WO1999012897A1 - A process for making epoxide intermediates - Google Patents
A process for making epoxide intermediates Download PDFInfo
- Publication number
- WO1999012897A1 WO1999012897A1 PCT/US1998/018593 US9818593W WO9912897A1 WO 1999012897 A1 WO1999012897 A1 WO 1999012897A1 US 9818593 W US9818593 W US 9818593W WO 9912897 A1 WO9912897 A1 WO 9912897A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- ring
- aliphatic ring
- lower alkyl
- prostaglandin
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- CVQCOFJXLUFZDE-PILWYADHSA-N COC(CCCCCCC([C@@H](CCC(CSc1ccccc1)O)[C@@H](C1)O)[C@H]1O)=O Chemical compound COC(CCCCCCC([C@@H](CCC(CSc1ccccc1)O)[C@@H](C1)O)[C@H]1O)=O CVQCOFJXLUFZDE-PILWYADHSA-N 0.000 description 1
- MNTFCAXCYYAWKV-QTRBPXOWSA-N OC(CC[C@H](C(CCCCCCC(O)=O)[C@H](C1)O)[C@@H]1O)COc(cccc1)c1F Chemical compound OC(CC[C@H](C(CCCCCCC(O)=O)[C@H](C1)O)[C@@H]1O)COc(cccc1)c1F MNTFCAXCYYAWKV-QTRBPXOWSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C405/00—Compounds containing a five-membered ring having two side-chains in ortho position to each other, and having oxygen atoms directly attached to the ring in ortho position to one of the side-chains, one side-chain containing, not directly attached to the ring, a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, and the other side-chain having oxygen atoms attached in gamma-position to the ring, e.g. prostaglandins ; Analogues or derivatives thereof
Definitions
- the present invention describes a process for making a novel epoxide intermediate useful for making 13,14-dihydro prostaglandin A, E and F derivatives.
- the present invention describes a novel process for making a novel epoxide intermediate useful for making 13,14-dihydro prostaglandin A, E and F derivatives.
- Naturally occurring prostaglandins (PGA, PGB, PGD, PGE, PGF, and PGI) are C-20 unsaturated fatty acids.
- Prostaglandin A, E, and F derivatives are distinguishable as such by the substituents on the alicyclic ring.
- PGA derivatives are characterized by a ketone at C 9 and a double bond between C 10 and C 1
- PGE derivatives are characterized by a ketone at C 9 and a hydroxyl at Cn.
- PGF derivatives are characterized by hydroxyl groups at both C 9 and at C ⁇ .
- Such derivatives are useful for the treatment of many medical disorders including, for example, ocular disorders, hypertension, fertility control, and osteoporosis.
- disclosed in U.S. Patent No. 3,776,938 (1973) by Bergstrom, S., and Sjovall.J. of the Kemiska Institutionen, Karolinska Institute, Sweden has a stimulatory effect on smooth muscle contraction as shown by test strips of guinea pig ileum, rabbit duodenum, or gerbil colon.
- Further information regarding the biological effects of 13,14-dihydro PGA, PGE and PGF derivatives are disclosed in the following references: U.S. Patent No.
- prostaglandin E derivatives have generally been assembled through the common Corey aldehyde intermediate via introduction of the omega side-chain through Wadsworth-Homer-Emmons phosphonate chemistry, reduction and protection of the C 15 position, introduction of the top chain via Wittig chemistry, oxidation of the C 9 position with Jones reagent, and finally, removal of the various protecting groups with the appropriate reagent(s).
- Prostaglandins of the A series have generally been assembled from the PGE series by acid or base induced elimination of the C11 hydroxyl group.
- Methods for conversion of PGE derivatives to PGA derivatives include those described in the following references: Stork et al., J. Amer. Chem. Soc. 1976, 98, p. 1583; Stork et al., J. Amer. Chem. Soc. 1978, 700, p. 8272.
- the prostaglandin F2 ⁇ skeleton is prepared in a variety of ways; generally from the condensation of the Corey aldehyde (see for example: Corey, E.J.; Weinshenker, N.M.; Schaaf, T.K.; Huber, W. "Stereo-Controlled Synthesis of Prostaglandins F2 ⁇ and E2 (dl)" J. Am. Chem. Soc. 1969, 91(20), p.5675-5677] with the appropriate oxophosphonate, followed by reduction at C15 (prostaglandin numbering)[see, for example: Noyori, R,; Tomino, I.; Yamada, M.; Nishizawa, M.
- This novel intermediate can be coupled with oxygen, carbon, sulfur, and nitrogen nucleophiles, in the presence of a base or a Lewis acid, in a ring-opening process to provide 13,14-dihydro prostaglandin A, E, and F derivatives.
- the present invention is directed to a process for making a novel Methyl 7-(2- hydroxy-5-(2-(2-oxiranyl)ethyl)-4-(1 ,1 ,2,2 tetramethyl-1-silapropoxy)cyclopentyl) heptanoate intermediate (the "epoxide intermediate").
- This epoxide intermediate is useful for making 13,14-dihydro prostaglandin A, E and F derivatives.
- the invention is further directed to a process for making 13,14-dihydroprostaglandin A, E and F derivatives.
- Alkyl is a saturated or unsaturated hydrocarbon chain having 1 to 18 carbon atoms, preferably 1 to 12, more preferably 1 to 6, more preferably still 1 to 4 carbon atoms. Alkyl chains may be straight or branched. Preferred branched alkyl have one or two branches, preferably one branch. Preferred alkyl are saturated. Unsaturated alkyl have one or more double bonds and/or one or more triple bonds. Preferred unsaturated alkyl have one or two double bonds or one triple bond, more preferably one double bond. Alkyl chains may be unsubstituted or substituted with from 1 to about 4 substituents. Preferred alkyl are unsubstituted.
- Preferred substituted alkyl are mono-, di-, or trisubstituted.
- Preferred alkyl substituents include halo, hydroxy, aryl (e.g., phenyl, tolyl, alkyloxphenyl, alkyloxycarbonylphenyl, halophenyl), heterocyclyl, and heteroaryl.
- Aromatic ring is an aromatic hydrocarbon ring system.
- Aromatic rings are monocyclic or fused bicyclic ring systems. Monocyclic aromatic rings contain from about 5 to about 10 carbon atoms, preferably from 5 to 7 carbon atoms, and most preferably from 5 to 6 carbon atoms in the ring. Bicyclic aromatic rings contain from 8 to 12 carbon atoms, preferably 9 or 10 carbon atoms in the ring.
- Aromatic rings may be unsubstituted or substituted with from 1 to 4 substituents on the ring.
- Preferred aromatic ring substituents include: halo, cyano, alkyl, heteroalkyl, haloalkyl, phenyl, phenoxy or any combination thereof. More preferred substituents include halo and haloalkyl.
- Preferred aromatic rings include naphthyl and phenyl. The most preferred aromatic ring is phenyl.
- Biohydrolyzable ester is an ester moiety that does not interfere with the therapeutic activity of the compound, or that is readily metabolized by a human or mammal.
- Carbocyclic aliphatic ring is a saturated or unsaturated hydrocarbon ring. Carbocyclic aliphatic rings are not aromatic. Carbocyclic aliphatic rings are monocyclic, or are fused, spiro, or bridged bicyclic ring systems. Monocyclic carbocyclic aliphatic rings contain from about 4 to about 10 carbon atoms, preferably from 4 to 7 carbon atoms, and most preferably from 5 to 6 carbon atoms in the ring. Bicyclic carbocyclic aliphatic rings contain from 8 to 12 carbon atoms, preferably from 9 to 10 carbon atoms in the ring.
- Carbocyclic aliphatic rings may be unsubstituted or substituted with from 1 to 4 substituents on the ring.
- Preferred carbocyclic aliphatic ring substituents include: halo, cyano, alkyl, heteroalkyl, haloalkyl, phenyl, phenoxy or any combination thereof. More preferred substituents include halo and haloalkyl.
- Preferred carbocyclic aliphatic rings include cyclopentyl, cyclohexyl, cyclohexenyl, cycloheptyl, and cyclooctyl. More preferred carbocyclic aliphatic rings include cyclohexyl, cycloheptyl, and cyclooctyl. The most preferred carbocyclic aliphatic ring is cycloheptyl.
- Halo is fluoro, chloro, bromo or iodo. Preferred halo are fluoro, chloro and bromo; more preferred are chloro and fluoro, especially fluoro.
- Haloalkyl is a straight, branched, or cyclic hydrocarbon substituted with one or more halo substituents. Preferred haloalkyl are C1-C-12; mo
- Heteroalkyl is a saturated or unsaturated chain containing carbon and at least one heteroatom, wherein no two heteroatoms are adjacent. Heteroalkyl chains contain from 1 to 18 member atoms (carbon and heteroatoms) in the chain, preferably 1 to 12, more preferably 1 to 6, more preferably still 1 to 4. Heteroalkyl chains may be straight or branched. Preferred branched heteroalkyl have one or two branches, preferably one branch. Preferred heteroalkyl are saturated. Unsaturated heteroalkyl have one or more double bonds and/or one or more triple bonds. Preferred unsaturated heteroalkyl have one or two double bonds or one triple bond, more preferably one double bond.
- Heteroalkyl chains may be unsubstituted or substituted with from 1 to 4 substituents.
- Preferred heteroalkyl are unsubstituted.
- Preferred heteroalkyl substituents include halo, hydroxy, aryl (e.g., phenyl, tolyl, alkyloxyphenyl, alkyloxycarbonylphenyl, halophenyl), heterocyclyl, heteroaryl.
- alkyl substituted with the following substituents are heteroalkyl: alkoxy (e.g., methoxy, ethoxy, propoxy, butoxy, pentoxy), aryloxy (e.g., phenoxy, chlorophenoxy, tolyloxy, methoxyphenoxy, benzyloxy, alkyloxycarbonylphenoxy, acyloxyphenoxy), acyloxy (e.g., propionyloxy, benzoyloxy, acetoxy), carbamoyloxy, carboxy, mercapto, alkylthio, acylthio, arylthio (e.g., phenylthio, chlorophenylthio, alkylphenylthio, alkoxyphenylthio, benzylthio, alkyloxycarbonylphenylthio), amino (e.g., amino, mono- and di- C1-C3 alkanylamino, methylphenylamino, methyl
- Heteroatom is a nitrogen, sulfur, or oxygen atom. Groups containing more than one heteroatom may contain different heteroatoms.
- Heterocyclic aliphatic ring is a saturated or unsaturated ring containing carbon and from 1 to about 4 heteroatoms in the ring, wherein no two heteroatoms are adjacent in the ring and no carbon in the ring that has a heteroatom attached to it also has a hydroxyl, amino, or thiol group attached to it. Heterocyclic aliphatic rings are not aromatic. Heterocyclic aliphatic rings are monocyclic, or are fused or bridged bicyclic ring systems. Monocyclic heterocyclic aliphatic rings contain from about 4 to about 10 member atoms (carbon and heteroatoms), preferably from 4 to 7, and most preferably from 5 to 6 member atoms in the ring.
- Bicyclic heterocyclic aliphatic rings contain from 8 to 12 member atoms, preferably 9 or 10 member atoms in the ring. Heterocyclic aliphatic rings may be unsubstituted or substituted with from 1 to 4 substituents on the ring. Preferred heterocyclic aliphatic ring substituents include: halo, cyano, alkyl, heteroalkyl, haloalkyl, phenyl, phenoxy or any combination thereof. More preferred substituents include halo and haloalkyl. Preferred heterocyclic aliphatic rings include piperzyl, morpholinyl, tetrahydrofuranyl, tetrahydropyranyl and piperdyl.
- Heteroaromatic ring is an aromatic ring system containing carbon and from 1 to about 4 heteroatoms in the ring. Heteroaromatic rings are monocyclic or fused bicyclic ring systems. Monocyclic heteroaromatic rings contain from about 5 to about 10 member atoms (carbon and heteroatoms), preferably from 5 to 7, and most preferably from 5 to 6 in the ring. Bicyclic heteroaromatic rings contain from 8 to 12 member atoms, preferably 9 or 10 member atoms in the ring. Heteroaromatic rings may be unsubstituted or substituted with from 1 to 4 substituents on the ring.
- Preferred heteroaromatic ring substituents include: halo, cyano, alkyl, heteroalkyl, haloalkyl, phenyl, phenoxy or any combination thereof. More preferred substituents include halo, haloalkyl, and phenyl.
- Preferred heteroaromatic rings include thienyl, thiazolo, purinyl, pyrimidyl, pyridyl, and furanyl. More preferred heteroaromatic rings include thienyl, furanyl, and pyridyl. The most preferred heteroaromatic ring is thienyl.
- “Lower alkyl” is an alkyl chain radical comprised of 1 to 6, preferably 1 to 4 carbon atoms.
- Phenyl is a six-membered monocyclic aromatic ring which may or may not be substituted with from about 1 to about 4 substituents.
- the substituents may be substituted at the ortho, meta or para position on the phenyl ring, or any combination thereof.
- Preferred phenyl substituents include: halo, cyano, alkyl, heteroalkyl, haloalkyl, phenyl, phenoxy or any combination thereof. More preferred substituents on the phenyl ring include halo and haloalkyl.
- the most preferred substituent is halo.
- the preferred substitution pattern on the phenyl ring is ortho or meta.
- the most preferred substitution pattern on the phenyl ring is ortho.
- the present invention is directed to a process for making a novel Methyl 7-(2- hydroxy-5-(2-(2-oxiranyl)ethyl)-4-(1 ,1 ,2,2 tetramethyl-1 -silapropoxy) cyclopentyl) heptanoate intermediate (the "epoxide intermediate”) having the following general formula:
- R is lower alkyl, carbocyclic aliphatic ring, heterocyclic aliphatic ring, aromatic ring, or heteroaromatic ring;
- R' is hydrogen, lower alkyl, carbocyclic aliphatic ring, heterocyclic aliphatic ring, aromatic ring, or heteroaromatic ring provided the carbon at C 15 (prostaglandin numbering) has only one heteroatom attached to it;
- Q is a suitable protecting group.
- Suitable protecting groups include tert-butyl dimethylsilyl, trimethylsilyl, benzyl, C r C 8 alkyl, or aromatic ether, or a benzoyl or acetyl ester.
- Preferred protecting groups include tert-butyl dimethylsilyl, trimethylsilyl, and benzyl ethers. The most preferred protecting group is a tert-butyl dimethylsilyl ether.
- This epoxide intermediate above is useful for making 13,14-dihydro prostaglandin A, E and F derivatives.
- the invention is further directed to a process for making 13,14-dihydro prostaglandin A, E and F derivatives having the following general formula:
- R is C0 2 H, C(0)NHOH, CO2R5, CH 2 OH, S(0) 2 Rs, C(0)NHR 5 , C(0)NHS(0)2R5, or tetrazole; wherein R5 is alkyl, heteroalkyl, carbocyclic aliphatic ring, heterocyclic aliphatic ring, aromatic ring, or heteroaromatic ring;
- R 2 is hydrogen, lower alkyl carbocyclic aliphatic ring, heterocyclic aliphatic ring, aromatic ring, or heteroaromatic ring;
- each R 3 is independently selected from the group consisting of: hydrogen, lower alkyl, alkoxy, haloalkyl, carbocyclic aliphatic ring, heterocyclic aliphatic ring, aromatic ring, and heteroaromatic ring;
- Y is NR 4 , S, S(O), S(0) 2 , O, or a bond wherein R 4 is hydrogen or lower alkyl;
- p is 0-5, q is 0-5, and p+q is 0-5 provided that when Y is a bond p is at least 1 ;
- Z is hydrogen, methyl, carbocyclic aliphatic ring, heterocyclic aliphatic ring, aromatic ring, or heteroaromatic ring provided that when Y is NR 4 , S, S(O), or S(0) 2 and q is 0, Z is not hydrogen;
- the 13,14-dihydro prostaglandin A, E and F derivatives described directly above may themselves be used as intermediates in the preparation of other 13,14-dihydro prostaglandin A, E or F derivatives. That is, the compounds prepared may be reacted further, using known chemistry, to yield other active derivatives, such as other PGA, PGE and PGF derivatives.
- the next step in the process is modifying the compound according to Formula III to yield a compound according to Formula IV.
- the compound according to Formula III is treated with a hydride reducing agent, such as those reported in the art for PGF derivatives (see for example Davis et al., "A Convergent Total Synthesis of ( + -)- Prostaglandin F2 ⁇ via Conjugate Addition and Regiospecific Enolate Trapping" J. Org. Chem. 1979, 44(22), p.3755-3759).
- the ketone is reacted with a hydride reducing agent in a polar protic solvent to give the Cg alcohol.
- Hydride reducing agent refers to any agent capable of delivering a hydride ion in a reaction.
- Preferred hydride reducing agents include L-selectride and sodium borohydride. The most preferred hydride reducing agent is sodium borohydride.
- Preferred polar protic solvents include methanol, ethanol, and butanol. The most preferred polar protic solvent is methanol.
- the preferred temperature range for the reduction is between -100°C and 23°C. More preferred still is between -60°C and 0°C. The most preferred temperature range is between -45°C and -20°C.
- the product alcohol so obtained can be isolated using methods known to those skilled in the art. Such methods include extraction, solvent evaporation, distillation, and crystallization procedures. Most preferably, the product is purified by flash chromatography on silica gel (Merck, 230-400 mesh) using 20% EtOAc/hexanes as the eluent.
- Epoxidizing agent refers to a chemical capable of producing a 3-membered ring possessing one oxygen atom from a carbon-carbon double bond.
- Preferred epoxidizing agents include meta-chloroperbenzoic acid and peracetic acid. More preferred epoxidizing agents include meta-chloroperbenzoic acid and peracetic acid. The most preferred epoxidizing agent is meta-chloroperbenzoic acid.
- Halocarbon solvent refers to a solvent which has one or more halogens attached to a carbon chain.
- Preferred halocarbon solvents include dichloromethane, dichloroethane, carbon tetrachloride, and chloroform. More preferred halocarbon solvents include dichloromethane and chloroform. The most preferred halocarbon solvent is dichloromethane.
- the epoxide intermediates according to Formula I can be isolated using methods known to those skilled in the art. Such methods include extraction, solvent evaporation, distillation, or crystallization procedures. Most preferably, the product is purified by flash chromatography on silica gel (Merck, 230-400 mesh) using 20% EtOAc/hexanes as the eluent.
- nucleophile HYZ carbon, oxygen, sulfur and nitrogen containing nucleophiles
- Nucleophile HYZ refers to any chemical agent suitable for adding to an epoxide to form a covalent bond in a ring-opening process.
- Preferred nucleophiles include 2-thienyl mercaptan, o,m,p-chlorophenol, ethyl mercaptan, o,m,p-lithio chlorobenzene, morpholine, thiophenol, aniline, o,/77,p-toluidine, o,m,p-chloro thiophenol, o,tr7,p-fluoro thiophenol, o,o-dichloro thiophenol, phenylurethane, o,m,p-trifluoromethyl thiophenol, furfuryl amine, benzyl amine, furfuryl alcohol, and 2-amino pyridine. More preferred nucleophiles include thiophenol, o-chloro thiophenol, and aniline. The most preferred nucleophile is o-F-thiophenol.
- Deprotection at C ⁇ can then be carried out when the compound according to Formula I is intended to be a PGF derivative.
- “Deprotection” refers to the removal of protecting groups used to protect sensitive functional groups. Deprotection includes the removal of silyl ethers of alcohols or alkyl esters of carboxylic acids.
- Conversion of the R ester of the Formula V compound to the desired R 1 of Formula II can be carried out using methods known to those skilled in the art. Such methods include, but are not limited to, deprotection of C 11 t deprotection of C 1 ? selective oxidation of C 9 , reduction of C,, base catalyzed elimination of the C ⁇ alcohol, condensation of C ⁇ with amines, and condensation of C, with hydroxylamines.
- Conversion to a PGE derivative from the corresponding PGF derivative according to Formula II can be carried out by oxidization at C 9 using methods known to those skilled in the art. Conversion to a PGA derivative from the corresponding PGE derivative can be carried out by elimination of the C ⁇ alcohol using methods known to those skilled in the art.
- Base means a basic reagent which is added to the reaction mixture to facilitate covalent bond formation and ring-opening of the epoxide and the nucleophile.
- Bases include nitrogen bases.
- Preferred bases include those which are soluble in organic solvents and are volatile.
- preferred bases include N,N diisopropylethylamine, triethylamine, trimethylamine, butylamine, pyridine, and 2,6- lutidine. The more preferred bases are 2,6-lutidine, triethylamine, and pyridine. The most preferred base is triethylamine.
- the reaction is carried out preferably at between 150°C and 0°C, more preferably between 120°c and 20°C and most preferably between 80°C and 50°C.
- the preferred organic solvents for the reaction are aromatic hydrocarbon solvents. More preferred organic solvents include xylenes, toluene, and benzene. The most preferred organic solvent is benzene.
- Lewis acid refers to any non-protic acid which is added to the reaction mixture to facilitate covalent bond formation and ring- opening of the epoxide with the nucleophile.
- the preferred Lewis acids include magnesium perchlorate, boron trifluoride etherate, titanium tetrachloride and triethylaluminum. The most preferred Lewis acid is magnesium perchlorate.
- Polar aprotic acids include N,N dimethylformamide and ethereal solvents.
- “Ethereal solvent” refers to a solvent which has two alkyl groups bonded to an oxygen including those in which the alkyl group and oxygen are part of a ring.
- Preferred ethereal solvents include diethyl ether and tetrahydrofuran.
- the most preferred ethereal solvent is tetrahydrofuran.
- the most preferred polar aprotic solvent is N,N dimethylformamide.
- the preferred reaction temperature is between 150°C and 23°C. The more preferred reaction temperature is between 125°C and 40°C. The most preferred temperature is between 100°C and 75°C.
- Addition of carbon nucleophiles generated from the anion is carried out in the presence of a Lewis acid and an ethereal solvent.
- Preferred ethereal solvents include diethyl ether and tetrahydrofuran.
- the most preferred ethereal solvent is tetrahydrofuran.
- the most preferred Lewis acid with carbon nucleophiles is boron trifluoride-etherate.
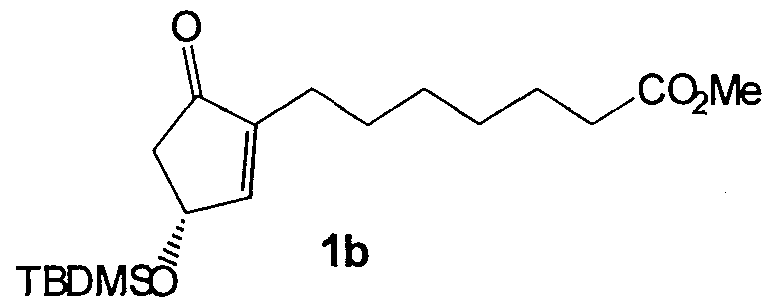
- the ketone 2a (1 equiv.) is dissolved in MeOH and cooled to -40°C.
- Sodium borohydride (0.9 equiv.) is added portionwise over 10 minutes. After the addition is complete the reaction is stirred for 13 hours at -40°C and then 12 hours at -78°C.
- the reaction is quenched with water, partitioned between brine and CH2CI2 and the layers separated.
- the aqueous layer is back-extracted with CH2CI2 and the organic layers combined and dried (Na2S ⁇ 4).
- the solvent is removed in vacuo and the residue chromatographed on Si ⁇ 2 (30 % EtOAc/hexanes) to give 75% of the alcohol 2b.
- the alcohol 2b (1 equiv.) is dissolved in CH2CI2 and cooled to 0°C. Sodium bicarbonate is added, followed by m-CPBA (57%-85% purity) (3 equiv.) portionwise over 15 minutes. After the addition is complete the reaction is stirred for 20 hours at room temperature. The reaction is poured onto water, partitioned between brine and CH2CI2 and the layers separated. The aqueous layer is back-extracted with CH2CI2 and the organic layers combined and dried (Na2S04). The solvent is removed in vacuo and the residue chromatographed on Si ⁇ 2 (20% EtOAc/hexanes) to give 73% of the epoxide diasteriomers 2c.
- aqueous layer is extracted three times with CH2CI2, the organic layers are combined and washed three time with saturated NaHC03, brine, and dried (Na2S04). After column (95% CH2CI2, 5% MeOH) 5a is recovered in 50% yield.
- the epoxide 2c is treated with pig liver esterase to remove the methyl ester. Then, to a 10 ml round bottomed flask at -78 °C, the acid and BF 3 Et 2 0 are stirred, then the lithio anion of o-bromotoluene(1.5 equiv.), in THF are added. After the reaction is stirred at -30°C under nitrogen for several hours, the reaction is done. The reaction is quenched with saturated NH 4 CI, and the solvent removed in vacuo. Without further purification to this crude reaction mixture, CH3CN and HF/Pyridine (0.6 equiv.) are added while the flask is kept at 0°C.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Epoxy Compounds (AREA)
Abstract
Description























































Claims







Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2000510709A JP2001515884A (en) | 1997-09-09 | 1998-09-04 | Method for producing epoxide intermediate |
| EP98945917A EP1012138A1 (en) | 1997-09-09 | 1998-09-04 | A process for making epoxide intermediates |
| PL98339220A PL339220A1 (en) | 1997-09-09 | 1998-09-04 | Method of obtaining intermediate epoxy compounds |
| IL13486498A IL134864A0 (en) | 1997-09-09 | 1998-09-04 | A process for making epoxide intermediates |
| BR9811771-8A BR9811771A (en) | 1997-09-09 | 1998-09-04 | A process for preparing epoxide intermediates |
| AU93057/98A AU9305798A (en) | 1997-09-09 | 1998-09-04 | A process for making epoxide intermediates |
| SK339-2000A SK3392000A3 (en) | 1997-09-09 | 1998-09-04 | A process for making epoxide intermediates |
| CA002303797A CA2303797A1 (en) | 1997-09-09 | 1998-09-04 | A process for making epoxide intermediates |
| NO20001140A NO20001140L (en) | 1997-09-09 | 2000-03-06 | Process for the preparation of epoxide intermediates |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US5825497P | 1997-09-09 | 1997-09-09 | |
| US60/058,254 | 1997-09-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999012897A1 true WO1999012897A1 (en) | 1999-03-18 |
Family
ID=22015649
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/018593 Ceased WO1999012897A1 (en) | 1997-09-09 | 1998-09-04 | A process for making epoxide intermediates |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US6066751A (en) |
| EP (1) | EP1012138A1 (en) |
| JP (1) | JP2001515884A (en) |
| CN (1) | CN1269783A (en) |
| AU (1) | AU9305798A (en) |
| BR (1) | BR9811771A (en) |
| CA (1) | CA2303797A1 (en) |
| HU (1) | HUP0003578A2 (en) |
| ID (1) | ID24840A (en) |
| IL (1) | IL134864A0 (en) |
| NO (1) | NO20001140L (en) |
| PE (1) | PE121499A1 (en) |
| PL (1) | PL339220A1 (en) |
| SK (1) | SK3392000A3 (en) |
| TR (1) | TR200000672T2 (en) |
| WO (1) | WO1999012897A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004526802A (en) * | 2001-05-08 | 2004-09-02 | ヨンスン ファイン ケミカル コーポレーション | Method for producing prostaglandin derivative and stereospecific starting material |
| US6894175B1 (en) | 1999-08-04 | 2005-05-17 | The Procter & Gamble Company | 2-Decarboxy-2-phosphinico prostaglandin derivatives and methods for their preparation and use |
| US7388029B2 (en) | 2000-03-31 | 2008-06-17 | Duke University | Compositions and methods for treating hair loss using non-naturally occurring prostaglandins |
| US7407987B2 (en) | 2000-03-31 | 2008-08-05 | Duke University | Compositions and methods for treating hair loss using C16-C20 aromatic tetrahydro prostaglandins |
| USRE43372E1 (en) | 1999-03-05 | 2012-05-08 | Duke University | C16 unsaturated FP-selective prostaglandins analogs |
| US9346837B2 (en) | 2000-03-31 | 2016-05-24 | Duke University | Cosmetic and pharmaceutical compositions and methods using 2-decarboxy-2-phosphinico derivatives |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3776938A (en) * | 1958-05-28 | 1973-12-04 | S Bergstrom | Dihydro-pge1 |
| US3505386A (en) * | 1965-12-29 | 1970-04-07 | Upjohn Co | Compounds related to prostaglandins |
| US3435053A (en) * | 1966-06-06 | 1969-03-25 | Upjohn Co | Cyclopenta(b)pyrans |
-
1998
- 1998-09-04 SK SK339-2000A patent/SK3392000A3/en unknown
- 1998-09-04 PL PL98339220A patent/PL339220A1/en not_active Application Discontinuation
- 1998-09-04 BR BR9811771-8A patent/BR9811771A/en not_active Application Discontinuation
- 1998-09-04 WO PCT/US1998/018593 patent/WO1999012897A1/en not_active Ceased
- 1998-09-04 CA CA002303797A patent/CA2303797A1/en not_active Abandoned
- 1998-09-04 HU HU0003578A patent/HUP0003578A2/en unknown
- 1998-09-04 AU AU93057/98A patent/AU9305798A/en not_active Abandoned
- 1998-09-04 TR TR2000/00672T patent/TR200000672T2/en unknown
- 1998-09-04 IL IL13486498A patent/IL134864A0/en unknown
- 1998-09-04 EP EP98945917A patent/EP1012138A1/en not_active Ceased
- 1998-09-04 CN CN98808986A patent/CN1269783A/en active Pending
- 1998-09-04 ID IDW20000642A patent/ID24840A/en unknown
- 1998-09-04 US US09/148,539 patent/US6066751A/en not_active Expired - Fee Related
- 1998-09-04 JP JP2000510709A patent/JP2001515884A/en not_active Withdrawn
- 1998-10-02 PE PE1998000932A patent/PE121499A1/en not_active Application Discontinuation
-
2000
- 2000-03-06 NO NO20001140A patent/NO20001140L/en not_active Application Discontinuation
Non-Patent Citations (2)
| Title |
|---|
| A.L. WATERHOUSE ET AL.: "STRUCTURAL ASPECTS OF RYANODINE ACTION AND SELECTIVITY.", JOURNAL OF MEDICINAL CHEMISTRY, vol. 30, 1987, WASHINGTON US, pages 710 - 716, XP002085203 * |
| D. DESMAELE ET AL.: "INFLUENCE DE LA SUBSTITUTION EN 1 SUR LA STEREOSELECTIVITE DE L' HYDROLYSE DE N,N DIETHYLAMINO-7 DIALKYL-1,6 BICYCLO (3,2,0) HEPTENE-6-ONES-2; UNE NOUVELLE VOIE D'ACCESS AU CONTROLE DES CENTRES C17 ET C20 DE STEROIDES.", TETRAHEDRON LETTERS, vol. 24, no. 30, 1983, OXFORD GB, pages 3079 - 3083, XP002085204 * |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE43372E1 (en) | 1999-03-05 | 2012-05-08 | Duke University | C16 unsaturated FP-selective prostaglandins analogs |
| US6894175B1 (en) | 1999-08-04 | 2005-05-17 | The Procter & Gamble Company | 2-Decarboxy-2-phosphinico prostaglandin derivatives and methods for their preparation and use |
| US7074942B2 (en) | 1999-08-04 | 2006-07-11 | The Procter & Gamble Company | 2-decarboxy-2-phosphinico prostaglandin derivatives and methods for their preparation and use |
| US7115659B2 (en) | 1999-08-04 | 2006-10-03 | The Procter & Gamble Company | Method of treating a condition by administering a prostaglandin derivative |
| US7388029B2 (en) | 2000-03-31 | 2008-06-17 | Duke University | Compositions and methods for treating hair loss using non-naturally occurring prostaglandins |
| US7407987B2 (en) | 2000-03-31 | 2008-08-05 | Duke University | Compositions and methods for treating hair loss using C16-C20 aromatic tetrahydro prostaglandins |
| US8906962B2 (en) | 2000-03-31 | 2014-12-09 | Duke University | Compositions and methods for treating hair loss using non-naturally occurring prostaglandins |
| US9346837B2 (en) | 2000-03-31 | 2016-05-24 | Duke University | Cosmetic and pharmaceutical compositions and methods using 2-decarboxy-2-phosphinico derivatives |
| US9579270B2 (en) | 2000-03-31 | 2017-02-28 | Duke University | Compositions and methods for treating hair loss using non-naturally occurring prostaglandins |
| US9675539B2 (en) | 2000-03-31 | 2017-06-13 | Duke University | Cosmetic and pharmaceutical compositions and methods using 2-decarboxy-2-phosphinico derivatives |
| JP2004526802A (en) * | 2001-05-08 | 2004-09-02 | ヨンスン ファイン ケミカル コーポレーション | Method for producing prostaglandin derivative and stereospecific starting material |
Also Published As
| Publication number | Publication date |
|---|---|
| PE121499A1 (en) | 2000-02-03 |
| HUP0003578A2 (en) | 2001-02-28 |
| SK3392000A3 (en) | 2000-09-12 |
| JP2001515884A (en) | 2001-09-25 |
| ID24840A (en) | 2000-08-24 |
| CN1269783A (en) | 2000-10-11 |
| IL134864A0 (en) | 2001-05-20 |
| EP1012138A1 (en) | 2000-06-28 |
| PL339220A1 (en) | 2000-12-04 |
| NO20001140L (en) | 2000-05-04 |
| CA2303797A1 (en) | 1999-03-18 |
| BR9811771A (en) | 2000-08-29 |
| NO20001140D0 (en) | 2000-03-06 |
| AU9305798A (en) | 1999-03-29 |
| US6066751A (en) | 2000-05-23 |
| TR200000672T2 (en) | 2000-07-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU731153B2 (en) | Aromatic C16-C20-substituted tetrahydro prostaglandins useful as FP agonists | |
| CA2303763C (en) | Aromatic c16-c20-substituted tetrahydro prostaglandins useful as fp agonists | |
| CS228916B2 (en) | Method of preparing new derivatives of 9 alpha,6-nitril | |
| US4178367A (en) | Prostaglandin I2 analogues | |
| IL43589A (en) | 16-phenoxy-15-hydroxy derivatives of 17,18,19,20-tetranorprostaglandins and their preparation | |
| US6066751A (en) | Process for making epoxide intermediates | |
| AU749540B2 (en) | C11oxymyl and hydroxylamino prostaglandins useful as FP agonists | |
| US4294849A (en) | Prostaglandin analogues | |
| US6444840B1 (en) | C11 oxymyl and hydroxylamino prostaglandins useful as FP agonists | |
| WO1999012899A1 (en) | A process for making prostaglandin f analogs | |
| HU176144B (en) | Process for producing new prostaglandine analogues | |
| GB2048254A (en) | 19,20-didehydro, 19-hydroxy and 19-oxo-prostaglandin derivatives | |
| WO2000051977A1 (en) | Aldehyde intermediates for the preparation of prostaglandin derivatives | |
| US4078021A (en) | Dimethyl 2-oxo-6-cyanohexyl-phosphonate | |
| JPS6126970B2 (en) | ||
| US4180675A (en) | 15-Cycloalkyl-prostaglandin derivatives | |
| CA1098124A (en) | Process for the preparation of prostaglandin analogues | |
| JPS6022707B2 (en) | Prostaglandin-like compounds | |
| US4110341A (en) | Dithio prostaglandin derivatives | |
| US4165437A (en) | Δ3 -Prostaglandin analogs | |
| KR810000412B1 (en) | Process for the preparation of new prostaglandines | |
| CZ2000853A3 (en) | Process for preparing epoxy intermediates | |
| IE45491B1 (en) | Prostagladin analogues | |
| MXPA00002430A (en) | Aromatic c16 | |
| GB1580201A (en) | Prostaglandin analogues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 134864 Country of ref document: IL Ref document number: 1200000181 Country of ref document: VN Ref document number: 98808986.6 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AT AU AZ BA BB BG BR BY CA CH CN CU CZ CZ DE DE DK DK EE EE ES FI FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2303797 Country of ref document: CA Ref document number: 2303797 Country of ref document: CA Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2000-853 Country of ref document: CZ Ref document number: 3392000 Country of ref document: SK Ref document number: 93057/98 Country of ref document: AU Ref document number: 2000/00672 Country of ref document: TR |
|
| ENP | Entry into the national phase |
Ref document number: 2000 510709 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007002518 Country of ref document: KR Ref document number: PA/A/2000/002435 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 503739 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998945917 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998945917 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1020007002518 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2000-853 Country of ref document: CZ |
|
| WWR | Wipo information: refused in national office |
Ref document number: 1998945917 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998945917 Country of ref document: EP |