalkyl group is, for example, a lower cycloalkyl group.
- the lower alkyl group is, for example, a lower cycloalkenyl group as a cycloalkenyl group
- the lower alkyl group is, for example, a haloalkyl group
- the lower alkyl group is, for example, a lower alkyl group as an alkylene group.
- the alkylene group is an alkylhydrazino group, for example, a lower alkylhydrazino group is an acylhydrazino group.
- Examples of the no group include a lower alkanol hydrazino group
- examples of the acylamino group include a lower alkanoylamino group.
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom.
- Examples of the lower alkyl group include a methyl group, an ethyl group, a propyl group, a butyl group, a pentyl group, a hexyl group, an 111-methylethyl group, a 1-methylpropyl group, and a 2-methylethyl group.
- Pill group 1-ethylpropyl group, 2-ethylpropyl group, 1-methylbutyl group, 2—methylbutyl group, 3—methylbutyl group, 1-ethylbutyl group, 2-ethylbutyl group, 3-ethylbutyl group, 1— Examples thereof include an alkyl group having 1 to 6 carbon atoms such as a methylpentyl group, a 2-methylpentyl group, a 3-methylpentyl group, and a 4-methylpentyl group.
- Examples of the lower alkoxy group include an alkoxy group having 1 to 6 carbon atoms such as a methoxy group, an ethoxy group, a propoxy group and a butoxy group. .
- Examples of the lower alkylthio group include an alkylthio group having 1 to 6 carbon atoms such as a methylthio group, an ethylthio group, a propylthio group and a butylthio group.
- Examples of the lower alkanol group include alkanol groups having 1 to 6 carbon atoms such as a formyl group, an acetyl group, a propanol group, and a butyl group.
- aryl group examples include, for example, an aryl group having 11 or less carbon atoms.
- a lower alkoxycarbonyl group such as a benzoyl group, a 11-naphthyl group or a 2-naphthyl group.
- alkoxycarbonyl groups having 1 to 6 carbon atoms such as methoxycarbonyl, ethoxycarbonyl, propoxypropyl, butoxycarbonyl, and the like.
- a cycloalkyl group having 3 to 6 carbon atoms such as a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc.
- an alkyl group such as a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc.
- an alkyl group for example, a cycloalkyl group having 3 to 6 carbon atoms such as a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl group, etc.
- an alkyl group for example, a cycloalkyl group having 3 to 6 carbon atoms such as a cyclopropyl group, a cyclobutyl group, a cyclopentyl group, a cyclohexyl
- Examples of the lower cycloalkenyl group include cycloalkenyl having 3 to 6 carbon atoms, such as 1-cyclohexenyl, 2-cyclohexenyl, and 3-cyclohexenyl. Groups are listed.
- halo-lower alkyl group examples include a trifluoromethyl group, a 1,1,1—trifluoroethyl group, a 1,1,1,1,2,2—a penfluorofluoroethyl group, and the like. And a haloalkyl group having 1 to 6 carbon atoms, which is substituted by a hydrogen atom.
- Examples of the substituted phenyl group include an alkyl group, an alkoxy group, a halogen atom, an alkylthio group, and a phenyl group substituted with an acyl group.
- Examples of the phenyl group include a 2-phenyl group and a 3-phenyl group.
- Examples of the furyl group include a 2-furyl group and a 3-furyl group.
- Lower alkylene groups include, for example, methylene, ethylene, trimethylene, tetramethylene, 1-methylene, 2-methylene Ruethylene, 1, 2—Dimethylethylene, 1-ethylethylene, 2_ethylethylene, 1—Methyltrimethylene, 2—Methyltriene Examples thereof include alkylene groups having 1 to 4 carbon atoms, such as methylene and 3 -methyltrimethylene.
- Examples of the lower alkylhydrazino group include N—methylhydrazino group, N—ethylhydrazino group, and N—propylhydrazino group.
- Examples of the lower alkyl hydrazino group include N—holmyl hydrazino group, N—acetyl hydrazino group, N—prono, . Nylhydrazino group, N—Busylhydrazino group, N'—Holmilhydrazino group, N'—Acetylhydrazino group Group, N'-propanol group, N'-group
- An alkanol hydrazino group having a lower alkanol group having 1 to 6 carbon atoms such as a tanyl hydrazino group is exemplified.
- Examples of the substituted aminocarbonyl group include an aminocarbonyl group in which one or two alkyl groups have been substituted, and examples of the substituted alkyl group include a lower alkyl group.
- Examples of the substituted aminocarbonyl group include an aminocarbonyl group in which one or two alkyl groups have been substituted, and examples of the substituted alkyl group include a lower alkyl group.
- a dialkylamine having two alkyl groups which may be the same or different from each other and have 1 to 6 carbon atoms, such as a rubonyl group, a acetylaminocarbonyl group, and an ethylmethylaminocarbonyl group. And a nocarbonyl group.
- the substituted aminocarbonyl group includes a cyclic aminocarbonyl group
- the cyclic aminocarbonyl group includes, for example, a 5-membered ring or a 6-membered ring containing a nitrogen atom.
- a cyclic amino carbonyl group having 5 to 6 carbon atoms which is a membered ring More specifically, examples include a 1-pyrrolidinocarbonyl group, a 1-piperidinocarbonyl group, and the like.
- Examples of the substituted amino group include an amino group in which one or two alkyl groups have been substituted, and examples of the substituted alkyl group include a lower alkyl group. More specifically, for example, an alkylamino group having an alkyl group having 1 to 6 carbon atoms, such as a methylamino group, an ethylamino group, a dimethylamino group, a getylamino group, and an ethyl group Even if it is the same with 1 to 6 carbon atoms such as methylamino group, And a dialkylamino group having two alkyl groups. Further, the substituted amino group includes a cyclic amino group.
- Examples of the cyclic amino group include carbon atoms in which the ring containing a nitrogen atom is a 5- or 6-membered ring.
- the cyclic amino group of the number 4 to 5 is exemplified. More specifically, examples include a 11-pyrrolidino group and a 1-piperidino group.
- Examples of the lower alkanoamino group include a formylamino group, an acetylamino group, and a prono group. Examples thereof include alkanolamino groups having 1 to 6 carbon atoms, such as a nutylamino group and a bushylamino group.
- heterocyclic structure having 2 to 4 nitrogen atoms examples include a 5-membered ring having an oxo group or an alkyl group on the ring.
- R 8 represents a hydrogen atom or an alkyl group
- R 3 and R 4 together represent a bond
- R 2 is a phenyl group, a substituted phenyl group, A compound or a salt thereof, which is a phenyl group, a furyl group, a cycloalkyl group or a cycloalkenyl group, or a compound of the formula (1) in which R 6 and R 7 are R 5 represents a amino group, a hydroxyamino group, a hydrazino group, an alkylhydrazino group, an acylhydrazino group, or A compound having an acylamino group or a salt thereof is preferred.
- Examples of the salt of the quinazoline compound included in the present invention include salts with mineral acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, and phosphoric acid, formic acid, acetic acid, and fumaric acid. , Maleic acid, lingic acid, tartaric acid, salts with organic carboxylic acids such as aspartic acid or glutamic acid, methansulfonate, benzenesulfonate, p — Salts with sulfonates such as toluenesulfonate, hydroxybenzensulfonate or dihydroxybenzensulfonate, sodium or potassium urea Salts with alkaline metals, salts with alkaline earth metals such as cane or magnesium, trimethylammine, triethylammine, or pyridine Salt or ammonia with an organic base such as Salt and the like.
- mineral acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, and phosphoric acid, formic acid
- the compound of the present invention includes optically active forms and tautomers, and further includes all hydrates and crystal forms.
- the compound of the present invention is a known compound and can be synthesized, for example, by the following method.
- (A) 3,4—Dihydro 2 (1H) -quinazolinone derivative can be synthesized, for example, by the following method.
- the compound represented by the following general formula (5) is obtained by, for example, dehydrating and condensing a compound represented by the following general formula (3) and an aldehyde represented by the following general formula (4) in an inert solvent in the presence of an acid.
- it can be synthesized (Japanese Patent Application Laid-Open Nos. 52-51739, JP-B-49-47979, JP-A-48-34998, No. 7-728, No. 47-No. 3 295).
- the compound represented by the general formula (3) can be synthesized, for example, by the method described in the above publication.
- R 1 and R 7 have the same meanings as described above, and R 1 ′ represents a phenyl group, a substituted phenyl group, a phenyl group, a furyl group, a cycloanoleyl group, Represents a chloroalkenyl group or an alkyl group, and X 1 represents an oxygen atom or a zeo atom.
- the compound represented by the following general formula (17) can be synthesized, for example, by reducing the compound represented by the following general formula (16) (Japanese Patent Application Laid-Open No. Sho. P arm. Bui 1., _2_9_ (8) 2 1 3 5-2 1 5 6 (1 9 8 1)).
- the compound represented by the general formula (16) can be synthesized, for example, by the method described in the above publication.
- R 1 , R 7 , and R 11 have the same meanings as described above, R 1 ° represents a hydrogen atom or an alkyl group, and X 2 represents a halogen atom.
- the compound represented by the following general formula (6) can be synthesized, for example, by a method of oxidizing a compound represented by the following general formula (5) in an inert solvent using an oxidizing agent (particularly).
- Japanese Patent Publication Nos. 51-82 287, 52-71 483, and JP-B 47-48 396 Japanese Patent Publication Nos. 51-82 287, 52-71 483, and JP-B 47-48 396.
- the compound represented by the following general formula (8) is, for example,
- the compound represented by the general formula (7) can be synthesized by a method of reacting with chlorosulfonylysocyanate, urea or tiorea in an inert solvent. (Synthetic Communications, 10 (10), 799-804 (1980), U.S. Pat. No. 3,055,553).
- the compound represented by the general formula (7) can be synthesized, for example, by the method described in the above publication.
- the compound represented by the following general formula (19) can be obtained, for example, by reacting a compound represented by the following general formula (18) in an inert solvent with phenyllithium, phenylmagnesium hydroxide, methylphenyl Reacts with nucleophiles such as organometallic reagents such as zinc and methylmagnesium hydride, or amide reagents such as ammonia, hydroxymine and hydrazine. It can be synthesized by the method (Japanese Patent Publication No. 48-21956).
- the compound represented by the general formula (18) can be synthesized, for example, by the method described in the above publication.
- the 2-substituted quinazoline derivative can be synthesized, for example, by the following method.
- the compound represented by the following general formula (10) can be obtained, for example, by reacting a compound represented by the following general formula (9) with a nucleophile such as ammonia, hydroxyamine, hydrazine or the like in an inert solvent. It can be synthesized by reacting (US Pat. No. 3,355,533, Japanese Patent Application Laid-Open No. 51-1998, and Japanese Patent Publication No. 45-150). No. 2 2 1 3 5).
- the compound represented by the general formula (9) can be synthesized, for example, by the method described in the above publication.
- the compound represented by (20) can be synthesized by reacting with ammonia in an inert solvent (Ber. Deut. Chem. Ges., 26, 1384-1399 ( Ber. 98, 1049-1059 (1965), Chem. Parm. Bull. 26 (6) 16 33-1651 (1978)).
- the compound represented by the general formula (20) can be synthesized, for example, by the method described in the above publication.
- R 1 and R 11 have the same meanings as described above, and X 4 represents a hydrogen atom, an alkyl group, or a trifluoromethyl group.
- a tetrazoquinazolin derivative represented by the following general formula (11) is obtained by reacting a compound represented by the following general formula (9) with sodium azide in an inert solvent.
- the compound can be synthesized (JP-A-53-12893).
- An imidazoquinazoline derivative (12) represented by the following general formula (12) is synthesized by reacting a compound represented by the following general formula (22) in an inert solvent in the presence of an acid.
- the compound represented by the general formula (22) can be synthesized, for example, by the method described in the above publication.
- a triazoquinazoline derivative represented by the following general formula (14) is prepared by heating a compound represented by the following general formula (13) in an inert solvent in the presence or absence of an acid.
- the compound can be synthesized by reacting (Japanese Patent Application Laid-Open No. 51-1998).
- the compound represented by the general formula (13) can be synthesized, for example, by the method described in the above publication.
- the substituent R 1 or R 7 has a functional group such as an amino group, a substituted amino group, a hydroxy group, etc.
- the above reaction is performed, and then the target compound can be synthesized by deprotection.
- the protecting group that can be used include an amino group, a substituted amino group includes, for example, an alkenyl group such as an acetyl group and a benzyl group, and an aryl group.
- the hydroxy group is, for example, an alkenyl group such as an acetyl group or a benzoyl group, or an aryl group, a benzyl group, a methyl group, a methoxymethyl group. Or trimethylsilyl group (TW Greene, "Protective Groups in Organic Synthesis, John Wiley & Sons Inc., 1989”) o
- the active ingredient of the present invention can be administered orally or parenterally. That is, it can be orally administered in the form of commonly used dosage forms, for example, tablets, capsules, syrups, suspensions, etc., or in the form of liquids, such as solutions, emulsions, suspensions, etc. It can be administered as an injection. It can also be administered rectally in the form of suppositories.
- Such a dosage form can be produced according to a general method by mixing an active ingredient with a usual carrier, excipient, binder, stabilizer and the like. When used in an injection preparation, a buffer, a solubilizing agent, an isotonic agent and the like can be added.
- the active ingredient is usually 10 to 500 mg per adult per day.
- the active ingredient can be administered in 1 to 100 mg once or in several divided doses.
- Tablets can be produced, for example, by the following method. Amount (mgZ tablet) Test Example 1 described below Compound No. 10
- An injection can be produced, for example, as follows.
- Physiological saline solution 10 ml The solution of the above components is sterilized by filtration, filled into a washed and sterilized vial bottle, sealed with washed and sterilized rubber, and fastened with a flip-off cap. To produce injections.
- mice (5 weeks old, female, Charles Rino K.K.) were intraperitoneally administered 3% thioglycollate medium 1 and 4 days after rearing for 4 days (final concentration 5 UZ)-Minimum essential medium (hereinafter abbreviated as MEM) supplemented with fetal bovine serum (FBS, GIBC0, 1% final concentration), Osaka University
- MEM fetal bovine serum
- FBS fetal bovine serum
- PEC peritoneal infiltrating cells
- the powder of the TNF production inhibitor of the present invention is dissolved in dimethyl sulfoxide to a concentration of 30 mM, and FBS (final concentration of 10%) to a final concentration of 30 M or 30 ⁇ M. ) was added and diluted with MEM to which the solution had been added, and 5001 of each gel was added to the above intraperitoneal macrophage to give a total volume of 1001. Further, a lipopolysaccharide slide (hereinafter, abbreviated as LPS, E. coli 0111B4, manufactured by DIFCOUSA) was added to each well to a final concentration of 10 ⁇ gZ. 1 0 0 ⁇ Added in 1, 3 7 ° (, 5 % C 0 2 presence, after 1 8 h culture, supernatants were harvested 2 5 a 1 in each ⁇ El.
- LPS lipopolysaccharide slide
- the TNF activity in the collected supernatant was measured in a bioassay using a TNF-sensitive mouse fibroblast cell line L9299. That is, MEM containing FBS (final concentration: 10%) was added to 96-well microplates at a ratio of 1001 to each well, and 25 pi 1 of the collected supernatant was used as a 5-fold series. The final concentration (the concentration after the addition of the next L929 cell solution) was adjusted to 10%, 2% .0.4%, and 0.08%.
- the MTT method developed by Monosann et al. (Onosann, T., J. Immunol.Method, 65, 55-63, 19983) ) was used to measure the number of living cells.
- TNFa (TNF-M, manufactured by Genzyme) was used as a standard substance, and it was determined as a unit (U) / 7 ⁇ from a calibration curve of absorbance for TNF activity obtained.
- the TNF production inhibitory activity of each compound was determined according to the following formula.
- TNF production inhibitory activity (TNF activity in culture supernatant of 1-compound-added group) TNF activity in culture supernatant of non-compound-added group) X 100
- D-galactosamine hydrochloride manufactured by Nakarai Tesk Co., Ltd .; hereinafter, abbreviated as D-gaIN
- LPS LPS
- the final concentration of the hydrochloride of the compound of Compound No. 33 of Example 1 in an aqueous solution of 5% dimethyl sulfoxide 10% nickel manufactured by Nihon Safa Factant Industry Co., Ltd.
- the components were dissolved at a concentration of 0, 0.25, 0.5, and 1 mgZ.
- Test Example 1 The hydrochloride salt of Compound No. 33 was used as an endotoxin for galactosamin-loaded mice at a dose of 5 mgZ kg or more. Shock lethality was significantly suppressed (Student t test was performed between the compound non-administration group and the compound administration group). Test example 3
- D-ga1N and LPS were dissolved in water such that the final concentrations were 75 mg ⁇ and 0.2 ⁇ gZ, respectively.
- the hydrochloride of Compound No. 33 of Test Example 1 was added to a 10% aqueous solution of 5% dimethyl sulfoxide at a final concentration of 0.5, 1, 2.5 mg / ⁇ . Dissolved.
- mice 63 mice divided into 4 groups of 18, 15, 15, 15 and 15 animals (group A, B, C, D in order), D_ga IN and LPS in all groups was administered intravenously at a dose of 200/1 per 20 g body weight, and immediately thereafter, in group A, a 5% dimethylsulfoxide- 10% solution in Nicol was used as a control.
- groups B, C, and D the hydrochlorides of the compound of Compound No. 33 of Test Example 1 described above were 0.5, 1,
- a 2.5 mg / rnS solution was administered intravenously at 200 g per 20 g body weight.
- Group A then received 3% each immediately after administration of the 5% dimethyl sulfoxide—10% nickel solution and at 0.5, 1, 1.5, 3, and 5 hours later. Blood was collected from each mouse by cardiac blood sampling, and the serum was separated. In groups B, C, and D, as in group A, 0.5, 1, 1.5,
- TNF activity in the collected serum was measured in a bioassay using L929 cells by the same method as in Example 1. That is, the above serum was diluted in a 3-fold dilution series using MEM supplemented with FBS (final concentration 10%) in a test tube, and the final concentration (the concentration after the addition of the next L929 cell solution) was determined. 5 1.7, 0.6, and 0.2%. The diluted serum was added to a 96-well microphone plate at a rate of 1001 per 1 ⁇ l.
- FBS final concentration: 10%
- actinomycin D final concentration: 1 gZ
- test Example 1 Compound No. 33
- the hydrochloride salt of compound 3 significantly inhibited the increase in blood TNF level in a dose-dependent manner at a dose of 1 mgZ kg or more (Student t test ) was performed between the TNF activity of the control group and the compound-treated group at the same elapsed time).
- Industrial applicability The compound represented by the general formula (1) or the general formula (2) has an effect of significantly inhibiting TNF production or secretion. Therefore, these compounds are effective as therapeutic agents for diseases in which TNF is considered to be involved in the pathogenesis, such as cachexia, sepsis, and multiple organ failure.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description






















Claims


Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/411,595 US5646154A (en) | 1992-10-07 | 1993-10-07 | Pharmaceutical compositions for inhibiting the formation of tumor necrosis factor |
| EP93922048A EP0664128A4 (en) | 1992-10-07 | 1993-10-07 | PHARMACEUTICAL COMPOSITION USED TO INHIBIT THE PRODUCTION OF TUMOR NECROSIS FACTORS. |
| CA002146126A CA2146126A1 (en) | 1992-10-07 | 1993-10-07 | Pharmaceutical compositions for inhibiting the formation of tumor necrosis factor |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP29645792 | 1992-10-07 | ||
| JP4/296457 | 1992-10-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994007498A1 true WO1994007498A1 (fr) | 1994-04-14 |
Family
ID=17833803
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1993/001443 Ceased WO1994007498A1 (fr) | 1992-10-07 | 1993-10-07 | Composition pharmaceutique utilisee pour inhiber la production des facteurs de necrose tumorale |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US5646154A (ja) |
| EP (1) | EP0664128A4 (ja) |
| CA (1) | CA2146126A1 (ja) |
| WO (1) | WO1994007498A1 (ja) |
Families Citing this family (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR19990044618A (ko) * | 1995-09-13 | 1999-06-25 | 다케다 야쿠힌 고교 가부시키가이샤 | 면역억제제 |
| US6096336A (en) * | 1996-01-30 | 2000-08-01 | The Stehlin Foundation For Cancer Research | Liposomal prodrugs comprising derivatives of camptothecin and methods of treating cancer using these prodrugs |
| AU3130597A (en) | 1996-05-16 | 1997-12-05 | Duke University | Tristetraprolin |
| CZ296959B6 (cs) * | 1997-04-25 | 2006-08-16 | Janssen Pharmaceutica N. V. | Chinazolon, zpusob a meziprodukt pro jeho výrobu a farmaceutický prostredek s jeho obsahem |
| JPH11209350A (ja) | 1998-01-26 | 1999-08-03 | Eisai Co Ltd | 含窒素複素環誘導体およびその医薬 |
| US6184246B1 (en) | 1999-07-30 | 2001-02-06 | The United States Of America As Represented By The Secretary Of Agriculture | Inhibition of cytokine production by polymethoxylated flavones |
| US6228855B1 (en) | 1999-08-03 | 2001-05-08 | The Stehlin Foundation For Cancer Research | Aromatic esters of camptothecins and methods to treat cancers |
| DE19951702A1 (de) * | 1999-10-27 | 2001-05-03 | Aventis Pharma Gmbh | Verwendung von 2-Amino-3,4-dihydro-chinazolinen zur Herstellung eines Medikaments zur Behandlung oder Prophylaxe von durch ischämischen Zuständen bewirkten Krankheiten |
| US6808902B1 (en) | 1999-11-12 | 2004-10-26 | Amgen Inc. | Process for correction of a disulfide misfold in IL-1Ra Fc fusion molecules |
| DE60118953T2 (de) * | 2000-11-28 | 2007-01-11 | Janssen Pharmaceutica N.V. | Farnesyl-protein-transferasehemmer zur behandlung der entzündlichen darmerkrankung |
| NZ530765A (en) | 2001-06-26 | 2006-11-30 | Amgen Fremont Inc | Antibodies that bind OPGL and compositions and methods for the treatment of bone diseases |
| WO2004060911A2 (en) | 2002-12-30 | 2004-07-22 | Amgen Inc. | Combination therapy with co-stimulatory factors |
| CA2524221A1 (en) * | 2003-04-30 | 2004-11-18 | The Institutes For Pharmaceutical Discovery, Llc | Substituted heteroaryls as inhibitors of protein tyrosine phosphatases |
| DE102005022977A1 (de) * | 2005-05-19 | 2006-12-07 | Merck Patent Gmbh | Phenylchinazolinderivate |
| KR101486090B1 (ko) * | 2006-11-22 | 2015-01-23 | 스미또모 가가꾸 가부시끼가이샤 | 사이토카이닌 신호전달을 저해할 수 있는 물질 |
| CA2993967A1 (en) | 2008-07-31 | 2010-02-04 | Senomyx, Inc. | Processes and intermediates for making sweet taste enhancers |
| HUE042011T2 (hu) | 2010-02-11 | 2019-06-28 | Celgene Corp | Arilmetoxi-izoindolin-származékok, azok készítményei és alkalmazási módszereik |
| WO2015120059A1 (en) * | 2014-02-04 | 2015-08-13 | Yates Charles Ryan | Inhibitors of paxillin function and related compositions and methods |
| WO2017149469A1 (en) | 2016-03-03 | 2017-09-08 | Emcure Pharmaceuticals Limited | Heterocyclic compounds useful as ido and/or tdo modulators |
| CN109111426B (zh) * | 2017-06-23 | 2021-12-14 | 中国科学院上海药物研究所 | 一类稠合双环杂芳基或芳基化合物,及其用途 |
| JP7453210B2 (ja) | 2018-08-07 | 2024-03-19 | フィルメニッヒ インコーポレイテッド | 5-置換4-アミノ-1H-ベンゾ[c][1,2,6]チアジアジン2,2-ジオキシド並びにその配合物及び使用 |
| MX2021006841A (es) | 2018-12-10 | 2021-07-02 | Ideaya Biosciences Inc | Derivados de 2-oxoquinazolina como inhibidores de metionina adenosiltransferasa 2?. |
| SI4081305T1 (sl) * | 2019-12-24 | 2025-03-31 | Carna Biosciences, Inc. | Spojine, ki modulirajo diacilglicerol kinazo |
| WO2021252680A1 (en) * | 2020-06-10 | 2021-12-16 | Ideaya Biosciences, Inc. | 4-arylquinazoline derivatives as methionine adenosyltransferase 2a inhibitors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5394040A (en) * | 1976-12-06 | 1978-08-17 | Sandoz Ag | Coagolation controlling agent for blood platelet |
| JPS5888369A (ja) * | 1981-10-21 | 1983-05-26 | サノフイ・ソシエテ・アノニム | 新規な4−フエニルキナゾリン誘導体、その製法及びその医薬品としての利用 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3305553A (en) * | 1965-10-18 | 1967-02-21 | Parke Davis & Co | 2-aminoquinazoline derivatives |
| IL102764A0 (en) * | 1991-08-16 | 1993-01-31 | Merck & Co Inc | Quinazoline derivatives,and pharmaceutical compositions containing them |
| WO1993004047A1 (en) * | 1991-08-16 | 1993-03-04 | Merck & Co., Inc. | Quinazoline derivatives as inhibitors of hiv reverse transcriptase |
-
1993
- 1993-10-07 EP EP93922048A patent/EP0664128A4/en not_active Withdrawn
- 1993-10-07 CA CA002146126A patent/CA2146126A1/en not_active Abandoned
- 1993-10-07 WO PCT/JP1993/001443 patent/WO1994007498A1/ja not_active Ceased
- 1993-10-07 US US08/411,595 patent/US5646154A/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5394040A (en) * | 1976-12-06 | 1978-08-17 | Sandoz Ag | Coagolation controlling agent for blood platelet |
| JPS5888369A (ja) * | 1981-10-21 | 1983-05-26 | サノフイ・ソシエテ・アノニム | 新規な4−フエニルキナゾリン誘導体、その製法及びその医薬品としての利用 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0664128A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0664128A4 (en) | 1997-12-17 |
| CA2146126A1 (en) | 1994-04-14 |
| US5646154A (en) | 1997-07-08 |
| EP0664128A1 (en) | 1995-07-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1994007498A1 (fr) | Composition pharmaceutique utilisee pour inhiber la production des facteurs de necrose tumorale | |
| JP3199265B2 (ja) | N▲上6▼−(ヒドラジノイミノメチル)リジン及び生体における一酸化窒素形成の阻止法 | |
| AU2014277506B2 (en) | Bisulfate of Janus kinase (JAK) inhibitor and preparation method therefor | |
| TW200402422A (en) | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| JP5279987B2 (ja) | アミド誘導体 | |
| KR20010083874A (ko) | 약제용 퀴나졸린 유도체 | |
| JP3712209B2 (ja) | 短時間作用型ジヒドロピリジン類 | |
| TW200413370A (en) | Methods for effecting chiral salt resolution from racemic mixtures of enantiomers used in making pyrrolo[2,3-d]pyrimidine compounds | |
| KR100354654B1 (ko) | 피리미디논 화합물, 이를 함유하는 약제학적 조성물 및이의 제조 방법. | |
| NZ615228A (en) | Protein kinase inhibitors | |
| CN1353690A (zh) | 缓激肽受体拮抗剂 | |
| RU2350354C2 (ru) | ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ АНТАГОНИСТ Р2Х7 РЕЦЕПТОРА И ФАКТОР НЕКРОЗА ОПУХОЛИ α | |
| CA2196215C (en) | 1,4-dihydropyridine compound and pharmaceutical composition containing the same | |
| JP2018538355A (ja) | ベンズアミド誘導体 | |
| JP2008179554A (ja) | ピロール縮合モルヒナン誘導体およびその医薬用途 | |
| JPH0688973B2 (ja) | 1,4−ジヒドロピリジン誘導体 | |
| JP4548884B2 (ja) | 4,5,6,7−テトラヒドロチエノ〔2,3−c〕ピリジン誘導体 | |
| JPH06192099A (ja) | 腫瘍壊死因子産生阻害剤 | |
| US5648359A (en) | Tumor necrosis factor production inhibitors | |
| JP4548882B2 (ja) | 4,5,6,7−テトラヒドロチエノ〔2,3−c〕ピリジン化合物 | |
| JPWO2004039806A1 (ja) | 複素環化合物 | |
| JPS6245864B2 (ja) | ||
| TW577877B (en) | Improved methods of preparing 4-cyano-4-(substituted indazole)cyclohexane-carboxylic acids useful as PDE4 inhibitors | |
| WO2025256633A1 (en) | Method of treating moderate to severe active rheumatoid arthritis | |
| US5760053A (en) | γ-Diketone compounds with inhibitory activity against platelet aggregation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2146126 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993922048 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08411595 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1993922048 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1993922048 Country of ref document: EP |