WO1999005282A1 - Non-b non-c non-g hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus - Google Patents
Non-b non-c non-g hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus Download PDFInfo
- Publication number
- WO1999005282A1 WO1999005282A1 PCT/JP1998/003340 JP9803340W WO9905282A1 WO 1999005282 A1 WO1999005282 A1 WO 1999005282A1 JP 9803340 W JP9803340 W JP 9803340W WO 9905282 A1 WO9905282 A1 WO 9905282A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- seq
- gene
- nucleotide sequence
- sequence
- oligonucleotide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/11—DNA or RNA fragments; Modified forms thereof; Non-coding nucleic acids having a biological activity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/005—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2770/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA ssRNA viruses positive-sense
- C12N2770/00011—Details
- C12N2770/24011—Flaviviridae
- C12N2770/24211—Hepacivirus, e.g. hepatitis C virus, hepatitis G virus
- C12N2770/24222—New viral proteins or individual genes, new structural or functional aspects of known viral proteins or genes
Definitions
- Non-B non-C non-G hepatitis virus gene polynucleotide, polypeptide, virus particle, virus particle separation method and virus detection
- the present invention relates to genes, polynucleotides, polypeptides, antibodies, antigens, and the like that can be used by discovering an unknown causative virus of blood-borne hepatitis for which the cause could not be identified by conventional diagnostic methods. Production and detection of viral genes, antibodies, and antigens used. Background art
- HBV hepatitis B virus
- HCV hepatitis C virus
- Diagnostic methods have been established for both viruses, and as a result of being introduced into blood transfusion screening in Japan early on, it has become possible to eliminate almost no new infections between recipients of both hepatitis viruses. Became.
- the development of diagnostic and therapeutic methods for unexplained viral hepatitis includes elucidation of the genetic information of the virus, determination of the virus-specific gene sequence and amino acid sequence, identification of the location of the epitope, and epitope.
- Establish a method for producing biological materials establish a method for producing antigens that react specifically with antiviral antibodies, provide specific antibodies against viruses, establish a method for separating and recovering virus particles, and neutralize the biological activity
- Genetic testing method, antibody testing method, antigen testing method, and method for producing neutralizing antibody-derived vaccine using the biological material derived from the virus obtained in this way are expected. Have been. Disclosure of the invention
- An object of the present invention is to establish a new diagnostic method and therapeutic method for unknown viral hepatitis for which the causative virus has not been identified to date, and therefore, the means of diagnosis, prevention and treatment have not been elucidated.
- An object of the present invention is to provide an antiviral antibody and a method for detecting a virus.
- the present inventors presumed that unknown hepatitis virus particles causing hepatitis or a part thereof were present in the blood of a hepatitis patient whose cause could not be determined by the existing diagnostic method for hepatitis virus. Based on this, the virus gene is isolated from the blood. Tried to. Specifically, the viral gene is present in the blood of hepatitis patients, but not in the human genome, nor in the blood of many normal people, or not before the onset of hepatitis. Based on this hypothesis, we searched for candidate genes in the blood of hepatitis patients based on this hypothesis and isolated them. The following points were further examined with respect to the candidate genes thus obtained, and genes meeting all of the following criteria were finally determined to be novel virus genes. That is,
- the obtained gene sequence is not homologous to the gene sequences of known hepatitis virus and other viruses;
- gene-positive blood is analyzed by density gradient centrifugation, which is widely used as a method for separating and collecting virus particles, gene-positive fractions are found in general virus particle fractions.
- hepatitis virus infection Known cases of hepatitis virus infection are known to be asymptomatic carriers, i.e., even if the virus is positive, there is no indication of hepatic inflammation and the condition is considered to be healthy. It was determined that this could be enough. For this reason, "the gene cannot be found in normal individuals" was not added to the criteria.
- the gene of the present invention was separated according to the above criteria, the sequence of the separated gene was determined, and an oligonucleotide sequence usable as a primer of PCR was searched. A gene detection method was established using the obtained primers. Furthermore, the open reading frame existing in the gene sequence was identified, and the amino acid sequence was identified. The present invention was completed by determining the density by density gradient centrifugation and establishing a virus recovery method.
- the present invention relates to PCR using, as a primer, an oligonucleotide having the nucleotide sequence of SEQ ID NO: 57 and an oligonucleotide having the nucleotide sequence of SEQ ID NO: 60, or the nucleotide sequence of SEQ ID NO: 57.
- Oligo with sequence Non-B having a base sequence capable of amplifying a sequence having a length of about 350 to 400 bases by PCR using nucleotides and oligonucleotides having the base sequence of SEQ ID NO: 61 as primers
- a non-C non-G hepatitis virus gene hereinafter, also referred to as the gene of the present invention.
- the gene of the present invention preferably has a base sequence capable of amplifying a sequence of about 360 bases and 3900 bases in length by the PCR.
- the gene of the present invention has a nucleotide sequence at the 5 ′ end and the 3 ′ end of the fragment amplified by the PCR, wherein the nucleotide sequences of nucleotides 3 to 300 and nucleotides 2402 to Each of them has 70% or more homology to the 3739 base sequence. More preferably, the homology is at least 80%.
- the present invention also provides the following non-B non-C non-G hepatitis virus genes (a) and (b).
- the gene of the present invention is preferably a non-B non-C non-G hepatitis virus gene having the nucleotide sequence of SEQ ID NO: 1 or a homologous mutant gene thereof.
- a gene include a non-B non-C non-G hepatitis virus gene (genotype 1) having the nucleotide sequence of SEQ ID NO: 45, a non-B non-C non-hepatitis virus having the nucleotide sequence of SEQ ID NO: 46, Non-B non-C non-G hepatitis virus gene (genotype 3) having the nucleotide sequence of SEQ ID NO: 47, having the nucleotide sequence of SEQ ID NO: 48 A non-B non-C non-G hepatitis virus gene (genotype 4), a non-B non-C non-G hepatitis virus gene having a nucleotide sequence described in SEQ ID NO: 49 (genotype 5), a nucleotide sequence
- the present invention also provides a polynucleotide having a nucleotide sequence complementary to the nucleotide sequence of the gene of the present invention.
- the present invention also provides an oligonucleotide comprising a nucleotide sequence specific to the gene or a nucleotide sequence complementary thereto, which is found in the nucleotide sequence of the gene of the present invention (hereinafter, also referred to as the oligonucleotide of the present invention).
- an oligonucleotide include a polynucleotide having the nucleotide sequence of SEQ ID NO: 2 (RD037), a polynucleotide having the nucleotide sequence of SEQ ID NO: 3 (RD038), and a polynucleotide having the nucleotide sequence of SEQ ID NO: 3.
- the present invention further provides a method for detecting a non-B non-C non-G hepatitis virus gene, which comprises performing PCR using the oligonucleotide of the present invention as a primer.
- the detection method includes an oligonucleotide having the nucleotide sequence of SEQ ID NO: 2 and an oligonucleotide having the nucleotide sequence of SEQ ID NO: 3, or an oligonucleotide having the nucleotide sequence of SEQ ID NO: 4, and A non-B non-C non-G hepatitis virus gene detection method (first detection method), wherein PCR is performed using an oligonucleotide having the nucleotide sequence of SEQ ID NO: 5 as a primer; Oligonucleotide having the base sequence of SEQ ID NO: 8 and the oligonucleotide having the base sequence of SEQ ID NO: 8, or oligonucleotide having the base sequence of SEQ ID NO: 7
- the present invention also provides a non-B non-C non-G non-G sample, wherein both the first detection method and the second detection method are performed on one sample, and the results obtained by the two gene detection methods are compared.
- a non-B non-C non-G method characterized by performing hepatitis B virus genotyping, and performing hybridization using oligonucleotides having genotype-specific sequences in genotypes 1 to 6 genes. Hepatitis B virus genotyping is provided.
- the present invention provides a polypeptide having an amino acid sequence encoded in the open reading frame found in the nucleotide sequence of the gene of the present invention (hereinafter, also referred to as the polypeptide of the present invention).
- polypeptide of the present invention examples include a polypeptide having the amino acid sequence of SEQ ID NO: 9 and a polypeptide having the amino acid sequence of SEQ ID NO: 10.
- the peptide of the present invention includes a polypeptide consisting of a non-B non-C non-G hepatitis virus-specific amino acid sequence found in the amino acid sequence of SEQ ID NO: 9 or 10.
- the polypeptide of the present invention preferably comprises a non-B non-C non-G hepatitis virus-specific epitope.
- the present invention provides a method for separating non-B non-C non-G hepatitis virus particles, comprising separating the non-B non-C non-G hepatitis virus particles based on the density of the non-B non-C non-G hepatitis virus particles.
- a virus particle isolated by this method, and a non-B non-C non-G hepatitis virus peptide obtained from the virus particle are provided.
- a recombinant gene expression vector that incorporates all or part of the nucleotide sequence encoding the amino acid sequence described in SEQ ID NO: 9 or 10, a nucleotide encoding the amino acid sequence described in SEQ ID NO: 9 or 10 A transformed cell that retains all or part of the sequence; a non-B non-C non-G hepatitis virus antigen peptide or a fragment thereof expressed by the transformed cell; Provided is a method for producing a non-B non-C non-G hepatitis virus antigen peptide, comprising culturing under conditions in which the non-hepatitis G virus antigen peptide is expressed, and collecting the expressed peptide.
- the present invention relates to a non-B non-C non-G hepatitis virus antibody using the polypeptide of the present invention (including those obtained from virus particles) or the above-mentioned virus particles as an antigen.
- Non-B non-C non-G hepatitis virus characterized in that an animal is immunized using the method of immunological detection of the body, the polypeptide of the present invention (including those obtained from virus particles) or the above-mentioned virus particles as an immunogen.
- the present invention relates to a non-B non-C non-G hepatitis virus neutralizing antibody having an amino acid sequence contained in the amino acid sequence encoded by the open reading frame in the nucleotide sequence of SEQ ID NO: 1.
- a vaccine comprising a polypeptide comprising a polypeptide sequence; and a vaccine comprising the virus particle.
- the gene of the present invention is preferably a non-B non-C non-G hepatitis virus gene having the nucleotide sequence set forth in SEQ ID NO: 62 or a homologous mutant gene thereof.
- This gene also comprises a polynucleotide having a nucleotide sequence complementary to the nucleotide sequence of the gene of the present invention, a nucleotide sequence specific to the gene or a nucleotide sequence complementary thereto, which is found in the nucleotide sequence of the gene of the present invention.
- An oligonucleotide a method for detecting a non-B non-C non-G hepatitis virus gene, which comprises performing PCR using this oligonucleotide as a primer, encoded in an open reading frame recognized in the nucleotide sequence of the gene of the present invention.
- a polypeptide having an amino acid sequence, a recombinant gene expression vector, a transformed cell, a non-B non-C non-G hepatitis virus antigen polypeptide or a fragment thereof expressed by the transformed cell, a non-B non-C non- Production method of hepatitis G virus antigen peptide, non-B non-C non-immunologic detection method of non-G hepatitis virus antibody, non-B A method for producing a B non-C non-G hepatitis virus antibody, an antibody, a method for immunologically detecting a non-B non-C non-G hepatitis virus antigen, and a vaccine are provided.
- the present invention will be described in detail.
- the present invention relates to an unknown virus different from all known viruses.
- the “non-B non-C non-G hepatitis virus” of the present invention means a virus that is infected by blood transmission and exhibits hepatic inflammation, and is the same blood-transmitting virus type B, C, It is an unknown virus different from hepatitis G virus. It is also different from non-blood-transmitting hepatitis A, Hepatitis E, Hepatitis F, and hepatitis D viruses, which are deficient viruses. F non Synonymous with hepatitis G virus.
- Hepatic inflammation generally refers to symptoms that indicate hepatitis, such as abnormal liver function values and jaundice.
- the virus of the present invention is regarded as one of the hepatitis viruses based on the origin of the discovery of the virus of the present invention. However, it does not always mean that the main disease caused by the virus of the present invention is hepatitis.
- HNT22 virus (hereinafter, abbreviated as "HNT22").
- HNT22 can also be defined as a non-B non-C non-G hepatitis virus having the following non-B non-C non-G hepatitis virus genes (a) and (b).
- PCR using an oligonucleotide having the nucleotide sequence of SEQ ID NO: 6 and an oligonucleotide having the nucleotide sequence of SEQ ID NO: 8 as a primer has a length of about 200 to 350 bases.
- TUS01 virus a gene that is not included in the above genes, that is, a non-B non-C non-G hepatitis virus gene having the nucleotide sequence of SEQ ID NO: 62 and a gene that is recognized as a homologous variant thereof is referred to as a “TUS01 virus”. (Hereinafter abbreviated as “TUS 01”).
- HNT22 and TUS01 are unknown viruses isolated from non-A non-B non-C non-D non-E non-F non-G hepatitis virus hepatitis patients, and hepatitis patients previously considered to be of unknown cause More isolated and all known viruses are viruses that are not virologically or molecularly homologous.
- HNT22 and TUS01 collectively comprise, at the end, the base sequence of base numbers 3 to 300 of the base sequence of SEQ ID NO: 1 or a base sequence having high homology thereto It has a column 35 having a nucleotide sequence having a nucleotide sequence or to the high homology of the nucleotide numbers 2902 to 373 8 of the nucleotide sequence of the end side in SEQ ID NO: 1, the 5 'end nucleotide sequence and 3' It can be defined as a gene having a length of about 3500 bases to about 4000 bases including the terminal base sequence, preferably about 3600 bases to about 3900 bases.
- HNT 22 hepatitis hepatitis that is positive for HNT 22
- TUS01 hepatitis Hepatitis for which TUS01 is positive
- Viruses are generally known to be more susceptible to mutation than other higher organisms, and that they are easier to fix in genes. Therefore, there are many strains (homogeneous mutants) within the same species, and there are genotypes that have a common gene sequence.
- the term HNT22 or TUS01 in the present invention is used as a generic term for viruses including these many strains and genotypes.
- the frequency of mutation in a virus is high and the frequency of the mutation being fixed in a gene is high. Therefore, when a virus is defined by its gene sequence or amino acid sequence, it is recognized that the virus includes not only the exemplified sequences but also those having a gene sequence and an amino acid sequence in a substantially homologous range.
- the range of substantial homology Lffl can be determined by the percentage of sequence homology that can be clearly distinguished from the virus when the gene sequence or amino acid sequence is compared with other viruses.
- the range of substantially homology can be determined by referring to the sequence diversity within a known species of a known virus of the same species.
- a gene having a homologous nucleotide sequence is substantially homologous to the gene of the present invention, and is considered to be included in the present invention.
- the homology (conserved) in the HNT22 gene was 55% or more, and the highest homology was found.
- the homology with other virus cases showing less than 60% was less than 60%.
- the homology means homology with respect to a base sequence and an amino acid sequence. Specifically, the sequences to be compared most closely match each other, including deletion, if necessary. The percentage of the number of matching bases or amino acids in the total number of bases or amino acids to be compared is expressed as a percentage.
- HCV hepatitis C virus
- HCV hepatitis C virus
- the gene sequence within the species showed 60% or more homology.
- the HCV that showed the same genotype had 80% or more homology in gene sequence except for some highly mutated regions. From these facts, it is considered appropriate to determine the range of genes included in the present invention with the above homology.
- homologous examples are substantially the same as the virus of the present invention. Homologous and included.
- HNT having the nucleotide sequence of SEQ ID NO: 45 22 genes (genotype 1), the HNT22 gene having the base sequence described in SEQ ID NO: 46 (genotype 2), the HNT22 gene having the base sequence described in SEQ ID NO: 47 (genotype 3), described in SEQ ID NO: 48 HNT22 gene having the nucleotide sequence of SEQ ID NO: 49 (genotype 4), HNT22 gene having the nucleotide sequence of SEQ ID NO: 49 (genotype 5), and HNT22 gene having the nucleotide sequence of SEQ ID NO: 50 (genotype) 6) and HNT22 gene (genotype 7) having the nucleotide sequence of SEQ ID NO: 51.
- HNT22 virus of a specific genotype By using such a nucleotide sequence, HNT22 virus of a specific genotype can be detected or distinguished.
- a method for discrimination not only a method of amplifying and sequencing according to the method described in Examples described later but also a method of using the oligonucleotide having a sequence specific to each genotype as a primer, A method of amplifying only the sequence, a method of selectively detecting the gene sequence using this oligonucleotide as a probe, or a combination of these methods.
- Selection of oligonucleotides that can be used in these methods and setting of reaction conditions can be performed by methods known to those skilled in the art.
- genotype-specific sequence a sequence portion characteristic of each genotype (hereinafter, referred to as a genotype-specific sequence) by comparing the genotype sequences with each other.
- the oligonucleotides thus selected can be used as primers or probes for HNT22 genotype-specific amplification or detection.
- the above-described HNT22 gene uses, as a primer, an oligonucleotide (NG059) having the nucleotide sequence of SEQ ID NO: 6 and an oligonucleotide (NG063) having the nucleotide sequence of SEQ ID NO: 8 PCR using a PCR and / or an oligonucleotide (NG061) having the nucleotide sequence of SEQ ID NO: ⁇ and an oligonucleotide (NG063) having the nucleotide sequence of SEQ ID NO: 8 as a primer Can be detected as having a base sequence that can be amplified.
- NG059 having the nucleotide sequence of SEQ ID NO: 6
- NG063 having the nucleotide sequence of SEQ ID NO: 8 PCR using a PCR and / or an oligonucleotide (NG061) having the nucleotide sequence of SEQ ID NO: ⁇ and an oligonucleotide
- the HNT22 gene having the nucleotide sequence of SEQ ID NO: 1 was prepared by using the oligonucleotide (RD037) having the nucleotide sequence of SEQ ID NO: 2 and the oligonucleotide (RD038) having the nucleotide sequence of SEQ ID NO: 3 as a primer.
- PCR and / or an oligonucleotide having the nucleotide sequence of SEQ ID NO: 4 (RD051) and an oligonucleotide having the nucleotide sequence of SEQ ID NO: 5 (RD052) are used as primers It can be detected as having a base sequence that can be amplified by PCR.
- the HNT22 gene of the present invention was obtained by the present inventors according to the following procedure.
- the present inventor attempted to screen for a gene that is present in hepatitis cases of unknown cause and that is not present in healthy individuals or patients before the onset of hepatitis, in order to search for an unknown hepatitis virus.
- a well-known representational difference analysis (Science 259, 946-950, 1993; hereinafter abbreviated as "RDA method”). To).
- the present inventors confirmed that the sequence was not homologous to the previously known sequence of the virus.
- the gene was not found in many healthy people. It was also confirmed that some of the gene-positive cases were infected by transfusion of the gene-positive blood while the gene was negative before blood transfusion, and were persistently positive thereafter. It was also proved that the gene was not a host-derived gene.
- the virus of the present invention is an unknown hepatitis virus gene.
- the above gene was inferred from its structure to be a part of the virus gene.
- the inventor obtained the gene of the present invention by the known Gene Walking method using the gene sequence as a clue.
- the embodiment of the present invention will be described by taking the HNT22 gene as an example, but it is easily understood that the TUS01 gene can be used in the same manner.
- the present invention provides oligonucleotides and polynucleotides having a sequence specific to the HNT22 gene and capable of complementarily hybridizing to the HNT22 gene under stringent conditions. Depending on their characteristics, these oligonucleotides / polynucleotides can be used effectively as a primer to amplify the HNT22 gene, or as a probe to capture or detect the HNT22 gene in a sample. it can.
- the term “specific sequence” refers to a characteristic sequence in the nucleotide sequence of the gene in question or an amino acid sequence deduced from the nucleotide sequence that can be distinguished from a sequence other than the sequence in question. means. Whether or not the sequence is characteristic can be determined based on sequence homology. Specifically, when the sequence is searched and compared using a known technique such as a database with a known sequence other than the sequence of the subject gene having the same length, the homology with the known sequence is determined. This means an array with a length of about 10% or less. Alternatively, the determination can be made based on whether the gene in question can be detected, identified, and amplified using the sequence.
- a gene detection method or gene amplification known to those skilled in the art as an oligonucleotide or polynucleotide having the sequence or an oligonucleotide or polynucleotide having a complementary sequence thereof as a probe or primer.
- the sequence in which the gene in question is detected, identified, or amplified by distinguishing it statistically from other genes is determined to be specific to the gene in question.
- the amino acid sequence can be determined based on the homology of the sequence to determine whether it is a specific sequence. Specifically, it is an amino acid sequence having a homology of about 10% or less when compared with a known sequence.
- amino acid sequence encoded by the gene in question or its amino acid sequence
- an antibody prepared using a peptide containing a sequence as an antigen and proven to bind to the amino acid sequence does not bind to other antigens not derived from the gene in question, or when the binding is statistically significantly weaker
- the amino acid sequence can be a sequence specific to the gene in question.
- polynucleotide and “oligonucleotide” refer to nucleotide polymers of any length, including ribonucleotides and deoxyribonucleotides, single-stranded DNA, Includes double-stranded DNA, as well as single- and double-stranded RNA.
- the term includes unmodified polynucleotides as well as modified polynucleotides and oligonucleotides, such as methylated, capping or enzyme labels, fluorescent labels.
- polynucleotides and oligonucleotides of the present invention include not only those derived from genomic DNA, but also polynucleotides and oligonucleotides obtained by synthesis, replication, transcription or amplification by known methods.
- the polynucleotide in the present invention includes the exemplified sequence, a sequence substantially homologous thereto, and a polynucleotide having a sequence complementary thereto.
- the oligonucleotide / polynucleotide of the present invention can be used as a primer for amplification of HNT22 gene by PCR. With this method, it is possible to amplify the HNT22 gene portion flanked by the primers, whereby the presence of the HNT22 gene portion in the subject, that is, the presence of HNT22 Can be confirmed.
- the present invention also provides an oligonucleotide probe or a polynucleotide probe useful for capturing the HNT22 gene by hybridization and detecting the same.
- Oligonucleotides of the invention are usually nucleotides having relatively short sequences that are contiguous in the polynucleotide, including those having sequences in homologous and complementary polynucleotides. Its length is usually 6 to 50 nucleotides, preferably 10 to 30 nucleotides, more preferably 15 to 20 nucleotides.
- nucleotide sequence in a region where the nucleotide sequence is relatively conserved among virus strains can be a gene-specific nucleotide sequence. It is easy for those skilled in the art to select such a nucleotide sequence based on the nucleotide sequence described in the present specification.
- oligonucleotide examples include a polynucleotide having the nucleotide sequence of SEQ ID NO: 2 (RD037), a polynucleotide having the nucleotide sequence of SEQ ID NO: 3 (RD038), and a polynucleotide having the nucleotide sequence of SEQ ID NO: 3;
- the present invention further provides a method for detecting the HNT22 gene, wherein PCR is performed using the oligonucleotide of the present invention as a primer.
- the detection method include an oligonucleotide having the nucleotide sequence of SEQ ID NO: 2 (RD 037) and an oligonucleotide having the nucleotide sequence of SEQ ID NO: 3 (RD 038), or the nucleotide sequence of SEQ ID NO: 4.
- An oligonucleotide having a nucleotide sequence (NG061) and an oligonucleotide having a nucleotide sequence described in SEQ ID NO: 8 (NG063) were used as primers. And have use and performing PCR HNT 22 gene detection method (the second detection method) elevation I can do it.
- the sample used for PCR is prepared by extracting nucleic acid or DNA from a biological sample such as plasma or serum collected from a sample by a known method. This preparation can be performed using a commercially available extraction kit.
- the conditions for PCR are appropriately selected according to a known PCR method using a thermostable DNA polymerase.
- the amplification product generated by the PCR can be detected by a known method such as electrophoresis, and the HNT22 gene can be detected based on the presence or absence of the amplification product.
- the present invention provides an HNT22 genotyping method comprising performing both the first detection method and the second detection method on one sample and comparing the results obtained by both gene detection methods. And an HNT22 genotyping method comprising performing hybridization using oligonucleotides having genotype-specific sequences in genotype 1 to 6 genes.
- Hybridization can be performed according to a known method. Specifically, a nucleic acid is prepared from a biological sample, the prepared nucleic acid is hybridized with the oligonucleotide under stringent conditions, and a hybridized product containing the oligonucleotide is detected.
- oligonucleotides listed above are naturally one of the examples, and are combined with the above by combining the nucleotide sequence of the HNT22 gene disclosed in the specification with a technique known to those skilled in the art. It is easy to select an oligonucleotide that achieves the same purpose. Further, by combining the nucleotide sequence of the HNT22 gene disclosed in the specification, the nucleotide sequence of the TUS01 gene, and a technique known to those skilled in the art, an oligonucleotide that can be used to detect both genes is selected. It is easy.
- Oligonucleotide having base sequence (NG054) and the oligonucleotide (NG021) having the nucleotide sequence set forth in SEQ ID NO: 61 The PCR is performed using these as primers to detect the HNT22 gene and the TUS01 gene. it can.
- the present invention provides a polypeptide having an amino acid sequence encoded in the open reading frame found in the nucleotide sequence of the gene of the present invention (hereinafter, also referred to as the polypeptide of the present invention).
- polypeptide refers to a linear union of amino acids, and does not specify a length. Therefore, the polypeptide of the present invention includes oligopeptides, proteins, and peptides which are generally referred to, and their origin is not only naturally occurring ones, but also fragments or various means such as chemical synthesis and recombinant expression.
- the polypeptide to be prepared is included.
- the sequence as described above, it is known that there is a difference in the gene sequence even within the same species in the virus, and as a result, a similar difference occurs in the amino acid sequence. Therefore, the homology of the amino acid sequence determined to be that of the virus of the present invention is 65% or more, preferably 70% or more, more preferably 80% or more, and more preferably 90% or more.
- ORF open reading frame
- ORF means a nucleotide sequence region encoding a polypeptide or a part thereof. This sequence is transcribed and translated into a polypeptide under the control of appropriate regulatory sequences. The boundaries of the coding sequence are a start codon at the 5 'end and a stop codon at the 3' end.
- the ORF can be determined as follows.
- the polypeptide of the present invention preferably comprises an amino acid sequence specific to HNT22.
- Those skilled in the art can determine the amino acid sequence specific to HNT22 by an amino acid sequence encoded by the nucleotide sequence described herein or the nucleotide sequence of a gene detected based on the above-described gene detection method. Can be selected based on the amino acid sequence encoded in
- polypeptide of the present invention further preferably contains an HNT22-specific epitope.
- Epitope in the present invention means an antigenic determinant.
- An antigenic determinant is a site in an antigen molecule that directly binds to an antibody, and is composed of at least three amino acids in a three-dimensional structure, and is generally composed of 5 to 10 amino acids. Methods for determining the position of the epitope and for determining the arrangement of the peptides in the three-dimensional structure are known in the art and can be carried out by those having ordinary skill in the art. Therefore, the “HNT22-specific epitope” in the present invention means an antigenic determinant specific to HNT22, and is an epitope composed of an amino acid sequence unique to HNT22. This ebitope is usually composed of eight or more consecutive amino acids.
- epitope means that the above-described epitope sequence is contained as a part thereof, and a polypeptide containing the epitope is specifically a carrier peptide or linker in the epitope sequence. Includes peptides combined.
- the polypeptide of the present invention can be effectively used as an antigen for antibody testing or as an immunogen for preparing antiviral antibodies.
- Some viral polypeptides do not have an amino acid sequence specific to the virus, and if this portion is used as an antigen, there may be a sequence that causes a non-specific antibody reaction. is expected. Therefore, also in the present invention, it is important to specify a sequence suitable for antibody testing in the entire amino acid sequence.
- a method for finding such a specific sequence that is, an epitope sequence
- a peptide of a certain length is synthesized so as to cover the entire sequence, and the peptide is infected with an antiviral antibody, ie, the virus.
- Anti with patient serum There is a method that uses the presence / absence of response and strength. Also, instead of the above synthesis method,
- the present invention also provides a method for separating non- ⁇ non-C non-G hepatitis virus particles, comprising separating the non- ⁇ non-C non-G hepatitis virus particles based on the density of the non- C non-C non-G hepatitis virus particles.
- Provided are isolated virus particles and a non-non-C-non-G hepatitis virus peptide obtained from the virus particles.
- “Viral particles” in the present invention means the gene-positive fraction of the present invention, which is collected into a specific density fraction by a density gradient centrifugation method used as a usual method for separating viral particles. It is not limited to those having a specific particle structure or infectious particles.
- Viruses have a density that depends on their particle structure, and the density can be used to separate other co-existing substances.
- the most common method for separating virus particles using density is to use density gradient centrifugation.
- a stepwise density carrier layer is formed in a centrifuge tube using a substance such as sucrose, and a virus-containing fraction is overlaid on the upper layer, followed by ultracentrifugation.
- This analysis method uses the phenomenon that when a virus that has moved in the direction of centrifugal force due to centrifugal force reaches the same density layer as the virus, the centrifugal force and buoyancy reach an equilibrium state and stop moving, and therefore concentrate on that fraction It is.
- the polypeptide can be obtained from the virus particles obtained by the above method by a known method.
- the virus particles are treated with a denaturing agent to separate the polypeptide from other components.
- the present invention also provides a gene expression vector incorporating the ⁇ 22 gene or a partial sequence thereof.
- the term "recombinant expression vector” refers to a vector into which an exogenous polynucleotide is inserted into a host cell gene in a state where it can be expressed. Specifically, it is a vector having a control sequence capable of incorporating the polynucleotide.
- the term "part” refers to a part that encodes a peptide sufficient to exhibit antigenicity.
- This expression vector provides a viral peptide with immunological or biological activity. It can be used effectively for At present, various vectors are known for integrating a gene desired to be expressed, and various host cells for integrating a gene using a vector and finally expressing a peptide are also known. It has been done.
- the gene expression vector provided by the present invention is a recombinant expression vector having the HNT22 genome or its ORF, and the ORF can operate on a control sequence compatible with a desired host. It is a recombinant expression vector that is linked in this manner. Using this expression vector, a recombinant gene expression system can be constructed.
- transformed cell means that all or a part of the gene of the present invention is incorporated into the gene and expresses all or a part of the polypeptide encoded by the gene of the present invention. Means cells that can be used.
- the transformed cell provided by the present invention can be obtained by directly introducing a polynucleotide containing all or a part of the gene of the present invention, or by introducing the above-described recombinant expression vector incorporating the polynucleotide into a host cell. Can be obtained by transforming a host cell. In the present invention, known transfer vectors and host cells can be used.
- the polypeptide (HNT22 virus antigen peptide) produced by the transformed cells is obtained by culturing the transformed cells under conditions in which the HNT22 virus antigen peptide is expressed, and collecting the expressed polypeptide.
- the present invention provides a method for immunologically detecting an HNT22 antibody using an HNT22 particle, an HNT22 polypeptide, an HNT22 peptide, or a polypeptide containing the HNT22 epitope as an antigen. Examples of a method for detecting an antibody using a peptide antigen include immunoturbidimetry, enzyme immunoassay, radiometric immunoassay, and agglutination, and these methods can be used in the present invention.
- the present invention also relates to a method for preparing an HNT22 antibody using a purified or partially purified HNT22 particle, an HNT22 polypeptide, an HNT22 epitope, or a polypeptide containing the HNT22 epitope as an immunogen, and an HNT22 antibody.
- a purified or partially purified HNT22 particle an HNT22 polypeptide, an HNT22 epitope, or a polypeptide containing the HNT22 epitope as an immunogen, and an HNT22 antibody.
- Known methods can be used to prepare antibodies using purified or partially purified virus particles or polypeptides.
- the present invention provides an immunoassay method and a kit for HNT22 antigen using an antibody that specifically binds to HNT22. As an example of a method for measuring an antigen using an antibody,
- viral antigens such as agglutination, reverse passive agglutination, enzyme immunoassay, radiometric immunoassay, and fluorescence polarization assay, can also be used in the practice of the present invention. it can.
- the present invention relates to purified HNT22 particles, polypeptides including neutralizing antibody epitopes obtained from purified virus particles, HNT22 particles obtained using recombinant expression, and neutralizing antibody epitopes.
- a vaccine comprising polypeptides including pitope is provided.
- the preparation of the vaccine of the present invention is based on HNT 22
- the protein encoding the antigenically active region can be obtained by the above-mentioned synthesis method or recombinant expression method and used.
- a peptide antigen or a recombinant antigen containing the epitope of the envelope antigen has the activity.
- other structural protein antigens have the activity alone or in combination with other antigens.
- a peptide antigen containing the envelope antigen peptide or a recombinant antigen contains the epitope.
- Other structural protein antigens, alone or in combination with other antigens contain a neutralizing antibody epitope.
- a multivalent vaccine containing a plurality of neutralizing antibody webs can be obtained. Can be.
- the separation and purification particles it is necessary to inactivate the virus, but this can be achieved by a known method such as a formalin treatment method.
- a vaccine containing an immunogenic polypeptide as an active ingredient is prepared as a liquid or suspension as an injection solution, or as a solid form suitable for dissolving and suspending in a liquid before injection.
- This immunologically active component is mixed with a suitable excipient.
- Excipients include water, saline, dextrose, glycerol, ethanol and the like.
- small amounts of auxiliary substances may be included if necessary. These include humectants, emulsifiers, pH buffers, or adjuvants. Examples of adjuvants include aluminum hydroxide, N-acetimulum-l-L-threonyl-D-isoglumin, N-acetyl-muramyl-L-aranyl-D-isoglumamine.
- the prescription for achieving the effect is appropriately determined.
- a single dose ranges from 5 to 250 mg / g of antigen, depending on the weight of the individual to be administered, the immunological ability, and the desired antibody induction. Decided. The number of times of administration is selected based on the same criteria.
- Figures 1 to 5 show that the genes obtained from 75 HNT22 positive samples were sandwiched between sequences complementary to NG001 (SEQ ID NO: 40) and RD052 (SEQ ID NO: 5).
- 3 9 6 salt 3 shows a comparison of base length sequences (corresponding to ntl862-2257 in SEQ ID NO: 1).
- the top row of the sample sequence shows the entire sequence of this region (nt78-299) of # 22 (SEQ ID NO: 11).
- base positions different from # 22 in samples 1 to 75 are indicated by alpha vectors indicating bases. .
- the same base is indicated by a dot (•). From the homology of the sequence (corresponding to ntl939-2160 in SEQ ID NO: 1 and nt78-299 in SEQ ID NO: 11),
- genotype I 1 to 49 groups (named genotype I), 50 to 73 (named genotype II), specimen 74 and specimen 75.
- the top row shows the positions of the primers used in Examples 2 and 3.
- RD 037 SEQ ID NO: 2
- RD 051 SEQ ID NO: 2
- FIG. 6 shows the extension of the # 22 clone sequence using the Gene Walking method (Example 4).
- Figure 7 shows an example of HNT 22 infection by blood transfusion (Example 7).
- HNT 22 gene sequence in transfused blood SEQ ID NO: 11
- FIG. 8 shows the results of a search for the open reading frame (ORF) of the HNT22 gene (Example 10).
- Upper 3 frames candidate start and stop codon positions in the nucleotide sequence of SEQ ID NO: 1.
- a short vertical line indicates the start codon and a long vertical line indicates the stop codon.
- Each frame shows the case where the reading frame is shifted by one base. Long ORFs were found in the first and second frames.
- Lower 3 frames candidate start and stop codon positions for the 3 frames in the sequence complementary to the base sequence of SEQ ID NO: 1. Long 0 R F was not observed in any of the frames.
- FIG. 9 shows the hydrophilicity / hydrophobicity score of the polypeptide based on the amino acid sequence encoded in reading frames 1 and 2.
- FIG. 10 shows a molecular phylogenetic tree based on the nucleotide sequence of the gene from the HNT22-positive case obtained in Example 12.
- FIG. 13 shows the positional relationship between primers and clones used for sequencing the full length of the TUS01 gene.
- the names of the clones sequenced are shown in squares.
- the names of the primers used for amplification are shown on the left and right sides of the square, and the numbers in parentheses indicate the base numbers when the first base at the 5 'end is 1.
- FIG. 14 shows the results of a search for the open reading frame (ORF) of the TUS01 gene.
- FIG. 15 shows a comparison of the sequence of the 5 ′ end region between the HNT22 gene and the TUS01 gene.
- FIG. 16 shows a comparison of the sequence of the 3 ′ end region of the HNT22 gene and the TUS01 gene.
- the RDA method is a method of detecting a gene present in only one of the comparison groups, and a method of efficiently searching for a different gene between a tester as a test subject and a driver as a control. is there.
- the post-transfusion sample of hepatitis in case 2 (see Example 6 for details) of the above three cases with post-transfusion hepatitis of clinical unknown origin was designated as Tess Yuichi (A).
- Tess Yuichi A
- B1 sample before hepatitis onset
- B2 acute hepatitis B patient sample from another patient
- nucleic acids were extracted from Tess Yuichi and the sera of each driver using a commercially available nucleic acid extraction kit (IS0GEN-LS, manufactured by Nippon Gene). That is, 100 l of serum and 300 l of nucleic acid extract were placed in a 1.5 ml eppendorf tube, mixed with stirring for 1 minute, and left at room temperature for 5 minutes.
- the nucleic acid precipitate obtained by the above operation was dissolved using DEPC-water (deionized water treated with getyl pyrocarbonate (Sigma)) 101. To this solution was added 1 ⁇ 1 of a random hexamer (5 Ong / ⁇ l), heated at 70 ° C. for 5 minutes, and then immediately cooled with ice.
- the lysate contains 5 ⁇ first-strand (1 ststr and) buffer, 41, 0.1 M DTT 2 ⁇ 1, 10 mM dNTP 1 ⁇ 1, ribonuclease inhibitor 1 1 for 40 U / ⁇ l (RNasin, PROMEGA) and 1 for 200 ⁇ / ju 1 reverse transcriptase (Superscript II, GIBCO-BRL) 1 was added and reacted at 37 ° C for 60 minutes to synthesize the first strand cDNA.
- Glycogen (2 Omg / ml) lzl, 7.5 M ammonium acetate (manufactured by Wako Pure Chemical Industries), 78/1, cold ethanol (manufactured by Wako Pure Chemical Industries, Ltd.) 2 ⁇ 1 was added, and the mixture was immediately centrifuged at 1500 rpm / min at room temperature for 20 minutes to obtain a nucleic acid as a precipitate fraction. The precipitate was washed with cold 70% ethanol 600 z1 and air-dried, and then dissolved in 5 Oj1 TE buffer (10 mM Tris-HCl, H7.5-1 mM EDTA).
- this solution was subjected to gel filtration using a Microspin S-400 HR Column (manufactured by Pharmacia).
- a Microspin S-400 HR Column manufactured by Pharmacia.
- adapters R—Bam24 and R—Bam12 that were compatible with the base sequence of the cleavage point of the restriction enzyme Sau3AI were bound, and adapters were introduced at both ends of the fragment.
- g Dissolve the nucleic acid fraction precipitate obtained in the above procedure in 16.11 water, add 10 times the concentration of T4 Reiges buffer (NEB) 3 ⁇ 1 and adapter R-B a m24 (10 OD / ml) 6.0 ⁇ 1, Adapter R—Bam 12 (10 OD / ml) 3.0 1 and then reduce the ambient temperature from 50 ° C to 10 ° C over 1 hour Then, T4 ligase (400U / ⁇ 1) (NEB) was added in 1.5 ⁇ 1 and allowed to react at 16 ° C.
- DNA polymerase was allowed to act on the gene fragment to obtain a complete double-stranded gene fragment over the entire length, and the gene fragment was subjected to PCR using RBam24 as a primer.
- the gene fragment was amplified. That is, the nucleic acid fraction into which the adapter was introduced by the above operation was heated at 70 ° C. for 15 minutes to inactivate the T 4 -ligase in the reaction solution.
- TaKaRa EX Taq Polymerase buffer (Takara Shuzo) 20 ⁇ 1 and 2.5 mM dNTP24 ⁇ l, water 149.3 ⁇ 1, RB am24 (10 OD / ml) 5.2 1
- TaKaRa EX Taq Polymerase 1 JJL1 was added and PCR was performed under the following conditions. After treatment at 72 ° C for 5 minutes, a reaction was performed for 30 cycles with 1 cycle of 95 ° C for 1 minute-72 ° C for 3 minutes, followed by treatment at 72 ° C for 7 minutes and cooling to 4 ° C.
- the nucleic acid was extracted with phenol and chloroform in the same manner as described above, and precipitated with ethanol. Further, the precipitates of eight PCR products were combined, dissolved in 100 1 of TE buffer, and subjected to gel filtration in the same manner as described above to obtain an eluate, which was precipitated with ethanol.
- the gel was filtered and precipitated with ethanol:
- the PCR product was dissolved by adding 90% water.
- To this lysate was added 10 ⁇ 1 of a 10-fold concentration of H buffer, and further Sau3AI was added and reacted at 37 ° C for 15 minutes.
- Then precipitate the nucleic acid in the same manner as above.
- gel filtration was performed using a MicroSpin S-400 HR Column in the same manner as described above, and the precipitate was recovered with ethanol. After re-dissolution, the absorbance at 260 nm was measured to confirm the yield.
- adapters J-Bam24 and J-Baml2 adapted to the Sau3AI sequence were connected only to the amplified gene fragment of Tess Yuichi. That is, among the amplification products from which R-Bam24 / R-Baml2 test was removed by the above operation, the PCR product of 10-fold concentration of the T4 product was added to the PCR product 1.0 ⁇ 3 ⁇ 1 and add the adapters J-Bam24 13.2 ⁇ 1 and J-Baml2 6.6 ⁇ 1 and water 4.9 ⁇ 1 to increase the ambient temperature from 50 ° C to 10 ° C 1 It was lowered and annealed over time.
- T4 DNA ligase 400 U // 1) (manufactured by NEB) was added, and the mixture was allowed to react at 16 ° C., and an adapter was connected to the cut end of Sau3AI. Thereafter, the mixture was reacted at 70 ° C. for 10 minutes to inactivate the ligase.
- the amplification product from the tester to which the adapter 1 was connected and the amplification product from the driver that had the adapter 1 removed in the previous operation were heat denatured to make all single-stranded DNA and then re- We met. That is, first, 30: 1 of a phenol: chloroform mixed solution (1: 1) was added to the reaction solution to remove proteins. To the resulting supernatant 17z1 was added 40 ⁇ g of the driver gene amplification product from which the adapter had been removed, followed by ethanol precipitation. The precipitate was dissolved by adding 3 times the concentration of EE buffer (14 ⁇ 1) and layered with 30/1 mineral oil (manufactured by Sigma), followed by reaction at 98 ° C for 10 minutes to denature the DNA.
- EE buffer 14 ⁇ 1
- 30/1 mineral oil manufactured by Sigma
- J-Bam-24 100 D / ml
- the PCR was performed under the conditions that the temperature was kept at 70 ° C for 7 minutes and then left at 4 ° C.
- the nucleic acid was extracted from the amplified sample using a phenol: chloroform mixture in the same manner as described above, and then ethanol-precipitated using glycogen as a coprecipitant. Under these conditions, only the tester-derived gene to which J-Bam24 is connected as an adapter can be amplified using the added J-Bam24 as a primer.
- DNA derived from the homo-double-stranded DNA derived from the Tesyuichi-specific gene can be amplified together, it can be amplified logarithmically.
- DNA derived from hetero-double-stranded DNA composed of genes common to Tesyuichi / Driver has only a multiple increase in the number of DNA strands that have the driver gene sequence with tester type II. Not just.
- the DNA chain derived from the homo-double-stranded DNA comprising only the driver gene cannot be amplified because the added primer cannot hybridize.
- a large number of homo-double-stranded DNAs having a tester-specific gene sequence occupy a large amount in the reaction solution after the amplification.
- Mung Bean Nuclease treatment To obtain only test-specific genes except for the single-stranded dryno-gene sequence DNA present in the reaction solution (exactly The double-stranded DNA derived from the driver was subjected to Mung's Bean nuclease (hereinafter abbreviated as “MBN”) treatment. That is, to each of the nucleic acid fractions obtained by precipitation from a sample hybridized with Tester A and Driver B 1 or Tester A and Driver B 2 Dissolved.
- MBN Mung's Bean nuclease
- PCR was carried out under the following conditions by adding mM dNTP24 / l, primer J-Bam24 and TaKaRa Ex Taq DNA Polymerase 1 zl, and further adding water to make the total amount 2001. That is, a reaction cycle consisting of 95 ° C, 1 minute-70 ° C, 3 minutes was repeated 20 times, and the reaction was further performed at 70 ° C, 7 minutes, and then the reaction temperature was lowered to 4 ° C. From this PCR product, 10 ⁇ 1 was taken and subjected to gel electrophoresis (1 ⁇ TBE buffer) of 2.5% NuSieve 3: 1 agarose (FMC BioProducts, USA). The results obtained were the same for the combination of A and B1 and for the combination of A and B2, so only the combination of A and B1 will be described hereafter.
- Nucleic acids were extracted from the remaining amplification products using the above-mentioned extraction method using a mixture of a phenol and chloroform, and recovered by precipitation with ethanol. To this precipitate was added TE buffer—45 ⁇ 1 to dissolve. This solution was subjected to gel filtration using the aforementioned MicroSpin S-400 HR Column, and the eluate was precipitated with ethanol.
- the cells were dissolved in TE buffer (5 mM Tris-HCl, pH 7.5-0.5 mM EDTA) at a concentration of 1/2 of 10 ⁇ .
- TE buffer 5 mM Tris-HCl, pH 7.5-0.5 mM EDTA
- the yield was determined by measuring the absorbance of this lysate at 260 nm. It is considered that this fraction contains a Tes-Yuichi-specific gene.
- the mixture was ice-cooled, 5M sodium chloride (manufactured by Wako Pure Chemical Industries, Ltd.) was added, and the mixture was reacted at 67 ° C for about 21 hours to perform hybridization.
- the nucleic acid was ethanol-precipitated from the sample obtained by the hybridization in the same manner as described above. To this precipitate, add 20 ⁇ 1 of TE buffer (1/2 concentration) to dissolve, and then add 2 ⁇ 1 of this solution to EX Taq DNA polymerase buffer 201, 2.5 mM dNTPs (240 l, water). 147 l of TaKaRa Ex Taq DNA polymerase (1 ⁇ .1) was added, and the mixture was reacted at 72 ° C.
- nucleic acids were extracted in the same manner using a phenol: chloroform mixture, and after adding glycogen, the nucleic acids were precipitated with ethanol. The nucleic acid fraction was dissolved by adding TE buffer at a concentration of 1/2 of 101.
- the nucleotide sequence of the tester-specific gene candidate obtained in the above procedure was determined by the following method, and a novel virus gene clone # 22 was isolated. That is, both # 22 have Sau3AI cleavage sequences at both ends. Using this sequence, it was cloned into pT7BlueTec Yuichi (Navagen). This clone was transfected into E. coli TG-1 and transformed cells were screened. Furthermore, the plasmid DNA of the obtained transformed cells was analyzed. That is, plasmid DNA was prepared for a total of 60 clones, and for each of the DNAs, a forward and reverse direction was determined using a Thermo Sequencer Fluorescent-labelled primer cycle sequencing kit (Amersham International pic.
- a plurality of oligonucleotides having a length of 20 bases constituting the nucleotide sequence of HNT22 clone # 22 obtained in Example 1 were prepared, and various combinations were examined for the usefulness as a primer for gene amplification.
- the gene is isolated
- PCR was performed on the gene-positive sample to search for sequences that efficiently amplify the gene and combinations thereof.
- SEQ ID NO: 2 (primer: RD 037) and SEQ ID NO: 3 (primer: RD 038), and SEQ ID NO: 4 (primer: RD 051) and SEQ ID NO: 5 (primer: RD 052)
- RD037 and RD051 are sense primers
- RD038 and RD052 are antisense primers. Amplification is performed using a pair of sense primer and antisense primer.
- Nucleic acids were extracted from 100% serum or plasma using a commercially available nucleic acid extraction kit (EX R & D, manufactured by Sumitomo Metal Industries). As described later, since the HNT22 virus was found to be a DNA virus (Example 8), this work treated the virus gene as DNA.
- the extracted DNA was dissolved in TE buffer 10/1, and the whole amount was used as a sample.
- 0.5 / 1 (10 OD) of 10 times concentration of AmpliTaq DNA polymerase buffer (Perkin Elmer) 5.0 / 1, 10 mM dNTP l. 0 / Is primer 1 RD 037 and RD 038 in a PCR tube. / m 1), 0.25 ⁇ 1 of a heat-resistant DNA polymerase (AmpliTaq DNA Polymerase: manufactured by Perkin Elmer), and further distilled water to make the total amount 50 ⁇ 1.
- This reaction solution was prepared for business use.
- the extracted DNA sample 1 described above was added to the tube containing the reaction solution. Further, 501 mineral oil was overlaid and stirred, followed by centrifugation at 6000 rpm for 30 seconds in a cooling centrifuge. After centrifugation, the tubes were mounted on a thermal cycler and PCR was performed. The conditions of the PCR were 95 ° C, 2 minutes and 30 seconds, and then a cycle of 94 ° C, 30 seconds—55 ° C, 30 seconds-72 ° C, 45 seconds was repeated 35 times. The reaction was carried out at 72 ° C for 7 minutes and completed.
- 501 mineral oils were layered and stirred, and the mixture was centrifuged at 6000 rpm for 30 seconds in a cooling centrifuge, and then the tube was mounted on a thermal cycler to perform PCR.
- the PCR conditions were as follows: a cycle of 94 ° C, 30 seconds-55 ° (30 seconds-72 ° C, 30 seconds) was repeated 25 times, and finally a reaction was performed at 72 ° C, 7 minutes.
- the HNT22 gene detection method of Example 2 was sufficient to detect HNT22 virus infection in the individual.However, for the purpose of examining the preservation of the broad band, the HNT22 gene
- the sequences in the included range were amplified and studied by the method using the primers NG001 / RD038 (1st PCR) and NG001 / RD052 (2nd PCR) described in Example 7 below.
- the virus was found to have multiple genotypes, including the two main genotypes I and II (named by the inventors as described in Example 12 below). .
- genotype II mismatches were scattered in the sequences of primers RD037 and RD051 used in Example 2, and it was found that the mismatches were present particularly on the 3 'side (see FIG. 1 to 5).
- a highly conserved sequence was searched for an oligonucleotide having the sequence of SEQ ID NO: 6 (primer: NG059), SEQ ID NO: 7 (NG061), SEQ ID NO: 8 (NG063) ( Figures 1 to 5).
- DNA was extracted from 50% of serum or plasma in the same manner as in Example 2.
- the extracted DNA was dissolved in 20 ⁇ 1 of distilled water, treated at 95 ° C for 15 minutes, and immediately cooled in ice for 2 minutes to prepare a sample for measurement.
- a PCR tube add 10 times concentration of AmpliTaq DNA Polymerase buffer (Perkin Elmer) 5.0 ⁇ 1, 2.5 mM dNTP 4 zl, primers NG 059 and NG 063 (10 OD / m 1) Heat-resistant DNA polymerase (AmpliTaq DNA Polymerase: Perkin Elmer) 0.25 ⁇ 1 was added, and distilled water 29.751 was further added to make the total amount 40/1.
- a PCR was performed.
- the PCR conditions were as follows: a cycle consisting of 94 ° (30 seconds – 60 ° 60, 45 seconds – 72 ° 45 seconds) was repeated 25 times, and the reaction was completed at 72 ° C for 7 minutes. Detection was performed in the same manner as in the agarose electrophoresis method described in Example 2. However, in this example The HNT22 gene, which is amplified by PCR using the first primers NG059 and NG063, is detected as a band at 286 bp, and further detected as a band at 271 bp by PCR using primers NG061 and NG063. it can. Needless to say, each PCR constituting the two-step PCR performed in this example may be performed independently.
- Example 3 can be used to amplify the HNT22 gene of many different genes including genotypes I and II, and the gene amplification method described in Example 2 makes the genotype 1 specific. Since the amplification is possible, the gene detection described in Examples 2 and 3 can be attempted for the same specimen, and the genotype can be classified based on the detection pattern (FIGS. 1 to 5).
- Example 4 Extension of the sequence of the elucidated gene
- the elucidated gene sequence was extended based on clone # 22. That is, a two-step combination of the PCR established in Examples 2 and 3 and having RD 037 and RD 038 as a primer pair and the RD 051 and PCR having D 052 as a primer pair.
- HNT22 virus virus is measured by a two-stage PCR that combines PCR with NG059 and NG063 as a pair of primers and PCR with NG061 and NG063 as a pair.
- the # 22 sequence was further extended in the 5, 5 and 3 'directions by walking using samples (plasma) with a value of 106 international units).
- the virus strain having the obtained nucleotide sequence was named T278.
- the first stage PCR (lst- ⁇ 10 is 94 ° 30 seconds-42 ° (45 seconds-72, 2 minutes), repeated 5 times, and the reaction product is subjected to SizeSep-400 Column (Pharmacia Biotechnology). After purification, a reaction cycle consisting of 94 ° C, 30 seconds—55 ° 45 seconds_72 ° 2 minutes was repeated 35 times, and the second-stage PCR (2nd PCR) was performed for 1st PCR product. A reaction cycle consisting of 94 ° C, 30 seconds-55 ° (, 30 seconds-72 ° (:, 2 minutes) was repeated 30 times using the / 10 amount.
- SEQ ID NO: 16 was obtained from the PCR amplification product obtained by combining primers SSP-A and RD052, and the sequence described in SEQ ID NO: 17 was obtained from primers RD051 and SS P_C Obtained from amplification products of the combination
- the first stage PCR was performed using a combination of any one of the antisense primer NGO12 and the above non-specific primer. ? ⁇ 1
- the conditions were 94 ° 30 seconds-55 ° 30 seconds_72, 2 minutes, 5 times, and the reaction product was repeated 5 times in a SizeSep-400 Column (Pharmaci a Biotechnology) and further 94.
- a reaction cycle consisting of 30 seconds—55 ° C., 45 seconds—72 ° C. and 2 minutes was repeated 35 times.
- the second-stage amplification is performed at 94 ° C for 30 seconds-55 ° 30 seconds-72, using 1/10 amount of the above-mentioned first-stage amplification product and NG013 as a specific antisense primer.
- an oligonucleotide having the nucleotide sequence of SEQ ID NO: 27 (primer name: NG031) was used as an antisense primer for single-sided side PCR at the first step, and had the nucleotide sequence of SEQ ID NO: 28 Oligonucleotides (primer name: NG032) were used as antisense primers for the second stage single'side PCR.
- an oligonucleotide having the nucleotide sequence of SEQ ID NO: 29 (primer name: NG045) was used as the first-stage single-sided PCR antisense primer, and the oligonucleotide of SEQ ID NO: 30 was used. Oligonucleotides (primer name: NG039) were used as the second stage single-sided PCR antisense primer.
- Non-specific primers include SEQ ID NOS: 12 to 15 and The oligonucleotide described in SEQ ID NO: 20 was used (FIG. 6).
- ntll09-1575 SEQ ID NO: 31: clone T10
- ntl2-759 SEQ ID NO: 32: clone T14
- nt3119-3315 SEQ ID NO: 31: clone T10
- SEQ ID NO: 33 For the sequence corresponding to clone T8), an oligonucleotide having the nucleotide sequence of SEQ ID NO: 34 (primer: NG 013) and 35 (primer: NG 025), respectively, SEQ ID NO: Oligonucleotides having the base sequences described in 36 (one primer: NG040) and 37 (one primer: NG046), SEQ ID NOs: 38 (one primer: RD 057) and 39 (one primer: RD) [058] An oligonucleotide having the described nucleotide sequence was amplified as a primer pair, the amplified product was clone
- sequences of the obtained clones were combined to obtain a total length of 3739 bp shown in SEQ ID NO: 1.
- the base with the highest frequency of occurrence was prioritized.
- BLAST and FAST A National Institute of Genetics, search programs: BLAST and FAST A
- Example 5 Detection of HNT22 Gene from Non-B Non-C Non-G Hepatitis Virus Marker-Negative Alanine Aminotransferase Level Donors and Chronic Hepatitis Patients Known Hepatitis Virus Markers Found in Japanese Donors
- the activity of alanintotransferase (ALT) was ⁇ 100 international units (IU / 1) for 207 persons and the activity of ⁇ -gamma-mil transbeptidase ( ⁇ -GTP) was 5
- the presence or absence of the HNT22 gene was examined using the gene detection method described in Example 2 for 26 patients who had 00 IU / 1 or more and 15 non-B non-C chronic hepatitis cases. As controls, 88 donors with normal liver function and 22 patients with chronic hepatitis C were also examined
- HNT22 gene was found in 3 out of 88 (3.4%) donors with normal liver function and 2 out of 22 (9.1%) in chronic hepatitis C cases.
- Example 6 Detection of HNT22 gene from post-transfusion hepatitis cases
- Example 2 For the post-transfusion hepatitis cases for which the known hepatitis virus marker was negative, the method of Example 2 before transfusion, before post-transfusion hepatitis, after hepatitis, and (where possible) blood transfusion was used to examine the presence and appearance time of the HNT22 gene.
- HB V, HCV, and GB V—C / HGV markers were negative at all times during the study period.
- ALT a liver function marker, showed normal values before blood transfusion, but showed abnormal values at 8 and 9 weeks after blood transfusion.
- the HNT22 gene was measured in the patient's blood before transfusion (labeled 0 weeks after transfusion) and 6, 8, 9, 10, 11, 12, 15, and 24 weeks after transfusion in this case. As a result, the HNT22 gene was detected in patient sera before transfusion (Table 1). The blood transfused in this case was 4 units. When the presence or absence of the HNT22 gene was confirmed in this transfused blood, the HNT22 gene was detected in one of the units. table 1
- ALT a liver function marker, showed normal values before blood transfusion, but showed abnormal values around 10 weeks after blood transfusion.
- the HNT22 gene was measured in the patient's blood at 2, 6, 8, and 10 weeks after transfusion in this case. As a result, HNT22 gene was detected in blood at 6 weeks, 8 weeks and 10 weeks after transfusion in the 1 st PCR with one-step amplification (Table 2). On the other hand, at the second week, the titer was low and was detected only in the 2nd PCR. In this case, the transfused blood was not stored, and therefore, it was not possible to confirm the presence or absence of the HNT22 gene in the transfused blood.
- HNT22 clone # 22 was isolated. The virus was found to be present in the blood of the second week used as the driver in the separation, but it was detected only during high-sensitivity two-step amplification. Is extremely low, and it is considered that this functioned substantially as the driver. Table 2
- ALT a function of liver function, showed normal values before blood transfusion, but showed abnormal values around 6 weeks after blood transfusion.
- PCR was performed to perform stepwise amplification, and the presence or absence of the HNT22 gene was confirmed.
- a series was prepared by diluting the sample 10 times, and the same measurement was performed.
- the HNT22 gene was detected in the blood at each point from 4 weeks to 6 weeks after the transfusion, although the results were negative at 2 weeks after the transfusion.
- ALT a function of liver function
- the presence or absence of the HNT22 gene and the titer of the viral gene were also determined (similar results were obtained with any of the methods of Examples 2 and 3). The results were negative at 4 weeks post-transfusion, but increased from 6 weeks post-transfusion, with the most pronounced liver dysfunction.
- the HNT22 gene of the present invention was detected from a post-transfusion hepatitis case in which the known hepatitis marker was negative and the cause was unknown in Example 6, and the change in the abnormal value of the liver function marker and the change in virus were well matched. This strongly suggested that HNT22 was responsible for the hepatitis.
- the purpose of this example was to demonstrate more clearly that HNT 22 is transmitted by blood transfusion. In other words, in the case where the HNT22 gene was detected in blood after transfusion and after blood transfusion and all transfused blood pilots were secured, the blood after transfusion and the transfused blood pilot were used.
- gene measurement was performed by the method described below to identify the gene-positive transfusion pilot, which is considered to be the source of infection, and the sequences of the gene obtained from the patient's blood and the pilot were determined and compared, and the The homology was examined.
- HNT22 gene measurement was performed on all 10 blood pilots used. As a result, HNT22 gene was detected in one of them.
- Nucleic acids were extracted from all HNT22-positive specimens according to the method described in Example 2.
- the 1st PCR sense primer RD037 of Example 2 was changed to a primer NG001 having the nucleotide sequence shown in SEQ ID NO: 40, and the PCR conditions were changed to 94 ° C and 30 seconds—55. ° 30 seconds—72 ° (changed to 1 minute reaction cycle and repeated 35 times.
- a positive example yields a 415 bp DNA fragment.
- Sense primer RD 05 1 was changed to NG 001, and the ⁇ 1 condition was set to 94 ° 30 seconds_55 ° (30 seconds-72 ° 1 minute reaction cycle repeated 25 times.
- Amplification yields a 396 bp DNA fragment in positive cases, inserts the 396 bp amplification product into a plasmid vector and clones it as in Example 1, and then bases three clones on each amplified example. Were determined and compared for homology.
- the sequence of the clone obtained from the HNT22-positive transfused blood pilot was completely identical to the HNT22 gene sequence obtained from the patient transfused from the pilot (Fig. 7). This indicated that the HNT22 virus was transmitted by blood transfusion and persisted thereafter.
- this gene sequence was compared with the sequence of clone # 22, three out of 356 bases other than the primer-derived sequences at both ends were different.
- Example 8 Proof that the virus of the present invention is a DNA virus
- HBV hepatitis viruses
- HCV RNA viruses
- Example 1 the sample from which the virus gene was isolated underwent a reverse transcription reaction step after nucleic acid extraction. Therefore, the DNA that originally existed in the sample after the reverse transcription reaction and the DNA that had existed as RNA before the reaction but became DNA by the reverse transcription reaction were mixed. Therefore, if the virus of the present invention is an RNA virus, omitting the reverse transcription reaction makes detection of the virus gene impossible or extremely difficult. For this reason, the present inventors carried out PCR based on Example 2 in which the reverse transcription reaction was omitted, and examined the possibility described above. As a result, it was confirmed that the HNT22 gene could be detected at the same level even when the reverse transcription reaction was omitted.
- HNT 22 was determined to be a DNA virus.
- the present inventors in addition to the examination in (1) above, treated the nucleic acid fraction extracted from the sample in which the presence of the HNT22 gene was confirmed by the method of Example 2 with DNA degrading enzyme, and reversed the treatment. A transcription reaction was performed, and the HNT22 gene measurement was performed by the method of Example 2 using the obtained DNA as a sample.
- the nucleic acid fraction extracted using the same commercially available kit (EX MD, manufactured by Sumitomo Metals) as in Example 2 was treated with DNase I (Takara Shuzo) at 37 ° C for 30 minutes, and then D Nase I activity was inhibited.
- DNase I Takara Shuzo
- an operation after the reverse transcription reaction was performed under the same conditions as described in Example 1 to try to detect the HNT22 gene. As a result, it was found that the HNT22 gene was not detected when treated with DNaseI.
- the inventors of the present invention assumed that the HNT22 gene was a double-stranded chain, a restriction enzyme E c 0 RI in which a specific sequence was recognized in the sequence, and a restriction enzyme in which no specific sequence was recognized as a control.
- B g 1 II was examined for cleavage.
- DNA was obtained using the method described in Example 1 from plasma 800-1 obtained from a patient whose HNT22 virus was confirmed to be positive by the method described in Examples 2 and 3 (90 1 ). From each of them, 15 ⁇ 1 was taken, and each was added with 2 c1 of EcoRI (Takara Shuzo) or Bglll (Takara Shuzo) and treated at 37 ° C for 2.5 hours. At the same time, the same reaction was performed by adding a phosphate buffer instead of the restriction enzyme. After restriction enzyme digestion, remove 3 ⁇ 1 from each sample and prepare 10-fold and 100-fold diluted samples, with primers NG 001 containing the EcoR I restriction enzyme specific sequence inside. Using RD038, the HNT22 gene region containing the EcoRI restriction enzyme specific sequence was amplified. As a result, no cleavage by the restriction enzyme was observed in any of the samples.
- Mung Bean Nuclease is known to specifically cleave single-stranded DNA. Therefore, if HNT22 is present in a single strand, it is considered to be sensitive to Mung's Bean nuclease.
- the present inventors obtained DNA from the same patients as those used in the above-mentioned restriction enzyme treatment experiment and treated them with Mung's Bean nuclease.
- a partial region of HNT22 amplified by the method of Example 2 was incorporated into M13 phage to prepare a double-stranded form and a single-stranded form.
- the HNT22 gene was amplified according to the method of Example 2.
- DNA obtained from HNT22-positive patients and single-stranded phage-derived DNA were not amplified, and were sensitive to Mung 'Bean Nuclease.
- HNT22 gene was present as a single-stranded DNA, and that the HNT22 virus was a single-stranded DNA virus.
- Example 10 Amino acid sequence encoded by HNT22 gene
- the amino acid sequence encoded by the HNT22 gene was analyzed.
- the start codon sequence and the stop codon sequence are searched for in the obtained base sequence, and the ORNT of the HNT22 gene is determined based on which combination gives the normally readable open reading frame (ORF). Searched for F. As a result, it was found that there were two ORFs (OR F 1: nt589-nt2898 (770 amino acid residue) and 0 RF 2: ntl07-nt712 (202 amino acid residue)) (FIG. 6 and FIG. 8). This suggests that the HNT22 gene encodes a polypeptide consisting of the amino acid sequence of SEQ ID NOS: 9 and 10.
- the HNT22 gene was amplified by the method of Example 2, and the HBV gene was amplified and detected by the following method.
- a DNA sample 5 ⁇ 1 was prepared from primers S2-11 having the nucleotide sequence of SEQ ID NO: 41 selected from sequences in the HBV surface antigen gene region and primers S1-2 having the base sequence of SEQ ID NO: 42.
- 0.5 ⁇ 1 (10OD / ml) Much, 2.5mMdNTP4 zl, 10 times concentration of heat-resistant DNA polymerase buffer (Ampl iTaq DNA Polymerase included: Perkin Elmer) 5 ⁇ 1, Heat resistant DNA polymerase (Amp 1 iT aq DNA Polymerase: Perkin Elmer) 0.25 jul Added to PCR tube containing amplification reaction solution containing 34.75 1 Thereafter, 501 mineral oil was overlaid, stirred, and further centrifuged at 5000 rpm for 30 seconds, and then mounted on a thermal cycler to perform PCR.
- 501 mineral oil was overlaid, stirred, and further centrifuged at 5000 rpm for 30 seconds, and then mounted on a thermal cycle
- the PCR reaction conditions were 94 ° C, 30 sec-55 ° (30 sec-72 ° 75 sec), and this cycle was performed 35 times. In this method, the HBV gene was 250 bp long in the electrophoresis gel. If more sensitive detection is required, a second stage PCR was performed using a portion of the above amplification product under the following conditions: The reaction solution was prepared under the same conditions as above except that the primer used was changed to primer $ 088 having the nucleotide sequence of SEQ ID NO: 43 and primer S2-2 having the nucleotide sequence of SEQ ID NO: 44.
- PCR was carried out after the same procedure as described above, and the PCR conditions were 94 ° (30 seconds-55 ° (30 seconds-72 ° 60 seconds), and this cycle was repeated 25 times.
- the amplification product obtained by the second stage PCR is detected in the electrophoresis gel as a 228 bp band.
- the HNT22 gene had a sucrose density of 54.5% and a density of 1.26 g. / to cm 3 exist with beak, the HBV as the other control similar to tell conventional distribution, be present with a peak density 1.. 26 to 1. 20 g / cm 3 was found (Table 5).
- This density analysis showed that the HNT22 gene was present in the virus particle fraction, indicating that the density fraction centrifugation method could separate and collect the above-mentioned density fraction.
- Table 5 Distribution of sucrose gradient centrifugation fraction of HNT 22 gene Sucrose density Density HNT 22 -DN A HB V-DNA
- HNT 22 has genetic inheritance
- the present inventors considered that HNT 22-positive cases that were positive by the gene detection method described in Example 3 were: The amplified gene was analyzed, a molecular phylogenetic tree was created based on the gene sequence, and the genotype of HNT22 was searched.
- the inventors examined the presence or absence of the HNT22 gene by the method described in Example 3 for Japanese, Americans, and French, and as a result, a total of 199 cases (187 Japanese, 8 Americans, French (4) positive cases were obtained.
- the amplified genes were cloned according to the method described in Example 1, the nucleotide sequence of at least three clones was determined, and a molecular phylogenetic tree was created using commercially available software.
- Example 11 It was confirmed that the HNT22 particles could be separated and recovered by the method described in Example 11.
- the present inventors have studied the detection of antibodies by immunoprecipitation using this.
- stool was collected from five HNT-infected patients for which the presence of the HNT22 gene was found in the blood, and suspended in an appropriate solution to prepare a sample.
- the presence or absence of the HNT22 gene in stool was examined according to the method described.
- the presence of the HNT22 gene in the stool, its flotation density of 1.35 g / cm 3 , and its gene sequence in the HNT22 gene sequence obtained from the blood of the same patient Turned out to be identical. From this, it was decided to collect HNT 22 particles from stool.
- Distilled water was added to the stool obtained from the patient to prepare a 15% (weight concentration) suspension.
- This suspension was centrifuged at 3000 rpm for 30 minutes using a cooling centrifuge (manufactured by Hitachi) to obtain a supernatant.
- This supernatant was further centrifuged at 10,000 rpm for 5 minutes using a micro-refrigerated centrifuge (High Apeed Micro Refrigerated Centrifuge: manufactured by Tomi Issei). The supernatant was collected to obtain a HNT 22 particle solution.
- DN A titer of particle liquid was 10 5 / m 1.
- the supernatant was transferred to another Eppendorf tube, and DNA was extracted using a commercially available nucleic acid extraction kit (EX R & D, manufactured by Sumitomo Metals).
- EX R & D a commercially available nucleic acid extraction kit
- a saline solution of 110 11 was gently added to the precipitate, centrifuged at 10,000 rpm for 5 minutes, the supernatant was discarded, and 110 ⁇ 1 of saline solution was added dropwise to suspend. DNA was similarly extracted from this suspension. Dissolve the obtained DNA fraction in 20 ⁇ 1 of distilled water, The mixture was treated at 95 ° C for 5 minutes, and half the amount was used as a sample for detecting the HNT22 gene. Detection of the HNT22 gene was performed in the same manner as in Example 3. Controls were the same serum or plasma without addition of the HNT22 particle solution and the same work, and the same work with HNT22-negative serum or plasma.
- HNT22 gene-positive serum or plasma to which the particle liquid was added showed the HNT22 gene in the sediment fraction, but the HNT22 gene was not added when the particle liquid was added and when the HNT-negative serum or plasma was used. No HNT22 gene was found in the painting. From this, it is considered that only when the HNT22 antibody is present, the HNT22 particles aggregate with this antibody via an anti-human IgG antibody to form a precipitate fraction. In cases where the gene is found, it is considered that an anti-HNT antibody is present.
- Example 14 Measurement of anti-HNT 22 antibody in post-transfusion hepatitis cases and fulminant hepatitis cases by anti-HNT 22 antibody detection method using HNT particle liquid
- Example 13 The inventors used the method of Example 13 to transiently infect HNT 22 after blood transfusion and showed liver dysfunction in Case 4 of Example 6, and a non-A- The fate of anti-HNT22 antibody was investigated in the recovery period of sporadic fulminant hepatitis G cases
- the anti-HNT22 antibody appeared in both cases after the disappearance of the virus, and showed a pattern similar to that of the neutralizing antibody in virus infection.
- the HNT 22 virus was shown to have multiple genotypes (Example 12), and other viruses, such as hepatitis B and C, have regional genotypic bias, indicating that Because the major HNT 22 genotype in US may differ from the major HNT 22 type in Japan, HNT22-positive samples obtained from Americans were examined. From 100 ⁇ 1 of each of U.S.-derived sera confirmed to be positive for HNT22 virus by the detection method described in Example 3 (referred to as UM3-17, UM3_34, UM3_56, and UM3-73), Using a commercially available diffusion extraction kit (Behringer 'Mannheim, High Pure Viral Nucleic Acid Kit), nucleic acids were extracted according to the instruction manual to obtain samples.
- UM3-17, UM3_34, UM3_56, and UM3-73 Using a commercially available diffusion extraction kit (Behringer 'Mannheim, High Pure Viral Nucleic Acid Kit), nucleic acids were extracted according to the instruction manual to obtain samples.
- oligonucleotide having the nucleotide sequence of SEQ ID NO: 55 (NG 055) and an oligonucleotide having the nucleotide sequence of SEQ ID NO: 8 (NG 063) selected based on the sequence of the HNT 22 virus known so far Using the above as a primer and the above sample as type III, PCR was performed under the following conditions. Figure 13 shows the positional relationship of the primers used. PCR conditions
- oligonucleotide (NT01) having the nucleotide sequence of SEQ ID NO: 56 was prepared as a primer for amplifying the nucleotide sequence of SEQ ID NO: 56.
- This oligonucleotide NT01 and an oligonucleotide (NG054) having the nucleotide sequence of SEQ ID NO: 57 prepared based on the sequence of the HNT22 virus known so far as a primer were used as primers to produce UM3--17.
- nucleic acid of origin as type ⁇ , PCR was performed under the following conditions, and the obtained amplified fragment was sequenced in the same manner as described above. The positions of the primers are shown in FIG.
- Oligonucleotides (primers NT03 and NT04) having the nucleotide sequences of SEQ ID NOS: 58 and 59 were prepared as primers for amplification.
- an oligonucleotide having the base sequence of SEQ ID NO: 60 or 61 as a primer for amplification was prepared.
- the 3′-side gene was amplified using NT03 and NG065, and a portion of the resulting amplified fragment was amplified using NT04 and NG065 or NGO21.
- Figure 13 shows the position of the primers. Amplification was performed using type UM3-17-derived nucleic acid under the following conditions. The nucleotide sequence of the amplified fragment was determined in the same manner as described above. CRC condition
- Example 1 Compared to the nucleotide sequence of Example 1, although extremely high homology was observed in the 5 ′ and 3 ′ regions, the homology in the other regions was as low as about 30%, and in Examples 2 and 3, It turned out that this sequence could not be detected by the detection method.
- the virus carrying this gene was named TUS01.
- ORF 1 nt590-nt2872 (76 amino acid residues)
- ORF 2 nt258-nt725 (156 amino acid residues)
- Table 7 summarizes the homology of each region. Table 7 Homology between HNT22 gene and TUS01 gene
- HNT 22 TUS 0 1 Sequence homology Total length 3739 nt 3722 nt 63.7%
- HNT 22 The sequence used for HNT 22 is that of TA278.
- Figures 15 and 16 show the results of comparison of the nucleotide sequences of the highly homologous 5 'untranslated region (5' terminal region) and 3 'untranslated region (3, terminal region).
- the TUS01 virus is estimated to be a variant of the HNT22 virus.
- Example 16 Simultaneous detection of HNT 22 gene and TUS 01
- NG054 and NG065 were selected as oligonucleotides that had a common sequence and were considered to be suitable primers for PCR.
- Nucleic acids were extracted from serum 100 z 1 according to the method described in Example 2. The extracted nucleic acid was dissolved in distilled water 101. NG054 and NG 065 (both 10 OD / ml) solution 1 ⁇ 1, 10 times concentration of Ex. Taq buffer (TaKaRa) 5 ⁇ 1, 2.5 mM dNTP 4 ⁇ 1, distilled water 29.5 ⁇ 1 Add 10/1 of the dissolved nucleic acid to the tube containing PCRW, immediately add heat-resistant DNA polymerase (Ex.
- Taq Polymerase TaKaRa
- treat at 95 ° C for 2 minutes and then heat at 95 ° C for 45 seconds.
- a cycle of 55 ° C for 30 seconds and 72 ° C for 120 seconds was repeated 35 times, and finally a reaction was added at 72 ° C for 7 minutes to amplify.
- the amplification product was separated by agarose electrophoresis, and the presence of a band having a length predicted from the nucleotide sequence was confirmed.
- the sequence of the amplified product for which a band was confirmed was determined. As a result, it was found that the HNT22 gene and the TUS01 gene can be simultaneously detected by this method.
- the present invention has identified an unknown cause virus for blood-borne hepatitis whose cause has been previously unknown. As a result, it is possible to establish gene measurement, antigen measurement, and antibody measurement methods, and to provide a kit for the hepatitis test and a vaccine for the prevention.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Genetics & Genomics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biomedical Technology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Microbiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Plant Pathology (AREA)
- Virology (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicinal Chemistry (AREA)
- Analytical Chemistry (AREA)
- Immunology (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Peptides Or Proteins (AREA)
Description


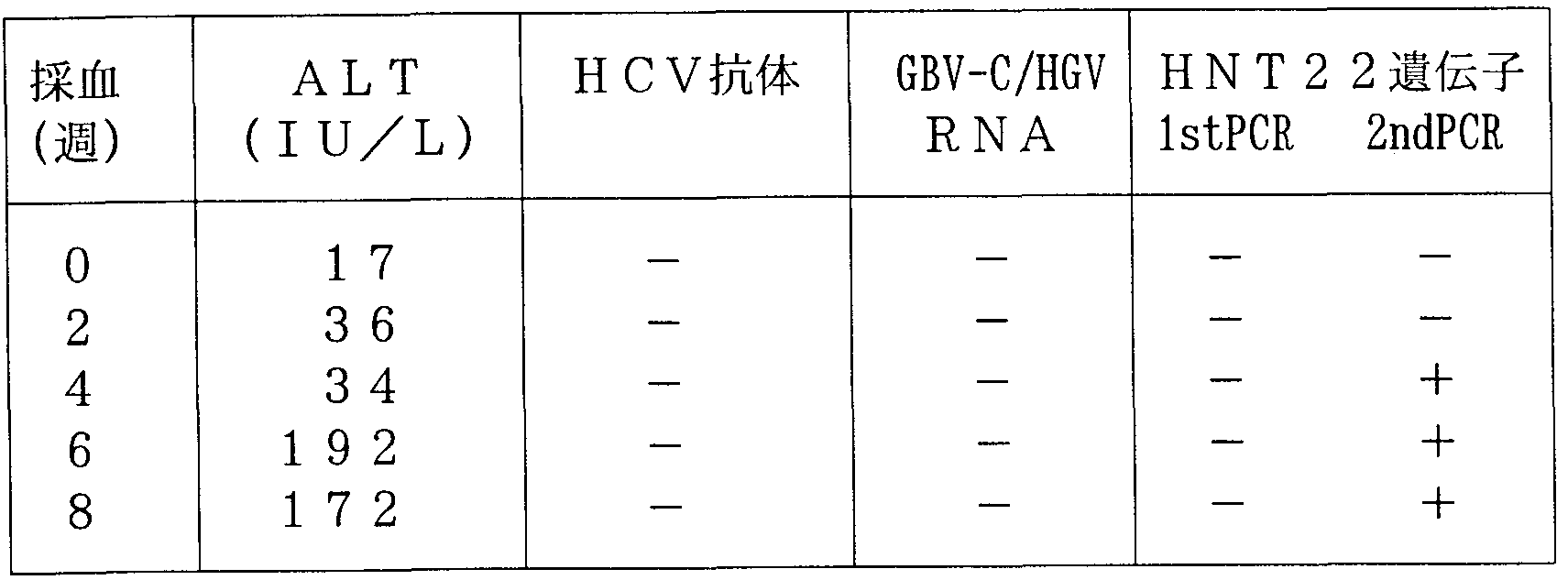



Claims
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002298103A CA2298103A1 (en) | 1997-07-25 | 1998-07-27 | Non-b, non-c, non-g hepatitis virus gene, polynucleotide, polypeptide, virus particle, method for isolating virus particle, and method for detecting virus |
| DE69837437T DE69837437D1 (de) | 1997-07-25 | 1998-07-27 | Gen, polynukleotid, polypeptid und virion des non-b, non-c, non-g-hepatitis virus, eine methode zur abtrennung dieses virions und methoden für die virusdetektion. |
| EP98933947A EP1010759B1 (en) | 1997-07-25 | 1998-07-27 | Non-b non-c non-g hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus |
| AU83586/98A AU741656B2 (en) | 1997-07-25 | 1998-07-27 | Non-B non-C non-G hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus |
| KR1020007000793A KR20010022220A (ko) | 1997-07-25 | 1998-07-27 | 비-b형 비-c형 비-g형 간염 바이러스 유전자,폴리뉴클레오티드, 폴리펩티드, 바이러스 입자, 바이러스입자의 분리 방법, 및 바이러스의 검출 방법 |
| US11/018,177 US20050158343A1 (en) | 1997-07-25 | 2004-12-21 | Non-B, non-C, non-G hepatitis virus gene, polynucleotide, polypeptide, virus particle, method for isolating virus particle, and method for detecting virus |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP23324697 | 1997-07-25 | ||
| JP9/233246 | 1997-07-25 | ||
| JP9/314196 | 1997-10-09 | ||
| JP31419697 | 1997-10-09 | ||
| JP8296298 | 1998-03-13 | ||
| JP10/82962 | 1998-03-13 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09463488 A-371-Of-International | 2000-05-01 | ||
| US10/781,599 Continuation US6849431B2 (en) | 1997-07-25 | 2004-02-18 | Non-B, non-C, non-G hepatitis virus gene, polynucleotide, polypeptide, virus particle, method for isolating virus particle, and method for detecting virus |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999005282A1 true WO1999005282A1 (en) | 1999-02-04 |
Family
ID=27304063
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1998/003340 Ceased WO1999005282A1 (en) | 1997-07-25 | 1998-07-27 | Non-b non-c non-g hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1010759B1 (ja) |
| KR (1) | KR20010022220A (ja) |
| AU (1) | AU741656B2 (ja) |
| CA (1) | CA2298103A1 (ja) |
| DE (1) | DE69837437D1 (ja) |
| WO (1) | WO1999005282A1 (ja) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000028039A3 (en) * | 1998-11-10 | 2000-10-05 | Diasorin International Inc | Identification of senv genotypes |
| WO2000066621A1 (en) * | 1999-05-04 | 2000-11-09 | Tripep Ab | Peptides from the tt virus sequence and monospecific antibodies binding to the tt virus |
| FR2808277A1 (fr) * | 2000-04-28 | 2001-11-02 | Bio Merieux | Polypeptide du ttv, acide nucleique codant pour ce polypeptide et utilisations |
| WO2000046407A3 (en) * | 1999-02-05 | 2002-05-10 | Abbott Lab | Methods of utilizing the tt virus |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1536013A4 (en) * | 2002-02-13 | 2005-11-30 | Terukatsu Arima | DNA SEQUENCE USED IN THE DIAGNOSIS OF HEPATITIS AND POLYPEPTIDE SO CODE |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01279844A (ja) * | 1988-04-28 | 1989-11-10 | Yasuo Moritsugu | A型肝炎ワクチン |
| JPH09299078A (ja) * | 1996-05-14 | 1997-11-25 | Niatsuku:Kk | 非a非b非c型肝炎ウィルス粒子の分離法、該ウィルスコア粒子の精製法、ならびに該ウィルスおよびコア抗体等の検出法 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5437974A (en) * | 1992-02-04 | 1995-08-01 | Dade International Inc | DNA sequence and encoded polypeptide useful in the diagnosis of hepatitis disease |
-
1998
- 1998-07-27 WO PCT/JP1998/003340 patent/WO1999005282A1/ja not_active Ceased
- 1998-07-27 EP EP98933947A patent/EP1010759B1/en not_active Expired - Lifetime
- 1998-07-27 CA CA002298103A patent/CA2298103A1/en not_active Abandoned
- 1998-07-27 KR KR1020007000793A patent/KR20010022220A/ko not_active Withdrawn
- 1998-07-27 DE DE69837437T patent/DE69837437D1/de not_active Expired - Lifetime
- 1998-07-27 AU AU83586/98A patent/AU741656B2/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01279844A (ja) * | 1988-04-28 | 1989-11-10 | Yasuo Moritsugu | A型肝炎ワクチン |
| JPH09299078A (ja) * | 1996-05-14 | 1997-11-25 | Niatsuku:Kk | 非a非b非c型肝炎ウィルス粒子の分離法、該ウィルスコア粒子の精製法、ならびに該ウィルスおよびコア抗体等の検出法 |
Non-Patent Citations (6)
| Title |
|---|
| BISPEN T. A., ET AL.: "PREPARATION AND PROPERTIES OF SOME POLYFLUORINATED PENTANES.", ZHURNAL PRIKLADNOI KHIMII, MAIK NAUKA: ROSSIISKAYA AKADEMIYA NAUK, RU, vol. 68., no. 05., 1 January 1995 (1995-01-01), RU, pages 793 - 796., XP002918171, ISSN: 0044-4618 * |
| NISHIZAWA T., ET AL.: "NOVEL DNA VIRUS (TTV) ASSOCIATED WITH ELEVATED TRANSAMINASE LEVELS IN POSTTRANSFUSION HEPATITIS OF UNKNOWN ETIOLOGY.", BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS, ELSEVIER, AMSTERDAM, NL, vol. 241., 18 December 1997 (1997-12-18), AMSTERDAM, NL, pages 92 - 97., XP002917290, ISSN: 0006-291X, DOI: 10.1006/bbrc.1997.7765 * |
| OKAMOTO H., ET AL.: "FECAL EXCRETION OF A NONENVELOPED DNA VIRUS (TTV) ASSOCIATED WITH POSTTRANSFUSION NON-A-G HEPATITIS.", JOURNAL OF MEDICAL VIROLOGY, JOHN WILEY & SONS, INC., US, vol. 56., no. 02., 1 October 1998 (1998-10-01), US, pages 128 - 132., XP002917300, ISSN: 0146-6615, DOI: 10.1002/(SICI)1096-9071(199810)56:2<128::AID-JMV5>3.0.CO;2-A * |
| OKAMOTO H., ET AL.: "MOLECULAR CLONING AND CHARACTERIZATION OF A NOVEL DNA VIRUS (TTV) ASSOCIATED WITH POSTTRANSFUSION HEPATITIS OF UNKNOWN ETIOLOGY.", HEPATOLOGY RESEARCH, AMSTERDAM, NL, vol. 10., 1 January 1998 (1998-01-01), NL, pages 01 - 16., XP002918456, ISSN: 1386-6346, DOI: 10.1016/S1386-6346(97)00123-X * |
| See also references of EP1010759A4 * |
| SIMMONDS P., ET AL.: "DETECTION OF A NOVEL DNA VIRUS (TTV) IN BLOOD DONORS AND BLOOD PRODUCTS.", THE LANCET, THE LANCET PUBLISHING GROUP, GB, vol. 352., 18 July 1998 (1998-07-18), GB, pages 191 - 195., XP002917299, ISSN: 0140-6736, DOI: 10.1016/S0140-6736(98)03056-6 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000028039A3 (en) * | 1998-11-10 | 2000-10-05 | Diasorin International Inc | Identification of senv genotypes |
| WO2000046407A3 (en) * | 1999-02-05 | 2002-05-10 | Abbott Lab | Methods of utilizing the tt virus |
| US6395472B1 (en) | 1999-02-05 | 2002-05-28 | Abbott Laboratories | Methods of utilizing the TT virus |
| WO2000066621A1 (en) * | 1999-05-04 | 2000-11-09 | Tripep Ab | Peptides from the tt virus sequence and monospecific antibodies binding to the tt virus |
| FR2808277A1 (fr) * | 2000-04-28 | 2001-11-02 | Bio Merieux | Polypeptide du ttv, acide nucleique codant pour ce polypeptide et utilisations |
| WO2001083757A1 (fr) * | 2000-04-28 | 2001-11-08 | Bio Merieux | Polypeptide du ttv, acide nucleique codant pour ce polypeptide et utilisations |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1010759A1 (en) | 2000-06-21 |
| DE69837437D1 (de) | 2007-05-10 |
| CA2298103A1 (en) | 1999-02-04 |
| EP1010759B1 (en) | 2007-03-28 |
| AU8358698A (en) | 1999-02-16 |
| KR20010022220A (ko) | 2001-03-15 |
| EP1010759A4 (en) | 2002-05-02 |
| AU741656B2 (en) | 2001-12-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4541712B2 (ja) | Ptlvエンベロープタンパク質の表面成分をコードする配列を起源とするオリゴヌクレオチド及びそれらの使用 | |
| EP0521318A2 (en) | Hepatitis C diagnostics and vaccines | |
| AU4046489A (en) | Post-transfusion, non-a, non-b hepatitis virus and antigens | |
| NL9002779A (nl) | Viraal middel. | |
| US5218099A (en) | Post-transfusion, non-A, non-B hepatitis virus polynucleotides | |
| LV10726B (en) | Hepatitis c diagnostics and vaccines | |
| JP2008178416A (ja) | 診断と治療に用いるhcvゲノム配列 | |
| Thomson et al. | Identification of Zucchini yellow mosaic potyvirus by RT-PCR and analysis of sequence variability | |
| BG61615B1 (bg) | Nanbv диагностика: полинуклеотиди за изследване на вируса нахепатит с | |
| EP0633320A1 (en) | Oligonucleotides of HCV, primers ND probes therefrom, method of determining HCV genotypes and method of detecting HVC in samples | |
| JP2007513604A (ja) | 新規エンテロウイルス、ワクチン、医薬および診断キット | |
| Carrick et al. | Examination of the buoyant density of hepatitis C virus by the polymerase chain reaction | |
| JP3809502B2 (ja) | ハンタウィルス抗原蛋白質およびモノクローナル抗体 | |
| WO1999005282A1 (en) | Non-b non-c non-g hepatitis virus gene, polynucleotide, polypeptide, virion, method for separating virion, and method for detecting virus | |
| Lampe et al. | Infection with GB virus C/hepatitis G virus in Brazilian hemodialysis and hepatitis patients and asymptomatic individuals | |
| EP0551275B1 (en) | Non-a non-b sequences | |
| US6849431B2 (en) | Non-B, non-C, non-G hepatitis virus gene, polynucleotide, polypeptide, virus particle, method for isolating virus particle, and method for detecting virus | |
| JPH06319563A (ja) | C型肝炎ウイルス遺伝子、オリゴヌクレオチド、並びにc型 肝炎ウイルス遺伝子型判定方法 | |
| US7479386B2 (en) | HXHV virus, nucleic material, peptide material and uses | |
| JPWO1999005282A1 (ja) | 非b非c非g型肝炎ウイルス遺伝子、ポリヌクレオチド、ポリペプチド、ウイルス粒子、ウイルス粒子分離法およびウイルス検出法 | |
| JP2542994B2 (ja) | オリゴヌクレオチド、並びにc型肝炎ウイルスのジェノタイプ鑑別方法 | |
| JP3373549B2 (ja) | 肝炎ウイルス遺伝子 | |
| JPH03266987A (ja) | 非a非b型肝炎ウイルス遺伝子断片およびウイルスの検出方法 | |
| JPH05176773A (ja) | オリゴヌクレオチド、オリゴペプタイド、ならびに抗c型肝 炎ウイルス抗体検出・鑑別方法 | |
| US20110195407A1 (en) | Nucleic acid occurring in blood from hepatitis patient, polypeptide encoded by the nucleic acid, and uses of the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BR CA CN JP KR MX NZ RU US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 83586/98 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2298103 Country of ref document: CA Ref country code: CA Ref document number: 2298103 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007000793 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998933947 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09463488 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998933947 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007000793 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 83586/98 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1020007000793 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1998933947 Country of ref document: EP |