WO1996026930A1 - Process for the preparation of 3-(substituted phenyl)-5-alkylidene-1,3-oxazolidine-2,4-dione derivatives - Google Patents
Process for the preparation of 3-(substituted phenyl)-5-alkylidene-1,3-oxazolidine-2,4-dione derivatives Download PDFInfo
- Publication number
- WO1996026930A1 WO1996026930A1 PCT/JP1996/000430 JP9600430W WO9626930A1 WO 1996026930 A1 WO1996026930 A1 WO 1996026930A1 JP 9600430 W JP9600430 W JP 9600430W WO 9626930 A1 WO9626930 A1 WO 9626930A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- carbon atoms
- alkyl group
- general formula
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/30—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D263/34—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/44—Two oxygen atoms
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/06—Halogens; Compounds thereof
- B01J27/128—Halogens; Compounds thereof with iron group metals or platinum group metals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/31—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by introduction of functional groups containing oxygen only in singly bound form
Definitions
- the present invention relates to a method for producing a 3-substituted funinyl-5-alkylidene-1,3-oxazolidin-2,4-dione derivative having a strong herbicidal effect.
- a substituted phenylisocyanate unit and a 2-hydroxy-3-argenate ester are used as a method for producing a 3-substituted phenyl-5-alkylidene-1,3-oxazolidin-2,4-dione derivative.
- a method is known in which the reaction is carried out in the presence and then the cyclization and the double bond are isomerized [WO87 / 02357 (EP-A-0241559, USP-4818272, USP-4983751)].
- phosgene or phosgene dimer which is very dangerous in the production process of the isocyanate as a raw material, and this is not always satisfactory as an industrial production method. Disclosure of the invention
- the present inventors have proposed a method for efficiently producing a 3-substituted phenyl-1,3-oxazolidin-2,4-dione derivative having an alkylidene group at the 5-position without using dangerous phosgene or phosgene dimer. Diligently examined.
- the N-substituted phenylcarbamate and the 2-hydroxy-3-butenoate can be heated and reacted in the presence of a catalyst, if necessary, to give a 3-substituted phenyl-5-alkylidene-1,3- Oxazolidine-2,4-dione derivative can be produced in good yield, and N-substituted phenylcarbamic acid ester and 3-alkoxy-2-hydroxycarboxylic acid ester can be prepared in the presence of a metal compound catalyst.
- 3-substituted phenyl-5-alkylidene-1,3-oxazoly..gin-2,4-dione derivative can be produced at once, and it can be used as a raw material for production.
- the inventors have found that -alkoxy-2-hydroxyalkanoic acid esters can produce glycidic acid esters and alcohols in the presence of an acid catalyst, and have completed the present invention.
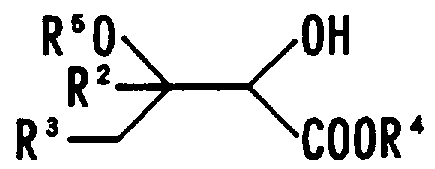
- the present invention provides a compound represented by the general formula (I):
- R 2 and R 3 are each independently a hydrogen atom or an atom having 1 to 12 carbon atoms.
- the present invention relates to a process for producing 1,3-oxazolidin-2,4-dione derivatives.
- the present invention is based on the general formula (I)
- R 2 and R 3 independently represent a hydrogen atom or an alkyl group having 1 to 12 carbon atoms
- R 4 and R s independently represent an alkyl group having 1 to 6 carbon atoms.
- the 3-alkoxy-2-hydroxyalkanoic acid ester of the formula (H) is reacted under heating in the presence of a metal compound catalyst.
- R 2 and R 3 independently represent a hydrogen atom or an alkyl group having 1 to 12 carbon atoms, and R 4 represents an alkyl group having 1 to 6 carbon atoms.
- R 5 represents an alkyl group having 1 to 6 carbon atoms.
- R 5 represents an alkyl group having 1 to 6 carbon atoms.
- an acid catalyst to give a 3-alkoxy group represented by the above general formula (IV).
- a 2-hydroxyl carboxylic acid ester was produced, and then the N-substituted phenylcarbamic acid ester represented by the above general formula (I) and the obtained 3-alkoxy-2-hydroxyl carboxylic acid ester (W) were added.
- the 3-substituted fuynyl-5-alkylidene-1,3-oxazolidin-2,4-dione represented by the above general formula (1) characterized in that the reaction is carried out under heating in the presence of a metal compound catalyst.
- the present invention relates to a method for producing a derivative.
- the present invention is characterized in that a glycidic acid ester represented by the general formula (V) is reacted with an alcohol represented by the general formula (VI) in the presence of an acid catalyst.
- 3-alkoxy represented by (IV) The present invention relates to a method for producing ci-2-hydroxyl carboxylic acid ester.
- Bases include, for example, trimethylamine, triethylamine, tripropylamine, triisopropylamine, tributylamine, triisobutylamine, tripentylamine, trihexylamine, triheptylamine, trioctylamine, trioctylamine, triethylamine.
- the amount of the base used is not particularly limited, but is preferably 0.001 to 0.5 equivalent, more preferably 0.01 to 0.1 equivalent, based on the substrate.
- the reaction can be carried out in the presence of a catalyst.
- a metal compound, a quaternary ammonium salt, or the like can be used as a catalyst, and in this case, the above base can coexist.
- the metal compound when used alone as a catalyst, or in some cases, in combination with a base, the target compound can be efficiently obtained in a short time.
- Metal compounds include iron, ruthenium, osmium, cobalt, rhodium, iridium, nickel, and c.
- fluoride, chloride, bromide, iodide, and carboxylate of radium, platinum, copper, silver, gold, zinc, potassium, mercury, tin or lead such as iron fluoride (E), Iron fluoride (m), iron chloride (E), iron chloride (11), iron bromide (E), iron bromide (m).
- a quaternary ammonium salt can be used as a catalyst.
- pyridinium whose counter anion is fluorine ion, chloride ion, bromide ion, iodine ion, BF 4 —, C 10 4 —, or sulfonate
- quaternary ammonium salts including ammonium salts, and more specifically, tetramethylammonium chloride, tetramethylammonium bromide, tetramethylammonium iodide, and borofluoride.
- Nmoniumu salt Nmoniumu salt.
- pyridine salts such as pyridine hydrochloride and pyridine paratoluenesulfonate can be exemplified.
- the amount of the catalyst to be used is preferably 0.0001 to 0.5 equivalent, more preferably 0.001 to 0.1 equivalent to the substrate.
- This method can be carried out without a solvent, but can also be carried out in a suitable organic solvent.
- the organic solvent include aliphatic hydrocarbon solvents such as decane, dodecane, tridecane, and tetradecane; aromatic solvents such as toluene, xylene, benzene, dichlorobenzene, and tetralin; and mixed solvents thereof.
- the reaction can be carried out in any other solvent that does not adversely affect the reaction.
- the reaction temperature varies depending on the catalyst or solvent used, etc., but the reaction can be carried out at a temperature at which the compound does not decompose. It is selected from the range of 100-250. Also, the 3-substituted phenyl-5-alkylidene-1.3-oxazolidin-2,4-dione using the 3-alkoxy-2-hydroxyalkanoate (IV) of the present invention as a part of the production raw material.
- the reaction is carried out in the presence of a metal compound catalyst.
- Gold compounds include iron, ruthenium, osmium, cobalt, rhodium, iridium, nickel, and nickel.
- Fluoride, chloride, bromide, iodide, and carboxylate of radium, platinum, copper, silver, gold, zinc, cadmium, mercury, tin, or lead are listed.
- compounds of tin are particularly preferred, for example, iron fluoride ( ⁇ ), iron fluoride (dish iron chloride ( ⁇ ), iron chloride ( ⁇ ), iron bromide ( ⁇ ), iron bromide (m), Iron iodide ( ⁇ ), iron acetate (ox), iron (m) acetyl acetatetonate, tin fluoride ( ⁇ ), tin fluoride (W), tin chloride ( ⁇ ), tin chloride (IV), odor Examples include tin (II) bromide, tin (IV) bromide, tin (II) iodide, tin acetate (II), and hydrates of these metal compounds may also be used as catalysts.
- the amount of the catalyst used is preferably from 0.0001 to
- the target compound can be obtained more efficiently and in a shorter time by combining a base with the catalyst of the metal compound.
- the base include, for example, trimethylamine, triethylamine, tripropylamine, triisopropylamine, tributylamine, triisobutylamine, tripentylamine, trihexylamine, triheptylamine, trioctylamine, tridecylamine.
- Triphenylamine, tribenzylamine, 4-methylmorpholine, ⁇ , N-dimethylaniline, pyridine, 4-t-butylpyridine and the like can be mentioned, but any other amines can be used in the reaction as long as they do not adversely affect the reaction.
- the amount of the base used is not particularly limited, but is preferably 0.001 to 0.5 equivalent, more preferably 0.01 to 0.1 equivalent, relative to the substrate.
- This method can be carried out without a solvent, but can also be carried out in a suitable organic solvent.
- organic solvent include aliphatic hydrocarbon solvents such as decane, dodecane, and tridecane; aromatic solvents such as toluene, cumene, xylene, mesitylene, benzene, dichlorobenzene, dibenzene, tetralin, and the like; Although a mixed solvent can be mentioned, the reaction can be performed in any other solvent that does not adversely affect the reaction.
- the reaction temperature varies depending on the catalyst, base or solvent used, etc., but it can be carried out at a temperature of about L, at which the compound does not decompose, and is preferably selected from the range of 100 to 250, especially 180 to 220. The range is more preferred.
- the acid catalyst that can be used includes: Protic acids such as sulfuric acid, P-toluenesulfonic acid, trifluoromethanesulfonic acid, phosphoric acid, and perchloric acid can be exemplified.
- the amount of the catalyst used is not particularly limited, and the desired 3-alkoxy-2-hydroxyl carboxylic acid ester can be obtained in good yield by using 0.0001 to 0.5 equivalent to the glycidic acid ester as a substrate. (IV) can be obtained.
- the alkyl group having 1 to 6 carbon atoms is a straight-chain or branched-chain alkyl group, for example, a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, a sec group. -Butyl, butyl, pentyl, neopentyl, 3-pentyl, hexyl, 3-hexyl, 3,3-dimethyl-2-butyl and the like.
- the alkyl groups having 12 carbon atoms represented by R 2 and R 3 are linear or branched alkyl groups unless otherwise specified.
- Groups such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, t-butyl, pentyl, neopentyl, 3-pentyl, hexyl, 3-hexyl, 3,3-dimethyl-2-butyl, heptyl, 2-heptyl, 3-heptyl, octyl, t-octyl, 2-octyl, 3-octyl, nonanyl Group, decyl group, dodecyl group and the like.
- Examples of the substituted phenyl group represented by Ar in the general formulas (I) and (1E) include a halogen atom, an alkyl group, an alkyloxy group, a cycloalkyloxy group, an alkenyloxy group, an alkynyloxy group, and a lower alkyloxycarbonyl. It is a phenyl group substituted by one or more substituents such as a oxy group, a nitro group and a cyano group.
- Examples of the halogen atom include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom. Wear.
- alkyl group examples include a linear or branched alkyl group having 1 to 12 carbon atoms, such as a methyl group, an ethyl group, a propyl group, an isopropyl group, a butyl group, an isobutyl group, and a sec-butyl group.
- alkyloxy group examples include a linear or branched alkyloxy group having 1 to 12 carbon atoms, such as a methoxy group, an ethoxy group, a propyloxy group, an isopropyloxy group, a butyloxy group, and an isobutyloxy group.
- Sec-butyloxy pentyloxy, neopentyloxy, hexyloxy, hexyl-3-oxy, heptyloxy, octyloxy, octyl-2-oxy, dodecyloxy, etc. i can.
- Examples of the cycloalkyloxy group include a cycloalkyloxy group having 3 to 12 carbon atoms, and examples thereof include a cyclopropyloxy group, a cyclobutyloxy group, a cyclopentyloxy group, a cyclohexyloxy group, and a cyclohexyl group.
- Examples include a butyloxy group, a cyclooctyloxy group, a cyclodecyloxy group, and a cyclododecyloxy group.
- alkenyloxy group examples include a linear or branched alkenyloxy group having 2 to 12 carbon atoms having a double bond, such as a vinyloxy group, an aryloxy group, a methallyloxy group, a 2-butenyloxy group, 3-butenyloxy, 3-methyl-2-butenyloxy, 2-methyl-3-butenyloxy, 1-pentenyl-3-oxy, 2-pentenyloxy, 3-pentenyl-2-oxy, 4-pentenyl Examples thereof include a hydroxy group, a 3-hexenyloxy group, a 3-heptenyloxy group, a 3-octenyloxy group, a 3-decanyloxy group, and a 3-dodecanyloxy group.
- An alkynyloxy group has a triple bond
- Examples thereof include linear or branched alkynyloxy groups having 3 to 8 carbon atoms, such as propargyloxy, 1-methyl-propargyloxy, 1,1-dimethylpropargyloxy, and 2 -Butynyloxy, 1-methyl-2-butynyloxy, 3-butynyloxy, 2-pentynyloxy, 3-pentynyloxy, 3-octynyloxy and the like.
- Examples of the lower alkyloxycarbonyloxy group include a methoxycarbonyloxy group, a propyloxycarbonyloxy group, and a butyloxycarbonyloxy group.
- More specific substituted phenyl groups include a 2-fluorophenyl group,
- the raw acid ester of general formula (I), which is a raw material of the present invention, can be synthesized, for example, by the method described in JP-A-5-17427, EP-A-0496347 (USP-5281742). It can be easily produced by reacting an aniline derivative with a chloroformate in the presence of a base. Further, the 2-hydroxy-3-argenic acid ester represented by the general formula (E), which is a raw material of the present invention, can be easily synthesized, for example, by the method described in EP-A-0153692 (USP-4621150). Further, the glycidic acid ester represented by the general formula (V), which is a raw material of the present invention, can be easily synthesized by the method described in, for example, Organic Reactions, Vol. V, p413. (1960).
- Ethyl N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) carbamate (3. Olg, lOmmol) and 2-hydroxy-3- were placed in a flask (100cc) equipped with an air-cooled tube (20cm).
- methyl 3-ethyl butenoate (4.32 g, 30 mmo 1) and tributylamine (92.7 mg, 0.5 ⁇ 1) were placed under reduced pressure such that the ethyl butene refluxes below half the height of the air-cooled tube. The reaction was performed for 15 hours.
- Ethyl N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) carbamate (3.Olg, lOmmol) and 2-hydroxy-3-methyl were placed in a flask (25 cc) equipped with an air-cooled tube (20 cm).
- 3-Ethyl butenoate (2.02 g, Hmmol) and stannous acetate (23.7 mg, 0.1 ⁇ ) are added, and the pressure is reduced so that the ethyl butylate refluxes below half the height of the air-cooled tube.
- the reaction was performed at 200 ° C for 2.5 hours.
- reaction solution was allowed to cool to 70 to 80 ° C., mixed with a mixed solution of methanol (10 m and 6N ′ hydrochloric acid (0.5 mL)), and allowed to stand at room temperature.
- the furnace is washed, washed with a mixed solution of methanol (20 DIL) and 6N hydrochloric acid (lmU, then with water (20 inL), and sufficiently dried under reduced pressure to obtain 3- (4-chloro-5-cyclopentyloxy).
- 2-fluorophenyl) -5-isopropylidene-1,3-oxazolidin-2,4-dione (4,77 g, yield 67.4%) was obtained c :
- N- (4-chloro-5-cyclopentylquino-2-fluorophenyl) -ethyl butylamine (3. Olg, lOmmo 1) and 2-hydroxy-3-methyl- Isobutyl 3-butenoate (4.22 g, 24.5 mmol) and tetrabutylammonium chloride (278 mg, 1.0 bandol) were added, and the mixture was reacted at 200 and 9 hours.
- Ethyl N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) carbamate (3. Olg, lOmio 1) and 2-hydroxy-3-methyl-3-butenoic acid are placed in a flask (25 cc) equipped with a distillation apparatus. After adding ethyl (2.16 g, 15 ⁇ 1) and tetrabutylammonium chloride (278 mg, 1.0 mmol), the mixture was reacted at 200 for 1.5 hours. After the reaction is completed, the reaction solution is The mixture was allowed to cool to room temperature, and a mixed solution of methanol (5 mL) and 6N hydrochloric acid (0.25 mL) was added to make it uniform.
- N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) -powered ethyl ester (3.01 g, lOmmo 1) and 2-hydroxy-3-methyl-3 -Ethyl butenoate (1.16 g, l.-2 mmol) and tetraethylammonium p-toluenesulfonate (151 rag, 0.50 mmol) were added and reacted at 200 for 3.5 hours.
- N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) N- (4-cyclopentyloxy) -fluoroethyl ester (3. Olg, 10C1), ferric chloride Hexahydrate (27 mg, 0.1 excitation 1) and triethylamine (129 mg. 1.27 ⁇ 1) were added, and the mixture was heated to 200 with stirring to obtain a homogeneous solution. Next, ethyl 2-hydroxy-3-methyl-3-butenoate (4.33 g, 30 mmol) was added dropwise, and the mixture was stirred for 1 hour after the addition.
- N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) -powered ethyl ethyl ester (674 g, 2.24 mol), ferric chloride ( 3.7 g, 22.8 ramol) and tributylamine (21 g, 0.113 mol) were added and the mixture was heated to 200 with stirring to form a homogeneous solution. Then, 2-hydroxy-3-methyl-3-ethylbutenoate (484 g, 3.51raol) was added dropwise over 5.5 hours, and after the addition, the mixture was further stirred for 2.5 hours.
- N '-(4-chloro-5-cyclohexylpentyloxy-2-fluorophenyl) carba'ethyl ester (10. Og, 33. lmraol) in a nickel-mouthed flask (50 cc) equipped with Din Stark Methyl 3-ethoxy-2-hydroxy-3-methylbutanoate (11.7 g, 66.4 mmol), ferric chloride (5.4 mg, 0.331 mmo 1), triptylamin (0.308 g, 1. 66ramol) and stirred while heating at 200 for 4 hours. After completion of the reaction, low-boiling compounds were removed under reduced pressure, and the mixture was cooled to room temperature.
- N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) N- (4-chloro-5-pentyloxy-2-fluorophenyl) -potassium ethyl ester (2.00 g, 6.63 mmol) was added to a Nikon eggplant-shaped flask (50 cc) equipped with Dean Stark. Methyl 2-hydroxy-3-methoxy-3-methylbutanoate (2.69 g, 16.6 iraol) and stannous acetate (0.20 g) were added, and the mixture was stirred while heating at 195 for 3 hours. After the reaction was completed, hexane (10 mL) was added, and unnecessary substances were separated by filtration.
- Nylon-type flask 200cc equipped with Dean Stark and Dimroth, and ethyl N- (4-chloro-5-cyclopentyloxy-2-fluorophenyl) carbamate (10.0g, 33.1 maraudal ol ), 3-ethoxy-2-hydroxy-3-Atil.
- Ethyl butyl citrate (9.44 g, 49.7 mmol)
- ferric chloride hexahydrate 9 (L2 mg, 0.03 mniol) and Tributylamine (309.5 mg, 1.7 mmol) was added, and the mixture was stirred with heating in an oil bath at 205.
- Ethyl 3,3-dimethylglycidate (73.8 g, 0.512 mol) and ethanol (250 mL) are placed in an eggplant-shaped flask (100 cc), and 703 ⁇ 4 perchloric acid (90 ing) is slowly added with stirring at room temperature. then, after the end 60 in 5 hours ⁇ the c the reaction, the reaction solution was concentrated under reduced pressure, toluene was added (100 mL), while at Kuwawae room temperature and sulfuric acid Na Bok helium and a small amount of carbonic anhydride Natoriumu I left it. The solid is removed by filtration, and the solvent is removed from the filtrate under reduced pressure. By removal, ethyl 3-ethoxy-2-hydroxy-3-methylbutyrate (82.2 g, yield 84.43 ⁇ 4) was obtained as a transparent liquid.
- the present invention provides a method for efficiently synthesizing a 3-substituted fuynyl-5-alkylidene-1,3-oxazolidine-2.4-dione derivative having a strong herbicidal effect without using dangerous phosgene or phosgene dimer. Provide possible industrial processes.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
































Claims








Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE69613460T DE69613460T2 (de) | 1995-02-27 | 1996-02-26 | Verfahren zur herstellung von 3-(substituiert phenyl)-5-alkyliden-1,3-oxazolidin-2,4-dion-derivaten |
| US08/894,655 US6090946A (en) | 1995-02-27 | 1996-02-26 | Process for the preparation of 3-(substituted phenyl)-5-alkylidene-1,3-oxazolidine-2,4-dione derivatives |
| EP96903234A EP0812834B1 (en) | 1995-02-27 | 1996-02-26 | Process for the preparation of 3-(substituted phenyl)-5-alkylidene-1,3-oxazolidine-2,4-dione derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP7/37843 | 1995-02-27 | ||
| JP3784395 | 1995-02-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996026930A1 true WO1996026930A1 (en) | 1996-09-06 |
Family
ID=12508822
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1996/000430 Ceased WO1996026930A1 (en) | 1995-02-27 | 1996-02-26 | Process for the preparation of 3-(substituted phenyl)-5-alkylidene-1,3-oxazolidine-2,4-dione derivatives |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6090946A (ja) |
| EP (1) | EP0812834B1 (ja) |
| KR (1) | KR100401275B1 (ja) |
| CN (1) | CN1083836C (ja) |
| DE (1) | DE69613460T2 (ja) |
| ES (1) | ES2159719T3 (ja) |
| WO (1) | WO1996026930A1 (ja) |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62167713A (ja) * | 1985-10-11 | 1987-07-24 | Sagami Chem Res Center | 除草剤 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE756350A (fr) * | 1969-09-18 | 1971-03-18 | Basf Ag | Procede pour la preparation de 5-methylene-2, 4-oxazolidine-diones et composes ainsi obtenus |
| DE2813873A1 (de) * | 1978-03-31 | 1979-10-11 | Basf Ag | Verfahren zur herstellung von oxazolidin-2,4-dionen |
| DE2913522A1 (de) * | 1979-04-04 | 1980-10-23 | Basf Ag | Verfahren zur herstellung von n-aryloxazolidin-2,4-dionen |
| WO1987002357A1 (fr) * | 1985-10-11 | 1987-04-23 | Sagami Chemical Research Center | Derives d'oxazolidinedione, procede de preparation et herbicides les contenant |
| DE69226813T2 (de) * | 1991-06-07 | 1999-02-18 | Ube Industries, Ltd., Ube, Yamaguchi | Pyrimidin- oder Triazin-Derivate, Verfahren zu ihrer Herstellung und diese enthaltende Herbizide |
| US5739337A (en) * | 1996-03-08 | 1998-04-14 | Neurogen Corporation | Process for preparing dibenzo-1-carboxamido-1,4-azabicyclo 3.2.1!octanes |
-
1996
- 1996-02-26 US US08/894,655 patent/US6090946A/en not_active Expired - Fee Related
- 1996-02-26 DE DE69613460T patent/DE69613460T2/de not_active Expired - Fee Related
- 1996-02-26 KR KR1019970705934A patent/KR100401275B1/ko not_active Expired - Lifetime
- 1996-02-26 EP EP96903234A patent/EP0812834B1/en not_active Expired - Lifetime
- 1996-02-26 WO PCT/JP1996/000430 patent/WO1996026930A1/ja not_active Ceased
- 1996-02-26 ES ES96903234T patent/ES2159719T3/es not_active Expired - Lifetime
- 1996-02-26 CN CN96191993A patent/CN1083836C/zh not_active Expired - Lifetime
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS62167713A (ja) * | 1985-10-11 | 1987-07-24 | Sagami Chem Res Center | 除草剤 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0812834B1 (en) | 2001-06-20 |
| CN1175946A (zh) | 1998-03-11 |
| KR100401275B1 (ko) | 2003-12-31 |
| EP0812834A1 (en) | 1997-12-17 |
| US6090946A (en) | 2000-07-18 |
| EP0812834A4 (en) | 1998-05-06 |
| DE69613460T2 (de) | 2001-11-22 |
| CN1083836C (zh) | 2002-05-01 |
| ES2159719T3 (es) | 2001-10-16 |
| KR19980702531A (ko) | 1998-07-15 |
| DE69613460D1 (de) | 2001-07-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6803469B2 (en) | Process for preparing quinolone antibiotic intermediates | |
| EA007965B1 (ru) | Способ получения комбретастатинов | |
| HU207279B (en) | Process for producing naphtalene derivatives | |
| JPH09216881A (ja) | トリアゾロン化合物の製法及びトリアゾロン化合物 | |
| WO1996026930A1 (en) | Process for the preparation of 3-(substituted phenyl)-5-alkylidene-1,3-oxazolidine-2,4-dione derivatives | |
| JP3002791B2 (ja) | ベンジルフェニルケトン誘導体 | |
| JPH01305071A (ja) | オキサゾロン誘導体の製造方法 | |
| JP3697045B2 (ja) | β−ヒドラジノエステル類並びにピラゾリジノン類、ピラゾロン類およびβ−アミノ酸誘導体の製造方法 | |
| JP3863589B2 (ja) | 3−置換フェニル−5−アルキリデン−1,3−オキサゾリジン−2,4−ジオン誘導体の製造方法 | |
| JPH0784427B2 (ja) | 医・農薬中間原料の製造方法 | |
| JPH09227535A (ja) | 3−置換フェニル−5−アルキリデン−1,3−オキサゾリジン−2,4−ジオン誘導体の製造法 | |
| US5476940A (en) | 3-substituted quinoline-5-carboxylic acids | |
| JPH0692913A (ja) | アニリン誘導体の製造方法 | |
| JPH0676361B2 (ja) | アテローム性動脈硬化症の治療または予防のための医薬組成物 | |
| JP3385353B2 (ja) | 官能基を有する環状ケイ素化合物 | |
| JPH11171876A (ja) | 2,4−オキサゾリジンジオン類の製造方法 | |
| JP2726337B2 (ja) | ジフェニルカーボネート誘導体及びその製造方法 | |
| JP2751970B2 (ja) | アゾリジン及びその製法 | |
| JP3323917B2 (ja) | インドール化合物の製造方法 | |
| JPH08225483A (ja) | ブロモメチル−置換フェニルケトンの製造方法 | |
| JPH03264557A (ja) | フッ素含有化合物 | |
| EA015450B1 (ru) | Фармацевтические промежуточные соединения в синтезе ингибиторов ацетилхолинэстеразы (асе) и их применение | |
| JP2004359583A (ja) | 鎖状乳酸オリゴマー誘導体およびその製造方法 | |
| JP2002363171A (ja) | 4−置換−3−アミノイソオキサゾール誘導体の製法 | |
| JPH1135563A (ja) | アゾール−1−イルアルキルニトリル類の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96191993.0 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CN KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1019970705934 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1996903234 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08894655 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996903234 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1019970705934 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1996903234 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1019970705934 Country of ref document: KR |