WO1998024792A1 - Anticoagulant glycosides and pharmaceutical compositions thereof - Google Patents
Anticoagulant glycosides and pharmaceutical compositions thereof Download PDFInfo
- Publication number
- WO1998024792A1 WO1998024792A1 PCT/HU1997/000078 HU9700078W WO9824792A1 WO 1998024792 A1 WO1998024792 A1 WO 1998024792A1 HU 9700078 W HU9700078 W HU 9700078W WO 9824792 A1 WO9824792 A1 WO 9824792A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- anhydro
- dithio
- mannopyranoside
- mixture
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H15/00—Compounds containing hydrocarbon or substituted hydrocarbon radicals directly attached to hetero atoms of saccharide radicals
- C07H15/02—Acyclic radicals, not substituted by cyclic structures
- C07H15/14—Acyclic radicals, not substituted by cyclic structures attached to a sulfur, selenium or tellurium atom of a saccharide radical
Definitions
- This invention relates to novel 2,6-anhydro-1 ,2-dithio-pyranosides of the formula (I), more particularly to D-manno- and D-altropyranosides of formula (la-Id)
- R ⁇ represents a hydroxy or an azido group
- the compounds of the invention possess valuable pharmaceutical properties, especially anticoagulant activity, even when administered by the oral route.
- the aim of the invention was to synthesize such new carbohydrate derivatives which are stronger inhibitors of the coagulation process than the known ones and are orally active too.
- the compounds of the invention can be synthesized by different known methods.
- represents a hydroxy group and R2 represents a cyano group, can be prepared e.g. by treatment of an anomeric mixture of glycosides of formula (III),
- the above reaction can preferably be carried out by hydrolyzing the anomeric mixture of glycosides of formula (III) by using sodium methoxide in methanolic solution, and separating the obtained a, b-anomers (la and lb) by crystallization and/or column chromatography.
- glycosides of formula (III), which are new compounds, can be prepared e.g. by acetylating the known [I.I. Cubero et al.: Carbohydr. Res., 242,
- glycosides of formula (III) can preferably be carried out by using sulfuric acid in acetic anhydride for the methoxy acetoxy exchange reaction, and trimethylsilyl triflate as promoter for the condensation with 4- cyanothiophenol.
- represents a hydroxy group and R2 represents an aminothiocarbonyl group, can be prepared e.g. by treatment of a compound of formula (lb), wherein the meaning of R-j is as defined above and R2 is a cyano group, with hydrogen sulfide using an organic base as solvent.
- the above reaction can preferably be carried out, using acetone as solvent and methyl iodide as reagent at reflux temperature.
- the above reaction can preferably be carried out, using methanol or ethanol as solvent at reflux temperature.
- the above reaction sequence can preferably be carried out by using ethanol as solvent and sodium borohydride - nickel(ll) chloride as reagent for the reduction, pyridine as a base for the acetylation and sodium methoxide in methanol for removing the ester groups.
- the above reaction sequence can preferably be carried out by using sodium methoxide in methanolic solution for the hydrolysis of the anomeric mixture of glycosides of formula (VI), and separating the so obtained a, b-anomers (lc and Id) by crystallization and/or column chromatography.
- glycosides of formula (VI), which are new compounds, can be prepared e.g. by reacting the known [K. Toshima et al.: Tetrahedr. Lett., 33, 1491 (1992)] triacetate of formula (VII)
- glycosides of formula (VI) can preferably be carried out at low temperature, preferably at 0 °C, using trimethylsilyl triflate as promoter.
- the compounds of formula (la and lb), in which R-j represents an azido group and R2 represents a nitro group, can be prepared e.g. by reacting the diacetate of formula (VIII) with 4-nitrothiophenol in the presence of a promoter, removing the acetyl groups of the obtained anomeric glycosides in a lower aliphatic alcohol by treatment with base, and optionally separating the anomers.
- the above reaction sequence can preferably be carried out by reacting the diacetate of formula (VIII) with 4-nitrothiophenol in dichloromethane at low temperature, preferably at 0 °C, using trimethylsilyl triflate as promoter, or in 1 ,2-dichloroethane at 20 °C, using boron trifluorid etherate as promoter, hydrolyzing the obtained anomeric glycosides by sodium methoxide in methanol, and optionally separating the obtained a, b-anomers by crystallization and/or column chromatography.
- the compounds of formula (I) of the invention possess valuable anticoagulant activity.
- the compounds of the present invention as well as their pharmaceutically acceptable salts can be used as such or suitably in the form of pharmaceutical compositions. These compositions also fall within the scope of the present invention.
- compositions contain an amount required to excert the therapeutical effect of a compound of formula (I) or its pharmaceutically acceptable salt, in admixture with known carriers, excipients, diluents and/or other additives commonly used in the pharmaceutical practice.
- the antithrombotic compound is formulated in capsules or tablets which may contain excipients such as binders, lubricants, disintegration agents and the like.
- the antithrombotic compound is formulated in a pharmaceutically acceptable diluent, e.g. physiological saline (0.9 %), 5% dextrose, Ringer's solution and the like.
- the doses required to excert the therapeutical effect of the compounds according to the invention may be varied depending on the individual condition and age of the patient to be treated and finally these doses are determined by the attending physician. However, for the prevention and/or treatment of diseases, where the application of an anticoagulant is desirable, daily doses of these compounds falling between about 0.01 mg/kg of body weight and about 100 mg/kg of body weight and preferably between about 0.1 mg/kg of body weight and about 10 mg/kg of body weight are used by the oral or parenteral, e.g. intravenous, route.
- the compounds according to the invention and the process for the preparation thereof are illustrated in detail by the following not limiting Examples.
- Multiplicities of the 1 3 C NMR spectra were obtained from DEPT experiments. The assignment of the protons were based on homonuclear decoupling and DNOE experiments. Connectivities between identified protons and protonated carbons were determined by HETCOR experiments. MS spectra were recorded with a Finnigan MAT 8430 mass spectrometer. In the case of FAB spectra samples were dissolved in 3-nitrobenzaldehyde or in glycerin.
- the starting anomeric mixture of formula (III) can be prepared the following way:
- the starting compound of formula (VIII) can be prepared the following way:
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Biotechnology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
This invention relates to novel 2,6-anhydro-1,2-dithio-D-manno- and D-altropyranosides of formula (I) wherein R1 represents a hydroxy or an azido group, R2 representa a nitro, cyano, amidino, aminothiocarbonyl, -C(=NH)-OCH3, -C(=NH)-NH-NH2, -C(=NH)-SCH3 or an acetamido group, and the acid addition salts thereof formed with organic or inorganic acids, if possible as well as pharmaceutical compositions containing them. The compounds of the invention possess valuable therapeutic, particularly anticoagulant properties.
Description
ANTICOAGULANT GLYCOSIDES AND PHARMACEUTICAL COMPOSITIONS THEREOF
This invention relates to novel 2,6-anhydro-1 ,2-dithio-pyranosides of the formula (I), more particularly to D-manno- and D-altropyranosides of formula (la-Id)


β-D-maπnoside

" 1 α-D-altroside

R . β-D-altroside wherein
R^ represents a hydroxy or an azido group,
R2 represents a nitro, cyano, amidino, aminothiocarbonyl, -C(=NH)-OCH3, -C(=NH)-NH-NH2, -C(=NH)-SCH3 or an acetamido group, and the acid addition salts thereof formed with organic or inorganic acids, if possible as well as pharmaceutical compositions containing them.
The compounds of the invention possess valuable pharmaceutical properties, especially anticoagulant activity, even when administered by the oral route.
Particularly valuable representatives of formula (I) according to the invention are the following ones:
4-cyanophenyl 2,6-anhydro-1 ,2-dithio-a-D-altropyranoside 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-b-D-altropyranoside 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-a-D-mannopyτanoside 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-b-D-mannopyraπoside 4-(hydraziπo)(imino)methylphenyl 2,6-anhydro-1 ,2-dithio-b-D-mannopyranoside 4-nitropheπyl 2,6-anhydro-1 ,2-dithio-b-D-mannopyranoside 4-acetamidophenyl 2,6-anhydro-1 ,2-dithio-b-D-mannopyranoside 4-(imino)(methoxy)methylphenyl 2,6-aπhydro-1 ,2-dithio-β-D-mannopyranoside 4-(aminothiocarbonyl)pheπyl 2,6-anhydro-1 ,2-dithio-β-D-mannopyranoside 4-(imino)(methylthio)methylphenyl 2,6-anhydro-1 ,2-dithio-β-D-mannopyranoside 4-amidinophenyl 2,6-anhydro-1 ,2-dithio-β-D-mannopyranoside 4-cyanophenyl 2,6-anhydro-3-azido-3-deoxy-1 ,2-dithio-β-D-mannopyranoside
4-nitrophenyl 2,6-anhydro-3-azido-3-deoxy-1 ,2-dithio-β-D-maπnopyranoside
Derivatives of the formula (I), are not known from the literature. However some 1 ,5-dithio-D-xylopyranosides of formula (II)

(in which the oxygen of the carbohydrate moiety is replaced by sulfur) possess advantageous anticoagulant activity even when administered orally. This biological activity decreased significantly when the oxygen of the carbohydrate moiety was not replaced by sulfur. A similar decrease in activity was observed, when the chirality of the most active b-D-xylopyranosides was changed, i.e. a sugar with a configuration different from D-xylose, or an a-D-anomer was investigated. Furthermore the activity decreased also when instead of pentose derivatives, hexose derivatives were synthesized [F. Bellamy et al.: Eur. J. Med. Chem. 30 (1995) 101],
The aim of the invention was to synthesize such new carbohydrate derivatives which are stronger inhibitors of the coagulation process than the known ones and are orally active too.
Surprisingly it was found, that the antithrombotic activity of carbohydrate de vatives can be substantially increased, by using hexoses instead of pentoses in which the ring oxygen atom is still present, but at the same time a 2,6-thioanhydro bridge is introduced into the molecule. Both the a- as well as the b-anomers of the so obtained thioglycosides possess a beneficial biological activity. This activity could be further increased by exchanging the 3-OH group of the carbohydrate moiety by azide. A similar increased activity was found for
those thioglycosides, in which the cyano substituent at C-4 of the aglycon was transformed into a carboxylic acid derivative.
The compounds of the invention can be synthesized by different known methods. a) The compound of formula (la and lb), in which R-| represents a hydroxy group and R2 represents a cyano group, can be prepared e.g. by treatment of an anomeric mixture of glycosides of formula (III),

wherein Ac represents an acetyl group, with base in a lower aliphatic alcohol, and separating subsequently the anomers.
The above reaction can preferably be carried out by hydrolyzing the anomeric mixture of glycosides of formula (III) by using sodium methoxide in methanolic solution, and separating the obtained a, b-anomers (la and lb) by crystallization and/or column chromatography.
The glycosides of formula (III), which are new compounds, can be prepared e.g. by acetylating the known [I.I. Cubero et al.: Carbohydr. Res., 242,
109 (1993)] methyl 2,6-anhydro-2-thio-a-D-mannopyranoside, exchanging the methoxy group of the obtained diacetate of formula (IV),

The above preparation of glycosides of formula (III) can preferably be carried out by using sulfuric acid in acetic anhydride for the methoxy acetoxy exchange reaction, and trimethylsilyl triflate as promoter for the condensation with 4- cyanothiophenol.
b) The compound of formula (lb), in which R-| represents a hydroxy group and R2 represents a -C(=NH)-OCH3 group, can be prepared e.g. by treatment of a compound of formula (lb), wherein the meaning of R-j is as defined above and R2 is a cyano group, with sodium methoxide in methanol.
c) The compound of formula (lb), in which R-| represents a hydroxy group and R2 represents an aminothiocarbonyl group, can be prepared e.g. by treatment of a compound of formula (lb), wherein the meaning of R-j is as defined above and R2 is a cyano group, with hydrogen sulfide using an organic base as solvent.
The above reaction can preferably be carried out at room temperature, using a 1 :1 mixture of triethylamine-pyridine as solvent.
d) The compound of formula (lb), in which R-| represents a hydroxy group and R2 represents a -C(=NH)-SCH3 group, can be prepared e.g. by treatment of a compound of formula (lb), wherein the meaning of R-| is as defined above and R2 is an aminothiocarbonyl group, with a methylating agent.
The above reaction can preferably be carried out, using acetone as solvent and methyl iodide as reagent at reflux temperature.
ό
e) The compound of formula (lb), in which R-| represents a hydroxy group and R2 represents an amidino group, can be prepared e.g. by treatment of a compound of formula (lb), wherein the meaning of R-i is as defined above and R2 is a -C(=NH)-SCH3 group, with ammonium acetate or with ammonia using a lower aliphatic alcohol as solvent.
The above reaction can preferably be carried out, using methanol or ethanol as solvent at reflux temperature.
f) The compound of formula (lb), in which R-j represents a hydroxy group and R2 represents a -C(=NH)-NH-NH2 group, can be prepared e.g. by treatment of a compound of formula (lb), wherein the meaning of R^ is as defined above and R2 is a -C(=NH)-SCH3 group, with hydrazine hydrate, using a lower aliphatic alcohol as solvent.
The above reaction can preferably be carried out at room temperature, using 98% hydrazine hydrate as reagent and ethanol as solvent.
g) The compound of formula (I), in which R-| represents a hydroxy group, and R2 represents a πitro group, can be prepared e.g. by reacting the triacetate of formula (IV), in which R represents an acetyl group, with 4-nitrothiophenol in the presence of a promoter and removing the acetoxy groups from the obtained anomeric mixture of formula (V)

with base in a lower aliphatic alcohol.
The above reaction can preferably be carried out by reacting the triacetate of formula (IV, R = Ac) with 4-nitrothiophenol in dichloromethane at low temperature, preferably at -10 °C using trimethylsilyl triflate as promoter, or in 1 ,2-dichloroethane at 20 °C using boron trifluo d etherate as promoter and subsequently deacetylating the obtained anomeric mixture of glycosides with sodium methoxide in methanolic solution.
h) The compound of formula (lb), in which R-j represents a hydroxy group and R2 represents an acetamido group, can be prepared e.g. by reducing the nitro group of the compound of formula (lb), in which R-] represents a hydroxy group and R2 represents a nitro group using a lower aliphatic alcohol as solvent, and treating the so obtained compound of formula (lb), in which R-i represents a hydroxy group and R2 represents an amino group, with acetic anhydride in the presence of a base, and removing subsequently the formed ester groups in the presence of a base using a lower aliphatic alcohol as solvent.
The above reaction sequence can preferably be carried out by using ethanol as solvent and sodium borohydride - nickel(ll) chloride as reagent for the reduction, pyridine as a base for the acetylation and sodium methoxide in methanol for removing the ester groups.
i) The compounds of formula (lc and Id), in which R-| represents a hydroxy group and R2 represents a cyano group, can be prepared e.g. by hydrolyzing the anomeric mixture of glycosides of formula (VI)

The above reaction sequence can preferably be carried out by using sodium methoxide in methanolic solution for the hydrolysis of the anomeric mixture of glycosides of formula (VI), and separating the so obtained a, b-anomers (lc and Id) by crystallization and/or column chromatography.
The glycosides of formula (VI), which are new compounds, can be prepared e.g. by reacting the known [K. Toshima et al.: Tetrahedr. Lett., 33, 1491 (1992)] triacetate of formula (VII)

with 4-cyanothiophenol in the presence of a promoter.
The above preparation of glycosides of formula (VI) can preferably be carried out at low temperature, preferably at 0 °C, using trimethylsilyl triflate as promoter.
j) The compounds of formula (la and lb), in which R-| represents an azido group and R2 represents a cyano group, can be prepared e.g. by reacting the diacetate of formula (VI 11)

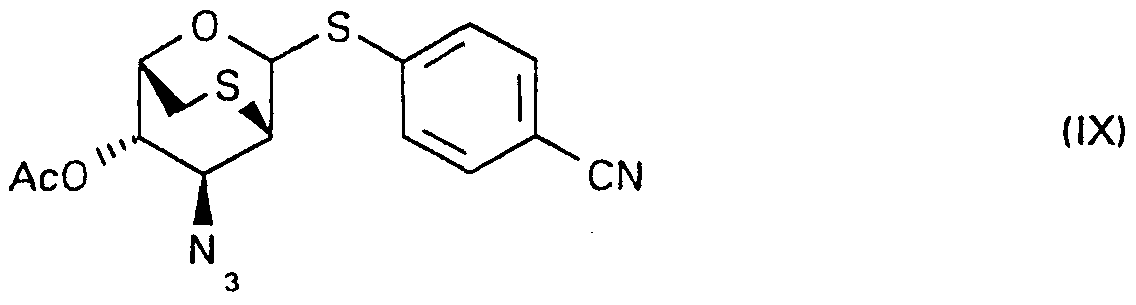
with base in a lower aliphatic alcohol, and separating subsequently the anomers.
The above reaction sequence can preferably be carried out by using trimethylsilyl triflate as promoter for the condensation of diacetate of formula (VIII) with 4-cyanothiophenol at low temperature, preferably at -10 °C, sodium methoxide in methanol for the hydrolysis of the so obtained glycosides, and separating subsequently the a,b-anomers (la and lb) by crystallization and/or column chromatography.
The diacetate of formula (VIII), which is new compound, can be prepared e.g. by converting the free hydroxy group of the known [R. D. Guthrie and D.
Murphy, J. Chem. Soc, (1963) 5288] glucopyranoside of formula (X),
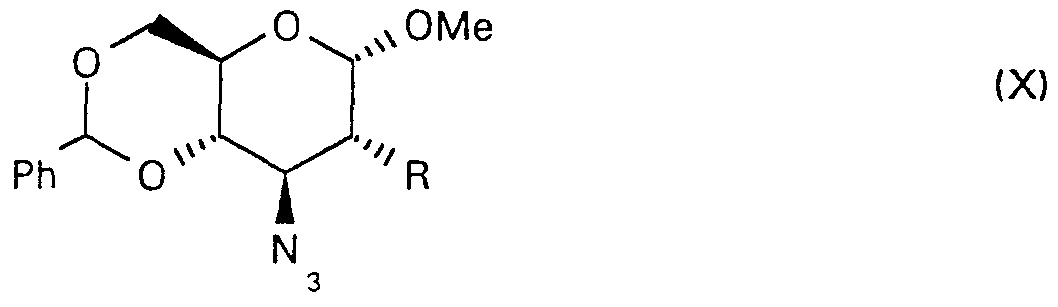
wherein R represents a hydroxy group, into an active ester, and subsequently into a thioester with inversion of configuration. The 4,6-O-benzylidene group of the so obtained mannoside of formula (XI)

is split by N-bromosuccinimide in carbon tetrachloride and the obtained 6- bromo compound of formula (XII)

is treated with base to afford the 2,6-thioanhydro compound of formula (XIII),

wherein R represents hydrogen. After acetylation of the latter the methoxy group is exchanged by an acetoxy group to afford the diacetate of formula (VIII). The above reaction sequence for the preparation of the diacetate of formula (VIII) can preferably be carried out by converting the free hydroxy group of the glucoside of formula (X, R = OH) into a thfluoromethanesulfonyl ester of formula (X, R = OTf) and the latter is transformed into the thioester of formula (XI) by using potassium thiobenzoate. For the deacylation and ringclosure of the 6-bromo compound of formula (XII) obtained from the latter, sodium methoxide is used. The free hydroxy group of the obtained mannose derivative
of formula (XIII, R = H) is acetylated by acetic anhydride in pyridine (XIII, R = Ac), and subsequently the methoxy group is exchanged by an acetoxy group in acetic anhydride in the presence of sulfuric acid.
k) The compounds of formula (la and lb), in which R-j represents an azido group and R2 represents a nitro group, can be prepared e.g. by reacting the diacetate of formula (VIII) with 4-nitrothiophenol in the presence of a promoter, removing the acetyl groups of the obtained anomeric glycosides in a lower aliphatic alcohol by treatment with base, and optionally separating the anomers.
The above reaction sequence can preferably be carried out by reacting the diacetate of formula (VIII) with 4-nitrothiophenol in dichloromethane at low temperature, preferably at 0 °C, using trimethylsilyl triflate as promoter, or in 1 ,2-dichloroethane at 20 °C, using boron trifluorid etherate as promoter, hydrolyzing the obtained anomeric glycosides by sodium methoxide in methanol, and optionally separating the obtained a, b-anomers by crystallization and/or column chromatography.
As mentioned in the introduction, the compounds of formula (I) of the invention possess valuable anticoagulant activity.
This anticoagulant activity of the compounds of formula (I) of the invention was determined on male SPRD rats, using the Pescador's venous thrombosis model [D. Bagdy et al.: Thromb. Haemost. 68 (1992) 125]. Accordingly 12.5 mg of the individual compounds was dissolved in 300 μl DMSO and this solution was diluted to 1 ml with physiological saline. From this solution a dose of 12.5 mg/kg was administered orally to the animals 3h prior to provoking the thrombus.
In Table 1 the antithrombotic activity of several representatives of the compounds of the invention is given in percentage of the inhibition. Beciparcil (4-cyanophenyl 1 ,5-dithio-β-D-xylopyranoside, EP 365.397) was used as reference compound.
Table 1.
The oral antithrombotic activity of compounds of formula (I) in rats at a dose of
12.5 mg/kg
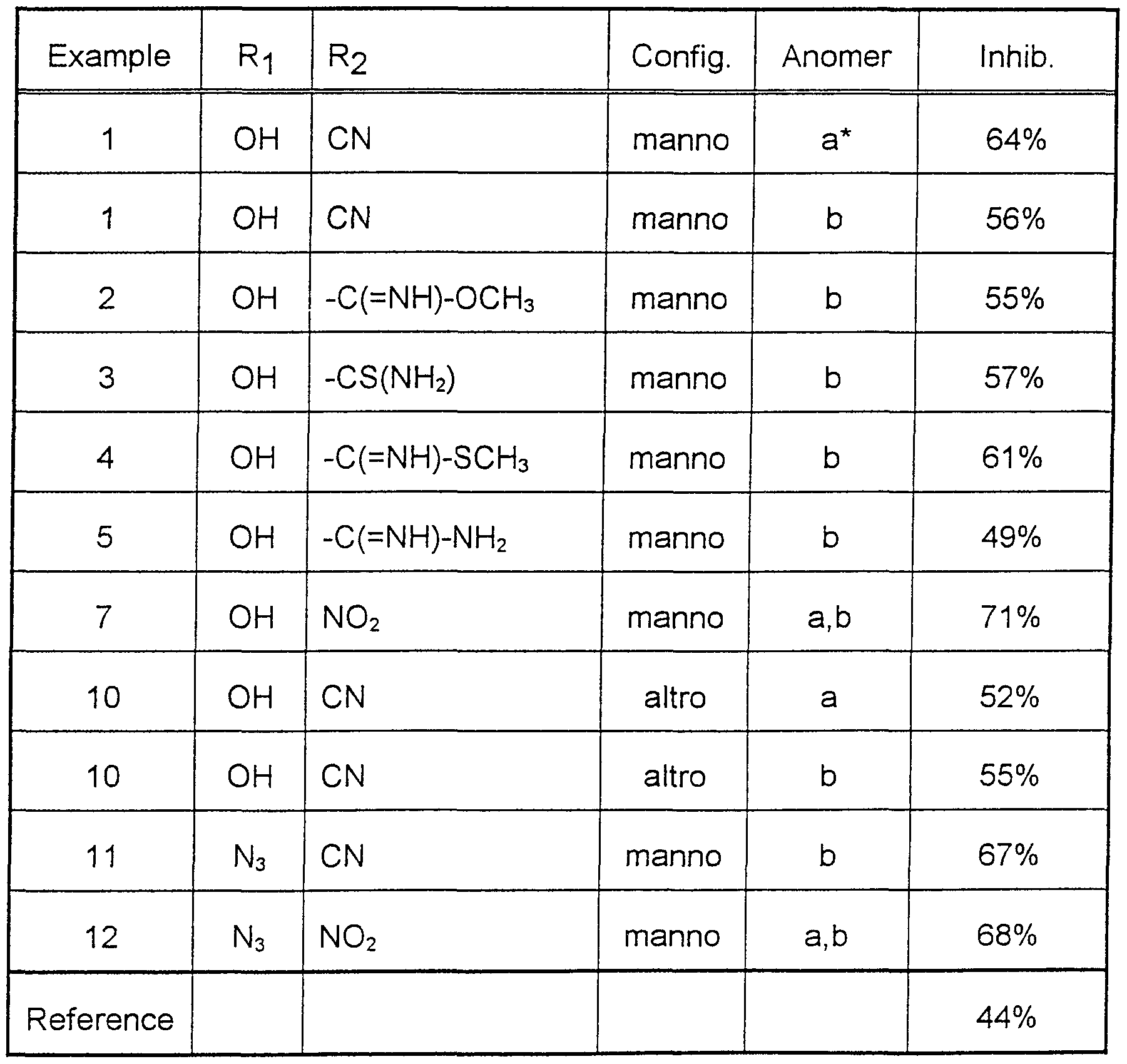
3:1 mixture of a, b-anomers
As can be seen from Table 1. the antithrombotic activity of several representatives of the compounds of formula (I) of the present invention exceeds that of the reference, in certain cases to a significant degree.
For therapeutical purposes, the compounds of the present invention as well as their pharmaceutically acceptable salts can be used as such or suitably in the form of pharmaceutical compositions. These compositions also fall within the scope of the present invention.
These pharmaceutical compositions contain an amount required to excert the therapeutical effect of a compound of formula (I) or its pharmaceutically acceptable salt, in admixture with known carriers, excipients, diluents and/or other additives commonly used in the pharmaceutical practice.
For oral administration the antithrombotic compound is formulated in capsules or tablets which may contain excipients such as binders, lubricants, disintegration agents and the like. For parenteral administration the antithrombotic compound is formulated in a pharmaceutically acceptable diluent, e.g. physiological saline (0.9 %), 5% dextrose, Ringer's solution and the like.
The doses required to excert the therapeutical effect of the compounds according to the invention may be varied depending on the individual condition and age of the patient to be treated and finally these doses are determined by the attending physician. However, for the prevention and/or treatment of diseases, where the application of an anticoagulant is desirable, daily doses of these compounds falling between about 0.01 mg/kg of body weight and about 100 mg/kg of body weight and preferably between about 0.1 mg/kg of body weight and about 10 mg/kg of body weight are used by the oral or parenteral, e.g. intravenous, route.
The compounds according to the invention and the process for the preparation thereof are illustrated in detail by the following not limiting Examples.
The Rf values given in the experimental part were determined by TLC, using E. Merck precoated Silica Gel 60 F254 plates, with the following solvents:
(A) benzene - methanol (9:1)
(B) benzene - methanol (4:1)
(C) benzene - acetone (9:1)
(D) hexane - ethyl acetate (1 :1) (E) hexane - ethyl acetate (2: 1 )
(F) hexane - ethyl acetate (4:1)
(G) carbon tetrachloride - ethyl acetate (4:1)
(H) ethyl acetate - pyridine - water - acetic acid (60:20:11 :6)
Spots were detected by spraying the plates with a 0.02 M solution of iodine and a 0.30 M solution of potassium iodide in 10% aq sulfuric acid solution followed by heating at ca. 200 °C. For column chromatography, Kieseigel 60 was used. Melting points are uncorrected. Optical rotations were determined at 20 °C. NMR spectra were recorded with a Varian XL-400 spectrometer at 400 MHz (1 H) and 100 MHz ( 3C) or with a Bruker AC 250 spectrometer at 250 MHz \-\) and 62.9 MHz (^ 3C) (Me4Si was used as internal standard). Multiplicities of the 13C NMR spectra were obtained from DEPT experiments. The assignment of the protons were based on homonuclear decoupling and DNOE experiments. Connectivities between identified protons and protonated carbons were determined by HETCOR experiments. MS spectra were recorded with a Finnigan MAT 8430 mass spectrometer. In the case of FAB spectra samples were dissolved in 3-nitrobenzaldehyde or in glycerin.
The "usual processing" during the work-up of acylation reactions, carried out in pyridine means, that if the product did not crystallize on pouring the reaction mixture on ice-water, it was extracted with dichloromethane and the organic solution was washed with 1 M sulfuric acid until a pH of ~3 was reached, then with water, with 5% aq sodium hydrogen carbonate and finally with water. Organic solutions were dried over sodium sulfate prior to concentration which was carried out under diminished pressure.
Example 1 4-Cyanophenyl 2,6-anhydro-1,2-dithio-α-D-mannopyranoside and 4- cyanophenyl 2,6-anhydro-1,2-dithio-β-D-mannopyranoside (la and lb, R- - OH, R2 = CN)
To a solution of 1.7 g 1 :4 mixture of c_,β-anomers of formula (III) in 20 ml of methanol 0.1 ml of 1 M methanolic sodium methoxide was added and the reaction mixture was stirred at room temperature for 1 h. Thereafter the solution was neutralized with carbon dioxide, the precipitated crystals were filtered off and washed with methanol to yield 0.5 g (38%) of the title β-anomer (lb). Mp:
181-184 °C, Rf (A) = 0.3, [α]p = -171° (c = 0.3, methanol). Concentration of the mother liquor and purification by column chromatography (solvent A) gave further 0.44 g (33%) of the β-anomer of formula (lb). Concentration of the mother liquor gave 0.35 g (26%) of the title anomeric mixture of formula (la and lb), which contains the α- and β-anomers in a 3:1 ratio. NMR (DMSO-dg), (la)
1 H: 5.99 (H-1), 2.98 (H-2), 4.15 (H-3), 3.55 (H-4) 4.08 (H-5), 3.09 (H-6a), 2.81 (H-6b), 5.28 and 5.34 (OH), 7.58 and 7.78 (aromatic H); J 2 1 -3, J2,3 3.1 , ^3,4
3.1 , J4,5 < 1 , ^5,6a 3.6, J5,6b 2.0, J6aβb 11.6 Hz; 13c: 87.4 (C-1 ), 43.0 (C-2),
76.4, 73.6 and 73.3 (C-3,4,5), 29.1 (C-6), 143.1, 132.8, 128.8, 108.4 (aromatic
C), 118.9 ppm (CN). (lb) H: 6.03 (H-1), 3.00 (H-2), 3.94 (H-3), 3.56 (H-4) 4.02 (H-5), 3.26 (H-6a), 2.80 (H-6b), 5.24 and 5.38 (OH), 7.60 and 7.76 (aromatic H); ^1 ,2 2.8, J2,3 3.2, J3 3.2, J4ι5 < 1 , J5βa 3.1 , J5|6b 2.5, 6a)6b 11.8 Hz; 13C: 87.6 (C-1), 44.3 (C-2), 76.7, 76.1 and 73.1 (C-3,4,5), 29.2 (C-6), 144.6, 132.7, 128.5, 108.0 (aromatic C), 119.0 ppm (CN).
The starting anomeric mixture of formula (III) can be prepared the following way:
Step a)
Methyl 3,4-di-0-acetyl-2,6-anhydro-2-thio-a-D-mannopyranoslde (IV, R = Me)
To a solution of 1.3 g methyl 2,6-anhydro-2-thio- -D-mannopyranoside [I.I. Cubero et al., Carbohydr. Res., 242, 109 (1993)] in 3 ml of py dine 3 ml of acetic anhydride was added. The reaction mixture was kept overnight at room temperature to give after usual processing 1.55 g (83%) of the title compound. Rf (D) = 0.7, [α]p = -31 ° (c = 0.5, chloroform). NMR (CDCI3), 1 H: 5.12 (H-1), 3.10 (H-2), 5.47 (H-3), 4.81 (H-4) 4.30 (H-5), 3.05 (H-6a), 2.83 (H-6b), 3.46 (OMe), 2.11 and 2.09 (OAc); J1 2 2.0, J2,3 3.4, 3)4 3.4, 4,5 - 1 , 5|6a -2, J5ι6b .0, J6aι6b 11.5 Hz.
Step b)
1,3,4-Tή-0-acetyl-2,6-anhydro-2-thio-D-mannopyranoside (IV, R = Ac)
To a stirred solution of the product obtained in the previous step (1.55 g) in 3.5 ml of acetic anhydride 0.01 ml of concentrated suifuric acid was added at 0 °C and stirring was continued for 15 min. The pH of the solution was adjusted
to ~4 with sodium acetate, 20 ml of ice-cold 6% aq sodium hydrogen carbonate was added and stirring was continued for 2 h at room temperature. Then the mixture was extracted with chloroform, washed with water, dried over sodium sulfate and concentrated. The residue was purified by column chromatography (solvent £) to yield 1.4 g (82%) of the title compound, which is a 1 :2 mixture of the - and β-anomers. Rf (£) = 0.4; NMR (CDCI3), (IVα) 1 H: 6.36 (H-1), 3.14 (H-2), 5.46 (H-3), 4.84 (H-4) 4.38 (H-5), 3.13 (H-6a), 2.85 (H-6b), 2.18-2.08 (OAc); J1 (2 2.3, J2,3 3.4, _ 3.4, J4 1.1 , J5 6a 4.3, J5τ6b 1.9, J6aβb 11.7 Hz. 1 3C: 94.5 (C-1), 36.8 (C-2), 75.1 , 71.6 and 70.2 (C-3,4,5), 28.1 (C-6), 170.5-169.5 (CO), 21.2-21.9 ppm (OAc). (IVβ) 1 |-l: 6.28 (H-1 ), 3.25 (H-2), 5.19
(H-3), 5.05 (H-4) 4.25 (H-5), 3.29 (H-6a), 2.91 (H-6b), 2.18-2.08 (OAc); J1 2 3.5, 2,3 2-7, J3ι4 4.1 , J4 0.5, J5βa 2.9, J5βb 3.0, J6aβb 12.0 Hz. 13Q: 92.4 (C- 1 ), 39.0 (C-2), 75.2, 74.5 and 70.0 (C-3,4,5), 29.0 (C-6), 170.5-169.5 (CO), 21.2-21.9 ppm (OAc).
Step c)
4-Cyanophenyl 3,4-di-0-acetyl-2, 6-anhydro-1,2-dithio-D-mannopyranoside (III)
Under argon, to a stirred solution of 1.8 g of 1 ,3,4-th-0-acetyl-2,6- anhydro-2-thio-D-mannopyranose (IV, R = Ac) in 65 ml of dry dichloromethane 1.7 g of 4-cyanothiophenol was added at -10 °C and thereafter 1.2 ml of trimethylsilyl triflate. The reaction mixture was stirred at room temperature for 2 h, neutralized with triethylamine, concentrated and the residue was purified by column chromatography (solvent E) to yield 1.7 g (76%) of the title compound, which is 1 :4 mixture of the α- and β-anomers [Rf (E) = 0.5]. When the reaction was carried out at -10 °C for 1.5 h, a 1 :1 mixture of the α- and β-anomers was formed. NMR (CDCI3), (illct) "Η: 5.93 (H-1), 3.24 (H-2), 5.54 (H-3), 4.98 (H-4)
4.35 (H-5), 3.06 (H-6a), 3.01 (H-6b), 7.60-7.50 (aromatic H) 2.13 and 2.12 ppm (OAc); J1 )2 <1 , J2,3 3.4, J3|4 3.4, J4 < 1 , J5,6a 3.7, J5βb 2.4, J6aβb 11.8 Hz; 3C: 87.8 (C-1), 38.8 (C-2), 75.8, 73.4 and 70.3 (C-3,4,5), 28.9 (C-6), 170.4 and 170.3 (CO), 142.8-110.0, (aromatic C), 1 18.6 (CN), 20.9 ppm (OAc). (Illβ) 1 H: 5.02 (H-1), 3.39 (H-2), 5.20 (H-3), 5.08 (H-4) 4.31 (H-5), 3.60 (H-6a), 2.98 (H-6b), 7.60-7.50 (aromatic H) 2.14 and 2.1 1 ppm (OAc); 2 3.1 , 2,3 2.9, J3,4 4.2, J4ι5 < 1 , J5βa 3.0, J5βb 3.0, J6aβb 12.0 Hz; C: 88.0 (C-1), 40.6 (C-2), 75.9, 75.6 and 70.2 (C-3,4,5), 28.9 (C-6), 170.4 and 170.3 (CO), 142.8- 110.0, (aromatic C), 118.6 (CN), 20.9 ppm (OAc).
Example 2
4-(lmino) (methoxy) methyl phenyl 2, 6-anhydro- 1, 2-dithlo-b-D-mannopyranoside
(lb, R1 = OH, R2 = -C(=NH)-OCH3)
To a solution of 380 mg of 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-β-D- mannopyranoside (lb, R-| = OH, R2 = CN) in 40 ml of dry methanol 0.1 ml of 1 M methanolic sodium methoxide was added. After 24 h at room temperature, the mixture was neutralized with carbon dioxide and concentrated. The residue was submitted to column chromatography (solvent A) to give, on concentration of the first fraction [Rf (A) = 0.3], the unchanged starting material (250 mg). Concentration of the second fraction [Rf (A) - 0.2 ] yielded 120 mg (83% counted on the recovered starting material) of the title compound. Mp: 164-168 °C, [α]D = -157° (c = 0.5, methanol). NMR (DMSO-d6), 1 H: 5.90 (H-1), 3.00 (H- 2), 3.93 (H-3), 3.54 (H-4) 4.01 (H-5), 3.18 (H-6a), 2.78 (H-6b), 5.38 and 5.25 (OH), 7.50 and 7.80 (aromatic H), 8.95 (NH), 3.78 ppm (OMe); J ]2 2.7, J2,3 2.7, J3 4 3.5, 4j5 <1 , J5βa 3.0, J5βb 2.4, J6aβb 1 1.7 Hz.
Example 3
4-(Amino-thiocarbonyl)phenyl 2, 6-anhydro- 1, 2-dithio-b-D-mannopyranoside (lb,
R1 = OH, R2 = -CS(NH2))
A solution of 0.75 g of 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-β-D- mannopyranoside (lb, R-| = OH, R2 = CN) in 15 ml of pyhdine and 15 ml of triethylamine was saturated with a slow stream of dry hydrogen sulfide for 5 h. The mixture was kept at room temperature overnight and was then concentrated. The residue was recrystallized from methanol to yield 0.74 g (88%) of the title compound. Mp: 189-194 °C, Rf (S) = 0.3, [α]p = -179° (c = 0.5, methanol). NMR (DMSO-d6), 1 H: 5.92 (H-1), 3.01 (H-2), 3.91 (H-3), 3.55 (H-4) 4.01 (H-5), 3.26 (H-6a), 2.78 (H-6b), 5.35 and 5.24 (OH), 7.45 and 7.88 (aromatic H), 9.45 and 9.80 (NH2); J| ,2 2-9, 2,3 2-8, J_ 4 4.0, J4 5 <1 , J5 βa 3.0, J5βb 2.6, J6a)6b 11.7 Hz; 1 3C: 88.1 (C-1), 44.6 (C-2), 76.8 76.1 and 73.0 (C-3,4,5), 29.2 (C-6), 199.2 (CSNH2), 140.9, 136.9, 128.1 and 128.0 ppm
(aromatic C).
Example 4
4-(lmino)(methylthio)methylphenyl 2, 6-anhydro-1,2-dithio-b-D-mannopyranoside (lb, ?,= OH, R2 = -C(=NH)-SCH3)
To a solution of 450 mg of 4-(aminothiocarbonyl)phenyl 2,6-anhydro-1 ,2- dithio-β-D-maπnopyranoside (lb, R^ = OH, R2 = -CS(NH2) ) in 65 ml of acetone 0.5 ml of methyl iodide was added and the reaction mixture was refluxed for 3 h. The precipitated product was filtered after cooling and was washed with acetone to yield 600 mg (93%) of the title compound as its hydroiodide. Mp: 203-205 °C, Rf (B) = 0.5.[C_]D = -132° (c = 0.58, pyhdine). NMR (DMSO-d6), 1 H:
6.07 (H-1 ), 3.03 (H-2), 3.08 (H-3), 3.58 (H-4) 4.04 (H-5), 3.26 (H-6a), 2.82 (H- 6b), 4.00 and 3.85 (OH), 7.65 and 7.82 (aromatic H), 12.0 (NH), 2.85 ppm (SMe); J1 ι2 2.6, J2β 2.8, J3|4 3.3, J4β <1 , 5 6a 2.0, J5βb 1.5, J6aβb 11.8 Hz; 1 3C: 87.3 (C-1), 44.2 (C-2), 76.6 76.0 and 73.1 (C-3,4,5), 20.2 (C-6), 188.0 (CSMeNH), 147.3, 128.6, 128.2 es 127.0 (aromatic C), 15.7 ppm (SMe).
Example 5
4-Amidinophenyl 2,6-anhydro-1,2-dithio-b-D-mannopyranoside (lb, R, = OH, R2
-C(=NH)-NH2)
To a stirred solution of 200 mg of 4-(imino)(methylthio)methylphenyl 2,6- anhydro-1 ,2-dithio-β-D-mannopyranoside hydroiodide (lb, R-j = OH, R2 =
-C(=NH)-SCH3) in 10 ml of dry ethanol 120 mg of ammonium acetate was added and stirring was continued at 60 °C for 4 h. The reaction mixture was cooled and the precipitated product was filtered off to yield 100 mg (63%) of the title compound as its acetate. Mp: 208-210 °C, Rf (H) = 0.7, [ ]p = -192° (c =
0.28, DMSO). TS: 312 (M)+
Example 6 4-(Hydrazino) (imino)methylphenyl 2, 6-anhydro-1, 2-dlthio-b-D-mannopyranoside (lb, R1 = OH, R2 = -C(=NH)-NHNH2)
To a stirred solution of 300 mg of 4-(imino)(methylthio)methylphenyl 2,6- anhydro-1 ,2-dithio-β-D-mannopyranoside hydroiodide (lb, R-| = OH, R2 = -C(=NH)-SCH3) in 30 ml of dry ethanol 1.1 ml of 98% hydrazine hydrate was added and stirring was continued at 20 °C for 24 h. The precipitated product was filtered off and washed with ethanol to yield 180 mg (86%) of the title
compound. Mp: 171-175 °C, Rf (H) = 0.8, [α]p = -159° (c = 0.5, DMSO). NMR (DMSO-d6), 1 H: 5.78 (H-1), 2.98 (H-2), 3.88 (H-3), 3.54 (H-4) 4.00 (H-5). 3.28 (H-6a), 2.76 (H-6b), 4.5-6.0 (OH, NH), 7.62 and 7.38 (aromatic H); J^2 2.7, ^2,3 2.5, J3ι4 3.2, J4 <1 , J5 6a 2.8, J5βb 2.0, J6aβb 11.5 Hz; 13c: 88.9 (C- 1), 44.8 (C-2), 76.8 76.2 and 72.9 (C-3,4,5), 29.3 (C-6), 145.5 [C(=NH)NHNH2],
136.1 , 133.6, 129.3 and 125.8 (aromatic C).
Example 7
4-Nitrophenyl 2,6-anhydro-1,2-dithio-D-mannopyranoside (la and lb, Ri - OH, R2 N02)
To a solution of a 1 :2 ,β-anomeric mixture of 270 mg of 4-nitrophenyl 3,4-di-0-ace.yl-2,6-anhydro-1 ,2-dithio-D-mannopyranoside (V) in 15 ml of dry methanol 0.1 ml of 1 M methanolic sodium methoxide was added and the reaction mixture was stirred at room temperature for 1 h. Thereafter the solution was neutralized with Dowex 50 WX (H+) resin and concentrated to yield 200 mg (94%) of the title compound, which is a 1 :2 mixture of the α.β-anomers. Rf (A) = 0.3. NMR (DMSO-d6), (la) 1 H: 6.03 (H-1), 2.98 (H-2), 4.14 (H-3), 3.54 (H- 4) 4.08 (H-5), 3.08 (H-6a), 2.80 (H-6b), 5.44 and 5.35 (OH), 7.58 and 8.14 ppm (aromatic H); J1 [2 1.3, J2)3 3.1 , J_, 3.0, J4β <1 , J5 6a 3.8, J5βb 2.5, J6aβb
11.7 Hz. (lb) H: 6.05 (H-1 ), 3.03 (H-2), 3.94 (H-3), 3.58 (H-4) 4.03 (H-5), 3.25 (H-6a), 2.78 (H-6b), 5.48 and 5.36 (OH), 7.60 and 8.12 ppm (aromatic H); 1 2 2.9, J2,3 3.0, J3]4 3.0, J4 <1 , J5 6a 3.1, J5ι6b 2.5, J6aβb 11.8 Hz.
The starting anomeric mixture of formula (V) can be prepared the following way:
4-Nitrophenyl 3, 4-di-0-acetyl-2, 6-anhydro- 1, 2-dithio-D-mannopyranoside
Under argon, to a stirred solution of 0.4 g of 1 ,3,4-tri-0-acetyl-2,6- anhydro-2-thio-β-D-mannopyranose (IV, R = Ac) in 20 ml of dry dichloromethane 0.4 g of 4-nitrothiophenol and 0.25 ml of trimethylsilyl triflate were added at -10 °C, then the reaction mixture was stirred for 1 h at room 5 temperature. After neutralizing with triethylamine the mixture was washed with water, dried and concentrated. The residue was submitted to column chromatography (solvent E) to yield 270 mg (51 %) of the title compound, which is a 1 :2 mixture of the α- and β-anomers. Rf (E) = 0.5. NMR (CDCI3), (Vα) 1 H: 5.98 (H-1), 3.26 (H-2), 5.54 (H-3), 4.99 (H-4) 4.47 (H-5), 3.28 (H-6a), 3.02 OHI O 6b), 2.16 and 2.12 (OAc), 7.62 and 8.12 ppm (aromatic H); >2 1 -6, J2,3 3.4, J3,4 3.4, J4β 1.0, J5.6a 3.7, 5|6b 2.3, J6aβb 12.0 Hz. (Vβ) H: 5.97 (H-1), 3.40 (H-2), 5.22 (H-3), 5.09 (H-4) 4.32 (H-5), 3.59 (H-6a), 3.00 (H-6b), 2.14 and 2.07 (OAc), 7.62 and 8.18 ppm (aromatic H); J 2 3-°> 2,3 3.0, 3 4 4.2, J4 5 <1 , J5.6a 3.0, J5,6b 2-9, 6a,6b 12.0 Hz. 15
Example 8
4-Nitrophenyl 2,6-anhydro-1,2-dithio-b-D-mannopyranoside (lb, Rη = OH, R2 =
Λ/O2J
0 Under argon, to a stirred solution of 1.0 g of 1 ,3,4-tri-0-acetyl-2,6- aπhydro-2-thio-β-D-mannopyranose (IV, R = Ac) in 20 ml of dry 1 ,2- dichloroethane 0.7 g of 4-nitrothiophenol and 0.4 ml of boron trifluoride etherate were added at 20 °C. After 24 h at room temperature the reaction mixture was poured into 20 ml of ice-cold 6% aq sodium hydrogen carbonate, 5 separated and the organic layer was washed with 6% aq sodium hydrogen carbonate and water, dried over sodium sulfate and concentrated to yield 1.3 g of a syrup, which is a 1 :4 mixture of the a, b-anomers of formula (V) [Rf (E) =
0.5]. The so obtained mixture was dissolved in a mixture of 30 ml of dry methanol and 20 ml of dry dichloromethane and 0.1 ml of 1 M methanolic sodium methoxide was added. After 1 h at room temperature the reaction mixture was neutralized with carbon dioxide, the precipitated crystals were filtered off, washed with methanol and ether to yield 450 mg (43%) pure b- anomer (lb, R-j = OH, R2 = N02). Mp: 202-204 °C, Rf (A) = 0.3, [α]D = -243° (c = 0.5, pyhdine).
Example 9 4-Acetamidophenyl 2,6-anhydro-1 ,2-dithio-b-D-mannopyranoside (lb, R-j - OH, R2 = NHCOCH3)
To a stirred solution of 180 mg of 4-nitrophenyl 2,6-anhydro-1 ,2-dithio-b- D-mannopyranoside (lb, R-i = OH, R2 = NO2) in 45 ml of ethanol 90 mg of sodium borohydride and 10 mg of πickel(ll) chloride hexahydrate were added and stirring was continued for 30 min. The reaction mixture was neutralized with aq HCI, filtered and concentrated. The residue was dissolved in a mixture of 8 ml of pyhdine and 4 ml of acetic anhydride and was kept at room temperature for 24 h. After usual processing the residue was dissolved in 10 ml of dry methanol and 0.1 ml of 1 M methanolic sodium methoxide was added. After 1 h at room temperature the reaction was neutralized with carbon dioxide, concentrated and submitted to column chromatography (solvent B) to yield 100 mg (54%) of the title compound. Mp: 246-249 °C, Rf (A) = 0.2, [ ]o = -169° (c = 0.5, pyhdine). NMR (DMSO-d6), 1 H: 5.62 (H-1), 2.98 (H-2), 3.85 (H-3), 3.54 (H- 4) 3.96 (H-5), 3.28 (H-6a), 2.76 (H-6b), 5.0-5.5 (OH), 10.00 (NH), 2.00 (Ac),
7.54 and 7.40 (aromatic H); J ^2 2.7, J2|3 2.5, J3 4 3.2, J4 5 <1 , J5 6a 2.9, ^5,6b 2.1 , 6a,6b H -2 Hz.
Example 10
4-Cyanophenyl 2,6-anhydro-1,2-dithio-α-D-altropyranoside and 4-cyanophenyl
2,6-anhydro-1,2-dlthio-β-D-altropyranoside (Ic and Id, Rη = OH, R2 = CN)
A 2:3 anomeric mixture of 730 mg of the compound of formula (VI) was dissolved in 30 ml of dry methanol and 0.1 ml of 1 M methanolic sodium methoxide was added to the solution at room temperature. After 1 h the reaction was neutralized with carbon dioxide, concentrated and the residue was submitted to column chromatography (solvent A). Concentration of the first fraction [Rf (A) = 0.3] gave 300 mg (53%) of a mixture of α,β-anomers (Ic + Id), while concentration of the second fraction [Rf (A) = 0.2] yielded 170 mg (30%) of pure β-anomer of formula (Id). Mp: 160-165 °C, Rf (A) = 0.2, [α]p = -1 14° (c = 0.5, methanol). Repeated column chromatography of the first fraction gave 130 mg (20%) of the pure α-anomer of formula (Ic). Mp: 144-148 °C, Rf (A) = 0.3, [CX]D = +273° (c = 0.5, methanol). NMR (DMSO-d6), (Ic) 1 H: 5.88 (H-1), 3.13 (H- 2), 4.19 (H-3), 3.92 (H-4) 4.15 (H-5), 3.12 (H-6a), 2.83 (H-6b), 5.10 and 5.56 (OH), 7.55 and 7.75 ppm (aromatic H); J-| 2 ~0. ^2,3 3-4. J3,4 8-8> 4,5 1 -°> ^5.6a 3.8, J5βb 1.9, 6aι6b 11.7 Hz. (Id) 1 H: 5.98 (H-1 ), 3.14 (H-2), 4.20 (H-3), 3.88 (H-4), 4.07 (H-5), 3.25 (H-6a), 2.80 (H-6b), 5.06 and 5.60 (OH), 7.58 and 7.77 ppm (aromatic H); J1 >2 2.9, J2j3 3.4, J3 4 8.1 , J4 0.8, J5 6a 3.1 , J5βb 2.2, J6aι6b 1 1.7 Hz.
The starting anomeric mixture of formula (VI) can be prepared the following way:
4-Cyanophenyl 3, 4-di-0-acetyl-2, 6-anhydro- 1, 2-dithio-a,β-D-altropyranoside
Under argon, to a stirred solution of 700 mg of 1 ,3,4-tri-0-acetyl-2,6- anhydro-2-thio-β-D-altropyranose (VII) [K. Toshima et al., Tetrahedron Letters, 33, 1491 (1992)] in 25 ml of dry dichloromethane 0.6 g of 4-cyanothiophenol and 0.44 ml of trimethylsilyl triflate were added at 0 °C and stirring was continued for 30 min at 0 °C. After neutralizing with t ethylamine the reaction mixture was washed with water, dried, filtered and concentrated. The residue was submitted to column chromatography (solvent E) to yield 730 mg (51 %) of the title compound [Rf (E) = 0.4], which is a 2:3 mixture of the - and β- anomers. NMR (CDCI3), (Via) 1 H: 5.85 (H-1), 3.25 (H-2), 5.52 (H-3), 5.12 (H-4), 4.44 (H-5), 3.30 (H-6a), 3.90 (H-6b), 2.14 and 2.18 (OAc), 7.45 - 7.65 ppm
(aromatic H); J1 2 ~0, 2j3 3.7, J3| 8.5, 4 5 1.0, J5 6a 2.7, J5βb 2.7, J6aβb 12.0 Hz. 13(3: 86.8 (C-1), 36.8 (C-2), 67.0, 68.3 and 69.8 (C-3,4,5), 27.8 (C-6), 170.2 - 169.5 (CO), 144.3 - 109.8 (aromatic C), 118.5 (CN), 20.8 - 20.6 ppm (OAc). (Vlβ) 1 H: 5.96 (H-1), 3.26 (H-2), 5.58 (H-3), 5.15 (H-4), 4.35 (H-5), 3.61 (H-6a), 2.84 (H-6b), 2.10 and 2.12 (OAc), 7.45 - 7.65 ppm (aromatic H); J 2 3.2, J2,3 4.1 , J3ι4 8.4, J4β 0.5, J5 6a 2.7, J5βb 2.7, 6aj6b 12-0 Hz. 13c: 84.6 (C-1), 37.4 (C-2), 68.0, 68.5 and 69.9 (C-3,4,5), 28.1 (C-6), 170.2 - 169.5 (CO), 144.3 - 109.8 (aromatic C), 118.6 (CN), 20.8 - 20.6 ppm (OAc).
Example 11
4-Cyanophenyl 2, 6-anhydro-3-azido-3-deoxy-1,2-dithio-α-D-mannopyranoside and 4-cyanophenyl 2, 6-anhydro-3-azido-3-deoxy-1 , 2-dithio-β-D-mannopyranoside (la and lb, Rή = Λ/3, R2 = CN)
Under argon, to a stirred solution of 400 mg of 1 ,4-di-0-acetyl-2,6- anhydro-3-azido-3-deoxy-2-thio-D-mannopyranose (VIII) in 15 ml of dry dichloromethane 0.4 g of 4-cyanothiophenol and 0.3 ml of trimethylsilyl triflate
were added at -10 °C. The reaction mixture was stirred at room temperature for 2 h, then quenched with triethylamine, concentrated and the residue was submitted to column chromatography (solvent F) to yield the compound of formula (IX) (450 mg) as a 1 :1 mixture of the - and β-anomers [Rf (F) = 0.4]. The so obtained mixture was dissolved in 15 ml of dry methanol and 0.1 ml of 1 M methanolic sodium methoxide was added. After 1 h at room temperature the reaction was neutralized with carbon dioxide, concentrated and the residue was submitted to column chromatography (solvent A) to yield 250 mg (56%) of a 1 :1 mixture of the title compounds (la + lb). This was purified by a second column chromatography (solvent G). Concentration of the first fraction [Rf (G) = 0.25] gave the pure α-anomer (la); Mp: 117-110 °C, {a r_ = +133° (c = 0.4, methanol). Concentration of the second fraction [Rf (G) = 0.2] yielded the pure β-anomer (lb); Mp: 143-145 °C, [α]o = -118° (c = 0.5, methanol). NMR (DMSO-d6), (la) 1 H: 6.00 (H-1), 4.08 (H-3), 4.17 (H-5), 3.15 (H-6a), 2.03 (H-6b); J1 j2 1.2, 2,3 2.8, J3 4 3.9, J5.6a 3.3, J6a,6b H -8 Hz; (lb) 1 H: 6.08 (H-1 ), 3.36 (H-2), 3.98
(H-3), 3.76 (H-4) 4.15 (H-5), 3.34 (H-6a), 2.92 (H-6b), 5.76 (OH), 7.60 and 7.80 ppm (aromatic H); J^ 2 3-0, J2β 2-5, J3 4 4.1 , J4 5 <1 , J_ Qa 3.0, J_βb 2.8, ^6a,6b H -8 Hz: 1 3C: 87.8 (C-1), 41.4 (C-2), 68.1 (C-3), 73.1 and 72.9 (C-4,5), 29.1 (C-6), 119.0 (CN), 143.9, 132.8, 128.7 and 108.3 ppm (aromatic C).
The starting compound of formula (VIII) can be prepared the following way:
Step a) Methyl 3-azido-4, 6-0-benzylidene-2-S-benzoyl-3-deoxy- -D-mannopyranoside (XI)
Under argon, to a stirred solution of 5.8 g of methyl-3-azido-4,6-0- benzylidene-3-deoxy-α-D-glucopyranoside (X, R = OH) [R. D. Guthrie and D. Murphy, J. Chem. Soα, (1963) 5288] in 60 ml of dry dichloromethane and 60 ml of pyhdine 5 ml of trifluoromethanesulfonic anhydride was added at -20 °C. The mixture was stirred for 1 h at room temperature to give after usual processing 6 g of the ester of formula (X, R = OTf). This was dissolved in 100 ml of dry N,N- dimethylformamide, 5 g of potassium thiobenzoate was added to the solution and the reaction was stirred at room temperature for 20 h. After concentration the residue was dissolved in dichloromethane, washed with water, dried and concentrated. The so obtained mixture was purified by column chromatography (solvent F) to yield 3.8 g (47%) of the title compound. Rf (F) = 0.5, [α]p = -28° (c = 0.5, chloroform). NMR (CDCI3), 1 H: 4.78 (H-1), 4.45 (H-2), 4.52 (H-3), 3.78 (H-4) 3.97 (H-5), 4.30 (H-6a), 3.84 (H-6b), 5.66 (PhCH), 7.35-7.65 and 8.02 (aromatic H), 3.42 ppm (OMe); J1 2 1.1 , J β 4.8, J3 4 10.0, J4 5 9.5, J5 6a 4.4, J5ι6b 9.8, J6aι6b 10-9 Hz.
Step b)
Methyl 2, 6-anhydro-3-azido-3-deoxy-2-thio-a-D-mannopyranoslde (XIII, R = H)
To a stirred solution of 2.4 g of the product of formula (XI) obtained in the previous step in 70 ml of carbon tetrachloride 1.1 g of N-bromosuccinimide and 4 g of barium carbonate were added and the mixture was stirred at reflux temperature for 3 h. After cooling to room temperature the mixture was filtered, the filtrate was concentrated, the residue was dissolved in ether, washed with water, dried and concentrated. The so obtained thiobenzoate of formula (XII) was dissolved in 100 ml of dry methanol and 3.5 ml of 3 M methanolic sodium methoxide was added. After standing at room temperature overnight, the
reaction was neutralized with carbon dioxide, concentrated and the residue was submitted to column chromatography (solvent E) to yield 0.65 g (46%) of the title compound. Rf (E) = 0.3, [α]β = -68° (c = 0.37, chloroform). NMR (CDCI3), H: 5.08 (H-1 ), 2.98 (H-2), 4.20 (H-3), 3.55 (H-4) 4.28 (H-5), 3.05 (H-6a), 2.68 (H-6b), 3.18 (OH), 3.50 ppm (OMe); J1 j2 2.4, J2β 4.0, J3 4 2.2, J4 2.0, J5 6a
5-0, J5,6b 1 -5, ^6a,6b H -6 Hz; 13C: 101.9 (C-1), 38.4 (C-2), 63.2, (C-3), 73.0 and 71.4 (C-4,5), 27.3 (C-6), 55.6 ppm (OMe).
Step c) Methyl 4-0-acetyl-2, 6-anhydro-3-azido-3-deoxy-2-thio-c-D-mannopyranoside (XIII, R = Ac)
To a stirred solution of 0.6 g of the product obtained in the step b) in 6 ml of pyhdine 3 ml of acetic anhydride was added and the reaction was kept at room temperature for 1 h. Usual processing yielded 0.6 g (96%) of the title compound, Rf (£) = 0.3, [α]p = -21° (c = 0.5, chloroform). NMR (CDCI3), 1 H: 5.12 (H-1), 2.90 (H-2), 4.32 (H-3), 4.70 (H-4) 4.30 (H-5), 3.05 (H-6a), 2.82 (H- 6b), 2.14 (OAc), 3.46 ppm (OMe); J1 ]2 1 -7, J2,3 3.1 , ^3,4 -0, J4 0.7, 5.6a 4.0, J5βb 2.0, Jβa,6b H -6 Hz; 13C: 102.0 (C-1), 39.3 (C-2), 60.4, (C-3), 75.8 (C-4), 69.3 (C-5), 28.6 (C-6), 170.4 (CO), 21.0 (OAc) 55.3 ppm (OMe).
Step d)
1, 4-Di-0-acetyl-2, 6-anhydro-3-azido-3-deoxy-2-thio-D-mannopyranose (VIII)
To a stirred solution of 0.6 g of the methyl glycoside obtained in the previous step in 7 ml of acetic anhydride, 0.1 ml of concentrated sulfuric acid was added at 0 °C, and the mixture was stirred for 15 min at 0 °C. Then the pH
was adjusted to 4 by adding sodium acetate, thereafter 50 ml of ice-cold 6% aq sodium hydrogen carbonate was added to the mixture and stirring was continued at room temperature for 2 h. Then the mixture was extracted with chloroform, washed with water, dried and concentrated. The residue was submitted to column chromatography (solvent £) to yield 0.46 g (69%) of the title compound as a 1 :1 mixture of the α and β-anomers. Rf (E) = 0.6. NMR (CDCI3), (Villa) 1 H: 6.35 (H-1), 2.96 (H-2), 4.30 (H-3), 4.77 (H-4) 4.40 (H-5), 3.14 (H-6a), 2.85 (H-6b), 2.14 and 2.13 ppm (OAc); 1 |2 2.2, J2_3 3.2, J3 4 3.9, ^4,5 1 -2, v/5.6a 4.1 , J5βb 2.2, J6aβb 11.7 Hz; 18C: 94.2 (C-1), 38.2 (C-2), 60.2, (C-3), 75.3 and 70.1 (C-4,5), 28.3 ppm (C-6). (Vlllβ) H: 6.19 (H-1), 3.21
(H-2), 3.95 (H-3), 4.98 (H-4) 4.30 (H-5), 3.30 (H-6a), 2.91 (H-6b), 2.18 and 2.16 ppm (OAc); J-| _2 3.4, J2,3 2.4, J3_4 4.9, J4] 5 <1 , J5 6a 2.7, J5βb 3.2, J6aβb 12.0 Hz; 13C: 92.9 (C-1 ), 40.0 (C-2), 63.3, (C-3), 75.4 and 70.2 (C-4,5), 29.1 ppm (C-6).
Example 12
4-Nitrophenyl 2, 6-anhydro-3-azido-3-deoxy- 1, 2-dithio- ,β-D-mannopyranoside
(la and lb, Rη = Λ/3, R2 = CN)
Under argon, to a stirred solution of 500 mg of 1 ,4-di-0-acetyl-2,6- anhydro-3-azido-3-deoxy-2-thio-D-mannopyranose (VIII) in 15 ml of dry dichloromethane 540 mg of 4-nitrothiophenol and 0.35 ml of trimethylsilyl triflate were added at 0 °C, and stirring was continued at room temperature for 30 min. After neutralizing with triethylamine, the mixture was concentrated and the residue was submitted to column chromatography (solvent F) to yield a syrup (450 mg, 68%), which is a 1 :1 mixture of the α- and β-anomers [Rf (F) = 0.4]. This mixture was dissolved in 30 ml of dry methanol and 10 ml of dry
dichloromethane and 0.1 ml of 1 M methanolic sodium methoxide was added. After 1 h at room temperature the reaction was neutralized with Dowex 50 WX (H+) resin, filtered and concentrated to yield 370 mg (02%) of the title compound as a 1 :1 mixture of the α,b-anomers. Rf (A) = 0.3. NMR (DMSO-de), (la) 1 H: 6.10 (H-1), 3.35 (H-2), 4.12 (H-3), 3.76 (H-4), 4.20 (H-5), 3.20 (H-6a), 2.94 (H-6b), 5.81 (OH); J1 (2 1.4, J2,3 2.9, J3,4 3.9, J4,5 L ^5.6a 3-4, J5.6b 2-5, Jβa.δb 11 -7 Hz; (lb) 1 H: 6.15 (H-1), 3.38 (H-2), 4.00 (H-3), 3.75 (H-4) 4.16 (H-5), 3.32 (H-6a), 2.92 (H-6b), 5.79 (OH), 7.65 and 8.20 ppm (aromatic H); J >2 2.9, J2τ3 2.5, J3|4 4.4, J4 <1 , J5.6a 2.7, J5βb 2.2, J6a,6b 11 -7 Hz.
Example 13
4-Nitrophenyl 2, 6-anhydro-3-azido-3-deoxy- 1, 2-dithio-β-D-mannopyranoside
(lb, Rή = Λ/3, R2 = CN)
To a solution of 350 mg of 4-nitrophenyl 4-0-acetyl-2,6-anhydro-3-azido-
3-deoxy-1 ,2-dithio-β-D-mannopyranoside in 30 ml of dry methanol and 10 ml of dry dichloromethane 0.1 ml of 1 M methanolic sodium methoxide was added. After 1 h at room temperature the mixture was neutralized with Dowex 50 WX (H+) resin, filtered and concentrated to yield 295 mg (95%) of the title compound. Mp: 100-102 °C, Rf (A) = 0.3, [a]p = -143° (c = 0.5, methanol).
The starting material can be prepared the following way:
4-Nitrophenyl 4-0-acetyl-2, 6-anhydro-3-azido-3-deoxy- 1, 2-dithio-β-D-manno- pyranoside
To a stirred solution of 500 mg of 1 ,4-di-0-acetyl-2,6-anhydro-3-azido-3- deoxy-2-thio-D-mannopyranose (VIII) in 10 ml of dry 1 ,2-dichloroethane 300 mg
I
of 4-nitrothiophenol and 0.2 ml boron thflouride etherate were added. After 24 h at room temperature the reaction mixture was poured into 15 ml of ice-cold 6% aq sodium hydrogen carbonate, separated and the organic layer was washed with 6% aq sodium hydrogen carbonate and water, dried and concentrated.
5 The residue was submitted to column chromatography (solvent F) to yield 0.55 g (82%) of a syrup, which is a 1 :9 mixture of the a- and b-anomers [Rf (F) = 0.5]. This was recrystallized from ether to give 350 mg (52%) of the title compound. Mp: 150-153 °C, Rf (F) = 0.5, [a]p = -194° (c = 0.5, chloroform). NMR (CDCI3), 1 H: 5.88 (H-1), 3.29 (H-2), 4.03 (H-3), 4.98 (H-4), 4.35 (H-5),
I 0 3.60 (H-6a), 2.98 (H-6b), 2.15 (OAc), 7.60 and 8.15 ppm (aromatic H); J 2 2.2,
J2,3 2.5, J3|4 4.8, J4 <1 , J5.6a 2.8, J5βb 2.9, J6aβb 12.0 Hz.
Claims
1. 2,6-anhydro-1 ,2-dithio-pyranosides of the formula (I), more particularly the D-manno- and D-altropyranosides of formula (la-Id)



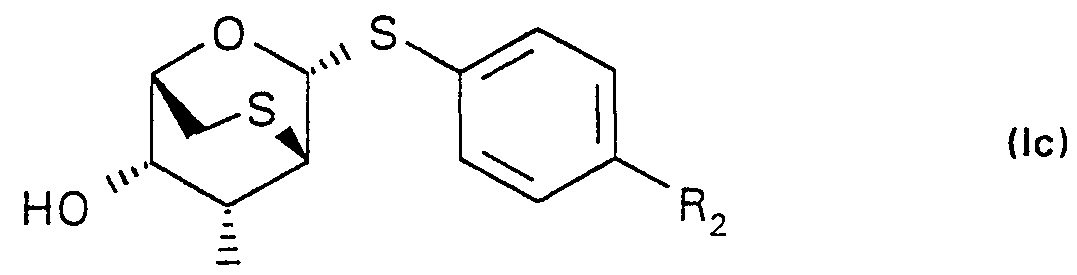
• 1 α-D-altroside 
R . β-D-altroside
wherein
R-\ represents a hydroxy or an azido group, R2 represents a nitro, cyano, amidino, aminothiocarbonyl,
-C(=NH)-OCH3, -C(=NH)-NH-NH2, -C(=NH)-SCH3 or an acetamido group, and if possible, the acid addition salts thereof formed with organic or inorganic acids. ( 12. 1997) 2. 2,6-anhydro-1 ,
2-dithio-pyranosides of the formula (I), more particularly the D-manno- and D-altropyranosides of formula (la-Id) wherein
R-I represents a hydroxy or an azido group,
R2 represents a nitro, cyano, amidino, aminothiocarbonyl, -C(=NH)-OCH3 or -C(=NH)-SCH3 group, and if possible, the acid addition salts thereof formed with organic or inorganic acids. (04. 12. 1996)
3. 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-D-altropyranoside
4. 4-cyanophenyl 2,6-anhydro-1 ,2-dithio-D-mannopyranoside
5. 4-(imino)(methoxy)methylphenyl 2,6-anhydro-1 ,2-dithio-D- mannopyranoside and the acid addition salts thereof
6. 4-(aminothiocarbonyl)phenyl 2,6-anhydro-1 ,2-dithio-D- mannopyranoside
7. 4-(imiπo)(methylthio)methylphenyl 2,6-anhydro-1 ,2-dithio-D- mannopyranoside and the acid addition salts thereof
8. 4-nitrophenyI 2,6-anhydro-1 ,2-dithio-D-mannopyranoside
9. 4-cyanophenyl 2,6-anhydro-3-azido-3-deoxy-1 ,2-dithio-D- mannopyranoside
10. 4-amidinophenyl 2,6-anhydro-1 ,2-dithio-D-mannopyranoside and the acid addition salts thereof
11. Pharmaceutical composition comprising as active ingredient a compound of general formula (I), more particularly of formula (la-Id), wherein R-1 and R2 have the same meaning as in claim 1 , or, if possible, a pharmaceutically acceptable salt thereof and solvents, diluents, carriers and filling materials usually applied in pharmaceuticals. ( 12. 1997)
12. Pharmaceutical composition comprising as active ingredient a compound of general formula (I), more particularly of formula (la-Id), wherein R<l and R2 have the same meaning as in claim 2, or, if possible, a pharmaceutically acceptable salt thereof and solvents, diluents, carriers and filling materials usually applied in pharmaceuticals. (04. 12. 1996)
13. Use of compounds of formula (I), more particularly of formula (la- Id), wherein R-j and R2 have the same meaning as in claim 1 , as pharmaceuticals. ( 12. 1997) 14. Use of compounds of formula (I), more particularly of formula (la-
Id), wherein R-j and R2 have the same meaning as in claim 2, as pharmaceuticals. (04. 12. 1996)
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU50646/98A AU5064698A (en) | 1996-12-04 | 1997-12-01 | Anticoagulant glycosides and pharmaceutical compositions thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HUP9603341 | 1996-12-04 | ||
| HU9603341A HUP9603341A3 (en) | 1996-12-04 | 1996-12-04 | Glucosides as blood-clotting-inhibitors and pharmaceutical compositions containing them |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1998024792A1 true WO1998024792A1 (en) | 1998-06-11 |
Family
ID=89994513
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/HU1997/000078 Ceased WO1998024792A1 (en) | 1996-12-04 | 1997-12-01 | Anticoagulant glycosides and pharmaceutical compositions thereof |
Country Status (3)
| Country | Link |
|---|---|
| AU (1) | AU5064698A (en) |
| HU (1) | HUP9603341A3 (en) |
| WO (1) | WO1998024792A1 (en) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0290321A1 (en) * | 1987-05-04 | 1988-11-09 | Fournier Industrie Et Sante | Beta-D-phenyl-thioxylosides, process for their preparation and their use in therapy |
| EP0365397A2 (en) * | 1988-10-18 | 1990-04-25 | Fournier Industrie Et Sante | Beta-D-phenyl thioxylosides, method for their preparation and their use in therapy |
| WO1995005182A1 (en) * | 1993-08-13 | 1995-02-23 | Glycomed Incorporated | Bridged oligosaccharides and sulfated derivatives thereof |
-
1996
- 1996-12-04 HU HU9603341A patent/HUP9603341A3/en unknown
-
1997
- 1997-12-01 WO PCT/HU1997/000078 patent/WO1998024792A1/en not_active Ceased
- 1997-12-01 AU AU50646/98A patent/AU5064698A/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0290321A1 (en) * | 1987-05-04 | 1988-11-09 | Fournier Industrie Et Sante | Beta-D-phenyl-thioxylosides, process for their preparation and their use in therapy |
| EP0365397A2 (en) * | 1988-10-18 | 1990-04-25 | Fournier Industrie Et Sante | Beta-D-phenyl thioxylosides, method for their preparation and their use in therapy |
| WO1995005182A1 (en) * | 1993-08-13 | 1995-02-23 | Glycomed Incorporated | Bridged oligosaccharides and sulfated derivatives thereof |
Non-Patent Citations (3)
| Title |
|---|
| F BELLAMY ET AL: "Thioxyloside derivatives as orally active venous antithrombotics", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 30, 1995, pages 101 - 115, XP002056034 * |
| K TOSHIMA: "The use of 2,6-anhydro-2-thio sugar for a highly stereocontrolled glycosylation", TETRAHEDRON LETTERS, vol. 31, no. 23, 1990, pages 3339 - 3342, XP002056035 * |
| MASSON P J ET AL: "The effect of the.beta.-D-xyloside naroparcil on circulating plasma glycosaminoglycans. An explanation for its known antithrombotic activity in the rabbit", J. BIOL. CHEM. (JBCHA3,00219258);95; VOL.270 (6); PP.2662-8, LABORATOIRES FOURNIER S.C.A.;CENTRE DE RECHERCHE ET DEVELOPPEMENT; DAIX; 21121; FR. (FR), XP002056036 * |
Also Published As
| Publication number | Publication date |
|---|---|
| HUP9603341A3 (en) | 1999-05-28 |
| HU9603341D0 (en) | 1997-01-28 |
| AU5064698A (en) | 1998-06-29 |
| HUP9603341A2 (en) | 1998-10-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Jacquinet et al. | Synthesis of heparin fragments. A chemical synthesis of the trisaccharide O-(2-deoxy-2-sulfamido-3, 6-di-O-sulfo-α-d-glucopyranosyl)-(1→ 4)-O-(2-O-sulfo-α-l-idopyranosyluronic acid)-(1→ 4)-2-deoxy-2-sulfamido-6-O-sulfo-d-glucopyranose heptasodium salt | |
| ES2423888T3 (en) | Synthetic heparin pentasaccharides | |
| EP1489090A1 (en) | Novel pseudoerythormycin derivatives | |
| EP0587364A1 (en) | A process for anomerizing nucleosides | |
| US12421270B2 (en) | Oligosaccharide, preparation method therefor, composition thereof and use thereof | |
| EP3415522A1 (en) | Novel hybrid galactoside inhibitor of galectins | |
| HU215152B (en) | Process for producing carbohydrate derivatives containing pentasaccharide unit and pharmaceutical compositions containing them | |
| JP3594990B2 (en) | 3-deoxyoligosaccharide, method for producing the same, and pharmaceutical composition containing the same | |
| JP4364959B2 (en) | Carbohydrate derivatives | |
| DE69900718T2 (en) | SYNTHETIC POLYSACCHARIDES, METHOD FOR THE PRODUCTION THEREOF AND PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME | |
| US5332756A (en) | 3-deoxy-mannosamine derivatives | |
| US3781267A (en) | O-esters of monosaccharides having ether groupings | |
| Van Boom et al. | Synthesis of a Conformationally Constrained Heparin‐like Pentasaccharide | |
| US6680304B2 (en) | Disaccharides with anti-arthrosic properties | |
| Juetten et al. | Stereoselective. alpha.-glycosylation of nitro sugar evernitrose: synthesis of the terminal AB unit of everninomicin antibiotics | |
| WO1997049716A1 (en) | Novel anticoagulant glycosides and pharmaceutical compositions thereof | |
| US4891425A (en) | N-glycosylamide derivatives, processes for their preparation and their use as medicaments | |
| WO1998024792A1 (en) | Anticoagulant glycosides and pharmaceutical compositions thereof | |
| Bozó et al. | Synthesis of 4-cyanophenyl 2-azido-2-deoxy-and 3-azido-3-deoxy-1, 5-dithio-β-d-xylopyranosides | |
| WO1999028312A1 (en) | Novel anticoagulant glycosides and pharmaceutical compositions thereof | |
| US4220643A (en) | Nitrosourea pentose compounds | |
| CN110041383A (en) | Chondroitin sulfate oligosaccharides, preparation method and application | |
| EP2857411B1 (en) | Method for preparing fully protection heparin pentasaccharide and intermediate thereof | |
| ES2202212T3 (en) | BENZOFENONA-A-D.GLICOPIRANOSIDOS, PREPARATION AND THERAPEUTIC USE. | |
| CA2350755A1 (en) | Benzylglycosylamides as inhibitors of smooth muscle cell proliferation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH HU IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZW AM AZ BY KG KZ MD RU TJ TM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH KE LS MW SD SZ UG ZW AT BE CH DE DK ES FI FR GB GR IE IT LU MC NL |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| 122 | Ep: pct application non-entry in european phase |